Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
WO 95/09173 PCT/EP94/03201
- 1 -
Anthracycline disaccharides, process for their preparation, and
pharmaceutical compositions containing them
Field of the invention
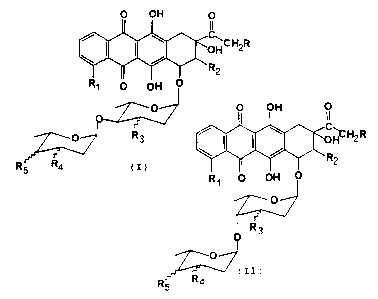
The present invention is referred to compounds of general formula (I) and
(II), respectively
p OH O
R1 O OH V 2R
TO~
R~ O OH V
O R3
O
R5 R4 ~I~ R3
(II)
R5 R4
where:
R is H or OH or the ORS group where R~ - CHO or COCH3 or the acylic
residue of a carboxylic acid containing up to 6 carbon atoms;
R1 is H or OH or OCH3;
R2 is H or F;
R3 is H or OH;
R4 and R5, identical or different, are each H or OH or NH2;
and bond symbol (~..) indicates that substituents R3, R~, and R5
may be either in axial or equatorial position; and their pharmaceutically
acceptable salts having anticancer properties.
As may be seen in the above formulas, compound:a (I) and (II) differ
'C H"R _
z~ 7~~ ~~
- 2 -
exclusively in the apace arrangement of the glycosidic groups and.
therefore. map be represented by formula iA)
0 off , o
'C H2R
(A)
r
R4
where symbol ice) indicates that the second carbohydrate residue may
be bound to carbon atom 4' of the first sugar either in axial or in
equatorial position.
The present invention is also referred to the process for the
preparation of said compounds. their phaz~aceuti.cally acceptable salts.
and the pharmaceutical compositions caata~ n~ ng ~th ~..
State of the art
Daunorubicir_ and daaorubicin are well-known ~atibiotics that are
currently used in the clinical practic= for the treatment of a variety of
solid tumours and leukaemia (F. Arc3mone in "Doxorubicin: Anticancer
Antibiotics". A.C. Sartorelli. Ed.. Academic Press. N. Y.. 1981).
A~NDED SHEEN
8
- 2bis -
Compounds having a structure similar to~those described in the
present application buk presenting only one glycosidic group are
described in EP-457215, WO 80/00305 ana WO 90/'07589. Compounds showing
two or more sugar soieties wherein the sugar directly linked to the
aglycone moiQty is amino-substituted ire descried for example in The
Journal of Antibiotics p. 1720-1730 Nov. 93; Tetrahedron Vol. 37, No.
24, 4219-422E (1981): flE 27 51 395; Carbohydrate Research, 228. 171-90
!1992) and DE-3641833.
Compounds h~-ing three glycoside moieties, and for which no activity
data are repcrted, are described in WO 92/07862.
As known. h.awever, the severe side effects caused by the anticancer
agents used nt present impose limits on the use of same in a good number
of patients ~aho, otbEr~aise. Would benefit from the treatment. Moreover.
AMENDED SHEET
2173158 a
-3-
remarkable advances are needed in the treatment of some important solid
tumours, e.g. pulmonary and ovarial, that do not adequately respond to
any current treatment.
It follows that there is an urgent need for the coming onto the market of
drugs highly selective in their inhibitory action against the
proliferation of diseased cells compared with the normal ones.
Detailed description of the invention
It is an object of the present invention to provide new anticancer
compounds, in particular anthracycline analogues, in which the
carbohydrate portion consists of a disaccharide residue.
It has surprisingly been found that the claimed anthracycline
disaccharides, in which the sugar directly bound to aglycone never
contains amino groups, exhibit higher anticancer acaivity and selectivity
than the anthracycline previously known. It is worth noting that in the
known anthracyclines, having similar structure and which contain two
carbohydrate residues, the sugar bound to aglycone always contains a free
or substituted amino group.
The compounds according to the present invention are the compounds of
general formula {I) and (II;, as reported above, and their
pharmaceutically acceptable salts where R, R1, R2, R3, R4, and R5 are as
described above.
The present invention is also referred t:o pharmaceutical compositions
containing said compounds, or salts thereof with pharmaceutically
acceptable acids, preferably hydrochloric acid.
Particularly preferred are the following compounds:
a) 7-0-[2,6-dideoxy-4-0-(2,3,6-trideoxy-3-amino-a-L-lyxo-exopyranosyl)-a-
L-lyxo-exopyranosyll daunorubicinone chlorhydrate;
b) '7-0-[2,6-dideoxy-4-0-(2,3,6-trideoxy-3-amino-a-L-lyxo-exopyranosyl)-n-
A
WO 95/09173 PCT/EP94/03201
~1~ ~~,5
- 4 -
L-arabino-exopyranosyl] daunorubicinone chlorhydrate;
c) '7-0-[2,6-dideoxy-4-0-(2,3,6-trideoxy-3-amino-a-L-lyxo-exopyranosyl)-a-
L-lyxo-exopyranosyl] doxorubicinone chlorhydrate;
d) '7-0-[2,6-dideoxy-4-0-(2,3,6-trideoxy-3-amino-a-L-lyxo-exopyranosyl)-a-
L-arabino-exopyranosyl] doxorubicinone chlorhydrate;
e) '7-0-[2,6-dideoxy-4-0-(2,3,6-trideoxy-3-amino-a-L-lyxo-exopyranosyl)-a-
L-arabino-exopyranosyl]-4-demethoxy-daunorubicinone chlorhydrate;
f) '7-0-[2,6-dideoxy-4-0-(2,3,6-trideoxy-3-amino-a-L-lyxo-exopyranosyl)-a=
L-lyxo-exopyranosyl]-4-demethoxy-daunorubicinone chlorhyrate;
g) 7-0-[2,6-dideoxy-4-0-(2,3,6-trideoxy-3-amino-a-L-lyxo-exopyranosyl)-a-
L-lyxo-exopyranosyl]-4-demethoxy-doxorubicinone chlorhydrate;
h) '7-0-[2,6-dideoxy-4-0-(2,3,6-trideoxy-3-amino-a-L-lyxo-exopyranosyl)-a-
L-arabino-exopyranosyl]-4-demethoxy-doxorubicinone chlorhydrate;
i) '7-0-[2,6-dideoxy-4-0-(2,3,4,6-tetradeoxy-4-amino-a-L-erythro-
exopyranosyl)-a-L-lyxo-exopyranosyl] daunorubicinone chlorhydrate;
j) ~-0-[2,6-dideoxy-4-0-(2,3,4,6-tetradeoxy-4-amino-a-L-erythro-
exopyranosyl)-a-L-lyxo-exopyranosyl]-4-demethoxy-daunorubicinone
chlorhydrate;
k) 7-0-[2,6-dideoxy-4-0-(2,3,4,6-tetradeoxy-4-amino-a-L-erythro-
exopyranosyl)-a-L-lyxo-exopyranosyl] doxorubicinone chlorhydrate;
1) '7-0-[2,6-dideoxy-4-0-(2,3,4,6-tetradeoxy-4-amino-a-L-erythro-
exopyranosyl)-a-L-lyxo-exopyranosyl]-4-demethoxy-doxorubicinone
chlorhydrate;
m) '7-0-[2.6-dideoxy-4-0-(2,3,6-trideoxy-3-amino-a-L-lyxo-exopyranosyl)-a-
L-lyxo-exopyranosyl]-4-demethoxy-8-fluoro-daunorubicinone chlorhydrate;
n) '7-0-[2.6-dideoxy-4-0-(2,3,6-trideoxy-3-amino-a-L-lyxo-exopyranosyl)-a-
L-lyxo-exopyranosyl]-4-demethoxy-8-fluoro-doxorubicinone chlorhydrate.
The compounds of general formula (I) and (II) can be prepared by a
WO 95/09173 PCT/EP94/03201
-5- ~ 21'~315g
process consisting of the following steps:
a) condensation of a compound of formula (III)
(III)
OH
H2 RO
R ~ O OH OH
where R1 and R2 are as defined above and R6 is H or the ORS group where
R7 is a protective group for an alcoholic function, preferably selected
among the acetyl-, dimethylterbutylsilyl- or
prmethoxyphenyldiphenylmethyl- groups, with a compound of formula (IV) or
(V) X
r~
R10
_-m rc9
(IV) ('V)
where R8 is H or a protected -OH group, preferably p-nitrobenzoate; R9
and R10, identical or different, are each H or a protected OH group,
preferably p-nitrobenzoate or a protected N'H2 group, preferably
trifluoroacetamide or allylcarboxyamide and X :is a group capable of
generating, under the condensation conditions, a stable carbo-cation that
may bind itself to a hydroxyl group in position C-'7 of the compound of
formula (III), said group X being conveniently selected among the groups
used in glycosidation reactions, e.g. a halogen such as chlorine or
WO 95/09173 PCT/EP94/03201
~1
6
O OH p
bromine, preferably chlorine, or a p-nitrobenzo~-loxy group. Compounds of
formula (VI) or (VII) are thus obtained:
II
\ OH C H2R 6
R2
R~ O OH O
O
O / ,r ( v1 )
R8
R10 Rg O OH O
_ Il I 11
H2 Rfi
R2
R ~ O OH O
O
R8 !vTy)
~O
~0 R9
where R1, R2, R6, R8, R9. R10 and symbol (.~ are as defined above;
b) one or more reactions of removal of the protective groups of OH
and/or NH2 functions from compounds of formula (VI) and (VII) to give
compounds of formula (I) and (II), where R, R1, R2, R3, R4, R5 and symbol
(~ are as defined above;
c) conversion, if any, of the aforesaid glycosides of formula (I) and
(II) into a pharmaceutically acceptable salt thereof, preferably
chlorhydrate.
The reaction conditions for the glycosidation of a compound of formula
~'1"~3.~~8 -
(III) with a compound of formula (IV) or (V) to give a compound of
formula (VI) or (fII~ may vary depending on the type of substituents
present i~ tie compounris of formula ( If ) ar (V) .
Glycosidatioa is carried out in an inert arganic solvent in the presence
of a condensing agent.
The condensing agents used are. e.g., silver triftnoromethane sulphonate.
silver perch3orate, fixtures of mercury Gti.de and mercury bromide, boron
halides . tit3aium or tin tetrachloride or ion. Eschange resins , such as
Amberlite ~, silver triflate, trimethylsilyltriflate, p-toluensulphonic
acid, trifluoroacetic acid.
Glycosidation is preferably carried out with 1.:1 to 1:3 molar ratios in
an inert organic solvent, such as for example benzene, toluene, ethyl
ether, tetrahydrofuran, dioxane, chloroform, methylene chloride or
dichloroethane and ~ctures thereof.
The reaction tempera~are may range from -~+0'C to ~0'C, preferably from -
20'C to 20'C, and tre reaction time from 15 min to 3 hrs.
The reaction mixture may include a cehydrating substance. such as an
activated molecular sieve.
In the course of +3~e reaction or at the reaction end, the reaction
mixture may be added with an organic base, such as pyridine, collidine.
N.N-dimethyiaminopyridine, triethylaaine or 1.8-bis-(dimethylamino)
naphthalene.
According to the present invention, the removal of the protective groups
for OH and/or NH2 functions from compounds of far~u3a (VI) and (VII) to
give compounds of formula (I) may be carristl out under different
conditions depending on the type of protective ~rua:p used.
When R9 and/or R10, Ldentical or different. ;~~e each a protected NH2
group such as trifluoroacetamide or a protected- OH group such as p
' nitrobenzoate, and/rr R8 is a prmtected ~g group such as p
~IpED SHEET
CA 02173158 2004-09-20
nitrobenzoate, and/or R6 is a protected OH group such as acetate,
deprotection reactions are carried out in a polar solvent, such as water,
methanol, ethanol, pyridine, dimethylformamide or mixtures thereof and in
the presence of an inorganic base, in a stoichiometric amount or in
excess of the stoichiometric, such as sodium, potassium, lithium or
barium hydroxide or carbonate.
The reaction temperature may range from 0°C to 50°C and the
reaction time
from 3 hrs to 48 hrs.
When R9 and/or R10 are each a protected NH2 group such as
allylcarboxyamide, deprotection is carried out in an inert solvent and in
the presence of a metal complex such as
tetrakis(triphenylphosphine)palladium, as disclosed, e.g., in Tetrahedron
Letters, 30, 3773 (1989), or tetracarbonyl nickel, as disclosed, e.g., in
J. Org. Chem., 38. 3233 (1973).
When R6 is a protected OH group such as dimethylterbutylsilylether,
deprotection is carried out in an inert solvent and in the presence of
tetrabutylammonium fluoride, as disclosed, e.g. in J. of Antibiot., 37,
853 (1984).
When R6 is a protected OH group such as
p-methoxyphenyldiphenylmethylether, deprotection is carried out in an
acid medium, e.g. in aqueous acetic acid, as disclosed, e.g. in J. Org.
Chem., 42, 3653 (1977).
Compounds of formula (III) are either known or may be prepared according
to methods and processes known in organic chemistry, as disclosed, e.g.
in Gazz. Chim. Ital., 114, 517 (1984), in Bull. Chem. Soc. Jpn., 59, 423
(1986), and in Italian Patent No. 1241927. Compounds of formula (IV) or
(V) are either known or may be prepared
WO 95109173 217 31 ~ 8 P~~~4103201
_ 9 _
according to methods and processes for the synthesis of disaccharides
known in organic chemistry [J. Carbohydr. Chem., 10, 833 (1991);
Carbohydr. Res.. 74, 199 (1979); Carbohydr. Res.. 20$, 111 (1980);
Tetrahedron, 46, 103 (1990)].
Alternatively, if so desired, anthracycline glycosides of formula (I) and
(II), where R1, R2, R3, R4, R5 are as defined above and R is an OH group,
may be prepared from glycosides of formula (I) and (II) or from
pharmaceutically acceptable salts thereof, where R1, R2. R3, R4, R5 and
symbol (wv~.) are as defined above and R is H, by bromination of the
carbon in position 14 with bromine in chloroform followed by hydrolysis.
at room temperature for a period of 48 hrs, of the resulting 14-
bromoderivatives with sodium formate.
If so desired, glycosides of formula (I) and (II.) may be converted into
their pharmaceutically acceptable salts, e.g. chlorhydrates, by treatment
with hydrochloric acid in methyl alcohol.
The present invention also relates to pharmaceutical compositions
containing, as active ingredient. a compound of formula (I) or (II) or a
pharmaceutically acceptable salt thereof combined with a pharmaceutically
acceptable diluent or carrier.
:according to the present invention, a therapeutically effective dose of a
compound of formula (I) or (II) is combined with an inert carrier.
The compositions may be formulated in a conventional manner using common
carriers.
The claimed compounds are useful for the therapeutic treatment on humans
and other mammals. In particular, said compounds are good anticancer
agents when administered in therapeutically effective doses.
The following examples illustrate the present invention in more detail.
WO 95/09173 PCT/EP94/03201
~1
- 10 -
Example 1
'7-0-[2,6-dideoxy-4-0-(2,3,6-trideoxy-3-amino-a-L-lyxo-exopyranosyl)-a-L-
lyxo-exopyranosyl]-4-demethoxy-daunorubicinone chlorhydrate (compound of
formula II, R~R1=R2=H, R3=R5=OH, R4=NH2).
A mixture of 4-demethoxydaunorubicinone (compound of formula III,
R1=R2=R6=H) (300 mg, 0.81 mmol) and 2,6-dideoxy-4-0-(2,3,6-trideoxy-4-0-
p-nitrobenzoyl-3-trifluoroacetamido-a-L-lyxo-exopyranosyl)-3-0-p-
nitrobenzoyl-a-L-lyxo-exopyranosyl-p-nitrobenzoate (compound of formula
=p-
IV, R3=R5=p-nitrobenzoyl-oxy-, R4=trifluoroacetamido-. X
nitrobenzoyloxy-) (600 mg, 0.'72 mmol) in methyl chloride ('72 ml) and
ethyl ether (24 ml) in the presence of molecular sieves (A4) at -20°C
was
treated with trimethylsilyltriflate (266 ul; 1.44 mmol). The reaction
mixture was stirred for 1 hr, then it was diluted with methylene
chloride, washed with a saturated sodium bicarbonate solution, and
evaporated to dryness. The residue was separated by chromatography on
silica gel (eluent CH2C12-EtOH, 99/1) yielding 360 mg of '7-0-[2,6-
dideoxy-4-0-(2,3.6-trideoxy-4-0-p-nitrobenzoyl-3-fluoroacetamido-a-L-
lyxo-exopyranosyl)-3-0-p-nitrobenzoyl-a-L-lyxo-exopyranosyl]-4-
demethoxydaunorubicinone (compound of formula VII, R1=R2=R6=H, R8=R10=p
nitrobenzoyloxy-. R9=trifluoroacetamido-).
A protected diglycoside suspension of compound of formula (VII)
(R1=R2=R6=H, R8=R10=p-nitrobenzoyloxy-, R9=trifluoroacetamido-) (120 mg;
0.11 mmol) in 1~.6 ml of a 0.1 M solution of Ba(OH)2 in H20/MeOH, 1/l,
was maintained under stirring at room temperature for a period of 3 hrs.
The reaction mixture was neutralized with a 0.2 M potassium bisulphate
solution and extracted with chloroform; the organic extracts were
collected together, dried over anhydrous sodium sulphate, evaporated to
dryness, and taken up with 0.002 M HC1 solution. The acid aqueous
WO 95/09173 PCT/EP94/03201
X173158
_ ~1 _
solution was washed with chloroform and freeze-dried to give 62 mg of the
desired product (compound of formula II, (R=R1=R2=H, R3=R5=OH, R4=NH2).
Yield 39%.
The NMR data obtained are reported below:
1H-NMR (DMSO-d6), d 1.05 (d,3H), 1.15 (d,3H)" 1.5-1.95 (m,4H), 2.1
(m,2H). 2.25 (s.3H), 2.95 (dd.2H). 3.55 (s,2H), 3.8 (m,lH). 3.95 (m,lH),
4.15 (q,lH). 4.35 (q,lH). 4.6 (d,lH). 4.9 (bs.2H), 5.25 (bs.lH), 5.35
(d,lH). 5.55 (s.lH). x.95 (bs.2H), 8.25 (bs,2H).
According to an analogous process, also the following compounds of
formula (I) and (II) were obtained:
'7-0-[2,6-dideoxy-4-0-(2,3,6-trideoxy-3-amino-a-L-lyxo-exopyranosyl)-a-L-
lyxo-exopyranosyl] daunorubicinone chlorhydrate (compound of formula II,
R=R2=H, R1=OCH3, R3=R5=OH. R4=NH2).
1H-NMR (DMSO-d6), a 1.05 (d.3H), 1.15 (d,3H), 1.35-2.15 (m,6H). 2.25
(s.3H). 2.95 (dd.2H), 3.55 (bs.2H). 3.8 (m,lH), 3.95 (s.3H). 4.05-4.2
(m+q,2H). 4.35 (q,lH), 4.9 (bs.2H), 5.25 (d,lH), 7.65 (m,lH), 7.9 (d,2H).
'7-0-[2,6-dideoxy-4-0-(2,3,6-trideoxy-3-amino-a-L-lyxo-exopyranosyl)-a-L-
arabino-exopyranosyl] daunorubicinone chlorhydrate (compound of formula
I; R=R2=H, R1=OCH3. R3=R5=OH. R4=NH2).
1H-NMR (DMSO-d6), 8 1.13 (d.3H), 1.15 (d,3H), 1.45-1.85 (m.4H), 2:05
(m.2H) , 2.15 (s,3H) , 2.8'7 (dd,2H) , 2.98 (m,lH) , 3.5 (m.lH) , 3.6 (m.lH) ,
3.85 (q,lH), 3.9 (q.lH). 3.9 (s.3H). 4.84 (m.a>.H), 5.13 (bs.lH). 5.28
(s,lH), 5.32 (d.lH), 5.55 (s,lH). 7.55 (m, 1H). '7.8 (m.2H).
~-0-[2,6-dideoxy-4-0-(2,3,6-trideoxy-3-amino-a-L-lyxo-exopyranosyl)-a-L-
arabino-exopyranosyl]-4-demethoxy-daunorubicinone chlorhydrate (compound
of formula I; R=R1=R2=H. R3=R5=OH, R4=NH2).
1H-NMR (DMSO-d6), d 1.1 (d,3H). 1.2 (d,3H), 1.5-1.95 (m,4H). 2.05-2.2
(m,2H) , 2.25 (s,3H) , 2.95 (dd,2H) , 3.1 (t,lH), :;.4 (m,lH) , 3'6 (bs.lH) ,
WO 95/09173 PCT/EP94/03201
- 12-
3.65 (m.lH), 3.85-4.00 (q+q,2H), 3.9 (m.lH). 4.95 (d.lH), 5.2 (d,lH), 5.4
(bs.2H), 5.'7 (s,lH). 7.95 (m.2H), 8.25 (m. 2H).