Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02244838 1998-07-27
W O97/32017 PCT~EP97100931
NOVE~ METHOD FOR THE IDENTIFICATION OF NUCLEIC ACID SEQUENCES
ENCOC~ING TWO OR MORE INTERACTING (POLY)PEPTIDES
The present invention relates to methods for identifying nucleic acid sequences which
encode two or more specific interacting peptides or proteins. Furthermore, the present
invention relates to kits which may be used for the ide~ icalion of nucleic acidsequences in accordance with the method of the present invention.
r,~Lein-protein interactions play an important role in all biological processes, from the
replication and expression of genes to the morphogenesis of organisms (Lewin, B.1994, Genes V. Oxford University Press). Methods for detecting protein-protein
interactions have proved useful in understanding the basic mechanisms of different
biological processes and the development of therapeutics. Detection of protein-protein
interactions can be divided into two main categories: (i) physico-chemical based and (ii)
genetic approaches (Phizicky, E.,M. & Fields, S. Microbiological Reviews 59 (1995) 94-
123). Detection of protein-protein interactions by physico-chemical methods usually
requires significant amounts of material, and more importantly, the identity of the
proteins to be studied must be known. Recent developments in methods of mass
spe~l,u.l)el"/ circumvent this problem but such suffer the disadvantage of requiring
sophisticated equipment and expertise (Wang, R. & Chait, B.T., Current Opinion in
Biotech. 5 (1994) 77-84). In contrast, genetic approaches provide an easy and powerful
method of identifying protein-protein interactions without the need for pure material and
speci ~';7ed equipment, with the added advantage of higher throughput.
Different genetic approaches have been used to identify protein-protein interactions.
The current method of choice is the yeast 2-hybrid system (Fields, S. & Song, O.~C.,
-
CA 02244838 1998-07-27
WO g7/32017 rCTtEP97/00931
Nature ~London) 340, (1989) 245-246) which allows the identification of novel proteins
that interact with a known protein.
Another popular genetic approach is the phage display system (Patent ApplicationW090/02809) whereby proteins are fused to a component of a surface protein of
filamentous phage to allow selection for binding to a ligand of interest. The gene
encoding the protein displayed on the surface of the phage is packaged inside the
phage allowing the coupling of genetic information with the gene product. This allows
the screening of "libraries" of proteins whereby the identity of the screened protein is
deduced from the nucleic acid sequence of the phage. This technique has been
extended by Winter et al. (Patent Applic~tion WO 9~/20791) to produce libraries of
multimeric members of a specific binding pair (e.g. combinations of VH and VL chains of
an antibody) and select for functional specific binding pair members that can bind to the
complementary specific binding pair member (e.g. antigen). Said libraries are
constructed by combining two sub-libraries each encoding a collection of corresponding
sub-units of said multimeric members (e.g. a library of VH chains is combined with a
library of VL chains) wherein in principle each sub-unit out of the first sub-library is able
to bind to each sub-unit out of the second sub-library non-specifically. Although this
method has led to the identification of unique antibodies against particular antigens, it
fails to provide a method for identifying two partners of a specific binding pair when both
are unknown.
A unique version of phage display which relies on non-infective phage has recently
been proposed (Duenas, M. & Borrebaeck, C. A. K., BiolTechnology 12 (1994) 999-
1002; EP 0 614 989). A version of this system that led to the identification of proteins
from a cDN~ library that interacts with the jun protein has been described (Gramatikoff
et al., Nucleic. Acids Res. ~ (1994) 5761-5762). The same principle has been also
shown to work with an antibody-antigen system (Krebber et al., FEBS Letters ;~
(1995) 227-231).
CA 02244838 1998-07-27
W O97/32017 PCTMEP97100931
In spite of the power of all the aforementioned genetic selection approaches, they are
limited to the selection of interacting binding entities from only a single genetically-
diverse population (library vs. individual).
It would, however, be highly desirable to simultaneously identify binding entities and
their specific binding partners in a library vs. Iibrary setting, wherein preferably at least
two genetically diverse populations are involved. A solution to this technical problem, i.e.
the ide,lliric~lion of interacting entities and the respective nucleic acid sequences from
more than one genetically diverse population (library vs. Iibrary) is neither provided nor
suggested by the prior art. The present invention solves the above technical problem by
providing the embodiments characterized in the claims. By using these embodiments, it
has become possible to increase exponentially the rate at which (poly)peptide-
(poly)peptide interactions are detected. The present invention may find applications in
the field of functional genomics, whereby different proteins of unknown functions can be
related with other proteins.
Accordingly, the present invention relates to a method for identifying a plurality of
nucleic acid sequences, said nucleic acid sequences each encoding a (poly)peptide
capable of interacting with at least one further (poly)peptide encoded by a different
member of said plurality of nucleic acid sequences, comprising the steps of:
(a) providing a first library of recombinant vector molecules containing
genetically diverse nucleic acid sequences corllprisi"g a variety of nucleic
acid sequences encoding (poly)peptides;
(b) providing a second library of recombinant vector molecules containing
genetically diverse nucleic acid sequences comprising a variety of nucleic
acid sequences encoding (poly)peptides capable of interacting with further
~, (poly)peptides as mentioned in step (a), wherein the vector rnolecules
CA 02244838 1998-07-27
W O97/32017 PCT~EP97/00931
employed for the production of said recombinant vector molecules and/or
the recombinant inserts display properties that are phenotypically
distinguishable from those of the vector molecules and/or the recombinant
inserts used in step (a) and wherein at least one of said properties
displayed by each of said vector molecules and/or the recombinant inserts
used in steps (a) and (b), upon the interaction of a (poly)peptide from said
first library with a (poly)peptide from said second library together generate
a screenable or selectable property;
(c) optionally, providing additional libraries of recombinant vector molecules
containing genetically diverse nucleic acid sequences com,ulising a variety
of nucleic acid sequences encoding (poly)peptides capable of interacting
with or causing interaction of (a) further (poly)peptide(s) as mentioned in
step (a) andfor step (b), wherein the vector molecules employed for the
production of said recombinant vector molecules and/or the recombinant
inserts display properties that are phenotypically distinguishable from
those of the vector molecules and/or the recombinant inserts used in steps
(a) and (b) and, optionally, at least one of said properties displayed by said
vector molecule and/or the recombinant inserts used in step (c) together
with at least one of said properties displayed by either said vector
molecule and/or said recombinant insert used in steps (a) and/or (b), upon
the interaction of a (poly)peptide from said additional library with either a
(poly)peptide from said first library and/or a (poly)peptide from said second
library generate a screenable or selectable property;
(d) expressing members of said libraries of recombinant vectors or nucleic
acid sequences mentioned in steps (a), (b) and optionally (c), in
appropriate host cells so that at least one interaction is established;
~e) selecting for the generation of said screenable or selectable property
representing the interaction of said (poly)peptides;
CA 02244838 1998-07-27
W O 97/32017 PCTrEP97/00931
optionally, carrying out further selection, screening and/or purification
steps; and
(g) identifying said nucleic acid sequences encoding said (poly)peptides
Thus, in the context of the present invention, the term "properties that are phenotypically
distinguishable" relates alternatively to properties that are encoded by the vector
molecule or to properties that are encoded by the recornbinant insert or to both types of
properties. As regards the vector-encoded properties, these may e.g. be resistance
markers or requirements for special nutrients. It should be noted that the recombinant
insert may comprise a nucleic acid portion encoding said property in addition to the
nucleic acid portion responsible for the interaction.
In the context of the present invention, the term "different member " denotes a different
entity which may be, but is not necessarily, structurally different.
Further, in the context of the present invention, the term "plurality" bears the meaning of
"at least two".
The novel properties generated by the at least two recombinant inserts reflect the
inventive principle of the present invention. That is, only if two (or more) (poly)peptides
interact, for example, in a homo-dimeric or hetero-dimeric fashion, a screenable or
selectable property is generated. The interaction between the two or more molecules
may be a direct one or may be mediated indirectly. Examples for a direct interaction are
the binding of an antibody encoded by a nucleic acid sequence from library 1 to a cDNA
protein from library 2, the binding of a protein encoded by a nucleic acid sequence from
cDNA library 1 to a protein from a cDNA library 2, as well as of an anti-idiotypic antibody
encoded by a nucleic acid sequence from one of the libraries to a corresponding
antibody encoded by a nucleic acid sequence from the other library. The nucleic acid
sequences are preferably DNA and most preferably genes or parts thereof.
CA 02244838 1998-07-27
W O97132017 PCT~EP97/00931
An example of an indirect interaction is the bridging of two (poly)peptides encoded by
the two libraries which is mediated by a phosphorylating enzyme. Once the
phosphorylation of one (poly)peptide encoded e.g. by library 1 is effected by the
respective kinase, then this protein is capable of interacting with the second
(poly)peptide encoded by library 2. The phosphorylating enzyme exemplifying this type
of interaction may be encoded by a nucleic acid from (one ofl the additional libraries
and/or may be encoded by the genome of the host cell. Typically, the interaction of the
two (poly)peptides forms a "bridge" of molecules, said "bridge" being detectable using
an appropriate detection process. Conveniently, said bridge is detectable by a tag
molecule that is associated with, encoded by or attached to one of the (poly)peptides
encoded by library 1 or preferably 2.
Furthermore, the present invention relates to a method for identifying a plurality of
nucleic acid sequences, said nucleic acid sequences each encoding a ~poly)peptide
capable of interacting with at least one further (poly)peptide encoded by a different
member of said plurality of nucleic acid sequences, comprising the steps of:
(a) expressing in appropriate host cells
~aa) nucleic acid sequences contained in a first library of recombinant
vector molecules containing genetically diverse nucleic acid
sequences co""u,i:,i"g a variety of nucleic acid sequences encoding
(poly)peptides;
(ab) nucleic acid sequences contained in a second library of recombinant
vector molecules containing genetically diverse nucleic acid
sequences comprising a variety of nucleic acid sequences encoding
(poly)peptides capable of interacting with further (poly)peptides as
.
CA 02244838 l998-07-27
W 097f32017 PCT/EP97/00931
mentioned in step (aa), wherein the vector molecules employed for
the production of said recombinant vector molecules and/or the
recombinant inserts display properties that are phenotypically
distinguishable from those of the vector molecules and/or the
" recombinant inserts used in step (aa) and wherein at least one of
said properties displayed by each of said vector molecules and/or the
recombinant inserts used in steps (aa) and (ab), upon the interaction
of a (poly)peptide from said first library with a (poly)peptide from said
second li~rary together generate a screenable or selectable property;
(ac) optionally, nucleic acid sequences contained in additional libraries of
recombinant vector molecules containing genetically diverse nucleic
acid sequences comprising a variety of nucleic acid sequences
encoding (poly)peptides capable of interacting with or causing
interaction of (a) further (poly)peptide(s) as mentioned in step ~aa)
and/or step (ab), wherein the vector molecules employed for the
production of said recombinant vector molecules and/or the
recombinant inserts display properties that are phenotypically
distinguishable from those of the vector molecules andlor the
recombinant inserts used in steps (aa) and (ab) and, optionally, at
least one of said properties displayed by said vector molecule andtor
the recombinant inserts used in step (ac) together with at least one of
said properties displayed by either said vector molecule and/or said
recombinant inserts used in steps (aa) and/or (ab), upon the
interaction of a (poly)peptide from said additional library with either a
(poly)peptide from said first library and/or a (poly)peptide from said
second library generate a screenable or selectable property;
.~
so that at least one interaction is established;
CA 02244838 1998-07-27
W O97/32017 rCTJEP97100931
(b) selecting for the generation of said screenable or selectable property
representing the interaction of said (poly)peptides;
(c) optionally, carrying out further screening, selection and/or purification
steps; and
(d) identifying said nucleic acid sequences encoding said (poly)peptides.
In a preferred embodiment of the method of the present invention, said screenable or
selectable property is expressed ekL.~ce'lt)l~rly.
This embodiment is conveniently employed in a number of laboratories which wouldmake use of rather conventional methodology of the extracellular detection of such
properties, e.g. by column chromatography wherein the e.g. screenable tag is retained,
in combination with e.g. plaque purification techniques, which allow the furtherpurification of the cells that were originally enriched by e.g. the column chromatography
step.
In a further preferred embodiment of the method of the present invention, said
recombinant vector molecule in step (a)/(aa) (the step identified after the slash refers to
the corresponding step of the second embodiment of the method of the invention
identified hereinabove) gives rise to a replicable genetic package (RGP) displaying said
~poly)peptides at its surface. In this context, the term replicable genetic package (RGP)
refers to an entity, such as a virus or bacteriophage, which can be replicated following
infection of a suitable host cell. In the case of bacteriophage, for example, the collection
of nucleic acid sequences can be inserted into either a phage or phagemid vector in
frame with a component of the phage coat, such as gene 111, resulting in display of the
encoded binding entities on the surface of the phage. Particularly preferred as a
CA 02244838 1998-07-27
W O 97~32017 P~TnEP971~0931
q
recombinant vector molecule is a recombinant phage, phagemid or virus, wherein said
phage is most preferabiy
(a) one of the class I phage fd, M13, If, Ike, ZJ/2, Ff;
(b) one of the class ll phage Xf, Pf1, and Pf3;
(c) one of the lambdoid phages, lamda,434, P1;
(d) one of the class of enveloped phages, PRD1; or
~e) one of the class paramyxo-viruses, orthomyxo-viruses, baculo-viruses, retro- viruses, reo-viruses and alpha-viruses.
In a further preferred embodiment of the method according to the invention, saidselection step (e)/(b) is carried out by selecting polyphage comprising the interacting
(poly)peptides. Polyphage contain more than one copy of phage genomic DNA. They
occur naturally at a low to moderate frequency when a newly forming phage coat
encapsulates two or more single-stranded DNA molecules. In the case of the present
invention, the polyphage which are formed will contain at least two phage genomes,
which may either (i) both be representatives of library 1, or (ii) both be representatives of
library 2, or (iii) be representatives of each of library 1 and library 2, or (iv) be a
combination of (i) to (iii) with at least one member of one of the additionai libraries. The
efficiency of polyphage production can be increased by the introduction of appropriate
mutations into the phage genome, as is well known to those skilled in the art (see, for
example, Lopez, J. and Webster, ~.E., Virology 127 (1983), 177-193, Bauer, M. and
Smith, G.P., Virology 167 (1988) 166-175, or Gailus, V. et al., Res. Microbiol. 14S
~1994) 699-709).
In a further preferred embodiment of the method of the invention, said screenable or
selectable property is connected to the infectivity of said RGP.
In this embodiment, use is made of the possibility that the infectivity of e.g. a
bacteriophage can be manipulated, said infectivity being directly correlated with the
interaction of said (poly)peptides.
CA 02244838 1998-07-27
W O97/32017 PCT/EP97/00931
In a most preferred embodiment of the method of the present inventlon, said RGP is
encoded by said recombinant vector used in step (a~l(aa) and rendered non-infective
and infectivity of said RGP is restored by interaction of said (poly)peptide of step (a~/(aa)
with the (poly)peptide of step (b)/(ab) and/or (c)/(ac), said (poly)peptide of step (b)/(ab)
and/or (c)l(ac) being fused to a domain that confers infectivity to said RGP.
In a further most preferred embodiment of the method of the invention, said F~GP is
rendered non-infective by modification of a genetic sequence which encodes a surface
protein necessar,v for the RGP's binding to and infection of a host cell.
These preferred and most preferred embodiments of the method of the present
invention relating to the infectivity of the RGP serve as an alternative to the use of the
screenable tag. In these embodiments, advantage can be taken of the phenomenon of
selective infection (Krebber et al., FEBS ~etters ~77 (1995) 227-239). While thescreenable tag enables physical separation of molecules from others in the population,
the use of selective infection enables positive selection for the interacting pair. This
phenomenon relies on the use of a construct which can selectively restore infectivity to
phage which have been rendered non-infective by, for example, deletion of all but the
C-terminus of the gene lll protein. Use of such phage for displaying library 1 gives non-
infectious phage carrying the binding entity. Co-expression with library 2 allows
interactions between binding entities and binding partners to be established, asdescribed above. Although the phage which carry the binding entity-binding partner pair
are non-infective, infectivity can be restored if, in place of the screenable tag referred to
above, an infectivity protein is used. In this context, the term infectivity protein refers to a
sllbst~nce which, when associated with the phage, can enable it to penetrate a bacterial
host, where it is subsequently replicated. An example of an infectivity protein is the N-
terminus ~at least the first 220 amino acids) of gene lll protein of the filamentous
bacteriophage.
- . -
CA 02244838 1998-07-27
W O g7J32017 PCT~EP97100931
~ I
The infectivity protein confers on those phage which carry it, the ability to be replicated.
Thus, only those phage which carry the binding entity/partner pair are replicated.
Purification of hybrid phage containing genes from both libraries 1 and 2 then reJies e.g.
on the use of two selectable markers as indicated above. The genes in the phage can
then be identified using methodology well known to those skilled in the art.
,
An additional preferred embodiment of the present invention relates to a method,wherein said recombinant vector molecules in step (a)/(aa) give rise to a fusion protein
which is expressed on the surface of a cell, =J,eferably a bacterium.
These fusion proteins, upon interaction with a suitable binding partner from library 2
connected e.g. with a screenable tag can be detected on the surface of host cells which
may be, for example, bacteria, yeast, insect cells or mammalian cells. ~he display of
fusion proteins on bacterial surfaces per se is well known in the art. Thus, lipoproteins
(Lpp), outer membrane proteins A (OmpA), and flagella have been used to target
antibodies and peptides to the cell surface of E.coli. Fuchs et al., Bio/Technology 9
~1991) 1369-1372, WO93/01287, presented a single chain antibody on the surface of
E.coli as a fusion protein with the N-terminus of the peptidoglycan-associated
lipoprotein. The antibody was visualized by the binding of fluorescently labeled antigen
and fluorescently labeled antibodies directed to the linker peptide of the displayed single
chain antibody. Francisco et al., Proc. Natl. Acad. Sci. USA 9û (1993) 10444-10448,
and t~eorgiu, G. et al., WO93/10214, displayed antibodies on the E.coli surface by
fusing the N-terminus of a single chain antibody to the C-terrninus of OmpA while the N-
terminus of OmpA was fused to the signal sequence and the first nine amino acids of
Lpp. Binding of a fluorescently labeled antigen to the OmpA-antibody fusion protein was
detected by F~CS. Klauser (WO 95/17509) transferred the IgA protease system fromNeisseria to E.coli to faciiitate display of antibodies. Integration of the beta-domain of
the IgA protease precursor into the outer membrane lead to the transport of the
CA 02244838 1998-07-27
W O97J32017 PCT~EP97/0~931
l~
protease domain across the membrane foltowed by autoproteolytic release into themedium. Antibodies linked to the beta-domain of IgA protease are therefore presented
on the surface of bacteria. Further, ~u, Z. et al., Bio/Technology 13 (19~4) 366-371,
described a system for displaying peptides on the surface of the bacterium by fusing it
to thioredoxin and the bacterial flagella, to screen for peptide mimics of the epitope for
an anti-lL-8 antibody.
The further identification of the desired nucleic acid molecule encoding the interacting
(poly)peptides may then be effected by methods known in the art, e.g. by purifying host
cells displaying a tag on their surface and further by antibioticum-based selection
techniques, DNA puriric~lion and sequencing.
In a particularly preferred embodiment of the method of the present invention, said
bacterium is Neisseria gonorrhoe or E.coli and said fusion protein consists of at least a
part of a flagellum, lam B, peptidoglycan-associated lipoprotein or the Omp A protein
and said ~poly)peptide.
As has been repeatedly pointed out hereinabove, a tag connected to the (poly)peptide
encoded by library 2 can conveniently be used in the identification strategy of the
desired nucleic acid sequences. Accordingly, in a further preferred embodiment of the
method of the invention, said (poly)peptides encoded by said recombinant vector
molecules of step ~b)/(ab) or (c)/(ac) are linked to at least one screenable or selectable
tag. In this context, the term screenable or selectable tag refers to a short sequence of
amino acids which can be recognized and bound by a particular substance. Tags are
commonly used for the purification of biomolecules: examples are His(n), where n = 4-6
which can be bound either by Ni, or a specific antibody, and the flag and myc tags which
are recognized by appropriate antibodies. In either of these cases, the tag can be
encoded as a C-terminal fusion to all binding partners in library 2. In accordance with
the present invention, the tag can be used to isolate e.g. the polyphage referred to
CA 02244838 1998-07-27
WO 97/32Q17 PCT~P97JOl~g31
13
above. Thus, the interaction between the phage-bound binding entity, and its interacting
binding partner, establishes a connection between the phage particle and the
screenable or selectable tag. This feature can be exploited in a step which relies on e.g.
affinity chromatography to isolate the polyphage carrying the interacting molecules. In a
final step, those polyphage which carry two distinct nucleic acid molecules and
preferably genes (encoding binding entity and binding partner) can be separated from
those carrying only one of the two genes e.g. by selection based on transduction or
diflerent selectable markers (e.g. antibiotic resistance) present in the individual
genomes. In this way, the genes which encode the two interacting molecules can be
identified.
A most preferred embodiment of the present invention relates to a method wherein said
screenable or selectable tag is encoded by said recombinant vector of step (b)/(ab) or
(c)/(ac).
A further most preferred embodiment of the present invention relates to a methodwherein said screenable or selectable tag is selected from the list His(n), myc, FLAG,
malE, thioredoxin, GST, ~ ota,/idin, beta-galactosidase, alkaline phosphatase T7 gene
10, Strep-tag and calmodulin. These screenable tags are all well known in the art and
are fully available to the person skilled in the art.
In an additional particularly preferred embodiment of the method of the invention, said
screenabie or selectable tag is encoded by the genome of the host cell.
An example for this embodiment is an anti-Fc-receptor specific antibody that is
expressed by the host cell and could function as an additional bridge in e.g. purification
~y column chromatography. Another example of this embodiment is an enzyme
produced by the host cell that creates a tag such as a phosphorylation on (poly)peptides
of the second library without destroying the interaction of (poly)peptides of step (b)/(ab~
,.
CA 02244838 1998-07-27
W O ~7~32017 PCT~EP97/00931
l~
with (a)J(aa) so that the modification caused by the enzyme is now the screenable or
selectable tag.
In a further preferred embodiment of the method of the invention, said (poly)peptides
encoded by the nucleic acid sequences of said additional libraries of step (c)l~ac) cause
the interaction of said (poly)peptides of steps (a)/(aa) and (b)/(ab) via phosphorylation,
glycosylation, methylation, lipidation or farnesylation of at least one of said
(poly)peptides of steps (a)/(aa) and (b)/(ab).
An additional preferred embodiment of the invention relates to a method wherein said
host celis in step (d)/(a) are spatially addressable, and the nucleic acid sequences
mentioned in step (g)/(d) are retrieved from the corresponding sp~ti~lly addressable
host cell.
In the context of the present invention, the term "spatially addressable" refers to a
situation where the individual cells harboring one of the potential combinations of
members of the first, second and optionally additional libraries are identifiable by their
relative position, e.g. by their position on a master plate. The screening or selection
may, for example, be performed either with single clones derived from the master plate,
or on a replica plate, thus maintaining the connection between the screenable orselectable property and the infc; n,l~lion contained in the host cell on the master plate.
An additional preferred embodiment of the invention relates to a method wherein said
screenabie or selectable property is expressed intracellularly.
Particularly preferred is a method wherein said screenable property is the
transactivation of the transcription of a reporter gene such as beta-galactosldase,
alkaiine phosphatase or nutritional markers such as his3 and leu or resistance genes
CA 02244838 1998-07-27
WO97132017 PCT~EP97100931
giving resistance to an antibiotic such as ampicillin, chloramphenicol, kanamycin,
zeocin, neomycin, tetracycline, or streptomycin.
Furthermore, use can be made of the yeast 2-hybrid system referred to hereinabove or
the interaction trap system (Brent et al., EP-A 0 672 131) or of a prokaryotic version
analogous to the above recited systems, utilizing the toxR system of Vibrio cholerae
(Fritz, H.-J. et al., EP-A 0 630 968). It is within the skills of the person skilled in the art to
combine further screening systems known in the art with the method of the present
invention.
In a further prefer"3d method of the present invention, said recombinant vectors of step
(a)/(aa), (b)t(ab) and ~c)/(ac) comprise r~coml,i.,~lion promoting sites and in said step
(e)J(b) recombination events are selected for, wherein said nucleic sequences encoding
said (poly)peptides of step (ay(aa), said nucleic acid sequences encoding said
(poly)peptides of step (b)/(ab) and optionally said nucleic acid sequences encoding said
(poly)peptides of step (c)/(ac) are contained in the same vector. In this approach, the
two genes can be coupled in a single vector, and packaged in a phage of standard size,
if appropriate recombination sites are incorporated in the vectors carrying libraries 1 and
2. Again, the phage which carry both nucleic acid sequences and genes are purified
with the use of e.g. the screenable tag. If reco,nbi.,ation is used to couple the genes
from the two libraries, some of the hybrid progeny phage will contain nonrecombinant
genomes, since site-specific recombination is not very efficient. However, the hybrid
phage can be selected by re-infection of host cells that do not contain library 2 followed
by a,)olller round of selection of the screenable tag.
In a particularly preferred embodiment of the method of the invention, said
recombination events are mediated by the site-specific recombination mechanisms Cre-
lox, attP-attB, Mu gin or yeast flp.
CA 02244838 1998-07-27
W O 97/32017 PCT~EP97/00931 1~
In a further particulariy preferred embodiment of the method of the invention, sa~d
recombination promoting sites are restriction enzyme recognition sites and said
recombination event is achieved by cutting the recombinant vector molecules mentioned
in steps (a)/(aa), (b)/tab) and optionally (c)/(ac) with at least two different restriction
enzymes and effecting recombination of the nucleic acid sequences contained in said
vectors by ligation.
The invention relates in an additional preferred embodiment to a method wherein said
identification of said nucleic acid sequences is effected after the selection step (e)/(b)
via PCR and preferably sequencing of said nucleic acid sequences after said PCR.After said selection step (e)/(b), PCR can be carried out with the enriched desired
product, conveniently using primers that hybridize to the vector portion of the
recombinant vector molecule. Sequencing of the PCR-product may then be carried out
according to conventional methods.
In a further preferred embodiment of the method according to the invention, saidrecombinant vectors of step (a)/(aa), (b)/(ab) and/or (c)/(ac) comprise at least one gene
encoding a selection marker.
Said genes encoding said selection markers are preferably different in each of the
vectors of step (a)/(aa), (b)/(ab) and/or (c)/(ac), i.e. said vectors comprise genes
encoding different selection markers. Said selection markers can conveniently be used
for the further purification envisaged in step (f)/(c). For example, a polyphageCCi~ ill9 two members of each library 1 and 2 can be selected for on the basis of a
double resistance to antibiotics. Also, a successful recombination event may create a
new recombinant vector carrying both nucleic acid molecules from library 1 and 2 as
well as genes encoding different selection markers. Again, the selection for a twofold
resistance will assist in the identification of the desired product.
CA 02244838 l998-07-27
W O 97~32017 PCTnEP97/~0931
17
In a particularly preferred embodiment of said method, said selection marker is a
resistance to an antibiotic, preferably to ampicillin, chloramphenicol, kanamycin, zeocin,
neomycin, tetracycline or streptomycin.
A further preferred embodiment of the present invention relates to a method wherein
said host cells are F' and preferably E.coli XL-1 Blue, K91 or its derivatives, TG1,
XL1 kan or TOP1 ûF.
In a particularly preferred embodiment of the present invention, said RGPs are
produced wi~h the use of helper phage taken from the list R408, M13kO7 and VCSM13,
M13de13, fCA55 and fKN16 or derivatives thereof.
~urther preferred is a method wherein at least one of said genetically diverse nucleic
acid sequences encode members of the immunoglobulin superfamily.
Said method is particularly preferred, if said genetically diverse nucleic acid sequences
encode a repertoire of immunoglobulin heavy or light chains.
In an additional pre~erred embodiment of the present invention, in said method said
genetically diverse nucleic acid sequences are generated by a mutagenesis method.
Various mutagenesis methods are well known to the person skilled in the art and need
not be described in here in any further detail.
The present invention relates in an additional preferred embodiment to a method in
which said genetically diverse nucleic acid sequences are generated from a cDNA
library.
In a final preferred embodiment of the method of the invention, said nucleic acid
sequences are genes or parts thereof.
CA 02244838 1998-07-27
W O97132017 PCT~EP97/00931
1~
As used herein, the terrn "parts thereo~' relates to parts of genes that encode a product
that is capable of interacting with a product encoded by any of the other libraries. Thus,
it is well known that various proteins are comprised of different domains. Only one of
said domains may be capable of interacting with a dif~erent (poly)peptide. Such a
domain might be encoded by a part of said gene in accordance with the present
invention.
The invention also provides for identi~ing genes encoding more than two interacting
peptides or proteins. This can be achieved by using additional vectors encoding
geneticalty diverse additional nucleic acids by an extension of the method described
above. As previously, the presence of either a screenable tag or an infectivity protein is
used to puri~y phage carrying genes which encode the components of the complex.
Again, the genes in the phage can then be sequenced using methodology well known to
those skilled in the art.
Additionally, the present invention relates to a kit comprising at least
(a~ a recombinant vector molecule as described in step (a)/(aa) or a corresponding
vector molecule;
(b) a recombinant vector molecule as described in step (b~/(ab) or a corresponding
vector molecule; and, optionally,
(c) at least one further recombinant vector molecule as described in step (c)/(ac) or a
corresponding vector molecule.
As a rule, if recombinant vector molecules are comprised in said kit, they will comprise a
li~rary of nucteic acid molecules. In other words, the kit of the invention will contain a
plurality of different recombinant vector molecules.
CA 02244838 l998-07-27
W O g7132017 PCT~EP97J00931
Legends to Figures and Tables
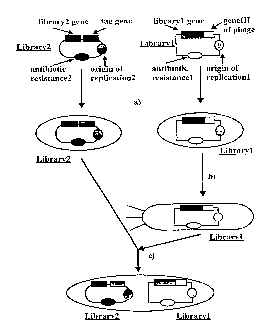
~igure 1: General description of the polyphage principle
a) llan~ro"" to E. coli hosts
b) infect host containing library1 with helper-phage to package li~rary1
into phage
c) infect ceils con~ai, ling library2 with phages containing library1 leading tocells harboring men,ber~ of libra,-y1 and library2; the presence of library1
and library2 is selected by the presence of the 2 antibiotic resistance
markers
d) expression of library1 and library2-tag gene products
e) infect cells with engineered helper-phage to induce polyphage
production
Note 1: Polyphage does not dis.;,i,llinale which genome to package
therefore the possibilities resulting from step e) arise in an infected cell. Toselect for the polyphage containing the right packaged genomes the
subsequent step is required
f) select for tag e.g., infectivity-mediating protein, in which case ability to
infect is selected and
g) select for ability to confer resistance to 2 antibiotics to infected cells
Note 2: Only polyphages that satisfy fl + ~) represent phages that display
the correct interacting pair and the corresponding genetic information~igure 2: Co-llal,~ror~ lion of twc phagemids, polyphage formation and selection
vfa His-tag: general ~esc, iplioll
A, B: libraries of phagemids, preferably with dirr~re"t resistance markers;
A: fusions to glllp; B: fusions to tag (His); after co-transformation phage
production ieading to a phage population displaying cog,lale pairs (left
part of the Figure) or not (right part), after selection infection of host cells,
selection for double-resistance
RE~TIFIED SHEET (R~J~E
ISA/EP
CA 02244838 1998-07-27
W O97/32017 PCTAEP97/00931
~0
Alternative methods include the infection o~ cells harbouring a piasmid- or
phagemid-based libraty B with a phage library A (prerequisite again:
interference-resistant constructs).
Figure 3: pBS vector series: functional map and sequence of pBS13~igure 4: Co-existence of phagemids: results of restriction digest
Restriction analysis of clones of double resistances (Amp/Cm). R1:
plG10.3, Xba/Scal; R2: pBS13, Xba/Scal, R1+R2: R1 and R2 are mixed in
approx. equal proportion; M1: marker ~: ~stEII; M2: rnarker pBR322: Mspl;
1 to 10: randomly picked clones: Xba/ScaJ~igure 5: Phagemid vector pYlNG1-C1: functional map
containing the fos peptide. The corresponding vectors pYlNG1-C2 and
pYlNG1-C3 cc",lain instead of fos the p75 and the IL16 peptides,
respectively~igure 6: Phagemid vector pYANG3-A: f~ll ,c~ional map
containing the jun peptide. The corresponding vectors pYANG3-Ape2,
pYANG3-Ape3, and pYANG3-Ape10 contain instead of jun the p7~-
binding peptides pe2, pe3, and pe10, respectively~:igure 7: Analysis of selected clones (see Table 2):
7.a: Restriction digest of clones before and after selection
R: pYANG3-Ape2: X~al; M1: marker ~: BsfEII; M2: marker pBR322: Mspl;
a/1 to 10: ra~du~lly picked clones before selection: XbaVHindlll; ,B/1 to 10:
randomly picked clones after selection: Xbai~rflndlll; size expected: jun-
9111: 74~; bp; fos: 256 bp; p75: 577 bp; IL-16: 502 bp
7.b: PCR reaction of clones after selection with primers OPEP~L and
OGI113
R1: pYANG3-A as tempiate; R2: pYANG3-Ape2 as template; i\A: market ~:
BstEII; ~/1 to 10: randomly picked clones after selection as templates~igure 8: Phagemid vector plNG1-C1: functional map
containing the His-tag peptide. The corresponding vector plNG3-C1
contains an additional Fi AG epitope; plNG1-C2 and plNG3-C2 contain
P~ECTI=IED SHEET (RULE 91
ISA/EP
CA 02244838 1998-07-27
W O97f32017 PCTAEP971nO93~
the Strep-tag instead of His-tag, with plNG3-C2 containing an additional
FLAG epitope.
Figure 9: Phagemid vector pONG3-A: functional map
for the generation of phage-display libraries (9111 fusions)
Fi~ure 10: Co-transformation of phage and plasmid, polyphage formation and
selection via SIP: general desc,ipLiol,
fA~ rary A in phage construct; ~: library B, library members fused to IMP;
preferably different resistance markers on phage and plasmid; after co-
transformation production of phages; in the case of cognate-pair
interaction formation of infectious phages; selection; by plating on double-
resistance identification of poiyphage particies.
Figure 11: Phagevectorfhag1A:functionalmap
for phage-display of the a-HAG scFv
Figure 11a: CAT gene module: funcliol1al map and sequence
Figure 12: Phage vector fjun1A: functional map
for phage-display of the jun peptide
Figure 13: Phage vector fjun1 B: functional map
for phage-display of the jun peptide
Figure 14: Pha~e vector fpep3_1 B: functional map
for phage-display of the peptide pe3 binding to the intracellular domain of
p75
Figure ~5: Phage vector fNGF_1 B: functional map
for phage-display of NGF
~iigure S6: Plasmid pUC1911MPhag: functional map
containing fusion of HAG peptide to the N-terminal domains of glllp (IMP)
Figure 17: Plasmid pUC1811MPp75: functional map
containing fusion of the intracellular domain of p75 to the N-terminal
domains of glllp ~IMP); pUC18/lMPfos contains the fos peptide instead of
the intracellular domain of p75
r~
RECTIFIED SHEET (RULE 91)
ISA/EP
CA 02244838 1998-07-27
W O 97/320l7 PCTAEP97/00931
2~
;3ure 18: Plasmid pUC18/lMPlL16: functional map
containing fusion of IL16 to the N-terminal domains of glllp (IMP)
Figure 19: Analysis of selected clones (see Table 3)
Lane 1: marker ~: BsfEII; lanes 2 to 20: polyphage transduct~nt clones ~ l
to $~19 digested with Xba~lindlll; f.._1b: fragment of phage vector after
digest; pUC18: r, a~ enl of plasmid after digest; a-HAG: fragmen!t
containing anti-HAG scFv fused to glllc; IMP-p7~ and IMP-HAG: fragment
conlai~,ing IMP fused to p75, and IMP-HAG peptide, respectively; pep3-
gllls: fragment containing pep3 fused to glllc (s: short version)~igure 20: Co-L,d"~ron"~lion of phagemids, in vivo recombination and selection via
His-tag: general description
A, B: libraries of phagemids; preferably with different resistance markers;
A: fusions to glllp; B: fusions to tag (His); both constructs containiny
recombination-promoting sites (*) such as lox/loxP; after co-LI~nsrur,,,~liorl
and recombination production of phages; selection via Ni-NTA; re-infection
of host cells, selection for double-resistance
Figure 21: In vifro recombination and selection via His-tag: general desc, iplion
A, B: libraries of phagemids; prerc~rably with clirrerenl resisLance markers;
A: fusions to glllp; B: fusions to tag (His); both constructs containin~3
corresponding recognition sites for restriction enzymes (+/o); after digest
and co-ligation l,d,,~ru,,,,~lio,, and production of phages; selection via Ni-
NTA; re-infection of host cells, selection for double-resistance
Figure 22: Phage vector fjunhag: f~ ,cliûnal map for phage display of the jun peptide
~igure 23: Spatial in vivo SIP: general desc, ilulion
After l,~"sror"lation or co-lransrur,,,~lion accordi-,g to any of the methods
described above, a master plate is made. From that phages secreted from
individual clones can be analyzed individually (top), or a replica (migration
of secreted phages through filter disc~ can be made whereon selection for
the presence of a tag or infectivity can be performed. By going back to the
ktl~! IEl~U SHEET (RULE 91)
ISA / EP
CA 02244838 1998-07-27
W ~ 97/32017 PCT~P97100931
~3
master-plate the information for selected cognate interacting pairs can be
retrieved without requiring recombination and/or potyphage production.~igure 24: E coli display: general description
A B: libraries of phagemids; preferably with different resistance markers;
A: fusions to E.coli surface-dispiay protein; B: fusions to tag (His); after co-transformation expression of constructs; surface-display; in the case of
cognate interaction taking place display of tag on the surface of the host
cell; selection
Figure 2~: pTERMsc2~i10myc3sCAM: functional map and sequence
Table 1: Phagemids constructed for Experiments 2 and 3
Table 2: Results of Experimenl 2 (see Figure 7)
2.a: Combination of phagemids present in initial library (a)
2.b: Combination of pnagemids present after selection (B)
Table 3: Results of Experiment 4 (see Figure 19)
3.a: Identification of phagelplasmid present in individual clones
3.b: Test for infectivity of individual clones
~he examples iilustrate the invention.
RECTIFIEO SHEET (RUL~ 91)
ISA/EP
CA 02244838 1998-07-27
W O97132017 PCTAEP97/00931
Example 1: General description o~ the polyphage principle (Figure 1)
The bindlng entities which comprise library 1 may be peptides or proteins, and are
encoded by a genetically diverse collection of first nucleic acid se~uences. These
nucleic acid sequences are inserted into a first vector which allows for display of the
encoded binding entities on the surface of a replicable genetic package. For thepurposes of subsequent selection, the first vector should also carry a gene encoding a
selectable marker. such as an antibiotic resistance. The binding partners which
comprise library 2 may be peptides or proteins, and are encoded by a geneticallydiverse collection of second nucleic acid sequences which are inserted into a second
vector. By way of example, this second vector may be a plasmid, or even a phage or
phagemid, in which case the origin of replicalion should be distinct from that of the first
vector. For the purposes of subsequent selection, the second vector should also carry a
gene encoding a selectable marker, such as an antibiotic resistance, preferably distinct
from that present in the first vector. To facilitate purification of the complex to be formed
between any binding entity-binding partner pair, a screenable tag can be conveniently
attached to members of library 2.
The two genetically diverse collections of nucleic acids are then introduced into a
population of host cells in such a way that encoded libraries 1 and 2 can be expressed.
This can be achieved by either (i) co-transr~"",dLion of the two vectors, or, as actually
shown in the figure, (ii) packaging one of the collections of nucleic acids into a vector
(such as a bacteriophage~ which can be used to infect with high efficiency a population
of cells into which the complementary collection of nucleic acid has been introduced.
The result is a population of cells in which individual cells carry representatives of each
library.
Expresslon of the two collections of nucleic acids results in the production of pairs of
molecules, one from each library, in the host cells. In each case, one or more members
CA 02244838 1998-07-27
W O97J32017 PCT~EP97/00931
of the library of binding entities is incorporated into the coat of an RGP. In some cells,
an interaction will be established between a binding partner on the surface of the RGP
and a binding partner expressed from library 2. When such an interaction is established,
the RGP therefore carries both the binding entity and the binding partner.
The RGPs displaying such an interaction can then be further purified with the help of
polyphage and differing selection markers, as has been discussed hereinabove. After
such selection, the nucleic acid sequences encoding one or both binding partners can
be conveniently identified by methodology known in the art, such as DNA sequencing.
Example 2: Co-transformation of phagemids with same E. coli origin
of replication, polyphage formation, and selection of correct pairlng
interactions via His-tag
2~1: Principle (see F!igure 2)
To demonstrate that polyphage formation allows the retrieval of the genetic information
for cognate protein pairs selected using a tag fused to one member of the protein pair,
two separate, small libraries in phagemid vectors are constructed.
2.2: Test of co-existence of phagemids with the same E coli ori~in of replication:
Prerequisite for the formation of polyphage particles containing two different phagemids
is that the different phagemid vectors can co-exist in the host cell.
The vector pBS13 is a derivative of the vector ~Krebber et al., 1996~ containing a
chloramphenicol-resistance gene instead of the kanamycin-resistance gene and a beta-
lactamase gene cassette instead of the 2H10-glll fusion gene, and can be assembled
by standard methods starting from pto2H10a3s. Figure 3 contains the functional map
and the sequence of pBS13. plGHAG1A (see Example 4.2.1.f) is digested with Xbal
and Hindlll. The 1.3 kb fragment containing the anti-HAG gene fused with the C-
CA 02244838 1998-07-27
W O 97/32017 PCT/EP97/00931 ~6
terminal domain of filamentous phage plll protein is isolated and ligated with a pre-
digested phagemid vectors plG10.3, and pBS13 (Xbal-Hindlll) to create the vectors
plG10.3-scFv(anti-HAG) (ApR) and pBS13-scFv(anti-HAG) (CmR), respectively. The
vectors are used to transform competent XL-1 Blue cells and selected on LB plates
containing Amp/Cm/Tet and glucose (20 mM).
The phagemids from clones of double-resistant colonies (Amp/Cm) are isolated. The
restriction digestions indicate the ~o-isolation of both phagemids from the single
colonies (Figure 4).
2.3: DQsign of liLr~ri~s A and B:
Library A contains three cyclic peptides each binding to the intracellular domain of the
low affinity nerve growth factor (NGF) receptor (see Example 4), and a leucine zipper
domain derived from the jun transcription factor, all N-terminally fused to the C-terminal
domain of 9111 from filamentous phage.
Library B encodes 3 members, namely the leucine zipper domain of the fos
transcriplion factor which heterodimerizes with jun via this domain, the intracellular
domain of the NGF receptor p7~, and, as a negative control which does not interact with
library A members, l~ -16, all fused at the N-terminus with a His6-peptide as tag (Hochuli
ef al., 1988; Lindner ef al., 1992).
The cognate pairings are from the interaction between jun and fos (Crameri and Suter,
1993), and p75 and selected cyclic peptides (see Example 4). A non-cognate pairing
would occur among the non-cognate pairs mentioned and among jun, or one of the
cyclic peptides, and IL-16.
2.4: PCR amplification of the individual constructs
Fos, N-terminus fused to His6, is PCR amplified using pOK1 (Gramatikoff e~ a/., 1994)
as template and oligonucleotides OFOS-5 and OFOS-3 as primers, where His6 is
CA 02244838 1998-07-27
W O9~J32017 PCTnEP97/00931
~7
encoded in the OFOS-5 primer. Jun is PCR amplified using pOK1 as template and
oligonucleotides OJUN-5 and OJUN-3 as primers.
OFOS-5 5'- GGG GA TA TCCACCACCACCACCACCACCTGCGGTGGTCTGACC
OFOS-3 5'- GGGGAATTCCMCCACCGTGTGCCG
OJUN-5 5'- GGGGATATCGGTGGTCGGATCGCC
OJUN-3 5'- GGGGAATTCACCACCGTGGTTCATGAC
The hot-start procedure is used. A step-wise touch-down PCR is applied: 92~C, 1 min;
58-52~C, ~T = 2~C, 1 min; 72~C, 1 min. This is followed by 26 cycles (92~C, 1 min; 52~C,
1 min; 72~C, 1 min).
The PCR products are purified using QlAquick kit (Qiagen) and eluted in ddH20. They
are then overnight digested with EcoRI and EcoRV.
The p75 fragment is also PCR amplified using pUC18-lMPp75 tsee Example 4) as
template and oligonucleotides OP75-5 (where His6 is encoded) and OP75-3 as primers:
OP75-5 5'- GGGGATATCCACCACCACCACCACCACMGAGGTGGMCAGC
OP75-3 5'- GGGGAATTCCACTGGGGATGTGGCAG
The same PCR and restriction digestion conditions as above are applied.
The IL-16 fragment is amplified from the cDNA clone pcDNA3-lLHu1 (M. Baier, Paul
Ehrlich Institute, Germany; Baier et al., 1995; Bannert et al., 1996), using OIL16-5
(where His6 is encoded) and OIL16-3 as primers.
OIL16-5 5'- GGGGATATCCACCACCACCACCACCACCCCGACCTCMCTCCTC
OIL16-3 5'-GGGGAA7~CGGAGTCTCCAGCAGCTG
The same PCR and restriction digestion conditions as above are ~r)plied.
In all cases, the fragments are readily amplified and digested.
-
2.5: Cloning into intermediate vectors
CA 02244838 1998-07-27
W O 97/32017 PCTAEP97100931
The digested PCR fragments are gel-purified (QlAquick kit, Qiagen) and eluted into TE
buffer. The EcoRV/EcoRI fragment of plG1 vector (Ge et al., 1995) is also isolated. The
digested PCR fragments of fos, p7~, and IL-16 are ligated into the vector fragment, and
the ligated vectors transformed into TG1 cells.
The constructs in the plG1 vector contains the OmpA signal sequence fused in-frame
with the constructs.
The correct clones are screened and collrlr~ed by sequencing. They are then
XbalfHindlll digested, and the fragments are isolated.
2.6: Cloning into the e~ ,csion v~ct~.~
The isolated fragments from 2.3 are inserted into pBS13 also excised with Xbal/Hindlll,
resulting in vectors pYlNG1-C1 (Fos), pYlNG1-C2 (p7~), pYlNG1-C3 (IL-16) (see
Figure ~). The fragment containing jun is cloned into plG10.3 vector via EcoRVlEcoRI
resulting in pYANG3-A (see Figure 6). The anti-p75 peptides pe2, pe3 and pe10 (see
Example 4) are cloned into plG10.3 via Xbal/Hindlll, resulting in vectors pYANG3-
Ape2, -Ape3 and -Ape10, respectively (see Figure 6).
2.7: -Selection of correct pairing via His-tag
TG1 cells are transformed with the combination of pYANG3-A + pYlNG1-C1, or
pYANG3-A ~ pYlNG1-C2, or pYANG3-A + pYlNG1-C3, or (pYANG3-Ape2, -Ape3 and -
Ape10) i pYlNG1-C1, or (pYANG3-Ape2, -Ape3 and -Ape10) + pYlNG1-C2, or
(pYANG3-Ape2, -Ape3 and -Ape10) + pYlNG1-C3, thus creating all possible
co~ inalions separately to ensure the presence of each of them in the selection
experiment. The transformed cells are plated on ampicillin/chl~r~mphenicol-containing
LB agar plates, and colonies with double resistance (ApR/CmR) are selected.
The colonies are scraped off the plates and used to inoculate 2xYT medium (Amp/Cm)
and shaken at 37~C for 3 hrs. The cultures are induced (1 mM IPTG) at 30~C for 1 hr
and infected with R408 (Stratagene) at 37~C for 30 min. The cultures are shaken at RT
for 3 hrs, kanamycin is added and shaking continued at RT overnight.
CA 02244838 1998-07-27
WO g7/32017 PCT/EP97/nO931
~9
The phage particles are harvested from the overnight cultures, mixed and PEG-
precipitated. The phages are directly selected on immobilized Ni-NTA (Nl-NTA HisSorb
Strips, Qiagen). The eluted phages are used to infect TG1 cells, which are plated on
ampicillin/chloramphenicol-containing LB agar plates, and colonies with double
resistance (ApR/CmR) are selected.
The phagemids of selected clones are isolated and analyzed by restriction digest (see
Figure 7.a) and used as templates for PCR screening. Primer OPEP5L is used to
amplify the pYANG3-Ape2, -Ape3 and -Ape10 constructs specifically (see Figure 7.b).
OPEP5L 5'- GACTACAAAGATGTCGACTG
There is a specific enrichment of constructs of correct pairing (Table 2).
Example 3: Interactive screening of E. coli genomic DNA libraries
(Polyphage/tag system~
3.1: Principle (see }igure 2)
Instead of using two model libraries as in Example 2, a genomic DNA library of E. coli is
prepared to be screened against itseif to identify interacting E. coli peptides or proteins.
3.2: Construction of display and e~,r~ssion vectors for genomic DNA
Exp,ession vectors are constructed having a blunt-end restriction site Smal inserted
either in front of His-tag, Strep-tag (Schmidt and Skerra, 1994) or the C-terminal domain
of glll (glllc) via oligonucleotide cassettes or PCR.
The self-complementary oligonucleotides OHIS5 & OHIS3, and OSTREP5 & OSTREP3,
are used to create ds DNA cassettes encoding the His-tag, and the Strep-tag,
respectively.
OHIS5 5'- AATTCCCCGGGCACCACCACCACCACCACTGATA
OHIS3 5'- AGCTTATCAGTGGTGGTGGTGGTGGTGCCCGGGG
CA 02244838 1998-07-27
W O97/32017 PCT~EP97/00931
OSTREP5 5'- AATTCCCCGGGTCTGCTTGGCGTCACCCGCAGTTCGGTGGT-
TGATA
OSTREP3 5' - AGCTTATCAACCACCGAACTGCGGGTGACGCCAAGCAGACC-
CGGGG
The cassettes upon phosphorylation and anneaiing recreate the EcoRI and Hindlll sites.
The cassettes are inserted into plG1 and plG3 vectors (Ge et al., 1995) cut by the same
restriction enzymes. The resulting vectors are plNG1-A1, plNG3-A1 (for His tag in plG1
and plG3 vectors) and plNG1-A2, plNG3-A2 (for Strep-tag), respectively. The correct
vectors are screened for the presence of Xmal site (isoschizomer of Smal) and the
constructs are cGnfi"ned by sequencing. The Xbal/Hindlll fragments of these vectors
are inserted into pBS13 vector, linearized with the same enzymes, resulting in vectors
plNG1-C1, plNG3-C1 and plNG1-C2, plNG3-C2, respectively (see Figure 8).
The glllc fragment containing the Smal site is generated from PCR amplification of
plG10.3 vector using primers OG1115 ancl OGI113, where OG1113 anneals 3' of the gene 111
in the vector:
OGI115 5'- CGGM7~CCCCGGGGAGCAGAAGCTGATC
OG1113 5'- 1 1 1 1 ICACTTCACAGGTC
Three rounds of PCR are performed with a hot-start: 92 C, 1 min; 46 C, 1 min; 72 C, 1.5
min. This is followed by 30 rounds of: 92 C, 1 min; 50 C, 1 min; 72 C, 1.5 min.
The PCR product is purified (QlAquick) and digested with EcoRI and Hindill. The
fragment is gel-purified (QlAquick) and ligated into plG10.3. The sequence of the
resulting vector, pONG3-A (see Figure 8), is confirrned by ~sl~il;tion analysis and by
sequencing.
3.2: ~Selecffon of Interacting Pairs from E. coli Genomic DNA via His-taq
Genomic DNA of E. coli strain XL-1 Blue (Stratagene) is isolated using the Blood & Cell
Culture DNA Maxi kit (Qiagen) and eluted in TE buffer (pH 8.0). 200 ~lg of the DNA is
CA 02244838 1998-07-27
W 097/32017 PCT~EP971~0931
3l
taken and sonicated (50 cycles, 27û mA, 0.~ s/stroke3. The fragmented DNA (average
size: max. 0.7 kB) is blunt-ended by a fill-in reaction with T4 DNA polymerase.
Vectors plNG1-C1 and pONG3-A are digested with EcoRV and Smal, the vector
fragments are gel-purified (Qiagen). The vector fragments are then ligated with the
blunt-ended genomic DNA at 1 6~C overnight. The ligation mixtures are taken to
transform TG 1 cells.
The plNG1-C1 and pONG3-A transformants are scratched from the plate and used to
inocul~te 2xYT medium containing Cm/glucose or Amp/glucose, respectively. The
plNG1-C1 culture is infected with helper-phage (\/CSM13 or M13kO7) and phage
particles are isolated. These phage particles are used to infect log-phase cellscontaining the pONG3-A library. The resulting culture is plated out on large
Amp/Cm/glucose plates.
The colonies are scratched from the surface of the plates above and transferred to 2xYT
medium containing Amp/Cm. After 30 min shaking at 37~C, the culture is then induced
(1 mM IPTG) for 30 min, infected with helper-phage at 37~C for 30 min and shaken at
RT overnight.
The phage particles are harvested from the ovemight culture and PEG-precipitated.
They are selected on immobilized Ni-NTA (Nl-NTA HisSorb Strips, Qiagen). The eluted
phages are used to infect log-phase TG1 cells. Selected protein pairs are characterised
by del~rn,il,ation of their corresponding DNA sequences.
Example 4: Polyphages and Selection of Correct Pairing Interactions
via SIP
4.1: Principle (see Figure 10~
The purpose of this experiment is to show that from a combination of 2 libraries one can
isolate and identify the correct interacting pairs using the SIP (Selectively Infective
Phage: Krebber ef al., 1995; the term "IMP" used in the experimental section denotes
"Infectivity mediating particle" comprising the N-terminal domains of the gene lll protein
CA 02244838 1998-07-27
W O97/32017 PCTAEP97100931
32,
of filamentous phage) selectlon system, and recover the i. ,rur" ,~llon about bo~h
interacting partners via the formation and selection of polyphage particles. The library
members forming interacting pairs with members of the corresponding library are being
'doped' with library members that do not interact with members of the corresponding
library, and thus should not give a positive SIP selection.
4.2: Construction of v~ctor~
4.2.1: fhag1A (see Figure 11)
a. The phage vector f1719-hag (Krebber et al., 1995) is digested with EcoRV and Xmnl.
The 1.1 kb fragment containing the anti-HAG Ab gene is isolated by agarose gel
electrophoresis and purified with a Qiagen gel extraction kit. This fragment is ligated
into a pre-digested plG10.3 vector (EcoRV-Xmnl). Ligated DNA is transformed intoDH5a cells and positive clones are verified by restriction analysis. The recombinant
clone is called plGhag1A. All cloning described above and subsequently are
according to standard protocols (Sambrook et al., 1989)
b. The vector f17/9-hag (Krebber et al., 1995) is digested with EcoRV and Stul. The 7.9
kb fragment is isolated and self-ligated to form the vector fhag2.
c. The chloramphenicol resistance gene (CAT) assembled via assembly PCR (Ge and
Rudolph, 1997) using the the template pACYC (Cardoso and Schwarz, 1992) (Figure
11 a shows the functional map and the sequence of the CAT gene) is amplified by the
polymerase chain reaction (PCR) with the primers-
CAT_BspEl(for): ~' GMTGCTCATCCGGAGTTC
CAT_Bsu361(rev): 5' mCACTGGCCTCAGGCTAGCACCAGGCG I I I AAG
d. The PCR is done following standard protocols (Sambrook et al., 1989). The amplifiedproduct is digested with BspEI and Bsu361 then ligated into pre-digested fhag2 vector
(BspEI-Bsu361; 7.2 kb fragment) to form fhag2C.
e. The vector fhag2C is digested with EcoRI and the ends made blunt by filling-in with
Klenow fragment. The flushed vector is self-ligated to form vector fhag2CdelEcoRI.
CA 02244838 1998-07-27
W O 97/32017 PCT/EP97/00931 33
f. plGHAG1A is digested with Xbal and Hindlll. The 1.3 kb fragment containing the anti-
HAG gene fused with the C-terminal domain of filamentous phage plll protein is
isolated and ligated with a pre-digested fhag2CdelEcoRI phage vector (Xba!-Hindlll;
6.4 kb) to create the vector fhag1A
-
4.2.2: ~un1A ~see Figure 12)
a. The EcoRV site of plG10.3 is converted to a Sall site by oligonucleotide site-directed
mutagenesis (Sambrook et a/., 1989) with primer:
Sall9-9primer(rev) 5'CTGAATGTCGACATC I I ~ GTAGTC3'
The mutated plG10.3 is called plG10.3 Sall.
b. The jun leucine-zipper dol.,ai" from pOK1 (Grammatikoff ef al., 1994) is amplified by
PCR with the primers:
jun2(for): 5'ACGCGTCGACGCCGGTGGTCGGATCGCCCGG3'
jun2(rev): 5'AATTCGGCACCACCGTGGTTCATGACT3'
c. The PCR is done following standard protocols (Sambrook et al. 1989). The amplifled
product is digested with Sall and EcoRI then ligated into pre-digested plG10.3Sall
vector (Sall-EcoRI) to form the vector jun-plG10.3Sall.
d. ~he vector jun-plG10.3Sall is digested with Xbal and EcoRI. The 0.14 kb fragment is
ligated into the pre-digested vector fhag1A (Xbal-EcoRI; 7kb) to form the vectorflun1A.
4.2.3: flun1B (see Figure 13)
a. The DNA encoding the C-terminal domain including the long linker separating it from
the amino terminal domain of the filamentous phage plll (glll short) is amplified by
PCR using pOK1 (Gral l ,r"~l;koff et a/. 1994) as template with the primers:
gll I short(for): 5'GCTTCCGGAGMTTCAATGCTGGCGGCGGCTCT3'
g 111 short(rev): 5'CCCCCCCAAGCT~ATCAAGACTCCTTATTACG3'
CA 02244838 1998-07-27
W O97/32017 PCT~EP97/00931
3 ~
b. The PCR is done following standard protocols (Sambrook et al., 1989). The amplified
product is digested with EcoRI and Hindlll, then ligated into pre-digested fhag1A
vector (EcoRI-Hindlll) to form the vectorfiun1B.
4.2.4: fpep2_1b, fpep3_1B, fpep10_1b (see Figure 14)
a. These constructs are obtained from a peptide library screened against the
intracellular domain of p75, the low affinity receptor of NGF, in a SIP experiment.
b. A peptide library cassette of cyclic peptides with length variants of 6-16 amino acids
is prepared from the oligos:
Groprim: 5'-CATGAATTCGGATCCTCC-3'
Gron10: 5'-CTATGGCGCGCCTGTCGACTGT(M)6 16TGTGGTGGTGGAGGATC-
CGMTTCATG-3'
where M is a mixture of 19 trinucleotide codons (Virnekas et al., 1994), excluding the
one coding for Cys. The length variation is achieved by coupling 6 trinucleotidepositions using the standard coupling procedure, and, for the next 10 coupling cycles,
by omitting the capping step during DNA synthesis and by diluting the trinucleotide
mixture to achieve stepwise coupling yields of 50%.
The oligos are annealed and filled in with the Klenow fragment of DNA polymerase I
to form a double-stranded DNA cassette with standard methods (Sambrook ef al.,
1989). The cassette is digested with Sall-EcoRI, purified with Qiaex DNA gel
extraction icit, and ligated to pre-digested fiun1B vector (Sall-EcoRI) to form the
peptide library. The ligated peptide library is transformed into competent DH5a cells
harboring pUC18/l~AP-p75 (see below) and plated on Luria Broth (LB) (30 ,ug/ml
chloramphenicol + 100 ~Lg/ml ampicillin) and incubated overnight at ambient
temperature.
c. The Ampr Cmr colonies are scraped with LB, and 1 ml of suspension is used to
inoculate 25 ml LB (30 ,ug/ml chloramphenicol ~100 ,ug/ml ampicillin + 1 mM IPTG).
The culture is incubated overnight at room temperature.
CA 02244838 1998-07-27
W Q 97132017 P~ 57100931
3 ~
d. The supernatant is separated from the cells by centrifugation (10,000 RPM, 10 min.,
4~C). 5 ml of 30% PEG/3M NaCI are added to the supernatant and mixed 100 times.
After 1 hour on ice, the phage precipitate is collected by centrifugation (10,000 RPM,
10 min., 4C). The pellet is resuspended in 1 ml TBS buffer. The suspension is filtered
with a 0.45 micron filter (Sartorius).
e. 100 ,ut of log phase K91 cells (or any male E. coli cells (F-pilus containing)) are
infected with 10 ~LI of phage supernatant, plated on LB (30 ~Lg/ml chloramphenicol)
and incubated overnight at ambient temperature.
f. Chloramphenicol-resistant transductants are picked, and over, ~ l cultures are
prepared to isolate DNA for sequencing. From the sequencing, fpep2_1b, fpep3_1B,fpep10_1b containing peptides pe2, pe3, and pe10 are identified.
pe2: 5'-TG I I I I I I I CGTGGTGG I I I I I I I AATCATAATCCTCGrrATTGT-3'
(CysPhePheArgGlyGlyPhePheAsnHisAsnProArgTyrCys3
pe3: 5'-TGTATTGmATCATGCTCATTATCTTGTTGCTAAGTGT-3'
(CyslleValTyrHisAlaHisTyrLeuValAlaLysCys)
pe10: 5'-TGTTCTTATCATCGTC I I l-CTACTCGTG I I I GT-3'
(CysSerTyrHisArgLeuSerThrArgValCys)
4.2.5: fNGF1B (see Figure 15)
a. The DNA encoding the nerve growth factor (NGFI) gene is amplified from pXM NGF
(Ibanez e~ al., 1992) as template with the primers:
NGF(for): 5'AAMAAGTCGACTCATCCACCCACCCAGTC3'
NGF(rev): 5'AGGAATTCGCCTCTTCTTGCAGCCTT3'
b. The PCR is done following standard protocols (Sambrook et al., 1989). The amplified
product is digested with Sall and EcoRI, then ligated into pre-rligested fjun1B vector
(Sall-EcoRI) to form the vector fNGF1B.
4.2.6: pUC19/lMP-HAG (see Figure 16)
CA 02244838 1998-07-27
W O97/32017 PCT~EP97/00931
36
a. The vector f1719-hag (Krebber et al., 1995) is digested with EcoRI and Hindlll. The
1.4 kb fragment containing the gene fusion of the IMP with the HAG peptide, is
isolated and cloned into pre-digested pUC19 (EcoRI-Hindlll) to form the vector
pUC1 9/lMP-HAG
4.2.7: pUC18/lMP~p75 (see Figure 17~
a. The intracellular domain of p75 containing the C-terminal 142 amino acids is
amplified from the cDNA clone of p75 (Chao et a/., 1986) as template with the
primers:
p75(for): 5' GCTGGCCCGTACGACAAGAGGTGGAACAGCTGC
p75(rev): 5' TCTCGAAGCTTATCACACTGGGGATGTGGC
b. The PCR is done foilowing standard protocols (Sambrook et a/., 1989). The amplified
product is digested with BsiWI and Hindlll, then ligated into pre-digested plJC19
vector (BsiWI-Hindlll) to form the vector pUC19/lMP-p75.
c. The vector pUC19/lMP-p75 is digested with Xbal and Hindlll. The 1 kb fragment is
isolate~ and cloned into the pre-digested pUC18 vector (Xbal-Hindlll) to form the
vector pUC18/lMP-p75.
4.2.8: pUC1811MP-lL16 (see Figure 18)
a. The IL16 gene is amplified from the clone pcDNA3-lLHu1 (M. Baier, Paul Ehrlich
Institute, Germany; Baier et al., 1995; Bannert et a/., 1996) as template with the
primers:
f1 Bsu361for: 5'AGACTGCCTCAGGCCAGCCCGACCTCMCTCC3'
~3Hindlllrev2: 5'ATATATAAGC I I I I AGGAGTCTCCAGCAGC3'
b. The PCR is done following standard protocols (Sambrook et al., 1989). The amplified
product is digested with Bsu361 and Hindlll, then ligated into pre-digested
pUC18/lMP-p75 vector (Bsu361-Hindlll) to form the vector pUC18/lMP-lL16.
4.3: In vivo SIP with co-transformation and polyphage
~ CA 02244838 1998-07-27
W 097/32017 PCT/EP97/00931 37
4.3.1: Combining 2 libraries (Library 1 is fused with glll while Library 2 is fused to the
IMP).
10 ng each of fiun1B, fjun1A, fpep3 1B, fhag1A, ~NGF1B with 500 ng each of
pUC18/lMP-p75, pUC18/lMP-HAG, pUC1811MP-lL16 are co-transformed into DH5a
cells by electroporation. The cells are plated on Luria Broth (LB) (30 ~lg/ml
chloramphenicol + ~ 00 ~g/ml ampicillin) and incubated overnight at ambient
temperature.
The Ampr Cmr colonies are scraped with LB and 1 ml of suspension is used to inoculate
25 ml LB (30 ~lg/ml chloramphenicol + 100 ~g/ml ampicillin + 1 mM IPTG) followed by
incubation overnight at room temperature.
4.3.2: In vivo SIP. The supernatant from the cells is separated by centrifugation (10,000
RPM, 10 min., 4~C). ~ ml of 30% PEG/3M NaCI are added to the supematant and mixed
100 times. After 1 hour on ice, the phage precipitate is collected by centrifugation
(10,000 R~'M, 10 min., 4~C). The pellet is resuspended in 1 ml TBS buffer, and the
suspension is filtered through a 0.45 micron filter (Sartorius).
2~)0 ~LI of phage supernatant are used to infect 1.8ml of log phase K91 cells (or any
ma~e E. coli cells (F-pilus containing)), and the cells are plated on LB (30 ,ug/ml
chloramphenicol + 100 ~lg/ml ampicillin) and incubated overnight at ambient
temperature.
4.3.3: Testing of infectious polyphage DNA patterns and infectity. Twenty individual
pr Cmr colonies are used to inoc~llate 5 ml LB (30 ,ug/ml chloramphenicol + 100
~Lg/ml ampicillin) in each case and incubated at ambient temperature overnight. Plasmid
and RF DNA are isolated from each clone with a Qiagen Miniprep DNA kit. Clones are
analysed by restriction analysis with restriction enzymes Xbal and Hindlll together with
appropriate buffers as supplied and instructed by the manufacturer. The restriction
CA 02244838 1998-07-27
W O 97/32017 PCT/EP97/00931 3~
digests are run in a 0.8% TBE agarose gel at constant voltage of 100V for 1.5 hours.
The restriction patterns, together with the relative intensity of the bands (because the
phage vectors (~un1B, fjun1A, fpep3_1B, fNGF1B, fhag1A) have significantly lowercopy numbers than the plasmid vectors) allow to identify correctly interacting pairs. For
the pair fhag1A+pUC19/lMP-HAG, an Xbal-Hindlll digest will yield a 6.~ kb, 3.3 kb, 1.3
kb, and 0.7 kb fragments, while for the pair fpep3_1 B+pUC1 8/lMP-p75, the same digest
will yield 6.3 kb, 2.8 kb, 1kb, and 0.7kb fragments. A problem though is to distinguish
the potential non-cognate combinations of ijun1B or fiun1A with pUC18/lMP-p7~
bec~use they would give similar patterns as the fpep3_1B+pUC18/lMP-p75. To further
resolve this, the clones containing identical patterns can be re-digested with BamHI-
Hindlll. The Ijun1A or fjun1B in combination with pUC18/lMP-p75 would yield only 4
~ragments - 4.1 kb and 2.9 kb, 2.6 kb, 1.2 kb fragments - while the cognate pairfpep3_1B+pUC18/lMP-p75 will yield 5 fragments - 3.5 kb, 2.9 kb, 2.6 kb, 1.2 kb, 0.~ kb.
To further prove that cognate interacting pairs have been selected, the ability of the
clones to form selectively-infective phage particles is tested. Only clones with a cognate
pair can form infectious phages. The supernatant from the overnight culture of the
individual clones is filtered with a 0.45 micron filter (Sartorius). Ten microliters of phage
supernatant are mixed with 100 ~11 of log phase K91 cells (or any male E. coli cells (F-
pilus containing)) for 10 minutes at 37~C. The suspension is plated on LB (30 llg/mi
chloramphenicol) and incubated overnight at 37~C. The result is shown in Table 3.b.
in s~mmary (see Figure 19), the results from the above example indicate that among 19
clones analyzed, 8119 have the cognate pair fpep3_1B+pUC18/lMP-p7~ and produce
selectively-infective phage; 1/19 has the fhag1A-lpUC19/lMP-HAG combination and
produces selectively-infective pha~e.
Example 5: Combination of Multiple Libraries into a Single Phagemid
Vector through Recombination, Screening via tag system
5.~: Principle (see Figure 20~
CA 02244838 1998-07-27
W O 97/32017 PCT~EP97100931
3q
To be able to retrieve the genetic il,ru""~lion for cognate protein pairs seJected via a tag
fused to one of the partners, two separate libraries in phagemid vectors are constructed
containing the lox recombination promoting sites and recombined on one phagemid by
action of the cre recombinase in an in vivo recombination.
-
5.2: Vector construction
Both loxP and loxP511sites (Hoess etal., 1986) are inserted in tandem into the regionflanked by the ColE1 ori and ~B-Iactamase in vector plNG1-C1, whereas in vector
pONG3-A, the loxP site is cloned upstream of the Xbal site and the loxP511
downstream of the Hindlll site. Therefore, the genomic DNAs to be cloned are flanked
by the loxP and /oxP511 sites.
5.3: Library construction and recombination
Tl~e libraries are prepared as in Example 3. The phagemids in the dou~le-resistant
clones are recombined through the cre recombinase which either is encoded in thephagemid being inducible (Tsurushita et al., 1996~, or is transferred through P1 phage
infection (Rosner, 1972; Waterhouse et al., 1993). Phages are prepared from the
recombined clones by helper phage infection and used to infect new E. coli cells (cre~).
5.4: -Selection
The phage particles are prepared from the CmR clones and subjected to His-tag
selection as in Examples 2 and 3. The sequences encoded in each phagemid, which
now contains members of both libraries, can be determined by sequencing using
primers specific for myc-tag region (library 1 ) and His-tag region (library 2).
Example 6: SlP-based library vs~ library screening via in vitro
recombination of separately constructed libraries into one phage
vector
CA 02244838 1998-07-27
W O 97/32017 PCT~EP97/~0931
6.1: Princ~ple (se~ Figure 21)
To be able to retrieve the genetic information for cognate protein pairs seiected by SIP
interaction in vivo, t~No separate libraries in phage and plasmid vectors are constructed
and recombined by co-ligation in an in vitro recombination.
6.2: Construction of Libraries A and B
Library A encodes 2 mem~ers, namely a single chain Fv antibody against a peptidederived from hemagglutinin (fahag) and the leucine zipper domain derived from the jun
,sc,i,ulion factor (fi,un), both N-terminally fused to the C-terminal domain of glll from
filamentous phage and preceded by the ompA signal sequence ~"~wed by the Flag
epitope.
Library B encodes 3 members on plasmid vectors of the pUC series, namely the
hemagglutinin peptide to which the above ahag antibody binds (pUC19-lMPhag), theleucine zipper domain of the fos transcription factor (pUC1 8-lMPfos) which
heterodimerizes with jun via this domain, and the intracellular domain of the low affinity
nerve growth factor receptor (pUC18-lMPp75), as a negative control which does not
interact with library A members, all fused to the infectivity-mediating N-terminal domains
of phage glll protein, preceded by the 9111 signal sequence.
~ibrary A members are cioned into a ~d phage vector which also contains downstream
of the library A insertion site the N-terminal domains (N1-N2) of glll, followed by the
cloning sites BslWI and Hindlll to allow in-frame insertion of library B members,
Library A construct fahag is identical to the f17/9-hag fd phage vector (Krebber et al,
1995) and serves as basis for construction of fiun The jun leucine zipper together with
amino acids 290 to 326 of the C-terrninal part of 9lll is PCR-amplified (primers FR620
and FR621, containing EcoRV and S~71 sites, respectively) from the construct fJun1B
(containing the jun leucine zipper fused to amino acids 290 to 493 of glll) generated in
Example 4. The resulting PCR fragment is ligated directionally into EcoRV/Sfil-digested
-
CA 02244838 1998-07-27
W Og7~32017 pCTAEP97/00931
f1719-hag vector in frame with amino acids 327 to 493 of the 9111 C-terminal domain
resulting in vector fiunhag (see Figure 22).
Generation of library B constructs pUC19-lMPhag and pUC18-lMPp75 is described inExample 4. To construct pUC18-lMPfos, amino acids 219 to 272 of the N-terminal part
of glll together with the fos leucine zipper are PCR-amplified (primers FR618 and
FR619, containing BsiWI and Hindlll sites, respectively) from the pOK1 phagemid
vector (G~ lihorr et al., 1994). The resulting PCR fragment is ligated directionally
into BsiWI/Hindlll-digested pUC18-lMPp75 to create pUC18-lMPfos (see Figure 17). Primers:
FR61 ~: 5'CGCCGTACGGCGGCTCTGGTGGTGGrrCTGGTGGC3'
FR61 9: 5'CCCAAGCmTAGACTAGCTGACTAGAAGATCTGC3'
FR620: 5'CGCGATATCGTCGACGCCGGTGGTCGGATCGCC3'
FR621: 5'CGCGGCCCCCGAGGCCCCACCACCGGAACCGCCTCCC3'
6~3: P.~,J~.alion and ~e:cc~ .lion of libraryA and B and selection of i"ler~ti-,g
proteiln pairs by SIP
Non-covalent, cognate interactions of ahag antibody with hag peptide ~Krebber ef al.
1995) and of fos and jun leucine zipper domains (Grammatikoff et al., 1994) generates
infective SIP phage~ Thus, from the six possible combinations of members of the model
llbraries A and B (fahag~hag, fahag-fos, fahag-p75, ~un-fos, fjun-hag, fjun-p75), only
two combinations (cognate pairs in bold) should be selected by in vivo SIP. To
recombine the library members in all possiL,le permutations, library A is linearized by
digestion with BsiWI/Hindlll to prepare it for random incorporation of library B members,
prepared by mass-excision with BsiWI/Hindlll from the construct B pool describedabove. After co-ligation of the mass-excised library B fragments into library A vectors,
the sample is transformed into competent E.coli cells, plated onto chloramphenicol-
containing LB agar plates and grown overnight at 37~C The recombined library size can
be determined by plating serial dilutions of the transformation and can be compared to
CA 02244838 1998-07-27
W O 97t32017 PCT/EP97/00931
the complexities of the individual libraries A and B. The total recombined library js
scraped from the plates in LB medium and used to inoculate an appropriate volume of
chloramphenicol-selective LB-medium supplemented with 1 mM IPTG. After growth at30~C overnight with constant shaking to allow production of SIP phages, the bacteria
are pelleted by centrifugation and phages present in the supernatant are precipitated on
ice for one hour by addition of 0.2~ volumes of 20% PEG/2.5 M NaCI. The phages are
pelleted by centrifugation for 30 min at 10 ~00 x 9 and 4~C. The pellet is resuspended in
an appropriate volume of 1 x TBS buffer and filtered through a 0.45 ~M filter. Serial
dilutions of this filtrate are used to infect F E.coli cells. The double-stranded, replicative
forrn phage DNA is prepared from resulting transductant colonies by standard methods
and analy~ed by r~ ;tion digest and sequencing for the presence and identity of
library A and B members. Furthermore, the supernatant of transductant colonies is
analyzed for the presence of infective SIP phages to confirm that protein-protein
interaction of a particular pair selected from the recombined libraries A and B is
responsible for SIP phage infectivity.
Alternatively, the model libraries A (2 members) and B (3 members) are used to
construct all possible combinations (listed above) individually, and equal amounts (50
ng) of each of the 6 combinations can be co-transformed into competent E. coli cells
followed by the steps listed above. The distribution of individual constructs after co-
transformation as well as the distribution of transductants resulting from the model
library can be analyzed as described above. The selective recovery of phage constructs
which co-encode cognate protein pairs der"G~ les the feasibility of SlP-based
selection of binding partners after an appropriate recombination event.
Example 7: 'Spatial' in vivo SIP
7.1: Principle (see Figure 23)
Coupling of information about members of interacting peptides or proteins is achieved
by having a spatial relationship between the particles displaying the sele~table or
-
CA 02244838 1998-07-27
W O97132017 PCTnEP971~931
4-3
screenabie property (in this example phages for the SIP experiment) and the package
containing the genetic information for the individual library members (in this example the
E. coli cell secreting the phage particle being screened), i. e. a correlation between the
phage being examined and the position of the corresponding E coli host on the master
plate.
7.2: Comblnlng 2 libraries (Library A is fused with glll while library B is fused to
the IMP)
10 ng each of fiun1B, fiun1A, fpep3_1B, fhag1A, fNGF1B are co~ n~fc.""ed with 500
ng each of pUC18/lMP-p75, pUC19/lMP-HAG, pUC18/lMP-lL16 into DH5a cells by
electroporation. The tra,,srur,,,ants are plated on LB (30 ~g/ml chloramphenicol + 100
~lg/ml ampicillin) and incubated overnight at ambient temperature.
7.3: Screening of co-tra,.src~r,..z.l.ls by SIP
From the master plate of co-transrunllallLs~ each of the co-l~dnsfo"~,anls are labelled
and inocl ~l~ted separately into 5 ml LB (30 ~Lg/ml clllord~ ~ Iphenicol + 100 ~g/ml
ampicillin) and incubated ove" liyhL at ambient temperature.
Plasmid and RF DNA are isolated from each clones with a Qiagen Miniprep DNA kit.Clones are analysed by restriction analysis with restriction enzymes Xbal and Hindlll
together with appropriate buffers as supplied and instructed by the manufacturer. The
restriction digests are run in a 0.8% TBE agarose gel at constant voltage of 100 V for 1
to 2 hours. Restriction patterns allow disc,i",;,lation of the particular clones.
The supernatant from the overnight culture of the individual clones is filtered with a 0.45
micron filter (Sartorius). Ten microliters of phage supernatant are mixed with 100 1ll of
log phase K91 cells (or any male E. coli cells (F-pilus containing)) for 10 minutes at
37~C. The suspension is plated on LB (30ug/ml chloramphenicol) and incubated
overnight at 37~C.
CA 02244838 1998-07-27
W O 97/32017 PCT~P97/00931
A positive co-transformant (i.e. conlains the correct interacting pair) has a
corresponding correct restridion pattern and is capable of producing infectious phages,
that are incapable of secondary or subsequent infections. Polyphage particles being
capable of such infections, and containing the genetic i.,rur,,,alion of an interacting pair
as well, can readily be identified by their restriction digest pattem.
Example 8: E. coli display
8.1: P,i,.ci~le (see Figure 24~
Two libraries are introduced into E.coli cells, with expressed members of library A (such
as antibody, peptide, or cDNA libraries) being presented at the surface of the cells. In
those cases where interacting pairs are formed, members of library B ~such as antibody,
peptide, or cDNA libraries) are transported in the complex with its cognate partner to the
surface of the cell as well, thus displaying a selectable or screenable property such as a
tag. Selected cells contain the information for both interacting partners.
82: Pr~ rdlion o~ Library A
A thioredoxin peptide library is prepared as fusions to the E. coli flagellin in the pFLlTRX
vector essentially as described (Lu et al., 1995).
8.3: Pre~uaralion of Library B
An cyclic, variable-length peptide library including a FLAG epitope ~Hopp et al., 1988;
Knappik and Pluckthun, 1994) is prepared essentially as described in Example 4.2.4,
and cloned in the pTERM vector, a modified version of the pto2H10a3s vector (Krebber
et al., 1996) containing a chloramphenicol-resistance gene instead of the kanamycin-
resislal-ce gene. The pT~RM vector can be assembled by standard methods startingfrom pto2H10a3s. This cyclic peptide library is packaged by infection with a helper
phage (M13K07 or VCSM13) by standard methods (Sambrook et al., 1989).
CA 02244838 1998-07-27
W 097/32017 PCT~P97J00931
4~
8.4: Combination of Library A and I ibrary B
An ali~uot of the E. coli cells containing Library A is used to inoculate 50 ml LB (100
~lg/ml ampicillin) and incubated at ambient temperature until the OD600 reached 0.4.
The cells are infected with phages containing Library B at a multiplicity of infection (MOI)
of 10. After 30 min of infection, the cells are collected by centrifugation (5000 RPM, 10
minutes, 4~C) and resuspended in 1 ml LB. The suspension is plated on M9 media (~ 1
mM MgCI2, supplemented with 0.5% glucose, 0.2% casamino acids, 100 ,ug/ml
ampicillin, 30 ~Lg/ml chloramphenicol).
8.5~ le ~ tlon of i..ter;~cti.~J pairs
The Ampr Cmr colonies are scraped with M9 media ( l 1 mM MgCI2, supplemented with
0.5% glucose, 0.2% casamino acids, 100 ~lg/ml ampicillin, 30 ~lg/ml chloramphenicol),
and an aliquot of the suspension is used to inoculate 25 ml M9 media (+ 1 mM MgCI2,
supplemented with 0.5% glucose, 0.2% casamino acids, 100 ,ug/ml ampicillin, 30 ~g/ml
chloramphenicol) and incùbated at 37~C until saturation. Selection is performed
essentially as described (Lu et al., 1995), the modification being that the antibody used
for selection is the M1 anti-FLAG antibody (Kodak).
Individual enriched Ampr Cmr colonies are isolated and the sequences of the
corresponding interacting peptide(s) and cyclic peptide(s) are determined by DNAsequencing. To confirm that the encoded peptide and cyclic peptide form a cognate
pair, each of the clones is tested for enrichment based on the selection method
described above, whereby the Ampr Cmr colonies bind to the M1 anti-FLAG antibody in
a single round of selection.
Literature:
Baier et a/., 1995, Nature 378, 563
Bannert et al., 1996, Nature 381, 30
Cardoso and Schwarz, J. Appl. Bacteriol. 72 (1992) 289-293
CA 02244838 1998-07-27
W O97/32017 PCT~EP97/00931
L~6
Crameri and Suter,1993, Gene 137, 69-75
Ge et a/., 1995, Antibody Engineering, 2 ed., 229-266
Ge and Rudolph, BioTechniques 22 (1997) 28-29
Gramatikoff et al., Nucleic Acids Res. 22 (1994) 5761-5762
Hochuli etal., 1988, Bio/7echnology6, 1321-1325
Hoess etal. 1986, Nucleic Acids Res. 14, 2287-2300
Hopp et al., 1988, Bio/Technology 6,1204-1210
Ibanez et al., 1992, Cell 69, 329-341
Knappik and Pllickthun,1994, BioTechniques 17, 754-761
Krebber et al., 1995, FEBS Leffers 377, 227-231
Krebberet~l., 1996, Gene 178, 71-74
Lindner et al., 1992, Methods: A companion fo Methods Enzymol. 4, 41 -56
Lu et al., 1995, Bio/Technology 13, 366-372
Rosner,1972, Virology 48, 679-689
Sambrook ef al.,1989, Molecular Cloning: a Laboratory Manual, 2 ed.
Schmidt and Skerra,1994, J. Chromatogr. A 676, 337-345
Tsurushita et al., 1996, Gene 172, 59-63
Virnek~s et al., 1994, Nucleic Acids Res. 22, 5600-5607
Waterhouse et al., 1993, Nucleic Acids Res. 21, 2265-2266