Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02258973 1999-O1-29
GLUCOSE REGULATED GENE
CROSS REFERENCE TO RELATED APPLICATION
This application claims priority from U.S. application no. 601069,352, which
is incorporated
by reference herein in its entirety.
FIELD OF THE INVENTION
The invention relates to an isolated glucose regulated gene and its protein
expression
product. The invention also relates to methods of modulating the gene for
treatment of
hyperglycemia, glomerulosclerosis and renal cell apoptosis.
BACKGROUND OF THE INVENTION
(i) Renal Failure as a Complication of Diabetes Mellitus
Renal failure caused by glomerulosclerosis is a major complication of insulin
dependent
("IDDM") and non insulin dependent ("NIDDM") diabetes (1, 2). Renal failure is
increasing in
Europe and North America (3-5), due to a variety of factors, including an
aging population, poor
dietary habits, and longer survival of juvenile diabetics. About 25% of
patients undergoing
treatment of end stage renal disease (ESRD) in the US and Canada, have kidney
failure
(nephropathy) caused by diabetes (1 ). Although renal failure in diabetes is
not well understood,
significant advances have been made recently. There still remains a clear need
to characterize
the processes that cause diabetes related kidney failure.
Renal disease occurs more frequently in IDDM than in NIDDM, and there is a
strong
genetic component associated with the former (1). So far, the genes involved
in this disease have
not been identified. There has been some suggestion that certain Angiotensin
Converting
Enzyme (ACE) polymorphisms predispose to development of diabetic nephropathy
(6). Only
about 30-40% of IDDM patients eventually develop ESRD (7). It would be helpful
if genetic factors
that protect the 60-70% majority of IDDM patients from progressive renal
failure could be
identified.
(ii) Hyperglycemia as a Cause of Diabetic Nephropathy - the Role of the
Mesangial Cell
Diabetics have chronically elevated blood glucose levels (hyperglycemia).
Hyperglycemia
contributes to development of microvascular and renal complications. There is
no doubt that
controlling blood sugar reduces these complications (8). Studies show that
diabetic
glomerulosclerosis is caused by expansion of the mesangial matrix (1 ). The
main product of the
mesangial matrix, collagen IV, is found throughout the expanded mesangium.
This is
CA 02258973 1999-O1-29
characteristic of diabetic glomerulosclerosis (16, 17). The mesangial cell is
now considered to be
involved in initiation of diabetic glomerulosclerosis. Current investigation
of renal failure centers
around mesangial cell ("MC") responses to hyperglycemia.
Hyperglycemia either directly (9) or indirectly (10) leads to the increased
production of
growth factors, accumulation of excess extracellular matrix ("ECM")and
creation of advanced
glycosylation end products. These findings have been reproduced and
corroborated in animal
models of diabetes.
There are factors in addition to hyperglycemia that contribute to
glomerulosclerosis and
microvasular changes. As mentioned above, most IDDM patients do not develop
diabetic renal
disease, despite the presence of life long elevated blood sugars.
Hyperglycemia is a necessary, but not sufficient condition for diabetic renal
complications.
Nevertheless, if hyperglycemia could be fully understood at the molecular
level, this would permit
targeted therapeutic intervention to prevent the hyperglycemia-induced
component of diabetic
complications. It would also help identify genes that afford cell protection
or establish cell
vulnerability to sustained, elevated glucose.
Recently, there has been a recognition that diabetic renal disease in the
presence of
hyperglycemia is associated with apoptosis.
(iii) Hyperglycemia-induced Alterations in ECM-MC Signaling
Although a number of different cellular metabolic pathways are known to be
altered by
exposure to elevated concentrations of glucose (17, 18), diacylglycerol
("DAG") induced protein
kinase C ("PKC") activation (especially its ~i2 isoform) is probably the most
important (13, 14, 17-
19). PKC inhibition reverses many of the acute and chronic effects of
hyperglycemia on MC by
blocking DAG binding to PKC (13). The sequence of events described below
occurs in
hyperglycemia. The model is derived from in vitro studies of MC response in
primary culture to
short term hyperglycemic conditions and in vivo investigations of early
changes in renal functional
parameters (increased glomerular filtration rate and urine protein excretion)
in animal models such
as streptozotocin treated rats.
High glucose enhances intracellular production of sorbitol via the aldose
reductase
pathway. This leads to an increase in intracellular osmolality (11). At the
same time (ii) high
glucose increases de novo synthesis of diacylglycerol (DAG) leading to
activation and
phosphorylation of protein kinase C (PKC). This is followed by a series of
"downstream" events,
including increased expression of various growth factors, most notably,
transforming growth factor
2
CA 02258973 1999-O1-29
beta (TGF~i). TGF(3, in an autocrine manner, stimulates MC production of
extracellular matrix
(ECM) elements, fibronectin and collagen IV, while at the same time reducing
ECM degradation by
increasing levels of the metalloproteinase inhibitor TIMP-2 (12). These
effects are prevented by
treatment with anti-TGF(3 antibodies. TGF(3 is critical in accumulation of ACM
following short term
exposure of MC to elevated glucose concentrations.
DAG-induced activation of MC PKC(32 is responsible for the acute and even
certain chronic
changes associated with diabetic microvascular and renal complications (13).
Administration of a
specific PKC~i2 inhibitor-LY333531, appears to prevent the in vivo and in
vitro sequelae of
hyperglycemia, described above (14).
PKC is a serine-threonine phosphorylation kinase. Many different PKC isoforms
exist, and
their specificity of action is attributable to their intracellular
compartmentalization, which varies
from cell to cell. All PKC isoforms contain 2 regulatory domains, C1 and C2,
which bind DAG and
Ca++, respectively, in addition to binding a kinase domain. Under resting
conditions, the kinase
domain is inactive due to its interaction with the C1 domain. When DAG binds
to C1, dissociation
occurs, allowing ATP to bind to the kinase region. This activates PKC. A drug
named LY333531
acts by competing with ATP for binding at the kinase domain. The effect of
this drug is to block
PKC phosphorylation without affecting intracellular DAG levels (14,15).
The PKC pathway is not well understood. Whether PKC activation is the dominant
dysfunction in diabetic glomerulopathy is undetermined. Also unknown is
whether other signaling
pathways stimulated by hyperglycemia are capable of interacting with and
modifying DAG induced
PKC activation.
It would be helpful if DAG activation of PKC (via binding to the C1 domain)
and its
interaction with other metabolic changes in glomeruli and microvasculature
during hyperglycemia
were characterized. This would lead to new treatments to control and prevent
damage to
glomerular and microvascular function caused by hyperglycemia and diabetes.
(iv) Signaling Proteins that Belong to the Same Superfamily as PKC
There has been also been growing interest in the characterization of a novel
class of
signaling proteins that belong to the same superfamily as protein kinase C,
but lack its kinase
activity. Unc-13, one of the members of this family, encodes a phorbol esterl
diacylglycerol-binding
protein in C. elegans. Initial evaluation suggested it had a role in
neurotransmitter release. (20-
23). Mammalian homologues (munc13s), munc13-1, -2, and -3, were originally
cloned from rat
brain and similar to Unc-13 in that both possess DAG and Ca2+ binding domains
(20). Syntaxin,
3
CA 02258973 1999-O1-29
synaptobrevin, SNAP 25 (24) and Doc2 (25) were found to coimmunoprecipitate
with munc13s,
consistent with the suggestion that this new family of DAG binding proteins is
involved in vesicle
trafficking and neurotransmitter release. It would be helpful if the role of
genes in this family was
characterized so that its role in metabolism was understood. No
characterization data to date has
linked this gene to hyperglycemia or kidney failure.
The function of these signaling proteins and related isoforms is largely
unknown.
Nevertheless there is emerging evidence that DAG activated munc13 is involved
in
neurotransmission (24).
In summary, there is recognition that non PKC DAG activated signaling pathways
regulate
important cellular functions. Since hypergelycemia results in increased
intracellular DAG
concentration, there is a need to identify and characterize the targets of DAG
that are involved in
the microvascular and renal complications of diabetes. This would lead to new
compounds and
methods for treatment of these complications.
SUMMARY OF THE INVENTION
We cloned a gene from human MC, Hmunc13, which is up-regulated by
hyperglycemia.
Hmunc13 mediates some of the acute and chronic changes in MC produced by
exposure to
hyperglycemia. These changes result in diabetic microvascular and renal
damage, such as
glomerulosclerosis and apoptosis.
We have established the following:
(a) Structure of Hmunc13 and biologically functional equivalent nucleotide
sequences:
Hmunc13 is a signaling molecule localized to the plasma membrane of renal
mesangial
cells, cortical epithelial cells and other cells, The topological organization
is illustrated
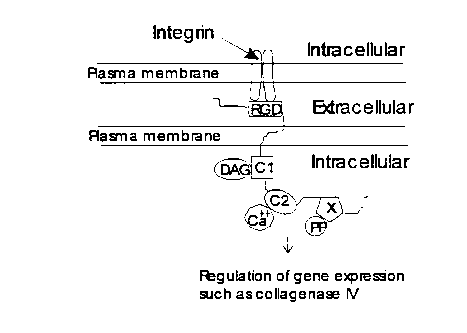
schematically in figure 7. There are functional extracellular RGD domains, and
intracellular
C1 and C2 domains. There is also an intracellular regulatory domain on Hmunc13
that
targets and activates a serine threonine catalytic phosphatase subunit to the
plasma
membrane
(b) Function of Hmunc13 and biologically functional equivalent nucleotide
sequences: The
functional role for Hmunc13 involves intracellular signal transduction and
regulation of cell
attachment and migration. Hmunc13 acts through modulation of phosphatase
activity. In
this way, Hmunc13 phosphatase activation opposes downstream serinelthreonine
phosphorylation initiated in response to PKC and integrin activation.
4
CA 02258973 1999-O1-29
(c) Disease Model & Therapeutic Intervention: Hmunc13 is activated in response
to
hyperglycemia-induced increases in DAG, causing (i) stimulation of phosphatase
activity
and, (ii) modulation of DAG-induced PKC(3 activation. We have identified a
model which
incorporates the two DAG activated pathways: (i) PKC dependent and (ii)
Hmunc13
dependent. These two pathways regulate two opposing cell phenotypes, PKC-
proliferation
and hmunc13-apoptosis. The over-expression of Hmunc13 under hyperglycemic
conditions and Hmunc13 DAG-induced apoptosisprove a role for Hmunc13 in
diabetic
renal cell injury. Modulation of Hmunc13 and biologically functional
equivalent nucleotide
sequences is particularly useful for treatment and prevention of renal cell
damage.
The invention is an isolated nucleotide sequence encoding a glucose regulated
munc
polypeptide. The nucleotide is preferably from a kidney cell, human cortical
epithelial cell or a cell
from testis, ovaries, prostate gland, colon, brain and heart, more preferably
a mesangial cell or a
kidney cortical epithelial cell. The nucleotide sequence preferably comprises
a Hmunc13
polypeptide and all or part of the amino acid sequence in sequence (a) in
Figure 1 [SEQ ID NO. 1].
The nucleotide sequence preferably comprises a Hmunc13 gene having all or part
of the
nucleotide sequence in Figure 8 [SEQ ID NO. 2]. The molecule preferably
comprises at least 40%
sequence identity to all or part of the nucleotide sequence of Figure 8. The
sequence is preferably
selected from a group consisting of mRNA, cDNA, sense DNA, anti-sense DNA,
single-stranded
DNA and double-stranded DNA. The nucleotide encodes an amino acid sequence of
the
invention. The nucleotide sequence that encodes all or part of a Hmunc13
polypeptide, preferably
hybridizes to the nucleotide sequence of all or part of Figure 8 under high
stringency conditions
(e.g. a wash stringency of 0.2X SSC to 2X SSC, 0.1 % SDS, at 65°C).
The invention also includes an isolated munc polypeptide, with the provisio
that the
polypeptide is not found in a mammalian central nervous system. The
polypeptide of preferably
has transmembrane ECM-cell signaling activity and DAG and Ca++ activated
phosphatase activity
and more preferably includes all or part of the Hmunc13 amino acid sequence in
sequence (a) in
Figure 1 [SEQ ID NO: 1]. The invention also includes amimetic of the purified
and isolated
polypeptide. The polypeptide preferably has at least 40% sequence identity to
all or part of the
amino acid sequence (a) in Figure 1 [SEQ ID NO: 1] . The polypeptide is
preferably from a
mammalian kidney cell. It is useful for inducing apoptosis and vesicle
trafficking.
The invention also includes a recombinant DNA comprising a DNA molecule the
invention
and a promoter region, operatively linked so that the promoter enhances
transcription of said DNA
molecule in a host cell. The invention also includes a system for the
expression of Hmunc13,
5
CA 02258973 1999-O1-29
comprising an expression vector and Hmunc13 DNA inserted in the expression
vector. The
expression vector preferably comprises a plasmid or a virus. The invention
also includes a cell
transformed by the expression vector. The invention also includes a method for
expressing
Hmunc13 polypeptide comprising: transforming an expression host with a Hmunc13
DNA
expression vector and culturing the expression host. The method preferably
also includes
isolating Hmunc13 polypeptide. The expression host is preferably selected from
the group
consisting of a plant, plant cell, bacterium, yeast, fungus, protozoa, algae,
animal and animal cell.
The invention also includes a pharmaceutical composition, including at least
all or part of
the polypeptide of the invention, and a pharmaceutically acceptable carrier,
auxiliary or excipient.
The invention also includes a pharmaceutical composition for use in gene
therapy, comprising all
or part of the nucleotide sequence of any of the invention and a
pharmaceutically acceptable
carrier, auxiliary or excipient. The pharmaceutical composition for use in
gene therapy, preferably
comprises all or part of an antisense sequence to all or part of the nucleic
acid sequence in Figure
8
Another embodiment of the invention is a kit for the treatment or detection of
a
disease, disorder or abnormal physical state, comprising all or part of the
nucleotide sequence of
the invention. A kit for the treatment or detection of a disease, disorder or
abnormal physical
state, preferably includes all or part of the polypeptide of the invention.
The invention also includes
a nucleic acid molecule detection kit including, preferably in a suitable
container means or
attached to a surface, a nucleic acid molecule of the invention encoding
Hmunc13, Mmunc13 or a
polypeptide having Hmunc13 or Mmunc13 activity and a detection reagent (such
as a detectable
label). Other variants of kits will be apparent from this description and
teachings in patents such
as U.S. Patent Nos. 5,837,472 and 5,801,233 which are Incorporated by
reference in their entirety.
The kit may also comprise an antibody to the polypeptide. The disorder is
preferably selected
from a group consisting of insulin dependent and independent diabetes,
glomerulopathy and renal
failure. The invention also includes a NH2-SQRSNDEVREFVKL-COOH specific
antibody,
preferably a polyclonal antibody.
The invention is also a method of medical treatment of a disease, disorder or
abnormal
physical state, characterized by excessive Hmunc13 expression, concentration
or activity,
comprising administering a product that reduces or inhibits Hmunc13
polypeptide expression,
concentration or activity. The product is preferably an antisense nucleotide
sequence to all or part
of the nucleotide sequence of Figure 8, the antisense nucleotide sequence
being sufficient to
reduce or inhibit Hmunc13 polypeptide expression. The antisense DNA is
administered in a
pharmaceutical composition comprising a carrier and a vector operably linked
to the antisense
6
CA 02258973 1999-O1-29
DNA.. The disease, disorder or abnormal physical state is preferably selected
from a group
consisting of insulin dependent diabetes and independent diabetes,
glomerulonephritis and
ischemic renal injuries.
The invention also includes a method of medical treatment of a disease,
disorder or
abnormal physical state, characterized by reduced Hmunc13 expression,
concentration or activity,
comprising administering a product that increases Hmunc13 polypeptide
expression,
concentration or activity. The product is preferably a nucleotide sequence
comprising all or part of
the nucleotide sequence of Figure 8, the DNA being sufficient to increase
Hmunc13 polypeptide
expression. The nucleotide sequence is preferably administered in a
pharmaceutical composition
comprising a carrier and a vector operably linked to the nucleotide sequence.
BRIEF DESCRIPTION OF THE DRAWINGS
Preferred embodiments of the invention will be described in relation to the
drawings in
which:
Figure 1. Protein sequence alignment of Hmunc13 [SEQ ID NO: 1] (GenBank
accession number
AF020202) with rat munc13s. (a) Alignment of all four proteins. Only a partial
(AA 251-2207) of
rat munc13-3 is shown. (b) Alignment of the first 100 amino acid at the N-
terminal of Hmunc13
and rat munc13-1. Identical residues are boxed. The dotted line above the
sequence indicates
the C1 domain and the continuous line indicates the C2 domain as proposed by
Brose et al. (7).
Figure 2. Expression of Hmunc13 in human MC culture in 5.5 mM D-glucose plus
9.5 mM L-
glucose (L(15)) or 19.5 mM L-glucose (L) or 15 mM (D(15)) 25 mM mM D-glucose
(D) and as
described in Methods. Increased expression of Hmunc13 after 25 mM D-glucose
treatment is
revealed by Relative RT-PCR (a) and Northern blot (b). All blots are
representative of at least 3
different experiments using different total RNA preparations.
Figure 3. Expression of Hmunc 13 (lane 7, 8) or munc13-2 (lane 9) in human
kidney MC (lane 7),
cortical epithelial cells (lane 8) or rat kidney MC (lane 9). RT-PCR was
performed using a pair of
primers for both Hmunc13 and rat munc13-2 indicated in the Methods which
amplified a segment
of 193 bp. A pair of primers for GAPDH generated a 453 fragment were used to
PCR no RT RNA
(lane 1-3) and RT products (lane 4-6) of human kidney MC (lane 1, 4), cortical
epithelial cells (lane
2, 5) and rat MC (lane 3, 6).
Figure 4. In vitro translation of Hmunc13. Note that a proportion of the
highest MW band (170
kDa) in the absence of microsomal membranes (lane 1) is shifted to higher MW
(180 kDa) in the
7
CA 02258973 1999-O1-29
presence of microsomal membranes (lane 2). Lane 3 is the supernatant of
derived from the in
vitro translation reaction with microsomal membranes as detailed in Methods.
Figure 5. Comparison of gene structure of Hmunc13 to various isoforms of rat
Munc13s.
Figure 6. Expression of rat munc13-2 in renal glomerulus of normal (A) or
streptozotocin-treated
(B) rats detected by in situ hybridization. A PCR fragment of rat munc13-2
(residues 5487-5669)
with a T7 promoter introduced in its sense primer was in vitro transcripted to
anti-sense cRNA with
DIG-labeled UTP. A section of normal and streptozotocin-treated rat kidneys on
the same slide
was hybridized with this probe and the signal was detected by Rodamine-
conjugated anti-DIG
antibody and observed by confocal microscopy. A negative control with sense
cRNA showed little
staining in all sections (data not shown). Note the morphological changes in
the glomerulus of
streptozotocin-treated rat and the higher staining of mesangila cells. This
study also confirms the
expression of munc13-2 in renal tubular epithelial cells.
Figure 7. Structure model of Hmunc13.
Figure 8. DNA sequence of Hmunc13 [SEQ ID NO: 2] (GenBank accession number
AF020202)
Figure 9. (i) Comparison of the structure of rat munc13s and Hmunc13. C1
represents the DAG
binding (C1) domain; C2 represents the Ca2~ binding (C2) domain. (ii)
Comparison of the
sequence of the C1 domain of rat munc13-1 and hmunc13. Continuous lines
indicate identical
amino acids and the dotted line indicates similar amino acids.
Figure 10. (i) Immunoblot of Hmunc13 and the C1 less mutant. Hmunc13-HA
(Hmunc13), C1 less
mutant (C1 less) or empty plasmid, pCMV.SPORT (pCMV), were transiently
transfected into OK
cells. Whole cell lysates were prepared and subjected to 6% SDS-PAGE. The blot
was detected
by anti-HA. Note the slightly decreased molecular weight of the C1 less
mutant. (ii)
Immunostaining of OK cells transiently transfected with hmunc13-HA (A-C, E-G)
and C1 less
mutant (D, H). Cells were stained with anti-HA then probed with anti-mouse IgG-
rhodamine for
detection of Hmunc13 (A-C) and C1 less mutant (D). The Golgi apparatus was
detected by
staining with WGA-FITC (E-H). Slides were observed by confocal microscopy
using a laser
scanning microscope with excitation wavelength at 568 nm for detecting
rhodamine (A-D) and 488
nm for detecting FITC (E-H). Cells were treated with vehicle (A, E), 0.1 NM
PDBu for 3 h (B, D, F,
H), 4 NM nocodazole + PDBu (C, G) as described in the Methods. Negative
controls obtained by
incubating with normal mouse IgG or immunostaining of cells transfected with
empty plasmid
(pCMV~SPORT) yielded very little or no staining (data not shown). Arrowheads
indicate co-
8
CA 02258973 1999-O1-29
localization of anti-HA and WGA staining. Note: Upper and lower panel pairs,
i.e. A and E, B and
F etc, represent anti-HA and WGA-FITC staining, respectively, of identical
fields.
(iii) Immunoblots of whole cell lysates (panel A) and Golgi membrane
preparations (panel B) from
Hmunc13 transfected OK cells with (+) or without (-) PDBu treatment for 3 h.
The whole cell
lysates represent small aliquots of cells for Golgi membrane preparations.
Equal amounts of
protein were loaded onto each lane of panel A or B. The blots were then
detected by anti-HA
antibody.
Figure 11. (i) Double labeling of apoptotic cells and expression of Hmunc13 or
C1 less mutant.
Hmunc13 (A-C, E-G) and C1 less mutant (D, H) transiently transfected cells
were subjected to
TUNEL labeled with fluorescein (E-H) and then subjected to anti-HA and anti-
mouse IgG-
rhodamine labeling for expression of Hmunc13 and C1 less mutant (A-D). Cells
were treated with
vehicle (A, E) or 0.1 p.M PDBu for 8 h (B, D, F, H) or 16 h (C, G). C1 less
mutant transfected cells
treated with vehicle exhibit a similar image as D and H (data not shown).
Negative controls of
TUNEL by incubating cells with labeling mix and no TdT yielded no staining of
fluorescein (data
not shown). Arrowheads indicate representative cells co-stained with anti-HA
(upper panels) and
TUNEL (lower panels) from identical fields. (ii) Graphic representation of the
percentage of
transfected (immunostaining positive) and apoptotic (TUNEL positive) cells in
Hmunc13 or C1 less
mutant (C1 less) transfected cells treated with or without PDBu for 8 or 16 h.
Cell numbers were
counted with an average of three low power views under the confocal
microscope. Bars are
representations of means ~ SD of three experiments.
Figure 12. Genomic DNA breakdown in Hmunc13 transfected cells by PDBu
treatment. Genomic
DNA obtained from empty plasmid (pCMV), Hmunc13 or C1 less mutant transfected
cells treated
with vehicle (-) or 0.1 p,M PDBu for 8 h or 16 h was subjected to 2 % agarose
gel electrophoresis.
Molecular size marker (M) is shown.
Figure 13. Expression of rat munc13-1 in kidney of normal (A) or STZ-treated
diabetic (B-D) rat
detected by in situ hybridization. Outer cortex (A, B), medulla (C) and a
higher power view of outer
cortex (D) from diabetic rat kidney are shown. Similar to diabetic rats,
staining in the renal medulla
for normal rat kidney is less than the cortex (data not shown). Note the
increased expression of
munc13-1 in the tubular epithelial cells as well as in certain glomerular
cells. Negative controls
with sense cRNA showed little staining in both normal and diabetic rat
sections (data not shown).
9
CA 02258973 1999-O1-29
Figure 14. Expression of munc13-1, munc13-2 and munc13-3 in the renal cortex
of the normal rat
and following 1 day (1d) and 11 day (11d) of hyperglycemia in STZ-treated
rats. 18S ribosome
RNA (18S) served as a housekeeping gene.
Figure 15. Schematic representation of DAG activated branched signaling
pathways involving
PKC and Hmunc13. DAG levels are increased by such factors as hyperglycemia,
phospholipase
C (PLC) pJy and phospholipase D (PLD) resulting in activation of both PKC and
Hmunc13 and
leading to two separate downstream signaling pathways, respectively resulting
in proliferation and
differentiation (PKC) or apoptosis (Hmunc13).
Figure 16. Sequence of mouse munc13 cDNA [SEQ ID N0:3) and its corresponding
translated
polypeptide sequence [SEQ ID N0:4].
Figure 17. Comparison of mouse and human munc13 protein sequence.
DETAILED DESCRIPTION OF THE INVENTION
Isolation and Identification of Hmunc13
We cloned a human munc13 gene (Hmunc13) and protein from kidney which has an
important role in cell signaling. This gene is regulated by glucose. Hmunc13
contributes to
the renal and microvascular complications associated with hyperglycemia in
diabetes mellitus,
through a variety of mechanisms including Hmunc13 linked apoptosis. We also
have identified
biologically functional equivalent nucleotide sequences and proteins.
We obtained the glucose regulated gene by differential display reverse
transcription
polymerase chain reaction (DDPT-PCR) of candidate genes differentially
expressed in human
MC exposed to hyperglycemic conditions, compared to controls. Using this
screening
procedure, we obtained a PCR product which was then used to clone the full
length cDNA.
This gene is similar to mammalian brain munc13s (it is a differentially
spliced isoform, munc
13-1 and munc 13-2). Hmunc13 is detectable in both MC, epithelial and other
cells. The
presence of a Hmunc13 gene in MC which has similarity to rat munc13 was very
unexpected
because rat munc13 is believed to be localized only in the brain (20).
We determined that Hmunc13 is a target for regulation by glucose in MC and
other
cells. For example, the expression of Hmunc13 is up-regulated by hyperglycemia
in cultured
kidney MC and epithelial cells. Hmunc13 protein is involved in the acute and
chronic effects of
hyperglycemia in MC and renal epithelial cells, and contributes to the
development of diabetic
glomerulopathy. Hmunc13 also interacts with the syntaxins. )
CA 02258973 1999-O1-29
We then used a full length cDNA clone of rat munc13-1 (a gene from rat brain
with
sequence similarity to Hmunc 13 and some similar functional domains) to show
how the gene
is regulated by glucose. In vitro experiments revealed that exposure of
fibroblasts transfected
with munc13-1 to phorbol esters caused translocation of munc-13-1 to the
plasma membrane.
We performed other in vitro experiments to show that, as a second messenger,
DAG can
activate either a PKC (proliferative) signaling pathway or alternatively, a
Hmunc13 (apoptosis)
signaling pathway. The combined action of these two pathways showed the
functional
responses of cells to stimuli such as hyperglycemia. Our results indicate that
hyperglycemic
activation of Hmunc13 and induction of apoptosis is a factor causing cell
injury in diabetic
nephropathy.
Localization of Hmunc13
We demonstrated the presence of Hmunc13 in primary cultured human MC and in a
human kidney cDNA library as well as munc13-2 in rat MC. A gene similar to
munc13s has never
previously been isolated outside the central nervous system. We also confirmed
that Hmunc13 is
expressed in the brain by PCR of a commercial human brain cDNA library (Gibco
BRL) In vitro
translation also indicates co-translational modification of Hmunc13. It is
unlikely that this initiates
N-glycosylation since addition of a competitive inhibitor of N-glycosylation,
Ac-Asn-Tyr-Thr-NH2
(26), did not shift the band to lower molecular weight.
11
CA 02258973 1999-O1-29
Hmunc13 Protein Three Dimensional Structure
Analysis of the hydropathy plot of Hmunc13 by Kyte-Doolittle analysis
indicates that there
are a few hydrophobic regions (residue 603-609, 817-825, 970-977, 1107-1111 )
with K-D values
from 139 to 172. However, these are not typical transmembrane segments. It is
possible that the
full-length protein can fold in such a way that hydrophobic loops can anchor
to the membrane but
that such folding is not possible for the partial length protein.
Functional Domains of Hmunc13 Protein
We reviewed the Hmuncl3 sequence and compared different segments of Hmunc13
with
other amino acid sequences.
Hmunc13 contains 1 C1 domain and 3 C2 domains. The N-terminal segment is more
similar to rat munc13-1 and the C-terminal segment is more similar to rat
munc13-2 which
contains 1 C1 and 2 C2 domains. After further analysis of the Hmunc13
nucleotide sequence, we
found that another AUG codon (residue 444-446) after the first C2 domain
contains an optimal
Kozak sequence (5'-CACCAUGG-3') (27). It is possible that Hmunc13 mRNA serves
as a
bifunctional mRNA (27) that encodes two open reading frames, one for an
isoform with 3 C2
domains (munc13-1 ) and the other with only 2 C2 domains (munc13-2).
We discovered that, in addition to C1 and C2 domains (fig.5), a segment of
Hmunc13 (aa
309-371 ) not present in rat munc13s, has similarity to a segment of the delta
isoform of the B'
subunit of protein phosphatase 2Ao - a serine threonine phosphatase (28). This
B' subunit has
been shown to be a regulatory subunit of the multimeric PP2Ao. The catalytic
subunit of PP2Ao
associates with specific proteins (B') that serve a targeting and regulatory
function. It is the
regulatory subunits that determine in vivo specificity of the phosphatase by
targeting the enzyme
to the subcellular location of their substrates, and also modulating
phosphatase activity by
reversible protein phosphorylation and binding of second messengers (29).
We have also identified two RGD binding domains at aa39-41 and 769-771 in
Hmunc13.
The presence of these motifs indicates that Hmunc13 interacts with ECM element
receptors-
integrins, such as vitronectin recetpor a~~ and fibronectin receptor a5(3~.
Such interaction is
important for cell survival. Over-expression of Hmunc13, in response to DAG
prevents
engagement of integrins to ECM resulting in apoptosis.
Taken together, the structural features of Hmunc13 described above, show a
multifunctional role that involves transmembrane ECM-cell signaling, as well
as DAG and Ca++
activated phosphatase activity.
12
CA 02258973 1999-O1-29
Our finding that MC Hmunc13 is regulated by glucose also indicates that it
modulates renal
cell responses to hyperglycemia either directly or through interaction with
PKC. We have also
confirmed that Hmunc 13 is upregulated in the streptozotocin treated diabetic
rat compared to
normal rats (Fig. 6). Thus Hmunc13 is implicated in the pathogenesis of
diabetic nephropathy.
Biologically Functional Equivalent Nucleotide Sequences
The invention also includes nucleotide sequences that are biologically
functional
equivalents of all or part of the sequence in Figure 8. Biologically
functional equivalent nucleotide
sequences are DNA and RNA (such as genomic DNA, cDNA, synthetic DNA, and mRNA
nucleotide sequences), that encode peptides, polypeptides, and proteins having
the same or
similar Hmunc13 activity as all or part of the Hmunc13 protein shown in Figure
1. Biologically
functional equivalent nucleotide sequences can encode peptides, polypeptides,
and proteins that
contain a region having sequence identity to a region of a Hmunc13 protein or
more preferably to
the entire Hmunc 13 protein.
Identity is calculated according to methods known in the art. The Gap program,
described
below, is most preferred. For example, if a nucleotide sequence (called
"Sequence A") has 90%
identity to a portion of the nucleotide sequence in Figure 8, then Sequence A
will be identical to
the referenced portion of the nucleotide sequence in Figure 8, except that
Sequence A may
include up to 10 point mutations, such as deletions or substitutions with
other nucleotides, per
each 100 amino acids of the referenced portion of the nucleotide sequence in
Figure 8.
Nucleotide sequences biologically functional equivalent to the Hmunc13
sequences can occur in a
variety of forms as described below.
A) Nucleotide sequences Encoding Conservative Amino Acid Changes in Hmunc13
Protein
The invention includes biologically functional equivalent nucleotide sequences
that encode
conservative amino acid changes within a Hmunc13 amino acid sequence and
produce silent
amino acid changes in Hmunc13.
B) Nucleotide Sequences Encoding Non-Conservative Amino Acid Substitutions,
Additions or
Deletions in Hmunc13 Protein
The invention includes biologically functional equivalent nucleotide sequence
that made
non conservative amino acid changes within the Hmunc 13 amino acid sequence to
the
sequences in Figure 8. Biologically functional equivalent nucleotide sequences
are DNA and RNA
that encode peptides, polypeptides, and proteins having non-conservative amino
acid substitutions
(preferably substitution of a chemically similar amino acid), additions, or
deletions but which also
13
CA 02258973 1999-O1-29
retain the same or similar Hmunc13 activity as all or part of the Hmunc13
protein shown in Figure
1 or disclosed in the application. The DNA or RNA can encode fragments or
variants of the
Hmunc13 of the invention. The Hmunc13 or Hmunc13 -like activity of such
fragments and variants
is identified by assays as described above. Fragments and variants of Hmunc13
encompassed by
the present invention should preferably have at least about 40%, 60%, 80% or
95% sequence
identity or preferably at least about 96%, at least about 97%, at least about
98% or at least about
99% sequence identity to the naturally occurring nucleotide sequence, or
corresponding region.
Most preferably, the fragments have at least 99.5% sequence identity to the
naturally occurring
nucleotide sequence, or corresponding region. Sequence identity (also known as
homology) is
preferably measured with the Gap program.
Nucleotide sequences biologically functionally equivalent to the Hmunc13 in
Figure 8 include:
(1 ) Altered DNA. For example, the sequence shown in Figure 8 may have its
length altered by
natural or artificial mutations such as partial nucleotide insertion or
deletion, so that when the
entire length of the coding sequence within Figure 8, is taken as 100%, the
biologically functional
equivalent nucleotide sequence preferably has a length of about 60-120%
thereof, more preferably
about 80-110% thereof. Fragments may be less than 60%.; or
(2) Nucleotide sequences containing partial (usually 80% or less, preferably
60% or less, more
preferably 40% or less of the entire length) natural or artificial mutations
so that some codons in
these sequences code for different amino acids, but wherein the resulting
protein retains the same
or similar Hmunc13 activity as that of a naturally occurring Hmunc13 protein.
The mutated DNAs
created in this manner should preferably encode a protein having at least
about 40%, preferably at
least about 60%, at least about 80%, and more preferably at least about 90% or
95%, and most
preferably 97%, 98% or 99% sequence identity (homology) to the amino acid
sequence of the
Hmunc13 protein in Figure 1. Sequence identity can preferably be assessed by
the Gap program.
C) Genetically Degenerate Nucleotide Sequences
Since the genetic code is degenerate, those skilled in the art will recognize
that the nucleic
acid sequence in Figure 8 is not the only sequences which may code for a
protein having
Hmunc13 activity. This invention includes nucleic acid sequences that have the
same essential
genetic information as the nucleotide sequence described in Figure 8.
Nucleotide sequences
(including RNA) having one or more nucleic acid changes compared to the
sequences described
in this application and which result in production of a polypeptide shown in
Sequence (a) in Figure
1 are within the scope of the invention.
14
CA 02258973 1999-O1-29
D) Biologically Functional Equivalent Nucleic Acid Sequences Detected by
Hybridization
Other biologically functional equivalent forms of Hmunc13 -encoding nucleic
acids can be
isolated using conventional DNA-DNA or DNA-RNA hybridization techniques. Thus,
the present
invention also includes nucleotide sequences that hybridize to one or more of
the sequences in
Figure 8 or its complementary sequence, and that encode expression for
peptides, polypeptides,
and proteins exhibiting the same or similar activity as that of the Hmunc13
protein produced by the
DNA in Figure 8 or its variants. Such nucleotide sequences preferably
hybridize to one or more of
the sequences in Figure 8 under moderate to high stringency conditions (see
Sambrook et al.
(1989) Molecular Cloning: A Laboratory Manual, Second Edition, Cold Spring
Harbor Laboratory
Press, Cold Spring Harbor, N.Y.). Preferable hybridization conditions are high
stringency, such as
42°C for a 20- to 30-mer oligonucleotide, 65°C for a 200-500 by
DNA probe or 70°C for a 200-400
by cRNA probe.
The present invention also encompasses nucleotide sequences that hybridize to
genomic
DNA, cDNA, or synthetic DNA molecules that encode the amino acid sequence of
the Hmunc13
protein, or genetically degenerate forms thereof due to the degeneracy of the
genetic code, under
salt and temperature conditions equivalent to those described in this
application, and that code on
expression for a peptide, polypeptide, or protein that has the same or similar
activity as that of the
Hmunc13 protein.
A nucleotide sequence described above is considered to possess a biological
function
substantially equivalent to that of the Hmunc13 genes of the present invention
if the protein
produced by the nucleotide sequence displays the following characteristics (i)
DAG activated
transloaction of the protein in vivo from the cytosol to Golgi (as measured by
immunocytochemistry, described in the Materials and Methods section), and (ii)
the protein
activates apoptosis (if the protein is expressed in vivo, the protein's
expression is preferably
induced by DAG).
Production of Hmunc13 in Eukaryotic and Prokaryotic Cells
The nucleotide sequences (also referred to as a DNA sequence or a nucleic acid
molecule;
these terms include either a full gene or a gene fragment.. It will be clear
to a person skilled in the
art whether it is appropriate to use a nucleotide fragment that includes all
or a fragment of a gene
when practicing the invention) of the invention may be obtained from a cDNA
library. The
nucleotide molecules can also be obtained from other sources known in the art
such as expressed
sequence tag analysis or in vitro synthesis. The DNA described in this
application (including
CA 02258973 1999-O1-29
variants that are biologically functional equivalents) can be introduced into
and expressed in a
variety of eukaryotic and prokaryotic host cells. A recombinant nucleotide
sequence for the
Hmunc13 contains suitable operatively linked transcriptional or translational
regulatory elements.
Suitable regulatory elements are derived from a variety of sources, and they
may be readily
selected by one with ordinary skill in the art (Sambrook, J, Fritsch, E.E. &
Maniatis, T. (1989).
Molecular Cloning: A laboratory manual. Cold Spring Harbor Laboratory Press.
New York;
Ausubel et al. (1989) Current Protocols in Molecular Biology, John Wiley &
Sons, Inc.). For
example, if one were to upregulate the expression of the gene, one could
insert the sense
sequence and the appropriate promoter into the vector. Promoters can be
inducible or
constitutive, environmentally - or developmentally-regulated, or cell - or
tissue-specific.
Transcription is enhanced with promoters known in the art such as CMV, RSV and
SV40.
If one were to downregulate the expression of the gene, one could insert the
antisense
sequence and the appropriate promoter into the vehicle. The nucleotide
sequence may be either
isolated from a native source (in sense or antisense orientations),
synthesized, or it may be a
mutated native or synthetic sequence or a combination of these.
Examples of regulatory elements include a transcriptional promoter and
enhancer or RNA
polymerase binding sequence, a ribosomal binding sequence, including a
translation initiation signal.
Additionally, depending on the vector employed, other genetic elements, such
as selectable
markers, may be incorporated into the recombinant molecule. Other regulatory
regions that may be
used include an enhancer domain and a termination region. The regulatory
elements may be from
animal, plant, yeast, bacterial, fungal, viral, avian, insect or other
sources, including synthetically
produced elements and mutated elements.
In addition to using the expression vectors described above, the polypeptide
may be
expressed by inserting a recombinant nucleotide sequence in a known expression
system derived
from bacteria, viruses, yeast, mammals, insects, fungi or birds. The
recombinant molecule may be
introduced into the cells by techniques such as Agrobacterium tumefaciens-
mediated
transformation, particle-bombardment-mediated transformation, direct uptake,
microinjection,
coprecipitation, transfection and electroporation depending on the cell type.
Retroviral vectors,
adenoviral vectors, DNA virus vectors and liposomes may be used. Suitable
constructs are inserted
in an expression vector, which may also include markers for selection of
transformed cells. The
construct may be inserted at a site created by restriction enzymes.
In one embodiment of the invention, a cell is transfected with a nucleotide
sequence of the
invention inserted in an expression vector to produce cells expressing the
nucleotide sequence.
16
CA 02258973 1999-O1-29
Another embodiment of the invention relates to a method of transfecting a cell
with a
nucleotide sequence of the invention, inserted in an expression vector to
produce a cell
expressing the Hmunc13 protein. The invention also relates to a method of
expressing the
polypeptides of the invention in a cell.
Probes
The invention also includes oligonucleotide probes made from the cloned
Hmunc13
nucleotide sequences described in this application or other nucleotide
sequences of the invention.
The probes may be 15 to 30 nucleotides in length and are preferably at least
30 or more
nucleotides. A preferred probe is 5'-CCTCTCCATTGTGTTCATCACCAC-3' or at least
15
nucleotides of this sequence. The invention also includes at least 30
consecutive nucleotides of
Hmunc13 in Figure 8. The probes are useful to identify nucleic acids encoding
Hmunc13 peptides,
polypeptides and proteins other than those described in the application, as
well as peptides,
polypeptides, and proteins biologically functionally equivalent to Hmunc13.
The oligonucleotide
probes are capable of hybridizing to one or more of the sequences shown in
Figure 8 or the other
sequences of the invention under stringent hybridization conditions. A
nucleotide sequence
encoding a polypeptide of the invention may be isolated from other organisms
by screening a
library under moderate to high stringency hybridisation conditions with a
labeled probe. The
activity of the polypeptide encoded by the nucleotide sequence is assessed by
cloning and
expression of the DNA. After the expression product is isolated the
polypeptide is assayed for
Hmunc13 activity as described in this application.
Biologically functional equivalent Hmunc13 nucleotide sequences from other
cells, or
equivalent Hmunc13 -encoding cDNAs or synthetic DNAs, can also be isolated by
amplification
using Polymerase Chain Reaction (PCR) methods. Oligonucleotide primers,
including degenerate
primers, based on the amino acid sequence of the sequences in Figures 8 can be
prepared and
used in conjunction with PCR technology employing reverse transcriptase (E. S.
Kawasaki (1990),
In Innis et al., Eds., PCR Protocols, Academic Press, San Diego, Chapter 3, p.
21) to amplify
biologically functional equivalent DNAs from genomic or cDNA libraries of
other organisms.
Alternatively, the oligonucleotides, including degenerate nucleotides, can be
used as
probes to screen cDNA libraries.
Biologically Functionally Equivalent Peptides, Polypeptides, and Proteins
The present invention includes not only the polypeptides encoded by sequences
presented
in this application, but also "biologically functional equivalent peptides,
polypeptides and proteins"
17
CA 02258973 1999-O1-29
that exhibit the same or similar Hmunc13 protein activity as proteins
described in this application.
The phrase "biologically functional equivalent peptides, polypeptides, and
proteins" denotes
peptides, polypeptides, and proteins that exhibit the same or similar Hmunc 13
protein activity
when assayed. Where only one or two of the terms peptides, polypeptides and
proteins is referred
to below, it will be clear to one skilled in the art whether the other types
of amino acid sequence
also would be useful. By "the same or similar Hmunc13 protein activity" is
meant the ability to
perform the same or similar function as the protein produced by Hmunc13. These
peptides,
polypeptides, and proteins can contain a region or moiety exhibiting sequence
identity (homology)
to a corresponding region or moiety of the Hmunc13 protein described in the
application, but this is
not required as long as they exhibit the same or similar Hmunc13 activity.
Identity refers to the
similarity of two polypeptides or proteins (or nucleotide sequences) that are
aligned so that the
highest order match is obtained. Identity is calculated according to methods
known in the art,
such as the Gap program, described below. For example, if a polypeptide
(called "Sequence A")
has 90% identity to a portion of the polypeptide in sequence (a) in Figure 1,
then Sequence A will
be identical to the referenced portion of the polypeptide in sequence (a) in
Figure 1, except that
Sequence A may include up to 10 point mutations, such as deletions or
substitutions with other
amino acids, per each 100 amino acids of the referenced portion of the
polypeptide in sequence
(a) in Figure 1. Peptides, polypeptides, and proteins biologically functional
equivalent to the
Hmunc13 proteins can occur in a variety of forms as described below.
A) Conservative Amino Acid Changes in Hmunc13 Sequences
Peptides, polypeptides, and proteins biologically functionally equivalent to
Hmunc13
protein include amino acid sequences containing amino acid changes in the
Hmunc13 sequence.
The biologically functional equivalent peptides, polypeptides, and proteins
have at least about 40%
sequence identity (homology), preferably at least about 60%, at least about
75%, at least about
80%, at least about 90% or at least about 95% sequence identity, to the
naturally occurring
polypeptide, or corresponding region. Most preferably, the biologically
functional equivalent
peptides, polypeptides, and proteins have at least 97%, 98% or 99% sequence
identity to the
naturally occurring protein, or corresponding region or moiety. "Sequence
identity" is preferably
determined by the Gap program. The algorithm of Needleman and Wunsch (1970 J
Mol. Biol.
48:443-453) is used in the Gap program. BestFit is also used to measure
sequence identity. It
aligns the best segment of similarity between two sequences. Alignments are
made using the
local homology algorithm of Smith and Waterman (1981) Adv. Appl. Math. 2:482-
489.
18
CA 02258973 1999-O1-29
B) Fragments and Variants of Hmunc13 Proteins
The invention includes peptides, polypeptides or proteins which retain the
same or similar
activity as all or part of Hmunc13. Such peptides preferably consist of at
least 5 amino acids. In
preferred embodiments, they may consist of 6 to 10, 11 to 15, 16 to 25 or 26
to 50, 50 to 150, 150
to 250, 250 to 500 or 500 to 750 amino acids of the Hmunc13. Fragments of the
Hmunc13 protein
can be created by deleting one or more amino acids from the N-terminus, C-
terminus or an
internal region of the protein (or combinations of these), so long as such
fragments retain the
same or similar Hmunc13 activity as all or part of the Hmunc13 protein
disclosed in the
application. These fragments can be natural mutants of the Hmunc13, or can be
produced by
restriction nuclease treatment of an encoding nucleotide sequence. Fragments
of the polypeptide
may be used in an assay to identify compounds that bind the polypeptide.
Methods known in the
art may be used to identify agonists and antagonists of the fragments.
Variants of the Hmunc13 protein may also be created by splicing. Variants can
also be
naturally occurring mutants of the Hmunc13 disclosed in the application. A
combination of
techniques known in the art may be used to substitute, delete or add amino
acids. For example, a
hydrophobic residue such as methionine can be substituted for another
hydrophobic residue such
as alanine. An alanine residue may be substituted with a more hydrophobic
residue such as
leucine, valine or isoleucine. An aromatic residue such as phenylalanine may
be substituted for
tyrosine. An acidic, negatively charged amino acid such as aspartic acid may
be substituted for
glutamic acid. A positively charged amino acid such as lysine may be
substituted for another
positively charged amino acid such as arginine. Modifications of the proteins
of the invention may
also be made by treating a polypeptide of the invention with an agent that
chemically alters a side
group, for example, by converting a hydrogen group to another group such as a
hydroxy or amino
group.
Peptides having one or more D-amino acids are contemplated within the
invention. Also
contemplated are peptides where one or more amino acids are acetylated at the
N-terminus.
Those skilled in the art recognize that a variety of techniques are available
for constructing peptide
mimetics (i.e. a modified peptide or polypeptide or protein) with the same or
similar desired
biological activity as the corresponding protein of the invention but with
more favorable activity
than the protein with respect to characteristics such as solubility,
stability, andlor susceptibility to
hydrolysis and proteolysis. See for example, Morgan and Gainor, Ann. Rep. Med.
Chem., 24:243-
252 (1989).
19
CA 02258973 1999-O1-29
The invention also includes hybrid genes and peptides, for example where a
nucleotide
sequence from the gene of the invention is combined with another nucleotide
sequence to
produce a fusion peptide. For example a nucleotide domain from a molecule of
interest may be
ligated to all or part of a Hmunc13 nucleotide sequence encoding Hmunc13
protein described in
this application. Fusion genes and peptides can also be chemically synthesized
or produced
using other known techniques.
The variants preferably retain the same or similar Hmunc13 activity as the
naturally
occurring Hmunc13 of the invention. The Hmunc13 activity of such variants can
be assayed by
techniques described in this application and known in the art of TUNEL and DNA
fragmentation
assay.
Variants produced by combinations of the techniques described above but which
retain the
same or similar Hmunc13 activity as naturally occurring Hmunc13 are also
included in the
invention (for example, combinations of amino acid additions, deletions, and
substitutions).
Fragments and variants of Hmunc13 encompassed by the present invention
preferably
have at least about 40% sequence identity, preferably at least about 60%, at
least about 75%, at
least about 80%, at least about 90% or at least about 95% sequence identity,
to the naturally
occurring protein, or corresponding region or moiety. Most preferably, the
fragments have at least
97%, 98% or 99% sequence identity to the naturally occurring polypeptide, or
corresponding
region. Sequence identity is preferably measured with either the Gap or
BestFit programs.
The invention also includes fragments of the polypeptides of the invention
which do not
retain the same or similar activity as the polypeptides but which can be used
as a research tool to
characterize the polypeptides of the invention.
Enhancement of Hmunc13 protein activity
The activity of the Hmunc13 protein is increased by carrying out selective
site-directed
mutagenesis. Using protein modelling and other prediction methods, we
characterize the binding
domain and other critical amino acid residues in the protein that are
candidates for mutation,
insertion and/or deletion. A DNA plasmid or expression vector containing the
Hmunc13 gene or a
nucleotide sequence having sequence identity is preferably used for these
studies using the
U.S.E. (Unique site elimination) mutagenesis kit from Pharmacia Biotech or
other similar
mutagenesis kits that are commercially available. Once the mutation is carried
out and confirmed
by DNA sequence analysis, the mutant protein is expressed using an expression
system and its
activity is monitored. This approach is useful not only to enhance activity,
but also to engineer
CA 02258973 1999-O1-29
some functional domains for other properties useful in the purification or
application of the proteins
or the addition of other biological functions. It is also possible to
synthesize a DNA fragment
based on the sequence of the proteins that encodes smaller proteins that
retain activity and are
easier to express. It is also possible to modify the expression of the cDNA so
that it is induced
under environmental conditions other than hyperglycemia or in response to
different chemical
inducers or hormones. It is also possible to modify the DNA sequence so that
the protein is
targeted to a different location. All these modifications of the DNA sequences
presented in this
application and the proteins produced by the modified sequences are
encompassed by the
present invention.
Pharmaceutical Compositions
Hmunc13 or its protein and biologically functional equivalent nucleotide
sequences or
proteins are also useful when combined with a carrier in a pharmaceutical
composition. Suitable
examples of vectors for Hmunc13 are described above. The compositions are
useful when
administered in methods of medical treatment of a disease, disorder or
abnormal physical state
characterized by insufficient Hmunc13 expression or inadequate levels or
activity of Hmunc13
protein. The invention also includes methods of medical treatment of a
disease, disorder or
abnormal physical state characterized by excessive Hmunc13 expression or
levels of activity of
Hmunc13 protein, for example by administering a pharmaceutical composition
comprising
including a carrier and a vector that expresses Hmunc13 antisense DNA.
The pharmaceutical compositions of this invention used to treat patients
having degenerative
diseases, disorders or abnormal physical states of tissue such as renal and
vascular tissue. There
is evidence that apoptosis plays a role in renal diseases related to (1)
glomerular inflammation (2)
tubular ischemia, toxins and ureteric obstruction (E.G. Neilson and W.G.
Couser, lmmunologic
Renal Disease, (1997, 309-329), 8), could include an acceptable carrier,
auxiliary or excipient. In
some diseases, apoptosis is protective. In other cases, apoptosis may
contribute to cell injury.
Regulation of apoptosis plays a critical role in many different renal disease
states including both
glomerular and tubulointerstitial types of injury. The conditions which may be
treated by the
compositions include microvascular and renal complications of diabetes and
disorders in which
renal apoptosis plays a role.
The pharmaceutical compositions can be administered to humans or animals by
methods
such as aerosol administration, intratracheal instillation and intravenous
injection. Dosages to be
administered depend on patient needs, on the desired effect and on the chosen
route of
administration. Nucleotide sequences and proteins may be introduced into cells
using in vivo delivery
21
CA 02258973 1999-O1-29
vehicles such as liposomes. They may also be introduced into these cells using
physical techniques
such as microinjection and electroporation or chemical methods such as
coprecipitation and
incorporation of DNA into liposomes.
The pharmaceutical compositions can be prepared by known methods for the
preparation of
pharmaceutically acceptable compositions which can be administered to
patients, and such that an
effective quantity of the nucleotide sequence or protein is combined in a
mixture with a
pharmaceutically acceptable vehicle. Suitable vehicles are described, for
example in Remington's
Pharmaceutical Sciences (Remington's Pharmaceutical Sciences, Mack Publishing
Company,
Easton, Pa., USA).
On this basis, the pharmaceutical compositions could include an active
compound or
substance, such as a Hmunc13 gene or protein, in association with one or more
pharmaceutically
acceptable vehicles or diluents, and contained in buffered solutions with a
suitable pH and isoosmotic
with the physiological fluids. The methods of combining the active molecules
with the vehicles or
combining them with diluents is well known to those skilled in the art. The
composition could include
a targeting agent for the transport of the active compound to specified sites
within tissue.
Heterologous overexpression of Hmunc13 as a Research Tool
Expression vectors are useful to provide high levels of protein expression.
Cell cultures
transformed with the nucleotide sequences of the invention are useful as
research tools. Cell
cultures are used in overexpression and research according to numerous
techniques known in the
art. A cell line (either an immortalized cell culture or a primary cell
culture) may be transfected
with a vector containing a Hmunc13 nucleotide sequence (or variants) to
measure levels of
expression of the nucleotide sequence and the activity of the nucleotide
sequence. A polypeptide
of the invention may be used in an assay to identify compounds that bind the
polypeptide.
Methods known in the art may be used to identify agonists and antagonists of
the polypeptides.
One may obtain cells that do not express Hmunc13 and use them in experiments
to assess
Hmunc13 gene expression. Experimental groups of cells may be transfected with
vectors
containing different types of Hmunc13 genes (or genes similar to Hmunc13 or
fragments of
Hmunc13 gene) to assess the levels of protein produced, its functionality and
the phenotype of the
cells produced. The polypeptides are also useful for in vitro analysis of
Hmunc13 activity. For
example, the protein produced can be used for microscopy or X-ray
crystallography studies.
Other expression systems can also be utilized to overexpress the Hmunc13 in
recombinant
systems.
22
CA 02258973 1999-O1-29
Hmunc13 is a useful research tool. For example, in one embodiment, Hmunc13
cDNA is
expressed after it is inserted in a mammalian cell expression plasmid
(pCMV~SPORT, Gibco BRL).
In a variation, Hmunc13 cDNA is inserted in an inducible mammalian cell
expression plasmid
(pIND, Invitrogen). Hmunc13 cDNA may also be positioned in reverse orientation
in pIND as a
negative control. One can also use N-terminal c-myc tag and C-terminal HA tag
Hmunc13 in pIND
and pCMV~SPORT. In a preferred embodiment, stable tansfected mouse mesangial,
NIH 3T3,
MDCK, HEK 293 and OK cell lines are created with an inducible Hmunc13 plasmid.
Gene Therapy
Since it is possible that certain diabetics may be protected from development
of renal
complications by either up or down regulation of Hmunc13, gene therapy to
replace or delete
Hmunc13 expression could also be used to modify the development/progression of
diabetic renal
and vascular complications. In addition, the use of anti-sense DNA that
inhibits the expression of
hmunc13 will allow treatment of diabetic nephropathy in humans.
The invention also includes methods and compositions for providing gene
therapy for
treatment of diseases, disorders or abnormal physical states characterized by
insufficient
Hmunc13 expression or inadequate levels or activity of Hmunc13 protein (see
the discussion of
phamaceutical discussions, above). The invention also includes methods and
compositions for
providing gene therapy for treatment of diseases, disorders or abnormal
physical states
characterized by excessive Hmunc13 expression or levels of activity of Hmunc13
protein
The invention includes methods and compositions for providing a nucleotide
sequence
encoding Hmunc13 or biologically functional equivalent nucleotide sequence to
the cells of an
individual such that expression of Hmunc13 in the cells provides the
biological activity or
phenotype of Hmunc13 protein to those cells. Sufficient amounts of the
nucleotide sequence are
administered and expressed at sufficient levels to provide the biological
activity or phenotype of
Hmunc13 protein to the cells. For example, the method can preferably involve a
method of
delivering a gene encoding Hmunc13 to the cells of an individual having a
disease, disorder or
abnormal physical state, comprising administering to the individual a vector
comprising DNA
encoding Hmunc13. The method may also relate to a method for providing an
individual with a
disease, disorder or abnormal physical state with biologically active Hmunc13
protein by
administering DNA encoding Hmunc13. The method may be performed ex vivo or in
vivo. Gene
therapy methods and compositions are explained, for example, U.S. Patent Nos.
5,672,344,
5,645,829, 5,741,486, 5,656,465, 5,547,932, 5,529,774, 5,436,146, 5,399,346
and 5,670,488,
5,240,846.
23
CA 02258973 1999-O1-29
The method may also relate to a method for producing a stock of recombinant
virus by
producing virus suitable for gene therapy comprising DNA encoding Hmunc13.
This method
preferably involves transfecting cells permissive for virus replication (the
virus containing
Hmunc12) and collecting the virus produced.
The invention also includes methods and compositions for providing a
nucleotide sequence
encoding an antisense sequence to Hmunc13 to the cells of an individual such
that expression of
the antisense sequence prevents Hmunc13 biological activity or phenotype. The
methods and
compositions can be used in vivo or in vitro. Sufficient amounts of the
nucleotide sequence are
administered and expressed at sufficient levels to prevent the biological
activity or phenotype of
Hmunc13 protein to the cells. Similar methods as described in the preceding
paragraph may be
used with appropriate modifications.
The methods and compositions can be used in vivo or in vitro. The evidence for
in vitro
usefulness is downregulation of Hmunc13 in hyperglycemia conditions can
inhibit hyperglycemia
induced renal cell injury.
The invention also includes compositions (preferably pharmaceutical
compositions for
gene therapy). The compositions include a vector containing Hmunc13 or a
biologically functional
equivalent molecule or antisense DNA. The carrier may be a pharmaceutical
carrier or a host cell
transformant including the vector. Vectors known in the art are adenovirus and
herpesvirus
vectors. The invention also includes packaging cell lines that produce the
vector. Methods of
producing the vector and methods of gene therapy using the vector are also
included with the
invention.
The invention also includes a transformed cell, such as an MC cell or other
cell described
in this application, containing the vector and recombinant Hmunc13 nucleotide
sequence or a
biologically functional equivalent molecule.
Identification of a Mouse munc13 ("Mmunc13") cDNA and Polypeptide
We identified a Mouse munc 13 gene by using a Genetrapper cDNA Positive
Selection
System (GIBCO BRL) using techniques similar to those previously reported (Song
et al., Kidney
International, 1998), we cloned a 3.5 kb cDNA which highly homologous to the
3' end of Hmunc13
from a mouse kidney cDNA library (GIBCO BRL) with a biotinylated oligo (5'-
GTGGTGATGAACACAATGGAGAGG-3'). To clone the 5' end of Mmunc13, we used nested
PCR
with gene specific primers (5'-GAGGTTGTTCCTGCAGCTATACTGG-3' and 5'-
AGTTCAAGCAGGCTTTCACACAGTCC-3') derived from the sequence obtained above and
24
CA 02258973 1999-O1-29
primers that targeted to an adapter (5'-
GCTATTTAGGTGACACTATAGAAGGTACGCCTGCAGGTACCGGTCCGGAATTCCCGGGTCGA
CCCACGCGTCCG-3' ) that introduced to the 5' end of the cDNA after reverse
transcription. PCR
was performed with a proof reading enzyme mix of Taq and Pfu (Elongase, GIBCO
BRL). The
Mmunc13 cDNA is shown in Figure 16.
The description of how modifications (e.g. to enhance activity), fragments and
variations
may be made to Hmunc13 nucleic acid molecules and polypeptides is also
applicable to
Mmunc13. The modified, fragmented and varied nucleic acid molecules and
polypeptides
preferably retain Mmunc13 functional activity. The description of Hmunc13
mimetics and their
preparation is also applicable to Mmunc13. The description of how to identify
sequences that
hybridize to the nucleotide sequence of Hmunc13 may also be adapted for
Mmunc13.
Recombinant DNA, systems for expression of Mmunc13 (eg. with plasmids and
virsues) and cells
transformed with the expression vector may also be adapted according to the
description in
relation to Hmunc13. Preferred methods for expressing Hmunc13 and isolating
the polypeptide
are also adaptable for Mmunc13. Pharmaceutical compositions including Mmunc13
gene or
polypeptide may also be prepared according to the description for Hmunc13 and
techniques
known in the art. Kits, antibodies (preferably monoclonal and polyclonal
antibodies) may be
prepared for Mmunc13 using techniques described with respect to Hmunc13.
Portions of the
Mmunc13 sequence are also useful as a probe. Mmunc13 may be used in methods of
medical
treatment (including gene therapy) of a disease, disorder or abnormal physical
state, characterized
by excessive or inadequate Hmunc13 expression, in the same manner as
techniques involving
Hmunc13. It will be apparent to those skilled in the art that other
description in relation to
Hmunc13 can be adapted and is applicable to Mmunc13.
Creation of a Mouse Knock-out Model for Mouse munc13
A probe of the 5' segment (400 bp) mouse munc13 (Mmunc13) was generated by
PCR.
Using this probe, a genomic DNA library prepared from mouse liver (129 svj) is
screened. A piece
of genomic DNA of Mmunc13 with its promoter (about 10 -12 kb) is isolated.
After characterizing
this gene, we construct a targeting vector containing a PGK-neo cassette
flanked by 5' and 3'
regions of homology totaling 10 kb, such that a homologous targeting event
results in the insertion
of PGK-neo into promoter region and exon 1 of Mmunc13. In addition, the vector
contains HSV-
TK at one end to allow the negative selection of non-homologous recombinant
events by
gancyclovir. The vector is introduced by electroporation into embryonic stem
(ES) cell (AB 2.2,
Stratagene) and dual-resistant clones are selected in 6418 and gancyclovir.
Homologous
CA 02258973 1999-O1-29
recombination clones are identified by PCR andlor Southern blot analysis.
Positive ES clones are
then be injected into wild-type blastocysts to generate chimeric mice, which
are then be used to
establish pedigrees carrying the mutant Mmunc13 allele. We characterize the
Mmunc 13
knockout mouse invention as a mouse model of "reduced apoptosis". The Mmunc 13
knockout will
not respond to endogenous diacylglycerol (DAG) by induction of apoptosis,
therefore, the DAG
induced proliferative signaling response mediated through PKC activation, will
go unchecked.
Such a mouse model is useful in research relating to a wide range of diseases,
most preferably
diabetes and cancer.
Preparation of Antibodies
The Hmunc13 protein is also useful as an antigen for the preparation of
antibodies that can
be used to purify or detect other munc13 or munc13-like proteins. Monoclonal
and polyclonal
antibodies are prepared according to other techniques known in the art. For
examples of methods
of the preparation and uses of monoclonal antibodies, see U.S. Patent Nos.
5,688,681, 5,688,657,
5,683,693, 5,667,781, 5,665,356, 5,591,628, 5,510,241, 5,503,987, 5,501,988,
5,500,345 and
5,496,705. Examples of the preparation and uses of polyclonal antibodies are
disclosed in U.S.
Patent Nos. 5,512,282, 4,828,985, 5,225,331 and 5,124,147. Antibodies
recognizing Hmunc13
can be employed to screen organisms containing Hmunc13 protein or Hmunc13-like
proteins. The
antibodies are also valuable for immuno-purification of Hmunc13 or Hmunc13-
like proteins from
crude extracts.
We prepare two peptide specific polyclonal antibodies against a C-terminal
segment
(preferably all or part of NH2-SQRSNDEVREFVKL-COOH) and an N-terminal segment
(preferably
all or part of NH2-TIRQSDEEGPGEW-COOH) of Hmunc13 which has ability to detect
rat munc13-
1, 13-2 and 13-3.
Screening for Agonists and Antagonists of Hmunc13 and Inhibitors of Hmunc13
Protein
As described above, munc13 is useful in a pharmaceutical preparation to treat
diabetes or
its complications. Hmunc13 is also useful as a target. Chemical libraries are
used to identify
pharmacophores which can specifically interact with Hmunc13 either in an
inhibitory or stimulatory
mode. The Hmunc13 targets that would be used in drug design include - e.g. the
DAG binding
site or some other functional domain specific to Hmunc13.
Modulation of Hmunc13 expression is commercially useful for identification and
development of drugs to inhibit and/or enhance Hmunc13 function directly. Such
drugs would be
targeted to any of the following sites: the DAG, Ca;+, phosphatase and RGD
domains.
26
CA 02258973 1999-O1-29
The invention also includes methods of screening a test compound to determine
whether it
antagonizes or agonizes Hmunc13 protein expression. For example, one method
involves testing
whether a compound inhibits the translocation of Hmunc13 from cytosol to Golgi
as well as its
apoptotic effect. The invention also includes methods of screening a test
compound to
determine whether it induces or inhibits Hmunc13 expression. For example, one
method involves
testing whether a compound inhibits the promoter activity of Hmunc13.
Expression of Hmunc13
Hmunc13 is expressed in MC, human cortical epithelial cells and cells from
testis, ovaries,
prostate gland, colon, brain and heart.. Experiments to determine where the
gene is expressed
were done with RT-PCR. The function of Hmunc13 in other cells will be similar
to that in renal
epithelial cells such as in translocation and apoptosis
Hmunc13 has a C1 domain. A region of the C1 domain from C. elegans unc-13
binds to
phorbol esters and DAG similar to PKC (21 ). We noted that the C1 domain is
similar among C.
elegans unc13, rat munc13s and Hmunc13 (Fig. 1), so the C1 domain in the
Hmunc13 can also
bind phorbol esters. Hmunc13 is also involved in cell signaling in response to
DAG binding.
Regulation of Hmunc13 in the Kidney
We found that expression of Hmunc13 in cultured MC was up-regulated by high-
glucose
treatment (25 mM D-glucose). Even 15 mM D-glucose is enough to stimulate the
over expression
of Hmunc13 as revealed by Northern blot. There are reports indicated that
hyperglycemia
increases PKC activity in MC (13, 14, 31). Furthermore, DAG levels are
increased when cultured
MC are exposed to hyperglycemia (17, 13). Since Hmunc13 and PKC share similar
binding
capacities for phorbol esters and DAG and both PKC contain C2 domains, Hmunc13
is part of an
alternative cascade following DAG binding. Thus Hmunc13 is activated in
response to
hyperglycemic induced increases in DAG. Even though Hmunc13 does not contain a
kinase
domain and cannot therefore serve as a downstream regulator by protein
phosphorylation (20,
30), nevertheless it is possible that Hmunc13 modulates intracellular events
through competitive
binding of PKC or by regulation of vesicle trafficking and exocytosis.
27
CA 02258973 1999-O1-29
Subcellular Localization of Hmunc13 in vitro
Expression of epitope-tagged hmunc13 in OK cells show that Hmunc13 has a
cytoplasmic
distribution under basal conditions, but with PDBu stimulation, Hmunc13 is
translocated to the
Golgi apparatus. This effect is unlikely to have taken place through
activation of endogenous
PKC, since the deletion mutant, C1 less mutant (without the DAG binding
domain), showed no
translocation. In a recent study reported by Betz et al. (24), munc13-1 was
localized to the
presynaptic region in rat brain by immunocytochemistry. In transfected HEK 293
cells, green
fluorescent protein tagged munc13-1, -2 and -3 are all translocated to plasma
membrane following
phorbol ester stimulation.
The fact that hmunc13 is translocated to the Golgi apparatus in response to
phorbol ester
activation compared to translocation of munc13-1, -2 and -3 to the plasma
membrane is proves
that Hmunc13 is a unique isoform of munc13s. This brings up the relationship
of the DAG
activated signaling pathways of munc13s and PKC. The multiplicity of PKC
isoforms and the
tissue specificity of PKC functional expression are well known (32). The
munc13 pathway is also
composed of tissue specific functionally different isoforms. However, unlike
PKC, the munc13
proteins have no kinase domain (20, 33).
The Golgi apparatus is involved in vesicular traffic. A number of SNARE
proteins, such as
yeast SedSp (34) and mVps45 (35), mammalian syntaxin 6 (36), VAMP4, Syntaxin
13 and mVtib
(36), have all been reported to be localized to the Golgi. Rat munc13-1 has
been shown to
interact with a number of proteins involved in vesicle docking and
trafficking, such as syntaxin (24)
and Doc2 (37). Interaction of munc13-1 and Doc2 was stimulated by DAG and has
been
suggested to be involved in Ca2+ dependent exocytosis (37). The finding in the
present study that
translocation of Hmunc13 to the Golgi after DAG stimulation is another
indication that Hmunc13 is
a protein that participates in DAG regulated vesicle trafficking and
exocytosis. Further studies are
required to investigate if Hmunc13 interacts with other Golgi localized SNARE
proteins or whether
some SNARE proteins co-translocate to the Golgi with Hmunc13 after DAG
stimulation. It has
also been suggested that PKC plays a role in Golgi budding (for review see
38). For example, a
study in S. Cerevisiae implicated DAG as playing an important role in the
formation of Golgi
budding involving Sec14 (39). Since Hmunc13 translocates to the Golgi after
DAG stimulation, it
would also be of interest to determine the role of Hmunc13 is involved in
Golgi budding and
interaction with Sec14L, the partial mammalian homologue of yeast Sec14 (40).
28
CA 02258973 1999-O1-29
Role of Hmunc13 in Apoptosis
We investigated the localization of Hmunc13 to determine whether exposure to
phorbol
esters had any effect on its intracellular translocation. In the course of
carrying out these studies,
we observed that cells transfected with Hmunc13 became rounded up and died
following
treatment with phorbol 12, 13-dibutyrate (PDBu), a phorbol ester analogue. We
examined the
mechanism of phorbol ester induced cell death in the transfected cells. We
showed that exposure
to phorbol ester causes apoptosis through activation of Hmunc13. This shows
the interaction
between the diabetic state, activation of Hmunc13 and cell damage.
The induction of apoptosis in Hmunc13 transfected cells after PDBu stimulation
was
unexpected. This effect is unlikely to have occurred through other DAG
activated pathways since
the C1 less mutant transfected cells were not apoptotic after PDBu treatment.
PDBu is a reagent
known to be a tumor promoter capable of stimulating cell proliferation through
PKC activation (41 ).
Although the role of PKC in apoptosis is not consistent in the literature (42,
43), the bulk of
evidence shows that PKC, especially PKCa, activated by phorbol esters such as
PMA and PDBu,
inhibits apoptosis (41-44). There is also a body of evidence suggesting that,
in the case of PKC
induced apoptosis, down-regulation rather than DAG activation of PKC
is~responsible for this
effect (43, 45).
Hmunc13 Participates in a Signaling Pathway and Counterbalances DAG Activated
PKC
Considering the functional characteristics of Hmunc13 as and the known
behavior of
munc13-1, -2, and -3 in rat brain, we determined a model for the cellular
activation of Hmunc13
and PKC isoforms. Since both munc13s and PKC have similar binding affinity to
phorbol esters,
our results showing that cells transfected with Hmunc13 become apoptotic after
DAG treatment
mean that Hmunc13 participates in a signaling pathway that serves to
counterbalance DAG
activated PKC. This concept is illustrated schematically in Figure 15. DAG
acts as a secondary
messenger to activate two alternate pathways - one pathway effected through
PKC results in
kinase activation and serinelthreonine phospholylaton of downstream targets
leading to cell
proliferation while the other pathway effected through Hmunc13 induces
apoptosis, preferably
through interaction involving vesicle trafficking.
Pathogenesis of the Microvascular and Renal Complications of Diabetes.
We have shown that in rat kidney, munc13-1 and munc13-2 are mainly localized
to cortical
tubular epithelial cells. Using both in situ hybridization and relative RT-
PCR, we have also
demonstrated that munc13-1 and munc13-2 are over-expressed in kidney of STZ-
treated diabetic
29
CA 02258973 1999-O1-29
rats. This result in rat kidney is consistent with our in vitro findings,
showing that expression of
Hmunc13 is up-regulated by high glucose treatment in cultured human mesangial
cells. It has
been reported that an increase in intracellular DAG levels is only detectable
after 2 days of high
glucose treatment (46). The fact that expression of both rat munc13-1 and
munc13-2 is found to
be increased after only 1 day of hyperglycemia shows that over-expression of
these genes is a
consequence of hyperglycemia and not secondary to stimulation by DAG.
Therefore, in diabetes,
there are two mechanisms acting to increase activity of Hmunc13: (i)
hyperglycemia itself, (ii)
hyperglycemia-induced increase in cellular DAG (47-49). The over-expression of
Hmunc13 is a
major contributor to cell injury in diabetic nephropathy by inducing
apoptosis. In this regard, it is
noteworthy that under hyperglycemic condition, renal tubular cells undergo
apoptosis (50-51 ).
Finally, since PKC inhibitors have been developed to treat diabetic
nephropathy (49), a potential
side effect of those inhibitors could result from overactivity of Hmunc13.
EXPERIMENTS
Experiment 1 - DDRT-PCR
DDRT-PCR carried out on RNA extracts from MC exposed to high vs. low glucose
conditions yielded 10 bands which exhibited differences between high glucose
treatment and
controls (both normal glucose and osmolarity controls) (data not shown). After
the bands had
been cut, reamplified, cloned and sequenced, the sequences were compared to
the GenBank
database. One of the cDNA sequences had identity to a segment (residues 3523-
3863) of rat
munc13-2 (20). Since rat munc13-2 is viewed as having a potential signaling
function particularly
in neurotransmission and in addition has not previously been reported in any
tissue outside the
brain, we elected to clone the full gene from human kidney and confirm the
nature of its regulation
by hyperglycemia.
Experiment 2 -Cloning of Hmunc13
As a first step we cloned a partial length cDNA from a commercial human kidney
cDNA
library using oligonucleotides derived from sequence information obtained from
DDRT-PCR
comparing cells at 25 mM D-glucose vs. 5.5 mM D-glucose and osmolarity control
(see Methods).
Then, using the sequence of the partial length clone, we designed another
oligonucleotide closer
to the 5' end and proceeded to clone a full-length cDNA (6.3kb,
pCMV~SPORTHmunc13), which
we have named Hmunc13. This cDNA encodes a protein with a predicted molecular
weight of
180.5 kDa. As shown in figure 1, kidney Hmunc13 contains 3 C2 domains and 1 C1
domain. The
CA 02258973 1999-O1-29
N-terminal segment of Hmunc13 (residues 1-100) is similar to to rat munc13-1
(Fig. 1 b). The next
segment (residues 101-391 ) exhibits considerable variation in Hmunc13
compared to rat munc13s
and unc-13 (7). The C-terminal segment of unc-13s is highly conserved among
human, rat and C.
elegans (Fig. 1, ref. 7). In particular, the protein segment from residue 392
to 1591 of Hmunc13 is
about 93% similar to rat munc13-2 (residue 766-1985), 79% similar to munc13-1
(residue 486-
1735) and 74% similar to munc13-3 (residue 1000-2207). In summary, the C
terminus of renal
Hmuncl3 has strongest identity to rat munc13-2 whereas the N-terminal of
Hmunc13 has
strongest identity to rat munc13-1.
Experiment 3 - Hyperglycemia Up-re4ulates Hmunc13 mRNA Expression in Kidne~r
MC
To confirm the differential expression of Hmunc13 under varying glucose
concentrations
two independent methods were employed. In a pilot study, by using ribonuclease
protection
assays, we have found that expression of Hmunc13 in human MC treated with 19.5
mM L-glucose
+ 5.5 mM D-glucose (osmolarity control) was not changed (data not shown).
Therefore, in the
following experiment, we only compared the difference of Hmunc-13 expression
between high D-
glucose and high L-glucose treated MC. We first used relative RT-PCR with 18S
rRNA as a
housekeeping gene. As shown in figure 2a, Hmunc13 was up-regulated in the high-
glucose
(25mM) treated MC compared to osmolarity controls. In a more quantitative way,
Northern blot
analysis was carried out on cells grown according to the same protocol. As
revealed by relative
RT-PCR, Hmunc13 expression was increased in MC after hyperglycemia (Fig. 2b).
Quantitative
desitometry analysis revealed 70% increase of Hmunc13 expression after
exposure to 25 mM D-
glucose treatment (p < 0.05, n = 5, student's t-test). As shown in figure 2b,
Hmunc13 expression
in MC following exposure to 15 mM D-glucose was also increased relative to
osmolarity control but
there was no statistically significant difference between 15 mM D-glucose and
25 mM D-glucose
treated cells.
Experiment 4 - Expression of Munc13 in Epithelial and Rat MC
To show that munc13 is also expressed in other cell types in the kidney
besides MC and
that it is expressed in the rat MC as well as human, RT-PCR was performed
using a pair of
primers specific for both Hmunc13 and rat munc13-2. As shown in figure 3,
Hmunc13 was
detected in cultured human kidney cortical epithelial cells and munc13-2 was
also expressed in
primary cultured rat MC. Genomic contamination is unlikely since no band was
observed in the no
RT control for the GAPDH housekeeping gene (Fig. 3).
31
CA 02258973 1999-O1-29
Experiment 5 - Hmunc13 is Expressed as a 180 kDa Protein in vitro and is
Membrane Associated
Using a cell free in vitro translation system, we have demonstrated that
Hmunc13 is
expressed as a 170 kDa protein (Fig. 4). This is close to the predicted MW
(180.5 kDa) from the
cDNA clone. A number of less prominent lower molecular weight bands is also
present following
in vitro translation because of either initiation of translation from internal
AUG codons rather than
the first interaction site or a premature termination of translation. Also
shown in figure 4 is that in
the presence of canine pancreatic microsomal membranes, a proportion of
Hmunc13 protein is
shifted to a higher molecular weight 0180 kDa) suggesting that it is membrane
associated and
undergoes co-translational processing. Only the full-length protein is
associated with the
membrane because the partial length in vitro translation products are not
observed in the
microsomal pellet (Fig. 4, lane 2).
Experiment 6 - Translocation of Hmunc13 to Golgi apparatus after DAG treatment
To study its cellular function, we elected to over-express Hmunc13 in opossum
kidney
(OK) cells, a cell line of renal epithelia origin and compare two constructs -
an HA tagged
Hmunc13 and an HA tagged Hmunc13 deletion mutant lacking the C1 domain (C1
less mutant).
Cells employed in the present study were grown on glass cover slips under
growth arrested
conditions with serum starvation. Transient transfection of OK cells was
confirmed by Western blot
analysis (Figure 10). As shown in Figure 10(i), an 180 kDa protein was
expressed in the
Hmunc13-HA transfected cells and a 175 kDa protein was detected in the C1 less
mutant
transfected cells. No band was detected in cells transfected with empty
plasmid, pCMV~SPORT.
Intracellular localization of Hmunc13-HA in transfected OK cells was monitored
by
immunocytochemistry (ICC) using cells doubly labeled with anti-HA antibody
(Fig. 10(ii), upper
panels) and wheat germ agglutinin (WGA) (Fig. 10(ii), lower panels). As
indicated in Figure 10(ii),
inspection of panel A reveals that Hmunc13 exhibits a cytosolic distribution
compared to the Golgi
apparatus stained with WGA shown in Panel E. But after exposure to 0.1 pM
PDBu, a DAG
analogue, Hmunc13 is translocated to the peri-nuclear area (panel B) and co-
localizes with WGA
at the Golgi apparatus (compare panels B and F). Translocation of Hmunc13 to
the Golgi after
PDBu treatment occurred in 15-30 min and became more obvious in 2-3 h. By
contrast, when
cells were transfected with the C1 less mutant, lacking a DAG binding domain,
there was no
translocation after PDBu treatment (refer to panels D and H) and Hmunc13
staining remained
cytosolic.
32
CA 02258973 1999-O1-29
When cells were treated with nocodazole, a drug that depolymerizes
microtubules, (52),
after PDBu treatment, the patterns of WGA and Hmunc13 staining became
identical and both
revealed a dispersed Golgi pattern (compare panels C and E of Fig. 10 (ii)).
Translocation of Hmunc13 from cytosol to the Golgi apparatus after PDBu
treatment was
also confirmed by immunoblot analysis of a Golgi membrane preparation,
following subcellular
fractionation. As shown in Figure 10 (iii), after PDBU treatment, Hmunc13 is
enriched in Golgi
membranes compared to whole cell lysates. .
Exaeriment 7 - Hmunc13 over-expressed cells are apoptotic after DAG treatment
The PDBu induced translocation from cytosol to Golgi suggests that Hmunc13 has
functional implications. While attempting to study the effect of prolonged
exposure to DAG
activation on Hmunc13 transfected cells, we noticed that the cells rounded up
and died. However,
Hmunc13 transfected cells without PDBu treatment and cells transfected with
the C1 less mutant,
with or without PDBu treatment, were relatively healthy. This finding was
somewhat unexpected
since DAG has long been known as a carcinogen and a promoter of cell growth,
and led us to
investigate the possibility and conclude that treatment with phorbol ester is
inducing apoptosis in
cells transfected with Hmunc13.
Using the TUNEL assay, we found that the number of apoptotic cells was
significantly
increased in hmunc13 transfected OK cells after 8 h and 16 h of PDBu
treatment. These results
are displayed in Figure 11 (i). The upper panels show the expression of
Hmunc13 in OK cells and
the lower panels demonstrate the presence of fluorescein labeled TUNEL on the
same cells.
Inspection of panel F (8 h of PDBu treatment) and panel G (16 h of PDBU
treatment) compared to
panel E (treatment with vehicle control) reveals evidence of DAG induced
increase in TUNEL
staining cells. This conclusion is further supported by the fact that cells
transfected with the C1
less mutant, exhibit almost no labeling with TUNEL following exposure to PDBu
for 16 h (compare
panel H with panels F and G). The above results are also summarized in fiugure
(ii). Finally, cells
transfected with empty plasmid also showed almost no TUNEL labeling with or
without PDBu
treatment (data not shown).
To further confirm, a DNA fragmentation assay was employed. Further evidence
of a
breakdown in genomic DNA is revealed by the "laddering" pattern shown in
Figure 12, obtained
after 8 and 16 h of PDBu treatment in Hmunc13 transfected cells.
Experiment 8 - Exaression of munc13s in normal and STZ-treated diabetic rat
kidney
33
CA 02258973 1999-O1-29
We have previously demonstrated that Hmunc13 is up-regulated by high glucose
treatment
in cultured human mesangial (33). Since the main thrust of the present study
was to investigate
the functional role of Hmunc13, we documented its in vivo expression.
Furthermore, confirmation
of up-regulation of Hmunc13 by hyperglycemia in an in vivo state is necessary
to show the role for
this gene in diabetic nephropathy. We characterized Hmunc13 expression in
human kidney. We
used an animal model of diabetes- the STZ treated rat (the relevant isoforms
being munc13-1, -2,
and -3). As shown in Figure 13, munc13-1 is expressed mainly in cortical
tubular epithelial cells of
both normal and STZ-treated diabetic rats. However, the expression level of
munc13-1 was
higher in STZ-treated diabetic rat after 11 days of hyperglycemia. Expression
of munc13-1 was
significantly higher in certain glomerular cells of diabetic animals. But it
is impossible to identify
these cells with any certainty at the resolution of confocal microscopy.
However, because of our
previous in vitro results (33), we determined that munc13-1 is up-regulated in
the mesangial cells.
Increased expression level of munc13-2 was also detected in diabetic rats with
similar expression
pattern as munc13-1. Possibly because of low basal expression, we could not
obtain satisfactory
in situ hybridization data for munc13-3 in rat kidney.
To confirm the over-expression of munc13-1 and munc13-2 in diabetic rat
kidney, we
performed relative RT-PCR on renal cortical RNA preparation. Relative RT-PCR
was chosen
because low expression of munc13s in the rat kidney and a very low signal was
detected in
Northern blot analysis. As shown in Figure 14, compared to the housekeeping
gene, 18S
ribosome RNA, expression of munc13-1 is over-expressed in the renal cortex of
the STZ-treated
diabetic rat after only 1 day of hyperglycemia whereas expression of munc13-2
is increased to a
much lesser extent. Interestingly, munc13-3 is down-regulated in the same
animal model. We
screen to detect a human homologue of rat munc13-3 in a commercial human
kidney cDNA library
(Gibco BRL) using PCR with primers targeted to different regions of munc13-3.
We determine the
role of munc13-3 in diabetic nephropathy.
MATERIALS AND METHODS
MC basal culture medium (MsBM) and renal epithelial basal medium (REBM) were
purchased from Clonetic, San Diego, CA. Fetal bovine serum (FBS), Dulbecco's
modified Eagle's
medium (DMEM), penicillin, streptomycin, human kidney cDNA library,
Superscript II RNase H-
reverse transcriptase, dNTP, E.coli RNase H, Taq DNA polymerase, Genetrapper
cDNA Positive
Selection System, 100 by DNA size markers, Klenow Fragment, m'G(5')ppp(5')G
RNA capping
analog, ElectroMAX DH10B cells and restriction enzymes were obtained from
Gibco BRL,
Burlington, ON, Canada. DNase I and T'Sequence kit were purchased from
Pharmacia Biotech,
34
CA 02258973 1999-O1-29
Uppsala, Sweden. TA cloning kit was from Invitrogen, San Diego, CA. RNeasy
total RNA
preparation kit, QIAshredder and QIAquick Gel Extraction kit were purchased
from Qiagen,
Chatsworth CA. SP6 RNA polymerase, human cyclophilin template, 18S rRNA
primers and
competimers were from Ambion, Austin, TX. Vent DNA polymerase was obtained
from New
England Biolab, Inc, Beverly, MA. Rapid hybridization buffer and a-[32P]-dATP
(specific activity
800 Cilmmol) were purchased from Amersham, Arlington Heights, IL. [35S]-
Methionine (specific
activity, 1000 Cilmmol) was from NEN Life Science Products, Boston, MA.
Duralon-UV
membranes was purchased from Stratagene, La Jolla, CA. Six percent denatured
polyacrylamide
solution was purchased from National Diagnostics, Somerville, NJ.
Oligonucleotides were
synthesized by Gibco BRL. X-ray film was from Kodak, Rochester, NY. Flexi
rabbit reticulocyte
lysate system and canine pancreatic microsomal membranes were purchased from
Promega,
Madison, WI. Other chemicals with cell culture or molecular biology grade were
obtained from
local suppliers.
Cell culture
Primary cultures of human kidney MC and cortical epithelial cells were
purchased from
Clonetic. Human MC were plated onto 25 cm2 culture flasks and incubated in
MsBM containing
5.5 mM D-glucose with 100UIml penicillin, 100 pg/ml streptomycin and 5% FBS.
Cells were
subcultured at 80-90 % confluence. Cortical epithelial cells were grown in
REBM supplement with
100UIml penicillin and 100 glml streptomycin. Rat renal MC were prepared and
cultured as
previously described (53,54).
Protocol for studying the effect of hyperglycemia on human kidney MC
Human MC between passage 5-9 were used in this study. Three parallel
experimental
conditions were employed: 25 mM D-glucose (hyperglycemia), 5.5 mM D-glucose
(low glucose
control) and 25 mM L-glucose (osmolarity control). The details are as follows:
for high glucose
treatment, subconfluent MC were growth-arrested in MsBM + 0.5% FBS overnight
and exposed to
5.5 mM or 25 mM D-glucose for 3 days with one change of medium on the second
day. In
parallel, L-glucose at the final concentration of 19.5 mM was added to the
culture medium to serve
as an osmolarity control. In order to investigate if any dose-dependency of
Hmunc13 expression
by D-glucose treatment, in Northern blot studies, we analyzed two more sets of
human MC
cultured in 15 mM D-glucose or 5.5 mM D-glucose + 9.5 mM L-glucose for 3 days.
We have
found that changing the medium every two days at 25 mM D-glucose is enough to
maintain
physiological pH in the medium (pH 7.4) (data not shown). At the end of the
experimental
treatment period, total RNA of the cells was prepared.
CA 02258973 1999-O1-29
Isolation of total RNA
Total RNA from human MC and cortical epithelial cells as well as rat MC was
prepared
using an RNeasy total RNA preparation kit according to manufacturer's
instructions. Cell lysates
were prepared following homogenization using a QIAshredder.
DDRT-PCR
DDRT-PCR was performed by modified methods published by Liang and Pardee (55)
and
Sokolov and Prockop (56). Total RNA from human kidney MC was incubated with
DNase I to
remove any contaminating genomic DNA prior to first strand DNA synthesis.
Reverse
transcription (RT) was carried out by incubating a 20 ~I reaction mixture
containing 1 pg total RNA,
100 ng fully degenerate hexamer, 500 pM each of dATP, dGTP, dCTP and dTTP and
200 units of
reverse transcriptase (Superscript II RNase H-) together with the buffer
provided by the
manufacturer. The reaction mixture was incubated at 42°C for 50 min.
The reaction was
terminated by heating at 70°C for 15 min. E. coli RNase H (2 units) was
then added to the
reaction mixture followed by incubation at 37°C for a further 20 min to
remove RNA
complementary to the cDNA. Demonstration that the RNA was free of genomic DNA
was
confirmed using a pair of GAPDH specific primers (5'-ACCACAGTCCATGCCATCAC-3'
and 5'-
GTCCACCACCCTGTTGCTGTA-3') to obtain PCR products before and after RT. We found
that
there was no amplification in the absence of RT but a strong band was present
in the presence of
RT (data not shown). PCR was carried out using two 10-mer oligonucleotides, 5'-
CAAGCGAGGT-3' and 5'-GTGGAAGCGT-3'. In a total of 12.5 ~I, the reaction
mixture contained
1 pl of RNA with RT, 100 wM of each of dNTP, 4 p.M of oligonucleotides, 1.5 mM
of MgCl2, 0.1
mCilml of a-[32P]-dATP and 1.25 unit of Taq DNA polymerase. PCR was carried
out using a
Perkin Elemer PCR System 2400 (Perkin Elemer, Foster City, CA) starting at
94°C for 1 min, 34°C
for 1 min and 72°C for 1 min for 45 cycles. The resulting PCR products
were subjected to 6%
denatured polyacrylamide gel electrophoresis (PAGE) using radiolabelled 100 by
ladder as size
markers. The gels were then dried and exposed to x-ray film overnight. Bands
which showed
clear cut differences in high (25 mM) compared to low (5.5 mM) D-glucose or
the osmolarity
control (25 mM L-glucose) were excised by aligning the film with the gel
followed by elution
overnight in 10 mM Tris-EDTA buffer (pH 8.0). Eluted DNA was purified and
subjected to a
second run of PCR by the same pair of 10-mer oligonucleotides under the same
experimental
conditions without radiolabelled dATP. Fresh PCR products from this last step
were cloned into
pCR2.1 using a TA cloning kit. Clones with inserts were sequenced by using a
"Sequencing kit
36
CA 02258973 1999-O1-29
with T7 promoter as a primer according to the manufacturer's instructions. The
resulting DNA
sequences were compared to the GenBank database using BLAST search.
Library Screening
Screening of Superscript human kidney cDNA library was achieved using a
Genetrapper
cDNA Positive Selection System. Captured cDNAs were transformed to ElectroMAX
DH10B
competent cells by electroporation with an electroporation system (BTX Inc.,
San Diego, Ca)
setting at 16.6 kV/cm. We first used an oligonucleotide (5'-
GTGGTGATGAACACAATGGAGAGG-
3') originally derived from sequence information following DDRT-PCR to capture
a partial length of
Hmunc13. According to this sequence information, we then designed another
oligonucleotide (5'-
TCCTGTTTGGGAGGAGAAGTTCC-3') closer to the 5' end of the sequence to capture a
full
length clone. The resulting clone (pCMV~SPORTHmunc13) was sequenced from both
strands
using standard techniques described above. The primers were SP6, T7 promoters
or synthetic
oligonucleotides derived from the sequence information. Alignment and analysis
of sequences
was performed with Genework 2.5.1 (Oxford Molecular Group, Campbell, CA) using
a Macintosh
computer. Comparisons of similarity were performed using the Gapped BLAST
search from
GenBank.
Relative RT-PCR and RT-PCR
For relative RT-PCR, RT products previously described were subjected to PCR
for 30
cycles using a pair of primers (5'-GGAGCAAATCAATGCCTTGG-3' and 5'-
TCGGATCCAATGTGCTCTGG-3') specific for Hmunc13, amplifying a 671 by fragment.
18S
rRNA was chosen as a housekeeping gene by using 18S rRNA primers and 18S rRNA
competimers with a ratio of 1:2. These primers amplify a 488 by fragment.
Resulting PCR
products were subjected to 1.2 % agarose gel electrophoresis.
To determine munc13 expression in epithelial and rat MC, we employed RT-PCR
with a
pair of primers (5'-GA(T)GTC(A)CTGAAGGAGCTCTGG-3' and 5'-
AGGACA(T)GCACACTGCTTTGG-3' ) targeted to Hmunc13 and rat munc13-2 both of
which yield
a 193 by fragment. RT were performed post DNase I treatment on total RNA
extracted from these
cells as described above.
Northern Blot Analysis
Total RNA (10 fig) extracted from human kidney MC was subjected to 1 %
denatured
formaldehyde agarose gel electrophoresis as described (36) then transferred to
Duralon-UV
membranes overnight and exposed to UV light for cross linking. An 32P-
radiolabelled probe of
37
CA 02258973 1999-O1-29
Hmunc13 were generated from a PCR fragment derived from pCMV~SPORTHmunc13
(4095 -
4288) with a-[32P]-dATP using a Klenow Fragment and random hexamers. Membranes
were pre-
incubated with rapid hybridization buffer at 65°C for 15 min and then
incubated with radiolabelled
probes at 65°C for 2 hours. After removal of the radiolabelled probes,
membranes were washed
first in 2 x SSPE (1 x SSPE contains 150 mM NaCI, 20 mM NaH2P04 and 1 mM EDTA,
pH, 7.4)
with 0.1 % SDS at room temperature for at least 20 min then twice with 0.1 x
SSPE with 0.1 % SDS
at 65°C for 30 min each. After exposure to the Phosphor screen
(Molecular Dynamics, Sunnyvale,
CA), the blots with Hmunc13 probe were stripped with a boiling solution of 0.1
x SSPE with 0.1
SDS. The stripped membranes were reprobed with a 32P-labelled human
cyclophilin template.
Radioactivity of each band in digital images was analyzed on a PC using
ImageQuant 4.0
(Molecular Dynamic).
In vitro Translation
In vitro translation was performed according to previously published method
(26). Plasmid
with Hmunc13 cDNA (pCMV~SPORTHmunc13) was linearlized with Hind III.
Linearlized DNA (1
pg) was transcribed with SP6 RNA polymerise and m'G(5')ppp(5')G RNA capping
analog .
Capped cRNA was extracted using an RNeasy total RNA preparation kit. Eluted
cRNA was
precipitated and resuspended in 5 pl diethylpyrocarbonated-treated water. In
the presence of 1 p,l
of this cRNA product, in vitro translation was achieved using a Flexi rabbit
reticulocyte lysate
system according to the method provided by supplier. Translation products were
detected by
incorporating 1 wCilpl of [35S] methionine in the reaction mixture. To
determine co-translational
processing, 1.5 equivalent of canine pancreatic microsomal membranes was added
to 10 wl of in
vitro translation reaction. The resulting reaction was centrifuged at 16,000 g
for 15 min to pellet
microsomes. In vitro translation products were subjected to 8% PAGE. The gel
was stained with
Commassie brilliant blue then destained. The stained gel was then dried and
exposed to x-ray
film.
Statistical Anal
Group differences in densitometry of the Northern blots were analyzed by
Student's t-test
using Systat 5.2.1. (Systat Inc., Evanston, IL) for the Macintosh.
Significance level was set at p <
0.05.
Construction of HA-taq<ged hmunc13 and truncated mutant without C1 domain
We constructed an HA-tagged hmunc13, by taking advantage of an EcoN I
restriction site
(nucleotide 3949) close to the 3' end of the open reading frame of hmunc13
constructed in
38
CA 02258973 1999-O1-29
pCMV~SPORT (Gibco, BRL, pCMV~SPORThmunc13), and used PCR to introduce the HA-
tag at
the C-terminal of hmunc13. A PCR fragment was generated with Vent DNA
polymerase, insert of
pCMV~SPORThmunc13 as a template and a pair of primers (5'-
GAATACGGTTCTGGATGAGCT-
3' and 5'-
gcggccgcTCAAGCGTAGTCTGGGACGTCGTATGGGTAGCTCCCCTCCTCCGTGGAACG -3')
where the HA tag sequence is underlined and a Not I site is shown in lower
case. A stop code (5'-
TCA-3') was placed between the HA tag and the Not I site. The PCR product was
then incubated
with 2 units of Taq DNA polymerase at 72 C for 15 min and extracted by
phenollchloroform and
ethanol precipitation. The resulting pellet was resuspended and ligated to
pCR2.1 by using a TA
cloning kit. This plasmid was then digested with Not l and EcoN l, subjected
to 1 % agarose gel
electrophoresis. The insert was purified and ligated to pCMV~SPORThmunc13
previously cut with
Not I and EcoN I. The resulting construct (hmunc13-HA) was sequenced to
confirm the addition of
the HA tag.
To construct a deletion mutant lacking the C1 domain (C1 less mutant), we
replaced the
entire C1 domain (AA 478-528) with two residues Ala and Arg. Primers 5'-
CGTTGGCGCGCCAGCGGGCTGCAGAAAAGAGC -3' (Asc 1 site is underline) and 5'-
CTGTCTCATCAAAGTACACC-3' were used to generate a PCR fragment with Vent DNA
polymerase and pCMV~SPORThmunc13 as a template. Another piece of PCR fragment
was
generated by primers of Sp6 promoter (5'-AGCTATTTAGGTGACACTATAG-3') and 5'-
GCTAGGCGCGCCGGAGTGGTGCACGAAATGG -3' (Asc I site is underline). The two PCR
fragments were digested with Asc l, ligated with T4 DNA ligase, and the
ligated product was
subjected to 1 % agarose gel electrophoresis to check the size and for
purification. The gel
purified ligated piece was further digested with Kpn l and BstZ17 I and
ligated to Kpn l and BstZ97
I digested pCMV~SPORThmunc13-HA.
Plasmids for cell transfection were prepared using a Midi plasmid preparation
kit according
to manufacturer's instructions.
Cells and transfection
OK cells were grown in MEM supplemented with 10% FBS and 100 Ulml penicillin
and 100
pglml streptomycin, and plated in 60 mm or 100 mm culture dishes or on glass
cover slips placed
in 24 wells culture plates. Cells were transiently transfected (transfected
rate 30-50%) with
hmunc13-HA or C1 less mutant by using Lipofectamine Plus according to the
manufacturer's
instruction, and maintained in serum free MEM overnight (3 h for apoptotic
experiments) after 24 h
39
CA 02258973 1999-O1-29
of transfection. Cell monolayers were washed and fresh medium containing PDBu
or the same
amount of vehicle (DMSO at a final concentration of 0.0001 %) was added to the
culture medium at
a final concentration of 0.1 NM and cells were analyzed at different time
points as indicated. For
nocodazole treatment experiments, nocodazole in DMSO was added to the medium
at a final
concentration of 4 ~M for 1 h and followed by addition of PDBu at a final
concentration of 0.1 NM.
Cells were subjected to immunostraining after 3 h of PDBu treatment. An
identical quantity of
DMSO was added to control cells.
Immunocytochemistry
Cells grown on cover slips were washed 3 times with iced cold Hank's solution,
fixed and
permeabilized with 100% methanol at -20 C for 5 min. The cover slips were then
air dried,
washed 3 times with PBS and incubated in blocking solution (PBS + 0.2% Tween-
20 (PBST)
containing 10% no-fat dry milk). Cells were then incubated with 0.02 mglml
anti-HA for 30 min at
room temperature followed by 0.02 mglml anti-mouse IgG-rhodamine for 30 min.
Cells were
washed at least 8 times with PEST between incubation of anti-HA and anti-mouse
IgG-rhodamine
or after anti-mouse IgG-rhodamine. Cover slips were then mounted on a glass
slide and observed
under a confocal scanning microscope. For labeling of the Golgi apparatus,
0.05 mglml WGA-
FITC was added to the anti-mouse IgG-rhodamine.
Immunoblot analysis and preparation of crude Golpi membrane
Cells grown on culture plates were washed 3 times with ice cold Hank's
solution and
scraped into 0.5 ml cell lysis buffer (50 mM Tris-HCI, 150 mM NaCI, 0.25%
sodium deoxycholate,
1 % NP-40, 1 mM EDTA and protease inhibitor cocktail, pH 7.5), and then rocked
at 4 C for 45 min.
The insoluble fraction was removed by centrifugation at 14,000 g for 5 min.
Supernatants were
subjected to 6% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-
PAGE) and
transferred to nitrocellulose. The membrane was washed twice with TBS, blocked
with TBS
containing 0.1 % Tween-20 (TBST) and 1 % normal horse serum for 30 min and
then incubated
with 0.5 uglml anti-HA in TBST. After washing with TBST for at least 4 times,
the membrane
fraction was incubated with 0.2 p,glml anti-mouse IgG-biotin, washed with TBST
and then
incubated with the A and B reagent mix in a Vector ABC staining kit according
to manufacturer's
instructions. The blot was detected by ECL according to the manufacturer's
instruction.
Golgi membranes were prepared by a sucrose density method reported previously
(57)
with a protease inhibitor cocktail presented in all buffer solution. The band
at the interface of 0.8M
and 1.2M sucrose was collected and subjected to 6% SDS PAGE and immunobloting
as
CA 02258973 1999-O1-29
described above. Protein concentration was determined by Lowry assay with
bovine serum
albumin as standard using a DC Protein Assay kit following its instruction.
Detection of apoatosis by DNA fragmentation
Cleaved genomic DNA during apoptosis for cells grown on cover slips was
detected by
terminal deoxynucleotidyl transferase (TdT) - mediated dUTP nick end labeling
(TUNEL) using a in
situ cell death detection kit following manufacturer's directions. Fluorescein
labels were
incorporated in nucleotide polymers. Negative controls were obtained by
incubating label solution
without TdT under the same conditions. After labeling for apoptosis, cells
were further subjected
to Immunocytochemistry as described above without fixation and
permeabilization to detect
expression of hmunc13 or its C1less mutant.
Genomic DNA fragmentation of cells grown on 60 mm culture dishes was analyzed
by 2%
agarose gel electrophoresis using the procedure described elsewhere (58).
Streptozotocin treated diabetic rat model
Rats received a single injection of STZ (65 mglkg body weight, i.p.) dissolved
in 20 mM
citric acid (pH 4.5). Blood glucose was monitored daily by tail blood sampling
with a Medisense
blood glucose sensor (Medisense Canada, Mississauga, ON, Canada). Blood
glucose was
maintained at a concentration of 15-20 mM with 2 U NPH insulin daily (s.c.)
after diabetes was
confirmed by elevated blood and urinary glucose. Rats were sacrificed after 1
or 11 days of
diabetes. Rat kidneys were collected as soon as possible, usually within 3-5
min, and processed
for total RNA preparation or tissue preparation for in situ hybridization as
described below. Control
rats were injected (i.p.) with the same amount of 20 mM citric acid and their
blood glucose levels
were also tested daily (< 5 mM).
Relative reverse transcriation polymerase chain reaction (RT-PCR)
Total RNA from rat kidney cortex was prepared using a TRlzol reagent according
to
instructions provided by the manufacturer and then treated with DNase I.
Confirmation of no
genomic DNA contamination in RNA preparations and relative RT-PCR were
performed as
described elsewhere (33). Primers for amplification of rat munc13-1 are 5'-
CGTGACCAAGATGAGTACTCC-3' (sense) and 5'-CGAAGTCGTGTAGTAAGGCG-3' (anti-sense)
yielded a fragment of 195 bp. Primers for rat munc13-2 are 5'-
GAGTCCTGAAGGAGCTCTGG-3'
(sense) and 5'-AGGACAGCACACTGCTTTGG-3' (anti-sense) yielded a fragment of 193
bp.
Primers for rat munc13-2 are 5'-
41
CA 02258973 1999-O1-29
AGATGACCTTGGCAAGTGC-3' (sense) and 5'-CGATACATCATGGATGGATGG-3' (anti-sense)
yielded a fragment of 198 bp. The sequence of PCR products was confirmed by
cloning PCR
fragments into pCR2.1 using a TA cloning kit and sequencing using a
"Sequencing kit with T7
promoter as a primer.
In situ hybridization
Templates for in vitro transcription were generated by PCR with primers
described above
for three different isoforms, except that for anti-sense cRNA, addition of T7
promoter (5'-
TAATACGACTCACTATAGGGA-3') was present in the sense strain and for sense cRNA,
T7
promoter was present in the anti-sense strain. Anti-sense and sense cRNA for
different isoforms
were obtained by in vitro transcription. PCR templates (200 ng) were incubated
with T7 RNA
polymerase (40U), its reaction buffer provided by the manufacturer and DIG RNA
labeling mix in a
total volume of 40 ~.I at 37 C for 90min. Twenty pl recombinant RNA was
purified by using a
RNeasy total RNA preparation kit and its yield was estimated by AZeo. The
remaining cRNA was
subjected to ethanol precipitation and resuspended in nuclease-free water.
All solutions used before the post-hybridization step were
diethylpyrocarbonate (DEPC)
treated or prepared in DEPC-treated water. Kidneys were quickly cut to 2 mm
thick blocks after
dissection then put in phosphate-buffered saline (PBS, pH 7.4) containing 4%
parafromaldehyde
for 4 h at 4 C. The tissue was soaked in PBS containing 30% sucrose overnight
at 4 C and then
stored in liquid nitrogen. Frozen tissues were sectioned (10 Nm) and placed on
a poly-L-lysine
coated glass slides. In order to ensure the same experimental conditions,
kidney sections from
control and diabetic rats were placed on the same slide. Tissue slides were
then dried at 40 C
overnight and stored at -80 C for less then a week. On the day of
hybridization, slides with tissue
sections were dried at 40 C for 2 h then washed twice with PBS. Slides were
then incubated with
0.3% Triton X-100 in PBS for 15 min at room temperature and washed twice with
PBS afterward.
Sections were incubated with 1 ~,glml RNase-free proteinase K in TE buffer
(100 mM Tris-HCI, 50
mM EDTA, pH 8.0) for 30 min at 37 C and then fixed by incubating with PBS
containing 4%
parafromaldehyde for 5 min at 4 C. Sections were then washed twice with PBS
and acetylated
with freshly prepared 0.1 M triethanolamine buffer (pH 8.0) containing 0.25%
acetic anhydride.
Slides were then incubated first with 4x SSPE (1x SSPE containing 150 mM NaCI,
20 mM
NaH2P04 and 1mM EDTA, pH 7.4) containing 50% formamide at 37 C for 20 min and
then
overlaid with 75 ul hybridization buffer (40% fromamide, 10% dextran sulfate,
0.02% Ficoll, 0.02%
polyvinylpyrolidone, 10 mg/ml bovine serum albumin, 4x SSPE, 10 mM DTT, 0.4
mg/ml yeast t-
RNA and 0.1 mglml poly(A) ) containing 50 ng of denatured DIG-labeled cRNA
probe. Slides
42
CA 02258973 1999-O1-29
were incubated in a humid chamber at 42 C overnight. After hybridization,
slides were washed at
least 4 times in 1x SSPE at 37 C. Sections were incubated with 20 p.glml RNase
A in NTE buffer
(500 mM NaCI, 10 mM Tris-HCI, 1 mM EDTA, pH 8.0) at 37 C for 30 min and washed
twice with
0.1x SSPE. Slides were washed and blocked in TBS (100 mM Tris-HCI and 150 mM
NaCI, pH
7.5) containing 1% blocking reagent and then incubated with 0.02 mglml anti-
DIG-rhodamine for 1
h. Slides were washed at least 5 x with TBS. Staining was assessed by a
confocal scanning
microscopy.
The present invention has been described in detail and with particular
reference to the
preferred embodiments; however, it will be understood by one having ordinary
skill in the art that
changes can be made thereto without departing from the spirit and scope
thereof.
All publications, patents and patent applications are herein incorporated by
reference in
their entirety to the same extent as if each individual publication, patent or
patent application was
specifically and individually indicated to be incorporated by reference in its
entirety.
43
CA 02258973 1999-O1-29
REFERENCES
1. S.M. Mauer. Structural-functional correlations of diabetic nephropathy.
Kidney Int. 45:
612-622, 1994
2. A.E. Thomson. Non-insulin dependent diabetes mellitus in nephrology: where
do we go
from here? Kidney Int. 52: S-83-S-84, 1997
3. T.F. Drury and A.L. Powell. Prevalence of known diabetes among black
Americans, in
Diabetes in America, US Dept. of Health & Human Services, NIH 87-1468, 1987
4. M.P. Stern, J.A. Knapp, H.P. Hazuda, S.M. Haffner, J.K. Patterson and B.D.
Mitchell.
Genetic and environmental determinants of type II diabetes in Mexican
Americans.
Diabetes Care 14: 649-659, 1991
5. J. Carter, J.A. Pugh and A. Monterrosa. Non-insulin-dependent diabetes
mellitus in
minorities in the United States. Ann. Int. Med. 125: 221-232, 1996
6. S-K. Ha and J-K. Seo. Insertionldeletion polymorphism in ACE gene as a
predictor for
progression of diabetic nephropathy. Kidney Int. 52: S28-S32, 1997
7. A.R. Andersen, J.S. Christiansen, J.K. Andersen, S. Kreiner and T. Deckert.
Diabetic
nephropathy in type I (insulin dependent) diabetes: an epidemiological study.
Diabetologia 25: 496-501, 1983
8. The effect of intensive treatment of diabetes on the development and
progression of long-
term complications in insulin-dependent diabetes mellitus. New Eng. J. Med.
329: 977-
986, 1993
9. S. Adler. Structure-function relationships in diabetic nephropathy: lessons
and limitations.
Kidney Int. 52: S43-S45, 1997
10. H.B. Lee, M.K. Cha, K.I. Song, J.H. Kim, E.Y. Lee, S.I. Kim, J. Kim and M.
H. Yoo.
Pathogenic role of advanced glycosylation end products in diabetic
nephropathy. Kidney
Int. 52: S60-S65, 1997
11. Kikkawa, R., Umemura, K., Haneda, M., Arimura, T., Ebata, K. and Shigeta,
Y. Evidence
for existence of polyol pathway in cultured rat mesangial cells. Diabetes 36:
240-243, 1987
12. F.N. Ziyadeh and D.C. Han. Involvement of transforming growth factor-~i
and its receptors
in the pathogenesis of diabetic nephropathy. Kidney Int. 52: S7-S11, 1997
44
CA 02258973 1999-O1-29
13. Koya D, Jirousek MR, Lin Y, Ishii H, Kuboki K, King GL: Characterization
of protein kinase
C ~3 isoform activation on the gene expression of transforming growth factor-
(3, extracellular
matrix components, and prostanoids in the glomeruli of diabetic rats. J Clin
Invest 100:
115-126, 1997
14. H. Ishii, M.R. Jirousek, D. Koya, C. Takagi, P. Xia, A. Clermont, S-E.
Bursell, T.S. Kern,
L.M. Ballas, W.F. Heath, L.E. Stratum, E.P. Feener and G.L. King. Amelioration
of
vascular dysfunctions in diabetic rats by an oral PKC(3 inhibitor. Science
272: 728-731,
1996
15. D. Mochly-Rosen. Localization of protein kinases by anchoring proteins: a
theme in signal
transduction. Science 268: 247-251, 1995
16. Ziyadeh FN: The extracellular matrix in diabetic nephropathy. Am J Kidney
Diseases 22:
736-744, 1993
17. Derubertis FR, Craven PA: Activation of protein kinase C in glomerular
cells in diabetes:
mechanisms and potential links to the pathogenesis of diabetic glomerulopathy.
Diabetes
43: 1-8, 1994
18. Porte D Jr., Schwartz MW: Diabetes complications: why is glucose
potentially toxic?
Science 272: 699-700, 1996
19. Fumo P, Kuncio GS, Ziyadeh FN: PKC and high glucose stimulate collagen a1
(IV)
transcriptional activity in a reporter mesangial cell line. Am J Physiol 267:
F632-F638,
1994
20. Brose N, Hofmann K, Hata Y, Sudhof C: Mammalian homologues of
Caenorhabditis
elegans unc-13 gene define novel family of C2-domain proteins. J Biol Chem
270: 25273-
25280, 1995
21. Maruyama IN, Brenner S: A phorbol ester/diacylglycerol-binding protein
encoded by the
unc-13 gene of Caenorhabditis elegans. Proc Natl Acad Sic USA 88: 5729-5733,
1991
22. Ahmed S, Maruyaam IN, Kozma R, Lee J, Brenner S, Lim L: The Caenorhabditis
elegans
unc-13 gene product is a phospholipid-dependent high-affinity phorbol ester
receptor.
Biochem J 287: 995-999, 199223
23. Kazanietz MG, Lewin NE, Bruns JD, Blumberg PM: Characterization of the
cysteine-rich
region of the Caenorhabditis elegans protein unc-13 as a high affinity phorbol
ester
receptor. J Biol Chem 270: 10777-10783, 199524
CA 02258973 1999-O1-29
24. Betz, A., U.Ashery, M. Rickmann, I. Augustin, E. Neher, T.C. Sudhof., J.
Rettig, and N.
Brose. 1998. Munc13 is a presynaptic phorbol ester receptor that enhances
neurotransmitter release. Neuron. 21:123-136.
25 Orita S, Naito A, Sakaguchi G, Maeda M, Igarashi H, Sasaki T, Takai Y:
Physical and
Functional Interactions of Doc2 and Munc13 in Ca2+-dependent Exocytotic
Machinery. J
Biol Chem 272: 16081-16084, 1997
24 Popov M, Tam LY, Li J, Reithmeier RA: Mapping the ends of transmembrane
segments in
a polytopic membrane protein: Scanning N-glycosylation mutagenesis of
extracytosolic
loops in the anion exchanger, Band 3. J Biol Chem 272:18325-18332, 1997
25 Kozak M: An analysis of vertebrate mRNA sequences: intimations of
translational control. J
Cell Biol 115: 887-903, 1991
26 C. Csortos, S. Zolnierowicz, E. Bako, S.D. Durbin and A.A. DePaoli-Roach.
High
complexity in the expression of the B' subunit of protein phosphatase 2A0. J.
Biol. Chem.
271: 2578-2588, 1996
27 D. Barford. Molecular mechanisms of the protein serineltherionine
phosphatases. TIBS
21: 407-412, 1997
28 Sudhof TC: The synaptic vesicle cycle: a cascade of protein-protein
interactions. Nature
375: 645-653, 1995
29 Williams B, Schrier RW: Glucose-induced protein kinase C activity regulates
arachidonic
acid release and eicosanoid production by cultured glomerular mesangial cells.
J Clin
I nvest 92:2889-2896, 1993
Blobe, G.C., Stribling, S., Obeid, L. M., and Hannum, Y.A. (1998). Protein
kinase C
isoenzymes: regulation and function. Cancer Surveys 27, 213-248.
31 Song, Y., Ailenberg, M., Silverman, M. (1998). Cloning of a novel gene in
the human
25 kidney homologous to rat munc13s: its potential role in diabetic
nephropathy. Kidney Int.
53, 1689-1695.
32 Banfield, D.K., Lweis, M.U., Rabouille, C., Warren, G., and Pelham, H.R.B.
(1994).
Localization of SedS, a putative vesicle targeting molecule, to the cis-Golgi
network
involves both its transmembrane and cytoplasmic domains. J. Cell Biol. 127,
357-371.
30 33 Tellam, J. T., Jams, D.E., Stevens, T.H., and Piper R.C. (1997).
Identification of a
mammalian Golgi Sec1 P-like protein, mVps45. J. Biol. Chem. 272, 6187-6193.
46
CA 02258973 1999-O1-29
34 Bock, J.B., Klumperman, J., Davanger, S., and Scheller, R.H. (1997).
Syntaxin 6 functions
in trans-Golgi network vesicle trafficking. Mol. Biol. Cell 8, 1261-1271.
35 Orita, S., Naito, A., Sakaguchi, G., Maeda, M., Igarashi, H., Sasaki, T.,
and Takai, Y.
(1997). Physical and functional interactions of Doc2 and Munc13 in Ca2+-
dependent
exocytotic machinery. J. Biol. Chem. 272, 1681-1684.
36 Martin, T.F.J. (1997). Greasing the Golgi budding machine. Nature 387, 21-
22.
37 Kearns, B.G., McGee, T.P., Mayinger, P., Gedvilaite, A., Phillips, S.E.,
Kagiwada, S., and
Bankaitis, V.A. (1997). Essential role for diacylglycerol in protein transport
from the yeast
Golgi complex. Nature 387, 101-105.
40 Chinen, K., Takahashi, E., and Nakamura, Y. (1996). Isolation and mapping
of a human
gene (SEC14L), partially homologous to yeast SEC14, the contains a variable
number of
tandem repeats (VNTR) site in its 3' untranslated region. Cytogene. Cell Gene.
73, 218-
223.
41 Mochly-Rosen, D., and Kauvar, L.M. (1998). Modulating protein kinase C
signal
transduction. Adv. Pharmacol. 44, 91-145.
42 Lavin, M.F., Watters, D., and Song, Q. (1996). Role of protein kinfise
activity in apoptosis.
Experientia 52, 979-994.
43 Deacon, E.M., Pongracz, J., Griffiths G., and Lord, J.M. (1997). Isoenzymes
of protein
kinase C: differential involvement in apoptosis and pathogenesis. Mol. Pathol.
50, 124-131.
44 Whitman, S.P., Civoli, F., and Daniel, L.W. (1997). Protein kinase Gill
activation by 1-(-D-
arabinofuranosylcytosine is antagonistic to stimulation of apoptosis and bcl-
2a down-
regulation. J. Biol. Chem. 272, 23481-23484.
45 Leszczynski, D. (1996). The role of protein kinase C in regulation of
apoptosis: a brief
overview of the controversy. Cancer J. 9, 308-313.
46 Xia, P., Inoguchi, T., Kern, T.S., Engerman, R.L., Oates, P.J., and King,
G.L. (1994).
Characterization of the mechanism for the chronic activation of diacyglycerol-
protein kinase
C pathway in diabetes and hypergalactosemia. Diabeties 43,1122-1129.
47
CA 02258973 1999-O1-29
47 Hise, M.K., and Mehta, P.S. (1988). Characterization and localization of
calciumlphospholipid-dependent protein kinase-C during diabetic renal growth.
Endocrinology 123: 1553-1558.
48 Derubertis, F.R., and Craven P.A. (1994). Activation of protein kinase C in
glomerular cells
in diabetes: mechanisms and potential links to the pathogenesis of diabetic
glomerulopathy. Diabetes 43, 1-8.
49 King, G.L., Ishii, H., and Koya, D. (1997) Diabetic vascular dysfunction: a
model of excessive
activation of protein kinase C. Kidney Int. 52 (suppl. 60), S-77-S-85.
50 Ishii, H., Jirousek, M.R., Koya, D., Takagi, C., Xia, P., Clermont, A.,
Bursell, S.E., Kern, T.S.,
Ballas, L.M., Heath, W.F., Stratum, L.E., Feener, E.P., and King, G.L. (1996).
Amelioration of
vascular dysfunctions in diabetic rats by an oral PKC (3 inhibitor. Science
272, 728-731.
51 Ortiz, A., Ziyadeh, F.N., and Neilson, E.G. (1997). Expression of apoptosis-
regulatory genes in
renal proximal tubular epithelia cells exposed to high ambient glucose and in
diabetic kidneys.
J. Invest. Med. 45, 50-56.
52 Morris, S.M., and Yu-Lee, L. (1998). Expression of RNUDC, a potential
nuclear movement
protein in mammalian cells: localization to the Golgi apparatus. Exp. Cell
Res. 238, 23-32.
53 Ailenberg M, Silverman M: Cellular activation of mesangial gelatinase A by
cytochalasin D is
accompanied by enhanced mRNA expression of both gelatinase A and its membrane-
associated gelatinase A activator (MT-MMP). Biochem J 313: 879-884, 1996
54 Zent R, Ailenberg M, Waddell TK, Downey GP, Silverman M: Puromycin
aminonucleoside
inhibits mesangial cell-induced contraction of collagen gels by stimulating
production of
reactive oxygen species. Kidney Int 47:811-817, 1995
55 Liang P, Pardee AB: Differential display of eukaryotic messenger RNA by
means of the
polymerase chain reaction. Science 257: 967-971, 1992
56 Sokolov BP, Prockop DJ: A rapid and simple PCR-based method for isolation
of cDNAs from
differentially expressed genes. Nucleic Acids Res 22: 4009-4015, 1994
57 Balch, W.E., Bunphy, W.G., Braell, W.A., and Rothman, J.E. (1994).
Reconstitution of the
transport of protein between successive compartments of the Golgi measured by
the coupled
incorporation of N-acetylglucosamine. Cell 39, 405-416.
48
CA 02258973 1999-O1-29
58 Eastman, A. (1995). Assays for DNA fragmentation, endonucleases, and
intracellular pH and
Ca2+ associated with apoptosis. In: Cell Death, Methods in Cell Biology, Vol.
46, L.M.
Schwartz and B.A. Osborne, eds. (San Diego, Academic Press), pp. !~1-55.
49