Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02274955 1999-06-14
WO 98/29431 PCT/US97/23553
-1-
METHOD TO DIAGNOSE AND TREAT PATHOLOGICAL CONDITIONS
RESULTING FROM DEFICIENT ION TRANSPORT
Technical Field
The invention relates to the fields of detecting and treating homozygous and
heterozygous genetic deficiencies in ion transport, particularly alterations
in nucleic
acid molecules and proteins that give rise to various forms of Bartter's
Syndrome and
Gitelman's Syndrome. More specifically, the invention provides compositions
and
methods for determining whether an individual is affected by or carriers a
mutation in
one or more genes involved in ion transport.
Background Art
In higher eukaryotes the maintenance of the proper ionic composition and
volume of the intravascular space is critical for normal neuromuscular
function and
delivery of oxygen and nutrients to tissues. The kidney plays a dominant role
in
determining the long-term set points of fluid and electrolyte balance,
maintaining
homeostasis despite wide variation in environmental exposure. Derangements in
these components of kidney function are likely to underlie a number of
clinical
disorders ranging from altered blood pressure due to changes in intravascular
volume
to abnormalities in electrolyte homeostasis. Examination of mendelian
disorders of
fluid and electrolyte homeostasis provides the opportunity to dissect the
fundamental
,, mechanisms governing this process. This effort provides insight into basic
physiology and also identify targets in which more subtle variation might
commonly
have effects in the population (Lifton, R.P., Proc. Nat. Acaa'. Sci. U.S.A.
92:8545-
8551 (1995)).
CA 02274955 1999-06-14
WO 98/29431 PCT/US97l23553
-2-
Bartter's Syndrome is an autosomal recessive disorder featuring hypokalaemic
metabolic alkalosis with salt wasting (Bartter, F.C., et al., Am. .I. Med.
33:811-828
( 1962)). Affected patients have been shown to have a diverse array of
additional
metabolic abnormalities, including elevated plasma renin activity (Bartter,
F.C., et al.,
Am. J. Med. 33:811-828 ( 1962)), hyperaldosteronism (Goodman, A.D., et al., N.
Eng.
J . Med. 281:1435-1439 (1969)), altered prostaglandin metabolism (Dune, M.J.,
Kid.
Int. 19:86-102 ( 1981 )), elevated levels of atrial natriuretic peptide (Imai,
M., et al., J .
Ped. 74:738-749 (1969)), Graham, R.M., et al., Hypertension 8:549-551 (1986)},
abnormal platelet function (Rodrigues Pereira, R., et al., Am. J. Med. Gen.
15:79-84
(1983)), and insensitivity to the vasoconstrictive effects of angiotensin II
and
norepinephrine (Bartter, F.C., et al., Am. J. Med. 33:811-828 (1962);
Silverberg, A.B.,
et al., Am. J. Med. 64:231-235 (1978)). Symptoms and signs of disease in
affected
patients reflect these diverse physiologic findings, and include signs of
intravascular
volume depletion (Bettinelli, A., et al., J. Pediatr. 120:38-43 ( I 992)),
seizures (Iwata,
F., et al., Acta Paed Japonica 35:252-257 (1993)), tetany (Bettinelli, A., et
al., J.
Pediatr. 120:38-43 (1992)), muscular weakness (Marco-Franco, J.E., et al.,
Clin.
Neph. 42:33-37 (1994)), paresthesias {Zarraga Larrondo, S., et al., Nephron
62:340-
344 ( 1992)), and joint pain with chondrocalcinosis (Smilde, TJ., et al., J.
of Rheum.
21:1515-1519 (1994)). Persistent abnormalities in electrolyte composition have
resulted in stunted growth and mental retardation in some affected subjects
(Simopoulos, A.P., et al., Nephron 23:130-135 ( 1979)). These profound
derangements in electrolyte homeostasis can lead to the misdiagnosis of
bulimia
and/or diuretic abuse in affected individuals (Okusa, M.D. and Bia, M.J.
Bartter's
Svndrome In H~tZ7lpne R?CiCtalll'P anrl Clthar Fnrinrrin' paradoxes, eds.
Cohen, P.
and Foa, P. 231-263 (Springer Verlag, New York, 1987)).
Bartter's Syndrome has been proposed to be a heterogeneous entity with at
least two subsets, Gitelman's Syndrome (Gitelman, H.J., Graham, J.B., and
Welt,
L.G. A new familial disorder characterized by hypokalaemia and hypomagnesemia.
Traps. Assoc. Am. Phys. 79:221-235 ( 1966)) and "true Bartter's Syndrome"
CA 02274955 1999-06-14
WO 98/29431 PCT/US97/23553
-3-
(Bettinelli, A., et al., J. Pediatr. 120:38-43 (1992)). Gitelman's Syndrome
refers to
the predominant subset of patients with hypokalaemic alkalosis in conjunction
with
hypocalciuria and hypomagnesemia, while true Bartter's Syndrome refers to
patients
with normal or hypercalciuria and typically normal magnesium levels. True
Bartter's
S patients are said to present clinically at early ages (less than S years)
with signs of
vascular volume depletion, while Gitelman's Syndrome patients typically
present at
older ages without overt hypovolemia (Bettinelli, A., et al., J. Pediatr.
120:38-43
( 1992)). Nonetheless, the overlapping features of these disorders has
resulted in
considerable confusion and controversy regarding their classification, with
many
patients having features of Gitelman's Syndrome being labeled as having
Banter's
Syndrome in the literature (Rudin, A., et al., Scand J. Urol. Nephrol. 22:35-
39
(1988)). The pathogenesis of these disorders has remained uncertain, with wide
speculation as to which observed abnormalities are primary and which are
secondary
consequences of underlying primary abnormalities (Clive, D.M. Am. J. Kid. Dis.
1 S 25:813-823 ( 199S)). Presently, there is not an easy method for
differentiating these
disorders; differentiation being based solely on evaluating the clinical
symptoms that
are presented.
Dissection of the physiology of renal electrolyte homeostasis has identified a
number of potential candidate genes for Gitelman's Syndrome and Bartter's
Syndrome; prior studies have investigated genes encoding atrial natriuretic
peptide
and the angiotensin II receptor (AT1 ) (Graham, R.M., et al., Hypertension
8:549-SS 1
( I 986), Yoshida, H., et al., Kid. Int. 46:1 SOS-1 S09 ( 1994)). Another
attractive
candidate gene is the thiazide-sensitive Na-CI cotransporter of the distal
convoluted
tubule (thiazide-sensitive cotransporter, TSC), which is believed to be the
principle
2S mediator of sodium and chloride reabsorption in this nephron segment,
accounting for
a significant fraction of net renal sodium reabsorption (Ellison, D.H., Ann.
Int. Med.
I 14:886-894 ( I 991 )). This cotransporter is the target of thiazide
diuretics, one of the
major classes of agents used in the treatment of high blood pressure. cDNAs
encoding the TSC have recently been cloned from flounder bladder and rat
kidney
CA 02274955 1999-06-14
WO 98/29431 PCT/US97/23553
-4-
(Gamba, G., et al., Proc. Natl. Acad. Sci. U.S.A. 90:2749-2753 (1993); Gamba,
G., et
al., J. Biol. Chem. 269:17713-17722 ( 1994)). The encoded protein from rat
comprises
1002 amino acids, and contains twelve putative transmembrane domains, with
long
intracellular amino and carboxy termini. Similarities in some features of
patients with
Gitelman's Syndrome and patients receiving thiazide diuretics raise the
possibility
that mutation in TSC causing loss of function could result in Gitelman's
Syndrome.
This consideration motivates examination of TSC as a candidate gene for
Gitelman's
Syndrome.
In Example 1, it is demonstrated that Giteiman's Syndrome is a genetically
homogeneous autosomal recessive trait caused by loss of function mutations in
the
thiazide-sensitive Na-Cl cotransponer protein (TSC) located in the renal
distal
convoluted tubule. The predominant clinical and physiologic abnormalities seen
in
these patients can be explained by the resultant salt wasting from this
nephron
segment.
These observations in patients with Gitelman's Syndrome Leave open the
question of whether Banter's Syndrome is an allelic variant of Gitelman's
Syndrome
or is due to mutation in a different gene. The occurrence of salt wasting,
impaired
urinary concentration and calcium wasting in Bartter's patients suggests a
primary
renal tubular defect in the thick ascending limb (TAL) of the loop of Henle
(Gill, J.R.,
et al., Am. J. Med. 65:766-772 (1978)). The absorptive variant of the
bumetanide-
sensitive Na-K-2C1 cotransponer (NKCCZ , also known as SLC I 2A 1 ) is the
primary
mediator of sodium and chloride reabsorption in this nephron segment (Greger,
R.,
Physiol. Rev. 65:760-797 (1985)), and loss of function of this cotransporter
could
produce many of the features seen in affected patients. Indeed, loop
diuretics, specific
antagonists of this cotransporter, can produce electrolyte disturbances very
similar to
those seen in patients with Banter's Syndrome (Greger, R., et al., Klin.
Woschenschr.
6/:1019-1027 (1991)).
cDNA's encoding NKCC2 have recently been cloned from rat (Gamba,:G. et
al., J. Biol. Chem. 26:17713-17722 (1994}), rabbit (Payee, J.A., et al., Proc.
Natl.
CA 02274955 1999-06-14
WO 98/29431 PCT/US97/23553
-5-
Acad Sci. (U.S.A.) 91:4544-4548 (1994)) and mouse (Igarashi, P. et al., Am. J.
- Physiol. 269:F405-F418 ( 1995)); a secretory variant of this cotransponer,
NKCC 1
(also known as SLC12A2) has also been cloned from shark (Xu, J.-C, et al.,
Proc.
Natl. Acad. Sci. (U.S.A.) 91:2201-2205 (1994)) and human (Payee, J.A. et al.,
J. Biol.
Chem. 270:17977-17985 {1995)). All members of this family have 12 putative
transmembrane spanning domains and also show structural and sequence
similarity to
TSC. By investigation of families with Banter's Syndrome, Example 2
demonstrates
that this variant of inherited hypokalaemic alkalosis is caused by mutations
in the
gene encoding NKCC2. These findings explain the molecular basis of this
disease
and suggest possible clinical features of the more common heterozygous carrier
state.
In Examples 1 and 2, evidence is presented that demonstrates that autosomal
recessive hypokalaemic alkalosis with salt wasting and low blood pressure can
be
caused by mutations in either of two genes. Mutations in TSC (locus symbol
SLC 12A3, sometimes referred to as NCCT), encoding the thiazide-sensitive Na-
Cl
cotransponer of the renal distal convoluted tubule, cause Gitelman's Syndrome,
featuring salt wasting and hypokalaemic alkalosis associated with marked
hypocalciuria and hypomagnesemia (Example 1 and Simon, D.B. et al., Nature
Genet.
12:24-30 { 1996)). Mutations in NKCC2 (locus symbol SLC 12A 1 ), encoding the
renal
bumetanide-sensitive Na-K-2Cl cotransporter of the thick ascending limb of
Henle's
loop (TAL), cause Banter's Syndrome, featuring salt wasting and hypokalaemic
alkalosis associated with marked hypercalciuria and frequently
nephrocalcinosis
(Example 2 and Simon, D.B., et al., Nature Genet. 13:183-188 (1996)). While
Banter's patients typically are born prematurely with polyhydramnios and show
marked dehydration in the neonatal period, Gitelman's patients typically
present at
older ages with neuromuscular signs and symptoms (Wang, W., et al., Ann. Rev.
Physiol. 54:81-96 {1992)).
Mutations in genes whose products regulate activity of either of these
cotransporters could potentially lead to similar clinical phenotypes. An
apical ATP-
sensitive K+ channel has been implicated as one such regulator of the Na-K-2C1
CA 02274955 1999-06-14
WO 98/29431 PCT/US97/23553
-6-
cotransporter in the TAL (Wang, W., et al., Ann. Rev. Physiol. 54:81-96 (
1992},
Giebisch, G., Kidneylnt. 48:1004-1009 (1995)). Since K+ levels in the TAL are
much
lower than levels of Na+ and Cl-, availability of tubular K+ is rate limiting
for
cotransporter activity; K+ entering the cell from the tubule must be
"recycled" to the
lumen in order to permit sustained cotransport activity. This key role of K+
channels
in the regulation of cotransporter activity is demonstrated by the ability of
potassium
channel antagonists to virtually abolish Na-K-2C1 cotransporter activity,
Giebisch, G.,
Kidneylnt. 48:1004-1009 (1995)}.
An inwardly rectifying K+ channel (IRK) bearing many features of this
regulatory channel (low single channel conductance, activation by low levels
of ATP
and protein kinase A (PKA), and insensitivity to voltage and calcium) has been
cloned
(Ho, K., et al., Nature 362:31-38 (1993)). This channel, ROMK (locus symbol
KCNJI ), is the prototype of the IRK family of potassium channels, comprising
two
transmembrane spanning domains, and a segment homologous to the characteristic
HS
pore domain. The channel contains PKA phosphorylation sites that are required
for
normal channel activity. Multiple ROMK isoforms encoded by the same
chromosome 11 locus are generated by alternative splicing (Yano, H., et al.,
Mol.
Pharmacology 45:854-860 (1994); Shuck, M.E. et al., J. Biol. Chent. 269:24261-
24270 ( 1994)); these isoforms have been shown to be expressed in the kidney,
specifically on the apical membrane of cells of the TAL as well as more distal
nephron segments (Lee, W.S., et al., Am. J. Physiol. (Renal Fluid Electrol.
Physiol.)
268:F1124-31 (1995); Boim, M.A. et al., Am. J. Physiol. (Renal Fluid Electrol.
Physiol.) 268:F1132-40 (1995); Hebert, S.C., Kidney Int. 48, 1010-1016
(1995)).
This channel has been proposed to be involved in potassium recycling in the
TAL, as
well as in net renal potassium secretion in the distal nephron.
The present invention provides compositions and methods that can be used to
differentiate and diagnose several types of ion transport deficiencies,
particularly
Bartter's Syndrome and Gitelman's Syndrome. The present invention further
provides methods and compositions that can be used to identify heterozygote
carriers
CA 02274955 1999-06-14
WO 98!29431 PCT/US97/23553
7-
for these disorders. Carriers, though not displaying severe clinical symptoms,
nonetheless display mild to moderate pathologies.
Summary of the Invention
The present invention is based, in part, on the identification of the roles of
the
human thiazide-sensitive Na-Cl cotransporter, TSC; the human ATP-sensitive K+
channel, ROMK; and the human Na-K-2CI cotransporter, NKCC2 in pathological
condition associated with abnormal ion transport, particularly Bartter's
Syndrome,
Gitelman's Syndrome, hypokalaemic alkalosis, hypokalaemic alkalosis with
hypercalciuria, kidney stones, high blood pressure, osteoporosis and
sensitivity to
diuretic-induced hyperkalaemia. The present invention specifically provides
the
amino acid sequences of several human wild-type and altered variants of the
TSC,
NKCC2 and ROMK proteins as well as the nucleotide sequence that encodes these
variants. These proteins and nucleic acid molecules can be used in diagnosing
ion
1 S transport disorders and in developing methods and agents for treating
these
pathologies.
Brief Descriptions of the Drawings
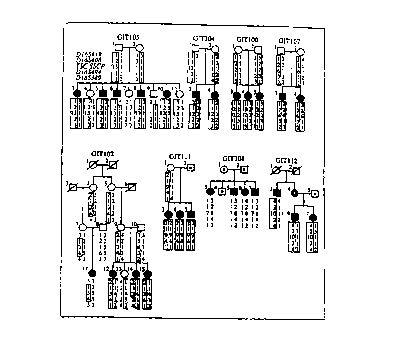
1 The
familial relationships of Gitelman's Syndrome kindreds used for linkage
studies are
shown. Individuals with Gitelman's Syndrome are indicated by filled symbols;
individuals who do not have Gitelman's Syndrome are indicated by unfilled
symbols;
deceased individuals are indicated by a diagonal line through the symbol.
Individuals
not sampled for genetic studies are indicated by a dot within the symbol. Each
kindred is given a unique kindred number, and each individual within the
kindred is
numbered above and to the left of the symbol. Below each symbol, genotypes at
loci
on chromosome 16 are shown in their map order (see Figure 3 for map of loci).
In
descending order, loci shown are D 165419, D 16S408, TSC SSCP, D 165494 and
CA 02274955 1999-06-14
WO 98129431 PCT/US97/23553
_g_
D 165389. TSC SSCP refers to variants identified in TSC; different variants
are given
different allele numbers and the nature of each variant is indicated in Table
1. SSCP
allele 1 represents the wild-type SSCP variant. Inferred haplotypes
cosegregating
with Gitelman's Syndrome are enclosed by boxes, with maternal and paternal
haplotypes distinguished by shaded or unfilled boxes, respectively.
A. Cosmid cnr,tig~g the T~r' ~~~"~ Thin horizontal bars represent
cloned human genomic DNA of cosmid clones; vertical bars indicate sites
cleaved by
restriction endonuclease EcoRI. Independent clones are drawn with their
overlaps
indicated, and the 5' to 3' orientation of transcription is shown.
~. Intron-exon organization of the TS g,~. Gray boxes indicate exons of
the TSC gene. The gene is composed of 26 coding exons, encoding a protein of
1021
amino acids. The first codon of each exon is indicated; the position in the
codon of
the first base of each exon is indicated by the subscript (e.g. 1672 indicates
that the
1 S first base of exon 4 is the second base in codon 167). The exact size of
each intron is
not known.
C.C. Seauence of the human TS protein. The sequence of the human TSC
protein is shown in single letter code. The corresponding sequence of the rat
and
flounder TSC is shown below the human sequence; amino acids that are identical
compared with the human sequence are indicated by dots, while different amino
acids
in the TSC of these species are indicated. The transmembrane domains proposed
from hydropathy plots are shaded and numbered M 1 to M 12. Amino acids that
are
mutated on Gitelman's Syndrome alleles are highlighted, appearing in white on
a
black background. These variants are numbered and correspond to those
indicated in
Figure 1, Table 1 and Figure 5.
Figure 3 Meltinoint lin_kag analy;zs of Gitelman's S3mdrome and loci nn
chromosome 16 Multipoint linkage analysis was performed, testing for linkage
of
Gitelman's Syndrome to a segment of chromosome 16 containing loci D 165419,
CA 02274955 1999-06-14
WO 98/29431 PCT/US97123553
-9-
D 165408, D 165494 and D 165389. These loci are shown in their map order, with
the
distance between adjacent loci indicated in centimorgans {Gyapay, G., et al.,
Nature
Genet. 7:246-339 { 1994); Shen, Y.S., et al., GenomicS 22:68-76 ( I 994)). The
. multipoint lod score for linkage of Gitelman's Syndrome across this interval
is shown,
revealing a lod score of 9.5 at a recombination fraction of zero with D
165408, and
showing odds of greater than i 000:1 favoring location of the trait locus in
the 11
centimorgan interval def ned by flanking loci D 165419 and D 165494. At the
top of
the figure, the lod-1 support intervals for the location of the TSC locus
defined in
CEPH kindreds and for the location of the Gitelman's locus in disease kindreds
are
shown, revealing that they overlap. Moreover, molecular variants in TSC show
linkage to Gitelman's Syndrome at a recombination fraction of zero (see Figure
I ).
FiQUre 4 Novel vat7antg In ~,l~leritS t,~yjth IltPlman'e C~rr,~ir ~~~ Variants
in
patients with Gitelman's Syndrome were identified by SSCP and subjected to DNA
sequence analysis as described in Methods. Representative examples are shown.
I 5 Autoradiograms of variants detected on non-denaturing gels (panels
A,B,C,E,F} or
denaturing gels (panel D} are shown at the left of each panel; patients are
identified as
in Figure l, and subjects with Gitelman's Syndrome are indicated by asterisks;
arrows
indicate variants specific for Gitelman's Syndrome kindreds. At the right of
each
panel the DNA sequences of the corresponding variant (top) and wild-type
(bottom)
are shown. Variant bases are indicated by an asterisk; in panel D, the 3 bases
in the
wild-type sequence that are deleted in the variant are indicated by a bracket.
All
sequences are shown in the antisense orientation with respect to the gene.
With the
exception of panels D and E, 9 bases, corresponding to the mutated codon and
the two
flanking codons, are shown. ~ Variant alters 8209 to W in G1T I02; ~ Variant
alters P349 to L in GIT107; ,~ Variant alters C421 to R in GIT102; p,, Three
base
. deletion changes sense sequence CCTTCA encoding PS561 to CCA, deleting codon
561 in GIT108; ~ Variant in sense orientation changes consensus 3' splice site
CAG
to CAT in intron 15 in GIT102. F Variant alters 8955 to Q in GIT111.
CA 02274955 1999-06-14
WO 98/29431 PCTIUS97/23553
-10-
i i '
~t~ndrome atient~ The TSC protein is represented as a 12 transmembrane domain
protein with intracytoplasmic amino and carboxy termini (Gamba, G., et al.,
Proc.
Natl. Acad. Sci. U.S.A. 90:2749-2753 (/993); Gamba, G., et al., .I. Biol.
Chem.
269:17713-17722.(1994)). The sites of mutations in exons identified in
Gitelman's
Syndrome patients are indicated; the numbers correspond to the numbered
variants in
Figure 1, 2c and Table 1. Mutations altering consensus splice sites are not
shown;
these are indicated in Tables 1, 3 and 4.
Figure 6. BartrPr's ~mdrome ~indrea;. Family relationships are shown.
Affected subjects, unaffected subjects, living unsampled subjects and deceased
subjects are indicated by filled symbols, unfilled symbols, dotted symbols and
diagonal lines, respectively. Index cases are indicated by an arrow. Genotypes
of loci
tightly linked to NKCC2 are indicated and are arranged in their chromosomal
order
(see Figure 7c); loci are identified to the left of kindred BAR152. Novel SSCP
variants detected in NKCC2 in each kindred are numbered, with the wild-type
SSCP
variant denoted by +. Affected offspring of consanguineous union are seen to
be
homozygous for all loci linked to NKCC2, and are homozygous far novel SSCP
variants.
Figure 7 Character;~atinn nfrhP h,~..,~" genomic NK r'2 l~rnc
A. Intron-exon organization. Gray boxes indicate the 26 exons encoding the
NKCC2 protein. The first codon in each exon is indicated; exons that begin
with the
second or third base of a codon are indicated by the subscript 2 or 3,
respectively.
B. Seauence of human NK C'? "rnrPin ~~$le letter co' ' The sequence of
the corresponding rat and shark sequence is shown beneath the human sequence.
Amino acids that are identical to human residues are indicated by dots while
residues
that are different in these species are indicated. Transmembrane domains
proposed
from hydropathy plots are shaded and numbered M 1 to M 12.
CA 02274955 1999-06-14
WO 98129431 PCT/US97/23553
C.Localization of NK on the human gene ~r map, Multipoint linkage
analysis of NKCC2 marker NKCGT7-3 and loci on I Sq is shown. 15q loci are
shown
in their map order, with 1 Sqter to the right, and the estimated genetic
distance between
adjacent loci in centimorgans is indicated; the lod score for linkage of NKCC2
to each
location on the map is plotted. The lod score peaks in the 3 cM interval
flanked by
D 1 SS I 32 and D I SS209, and location in this interval is supported by odds
of more
than 100:1 over any alternative interval.
' Variants
were identified by SSCP and subjected to DNA sequence analysis. Representative
examples of autoradiograms are shown at the top of each panel, and the
corresponding
DNA sequence of the sense strand of wild-type (left) and mutant alleles
(right) are
shown at the bottom of each panel. Patients are numbered as in Figure 6, and
subjects
with Bartter's Syndrome are indicated by asterisks. The symbol nl represents
unrelated normal subjects. Arrows indicate variants specific for Bartter's
Syndrome
kindreds. In panels a and b, brackets above the sequence figures indicate the
positions
of single base insertions or deletions. In panels d and e, variant bases are
indicated by
an asterisk above the sequence figures. ~, A single base insertion in codon
ATG
M195 results in a frameshift mutation. ~ A single base deletion produces a
frameshift in codon CGG 8302. ~ The last base of exon 14, representing the
first
base of codon 648, is mutated from G to A, changing D648 to N648. ~ G to T
transversion in the first base of codon 272 alters V272 to F272.
i
'o
A diagram of a nephron is shown. Plasma is filtered at the crescent-shaped
structure
representing the glomeruius) and sodium is reabsorbed as filtrate passes along
the
nephron. The physiologic mediators of sodium reabsorption are indicated, and
the
" fraction of filtered sodium that is normally reabsorbed by each pathway is
indicated.
Disorders resulting from mutations in specific mediators of sodium
reabsorption are
indicated. The principle mediators of sodium reabsorption are: Na+-H+ exchange
in
the proximal tubule; Na-K-2C1 cotransport in the thick ascending limb of
Henle; Na-
58814
CA 02274955 1999-06-14
WO 98/29431 PCT/US97/23553
-12-
Cl cotransport in the distal convoluted tubule; electrogenic sodium
reabsorption via
the epithelial sodium channel, composed of at least 3 different subunits, in
the distal
nephron. This last pathway is indirectly coupled to secretion of K+ and H+.
Figure 10. BaTttPr'c . ~drome ~indre~~. Family relationships are shown.
Affected subjects, unaffected subjects, living unsampled subjects and deceased
subjects are indicated by filled symbols, unfilled symbols, dotted symbols and
diagonal lines, respectively. Index cases are indicated by an arrow. Genotypes
of loci
tightly linked to NKCC2 are indicated and are arranged in their chromosomal
order;
these 5 loci are linked within a 3 cM interval; GT7-3 is present on the same
PAC
clones as NKCC23. Below these genotypes, novel SSCP variants detected in ROMK
in each kindred are numbered, and correspond to numbered variants in Figure
11; the
wild-type SSCP variant is denoted by +, Linkage to NKCC2 is seen to be
excluded in
the consanguineous kindreds BAR159 and BAR161. In all 4 kindreds, novel ROMK
variants are identified that cosegregate with the disease.
Fiuure 11. Novel varian s in Nx<'r~ ;" Bartter'~ ~vndrome ' nts. Variants
were identified by SSCP and subjected to DNA sequence analysis. Representative
examples of autoradiograms are shown at the top of each panel, and the
corresponding
DNA sequence of the sense strand of wild-type (left) and mutant alleles
(right} are
shown at the bottom of each panel. Patients are numbered as in Figure 10, and
subjects with Bartter's Syndrome are indicated by asterisks. The symbol nl
represents
unrelated normal subjects. Arrows indicate variants specific for Bartter's
Syndrome
kindreds, and are numbered as in Figure 10. In panels b and c, brackets above
the
sequence figures indicate the positions of base pair insertions or deletions.
In panels a
and d, variant bases are indicated by an asterisk above the sequence figures.
~ A
single base substitution changes codon TAC (Y60} to TAG (StopbO) in BAR159;
this
mutation is homozygous in affected, but not unaffected kindred members. ~ A
single
base insertion produces a frameshift in codons 13-14 in BAR161; this mutation
is
homozygous in the affected member of this kindred. ~,, A 4 base deletion
spanning
codons 313-314 results in a &ameshift mutation; the affected subject has
another
CA 02274955 1999-06-14
WO 98/29431 PCT/US97I23553
-13-
missense mutation (A195V) on the other ROMK allele (data not shown). 1~, A
single
base substitution changes codon AGC (S200) to AGG (R200); this substitution
eliminates a PKA phosphorylation site. The affected subject has a nonsense
mutation
on the other ROMK allele (W58Stop).
i w. ,
A schematic diagram of ROMK2 is shown and depicted as spanning the plasma
membrane twice with an HS domain containing the channel pore (Ho, K., et al.,
Nature 362:31-38 (1993}). The locations and consequences of mutations
identified in
Bartter's patients are identified.
IO
Modes of Carrying Out the Invention
I. General Description
The present invention is based, in part, on the identification of the roles of
the:
human thiazide-sensitive Na-Cl cotransporter, TSC; the human ATP-sensitive K+
channel, ROMK; and the human Na-K-2Cl cotransporter, NKCC2 in pathological
conditions associated with abnormal ion transport, particularly Bartter's
Syndrome,
Gitelman's Syndrome, hypokalaemic alkalosis, hypokalaemic alkalosis with
hypercalciuria, kidney stones, high blood pressure, osteoporosis and
sensitivity to
diuretic induced hyperkalaemia. The present invention specifically provides
the
amino acid sequences of several human wild-type and altered variants of the
TSC,
NKCC2 and ROMK proteins as well as the nucleotide sequence that encodes these
variants. These proteins and nucleic acid molecules can be used in diagnosing
ion
transport disorders and in developing methods and agents for treating these
pathologies.
CA 02274955 1999-06-14
WO 98129431 PCTIUS97l23553
- 14-
II. Specific Embodiments
A. TSC, NKCC2 or ROMK Protein
Prior to the present invention the art had identified: the amino acid sequence
of one allelic variant of a presumably wild-type rat and a wild-type flounder
thiazide-
sensitive Na-Cl cotransporter protein (TSC); the amino acid sequence of
several
allelic variants of a presumably wild-type human ATP-sensitive K+ channel
protein
(ROMK); and the amino acid sequence of one allelic variant of a presumably
wild-
type rat, a wild-type rabbit and a wild-type mouse Na-K-2C1 cotransporter
protein
(NKCC2). However, prior to the present invention, no one had identified that
alterations in the human variants (homologues) of these proteins result in
viable
individuals that suffer from pathologies caused by abnormal ion transport; no
one had
characterized naturally occurring human wild-type variants of the TSC and
NKCC2
proteins; no one had characterized human altered variants of the TSC, ROMK and
NKCC2 proteins; and no one had shown that pathological conditions that are a
result
1 S of abnormal ion transport, such as Gitelman's Syndrome and Banter's
Syndrome,
could be identified by analyzing a sample for the presence of a wild-type or
altered
variant of a TSC, NKCC2 or ROMK protein. The present invention provides, in
part,
the amino acid sequences of several allelic variants of wild-type human TSC
protein,
wild-type human NKCC2 protein, altered variants of the human TSC protein that
give
rise to ion transport def ciencies, and altered variants of the human NKCC2
protein
that give rise to ion transport deficiencies, altered variants of the human
ROMK
protein that give rise to ion transport deficiencies, as well as the
nucleotide sequence
of the encoding nucleic acid molecules.
In one embodiment, the present invention provides the ability to produce
previously unknown wild-type and altered variants of the human TSC, NKCC2 and
ROMK proteins using the cloned nucleic acid molecules herein described.
As used herein, a wild-type human TSC protein refers to a protein that has the
amino acid sequence of a wild-type allelic variant of human TSC. In Example 1,
CA 02274955 1999-06-14
WO 98/29431 PCT/US97/23553
- 15-
DNA sequencing was performed on DNA isolated from 50 unrelated healthy
individuals to identify wild-type TSC encoding DNA molecules. Figure 2 and
Table
3 provide the amino acid sequences of several wild-type allelic variants of
the human
~ TSC protein. The wild-type TSC proteins of the present invention include
those
S specifically identified and characterized herein as well as allelic variants
that can be
isolated and characterized without undue experimentation following the methods
outlined below. For the sake of convenience, all of the wild-type human TSC
proteins
of the present invention will be collectively referred to as the wild-type TSC
proteins
or the wild-type human TSC proteins of the present invention.
The term "wild-type human TSC proteins" includes all naturally occurring
allelic variants of the human TSC protein that posses normal TSC activity. In
general,
wild-type allelic variants of the TSC protein may/will have a slightly
different amino
acid sequence than that specifically provided in Seq. ID Nos - for the herein-
described wild-type TSC proteins. Allelic variants, though possessing a
slightly
different amino acid sequence than those recited above, will posses the
ability to
transport Na, Cl, Ca, and Mg at levels equivalent to the wild-type TSC
proteins herein
described. Typically, allelic variants of the wild-type TSC protein will
contain
conservative amino acid substitutions from the wild-type TSC sequences herein
described or will contain a substitution of an amino acid from a corresponding
position in a TSC homologue (a TSC protein isolated from an organism other
than
human such as the rat or flounder homologues). Figure 2 and Table 3 identify
conserved amino acid residues.
As used herein, a mutated or altered human TSC protein refers to a protein
that
has the amino acid sequence of a mutated or altered allelic variant of human
TSC.
Figure 4, Table l and Table 4 provide the amino acid sequences of several
mutated or
altered allelic variants of the human TSC protein. The mutated or altered TSC
proteins of the present invention include those specifically identified and
characterized herein as well as allelic variants that can be isolated and
characterized
without undue experimentation following the methods outlined below. For the
sake
CA 02274955 1999-06-14
WO 98/29431 PCT/US97123553
- lb -
of convenience, all of the mutated or altered human TSC proteins of the
present
invention will be collectively referred to as the mutated or altered TSC
proteins or the
mutated or altered human TSC proteins of the present invention.
The term "mutated or altered human TSC proteins" includes all naturally
occurring allelic variants of the human TSC protein that do not posses normal
TSC
activity. In general, mutated or altered allelic variants of the TSC protein
may/wiil
have a slightly to a radically different amino acid sequence than that
specifically
provided in Seq. ID Nos - for the herein-described wild-type TSC proteins.
Mutated or altered allelic variants will lack or have a reduced ability to
transport one
or more of the ions that are transported by wild-type TSC. Typically, allelic
variants
of the mutated or altered TSC protein contain: non-conservative amino acid
substitutions from the wild-type sequences herein described, a substitution of
an
amino acid other than the amino acid found in a corresponding position in a
TSC
homologue (a TSC protein isolated from an organism other than human), a frame
shift
mutation, an insertion of a stop codon, or a deletion or insertion of one or
more amino
acids into the TSC sequence.
As used herein, a wild-type human NKCC2 protein refers to a protein that has
the amino acid sequence of a wild-type allelic variant of human NKCC2. In
Example
2, DNA sequencing was performed on DNA isolated from 50 unrelated, healthy
individuals to identify wild-type. 1VKCC2 encoding DNA molecules. Figure 6
provides the amino acid sequences of the only wild-type allelic variant of the
human
NKCC2 protein thus far identified. Variations were seen in intron regions but
no
variation has been observed in the exon regions. The wild-type NKCC2 proteins
of
the present invention include the one specifically identified and
characterized herein
as well as allelic variants that can be isolated and characterized without
undue
experimentation following the methods outlined below. For the sake of
convenience,
all of the wild-type human NKCC2 proteins of the present invention will be
collectively referred to as the wild-type NKCC2 proteins or the wild-type
human
NKCC2 proteins of the present invention.
CA 02274955 1999-06-14
WO 98129431 PCT/US97/23553
-17-
The term "wild-type human NKCC2 proteins" includes all naturally occurring
allelic variants of the human NKCC2 protein that posses normal NKCC2 activity.
In
general, wild-type allelic variants of the NKCC2 protein may/wilI have a
slightly
different amino acid sequence than that specifically provided in Seq. ID Nos _
for
the herein-disclosed wild-type NKCC2 proteins. Allelic variants, though
possessing a
slightly different amino acid sequence than those recited above, will posses
the ability
to transport Na, Cl, K, and Ca at levels equivalent to the wild-type NKCCS
proteins
herein described. Typically, allelic variants of the wild-type NKCC2 protein
will
contain conservative amino acid substitutions from the wild-type sequences
herein
described or will contain a substitution of an amino acid from a corresponding
position in a NKCC2 homologue (a NKCC2 protein isolated from an organism other
than human).
As used herein, a mutated or altered human NKCC2 protein refers to a protein
that has the amino acid sequence of a mutated or altered allelic variant of
human
NKCC2. Figure 8 and Table 7 provide the amino acid sequences of several
mutated
or altered allelic variants of the human NKCC2 protein. The mutated or altered
NKCC2 proteins of the present invention include those specifically identified
and
characterized herein as well as allelic variants that can be isolated and
characterized
without undue experimentation following the methods outlined below. For the
sake
of convenience, all of the mutated or altered human NKCC2 proteins of the
present
invention will be collectively referred to as the mutated or altered NKCC2
proteins or
the mutated or altered human NKCC2 proteins of the present invention.
The term "mutated or altered human NKCC2 proteins" includes all naturally
occurring allelic variants of the human NKCC2 protein that do not posses
normal
NKCC2 activity. In general, mutated or altered allelic variants of the NKCC2
protein
may/will have a slightly to a radically different amino acid sequence than
that
specifically provided in Seq. ID Nos _ for the herein-described wild-type
NKCC2
proteins. Mutated or altered allelic variants will be not be able to transport
one or
more of the ions that are transported by wild-type NKCC2 or will transport
ions at a
CA 02274955 1999-06-14
WO 98/29431 PCT/US97I23553
- I8-
rate that is substantially lower than the wild-type proteins. Typically,
allelic variants
of the mutated or altered NKCC2 protein contain: non-conservative amino acid
substitutions from a wild-type sequences herein described, a substitution of
an amino
acid other than the amino acid found in a corresponding position in a NKCC2
homologue (a NKCC2 protein isolated from an organism other than human), a
frame
shift mutation, an insertion of a stop codon, or a deletion or insertion of
one or more
amino acids into the NKCC2 sequence.
As used herein, a wild-type human ROMK protein refers to a protein that has
the amino acid sequence of a wild-type allelic variant of human ROMK. In
Example
3, DNA from 50 unrelated, healthy individuals was sequenced to identify wild-
type
ROMK encoding DNA molecules. The amino acid sequences of the only wild-type
allelic variant of the human ROMK protein identified are disclosed in
Variations were seen in intron regions. However, no variation was observed in
the
exon regions of all ROMK encoding DNA molecules thus far examined. The wild-
1 S type ROMK proteins of the present invention include that specifically
identified and
characterized in the art as well as allelic variants that can be isolated and
characterized
without undue experimentation following the methods outlined below. For the
sake
of convenience, all of the wild-type human ROMK proteins of the present
invention
will be collectively referred to as the wild-type ROMK proteins or the wild-
type
human ROMK proteins of the present invention.
The term "wild-type human ROMK proteins" includes ali naturally occurring
allelic variants of the human ROMK protein that posses normal ROMK activity.
In
general, wild-type allelic variants of the ROMK protein will have a slightly
different
amino acid sequence than that specifically provided in Seq..ID Nos - Allelic
variants, though possessing a slightly different amino acid sequence than
those recited
above, will posses the ability to be an ATP sensitive K transporter.
Typically, allelic
variants of the wild-type ROMK protein will contain conservative amino acid
substitutions from the wild-type sequences herein described or will contain a
CA 02274955 1999-06-14
WO 98/29431 PCT/US97123553
- 19-
substitution of an amino acid from a corresponding position in a ROMK
homologue
(a ROMK protein isolated from an organism other than human).
As used herein, a mutated or altered human ROMK protein refers to a protein
that has the amino acid sequence of a mutated or altered allelic variant of
human
S ROMK. Figures 1 l and I2 and Table 10 provide the amino acid sequences of
several
mutated or altered allelic variants of the human ROMK protein. The mutated or
altered ROMK proteins of the present invention include those specifically
identified
and characterized herein as well as allelic variants that can be isolated and
characterized without undue experimentation following the methods outlined
below.
For the sake of convenience, all of the mutated or altered human ROMK proteins
of
the present invention will be collectively referred to as the mutated or
altered ROMK
proteins or the mutated or altered human ROMK proteins of the present
invention.
The telTn "mutated or altered human ROMK proteins" includes all naturally
occurring allelic variants of the human ROMK protein that do not posses normal
ROMK activity. In general, mutated or altered allelic variants of the ROMK
protein
may/will have a slightly to a radically different amino acid sequence than
that
specifically provided in Seq. ID Nos - for the herein-described wild-type ROMK
proteins. Mutated or altered allelic variants will be not be able to transport
one or
more of the ions that are transported by wild-type ROMK. Typically, allelic
variants
of the mutated or altered ROMK protein will contain: non-conservative amino
acid
substitutions from the wild-type sequences herein described, a substitution of
an
amino acid other than the amino acid found in a corresponding position in a
ROMK
homologue (a ROMK protein isolated from an organism other than human), a frame
shift mutation, an insertion of a stop codon, or a deletion or insertion of
one or more
amino acids into the ROMK sequence.
The TSC, NKCC2 and ROMK proteins of the present invention {wild-type
and mutated variants) are preferably in isolated from. As used herein, a
protein is said
to be isolated when physical, mechanical or chemical methods are employed to
remove the TSC, NKCC2 or ROMK protein from cellular constituents that are
CA 02274955 1999-06-14
WO 98129431 PCTIUS97/23553
-20-
normally associated with the protein. A skilled artisan can readily employ
standard
purification methods to obtain an isolated TSC, NKCC2 or ROMK protein. The
nature and degree of isolation will depend on the intended use.
The cloning of TSC, NKCC2 and ROMK encoding nucleic acid molecules
makes it possible to generate defined fragments of the TSC, NKCC2 and ROMK
proteins of the present invention. As discussed below, fragments of the TSC,
NKCC2
and ROMK proteins of the present invention are particularly useful in
generating
domain specific antibodies, in identifying agents that bind to a TSC, NKCC2 or
ROMK protein and in identifying TSC, NKCC2 or ROMK intra- or extracellular
binding partners.
Fragments of the TSC, NKCC2 and ROMK proteins can be generated using
standard peptide synthesis technology and the amino acid sequences disclosed
herein.
Alternatively, recombinant methods can be used to generate nucleic acid
molecules
that encode fragments of the TSC, NKCC2 and ROMK proteins. Figures 2, 5, 7 and
12 and Tables I, 3, 4, 7 and 10 identify amino acid residues that are altered
from wild-
type residues in the altered variants of the TSC, NKCC2 and ROMKI proteins
herein
described. Fragments containing these residues/alterations are particularly
useful in
generating altered variant specific anti-TSC, NKCC2 or ROMK antibodies.
As described below, members of the TSC, NKCC2 and ROMK family of
proteins can be used for, but are not limited to: 1) a target to identify
agents that
block or stimulate TSC, NKCC2 or ROMK activity, 2) a target or bait to
identify and
isolate binding partners that bind a TSC, NKCC2 or ROMK protein, 3)
identifying
agents that block or stimulate the activity of a TSC, NKCC2 or ROMK protein
and
4) an assay target to identify TSC, NKCC2 or ROMK mediated activity or
disease.
B. Anti-TSC, NKCC2 or ROMK Antibodies
The present invention further provides antibodies that selectively bind one or
more of the TSC, NKCC2 or ROMK proteins of the present invention. The most
preferred antibodies will bind to an altered variant of a TSC, NKCC2 or ROMK
58814
CA 02274955 1999-06-14
WO 98/29431 PCT/LTS97/23553
-21 -
protein but not to a wild-type variant or will bind to a wild-type variant of
a TSC,
NKCC2 or ROMK protein but not to an altered variant. Anti-TSC, NKCC2 or
ROMK antibodies that are particularly contemplated include monoclonal and
polyclonal antibodies as well as fragments containing the antigen binding
domain
and/or one or more complement determining regions.
Antibodies are generally prepared by immunizing a suitable mammalian host
using a TSC, NKCC2 or ROMK protein, or fragment, in isolated or
immunoconjugated variant (Harlow, Antibodies, Cold Spring Harbor Press, NY
(1989)). Figures 2, 5, 7 and 12 and Tables 1, 3, 4, 7 ad 10 identify several
regions of
the TSC, NKCC2 and ROMK proteins that have been shown to be mutated in various
altered variants of the TSC, NKCC2 and ROMK proteins described herein.
Fragments containing these residues are particularly suited in generating wild-
type or
mutated-variant specific anti-TSC, NKCC2 or ROMK antibodies.
Methods for preparing a protein for use as an immunogen and for preparing
1 S immunogenic conjugates of a protein with a carrier such as BSA, KLH, or
other
carrier proteins are well known in the art. In some circumstances, direct
conjugation
using, for example, carbodiimide reagents may be used; in other instances
linking
reagents such as those supplied by Pierce Chemical Co., Rockford, IL, may be
effective.
Administration of the TSC, NKCC2 or ROMK imlnunogen is conducted
generally by injection over a suitable time period and with use of a suitable
adjuvant,
as is generally understood in the art. During the immunization schedule,
titers of
antibodies can be taken to determine adequacy of antibody formation.
While the polyclonal antisera produced in this way may be satisfactory for
some applications, for pharmaceutical compositions, monoclonal antibody
preparations are preferred. Immortalized cell lines which secrete a desired
monoclonal antibody may be prepared using the standard method of Kohler and
Milstein or modifications which effect immortaiization of lymphocytes or
spleen
CA 02274955 1999-06-14
WO 98/29431 PCT/US97/23553
-22-
cells, as is generally known. The immortalized cell lines secreting the
desired
antibodies are screened by immunoassay in which the antigen is the TSC, NKCC2
or
ROMK protein or peptide fragment. When the appropriate immortalized cell
culture
secreting the desired antibody is identified, the cells can be cultured either
in vitro or
by production in ascites fluid.
The desired monoclonal antibodies are then recovered from the culture
supernatant or from the ascites supernatant. Fragments of the monoclonals or
the
polyclonal antisera which contain the immunologically significant portion can
be used
as antagonists, as well as the intact antibodies. Use of immunologically
reactive
fragments, such as the Fab, Fab', of F(ab')2 fragments is often preferable,
especially in
a therapeutic context, as these fragments are generally less immunogenic than
the
whole immunoglobuiin.
The antibodies or fragments may also be produced, using current technology,
by recombinant means. Regions that bind specifically to the desired regions of
the
transporter can also be produced in the context of chimeric or CDR grafted
antibodies
of multiple species origin.
As described below, anti-TSC, NKCC2 or ROMK antibodies are useful as
modulators of TSC, NKCC2 or ROMK activity, are useful in immunoassays far
detecting TSC, NKCC2 or ROMK expression/activity and for purifying wild-type
and
altered variants of the TSC, NKCC2 and ROMK proteins.
C. TSC, NKCC2 or ROMK Encoding Nucleic Acid Molecules
As described above, the present invention is based, in part, on isolating
nucleic
acid molecules from humans that encode wild-type or altered variants of the
TSC,
NKCC2 and ROMK proteins. Accordingly, the present invention further provides
nucleic acid molecules that encode the herein disclosed wild-type and altered
variants
of the TSC, NKCC2 and ROMK proteins as herein defined, preferably in isolated
variant. For convenience, all TSC, NKCC2 or ROMK encoding nucleic acid
molecules will be referred to as TSC, NKCC2 or ROMK encoding nucleic acid
CA 02274955 1999-06-14
WO 98/29431 PCT/US97/23553
- 23 -
molecules, the TSC, NKCC2 or ROMK genes, or TSC, NKCC2 or ROMK. The
nucleotide sequence of identified wild-type TSC encoding nucleic acid
molecules~are
provided in Figure 2 and Table 3. The nucleotide sequence of identified
altered TSC
encoding nucleic acid molecules are provided in Figures 2 and 4 and Tables I
and 4.
The nucleotide sequence of identified wild-type NKCC2 encoding nucleic acid
molecules are provided in Figure 7. The nucleotide sequence of identified
altered
NKCC2 encoding nucleic acid molecules are provided in Figure 8 and Table 7.
The
nucleotide sequence of identified altered ROMK encoding nucleic acid molecules
are
provided in Figure 11 and Table i 0.
As used herein, a "nucleic acid molecule" is defined as an RNA or DNA
molecule that encodes a peptide as defined above, or is complementary to a
nucleic
acid sequence encoding such peptides. Particularly preferred nucleic acid
molecules
will have a nucleotide sequence identical to or complementary to the human
cDNA
sequences herein disclosed. Specifically contemplated are genomic DNA, cDNA,
mRNA and antisense molecules, as well as nucleic acids based on an alternative
backbone or including alternative bases whether derived from natural sources
or
synthesized. Such nucleic acid molecules, however, are defined further as
being novel
and unobvious over any prior art nucleic acid molecules encoding non-human
homologues of TSC, NKCC2 or ROMK isolated from non-human organisms and
known human ROMK proteins.
As used herein, a nucleic acid molecule is said to be "isolated" when the
nucleic acid molecule is substantially separated from contaminant nucleic acid
encoding other polypeptides. A skilled artisan can readily employ nucleic acid
isolation procedures to obtain an isolated TSC, NKCC2 or ROMK encoding nucleic
acid molecule.
The present invention further provides fragments of the TSC, NKCC2 or ROMK
encoding nucleic acid molecules of the present invention. As used herein, a
fragment of
a TSC, NKCC2 or ROMK encoding nucleic acid molecule refers to a small portion
of
the entire protein encoding sequence. The size of the fragment will be
determined by
CA 02274955 1999-06-14
WO 98/29431 PCTIUS97123553
-24-
the intended use. For example, if the fragment is chosen so as to encode an
active
portion of the TSC, NKCC2 or ROMK protein, such an intracellular or
extracellular
domain, then the fragment will need to be large enough to encode the
functional
regions) of the TSC, NKCC2 or ROMK protein. If the fragment is to be used as a
nucleic acid probe or PCR primer, then the fragment length is chosen so as to
obtain a
relatively small number of false positives during probing/priming. Figures 2,
7, 11 and
Tables 1-4, 6, 7, 9 and 10 identify fragments of the TSC, NKCC2 and ROMK genes
that
are particularly useful as selective hybridization probes or PCR primers. Such
fragments contain regions that are conserved among wild-type or altered
variants of
TSC, NKCC2 or ROMK, regions of homology that are shared with the previously
identified TSC, NKCC2 and ROMK genes, and regions that are altered in altered
variants
of the TSC, NKCC2 and RDMK genes.
Fragments of the TSC, NKCC2 or ROMK encoding nucleic acid molecules of
the present invention (i.e., synthetic oligonucleotides) that are used as
probes or specific
I5 primers for the polymerase chain reaction (PCR), or to synthesize gene
sequences
encoding TSC, NKCC2 and ROMK proteins, can easily be synthesized by chemical
techniques, for example, the phosphotriester method of Matteucci) et al., JAm
Chem
.Soc (1981) 103:3185-3191 or using automated synthesis methods. In addition,
larger
DNA segments can readily be prepared by well known methods, such as synthesis
of a
group of oligonucleotides that define various modular segments of the TSC,
NKCC2 or
ROMK gene, followed by ligation of oligonucleotides to build the complete
modified
TSC, NKCC2 or ROMK gene.
The TSC, NKCC2 or ROMK encoding nucleic acid molecules of the present
invention may further be modified so as to contain a detectable label for
diagnostic
and probe purposes. As described above, such probes can be used to identify
nucleic
acid molecules encoding other allelic variants of wild-type or altered TSC,
NKCC2
and ROMK proteins and as described below, such probes can be used to diagnosis
the
presence of an altered variant of a TSC, NKCC2 or ROMK protein as a means for
diagnosing a pathological condition caused by abnormal ion transport. A
variety of
CA 02274955 1999-06-14
WO 98/29431 PCT/US97/23553
- 25 -
such labels are known in the art and can readily be employed with the TSC,
NKCC2
or ROMK encoding molecules herein described. Suitable labels include, but are
not
limited to, biotin, radioIabeled nucleotides, biotin, and the like. A skilled
artisan can
employ any of the art known labels to obtain a labeled TSC, NKCC2 or ROMK
encoding nucleic acid molecule.
D. Isolation of Other Wild-Type and Altered Forms of TSC, NKCC2
and ROMK Encoding Nucleic Acid Molecules
As described above, the identification of the role of the TSC, NKCC2 and
ROMK proteins in the pathology/severity of ion transport mediated deficiencies
has
made possible the identification of several allelic variants of the wild-type
TSC, NKCC2
and ROMK proteins as well as several altered variants of the TSC, NKCC2 and
ROMK
proteins that confer a pathology associated with abnormal ion transport. These
observations allows a skilled artisan to isolate nucleic acid molecules that
encode other
wild-type and altered variants of the TSC, NKCC2 and ROMK proteins, in
addition to
the sequence herein described.
Essentially, a skilled artisan can readily use the amino acid sequence of the
human TSC, NKCC2 and ROMK proteins to generate antibody probes to screen
expression libraries prepared from cells. Typically, polyclonal antiserum from
mammals such as rabbits immunized with the purified protein (as described
below) or
monoclonal antibodies can be used to probe a human cDNA or genomic expression
library, such as lambda gtll library, prepared from a normal or effected
individual, to
obtain the appropriate coding sequence for wild-type or altered variants of
the TSC,
NKCC2 or ROMK protein. The cloned cDNA sequence can be expressed as a fusion
protein, expressed directly using its own control sequences, or expressed by
constructions using control sequences appropriate to the particular host used
for
expression of the enzyme. Figures 2, 7 and 11 and Tables 1-4, 7, 9 and 10
identify
important operative domains and domains that have been shown to contain
alterations in
mutated variants of each of the TSC, NKCC2 and ROMK proteins. Such regions are
CA 02274955 1999-06-14
WO 98/29431 PCTIUS97/23553
-26-
preferred sources of antigenic portions of the TSC, NKCC2 or ROMK protein for
the
production of probe, diagnostic, and therapeutic antibodies.
Alternatively, a portion of the TSC, NKCC2 or ROMK encoding sequence
herein described can be synthesized and used as a probe to retrieve DNA
encoding a
member of the TSC, NKCC2 or ROMK family of proteins from individuals that have
normal ion transport or from individuals suffering fiom a pathological
condition that is a
result of abnormal ion transport. Oligomers containing approximately 18-20
nucleotides
(encoding about a 6-7 amino acid stretch) are prepared and used to screen
genomic
DNA or cDNA libraries to obtain hybridization under stringent conditions or
conditions
of sufficient stringency to eliminate an undue level of false positives. This
method can
be used to identify and isolate altered and wild-type variants of the TSC,
NKCC2 and
ROMK encoding sequences.
Additionally, pairs of oligonucleotide primers can be prepared for use in a
polymerase chain reaction (PCR) to selectively amplify/clone a TSC, NKCC2 or
ROMK-encoding nucleic acid molecule, or fragment thereof. A PCR
denature/anneal/extend cycle for using such PCR primers is well known in the
art and
can readily be adapted for use in isolating other TSC, NKCC2 or ROMK encoding
nucleic acid molecules. Figures 2, 7 and 1 l and Tables 1-4, 6, 7, 9 and 10
identify
regions of the human TSC, NKCC2 and ROMK genes that are particularly well
suited
for use as a probe or as primers. In general, the preferred primers will flank
one or
more exons of the TSC, NKCC2 or RQMK encoding nucleic acid molecule.
E. Methods for Identifying Pathological Conditions Involving
Abnormal Ion Transport
The present invention further provides methods for identifying cells and
individuals expressing active and altered variants of the Na-K-2C1
cotransporter
NKCC2, the renal thiazide-sensitive Na-Cl cotransporter, TSC, and the ATP-
sensitive
potassium channel, ROMK. Such methods can be used to diagnose biological and
pathological processes associated with altered ion transport, particularly
various
CA 02274955 1999-06-14
WO 98/29431 PCTIUS97/23553
-27-
variants of Banter's Syndrome and Gitelman's Syndrome, the progression of such
conditions, the susceptibility of such conditions to treatment and the
effectiveness of
treatment for such conditions. The methods of the present invention are
particularly
useful in identifying carriers of ion transport deficiencies, particularly
Gitelman's and
Banter's Syndromes, as well as in differentiating between Gitelman's and
Banter's
Syndromes. Specifically, the presence of wild-type or altered variants of the
TSC,
NKCC2 and ROMK proteins can be identified by determining whether a wild-type
or
altered variant of the TSC, NKCC2 or ROMK protein, or nucleic acid encoding
one or
more of these proteins, is expressed in a cell. The expression of an altered
variant, or
departure from the normal level of TSC, NKCC2 or ROMK expression, can be used
as a means for diagnosing pathological conditions mediated by abnormal TSC,
NKCC2 or ROMK activity/expression, differentiating between various ion
transport
deficiencies, and to identify carriers of ion transport deficiencies.
A variety of immunological and molecular genetic techniques can be used to
determine if a wild-type or an altered variant of a TSC, NKCC2 or ROMK protein
is
expressed/produced in a particular cell and/or the level at which the protein
is
expressed. The preferred methods will identify whether a wild-type or mutated
from
of the TSC, NKCC2 or ROMK protein is expressed.
In general, an extract containing nucleic acid molecules or an extract
containing proteins is prepared from cells of an individual. The extract is
then
assayed to determine whether a TSC, NKCC2 or ROMK protein, or a TSC, NKCC2
or ROMK encoding nucleic acid molecule, is produced in the cell. The type of
protein/nucleic acid molecule expressed or the degree/level of expression
provides a
measurement of the nature and degree of TSC, NKCC2 or ROMK activity.
For example, to perform a diagnostic test based on nucleic acid molecules, a
suitable nucleic acid sample is obtained and prepared from a subject using
conventional techniques. DNA can be prepared, for example, simply by boiling
the
sample in SDS. Most typically, for nucleic acid samples, a blood sample, a
buccal
swab, a hair follicle preparation or a nasal aspirate is used as a source of
cells to
CA 02274955 1999-06-14
WO 98/29431 PCTIUS97/23553
- 28 -
provide the nucleic acid molecules. The extracted nucleic acid can then be
subjected
to amplification, for example by using the polymerase chain reaction (PCR)
according
to standard procedures, to selectively amplify a TSC, NKCC2 or ROMK encoding
nucleic acid molecule or fragment thereof. The size of the amplified fragment
S (typically following restriction endonuclease digestion) is then determined
using gel
electrophoresis or the nucleotide sequence of the fragment is determined (for
example,
see Weber and May Am (l Hum Genet (1989) 44:388-339; Davies, J. et al. Nature
( 1994) 371:130-136)). The resulting size of the fragment or sequence is then
compared to the known wild-type, predicted wild-type, known altered variants
and
predicted altered variants of the protein in question. Using this method, the
presence
of wild-type or altered variants of the TSC, NKCC2 and ROMK proteins can be
differentiated and identified.
Alternatively, the presence or absence of one or more single base-pair
polymorphism(s) within the TSC, NKCC2 or ROMK encoding nucleic acid molecules
can be determined by conventional methods which included, but are not limited
to,
manual and automated fluorescent DNA sequencing, selective hybridization
probes,
primer extension methods (Nikiforov, T.T. et al. Nucl Acids Res ( 1994)
22:4167-
4175); oligonucleotide ligation assay (OLA) (Nickerson, D.A. et al. Proc Natl
Acad
Sci USA ( 1990) 87:8923-8927); allele-specific PCR methods (Rust, S. et al.
Nucl
Acids Res ( 1993) 6:3623-3629); RNase mismatch cleavage, single strand
conformation polymorphism (SSCP) (Orita, M. et al., Proc Natl Acad Sci USA
86:2766-2770 (1989)), denaturing gradient gel electrophoresis (DGGE) and the
like.
The present diagnosis method is particularly well suit for use in biochips
technologies
that are being developed to be used to identify whether one of many sequence
variations is present in a sample. A skilled artisan can readily adapt any
nucleic acid
analytical method for use in determining whether a sample contains nucleic
acid
molecules that encode a wild-type or altered variant of a TSC, NKCC2 or ROMK
protein.
CA 02274955 1999-06-14
WO 98/29431 PCT/US97/23553
-29-
To perform a diagnostic test based on protein, a suitable protein sample is
obtained and prepared from a subject using conventional techniques. Protein
samples
can be prepared, for example, simply by mixing the sample with SDS followed by
salt
precipitation of a protein fraction. Typically, for protein samples, a blood
sample, a
buccal swab, a nasal aspirate, or a biopsy of cells from tissues expressing a
TSC,
NKCC2 or ROMK protein is used as a source of cells to provide the protein
molecules. The extracted protein can then be analyzed to determine the
presence of a
wild-type or altered variant of a TSC, NKCC2 or ROMK protein using known
methods. For example, the presence of specific sized or charged variants of a
protein
can be identified using mobility in an electric filed. Alternatively, wild-
type or altered
variant specific antibodies can be used. A skilled artisan can readily adapt
known
protein analytical methods to determine if a sample contains a wild-type or
altered
variant of a TSC, NKCC2 or ROMK protein.
TSC, NKCC2 or ROMK expression can also be used in methods to identify
1 S disorders that occur as a result of an increase or decrease in the
expression of a
naturally occurring TSC, NKCC2 or ROMK gene. Specifically, nucleic acid probes
that detect mRNA can be used to detect cells or tissues that express a TSC,
NKCC2 or
ROMK protein and the level of such expression.
As provided above, the presence of only an altered variant of a TSC protein
(homozygous state) in a sample is diagnostic of Gitelman's Syndrome. Altered
variants
of the TSC protein, when present in sample that additionally contains a wild-
type
variant of TSC (heterozygous state), is diagnostic for carriers of Gitelman's
Syndrome
and individuals expressing lower levels of active TSC. Decreased levels of
active TSC
lead to decreased urinary calcium, increased bone density and a propensity for
deposition of calcium in the joints and diuretic induced hypokalaemia.
Elevated levels
of TSC expression are diagnostic for increased urinary calcium, decreased bone
density,
and a propensity for high blood pressure and kidney stones. The presence of
only an
altered variant of a NKCC2 protein (homozygous state) in a sample is
diagnostic of
several variants of Banter's Syndrome. Altered variants of the NKCC2 protein,
when
CA 02274955 1999-06-14
WO 98129431 PCT/US97123553
-30-
present in sample that additionally contains a wild-type variant of NKCC2
(heterozygous state), is diagnostic for carriers of Banter's Syndrome and
individuals
expressing lower levels of active NKCC2. Decreased NKCC2 activity leads to
increased urinary calcium, decreased bone-niass and a propensity for kidney
stones,
S osteoporosis and diuretic induced hypokalaemia. The presence of only an
altered
variant of a ROMK protein (homozygous state) in a sample is diagnostic of
several
variants of Bartter's Syndrome. Altered variants of the ROMK protein, when
present in
sample that additionally contains a wild-type variant of ROMK (heterozygous
state), is
diagnostic for carriers of Banter's Syndrome and individuals expressing lower
levels of
active ROMK. Decreased ROMK activity leads to increased urinary calcium,
decreased
bone mass and a propensity for kidney stones and osteoporosis.
Alternatively, TSC, NKCC2 or ROMK expression can also be used in
methods to identify agents that increase or decrease the level of expression
of a
naturally occurring TSC, NKCC2 or ROMK gene. For example, cells or tissues
1 S expressing a TSC, NKCC2 or RMOK protein can be contacted with a test agent
to
determine the effects of the agent on TSC, NKCC2 or ROMK expression. Agents
that
activate TSC, NKCC2 or ROMK expression can be used as an agonist of TSC,
NKCC2 or ROMK activity whereas agents that decrease TSC, NKCCZ or ROMK
expression can be used as an antagonist of TSC, NKCC2 or ROMK activity.
F. rDNA Molecules Containing a TSC, NKCC2 or ROMK Encoding
Nucleic Acid Molecule
The present invention further provides recombinant DNA molecules (rDNAs)
that contain one or more of the wild-type or altered TSC, NKCC2 or ROMK
encoding
sequences herein described, or a fragment of the herein-described nucleic acid
molecules. As used herein, an rDNA molecule is a DNA molecule that has been
subjected to molecular manipulation in vitro. Methods for generating rDNA
molecules
are well known in the an, for example, see Sambrook et al., Molecular Cloning
( 1989).
In the preferred rDNA molecules, a TSC, NKCC2 or ROMK encoding DNA sequence .
that encodes a wild-type or altered variant of the TSC, NKCC2 or ROMK protein
is
CA 02274955 1999-06-14
WO 98!29431 PCTIi1S97l23553
-31 -
operably linked to one or more expression control sequences and/or vector
sequences.
Most preferably, the TSC, NKCC2 or ROMK encoding nucleic acid molecules will
encode one of the novel altered or wild-type variants herein described.
The choice of vector and/or expression control sequences to which one of the
TSC, NKCC2 or ROMK encoding sequences of the present invention is operably
linked
depends directly, as is well known in the art, on the functional properties
desired, e.g.,
protein expression, and the host cell to be transformed. A vector contemplated
by the
present invention is at least capable of directing the replication or
insertion into the host
chromosome, and preferably also expression, of a TSC, NKCC2 or ROMK encoding
sequence included in the rDNA molecule.
Expression control elements that are used for regulating the expression of an
operably linked protein encoding sequence are known in the art and include,
but are not
limited to, inducible promoters, constitutive promoters, secretion signals,
enhancers,
transcription terminators and other regulatory elements. Preferably, an
inducible
promoter that is readily controlled, such as being responsive to a nutrient in
the host
cell's medium, is used.
In one embodiment, the vector containing a TSC, NKCC2 or ROMK encoding
nucleic acid molecule will include a prokaryotic replicon, i.e., a DNA
sequence having
the ability to direct autonomous replication and maintenance of the
recombinant DNA
molecute intrachromosomally in a prokaryotic host cell, such as a bacterial
host cell,
transformed therewith. Such replicons are well known in the art. In addition,
vectors
that include a prokaryotic replicon may also include a gene whose expression
confers a
detectable marker such as a drug resistance. Typical bacterial drug resistance
genes are
those that confer resistance to ampicillin or tetracycline.
Vectors that include a prokaryotic replicon can further include a prokaryotic
or
viral promoter capable of directing the expression (transcription and
translation) of the
TSC, NKCC2 or ROMK encoding gene sequence in a bacterial host cell, such as
E. coli. A promoter is an expression control element formed by a DNA sequence
that
CA 02274955 1999-06-14
WO 98/29431 PCTIUS97/23553
-32-
permits binding of RNA polymerase and transcription to occur. Promoter
sequences
compatible with bacterial hosts are typically provided in plasmid vectors
containing
convenient restriction sites far insertion of a DNA segment of the present
invention.
Typical of such vector plasmids are pUCB, pUC9, pBR322 and pBR329 available
from
Biorad Laboratories (Richmond, CA), pPL and pKK223 available from Pharmacia,
Piscataway, NJ.
Expression vectors compatible with eukaryotic cells, preferably those
compatible with vertebrate cells, can also be used to variant rDNA molecules
that
contain a TSC, NKCC2 or ROMK encoding sequence. Eukaryotic cell expression
vectors are well known in the art and are available from several commercial
sources.
Typically, such vectors are provided containing convenient restriction sites
for insertion
of the desired DNA segment. Typical of such vectors are PSVL and pKSV-10
(Pharmacia), pBPV-I/pML2d (International Biotechnologies, Inc.), pTDTI (ATCC,
#31255), the vector pCDM8 described herein, and the like eukaryotic expression
I S vectors.
Eukaryotic cell expression vectors used to construct the rDNA molecules of the
present invention may further include a selectable marker that is effective in
an
eukaryotic cell, preferably a dnzg resistance selection marker. A preferred
drug
resistance marker is the gene whose expression results in neomycin resistance,
i.e., the
neomycin phosphotransferase (neo) gene. Southern et al., JMoI Anal Genet
(1982)
1:327-341. Alternatively, the selectable marker can be present on a separate
plasmid,
and the two vectors are introduced by cotransfection of the host cell, and
selected by
culturing in the presence of the appropriate drug for the selectable marker.
G. Host Cells Containing an Exogenously Supplied TSC, NKCC2 or
ROMK Encoding Nucleic Acid Molecule
The present invention further provides host cells transformed with a nucleic
acid
molecule that encodes a human wild-type or altered TSC, NKCC2 or ROMK protein
of
the present invention. The host cell can be either prokaryotic or eukaryotic.
Eukaryotic
CA 02274955 1999-06-14
WO 98/29431 PCT/US97/23553
-33-
cells useful for expression of a TSC, NKCC2 or ROMK protein are not limited,
so long
as the cell line is compatible with cell culture methods and compatible with
the
propagation of the expression vector and expression of the TSC, NKCC2 or ROMK
gene product. Preferred eukaryotic host cells include, but are not limited to,
yeast, insect
and mammalian cells, preferably vertebrate cells such as those from a mouse,
rat,
monkey or human fibroblastic cell line, the most preferred being cells that do
not
naturally express a human TSC, NKCC2 or ROMK protein.
Any prokaryotic host can be used to express a TSC, NKCC2 or ROMK-
encoding rDNA molecule. The preferred prokaryotic host is E. coli.
Transformation of appropriate cell hosts with an rDNA molecule of the present
invention is accomplished by well known methods that typically depend on the
type of
vector used and host system employed. With regard to transformation of
prokaryotic
host cells, electroporation and salt treatment methods are typically employed,
see, for
example, Cohen et al., Proc Acad Sci USA (1972) 69:2110; and Maniatis et al.,
1 S Molecular Toning) A .ahnratpni ~r~.."~ l, Cold Spring Harbor Laboratory,
Cold Spring
Harbor, NY (1982). With regard to transformation of vertebrate cells with
vectors
containing rDNAs, electroporation, cationic lipid or salt treatment methods
are typically
employed, see, for example, Graham et al., virol {1973) 52:456; Wigler et al.,
Proc Natl
Acad Sci USA (1979) 76:1373-76.
Successfully transformed cells, i.e., cells that contain an rDNA molecule of
the
present invention, can be identified by well known techniques. For example,
cells
resulting from the introduction of an rDNA of the present invention can be
cloned to
produce single colonies. Cells from those colonies can be harvested, lysed and
their
DNA content examined for the presence of the rDNA using a method such as that
described by Southern, JMoI Biol ( 1975) 98:503, or Berent et al., Biotech (
1985) 3:208
or the proteins produced from the cell assayed via an immunological method.
CA 02274955 1999-06-14
WO 98/29431 PCTIUS97/23553
-34-
H. Production of a TSC, NKCC2 or ROMK Protein Using an rDNA
Molecule
The present invention further provides methods for producing a human wild-
type or altered TSC, NKCC2 or ROMK protein that uses one of the TSC, NKCC2 or
ROMK encoding nucleic acid molecules herein described. In general terms, the
production of a recombinant human wild-type or altered TSC, NKCC2 or ROMK
protein typically involves the following steps.
First, a nucleic acid molecule is obtained that encodes a TSC, NKCC2 or
ROMK protein, such as the nucleic acid molecule depicted in Figures 2 and 7.
If the
TSC, NKCC2 or ROMK encoding sequence is uninterrupted by introns, it is
directly
suitable for expression in any host. If not, then a spliced variant of the
TSC, NKCC2 or
ROMK encoding nucleic acid molecule can be generated and used or the intron
containing nucleic acid molecule can be used in a compatible eukaryotic
expression
system.
The TSC, NKCC2 or ROMK encoding nucleic acid molecule is then preferably
placed in an operable linkage with suitable control sequences, as described
above, to
vanant an expression unit containing the TSC, NKCC2 or ROMK encoding sequence.
The expression unit is used to transform a suitable host and the transformed
host is
cultured under conditions that allow the production of the TSC, NKCC2 or ROMK
protein. Optionally the TSC, NKCC2 or ROMK protein is isolated from the medium
or
fiom the cells; recovery and purification of the protein may not be necessary
in some
instances where some impurities may be tolerated.
Each of the foregoing steps can be done in a variety of ways. For example, the
desired coding sequences may be obtained from genomic fragments and used
directly in
an appropriate host. The construction of expression vectors that are operable
in a variety
of hosts is accomplished using an appropriate combination of replicons and
control
sequences. The control sequences, expression vectors, and transformation
methods are
dependent on the type of host cell used to express the gene and were discussed
in detail
CA 02274955 1999-06-14
WO 98/29431 PCT/US97I23553
-35-
earlier. Suitable restriction sites can, if not normally available, be added
to the ends of
the coding sequence so as to provide an excisable gene to insert into these
vectors. A
skilled artisan can readily adapt any host/expression system known in the art
for use
with TSC, NKCC2 or ROMK encoding sequences to produce a TSC, NKCC2 or
ROMK protein. Particularly well suited are expression systems that result in
the
production of lipid vesicles containing the expressed protein. Such lipid
containing
vesicles are well suited for identifying agonists and antagonists of the TSC,
NKCC2 or
ROMK protein.
I. Ion Transport
As provided above, alterations in the TSC, NKCC2 or ROMK protein cause
pathological conditions that are a result of abnormal ion transport.
Accordingly, the
wild-type and altered variants of the TSC, NKCC2 and ROMK proteins of the
present
invention can be used in methods to alter the extra or intracellular
concentration of
Na, Ca, Cl, Mg, and/or K. In general, the extra or intracellular concentration
of Na,
1 S Ca, Cl, Mg, and/or K can be altered by altering the expression of a TSC,
NKCC2 or
ROMK protein or the activity of a TSC, NKCC2 or ROMK protein.
There are a number of situation in which it is desirable to alter the extra or
intracellular concentration of Na, Ca, Cl, Mg, and/or K. Abnormal extra or
intracellular pH leads to water retention, increased blood pressure, chronic
respiratory
and metabolic acidosis, inflammation, sperm activation/inactivation,
hydroencephaly,
glaucoma, colitis, etc.
Hence, a TSC, NKCC2 or ROMK protein or TSC) NKCC2 or ROMK gene
expression can be used as a target for, or as means to alter extra or
intracellular Na,
Ca, C1, Mg and/or K concentration. For example, a TSC, NKCC2 or ROMK gene can
be introduced and expressed in cells to increase TSC, NKCC2 or ROMK
expression.
This provides a means and methods for altering extra and intracellular ion
levels.
There are pathological conditions characterized by inappropriate extra or
intracellular ion concentrations. For example, Bartter's Syndrome, Gitelman's
CA 02274955 1999-06-14
WO 98129431 PCT/US97/23553
-36-
Syndrome, hypokalaemic alkalosis, hypokalaemic alkalosis with hypercalciuria,
kidney stones, high blood pressure, osteoporosis and sensitivity to diuretic-
induced
hyperkalaemia, are all associated with abnormal intracellular or extracellular
ion
concentration. Accordingly, TSC, NKCC2 or ROMK activity/expression is targeted
as a means of treating these conditions. Various methods for regulating TSC,
NKCC2
or ROMK activitylexpression are discussed in detail below.
J. Identification of Agents that Bind to a TSC, NKCC2 or ROMK
Protein
Another embodiment of the present invention provides methods for identifying
agents that are agonists or antagonists of the TSC, NKCC2 and ROMK proteins
herein described. Specifically, agonists and antagonists of a TSC, NKCC2 or
ROMK
protein can be first identified by the ability of the agent to bind to one of
the wild-type
or altered variants of the TSC, NKCC2 and ROMK proteins herein described.
Agents
that bind to a TSC, NKCC2 or ROMK protein can then be tested for the ability
to
i 5 stimulate or block ion transport in a TSC, NKCC2 or ROMK expressing cell.
In detail, a TSC, NKCC2 or ROMK protein is mixed with an agent. After
mixing under conditions that allow association of TSC, NKCC2 or ROMK with the
agent, the mixture is analyzed to determine if the agent bound the TSC, NKCC2
or
ROMK protein. Agonists and antagonists are identified as being able to bind to
a
TSC, NKCC2 or ROMK protein.
The TSC, NKCC2 or ROMK protein used in the above assay can either be an
isolated and fully characterized protein, can be a partially purified protein,
can be a
cell that has been altered to express a TSC, NKCC2 or ROMK protein or can be a
fraction of a cell that has been altered to express a TSC, NKCC2 or ROMK
protein.
Further, the TSC, NKCC2 or ROMK protein can be the entire TSC, NKCC2 or
ROMK protein or a specific fragment of the TSC, NKCC2 or ROMK protein. It will
be apparent to one of ordinary skill in the art that so long as the TSC, NKCC2
or
CA 02274955 1999-06-14
WO 98129431 PCT/US97/23553
-37-
ROMK protein can be assayed for agent binding, e.g., by a shift in molecular
weight
or change in cellular ion content, the present assay can be used.
The method used to identify whether an agent binds to a TSC, NKCC2 or
ROMK protein will be based primarily on the nature of the TSC, NKCC2 or ROMK
S protein used. For example, a gel retardation assay can be used to determine
whether
an agent binds to a soluble fragment of a TSC, NKCC2 or ROMK protein whereas
patch clamping, voltage clamping, ion-sensitive microprobes or ion-sensitive
chromaphores can be used to determine whether an agent binds to a cell
expressing a
TSC, NKCC2 or ROMK protein and affects the activity of the expressed protein.
A
skilled artisan can readily employ numerous techniques for determining whether
a
particular agent binds to a TSC, NKCC2 or ROMK protein.
Once binding is demonstrated, the agent can be further tested for the ability
to
modulate the activity of a wild-type or altered variant of the TSC, NKCC2 or
ROMK
protein using a cell or oocyte expression system and an assay that detects
TSC,
NKCC2 or ROMK activity. For example, voltage or patch clamping, ion-sensitive
microprobes or ion-sensitive chromaphores and expression in Xenopus oocytes or
recombinant host cells can be used to determine whether an agent that binds a
TSC,
NKCC2 or ROMK protein can agonize or antagonize TSC, NKCC2 or ROMK
activity.
As used herein, an agent is said to antagonize TSC, NKCC2 or ROMK activity
when the agent reduces TSC, NKCC2 or ROMK activity. The preferred antagonist
will selectively antagonize TSC, NKCC2 or ROMK, not affecting any other
cellular
proteins, particularly other ion transport proteins. Further, the preferred
antagonist
will reduce TSC, NKCC2 or ROMK activity by more than 50%, more preferably by
more than ~90%, most preferably eliminating all TSC, NKCC2 or ROMK activity.
As used herein, an agent is.said to agonize TSC, NKCC2 or ROMK activity
when the agent increases TSC, NKCC2 or ROMK activity. The preferred agonist
will
selectively agonize altered variants of TSC, NKCC2 or ROMK, not affecting any
CA 02274955 1999-06-14
WO 98/29431 PCT/US97123553
-38-
other cellular proteins, particularly other ion transport proteins. Further,
the preferred
agonist will increase TSC, NKCC2 or ROMK activity by more than 50%, more .
preferably by more than 90%, most preferably more than doubling the level of
TSC,
NKCC2 or ROMK activity.
Agents that are assayed in the above method can be randomly selected or
rationally selected or designed. As used herein, an agent is said to be
randomly
selected when the agent is chosen randomly without considering the specific
sequences of the TSC, NKCC2 or ROMK protein. An example of randomly selected
agents is the use a chemical library or a peptide combinatorial library, or a
growth
broth of an organism.
As used herein, an agent is said to be rationally selected or designed when
the
agent is chosen on a nonrandom basis which takes into account the sequence of
the
target site and/or its conformation in connection with the agent's action.
Agents can
be rationally selected or rationally designed by utilizing the peptide
sequences that
make up the TSC, NKCC2 or ROMK protein. For example, a rationally selected
peptide agent can be a peptide whose amino acid sequence is identical to a
fragment
of a TSC, NKCC2 or ROMK protein.
The agents of the present invention can be, as examples, peptides, small
molecules, and vitamin derivatives, as well as carbohydrates. A skilled
artisan can
readily recognize that there is no limit as to the structural nature of the
agents of the
present invention. One class of agents of the present invention are peptide
agents
whose amino acid sequences are chosen based on the amino acid sequence of the
TSC, NKCC2 or ROMK protein.
The peptide agents of the invention can be prepared using standard solid phase
(or solution phase) peptide synthesis methods, as is known in the art. In
addition, the
DNA encoding these peptides may be synthesized using commercially available
oligonucleotide synthesis instrumentation and produced recombinantly using
standard
CA 02274955 1999-06-14
WO 98/29431
PCT/ITS97/23553
-39-
recombinant production systems. The production using solid phase peptide
synthesis
is necessitated if non-gene-encoded amino acids are to be included.
Another class of agents of the present invention are antibodies immunoreactive
with critical positions of the TSC, NKCC2 or ROMK protein. As described above,
antibodies are obtained by immunization of suitable mammalian subjects with
peptides, containing as antigenic regions, those portions of the TSC, NKCC2 or
ROMK protein intended to be targeted by the antibodies. Critical regions
include the
domains identified in Figures 5, 7 and 12.
K. Uses of Agents that Bind to a TSC, NKCC2 or ROMK Protein
As provided in the Background section, the TSC, NKCC2 and ROMK proteins
are involved in regulating intracellular and extracellular ion concentration.
Agents
that bind a TSC, NKCC2 or ROMK protein and act as an agonist or antagonist can
be
used to modulate biological and pathologic processes associated with TSC,
NKCC2
or ROMK function and activity. In detail, a biological or pathological process
I S mediated by TSC,1VKCC2 or ROMK can be modulated by administering to a
subject
an agent that binds to a TSC, NKCC2 or ROMK protein and acts as an agonist or
antagonist of TSC, NKCC2 or ROMK activity.
As used herein, a subject can be any mammal, so long as the mammal is in
need of modulation of a pathological or biological process mediated by TSC,
NKCC2
or ROMK. The term "mammal" means an individual belonging to the class
Mammalia. The invention is particularly useful in the treatment of human
subjects.
As used herein, a biological or pathological process mediated by TSC,
NKCC2 or ROMK refers to the wide variety of cellular events mediated by a TSC,
NKCC2 or ROMK protein. Pathological processes refer to a category of
biological
processes which produce a deleterious effect. For example, pathological
processes
mediated by TSC, NKCC2 or ROMK include hypokalaemic alkalosis, hypokalaemic
alkalosis with hypercalciuria, kidney stones, high blood pressure,
osteoporosis and
sensitivity to diuretic-induced hyperkalaemia. These pathological processes
can be
CA 02274955 1999-06-14
WO 98/29431
PCT/US97123553
-40-
modulated using agents that reduce or increase the activity of a TSC, NKCC2 or
ROMK protein. Preferably, the agent will act to activate an otherwise inactive
altered
variant of a TSC, NKCC2 or ROMK protein.
As used herein, an agent is said to modulate a pathological process when the
agent reduces the degree or severity of the process. For example, an agent is
said to
modulate Bartter's Syndrome when the agent contributes to normal intra and
extracellular ion concentrations.
L. Administration of Agonists and Antagonists of a TSC, NKCC2 or
ROMK Protein
Agonists and antagonists of the TSC, NKCC2 or ROMK protein can be
administered via parenteral, subcutaneous, intravenous, intramuscular,
intraperitoneal,
transdermal, or buccal routes. Alternatively, or concurrently, administration
may be
by the oral route. The dosage administered will be dependent upon the age,
health,
and weight of the recipient, kind of concurrent treatment, if any, frequency
of
1 S treatment, and the nature of the effect desired. For example, to treat
pathological
conditions resulting from abnormal ion transport, such as water retention,
increased
blood pressure, chronic respiratory and metabolic acidosis, inflammation,
etc., an
agent that modulates TSC, NKCC2 or ROMK activity is administered systemically
or
locally to the individual being treated. As described below, there are many
methods
that can readily be adapted to administer such agents.
The present invention further provides compositions containing an antagonist
or agonist of a TSC, NKCC2 or ROMK protein that is identified by the methods
herein described. While individual needs vary, a determination of optimal
ranges of
effective amounts of each component is within the skill of the art. Typical
dosages
comprise 0.1 to 100 ~tg/kg body wt. The preferred dosages comprise 0.1 to 10
~g/kg
p.g/kg body wt. The most preferred dosages comprise 0.1 to I ~g/kg body wt.
In addition to the pharmacologically active agent, the compositions of the
present invention may contain suitable pharmaceutically acceptable Garners
CA 02274955 1999-06-14
WO 98129431 PCTIUS97/23553
-41 -
comprising excipients and auxiliaries which facilitate processing of the
active
compounds into preparations which can be used pharmaceutically for delivery to
the
site of action. Suitable formulations for parenteral administration include
aqueous
solutions of the active compounds in water-soluble variant, for example, water-
soluble
S salts. In addition, suspensions of the active compounds and as appropriate,
oily
injection suspensions may be administered. Suitable lipophilic solvents or
vehicles
include fatty oils, for example, sesame oil, or synthetic fatty acid esters,
for example,
ethyl oleate or triglycerides. Aqueous injection suspensions may contain
substances
which increase the viscosity of the suspension and include, for example,
sodium
carboxymethyl cellulose, sorbitol, and/or dintran. Optionally, the suspension
may
also contain stabilizers. Liposomes can also be used to encapsulate the agent
for
delivery into the cell.
The pharmaceutical formulation for systemic administration according to the
invention may be formulated for enteral, parenteral or topical administration.
Indeed,
all three types of formulations may be used simultaneously to achieve systemic
administration of the active ingredient.
Suitable formulations for oral administration include hard or soft gelatin
capsules, pills, tablets, including coated tablets, elixirs, suspensions,
syrups or
inhalations and controlled release variants thereof.
M. Combination Therapy
The agents of the present invention that modulate TSC, NKCC2 or ROMK
activity can be provided alone, or in combination with another agents that
modulate a
particular biological or pathological process. For example, an agent of the
present
invention that reduces TSC, NKCC2 or ROMK activity can be administered in
combination with other agents that affect the target ion transporter, for
example, a
diuretic agent. As used herein, two agents are said to be administered in
combination
when the two agents are administered simultaneously or are administered
independently in a fashion such that the agents will act at the same time..
CA 02274955 1999-06-14
WO 98/29431 PCT/IJS97/23553
- 42 -
N. Animal Models and Gene Therapy
The TSC, NKCC2 and ROMK genes and the TSC, NKCC2 and ROMK
proteins can also serve as targets for gene therapy in a variety of contexts.
For
example, in one application, TSC, NKCC2 or ROMK-deficient non-human animals
can be generated using standard knock-out procedures to inactivate a TSC)
NKCC2 or
ROMK gene or, if such animals are non-viable, inducible TSC, NKCC2 or ROMK
antisense molecules can be used to regulate TSC, NKCC2 or ROMK
activity/expression. Alternatively, an animal can be altered so as to contain
a TSC,
NKCC2 or ROMK or antisense-TSC, NKCC2 or ROMK expression unit that directs
the expression of TSC, NKCC2 or ROMK or the antisense molecule in a tissue
specific fashion. In such uses, a non-human mammal, for example a mouse or a
rat, is
generated in which the expression of the TSC, NKCC2 or ROMK gene is altered by
inactivation or activation. This can be accomplished using a variety of art-
known
procedures such as targeted recombination. Once generated, the TSC, NKCC2 or
ROMK-deficient animal, the animal that expresses TSC, NKCC2 or ROMK in a
tissue specific manner, or an animal that expresses an antisense molecule can
be used
to 1 ) identify biological and pathological processes mediated by TSC, NKCC2
or
ROMK, 2) identify proteins and other genes that interact with TSC, NKCC2 or
ROMK, 3) identify agents that can be exogenously supplied to overcome TSC,
NKCC2 or ROMK deficiency and 4) serve as an appropriate screen for identifying
mutations within TSC, NKCC2 or ROMK that increase or decrease activity.
For example, it is possible to generate transgenic mice expressing the human
minigene for TSC, NKCC2 or ROMK in a tissue specific-fashion and test the
effect of
over-expression of the protein in cells and tissues that normally do not
contain TSC,
NKCC2 or ROMK. This strategy has been successfuiiy used for other proteins,
namely bcl-Z (Veis et al. Cell 75:229 (1993)). Such an approach can readily be
applied to the TSC, NKCC2 or ROMK protein and can be used to address the issue
of
a potential beneficial effect of TSC, NKCC2 or ROMK in a specific tissue area.
CA 02274955 1999-06-14
WO 98129431 PCT/LTS97/23553
- 43 -
In another embodiment, genetic therapy can be used as a means for
modulating a TSC, NKCC2 or ROMK-mediated biological or pathological processes.
For example, it may be desirable to introduce into a subject being treated a
genetic
expression unit that encodes a modulator of TSC, NKCC2 or ROMK expression,
such
as an antisense encoding nucleic acid molecule or a TSC) NKCC2 or ROMK
encoding nucleic acid molecule, or a functional TSC, NKCC2 or ROMK expression
unit. Such modulators can either be constitutively produced or inducible
within a cell
or specific target cell. This allows a continual or inducible supply of a
modulator of
TSC, NKCC2 or ROMK or the protein expression within a subject.
The following examples are intended to illustrate, but not to limit, aspects
of
the present invention.
EXAMPLE 1
1 1 w' i ' el ' f f r'
d a ~v N- 1 r
Recruitment of ,itelman'c S~m~ro;;;e ~r~n.~re~~. Eleven of the recruited
Gitelman's Syndrome kindreds have been previously reported (lwata, F., et al.,
Acta
Paed. Japonica 35:252-257 (1993); Marco-Franco, J.E., et al., Clin. Neph.
42:33-37
( 1994); Zarraga Larrondo, S., et al. , Nephron 62:340-344 ( 1992); Smilde,
TJ., et al. , J.
of Rheum. 21:1515-1519 ( 1994); Okusa, M.D. and Bia, M.J. Banter's , drom In:
Ho one Re icrance nd Other ndo crine Paradox ~c (eds. Cohen, P. and Foa, P.)
231-263 (Springer Verlag, New York, 1987); Gitelman, H.J., Trans. Assoc. Am.
Phys,
79:221-235 (1966); Cushner, H.M., et al., Amer. J. Kid. Dis. 16:495-500
(1990);
Sutton, R.A.L., et al., Miner. Elect. Metab. 18:43-51 (1992)). The remaining
kindred,
GIT102, was ascertained through an index case referred for evaluation of
CA 02274955 1999-06-14
WO 98/29431 PCT/US97123553
-44-
hypokalaemic metabolic alkalosis. Genomic DNA was prepared from venous blood
of members of Gitelman's Syndrome kindreds by standard procedures (Bell, G.,
et al.,
Proc. Natl. Acad. Sci. USA 78:5759-5763 (1981)).
Clonine and haracteri a ion of the Human ~~~nomic Tf . Partial human and
mouse cDNAs encoding the TSC protein were isolated by PCR using
oligonucleotide
primers corresponding to either flounder (Gamba, G., et al., Proc. Natl. Acad.
Sci.
U.S.A. 90:2749-2753 (/993)) or rat (Gamba, G., et al., J. Biol. Chem.
269:177/3-
17722 ( 1994)) TSC cDNA sequence and either mouse kidney cDNA (Obermiiller,
N.,
et al., Amer. J. Physiol., in press ( 1995)) or a human kidney cDNA library as
template. The resulting cDNA segments were radiolabeled (Feinberg, A.P., et
al.,
Anal. Biochem. 132:6-13 (1983)) and used to screen a human genomic cosmid
library
by hybridization; the library and screening procedures have been described
previously
(Shimkets, R.A., et al., Cell 79:407-414 (/994)). Maps of resulting clones
were
def ned by the overlapping products of digestion as well as by hybridization
to
specific exon or cDNA segments. Genomic fragments bearing exons of TSC were
subcloned by digestion of cosmid DNA to completion with Sau3AI and ligation of
products into BamHI-digested pBluescript; resultant clones hybridizing to
mouse or
human TSC cDNA were isolated and subjected to DNA sequence analysis by the
dideoxy chain termination method using an ABI 373 instrument and following a
standard protocol.
Marker Development, Genotypine any t inkage Anal,,; ~. Sau3A 1 subclones
of the cosmid contig were screened for (GT)n repeat sequences by hybridization
as
previously described (Shimkets, R.A., et al., Cell 79:407-414 ( 1994)). This
screen
identified clone phTSCGT-14; sequence analysis revealed an interrupted array
of
(GT) (Clive, D.M. Am. J. Kid. Dis. 25:813-823 (1995)); primers flanking this
simple
sequence repeat were designed and used to direct PCR from genomic DNA of
unrelated subjects (primer hTSCGT14-A: 5'-GTGAGCCACTGCGCTTAGCTG-3';
hTSCGT 14-B: 5'-CTGCTGAGCTCTGGTCTGGAG-3').
CA 02274955 1999-06-14
WO 98129431 PCT/US97/23553
- 45 -
Genotyping of CEPH and disease kindreds for loci indicated in the text was
performed by polymerase chain reaction using specific primers (Gyapay, G., et
al.,
Nature Genet. 7:246-339 (1994), Shen, Y.S., et al., Genomics 22:68-76 (1994))
as
described previously (Shimkets, R.A., et al., Cell 79:407-414 (1994)), except
that the
products were labeled by inclusion of 1 pCi of [I-32PjdCTP in the reaction
mixture.
All genotypes were scored independently by two investigators who were blinded
to
affection status.
Analysis of linkage was performed using the LINKAGE programs {Lathrop,
G.M., et al., Proc. Natl. Acad. Sci. USA 81:3443-3446 (1984)). Gitelman's
Syndrome
was specified as an autosomal recessive trait with 99% penetrance, a sporadic
prevalence of 0.001, and a mutant allele frequency of I in 200.
SSCP and DNA Seq ~ .n .in Single-strand conformational polymorphism
(C~rita, M., et al., Proc. Natl. Acad. Sci. USA 86:2766-2770 (1989)) was
performed by
using 27 sets of specific primers (Table 2) to separately direct PCR using
genomic
DNA of disease family members or CEPH controls as a template as previously
described (Shimkets, R.A., et al., Cell 79:407-414 (1994)). Amplified products
were
analyzed for molecular variants by electrophoresis under 3 different non-
denaturing
conditions as well as under denaturing urea gels as previously described
(Shinlkets,
R.A., et al., Cell 79:407-414 (1994)). Identified variants were eluted from
the gels,
reamplified by PCR and sequenced using an ABI 373 instrument as previously
described (Shimkets, R.A., et al., Cell 79:407-414 ( 1994)). In all cases, DNA
sequences were confirmed by sequencing both DNA strands.
Multiplex Gitelman' ~~drome ~=r~~~~~
_ _-___- 111L1 V
Thirty patients with Gitelman's Syndrome from 12 unrelated families were
recruited for the initial study; 11 of these are multiplex families, with 2 or
more
affected subjects, and 11 of these families have been reported previously (see
Methods). In 8 families, samples were obtained from two or more affected
members
CA 02274955 1999-06-14
WO 98/29431 PCT/US97123553
-46-
(Figure 1 ), and in 4 families a single affected subject was sampled. The
diagnosis of
hypokalaemic metabolic alkalosis was based on the finding of spontaneous
hypokalaemia (serum potassium < 3.0 meq/l; nl > 3.5 meq/1), with metabolic
alkalosis
(serum bicarbonate > 29 meq/1, nl < 26 meq/1) in the absence of intercurrent
illness,
hypertension and diuretic therapy. All patients studied had elevated plasma
renin
activity. Diuretic abuse was excluded by the absence of detectable diuretics
in urine.
Gitelman's Syndrome was prospectively distinguished from Banter's
Syndrome following the criteria of Bettinelli (Bettinelli, A., et al., J.
Pediatr. 120:38-
43 (1992)): index cases in ali of these families had marked hypocalciuria (<2
mg/kg/day, nl >4 mglkg/day), hypomagnesemia (<0.5 meq/l, nl 0.8-1.0 meq/1),
and
symptomatic presentation after age 8. Twenty-seven patients were symptomatic,
presenting with a variety of muscular symptoms including persistent muscular
weakness, recurrent cramping with exertion, carpopedal spasm, total body
tetany, and
periodic paralysis with respiratory compromise. Other manifestations included
seizures, paresthesias, polyuria/polydipsia, and joint pains attributable to
chondrocalcinosis. Subjects classified as unaffected within multiplex families
were
over age 20, clinically asymptomatic, and had normal serum potassium and
magnesium levels. Ail diagnoses were established prospectively. Affected
subjects
were the offspring of two clinically normal parents, consistent with recessive
inheritance, with the exception of kindred GIT112, in which the disease is
present in 2
successme generations; in addition, in kindred GIT102 the disease is found in
second
cousins, but is absent in the relatives linking these individuals (Figure 1 ).
Cloning and characterization of the hurr~an Tf . To permit testing TSC as a
candidate gene for Gitelman's Syndrome, we identified genomic cosmid clones
encoding the human homolog of TSC by hybridization to mouse or flounder TSC
cDNAs (see Methods). Five resulting clones were characterized, with their maps
revealing that they overlap and define a single contig of 55 kb (Figure 2a).
The
intron-exon organization of the gene was determined, demonstrating that the
human
TSC protein is encoded by 26 eXons (Figure 2b, 2c); all intron-exon boundaries
have
CA 02274955 1999-06-14
WO 98/29431 PCTIUS97/23553
-47-
conventional 5' GT and 3' AG consensus splice sites. The encoded human protein
shows 89% identity with rat TSC and 63% identity with flounder TSC (Figure
2c).
The human protein contains 17 amino acids that are not present in the rat;
this
additional segment is encoded in a separate exon, exon 20. Southern blotting
indicates the presence of a single gene encoding this protein in the human
genome
(data not shown).
A polymorphic genetic marker (TSCGT-1) at this locus was found by
identification of a GT dinucleotide repeat sequence in the contig.
Amplification of
this segment from genomic DNA of unrelated subjects using specific primers
(see
Methods) demonstrated that this marker is polymorphic, displaying 3 alleles
and
showing a heterozygosity of 48% in 45 unrelated Caucasian subjects. This
marker
was shown to be linked to the TSC locus by finding this same marker on 4
independent cosmids from the cloned contig, as well as by subsequent linkage
analysis of TSC variants with this marker in disease families (data not
shown).
This marker was used to localize TSC on the human genetic map by linkage in
CEPH pedigrees. Pairwise analysis revealed strong evidence for linkage to a
cluster
of markers on human chromosome 16. Multipoint linkage analysis confirmed
linkage
to chromosome 16, with a peak lod score of 10.1 in the 3 cM interval flanked
by
D 165408 and D 165494, with odds favoring location in this interval of 950 to
1 over
the next most likely interval (Figure 3). These findings localize the TSC on
the
human genetic map and identify highly in~ative genetic markers spanning the
TSC
locus, permitting us to test for linkage between this segment of chromosome I
6 and
Gitelman's Syndrome.
Linkage of IltPlman' yndrome an,~ Tc~~ Highly in~ative markers
spanning the TSC locus were genotyped in multiplex Gitelman's Syndrome
kindreds
(Figure 1 ). Linkage was analyzed under a prospectively specified model of
recessive
inheritance (see Methods). Pairwise linkage analysis revealed a maximum lod
score
of 6.3 for linkage of Gitelman's Syndrome and locus D 165408 at a
recombination
fraction of zero. Multipoint linkage analysis (Figure 3) confirmed strong
evidence for
CA 02274955 1999-06-14
WO 98129431 PCT/US9~/23553
-48-
linkage, demonstrating a maximum lod score of 9.5 for linkage of GiteIman's
Syndrome and D16S408 at a recombination fraction of zero (odds of more than 3
billion to 1 in favor of linkage). Flanking markers demonstrated recombinants
with
Gitelman's Syndrome (Figure 1 and Figure 3), localizing the gene with odds of
greater than 1000:1 to the 11 centimorgan interval flanked by loci D 165419
and
D 165494. Within this interval, the lod-1 support interval localizes the gene
to a 7 cM
segment that includes the location of the TSC gene (Figure 3). These findings
reveal
no recombinants between Gitelman's Syndrome and the segment of chromosome 16
containing TSC, and are consistent with both genetic homogeneity of the trait
and
autosomal recessive transmission in all families studied.
Mutations in TS . in Titelman'a Syndrome natiPnte The finding of complete
linkage of TSC and Gitelman's Syndrome motivated the search for mutations in
this
gene in affected subjects. Twenty-seven pairs of specific primers were used to
amplify 150-300 base pair segments of exons and intron-exon boundaries of the
gene
by polymerase chain reaction, using genomic DNA of affected subjects as a
template
as described in Methods; these primer sets cover the coding region and splice
sites of
the gene. Variants were identified by single strand conformational
polymorphism
(SSCP) and were subjected to DNA sequence analysis (Figure 4).
Seventeen different molecular variants inferred to alter the structure of the
encoded protein were identified on the 26 mutant alleles (Tables 1 and 4,
Figure 4),
while no such variants were detected on 26 alleles from control subjects.
Three
variants were found to be homozygous in Gitelman's patients, with inheritance
from
both parents (Tables 1 and 4). All of these variants cosegregated with the
disease in
multiplex families (Figure 1 ) and multipoint linkage of Gitelman's Syndrome
versus
D 165408, TSC variants and D 165494 yields a lod score of 10.6 at a
recombination
fraction of zero with both TSC and D16S408 (data not shown). None of these
specific
variants were detected in any of 80 alleles from unrelated Caucasian control
subjects.
These 17 variants are distributed throughout the gene (Figure 2c, Figure 5).
Thirteen of these variants are missense (Tables 1 and 4), and these all alter
residues
CA 02274955 1999-06-14
WO 98/29431
-49-
PCT1US97/23553
that are identical in normal humans and flounder (Figure 2C), species that
last shared
a common ancestor 400 million years ago. There is a strong bias for non-
conservative
amino acid substitutions ( 11/13 missense variants); 7 of these alter the
charge of the
encoded amino acid, and 2 introduce or remove a proline residue (Tables 1 and
4).
Two other variants alter splice site consensus sequences, changing the CAG 3'
consensus splice sequence to CAT at the junction of intron 15 and exon 16
(variant #5
in GIT102, Tables 1 and 4), and changing GT to TT at the junction of exon 24
and
intron 24 (#13 in GIT105). Both of these splice site variants are highly
unlikely to
give rise to normal proteins. One variant (#7 in GIT108) deletes 3 base pairs,
resulting in deletion of a serine residue at codon 56I; this serine residue is
a potential
protein kinase C phosphorylation site in the cytoplasmic carboxy terminus of
the
protein (Figure 2c). Finally, one variant (#10 in GITI 12) introduces a
premature
termination codon, deleting the last 54 amino acids from the encoded protein.
CA 02274955 1999-06-14
WO 98/29431
-SO-
PCT/US97/23553
Table 1. Mutations
in the thiazide-sensitive
Na-CI
cotransporter gene
(TSCI in Gitelman's
Syndrome
Kindred Location Mutation Consequence Homozy
ous V
g ariant
GiT100 Spain CTT -.> CCT L850P #
+ 2
GIT102 USA TGC -.~ CGC C421 R
_ 3
CGG ->TGG R209W
_ 4
CAG SCAT Intron 15
_ 5
3' splice site
GIT103 USA CGC -~ CAC R655H
_ 17
GIT104 USA CGC -~ CTC R653L
_ 12
GIT105 USA C~ -~GTT Intron 24
- 13
5' splice site
GIT106 Spain GCG -a GTG A588V
_ 18
GiT107 England CCC -~CTC P349L
+ 6
GIT108 Sweden CCTTCA -..) PS561 P
CCA
_ 8
GGC -~ GTC G630V
_ 14
GIT109 Canada GAC -~ AAC D486N
_ 14
GIT110 Philippines GCA -i A728T
ACA
_ 15
GGC --> TGC G496C
_ 16
GIT111 Japan CGG -~ CAG R955Q
+ 9
GIT1 Netherlands CGA --.~ R968stop
12 TGA
- 10
GGG --> AGG 6741 R
_ 1 1
Sequences are shown. Underlined sequences
of the indicate
sense
strand
of the
gene
consensus
splice
sites
at intron-exon
junctions;
bold
sequence
in GIT108
indicates
segment eleted in variant.
d Variant # refers to
variant numbers in
Figs 1 and 5.
CA 02274955 1999-06-14
WO 98/29431 PCT/US97/23553
-51 -
Table 2. PCR Primers for SSCP analysis of TSC
Primer name Exon Forward primer Reverse primer
hTSCexIA 1 TCCTGGCCCCTCCCTGGACAC ATAGAGCTCATATGTGGGCAC
hTSCex1B 1 CAGCACCTTCTGCATGCGCAC GGAAGTGGCCAGTCTTCTGAG
hTSCex2 2 CTACCTGCCTGACTTGTGGTC TCGACATCACGCACCACCCAC
hTSCex3 3 TGTCCACCCAGGTGGCCTCTG GCTGGGAAGAATGGGATTCAG
hTSCex4 4 GCCCTGCCTAAGCTTTGGGTG CTCGAGAGGAGGGCCTTGGTG
hTSCex5 5 TGGTTTCATGGTTCCCGGCTC ATCCCTCTACCCAGGGTCCAG
hTSCex6 6 CAGAGGGTGGCTTGCAGCCTG GCTTCTCCACGTGACCACCTC
hTSCex7 7 TACTGACCTCTGAGGTCCTTC AGAGCCATGGTCAGGGCCTTG
hTSCexB 8 AGTCTTACTCATCAGGCCTTG CGGCAGATGCCACTAGAGCAG
hTSCex9 9 CTCTCTCCCTCCCTCCTTCAG CTGCAGGGTGGAGGCCAGGTC
hTSCexlO 10 AGGACAGAGTAAGGAGGGAAG GTGTCTGGTGGGTCAGCTCTC
hTSCexl1 11 CAGTAGGGAATGAAGTGCCAC TTGTGCCTCTAGCCCAGGCTC
hTSCexl2 72 AGTGGCAGGTCCCAGCCTAAG AACAGGAGGCCAGGCCCTGTG
hTSCexl3 13 AGACTGTCCTCTCTCTCCCTG TGCCTCCTCCTGAGGTGGGTG
hTSCexl4 14 AGGCATGCCCACTGACTGGTG GCCGCCTGCATGGCTACCCTG
hTSCexl5 15 CGTGTCTGGTTTCCTCTAGTG GTGGAGCCATCACTGGCCCTG
hTSCexl6 16 AGGTGCCTTTCGCACCCAGAC TGCTGGGTTTACAGGCATGAG
hTSCexl7 17 GACATCACCAGCTGCCTTCAC GCCACCAAGCCGTAAGTCCTG
hTSCexl8 18 GTTCCCCATCTCACCCCTATC CACTTGCTCAAGGCCCAATGG
hTSCexl9 19 GGAGAAGCTGGACCTCACCTC AGAACTTTCTGGGAGTGGGTG
hTSCex20 20 ACGGTGCCCTCAGACAAGGAG GAGTGCCCTGAGCTCTGAGTG
hTSCex21 21 GGCTGCTGGCTCTGCTCTGAC GGGCAGGAGGGCTGATCCAAG
hTSCex22 22 CATAGTGCTCTGTCCTGAGTG AGATGACACTGGTCCCTGCAG
hTSCex23 23 GACAGAGCAAGACGCTGTCTC CACAGTTGGCCCTTCTGCCTG
hTSCex24 24 TCTCAGCCGGCCTCAACCCAC TCCCTGACCCAGTGATGTGTC
hTSCex25 25 CGTGAAGGATTGAGTGACCTC CACCTGACTCTGGACAGACTG
hTSCex26 26 ACTTTGCCCATAGGGAGGAAG AGAGCTGTGGACAGGGATGTC
Pri mers ( 5'-3' 1 are all within introns with the exception of: hTSCex 1 A-
reverse and
hTSCex1 B-forward, which lie within exon 1; hTSCex26-forward lies at the
beginning of
exon 26 and hTSCex26-reverse lies distal to the normal termination codon.
Table 3. Normal variants in the Na-CI cotransporter TSC
Codon Normal sequence i Normal sequence 2 Consequences of Variant
1017 ATC ATT I, o ~ ~ I
499 TAC TAT YassY
875 GGC GGT Ge~SG
628 TCG TCA SszeS
714 GCC GCT /y,4A
913 CGG CAG Rstaa
DNA sequence of each codon is shown. The consequence of each variant sequence
on
the encoded protein is shown. I~o"I denotes that the substitution of ATT for
ATC does
not change the isoleucine residue encoded at position 1017.
CA 02274955 1999-06-14
WO 98/29431 PCTlUS97123553
-52-
Table 4.
Summary
of mutations
in TSC
that cause
Gitelman's
Syndrome
PHENOTYPE MUTATION CONSEQUENCE HOMOZ.
GiTELMAN CTT -> CCT L859 -~ P
GITELMAN TGC ~ CGC C421 -~ R
CGG ~ TGG R2pg -~ W
CAG -a CAT Intron 15 - 3'
splice
GITELMAN CGC -~ CAC 8655 -.~ N
AAG --~ AGG K284 -~ R
GITELMAN CGC -~ CTC 8655 -~ L
GITELMAN GGT -~ GTT Intron 24 - 5'
splice
CGG -~ CAG 8964 .-> Q
GITELMAN GCG --> GTG A588 -r V
CTT -~ CCT L859 --> P
GITELMAN CCC -~ CTC P349 -~ L
GITELMAN CCTTCA -.> PS561 -~ P
CCA
GGC -~ GTC 6630 -~ V
GITELMAN CAC ~ AAC 0486 --> N
CGT -~ TGT 8928 -> C
GITELMAN GCA -a ACA A728 -> T
GGC --~ TGC G49fi -.~ C
GITELMAN CGG -> CAG 8964 -a Q
GITELMAN CGA ~ TGA 8977 -> TERM
GGG --~ AGG 6741 -.~ R
CGT -~ TGT 8928 -~ C
GITELMAN CGC -~ UGC 8861 -> C
GITELMAN UGC -> UAC C994 --~ Y
GITELMAN GGG -> AGG G741-~ R
GITELMAN GGT -~ GTT Intron 9 - 5' splice
GITELMAN GGG -r AGG 6741 -a R
GITELMAN GGC ~ AGC 6439 -~ S
CA 02274955 1999-06-14
WO 98129431
- 53 -
PCT/US97I23S53
PHENOTYPE MUTATION CONSEQUENCE HOMOZ.
CAC --> AAC H69 --~ N
G1TELMAN CGG -.~ CAG 8964 -~ Q
GITELMAN GGC -~ AGC 6439 --~ S
GITELMAN CGG -~ CAG 8964 -> Q
AAG -.~ AAC K734 -a N
GITELMAN 7 by deletion H696 -.> FS
GGT ~ GTT 6729 --~ V
GITELMAN AGG --~ TGG 8334 --~ W
GITELMAN 2 by deletion 6731 -.) FS
GGC -~ AGC 6439 -> S
GITELMAN 1 by deletion F765 -~ FS
GITELMAN AGG -~ TGG 8334 .-> W
CGT --~ TGT 8399 -.> C
GITELMAN CGC -~ TGC 8861 -.~ C
GITELMAN GGG --~ AGG 6741 -~ R
CGC -~ CAC 8642 .-~ H
G1TELMAN GGT -~ GTT intron 9 - 5' splice
GITELMAN 7 by insertionD400 -~ TERM
GGC -a GAC 6460 .-~ D
GITELMAN CGT --r CTT 8399 --~ L
GITELMAN CGG --~ TGG 8321 -.~ W
CTT -~ CCT L859 -.> P
GITELMAN CGG -~ CAG 8209 -~ Q
AAA -~ UAA K497 -~ TERM
GITELMAN CGC -~ GGC 8642 .-~ G
AGC --r ATC S448 ~ I
GITELMAN AUG -.> AAG M343 -3 K
G1TELMAN GGG -> AGG 6741 -) R
GITELMAN GTG -~ ATG V1024 --> M
ATG -i AAG M 581 -~ K
GITELMAN GGG -a AGG 6741 -> R
1
CA 02274955 1999-06-14
WO 98/29431 PCT/iJS97/23553
-54-
PHENOTYPE MUTATION CONSEQUENCE HOMOZ.
GITELMAN CGG -~ CAG 8964 -~ Q
GAC --~ AAC D486 ~ N
GITELMAN GGG --> AGG 6741 -~ R
GITELMAN CCC -+ CTC P643 -+ L
CGG -~ CAG 8964 --> Q
GITELMAN CTT -+ CCT L859 -~ P
CGT -> CTT 8399 -~ L
GITELMAN CGC -~ CAC 8655 -~ H
GT -~ AT Intron 5 - 5'SPLICE
GITELMAN CCC -+ CTC P643 -+ L
GITELMAN GGG -+ AGG 6989 -> R
GITELMAN TCC -> CCC S620 -a P
GITELMAN GAC --> AAC D486 --> N
GITELMAN CGG -> CAG 8964 --i Q
GITELMAN CGT ~ GT 8928 --> C
GITELMAN CAG ~ TAG E112 -~ TERM
GGT -> GTT 6729 -> V
GITELMAN GGT -.> GTT Intron 9 - 5' splice"+"
GITELMAN GGT .~ GTT Intron 24 - 5' splice"+"
GITELMAN CGC -i TGC 8861 -.~ C
CGA ->CAA 8896 --> Q
GITELMAN CGC -> TGC 8861 -> C
GITELMAN 7 BP DELETION AA 969
GITELMAN CGA --~ TGA 81018 --~ TERM "+"
GITELMAN 7 8P DELETION AA696
CGT -~ CCT 8871 --> P
GITELMAN CGT --> CCT 8871 -> P
GITELMAN GGT -) GTT Intron 24 - 5' splice"+"
CA 02274955 1999-06-14
WO 98/29431 PCT/LJS97/23553
-55-
PHENOTYPE MUTATION CONSEQUENCE HOMOZ.
GITELMAN 4 by InsertionL998 -> FS "+
GITELMAN GAC -~ AAC D486 -> N
GITELMAN GTG -~ ATG V647 ~ M +-'
GITELMAN TCG -a TTG S555 -~ L
CTT -> CCT L859 -> P
GITELMAN GGT -a GTT Intron 9 - 5' splicea+"
GITELMAN CTT -a CCT L859 -.> P
ACC -~ AUC T1026 -a I
GITELMAN GGT -> GTT Intron 9 - 5' splice
GGG --~ AGG 6741 -~ R
GITELMAN CGC --r CAC 8642 --~ N
CTC -~ CGC L1010 -a R
GITELMAN GCC -~ ACC A464 -) T
GITELMAN GGT -.> GTT Intron 24 - 5' splice
Discussion
The finding of complete linkage of Gitelman's Syndrome with TSC, coupled
with the finding of a large number of independent variants that are highly
likely to
disrupt TSC function and which are specific for Gitelman's Syndrome families,
constitute proof that mutations in TSC cause Gitelman's Syndrome, the
predominant
subset of patients with Banter's Syndrome. Whether the minority of patients
with
inherited hypokalaemic alkalosis and normal or hypercalciuria, so-called "true
Banter's" patients (Bettinelli, A., et al., J. Pediatr. 120:38-43 {1992)),
will prove to
have mutations in this same gene or a different gene will require further
study.
CA 02274955 1999-06-14
WO 98/29431 PCT/US97123553
-Sb-
The diverse physiologic features of Gitelman's Syndrome must all stem from
mutation in TSC. From the autosomal recessive inheritance and the known
physiology of the syndrome, we propose that mutant alleles result in loss of
normal
TSC function leading to defective sodium and chloride reabsorption in the
distal
convoluted tubule. This defect would be expected to result in sodium plus
chloride
wasting, resulting in some degree hypovolemia and metabolic alkalosis. The
reduced
vascular volume activates the renin-angiotensin system, elevating renin and
aldosterone levels. The elevated aldosterone levels lead to increased
electrogenic
sodium reabsorption via the epithelial sodium channel in an effort to defend
intravascular volume. Sodium reabsorption via this channel is counterbalanced
by
potassium and hydrogen ion excretion, resulting in hypokalaemia and
contributing to
metabolic alkalosis. These are all characteristic features of Gitelman's
Syndrome.
How this primary defect in TSC function results in loss of magnesium and
hypocalciuria remains a matter of speculation) however it is noteworthy that
these
same effects are seen in patients taking thiazide diuretics, specific
inhibitors of the
TSC; these observations further support the interpretation that TSC mutations
result in
loss of function. Importantly, it is now apparent that these latter
abnormalities must
be secondary consequences of primary defects in TSC.
Prevalence and transmissi n of itelman's all .iPs
Unusual segregation ratios and patterns of inheritance in some families have
confused the understanding of the inheritance of Gitelman's Syndrome. The
present
linkage findings are consistent with genetic homogeneity of transmission as an
autosomal recessive trait. The linkage findings support recessive inheritance
more
than 100,000-fold better than either dominant inheritance with incomplete
penetrance
or mitochondrial inheritance. The finding of affected subjects in different
branches of
some kindreds (e.g. GIT 102 and GIT 112) can now be explained in molecular
terms
by spouses in different branches introducing additional mutant alleles. For
example,
the 3 affected siblings of GIT102 (subjects I2, I4 and 15) are compound
CA 02274955 1999-06-14
WO 98/29431
PCTIUS97123553
-57-
heterozygotes for variants 8421 and W209 (variants #3 and #4, Figure 1, Tables
1
and 4). 8421 is also inherited by an affected second cousin, family member 11,
by
descent from their common ancestor; this subject is also a compound
heterozygote,
having inherited a third independent variant, altering a consensus splice site
variant
(variant #5; Tables l and 4, Figure 1) from her father.
Finding independent mutant alleles introduced into different branches of
kindreds suggests that mutant alleles are not rare in the population.
Moreover, the
findings that consanguineous marriage is not prominent in Gitelman's Syndrome
kindreds and that a high proportion of patients are compound heterozygotes are
consistent with this notion. Also consistent with this notion is the
remarkable
conservation of 629 amino acids of this protein between humans and flounder,
suggesting that many of these residues are required for normal protein
function.
These observations suggest that the potential target size for mutations
generating
Gitelman's Syndrome alleles will prove to be large, affording a relatively
high rate of
introduction of new mutant alleles into the population. The finding that thus
far only
one residue is mutated more than once on independent mutant alleles supports
the
contention of a large target size for Gitelman's mutations. While the true
prevalence
of this disease is unknown, minimum estimates of disease prevalence based on
clinical features suggest a prevalence of heterozygotes of at least 1 % in
Swedish and
Italian populations. Once the spectrum of mutations is defined, the true
prevalence of
mutant alleles in different populations can be determined independent of
phenotypic
effect.
Given the recessive inheritance of Gitelman's Syndrome, the high proportion
of affected offspring of heterozygous parents in the Gitelman's kindreds
studied to
date is striking (Figure 1 ). After excluding index cases in these kindreds,
22/33
offspring of two heterozygous parents have Gitelman's Syndrome, far more than
the
expected 8 affected subj ects. This high proportion of affected offspring
could
certainly result from ascertainment bias favoring identification of multiplex
families
with unusual segregation ratios. An alternative explanation would be
segregation
CA 02274955 1999-06-14
WO 98!29431
PCT/US97123553
-58-
distortion, with transmission of mutant alleles from heterozygous parents to
more than
the expected 50% of offspring. With the identification of mutant alleles, the
segregation ratio of Gitelman's alleles in extended kindreds can be determined
in
order to directly assess this possibility without ascertainment bias.
Potential implicati~nc in hP.rPrn~ygotes
The identification of mutations that cause relatively severe disease in
homozygotes raises the possibility that these alleles might have more modest
effects
in the much more common heterozygous state. One can imagine several possible
phenotypes in Gitelman's heterozygotes, including modestly reduced blood
pressure
and/or predisposition to diuretic-induced hypokalaemia. Hypertension is a
common
multifactoriai trait frequently associated with increased renal sodium
reabsorption and
sensitivity of blood pressure to the effects of dietary salt. Individuals
heterozygous
for Gitelman's mutations may be protected from development of hypertension by
having modestly reduced renal sodium reabsorption, leading to a reduced set
point of
sodium balance, reduced intravascular volume and lower blood pressure. The
potential impact of such alleles on blood pressure in the general population
would be
dependent on the true prevalence of such heterozygotes and their quantitative
effect
on blood pressure, both of which are unknown. Since the morbid clinical
consequences of hypertension typically occur well after reproductive age, it
seems
unlikely that these alleles would increase reproductive fitness.
Hypokalaemic alkalosis is a relatively common complication of diuretic
therapy of largely unknown cause. Mutant TSC alleles could contribute to
development of this complication by further augmenting delivery of sodium and
chloride to the distal nephron. This effect might be most likely in patients
taking loop
diuretics such as furosemide, and these mutations could consequently
predispose to a
particular complication of pharmacologic therapy.
Identification of specific mutations causing Giteiman's Syndrome permits
testing of these hypotheses by identifying cohorts of heterozygous carriers
and
CA 02274955 1999-06-14
WO 98/29431 PCTIUS97/23553
-59-
comparing their blood pressures and responses to pharmacologic intervention to
those
of their homozygous wild-type siblings or other controls. In addition,
subjects with
unexplained hypokalaemia or hypokalaemia complicating drug therapy may be
candidates for harboring Gitelman's mutations.
Finally, identification of the molecular basis of Gitelman's Syndrome provides
for the genetic diagnosis of this disorder. At initial presentation, some
patients with
this disease are incorrectly believed to be diuretic abusers or to have
bulimia. Indeed,
one of the patients described herein was committed to a locked psychiatric
ward and it
was only when her hypokalaemia persisted for two weeks in this setting that a
proper
diagnosis was made. The ability to make a molecular genetic diagnosis is
therefore of
practical clinical benefit in many settings.
EXAMPLE 2
r w~ i
by mutations in the Na-K-2C'1 Cntra"Snorter NKC(=2
Bartter , yndrome KinrjrPric BpRl38 has been previously reported (Di Pietro,
A. et al., Ped. Med. Chir. 13:279-280 ( 1991 )). The other kindreds were
recruited via
ascertainment of a severely affected index case. Genomic DNA was prepared from
venous blood of members of Bartter's Syndrome kindreds by standard procedures
(Bell, G., Karam, J., et al., Proc. Natl. Acad. Sci. (U.S.A.) 78:5759-5763
(1981)).
CIoninE and Characterisation of the Human ~C' ~nomi NK
Oligonucleotide primers corresponding to either rabbit (Payee, J.A., et al.,
Proc. Natl.
Acad. Sci. (U. S.A.) 91:4544-4548 ( 1994)) or rat (Gamba, G. et al., J. Biol.
Chem.
26:17713-I 7722 ( I 994)) NKCC2 cDNA sequence were used to direct PCR using a
human kidney cDNA library as template. The sequence of the corresponding human
cDNA encoding NKCC2 was determined; sequences were determined from both
CA 02274955 1999-06-14
WO 98129431 PCTIUS97/23553
-60-
DNA strands. Segments from the 5' and 3' ends of the human NKCC2 cDNA were
radiolabeled and used to screen a human genomic PAC library35 by hybridization
as
described previously (Shimkets, R.A. et al., Cell 79:407-414 ( 1994)). Intron-
exon
boundaries were determined by sequencing both strands of genomic DNA
fragments.
Genomic fragments bearing single introns and portions of adjacent exons of
NKCC2
were isolated by long-range PCR (ExpandTM Long PCR System; Boehringer-
Manheim) using PAC clone or genomic DNA as a template and primers likely to
Iie
in within each exon based on the organization of the related gene TSC. DNA
sequence analysis was by the dideoxy chain termination method using an ABI 373
instrument following a standard protocol.
Marker Development. enot~ ing~ and i ka~ na ,yes. Sau3AI subclones
of the NKCC2 PAC clone were screened for (GT)n repeat sequences by
hybridization
as previously described (Shimkets, R.A. et al., Cell 79:407-414 ( 1994)). This
screen
identified clone NKCGT7-3 which contained an array of (GT) (McCredie, D.A., et
al., Aust. Pied. J. 10:286-295 (1974)); primers flanking this simple sequence
repeat
were designed and used to direct PCR from genomic DNA of unrelated subjects
(NKCGT7-3F: CACTAGGCTATTGTGTGGCTC; NKCGT7-3R:
GTCTGTCCTCCACACTAG).
Genotyping of CEPH and disease kindreds for Ioci indicated in the text was
performed by polymerise chain reaction using specific primers (Gyapay, G. et
al.,
Nature Genet. 7:246-339 (1994)) as described previously (Shimkets, R.A. et
al., Cell
79:407-414 (1994)), except that the products were labeled by inclusion of 1
uCi of
[I-3zP]dCTP in the reaction mixture. All genotypes were scored independently
by two
investigators who were blinded to affection status. Analysis of linkage was
performed
using the LINKAGE programs Lathrop, G.M. et al., Proc. Natl. Acid. Sci.
(U.S.A.)
81:3443-3446 (1984)).
SSCP and DNA Sea ~ n in . Molecular variants in genes were sought using
single-strand conformational polymorphism (Orita, M. et al., Proc. Natl. Acid.
Sci.
(U. S.A.) 86:2766-2770 ( 1989)). Primers for TSC were as described previously
CA 02274955 1999-06-14
WO 98/29431
-61 -
PCTIUS97/23553
(Example 1 and Simon, D.B. et al., Nature Genet. 12:24-30 ( 1996)); exons of
NKCC2
were screened using 27 sets of specific primers {Table 6) based on the genomic
organization of the gene. Primer pairs were employed to separately direct PCR
using
genomic DNA of disease family members or CEPH controls as a template as
previously described (Shimkets, R.A. et al., Cell 79:407-414 (1994)).
Amplified
products were analyzed for molecular variants by electrophoresis under 3
different
non-denaturing conditions as well as under denaturing urea gels as previously
described (Shimkets, R.A. et al., Cell 79:407-414 (1994)). Identified variants
were
eluted from gel, reamplified by PCR, and DNA from both strands was sequenced
in
all cases. All SSCP genotypes were confirmed by independent amplifications.
~Svndrome kindr rls 4 kindreds with at least one subject diagnosed
with Bartter's Syndrome were studied. One of these cases has been reported
previously (Di Pietro, A. et al., Ped. Med. Chir. I 3:279-280 ( I 991 )). In
three of these
families, at least one affected subject is known to be the offspring of
consanguineous
union (Figure 6). In addition, in kindred BAR156 two second cousins of an
index
case are also affected. Index cases (Table 5) were all born prematurely with
documented polyhydramnios and birth weight below 2 kg, and presented in the
neonatal period with severe dehydration associated with marked hypokalaemia
and
hyperreninemic hyperaldosteronism. All had marked hypercalciuria and 3 had
ultrasonographic evidence of nephrocalcinosis; none of these patients had
hypomagnesemia. The early and severe presentation, hypercalciuria,
polyhydramnios,
and nephrocalcinosis all distinguish these patients from those previously
classified as
having Gitelman's Syndrome who were found to have mutations in TSC as the
cause
of their disease (Simon, D.B. et al., Nature Genet. 12:24-30 ( I996)).
Evaluation of T To determine whether Banter's Syndrome is due to
mutations in the thiazide-sensitive Na-CI cotransporter (TSC), we genotyped
markers
tightly linked to this locus on chromosome 16 in the consanguineous kindreds.
If
Banter's Syndrome were due to mutations in TSC, we would anticipate that
affected
CA 02274955 1999-06-14
WO 98/29431
-62-
PCT/US97/23553
subjects in these kindreds would be homozygous at the trait locus and at
closely
linked loci (Lander, E.S., et al., Science 236:1567-1570 (1987)). D16S408 is
linked
to TSC at a recombination fraction of 1 % (Simon, D.B. et al., Nature Genet.
12:24-30
(1996)). This marker proved to be heterozygous in all of these affected
subjects;
analysis of additional closely linked flanking markers confirmed that none of
these
subjects have inherited two copies of the same haplotype {data not shown).
These
findings do not support mutation in TSC as the cause of Bartter's Syndrome.
This
conclusion is further supported by results of screening all 26 exons encoding
the TS C
protein for molecular variants by single strand conformational polymorphism
(SSCP)
(Orita, M. et al., Proc. Natl. Acad. Sci. (U.S.A.) 86:2766-2770 (1989)). No
variants
altering the encoded protein were detected in any of these patients (data not
shown).
C'haracteri~at;on of h Oman NK In order to investigate NKCC2 for its
potential role in Banter's Syndrome, we characterized the human NKCCZ locus
(Figure 7). The predominant variant of human NKCCZ cDNA isolated from a human
kidney cDNA library encodes a protein of 1099 amino acids that shows strong
sequence similarity to NKCC2 cDNAs characterized from rabbit {Payne, J.A., et
al.,
Proc. Natl. Acad. Sci. (U.S.A.) 9I :4544-4548 ( 1994)) (95% amino acid
identity), and
rat (Gamba, G. et al., J. Biol. Chem. 26:17713-17722 (1994)) (93% identity).
In
addition, human NKCC2 shows considerable similarity in amino acid sequence to
NKCC1 from shark rectal gland (Xu, J.-C. et al., Proc. Natl. Acad. Sci.
(U.S.A.)
91:2201-2205 (1994)) (60% identity) and the human TSC (Simon, D.B. et al.,
Nature
Genet. 12:24-30 (1996)) (47% identity).
Plasmid artificial chromosome (PAC) (Ioannou, P.A. et al., Nature Genet.
6:84-89 (1994)) clones containing the human genomic NKCC2 locus were isolated,
and one of these, NKCC2-6A, was found to contain all the coding exons. This
permitted determination of the intron-exon structure of the gene (see
Methods). The
NKCC2 protein is encoded in 26 exons (Figure 7a ); introns range in length
from 120
base pairs to 15 kb, and the coding region spans a total of 80 kb in genomic
DNA.
The location of intron-exon boundaries is very similar to those seen in TSC
through
CA 02274955 1999-06-14
WO 98/29431
-63-
PCT/US97/23553
exon 19 (Simon, D.B. et al., Nature Genet. 12:24-30 (1996)). Three alternative
variants of exon 4 are encoded in genomic DNA, as reported in other species
(Igarashi, P.et al., Am. .l. Physiol. 269:F405-F418 ( 1995)). By using primers
from
genomic sequences to direct PCR using DNA of somatic cell hybrids as a
template,
NKCC2 was localized to human chromosome 15 (data not shown).
A (GT) dinucleotide repeat sequence was identified at the cloned NKCC2
locus; this repeat proved to be polymorphic in genomic DNA, with S alleles and
42%
heterozygosity in 50 unrelated subjects. This marker was genotyped in CEPH
reference kindreds in order to localize NKCC2 on the human genetic map;
analysis
revealed linkage to a cluster of loci at 1 Sq 1 S-21. Multipoint analysis
yielded a
maximum lod score of 13.3 for linkage to a 3 centimorgan interval flanked by
D 1 SS 132 and D 1 SS209 (Figure 7c); the odds favoring location in this
interval were
more than 100 times as likely as location in the next most likely interval
(Figure 7c).
Homozv~nsitv of NKCC''~ .n R~,ltnr'c >r:.,a a ~e genotyped members of
Bartter's Syndrome families for 4 markers in and flanking NKCC2. In each
family,
all affected offspring of consanguineous union were homozygous for alleles of
each
marker, and these haplotypes cosegregated with the disease, providing strong
evidence supporting linkage of Banter's Syndrome and the NKCC2 locus (Figure
6).
'Moreover, while the affected subject in BAR138 is not known to be the product
of
consanguineous union, she too is homozygous for all markers tested in this
segment.
Mutations in NK in Bartt~r's ymdro; ;~. These linkage data strongly
suggest that Bartter's Syndrome is attributable to mutation at the NKCCl
locus,
motivating a search for functional mutations at this locus in these patients.
Primers
within introns of NKCC2 were used to direct PCR from genomic DNA of affected
subjects and the products were analyzed by SSCP. A single novel variant was
found
in each kindred and each cosegregated with the disease phenotype (Figures 6,
8). Ali
these variants are homozygous in affected subjects with the exception of two
affected
siblings in,BAR156. In this kindred, the index case is homozygous for a novel
variant. Her affected second cousins have inherited one copy of the same
variant
CA 02274955 1999-06-14
WO 98/29431 PCT/US97l23553
-64-
along with a portion of her haplotype; these subjects have presumably
inherited a
second, and as yet undetected, variant on the maternally inherited allele.
None of
these variants have been observed in examination of 80 alleles from unrelated
subjects
who do not have Bartter's Syndrome, and no variants altering the amino acid
sequence have been detected in unrelated unaffected subjects.
Analysis of the DNA sequence of these variants reveals that each alters the
encoded protein (Figure 8). The variant in BAR152 is a 1 base pair insertion
introducing a frameshift mutation at codon M 19S in the first transmembrane
domain.
The variant in BAR138 represents a 1 base pair deletion in codon 8302 in the
fourth
transmembrane domain, also resulting in a frameshift mutation. Both of these
mutations are extremely unlikely to yield proteins with normal function.
The other two variants result in non-conservative missense mutations at
residues that are conserved among members of the NKCC family from distantly
related species. The variant in BAR 1 S6 substitutes phenylalanine for valine
at residue
272 in the third transmembrane domain. This valine residue is conserved in
NKCC2
from human, rabbit, mouse, and rat, as well as NKCC 1 in shark and human.
Similarly, the variant in BAR16S substitutes an asparagine residue for
aspartate at
amino acid 648, just distal to the 12th transmembrane domain; this aspartate
residue is
also conserved in every member of the NKCC family.
Table 5.
Clinical
characteristics
of index
cases with
Banter's
Syndrome
KindredLocation K+ HC03~ UCa/UCr Nephrocalcinosis
Preterm
BAR138 Italy i.7 37 0.9 No Yes
BAR Saudi Arabia3.1 27 0.7 Yes Yes
152
BAR156 Saudi Arabia2.2 33 0.6 Yes Yes
BART Saudi Arabia1 .9 27 0.5 Yes Yes
65
K+: serum potassium (mM, nl 3.5-5.0 mM); HC03-: serum bicarbonate (mM, nl 23-
26
mM); UCa/UCr: urinary calcium:creatinine ratio (mM:mM; nl ratio 0.2 - 0.4?.
Nephrocalcinosis determined by ultrasonography. Preterm: birth before 36
weeks'
gestation. ND, not done.
CA 02274955 1999-06-14
WO 98/29431 PCT/US97123553
-65-
Table 6. PCR Primers for SSCP Analysis of the Human NKCC2 Gene
Primer Name Exon Forward Primer RPVPrca Primor
m ~ HH~~ACAAAGTAGATAGCTCAG AACTGGCATCTGTTTTAGCAG
hNKCC2exlB 1 GGGAATCAGGAGTGCTATGAC AAGGGAGGAGACTTGCTTGTG
hNKCC2ex2 2 TGTTCATTGACCAACTACTGTG GCCTTGTTCACTCTTAATCCAC
hNKCC2ex3 3 CACTATCGTTTGTCCTGTCTC GTGACCTTCATCTCACATTCAG
hNKCC2ex4 4 GATGTTTACCCTAGACTTGCTG ACAAATGATGGTGCGGGTCAC
hNKCC2ex5 5 CTAGCAGTTCCTCAATGTGAAG ACTAAATTATGCTGCTTGGCAG
hNKCC2ex6 6 ATGCTGCAATAAGACTCACATG CAGGACCTGACCAGCCACTG
hNKCC2ex7 7 GAGTCTTTCTGCAGTGGACAC GAGGAGGGCAATGGAGAAG
hNKCC2ex8 8 GTAACTTAATCTCCTGTACTGTG TCCCAGGAATGCAAAGCAGAG
hNKCC2ex9 9 CTCTGTATTCTTCTACCTCCAC TGACATTCTGACACTGGAGAC
hNKCC2ex10 10 TAGAAAACCGTAAGGGACCAG TCAGAAATCTTACTGTATGTGAC
hNKCC2ex11 11 ATGAAACAGATTCCAAATCACAG AATAGGGAGAAGCACAAGCTG
hNKCC2ex12 12 CTAGAGAAAATGACTGTGCATAG GGAAAGCCCTATGAATAATCAG
hNKCC2ex13 13 GCTCATCACTCATACGTACATG CGTTTTATTGAGACAAACTAACTG
hNKCC2ex14 14 GTGCCATGATCATAGTAGAGTG TGGAAACGCTATTCCAGACAG
hNKCC2ex15 15 TGCACAGAGGAAAGGTCAGTG ACACCAGGATGCCTGAGACAC
hNKCC2ex16 16 CAGGCTTCTTGCAGGGGCAC GGAGGAAAAAGGACTTCCCTG
hNKCC2exi7 17 CAGCAATGTGATATATAATAGCAG GTGCTCATTCCCTCAATGCAG
hNKCC2ex18 18 AGTACGGTAAGGATTGCCCAC TATGTACTGCCCTGCTTAGTG
hNKCC2ex19 19 GTAATACTAGTCCAAAGCTTGAG TCAGGCACAAAGTAGGTGCTC
hNKCC2ex20 20 GTAGTTCTGAGTTAAGTAGGTG CATAGATGCTCAAATAGTGACTC
hNKCC2ex21 21 GCCCTCAAAAGCAAACAGATG CCCATATACCTTCTCATGCAG
hNKCC2ex22 22 CCATTTAGATATACTCTTTGTGTC TGAAATGACCTAACATGTGAGTG
hNKCC2ex23 23 AAGCTAAGCTGAAATAAGACGTG GTACCATGGGTAATCAATGTCTC
hNKCC2ex24 24 GTTTCCCACTGTGAGGCCTC CCTTTCTCAGCTAGTTAGACAG
hNKCC2ex25 25 CATAATTCTGGTAGAACTGTACTC TCCCACCTGAAGAGTCCCAAG
hNKCC2ex26 26 CTAGTGCCGTTACTACCTATAG GATCAC;ATTTnr~nnr-eTnnnr_Tnr~
~.G. ~ ar C au mrmn introns with the exception of: hNKCC2ex 1 A-reverse and
hNKCC2ex1 B-forward, which lie within exon 1; hNKCC2ex26-reverse lies distal
to the
normal termination colon.
Table 7. Summary of mutations in NKCC2 that cause Bartter's Syndrome
Phenotype Mutation Conseauenr:e Hnmn~~in~,~ ~~
warmer ATG --> AATG M195 -~ Frameshift +
Bartter CGG -.~ GG 8302 --> Frameshift +
Bartter GAC -.~AAC D648 -~ N
Bartter GTT -a TTT V272 -~ F +
Bartter CAG -~ CTAG Q823 -~ Frameshift
+
Bartter TCC -~ CCC S507 --~ P
CA 02274955 1999-06-14
WO 98!29431 PCT/C1S97123553
- 66 -
Discussion
Identification of independent mutations in NKCC2 that segregate with the
disease, show specificity for Banter's Syndrome kindreds, and result in
&ameshifts or
non-conservative amino acid substitutions in highly conserved residues,
constitutes
S proof that mutations in the renal absorptive Na-K-2C1 cotransporter cause
Bartter's
Syndrome. This finding establishes a molecular basis for this disease and
permits
genetic distinction of this disorder from Gitelman's Syndrome. These findings
expand knowledge of the molecular mechanisms underlying inherited diseases of
renal ion transport. Mutations in 5 different genes mediating renal sodium
reabsorption are now implicated in 4 different mendelian disorders (Figure 9)
(Shimkets, R.A. et al., Cell 79:407-414 ( 1994); Hansson, J.H. et al., Nature
Genet.
11:76-82 ( I 995); Chang, S. S. et al., Nature Genet. 12:248-253 ( 1996);
Simon, D.B. et
al., Nature Genet. 12:24-30 ( 1996)). In addition, mutations leading to
increased
activity of the mineraloconicoid receptor, including glucoconicoid-remediable
i 5 aldosteronism (Lifton, R.P. et al., Nature 355:262-265 ( 1992)} and the
syndrome of
apparent mineraloconicoid excess (Mune, T. et al., Nature Genet. 10:394-399
(1995)), act by increasing activity of the epithelial sodium channel of the
distal
nephron.
Disease 1 a~ thop~y iolo v Virtually all features of the pathophysiology of
Banter's Syndrome can be explained by mutations in the Na-K-2C1 cotransponer
leading to loss of function. This cotransponer is located in the thick
ascending limb
of the loop of Henle and accounts for reabsorption of approximately 30% of the
filtered load of sodium (Greger, R., Physiol. Rev. 65:760-797 (1985)) (Figure
9);
consequently, loss of function leads to marked salt wasting and volume
contraction,
accounting for the dehydration seen in affected patients. This results in
secretion of
renin and elevated aldosterone levels, producing increased electrogenic sodium
reabsorption via the amiloride-sensitive epithelial sodium channel of the
distal
nephron. Sodium reabsorption via this channel is indirectly coupled to
potassium and
hydrogen ion secretion, producing the characteristic hypokalaemic alkalosis.
CA 02274955 1999-06-14
WO 98/29431
PCT/US97/23553
-67-
The reabsorption of approximately 25% of filtered calcium also occurs in the
thick ascending limb (Sutton, R.A.R., et al., In The Kidney (eds. Brenner,
B.M. &
Rector, F.C.) 551-618 (Saunders, Philadelphia, 1981)) and this is coupled to
sodium
reabsorption in this nephron segment. Loss of sodium reabsorption here would
consequently be expected to result in the characteristic hypercalciuria and
nephrocalcinosis seen in these patients.
Urinary prostaglandin E2 levels have been reported to be elevated in patients
with Bartter's Syndrome (Seyberth, H. W. et al. , Pediatr. Nephrol. 1:491-497
( I 987);
Gill, J.R., Jr., et al., Am. J. Med. 61:43-51 ( I 976)); high PGE2 levels can
be produced
by administration of loop diuretics (Dunn, M.J.) Kidney Int. I 8:86-102 ( 198
/ )),
consistent with genetic loss of Na-K-2C1 function resulting in a secondary
increase in
PGE2 levels.
(classification of hvnokalaem» ~»LI~n~~ It is now apparent that inherited
hypokalaemic alkalosis is genetically heterogeneous and due to mutations in
genes
encoding at least two proteins -- the Na-K-2CI cotransporter of the thick
ascending
limb of the loop of Henle (NKCC2) and the Na-Cl cotransporter of the distal
convoluted tubule (TSC). Thus far, the clinical and physiologic features of
patients
with mutations in these two genes are readily distinguishable -- ail patients
ascertained
with hypokalaemic alkalosis plus neonatal presentation and hypercaiciuria have
mutations in NKCC2, while all patients ascertained with hypokalaemic alkalosis
plus
presentation after age 8 and hypocalciuria have had mutations in TSC (Simony
D.B. et
al., Nature Genet. 12:24-30 (/996)).
Whether all inherited hypokalaemic alkalosis with normal or low blood
pressure will prove due to mutation in one of these two genes or whether
further
genetic heterogeneity will be found is unresolved. A group of patients has
been
described with severe magnesium wasting, hypokalaemic alkalosis and
nephrocalcinosis (Gunner Syndrome) (Gullner, H.-G., et al., Am. J. Med. 71:578-
582
( 1981 )). The hypokalaemia in these patients has been reported to be
corrected with
magnesium replacement. These patients seem likely to be another distinct
subset of
CA 02274955 1999-06-14
WO 98/29431 PCT/LTS97/23553
-68-
the spectrum of inherited hypokalaemic alkalosis. The finding of high urinary
prostaglandin levels in some patients with, typically, neonatal hypokalaemic
alkalosis,
has led to suspicion that hyperprostaglandiuria might represent a distinct
syndrome
(hyperprostaglandinuria E syndrome) (Seyberth, H. W. et al., Pediatr. Nephrol.
1:491-
497 (1987)), however the present results suggest that this group of patients
will likely
prove to have mutations in NKCC2 with hyperprostaglandinuria as a secondary
phenomenon. At present, the evidence for two distinct clinical syndromes
supported
by mutations in two different genes supports a logical framework for
classification of
these patients as having either Gitelman's Syndrome or Bartter's Syndrome.
Conversely, the spectrum of clinical and physiologic features resulting from
mutations in these two genes also remains to be defined. Patients who have
been
studied thus far represent extremes of the phenotypic spectra. Some patients
with
hypokalaemic alkalosis have been reported to have normal urinary calcium;
these
patients could have mutations in NKCC2, TSC or another gene. Further
examination
of genotype-phenotype relationships and specific phenotypic consequences of
particular mutant alleles will consequently be of interest, and should permit
rigorous
classification based on analysis of mutations and clinical features.
Disease prevalence. The proteins encoded by NKCC2 and TSC are closely
related, with similar highly conserved sequence, structure and function. From
this,
one might expect the prevalence of mutant alleles in these genes, and the
prevalence
of clinical disease, to be similar. However, from our experience in recruiting
published patients with hypokalaemic alkalosis, it appears that patients with
Gitelman's Syndrome (defined by the presence of hypocalciuria) grossly
outnumber
those with Bartter's Syndrome (defined by the presence of hypercalciuria)
(Simon et
al., unpublished data). The greater morbidity and mortality associated with
Bartter's
Syndrome could reduce reproductive fitness, resulting in increased loss of
mutant
NKCC2 alleles from the population.
There may also be a difference in the rate of introduction of new mutant
alleles
at these loci. The most common site for single base substitution in mammals is
at
CA 02274955 1999-06-14
WO 98/29431
PCT/US97/23553
-69-
CpG dinucleotides owing to methylation at these cytosine residues and
subsequent
deamination to thymidine (Cooper, D.N., et al., Harm. Genet. 83:181-188
(1989)).
There is a striking difference in the prevalence of CpG dinucleotides in
coding regions
of these two genes. Cytosine residues of CpG dinucleotides occupy the first or
second
codon positions of 60 codons in TSC but only 30 such sites in NKCC2. This
difference is not merely due to differences in amino acid sequence, but is
reflected in
significant differences in codon usage. For example, while 30 of 48 arginine
codons
in TSC have CpG dinucleotides, only 15 of 43 arginine residues in NKCC2
utilize
such codons (c2,1 df = 6.99, p < 0.01 ). These differences may reflect
stronger
selection against CpG dinucleotides in NKCC2, and may in part account for the
large
difference in prevalence G-C base pairs in coding regions of these two genes
(59% in
TSC, 45% in NKCC2).
Potential imo~lications in hetero Ygo a . While mutant NKCC2 alleles have
severe effects when homozygous, they may also have detectable phenotypes in
the
more prevalent heterozygotes. Among potential phenotypes, lowered blood
pressure is
a reasonable possibility, owing to reduced sodium reabsorption in the thick
ascending
limb, leading to a reduced set point of sodium balance and decreased
intravascular
volume. The ability of loop diuretics to lower blood pressure by inhibition of
NKCC?
supports this possibility.
Another potential phenotype in heterozygotes is susceptibility to diuretic-
induced hypokalaemia. Thiazide diuretics are commonly used anti-hypertensive
agents that inhibit TSC in the distal convoluted tubule; thiazide-induced
hypokalaemia is a relatively common complication. Heterozygotes for NKCC2
mutations may be able to compensate for diminished sodium reabsorption in the
thick
ascending limb by increased distal reabsorption via TSC in the distal
convoluted
tubule (Ellison, D.H. Ann. Int. Med. 114:886-894 (1991)) (Figure 9).
Inhibition of
this latter cotransporter by thiazides could, however, result in marked salt
wasting
which could only be compensated by increased activity of the epithelial sodium
channel in the distal nephron at the expense of marked potassium loss.
Heterozygous
CA 02274955 1999-06-14
WO 98129431 PCTlUS97/23553
-70-
NKCC2 mutations could consequently predispose to this specific complication of
pharmacologic therapy, and such heterozygous carriers could constitute a
significant
fraction of patients developing this complication.
Homozygotes for NKCC2 mutations have marked urinary calcium wasting,
leading to both early and severe nephrocalcinosis and marked demineralization
of
bone. Salt wasting in heterozygotes for NKCC2 mutations might also result in
hypercalciuria, potentially increasing susceptibility to nephrolithiasis
(renal stones)
and osteoporosis (bone demineralization), multifactorial traits with known
inherited
components. Interestingly, mutations in an X-linked renal chloride channel
have been
shown to promote development of nephrolithiasis {Lloyd, S.E. et al., Nature
379:445-
449 (1996)}, consistent with the notion that primary abnormalities in renal
sodium and
chloride handling may underlie stone development in a significant fraction of
affected
subj ects.
In addition to these mutations believed to cause loss of transport function,
it is
I 5 possible that variants in this gene could also lead to gain of function,
and potentially
contribute to development of hypertension. A comparable situation has been
demonstrated for the amiioride-sensitive epithelial sodium channel in which
gain-of
function mutations cause the hypertensive disorder Liddle Syndrome (Shimkets,
R.A.
et al., Ce1179:407-414 (1994); Hansson, J.H. et al., Nature Genet. 11:76-82
(1995})
while loss of function mutations cause the salt wasting disease
pseudohypoaldosteronism type I (Chang, S.S. et al., Nature Genet. 12:248-253
( 1996)) (Figure 9).
CA 02274955 1999-06-14
WO 98129431
PCT/US97/23553
-71-
EXAMPLE 3
f '
hvnercalciuria re~Paled by mutations in rhP K~ channel ROMK
Methods
Bartter's Syndrome kinrlrPr~~, ~ndreds were recruited via ascertainment of
affected index cases. Genomic DNA was prepared from venous blood of members of
Bartter's Syndrome kindreds by standard procedures (Bell, G., et al., Proc.
Natl.
Acad. Sci. (U.S.A.) 78:5759-5763 (1981)).
Characterization of h n"a" genomic R~~~~~ ;n-ron ev~~ junctions. Genomic
fragments bearing single introns and portions of adjacent exons of ROMK were
isolated by long-range PCR (ExpandTM Long PCR System; Boehringer-Mannheim)
using genomic DNA as a template and primers within each exon based on
published
data (Shuck, M.E. et al., J. Biol. Chem. 269:24261-24270 (1994)). DNA sequence
1 S analysis of products was by the dideoxy chain termination method using an
ABI 373A
instrument following a standard protocol. Intron sequences were used to define
primers amplifying intron-exon boundaries and coding regions of ROMK (Table
9);
additional junction sequences have been submitted to GENBANK.
enoty~ing and linkage an~IvSi~, Genotyping of loci indicated in the text was
performed by polymerase chain reaction using specific primers as described
previously (Simon, D.B., et al., Nature Genet. 13:183-188 (1996)). All
genotypes
were scored independently by two investigators who were blinded to affection
status.
Analysis of linkage was performed using the LINKAGE programs (Lathrop, G.M. et
al., Proc. Natl. Acad. Sci. (U.S.A.) 81:3443-3446 (1984)), specifying
Bartter's
. 25 Syndrome as an autosomal recessive trait with 99% penetrance and a
prevalence of
phenocopies of 0.1 %.
SSCP and DNA SPrnIPnrinn, Molecular variants in genes were sought using
single-strand conformational polymorphism (Orita, M. et al., Proc. Natl. Acad.
Sci.
CA 02274955 1999-06-14
WO 98/29431 PCT/US97123553
-72-
(U.S.A.) 86:2766-2770 ( 1989)). Primers for NKCC2 and TSC were as described
previously (Examples l and 2 and Simon, D.B. et al., Nature Genet. 12:24-30
(1996),
Simon, D.B., et al., Nature Genet. 13:183-188 (1996)). Primer pairs were
employed
to direct PCR using genomic DNA of disease family members or controls as a
template as previously described (Simon, D.B. et al., Nature Genet. 12:24-30
(1996)).
All primer pairs generate products with sizes between 150 and 300 base pairs.
Amplified products were analyzed for molecular variants by electrophoresis
under 2
different non-denaturing conditions as well as on denaturing urea gels as
previously
described (Simon, D.B. et al., Nature Genet. 12:24-30 (1996)). Identified
variants
were eluted from gel, reamplified by PCR, and DNA from both strands was
sequenced in all cases. All SSCP variants were confirmed by independent
amplifications.
Results
Banter's Syndrome Kindr .ric
I S We investigated subjects from 9 previously unreported kindreds with
Banter's
Syndrome, defined by the presence of hypokalaemic metabolic alkalosis with
salt
wasting and low blood pressure in association with hypercalciuria and
presentation in
the neonatal period (Table 8). All patients had markedly elevated plasma
aldosterone
levels and plasma renin activities (data not shown). The clinical and
biochemical
features of these patients are indistinguishable from those found in patients
previously
found to have mutations in NKCC2, and are distinct from those found in
patients with
mutations in TSC.
Ar~alvsis of NKCC2
Affected subjects in 6 of these kindreds were known to be offspring of
marriage between first cousins (Table 8). In these kindreds, it is highly
likely that
affected subjects are homozygous for identical disease mutations as well as
flanking
markers by descent from a heterozygous great-grandparent. Testing for
homozygasity
of intragenic and flanking markers is consequently a test of linkage in such
kindreds
CA 02274955 1999-06-14
WO 98/29431 PCTIUS97/23553
-73-
(Lander, E.S. et al., Science 236:1567-1570 (1987)). Accordingly, we genotyped
highly polymorphic markers spanning a 3 cM interval containing NKCC2 in these
consanguineous kindreds wre genotyped; these loci have proved homozygous in 5
previously studied kindreds with Bartter's Syndrome due to mutation in NKCC2.
In 3
of these consanguineous kindreds (BAR157, BAR181, BAR182), all affected
subjects
are homozygous for NKCC2 haplotypes, consistent with Bartter's Syndrome in
some
or all of these kindreds being attributable to mutation in NKCC2, as reported
previously (Example 2 and Simon, D.B., et al., Nature Genet. 13:183-188
(1996))
(data not shown). In contrast, in kindreds BAR159 and BAR161, homozygosity by
descent at NKCC2 is excluded (Figure 10). Moreover, in BAR 159, two affected
siblings are discordant for NKCCl haplotypes, and an unaffected sib shares
NKCC2
haplotypes with an affected sib; traditional analysis of linkage in this
kindred under a
conservative model of the trait locus (see Methods) rejects linkage with a lod
score of
-3.4. In addition, affected members of neither kindred are homozygous for loci
tightly
linked to TSC (data not shown). These findings provide strong evidence of
genetic
heterogeneity of Bartter's Syndrome, and indicate that mutation in NKCC2 does
not
cause the disease in BAR159 and BAR161. This conclusion is supported by the
failure to identify mutations in NKCC2 in affected subjects of these families
by
screening all 26 exons encoding NKCC2 (Simon, D.B., et al., Nature Genet.
13:I83-
188 (1996)) by single-strand conformational polymorphism (SSCP) (Orita, M. et
al.,
Proc. Natl. Acad. Sci. (U.S.A.) 86:2766- 2770 ( 1989)) (data not shown).
These observations motivated consideration that loss of function mutations in
a proposed regulator of NKCC2 function, ROMK, could account for the disease in
some of these families. The gene encoding ROMK has been cloned, and intron-
exon
organization defined (Yano, H., et al., Mol. Pharmacology 45:854-860 ( 1994);
Shuck,
M.E. et al., J. Biol. Chem. 269:24261-24270 ( 1994)): there are 5 exons which
are
used in varying combinations to produce 3 distinct ROMK proteins that differ
at the
amino terminus and vary in length from 372 to 391 amino acids in humans
(Shuck,
CA 02274955 1999-06-14
WO 98/29431 PCT/US97123553
- 74 -
M.E. et al., J. Biol. Chem. 269:24261-24270 (1994)). All isoforms share a core
of
372 amino acids encoded by exon 5; this 372 amino acid protein corresponds to
isoform ROMK2. The published intron sequence data was supplemented with
additional intron sequence information disclosed herein to permit examination
of all 5
exons and intron-exon boundaries (see Methods).
Affected subjects of BAR159 and BAR161 were screened for ROMK
mutations by SSCP. Homozygous ROMK variants were found in affected subjects of
both kindreds, and these variants cosegregated with the disease (Figures 10
and 11 a,
b). These variants are not common in the population, as they are not detected
on 80
chromosomes from unaffected unrelated subjects from Saudi Arabia and the CEPH
reference pedigrees. By homozygosity mapping, specifying these variants as
rare, the
lod score for linkage of ROMK and Bartter's Syndrome in these two families is
3.2,
supporting linkage. Analysis of the DNA sequence of these variants
demonstrates
that the homozygous variant in BAR 159 (Figure 11 a) changes codon 60 (codon
1 S number in ROMK2) from TAC, encoding tyrosine, to TAG, specifying chain
termination and truncating the encoded protein prior to the first
transmembrane
domain (Figure 12). The homozygous variant in BAR161 results in insertion of a
single T-A base pair into a sequence of 6 consecutive T residues spanning
codons 13
and 14 (Figure l lb), resulting in a frameshift mutation changing the encoded
protein
from amino acid 1 S onward (Figure 12), resulting in premature termination at
codon
54. These findings provide strong evidence that mutations in ROMK cause
Bartter's
Syndrome in these two families.
The 7 additional Banter's kindreds were then screened for ROMK variants.
Two variants were found in each of 2 outbred kindreds that had revealed no
variants
in screening of NKCC2; none of these variants were found in 80 chromosomes
from
unaffected subjects. In BAR208, one variant results in a missense mutation,
substituting arginine for serine at codon 200 (Figures l ld, 12). This serine
is highly
conserved among members of the IRK family of K+ channels and represents a
protein
kinase A phosphorylation site in the cytoplasmic carboxy terminus of the
protein; this
CA 02274955 1999-06-14
WO 98/29431 PCT/US97/23553
-75-
site has been shown to be phosphorylated by PKA, and substitution of alanine
by site-
directed mutagenesis has shown that this site is required for normal K+
channel
activity, with mutant ROMK showing an approximately 50% reduction in channel
activity (Xu, Z-C., et al., J. Biol. Chem. 271:9313-9319 (1996)). The second
variant
in this kindred, present on the other ROMK allele, represents another
premature
termination codon, occurring at codon 58, again truncating the encoded protein
prior
to the first transmembrane domain (Figure 12). In BAR206, one variant
represents a 4
base pair deletion, spanning the last base of codon T313 and all of codon
K314,
resulting in a frameshift mutation and altering the encoded protein from amino
acid
315 onward, ending at a new stop codon at position 350 (Figures 11 c, 12). The
second variant in this kindred arises from substitution of valine for alanine
at amino
acid 195, in the cytoplasmic carboxy terminus of ROMK (Figure 12); this
alanine
residue is conserved in rat and human ROMK, and has not been observed in
unrelated
unaffected subjects. In one additional outbred kindred, BAR139 in which no
NKCC2
variants have been identified, a single ROMK variant, substituting threonine
for
methionine at amino acid M338 was identified (Figure 12); this variant has not
been
seen on 80 chromosomes from unaffected subjects. As yet, no mutation has been
identified on the other ROMK allele in this affected subject.
Table 8.
Clinical
characteristics
of index
cases with
Banter's
Syndrome
Kindred Ancestry K+ HC03' UCa/UCr NephrocalcinosisConsang.
BAR159 Saudi Arabia3.2 27 0.5 Yes Yes
BAR161 Saudi Arabia2.7 33 1.1 Yes Yes
BAR206 Spain 3.3 32 1.0 Yes No
BAR208 Spain 3.4 28 0.9 Yes No
BAR139 Italy 1.8 36 0.9 No No
BAR144 Pakistan 2.5 31 0.8 Yes No
8AR157 Yemen 1.1 52 1.0 Unknown Yes
BAR181 Saudi Arabia2.2 31 1.1 Yes Yes
BAR182 Saudi Arabia1.9 35 0.8 Yes Yes
K+: serum potassium (mM, nl 3.5-5.0 mM); HC03 : serum bicarbonate (mM, nl 23-
26
mM); UCa/UCr: urinary calcium:creatinine ratio (mM:mM, nl <0.4 ).
Nephrocalcinosis
determined by ultrasonography. Consang.: Offspring of consanguineous union.
CA 02274955 1999-06-14
WO 98129431 PCT/US97123553
- 76 -
Table 9. PCR Primers for SSCP Analysis of the Human ROMK Gene
Primer Name Exon Forward Primer Reverse Primer
hROMKex1 1 TGCCATACAGATGAGTTGGCAG CCTACAAAGAGACAATGAGGTG
hROMKex2 2 AAAGGCAAGAGTTAGCCAGAC CTGTAAGATACAGATATTGGGAG
hROMKex3 3 CTCCAGACAGTAGCCATATGTG TTTCTTGTAGCCTGGGGTGTC
hROMKex4 4 AGCGTCAGTCCACTGACTGTC GCCTGGCTTTCCAGAGAGGTG
hROMKexSA 5 CATGTGGGTCACCTAGTTCAC TACCGTTGTCCAGATGTCCAC
hROMKexSB 5 TGGCAATGTGGAGGCACAGTC TGGTCACTTGAGTCTCCAGAG
hROMKexSC 5 AATCACACTCCCTGTGTGGAG CTGAACGTAATGGTCTTGGCAC
hROMKexSD 5 CATCTTAGCCAAGATCTCCAG CTACAAAGTTGATATTGATCTGGTC
hROMKex5E 5 AGTCACTCCTGAAGGAGAGAC GCACTGGTGGACTCCACTGTG
hROMKexSF 5 ATGGCAGCGGAGACCCTTCTC GGGGTCTCCACTTCCACTGTC
hROMKexSG 5 AAAGGAAGGGAAATACCGAGTG AGGTACTAGGAGCTTTAGAGAC
Primers hROMKexI, hROMKex2, hROMKex3, hROMKex4, and hROMKexSA-forward are
within introns. All other primers lie within exon 5.8
Table 10. Summary of mutations
in ROMK that
cause Banter's
Syndrome
Phenotype Mutation Consequence Homozygous
Banter TAC --~ TAG Y60 -a TERM +
Sartter TTT -~ TTTT F13 -~ Frameshift +
Banter AGC -~ AGG S200 --~ R
Banter TGG -> TAG W58 -> TERM
Banter CAAAGG -> CG T313 -~ Frameshift
Banter GCT -~ GTT A195 -~ V
Banter ATG -i ACG M338 -s T
Discussion
The finding of independent ROMK mutations that drastically alter channel
structure and that cosegregate with Bartter's Syndrome demonstrates that
mutations in
ROMK cause Bartter's Syndrome. These findings establish genetic heterogeneity
of
Banter's Syndrome and have implications for genetic testing for this disease.
Moreover, they strongly support the role of ROMK in the regulation of renal Na-
K-
2C1 cotransport activity and net salt reabsorption in vivo by recycling K+
entering cells
of the TAL back to the lumen. Whether mutations in NKCC2 and ROMK are the sole
genes in which mutation causes Bartter's Syndrome, or whether additional genes
will
be identified remains an open question; of the total of 15 families harboring
independent Banter's mutations studied thus far (Simon, D.B., et al., Nature
Genet.
CA 02274955 1999-06-14
WO 98/29431 PCTIUS97I23553
_77_
13:183-188 ( 1996) and the present study), NKCC2 variants have been identified
or
implicated by linkage in 10 families, and ROMK mutations have been identified
in 5
families. In one outbred family mutation in either ROMK or NKCC2 have not been
found to date. This could reflect either incomplete sensitivity of mutation
detection or
alternatively could be accounted for by further genetic heterogeneity. The
present
findings bring to 6 the number of renal ion channels, subunits or transporters
in which
mutation has been shown to affect blood pressure in humans (Lifton R.P.,
Science
272:67b-680 ( 1996); Simon, D.B., et al., Nature Genet. 13:183-188 ( 1996)).
The finding of homozygous premature termination and frameshift mutations
early in the encoded protein (Figure 12) provides very strong evidence that
these
ROMK mutations result in loss of potassium channel activity; this suggestion
is
further supported by f nding mutation in a PKA phosphorylation site that has
been
shown by expression in Xenopus oocytes to be required for full ROMK activity
(Xu,
Z-C., et al., J. Biol. Chem. 271:9313-9319 ( 1996)). In addition, a distal
frameshift
mutation deleting the last 58 normal amino acids strongly suggests an
essential role of
this carboxyl terminus in normal potassium channel function. The functional
consequences of this mutation and two additional missense variants, A195V and
M338T, can be assessed by expression studies; these may provide new insight
into
channel structure and function. It should be noted that all the mutations
identified to
date are in the core peptide shared by all known ROMK isoforms, and
consequently
activity of all isoforms is expected to be affected by these mutations.
The pathophysiology seen in affected patients can be explained by the loss of
ROMK function resulting in inability to recycle K+ from cells of the TAL back
into
the renal tubule, resulting in K+ levels in the lumen that are too low to
permit
continued Na-K-2C1 cotransport activity, producing salt wasting from this
nephron
segment. This salt wasting results in activation of the renin-angiotensin
system,
increased aldosterone levels, and increased electrogenic sodium reabsorption
via the
epithelial sodium channel of the distal nephron in exchange for K+ and H+,
accounting
for the observed hypokalaemic alkalosis. Since calcium reabsorption is coupled
to
CA 02274955 1999-06-14
WO 98129431 PCT/US97/23553
- 78 _
Na+ reabsorption in the TAL, loss of Na+ transport in this segment leads to
the
characteristic hypercalciuria of these patients.
While at least one additional secretory potassium channel has been identified
in the TALS, the present results indicate that these two K+ channels are not
redundant
in function, as this other channel cannot substitute for loss of ROMK
function.
ROMK isoforms are also expressed at more distal sites in the nephron, and it
has been
proposed that isoforms of this same channel contribute to net renal potassium
secretion in the distal nephron (Lee, W.S., et al., Am. J. Physiol. (Renal
Fluid Electrol.
Physiol.) 268:F1124-31 (1995), Boim, M.A. et al., Am. J. Physiol. (Renal Fluid
Electrol. Physiol.) 268:F 1132-40 ( 1995), Heben, S.C., Kidney Int. 48:1010-1
O 16
( 1995)). Since distal secretion is the major determinant of net renal
potassium
secretion, loss of this secretory potassium channel would be expected to
result in
impaired potassium secretion in response to aldosterone. As a consequence, one
might expect to be able to distinguish Banter's patients with NKCC2 mutations
(and
unimpaired distal K+ secretion) from those with mutations in ROMK by higher
potassium levels in the latter patients. In retrospect, the 4 patients with
ROMK
mutations identified on both alleles show a trend toward higher K+ levels
{Table 2
and Simon, D.B., et al., Nature Genet. 13:183-188 ( 1996)}, however they alt
still are
well below the normal range. This finding suggests that ROMK isoforms do not
play
an indispensable role in distal renal potassium excretion in vivo in humans.
Further
work will be required to address this question and to determine whether the
clinical
features of patients with mutations in these two genes can be distinguished
clinically.
In addition, while ROMK isoforms are also expressed in brain, spleen, lung and
eye
(Heben, S.C., Kidneylnt. 48:1010-1016 (1995}), there are no obvious phenotypes
in
these patients that suggest an essential role for ROMK function in these
organs.
The finding that homozygous loss of ROMK function alters renal sodium
handling raises the possibility that heterozygous carriers of ROMK mutations
might
have less drastic phenotypes owing to haploinsufficiency. These potential
phenotypes
would be expected to be similar to those previously suggested for NKCC2 mutant
CA 02274955 1999-06-14
WO 98/29431 PCTIUS97123553
-79-
heterozygotes (Simon, D.B., et al., Nature Genet. 13:183-188 ( 1996)),
including
reduced blood pressure, and predisposition to osteoporosis and/or
nephrolithiasis:
With the ability to identify heterozygous carriers, these possibilities can be
tested.
Similarly, the finding that increased renal sodium reabsorption underlies many
S variants of hypertension raises the question of whether gain of function
mutations in
ROMK could result in increased renal sodium reabsorption and contribute to
elevated
blood pressure. Identification of ROMK as an important regulator of net renal
sodium
reabsorption motivates determination of whether variants in this gene as well
as in
regulators of ROMK activity play a role in the determination of blood pressure
in
humans. A skilled artisan can readily practice the inventions disclosed herein
following the methods and Examples provided herein.