Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02296319 2000-O1-17
WO 99/04026 PCT/US98/14996
LENTIVIRAL VECTORS
TECHNICAL FIELD
The present invention relates generally to pharmaceutical compositions
and methods, and more particularly, to lentiviral vectors which are suitable
for a wide
variety of gene therapy applications.
BACKGROUND OF THE INVENTION
Since the discovery of nucleic acids in the 1940's and continuing through
the most recent era of biotechnology, substantial research has been undertaken
in order
to realize the possibility that the course of disease may be affected through
interaction
to with the nucleic acids of living organisms. Most recently, a wide variety
of methods
have been described for altering or affecting genes, including for example,
viral vectors
derived from retroviruses, adenoviruses, vaccinia viruses, herpes viruses, and
adeno-
associated viruses (see Jolly, Ca~tcer Gene Thercrly l ( 1 ):51-64, 1994).
Of these techniques, recombinant retroviral gene delivery methods have
been most extensively utilized, in part due to: (1 ) the efficient entry of
genetic material
(the vector genome) into cells; (2) an active, efficient process of entry into
the target
cell nucleus; (3) relatively high levels of gene expression; (4) the potential
to target
particular cellular subtypes through control of the vector-target cell binding
and the
tissue-specific control of gene expression; (5) a general lack of pre-existing
host
2o immunity; and (6) substantial knowledge and clinical experience which has
been gained
with such vectors.
Briefly, retroviruses are diploid positive-strand RNA viruses that
replicate through an integrated DNA intermediate. In particular, upon
infection by the
RNA virus, the retroviral genome is reverse-transcribed into DNA by a virally
encoded
reverse transcriptase that is carried as a protein in each retrovirus. The
viral DNA is
then integrated pseudo-randomly into the host cell genome of the infecting
cell,
forming a "provirus" which is inherited by daughter cells.
One type of retrovirus, the murine leukemia virus, or "MLV", has been
widely utilized for gene therapy applications (see generally Mann et al. {Cell
33:153,
1983), Cane and Mulligan (Pr-oc. Nat'l. Acad. Sci. USA 81:6349, 1984), and
Miller
et al., Human Gene Therapy I:5-14,1990. One major disadvantage of MLV-based
vectors, however, is that the host range (i.e., cells infected with the
vector) is limited,
and the frequency of transduction of non-replicating cells is generally low.
CA 02296319 2000-O1-17
WO 99/04026 PCT/US98/14996
7
In order to improve lentiviral-based gene therapy vectors for certain
applications, the present invention provides improved lentiviral based vectors
and
packaging cell lines. The invention also provides other, related, advantages.
SUMMARY OF THE INVENTION
Briefly stated, within one aspect of the present invention lentiviral
vectors are provided, comprising a 5' lentiviral LTR, a tRNA binding site, a
packaging
signal, a promoter operably linked to one or more genes of interest, an origin
of second
strand DNA synthesis and a 3' lentiviral LTR, wherein the lentiviral vector
contains a
nuclear transport element. The nuclear transport element may be located either
l0 upstream (5') or downsteam (3') of the gene of interest. Within certain
embodiments,
the nuclear transport element is not RRE. Within one embodiment the packaging
signal
is an extended packaging signal. Within other embodiments the promoter is a
tissue
specific promoter, or, alternatively, a promoter such as CMV. Within further
embodiments, the lentiviral vector further comprises an internal ribosome
entry site.
Within various embodiments, the lentiviral vector expresses a gene of
interest (e.g., a heterologous sequence, although, within certain embodiments
lentivirus
sequences such as HIV env may also be expressed). Representative examples of
suitable genes of interest include cytokines, insulin, (i-gal, alkaline
phosphatase (e.g.,
placental alkaline phosphatase), green fluorescence protein, factor VIII,
factor IX, LDL
receptor, human growth hormone, EPO, TPO, prodrug activating enzymes, trans-
dominant negative viral or cancer-associated proteins, and tyrosine
hydroxylase.
A wide variety of lentiviruses may be utilized within the context of the
present invention, including for example, lentiviruses selected from the group
consisting of HIV, HIV-l, HIV-2 and SIV.
Within other aspects of the invention expression cassettes are provided.
Within one embodiment, gag/pol expression cassettes are provided comprising a
promoter and a sequence encoding gag/pol and at least one of vpr, vpu, nef or
vif,
wherein the promoter is operably linked to gag/pol and vpr, vpu, nef or vif.
Within
another embodiment, tat expression cassettes are provided comprising a
promoter and a
sequence encoding tat and at least one of vpr, vpu, nef or vif, wherein the
promoter is
operably linked to tat and vpr, vpu, ncf or vif. Within further embodiments
rev
expression cassettes are provided comprising a promoter and a sequence
encoding rev
and at least one of vpr, vpu, nef or vif, wherein the promoter is operably
linked to rev
and vpr, vpu, nef or vif. Within yet other embodiments, VSV-G expression
cassettes
are provided comprising a promoter and a sequence encoding VSV-G and at least
one
CA 02296319 2000-O1-17
WO 99/04026 PCT/US98/14996
3
of vpr, vpu, nef or vif, wherein the promoter is operably linked to VSV-G and
vpr, vpu,
nef or vif.
Within yet other aspects of the invention, host cells (e.g., packaging cell
lines) are provided within contain any of the expression cassettes described
herein. For
example, within one aspect packaging cell line are provided comprising an
expression
cassette that comprises a sequence encoding gag/pol, and a nuclear transport
element,
wherein the promoter is operably linked to the sequence encoding gag/pol.
Within
other aspects, packaging cell lines are provided comprising a promoter and a
sequence
encoding tat, rev, or an envelope (e.g., VSV-G), wherein the promoter is
operably
linked to the sequence encoding tat, rev, or, the envelope. Within further
embodiments,
the packaging cell line may further comprise a sequence encoding any one or
more of
nef, vif, vpu or vpr. Within another embodiment, the expression cassette is
stably
integrated. Within yet another embodiment, the packaging cell line, upon
introduction
of a lentiviral vector, produces particles at a concentration of greater than
10' cfu/ml.
Within further embodiments the promoter is inducible. Within certain preferred
embodiments of the invention, the packaging cell line, upon introduction of a
lentiviral
vector, produces particles that are free of replication competent virus.
Within yet other aspects of the present invention, packaging cell lines are
provided comprising an expression cassette which directs the expression of a
gaglpol
2o gene, an expression cassette which directs the expression of an eftv gene
(e.g., VSV-G,
or an amphotrophic envelope), an expression cassette which directs the
expression of
Tat, and expression cassette which directs the expression of Rev. Within
further
aspects, a lentiviral vector is introduced into the packaging cell line to
produce a vector
producing cell line.
Within further aspects, methods are provided for enhancing production
of infectious virus, comprising the step of infecting packaging cell lines
with a viral
vector, wherein a butyrate salt (e.g., sodium butyrate or potassium butyrate)
is added
subsequent to, or after infection of the packaging cell line. Within certain
embodiments, the butyrate salt is added prior to the step of harvesting
infectious virus.
3o These and other aspects of the present invention will become evident
upon reference to the following detailed description and attached drawings. In
addition,
various references are set forth below which describe in more detail certain
procedures
or compositions (e.g., plasmids, etc.), and are therefore incorporated by
reference in
their entirety as if each were individually noted for incorporation.
CA 02296319 2000-O1-17
WO 99/04026 PCT/US98/14996
4
BRIEF DESCRIPTION OF THE DRAWINGS
Figure 1 is a schematic illustration of one representative HIV-1 based
vector.
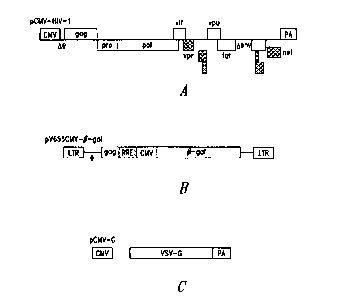
Figures 2A, 2B and 2C are schematic illustrations of pCMV-HIV-1,
pV653CMV-[3gal, and pCMU-G, respectively.
Figures 3A, 3B, 3C and 3D are schematic illustrations of pCH-GP-1,
pCH-GP-2, pCH-GP-3 and pCH-GP-4, respectively.
Figures 4A and 4B are schematic illustrations of pCMV-tat and pTetO-
rev.
Figure 5 is a flow chart of one representative method for packaging cell
line generation.
Figure 6 is a schematic illustration which shows a comparison of the
genome organizations of lenti- and oncoretroviruses.
Figure 7 is a table which shows the affected HIV-1 accessory proteins on
vector production.
Figure 8 is a table which shows the expression level of p24 in pCHGP
transfected cells.
Figure 9 is a bar graph which S110wS St1111uIat1017 Of vector production by
sodium butyrate. Briefly, vectors derived from either pCMV-HIV-1 (closed
boxes) or
PCHGP-2 (striped boxes) were generated in 293T cells in the presence of
various
concentrations of sodium butyrate as indicated. Titers were determined in
HT1080 cells
as described in materials and methods. Values are the ratios of titers with
sodium
butyrate over titers without sodium butyrate for each vector.
Figure 10 is a bar graph which shows transduction efficiency in HeLa
cells. Briefly, 300 ul of different viral preparations were used to transduce
actively
dividing or growth-arrested HeLa cells in 12 well plates. The cells were
harvested two
days after transduction and the total [3-galactosidase activity was determined
by blue
cell count after X-Gal staining. The results are presented as a percentage
value of
vector titer observed in HT1080 cells for each viral preparation ([Titer in
HeLa
(dividing or quiescent)/Titer in HT1080] X 100).
Figure 11 is a bar graph which shows the transduction efficiency in
human skin fibroblasts. Briefly, 10 pl of different types were used to infect
dividing
and quiescent fibroblast in a 12 well plate. Two days after transduction,
titer was
determined by blue cell counting after X-Gal staining. Data for transduction
of
quiescent and dividing fibroblasts, are presented as a percentage value of
titer observed
CA 02296319 2000-O1-17
WO 99/04026 PCT/US98/14996
in growing HT1080 for each viral preparation ([Titer in fibroblasts (growing
or
quiescent)/Titer in HT1080] X 100). The values are an average of four
experiments.
DETAILED DESCRIPTION OF THE INVENTION
Prior to setting forth the invention, it may be helpful to an understanding
5 thereof to set forth definitions of certain terms that will be used
hereinafter.
"Vector construct", "lentiviral vector", and "recombinant lentiviral
vector" refers to a nucleic acid construct which carries, and within certain
embodiments,
is capable of directing the expression of a nucleic acid molecule of interest.
The
lentiviral vector must include at least one transcriptional promoter/enhancer
or locus
1o defining element(s), or other elements which control gene expression by
other means
such as alternate splicing, nuclear RNA export, post-translational
modification of
messenger, or post-transcriptional modification of protein. Such vector
constructs must
also include a packaging signal, long terminal repeats (LTRs} or portion
thereof, and
positive and negative strand primer binding sites appropriate to the
retrovirus used (if
these are not already present in the retroviral vector). Optionally, the
recombinant
lentiviral vector may also include a signal which directs polyadenylation,
selectable
markers such as Neo, TK, hygromycin, phieomycin, histidinol, or DHFR, as well
as one
or more restriction sites and a translation termination sequence. By way of
example,
such vectors typically include a 5' LTR, a iRNA binding site, a packaging
signal, an
origin of second strand DNA synthesis, and a 3' LTR or a portion thereof.
"Lentiviral vector a~rtiele" as utilized within the present invention refers
to a lentivirus which carries at least one gene of interest. The retrovirus
may also
contain a selectable marker. The recombinant lentivirus is capable of reverse
transcribing its genetic material into DNA and incorporating this genetic
material into a
host cell's DNA upon infection. Lentiviral vector particles may have a
lentiviral
envelope, a non-lentiviral envelope (e.g., an ampho or VSV-G envelope), or a
chimeric
envelope.
"Nucleic acid expression vector" or "Expression cassette" refers to an
assembly which is capable of directing the expression of a sequence or gene of
interest.
3o The nucleic acid expression vector must include a promoter which, when
transcribed, is
operably linked to the sequences) or gene{s) of interest, as well as a
polyadenylation
sequence. Within certain embodiments of the invention, the nucleic acid
expression
vectors described herein may be contained within a plasmid construct. In
addition to
the components of the nucleic acid expression vector, the plasmid construct
may also
include a bacterial origin of replication, one or more selectable markers, a
signal which
CA 02296319 2000-O1-17
WO 99/04026 PCT/US98/14996
6
allows the plasmid construct to exist as single-stranded DNA (e.g., a M13
origin of
replication), a multiple cloning site, and a "mammalian" origin of replication
(e.g., a
SV40 or adenovirus origin of replication).
"Packa~ inn cell" refers to a cell which contains those elements necessary
for production of infectious recombinant retrovirus which are lacking in a
recombinant
retroviral vector. Typically, such packaging cells contain one or more
expression
cassettes which are capable of expressing proteins which encode gc~g, pol and
env
proteins.
"Producer cell" or "Vector producing cell" refers to a cell which contains
all elements necessary for production of recombinant retroviral vector
particles.
CONSTRUCTION AND PREPARATION OF LENT1V1RAL VECTORS
As noted above, the present invention provides lentiviral vectors which
are designed to carry or express a selected genes) or sequences) of interest.
Lentiviral
vectors may be readily constructed from a wide variety of lentiviruses (see
RNA Tumor
Viruses, Second Edition, Cold Spring Harbor Laboratory, 1985). Representative
examples of lentiviruses included HIV, HIV-1, HIV-2 and SIV. Such lentiviruses
may
either be obtained from patient isolates, or, more preferably, from
depositories or
collections such as the American Type Culture Collection (ATCC, Rockville,
MD), or
isolated from known sources using commonly available techniques.
Any of the above lentiviruses may be readily utilized in order to
assemble or construct lentiviral gene delivery vehicles given the disclosure
provided
herein, and standard recombinant techniques (e.g., Sambrook et ul. Molecz~lar
Cloning:
A Labor-ato~y Manual, 2d ed., Cold Spring Harbor Laboratory Press, 1989;
Kunkle,
PNAS 82:488, 1985). In addition, within certain embodiments of the invention,
portions of the lentiviral gene delivery vehicles may be derived from
different viruses.
For example, within one embodiment of the invention, recombinant lentiviral
vector
LTRs may be derived from an HIV, a packaging signal from SIV, and an origin of
second strand synthesis from HIV-2.
Within one aspect of the present invention, lentiviral vector constructs
3o are provided comprising a 5' lentiviral LTR, a tRNA binding site, a
packaging signal,
one or more heterologous sequences, an origin of second strand DNA synthesis
and a 3'
LTR, wherein said lentiviral vector contains a nuclear transport element that
is not
RRE. Briefly, Long Terminal Repeats ("LTRs") are subdivided into three
elements,
designated U5, R and U3. These elements contain a variety of signals which are
responsible for the biological activity of a retrovirus, including for
example, promoter
CA 02296319 2000-O1-17
WO 99104026 PCT/US98/14996
7
and enhancer elements which are located within U3. LTRs may be readily
identified in
the provirus (integrated DNA form) due to their precise duplication at either
end of the
genome. As utilized herein, a 5' LTR should be understood to include a 5'
promoter
element and sufficient LTR sequence to allow reverse transcription and
integration of
s the DNA form of the vector. The 3' LTR should be understood to include a
polyadenylation signal, and sufficient LTR sequence to allow reverse
transcription and
integration of the DNA form of the vector.
The tRNA binding site and origin of second strand DNA synthesis are
also important for a retrovirus to be biologically active, and may be readily
identified by
to one of skill in the art. For example, retroviral tRNA binds to a tRNA
binding site by
Watson-Crick base pairing, and is carried with the retrovirus genome into a
viral
particle. The tRNA is then utilized as a primer for DNA synthesis by reverse
transcriptase. The tRNA binding site may be readily identified based upon its
location
just downstream from the 5' LTR. Similarly, the origin of second strand DNA
synthesis
15 is, as its name implies, important for the second strand DNA synthesis of a
retrovirus.
This region, which is also referred to as the poly-purine tract, is located
just upstream of
the 3' LTR.
In addition to a 5' and 3' LTR, tRNA binding site, and origin of second
strand DNA synthesis, certain preferred recombinant retroviral vector
constructs which
2o are provided herein also comprise a packaging signal, as well as one or
more genes of
interest, each of which is discussed in more detail below. In addition, the
lentiviral
vectors have a nuclear transport element which, in preferred embodiments is
not RRE.
Representative examples of suitable nuclear transport elements include the
element in
Rous sarcoma virus {Ogert, et al., J. Tirol. 70, 3834-3843, 1996), the element
in Rous
25 sarcoma virus (Liu & Mertz, Genes & Dev., 9, 1766-1789, 1995) and the
element in the
genome of simian retrovirus type 1 (Zolotukhin, et al., J. Tirol. 68, 7944-
7952, 1994).
Other potential elements include the elements in the histone gene (Kedes,
Annu. Rev.
Biochem. 48, 837-870, 1970), the a interferon gene (Nagata et al., Nature 287,
401-408,
1980), the 13-adrenergic receptor gene (Koilka, et al., Natacre 329, 75-79,
1987), and the
30 c-Jun gene (Hattorie, et al., Proc. Natl. Acad. Sci. USA 85, 9148-9152,
1988).
Within one aspect of the invention, recombinant lentiviral vector
constructs are provided which lack both gaglpol and env coding sequences. As
utilized
herein, the phrase "lacks gaglpol or env coding sequences" should be
understood to
mean that the recombinant lentiviral vector does not contain at least 20,
preferably at
35 least 15, more preferably at least 10, and most preferably at least 8
consecutive
nucleotides which are found in gaglpol or env genes, and in particular, within
gaglpol or
CA 02296319 2000-O1-17
WO 99/04026 PCT/US98/14996
8
env expression cassettes that are used to construct packaging cell lines for
the
recombinant retroviral vector constructs are set forth in more detail below
and in
Example 1.
As an illustration, within one embodiment of the invention construction
of recombinant lentiviral vector constructs which lack gaglpol or env
sequences may be
accomplished by preparing vector constructs which lack an extended packaging
signal.
As utilized herein, the phrase "extended packaging signal" refers to a
sequence of
nucleotides beyond the minimum core sequence which is required for packaging,
that
allows increased viral titer due to enhanced packaging.
Within certain embodiments of the invention, lentiviral vectors are
provided wherein tissue-specific promoters are utilized to drive expression of
one or
more genes of interest. For example, lentiviral vector particles of the
invention can
contain a liver specific promoter to maximize the potential for liver specific
expression
of the exogenous DNA sequence contained in the vectors. Preferred liver
specific
promoters include the hepatitis B X-gene promoter and the hepatitis B core
protein
promoter. These liver specific promoters are preferably employed with their
respective
enhancers. The enhancer element can be linked at either the 5' or the 3' end
of the
nucleic acid encoding the therapeutic molecule. The hepatitis B X gene
promoter and
its enhancer can be obtained from the viral genome as a 332 base pair EcoRV-
NcoI
DNA fragment employing the methods described in Twu, 1987, J. Virol. 61:3448-
3453.
The hepatitis B core protein promoter can be obtained from the viral genome as
a 584
base pair BamHI-BgIII DNA fragment employing the methods described in Gerlach,
1992, Yirol 189:59-66. It may be necessary to remove the negative regulatory
sequence
in the BamHI-BgIII fragment prior to inserting it. Other liver specific
promoters
include the AFP (alpha fetal protein) gene promoter and the albumin gene
promoter, as
disclosed in EP Patent Publication 0 415 731, the -1 antitrypsin gene
promoter, as
disclosed in Rettenger, 1994, Proc. Natl. Acud. Sci. 91:1460-1464, the
fibrinogen
gene promoter, the APO-Al (Apolipoprotein Al) gene promoter, and the promoter
genes for liver transference enzymes such as, for example, SGOT, SGPT and -
3o glutamyle transferase. See also PCT Patent Publications WO 90/07936 and WO
91/02805 for a description of the use of liver specific promoters in
lentiviral vector
particles.
Within certain embodiments of the invention, the lentiviral vector
constructs provided herein may be generated such that more than one gene of
interest is
expressed. This may be accomplished through the use of di- or oligo-cistronic
cassettes
(e.g., where the coding regions are separated by 80 nucleotides or less, see
generally
CA 02296319 2000-O1-17
WO 99/04026 PCT/US98/14996
9
Levin et al., GerTe 108:167-174, 1991), or through the use of Internal
Ribosome Entry
Sites ("IRES").
PACKAGING / PRODUCER CELL LINES
Packaging cell lines suitable for use with the above described
recombinant retroviral vector constructs may be readily prepared given the
disclosure
provided herein. Briefly, the parent cell line from which the packaging cell
line is
derived can be selected from a wide variety of mammalian cell lines, including
for
example, human cells, monkey cells, dog cells, mouse cells, and the like.
Within one embodiment of the invention, potential packaging cell line
to candidates are screened by isolating the human placental alkaline
phosphatase (PLAP)
gene from pBAAP, and inserting the gene into pNL4-3. To generate infectious
virus,
the construct is co-transfected with pCMV-G into 293 cells, and the virus
harvested 48
hurs after transfection. The resulting virus can be utilized to infect
candidate host cells
(e.g." human cells such as HeLa, HY1080, 293, Jurkats, supTl and CEM), which
are
subsequently sourted using antibodies specific for PLAP. Production of p24 and
reverse transcriptase can also be analyzed in the assessment of suitable
packaging cell
lines.
After selection of a suitable host cell for the generation of a packaging
cell line, one or more expression cassettes are introduced into the cell line
in order to
2o complement or supply in traps components of the vector which have been
deleted (see
generally U.S. Serial No. 08/240,030, filed May 9, 1994; see also U.S. Serial
No. 07/800,921, filed November 27, 1991 ).
Representative examples of suitable expression cassettes include gag/pol
expression cassettes which comprise a promoter and a sequence encoding gag/pol
and at
least one of vpr, vpu, nef or vif, wherein the promoter is operably linked to
gag/pol and
vpr, vpu, nef or vif. Within another embodiment, tat expression cassettes are
provided
comprising a promoter and a sequence encoding tat and at least one of vpr,
vpu, nef or
vif, wherein the promoter is operabIy linked to tat and vpr, vpu, nef or vif.
Within
further embodiments rev expression cassettes are provided comprising a
promoter and a
3o sequence encoding rev and at least one of vpr, vpu, nef or vif, wherein the
promoter is
operably linked to rev and vpr, vpu, nef or vif. Within yet other embodiments,
VSV-G
expression cassettes are provided comprising a promoter and a sequence
encoding
VSV-G and at least one of vpr, vpu, nef or vif, wherein the promoter is
operably linked
to VSV-G and vpr, vpu, nef or vif.
CA 02296319 2000-O1-17
WO 99/04026 PCT/US98/14996
Utilizing the above-described expression cassettes, a wide variety of
packaging cell lines can be generated. For example, within one aspect
packaging cell
line are provided comprising an expression cassette that comprises a sequence
encoding
gag/pol, and a nuclear transport element, wherein the promoter is operably
linked to the
5 sequence encoding gag/pol. Within other aspects, packaging cell lines are
provided
comprising a promoter and a sequence encoding tat, rev, or an envelope (e.g.,
VSV-G),
wherein the promoter is operably linked to the sequence encoding tat, rev, or,
the
envelope. Within further embodiments, the packaging cell line may further
comprise a
sequence encoding any one or more of nef, vif, vpu or vpr. For example, the
packaging
1o cell line may contain only nef, vif, vpu, or vpr alone, nef and vif, nef
and vpu, nef and
vpr, vif and vpu, vif and vpr, vpu and vpr, nef vif and vpu, nef vif and vpr,
nef vpu and
vpr, vvir vpu and vpr, or, all four of nef vif vpu and vpr.
Within another embodiment, the expression cassette is stably integrated.
Within yet another embodiment, the packaging cell line, upon introduction of a
lentiviral vector, produces particles at a concentration of greater than
10',106, 10', 10~,
or, 109 cfu/ml. Within further embodiments the promoter is inducible. Within
certain
preferred embodiments of the invention, the packaging cell line, upon
introduction of a
lentiviral vector, produces particles that are free of replication competent
virus.
C'tENFS OF INTEREST / HETEROLOGOUS NUCLEIC ACID MOLEC1JLES
A wide variety of nucleic acid molecules may be carried and/or
expressed by the lentiviral vector particles of the present invention. As used
herein,
"pathogenic agent" refers to a cell that is responsible for a disease state.
Representative
examples of pathogenic agents include tumor cells, autoreactive immune cells,
hormone
secreting cells, cells which lack a function that they would normally have,
cells that
have an additional inappropriate gene expression which does not normally occur
in that
cell type, and cells infected with bacteria, viruses, or other intracellular
parasites. In
addition, as used herein "pathogenic agent" may also refer to a cell that has
become
tumorigenic due to inappropriate insertion of nucleic acid molecules contained
by the
lentiviral vector into a host cell's genome.
3o Examples of nucleic acid molecules which may be carried and/or
expressed by lentiviral vector particles of the present invention include
genes and other
nucleic acid molecules which encode a substance, as well as biologically
active nucleic
acid molecules such as inactivating sequences that incorporate into a
specified
intracellular nucleic acid molecule and inactivate that molecule. A nucleic
acid
molecule is considered to be biologically active when the molecule itself
provides the
CA 02296319 2000-O1-17
WO 99/04026 PCT/US98/1499b
11
desired benefit. For example, the biologically active nucleic acid molecule
may be an
inactivating sequence that incorporates into a specified intracellular nucleic
acid
molecule and inactivates that molecule, or the molecule may be a tRNA, rRNA or
mRNA that has a configuration that provides a binding capability.
Substances which may be encoded by the nucleic acid molecules
described herein include proteins (e.g., antibodies including single chain
molecules),
immunostimulatory molecules (such as antigens) immunosuppressive molecules,
blocking agents, palliatives (such as toxins, antisense ribonucleic acids,
ribozymes,
enzymes, and other material capable of inhibiting a function of a pathogenic
agent)
1o cytokines, various polypeptides or peptide hormones, their agonists or
antagonists,
where these hormones can be derived from tissues such as the pituitary,
hypothalamus,
kidney, endothelial cells, liver, pancreas, bone, hemopoetic marrow, and
adrenal. Such
polypeptides can be used for induction of growth, regression of tissue,
suppression of
immune responses, apoptosis, gene expression, blocking receptor-ligand
interaction,
immune responses and can be treatment for certain anemias, diabetes,
infections, high
blood pressure, abnormal blood chemistry or chemistries (e.g., elevated blood
cholesterol, deficiency of blood clotting factors, elevated LDL with lowered
HDL),
levels of Alzheimer associated amyloid protein, bone erosion/calcium
deposition, and
controlling levels of various metabolites such as steroid hormones, purines,
and
pyrimidines.
For palliatives, when "capable of inhibiting a function" is utilized within
the context of the present invention, it should be understood that the
palliative either
directly inhibits the function or indirectly does so, for example, by
converting an agent
present in the cells from one which would not nom~ally inhibit a function of
the
pathogenic agent to one which does. Examples of such functions for viral
diseases
include adsorption, replication, gene expression, assembly, and exit of the
virus from
infected cells. Examples of such functions for cancerous diseases include cell
replication, susceptibility to external signals (e.g., contact inhibition),
and lack of
production of anti-oncogene proteins. Examples of such functions for
cardiovascular
3o disease include inappropriate growth or accumulation of material in blood
vessels, high
blood pressure, undesirable blood levels of factors such as cholesterol or low
density
lipoprotein that predispose to disease, localized hypoxia, and inappropriately
high and
tissue-damaging levels of free radicals. Examples of such functions for
neurological
conditions include pain, lack of dopamine production, inability to replace
damaged
cells, deficiencies in motor control of physical activity, inappropriately low
levels of
various peptide hormones derived from neurological tissue such as the
pituitary or
CA 02296319 2000-O1-17
WO 99/04026 PCT/US98/14996
12
hypothalamus, accumulation of Alzheimer's Disease associated amyloid plaque
protein,
and inability to regenerate damaged nerve junctions. Examples of such
functions for
autoimmune or inflammatory disease include inappropriate production of
cytokines and
lymphokines, inappropriate production and existence of autoimmune antibodies
and
cellular immurxe responses, inappropriate disruption of tissues by proteases
and
collagenases, lack of production of factors normally supplied by destroyed
cells, and
excessive or aberrant regrowth of tissues under autoimmune attack.
Within one aspect of the present invention, methods are provided for
administration of a recombinant lentivirus which directs the expression of a
palliative.
1o Representative examples of palliatives that act directly to inhibit the
growth of cells
include toxins such as ricin (Lamb et al., Eur-. J. Biochem. 148:265-270,
1985), abrin
(Wood et al., Eur. J. Biochem. 198:723-732, 1991; Evensen et al., J. of Biol.
Cheat.
266:6848-6852, 1991; Collins et al., J. of Biol. Chem. 265:8665-8669, 1990;
Chen
et al., Fed. ofEur. Biochem Soc. 309:115-118, 1992), diphtheria toxin (Tweten
et al., J.
Biol. Chern. 260:10392-10394, 1985), cholera toxin (Mekalanos et al., Nature
306:551-
557, 1983; Sanchez & Holmgren, PNAS 86:481-485, 1989), gelonin (Stirpe et al.,
J.
Biol. Chem. 255:6947-6953, 1980), pokeweed (Irvin, Pharmac. Tlrer-. 21:371-
387,
1983), antiviral protein (Barbieri et al., Biochem. J. 203:55-59, 1982; Irvin
et al., Arch.
Biochem. & Biophys. 200:418-425, 1980; Irvin, Arch. Biochern. c~ BiopITys.
169:522-
528, 1975), tritin, Shigella toxin (Calderwood et al., PNAS 84:4364-4368,
1987;
Jackson et al., Microb. Path. 2:147-153, 1987), and Pseudomonas exotoxin A
(Carroll
and Collier, J. Biol. Cheot. 262:8707-8711, 1987). A detailed description of
recombinant retroviruses which express Russel's Viper Venom is provided in
U.S.
Serial No. 08/368,574, filed December 30, 1994
Within other aspects of the invention, the lentiviral vector carries a gene
specifying a product which is not in itself toxic, but when processed or
modified by a
protein, such as a protease specific to a viral or other pathogen, is
converted into a toxic
form. For example, recombinant retrovirus could carry a gene encoding a
proprotein
chain, which becomes toxic upon processing by the HIV protease. More
specifically, a
3o synthetic inactive proprotein form of the toxic ricin or diphtheria A
chains could be
cleaved to the active form by arranging for the HIV virally encoded protease
to
recognize and cleave off an appropriate "pro" element.
Within a related aspect of the present invention, lentiviral vectors are
provided which direct the expression of a gene products) that activates a
compound
with little or no cytotoxicity into a toxic product. Briefly, a wide variety
of gene
products which either directly or indirectly activate a compound with little
or no
CA 02296319 2000-O1-17
WO 99/04026 PCT/US98/14996
13
cytotoxicity into a toxic product may be utilized within the context of the
present
invention. Representative examples of such gene products include HSVTK and
VZVTK which selectively monophosphorylate certain purine arabinosides and
substituted pyrimidine compounds, converting them to cytotoxic or cytostatic
metabolites. More specifically, exposure of the drugs ganciclovir, acyclovir,
or any of
their analogues (e.g., FIAC, DHPG) to HSVTK, phosphorylates the drug into its
corresponding active nucleotide triphosphate form.
For example, within one embodiment of the invention, the lentiviral
vector directs the expression of the herpes simplex virus thymidine kinase
("HSVTK")
1o gene downstream, and under the transcriptional control of an HIV promoter
(which is
known to be transcriptionally silent except when activated by HIV tat
protein). Briefly,
expression of the tat gene product in human cells infected with HIV and
carrying the
recombinant retrovirus causes increased production of HSVTK. The cells (either
in vitro or in vivo) are then exposed to a drug such as ganciclovir, acyclovir
or its
analogues (FIAC, DHPG). As noted above, these drugs are known to be
phosphorylated by HSVTK (but not by cellular thymidine kinase) to their
corresponding active nucleotide triphosphate forms. Acyclovir triphosphates
inhibit
cellular polymerases in general, leading to the specific destruction of cells
expressing
HSVTK in transgenic mice (see Borrelli et al., Proc. Natl. Acad. Sci. USA
85:7572,
1988). Those cells containing the recombinant retrovirus and expressing HIV
tat
protein are selectively killed by the presence of a specific dose of these
drugs.
In a manner similar to the preceding embodiment, lentiviral vectors may
be generated which carry a gene for phosphorylation, phosphoribosylation,
ribosylation,
or other metabolism of a purine- or pyrimidine-based drug. Such genes may have
no
equivalent in mammalian cells, and might come from organisms such as a virus,
bacterium, fungus, or protozoan. Representative examples include: E. toll
guanine
phosphoribosyl transferase ("gpt"} gene product, which converts thioxanthine
into
thioxanthine monophosphate (see Besnard et al., Mol. Cell. Biol. 7:4139-4141,
1987);
alkaline phosphatase, which will convert inactive phosphorylated compounds
such as
3o mitomycin phosphate and doxorubicin-phosphate to toxic dephosphorylated
compounds; fungal (e.g., Fusarium oxysporum) or bacterial cytosine deaminase
which
will convert 5-fluorocytosine to the toxic compound 5-fluorouracil {Mullen,
PNAS
89:33, 1992); carboxypeptidase G2 which will cleave the glutamic acid from
para-N-bis
(2-chloroethyl) aminobenzoyl glutamic acid, thereby creating a toxic benzoic
acid
mustard; and Penicillin-V amidase, which will convert phenoxyacetabide
derivatives of
doxorubicin and melphalan to toxic compounds. Conditionally lethal gene
products of
CA 02296319 2000-O1-17
WO 99/04026 PCT/US98/14996
14
this type have application to many presently known purine- or pyrimidine-based
anticancer drugs, which often require intracellular ribosylation or
phosphorylation in
order to become effective cytotoxic agents. The conditionally lethal gene
product could
also metabolize a nontoxic drug, which is not a purine or pyrimidine analogue,
to a
cytotoxic form (see Searle et al., Brit. J. Cancer 53:377-384, 1986).
Additionally, in the instance where the target pathogen is a mammalian
virus, lentiviral vectors may be constructed to take advantage of the fact
that
mammalian viruses in general tend to have "immediate early" genes, which are
necessary for subsequent transcriptional activation of other viral promoter
elements.
to Gene products of this nature are excellent candidates for intracellular
signals (or
"identifying agents") of viral infection. Thus, conditionally lethal genes
transcribed
from transcriptional promoter elements that are responsive to such viral
"immediate
early" gene products could specifically kill cells infected with any
particular virus.
Additionally, since the human and interferon promoter elements are
transcriptionally
activated in response to infection by a wide variety of nonrelated viruses,
the
introduction of vectors expressing a conditionally lethal gene product like
HSVTK, for
example, from these viral-responsive elements (VREs) could result in the
destruction of
cells infected with a variety of different viruses.
In another embodiment of the invention, lentiviral vectors are provided
2o that produce substances such as inhibitor palliatives, that inhibit viral
assembly. In this
context, the recombinant retrovirus codes for defective gcrg, pol, ejiv or
other viral
particle proteins or peptides which inhibit in a dominant fashion the assembly
of viral
particles. Such inhibition occurs because the interaction of normal subunits
of the viral
particle is disturbed by interaction with the defective subunits.
One way of increasing the effectiveness of inhibitory palliatives is to
express inhibitory genes, such as viral inhibitory genes, in conjunction with
the
expression of genes which increase the probability of infection of the
resistant cell by
the virus in question. The result is a nonproductive "dead-end" event which
would
compete for productive infection events. In the specific case of HIV, a
recombinant
3o retrovirus may be administered that inhibits HIV replication (by expressing
anti-sense
tat, etc., as described above) and also overexpress proteins required for
infection, such
as CD4. In this way, a relatively small number of vector-infected HIV-
resistant cells
act as a "sink" or "magnet" for multiple nonproductive fusion events with free
virus or
virally infected cells.
In another embodiment of the invention, lentiviral vectors are provided
for the expression substances such as inhibiting peptides or proteins specific
for viral
CA 02296319 2000-O1-17
WO 99/04026 PCT/US98/14996
protease. Viral protease cleaves the viral gag and gaglpol proteins into a
number of
smaller peptides. Failure of this cleavage in all cases leads to complete
inhibition of
production of infectious retroviral particles. The HIV protease is known to be
an
aspartyl protease, and these are known to be inhibited by peptides made from
amino
5 acids from protein or analogues. Lentiviral vectors that inhibit HIV will
express one or
multiple fused copies of such peptide inhibitors.
Administration of the lentiviral vectors discussed above should be
effective against many virally linked diseases, cancers, or other pathogenic
agents.
In yet another aspect, lentiviral vectors are provided which have a
to therapeutic effect by encoding one or more ribozymes (RNA enzymes)
(Haseloff and
Gerlach, Nature 334:585, 1989) which will cleave, and hence inactivate, RNA
molecules corresponding to a pathogenic function. Since ribozymes function by
recognizing a specific sequence in the target RNA and this sequence is
normally 12 to
17 bp, this allows specific recognition of a particular RNA sequence
corresponding to a
15 pathogenic state, such as HIV tat, and toxicity is specific to such
pathogenic state.
Representative examples of suitable ribozymes include hammerhead ribozymes
(see
Rossi et al., Pharmac. Ther 50:245-254, 1991 ) and hairpin ribozymes (Hampel
et al.,
Nucl. Acids Res. 18:299-304, 1990; U.S. Patcnt No. 5,254,678) and Tetrahymena
based
ribozymes (U.S. Patent No. 4,987,071). Additional specificity may be achieved
in
2o some cases by making this a conditional toxic palliative, as discussed
above.
In still another aspect, lentiviral vectors are provided comprising a
biologically active nucleic acid molecule that is an antisense sequence (an
antisense
sequence may also be encoded by a nucleic acid sequence and then produced
within a
host cell via transcription). Briefly, antisense sequences are designed to
bind to RNA
transcripts, and thereby prevent cellular synthesis of a particular protein,
or prevent use
of that RNA sequence by the cell. Representative examples of such sequences
include
antisense thymidine kinase, antisense dihydrofolate reductase (Maker and
Dolnick,
Arch. Biochem. & Biophys. 253:214-220, 1987; Bzik et al., PNAS 84:8360-8364,
1987),
antisense HER2 (Coussens et al., Science ?30:1132-1139, 1985), antisense ABL
(Fainstein et al., Oncogene 4:1477-1481, 1989), antisense Myc (Stanton et al.,
Nature
310:423-42~, 1984) and antisense ras, as well as antisense sequences which
block any
of the enzymes in the nucleotide biosynthetic pathway. In other embodiments,
the
antisense sequence is selected from the group consisting of sequences which
encode
influenza virus, HIV, HSV, HPV, CMV, and HBV. The antisense sequence may also
be an antisense RNA complementary to RNA sequences necessary for
pathogenicity.
CA 02296319 2000-O1-17
WO 99/04026 PCT/US98/14996
16
Alternatively, the biologically active nucleic acid molecule may be a sense
RNA (or
DNA) complementary to RNA sequences necessary for pathogenicity.
Within a further embodiment of the invention antisense RNA may be
utilized as an anti-tumor agent in order to induce a potent Class I restricted
response.
Briefly, in addition to binding RNA and thereby preventing translation of a
specific
mRNA, high levels of specific antisense sequences are believed to induce the
increased
expression of interferons (including gamma-interferon), due to the formation
of large
quantities of double-stranded RNA. The increased expression of gamma
interferon, in
turn, boosts the expression of MHC Class I antigens. Preferred antisense
sequences for
use in this regard include actin RNA, myosin RNA, and histone RNA. Antisense
RNA
which forms a mismatch with actin RNA is particularly preferred.
In another embodiment, lentiviral vectors of the invention express a
surface protein that is itself therapeutically beneficial. For example, in the
particular
case of HIV, expression of the human CD4 protein specifically in HIV-infected
cells
may be beneficial in two ways:
I. Binding of CD4 to HIV env intracellularly could inhibit the
formation of viable viral particles much as soluble CD4 has been shown to do
for free
virus, but without the problem of systematic clearance and possible
immunogenicity,
since the protein will remain membrane bound and is structurally identical to
endogenous CD4 (to which the patient should be immunologically tolerant).
2. Since the CD4/HIV env complex has been implicated as a cause
of cell death, additional expression of CD4 (in the presence of excess HIV-env
present
in HIV-infected cells) leads to more rapid cell death and thus inhibits viral
dissemination. This may be particularly applicable to monocytes and
macrophages,
which act as a reservoir for virus production as a result of their relative
refractility to
HIV-induced cytotoxicity (which, in turn, is apparently due to the relative
lack of CD4
on their cell surfaces).
Still further aspects of the present invention relate to lentiviral vectors
capable of immunostimulation. Briefly, the ability to recognize and defend
against
foreign pathogens is essential to the function of the immune system. In
particular, the
immune system must be capable of distinguishing "self' from "nonself' (i.e.,
foreign),
so that the defensive mechanisms of the host are directed toward invading
entities
instead of against host tissues. Cytolytic T lymphocytes (CTLs) are typically
induced,
or stimulated, by the display of a cell surface recognition structure, such as
a processed,
pathogen-specific peptide, in conjunction with a MHC class I or class II cell
surface
protein.
CA 02296319 2000-O1-17
WO 99/04026 PCT/US98/14996
17
Diseases suitable to treatment include viral infections such as influenza
virus, respiratory syncytial virus, HPV, HBV, HCV, EBV, HIV, HSV, FeLV, FIV,
Hantavirus, HTLV I, HTLV II and CMV, cancers such as melanomas, renal
carcinoma,
breast cancer, ovarian cancer and other cancers, and heart disease.
In one embodiment, the invention provides methods for stimulating a
specific immune response and/or inhibiting viral spread by using lentiviral
vectors that
direct the expression of an antigen or modified form thereof in susceptible
target cells,
wherein the antigen is capable of either (1) initiating an immune response to
the viral
antigen or (2) preventing the viral spread by occupying cellular receptors
required for
1o viral interactions. Expression of the protein may be transient or stable
with time.
Where an immune response is to be stimulated to a pathogenic antigen, the
lentiviral
vector is preferably designed to express a modified form of the antigen which
will
stimulate an immune response and which has reduced pathogenicity relative to
the
native antigen. This immune response is achieved when cells present antigens
in the
is correct manner, i.e., in the context of the MHC class I and/or II molecules
along with
accessory molecules such as CD3, ICAM-l, ICAM-2, LFA-1, or analogs thereof
(e.g.,
Altmann et al., Nature 338:512, 1989).
An immune response can also be achieved by transferring to an
appropriate immune cell (such as a T lymphocyte) (a) the gene for the specific
T-cell
20 receptor that recognizes the antigen of interest (in the context of an
appropriate MHC
molecule if necessary), (b) the gene for an immunoglobulin which recognizes
the
antigen of interest, or (c) the gene for a hybrid of the two which provides a
CTL
response in the absence of the MHC context. Thus, recombinant retroviruses may
also
be used as an immunostimulant, immunomodulator, or vaccine, etc.
25 In the particular case of disease caused by HIV infection, where
immunostimulation is desired, the antigen generated from a recombinant
retrovirus may
be in a form which will elicit either or both an HLA class I- or class II-
restricted
immune response. In the case of HIV envelope antigen, for example, the antigen
is
preferably selected from gp 160, gp 120, and gp 41, which have been modified
to
3o reduce their pathogenicity. In particular, the selected antigen is modified
to reduce the
possibility of syncytia, to avoid expression of epitopes leading to a disease
enhancing
immune response, to remove immunodominant, but haplotype-specific epitopes or
to
present several haplotype-specific epitopes, and allow a response capable of
eliminating
cells infected with most or all strains of HIV. The haplotype-specific
epitopes can be
35 further selected to promote the stimulation of an immune response within an
animal
which is cross-reactive against other strains of HIV. Antigens from other HIV
genes or
CA 02296319 2000-O1-17
WO 99/04026 PCT/US98/14996
18
combinations of genes, such as gag, pol, rev, vif, nef, prot, gag/pol, gag
prot, etc., may
also provide protection in particular cases.
HIV is only one example. This approach may be utilized for many
virally linked diseases or cancers where a characteristic antigen (which does
not need to
be a membrane protein) is expressed. Representative examples of such "disease
associated" antigens all or portions of various eukaryotic (including for
example,
parasites), prokaryotic (e.g., bacterial) or viral pathogens. Representative
examples of
viral pathogens include the Hepatitis B Virus ("HBV") and Hepatitis C Virus
("HCV";
see U.S. Serial No. 08/102/132), Human Papiloma Virus ("HPV"; see WO 92/05248;
l0 WO 90/10459; EPO 133,123), Epstein-Ban Virus ("EBV"; see EPO 173,254; JP
1,128,788; and U.S. Patent Nos. 4,939,088 and 5,173,414), Feline Leukemia
Virus
("FeLV"; see U.S. Serial No. 07/948,358; EPO 377,842; WO 90/08832; WO
93/09238),
Feline Immunodeficiency Virus ("FIV"; U.S. Patent No. 5,037,753; WO 92/15684;
WO
90/13573; and JP 4,126,085), HTLV I and II, and Human Immunodeficiency Virus
("HIV"; see U.S. Serial No. 07/965,084).
In accordance with the immunostimulation aspects of the invention,
substances which are carried and/or expressed by the lentiviral vectors of the
present
invention may also include "immunomodulatory factors," many of which are set
forth
above. Immunomodulatory factors refer to factors that, when manufactured by
one or
2o more of the cells involved in an immune response, or, which when added
exogenously
to the cells, causes the immune response to be different in quality or potency
from that
which would have occurred in the absence of the factor. The factor may also be
expressed from a non-recombinant retrovirus derived gene, but the expression
is driven
or controlled by the recombinant retrovirus. The quality or potency of a
response may
be measured by a variety of assays known to one of skill in the art including,
for
example, in vitro assays which measure cellular proliferation (e.g., 3H
thymidine
uptake), and in vitro cytotoxic assays (e.g., which measure 51 Cr release)
(see, Warner
et al., AIDS Res. and Human Retroviruses 7:645-bSS, 1991 ). Immunomodulatory
factors may be active both in vivo and ex vivo.
Representative examples of such factors include cytokines, such as IL-1,
IL-2 (Karupiah et al., J. Immunology 144:290-298, 1990; Weber et al., J. Exp.
Med.
166:1716-1733, 1987; Gansbacher et al., J. Exp. Med. 172:1217-1224, 1990; U.S.
Patent No. 4,738,927), IL-3, IL-4 (Tepper et al., Cell 57:503-512, 1989;
Golumbek
et al., Science 254:713-716, 1991; U.S. Patent No. 5,017,691), IL-S, IL-6
(Brakenhof
et al., J. Irnmunol. 139:4116-4121, 1987; WO 90/06370), IL-7 (U.S. Patent
No.4,965,195), IL-8, IL-9, IL-10, IL-11, IL-12, IL-13 (Cvtokine Bulletin,
Summer
CA 02296319 2000-O1-17
WO 99/04026 PCT/US98/14996
19
1994), IL-14 and IL-15, particularly IL-2, IL-4, IL-6, IL-12, and IL-13, alpha
interferon
(Finter et al., Drugs 42(5):749-765, 1991; U.S. Patent No. 4,892,743; U.S.
Patent
No. 4,966,843; WO 85/02862; Nagata et al., Nature 284:316-320, 1980;
Familletti
et al., Methods in Enz. 78:387-394, 1981; Twu et al., Proc. Natl. Acad. Sci.
USA
86:2046-2050, 1989; Faktor et al., Oncogene 5:867-872, 1990), beta interferon
(Seif
et al., J. Virol. 65:664-671, 1991), gamma interferons (Radford et al., The
American
Society of Hepatology 2008-2015, 1991; Watanabe et al., PNAS 86:9456-9460,
1989;
Gansbacher et al., Cancer Research 50:7820-7825, 1990; Maio et al., Can.
Immunol.
Immunother. 30:34-42, 1989; U.S. Patent No.4,762,791; U.S. Patent
No.4,727,138),
G-CSF (U.S. Patent Nos. 4,999,291 and 4,810,643), GM-CSF (WO 85/04188), tumor
necrosis factors (TNFs) (Jayaraman et al., J. Immunolog>> 144:942-951, 1990),
CD3
(Krissanen et al., Immunogenetics 26:258-266, 1987), ICAM-1 (Altman et al.,
Nature
338:512-514, 1989; Simmons et al., Nature 331:624-627, 1988), ICAM-2, LFA-1,
LFA-3 (Wanner et al., J. Exp. Med. 166(4):923-932, 1987), MHC class I
molecules,
t5 MHC class II molecules, B7.1-.3, X32-microglobulin (Parnes et al., PNAS
78:2253-2257,
1981 ), chaperones such as calnexin, MHC linked transporter proteins or
analogs thereof
(Powis et al., Nature 354:528-531, 1991 ). Immunomodulatory factors may also
be
agonists, antagonists, or ligands for these molecules. For example soluble
forms of
receptors can often behave as antagonists for these types of factors, as can
mutated
forms of the factors themselves.
The choice of which immunomodulatory factor to include within a
lentiviral vector may be based upon known therapeutic effects of the factor,
or,
experimentally determined. For example, a known therapeutic effector in
chronic
hepatitis B infections is alpha interferon. This has been found to be
efficacious in
compensating a patient's immunological deficit, and thereby assisting recovery
from the
disease. Alternatively, a suitable immunomodulatory factor may be
experimentally
determined. Briefly, blood samples are first taken from patients with a
hepatic disease.
Peripheral blood lymphocytes (PBLs) are restimulated in vitro with autologous
or HLA
matched cells {e.g., EBV transformed cells) that have been transduced with a
recombinant retrovirus which directs the expression of an immunogenic portion
of a
hepatitis antigen and the immunomodulatory factor. These stimulated PBLs are
then
used as effectors in a CTL assay with the HLA matched transduced cells as
targets. An
increase in CTL response over that seen in the same assay performed using HLA
matched stimulator and target cells transduced with a vector encoding the
antigen alone,
indicates a useful immunomodulatory factor. Within one embodiment of the
invention,
the immunomodulatory factor gamma interferon is particularly preferred.
CA 02296319 2000-O1-17
WO 99/04026 PCT/US98/14996
- 20
The present invention also includes lentiviral vectors which encode
immunogenic portions of desired antigens including, for example, viral,
bacterial or
parasite antigens. For example, at least one immunogenic portion of a
hepatitis B
antigen can be incorporated into a lentiviral vector. The immunogenic
portions) which
are incorporated into the lentiviral vector may be of varying length, although
it is
generally preferred that the portions be at least 9 amino acids long, and may
include the
entire antigen. Immunogenicity of a particular sequence is often difficult to
predict,
although T cell epitopes may be predicted utilizing the HLA A2.1 motif
described by
Falk et al. (Nature 351:290, 1991 ). From this analysis, peptides may be
synthesized
to and used as targets in an in vitro cytotoxic assay. Other assays, however,
may also be
utilized, including, for example, ELISA which detects the presence of
antibodies against
the newly introduced vector, as well as assays which test for T helper cells,
such as
gamma-interferon assays, IL-2 production assays, and proliferation assays.
Within one embodiment of the present invention, at least one
immunogenic portion of a hepatitis C antigen can be incorporated into a
lentiviral
vector. Preferred immunogenic portion{s) of hepatitis C may be found in the C
and
NS3-NS4 regions since these regions are the most conserved among various types
of
hepatitis C virus (Houghton et al., Hepatolo~~ 14:381-388, 1991). Particularly
preferred immunogenic portions may be determined by a variety of methods. For
2o example, as noted above for the hepatitis B virus, identification of
immunogenic
portions of the polypeptide may be predicted based upon amino acid sequence.
Briefly,
various computer programs which are known to those of ordinary skill in the
art may be
utilized to predict CTL epitopes. For example, C'rL epitopes for the HLA A2.1
haplotype may be predicted utilizing the HI_A A2.1 motif described by Falk et
al.
(Nature 351:290, 1991). From this analysis, peptides are synthesized and used
as
targets in an in vitro cytotoxic assay.
Other disease-associated antigens which may be carried by the gene
delivery constructs of the present invention include, for example immunogenic,
non-
tumorigenic forms of altered cellular components which are normally associated
with
3o tumor cells (see U.S. Serial No. 08/104,424). Representative examples of
altered
cellular components which are normally associated with tumor cells include
ras*
(wherein ~~*~~ is understood to refer to antigens which have been altered to
be non-
tumorigenic), p53*, Rb*, altered protein encoded by Wilms' tumor gene,
ubiquitin*,
mucin, protein encoded by the DCC. APC, and MCC genes, as well as receptors or
receptor-like structures such as neu, thyroid hornlone receptor, Platelet
Derived Growth
CA 02296319 2000-O1-17
WO 99/04026 PCT/US98/14996
21
Factor ("PDGF") receptor, insulin receptor, Epidermal Growth Factor ("EGF")
receptor,
and the Colony Stimulating Factor {"CSF") receptor.
Immunogenic portions of the disease-associated antigens described
herein may be selected by a variety of methods. For example, the HLA A2.1/Kb
transgenic mouse has been shown to be useful as a model for human T-cell
recognition
of viral antigens. Briefly, in the influenza and hepatitis B viral systems,
the murine
T-cell receptor repertoire recognizes the same antigenic determinants
recognized by
human T-cells. In both systems, the CTL response generated in the HLA A2.1/Kb
transgenic mouse is directed toward virtually the same epitope as those
recognized by
to human CTLs of the HLA A2.1 haplotype (Vitiello et al., J. Exp. Med.
173:1007-1015,
1991; Vitiello et al., Abstract of Molecular Biology' of Hepatitis B Virus
Symposia,
1992).
Immunogenic proteins of the present invention may also be manipulated
by a variety of methods known in the art, in order to render them more
immunogenic.
Representative examples of such methods include: adding amino acid sequences
that
correspond to T helper epitopes; promoting cellular uptake by adding
hydrophobic
residues; by forming particulate structures; or any combination of these (see
generally,
Hart, op. cit., Milich et al., Proc. Natl. Acad. Sci. USA 85:1610-1614, 1988;
Willis,
Nature 340:323-324, 1989; Griffiths et al., J. Virol. 65:450-456, 1991 ).
2o Sequences which encode the above-described nucleic acid molecules
may be obtained from a variety of sources. For example, plasmids which contain
sequences that encode altered cellular products may be obtained from a
depository such
as the American Type Culture Collection (ATCC, Rockville, Maryland), or from
commercial sources such as Advanced Biotechnologies (Columbia, Maryland).
2s Representative examples of plasmids containing some of the above-described
sequences
include ATCC No. 41000 (containing a G to T mutation in the 12th codon of
ras), and
ATCC No. 41049 (containing a G to A mutation in the 12th codon).
Other nucleic acid molecules that encode the above-described
substances, as well as other nucleic acid molecules that are advantageous for
use within
30 the present invention, may be readily obtained from a variety of sources,
including for
example depositories such as the American Type Culture Collection (ATCC,
Rockville,
Maryland), or from commercial sources such as British Bio-Technology Limited
(Cowley, Oxford England). Representative examples include BBG 12 (containing
the
GM-CSF gene coding for the mature protein of 127 amino acids), BBG 6 (which
35 contains sequences encoding gamma interferon), ATCC No. 39656 (which
contains
sequences encoding TNF), ATCC No. 20663 (which contains sequences encoding
alpha
CA 02296319 2000-O1-17
WO 99/04026 PCT/US98/14996
22
interferon), ATCC Nos. 31902, 31902 and 39517 (which contains sequences
encoding
beta interferon), ATCC No 67024 (which contains a sequence which encodes
Interleukin-lb), ATCC Nos. 39405, 39452, 39516, 39626 and 39673 (which
contains
sequences encoding Interleukin-2), ATCC Nos. 59399, 59398, and 67326 (which
contain sequences encoding Interleukin-3), ATCC No. 57592 (which contains
sequences encoding Interleukin-4), ATCC Nos. 59394 and 59395 (which contain
sequences encoding Interleukin-S), and ATCC No. 67153 (which contains
sequences
encoding Interleukin-6).
Molecularly cloned genomes which encode the hepatitis B virus may be
obtained from a variety of sources including, for example, the American Type
Culture
Collection (ATCC, Rockville, Maryland). For example, ATCC No. 45020 contains
the
total genomic DNA of hepatitis B (extracted from purified Dane particles) (see
Figure 3
of Blum et al., TIG S(S):154-158, 1989) in the BamH I site of pBR322 (Moriarty
et al.,
Proc. Natl. Acad. Sci. USA 78:2606-2610, 1981 ). (Note that correctable errors
occur in
the sequence of ATCC No. 45020.)
Alternatively, eDNA sequences for use with the present invention may
be obtained from cells which express or contain the sequences. Briefly, within
one
embodiment mRNA from a cell which expresses the gene of interest is reverse
transcribed with reverse transcriptase using oligo dT or random primers. The
single
stranded cDNA may then be amplified by PCR (see LLS. Patent Nos.4,683,202,
4,683,195 and 4,800,159). See also PCR Techrtologl>: Principles ajtd
Applications for
DNA Amplification, Erlich (ed.), Stockton Press, 1989) utilizing
oligonucleotide
primers complementary to sequences on either side of desired sequences. In
particular,
a double stranded DNA is denatured by heating in the presence of heat stable
Taq
polymerase, sequence specific DNA primers, ATP, CTP, GTP and TTP. Double-
stranded DNA is produced when synthesis is complete. This cycle may be
repeated
many times, resulting in a factorial amplification of the desired DNA.
Nucleic acid molecules which are carried and/or expressed by the
lentiviral vectors described herein may also be synthesized, for example, on
an Applied
Biosystems Inc. DNA synthesizer (e.g., APB DNA synthesizer model 392 (Foster
City,
California).
METHODS FOR UTILIZING LENT1VIR S VECTOR PARTICLES
As noted above, the present invention also provides methods for
delivering a selected heterologous sequence to a vertebrate or insect,
comprising the
step of administering to a vertebrate or insect a lentiviral vector particle
as described
CA 02296319 2000-O1-17
WO 99!04026 PCT/US98/14996
23
herein which is capable of expressing the selected heterologous sequence. Such
lentiviral vector particles may be administered either directly (e.g.,
intravenously,
intramuscularly, intraperitoneally, subcutaneously, orally, rectally,
intraocularly,
intranasally), or by various physical methods such as lipofection (Felgner et
al., Proc.
Natl. Acad. Sci. USA 84:7413-7417, 1989), direct DNA injection (Fung et al.,
Proc.
Natl. Acad. Sci. USA 80:353-357, 1983; Seeger et al., Proc. Natl. Acad. Sci.
USA
81:5849-5852; Acsadi et al., Nature 352:815-818, 1991 ); microprojectile
bombardment
(Williams et al., PNAS 88:2726-2730, 1991 ); liposomes of several types (see,
e.g.,
Wang et al., PNAS 84:7851-7855, 1987); CaPOa (Dubensky et al., PNAS 81:7529-
7533,
l0 1984); DNA ligand (Wu et al, J. Biol. Cheat. 264:16985-16987, 1989);
administration
of nucleic acids alone (WO 90/11092); or administration of DNA linked to
killed
adenovirus (Curie! et al., Hum. Gene Ther. 3:147-154, 1992); via polycation
compounds such as polylysine, utilizing receptor specific ligands; as well as
with
psoralen inactivated viruses such as Sendai or Adenovirus. In addition, the
lentiviral
vector particles may either be administered directly (i.e., in vivo), or to
cells which have
been removed (ex vivo), and subsequently returned.
As discussed in more detail below, lentiviral vector particles may be
administered to a vertebrate or insect organism or cell for a wide variety of
both
therapeutic or productive purposes, including for example, for the purpose of
2o stimulating a specific immune response; inhibiting the interaction of an
agent with a
host cell receptor; to express a toxic palliative, including for example,
conditional toxic
palliatives; to immunologically regulate the immune system; to express
markers, for
replacement gene therapy and/or to produce a recombinant protein. These and
other
uses are discussed in more detail below.
1. Immunostimulation
Within one aspect of the present invention, compositions and methods
are provided for administering a lentiviral vector particle which is capable
of
preventing, inhibiting, stabilizing or reversing infectious, cancerous, auto-
immune or
immune diseases. Representative examples of such diseases include viral
infections
3o such as HIV, HBV, HCV, HTLV I, HTLV II, CMV, EBV and HPV, melanomas,
diabetes, graft vs. host disease, Alzheimer's disease and heart disease. More
specifically, within one aspect of the present invention, compositions and
methods are
provided for stimulating an immune response (either humoral or cell-mediated)
to a
pathogenic agent, such that the pathogenic agent is either killed or
inhibited.
CA 02296319 2000-O1-17
WO 99/04026 PCT/US98/14996
24
Representative examples of pathogenic agents include bacteria, fungi,
parasites, viruses
and cancer cells.
Within one embodiment of the invention the pathogenic agent is a virus,
and methods are provided for stimulating a specific immune response and
inhibiting
viral spread by using a lentiviral vector particle that directs the expression
of an antigen
or modified form thereof to susceptible target cells capable of either (1)
initiating an
immune response to the viral antigen or (2) preventing the viral spread by
occupying
cellular receptors required for viral interactions. Expression of the vector
nucleic acid
encoded protein may be transient or stable with time. Where an immune response
is to
1o be stimulated to a pathogenic antigen, the lentiviral vector is preferably
designed to
express a modified form of the antigen which will stimulate an immune response
and
which has reduced pathogenicity relative to the native antigen. This immune
response
is achieved when cells present antigens in the correct manner, i.e., in the
context of the
MHC class I and/or II molecules along with accessory molecules such as CD3,
ICAM-
1, ICAM-2, LFA-1, or analogues thereof (e.g., Altmann et al., Nature 338:512,
1989).
Cells infected with lentiviral vector particles are expected to do this
efficiently because
they closely mimic genuine viral infection and because they: (a) are able to
infect non-
replicating cells, (b) do not integrate into the host cell genome, (c) are not
associated
with any life threatening diseases, and (d) express high levels of
heterologous protein.
Because of these differences, lentiviral vectors can easily be thought of as
safe viral
vectors which can be used on healthy individuals for vaccine use.
This aspect of the invention has a further advantage over other systems
that might be expected to function in a similar manner, in that the presenter
cells are
fully viable and healthy, and low levels of viral antigens, relative to
heterologous genes,
are expressed. This presents a distinct advantage since the antigenic epitopcs
expressed
can be altered by selective cloning of sub-fragments of the gene for the
antigen into a
lentiviral vector particle, leading to responses against immunogenic epitopes
which may
otherwise be overshadowed by immunodominant epitopcs. Such an approach may be
extended to the expression of a peptide having multiple epitopes, one or more
of the
3o epitopes being derived from different proteins. Further, this aspect of the
invention
allows eff cient stimulation of cytotoxic T lymphocytes (CTL) directed against
antigenic epitopes, and peptide fragments of antigens encoded by sub-fragments
of
genes, through intracellular synthesis and association of these peptide
fragments with
MHC Class I molecules. This approach may be utilized to map major
immunodominant epitopes for CTL induction.
CA 02296319 2000-O1-17
WO 99104026 PCT/US98/14996
An immune response may also be achieved by transferring to an
appropriate immune cell (such as a T lymphocyte) the gene for the specific T
cell
receptor which recognizes the antigen of interest (in the context of an
appropriate MHC
molecule if necessary), for an immunoglobulin which recognizes the antigen of
interest,
5 or for a hybrid of the two which provides a CTL response in the absence of
the MHC
context. Thus, the lentiviral vector particles may be used as an
immunostimulant,
immunomodulator, or vaccine.
In another embodiment of the invention, methods are provided for
producing inhibitor palliatives wherein lentiviral vector particles deliver
and express
to defective interfering viral structural proteins, which inhibit viral
assembly. Such
lentiviral vector particles may encode defective gug, pol, env or other viral
particle
proteins or peptides and these would inhibit in a dominant fashion the
assembly of viral
particles. This occurs because the interaction of normal subunits of the viral
particle is
disturbed by interaction with the defective subunits.
15 In another embodiment of the invention, methods are provided for the
expression of inhibiting peptides or proteins specific for viral protease.
Briefly, viral
protease cleaves the viral gag and gaglpol proteins into a number of smaller
peptides.
Failure of this cleavage in all cases leads to complete inhibition of
production of
infectious retroviral particles. As an example, the HIV protease is known to
be an
20 aspartyl protease and these are known to be inhibited by peptides made from
amino
acids from protein or analogues. Lentiviral vectors to inhibit HIV will
express one or
multiple fused copies of such peptide inhibitors.
Another embodiment involves the delivery of suppressor genes which,
when deleted, mutated, or not expressed in a cell type, lead to tumorigenesis
in that cell
25 type. Reintroduction of the deleted gene by means of a lentiviral vector
particle leads to
regression of the tumor phenotype in these cells. Examples of such cancers are
retinoblastoma and Wilms Tumor. Since malignancy can be considered to be an
inhibition of cellular terminal differentiation compared with cell growth,
administration
of the lentiviral vector particle and expression of gene products which lead
to
differentiation of a tumor should also, in general, lead to regression.
In yet another embodiment, the lentiviral vector provides a therapeutic
effect by transcribing a ribozyme (an RNA enzyme) (Haseloff and Gerlach,
Nature
334:585, 1989) which will cleave and hence inactivate RNA molecules
corresponding
to a pathogenic function. Since ribozymes function by recognizing a specific
sequence
in the target RNA and this sequence is normally 12 to 17 bp, this allows
specific
recognition of a particular RNA species such as a RNA or a retroviral genomc.
CA 02296319 2000-O1-17
WO 99/04026 PCT/US98/14996
26
Additional specificity may be achieved in some cases by making this a
conditional toxic
palliative (see below).
One way of increasing the effectiveness of inhibitory palliatives is to
express viral inhibitory genes in conjunction with the expression of genes
which
increase the probability of infection of the resistant cell by the virus in
question. The
result is a nonproductive "dead-end" event which would compete for productive
infection events. In the specific case of HIV, lentiviral vector particles may
be
delivered which inhibit HIV replication (by expressing anti-sense tat, etc.,
as described
above) and also overexpress proteins required for infection, such as CD4. In
this way, a
relatively small number of vector-infected HIV-resistant cells act as a "sink"
or
"magnet" for multiple nonproductive fusion events with free virus or virally
infected
cells.
2. Blocking Agents
Many infectious diseases, cancers, autoimmune diseases, and other
diseases involve the interaction of viral particles with cells, cells with
cells, or cells with
factors produced by themselves or other cells. In viral infections, viruses
commonly
enter cells via receptors on the surface of susceptible cells. In cancers or
other
proliferative conditions (e.g., restenosis), cells may respond inappropriately
or not at all
to signals from other cells or factors, or specific factors may be mutated,
overexpressed,
or underexpressed, resulting in loss of appropriate cell cycle control. In
autoimmune
disease, there is inappropriate recognition of "self' markers. Within the
present
invention, such interactions may be blocked by producing, in vivo, an analogue
to either
of the partners in an interaction. Alternatively, cell cycle control may be
restored by
preventing the transition from one phase to another (e.g., G1 to S phase)
using a
blocking factor which is absent or underexpressed. This blocking action may
occur
intracellularly, on the cell membrane, or extracellularly, and the action of
the lentivirus
vector particle carrying a gene for a blocking agent, can be mediated either
from inside
a susceptible cell or by secreting a version of the blocking protein to
locally block the
pathogenic interaction.
In the case of HIV, the two agents of interaction are the gp 1201gp 41
envelope protein and the CD4 receptor molecule. Thus, an appropriate Mocker
would
be a lentiviral vector expressing either an HIV env analogue that blocks HIV
entry
without causing pathogenic effects, or a CD4 receptor analogue. The CD4
analogue
would be secreted and would function to protect neighboring cells, while the
gp 120/gp
41 is secreted or produced only intracellularly so as to protect only the
vector-
CA 02296319 2000-O1-17
WO 99/04026 PCT/US98/14996
27
containing cell. It may be advantageous to add human immunoglobulin heavy
chains or
other components to CD4 in order to enhance stability or complement lysis.
Administration of a lentiviral vector particle encoding such a hybrid-soluble
CD4 to a
host results in a continuous supply of a stable hybrid molecule. Efficacy of
treatment
can be assayed by measuring the usual indicators of disease progression,
including
antibody level, viral antigen production, infectious HIV levels, or levels of
nonspecific
infections.
In the case of uncontrolled proliferative states, such as cancer or
restenosis, cell cycle progression may be halted by the expression of a number
of
different factors that affect signaling by cyclins or cyclin-dependent kinases
(CDK).
For example, the cyclin-dependent kinase inhibitors, p16, p21, and p27 each
regulate
cyclin:CDK mediated cell cycle signaling. Overexpression of these factors
within a cell
by a lentiviral vector particle results in a cytostatic suppression of cell
proliferation.
Other factors that may be used therapeutically, as blocking agents or targets,
include,
for example, wild-type or mutant Rb, p53, Myc, Fos, Jun, PCNA , GAX, and p15.
3. Expression of Palliatives
Techniques similar to those described above can be used to produce
lentiviral vector particles which direct the expression of an agent (or
"palliative") which
is capable of inhibiting a function of a pathogenic agent or gene. Within the
present
invention, "capable of inhibiting a function" means that the palliative either
directly
inhibits the function or indirectly does so, for example, by converting an
agent present
in the cells from one which would not normally inhibit a function of the
pathogenic
agent to one which does. Examples of such functions for viral diseases include
adsorption, replication, gene expression, assembly, and exit of the virus from
infected
cells. Examples of such functions for a cancerous cell, cancer-promoting
growth factor,
or uncontrolled proliferative condition (e.g., restenosis) include viability,
cell
replication, altered susceptibility to external signals (e.g., contact
inhibition), and lack
of production or production of mutated forms of anti-oncogene proteins.
a. Inhibitor Palliatives
In one aspect of the present invention, the lentiviral vector particle
directs the expression of a gene which can interfere with a function of a
pathogenic
agent, for instance in viral or malignant diseases. Such expression may either
be
essentially continuous or in response to the presence in the cell of another
agent
associated either with the pathogenic condition or with a specific cell type
(an
CA 02296319 2000-O1-17
WO 99/04026 PCT/US98/14996
28
"identifying agent"). In addition, vector delivery may be controlled by
targeting vector
entry specifically to the desired cell type (for instance, a virally infected
or malignant
cell) as discussed above.
One method of administration is leukophoresis, in which about 20% of
an individual's PBLs are removed at any one time and manipulated in vitro.
Thus,
approximately 2 x 109 cells may be treated and replaced. Repeat treatments may
also be
performed. Alternatively, bone marrow may be treated and allowed to amplify
the
effect as described above. In addition, packaging cell lines producing a
vector may be
directly injected into a subject, allowing continuous production of
recombinant virions.
In one embodiment, lentiviral vector particles which express RNA
complementary to key pathogenic gene transcripts (for example, a viral gene
product or
an activated cellular oncogene) can be used to inhibit translation of that
transcript into
protein, such as the inhibition of translation of the H1V tat protein. Since
expression of
this protein is essential for viral replication, cells containing the
lentiviral vector particle
would be resistant to HIV replication.
In a second embodiment, where the pathogenic agent is a single-stranded
virus having a packaging signal, RNA complementary to the viral packaging
signal
(e.g., an HIV packaging signal when the palliative is directed against HIV) is
expressed,
so that the association of these molecules with the viral packaging signal
will, in the
case of retroviruses, inhibit stem loop formation or tRNA primer binding
required for
proper encapsidation or replication.
In a third embodiment, lentiviral vector particles may be introduced
which expresses a palliative capable of selectively inhibiting the expression
of a
pathogenic gene, or a palliative capable of inhibiting the activity of a
protein produced
by the pathogenic agent. In the case of HIV, one example is a mutant tat
protein which
lacks the ability to transactivate expression from the HIV LTR and interferes
(in a
transdominant manner) with the normal functioning of tat protein. Such a
mutant has
been identified for HTLV II tat protein ("XII Leus" mutant; see Wachsman et
al.,
Science 235:674, 1987). A mutant transrepressor tat should inhibit replication
much as
has been shown for an analogous mutant repressor in HSV-1 (Friedmann et al.,
Nature
335:452, 1988).
Such a transcriptional repressor protein can be selected for in tissue
culture using any viral-specific transcriptional promoter whose expression is
stimulated
by a virus-specific transactivating protein (as described above). In the
specific case of
HIV, a cell line expressing HIV tat protein and the HSVTK gene driven by the
HIV
promoter will die in the presence of ACV. However, if a series of mutated tat
genes are
CA 02296319 2000-O1-17
WO 99/04026 PCT/LJS98/14996
29
introduced to the system, a mutant with the appropriate properties (i.e.,
represses
transcription from the HIV promoter in the presence of wild-type tat) will
grow and be
selected. The mutant gene can then be reisolated from these cells. A cell line
containing multiple copies of the conditionally lethal vector/tat system may
be used to
assure that surviving cell clones are not caused by endogenous mutations in
these genes.
A battery of randomly mutagenized tat genes are then introduced into these
cells using a
"rescuable" lentivirus vector (i.e., one that expresses the mutant tat protein
and contains
a bacterial origin of replication and drug resistance marker for growth and
selection in
bacteria). This allows a large number of random mutations to be evaluated and
permits
to facile subsequent molecular cloning of the desired mutant cell line. This
procedure may
be used to identify and utilize mutations in a variety of viral
transcriptional
activator/viral promoter systems for potential antiviral therapies.
b. Conditional Toxic Palliatives
Another approach for inhibiting a pathogenic agent is to express a
palliative which is toxic for the cell expressing the pathogenic condition. In
this case,
expression of the palliative from the lentiviral vector should be limited by
the presence
of an entity associated with the pathogenic agent, such as a specific viral
RNA sequence
identifying the pathogenic state, in order to avoid destruction of
nonpathogenic cells.
In one embodiment of this method, lentiviral vector particles can be
2o utilized to express a toxic gene (as discussed above) from a cell-specific
responsive
vector. In this manner, rapidly replicating cells, which contain the RNA
sequences
capable of activating the cell-specific responsive vectors, are preferentially
destroyed by
the cytotoxic agent produced by the lentiviral vector particle.
In a similar manner to the preceding embodiment, the lentiviral vector
can carry a gene for phosphorylation, phosphoribosylation, ribosylation, or
other
metabolism of a purine- or pyrimidine-based drug. This gene may have no
equivalent
in mammalian cells and might come from organisms such as a virus, bacterium,
fungus,
or protozoan. An example of this would be the E. toll guanine phosphoribosyl
transferase gene product, which is lethal in the presence of thioxanthine (see
Besnard
3o et al., Mol. Cell. Biol. 7:4139-4141, 1987). Conditionally lethal gene
products of this
type (also referred to as "pro-drugs" or "prodrug activating enzymes") have
application
to many presently known purine- or pyrimidine-based anticancer drugs, which
often
require intracellular ribosylation or phosphorylation in order to become
effective
cytotoxic agents. The conditionally lethal gene product could also metabolize
a
CA 02296319 2000-O1-17
WO 99/04026 PCT/US98/14996
nontoxic drug which is not a purine or pyrimidine analogue to a cytotoxic form
(see
Searle et al., Brit. J. Cancer 53:377-384, 1986).
In another aspect of the present invention, lentiviral vectors are provided
which direct the expression of a gene product capable of activating an
otherwise
5 inactive precursor into an active inhibitor of the pathogenic agent. For
example, the
HSVTK gene product may be used to more effectively metabolize potentially
antiviral
nucleoside analogues such as AZT or ddC. The HSVTK gene may be expressed under
the control of a cell-specific responsive vector and introduced into these
cell types.
AZT (and other nucleoside antivirals) must be metabolized by cellular
mechanisms to
10 the nucleotide triphosphate form in order to specifically inhibit
retroviral reverse
transcriptase, and thus, HIV replication (Furnlam et al., Proc. Natl. Acad.
Sci. USA
83:8333-8337, 1986). Constitutive expression of HSVTK (a nucleoside and
nucleoside
kinase with very broad substrate specificity) results in more effective
metabolism of
these drugs to their biologically active nucleotide triphosphate form. AZT or
ddC
15 therapy will thereby be more effective, allowing lower doses, less
generalized toxicity,
and higher potency against productive infection. Additional nucleoside
analogues
whose nucleotide triphosphate forms show selectivity for retroviral reverse
transcriptase
but, as a result of the substrate specificity of cellular nucleoside and
nucleotide kinascs
are not phosphorylated, will be made more efficacious.
20 Administration of these lentiviral vector particles to human T cell and
macrophage/monocyte cell lines can increase their resistance to HIV in the
presence of
AZT and ddC compared to the same cells without retroviral vector treatment.
Treatment with AZT would be at lower than normal levels to avoid toxic side
effects
but still efficiently inhibit the spread of HIV. The course of treatment would
be as
25 described for the blocker.
In one embodiment, the lentiviral vector particle carries a gene
specifying a product which is not in itself toxic but, when processed or
modified by a
protein such as a protease specific to a viral or other pathogen, is converted
into a toxic
form. For example, the lentiviral vector could carry a gene encoding a
proprotein for
3o ricin A chain, which becomes toxic upon processing by the HIV protease.
More
specifically, a synthetic inactive proprotein form of the toxin ricin or
diphtheria A
chains could be cleaved to the active form by arranging for the HIV virally
encoded
protease to recognize and cleave off an appropriate "pro" element.
In another embodiment, the lentiviral vector particle may express a
"reporting product" on the surface of the target cells in response to the
presence of an
identifying agent in the cells (such as expression of a viral gene). This
surface protein
CA 02296319 2000-O1-17
WO 99104026 PCT/US98/14996
31
can be recognized by a cytotoxic agent, such as antibodies for the reporting
protein, or
by cytotoxic T cells. In a similar manner, such a system can be used as a
detection
system (see below) to simply identify those cells having a particular gene
which
expresses an identifying protein.
Similarly, in another embodiment, a surface protein could be expressed
which would itself be therapeutically beneficial. In the particular case of
HIV,
expression of the human CD4 protein specifically in HIV-infected cells may be
beneficial in two ways:
1. Binding of CD4 to HIV env intracellulariy could inhibit the
to formation of viable viral particles, much as soluble CD4 has been shown to
do for free
virus, but without the problem of systematic clearance and possible
immunogenicity,
since the protein will remain membrane bound and is structurally identical to
endogenous CD4 (to which the patient should be immunologically tolerant).
2. Since the CD4/HIV env complex has been implicated as a cause
of cell death, additional expression of CD4 (in the presence of excess HIV-env
present
in HIV-infected cells) leads to more rapid cell death and thus inhibits viral
dissemination. This may be particularly applicable to monocytes and
macrophages,
which act as a reservoir for virus production as a result of their relative
refractility to
HIV-induced cytotoxicity (which, in turn, is apparently due to the relative
lack of CD4
on their cell surfaces).
In another embodiment, the lentiviral vector particle can provide a
ribozyme which will cleave and inactivate RNA molecules essential for
viability of the
vector infected cell. By making ribozyme production dependent on a specific
RNA
sequence corresponding to the pathogenic state, such as HIV tat, toxicity is
specific to
the pathogenic state.
4. Expression of Markers
The above-described technique of expressing a palliative in a cell in
response to a specific RNA sequence can also be modified to enable detection
of a
particular gene in a cell which expresses an identifying protein (for example,
a gene
3o carried by a particular virus), and hence enable detection of cells
carrying that virus. In
addition, this technique enables the detection of viruses (such as HIV) in a
clinical
sample of cells carrying an identifying protein associated with the virus.
This modification can be accomplished by providing a genome coding
for a product, the presence of which can be readily identified (the "marker
product"), in
a lentiviral vector which responds to the presence of the identifying protein
in the
CA 02296319 2000-O1-17
WO 99/04026 PCT/US98/14996
32
infected cells. For example, HIV, when it infects suitable cells, makes tat
and rev. The
indicator cells can thus be provided with a genome (such as by infection with
an
appropriate lentivirus particle) which codes for a marker gene, such as the
alkaline
phosphatase gene, ~3-galactosidase gene, or the luciferase gene which is
expressed by
the lentivirus particle upon activation by the tat and/or rev RNA transcript.
In the case
of (3-galactosidase or alkaline phosphatase, exposing the cells to substrate
analogues
results in a color or fluorescence change if the sample is positive for HIV.
In the case of
luciferase, exposing the sample to luciferin will result in luminescence if
the sample is
positive for HIV. For intracellular enzymes such as ~3-galactosidase, the
viral titre can
to be measured directly by counting colored or fluorescent cells, or by making
cell extracts
and performing a suitable assay. For the membrane bond form of alkaline
phosphatase,
virus titre can also be measured by performing enzyme assays on the cell
surface using
a fluorescent substrate. For secreted enzymes, such as an engineered form of
alkaline
phosphatase, small samples of culture supernatant are assayed for activity,
allowing
continuous monitoring of a single culture over time. Thus, different forms of
this
marker system can be used for different purposes. These include counting
active virus,
or sensitively and simply measuring viral spread in a culture and the
inhibition of this
spread by various drugs.
Further specificity can be incorporated into the preceding system by
testing for the presence of the virus either with or without neutralizing
antibodies to that
virus. For example, in one portion of the clinical sample being tested,
neutralizing
antibodies to HIV may be present; whereas in another portion there would be no
neutralizing antibodies. If the tests were negative in the system where there
were
antibodies and positive where there were no antibodies, this would assist in
confirming
the presence of HIV.
Within an analogous system for an in vitro assay, the presence of a
particular gene, such as a viral gene, may be determined in a cell sample. In
this case,
the cells of the sample are infected with a suitable lentiviral vector
particle which
carnes the reporter gene which is only expressed in the presence of the
appropriate viral
3o RNA transcript. The reporter gene, after entering the sample cells, will
express its
reporting product (such as (3-galactosidase or luciferase) only if the host
cell expresses
the appropriate viral proteins.
These assays are more rapid and sensitive, since the reporter gene can
express a greater amount of reporting product than identifying agent present,
which
results in an amplifcation effect.
CA 02296319 2000-O1-17
WO 99/04026 PCT/US98/14996
33
5. Immune Down-Regulation
As described above, the present invention also provides lentiviral vector
particles capable of suppressing one or more elements of the immune system in
target
cells infected with the lentivirus. Briefly, specific down-regulation of
inappropriate or
unwanted immune responses, such as in chronic hepatitis or in transplants of
heterologous tissue such as bone marrow, can be engineered using immune-
suppressive
viral gene products which suppress surface expression of transplantation (MHC)
antigen. Group C adenoviruses Ad2 and Ad5 possess a 19 kd glycoprotein (gp 19)
encoded in the E3 region of the virus. This gp 19 molecule binds to class I
MHC
to molecules in the endoplasmic reticulum of cells, and prevents terminal
glycosylation
and translocation of class I MHC to the cell surface. For example, prior to
bone marrow
transplantation, donor bone marrow cells may be infected with a gp 19-encoding
lentiviral vector which, upon expression of the gp 19, inhibit the surface
expression of
MHC class I transplantation antigens. These donor cells may be transplanted
with low
risk of graft rejection and may require a minimal immunosuppressive regimen
for the
transplant patient. This may allow an acceptable donor-recipient chimeric
state to exist
with fewer complications. Similar treatments may be used to treat the range of
so-
called autoimmune diseases, including lupus erythromiatis, multiple sclerosis,
rheumatoid arthritis or chronic hepatitis B infection. In the context of
arthritis,
2o lentiviral vectors may be utilized to directly transduce synoviocytes,
either in vivo, ex
vivo.
An alternative method involves the use of anti-sense message, ribozyme,
or other specific gene expression inhibitor specific for T cell clones which
are
autoreactive in nature. These block the expression of the T cell receptor of
particular
unwanted clones responsible for an autoimmune response. The anti-sense,
ribozyme, or
other gene may be introduced using the viral vector delivery system.
6. Ret~lacement or Augmentation CTene Therapy
One further aspect of the present invention relates to transforming cells
of a vertebrate or insect with a lentiviral vector which supplies genetic
sequences
3o capable of expressing a therapeutic protein. Within one embodiment of the
present
invention, the lentiviral vector is designed to express a therapeutic protein
capable of
preventing, inhibiting, stabilizing or reversing an inherited or noninherited
genetic
defect in metabolism, immune regulation, hormonal regulation, enzymatic or
membrane
associated structural function. This embodiment also describes the lentiviral
vector
particle capable of transducing individual cells, whereby the therapeutic
protein is able
CA 02296319 2000-O1-17
WO 99/04026 PCT/US98/14996
34
to be expressed systemically or locally from a specific cell or tissue,
whereby the
therapeutic protein is capable of (a) the replacement of an absent or
defective cellular
protein or enzyme, or (b) supplement production of a defective of low
expressed
cellular protein or enzyme. Such diseases may include cystic fibrosis,
Parkinson's
disease, hypercholesterolemia, adenosine deaminase deficiency, 13-globin
disorders,
Hemophilia A & B, Gaucher's disease, diabetes and leukemia. Within certain
preferred
embodiments vectors as described herein are utilized in order to provide Iong-
term
expression of the desired gene of interest.
a. Treatment of Gaucher disease
As an example of the present invention, lentiviral vector particles can be
constructed and utilized to treat Gaucher disease. Briefly. Gaucher disease is
a genetic
disorder that is characterized by the deficiency of the enzyme
glucocerebrosidase. This
type of therapy is an example of a single gene replacement therapy by
providing a
functional cellular enzyme. This enzyme deficiency leads to the accumulation
of
glucocerebroside in the lysosomes of all cells in the body. However, the
disease
phenotype is manifested only in the macrophages, except in the very rare
neuronpathic
forms of the disease. The disease usually leads to enlargement of the liver
and spleen
and lesions in the bones. (For a review, see Science ZSG:794, 1992, and The
Metabolic
Basis of Inherited Disease, 6th ed., Scriver et aL, vol. 2, p. 1677).
b. Lentiviral vector particles Expressing Human Factor VIII and Factor IX
for Treatment of Hemophilia
Within one embodiment of the invention, lentiviral vector particles
expressing a B-domain deleted factor VIII protein are provided (see also PCT
WO
91/09122, and Attorney's Docket No. 1155.005 entitled "Methods for
Administration of
Recombinant Gene Delivery Vehicles for Treatment of Hemophilia and Other
Disorders")
Briefly, the B domain separates the second and third A domains of factor
FVIII in the newly synthesized single-chain molecule. The B domain extends
from
amino acids 712 to 1648 according to Wood et al., 1984, Nature 312:330-337.
Proteolytic activation of factor VIIII involves cleavage at specific Arg
residues located
at positions 372, 740, and 1689. Cleavages of plasma factor VIII by thrombin
or Factor
Xa at Arg 372 and Arg 1689 are essential for factor VIII to participate in
coagulation.
Therefore, activated factor VIII consists of a heterodimer comprising amino
acids
CA 02296319 2000-O1-17
WO 99/04026 PCT/US98/14996
residues 1-372 (containing the A1 domain) and residues 373-740 (containing the
A2
domain), and residues 1690-2332 (containing the A3-C1-C2 domain).
An important advantage in using the B domain deleted FVIII molecule is
that the reduced size appears to be less prone to proteolytic degradation and
therefor, no
5 addition of plasma-derived albumin is necessary for stabilization of the
final product.
The term "B domain deletion" as used herein with respect to factor VIII
protein refers to
a factor VIII protein in which some or all removal of some or all of the amino
acids
between residues 711 and 1694 have been deleted, and which still preserves a
biologically active FVIII molecule.
to A range of B domain deletions can exist depending on which amino acid
residues in the B domain is deleted and whereby the biological activity of the
FVIII
molecule is still preserved. A specific B domain deletion called the SQN
exists which
is created by fusing Ser 743 to Gln 1638 (Lied et crl., 1995, Eur J. Biochem
323:19-27,
and PCT WO 91/09122) This deletes amino acid residues 744 to 1637 from the B
15 domain creating a Ser-Glu-Asn (SQN) link between the A2 and A3 FVIII
domains.
When compared to plasma-derived FVIII, the SQN deletion of the B domain of
FVIII
did not influence its in vivo pharmacokinetics (Fijnvandraat, et al., P.
R.Schattauer
Vertagsgesellschatt mbH (Stuttgart) 77:298-302, 1997). The terms "Factor VIII
SQN
deletion" or "SQN deletion" as used herein refer to this deletion and to other
deletions
2o which preserve the single S-Q-N tripeptide sequence and which result in the
deletion of
the amino acids between the two B-domain SQN sequences (See PCT WO 91/09122
for
a description of this amino acid sequence).
There are number of other B-domain deleted forms of factor VIII.
cDNA's encoding all of these B-domain deleted factor VIII proteins can be
inserted into
25 lentiviral vector particles by using standard molecular biology techniques.
For example
cDNA molecules encoding the following B-domain factor VIII deletions can be
constructed as described below:
CA 02296319 2000-O1-17
WO 99/0402b PCT/US98/14996
- 36
Eaton (1986) Biochemistry des 797-1562 deletion
25:8343
Toole (1986) PNAS 83:5939 des 760-1639 (LA-FVIII)
Meutien (1988) Prot Eng 2:301des 771-1666 (FVIII del II: missing
one
thrombin site)
Sarver (1987) DNA 6:553 des 747-1560
Mertens (1993) Br J Haematoldes 868-1562
85:133
des 713-1637 {thrombin resistant)
Esmon (1990) Blood 76:1593 des 797-1562
Donath (1995) Biochem J 312:49des 741-1668
Webb (1993) BBRC 190:536 PCR cloned from mRNA
Lind (1995) Eur J Biochem des 748-1648 (partially processed)
232:19
des 753-1648(partially processed)
dcs 777-1648(partially processed)
des 744-1637 (FV1II-SQ)
des 748-1645 (FVIII-RH)
des B-domain + 0, 1 ,2 Arg (partially
processed)
desB,+3Arg (FVIIIR4)
desB, +4Arg {FVIIIRS)
Langner (1988) Behring Inst des 741-1689
Mitt 16-25
des 816-l 598
Cheung (1996) Blood 88:325a des 746-1639
Pipes (1996) Blood 88:441a des 795-1688 (thrombin sites
mutated)
A B domain deletion in which an 1gG hinge region has been inserted can
also be used. For instance, a deletion of this type can be obtained from
plasmid PSVF8-
t~32, which was designed to link the heavy and light chains with a short hinge
region
from immunoglobulin A. To obtain cleavage at the end of the heavy chain and to
release the light chain, some residues of the (3 domain are included on either
side of the
hinge sequence. The 5' untranslated leader and signal peptide are from the
human
Factor VIII:C cDNA, with the Kozak consensus sequence at the initiation codon
as in
pSVF8-302. A description of this vector is included in Chapman et al., U.S.
Patent No.
5,595,886. The 3' untranslated region is the same fused Factor VIII and tPA
sequence
as found in pSVFB-80K.
The construction may be completed in two steps: an oligomer with
cohesive ends for EcoRI and BeII (117 bp) was cloned into a transfer vector,
pF8GM7,
the DNA sequence of the oligomer was checked by ml3 subcloning and Sanger
sequencing.
Next, the final plasmid was assembled by ligation of the following three
fragments
CA 02296319 2000-O1-17
WO 99/04026 PC'T/US98/14996
37
(a) FspI-EcoRI fragment form pSVFB-92S;
(b) EcoRI-NdeI fragment of the transfer vector pF8GM7 with
oligomer; and
(c) FspI-NdeI fragment of pSVFB-80K.
Descriptions of pSVFB-925 and pSVFB-80K are included in Chapman et
al., U.S. Patent No. 5,595,886.
Three additional B domain-deleted factor VIII constructs of particular
interest for inclusion in the lentiviral vector particles of the invention can
be prepared as
follows. Plasmid pSVFB-500 encodes a factor VIII protein with amino acids 770
to
1656 of the full length Factor VIII deleted. In addition the threonine at
position 1672 of
the full-length factor VIII sequence was also deleted. The following is a
description of
the construction of the vector.
The pSVF8-500 plasmid is a derivative of pSVFB-302 in which the
regions coding for the 92K and 80K domains are fused with a small connecting
(3-
region of 21 amino acids, retaining the natural proteolytic processing sites.
This
plasmid was constructed in the following manner:
(1) A SaII-KpnI fragment of 1984 by containing the region coding
for the 92K protein (except for the carboxyl terminal end) and BstXI-SaII
fragment of
2186 by containing the region coding for the carboxyl end of the 80K protein
with 3'
2o end untranslated region were isolated by gel electrophoresis after
digestion of pSVFB-
302 with restriction enzymes.
{2) A BcII-BstXI fragment of 1705 by containing most of the region
coding for the 80K protein was isolated after gel electrophoresis of the BamHI-
XbaI
fragment of pUC12F8. (pUCF812 is prepared from pF8-102 which is described in
U.S.
Patent No. 5,045,455. pF8-102 is digested with Bam-XhaI and ligated into
vector
pUCl2 by in vitro mutagenesis at a BcII site using the following primer: 5'
ACT ACT
CTT CAA TCT GAT CAA GAG GAA 3' (Seq ID No. ~.
(3) A KpnI-EcoRI fragment containing the carboxyl end of the 92K
protein and part of the (3 region (4 amino acids) was obtained by digestion of
the SaII
cassette from pSVF8-302 with KpnI and EcoRI.
(4) Ligation of four pieces of synthetic DNA (shown in Figure 39) to
the fragments of steps (2) and (3) and digestion with KpnI.
(S) Final ligation of fragments from steps ( 1 ) and (4); digestion with
SaII and gel purification of the 6428 by SaII cassette.
(6) Ligation of the SaII cassette into pSV7d vector; transformation of
HB101 and colony hybridization to isolate pSVF8-500 (Figure 40). The sequence
of
CA 02296319 2000-O1-17
WO 99/04026 PCT/US98/14996
38
the junction region coding for 92K-(3-80K was verified by DNA sequence after
cloning
in M13.
The sequence was changed to incorporate unique NruI and MIuI
restriction sites without changing the amino acid sequence. These sites were
alsoused
to construct other two additional B-domain deleted vectors which are described
below.
pSV500BOThr was constructed from pSVFB-500. The threonine
deletion at position 16?2 was maintained. A synthetic linker was used to
construct
pSV500B~Thr. The linker extends from a unique NruI site at Ser(765) to a
unique
MIuI site at Ile(1659) in the pSVF8-500 vector. This linker was substituted
for the
corresponding region of pSVFB-500.
A third vector pSVFB-500B was constructed from pSV500BOThr. This
vector is identical to pSVFB-500B except that the codon for threonine 1672 was
re-
inserted using standard mutagenesis methods. The relationship between, pSVF8-
500B,
pSVFB-500B, is further illustrated in the table below. Amino acid sequence
numbers in
the table were determined by reference to full-length factor VIII sequence.
Name Amino Acids Deleted Thr at 1672 Deletion
pSVFB-500 770 to 1656 Yes
pSVFB-500B~Thr 779 to 1658 Yes
pSVF80-500B 779 to 1658 No
In all cases, the BgIII-PflI 1.35 kb fragments of each modified cDNA
listed above can be inserted into the lentiviral vector particles described
herein using
standard molecular biology procedures known to those of skill in the art and
described
herein.
The full-length factor VIII cDNA can also be inserted into the lentiviral
vector particles of the invention (see, e.g., WO 96/21035). A variety of
Factor VIII
deletions, mutations, and polypeptide analogs of Factor VIII can also be
introduced into
the lentiviral vector particles of the invention including lentiviral vector
particles by
modifications of the procedures described herein. These analogs include, for
instance,
those described in PCT Patent Publication Nos. WO 97/03193, WO 97/03194, WO
97/03195, and WO 97/03191, all of which are hereby incorporated by reference.
Hemophilia B can also be treated with systemically administered factor
IX-expressing lentiviral vector particles including lentiviral vector
particles. Human
factor IX deficiency (Christmas disease or Hemophilia B) affects primarily
males
because it is transmitted as sex-linked recessive trait. It affects about 2000
people in the
US. The human factor gene codes a 416 amino acids of mature protein.
CA 02296319 2000-O1-17
WO 99/04026 PCT/US98/14996
- 39
The human factor IX eDNA can be obtained for instance by constructing
plasmid pHfIXl, as described by Kurachi and Davie, 1982, PNAS 79(21):6461-
6464.
The cDNA sequence can be excised as a PstI fragment of about 1.5 kb, blunt
ended
using T4 DNA polymerase. The factor cDNA fragment can be readily inserted, for
example into a SrfI site introduced into a lentiviral vector particle.
c. Lentiviral vectorarticles expressing other clotting factors
i. Factor V.
Lentiviral vector particles can be constructed using molecular biology
techniques known to those of skill in the art. For instance, Factor V cDNA is
obtained
l0 from pMT2-V (Jenny, 1987, Proc. Natl. Acad. Sci. USA 84:4846; ATCC deposit
#40515) by digestion with SaII. The 7 kb cDNA band is excised from agarose
gels and
cloned into lentiviral vector particles, using standard molecular biology
techniques.
Either a full-length or a B-domain deletion or substitution of the factor
V cDNA can be expressed by the gene therapy vectors of the invention. Factor V
B
domain deletions such as those reported by Marquette, 1995, Blood 86:3026, and
Kane,
1990, Biochemistry 29:6762, can be made as described by these authors.
ii. Antithrombin III
Lentiviral vector particles capable of expressing ATIII cDNA can be
2o readily constructed using standard molecular biology techniques known to
those of skill
in the art. For instance a lentiviral vector particle expressing AT III can be
constructed
from the vector pKT218 (Prochownik, 1983, J. Biol. Chem. 258:8389; ATCC number
57224/57225) by excision with PstI. The 1.6 kb cDNA insert can be recovered
from
agarose gels and cloned into the PstI site of vector SK-. The insert can be
recovered by
restriction enzyme digestion and cloned into lentiviral veclor particles
described herein
by the restriction enzymes.
iii. Pr '
The lentiviral vector particles of the invention capable of expressing
Protein C can be made using a wide variety of techniques given the present
disclosure.
For instance, protein C cDNA will be obtained by restriction enzyme digestion
of
published vector (Foster, 1984, Proc. Natl. Acad. Sci. USA 81:4766; Beckmann,
1985,
Nucleic Acids Res 13:5233). The 1.6 kb cDNA insert can be recovered from
agarose
gels and cloned into the multiple cloning site of vector SK- under standard
conditions.
The insert can be recovered by restriction enzyme digestion and cloned into a
Ientiviral
CA 02296319 2000-O1-17
WO 99/04026 PCT/US98/14996
vector particle; for example, excision by XhoI/NotI digestion followed by
cloning into
XhoI/NotI digested lentiviral vector particle.
iv. Prothrombin
5 Lentiviral vector particles expressing prothrombin and its variants can be
constructed by methods known to those of skill in the art, by using variations
on the
methods described herein. For instance, prothrombin cDNA can be obtained by
restriction enzyme digestion of a published vector (Degen (1983) Biochemistry
22:2087). The 1.9 kb cDNA insert can be recovered from agarose gels and cloned
into
10 the multiple cloning site of vector SK-. The insert can be recovered by
restriction
enzyme digestion and cloned into a lentiviral vector panicle using restriction
enzyme
digestion
v. Thrombomodulin
15 Lentiviral vector particles expressing thrombomodulin and its variants
can be constructed using techniques Imown to those of skill in the art. For
instance,
thrombomodulin cDNA can be obtained from the vector puc I 9TM 15 (Jackman,
1987,
Proc. Natl. Acad. Sci. USA 84:6425; Shirai, 1988, J. f3iochem. 103:281; Wen ,
I 987,
Biochemistfy 26:4350; Suzuki, 1987, EMBO J 6:1891; ATCC number 61348,61349) by
20 excision with SaII. The 3.7 kb cDNA insert can be recovered from agarose
gels and
cloned into the SaII site of lentiviral vector particle.
d. Lentiviral vector particles treatment of hereditary disorders and other
conditions
There are a number of proteins useful for treatment of hereditary
25 disorders that can be expressed in vivo by the methods of invention. Many
genetic
diseases caused by inheritance of defective genes result in the failure to
produce normal
gene products, for example, thalassemia, phenylketonuria, Lesch-Nyhan
syndrome,
severe combined immunodeficiency (SCID), hemophilia, A and B, cystic fibrosis,
Duchenne's Muscular Dystrophy, inherited emphysema and familial
30 hypercholesterolemia (Mulligan et al., 1993, Science 260:926; Anderson et
al., 1992,
Science 256:808; Friedman et al., 1989, Science 244:1275). Although genetic
diseases
may result in the absence of a gene product, endocrine disorders, such as
diabetes and
hypopituitarism, are caused by the inability of the gene to produce adequate
levels of
the appropriate hormone insulin and human growth hormone respectively.
CA 02296319 2000-O1-17
WO 99/04026 PCT/US98/14996
41
Gene therapy by the methods of the invention is a powerful approach for
treating these types of disorders. This therapy involves the introduction of
normal
recombinant genes into somatic cells so that new or missing proteins are
produced
inside the cells of a patient. A number of genetic diseases can be treated by
gene
therapy, including adenine deaminase deficiency, cystic fibrosis, oc,-
antitrypsin
deficiency, Gaucher's syndrome, as well as non-genetic diseases. Other
representative
diseases include lactase for treatment of hereditary lactose intolerance, AD
for treatment
of ADA deficiency, and alpha-1 antitypsin for treatment of alpha-1 antitrypsin
deficiency. See F.D. Ledley, 1987, J. Pediatrics 110:157-174; I. Verma,
Scientific
American (Nov., 1987) pp. 68-84; and PCT Patent Publication WO 95/27512
entitled
"Gene Therapy Treatment for a Variety of Diseases and Disorders" for a
description of
gene therapy treatment of genetic diseases.
One such disorder is familial hypercholesterolemia is a disease
characterized clinically by a lifelong elevation of low density lipoprotein
(LDL), the
major cholesterol-transport lipoprotein in human plasma; Pathologically by the
deposition of LDL-derived cholesterol in tendons, skin and arteries leading to
premature
coronary heart disease; and genetically by autosomal dominant inherited trait.
Hetrozygotes number about 1 in 500 persons worldwide. Their cells are able to
bind
cholesterol at about half the rate of normal cells. Their plasma cholesterol
levels show
two fold elevation starting at birth. Homozygotes number 1 in 1 million
persons. They
have severe cholesterolemia with death occurring usually before age 20. The
disease
(Arteriosclerosis) depends on geography. It affects 15.5 per 100,000
individuals in the
U.S. (20,000 total) and 3.3 per 100,000 individuals in Japan. Lentiviral
vector particles
expressing the LDL receptor for treatment of disorders manifesting with
elevated serum
LDL can be constructed by techniques known to those of skill in the art. An
example of
a lentiviral vector particle expressing LDS receptor is shown in example 32
herein.
There are a variety of other proteins of therapeutic interest that can be
expressed in vivo by lentiviral vector particles using the methods of the
invention. For
instance sustained in vivo expression of tissue factor inhibitory protein
(TFPI) is useful
3o for treatment of conditions including sepsis and DIC and in preventing
reperfusion
injury. (See PCT Patent Publications Nos. WO 93/24143 ,WO 93/25230 and WO
96/06637. Nucleic acid sequences encoding various forms of TFPI can be
obtained, for
example, as described in US Patent Nos. 4,966,852; 5,106,833; and 5,466,783,
and can
be incorporated in lentiviral vector particles as is described herein.
Other proteins of therapeutic interest such as erythropoietin (EPO) and
leptin can also be expressed in vivo by lentiviral vector particles according
to the
CA 02296319 2000-O1-17
WO 99/04026 PCT/US98/14996
42
methods of the invention. For instance EPO is useful in gene therapy treatment
of a
variety of disorders including anemia (see PCT publication number WO 95/13376
entitled "Gene Therapy for Treatment of Anemia".) Sustained gene therapy
delivery of
Ieptin by the methods of the invention is useful in treatment of obesity. (See
WO
96/05309 entitled "Obesity Polypeptides able to modulate body weight" for a
description of the leptin gene and its use in the treatment of obesity.
Lentiviral vector
particle expressing EPO or leptin can readily be produced using the methods
described
herein and the constructs described in these two patent publications.
A variety of other disorders can also be treated by the methods of the
invention. For example, sustained in vivo systemic production of
apolipoprotein E or
apolipoprotein A by the lentiviral vector particles of the invention can be
used for
treatment of hyperlipidemia. (See Breslow, J. et al. Biotechnology 12, 365
(1994).) In
addition, sustained production of angiotensin receptor inhibitor (T.L.
Goodfriend, et al.,
1996, N. Engl. J. Med. 334:1469) can effected by the gene therapy methods
described
herein. As yet an additional example, the long terns ire vivo systemic
production of
angiostatin by the lentiviral vector particles of the invention is useful in
the treatment of
a variety of tumors. (See O'Reilly et al., 1996, Nuture Med. ?:689.
7. L ly~r phokines and L,~mphokine Receptors
As noted above, the present invention also provides lentiviral vector
2o particles which can, among other functions, direct the expression of one or
more
cytokines or cytokine receptors. Briefly, in addition to their role as cancer
therapeutics,
cytokines can have negative effects resulting in certain pathologicai
conditions. For
example, most resting T-cells, B cells, large granular lymphocytes and
monocytes do
not express IL-2R (receptor). In contrast to the lack of IL-2R expression on
normal
resting cells, IL-2R is expressed by abnormal cells in patients with certain
leukemias
(ATL, Hairy-cell, Hodgkins, acute and chronic granulocytic), autoimmune
diseases, and
is associated with allograft rejection. Interestingly, in most of these
patients the serum
concentration of a soluble form of IL-2R is elevated. Therefore, with certain
embodiments of the invention therapy may be effected by increasing the serum
3o concentration of the soluble form of the cytokine receptor. For example, in
the case of
IL-2R, a lentiviral vector can be engineered to produce both soluble IL-2R and
IL-2R,
creating a high affinity soluble receptor. In this configuration, serum IL-2
levels would
decrease, inhibiting the paracrine loop. This same strategy also may be
effective against
autoimmune diseases. In particular, because some autoimmune diseases (e.g.,
Rheumatoid arthritis, SLE) also are associated with abnormal expression of IL-
2,
CA 02296319 2000-O1-17
WO 99/04026 PCT/US98/14996
43
blocking the action of IL-2 by increasing the serum level of receptor may also
be
utilized in order to treat such autoimmune diseases.
In other cases inhibiting the levels of IL-1 may be beneficial. Briefly,
IL-1 consists of two polypeptides, IL-1 and IL-1, each of which has
plieotropic effects.
IL-1 is primarily synthesized by mononuclear phagocytes, in response to
stimulation by
microbial products or inflammation. There is a naturally occurring antagonist
of the
IL-1R, referred to as the IL-1 Receptor antagonist ("IL-1Ra"). This IL-IR
antagonist
has the same molecular size as mature IL-1 and is structurally related to it.
However,
binding of IL-1Ra to the IL-1R does not initiate any receptor signaling. Thus,
this
1o molecule has a different mechanism of action than a soluble receptor, which
complexes
with the cytokine and thus prevents interaction with the receptor. IL-I does
not seem to
play an important role in normal homeostasis. In animals, antibodies to IL-1
receptors
reduce inflammation and anorexia due to endotoxins and other inflammation
inducing
agents.
In the case of septic shock, IL-1 induces secondary compounds which
are potent vasodilators. In animals, exogenously supplied IL-1 decreases mean
arterial
pressure and induces leukopenia. Neutralizing antibody to IL-1 reduced
endotoxin-
induced fever in animals. In a study of patients with septic shock who were
treated with
a constant infusion of IL-1R for three days, the 28 day mortality was 16%
compared to
44% in patients who received placebo infusions. In the case of autoimmune
disease,
reducing the activity of IL-1 reduces inflammation. Similarly, blocking the
activity of
IL-I with recombinant receptors can result in increased allograft survival in
animals,
again presumably by decreasing inflammation.
These diseases provide further examples where lentiviral vector particles
may be engineered to produce a soluble receptor or more specifically the IL-
1Ra
molecule. For example, in patients undergoing septic shock, a single injection
of IL-
1Ra producing vector particles could replace the current approach requiring a
constant
infusion of recombinant IL-1 R.
Cytokine responses, or more specifically, incorrect cytokine responses
3o may also be involved in the failure to control or resolve infectious
diseases. Perhaps the
best studied example is non-healing forms of leishmaniasis in mice and humans
which
have strong, but counterproductive Tf,2-dominated responses. Similarly,
lepromotomatous leprosy is associated with a dominant, but inappropriate TH2
response.
In these conditions, lentiviral vector particles may be useful for increasing
circulating
levels of IFN gamma, as opposed to the site-directed approach proposed for
solid tumor
therapy. IFN gamma is produced by T,~-I T-cells, and functions as a negative
regulator
CA 02296319 2000-O1-17
WO 99/04026 PCT/US98/14996
44
of TH-2 subtype proliferation. IFN gamma also antagonizes many of the IL-4
mediated
effects on B-cells, including isotype switching to IgE.
IgE, mast cells and eosinophils are involved in mediating allergic
reaction. IL-4 acts on differentiating T-cells to stimulate TH-2 development,
while
inhibiting TE,-1 responses. Thus, lentivirus-based gene therapy may also be
accomplished in conjunction with traditional allergy therapeutics. One
possibility is to
deliver lentiviral vector particles which produces IL4R with small amounts of
the
offending allergen (i.e., traditional allergy shots). Soluble IL-4R would
prevent the
activity of IL-4, and thus prevent the induction of a strong T,,-2 response.
a. Lentiviral vector particles for treatment of viral hepatitis
The lentiviral vector particles including lentiviral vector particles and the
methods of administration described are useful for treatment of viral
hepatitis, including
hepatitis B and hepatitis C. For instance, the lentiviral vector particles of
the invention
can be used to express interferon-alpha for treatment of viral hepatitis.
While not
wishing to be bound by theory, lentiviral vector particles injected
intravenously
preferentially transduce liver cells. Thus, the methods of intravenous
delivery described
herein for lentiviral vector particles can be used for treatment of liver
diseases such as
hepatitis and in particular viral hepatitis, in which therapeutic proteins
expressed by the
lentiviral vector particles such as lentiviral vector particles can be
delivered
2o preferentially to the liver.
Currently, the only approved treatment for chronic hepatitis B, C and D
infections is the use of alpha interferon 2a and 2b. Alpha-interferon is a
secreted protein
induced in B lymphocytes, macrophages and null lymphocytes by foreign cells,
virus-
infected cells, tumor cells, bacterial cells and products and viral envelopes.
The
mechanism of antiviral action of interferon is by inducing the synthesis of
effector
proteins: two of the most important are 2', 5'-oligo-adenylate synthetase
(OAS) and
dsRNA-dependent protein kinase (RDPK). OAS synthesizes adenylate oligomers
that
activate RNAaseL, which degrades viral single stranded RNA. RDPK
phosphorylates
initiation factor eIF-2a which results in the inhibition of viral protein
translation. In
3o addition to the direct antiviral effect, alpha interferon has
immunomodulatory effects
that are important against viral infections. These immunomodulatory effects
are:
enhancement of the expression of both Class 1 and class II major
histocompatibility
complex (MHC) molecules, modulation of the expression of the interleukin-2
receptor,
TNF-a receptor, transferrin receptor, enhancement of spontaneous natural
killer (NK)
cell cytotoxicity and modulation of antibody production by B cells. In chronic
hepatitis
CA 02296319 2000-O1-17
WO 99!04026 PCT/US98/14996
B infection, the beneficial effect of interferon alpha appears to be from the
immunomodulatory effects, while in chronic hepatitis C infection, the
beneficial effect
is dependent on its antiviral activity. (Bresters, D., in Hepatitis C Virus,
pp121-136,
Reesink HW (ed), 1994). The mechanism of action in interferon alpha for
treatment of
5 chronic hepatitis D is poorly understood (Rizzetto, M. and Rosina, F. in
Viral
Hepatitis, pp. 363-369, Zuckerman, A. J. and Thomas H. C. (ed), 1993).
Localized expression of interferon alpha in the liver from a lentiviral
vector particle such as a lentivirai vector particle can be an effective
treatment for
hepatitis. While not wishing to bound by theory, delivery of alpha interferon
at the site
1o of infection by the gene therapy vectors of the invention, including
lentiviral vector
particles, results in high local concentration of the cytokine thereby
focusing the
antiviral and immunological effects to the adjacent infected hepatocytes. A
further
advantage of this treatment is that the current systemic mode of systemic
alpha
interferon therapy may either be unnecessary or be reduced in dose and
frequency of
15 treatment. This reduction can reduce the adverse side effects associated
with the
systemic delivery of alpha interferon. Thus, the gene therapy approaches
described
herein may be used in combination with administration of alpha-interferon
protein
formulations.
The construction of a number of different lentiviral vector particles
2o expressing interferon-alpha can be readily accomplished given the
disclosure provided
herein. There are at least 24 different human alpha interferon genes or
pseudogenes.
There are two distinct families (I and II); mature human alpha interferon (I)
are 166
amino acids long (one is 165 amino acids) whereas alpha interferon (II) have
172 amino
acids. Eighteen genes are in the alpha interferon I family, including at least
four
25 pseudogenes. Six genes are in the alpha interferon II family, including
five
pseudogenes {Callard, R., and Gearing, A., Cytokine Fucts Book, Academic
Press, 1994
pp. 148-154). In Example 33 herein, we use alpha interferon 2a, 2b, 2c, 54 and
76, all
members of the alpha interferon (I) family. Similar techniques can be used for
inserting
other members of the alpha interferon I family (such as alpha interferon F and
N) into
3o lentiviral vector particles. Thus other biologically active forms of alpha-
interferon in
addition to 2a, 2b, 2c, 54 and 76 as described herein can also be expressed by
the
lentiviral vector particles of the invention and used for treatment of viral
hepatitis.
Patients with viral hepatitis can be treated a combination gene therapy
approach. A lentiviral vector particle expressing a protein drug such as alpha-
interferon
35 can be administered intravenously or directly to the liver by methods
described herein.
This therapeutic approach can be combined with intramuscular delivery of a
lentiviral
CA 02296319 2000-O1-17
WO, 99104026 PCT/US98/14996
46
vector particle expressing a hepatitis B or hepatitis C antigen for inducing a
immune
response against the hepatitis virus. Specific hepatitis B and C antigens
useful in this
type of therapy and the construction of lentiviral vector particles expressing
such
antigens are described herein and in PCT Patent Publication No. WO 93/15207.
In
addition, molecularly cloned genomes which encode the hepatitis B virus may be
obtained from a variety of sources including, for example, the American Type
Culture
Collection (ATCC, Rockville, Maryland). For example, ATCC No. 45020 contains
the
total genomic DNA of hepatitis B (extracted from purified Dane particles) (see
Figure 3
of Blum et al., 1989, TIG 5(5):154-158) in the Bam HI site of pBR322 (Moriarty
et al.,
l0 1981, Pf-oc. Natl. Acad. Sci. USA 78:2606-2610). (Note that correctable
errors occur in
the sequence of ATCC No. 45020.)
8. Suicide Vectors
One further aspect of the present invention relates to the use of lentiviral
vector suicide vectors to limit the spread of wild-type lentivirus in the
packaginglproducer cell Lines. For example, within one embodiment the
lentiviral
vector particles contains a prodrug activating enzyme as discussed above
which, upon
administration of the prodrug (e.g., gancyclovir) results in the death of
cells containing
the vector particles.
9. Lentiviral vectors to Prevent the pread of Metastatic Tumors
One further aspect of the present invention relates to the use of lentiviral
vector particles for inhibiting or reducing the invasiveness of malignant
neoplasms.
Briefly, the extent of malignancy typically relates to vascularization of the
tumor. One
cause for tumor vascularization is the production of soluble tumor
angiogenesis factors
(TAF) (Paweletz et al., Crit. Rev. Oncol. Hematol. 9:197, 1989) expressed by
some
tumors. Within one aspect of the present invention, tumor vascularization may
be
slowed utilizing lentiviral vectors to express antisense or ribozyme RNA
molecules
specific for TAF. Alternatively, anti-angiogenesis factors (Moses et al.,
Science
248:1408, 1990; Shapiro et al., PNAS 84:2238, 1987) may be expressed either
alone or
in combination with the above-described ribozymes or antisense sequences in
order to
slow or inhibit tumor vascularization. Alternatively, lentiviral vector
particles can also
be used to express an antibody specific for the TAF receptors on surrounding
tissues.
CA 02296319 2000-O1-17
WO 99/04026 PCTIUS98/14996
47
10. Modulation of TranscrirJtion Factor ActivitX
In yet another embodiment, lentiviral vector particles may be utilized in
order to regulate the growth control activity of transcription factors in the
infected cell.
Briefly, transcription factors directly influence the pattern of gene
expression through
sequence-specific traps-activation or repression (Karin, New Biologist 21:126-
131,
1990). Thus, it is not surprising that mutated transcription factors represent
a family of
oncogenes. Lentiviral vector particles can be used, for example, to return
control to
tumor cells whose unregulated growth is activated by oncogenic transcription
factors,
and proteins which promote or inhibit the binding cooperatively in the
formation of
1o homo- and heterodimer traps-activating or repressing transcription factor
complexes.
One method for reversing cell proliferation would be to inhibit the
traps-activating potential of the c-rrtvclMax heterodimer transcription factor
complex.
Briefly, the nuclear oncogene c-myc is expressed by proliferating cells and
can be
activated by several distinct mechanisms, including retroviral insertion,
amplification,
and chromosomal translocation. The Max protein is expressed in quiescent cells
and,
independently of c-myc, either alone or in conjunction with an unidentified
factor,
functions to repress expression of the same genes activated by the myc/Max
heterodimer (Cole, Cell 65:715-716, 1991 ).
Inhibition of c-myc or c-myc/Max proliferation of tumor cells may be
2o accomplished by the overexpression of Max in target cells controlled by
lentiviral
vectors. The Max protein is only 160 amino acids (corresponding to 480
nucleotide
RNA length) and is easily incorporated into a lentiviral vector either
independently, or
in combination with other genes and/or antisense/ribozyme moieties targeted to
factors
which release growth control of the cell.
Modulation of homo/hetero-complex association is another approach to
control transcription factor activated gene expression. For example, transport
from the
cytoplasm to the nucleus of the traps-activating transcription factor NF-B is
prevented
while in a heterodimer complex with the inhibitor protein IB. Upon induction
by a
variety of agents, including certain cytokines, IB becomes phosphorylated and
NF-B is
3o released and transported to the nucleus, where it can exert its sequence-
specific
traps-activating function (Baeuerle and Baltimore, Science 242:540-546, 1988).
The
dissociation of the NF-B/IB complex can be prevented by masking with an
antibody the
phosphorylation site of IB. This approach would effectively inhibit the traps-
activation
activity of the NF-IB transcription factor by preventing its transport to the
nucleus.
Expression of the IB phosphorylation site specific antibody or protein in
target cells
may be accomplished with an lentivirus gene transfer vector. An approach
similar to
CA 02296319 2000-O1-17
WO 99/04026 PCTNS98/14996
48
the one described here could be used to prevent the formation of the traps-
activating
transcription heterodimer factor AP-1 (Turner and Tijan, Science 243:1689-
1694,
1989), by inhibiting the association between the jun and fos proteins.
FORMiILATIOI~i
Within other aspects of the present invention, methods are provided for
preserving an infectious lentiviral vector particle, such that the lentiviral
vector particle
is capable of infecting mammalian cells upon reconstitution (see U.S. Serial
No.08/153,342). Briefly, lentiviral vector particle which has been purified or
concentrated may be preserved or formulated into a pharmaceutical compound or
1o medicament by first adding a sufficient amount of a formulation buffer to
the media
containing the lentiviral vector particle, in order to form an aqueous
suspension. The
formulation buffer is an aqueous solution that contains a saccharide, a high
molecular
weight structural additive, and a buffering component in water. As utilized
within the
context of the present invention, a "buffering compound" or "buffering
component"
should be understood to refer to a substance that functions to maintain the
aqueous
suspension at a desired pH. The aqueous solution may also contain one or more
amino
acids.
The lentiviraI vector particle can also be preserved in a purified form.
More specifically, prior to the addition of the formulation buffer, the crude
lentiviral
2o vector particle described above may be clarified by passing it through a
filter, and then
concentrated, such as by a cross flow concentrating system (Filtron Technology
Corp.,
Nortborough, MA). Within one embodiment, DNase is added to the concentrate to
digest exogenous DNA. The digest is then dia(iltrated to remove excess media
components and establish the lentiviral vector particle in a more desirable
buffered
solution. The diafiltrate is then passed over a Senhadex S-500 gel column and
a
purified lentiviral vector particle is eluted. A sufficient amount of
formulation buffer is
added to this eluate to reach a desired final concentration of the
constituents and to
minimally dilute the lentiviral vector particle, and the aqueous suspension is
then
stored, preferably at -70°C or immediately dried. As noted above, the
formulation
buffer is an aqueous solution that contains a saccharide, a high molecular
weight
structural additive, and a buffering component in water. The aqueous solution
may also
contain one or more amino acids.
The crude lentiviral vector particle can also be purified by ion exchange
column chromatography (see U.S. Patent Application Serial No.08/093,436). In
general, the crude lentiviral vector particle is clarified by passing it
through a filter, and
CA 02296319 2000-O1-17
WO 99/04026 PCT/US98/14996
49
the filtrate loaded onto a column containing a highly sulfonated cellulose
matrix. The
lentiviral vector particle is eluted from the column in purified form by using
a high salt
buffer. The high salt buffer is then exchanged for a more desirable buffer by
passing
the eluate over a molecular exclusion column. A sufficient amount of
formulation
buffer is then added, as discussed above, to the purified lentiviral vector
particle and the
aqueous suspension is either dried immediately or stored, preferably at -
70°C.
The aqueous suspension in crude or purified form can be dried by
lyophilization or evaporation at ambient temperature. Specifically,
lyophilization
involves the steps of cooling the aqueous suspension below the glass
transition
to temperature or below the eutectic point temperature of the aqueous
suspension, and
removing water from the cooled suspension by sublimation to form a lyophilized
lentivirus. Briefly, aliquots of the formulated lentiviral vector particle are
placed into
an Edwards Refrigerated Chamber (3 shelf RC3S unit) attached to a freeze dryer
(Supermodulyo 12K). A multistep freeze drying procedure as described by
Phillips
et al. (Cryobiology 18:414, 1981) is used to lyophilize the formulated
lentiviral vector
particle, preferably from a temperature of -40°C to -45°C. The
resulting composition
contains less than 10% water by weight of the lyophilized lentivirus. Once
lyophilized,
the lentiviral vector particle is stable and may be stored at -20°C to
25°C.
Within the evaporative method, water is removed from the aqueous
2o suspension at ambient temperature by evaporation. Within one embodiment,
water is
removed through spray drying (EP 520,748). Within the spray drying process,
the
aqueous suspension is delivered into a flow of preheated gas, usually air,
whereupon
water rapidly evaporates from droplets of the suspension. Spray drying
apparatus are
available from a number of manufacturers (e.g., Drytec, Ltd., Tonbridge,
England; Lab
Plant, Ltd., Huddersfield, England). Once dehydrated, the lentiviral vector
particle is
stable and may be stored at -20°C to 25°C. Within the methods
described herein, the
resulting moisture content of the dried or lyophilized lentivirus may be
determined
through use of a Karl-Fischer apparatus (EM Science AquastarTM V1B volumetric
titrator, Cherry Hill, NJ), or through a gravimetric method.
The aqueous solutions uscd for formulation, as previously described, are
composed of a saccharide, high molecular weight structural additive, a
buffering
component, and water. The solution may also include one or more amino acids.
The
combination of these components act to preserve the activity of the lentiviral
vector
particle upon freezing and lyophilization, or drying through evaporation.
Although a
preferred saccharide is lactose, other saccharides may be used, such as
sucrose,
mannitol, glucose, trehalose, inositol, fructose, maltose or galactose. In
addition,
CA 02296319 2000-O1-17
WO 99/04026 PCT/US98/14996
combinations of saccharides can be used, for example, lactose and mannitol, or
sucrose
and mannitol (e.g., a concentration of lactose is 3%-4% by weight. Preferably,
the
concentration of the saccharide ranges from 1 % to 12% by weight.
The high molecular weight structural additive aids in preventing viral
5 aggregation during freezing and provides structural support in the
lyophilized or dried
state. Within the context of the present invention, structural additives are
considered to
be of "high molecular weight" if they are greater than 5000 m.w. A preferred
high
molecular weight structural additive is human serum albumin. However, other
substances may also be used, such as hydroxyethyl-cellulose, hydroxynlethyl-
cellulose,
10 dextran, cellulose, gelatin, or povidone. A particularly preferred
concentration of
human serum albumin is 0.1 % by weight. Preferably, the concentration of the
high
molecular weight structural additive ranges from 0.1 % to 10% by weight.
The amino acids, if present, function to further preserve viral infectivity
upon cooling and thawing of the aqueous suspension. In addition, amino acids
function
15 to further preserve viral infectivity during sublimation of the cooled
aqueous suspension
and while in the lyophilized state. A preferred amino acid is arginine, but
other amino
acids such as lysine, ornithine, serine, glycine, glutamine, asparagine,
glutamic acid or
aspartic acid can also be used. A particularly preferred arginine
concentration is 0.1 %
by weight. Preferably, the amino acid concentration ranges from 0.1% to 10% by
2o weight.
The buffering component acts to buffer the solution by maintaining a
relatively constant pH. A variety of buffers may be used, depending on the pH
range
desired, preferably between 7.0 and 7.8. Suitable buffers include phosphate
buffer and
citrate buffer. A particularly preferred pH of the lentiviral vector particle
formulation is
25 7.4, and a preferred buffer is tromethamine.
In addition, it is preferable that the aqueous solution contain a neutral
salt which is used to adjust the final formulated lentiviral vector particle
to an
appropriate iso-osmotic salt concentration. Suitable neutral salts include
sodium
chloride, potassium chloride or magnesium chloride. A preferred salt is sodium
3o chloride.
Aqueous solutions containing the desired concentration of the
components described above may be prepared as concentrated stock solutions.
One method of preserving lentiviral vector particles in a lyophilized state
for subsequent reconstitution comprises the steps of (a) combining an
infectious
35 lentiviral vector particle with an aqueous solution to form an aqueous
suspension, the
aqueous suspension including 4% by weight of lactose, 0.1 % by weight of human
CA 02296319 2000-O1-17
WO 99104026 PCT/US98/14996
51
serum albumin, 0.03% or less by weight of NaCI, 0.1% by weight of arginine,
and an
amount of tromethamine buffer effective to provide a pH of the aqueous
suspension of
approximately 7.4, thereby stabilizing the infectious lentiviral vector
particle;
(b) cooling the suspension to a temperature of from -40°C to -
45°C to form a frozen
suspension; and (c) removing water from the frozen suspension by sublimation
to form
a lyophilized composition having less than 2% water by weight of the
lyophilized
composition, the composition being capable of infecting mammalian cells upon
reconstitution. It is preferred that the lentiviral vector particle be
replication defective
and suitable for administration into humans upon reconstitution.
l0 It will be evident to those skilled in the art given the disclosure
provided
herein that it may be preferable to utilize certain saccharides within the
aqueous
solution when the lyophilized lentivirus is intended for storage at room
temperature.
More specifically, it is preferable to utilize disaccharides, such as lactose
or trehalose,
particularly for storage at room temperature.
The lyophilized or dehydrated Lentiviruses of the subject invention may
be reconstituted using a variety of substances, but are preferably
reconstituted using
water. In certain instances, dilute salt solutions which bring the final
formulation to
isotonicity may also be used. In addition, it may be advantageous to use
aqueous
solutions containing components known to enhance the activity of the
reconstituted
lentivirus. Such components include cytokines, such as IL-2, polycations, such
as
protamine sulfate, or other components which enhance the transduction
efficiency of the
reconstituted lentivirus. Lyophilized or dehydrated lentiviral vector particle
may be
reconstituted with any convenient volume of water or the reconstituting agents
noted
above that allow substantial, and preferably total solubilization of the
lyophilized or
dehydrated sample.
ADMINISTRATION
As noted above, recombinant lentiviral particles of the present invention
may be administered to a wide variety of locations including, for example,
into sites
such as the cerebral spinal fluid, bone marrow, joints, arterial endothelial
cells, rectum,
3o buccalJsublingual, vagina, the lymph system, to an organ selected from the
group
consisting of lung, liver, spleen, skin, blood and brain, or to a site
selected from the
group consisting of tumors and interstitial spaces. Within other related
embodiments,
the lentiviral vector particle may be administered intrarticularly,
intraocularly,
intranasally, sublinually, orally, topically, intravesically, intrathecally,
topically,
intravenously, intraperitoneally, intracranially, intramuscularly, or
subcutaneously.
CA 02296319 2000-O1-17
W0.99/04026 PCT/US98/14996
52
Other representative routes of administration include gastroscopy, ECRP and
colonoscopy, which do not require full operating procedures and
hospitalization, but
may require the presence of medical personnel.
Considerations for administering the compositions of the present
invention include the following:
Oral administration is easy and convenient, economical (no sterility
required), safe (over dosage can be treated in most cases), and permits
controlled release
of the active ingredient of the composition (the lentiviral vector particle).
Conversely,
there may be local irntation such as nausea, vomiting or diarrhea, erratic
absorption for
1o poorly soluble drugs, and the lentiviral vector particle will be subject to
"first pass
effect" by hepatic metabolism and gastric acid and enzymatic degradation.
Further,
there can be slow onset of action, efficient plasma levels may not be reached,
a patient's
cooperation is required, and food can affect absorption. Preferred embodiments
of the
present invention include the oral administration of lentiviral vector
particles that
express genes encoding erythropoietin, insulin, GM-CSF cytokines, various
polypeptides or peptide hormones, their agonists or antagonists, where these
hormones
can be derived from tissues such as the pituitary, hypothalamus, kidney,
endothelial
cells, liver, pancreas, bone, hemopoetic marrow, and adrenal. Such
polypeptides can be
used for induction of growth, regression of tissue, suppression of immune
responses,
2o apoptosis, gene expression, blocking receptor-ligand interaction, immune
responses and
can be treatment for certain anemias, diabetes, infections, high blood
pressure, abnormal
blood chemistry or chemistries (e.g., elevated blood cholesterol, deficiency
of blood
clotting factors, elevated LDL with lowered HDL), levels of Alzheimer
associated
amaloid protein, bone erosion/calcium deposition, and controlling levels of
various
metabolites such as steroid hormones, purines, and pyrimidines. Preferably,
the
lentiviral vector particles are first lyophilized, then filled into capsules
and
administered.
Buccal/sublingual administration is a convenient method of
administration that provides rapid onset of action of the active components)
of the
3o composition, and avoids first pass metabolism. Thus, there is no gastric
acid or
enzymatic degradation, and the absorption of lentiviral vector particles is
feasible.
There is high bioavailability, and virtually immediate cessation of treatment
is possible.
Conversely, such administration is limited to relatively low dosages
(typically about 10-
I S mg), and there can be no simultaneous eating, drinking or swallowing.
Preferred
embodiments of the present invention include the buccal/sublingual
administration of
lentiviral vector particles that contain genes encoding self and/or foreign
MHC, or
CA 02296319 2000-O1-17
WO 99/04026 PCT/US98/14996
53
immune modulators, for the treatment of oral cancer; the treatment of
Sjogren's
syndrome via the buccal/sublingual administration of such lentiviral vector
particles
that contain IgA or IgE antisense genes; and, the treatment of gingivitis and
periodontitis via the buccal/sublingual administration of IgG or cytokine
antisense
genes.
Rectal administration provides a negligible first pass metabolism effect
(there is a good blood/lymph vessel supply, and absorbed materials drain
directly into
the inferior vena cava), and the method is suitable of children, patients with
emesis, and
the unconscious. The method avoids gastric acid and enzymatic degradation, and
the
to ionization of a composition will not change because the rectal fluid has no
buffer
capacity (pH 6.8; charged compositions absorb best). Conversely, there may be
slow,
poor or erratic absorption, irritation, degradation by bacterial flora, and
there is a small
absorption surface (about O.OSm2). Further, lipidophilic and water soluble
compounds
are preferred for absorption by the rectal mucosa, and absorption enhancers
(e.g., salts,
EDTA, NSAID) may be necessary. Preferred embodiments of the present invention
include the rectal administration of lentiviral vector particles that contain
genes
encoding colon cancer antigens, self and/or foreign MHC, or immune modulators.
Nasal administration avoids first pass metabolism, and gastric acid and
enzymatic degradation, and is convenient. In a preferred embodiment, nasal
2o administration is useful for lentiviral vector particle administration
wherein the
lentiviral vector particle is capable of expressing a polypeptide with
properties as
described herein. Conversely, such administration can cause local irritation,
and
absorption can be dependent upon the state of the nasal mucosa.
Pulmonary administration also avoids first pass metabolism, and gastric
acid and enzymatic degradation, and is convenient. Further, pulmonary
administration
permits localized actions that minimize systemic side effects and the dosage
required
for effectiveness, and there can be rapid onset of action and self medication.
Conversely, at times only a small portion of the administered composition
reaches the
brochioli/alveoli, there can be local irritation, and overdosing is possible.
Further,
3o patient cooperation and understanding is preferred, and the propellant for
dosing may
have toxic effects. Preferred embodiments of the present invention include the
pulmonary administration of lentiviral vector particles that express genes
encoding IgA
or IgE for the treatment of conditions such as asthma, hay fever, allergic
alveolitis or
fibrosing alveolitis, the CFTR gene for the treatment of cystic fibrosis, and
protease and
collagenous inhibitors such as a-I-antitrypsin for the treatment of emphysema.
CA 02296319 2000-O1-17
WO 99/04026 PCT/US98/14996
54
Alternatively, many of the same types of polypeptides or peptides listed above
for oral
administration may be used..
Ophthalmic administration provides local action, and permit prolonged
action where the administration is via inserts. Further, avoids first pass
metabolism,
and gastric acid and enzymatic degradation, and permits self administration
via the use
of eye-drops or contact lens-like inserts. Conversely, the administration is
not always
efficient, because the administration induces tearing. Preferred embodiments
of the
present invention include the ophthalmic administration of lentiviral vector
particles
that express genes encoding IgA or IgE for the treatment of hay fever
conjunctivitis or
1o vernal and atomic conjunctivitis; and ophthalmic administration of
lentiviral vector
particles that contain genes encoding melanoma specific antigens (such as high
molecular weight-melanoma associated antigen), self and/or foreign MHC, or
immune
modulators.
Transdermal administration permits rapid cessation of treatment and
prolonged action leading to good compliance. Further, local treatment is
possible, and
avoids first pass metabolism, and gastric acid and enzymatic degradation.
Conversely,
such administration may cause local irritation, is particularly susceptible to
tolerance
development, and is typically not preferred for highly potent compositions.
Preferred
embodiments of the present invention include the transdermal administration of
lentiviral vector particles that express genes encoding IgA or IgE for the
treatment of
conditions such as atopic dermatitis and other skin allergies; and transdermal
administration of lentiviral vector particles encoding genes encoding melanoma
specific
antigens (such as high molecular weight-melanoma associated antigen), self
and/or
foreign MHC, or immune modulators.
Vaginal administration provides local treatment and one preferred route
for hormonal administration. Further, such administration avoids first pass
metabolism,
and gastric acid and enzymatic degradation, and is preferred for
administration of
compositions wherein the lentiviral vector particles express peptides.
Preferred
embodiments of the present invention include the vaginal administration of
lentiviral
vector particles that express genes encoding self and/or foreign MHC, or
immune
modulators. Other preferred embodiments include the vaginal administration of
genes
encoding the components of spernl such as histone, flagellin, etc., to promote
the
production of sperm-specific antibodies and thereby prevent pregnancy. This
effect
may be reversed, and/or pregnancy in some women may be enhanced, by delivering
lentiviral vector particles vectors encoding immunoglobulin antisense genes,
which
genes interfere with the production of sperm-specifc antibodies.
CA 02296319 2000-O1-17
WO 99/04026 PCT/US98/14996
Intravesical administration permits local treatment for urogenital
problems, avoiding systemic side effects and avoiding first pass metabolism,
and gastric
acid and enzymatic degradation. Conversely, the method requires urethral
catheterization and requires a highly skilled staff. Preferred embodiments of
the present
5 invention include intravesical administration of lentiviral vector particle
encoding
antitumor genes such as a prodrug activation gene such thymidine kinase or
various
immunomodulatory molecules such as cytokines.
Endoscopic retrograde cystopancreatography (FRCP) (goes through the
mouth; does not require piercing of the skin) takes advantage of extended
gastroscopy,
to and permits selective access to the biliary tract and the pancreatic duct.
Conversely, the
method requires a highly skilled staff, and is unpleasant for the patient.
Many of the routes of administration described herein (e.g., into the CSF,
into bone marrow, into joints, intravenous, intra-arterial, intracranial
intramuscular,
subcutaneous, into various organs, intra-tumor, into the interstitial spaces,
intra-
15 peritoneal, intralymphatic, or into a capillary bed) may be accomplished
simply by
direct administration using a needle, catheter or related device. In
particular, within
certain embodiments of the invention, one or more dosages may be administered
directly in the indicated manner at dosages greater than or equal to 105, 106,
10', 10g,
109, 10'° or 10" cfu.
2o Lentiviral vector particle may be delivered to the target from outside of
the body (as an outpatient procedure) or as a surgical procedure, where the
vector is
administered as part of a procedure with other purposes, or as a procedure
designed
expressly to administer the vector. Other routes and methods for
administration include
the non-traumatic routes disclosed within PCT/LJS95/169G7, as well as
administration
25 via multiple sites as disclosed within PCT/US95/16471.
The following examples are offered by way of illustration, and not by
way of limitation.
CA 02296319 2000-O1-17
WO 99/04026 PCT/US98/14996
56
EXAMPLES
EXAMPLE 1
CONSTRUCTION OF AN HIV-1 BASED VECTOR
A. Construction of a vector backbone
Plasmid pSK-OH3 is first constructed by digestion, filling and blunt end
ligation of the unique HindIII site of pBluescript SK(-) (Stratagene, La
Jolla, CA).
The HIV-1 LTR from the molecular clone pNL4-3 (Adachi et al., J.
Virol. 1986, 59, 284/ NIH AIDS Research and Reference reagent program catalog
#I 14)
is amplified by Polymerase Chain Reaction (PCR) using oligomers LTRBS : 5' TAG
GAT CCT GGA AGG GCT AAT TTG G 3' (Sequence ID No. ~ and LTRB3 : 5'
TAG GAT CCT TTC GCT TTC AAG TCC C 3' (Sequence ID No. ~, which both
display a BaniHI restriction site at their 5' end. The amplified fragment of
681 by in
length is cloned into the unique BamHI site of pSK-OH3. The resulting plasmid
is
named pHIV-LTR.
The 1181 by HindIII to HindIII fragment of pNL4-3, comprising the 3'
end of the S' LTR and the 5' part of the gag gene is cloned into the unique
HindIII site
of pbluescript SK(-) to create pHIV-H3F5'.
The 916 by HindIII to SphI fragment from pHIV-H3F5' is ligated with
the 3482 by HindIll to EcoRI fragment from pHIV-LTR, using a SphI-EcoRI
adaptor to
give pHIV-LTRS'.
In order to abolish the expression of the gcrg gene portion present in the
pHIV-LTRS' construct, the initiation codon region is modified from GAG ATG GGT
to
GAG AAC CGG T {Sequence ID No. ) by site directed mutagenesis (Muta-gene kit,
Biorad, Hercules, CA) using the mutated oligonucleotide 5' GGA GGC TAG AAG
GAG AGA GAA CCG GTG CGA GAG CGT CG 3' (Sequence ID No. ~. This
modification results in the destruction of the ATG codon, as well as a +1
frameshift in
the open reading frame of the HIV gag gene. The mutation also allows the
creation of a
unique Age I site (ACCGGT).
In order to delete undesirable restriction sites, the region comprised
between the EcoRI to Xho I sites of plasmid pHIV-LTR is destroyed by digestion
with
the corresponding enzymes, filling and subsequent religation to give pHIV-LTR-
ORI/Xho.
CA 02296319 2000-O1-17
WO 99/04026 PCT/US98/14996
57
The 719 by Xho I to HindIII fragment from pNL4-3 is Iigated with the
3067 by Xba I to HindIII fragment from pHIV-LTR-~RI/Xho using a Xba I-Xho I
adaptor to give pLTR-HIV3'.
To create the HIV-1-based vector backbone, named vHIV-I, the 1510 by
Not I to Xho I fragment from pHIV-LTRS' is inserted into the Not I and Xho I
sites of
pHIV-LTR3' (see Figure 1 ).
B. Addition of nuclear tr~~SnOrt elements
In order to allow the vector transcript to be efficiently translocated from
the cell nucleus to the cytoplasm, nuclear transport elements can be added to
vHIV.
1o The HIV-1 Rev-responsive element (RRE) is amplified by PCR from
pNL4-3 using the oligomers RRE1 : S' GCA AGC TTC TGC AGA GCA GTG GGA
ATA GG 3' (Sequence ID No.~ and RRE2 : 5' GCA AGC TTA CCC CAA ATC
CCC AGG AGC TG 3' (Sequence ID No. ), which harbor the Hind III site at their
5'
end. The resulting amplified fragment of 283 by in length is inserted into the
Bam HI
site of pSK(-) to create pRRE.
The RRE fragment is extracted from pRRE using the enzymes Eco RI
and Cla I and inserted into the Eco RI and Cla I sites of vHIV to give vHIVR.
The Hepatitis B Virus (HBV) posttranscriptional regulatory element
(PRE) (Liang and Huang, Mol. Cell. Biol. 13:7476, 1993) or the Mason-Pfizer
monkey
2o Virus (MPMV) constitutive transport element (CTE) (Bray et al., Proc. Natl.
Acad. Sci.
USA 91:1256, 1994) are amplified by PCR using specific primers which also
display
Hind III restriction sites at their 5' extremities and inserted in place of
the HIV-1 RRE
in the vHIVR into the Eco RI and Cla I sites to give vHIVP and vHIVC
respectively.
C. Addition of the gene of interest sequence to the vector backbone.
A 2kb XbaI-SstI fragment from pSP6-~igal (Shapira et al., Gene 25:71-
82, 1983) is ligated together with a 1.1 kb Sst I - Sma I fragment from pSP6-
~3ga1 and a
2.6 kb Xba I - Sma I digested pUCl9 DNA to form pUC-(gal. The beta-
galactosidase
gene is then extracted from pUC-beta-Gal by cutting with the restriction
enzymes Sal I
and Sma I. A 750 by fragment containing the human Cytomegalovirus (hCMV) early
3o genes promoter is also extracted from pCMV-G (Yee et al., Proc. Natl. Acad.
Sci. USA,
91:9564, 1994) by cutting with Xba I and Sal I. These two fragments are
ligated
together and inserted into the Xba I and Sma I site of pBluescript (SK-) to
give pCMV-
beta-Gal.
CA 02296319 2000-O1-17
WO 99/04026 PCT/US98/14996
58
A 3800 by Not I to Hind III fragment is extracted from pCMV-beta-Gal
and inserted into the Xho I site of vHIVR, after the sites are blunt-ended, to
give vHRC
beta-Gal. The ligation of the blunt-ended sites Not I and Xlao I allow to
recreate a Xho I
site. The same cloning strategy is applied to vHIVC to create vHCC-beta-Gal,
and to
vHIVP to create vHPC-beta-gal).
D. Tissue-specific promoters
To restrict expression of the gene of interest in certain cell-types, the
beta-galactosidase gene is put under the control of two different tissue-
specific
promoters.
to More specifically, in order to induce liver tissue specific expression of
the transgene, the 88 by Hirzc II to Rsa I fragment from the construct
pOCCAT7, (Yee,
Science, 1989, 246, 658), which contains the Hepatitis B Virus (HBV) liver
specific
enhancer, is cloned upstream the Herpes simplex virus (HSV) thymidine kinase
(TK)
gene promoter to drive the expression of the beta-galactosidase gene in place
of the
CM V promoter.
Similarly, the CD2 gene enhancer (Lake et al., EMBO J. 9:3129, 1990) is
cloned upstream the HSV TK gene promoter to induce lymphocyte-specific
expression.
E. Expression of a dicistronic cassette
In order to achieve the expression of two or more transgenes driven by
the same promoter from a single vector, two types of dicistronic constructs
are
designed.
In the first construct the 2.1 kb Barn HI to Hind III fragment encoding
the firefly luciferase gene sequence (luc) and the Neomycin phosphotransfcrase
gene
sequence (neo) distant of 78 nucleotides from pLL78NL (Levine et al., Gene
108:167,
1991), is inserted in the Sal I to Sal I backbone of VHRC-beta-gal to create
vHRCL78N. The 78 nucleotide linker between the two genes allows reinitiation
of
translation of the second open reading frame before the large ribosome subunit
detaches
from the mRNA.
In the second type of construct, the poliovirus internal ribosome entry
site (PO-IRES) is inserted in between the two gene sequences (Adam, .l.
Vif~ol. 65,
4985, 1991). The IRES allows initiation of the second open reading frame
independently of that of the first one.
CA 02296319 2000-O1-17
WO 99/04026 PCTNS98/14996
59
EXAMPLE 2
PACKAGING CELL LINE GENERATION
A. Construction of a ~agl~pression cassette: pCMV-HIV-1
To construct the packaging cassette pCMV-HIV-l, the 0.7-kilobase pair
(kb) BamHl Sphl fragment with a 19-base pair (bp) deletion in the putative
packaging
signal of pCMVDPIDenvpA ( Parolin et al., J. Virol. 68:3888-3895, 1994) was
fused
with the 8-kb Sphl Hindlll (from position 1447 to 9606) fragment of pNL4-3
(containing a full-length infectious HIV-1 genome, Adachi et al., J. Virol.
59:284, 1986,
obtained from NIH AIDS Reagent Program) and the 4-kb Sull-EcoRI fragment from
Io pCMV-G (Yee et al., Proc. Natl. Acad. Sci. USfI 91:9564-9568, 1994). In
addition, a
deletion of the 580-by Bglll (at position 7031 in pNL4-3)-Bglll (at position
7611 in
pNL4-3) fragment was created in the HIV-1 Env coding region to eliminate the
expression of this protein and reduce the potential of generating helper virus
during
vector production.
1. Generation of nef (_) mutants
To generate pCMV-HIVnef(-), the sequences between Hpul (at position
8650 in pNL4-3) and Hindlll (at position 9606 in pNL4-3) of pCMV-HIV-1 was
deleted.
2. Generation of vifl_-1 mutants
2o To generate pCMV-HIVvif(-), pCMV-HIV-1 was digested with Ndel (at
position 5123 in pNL4-3) and repaired with the Klenow fragment to create a 2-
by
insertion in the coding region of the viJgene.
3. Generation of vpu(-) mutants
To generate pCMV-HIVvpu(-), the initiation codon of Vpu was mutated
by site directed mutagenesis (Mutagene kit, Biorad, Hercules, CA) using the
oligonucleotide 5'TGCTACTATTATAGGTTGTACATGTACTACTTACTG3'.
CA 02296319 2000-O1-17
WO 99/04026 PCT/US98/14996
- 60
4. Generation of vp~-) mutants
To generate pCMV-HIVvpr(-), the pCMV-HIV-1 was digested with
EcoRl (at position 5747 in pNL4-3) and repaired with the Klenow fragment to
generate
a 4-by insertion in the coding region of the vpr gene.
5. Generation of vpr(,l nef(~ double mutant
The pCMV-HIVvpr(-)nef(-) double mutant was generated by digesting
the pCMV-HIVnef(-) with EcoRl and repaired with the Klenow fragment as
described
above.
4. Generation ofp H-GP-I
1o To generate pCH-GP-1, the 0.66-kb fragment between position 766 and
the Sphl site at position 1447 in pNL4-3 was amplified by polymerase chain
reaction
(PCR) using the oligonucleotides Gags' S'GAGGATCCTAGAAGGA-
GAGAGATGGGT3' and Gag3' S'GAGGATCCAATAGGCCCTGCATGCACTG3'.
The resulting fragment was ligated with the 3.7-kb Sphl-Nclel fragment from
pNL4-3
and the 4-kb Sall-EcoRI fragment from pCMV-G.
B. Addition of nuclear transDOrt ~,~nals
1. Addition of RRE
To insert the RRE sequence into pCH-GP-I for efficient transportation
of the gag/pol transcripts from nucleus to cytoplasm, the RRE (between
positions 7754
and 8013 in HXB-2 (33)) was amplified by PCR from pv653RSN (31) using the
oligonucleotides RRES 5'GCAAGCTTCTGCAGAGCA-GTGGGAATAGG3' and
RRE3 5' GCAAGCTTAC CCCAAATCCC CAGGAGCTG 3' and cloned immediately
after the gaglpol gene in pCHGP-1. This construct was designated pCH-GP-2
(Figure 3A).
2. Addition of PRE
To insert the HBV PRE sequence for efficient transportation of the
gag/pol transcripts, the 650-by StuI-HindIII HBV fragment spanning the PRE
region
(Liang & Huang, Mol. Cell. Biol. 13:7476, 1993; Huang & Yen, J, vir-ol.
68:3193,
1994; Donello et al., .I. Virol. 70:4345, 1996) in pCCAT-1 (Yee, Science
246:658,
3o 1989) is inserted into the EcoRV site of pBluescript SK(-) to form pSK-PRE.
The PRE
CA 02296319 2000-O1-17
WO 99/04026 PCT/US98/14996
61
fragment in pSK-PRE is isolated by CIaI and EcoRI digestion and cloned into
the CIaI
and EcoRI-digested pCH-GP-1 to form pCH-GP-3 (Figure 3B).
3. Addition of CTE
To insert the CTE sequence from Mason-Pfizer monkey virus (MPMV)
s (Bray et al., Proc. Natl. Acad. Sci. USA 91:1256, 1994) into pCH-GP-1, the
CTE
sequence in pGem7fz(-)MPMV is amplified by PCR using a pair of primers, 5'
GTCATCGATA GACTGGACAG CCAATG 3' (Sequence ID No. ~ and S'
CTAGAATTCC CAAGACATCA TCCGGG3' (Sequence ID No. ~, containing the
CIaI and the EcoRI, respectively, as described above. The PCR product is
inserted into
to the CIaI and EcoRI-digested pCH-GP-1 to form pCH-GP-4 (Figure 3C).
C. Generation of stable ap ekaein~ cell lines
To establish stable packaging cell lines, either the amphotropic env or
the VSV-G can be expressed instead of the HIV env to pseudotype the HIV
vectors.
The first strategy is to establish 293 cell lines expressing the HIV tat
necessary for
i5 efficient expression of the vector genomic RNA. To construct the tat
expression
plasmid, a 360-by SaII-BamHI fragment spanning the first exon of the tat gene
is
isolated from pCVI (Arya et al., Science ??9:69, 1985). A 1390-by XbaI-XhoI
fragment containing the CMV promoter and the rabbit ~3-globm gene splice
signal is
isolated from pCMV-G. A 3295-by BamH1-XbaI fragment containing the
2o polyadenylation site of the rabbit ~3-globin gene is also isolated from
pCMV-G. All
three fragments are ligated together to form pCMV-tat (Figure 4A). To
establish stable
tat-expressing cell lines, 293 cells are co-transfected with pCMV-tat and pSV2-
gpt
(Mulligan & Berg, Proc. Natl. Acad. Sci. USA 78:2072, 1981, obtained from
ATCC) at
a ratio of 10 to 1. The transfected cells are subjected to selection in
mycophenolic acid,
25 xanthine and HAT-containing medium. The surviving cells are pooled and
clones are
isolated by limiting dilution. To screen for tat expression, the culture
supernatant from
each clone is spotted on a nylon membrane and tat is detected by the ECL
Western
blotting system (Amersham, Arlington Heights, IL) using an anti-tat antibody
(Advanced Biotechnologies Inc., Columbia, MD).
3o To express the HIV gag/pol, pCH-GP-3 or pCH-GP-4 is co-transfected
with pFR400 (Proc. Natl. Acad. Sci. USA 80:2495, 1983) into 293/tat cells. The
transfected cells are selected in methotrexate (Mtx)-containing medium and
cloned by
limiting dilution. Mtx-resistant clones are picked and gag/pol expression is
assayed by
CA 02296319 2000-O1-17
WO 99/04026 PCT/US98/14996
62
HIV p24 in culture supernatant. The 293/tat/gag/pol clone expressing the
highest level
of p24 is expanded and used for stable expression of amphotropic env.
To express the amphotropic env gene, pCMVenva"'DraI (PCT #WO
92/05266; US patent #5,591,624) is co-transfected with pTK-puro (Chen et al.,
Proc.
Natl. Acad. Sci. USA 93:10057, 1996) into the 293/tat/gag/pol cells. The
293/tat/gag/pol/env clones are isolated from puromycin-resistant colonies by
limiting
dilution. Expression of the env gene is detected by Western blot analysis
using, RLV
gP69/71, the anti-env polyclonal antibody (Quality Biotech Inc., Camden, NJ).
The
clone expressing the highest level of env is identified and expanded.
Packaging cell lines expressing VSV-G instead of the amphotropic env
are also established. The VSV-G gene is under the control of the inducible
promoter of
the tet system (Gossen & Bujard, Proc. Natl. Acad. Sci. USA 89:5547, 1992,
obtained
from Bujard) in pTetO-G-2 (Chen et al., Proc. Ncrtl. Acad. Sci. USA 93:10057,
1996)
since stable VSV-G expression resulted in cell death. To express VSV-G, stable
293
clones expressing tTAER (Chen et al., Pros. Natl. Acacl. Sci. USA 93:10057,
1996), the
transactivator for the inducible promoter, need to be established first. The
293/tat cells
described above are transfected with phyg-CMV-tTAER (Chen et al., Proc. Natl.
Acad.
Sci. USA 93:10057, 1996) and hygromycin-resistant clones are isolated by
limiting
dilution. To identify the clone expressing the highest levels of tTAER, cells
derived
from these clones are transfected with pUHCl3-3 (Gossen & Bujard, Proc. Natl.
Acacl.
Sci. USA 89:5547, 1992, obtained from Bujard) in the presence of tetracycline
or ~-
estradiol and the luciferase (lux) activity is determined 48 hours after
transfection. The
293/tat/tTAER clone expressing the highest level of lux activity in the
presence of (3-
estradiol is identified and expanded.
To express the HIV gag/pol, pCH-GP-3 or pCH-GP-4 is co-transfected
with pFR400 into 293/tat/tTAER cells, and the 293/tat/tTAER/gag/pol clone
expressing
the highest level of p24 is isolated as described above. To introduce the VSV-
G gene,
the 293/tat/tTAER/gag/pol cells are transfected with pTetO-G-2 and selected in
puromycin-containing medium. The 293/tat/tTAER/gag/pol/G clone expressing low
level of VSV-G is isolated by limiting dilution of the puromycin-resistant
cells,
followed by ~i-estradiol induction of VSV-G expression and FACS analysis using
I1,
the monoclonal antibody specific for G (Burns et al., Proc. Natl. Acad. Sci.
USA
90:8033, 1993). Clones expressing low-level of VSV-G are isolated because high-
level
VSV-G expression leads to rapid cell death (within 4 to S days) whereas low-
level
VSV-G expression has little effect on cell growth, resulting in an extended
duration of
CA 02296319 2000-O1-17
WO 99/04026 PCT/US98/14996
63
virus harvest ( up to two weeks) (Chen et al., Proc. Natl. Acad. Sci. USA
93:10057,
1996).
The packaging cell lines described above express HIV gag and pol using
the CTE and PRE sequence which bypass the requirement of rev and RRE for
efficient
transportation of the gag/pol mRNA from nucleus into cytoplasm. To determine
whether rev and RRE can generate higher virus titers, 293/tat/tTAER cells are
co-
transfected with pCH-GP-2 and pFR400, and the transfected cells are cloned
selection
in Mtx-containing medium and limiting dilution. Mtx-resistant clones are
picked and
gag/pol expression is assayed by introducing the rev gene into these clones.
To
1o introduce rev, these clones are infected with the KT-1 virus (Chiron
Technologies, San
Diego, CA) containing the HIV rev gene in a MLV-based vector. HIV p24 in
culture
supernatant is assayed 48 hours after infection. The 293/tat/tTAER/ gag/pol
clone
expressing the highest level of p24 upon rev introduction will be expanded and
used for
stable expression of rev.
Since high-level expression of rev can be toxic to cells, the rev gene is
placed under the control of the inducible promoter of the tet system. The 722-
by
Bsu36I fragment containing the rev cDNA is isolated from pCV 1 and inserted
into the
unique BamHI site of pUHGlO-3 (Gossen & Bujard, Proc. Natl. Acad. Sci. USA
89:5547, 1992, obtained from Bujard) to fornl pTetO-rev (Figure 4B). To
introduce
the rev gene, pTetO-rev is co-transfected with pTK-phleomycin (Yee, personal
communication) into 293/tat/tTAER/gag/pol cells. The clone expressing the
highest
level of rev is identified by limiting dilution of phleomycin-resistant cells,
followed by
(3-cstradiol induction and p24 detection in the culture supernatant.
To express VSV-G or amphotropic env in 293/tat/tTAER/gag/pol/rev
am
cells, the VSV-G gene in pTetO-G-2 or the amphotropic env gene in pCMVenv DraI
is introduced into the 293/tat/tTAER/gag/pol/rev cells and clones expressing
either
VSV-G or env are isolated as described above.
D. Accessorv Proteins
Genes encoding the accessory protein can be PCR amplified from pNL4
3 and clonebd into pCMV-Bam for expression. The plasmid is co-transfected with
pCMUIV-K (Song et al., J. Biol. Chem. 269:7024, 1994, obtained from b er
Peterson,
The Scripps Research Institute, La Jolla, CA) containing the murine H-2K cDNA
into
one of the packaging cell lines described above. Transfecbted cells are
isolated by FRCS
sorting using Y3, the monoclonal antibody for H-2K . The clone expressing the
highest level of the accessory protein is identified using Western blot
analysis with the
CA 02296319 2000-O1-17
WO 99/04026 PCT/US98/14996
64
specific polycIonal antibody (obtained from NIH AIDS Reagent Program). In the
case
of Vpr expression which is know to block cell cycle at G2 phase (Planelles et
al., J.
Virol 70:2516, 1996), the PCR amplified Vpr cDNA is inserted into pUHGlO-3
under
the control of the tet inducible promoter as described for the rev expression.
The
construct is co-transfected with pCMUIV- Kb into the packaging cell lines and
the Vpr-
expressing clone is isolated as described above.
Besides 293-based packaging cell lines, human HT1080 and HeLa cells,
canine cf2 and D17-based packaging cell lines are established to test viral
titer potential
with the same strategy described above.
to To generate stable VCL, the HIV vector described above is co-
transfected with pCMV-G and pCMVOR9 (Naldini et al., Science 272:263, obtained
from Verma, The Salk Institute, La Jolla, CA) into 293 cells. Virus harvested
48 hours
after transfection is used to infect one of the packaging cell lines described
here and
VCL clones are isolated by the neo selection or FACS sorting with the antibody
specific
for the marker gene carried in the vector ((3-gal, alkaline phosphatase or
nerve growth
factor receptor). Titer potential for the clones is determined by infecting
HT1080 cells
with the harvested virus from each clone, followed by the neo selection or
FACS
analysis.
2o EXAMPLE 3
TRANSIENT PRODUCTION OF INFECTIOUS VECTOR PARTICLES
A. Protocols
To generate infectious HIV vectors. 293T cells were seeded at a density
of 4 x 106 cells per 10 cm-diameter culture dish. Infectious vector with all
the
accessory proteins was generated by cotransfecting lOpg pCMV-HIV-l, l0ug pCMV-
G and 20ug pv653CMVb-gal using the calcium phosphate co-precipitation method
(Graham and van der Eb, Virology 52:456-467, 1973). Culture medium was
replaced 6
to 8 hours later and the culture supernatant was collected 18 hours after
transfection,
filtered through 0.45 um filters and stored at -80°C. To generate the
vector without any
3o accessory protein, 293T cells were cotransfected with the following five
plasmids: 8pg
pCHGP plasmid series (the gag/pol plasmid), 8pg pCMV-G {VSV-G plasmid), 16~g
pv653CMV ~i-gal (HIV-vector), 4ug pCMV-Tat and 4pg pCMV-Rev. Within certain
experiments, 0 to 10 mM Sodium Butryate is added to the media during
transfection.
CA 02296319 2000-O1-17
WO 99/04026 PCTNS98/14996
Utilizing this five-plasmid transfection protocol, all accessory proteins have
been
eliminated. Moreover, the possibility of generating replication competent
virus is
greatly reduced.
To determine the vector titer, 5 x 104 HT1080 cells were plated in a 12-
5 well plate in the presence of 8 pg/ml polybrene 24 hours prior to infection.
The cells
were infected overnight with various dilutions of the vector and assayed for
the b-gal
activity 48 hours after infection.
To assay for (3-gal activity, cells were washed once with PBS, fixed in
1.25% glutaraldehyde for 15 min. and stained for 4 hours at 37°C in a
solution
to containing S mM potassium ferriferrocyanide, 400 pg/m1 X-Gal (GBT, ST
Louis, MO}
and 1 mM MgCl2.
B. lts
Results are shown in Figure 7. Brief~y> as can be seen by this table,
deletion of the accessory protein from the vector particle appears to have
little effect on
15 vector titer.
Results are also shown in Figure 8. Briefly, CTE from Mason Pfizer
Monkey Virus can substitute the function of RRE. But RRE and rev together
still
appear to generate the highest level of gag production.
Results of the five plasmid transfection protocol are shown in Figure 9.
2o Briefly, as noted above, utilizing the five-plasmid transfection protocol
all accessory
proteins can be eliminated. Moreover, the possibility of generating
replication
competent virus is greatly reduced.
Results of the use of sodium butyrate in the five plasmid transfection
protocol is shown in Figure 9. Briefly, addition of sodium butyrate in the
transfection
25 media can enhance vector particle production by greater than 10 fold.
EXAMPLE 4
EFFECT OF ACCESSORY PROTEINS ON VECTOR INFECTIVITY
30 To study the ability of the HIV vector to infect quiescent cells and the
effect of the accessory proteins on infectivity, HeLa cells were exposed to
gamma-
irradiatian to arrest cells at the G2 phase of the cell cycle. More
specifically,
proliferating or growth-arrested HeLa cells were transduced with either MLV-(3-
gal, a
CA 02296319 2000-O1-17
WO 99/04026 PCT/US98/14996
66
(3-gal gene-containing MLV vector or the HIV-1-based vector v653CMV~i-gal(+)
containing all four accessory proteins or v653CMVb-gal(-) containing no
accessory
protein. Positive cells were scored by X-Gal staining two days after
transduction.
Results in HeLa were expressed as the percentage of titers observed with
the same virus preparations in growing HT1080. As shown in Figure 10, no
significant
difference in titer was observed in proliferating or quiescent HeLa cells
transduced with
either the v653CMVj3-gal(+) or the v653CMV~3-gal(-) vector. In contrast, the
transduction efficiency of the MLV vector in quiescent cells was reduced more
than
2000 fold. Similar results were obtained with irradiated HT1080 cells,
transduced with
1o the three vectors (data not shown).
To ascertain the observed (3-gal activity is not due to pseudotransduction
of the b-gal activity present in the vector preparation, proliferating HeLa
cells
transduced with the vector were treated with increasing concentrations of 3'-
azido-3'-
deoxythymidine (AZT). Both the blue cell number and the b-activity in cell
extracts
decreased with increasing concentrations of AZT (data not shown). These
results
demonstrate that, in contrast to the MLV vector, H1V-1-based vectors can
transduce
quiescent cells efficiently and the HIV-1 encoded accessory proteins are not
required to
transduce these cells.
To test the infectivity of HIV-I-based vectors in other cell types, primary
human skin fibroblasts were allowed either to proliferate or to grow to
confluency and
then infected with the three retroviral vectors described above. Fibroblasts
grown to
confluency become contact inhibited and arrested in the GO~Gl phase of the
cell cycle
(data not shown). As shown in Figure 11, the three vectors exhibit similar
transduction
efficiency in dividing fibroblasts. However, in quiescent cells, MLV-~3-gal
vector
transduction dropped to barely detectable levels. The capacity of v653CMV(3-
gal(+)
remained unchanged. In contrast, v653CMV(3-gal(-), defective for the HIV-1
accessory
proteins showed a 4- to 7 fold decreased Level of efficiency to transduce the
contact
inhibited fibroblasts relative to the v653CMV~3-gal(+) vector. These results
suggest
that the requirement for accessory proteins for efficient transduction by HIV-
1-based
vectors is cell-type dependent.