Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02303482 2000-02-15
WO 99/10508 PCTlUS971152I9
CHD~RIC TRANSCRE''ftONAL ACTIVATORS AND C011~OS1TIONS AND USES RELATED
TEIFRETO
Background of the Invention
A large number of biological and clinical protocols, among others, gene
therapy,
production of biological materials, and biological research, depend on the
ability to elicit
specific and high-level expression of genes encoding RNAs or proteins of
therapeutic,
commercial, or experimental value. A variety of expression systems have been
developed,
including regulated expression systems, involving allosteric on switches
triggered by
tetracycline, RU486 and ecdysone, as well as dimerization based on-off
switches triggered
1o by FK1012, FK-CsA, rapamycin and analogs thereof. See e.g. Clackson,
"Controlling
mammalian gene expression with small molecules" Current Opinion in Chemical
Biology
1997, 1:210-218. Still, achieving a sufficiently high level of expression for
clinical or
other utility in genetically engineered cells in various contexts, including
within whole
organisms, has often been a limiting problem. Various approaches for
addressing this
~ 5 problem, including the search for stronger transcriptional promoters or
higher transfection
efficiencies, have in many cases not met with success. Meanwhile, in various
lines of
research with transcription factors, promising results in transient
transfection models have
not been borne out with chromosomally integrated reporter gene constructs.
Furthermore,
overexpression of transcription factors is commonly associated with toxicity
to the host
2o cell. Despite those precedents, this invention takes a novel approach to
the challenge of
optimizing gene expression through new uses of, and new designs for,
transcription factor
proteins which are expressed within the engineered cells containing the target
gene. The
invention provides improved methods and materials for achieving high-level
expression of
a target gene in genetically engineered cells, including genetically
engineered cells within
25 whole organisms.
Summary of the Invention
In the course of our research on transcription activation, we have quite
unexpectedly discovered an important phenomenon which may explain the absence
of
3o greater progress in the discovery and/or optimization of more powerful
transcription
activating domains. Briefly, we have made the unexpected discovery that in
conventional
transient transfection experiments of the sort typically used in this field to
study
transcription activation, as the number or apparent potency of activation
domains
increases, the observed level of transcription increases to a maximum and then
levels off
35 or decreases in a phenomenon termed "squelching". However, when analogous
experiments are conducted using stably incorporated reporter genes in place of
transiently
CA 02303482 2000-02-15
WO 99/10508 PCT/13S97/15219
-2-
transfected reporter genes, squelching is avoided, and instead, an unimpeded
structure-
activity relationship can be observed for various transcription activation
domains and
modifications thereof. With the blinders otherwise imposed by squelching now
removed,
we have made significant advances in the design and implementation of
transcription
activation domains in chimeric proteins and transcription systems.
This invention encompasses nucleic acid constructs encoding various chimeric
transcription activator proteins (also referred to below as "composite
activators") and in
some embodiments auxiliary chimeric proteins, as well as related materials and
methods,
as disclosed in greater detail below. A number of illustrative embodiments are
highlighted
1 o below.
For example, a number of embodiments of the invention involve a nucleic acid
encoding a chimeric transcription activator protein which activates the
transcription of a
gene to which the chimeric transcription activator protein is targeted.
Targeting may be
effected via one or more DNA-binding domains ("DBDs") which bind to a DNA
sequence
to which a target gene is operatively linked. The DBD(s) may be present within
the
chimeric transcription activator protein or may be provided by an auxiliary
chimeric
protein as disclosed in extensive detail below.
In certain embodiments of the invention the chimeric transcription activator
protein
contains at least one composite transcription activation domain ("TAD") and at
least one
2o DBD. The composite TAD comprises a continuous polypeptide region containing
two or
more component polypeptide regions (also referred to below as "activation
tags"), at least
two of which do not occur together in the same gene product in nature. In
certain of these
embodiments the chimeric protein comprises a ligand binding domain for
allosteric
activation, e.g. a receptor domain capable of binding a cell permeant ligand
such as
tetracycline or an analog thereof or a steroid such as RU486 or ecdysone, for
example. In
such cases, the chimeric transcription activator selectively activates
transcription of a
target gene in the presence of the respective ligand. In other embodiments the
chimeric
transcription activator is capable of activating target gene expression
constitutively, i.e.,
without the need for the presence of any such ligand.
3o In other embodiments, the chimeric transcription activator protein contains
at least
one composite TAD comprising three or more component polypeptide regions, at
least two
of which do not occur together in the same gene product in nature, and at
least one
additional domain which is heterologous with respect to at least one of the
component
polypeptide regions of the composite TAD. The additional domains) may include
a DBD
or a receptor domain for an oligomerizing ligand. Oligomerizing ligands are
multivalent,
preferably cell permeant, compounds, generally having a molecular weight below
about 5
kD, and preferably below about 2 kD, which mediate the formation of complexes
with
CA 02303482 2000-02-15
WO 99/10508 PCT/US97115219
-3-
proteins containing receptor domains to which the ligands binds. Non-limiting,
illustrative
examples of oligomerizing ligands include FK1012 (with respect to proteins
containing
FKBP domains), coumermycin (with respect to proteins containing DNA Gyrase
domains), fujisporin (with respect to proteins containing cyclophilin and FKBP
domains,
respectively) and rapamycin (with respect to proteins containing FKBP and FRAP
domains, respectively).
In still other embodiments, the chimeric transcription activator protein
contains at
least one composite TAD comprising a continuous polypeptide region containing
two or
more component polypeptide regions, at least two of which do not occur
together in the
t o same gene product in nature, or at least not in the same order,
arrangement or number as
found in the chimeric protein of this invention, and at least one additional
domain which is
heterologous with respect to at least one of the component polypeptide regions
of the
composite TAD. In some of these embodiments, the components are all of human
origin.
In other of these embodiments, at least one of the heterologous domains
comprises a
~ 5 domain derived from an immunophilin, cyclophilin, calcineurin, FRAP or DNA
gyrase
domain; a domain derived from a receptor for tetracycline or ecdysone or
another steroid;
or a composite DNA binding domain.
In still other embodiments, the chimeric transcription activator protein
contains at
least one transcription activation domain, which may be a previously known
transcription
2o activation domain such as VP16, a novel truncated p65-derived activation
domain
disclosed in detail below or a composite TAD as described herein, and at least
one
bundling domain. A bundling domain is a domain permitting assembly of
complexes of
two or more proteins each of which comprises a copy of the bundling domain or
a close
variant thereof. Non-limiting examples of bundling domains include domains
derived from
25 p53 or the E coli lac repressor as well as various leucine zipper domains.
Bundling
domains are distinguished from heterooligomerizing proteins such as the
FKBP/calcineurin pair, the FKBP/FRAP pair, the cyclophilin/calcineurin pair,
the
RXRlI'BP pair, for example, which form heterooligomers of proteins containing
very
different binding domains. In contrast, bundling domains permit
oligomerization between
3o proteins containing the same bundling domains or very similar variants
thereof, and do so
without the need for ligand-mediation.
In various embodiments, one or more of the DBD, receptor domain, oligomerizing
ligand binding domain or other additional heterologous domain is heterologous
with
respect to at least one of the component polypeptide regions of the composite
transcription
35 activation domain. In other embodiments, one or more of such additional
heterologous
domains is heterologous with respect to at least two of the component
polypeptide regions
of the composite transcription activation domain. In one embodiment, the
composite
CA 02303482 2000-02-15
WO 99/10508 PCT/US97/15219
-4-
activator protein includes at least two different activation tags from
heterologous sources,
e.g., activation tags which are derived from two or more different
transcription factors or
co-activators and which do not naturally occur together in the same protein.
While not
wishing to be bound by any particular mechanistic hypothesis, two or more of
the
activation tags of a composite activator, more preferably two which are
derived from
disparate proteins, may recruit TAFs which are not required for basal
activation of a gene.
Through the activation tags, the chimeric activator may recapitulate the
interaction of
several otherwise discrete factors with the polymerase complex, or provide
novel contacts
with that complex, which interactions may recruit and/or stabilize the
formation of
activator-dependent transcriptional complexes.
In this context, it has also been discovered that a variety of activation tags
can be
repeated multiple times in the same chimeric protein, or multiply associated
therewith,
with a concomitant increase in transcriptional activation. The repetitive
activation tag
embodiment can be combined with other activation tags, e.g., from the same or
heterologous sources as the repeated activation tag(s).
Another aspect of the invention relates to the identification of novel
activation tags.
In this regard, an activation tag has been identified in the NF-kB
transcription factor
subunit p65. The so-called "alanine/proline rich" or "AP" activation tag of
p65 extends
from about amino acids 361 to about amino acid 450 of that protein. See SEQ ID
No. 2.
2o Similar AP activation tags are also present in, e.g., the p53 and CTF
proteins. The present
invention also contemplates fragments of p65 (or homologous sequences thereto,
e.g.,
from p53 or CTF) which are about 75, 60, 50, 30 or even 20 amino acid residues
in length.
In other embodiments, the AP activation tag has an amino acid sequence at
least 95%,
90%, 80% or 70% identical to the AP activation tag of SEQ ID No. 2.
The subject chimeric activators can be used to drive high levels of
transcription
from naturally-occurring, or otherwise genomicaliy-integrated genes. The
chimeric
activators of the invention are particularly useful for activating
transcription of integrated
single copy genes, which in the past have not successfully transactivated at
appreciable
levels. In preferred embodiments the level of expression of a chromosomally-
integrated
3o target gene achieved with the novel and/or composite transcription
activation domains
disclosed herein is at least two-fold, preferably three-fold, more preferably
five-fold, and
optimally ten-fold or better greater than expression levels achieved using the
VP16 domain
as the transcription activation domain in the analgous expression system, as
measured in a
scientifically valid comparison.
This invention also encompasses nucleic acid compositions comprising a first
nucleic acid encoding a chimeric transcription activator protein which
contains one or
more ligand-binding domains for an oligomerizing ligand such as described
above and a
CA 02303482 2000-02-15
WO 99/10508 PCT/US97/15219
-5-
second nucleic acid encoding a chimeric DNA-binding protein which comprises at
least
one DNA-binding domain and at least one ligand binding domain for a cell
permeant
ligand. Thus, the chimeric proteins encoded by these nucleic acids each
contain one or
more ligand-binding domains for binding to an oligomerizing ligand which
mediates the
dimerization or higher-order hetero-oligomerization of the chimeric proteins.
In some
embodiments, one or both of the chimeric proteins contains two or more ligand-
biding
domains. The nucleic acid composition may fiurther comprise a target gene
construct
comprising a target gene operatively linked to a transcriptional regulatory
element which
includes a DNA sequence to which the chimeric DNA-binding protein binds.
This invention also encompasses nucleic acid compositions comprising a first
nucleic acid encoding a chimeric transcription activator protein which
contains one or
more composite TADS and one or more DBDs, such as described above, and a
second
nucleic acid comprising a target gene construct comprising a target gene
operatively linked
to a transcriptional regulatory element which includes a DNA sequence to which
the the
~ 5 chimeric transcription activator protein binds.
A nucleic acid encoding a chimeric protein of this invention may be operably
linked to a transcriptional regulatory element permitting expression of the
chimeric protein
in cells. The various nucleic acids may be provided in DNA vectors as
disclosed below.
The invention also encompasses methods for engineering cells for the regulated
or
2o constitutive expression of a heterologous target gene. That method involves
introducing
nucleic acids or nucleic acid compositions of this invention into the cells
using methods
and materials permitting uptake by the cells of the nucleic acids. In
embodiments of
particular interest, the target gene is integrated within the chromosomes of
the host cells.
In some cases the transfected cells are selected and separately recovered from
non-
25 transfected cells. In certain embodiments the cells are grown in culture.
In some
embodiments the cells are engineered in vivo (within a whole organism), while
in other
embodiments cells are transfected in vitro and the transfected cells and/or
their progeny
are subsequently introduced into whole organisms. In the latter case, the
engineered cells
may be encapsulated prior to introduction into the organism.
3o Engineered cells which contain one or more nucleic acids or nucleic ' acid
compositions of this invention are also encompassed. Again, engineered cells
in which the
target gene is stably integrated within the cell's chromosomes are of
particular interest.
This invention further encompasses methods for effecting expression of a
target
gene which comprises maintaining genetically engineered host cells as
described herein
35 under conditions suitable for gene expression. In regulated expression
embodiments, this
will involve contacting the cells, in vitro or in vivo, with the ligand or
oligomerizing agent
CA 02303482 2000-02-15
WO 99/10508 PCT/US97/15219
-6-
which binds to one or both of the chimeric proteins.
Applications of this invention include transcription of genes, constitutively
or in a
drug-dependent manner in vitro, e.g. for the production of a desired protein
which may be
separately recovered, for achieving higher levels of expression in
transcription based
s assays (including two-hybrid assays), and for the regulated expression of
required viral
genes in producer cells lines used for production of recombinant viruses (e.g.
for the
regulated expression of AAV rep and/or cap genes in host cells used for the
production of
recombinant AAV). Other applications include in vivo applications such as the
constitutive or regulated expression of a target gene of interest in an animal
model (e.g. for
research or veterinary purposes) as well as for the constitutive or regulated
expression of a
target gene of interest in a human subject, e.g. in the case of gene therapy.
In the case of
human gene therapy, it will often be preferred that the components of the
chimeric proteins
be of human origin and/or that the engineered cells be encapsulated.
Other features and advantages of the invention will be apparent from the
following
~ s detailed description and claims.
Brief Description of the Figures
Figure 1 A shows the level of reporter gene expression (in EAP Units) from the
plasmid SxGAL4-IL2-SEAP transiently transfected into HT1080 cells
cotransfected with
2o various amounts of GAL4-p65 (diamonds) or GAL4-VP16 (squares) expression
constructs
(Activator DNA). Mean values of SEAP activity secreted into the medium are
shown (+/-
S.D.).
Figure 1 B shows the level of reporter gene expression (in EAP Units) from a
clone
of HT1080 cells (HT1080B) having an integrated plasmid cotransfected with
various
2s amounts of GAL4-p65 (diamonds} or GAL4-VP16 (squares) expression
constructs. Mean
values of SEAP activity secreted into the medium are shown (+/- S.D.).
Figure 1 C shows the level of reporter gene expression (in EAP Units) in a
pool of
hundreds of independent HT1080 clones carrying an integrated plasmid pLH-
SxGAL4-
IL2-SEAP cotransfected with various amounts of GAL4-p65 (diamonds) or GAL4-
VP16
30 (squares) expression constructs. Mean values of SEAP activity secreted into
the medium
are shown (+/- S.D.).
Figure 2 shows the transcriptional activity of the integrated SEAP gene from
pLH-
SxGAL4-IL2-SEAP plasmid in the presence (squares) or absence (circles) of co-
transfected SxGAL4-IL2-hGH reporter plasmid, as well as the transcriptional
activity of
35 the transiently transfected SxGAL4-IL2-hGH reporter plasmid (triangles) in
HT1080 cells
CA 02303482 2000-02-15
WO 99/10508 PCT/US97/15219
cotransfected with various amounts of GAL1-p65 expression construct
(Activator). Mean
values of SEAP activity and hGH protein secreted into the medium are shown (+/-
S.D.).
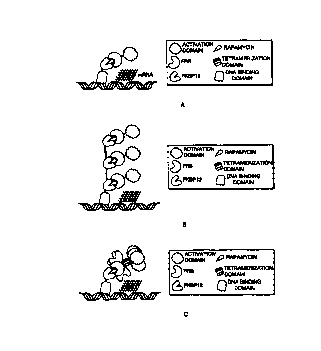
Figure 3A is a diagrammatic representation of rapamycin-induced dimerization
of
two fusion proteins, one containing the GAL4 DNA binding domain fused to FKBP
12,
s and the other containing the p65, activation domain fused to FRB, thereby
leading to
stimulation of target gene expression.
Figure 3B is a diagrammatic representation of rapamycin-induced dimerization
of
two fusion proteins, one containing a GAL4 DNA binding domain fused to the
three
copies of FKBP12, and the other containing the p65, activation domain fused to
FRB,
1 o thereby leading to association of three p65 activation domains with each
GAL4 mononer
in the presence of rapamycin.
Figure 3C is a diagrammatic representation of rapamycin-induced dimerization
of
two fusion proteins, one containing a GAL4 DNA binding domain fused to one
copy of
FKBP12, and the other containing the "bundled" fusion protein, RLS, which
contains the
~ 5 tetramerization domain of lactose repressor between FRB and the p65
activation domain,
thereby leading to association of four activation domains with each FKBP in a
rapamycin-
dependent manner.
Figure 4A represents the level of reporter gene expression (SEAP Units) of a
stably
integrated pLH-SxGAL4-IL2-SEAP plasmid in HT1080B cells cotransfected with a
2o plasmid encoding a GAL4DNA binding domain linked to I, 2, 3, or 4 FKBP12
proteins
(GFI, GF2, GF3, and GF4, respectively) and a either a plasmid encoding FRB
fused to
p65 activation domain (RS) or a plasmid encoding FRB fused to the E. coli
tetramerization
domain and p65 activation domain (RSL) in the presence of 10 nM rapamycin.
Mean
values of SEAP activity secreted into the medium following addition of 10 nM
rapamycin
25 are shown (+/- S.D.). AD/DBD ratio indicates the ratio between the amount
of plasmid
encoding a encoding an activation domain (AD) and a plasmid encoding a GAL4
DNA
binding domain (DBD).
Figure 4B represents the level of reporter gene expression (SEAP Units) of a
transiently transfected SxGAL4-IL2-SEAP plasmid in HTI080 cells cotransfected
with a
3o plasmid encoding a GAL4DNA binding domain linked to l, 2, 3, or 4 FKBP12
proteins
(GFI, GF2, GF3, and GF4, respectively) and a either a plasmid encoding FRB
fused to
p65 activation domain (RS) or a plasmid encoding FRB fused to the E. coli
tetramerization
domain and p65 activation domain (RSL) in the presence of 10 nM rapamycin.
Mean
values of SEAP activity secreted into the medium following addition of IO nM
rapamycin
35 are shown (+/- S.D.). AD/DBD ratio indicates the ratio between the amount
of plasmid
encoding a encoding an activation domain (AD) and a plasmid encoding a GAL4
DNA
CA 02303482 2000-02-15
WO 99!10508 PCT/US97/15219
_g_
binding domain (DBD).
Figure 5 shows the level of reporter gene expression (SEAP Units) of an
integrated
SEAP gene in HT1080B cells transiently transfected with varying amounts of
GAL4-p65
plasmid in the absence (square) or presence of either 120 nM trichostatin A
(diamond) 1
mM sodium butyrate (circle). Median values of SEAP activity secreted into the
medium
are shown (+/- S.D}.
Figure 6 shows the level of reporter gene expression (SEAP Units) of an
integrated
SEAP gene in HT1080B cells transiently transfected with expression vectors
encoding
GAL4 fusion proteins having an activation domain from various transcription
factors.
Figure 7 shows the level of reporter gene expression (SEAP Units) of an
integrated
SEAP gene in HT1080B cells transiently transfected with expression vectors
encoding
GAL4 fusion proteins having no activation domain (G only); 1, 2, or 4
activation domains
from VP16 (GVP16X1, GVP16X2, and GVP16X3, respectively); 1, 2, 3, or 4
activation
domains from p65 (Gp65X1, Gp65X2, Gp65X3, and Gp65X4, respectively); or a
combination of an activation domain from VP 16 and an activation domain from
p65
(Gp65 + VP16).
Figure 8A shows the level of reporter gene expression (SEAP Units) of an
integrated SEAP gene in HT1080B cells transiently transfected with expression
vectors
encoding GAL4 fusion proteins having 2, 4, 8, or 12 copies of the V8 (GV8X2,
GV8X4,
2o GV8X8, and GV8X12, respectively).
Figure 8B shows the level of reporter gene expression (SEAP Units) of an
integrated SEAP gene in HT1080B cells transiently transfected with expression
vectors
encoding GAL4 fusion proteins having 1, 2, 3, 4, 5, or 6 copies of the Vc
(GVCX2,
GVCX3, GVCX4, GVCXS, and GVCX6, respectively).
Figure 8C shows the level of reporter gene expression (SEAP Units) of an
integrated SEAP gene in HT1080B cells transiently transfected with expression
vectors
encoding GAL4 fusion proteins having 8 V8 copies (GV8X8), 5 Vc copies (GVCXS),
8
copies of V8 and 5 copies of Vc (GV8X8 + GVCXS), 8 copies of V8 and S copies
of Vc in
the reverse order (GVCXS + GV8X8), or VP16 (GVP16).
3o Figure 9A shows the level of reporter gene expression (SEAP Units) of an
integrated SEAP gene in HT1080B cells transiently transfected with expression
vectors
encoding GAL4 fusion proteins having amino acids 450-550 of p65 (Gp65 (450-
550)); I,
2, or 4 copies of amino acids 361-450 of p65; or 1, 2, or 4 copies of an
activation domain
from Spl (is this correct?).
Figure 9B shows the level of reporter gene expression (SEAP Units) of an
CA 02303482 2000-02-15
WO 99/10508 PCT/US9'7/15219
-9-
integrated SEAP gene in HT1080B cells transiently transfected with expression
vectors
encoding GAL4 fusion proteins having an Spl activation domain alone (GS) or
together
with one or two copies of the AP domain.
Figure l0A shows the level of reporter gene expression (SEAP Units) of an
integrated SEAP gene in HT1080B cells transiently transfected with expression
vectors
encoding GAL4 fusion proteins various activation domains or combinations
thereof.
Figure lOB shows the level of reporter gene expression (SEAP Units) of an
integrated SEAP gene in HT1080B cells transiently transfected with expression
vectors
encoding GAL4 fusion proteins various activation domains or combinations
thereof.
Figure 11 is a schematic representation of a composite activator.
Detailed Description of the Invention
I. General
1s Protein-encoding genes in eukaryotes are transcribed by RNA polymerise II
(pol
II), a multisubunit enzyme that is brought to an appropriate gene promoter
(pol II
promoter) through the assembly of a pre-initiation complex comprising a number
of
general transcription factors. The multisubunit protein complex TFIID is
required for
transcription by most, if not all, promoters targeted by pol II. Whereas the
TATA-box
2o binding protein (TBP} of TFIID is sufficient for basal transcription, pol
II transcription is
also regulated by gene-specific activator proteins. Activator-dependent
transcription
requires, inter aiia, TBP-associated proteins (TAFs) and other transcriptional
cofactors.
One of the important concepts to emerge from studies of eukaryotic gene
expression is that
activators of pol II-dependent transcription are composed of functional
modules whose
25 abilities to bind to subunits of the ultimate pol II complexes regulates
transcriptional
activity of a nearby gene.
The present invention pertains to nucleic acid molecules and proteins which
can be
used to regulate the expression of genes in eukaryotic cells.
One aspect of the present invention relates to chimeric transcriptional
activators
30 ("composite activators") which are derived to include a multiplicity of
heterologous
"activation tags" (further defined infra), e.g., polypeptide sequences capable
of affecting
transcriptional activation, as for example, affecting the assembly or
stability of an active
polymerise complex. It has been discovered that activation tags from disparate
proteins
can be combined in a single polypeptide, or artificially recruited by a
complex to a single
35 recognition element and retain the ability to synergistically activate
transcription. In its
CA 02303482 2000-02-15
WO 99/10508 PCT/US97/15219
-10-
simplest elaboration, the chimeric activator includes at least two different
activation tags
from heterologous sources, e.g., activation tags which are derived from two or
more
different transcription factors or co-activators and which do not naturally
occur together in
the same protein. As described further in the appended examples, constructs of
this type
s are observed to form potent transcriptional activators with a variety of
uses. Accordingly,
the invention provides composite transcriptional activators which are derived
with
activation tags from at least two separate proteins.
In this context, it has also been discovered that a variety of activation tags
can be
repeated multiple times in the chimeric protein, or multiply associated
therewith, with a
1o concomitant increase in transcriptional activation. This result was
unexpected in light of,
as an example, the belief that squelching observed in the systems of the prior
art was a
result of a rate limiting step involving the availability of general
transcription factors, e.g.,
those of the TFIID complex. Accordingly, another type of composite activator
of the
present invention is one wherein multiple copies of an activation tag are
repeated, e.g.,
15 from 2 to 20 times, in a given composite activator. The repetitive
activation tag
embodiment can be combined with other activation tags, e.g., from the same or
heterologous sources as the repeated activation tag(s). As described with
greater detail
herein, in either of the above embodiments, the activation tag can itself
induce activator
dependent transcription. Alternatively, the activation tag may be one which
alone is
2o insufficient to induce activator-dependent transcription, but rather
produces a synergistic
effect when provided with a second activation tag which itself has some
ability to induce
activator-dependent transcription. The composite activators of the present
invention may
be generated to include DNA binding domains, ligand binding domains (LBDs)
and/or
oligomerization domains (ODs).
25 Another aspect of the invention relates to the identification of novel
activation tags.
In this regard, an activation tag has been identified in the NF-kB
transcription factor
subunit p65. The so-called "alanine/proline rich" or "AP" activation tag of
p65 extends
from about amino acids 361 to about amino acid 450 of that protein. See SEQ ID
No. 2.
Similar AP activation tags are also present in, e.g., the p53 and CTF
proteins. As
3o described in the Examples, the presence of one or several copies of the AP
domain alone
in a protein does not provide the ability to induce activator-dependent
transcriptional
activation. However, when linked to activation tags which are themselves
capable of
inducing some level of activator-dependent transcription, e.g., another
portion of p65 or
VP16, the AP activation tag synergizes with the second activation domain to
induce an
35 increase in the level of activated transcription. The AP activation tag can
be used, for
example, to construct chimeric transcription factors, to generate drug
screening assays, or
as a competitive inhibitor of p65 or other transcription factors which utilize
the AP
CA 02303482 2000-02-15
WO 99/10508 PCT/US97/15219
-11-
activation tag to form transcriptional complexes.
Still another aspect of the present invention relates to the observation that,
in
contrast to episomal genes, the level of transcription of an integrated gene
is directly
related to the number of activation tags that can be delivered to the gene.
While not
wishing to be bound by any particular mechanistic theory, the inhibition of
transcription of
an episomal gene in the presence of multiple activation unit might result from
the
sequestration by the activator (unbound to DNA) of either (i) an adaptor
component
necessary to bridge the activator with the basal machinery, and/or (ii) a
basal component.
In contrast, transcription of an integrated gene appears to be enhanced by
delivery of
to additional activation units to the promoter sequences) of the gene. Thus,
the subject
chimeric activators can be used to drive high levels of transcription from
naturally-
occurnng, or otherwise genomically-integrated genes. The chimeric activators
of the
invention are particularly useful for activating transcription of integrated
single copy
genes, which in the past have not successfully transactivated at appreciable
levels.
II. Defini, 'ona
For convenience, the meaning of certain terms and phrases employed in the
specification, examples, and appended claims are provided below.
As used herein, the term "gene" or "recombinant gene" refers to a nucleic acid
2o molecule comprising an open reading frame and including at least one exon
and
(optionally) an intron sequence. The term "intron" refers to a DNA sequence
present in a
given gene which is not translated into protein and is generally found between
exons.
As used herein, the term "nucleic acid" refers to polynucleotides such as
deoxyribonucleic acid (DNA), and, where appropriate, ribonucleic acid (RNA).
The term
should also be understood to include, as equivalents, derivatives, variants
and analogs of
either RNA or DNA made from nucleotide analogs, and, as applicable to the
embodiment
being described, single (sense or antisense) and double-stranded
polynucleotides.
The term "operably linked" when referring to a transcriptional regulatory
sequence
and a coding sequence is intended to mean that the regulatory sequence is
associated with
3o the coding sequence in such a manner as to facilitate transcription of the
coding sequence
in an activator-dependent fashion.
The terms "protein", "polypeptide" and "peptide" are used interchangeably
herein
when referring to a gene product, e.g., as may be encoded by a coding
sequence.
"Transcriptional regulatory sequence", also termed herein "regulatory
element'',
"regulatory sequence" or "regulatory element", are generic terms used
throughout the
CA 02303482 2000-02-15
WO 99/10508 . PGTNS97/15219
-12-
specification to refer to DNA sequences, such as initiation signals,
enhancers, and
promoters, which induce or control transcription of protein coding sequences
with which
they are operably linked. The term "enhancer", also referred to herein as
"enhancer
element", is intended to include regulatory elements capable of increasing,
stimulating, or
enhancing transcription from a basic promoter. The term "silencer", also
referred to
herein as "silencer element" is intended to include regulatory elements
capable of
decreasing, inhibiting, or repressing transcription from a basic promoter.
Regulatory
elements can also be present in genes other than in 5' flanking sequences.
Thus, it is
possible that regulatory elements of a gene are located in introns, exons,
coding regions,
and 3' flanking sequences.
The terms "basic promoter" or "minimal promoter", as used herein, are intended
to
refer to the minimal transcriptional regulatory sequence that is capable of
initiating
transcription of a selected DNA sequence to which it is operably linked. This
term is
intended to represent a promoter element providing basal transcription. A
basic promoter
frequently consists of a TATA box or TATA-like box and is bound by an RNA
polymerase and by numerous transcription factors, such as GTFs and TATA box
Binding
Proteins (TBPs).
The terms "basic promoter" and "regulatory element" further encompass "tissue
specific" promoters and regulatory elements, i.e., promoters and regulatory
elements
2o which effect expression of the selected DNA sequence preferentially in
specific cells (e.g.,
cells of a specific tissue). Gene expression occurs preferentially in a
specific cell if
expression in this cell type is significantly higher than expression in other
cell types. The
terms "promoter" and "regulatory element" also encompass so-called "leaky"
promoters
and "regulatory elements", which regulate expression of a selected DNA
primarily in one
tissue, but cause expression in other tissues as well. The terms "promoter"
and "regulatory
element" also encompass non-tissue specific promoters and regulatory elements,
i.e.,
promoters and regulatory elements which are active in most cell types.
Furthermore, a
promoter or regulatory element can be a constitutive promoter or regulatory
element, i.e., a
promoter or regulatory element which constitutively regulates transcription,
as opposed to
3o a promoter or regulatory element which is inducible, i.e., a promoter or
regulatory element
which is active primarily in response to a stimulus. A stimulus can be, e.g.,
a molecule,
such as a hormone, a cytokine, a heavy metal, phorbol esters, cyclic AMP
(cAMP), or
retinoic acid.
The term "core promoter element" is intended to include the TATA box and the
initiator element.
"DNA recognition sequence" or "DNA recognition element", as those phrases are
used herein, mean a DNA sequence which is capable of binding to one or more
DNA-
CA 02303482 2000-02-15
WO 99/10508 PCTNS97/15219
-13-
binding domains, e.g., of a transcription factor.
The term "initiator" refers to a short, weakly conserved element that
encompasses
_ the transcription start site and which is important for directing the
synthesis of properly
initiated transcripts. While not wishing to be bound by any one theory, it is
thought that
TFIID contacts the initiator.
The term "transcription factor" refers to any protein or modified form thereof
that
is involved in the initiation of transcription but which is not itself a part
of the polymerise.
Transcription factors are proteins or modified forms thereof, which interact
preferentially
with specific nucleic acid sequences, i.e., regulatory elements, and which in
appropriate
conditions stimulate transcription ("transcriptional activators") or repress
transcription
("transcriptional repressors"). Some transcription factors are active when
they are in the
form of a monomer. Alternatively, other transcription factors are active in
the form of
oligomers consisting of two or more identical proteins or different proteins
(heterodimer).
The factors have different actions during the transcription initiation: they
may interact with
other factors, with the RNA polymerise, with the entire complex, with
activators, or with
DNA. The factors are generally classifiable into two groups: (i) the general
transcription
factors, and (ii) the transcription activators. Transcription factors usually
contain one or
more regulatory domains.
The term "regulatory domain" refers to any domain which regulates
transcription,
2o and includes both activation and repression domains. The term "activation
domain"
denotes a domain in a transcription factor which positively regulates
(increases) the rate of
gene transcription. The term "repression domain" denotes a domain in a
transcription
factor which negatively regulates (inhibits or decreases) the rate of gene
transcription.
The term "general transcription factor" used interchangeably herein with the
term
"GTF" and with "basic transcription factor" refers to proteins or protein
complexes which
work in concert with RNA Polymerise II to bring about promoter recognition and
accurate
transcription initiation. These proteins constitute, together with the RNA
polymerise II,
the Transcription Initiation Complex. GTFs include TFIIA, TFIIB, TFIID, TFIIE,
TFIIF,
and TFIIH. These GTFs are usually sufficient to direct basal levels of
transcription in
3o vitro from strong promoters (i.e., those containing TATA boxes). Several
GTF interact
with one another and/or with RNA Polymerise II. For example, TFIIE interacts
with
TFIIH and RNA Polymerise II, TFII F interacts with RNA Polymerise II and with
TFIIB,
and TFII B interacts with TBP from TFIID and RNA Polymerise II.
The term "transcriptional activator" as used herein refers to a protein or
protein
complex which is capable of enhancing the efficiency with which the basal
transcription
complex performs, i.e., activating transcription. Thus, as used herein, a
transcriptional
CA 02303482 2000-02-15
WO 99/10508 PCT/US97/15219
-14-
activator can be a single protein or alternatively it can be composed of
several units at least
some of which are not covalently linked to each other. A transcriptional
activator typically
has a modular structure, i.e., comprises various domains, such as a DNA
binding domain,
and one or more transcriptional activation tags. Some transcriptional
activators may
engage a subset of GTFs. For example, some transcriptional activators may
include
activation tags which collectively contact both TFIID and TFIIB. Other
transcriptional
activators may contact a single GTF.
The term "cofactor" which is used interchangeably herein with the terms "co-
activator", "adaptor" and "mediator" refers to proteins which either enhance
or repress
1o transcription in a non-gene specific manner, e.g., which lack intrinsic DNA
binding
specificity. Thus, cofactors are general effectors. Positively acting
cofactors do not
stimulate basal transcription, but enhance the response to an activator.
Positively acting
cofactors include PC1, PC2, PC3, PC4, and ACF. TAFs which interact directly
with
transcriptional activators are also referred to as cofactors.
The term "transcriptional activation tag", also referred to herein as
"activation tag",
"transcriptional activation unit" and "activation unit", refers to a peptide
sequence which is
capable of inducing or otherwise potentiating activator-dependent
transcription, either on
its own or when linked covalently or non-covalently to another transcriptional
activation
unit. As opposed to a transcriptional activator generally, an activation tag
corresponds to a
2o minimal polypeptide sequence which retains the ability to interact directly
or indirectly
with a transcription factor. Of course, unless otherwise clear from the
context, where a
chimeric protein is referred to as "including" or "comprising" an activation
tag, it will be
understood that other portions of the protein from which the tag is derived
can be included.
Transcriptional activation tags can be rich in certain amino acids. For
example, a
transcriptional activation unit can be a peptide rich in acidic residues,
glutamine, proline,
or serine and threonine residues. Yet other transcriptional activators can be
rich in
isoleucine or basic amino acid residues (see, e.g., Triezenberg (1995) Cur.
Opin. Gen.
Develop. 5:190, and references therein). For instance, an activation tag can
be a peptide
motif of at least about 6 amino acid residues associated with a transcription
activation
3o domain, including the well-known "acidic", "glutamine-rich" and "proline-
rich" motifs
such as the K13 motif from p65, the OCT2 Q domain and the OCT2 P domain,
respectively.
A "dimerization domain" is defined as a domain that induces formation of
dimers
between two proteins having that domain, while a "tetramerization domain" is
defined as a
domain that induces formation of tetramers amongst proteins containing the
tetramerization domain. An "oligomerization domain", generic for both
dimerization and
tetramerization domains, facilitates formation of oligomers, which can be of
any subunit
CA 02303482 2000-02-15
WO 99/10508 PCT/US97/15Z19
-15-
stoiechiometry (of course greater than one).
The term "interact" as used herein is meant to include detectable interactions
- between molecules, such as can be detected using, for example, a yeast two
hybrid assay
or by immunoprecipitation. The term interact is also meant to include
"binding"
interactions between molecules. Interactions may be, for example, protein-
protein,
protein-nucleic acid, protein-small molecule or small molecule-nucleic acid in
nature.
The term "holoenzyme complex" refers to RNA Polymerase II-containing
complexes.
The term "squelching" which is used interchangeably herein with the term
"activator interference" refers to the inhibition of transcription observed
when an activator
is present at artificially high concentrations (Ptashne and Gann (1990) Nature
346:329).
While not bound by any particular theory, this inhibition is understood to
result from the
sequestration by the activator (unbound to DNA) of either (i) an adaptor
component
necessary to bridge the activator with the basal machinery, and/or (ii) a
basal component.
The term "subunit", when referring to the subunit of a transcriptional
activator,
refers to any unit of the transcriptional activator, e.g., a transcriptional
activation unit, a
DNA binding domain, or a ligand binding domain.
The term "unit", when referring to a unit of a transcription factor, refers
generally
to a minimum portion of a transcription factor having a specific activity,
e.g.,
2o transcriptional activation, transcriptional repression, DNA binding, or
ligand binding.
As used herein, the term "transfection" means the introduction of a nucleic
acid,
e.g., an expression vector, into a recipient cell by nucleic acid-mediated
gene transfer. The
term "transduction" is generally used herein when the transfection with a
nucleic acid is by
viral delivery of the nucleic acid. "Transformation", as used herein, refers
to a process in
which a cell's genotype is changed as a result of the cellular uptake of
exogenous DNA or
RNA, and, for example, the transformed cell expresses a recombinant form of a
polypeptide or, in the case of anti-sense expression from the transferred
gene, the
expression of a naturally-occurring form of the recombinant protein is
disrupted.
As used herein, the term "transgene" refers to a nucleic acid sequence which
has
3o been introduced into a cell. Daughter cells deriving from a cell in which a
transgene has
been introduced are also said to contain the transgene (unless it has been
deleted). A
transgene can encode, e.g., a polypeptide, partly or entirely heterologous,
i.e., foreign, to
the transgenic animal or cell into which it is introduced, or, is homologous
to an
endogenous gene of the transgenic animal or cell into which it is introduced,
but which is
designed to be inserted, or is inserted, into the animal's genome in such a
way as to alter
the genome of the cell into which it is inserted (e.g., it is inserted at a
location which
CA 02303482 2000-02-15
WO 99/10508 PCTNS97/15219
- 16-
differs from that of the natural gene). Alternatively, a transgene can also be
present in an
episome. A transgene can include one or more transcriptional regulatory
sequences and
any other nucleic acid, (e.g. intron), that may be necessary for optimal
expression of a
selected coding sequence.
As used herein, the term "vector" refers to a nucleic acid molecule capable of
transporting another nucleic acid to which it has been linked. One type of
preferred vector
is an episome, i.e., a nucleic acid capable of extra-chromosomal replication.
Preferred
vectors are those capable of autonomous replication and/or expression of
nucleic acids to
which they are linked. Vectors capable of directing the expression of genes to
which they
1 o are operatively linked are referred to herein as "expression vectors". In
general, expression
vectors of utility in recombinant DNA techniques are often in the form of
"plasmids"
which refer generally to circular double stranded DNA loops which, in their
vector form
are not bound to the chromosome. In the present specification, "plasmid" and
"vector" are
used interchangeably as the plasmid is the most commonly used form of vector.
However,
the invention is intended to include such other forms of expression vectors
which serve
equivalent functions and which become known in the art subsequently hereto.
"Derived from" as that phrase is used herein indicates a peptide or nucleotide
sequence selected from within a given sequence. A peptide or nucleotide
sequence derived
from a named sequence may contain a small number of modifications relative to
the parent
2o sequence, in most cases representing deletion, replacement or insertion of
less than about
1 S%, preferably less than about 10%, and in many cases less than about 5%, of
amino
acid residues or base pairs present in the parent sequence. In the case of
DNAs, one DNA
molecule is also considered to be derived from another if the two are capable
of selectively
hybridizing to one another.
The terms "chimeric", "fusion" and "composite" are used to denote a protein,
peptide domain or nucleotide sequence or molecule containing at least two
component
portions which are mutually heterologous in the sense that they are not,
otherwise, found
directly (covalently) linked in nature. More specifically, the component
portions are not
found in the same continuous polypeptide or gene in nature, at least not in
the same order
or orientation or with the same spacing present in the chimeric protein or
composite
domain. Such materials contain components derived from at least two different
proteins or
genes or from at least two non-adjacent portions of the same protein or gene.
Composite
proteins, and DNA sequences which encode them, are recombinant in the sense
that they
contain at least two constituent portions which are not otherwise found
directly linked
(covalently) together in nature.
CA 02303482 2000-02-15
WO 99/10508 PCTNS97I15219
-17_
III ExemularY Chimeric Activators
In one aspect, the invention provides composite activators comprised of a
multiplicity of activation tags fused to, e.g., a DNA-binding domain, a ligand
binding
domain and/or an oligomerization domain.
A composite transcription activation region consists of a continuous
polypeptide
region containing two or more reiterated or otherwise heterologous activation
tags. The
activation tags comprise polypeptide sequences derived from at least two
different
proteins, polypeptide sequences which do not ordinarily occur in the same
orientation
relative to one another (including reiterated copies of a polypeptide
sequence), or
1o polypeptide sequences which do not occur in nature.
In one embodiment, at least one of the activation tags recruits TFIIA to a
TFIID
complex to form a "DA" complex, and/or stabilized the formation of a DA
complex.
Other activation tags recruit, or stabilize, complexes including other TAFs
and co-
activators required for activator-dependent transcription. For instance, in
certain cases the
composite activator sequence may recruit TFIIA and TFIIB, TFIIE, TFIIF, or
TFIIH. In
a preferred embodiment, the composite activator includes at least two
different activation
tags from the group of acidic activation tags, proline-rich transcription
activation tags,
serine/threonine-rich activation tags, glutamine-rich activation tags, and AP
activation
tags, and even more preferably, at least two of those activation tags are
selected from
2o disparate proteins (i.e., that do not naturally occur together in the same
protein).
Where multiple copies of a particular activation tag are included in the same
contiguous polypeptide, the composite activator preferably includes at least 3
copies of the
activation tag, but more preferably at least 5,10, I 5, or even at least 20
copies of the tag.
Shown in Figure 11 is a representative example of the subject composite
activators.
In the exemplary composite activator, two or more of the activation tags are
from different
proteins. The AP activation sequence and the p65 (450-550) activation sequence
are
derived from the human NF-(B p65 subunit, whereas the V8 domain is an acidic
activation
tag from the N-terminus of VP16, and the Vc activation tag is a C-terminal
portion of
VP16. Each of the p65(450-550), V8, and Vc sequences are capable of inducing
activator-
3o dependent transcription. Presented in the same polypeptide sequence, these
activation tags
induce expression of a responsive gene in a synergistic fashion. On the other
hand, the AP
activation tag on its own is insufficient to induce activator-dependent
transcription, but
when combined with an activation tag such as the V 8 activation tag, the
presence of the
AP activation tag increases transactivation of the gene relative to the V8
activation tag
alone.
Another feature of the illustrated composite activator is the repetition of
certain of
CA 02303482 2000-02-15
WO 99/10508 PCT/US97/15219
-18-
the activation tag sequences. Reiterating the V8 and Vc activation tags, for
example,
increases the fortitude of the resulting protein as a transcriptional
activator, particularly of
genomically integrated genes.
As described in more detail below, the ideal number of activation tags to
include,
and the relative orientations of the various tags to one another in the
composite activators,
can be readily determined by techniques known in the art, including high
throughput
screening using reporter gene-based assays.
Based at least in part on the lack of squelching observed from the instant
composite
activators, the subject fusion proteins can include one or more
oligomerization sequences
t o which permits non-covalent oligomerization of multiple composite
activators. For
instance, as Figure 3C shows, including a tetramerization domain permits the
formation of
multimerized complexes of the composite activator sequences. Reiterating from
above,
this embodiment derives in part from the discovery that squelching due to
abundance of
activation tags, particularly for forming the DA complex, is not a hindrance
in expression
from genomic sequences.
As desired, the composite activation sequence can be provided as part of a
fusion
protein including a DNA binding domain. In other embodiments, such as
illustrated in
Figure 3, the composite activation sequence can be fused with a ligand binding
domain
which, in the presence of a multivalent ligand, can facilitate recruitment of
the composite
2o activator to a DNA-bound complex. The complex can be loaded with multiple
activators,
in a ligand-dependent manner, by inclusion of multiple ligand binding domains.
In the instance where the composite activator contains one or more
oligomerization
domains and/or ligand binding domains, but is not contiguous with a DNA
binding
domain, the composite activator can be coexpressed in cells with a second
protein
including a DNA binding domain and appropriate oligomerization or ligand
binding
domains to form complexes with the composite activator proteins. Thus,
composite
activator proteins can be recruited to a site of transcriptional regulation by
interaction with
a DNA binding protein by oligomerization, which may be constitutive or
inducible.
Techniques for making the subject fusion proteins are adapted from well-known
3o procedures. Essentially, the joining of various DNA fragments coding for
different
polypeptide sequences is performed in accordance with conventional techniques,
employing blunt-ended or stagger-ended termini for ligation, restriction
enzyme digestion
to provide for appropriate termini, filling in of cohesive ends as
appropriate, alkaline
phosphatase treatment to avoid undesirable joining, and enzymatic ligation.
Alternatively,
the fusion gene can be synthesized by conventional techniques including
automated DNA
synthesizers. In another method, PCR amplification of gene fragments can be
carried out
CA 02303482 2000-02-15
WO 99/10508 PCT/US97/15219
-19-
using anchor primers which give rise to complementary overhangs between two
consecutive gene fragments. Amplification products can subsequently be
annealed to
generate a chimeric gene sequence (see, for example, Current Protocols in
Molecular
Biology, Eds. Ausubel et al. John Wiley & Sons: 1992).
s
A. Activation Tags
The activation tags useful in the composite activators of the present
invention can
be derived from one or more transcription factors. Polypeptides which can
fi,uiction to
activate transcription in eukaryotic cells are well known in the art. In
particular,
transcriptional activation domains which contain suitable activation tags have
been
described for many DNA binding proteins and have been shown to retain their
activation
function when the transcriptional activation domain, or a suitable fragment
thereof, is
transferred to a heterologous protein.
Activation tags can be naturally occurring or can be synthetic, so long as,
either
t 5 alone or in combination with other activation tags, they are capable of
enhancing the
efficiency with which the basal and/or activator-dependent transcription
complex
performs, i.e., so long as they are capable of interacting with a
transcription factor or co-
activator protein. Any particular activation tag is preferably at least 6
amino acids in
ti length, and preferably contains no more than about 300 amino acid residues,
though even
2o more preferably, less than 200 or even less than 100 residues.
Naturally occurring activation units include portions of transcription
factors, such as a
thirty amino acid fragment of the C-terminus of VP16 (amino acids 461-490),
referred to
herein as "Vc".
Other activation units are derivatives of naturally occurring peptides. For
example,
25 the replacement of one amino acid of a naturally occurring activation unit
by another may
further increase activation. An example of such an activation unit is a
derivative of an
eight amino acid peptide of VP16, the derivative having the amino acid
sequence
DFDLDMLG.
Yet other activation units axe entirely synthetic. It is known, for example,
that
3o certain random alignments of acidic amino acids are capable of activating
transcription.
It is well known in the art that certain transcription factors are active only
in specific cell
types, i.e., that transcription factors can act in a tissue specific manner.
Without wanting
to be limited to a specific mechanism of action, it is possible that this
tissue specificity
results from the fact that the transcription factor interacts with specific
factors, e.g,
35 cofactors, which are present only in certain cell types. This tissue
specificity can be
CA 02303482 2000-02-15
WO 99/10508 PCT/US97/15219
-20-
localized to a specific portion of the transcription factor. In certain
transcription factors,
this portion is located outside activation domains, whereas in other
transcription factors,
_ this portion may be localized within an activation domain. Thus, by using
activation tags
which are functional essentially in specific cells, it is possible to design a
transcriptional
activator of the invention having a certain tissue specificity.
A preferred polypeptide for use in the fusion protein of the invention is the
herpes simplex
virus virion protein 16 (referred to herein as VP 16, the amino acid sequence
of which is
disclosed in Triezenberg, S.J. et al. (1988) Genes Dev. 2:718-729). In one
embodiment, an
activation tag corresponding to about 127 of the C-terminal amino acids of
VP16 is used.
1o For example, a polypeptide having an amino acid sequence shown in SEQ ID
NO: X
(positions 208-335) can be used as the second polypeptide in the fusion
protein. In
another embodiment, at least one copy of about 11 amino acids from the C-
terminal region
of VP 16 which retain transcriptional activation ability is used as the
activation tag.
Preferably, an oligomer of this region (i.e., about 22 amino acids) is used.
Suitable C-
t5 terminal peptide portions of VP16 are described in Seipel, K. et al. (EMBO
J. (1992)
13:4961-4968).
Another example of an acidic activation tag is provided in residues 753-881 of
GAL4.
One particularly important source of transcription activation tags which are
20 featured in a number of embodiments of the invention is is the (human) NF-
kB subunit
p65. In one embodiment the chimeric activator contains one or more copies of a
peptide
sequence comprising all or part of the p65 sequence spanning residues 450-550,
or a
peptide sequence derived therefrom. In certain embodiments, it has been found
that
extending the p65 peptide sequence to include sequence spanning p65 residues
361-450,
25 e.g., including the "AP activation tag", leads to an unexpected increase in
transcription
activation. Moreover, a peptide sequence comprising all or a portion of
p65(361-550), or
peptide sequence derived therefrom, in combination with heterologous
activation tags, can
yield surprising additional increases in the level of transcription
activation. p65-based
activation domains function across a broad range of promoters and have yielded
increases
3o in transcription levels six-fold, eight-fold and even 14-15-fold higher
than obtained with
tandem copies of VP16 which itself is widely recognized as a very potent
activation
domain.
While the resultant increases in activation potency are dramatic, p65-based
transcription factors possess additional and unexpected characteristics. For
instance, unlike
35 VP16, the subject p65-based activators do not appear to be toxic to the
engineered cells.
This is clearly of profound practical significance in many applications. It is
expected that
recombinant DNA molecules encoding chimeric .proteins which contain a p65
activation
CA 02303482 2000-02-15
WO 99/10508 PCT/US97/15219
-Z1 -
tag, or peptide sequence derived therefrom, will provide significant
advantages for
heterologous gene expression in its various contexts, including constitutive
systems such
_ as described herein, as well as in regulated systems such as described in
International
patent applications PCT/US94/01617, PCT/(JS95/10591, PCT/L1S96/09948 and the
like,
as well as in other heterologous transcription systems such as those involving
tetracyclin
based regulation reported by Bujard et al. and those involving steroid or
other hormone-
based regulation.
One class of p65-based transcription factors contain more than one copy of a
p65-
derived domain. Such proteins will typically contain two to about six copies
of a peptide
1o sequence comprising all or a portion of p65(361-550), or peptide sequence
derived
therefrom.
Other polypeptides with transcriptional activation ability in eukaryotic cells
can be
used to provide activation tags for the fusion protein of the invention.
Transcriptional
activation domains found within various proteins have been grouped into
categories based
1 s upon similar structural features. Types of transcriptional activation
domains include, in
addition to the acidic transcription activation domains, proline-rich
transcription activation
domains, serine/threonine-rich transcription activation domains and glutamine-
rich
transcription activation domains. Examples of proline-rich activation domains
include
amino acid residues 399-499 of CTF/NF1 and amino acid residues 31-76 of AP2.
2o Examples of serine/threonine-rich transcription activation domains include
amino acid
residues 1-427 of ITFI and amino acid residues 2-451 of ITF2. Examples of
gluta~ine-
rich activation domains include amino acid residues 175-269 of Octl and amino
acid
residues 132-243 of Spl. The amino acid sequences of each of the above
described
regions, and of other useful transcriptional activation domains, are disclosed
in Seipel, K.
25 et al. (EMBO J. (1992) 13:4961-4968).
Still other illustrative activation domains and motifs of human origin include
the
activation domain of human CTF, the 18 amino acid (NFLQLPQQTQGALLTSQP)
glutamine rich region of Oct-2, the N-terminal 72 amino acids of p53, the
SYGQQS repeat
in Ewing sarcoma gene and an 11 amino acid (535-545) acidic rich region of Rel
A
3o protein.
In addition to previously described transcriptional activation domains, novel
transcriptional activation tags, which can be identified by standard
techniques, are within
the scope of the invention. The transcriptional activation ability of a
polypeptide can be
assayed by linking the polypeptide to another polypeptide having DNA binding
activity
35 and determining the amount of transcription of a target sequence that is
stimulated by the
fusion protein. For example, a standard assay used in the art utilizes a
fusion protein of a
putative activation tag and a GAL4 DNA binding domain (e.g., amino acid
residues 1-93).
CA 02303482 2000-02-15
WO 99/10508 PCT/US97/15219
-22-
This fusion protein is then used to stimulate expression of a reporter gene
linked to GAL4
binding sites (see e.g., Seipel, K. et al. (1992) EMBO J. 11:4961-4968 and
references cited
therein).
As set out above, transcriptional activators can also comprise activation
tags, which
in the absence of other activation tags are incapable of activating
transcription. For
example, the activation tag AP from VP 16 does not activate transcription in
the absence of
other activation units, even when present in multiple copies.
Accordingly, the invention provides an AP activation tag, or derivative
thereof,
which may interact with TFIIA, but which on its own is incapable of activating
1 o transcription. In one embodiment, the AP polypeptide has the amino acid
sequence of
residues 361-450 of SEQ ID N0.2, or a fragment thereof. Functionally
equivalent
derivatives of the AP activation tag can be obtained, for instance, by
screening derivatives
of AP for binding to TFIIA and measuring transcriptional activity of such a
derivative in a
co-transfection assay, as described above. Such equivalents are expected to
include forms
~ 5 of the activation tag which are tuncated at either the N-terminus or C-
terminus or both,
e.g., fragments of p65 (or homologous sequences thereto) which are about 75,
60, 50, 30
or even 20 amino acid residues in length (e.g., ranging in length from 20-89
amino acids).
Likewise, it is expected that the AP activation tag sequence from p65 can
tolerate amino
acid substitutions, e.g., to produce AP tags of at least 95%, 90%, 80% and
even 70%
2o identity with the AP activation tag sequence of SEQ ID No. 2.
The multiple activation units and other domains of the transcriptional of the
invention can be from any eukaryotic species, and it is not necessary that
every unit or
domain be from the same species. Preferred species include vertebrates, such
as
mammals. Even more preferred units or domains are from humans. For use of the
25 transcriptional activators of the invention in gene therapy in a subject of
a specific species,
e.g., human, it is preferable to use units and domains from the same species
to avoid
immune reactions against the transcriptional activator or complex.
The activation units of a transcriptional coactivator can be covalently linked
to
each other in a linear array, i.e., the NH2-terminus of one activation unit is
linked to the
3o COOH-terminus of another activation unit. The activation units can be
arranged in any
order. However, as described herein, it has been observed, that certain
arrangements of
activation units results in higher levels of transcriptional activation than
other
arrangements. The order in which the activation units should be arranged will
depend on
the result desired, i.e., the degree of transactivation that one desires to
achieve, and can be
35 determined, e.g., by performing cotransfection experiments, as described in
the Examples.
Briefly, expression vector encoding the activation units in various
arrangements linked to a
DNA binding domain are cotransfected together with a reporter construct
containing a
CA 02303482 2000-02-15
WO 99/10508 PCT1US97/15219
-23-
reporter gene operably linked to a promoter containing a DNA site recognized
by the DNA
binding domain, and expression of the reporter gene is measured. For such
assays, it is
preferable to use a cell line in which the activation units are known to be
active.
In order to optimize the transcriptional activity of a composite activator of
the
subject invention, it can be advantageous to shuffle the activation tags
relative to one
another in polypeptide. There are a variety of combinatorial techniques
available which
can be adapted for creating combinatorial libraries of the subject composite
activators by
creating and sampling libraries of proteins in which the activation tags are
shuffled in the
polypeptide to some degree relative to one another.
io In an exemplary embodiment, the coding sequences for individual activation
tags
are synthesized to include a NotI cleavage site at both the Si and 3i end by
addition of the
sequence GCGGCCGCN, which also encodes (Ala)3, an unstructured linker. The
benefits
of unstructured linkers are described below. A variety of nucleic acids
encoding such
activation tag sequences can be treated with NotI, admixed with one another,
and religated
to form a combinatorial library representing various positional combinations
of the
different activation tag sequences relative to each other. Those combinations)
of
activation tags producing the greatest level of transcriptionaI activation can
be readily
identified in the resulting combinatorial library, e.g., by selections based
on expression of
a quantifiable reporter gene.
2o In another embodiment, combinatorial intron splicing can be used to
generate a
diverse library of composite activation sequences. U.S. Patent 5,498,531
describes a
means for carrying out the equivalent of "exon shuffling" by intron-mediated
trans-
splicing. Briefly, the traps-splicing system of the X531 patent provides an
active set of
transcripts for traps-splicing wherein flanking intronic sequences can
interact in an
intermolecular reaction to reconsitute a reactive complex which promotes
transesterification of two or more transcripts. RNA transcripts are derived
which include
an "exon" sequence, e.g., encoding an activation tag, which is flanked on each
side by
intron sequences that can direct traps-splicing of the exon sequences to each
other. By
admixing these RNA constructs, intermolecular complementation between the
flanking
3o intron sequences of two different constructs forms a functional intron
which mediates the
transesterification reactions necessary to ligate two discontinuous activation
tag sequences
to one another, and thereby generate a elongated transcript comprising these
concatenated
RNA sequences encoding activation tag sequence. In one embodiment, the
activation tag
"exons" are flanked by portions of one of a group I or group II intron, such
that the
interaction of the flanking intronic sequences is sufficient to produce an
autocatalytic core
capable of driving ligation of the exons in the absence of any other factors.
In the present
context, the term "exon" merely denotes nucleic acid sequences encoding
polypeptides
CA 02303482 2000-02-15
WO 99/10508 PCT/US97/15219
-24-
including an activation tag, and can, for instance, correspond to naturally
and non-
naturally occurnng exon sequences. The resulting RNA transcripts are reverse
transcribed, and as above, the positional permutations of greatest
transcriptional activity
are easily isolated.
B. DNA binding domain
In certain embodiments of the invention, the composite activator is provided
as
part of a chimeric protein which fiu~ther comprises a DNA-binding domain. In
other
embodiments, the composite activator is localized to a DNA sequence by virture
of a
constitutive or inducible oligomerization with chimeric DNA binding domain. In
such
instances, the DNA binding domain can be provided in a fusion protein which
one or more
oligomerization domains or ligand binding domains. The choice of component DNA-
binding domains may be influenced by a number of considerations, including the
species,
system and cell type to which is targeted; the feasibility of its
incorporation into a chimeric
~ 5 protein, as may be shown by modeling; and the desired application or
utility.
The DNA binding domain can be a naturally occurring DNA-binding domain from
a transcription factor. Alternatively, the DNA binding domain can be an
artificial (or
partially artificial) polypeptide sequence having DNA binding activity. For
example, the
DNA-binding domain can be a naturally occurring DNA binding domain that has
been
2o modified to recognize a different DNA binding site. The particular DNA-
binding domain
chosen will depend on the target promoter. For example, if the gene to be
transcriptionally
activated by the subject method is an endogenous gene, the DNA-binding domain
must be
able to interact with the promoter of the endogenous gene (endogenous
promoter).
Alternatively, as described in greater detail below, the endogenous promoter
could be
25 replaced, e.g., by homologous recombination, with a heterologous promoter
for which the
DNA binding domain is selected. Such a substitution may be necessary if no
transcription
factor is known to bind the endogenous promoter of interest. Alternatively, in
such a
situation, it is also possible to clone a DNA-binding domain interacting
specifically with a
sequence in the promoter of interest. This can be done, e.g., by phage display
screening
3o with a DNA molecule comprising at least a portion of the promoter of
interest.
Desirable properties of DNA binding domains include high affinity for specific
nucleotide sequences, termed herein "target sequences", low affinity for most
other
sequences in a complex genome (such as a mammalian genome), low dissociation
rates
from specific DNA sites, and novel DNA recognition specificities distinct from
those of
35 known natural DNA-binding proteins. Preferably, binding of a DNA-binding
domain to a
specific target sequence is at least two, more preferably three and even more
preferably
CA 02303482 2000-02-15
WO 99110508 PCT/US97115219
-25-
more than four orders of magnitude greater than binding to any one alternative
DNA
sequence, as may be measured by relative Kd values or by relative rates or
levels of
transcription of genes associated with the selected and any alternative DNA
sequences. It
is also preferred that the selected DNA sequence be recognized to a
substantially greater
degree by the DNA binding domain of the trancriptional activator of the
invention than by
an endogenous protein. Thus, for example, target gene expression in a cell is
preferably
two, more preferably three, and even more preferably more than four orders of
magnitude
greater in the presence of the transcriptional activator of the invention
containing a DNA-
binding region than in its absence.
to Preferred DNA binding domains have a dissociation constant for a target
sequence
below 10-8 M, preferably 10-9 M, more preferably below 10-1 o M, even more
preferably
below 10-> > M. Far gene therapy applications, they are preferably derived
from human
proteins.
From a structural perspective, DNA-binding that can be used in the invention
may
be classified as DNA-binding proteins with a helix-turn-helix structural
design, such as,
but not limited to, Myb, Ultrabithorax, Engrailed, Paired, Fushi tarazu, HOX,
Unc86, the
Ets and homeobox families of transcription factors, and the previously noted
Octl, Oct2
and Pit; zinc f nger proteins, such as Zifl68, SWIS, Kr,ppel and Hunchback;
steroid
receptors; DNA-binding proteins with the helix-loop-helix structural design,
such as
2o Daughterless, Achaete-scute (T3), MyoD, E12 and E47; and other helical
motifs like the
leucine-zipper, which includes GCN4, C/EBP, c-Fos/c-Jun and Jung. The amino
acid
sequences of the component DNA-binding domains may be naturally-occurring or
non-
naturally-occurnng (or modified). DNA-binding domains and their target sites
can be
found at TF SEARCH (http://www.genome.ad jp/SIT/TFSEARCH html). Another
publicly available database of transcription factors and the sequences to
which they bind is
available from the National Library of Medicine in the "Transcription Data
Base".
One strategy for obtaining component DNA-binding domains with properties
suitable for this invention is to modify an existing DNA-binding domain to
reduce its
affinity for DNA into the appropriate range. For example, a homeodomain such
as that
3o derived from the human transcription factor Phoxl, may be modified by
substitution of the
glutamine residue at position 50 of the homeodomain. Substitutions at this
position
remove or change an important point of contact between the protein and one or
two base
pairs of the 6-by DNA sequence recognized by the protein. Thus, such
substitutions
reduce the free energy of binding and the affinity of the interaction with
this sequence and
may or may not simultaneously increase the affinity for other sequences. Such
a reduction
in affinity is sufficient to effectively eliminate occupancy of the natural
target site by this
protein when produced at typical levels in mammalian cells. But it would allow
this
CA 02303482 2000-02-15
WO 99/10508 PCT/US97/15219
-26-
domain to contribute binding energy to and therefore cooperate with a second
linked
DNA-binding domain. Other domains that amenable to this type of manipulation
include
the paired box, the zinc-finger class represented by steroid hormone
receptors, the myb
domain, and the ets domain.
In another embodiment, the DNA binding domain is created from the assembly of
DNA binding domains from various transcription factors, resulting in a DNA
binding
domain having a novel DNA binding specificity. Such DNA binding domains,
referred to
herein as "composite DNA binding domains" can be designed to specifically
recognize
unique binding sites. For example, a DNA binding domain can be constructed
that
1o comprises DNA binding regions from a zinc finger protein and a homeobox
protein. One
such DNA binding domain is ZFHD1, a composite DNA binding domain comprising an
Oct-1 homeodomain and zinc fingers l and 2 of Zif268, which is further
described in PCT
Application WO 96/20951 by Pomerantz et al.
The DNA sequences recognized by a chimeric protein containing a composite
~ 5 DNA-binding domain can be determined experimentally, as described below,
or the
proteins can be manipulated to direct their specificity toward a desired
sequence. A
desirable nucleic acid recognition sequence consists of a nucleotide sequence
spanning at
least ten, preferably eleven, and more preferably twelve or more bases. The
component
binding portions (putative or demonstrated) within the nucleotide sequence
need not be
2o fully contiguous; they may be interspersed with "spacer" base pairs that
need not be
directly contacted by the chimeric protein but rather impose proper spacing
between the
nucleic acid subsites recognized by each module. These sequences should not
impart
expression to linked genes when introduced into cells in the absence of the
engineered
DNA-binding protein.
25 To identify a nucleotide sequence that is recognized by a transcriptional
activator
protein containing the composite DNA-binding region, preferably recognized
with high
affinity (dissociation constant 10-1 ~ M or lower are especially preferred),
several methods
can be used. If high-affinity binding sites for individual subdomains of the
composite
DNA-binding region are already known, then these sequences can be joined with
various
3o spacing and orientation and the optimum configuration determined
experimentally (see
below for methods for determining affinities). Alternatively, high-affinity
binding sites
for the protein or protein complex can be selected from a large pool of random
DNA
sequences by adaptation of published methods (Pollock, R. and Treisman, R.,
1990, A
sensitive method for the determination of protein-DNA binding specificities.
Nucl. Acids
35 Res. 18, 6197-6204). Bound sequences are cloned into a plasmid and their
precise
sequence and affinity for the proteins are determined. From this collection of
sequences,
individual sequences with desirable characteristics (i.e., maximal affinity
for composite
CA 02303482 2000-02-15
WO 99/10508 PCT/US97/15219
_27_
protein, minimal affinity for individual subdomains) are selected for use.
Alternatively, the
collection of sequences is used to derive a consensus sequence that carries
the favored base
pairs at each position. Such a consensus sequence is synthesized and tested
(see below) to
confirm that it has an appropriate level of affinity and specificity.
A number of well-characterized assays are available for determining the
binding
affinity, usually expressed as dissociation constant, for DNA-binding proteins
and the
cognate DNA sequences to which they bind. These assays usually require the
preparation
of purified protein and binding site (usually a . synthetic oligonucleotide)
of known
concentration and specific activity. Examples include electrophoretic mobility-
shift assays,
DNaseI protection or "footprinting", and filter-binding. These assays can also
be used to
get rough estimates of association and dissociation rate constants. These
values may be
determined with greater precision using a BIAcore instrument. In this assay,
the synthetic
oligonucleotide is bound to the assay "chip," and purified DNA-binding protein
is passed
through the flow-cell. Binding of the protein to the DNA immobilized on the
chip is
~5 measured as an increase in refractive index. Once protein is bound at
equilibrium, buffer
without protein is passed over the chip, and the dissociation of the protein
results in a
return of the refractive index to baseline value. The rates of association and
dissociation
are calculated from these curves, and the affinity or dissociation constant is
calculated
from these rates. Binding rates and affinities for the high affinity composite
site may be
2o compared with the values obtained for subsites recognized by each subdomain
of the
protein. As noted above, the difference in these dissociation constants should
be at least
two orders of magnitude and preferably three or greater.
The invention further provides composite activators of the present invention
provided as a fusion protein with an inducible DNA binding domain(s). In one
25 embodiment, the inducible DNA binding domain is the E. coli tet repressor
(TetR), which
binds to tet operator (tet0) sequences upstream of target genes. In the
presence of
tetracycline, or an analog, which bind to tetR, DNA binding is abolished and
thus
transactivation is abolished. This system, in which the TetR had previously
been linked to
transcription activation domains, e.g, from VP16, is generally referred to as
an allosteric
30 "ofd switch" described by Gossen and Bujard (Proc. Natl. Acad. Sci. U.S.A.
(1992)
89:5547) and in U.S. Patents 5,464,758; 5,650,298; and 5,589,362 by Bujard et
al.
Furthermore, depending on the concentration of the antibiotic in the culture
medium (0-1
mu g/ml), target gene expression can be regulated over concentrations up to
several orders
of magnitude. Thus, the system not only allows differential control of the
activity of an
35 individual gene in.eukaryotic cells but also is suitable for creation of
"on/off' situations for
such genes in a reversible way. This system provides low background and
relatively high .
target gene expression in the absence of tetracycline or an analog. Thus, the
invention
CA 02303482 2000-02-15
WO 99/10508 ~ PCT/US97/15219
-28-
described herein provides a method for obtaining even stronger transcriptional
induction of
a target gene, which is regulatable by the tetracycline system or other
inducible DNA
binding domain. For example, a TetR can be linked to a multiplicity of
transcription
activation units, such that high levels of transcription occur in the absence
of tetracycline
or analog thereof and that transcription is repressed in the presence of
tetracycline.
In another embodiment, a "reverse" Tet system is used, again based on a DNA
binding domain that is a mutant of the E. coli TetR, but which binds to TetO
in the
presence of Tet. Thus, the invention described herein provides a method for
obtaining even
stronger transcriptional induction of a target gene in the presence of
tetracycline or an
1o analog thereof from a very low background in the absence of tetracycline.
C. Oligomerization domains
As set out above, in various embodiments of the composite activators, the
fusion
proteins can also include at least one oligomerization domain. Such a domain
can be a
~ 5 constitutive oligomerization domain, or an inducible oligomerization
domain, i.e., a
domain mediating oligomerization only in the presence of a third molecule,
such as a
small organic molecule. Examples of constitutive oligomerization domains
include
leucine zippers.
Example of inducible oligomerization domains include FK506 and cyclosporin
20 binding domains of FK506 binding proteins and cyclophilins, and the
rapamycin binding
domain of FRAP (tort). Such inducible oligomerization domains are referred to
herein as
"ligand binding domains" and are further described herein under the section
entitled
accordingly.
In one embodiment of the invention, at least one activation tag or ligand
binding
25 domain or DNA binding domain is linked to a constitutive oligomerization
domain, e.g., a
dimerization or tetramerization domain. A dimerization domain is defined
herein as a
sequence of amino acids capable of forming homodimers or heterodimers. One
example
of a dimerization domain is the leucine zipper (LZ) element. Leucine zippers
have been
identified, generally, as stretches of about 35 amino acids containing 4-5
leucine residues
30 separated from each other by six amino acids (Maniatis and Abel (1989)
Nature 341:24-
25). Exemplary leucine zippers occur in a variety of eukaryotic DNA binding
proteins,
such as GCN4, C/EBP, c-Fos, c-Jun, c-Myc and c-Max. Other dimerization domains
include helix-loop-helix domains (Murre, C. et al. (1989) Cell 58:537-544).
Dimerization
domains may also be selected from other proteins, such as the retinoic acid
receptor, the
35 thyroid hormone receptor or other nuclear hormone receptors (Kurokawa et
al. (1993)
Genes Dev. 7:1423-1435) or from the yeast transcription factors GAL4 and HAP1
CA 02303482 2000-02-15
WO 99/10508 PCT/US97/15219
-29-
(Marmonstein et al. (1992) Nature 356:408-414; Zhang et al. (1993) Proc. Natl.
Acad. Sci.
USA 90:2851-2855). Dimerization domains are further described in U.S. Patent
No.
5,624,818 by Eisenman.
In another embodiment, the oligomerization domain is a tetramerization domain.
For example, four activation units can be linked to a single DNA binding
domain or a
Iigand binding domain by covalently linking the activation units to a
tetramerization
domain. In a preferred embodiment, the tetramerization domain is the E. coli
lactose
repressor tetramerization domain (amino acids 46-360; Chakerian et al. ( 1991
) J. Biol.
Chem. 266:1371; Alberti et al. (1993) EMBO J. 12:3227; and Lewis et al. (1996)
Nature
t o 271:1247), as described in the Examples. Thus, the inclusion of a
tetramerization domain
in a transcriptional activator allows four activation domains to be complexed
together and
form a transcriptional activator complex. Furthermore, more than one
activation unit can
be linked to one tetramerization domain, to thereby form a transcriptional
activator
complex comprising more than 4 activation units.
1 s In another embodiment, the tetramerization domain is that from a p53
protein. The
p53 tetramerization domain maps to residues 322-355 of p53 (Wang et al. (1994)
Mol.
Cell. Biol. 14:5182; Clore et al. (1994) Science 265:386) and is further
described in LT.S.
Pat. No. 5,573,925 by Halazonetis.
The invention also provides for transcriptional activators containing at least
one
2o modified oligomerization domain. Modifications in the oligomerization
domain may
increase the stability of tetramer formation, for example, substitutions that
stabilize
oligomerization driven by leucine zippers are known (Krylov et al. ( 1994)
cited above;
O'Shea et al. (1992) cited above). As an exemplary modification of this type,
residues 174
or 175 of human p53 are substituted by glutamine or Ieucine, respectively, in
a p53
2s chimeric protein of this invention.
In other embodiments, the oligomerization domain can be an altered p53
tetramerization domain which is incapable of forming hetero-tetramers with p53
proteins
that have a wild-type p53 tetramerization domain, such as wild-type p53 or
tumor-derived
p53 mutants. Such altered p53 tetramerization domains are further described in
U.S. Pat.
3o No. 5,573,925 by Halazonetis.
These altered p53 tetramerization domains are characterized by disruption of
the
native p53 tetramerization domain and insertion of a heterologous
oligomerization
domain in a way that preserves tetramerization. According to this invention, a
disruption
of the p53 tetramerization domain, involving residues 335-348 or a subset of
these
35 residues, sufficiently disrupts the function of this domain so that it can
no longer drive
tetramerization with wild-type p53 or tumor-derived p53 mutants. At the same
time,
CA 02303482 2000-02-15
WO 99/10508 PCT/US97/15219
-30-
however, introduction of a heterologous dimerization domain reestablishes the
ability to
form tetramers, which is mediated both by the heterologous dimerization domain
and by
the residual tetramerizadon domain of p53.
Other exemplary suitable tetramerization domains include artificial
tetramerization domains, such as variants of the GCN4 leucine zipper that form
tetramers
(Alberti et al. (1993) EMBO J. 12:3227-3236; Harbury et al. (1993) Science
262:1401-
1407; Krylov et al. (1994) (1994) EMBO J. 13:2849-2861). One of skill in the
art could
readily select alternate tetramerization domains. For example, the tetrameric
variant of
GCN4 leucine zipper described in Harbury et al. (1993), supra, has isoleucines
at positions
1o d of the coiled coil and leucines at positions a, in contrast to the
original zipper which has
leucines and valines, respectively.
The GCN4 leucine zipper drives parallel subunit assembly [Harbury et al.
(1993),
cited above], while the native p53 tetramerization domain drives andparallel
assembly
[Clore et al. (1994) cited above; Sakamoto et al. (1994) Proc. Natl. Acad.
Sci. USA
91:8974-8978]. Thus, various conformations of activation unit complexes can be
obtained
by choosing various tetramerization domains.
In addition, the art also provides a variety of techniques for identifying
other
naturally occurring oligomerization domains, as well as oligomerization
domains derived
from mutant or otherwise artificial sequences. See, for example, Zeng et al. (
1997) Gene
185:245; O'Shea et al. (1992) Cell 68:699-708; Krylov et al. [cited above].
The distance between the oligomerization domain and other components of the
fusion proteins can be varied. In one embodiment, there is no Linker between
an activation
unit and a tetramerization domain, e.g, an altered GCN4 leucine zipper. In
other
embodiments however, there are glutamic acid or asparagine or isoleucine
linkers,
respectively. Linkers may be present for cloning convenience or to confer some
useful
property. For example, residues that stabilize specific secondary structure
elements, such
as alpha -helices, are known (Richardson et al. (1988) Science 240:1648-1652].
Such
residues can be introduced in the linkers to stabilize the oligomerization
domains. For
example the linkers glycine-asparagine, arginine-glycine- asparagine, arginine-
glycine-
3o glycine-asparagine-proline-glutamic acid, glycine-glycine- asparagine-
glutamine-alanine,
are all designed to stabilize the N-terminus of the alpha -helical
oligomerization domain.
In one embodiment, the chimeric protein comprises an activation unit fused to
an
asparagine linker and then to a tetrameric variant of GCN4 residues 249-281.
Alternatively, the linker can be an arginine-glycine-asparagine linker, an
arginine-glycine-
glycine-asparagine- proline-glutamic acid linker, a glycine-glycine-asparagine-
glutamine-
alanine linker.
CA 02303482 2000-02-15
WO 99/10508 PCT/US97/15219
-31 -
A variety of other amino acid or peptide linkers may be used for the reasons
discussed above, provided they do not interfere with the function of the
activation units
and ligand binding domain or DNA binding domain.
D. Ligand binding domain
In another embodiment of the invention, components of the subject system
include
one or more ligand binding domains for mediating oligomerization of fusion
proteins in a
ligand-dependent fashion. In a preferred embodiment, the ligand is capable of
interacting
with two ligand binding domains. In an exemplary embodiment, formation of
to transcriptional complexes is regulated by addition of a ligand, and
comprises introducing
into the cell a fusion protein including a composite activator and a ligand
binding domain,
as well as a second fusion protein including a DNA binding domain and a ligand
binding
domain, such that in the presence of the ligand, a transcriptional activator
complex is
formed between the two fusion proteins. Preferred ligands include macrolides
such as
I5 rapamycin, cyclosporin A, FK506, FK1012, and analogs thereof, and other
synthetic
dimerizers or oligomerizers. Ligand binding domains include the FK506 binding
domain
of FKBP, the cyclosporin-binding domain of calcineurin, and the raparnycin-
binding
domain of FRAP. These binding domains and ligands are further disclosed, e.g.,
in
PCTlUS93/01617. Such fusion proteins permit control of the expression of a
target gene to
2o be dependent on addition of an appropriate ligand, e.g., one which is
capable of interacting
simultaneously with the two ligand binding domains.
In general, the ligand binding domain of a chimeric protein of this invention
can be
any convenient domain which will allow for ligand-dependent oligomerization of
fusion
proteins using a natural or unnatural ligand, preferably an unnatural
synthetic ligand. Of
25 particular interest are binding proteins for which ligands (preferably
small organic ligands)
are known or may be readily produced. These receptors or ligand binding
domains include
the FKBPs and cyclophilin receptors, the steriod receptors, the tetracycline
receptor, the
other receptors indicated above, and the like, as well as "unnatural"
receptors, which can
be obtained from antibodies, particularly the heavy or light chain subunit,
mutated
3o sequences thereof, random amino acid sequences obtained by stochastic
procedures,
combinatorial syntheses, and the like.
For the most part, the receptor domains will be at least about 50 amino acids,
and
fewer than about 350 amino acids, usually fewer than 200 amino acids, either
as the
natural domain or truncated active portion thereof. Preferably the binding
domain will be
35 small (<25 kDa, to allow effcient transfection in viral vectors), monomeric
(this rules out
the avidin-biotin system), nonimmunogenic, and should have synthetically
accessible, cell
CA 02303482 2000-02-15
WO 99/10508 PCT/US97/15219
-32-
permeable, nontoxic ligands that can be configured for dimerization.
Multimerizing ligands useful in practicing this invention are multivalent,
i.e.,
capable of binding to, and thus multimerizing, two or more of chimeric protein
molecules
having a ligand binding domain. The multimerizing ligand may bind to the
proteins
containing such ligand-binding domains, in either order or simultaneously,
preferably with
a Kd value below about 10-6, more preferably below about 10-x, even more
preferably
below about I0-8, and in some embodiments below about 10-9 M. The ligand
preferably is
not a protein or polypeptide and has a molecular weight of less than about 5
kDa,
preferably below 2 kDa. The ligand-binding domains of the chimeric proteins so
multimerized may be the same or different. Ligand binding domains include
among
others, various immunophilin domains. One example is the FKBP domain which is
capable of binding to dimerizing ligands incorporating FK506 moieties or other
FKBP-
binding moieties. See e.g. PCT/L1S93/01617, the full contents of which are
hereby
incorporated by reference.
The portion of the construct encoding the ligand binding domain can be
subjected
to mutagenesis for a variety of reasons. The mutagenized domain can provide
for higher
binding affinity, allow for discrimination by a ligand between the mutant and
naturally
occurring forms of the ligand binding domain, provide opportunities to design
a ligand-
ligand binding domain pairs, or the like. The change in the ligand binding
domain can
2o involve changes in amino acids known to be at the binding site, random
mutagenesis using
combinatorial techniques, where the codons for the amino acids associated with
the
binding site or other amino acids associated with conformational changes can
be subject to
mutagenesis by changing the codon{s) for the particular amino acid, either
with known
changes or randomly, expressing the resulting proteins in an appropriate
prokaryotic host
and then screening the resulting proteins for binding. Illustrative of this
situation is to
modify FKBP12's Phe36 to Ala and/or Asp37 to Gly or Ala to accommodate a
substituent
at positions 9 or 10 of FK506 or FK520. In particular, mutant FKBP12 moieties
which
contain Val, Ala, Gly, Met or other small amino acids in place of one or more
of Tyr26,
Phe36, Asp37, Tyr82 and Phe99 are of particular interest as receptor domains
for FK506-
3o type and FK-520-type ligands containing modifications at C9 and/or C 10.
Illustrative examples of rapamycin-binding domains are those which include an
approximately 89-amino acid rapamycin-binding domain from FRAP, e.g.,
containing
residues 2025-2113 of human FRAP. Similar considerations apply to the
generation of
mutant FRAP-derived domains which bind preferentially to rapamycin analogs
(rapalogs)
containing modifications (i.e., are 'bumped') relative to rapamycin in the
FRAP-binding
effector domain. For example, one may obtain preferential binding using
rapalogs bearing
substituents other than -OMe at the C7 position with FRBs based on the human
FR.AP
CA 02303482 2000-02-15
WO 99/10508 PCT/US97115219
-33-
FRB peptide sequence but bearing amino acid substitutions for one of more of
the residues
Tyr2038, Phe2039, Thr2098, G1n2099, Trp2101 and Asp2102. Exemplary mutations
include Y2038H, Y2038L, Y2038V, Y2038A, F2039H, F2039L, F2039A. F2039V,
D2102A, T2098A, T2098N, andT2098S. Rapalogs bearing substituents other than -
OH at
C28 and/or substituents other than =O at C30 may be used to obtain
preferential binding to
FRAP proteins bearing an amino acid substitution for GIu2032. Exemplary
mutations
include E2032A and E2032S. Proteins comprising an FRB containing one or more
amino
acid replacements at the foregoing positions, libraries of proteins or
peptides randomized
at those positions (i.e., containing various substituted amino acids at those
residues),
libraries randomizing the entire protein domain, or combinations of these sets
of mutants
are made using the procedures described .above to identify mutant FRAPs that
bind
preferentially to bumped rapalogs.
Other macrolide binding domains useful in the present invention. including
mutants thereof, are described in the art. See, for example, W096/41865,
W096/13613,
~ 5 W096/06111, W096/06110, W096/06097, W096/12796, W095/05389, W095/02684,
W094/18317, each of which is expressly incorporated by reference herein.
The ability to employ in vitro mutagenesis or combinatorial modifications of
sequences encoding proteins allows for the production of libraries of proteins
which can be
screened for binding affinity for different ligands. For example, one can
totally randomize
20 a sequence of 1 to 5, 10 or more codons, at one or more sites in a DNA
sequence encoding
a binding protein, make an expression construct and introduce the expression
construct
into a unicellular microorganism, and develop a library. One can then screen
the library
for binding affinity to one or desirably a plurality of ligands. The best
affinity sequences
which are compatible with the cells into which they would be introduced can
then be used
25 as the ligand binding domain. The ligand would be screened with the host
cells to be used
to determine the level of binding of the ligand to endogenous proteins. A
binding profile
could be defined weighting the ratio of binding affinity to the mutagenized
binding
domain with the binding affinity to endogenous proteins. Those ligands which
have the
best binding profile could then be used as the ligand. Phage display
techniques, as a non-
30 limiting example, can be used in carrying out the foregoing.
In other embodiments, antibody subunits, e.g. heavy or light chain,
particularly
fragments, more particularly all or part of the variable region, or fusions of
heavy and light
chain to create single chain antibodies, can be used as the ligand binding
domain.
Antibodies can be prepared against haptenic molecules which are
physiologically
35 acceptable and the individual antibody subunits screened for binding
affinity. The cDNA
encoding the subunits can be isolated and modified by deletion of the constant
region,
portions of the variable region, mutagenesis of the variable region, or the
like, to obtain a
CA 02303482 2000-02-15
WO 99/10508 PCT/US97/15219
-34-
binding protein domain that has the appropriate affinity for the ligand. In
this way, almost
any physiologically acceptable haptenic compound can be employed as the ligand
or to
provide an epitope for the ligand. Instead of antibody units, natural
receptors can be
employed, where the binding domain is known and there is a useful ligand for
binding.
In yet another embodiment of the invention, the DNA binding unit is linked to
more than one ligand binding domain. For example, a DNA binding domain can be
linked
to at least 2, 3, 4, or 5 ligand binding domains. A DNA binding domain can
also be linked
to at least 5 ligand binding domains or any number of ligand binding domains.
In such
embodiments, the ligand binding domains can be, by illustration, linked to
each other in a
linear array, by linking the NH2-terminus of one ligand binding domain to the
COOH-
terminus of another ligand binding domain, e.g., as shown in Figure 3. Thus,
numerous
composite activators can be linked to a single DNA binding domain in the
presence of a
ligand.
The invention further provides additional induction systems. In one
embodiment,
~ 5 the invention uses an alten;iative allosteric on-switch for transcription
which employs a
deletion mutant of the human progesterone receptor, i.e., which no longer
binds
progesterone or any known endogenous steroid but can be activated by the
orally active
progesterone antagonist RU486, described, e.g, in Wang et al. (1994) Proc.
Natl. Acad.
Sci. U.S.A. 91:8180. Activation was demonstrated, e.g, in cells transplanted
into mice
2o using doses of RU486 (5-50 pg/kg) considerably below the usual dose for
inducing
abortion in humans (10 mg/kg). However, according to the art describing this
system, the
induction ratio in culture and in animals was rather low. Applying the
invention
described herein in this system would provide an inducible system having a
higher
induction ratio. Thus, the invention provides a transcriptional activator
comprising at least
25 one subunit which is covalently linked to a mutant steroid binding domain
to yield a
transcriptional activator which transactivates in a RU486-dependent manner,
resulting in
high induction ratios.
The invention can be adapted to an ecdysone inducible system. Early work
demonstrated that fusing the Drosophila steroid ecdysone (Ec) receptor (EcR)
Ec- binding
3o domain to heterologous DNA binding and activation domains, such as E. coli
lexA and
herpesvirus VP16 permits ecdysone-dependent activation of target genes
downstream of
appropriate binding sites (Christopherson et al. (1992) Proc. Natl. Acad. Sci.
U.S.A.
89:6314). An improved ecdysone regulation system has been developed, using the
DNA
binding domain of the EcR itself. In this system, the regulating transcription
factor is
35 provided as two proteins: (1) a truncated, mutant EcR fused to herpes VP16
and (2) the
mammalian homolog (RXR) of Ultraspiracle protein (USP), which heterodimerizes
with
the EcR (No et al. (1996) Proc. Natl. Acad. Sci. U.S.A. 93:3346). In this
system, because
CA 02303482 2000-02-15
WO 99/10508 PCTNS97/15219
-35-
the DNA binding domain was also recognized by a human receptor (the human
farnesoid
X receptor), it was altered to a site recognized only by the mutant EcR. Thus,
the
invention provides an ecdysone inducible system, in which a truncated mutant
EcR is
fused to at least one subunit of a transcriptional activator of the invention.
The
transcriptional activator further comprises USP, thereby providing high level
induction of
transcription of a target gene having the EcR target sequence, dependent on
the presence
of ecdysone.
The invention can also be applied to any other inducible system, thereby
providing
an inducible system having a higher ratio of background to induction due to
the potent
1 o transcriptional activity of the transcriptional activators of the
invention.
E. Additional domains and linkers
Additional domains may be included in the transcriptional activators of this
invention. For example, the transcriptional activators may contain a nuclear
localization
~ 5 sequence which provides for the protein to be translocated to the nucleus.
Accordingly, in
one embodiment of the invention, at least one of the subunits of the
transcriptional
activator of the invention, e.g., activation unit, DNA binding domain, or
ligand binding
domain, further comprises a nuclear localization signal (NLS). A NLS can be
fused to the
N-terminus, or the C-terminus of a subunit, e.g., an activation unit, or can
be inserted at
2o the junction of one subunit with another subunit, e.g., between an
activation domain and a
DNA binding or ligand binding domain or oligomerization domain or elsewhere in
the
protein, as long as the function of the subunits is not disrupted by insertion
of the NLS.
Typically a nuclear localization sequence has a plurality of basic amino
acids, referred to
as a bipartite basic repeat (reviewed in Garcia-Bustos et al. (1991)
Biochimica et
25 Biophysics Acta 1071:83-101). The NLS may be that of SV40 large T antigen
which is
comprised of amino acids proline-lysine-lysine-lysine-arginine-lysine-valine
(Kalderon et
al. (1984) Cell 39:499-509). The NLS may also be from a p53 protein. Wild-type
p53
contains three nuclear localization signals (NLS), all of which map to the C-
terminus of
wild-type p53 and specifically to residues 316-325, 369-375 and 379-384 of p53
30 (Shaulsky et al. (1990) Mol. Cell. Bio1.10:6565-6577). Additional
heterologous NLS are
described by Shaulsky et al (1990) supra and Shaulsky et al.(1991) Oncogene
6:2056.
The chimeric proteins may include domains that facilitate their purification,
e.g.
"histidine tags" or a glutathione-S-transferase domain. They may include
"epitope tags"
encoding peptides recognized by known monoclonal antibodies for the detection
of
3s proteins within cells or the capture of proteins by antibodies in vitro.
It may be necessary in some instances to introduce an unstructured polypeptide
CA 02303482 2000-02-15
WO 99/10508 PCT/IJS97/15219
-36-
linker region between an activation tag or tags and other portions of the
chimeric activator.
Where the fusion protein also includes, for example, oligomerization
sequences, it may be
preferable to situate the linker between the oligomerization sequences and the
activation
tags. The linker can facilitate enhanced flexibility of the fusion protein,
while the
oligomerization sequences are relatively free to make other inter-protein
contacts, e.g.,
with other chimeric activators. The linker can also reduce steric hindrance
between any
two fragments of the fusion protein. The linker can also facilitate the
appropriate folding
of .each fragment to occur. The linker can be of natural origin, such as a
sequence
determined to exist in random coil between two domains of a protein. An
exemplary
linker sequence is the linker found between the C-terminal and N-terminal
domains of the
RNA polyrnerase a subunit. Other examples of naturally occurring linkers
include linkers
found in the 1cI and LexA proteins. Alternatively, the linker can be of
synthetic origin.
For instance, the sequence (Gly4Ser)3 can be used as a synthetic unstructured
linker.
Linkers of this type are described in Huston et al. (1988) PNAS 85:4879; and
U.S. Patent
~5 No. 5,091,513, both incorporated by reference herein.
In some embodiments it is preferable that the design of a linker involve an
arrangement of domains which requires the linker to span a relatively short
distance,
preferably less than about 10 ~. However, in certain embodiments, depending,
e.g., upon
the selected DNA-binding domains and the configuration, the linker may span a
distance
of up to about 50 ~.
Within the linker, the amino acid sequence may be varied based on the
preferred
characteristics of the linker as determined empirically or as revealed by
modeling. For
instance, in addition to a desired length, modeling studies may show that side
groups of
certain amino acids may interfere with the biological activity, e.g. DNA
binding or
transcriptional activation, of the protein. Considerations in choosing a
linker include
flexibility of the linker, charge of the linker, and presence of some amino
acids of the
linker in the naturally-occurring subunits. The linker can also be designed
such that
residues in the linker contact DNA, thereby influencing binding affinity or
specificity, or
to interact with other proteins. For example, a linker may contain an amino
acid sequence
3o which can be recognized by a protease so that the activity of the chimeric
protein could be
regulated by cleavage. in some cases, particularly when it is necessary to
span a longer
distance between subunits or when the domains must be held in a particular
configuration,
the linker may optionally contain an additional folded domain.
Most of the subject fusion proteins can be tested for activity in vivo using a
simple
assay (F.M. Ausubel et al. Eds. Current Protocols in Molecular Biology, John
Wiley &
Sons, New York, 1994; de Wet et al. (1987) Mol. Cell Biol. 7:725). The in vivo
assay
requires an expression construct containing and capable of directing the
expression of a
*rB
CA 02303482 2000-02-15
WO 99/10508 PCT/US97/15219
-37-
recombinant DNA sequence encoding the composite activator, and as appropriate,
other
proteins required for DNA localization of the activator. The assay also
requires a plasmid
containing a reporter gene , e.g., the luciferase gene, the chloramphenicol
acetyl
transferase (CAT) gene, secreted alkaline phosphatase or the human growth
hormone
(hGH) gene, linked to a binding site for the transcription factor. The
expression constructs
are introduced into host cells which normally do not produce interfering
levels of the
reporter gene product. A second group of cells, which lacks the composite
activator or the
means for localizing the activator to the reporter gene can serve as the
control.
The production of mRNA or protein encoded by the reporter gene is measured. An
1o increase in reporter gene expression not seen in the controls indicates
that the transcription
factor is a positive regulator of transcription. If reporter gene expression
is less than that of
the control, the transcription factor is a negative regulator of
transcription.
Optionally, the assay may include a transfection efficiency control plasmid.
This
plasmid expresses a gene product independent of the test gene, and the amount
of this gene
product indicates roughly how many cells are taking up the plasmids and how
efficiently
the DNA is being introduced into the cells. Additional guidance on evaluating
chimeric
proteins of this invention is provided below.
III. Nucleic Acid Compositions
2o In another aspect of the invention, the proteins described herein are
provided in
expression vectors. For instance, expression vectors are contemplated which
include a
nucleotide sequence encoding a polypeptide containing a composite activator of
the
present invention, which coding sequence is operably linked to at least one
transcriptional
regulatory sequence. Regulatory sequences for directing expression of the
instant fusion
proteins are art-recognized and are selected by a number of well understood
criteria.
Exemplary regulatory sequences are described in Goeddel; Gene Expression
Technology:
Methods in Enzymology, Academic Press, San Diego, CA (1990). For instance, any
of a
wide variety of expression control sequences that control the expression of a
DNA
sequence when operatively linked to it may be used in these vectors to express
DNA
3o sequences encoding the fusion proteins of this invention. Such useful
expression control
sequences, include, for example, the early and late promoters of SV40,
adenovirus or
cytomegalovirus immediate early promoter, the lac system, the trp system, the
TAC or
TRC system, T7 promoter whose expression is directed by T7 RNA polymerase, the
promoter for 3-phosphoglycerate kinase or other glycolytic enzymes, the
promoters of acid
phosphatase, e.g., PhoS, and the promoters of the yeast a-mating factors and
other
sequences known to control the expression of genes of prokaryotic or
eukaryotic cells or
CA 02303482 2000-02-15
WO 99/10508 PCT/US97/15219
-38-
their viruses, and various combinations thereof. It should be understood that
the design of
the expression vector may depend on such factors as the choice of the host
cell to be
transformed. Moreover, the vector's copy number, the ability to control that
copy number
and the expression of any other protein encoded by the vector, such as
antibiotic markers,
should also be considered.
As will be apparent, the subject gene constructs can be used to cause
expression of
the subject fusion proteins in cells propagated in culture, e.g. to produce
proteins or
polypeptides, including fusion proteins, for purification.
This invention also pertains to a host cell transfected with a recombinant
gene in
to order to express one of the subject polypeptides. The host cell may be any
prokaryotic or
eukaryotic cell. For example, a fusion proteins of the present invention may
be expressed
in bacterial cells such as E. coli, insect cells (baculovirus), yeast, or
mammalian cells.
Other suitable host cells are known to those skilled in the art.
Accordingly, the present invention further pertains to methods of producing
the
subject fusion proteins. For example, a host cell transfected with an
expression vector
encoding a protein of interest can be cultured under appropriate conditions to
allow
expression of the protein to occur. The protein may be secreted, by inclusion
of a
secretion signal sequence, and isolated from a mixture of cells and medium
containing the
protein. Alternatively, the protein may be retained cytoplasmically and the
cells harvested,
lysed and the protein isolated. A cell culture includes host cells, media and
other
byproducts. Suitable media for cell culture are well known in the art. The
proteins can be
isolated from cell culture medium, host cells, or both using techniques known
in the art for
purifying proteins, including ion-exchange chromatography, gel filtration
chromatography,
ultrafiltration, electrophoresis, and immunoaffinity purification with
antibodies specific for
2s particular epitopes of the protein.
Thus, a coding sequence for a fusion protein of the present invention can be
used to
produce a recombinant form of the protein via microbial or eukaryotic cellular
processes.
Ligating the polynucleotide sequence into a gene construct, such as an
expression vector,
and transforming or transfecting into hosts, either eukaryotic (yeast, avian,
insect or
3o mammalian) or prokaryotic (bacterial cells), are standard procedures.
Expression vehicles for production of a recombinant protein include plasmids
and
other vectors. For instance, suitable vectors for the expression of the
instant fusion
proteins include plasmids of the types: pBR322-derived plasmids, pEMBL-derived
plasmids, pEX-derived plasmids, pBTac-derived plasmids and pUC-derived
plasmids for
35 expression in prokaryotic cells, such as E. coli.
A number of vectors exist for the expression of recombinant proteins in yeast.
For
CA 02303482 2000-02-15
WO 99/10508 PCT/US97115219
-39-
instance, YEP24, YIPS, YEP51, YEP52, pYES2, and YRP17 are cloning and
expression
vehicles useful in the introduction of genetic constructs into S. cerevisiae
(see, for
_ example, Broach et al., (1983) in
ExperimentalfiManipulationtoffGene'hExpression, ed.
M. Inouye Academic Press, p. 83, incorporated by reference herein). These
vectors can
replicate in E. coli due the presence of the pBR322 ori, and in S. cerevisiae
due to the
replication determinant of the yeast 2 micron plasmid. In addition, drug
resistance
markers such as ampicillin can be used.
The preferred mammalian expression vectors contain t~th prokaryotic sequences
to facilitate the propagation of the vector in bacteria, and one or more
eukaryotic
to transcription units that are expressed in eukaryotic cells. The pcDNAI/amp,
pcDNAI/neo,
pRc/CMV, pSV2gpt, pSV2neo, pSV2-dhfr, pTk2, pRSVneo, pMSG, pSVT7, pko-neo and
pHyg derived vectors are examples of mammalian expression vectors suitable for
transfection of eukaryotic cells. Some of these vectors are modified with
sequences from
bacterial plasmids, such as pBR322, to facilitate replication and drug
resistance selection
~ s in both prokaryotic and eukaryotic cells. Alternatively, derivatives of
viruses such as the
bovine papilloma virus (BPV-1), or Epstein-Barr virus (pHEBo, pREP-derived and
p205)
can be used for transient expression of proteins in eukaryotic cells. Examples
of other
viral (including retroviral) expression systems can be found below in the
description of
gene therapy delivery systems. The various methods employed in the preparation
of the
2o plasmids and transformation of host organisms are well known in the art.
For other
suitable expression systems for both prokaryotic and eukaryotic cells, as well
as general
recombinant procedures, see Molecular Cloning:fiA LaboratorytManual, 2nd Ed.,
ed. by
Sambrook, Fritsch and Maniatis (Cold Spring Harbor Laboratory Press, 1989)
Chapters 16
and 17. In some instances, it may be desirable to express the recombinant
fusion proteins
2s by the use of a baculovirus expression system. Examples of such baculovirus
expression
systems include pVL-derived vectors (such as pVL1392, pVLI393 and pVL941),
pAcUW-derived vectors (such as pAcUW 1 ), and pBlueBac-derived vectors (such
as the -
gal containing pBlueBac III).
In yet other embodiments, the subject expression constructs are derived by
3o insertion of the subject gene into viral vectors including recombinant
retroviruses,
adenovirus, adeno-associated virus, and herpes simplex virus-1, or recombinant
bacterial
or eukaryotic plasmids. As described in greater detail below, such embodiments
of the
subject expression constructs are specifically contemplated for use in various
in vivo and
ex vivo gene therapy protocols.
3s Retrovirus vectors and adeno-associated virus vectors are generally
understood to
be the recombinant gene delivery system of choice for the transfer of
exogenous genes in
vivo, particularly into humans. These vectors provide effcient delivery of
genes into
CA 02303482 2000-02-15
WO 99/10508 PCTNS97/15219
-40-
cells, and the transferred nucleic acids are stably integrated into the
chromosomal DNA of
the host. A major prerequisite for the use of retroviruses is to ensure the
safety of their
use, particularly with regard to the possibility of the spread of wild-type
virus in the cell
population. The development of specialized cell lines (termed "packaging
cells") which
produce only replication-defective retroviruses has increased the utility of
retroviruses for
gene therapy, and defective retroviruses are well characterized for use in
gene transfer for
gene therapy purposes (for a review see Miller, A.D. (1990) Blood 76:271).
Thus,
recombinant retrovirus can be constructed in which part of the retroviral
coding sequence
(gag, pol, env) has been replaced by nucleic acid encoding a fusion protein of
the present
to invention, e.g., a composite activator, rendering the retrovirus
replication defective. The
replication defective retrovirus is then packaged into virions which can be
used to infect a
target cell through the use of a helper virus by standard techniques.
Protocols for
producing recombinant retroviruses and for infecting cells in vitro or in vivo
with such
viruses can be found in Current Protocols in Molecular Biology, Ausubel, F.M.
et al.,
t5 (eds.) Greene Publishing Associates, (1989), Sections 9.10-9.14 and other
standard
laboratory manuals. Examples of suitable retroviruses include pLJ, pZIP, pWE
and pEM
which are well known to those skilled in the art. Examples of suitable
packaging virus
lines for preparing both ecotropic and amphotropic retroviral systems include
SYMBOL
121 \f "Symbol"Crip, SYMBOL 121 \f "Symbol"Cre, SYMBOL 121 \f "Symbol"2 and
2o SYMBOL 121 \f "Symbol"Am. Retroviruses have been used to introduce a
variety of
genes into many different cell types, including neural cells, epithelial
cells. endothelial
cells, lymphocytes, myoblasts, hepatocytes, bone marrow cells, in vitro and/or
in vivo (see
for example Eglitis et al., (1985) Science 230:1395-1398; Danos and Mulligan,
(1988)
PNAS USA 85:6460-6464; Wilson et al., (1988) PNAS USA 85:3014-3018; Armentano
et
25 al., (1990) PNAS USA 87:6141-6145; Huber et al., (1991) PNAS USA 88:8039-
8043;
Ferry et al., (1991) PNAS USA 88:8377-8381; Chowdhury et al., (1991) Science
254:1802-1805; van Beusechem et al., (1992) PNAS USA 89:7640-7644; Kay et al.,
(1992) Human Gene Therapy 3:641-647; Dai et al., (1992) PNAS USA 89:10892-
10895;
Hwu et al., (1993) J. Immunol. 150:4104-4115; U.S. Patent No. 4,868,116; U.S.
Patent
3o No. 4,980,286; PCT Application WO 89/07136; PCT Application WO 89/02468;
PCT
Application WO 89/05345; and PCT Application WO 92/07573).
Furthermore, it has been shown that it is possible to limit the infection
spectrum of
retroviruses and consequently of retroviral-based vectors, by modifying the
viral
packaging proteins on the surface of the viral particle (see, for example PCT
publications
35 W093/25234, W094/06920, and W094/11524). For instance, strategies for the
modification of the infection spectrum of retroviral vectors include: coupling
antibodies
specific for cell surface antigens to the viral env protein (Roux et al.,
(1989) PNAS USA
86:9079-9083; Julan et al., (1992) J. Gen Viml 73:3251-3255; and Goud et al.,
(1983)
CA 02303482 2000-02-15
WO 99/10508 PCT/US97/15219
-41 -
Virology 163:251-2S4); or coupling cell surface ligands to the viral env
proteins (Veda et
al., (1991) J. Biol. Chem. 266:14143-14146). Coupling can be in the form of
the chemical
cross-linking with a protein or other variety (e.g. lactose to convert the env
protein to an
asialoglycoprotein), as well as by generating fusion proteins (e.g. single-
chain
antibody/env fusion proteins). This technique, while useful to limit or
otherwise direct the
infection to certain tissue types, and can also be used to convert an
ecotropic vector in to
an amphotropic vector.
Another viral gene delivery system useful in the present invention utilizes
adenovirus-derived vectors. The genome of an adenovirus can be manipulated
such that it
encodes a gene product of interest, but is inactivate in terms of its ability
to replicate in a
normal lytic viral life cycle {see, for example, Berkner et al., ( 1988)
BioTechniques 6:616;
Rosenfeld et al., (1991) Science 252:431-434; and Rosenfeld et al., (1992)
Cell 68:143-
1SS). Suitable adenoviral vectors derived from the adenovirus strain Ad type S
d1324 or
other strains of adenovirus (e.g., Ad2, Ad3, Ad7 etc.) are well known to those
skilled in
~ 5 the art. Recombinant adenoviruses can be advantageous in certain
circumstances in that
they are not capable of infecting nondividing cells and can be used to infect
a wide variety
of cell types, including airway epithelium (Rosenfeld et al., {1992) cited
supra),
endothelial cells (Lemarchand et al., ( 1992) PNAS USA 89:6482-6486),
hepatocytes (Hen
and Gerard, (1993) PNAS USA 90:2812-2816) and muscle cells (Quantin et al.,
(1992)
2o PNAS USA 89:2581-2584). Furthermore, the virus particle is relatively
stable and
amenable to purif cation and concentration, and as above, can be modified so
as to affect
the spectrum of infectivity. Additionally, introduced adenoviral DNA (and
foreign DNA
contained therein) is not integrated into the genome of a host cell but
remains episomal,
thereby avoiding potential problems that can occur as a result of insertional
mutagenesis in
25 situations where introduced DNA becomes integrated into the host genome
(e.g., retroviral
DNA). Moreover, the carrying capacity of the adenoviral genome for foreign DNA
is
large (up to 8 kilobases) relative to other gene delivery vectors (Berkner et
al., supra; Haj-
Ahmand and Graham (1986) J. Virol. 57:267). Most replication-defective
adenoviral
vectors currently in use and therefore favored by the present invention are
deleted for all or
3o parts of the viral E1 and E3 genes but retain as much as 80% of the
adenoviral genetic
material (see, e.g., Jones et al., (1979) Cell 16:683; Berkner et al., supra;
and Graham et
al., in Methods in Molecular Biology, E.J. Murray, Ed. (Humans, Clifton, NJ,
1991) vol.
7. pp. 109-127). Expression of the inserted chimeric gene can be under control
of, for
example, the E 1 A promoter, the major late promoter (MLP) and associated
leader
35 sequences, the viral E3 promoter, or exogenously added promoter sequences.
Yet another viral vector system useful for delivery of the subject chimeric
genes is
the adeno-associated virus (AAV). Adeno-associated virus is a naturally
occurring
CA 02303482 2000-02-15
WO 99/10508 PC'TNS97/15219
-42-
defective virus that requires another virus, such as an adenovirus or a herpes
virus, as a
helper virus for effcient replication and a productive life cycle. (For a
review, see
Muzyczka et al., Curr. Topics in Micro. and Immunol. (1992) 158:97-129). It is
also one
of the few viruses that may integrate its DNA into non-dividing cells, and
exhibits a high
frequency of stable integration (see for example Flotte et al., (1992) Am. J.
Respir. Cell.
Mol. Biol. 7:349-356; Samulski et al., (1989) J. Viml. 63:3822-3828; and
McLaughlin et
al., (1989) J. Virol. 62:1963-1973). Vectors containing as little as 300 base
pairs of AAV
can be packaged and can integrate. Space for exogenous DNA is limited to about
4.5 kb.
An AAV vector such as that described in Tratschin et al., (1985) Mol. Cell.
Biol. 5:3251-
3260 can be used to introduce DNA into cells. A variety of nucleic acids have
been
introduced into different cell types using AAV vectors (see for example
Hermonat et al.,
(1984) PNAS USA 81:6466-6470; Tratschin et al., (1985) Mol. Cell. Biol. 4:2072-
2081;
Wondisford et al., (I988) Mol. Endocrinol. 2:32-39; Tratschin et al., (1984)
J. Virol.
51:611-619; and Flotte et al., (1993) J. Biol. Chem. 268:3781-3790).
Other viral vector systems that may have application in gene therapy have been
derived fxom herpes virus, vaccinia virus, and several RNA viruses. In
particular, herpes
virus vectors may provide a unique strategy for persistence of the recombinant
gene in
cells of the central nervous system and ocular tissue (Pepose et al., ( 1994)
Invest
Ophthalmol Vis Sci 35:2662-2666)
2o In addition to viral transfer methods, such as those illustrated above, non-
viral
methods can also be employed to cause expression of a protein in the tissue of
an animal.
Most nonviral methods of gene transfer rely on normal mechanisms used by
mammalian
cells for the uptake and intracellular transport of macromolecules. In
preferred
embodiments, non-viral gene delivery systems of the present invention rely on
endocytic
2s pathways for the uptake of the gene by the targeted cell. Exemplary gene
delivery systems
of this type include liposomal derived systems, poly-lysine conjugates, and
artificial viral
envelopes.
In a representative embodiment, a gene encoding a composite activator can be
entrapped in liposomes bearing positive charges on their surface (e.g.,
lipofectins) and
30 (optionally) which are tagged with antibodies against cell surface antigens
of the target
tissue (Mizuno et al., (1992) No Shinkei Geka 20:547-551; PCT publication
W091/06309;
Japanese patent application 1047381; and European patent publication EP-A-
43075). For
example, lipofection of neuroglioma cells can be carried out using liposomes
tagged with
monoclonal antibodies against glioma-associated antigen (Mizuno et al., (
1992) Neurol.
35 Med. Chir. 32:873-876).
In yet another illustrative embodiment, the gene delivery system comprises an
antibody or cell surface ligand which is cross-linked with a gene binding
agent such as
CA 02303482 2000-02-15
WO 99/10508 PCTlUS97/15219
-43-
poly-lysine (see, for example, PCT publications W093/04701, W092/22635,
W092/20316, W092/19749, and W092/06180). For example, any of the subject gene
constructs can be used to transfect specific cells in vivo using a soluble
polynucleotide
carrier comprising an antibody conjugated to a polycation, e.g. poly-lysine
(see U.S. Patent
5,166,320). It will also be appreciated that et~ective delivery of the subject
nucleic acid
constructs via -mediated endocytosis can be improved using agents which
enhance escape
of the gene from the ~endosomal structures. For instance, whole adenovirus or
fusogenic
peptides of the influenza HA gene product can be used as part of the delivery
system to
induce efficient disruption of DNA-containing endosomes (Mulligan et al.,
(1993) Science
l0 260-926; Wagner et al., (1992) PNAS USA 89:7934; and Christiano et al.,
(1993) PNAS
USA 90:2122).
In clinical settings, the gene delivery systems can be introduced into a
patient by
any of a number of methods, each of which is familiar in the art.
For instance, a pharrmaceutical preparation of the gene delivery system can be
I5 introduced systemically, e.g. by intravenous injection, and specific
transduction of the
construct in the target cells occurs predominantly from specificity of
transfection provided
by the gene delivery vehicle, cell-type or tissue-type expression due to the
transcriptional
regulatory sequences controlling expression of the gene, or a combination
thereof. In
other embodiments, initial delivery of the recombinant gene is more limited
with
2o introduction into the animal being quite localized. For example, the gene
delivery vehicle
can be introduced by catheter (see U.S. Patent 5,328,470) or by stereotactic
injection (e.g.
Chen et al., (1994) PNAS USA 91: 3054-3057).
IV. Target gene
25 As used herein, the term "target gene" refers to a gene, whose
transcription is
stimulated according to the method of the invention. In a preferred
embodiment, the gene
is integrated in the chromosomal DNA of a cell. A cell comprising a target
gene is
referred to herein as a "target cell".
In a preferred embodiment of the invention, the target gene is an endogenous
gene.
3o As used herein, the term "endogenous gene" refers to a gene which is
naturally present in a
cell, in its natural environment, i.e., not a gene which has been introduced
into the cell by
genetic engineering. The endogenous gene can be any gene having a promoter
that is
recognized by at least one transcription factor. In a preferred embodiment,
the promoter or
any regulatory element thereof, of the endogenous gene ("endogenous promoter"
and
35 "endogenous regulatory element", respectively), is recognized by a known,
preferably
cloned, DNA binding protein, whether it is a transcriptional activator or
repressor.
CA 02303482 2000-02-15
WO 99/I0508 PCTNS97/15219
- 44 -
Alternatively, if no DNA binding protein is known to interact with a target
promoter, it is
possible to clone such a factor using techniques well known in the art without
undue
_ experimentation, such as screening of expression libraries with at least a
portion of the
target promoter. Furthermore, the affinity of binding of a DNA binding domain
to a target
sequence can be improved according to methods known in the art. Such methods
comprise, e.g., introducing mutations into the DNA binding domain and
screening for
mutants having increased DNA binding affinity.
In another embodiment of the invention, the target gene is an endogenous gene,
which contains an exogenous target sequence. The exogenous target sequence can
be
1o inserted into the endogenous promoter or substitute at least a portion of
the endogenous
promoter. In preferred embodiments, the exogenous promoter or regulatory
element
introduced into the endogenous target promoter is recognized by a DNA binding
protein,
capable of binding with high affinity and specificity to a target sequence. In
a preferred
embodiment, the DNA binding protein is human. However, the DNA binding protein
can
be from any other species. For example, the DNA binding protein can be from
the yeast
GAL4 protein.
In yet another embodiment, the target gene is an exogenous gene. In a
preferred
embodiment, the exogenous gene is integrated into the chromosomal DNA of a
cell. The
exogenous gene can be inserted into the chromosomal DNA, or the exogenous gene
can
2o substitute for at least a portion of an endogenous gene. The target gene
can be present in a
single copy or in multiple copies. In view of the experimental results
described herein, it
is not necessary that the target gene be present in more than one copy.
However, if even
higher levels of protein encoded by the target gene is desired, multiple
copies of the gene
can be used.
In one embodiment, the taget gene construct enables transcription of a target
gene
to be regulated by a transcription factor in accordance with this invention
comprises a
DNA molecule which includes a synthetic transcription unit typically
consisting of (1)
one copy or multiple copies of a DNA sequence recognized with high-affinity by
the DNA
binding domain of a fusion protein which includes a composite activator, or of
a protein
3o which recruits the composite activator; (2) a promoter sequence consisting
minimally of a
TATA box and initiator sequence but optionally including other transcription
factor
binding sites; (3) a coding sequence for a desired gene product, including
sequences that
pmmote the initiation and termination of translation, if appropriate; (4) an
optional
sequence consisting of a splice donor, splice acceptor, and intervening intron
DNA; and
(5) a sequence directing cleavage and polyadenylation of the resulting RNA
transcript.
A wide variety of genes can be employed as the target gene, including genes
that encode a
therapeutic protein, antisense sequence or ribozyme of interest. The target
gene can be any
CA 02303482 2000-02-15
WO 99/10508 PCTNS9'7/15219
-45-
sequence of interest which provides a desired phenotype. It can encode a
surface
membrane protein, a secreted protein, a cytoplasmic protein, or there can be a
plurality of
target genes encoding different products. The target gene may be an antisense
sequence
which can modulate a particular pathway by inhibiting a transcriptional
regulation protein
or turn on a particular pathway by inhibiting the translation of an inhibitor
of the pathway.
The target gene can encode a ribozyme which may modulate a particular pathway
by
interfering, at the RNA level, with the expression of a relevant
transcriptional regulator or
with the expression of an inhibitor of a particular pathway. The proteins
which are
expressed, singly or in combination, can involve homing, cytotoxicity,
proliferation,
immune response, inflammatory response, clotting or dissolving of clots,
hormonal
regulation, etc. The proteins expressed may be naturally-occurring proteins,
mutants of
naturally-occurnng proteins, unique sequences, or combinations thereof.
Various secreted products include hormones, such as insulin, human growth
hormone, glucagon, pituitary releasing factor, ACTH, melanotropin, relaxin,
etc.; growth
factors, such as EGF, IGF-1, TGF-a, -(3, PDGF, G-CSF, M-CSF, GM-CSF, FGF,
erythropoietin, thrombopoietin, megakaryocytic stimulating and growth factors,
etc.;
interleukins, such as IL-1 to -13; TNF-a and -(3, etc.; and enzymes and other
factors, such
as tissue plasminogen activator, members of the complement cascade, performs,
superoxide dismutase, coagulation factors, antithrombin-III, Factor VIIIc,
Factor VIIIvW,
2o Factor IX, a-antitrypsin, protein C, protein S, endorphins, dynorphin, bone
morphogenetic
protein, etc.
The gene can encode a naturally-occurring surface membrane protein or a
protein
made so by introduction of an appropriate signal peptide and transmembrane
sequence.
Various such proteins include homing receptors, e.g. L-selectin (Mel-14),
blood-related
proteins, particularly having a kringle structure, e.g. Factor VIIIc, Factor
VIIIvW,
hematopoietic cell markers, e.g. CD3, CD4, CDB, B-cell receptor, TCR subunits
a, (i, y, b,
CD10, CD19, CD28, CD33, CD38, CD41, etc., receptors, such as the interleukin
receptors
IL-2R, IL-4R, etc., channel proteins for influx or efflux of ions, e.g. Ca+2,
K+, Na+, Cl-
and the like; CFTR, tyrosine activation motif, zap-70, etc.
3o Proteins may be modified for transport to a vesicle for exocytosis. By
adding the
sequence from a protein which is directed to vesicles, where the sequence is
modified
proximal to one or the other terminus, or situated in an analogous position to
the protein
source, the modified protein will be directed to the Golgi apparatus for
packaging in a
vesicle. This process in conjunction with the presence of the chimeric
proteins for
exocytosis allows for rapid transfer of the proteins to the extracellular
medium and a
relatively high localized concentration.
Also, intracellular proteins can be of interest, such as proteins in metabolic
CA 02303482 2000-02-15
WO 99/10508 PC'T/US97/15219
-46-
pathways, regulatory proteins, steroid receptors, transcription factors, etc.,
depending upon
the nature of the host cell. Some of the proteins indicated above can also
serve as
intracellular proteins.
By way of further illustration, in T-cells, one may wish to introduce genes
encoding one or both chains of a T-cell receptor. For B-cells, one could
provide the heavy
and light chains for an immunoglobulin for secretion. For cutaneous cells,
e.g.
keratinocytes, particularly stem cell keratinocytes, one could provide for
protection against
infection, by secreting a-, [3- or y-interferon, antichemotacdc factors,
proteases specific for
bacterial cell wall proteins, etc.
In addition to providing for expression of a gene having therapeutic value,
there
will be many situations where one may wish to direct a cell to a particular
site. The site
can include anatomical sites, such as lymph nodes, mucosal tissue, skin,
synovium, lung or
other internal organs or functional sites, such as clots, injured sites, sites
of surgical
manipulation, inflammation, infection, etc. By providing for expression of
surface
membrane proteins which will direct the host cell to the particular site by
providing for
binding at the host target site to a naturally-occurring epitope, localized
concentrations of a
secreted product can be achieved. Proteins of interest include homing
receptors, e.g. L-
selectin, GMP140, CLAM-1, etc., or addressins, e.g. ELAM-1, PNAd, LNAd, etc.,
clot
binding proteins, or cell surface proteins that respond to localized gradients
of chemotactic
2o factors. There are numerous situations where one would wish to direct cells
to a particular
site, where release of a therapeutic product could be of great value.
For use in gene therapy, the target gene can encode any gene product that is
beneficial to a subject. The gene product can be a secreted protein, a
membraneous
protein, or a cytoplasmic protein. Preferred secreted proteins include growth
factors,
differentiation factors, cytokines, interleukins, tPA, and erythropoietin.
Preferred
membraneous proteins include receptors, e.g, growth factor or cytokine
receptors or
proteins mediating apoptosis, e.g., Fas receptor. Other candidate therapeutic
genes are
disclosed in PCT/US93/01617.
In yet another embodiment, a "gene activation" construct which, by homologous
3o recombination with a genomic DNA, alters the transcriptional regulatory
sequences of an
endogenous gene, can be used to introduce recognition elements for a DNA
binding
activity of one of the subject engineered proteins. A vareity of different
formats for the
gene activation constructs are available. See, for example, the Transkaryotic
Therapies, Inc
PCT publications W093/09222, W095/31560, W096/29411, W095/31560 and
W094/12650.
CA 02303482 2000-02-15
WO 99/10508 PCT/US97/15219
-47-
V.. Methods of the invention
The invention provides methods for stimulating transcription of a target gene,
in
particular, an integrated gene. The gene can be an endogenous gene, an
exogenous gene,
or a combination of both. The method of the invention comprises introducing
into a target
cell a multiplicity of transcriptional activation units linked to a DNA
binding domain
and/or a Iigand binding domain. If the transcriptional activation units are
linked to a
ligand binding domain, then the method further comprises introducing into the
cell a DNA
binding domain linked to at least one ligand binding domain and a ligand
capable of
interacting with the ligand binding domains, such that the transcriptional
activation units
and DNA binding domain are interacting, thereby stimulating transcription of a
target
gene.
In another embodiment, the method of the invention comprises introducing into
a
target cell a multiplicity of ligand binding domains linked to a DNA binding
domain
together with at least one transcriptional activity unit linked to a ligand
binding domain
and a ligand which is capable of simultaneously binding to the two ligand
binding
domains. Thus, exposure of a target cell with an appropriate ligand will
result in
formation of a transcriptional activator complex comprising a multiplicity of
activation
units. Furthermore, since, as shown herein, the level of transcription of a
target gene is
directly correlated to the number of activation units that can be delivered to
the target
2o gene, the level of transcription of a target gene can be controlled by the
level of ligand
used to treat the target cell.
In a preferred embodiment, the method of the invention comprises introducing
into
a cell a target gene, a nucleic acid encoding a polypeptide having a
multiplicity of
activation units and a DNA binding domain or a ligand binding domain. If the
polypeptide
comprises a ligand binding domain, then the method further comprises
introducing into the
cell a nucleic acid encoding a polypeptide comprising a DNA binding domain and
a ligand
binding domain. The nucleic acids and vectors comprising such can be prepared
as
described above. These can be introduced into a cell comprising a target gene,
i.e., target
cell, according to methods known in the art. In embodiments in which the
target cell is in
3o vitro, the nucleic acids can be introduced into the target cell by
transfection involving
electroporation, chemical transformation (e.g., calcium chloride), liposomes,
or viral
transformation. In embodiments in which the target cell is in vivo, the
nucleic acids can
be introduced into the cell by direct injection of naked DNA, Iiposomes, or
any other
method of administration of nucleic acids to a subject.
*rB
CA 02303482 2000-02-15
WO 99/10508 PCT/US97/15219
-48-
VI Kits
This invention further provides kits useful for the foregoing applications.
One
_ such kit contains one or more nucleic acids encoding a transcriptional
activator or subunits
thereof. The kit may further comprise an additional nucleic acid containing a
target gene
linked to a DNA sequence to which the transcriptional activator is capable of
binding.
Alternatively, the additional nucleic acid may contain a cloning site for
insertion of a
desired target gene by the practitioner. For regulatable applications, i.e.,
in cases in which
the recombinant protein contains a ligand binding domain or inducible domain,
the kit may
further contain an oligomerizing agent, such as the macrolide dimerizers
discussed above.
Such kits may for example contain a sample of a dimerizing agent capable of
dimerizing
the two recombinant proteins and activating transcription of the target gene.
VII. Exemplary Uses
The invention provides methods for stimulating transcription of a target gene
at
high levels, in particular of target genes integrated into chromosomal DNA.
Strong
transcriptional activators had not been prepared in the past, since it was
known that potent
transcriptional activators would induce squelching. However, as shown herein,
transcription of an integrated gene can be stimulated at significantly higher
levels than
non-integrated genes due to the absence of squelching on integrated genes.
This
observation is important in the context of certain gene therapy applications
because it
suggests that highly potent transcriptional activators can be used to drive
therapeutic gene
expression to very high levels without general toxicity to the cell and that
efficacious
levels of secreted therapeutic proteins may be attained with fewer engineered
cells than
previously thought. Furthermore, the invention provides a method for
controlling the level
of transcription, by controlling the amount of transcriptional activation
units delivered to
the target gene by use of varying amounts of the oligomerizer.
In one embodiment, the invention is used to produce higher levels of a desired
protein ex vivo. Production of recombinant therapeutic proteins for commercial
and
investigational purposes is often achieved through the use of mammalian cell
lines
3o engineered to express the protein at high level. The use of mammalian
cells, rather than
bacteria or yeast, is indicated where the proper function of the protein
requires post-
translational modifications not generally performed by heterologous cells.
Examples of
proteins produced commercially this way include erythropoietin, tissue
plasminogen
activator, clotting factors such as Factor VIII:c, antibodies, etc. The cost
of producing
proteins in this fashion is directly related to the level of expression
achieved in the
engineered cells. Thus, because the invention described herein can achieve
considerably
CA 02303482 2000-02-15
WO 99/10508 PCT/US97/15219
-49-
higher expression levels than conventional expression systems, it may greatly
reduce the
cost of protein production. A second limitation on the production of such
proteins is
toxicity to the host cell: Protein expression may prevent cells from growing
to high
density, sharply reducing production levels. Therefore, the ability to tightly
control
protein expression, as described for regulated gene therapy, permits cells to
be grown to
high density in the absence of protein production. Only after an optimum cell
density is
reached, is expression of the gene activated and the protein product
subsequently
harvested.
A similar problem is encountered in the construction and use of "packaging
lines"
1o for the production of recombinant viruses for commercial (e.g., gene
therapy) and
experimental use. These cell lines are engineered to produce viral proteins
required for the
,_," assembly of infectious viral particles harboring defective recombinant
genomes. Viral
vectors that are dependent on such packaging lines include retrovirus,
adenovirus, and
adeno-associated virus. In the latter case, the titer of the virus stock
obtained from a
packaging line is directly related to the level of production of the viral rep
and core
proteins. But these proteins are highly toxic to the host cells. Therefore, it
has proven
difficult to generate high-titer recombinant viruses. This invention provides
a solution to
this problem, by allowing the construction of packaging lines in which the rep
and core
genes are placed under the control of regulatable transcription factors of the
design
described here. The packaging cell line can be grown to high density, infected
with helper
virus, and transfected with the recombinant viral genome. Then, expression of
the viral
proteins encoded by the packaging cells is induced by the addition of
dimerizing agent to
allow the production of virus at high titer.
In other embodiments, the subject constructs are used as part of a therapeutic
treatment program from an animal. In one embodiment, the constructs of the
invention are
used to stimulate transcription of an endogenous gene of a subject. The
endogenous gene
can be any gene of the genome, increased expression of which is beneficial to
a subject.
For example, a subject may not produce sufficient amounts of a specific
protein, due to a
defect in a protein regulating the expression of the gene encoding the
protein. In other
3o embodiments, expression of a target gene is desired to compensate for the
deficiency of
expression of another gene. In yet other embodiments, the method of the
invention is used
to stimulate expression of an endogenous gene to compensate for a loss of the
protein
encoded by the endogenous gene. For example, the number of cells producing a
specific
secreted protein may be reduced in a subject, e.g, as the result of a disease
or condition,
thus resulting in reduced production of the specific factor in the subject.
In a preferred embodiment, the method of the invention is used to stimulate
production of a factor which is necessary for the proliferation and/or
differentiation of one
CA 02303482 2000-02-15
WO 99/10508 PCT/US97/15219
-50-
or more specific cell types. For example, it may be desirable to stimulate the
expression of
growth factors and lymphokines in a subject in which at least some of the
blood cells have
been destroyed, e.g., by radiotherapy or chemotherapy. Accordingly, in a
preferred
embodiment, expression of the erythropaietin gene is stimulated in a subject,
such that
s higher levels of erythropoietin are produced in the subject and that
production of red blood
cells is stimulated. Similarly in diseases or conditions in which one or more
specific cell
types are destroyed by the disease process, e.g., in autoimmune diseases, the
specific cells
can be replenished by stimulating expression of one or more genes encoding
factors
stimulating proliferation of these cells. The method of the invention can also
be used to
1o increase the number of lymphocytes in a subject having AIDS, such as by
stimulating
expression of lymphokines, e.g., IL-4, which stimulates proliferation of
certain T helper
(Th) cells.
At least one advantage of increasing the production of a specific protein in a
subject by stimulating expression of the endogenous gene encoding the protein
is the
15 absence of an immune reaction against the protein, thus resulting in a more
efficient
treatment of the subject. Furthermore, for the same reason, it is desirable
that the
transcriptional activator or nucleic acid encoding such administered to a
subject originate
from the same species as that of the subject to which it is administered. In
particular, it is
preferable to administer a transcriptional activator of human origin to a
human subject.
2o However, transcriptional activators having activation tags which are not of
human origin
can also be used according to the methods of the invention. In such
conditions, it may be
preferable to simultaneously administer to the subject an immunosuppressant
drug, e.g.,
cyclosporin A, or other compound which represses immune reactions.
Immunosuppressive drugs are well known in the art.
25 Cells which have been modified ex vivo with the DNA constructs of the
present
invention may be grown in culture under selective conditions and cells which
are selected
as having the desired constructs) may then be expanded and further analyzed,
using, for
example, the polymerase chain reaction for determining the presence of the
construct in
the host cells and/or assays for the production of the desired gene pmduct(s).
Once
3o modified host cells have been identified, they may then be used as planned,
e.g. grown in
culture or introduced into a host organism.
Depending upon the nature of the cells, the cells may be introduced into a
host
organism, e.g. a mammal, in a wide variety of ways. Hematopoietic cells may be
adnainister~ by injection into the vascular system, there being usually at
least about 104
35 cells and generally not more than about 1010 cells. The number of cells
which are
employed will depend upon a number of circumstances, the purpose for the
introduction,
the lifetime of the cells, the protocol to be used, for example, the number of
*rB
CA 02303482 2000-02-15
WO 99/105118 PCT/US97/15219
-51-
administrations, the ability of the cells to multiply, the stability of the
therapeutic agent,
the physiologic need for the therapeutic agent, and the like. Generally, for
myoblasts or
fibroblasts for example, the number of cells will be at least about 104 and
not more than
about 109 and may be applied as a dispersion, generally being injected at or
near the site
of interest. The cells will usually be in a physiologically-acceptable medium.
Cells engineered in accordance with this invention may also be encapsulated,
e.g.
using conventional biocompatible materials and methods, prior to implantation
into the
host organism or patient for the production of a therapeutic protein. See e.g.
Hguyen et al,
Tissue Implant Systems and Methods for Sustaining viable High Cell Densities
within a
to Host, US Patent No. 5,314,471 (Baxter International, Inc.); Uludag and
Sefton, 1993, J
Biomed. Mater. Res. 27(10):1213-24 (HepG2 cells/hydroxyethyl methacrylate-
methyl
methacrylate membranes); Chang et al, 1993, Hum Gene Ther 4(4):433-40 (mouse
Ltk-
cells expressing hGH/immunoprotective perm-selective alginate microcapsules;
Reddy et
al, 1993, J Infect Dis 168(4):1082-3 (alginate); Tai and Sun, 1993, FASEB J 7(
11 ):1061-9
(mouse fibroblasts expressing hGH/alginate-poly-L-lysine-alginate membrane);
Ao et al,
1995, Transplanataion Proc. 27(6):3349, 3350 (alginate); Rajotte et al, 1995,
Transplantation Proc. 27(6):3389 (alginate); Lakey et al, 1995,
Transplantation Proc.
27(6):3266 (alginate}; Korbutt et al, 1995, Transplantation Proc. 27(6):3212
(alginate);
Dorian et al, US Patent No. 5,429,821 (alginate); Emerich et al, 1993, Exp
Neurol
122(1):37-47 (polymer-encapsulated PC12 cells); Sagen et al, 1993, J Neurosci
13(6):2415-23 (bovine chromaffn cells encapsulated in semipermeable polymer
membrane and implanted into rat spinal subarachnoid space); Aebischer et al,
1994, Exp
Neurol 126(2):151-8 (polymer-encapsulated rat PC12 cells implanted into
monkeys; see
also Aebischer, WO 92/19595); Savelkoul et al, 1994, J Immunol Methods
170(2):185-96
(encapsulated hybridomas producing antibodies; encapsulated transfected cell
lines
expressing various cytokines); Winn et al, 1994, PNAS USA 91 (6):2324-8
(engineered
BHK cells expressing human nerve growth factor encapsulated in an
immunoisolation
polymeric device and transplanted into rats); Emerich et al, 1994, Prog
Neuropsychopharmacol Biol Psychiatry 18(5):935-46 (polymer-encapsulated PC12
cells
3o implanted into rats); Kordower et al, 1994, PNAS USA 91(23):10898-902
(polymer-
encapsulated engineered BHK cells expressing hNGF implanted into monkeys) and
Butler
et al WO 95/04521 (encapsulated device). The cells may then be introduced in
encapsulated form into an animal host, preferably a mammal and more preferably
a human
subject in need thereof. Preferably the encapsulating material is
semipermeabie, permitting
release into the host of secreted proteins produced by the encapsulated cells.
In many
embodiments the semipermeable encapsulation renders the encapsulated cells
immunologically isolated from the host organism in which the encapsulated
cells are
introduced. In those embodiments the cells to be encapsulated may express one
or more
CA 02303482 2000-02-15
WO 99/10508 PCT/US97/15219
-52-
chimeric proteins containing component domains derived from proteins of the
host species
and/or from viral proteins or proteins from species other than the host
species. For
example in such cases the chimeras may contain elements derived from GAL4 and
VP16.
The cells may be derived from one or more individuals other than the recipient
and may be
s derived from a species other than that of the recipient organism or patient.
Instead of ex vivo modification of the cells, in many situations one may wish
to
modify cells in vivo. For this purpose, various techniques have been developed
for
modification of target tissue and cells in vivo. A number of viral vectors
have been
developed, such as described above, which allow for transfection and, in some
cases,
1 o integration of the virus into the host. See, for example, Dubensky et al.
( 1984) Proc. Natl.
Acad. Sci. USA 81, 7529-7533; Kaneda et al., (1989) Science 243,375-378;
Hiebert et al.
(1989} Proc. Natl. Acad. Sci. USA 86, 3594-3598; Hatzoglu et al. (1990) J.
Biol. Chem.
265, 17285-17293 and Ferry, et al. (1991) Proc. Natl. Acad. Sci. USA 88, 8377-
8381. The
vector may be administered by injection, e.g. intravascularly or
intramuscularly,
t 5 inhalation, or other parenteral mode. Non-viral delivery methods such as
administration of
the DNA via complexes with liposomes or by injection, catheter or biolistics
may also be
used.
In accordance with in vivo genetic modification, the manner of the
modification
will depend on the nature of the tissue, the efficiency of cellular
modification required, the
2o number of opportunities to modify the particular cells, the accessibility
of the tissue to the
DNA composition to be introduced, and the like. By employing an attenuated or
modified
retrovirus carrying a target transcriptional initiation region, if desired,
one can activate the
virus using one of the subject transcription factor constructs, so that the
virus may be
produced and transfect adjacent cells.
25 The DNA introduction need not result in integration in every case. In some
situations, transient maintenance of the DNA introduced may be sufficient. In
this way,
one could have a short term effect, where cells could be introduced into the
host and then
turned on after a predetermined time, for example, after the cells have been
able to home
to a particular site.
3o In another embodiment of the invention, the transcriptional activator of
the
invention recognizes a target endogenous gene, in which the promoter and/or
one or more
other regions of the gene has been modified to include a target sequence that
is specifically
recognized by the DNA binding domain of a known transcription factor and the
transcriptional activator contains this DNA binding domain. Thus, the target
endogenous
35 gene is modified to be specifically recognized by a desired transcription
factor. Such an
embodiment can be useful in situations in which no DNA binding protein is
known to
specifically bind to a regulatory region of the target gene. Thus, in one
embodiment, a cell
CA 02303482 2000-02-15
WO 99/I0508 PCT/US97/15219
-53-
is obtained from a subject and the cell is genetically engineered in vitro to
insert a desired
regulatory sequence into the promoter of the target gene. The cell can then be
further
administered to the subject. Alternatively, prior to administration of the
cell to the subject,
the cell can further be modified to include a nucleic acid encoding a
transcriptional
s activator comprising a DNA binding domain which is capable of interacting
specifically
with the regulatory element introduced into the target gene. In another
embodiment, an
endogenous gene is modified in vivo by, e.g., homologous recombination.
Modification of a gene in a cell can be done, e.g., by homologous
recombination, a
technique well known in the art, and described, e.g., in Thomas and Capecchi
(1987) Cell
51:503; Mansour et al. (1988) Nature 336:348; and Joyner et al. (1989) Nature
338:153.
In another embodiment, the transcriptional activator is used to stimulate
transcription of an exogenous gene integrated into chromosomal DNA of a
subject. An
exogenous target gene can be introduced into a subject, by obtaining a cell
from a subject,
introducing the target gene and optionally a nucleic acid encoding a
transcriptional
~5 activator into the cell and administering the cell to the subject. This
embodiment is useful
in situations in which in which no DNA binding protein is known to
specifically bind to a
regulatory region of the target gene or in situations in which the target gene
encodes a
protein which is not naturally produced by a cell. For example, the target
gene can be a
tumor antigen, which is not produced by the subject under normal conditions,
but which
20 one desires to express in the subject as a vaccine antigen to prevent
development of a
tumor expressing the tumor antigen.
Exogenous genes can also encode antisense RNA or ribozymes or other RNA
molecules which are not translated. For example, the method of the invention
can be used
to inhibit production of one or more specific proteins in a cell of a subject.
The
25 availability of potent transcriptional activators provided by the invention
will ensure that
high levels of RNA, e.g., antisense RNA, are produced in a cell.
In a preferred embodiment of the invention, the transcriptional activator is a
complex comprising a first fusion protein having multiple activation units and
a ligand
binding domain, a second fusion protein having a DNA binding domain and a
ligand
3o binding domain, and a ligand which interacts simultaneously with both
ligand binding
domains. Thus, activation of transcription of a target gene is stimulated only
in the
presence of the ligand, e.g., dimerizing agent. Accordingly, expression of the
target gene
in a subject is stimulated only upon administration of the ligand to the
subject.
The dimerizing ligand may be administered to the patient as desired to
activate
35 transcription of the target gene. Depending upon the binding affinity of
the ligand, the
response desired, the manner of administration, the half life, the number of
cells present,
CA 02303482 2000-02-15
WO 99/10508 PCTlUS97/15219
-54-
various protocols may be employed. The Iigand may be administered parenterally
or
orally. The number of administrations will depend upon the factors described
above. The
ligand may be taken orally as a pill, powder, or dispersion; bucally;
sublingually; injected
intravascularly, intraperitoneally, subcutaneously; by inhalation, or the
like. The ligand
s (and monomeric antagonist compound) may be formulated using conventional
methods
and materials well known in the art for the various routes of administration.
The precise
dose and particular method of administration will depend upon the above
factors and be
determined by the attending physician or human or animal healthcare provider.
For the
most part, the manner of administration will be determined empirically.
In the event that transcriptional activation by the ligand is to be reversed
or
terminated, a monomeric compound which can compete with the dimerizing ligand
may be
administered. Thus, in the case of an adverse reaction or the desire to
terminate the
therapeutic effect, an antagonist to the dimerizing agent can be administered
in any
convenient way, particularly intravascularly, if a rapid reversal is desired.
Alternatively,
15 one may provide for the presence of an inactivation domain (or
transcriptional silencer)
with a DNA binding domain. In another approach, cells may be eliminated
through
apoptosis via signaling through Fas or TNF receptor as described elsewhere.
See
International Patent Applications PCT/1JS94/01617 and PCT/US94/08008.
The particular dosage of the ligand for any application may be determined in
20 accordance with the procedures used for therapeutic dosage monitoring,
where
maintenance of a particular level of expression is desired over an extended
period of times,
for example, greater than about two weeks, or where there is repetitive
therapy, with
individual or repeated doses of ligand .over short periods of time, with
extended intervals,
for example, two weeks or more. A dose of the ligand within a predetermined
range
25 would be given and monitored for response, so as to obtain a time-
expression level
relationship, as well as observing therapeutic response. Depending on the
levels observed
during the time period and the therapeutic response, one could provide a
larger or smaller
dose the next time, following the response. This process would be iteratively
repeated
until one obtained a dosage within the therapeutic range. Where the ligand is
chronically
3o administered, once the maintenance dosage of the ligand is determined, one
could then do
assays at extended intervals to be assured that the cellular system is
providing the
appropriate response and level of the expression product.
It should be appreciated that the system is subject to many variables, such as
the
cellular response to the ligand, the efficiency of expression and, as
appropriate, the level of
35 secretion, the activity of the expression product, the particular need of
the patient, which
may vary with time and circumstances, the rate of loss of the cellular
activity as a result of
loss of cells or expression activity of individual cells, and the like.
Therefore, it is
CA 02303482 2000-02-15
WO 99/10508 PCT/US97/15219
-55-
expected that for each individual patient, even if there were universal cells
which could be
administered to the population at large, each patient would be monitored for
the proper
dosage for the individual.
In one embodiment of the invention, methods of introduction of an iigand,
e.g.,
dimerizing agent, and/or a cell modified according to the method of the
invention to
synthesize high levels of protein from a target gene may be provided by
rechargable or
biodegradable devices. Various slow release polymeric devices have been
developed and
tested in vivo in recent years for the controlled delivery of drugs, including
proteinacious
biopharmaceuticals. A variety of biocompatible polymers (including hydrogels),
1 o including both biodegradable and non-degradable polymers, can be used to
form an
implant for the sustained release of a dimerizer or a protein produced by a
cell modified
according to the method of the invention at a particular target site. Such
embodiments of
the present invention can be used for the delivery of an exogenously purified
protein
produced according to the method of the invention, which has been incorporated
in the
polymeric device, or for the delivery of a protein produced by a cell
encapsulated in the
polymeric device.
An essential feature of certain embodiments of the implant can be the linear
release
of the dimerizer or protein produced by the encapsulated cell which can be
achieved
through the manipulation of the polymer composition and form. By choice of
monomer
2o composition or polymerization technique, the amount of water, porosity and
consequent
permeability characteristics can be controlled. The selection of the shape,
size, polymer,
and method for implantation can be determined on an individual basis according
to the
disorder to be treated and the individual patient response. The generation of
such implants
is generally known in the art. See, for example, Concise Encylopedia of
Medical & Dental
Materials, ed. by David Williams (MIT Press: Cambridge, MA, 1990); and the
Sabel et al.
U.S. Patent No. 4,883,666. In another embodiment of an implant, a source of
cells,
modified according to the method of the invention, producing a desired
protein, or a
solution of hydogel matrix containing purifed protein or dimerizer, is
encapsulated in
implantable hollow fibers. Such fibers can be pre-spun and subsequently loaded
with the
3o protein source or dimerizer (Aebischer et al. U.S. Patent No. 4,892,538;
Aebischer et al.
U.S. Patent No. 5,106,627; Hoffman et al. (1990) Expt. Neurobiol. 110:39-44:
Jaeger et al.
( 1990) Prog. Brain Res. 82:41-46; and Aebischer et al. ( 1991 ) J. Biomech.
Eng. 113 :178-
183), or can be co-extruded with a polymer which acts to form a polymeric coat
about the
cell, protein or dimerizer (Lim U.S. Patent No. 4,391,909; Sefton U.S. Patent
No.
4,353,888; Sugamori et al. (1989) Trans. Am. Artif. Intern. Organs 35:791-799;
Sefton et
al. (1987) Biotehnol. Bioeng. 29:1135-1143; and Aebischer et al. .(1991)
Biomaterials
12:50-5 5).
i
CA 02303482 2000-02-15
WO 99/10508 PCT/US97/15219
-56-
Thus, the method of the invention can broadly be applied to any situation,
e.g., for
treating or preventing any disease or condition, in which transcriptional
activation of an
_ integrated target gene is desired. Depending on the specific embodiment of
the invention,
a transcriptional activator, nucleic acid encoding such, target exogenous
gene, and/or
oligomerizer is administered to a subject. These an be administered as such or
together
with a delivery vehicle, e.g., liposomes. Whether with or without a delivery
vehicle, these
compounds are preferably administered together with a pharmaceutically
acceptable
carrier. Methods of administration of these compounds are known in the art and
are
briefly disclosed below.
1 o Toxicity and therapeutic efficacy of such compounds can be determined by
standard pharmaceutical procedures in cell cultures or experimental animals,
e.g., for
determining the LDADVANCE \d 1 SOADVANCE \u 1 (the dose lethal to 50% of the
population) and the EDADVANCE 1d 1 SOADVANCE \u 1 (the dose therapeutically
effective in 50% of the population). The dose ratio between toxic and
therapeutic effects
is the therapeutic index and it can be expressed as the ratio LDADVANCE \d 1
SOADVANCE \u 1 /EDADVANCE \d 1 SOADVANCE 1u 1 . Compounds which exhibit
large therapeutic indices are preferred. While compounds that exhibit toxic
side effects
may be used, care should be taken to design a delivery system that targets
such compounds
to the site of affected tissue in order to minimize potential damage to
uninfected cells and,
thereby, reduce side effects.
The data obtained from the cell culture assays and animal studies can be used
in
formulating a range of dosage for use in humans. The dosage of such compounds
lies
preferably within a range of circulating concentrations that include the
EDADVANCE \d 1
SOADVANCE \u 1 with little or no toxicity. The dosage may vary within this
range
depending upon the dosage form employed and the route of administration
utilized. For
any compound used in the method of the invention, the therapeutically
effective dose can
be estimated initially from cell culture assays. A dose may be formulated in
animal
models to achieve a circulating plasma concentration range that includes the
ICADVANCE \d 1 SOADVANCE \u 1 (i.e., the concentration of the test compound
which
3o achieves a half maximal inhibition of symptoms) as determined in cell
culture. Such
information can be used to more accurately determine useful doses in humans.
Levels in
plasma may be measured, for example, by high performance liquid
chromatography.
Pharmaceutical compositions for use in accordance with the present invention
may
be formulated in conventional manner using one or more physiologically
acceptable
Garners or excipients. Thus, the compounds and their physiologically
acceptable salts and
solvates may be formulated for administration by, for example, injection,
inhalation or
insufflation (either through the mouth or the nose) or oral, buccal,
parenteral or rectal
CA 02303482 2000-02-15
WO 99/10508 PCT/US97/15219
-57-
administration.
For such therapy, the compounds of the invention can be formulated for a
variety
of loads of administration, including systemic and topical or localized
administration.
Techniques and formulations generally may be found in Remmington's
Pharmaceutical
Sciences, Meade Publishing Co., Easton, PA. For systemic administration,
injection is
preferred, including intramuscular, intravenous, intraperitoneal, and
subcutaneous. For
injection, the compounds of the invention can be formulated in liquid
solutions, preferably
in physiologically compatible buffers such as Hank's solution or Ringer's
solution. In
addition, the compounds may be formulated in solid form and redissolved or
suspended
io immediately prior to use. Lyophilized forms are also included.
For oral administration, the pharmaceutical compositions may take the form of
for
example, tablets or capsules prepared by conventional means with
pharmaceutically
acceptable excipients such as binding agents (e.g., pregelatinised maize
starch,
polyvinylpyrrolidone or hydroxypropyl methylcellulose); fillers (e.g.,
lactose,
microcrystalline cellulose or calcium hydrogen phosphate); lubricants (e.g.,
magnesium
stearate, talc or silica); disintegrants (e.g., potato starch or sodium starch
glycolate); or
vretting agents (e.g., sodium lauryl sulphate). The tablets may be coated by
methods well
known in the art. Liquid preparations for oral administration may take the
form of, for
example, solutions, syrups or suspensions, or they may be presented as a dry
product for
2o constitution with water or other suitable vehicle before use. Such liquid
preparations may
be prepared by conventional means with pharmaceutically acceptable additives
such as
suspending agents (e.g., sorbitol syrup, cellulose derivatives or hydrogenated
edible fats);
emulsifying agents (e.g., lecithin or acacia); non-aqueous vehicles (e.g.,
almond oil, oily
esters, ethyl alcohol or fractionated vegetable oils); and preservatives
(e.g., methyl or
propyl-p-hydroxybenzoates or sorbic acid). The preparations may also contain
buffer
salts, flavoring, coloring and sweetening agents as appropriate.
Preparations for oral administration may be suitably formulated to give
controlled
release of the active compound. For buccal administration the compositions may
take the
form of tablets or lozenges formulated in conventional manner. For
administration by
3o inhalation, the compounds for use according to the present invention are
conveniently
delivered in the form of an aerosol spray presentation from pressurized packs
or a
nebuliser, with the use of a suitable propellant, e.g.,
dichlorodifluoromethane,
trichlorofluoromethane, dichlorotetrafluoroethane, carbon dioxide or other
suitable gas. In
the case of a pressurized aerosol the dosage unit may be determined by
providing a valve
to deliver a metered amount. Capsules and cartridges of e.g. gelatin for use
in an inhaler
or insui~lator may be formulated containing a powder mix of the compound and a
suitable
powder base such as lactose or starch.
CA 02303482 2000-02-15
WO 99/10508 PCT/US97/15219
-58-
The compounds may be formulated for parenteral administration by injection,
e.g.,
by bolus injection or continuous infusion. Formulations for injection may be
presented in
unit dosage form, e.g., in ampoules or in multi-dose containers, with an added
preservative. The compositions may take such forms as suspensions, solutions
or
emulsions in oily or aqueous vehicles, and may contain formulatory agents such
as
suspending, stabilizing and/or dispersing agents. Alternatively, the active
ingredient may
be in powder form for constitution with a suitable vehicle, e.g., sterile
pyrogen-free water,
before use.
The compounds may also be formulated in rectal compositions such as
1o suppositories or retention enemas, e.g., containing conventional
suppository bases such as
cocoa butter or other glycerides.
In addition to the formulations described previously, the compounds may also
be
formulated as a depot preparation. Such long acting formulations may be
administered by
implantation (for example subcutaneously or intramuscularly) or by
intramuscular
injection. Thus, for example, the compounds may be formulated with suitable
polymeric
or hydrophobic materials (for example as an emulsion in an acceptable oil) or
ion
exchange resins, or as sparingly soluble derivatives, for example, as a
sparingly soluble
salt.
Systemic administration can also be by transmucosal or transdermal means. For
2U transmucosal or transdermal administration, penetrants appropriate to the
barrier to be
permeated are used in the formulation. Such penetrants are generally known in
the art, and
include, for example, for transmucosal administration bile salts and fusidic
acid
derivatives. In addition, detergents may be used to facilitate permeation.
Transmucosal
administration may be through nasal sprays or using suppositories. For topical
administration, the oligomers of the invention are formulated into ointments,
salves, gels,
or creams as generally known in the art.
In clinical settings, the gene delivery systems for the genes encoding
transcriptional activators and optionally target gene can be introduced into a
patient by any
of a number of methods, each of which is familiar in the art. For instance, a
3o pharmaceutical preparation of the gene delivery system can be introduced
systemically,
e.g. by intravenous injection, and specific transduction of the protein in the
target cells
occurs predominantly from specificity of transfection provided by the gene
delivery
vehicle, cell-type or tissue-type expression due to the transcriptional
regulatory sequences
controlling expression of the receptor gene, or a combination thereof. In
other
embodiments, initial delivery of the recombinant gene is more limited with
introduction
into the animal being quite localized. For example, the gene delivery vehicle
can be
introduced by catheter (see U.S. Patent 5,328,470) or by stereotactic
injection (e.g. Chen et
CA 02303482 2000-02-15
WO 99/10508 PCT/US97/15219
-59-
al. (1994) PNAS 91: 3054-3057).
The pharmaceutical preparation of the gene therapy construct can consist
_ essentially of the gene delivery system in an acceptable diluent, or can
comprise a slow
release matrix in which the gene delivery vehicle is imbedded. Alternatively,
where the
complete gene delivery system can be produced intact from recombinant cells,
e.g.
retroviral vectors, the pharmaceutical preparation can comprise one or more
cells which
produce the gene delivery system.
The compositions may, if desired, be presented in a pack or dispenser device
which
may contain one or more unit dosage forms containing the active ingredient.
The pack
to may for example comprise metal or plastic foil, such as a blister pack. The
pack or
dispenser device may be accompanied by instructions for administration.
The present invention is further illustrated by the following examples which
should
not be construed as limiting in any way. The contents of all cited references
including
literature references, issued patents, published patent applications as cited
throughout this
~ 5 application are hereby expressly incorporated by reference. The practice
of the present
invention will employ, unless otherwise indicated, conventional techniques of
cell biology,
cell culture, molecular biology, transgenic biology, microbiology, recombinant
DNA, and
immunology, which are within the skill of the art. Such techniques are
explained fully in
the literature. See, for example, Molecular CloningfiA LaboratoryfiManual, 2nd
Ed., ed. by
20 Sambrook, Fritsch and Maniatis (Cold Spring Harbor Laboratory Press: 1989);
DNA
Cloning, Volumes I and II (D. N. Glover ed., 1985); Oligonucleotide Synthesis
(M. J. Gait
ed., 1984); Mullis et al. U.S. Patent No: 4,683,195; Nucleic Acid
Hybridization (B. D.
Hames & S. J. Higgins eds. 1984); Transcription And Translation (B. D. Hames &
S. J.
Higgins eds. 1984); Culture Of Animal Cells (R. I. Freshney, Alan R. Liss,
Inc., 1987);
25 Immobilized Cells And Enzymes (IRL Press, 1986); B. Perbal, A Practical
Guide To
Molecular Cloning (1984); the treatise, Methods In Enzymology (Academic Press,
Inc.,
N.Y.); Gene Transfer Vectors For Mammalian Cells (J. H. Miller and M. P. Calos
eds.,
1987, Cold Spring Harbor Laboratory); Methods In Enzymology, Vols. 154 and 155
(Wu
et al. eds.), Immunochemical Methods In Cell And Molecular Biology (Mayer and
3o Walker, eds., Academic Press, London, 1987); Handbook Of Experimental
Immunology,
Volumes I-IV (D. M. Weir and C. C. Blackwell, eds., 1986); Manipulating the
Mouse
Embryo, (Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y., 1986).
Egemplific$tion
35 The invention now being generally described, it will be more readily
understood by
reference to the following examples which are included merely for purposes of
illustration
CA 02303482 2000-02-15
WO 99/10508 PCT/US97/15219
-60-
of certain aspects and embodiments of the present invention, and are not
intended to limit
the invention.
Example 1: Stimulation of High Level Transcription of an Integrated Gene
This Example shows that a gene integrated into chromosomal DNA is immune
from the effect of squelching and that high level transcription of an
integrated gene can be
obtained by delivering multiple activation domains to the gene.
First, the effect of increasing concentrations of a transcriptional activator
on an
integrated versus a non-integrated reporter gene was compared as follows. Two
constructs
1 o encoding chimeric transcriptional activators under the control of the CMV
enhancer/promoter were used. The first construct (pCG-GV) encodes a chimeric
transcription factor composed of the yeast GAL4 DNA-binding domain (amino
acids 1-94)
and activation domain (amino acids 410-490) derived from the herpes simplex
virus
protein VP16 (Sadowski, L, et al. (1988) Nature 335:563-4). The second
construct (pCG-
CS) encodes a chimeric transcription factor composed of the yeast GAL4 DNA-
binding
domain (amino acids 1-94) and the activation domain (amino acids 361-550) from
the NF-
B ~ p65 protein (Ballard, D. W., et al. ( 1991 ) Proc. Natl. Acad. Sci 89:1875-
1879;
Schmitz, M.L. and Baeuerle, P.A. (1991) EMBO J. 10:3805-3817). The effect of
these
transcription factors was tested on a target gene composed of a secreted
alkaline
2o phosphatase (SEAP) reporter under the control of a minimal human IL-2 gene
promoter
flanked by five GAL4 binding sites.
HT 1080 cells were transiently transfected with the reporter construct and/or
a
construct encoding a chimeric transcription factor as follows. HT1080 cells
were grown at
37 C in MEM medium containing 10% fetal calf serum, non-essential amino acids
and
penicillin-streptomycin. Twenty-four hours before transfection, approximately
2X105
cells were seeded in each well in a 12-well plate. Cells were transfected
using
Lipofectamine as recommended (Gibco BIRI,). Cells in each well received the
amount of
plasmids indicated in the figure, with or without 400 ng of reporter plasmid,
with the total
amount of DNA being adjusted to 1.25 pg with pUCl9. Five hours later, the
medium was
3o removed and 1 ml of fresh medium added. 18-24 hours later, 100 ~l medium
was removed
and assayed for SEAP activity using a Luminescence Spectrometer (Perkin Elmer)
at 350
nm excitation and 450 nm emission.
HT1080 cells were stably transfected with the reporter construct pLH-SxGAL4-
IL2-SEAP as follows. The retroviral vector pLH-SxGal4-IL2-SEAP was constructed
by
3s cloning the SxGAL4-IL2-SEAP fragment described above into the vector pLH (
lzivera,
V.M., et al. (1996) Nature Medicine 2:1028-1032), which also contains the
hygromycin B
CA 02303482 2000-02-15
WO 99/10508 PCT/US97/15219
-61 -
resistance gene driven by the Moloney marine leukemia virus long terminal
repeat.
Helper-free retrovirus, generated as described (Rivers, V.M., et al. Nature
Medicine
2,1028-1032 (1996)) was used to infect HT1080 cells. Hundreds of hygromycin B
(300
~g/ml) resistant clones were pooled (HT1080 B pool) and individual clones
screened by
transient transfection with pCG-GS. The most responsive clone, HT1080B, was
seiected
for further analysis.
When the reporter gene and expression plasmids encoding the transcription
factors
were both introduced transiently into HT1080 human fibrosarcoma cells, peak
reporter
gene expression at relatively low levels of input activator plasmid was
observed, while
1 o higher levels of activator led to a sharp reduction in reporter gene
expression (Fig. 1 A).
This inhibition of gene expression in the presence of high levels of activator
has been
widely observed, and the phenomenon has been termed "squelching." On the
contrary,
expression of the integrated reporter gene was not inhibited by levels of
activator that
sharply squelched a transiently transfected reporter (Fig. 1 B).
To rule out the possibility that the immunity of the integrated gene to
squelching
reflects the effect of a single unusual integration site, this experiment was
repeated using a
pool of several hundred HT1080-derived clones harboring independent reporter
gene
insertions. As shown in Figure 1 C, expression of this population of inserted
reporter genes
was not inhibited by high levels of activator. A second reporter construct in
which the
2o same GAL4-driven IL-2 promoter was fused to a human growth hormone (hGH)
reporter
gene was constructed by replacing the SEAP gene with the hGH gene. This
plasmid was
cotransfected with the GAL4-p65 expression plasmid into cells containing an
integrated
SEAP reporter gene, thus allowing both reporter genes to be assayed in the
same cell
population. Transfections and reporter gene expression were measured as
described
above. For determining expression of the hGH reporter construct, 2-5 ~.l of
medium was
assayed for hGH protein as recommended (Nichols Diagnostic).
If, in the presence of a high-copy episomal template, GAL4-p65 titrates GTFs
necessary for the activity of this promoter, then one would expect to observe
inhibition of
both the episomal hGH gene and the integrated SEAP gene. However, the results
indicate
3o that, whereas expression of the episomal hGH gene was inhibited at high
activator
concentrations, the integrated SEAP gene responded identically whether or not
the
episomal gene was present (Fig. 2). Similar results were obtained in parallel
experiments
with GAL4-VP16. This observation indicates that one or more mechanistic steps
in the
transcription cycle differ between episomal and integrated genes.
Example 2: Transcription of an Integrated Gene Increases Proportionally to the
*rB
CA 02303482 2000-02-15
WO 99/10508 ~ PCT/US97/15219
-62-
Number of Activation Domains Delivered to the Gene
This Example shows that expression of an integrated gene can be driven to very
high levels by delivering many potent activation domains to the promoter of
the gene.
A modular strategy was designed to deliver different numbers of activation
domains to a single integrated target gene. This strategy was based on the
ability of a
small-molecule "dimerizer" to recruit activation domains to a DNA-bound
receptor
(Belshaw, P.J., et al. (1996) Proc. Natl. Acad. Sci 93:4604-4607; Rivers,
V.M., et al.
(1996) Nature Medicine 2:1028-1032; Ho, S.N., et al. (1996) Nature 382:822-
824). The
basic system, which is diagrammed in Fig. 3A, was composed of a GAL4 DNA-
binding
o domain fused to a single copy of human FKBP12 and a p65 activation domain
fused to the
FRB domain of FRAP (Ho, S.N., et al. , supra; Sabatini, D.M., et al. (1994)
Cell 78:35-
43). In the presence of the natural-product immunosuppressive compound
rapamycin, the
FRB-p65 fusion protein is efficiently recruited to the GAL4-FKBP fusion
protein. This
basic system results in the delivery of a maximum of one p65 activation domain
per GAL4
monomer. The number of deliverable activation domains was increased in two
ways: (i)
by increasing the number of FKBP moieties fused to GAL4, as indicated in Fig.
3B; and
(ii) through the use of a tetramerization domain derived from the E. coli lac
repressor
(Chakerian, A.E., et al. (1991) J Biol Chem 266:1371-4; Alberta, S., et al.
(1993) EMBO
J. 12: 3227-36; Lewis, M., et al. (1996) Nature 271:1247-1254) to deliver
"bundles" of
2o four activation domains to each FKBP moiety, as shown in Fig. 3C. These
different
configurations allowed recruitment of up to sixteen p65 activation domains to
a single
GAL4 monomer.
These expression constructs were prepared as follows. pCGNN-G expression
vector was made by inserting a PCR fragment containing the GAL4 DNA binding
domain
(amino acids 1-94) flanked by upstream Xbal and downstream SpeI and BamHI
sites into
XbaI- and BamHI- digested pCGNN (Ricardo Attar, please provide a reference).
FKBP12
coding sequence (amino acids 1-107), described in....., flanked by upstream
XbaI and
downstream SpeI and BamHI sites was inserted between the SpeI and BamHI sites
of
pCGNN-G to generate pCGNN-GF 1. Plasmids pCGNN-GF2, GF3 and GF4 were made
3o by the sequential insertion of FKBPI2 coding sequences into SpeI- and BamHI
digested
pCGNN-GF1, 2 and 3 plasmids, respectively. PCGNN-RL was constructed by cloning
an
XbaI-BamHI fragment containing a portion of the E. coli lactose repressor
(amino acids
46-360) into SpeI- and BamHI-digested pCGNN-R (Rivers, V.M., et al. supra) to
fuse it to
the carboxy terminus of the FRB domain. The p65 activation domain was fused to
the
carboxy terminus of this chimera by inserting an XbaI-BamHI fragment into SpeI-
and
BamHI digested pCGNN-RL to create pCGNN-RLS.
Various combinations of expression constructs, i.e., 10 ng of plasmid
expressing
CA 02303482 2000-02-15
WO 99/10508 PCT/US97/15219
-63-
DNA binding domain fusions and increasing amounts of plasmid expressing p65
activation domain fusions) were transfected into HT1080B cells carrying an
integrated
reporter gene. The cells were then treated with 10 nM rapamycin to deliver the
activation
domains to the target gene and the amount of SEAP was measured in the
supernatant. As
shown in Figure 4B and 4C, the results indicate a roughly linear increase in
reporter gene
expression as a function of the number of activation domains that are
delivered to a single
GAL4 monomer. In contrast, except for the increment from one to two activation
domains
per GAL4 monomer, increasing the number of activation domains that can be
delivered to
an episomal target gene (introduced by transient transfection) failed to
enhance the peak
o level of gene expression achieved before squelching occurred (Figures 4B and
4C).
These results indicate that expression of the episomal reporter gene can be
driven
only to a fixed level before squelching sets in, effectively establishing a
ceiling for
expression from the episomal gene. Because squelching does not impose such a
limitation
on the integrated gene, expression can be substantially augmented by
delivering more
activation domains, indicating that expression of an integrated chromatin-
embedded gene
is limited largely by,the number (or strength) of activation domains bound at
the promoter.
Furthermore, the level of expression reached by the integrated gene is
considerably higher
than that attained by the episomal gene, especially when considered on a per-
template
basis, as shown in Figure 4C.
Example 3: Transcriptional Activation Units can Synergize to Activate
Transcription of an Integrated Target Gene
This Example shows that specific combinations of transcriptional activation
domains can result in potent transcriptional activators capable of strong
transactivation of
integrated single copy genes.
The effect of various constructs encoding GAL4 DNA binding domain fusion
proteins on transcription of an integrated pLH-SxGal4-IL2-SEAP vector in
HT1080 cells
(clone HT1080B) was determined. The activation domains fused to the GAL4 DNA
binding domain (amino acids 1-94) consisted of amino acids 18x4 of QIII
(GQIII), amino
3o acids 1-92 of p53 (Gp53), amino acids 417-490 of VP16 (GVP16), amino acids
450-550
of p65 (Gp65), amino acids 399-499 of CTF (GCTF), amino acids 411-508 of SRF
(GSRF), or amino acids 263-499 of SP 1 (GSP 1 ).
Various amounts of these constructs were transiently transfected into the
HT1080B
cell line having a single integrated copy of the vector pLH-SxGal4-IL2-SEAP
and the
level of expression of the reporter gene was measured as described above. The
results.
which are represented in Figure 6, indicate that among the various fusion
proteins, only
CA 02303482 2000-02-15
WO 99/10508 PCTNS97/15219
GAL4-VP 16 and GAL4-p65 activate transcription of the integrated single copy
reporter
gene.
Next, the transactivation potential of combinations of transcriptional
activation
domains were tested on a single copy integrated gene. Expression constructs
encoding a
GAL4 DNA binding domain linked to l, 2, or 4 VP16 activation domains (amino
acids
417-490) or 1, 2, 3, or 4 p65 activation domains (amino acids 450-550) or a
p65 activation
domain fused to a VP 16 activation domain. The results are presented in Figure
7 and
indicate that a multiplicity of p65 activation domains or VP 16 activation
domains do not
provide higher levels of transcription relative to a transcriptional activator
containing a
to single p65 or VP16 activation domain. In contrast, the activation domains
of p65 and
VP16 are capable of cooperating in transcriptional activation of an integrated
target gene.
In another experiment, the transcriptional potential of combinations of
transcriptional activation domains of VP16 on a single copy integrated gene
was
determined. Expression constructs encoding the following fusion proteins
containing a
GAL4 DNA binding domain (amino acids 1-94) were prepared and tested in
transient
transfections of the HT1080 cell clone containing a single integrated vector
pLH-SxGal4-
IL2-SEAP: fusion proteins containing 2, 4, 8, or 12 copies (GV8X2, GV8X4,
GV8X8, and
GV8X12, respectively) of an eight amino acid variant of a transactivation
domain from
VP16 having the amino acid sequence DFDLDMLG (referred to herein as "V8"
peptide);
2o fusion proteins containing 1, 2, 3, 4, 5, or 6 copies (GVCX1, GVCX2, GVCX3,
GVCX4,
GVCXS, GVCX6, respectively) of a transactivation domain from the C-terminus of
VP16
corresponding to amino acids 461-490 (referred to herein as "Vc peptide); and
fusion
proteins containing 8 copies of V8 and 5 copies of Vc, wherein the V8 copies
are fused
either to the N-terminus or the C-terminus of Vc. The results, which are
presented in
Figure 8, panels A and B, indicate cooperativity between V8 peptides up to
about 8 copies
of V8 and up to 5 copies of Vc. Higher numbers of copies of V8 and Vc result
in reduced
transcriptional activity of the reporter gene. However, as shown in Figure 8,
panel C, a
combination of 8 copies of V8 fused to the N-terminus of 5 copies of Vc,
results in
stronger transcriptional levels than that obtained with either 8 copies of V8
or 5 copies of
3o Vc. This transcriptional level is even higher than that obtained with full
length VP16.
Interestingly however, a fusion protein containing 8 copies of V8 fused to the
C-terminus
of 5 copies of Vc does not result in higher transcriptional activity of the
reporter
constructs, relative to 8 copies of V8 and 5 copies of Vc. Thus, specific
arrangements of
transcriptional activation domains are more potent in activating transcription
than other
arrangements.
Thus, this example demonstrates that combination of multiple activation
domains
can result in potent transcriptional activators capable of stimulating
transcription of a
CA 02303482 2000-02-15
WO 99/10508 PCT/US97/15219
-65-
single copy integrated gene.
Example 4: An Alanine/Proline Rich Domain of p65 is Capable of Strongly
Transactivating a Target Gene, when in the Presence of Another Transactivation
Domain
This Example demonstrates that a specific domain of gp65 is incapable to
stimulate
transcription of a target gene on its own, but can synergize with other
transcriptional
activation domains to stimulate transcription of a target gene.
Analysis of the p65 amino acid sequence revealed the presence of two
Alanine/Proline rich regions in p65. The first domain, i.e., domain I,
consists of amino
acids 376-401 and has the amino acid sequence SALALAPAPPQVLPQAPAPAPAPAMV
(SEQ ID NO. ). The second domain, i.e., domain II, consists of amino acids 402-
427 and
has the amino acid sequence SALAQAPAPVPVLAPGPPQAVAPPAP (SEQ ID NO...).
The transcriptional potential of these sequences was investigated as follows.
Constructs
encoding fusion proteins containing GAL4 DNA binding domain (amino acids I-92)
and
the following domains were prepared for use in transient transfection: fusion
proteins
containing 1, 2, or 3 copies of amino acids 361-450 of p65 containing the two
above-
described alanine/proline rich regions and referred to herein as the AP
domain; fusion
proteins containing I, 2, or 4 copies of the SPI activation domain; and fusion
proteins
containing an SPI activation domain fused at its N-terminal or C-terminal
region to one or
two AP domains.
The amount of reporter gene expression obtained upon transfection of these
constructs into the HT1080B cell line containing an integrated copy of the
vector pLH-
SxGal4-IL2-SEAP is shown in Figures 9A and B. These results indicate that 2
and 4
copies of the AP domain does not result in significant transactivation of the
reporter gene,
in contrast to an increase in transcription proportional to the number of Spl
domains added
(Figure 9A). However, the combination of one or more AP domains with an Sp 1
activation domain synergistically activated transcription of the reporter
construct (Figure
9B). Interestingly, the synergy appears to be stronger when the AP domain is
linked to the
N-terminus of the Spl activation domain. Thus, a combination of
transcriptional
activation domains results in transcriptional activators which can be much
more potent
than naturally occurring transcription factors.
Yet other combinations of transactivation domains were tested for their effect
on
transcription of an integrated single copy gene. The fusion proteins tested
comprised a
GAL4 DNA binding domain and one or more V8 domains, one or more Vc domains,
and/or one or more AP domains. Expression constructs encoding these fusion
proteins
CA 02303482 2000-02-15
WO 99/1OS08 PCTNS97/15219
-66-
were transiently transfected into the HT1080 cell line containing an
integrated copy of the
vector pLH-SxGal4-IL2-SEAP. The results are shown in Figures l0A and i OB.
These
results show that the AP domain potentiates the transcriptional activation of
various
combinations of transcriptional activation domains. However, the AP domain has
a
stronger potentiating effect on some activation domains than on others. In
particular, as
shown in Figure 10A, AP synergizes with V8, but increases only slightly
transcription by
Vc. While not wishing to be bound by any particular theory, we note that one
possible
explanation for this phenomenon is that both the AP domain and Vc interact
with the same
GTF, i.e., TFIIA, whereas V8 interacts with a different GTF. Thus, it is
possible that
to synergy between different transcriptional activation domains is induced
when the
activation domains interact with different GTFs. Figure l0A further shows that
certain
combinations of activation domains, such as AP-V8x8-Vcx2 and AP-V8x8-AP-Vcx2,
are
capable of stimulating transcription of the reporter gene to higher levels
than VP 16 and
even p65, which is one of the most potent naturally occurnng transcription
factors known.
~ 5 Figure 1 OB shows the results of transfections with yet other fusion
proteins having
various combination, of transactivating domains. In particular, Figure l OB
shows that the
transcriptional activity of a transcriptional activation domain of p65,
corresponding to
amino acids 361-550 (containing the AP domain) is further potentiated by the
addition of
one or more other transa.ctivation domains, such as an additional AP domain
and/or V 8 or
2o Vc domains.
In yet another set of transfections, constructs encoding fusion proteins
having
activation domains fused to three copies of the ligand binding domain FKBP,
DNA
binding domains fused to ligand binding domains (FKBP or FRB in a single or
mutliple
copies) were used. In these transfections, transcription was induced by
addition of a
25 ligand, e.g., rapamycin, FK1012, AP1510, or other synthetic dimerizer
(depending on the
ligand binding domain). The results obtained were similar to those obtained
with the
covalent system.
Thus, this Example shows that potent transcriptional activators can be created
by
the combination of various transcriptional activation domains and that these
transcriptional
3o activators stimulate transcription of an integrated single copy gene.
CA 02303482 2000-02-15
WO 99/10508 PCT/US97/I5219
-6?-
All of the above-cited references and publications are hereby incorporated by
reference.
Equivalents
Those skilled in the art will recognize, or be able to ascertain using no more
than
routine experimentation, numerous equivalents to the specific polypeptides,
nucleic acids,
methods, assays and reagents described herein. Such equivalents are considered
to be
within the scope of this invention.
t0
*rB
CA 02303482 2000-02-15
WO 99/10508 PCT/US97/15219
-68-
SEQUENCE LISTING
(2) INFORMATION FOR SEQ ID NO:1:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 2444 base pairs
(B) TYPE: nucleic acid
(C) STRANDEDNESS: both
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: cDNA
(ix) FEATURE:
(A) NAME/KEY: CDS
(B) LOCATION: 39..1649
(xi)SEQUENCE SEQ ID
DESCRIPTION: NO:1:
GGCACGAGGC GGGGCCGGGT 53
CGCAGCTGGG
CCCGCGGC
ATG
GAC
GAA
CTG
TTC
Met lu
Asp Leu
G Phe
1 5
CCCCTCATCTTCCCGGCAGAGCAGCCC AAGCAGCGGGGCATGCGCTTC 101
ProLeuIlePheProAlaGluGlnPro LysGlnArgGlyMetArgPhe
10 15 20
'
CGCTACAAGTGCGAGGGGCGCTCCGCG GGCAGCATCCCAGGCGAGAGG 149
ArgTyrLysCysGluGlyArgSerAla GlySerIleProGlyGluArg
25 30 35
AGCACAGATACCACCAAGACCCACCCC ACCATCAAGATCAATGGCTAC 197
SerThrAspThrThrLysThrHisPro ThrIleLysIleAsnGlyTyr
40 45 50
ACAGGACCAGGGACAGTGCGCATCTCC CTGGTCACCAAGGACCCTCCT 245
ThrGlyProGlyThrValArgIleSer LeuValThrLysAspProPro
55 60 65
CAC CGG CCT CAC CCC CAC GAG CTT GTA GGA AAG GAC TGC CGG GAT GGC 293
His Arg Pro His Pro His Glu Leu Val Gly Lys Asp Cys Arg Asp Gly
70 75 80 85
TTC TAT GAG GCT GAG CTC TGC CCG GAC CGC TGC ATC CAC AGT TTC CAG 341
Phe Tyr Glu Ala Glu Leu Cys Pro Asp Arg Cys Ile His Ser Phe Gln
90 95 100
AAC CTG GGA ATC CAG TGT GTG AAG AAG CGG GAC CTG 389
GAG CAG GCT ATC
Asn Leu Gly Ile Gln Cys Val Lys Lys Arg Asp Leu
Glu Gln Ala Ile
105 110 115
AGT CAG CGC ATC CAG ACC AAC AAC AAC CCC TTC CAA 437
GTT CCT ATA GAA
Ser Gln Arg Ile Gln Thr Asn Asn Asn Pro Phe Gln
Val Pro Ile Glu
120 125 130
GAG CAG CGT GGG GAC TAC GAC CTG AAT GCT GTG CGG 485
CTC TGC TTC CAG
Glu Gln Arg Gly Asp Tyr Asp Leu Asn Ala Val Arg
Leu Cys Phe Gln
135 140 145
GTG ACA GTG CGG GAC CCA TCA GGC AGG CCC CTC CGC 533
CTG CCG CCT GTC
Val Thr Val Arg Asp Pro Ser Gly Arg Pro Leu Arg
Leu Pro Pro Val
150 155 160 165
CTT TCT CAT CCC ATC TTT GAC AAT CGT GCC CCC AAC 581
ACT GCC GAG CTC
Leu Ser His Pro Ile Phe Asp Asn Arg Ala Pro Asn
Thr Ala Glu Leu
170 175 180
AAG ATC TGC CGA GTG AAC CGA AAC TCT GGC AGC TGC 629
CTC GGT GGG GAT
Lys Ile Cys Arg Val Asn Arg Asn Ser Gly Ser Cys
Leu Gly Gly Asp
i
CA 02303482 2000-02-15
WO 99/10508 PCT/US97/15219
-69-
I85 190 195
GAGATCTTCCTACTGTGTGACAAGGTGCAGAAAGAGGAC ATTGAGGTG 677
GluIlePheLeuLeuCysAspLysValGlnLysGluAsp IleGluVal
200 205 210
TATTTCACGGGACCAGGCTGGGAGGCCCGAGGCTCCTTT TCGCAAGCT 725
TyrPheThrGlyProGlyTrpGluAlaArgGlySerPhe SerGlnAla
215 220 225
GATGTGCACCGACAAGTGGCCATTGTGTTCCGGACCCCT CCCTACGCA 773
AspValHisArgGlnValAlaIleValPheArgThrPro ProTyrAla
230 235 290 295
GACCCCAGCCTGCAGGCTCCTGTGCGTGTCTCCATGCAG CTGCGGCGG 821
AspProSerLeuGlnAlaProValArgValSerMetGln LeuArgArg
250 255 260
CCTTCCGACCGGGAGCTCAGTGAGCCCATGGAATTCCAG TACCTGCCA 869
ProSerAspArgGluLeuSerGluProMetGluPheGln TyrLeuPro
265 270 275
GATACAGACGATCGTCACCGGATTGAGGAGAAACGTAAA AGGACATAT 917
AspThrAspAspArgHisArgIleGluGluLysArgLys ArgThrTyr
280 285 290
GAGACCTTCAAGAGCATCATGAAGAAGAGTCCTTTCAGC GGACCCACC 965
GluThrPheLysSerIleMetLysLysSerProPheSer GlyProThr
295 300 305
GACCCCCGGCCTCCACCTCGACGCATTGCTGTGCCTTCC CGCAGCTCA 1013
AspProArgProProProArgArgIleAlaValProSer ArgSerSer
310 315 320 325
GCTTCTGTCCCCAAGCCAGCACCCCAGCCCTATCCCTTT ACGTCATCC 1061
AlaSerValProLysProAlaProGlnProTyrProPhe ThrSerSer
330 335 390
CTGAGCACCATCAACTATGATGAGTTTCCCACCATGGTG TTTCCTTCT 1109
LeuSerThrIleAsnTyrAspGluPheProThrMetVal PheProSer
345 350 355
GGGCAGATCAGCCAGGCCTCGGCCTTGGCCCCGGCCCCT CCCCAAGTC 1157
GlyGlnIleSerGlnAlaSerAlaLeuAlaProAlaPro ProGlnVal
360 365 370
CTGCCCCAGGCTCCAGCCCCTGCCCCTGCTCCAGCCATG GTATCAGCT 1205
LeuProGlnAlaProAlaProAlaProAlaProAlaMet ValSerAla
375 380 385
CTGGCCCAGGCCCCAGCCCCTGTCCCAGTCCTAGCCCCA GGCCCTCCT 1253
LeuAlaGlnAlaProAlaProValProValLeuAlaPro GlyProPro
390 395 400 905
CAGGCTGTGGCCCCACCTGCCCCCAAGCCCACCCAGGCT GGGGAAGGA 1301
GlnAlaValAlaProProAlaProLysProThrGlnAla GlyGluGly
910 915 420
ACGCTGTCAGAGGCCCTGCTGCAGCTGCAGTTTGATGAT GAAGACCTG 1349
ThrLeuSerGluAlaLeuLeuGlnLeuGlnPheAspAsp GluAspLeu
925 930 935
GGGGCCTTGCTTGGCAACAGCACAGACCCAGCTGTGTTC ACAGACCTG 1397
GlyAlaLeuLeuGlyAsnSerThrAspProAlaValPhe ThrAspLeu
440 495 950
GCATCCGTCGACAACTCCGAGTTTCAGCAGCTGCTGAAC CAGGGCATA 1445
AlaSerValAspAsnSerGluPheGlnGlnLeuLeuAsn GlnGlyIle
455 960 465
CCTGTGGCCCCCCACACAACTGAGCCCATGCTGATGGAG TACCCTGAG 1493
I
CA 02303482 2000-02-15
WO 99/10508 PCTNS97/15219
-70-
Pro Val Ala Pro His Thr Thr Glu Met Glu Tyr Pro
Pro Met Leu Glu
470 975 980 485
GCT ATA ACT CGC CTA GTG ACA GCC CCC GAC CCA GCT 1541
CAG AGG CCC CCT
Ala Ile Thr Arg Leu Val Thr Ala Pro Asp Pro Ala
Gln Arg Pro Pro
490 995 500
GCT CCA CTG GGG GCC CCG GGG CTC CTC CTT TCA GGA 1589
CCC AAT GGC GAT
Ala Pro Leu Gly Ala Pro Gly Leu Leu Leu Ser Gly
Pro Asn Gly Asp
10505 510 515
GAA GAC TTC TCC TCC ATT GCG GAC TCA GCC CTG CTG 1637
ATG GAC TTC AGT
Glu Asp Phe Ser Ser Ile Ala Asp Ser Ala Leu Leu
Met Asp Phe Ser
520 525 530
CAG ATC AGC TCC TAAGGGGGTG ACGCCTGCCC 1689
TCCCCAGAGC ACTGGTTGCA
Gln Ile Ser Ser
535
20GGGGATTGAA GCCCTCCAAA AGCACTTACG GGGTGTGTTC CAACTGCCCC1749
GATTCTGGTG
CAACTTTGTG GATGTCTTCC TTGGAGGGGG TTATTCTTTT ATTGTCAGTA1809
GAGCCATATT
TCTGTATCTC TCTCTCTTTT TGGAGGTGCT CATTAACTTC TCTGGAAAGG1869
TAAGCAGAAG
GGGGAGCTGG GGAAACTCAA ACTTTTCCCC GTCAGCTCCC TTCTCTGTAG1929
TGTCCTGATG
GGAACTGTGG GGTCCCCCAT CCCCATCCTC TACTCTCCTA GAGACAGAAG1989
CAGCTTCTGG
30CAGGCTGGAG GTAAGGCCTT TGAGCCCACA AAGTGTCTTC CATCATGGAT2049
AAGCCTTATC
TCATTACAGC TTAATCAAAA TAACGCCCCA CCTGTATGGC ACTGGCATTG2109
GATACCAGCC
TCCCTGTGCC TAACACCAGC GTTTGAGGGG GCCCTACAGA GGTCTCTGCC2169
CTGCCTTCCT
GGCTCTTTCC TTGCTCAACC ATGGCTGAAG AACAGCACTG GCTCTCTCCA2229
GAAACAGTGC
GGATCCAGAA GGGGTTTGGT CTGGACTTCC CTCTTCTCAA GTGCCTTAAT2289
TTGCTCTCCC
40AGTAGGGTAA GTTGTTAAGA GTGGGGGAGA AGCTCTCCAG TCAGGAGGCA2349
GCAGGCTGGC
TAGTTTTTAG TGAACAATCA AAGCACTTGG TTTCTACTCT GAACTAATAA2409
ACTCTTGCTC
AGCTGTTGCC AAGCTGGACG GCACGAGCTC 2444
GTGCC
(2) INFORMATION FOR SEQ ID N0:2:
(i) SEQUENCE CHARACTERISTICS:
50(A) LENGTH: 537 amino acids
(B) TYPE: amino acid
(D) TOPOLOGY: linear
(ii) MOLECULE TYPE: protein
(xi) SEQUENCE DESCRIPTION: SEQ
ID N0:2:
Met Asp Glu Leu Phe Pro Leu Ile Glu Gln Pro Lys
Phe Pro Ala Gln
1 5 10 15
Arg Gly Met Arg Phe Arg Tyr Lys Arg Ser Ala Gly
Cys Glu Gly Ser
20 25 30
Ile Pro Gly Glu Arg Ser Thr Asp Thr His Pro Thr
Thr Thr Lys Ile
6535 40 45
Lys Ile Asn Gly Tyr Thr Gly Pro Arg Ile Ser Leu
Gly Thr Val Val
SO 55 60
70Thr Lys Asp Pro Pro His Arg Pro Glu Leu Val Gly
His Pro His Lys
70 75 80
i
CA 02303482 2000-02-15
WO 99/10508 PCT/US97/15219
_71 _
Asp Cys Arg Asp Gly Phe Tyr Glu Ala Glu Leu Cys Pro Asp Arg Cys
85 90 95
Ile His Ser Phe Gln Asn Leu Gly Ile Gln Cys Val Lys Lys Arg Asp
100 105 110
Leu Glu Gln Ala Ile Ser Gln Arg Ile Gln Thr Asn Asn Asn Pro Phe
115 120 225
Gln Val Pro Ile Glu Glu Gln Arg Gly Asp Tyr Asp Leu Asn Ala Val
130 135 140
Arg Leu Cys Phe Gln Val Thr Val Arg Asp Pro Ser Gly Arg Pro Leu
145 150 155 160
Arg Leu Pro Pro Val Leu Ser His Pro Ile Phe Asp Asn Arg Ala Pro
165 170 175
Asn Thr Ala Glu Leu Lys Ile Cys Arg Val Asn Arg Asn Ser Gly Ser
180 185 190
Cys Leu Gly Gly Asp Glu Ile Phe Leu Leu Cys Asp Lys Val Gln Lys
195 200 205
Glu Asp Ile Glu Val Tyr Phe Thr Gly Pro Gly Trp Glu Ala Arg Gly
210 215 220
Ser Phe Ser Gln Ala Asp Val His Arg Gln Val Ala Ile Val Phe Arg
225 230 235 240
Thr Pro Pro Tyr Ala Asp Pro Ser Leu Gln Ala Pro Val Arg VaI Ser
245 250 255
Met Gln Leu Arg Arg Pro Ser Asp Arg Glu Leu Ser Glu Pro Met Glu
260 265 270
Phe Gln Tyr Leu Pro Asp Thr Asp Asp Arg His Arg Ile Glu Glu Lys
275 280 285
Arg Lys Arg Thr Tyr Glu Thr Phe Lys Ser Ile Met Lys Lys Ser Pro
290 295 300
Phe Ser Gly Pro Thr Asp Pro Arg Pro Pro Pro Arg Arg Ile Ala Val
305 310 315 320
Pro Ser Arg Ser Ser Ala Ser Val Pro Lys Pro Ala Pro Gln Pro Tyr
325 330 335
Pro Phe Thr Ser Ser Leu Ser Thr Ile Asn Tyr Asp Glu Phe Pro Thr
340 345 350
Met Val Phe Pro Ser Gly Gln Ile Ser Gln Ala Ser Ala Leu Ala Pro
355 360 365
Ala Pro Pro Gln Val Leu Pro Gln Ala Pro Ala Pro Ala Pro Ala Pro
370 375 380
Ala Met Val Ser Ala Leu Ala Gln Ala Pro Ala Pro Val Pro Val Leu
385 390 395 400
Ala Pro Gly Pro Pro Gln Ala Val Ala Pro Pro Ala Pro Lys Pro Thr
405 410 415
Gln Ala Gly Glu Gly Thr Leu Ser Glu Ala Leu Leu Gln Leu Gln Phe
920 925 930
Asp Asp Glu Asp Leu Gly Ala Leu Leu Gly Asn Ser Thr Asp Pro Ala
935 440 495
Val Phe Thr Asp Leu Ala Ser Val Asp Asn Ser Glu Phe Gln Gln Leu
CA 02303482 2000-02-15
WO 99/10508 PCTNS97/15219
-72-
450 455 960
Leu Asn Gln Gly Ile Pro Val Ala Pro His Thr Thr Glu Pro Met Leu
465 970 475 980
Met Glu Tyr Pro Glu Ala Ile Thr Arg Leu Val Thr Ala Gln Arg Pro
485 490 495
Pro Asp Pro Ala Pro Ala Pro Leu Gly Ala Pro Gly Leu Pro Asn Gly
500 505 510
Leu Leu Ser Gly Asp Glu Asp Phe Ser Ser Ile Ala Asp Met Asp Phe
515 520 525
Ser Ala Leu Leu Ser Gln Ile Ser Ser
530 535