Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02305780 2000-03-30
WO 99/16886 PCT/EP98/06134
Process for the Production of Ergosterol and its Intermediate
Products using Recombinant Yeasts
This invention relates to a process for the production of
ergosterol and its intermediate products using recombinant yeasts
and plasmids for the transformation of yeasts.
Ergosterol is the end product of sterol synthesis in yeasts
and fungi. The economic importance of this compound lies, on the
one hand, in obtaining vitamin DZ from ergosterol with W
irradiation, and, on the other hand, in obtaining steroid
hormones with biotransformation, starting from ergosterol.
Squalene is used as a synthesis component for the synthesis of
terpenes. In hydrogenated form, it is used as squalene in
dermatology and cosmetics and in various derivatives as
components of skin and hair cleansers. Also of economic
importance are the intermediate products of the ergosterol
metabolic process. Farnesol, geraniol and squalene can be named
as most important here. In addition, sterols, such as, e.g.,
zymosterol and lanosterol, can be used economically, whereby
lanosterol is pivotal in terms of crude and synthesis for the
chemical synthesis of saponins and steroid hormones. Because of
its good skin penetration and spreading properties, lanosterol is
used as an emulsion adjuvant and active ingredient for skin
creams.
CA 02305780 2000-03-30
2
The genes of the ergosterol metabolism in yeast are largely
known and cloned, e.g., the HMG-CoA reductase (HMG1) (Basson et
al. (1988)), the squalene synthetase (ERGS) (Fegueur et al.
(1991)), the acyl-CoA: sterol-acyl transferase (sATi) (Yu et al.
(1996)), and the squalene epoxidase (ERG1) (Jandrositz et al.
(1991)). Squalene synthetase catalyzes the reaction of farnesyl
pyrophosphate on presqualene pyrophosphate to squalene. The
reaction mechanisms of sterol-acyl transferase are not fully
determined. An over-expression of genes of these above-mentioned
enzymes was already attempted, but it did not result in any
significant increase in the amount of ergosterol. In the case of
the 8MG1 over-expression, the overproduction of squalene was
described; moreover, additional mutations were introduced to
interrupt the route following squalene (EP-0 486 290).
The overproduction of geraniol and farnesol was also
described, but here no over-expression of genes of the ergosterol
metabolism took place, rather an interruption of the reaction
process as regards geraniol and farnesol formation (EP-0313 465).
Specific inhibitors of the ergosterol biosynthesis can also
result in the accumulation of larger amounts of certain
intermediate products, e.g., allylamines, which prevent the
conversion of squalene into squalene epoxide. As a result, large
amounts (up to 600 times the normal level) of squalene are
accumulated (Jandrositz et al., (1991)).
Although the use of inhibitors leads to a major accumulation
of, e.g., squalene, the addition of these substances may yet turn
out to be disadvantageous since only small amounts exert the same
CA 02305780 2000-03-30
3
action in the organism, so that a production of the products of
ergosterol biosynthesis in the process of overproduction is
advantageous.
The object of this invention is to synthesize a
microbiological process for the production of ergosterol and its
intermediate products, the microorganisms that are necessary for
this purpose, such as yeast strains, the increased amounts of
ergosterol or intermediate products that are necessary for this
purpose, and to prepare the plasmids that are necessary for the
transformation of the yeast strain.
It was now found that the amount of ergosterol and its
intermediate products can be increased, if the genes of HMG1
(Basson et al., (1988)), ERGS (Fegueur et al., (1991)), Current
Genetics 20: 365-372), SAT1 (Yu et al., (1996)) and ERG1
(Jandrositz et al. (1991)) are introduced in altered form into
microorganisms such as, e.g., yeasts, whereby the genes are
located either individually on a plasmid or in combination on one
or more plasmids and can be brought to the host simultaneously or
in succession.
The subject of this invention is thus a process that is
characterized in that
a) first a plasmid is designed, into which several
suitable genes of the ergosterol metabolic process are
inserted in altered form,
or
CA 02305780 2000-03-30
4
b) first plasmids are designed, into which in each case
one of the genes of the ergosterol metabolic process is
inserted in altered form,
c) microorganisms are transformed with the thus produced
plasmids, whereby the microorganisms are transformed
with a plasmid under a) or they are transformed
simultaneously or in succession with several plasmids
under b) ,
d) fermentation into ergosterol is performed with the thus
produced microorganisms,
e) after fermentation has ended, the ergosterol and its
intermediate products are extracted from the cells and
analyzed, and finally,
f) the thus obtained ergosterol and its intermediate
products are purified using column chromatography and
isolated.
The subject of this invention is especially a process which
is characterized in that
a-i) first a plasmid is designed, into which the following
genes are inserted:
i) the gene of HMG-Co-A-reductase (t-HMG),
ii) the gene of squalene synthetase (ERGS),
iii) the gene of Acyl-CoA: sterol-acyl transferase
(sATi),
and
iv) the gene of squalene epoxidase (ERG1),
or
CA 02305780 2000-03-30
a-ii) first a plasmid is designed, into which the following
genes are inserted:
i) the gene of HMG-Co-A-reductase (t-HMG),
and
ii) the gene of sgualene synthetase (ERGS),
or
a-iii) first a plasmid is designed, into which the following
genes are inserted:
i) the gene of HMG-Co-A-reductase (t-HMG),
and
iii) the gene of acyl-CoA: sterol-acyl transferase
(SAT1),
or
a-iv) first a plasmid is designed, into which the following
genes are inserted:
i) the gene of the HMG-Co-A-reductase (t-HMG),
and
iv) the gene of squalene epoxidase (ERG1),
or
a-v) first a plasmid is designed, into which the following
genes are inserted:
ii) the gene of squalene synthetase (ERG9),
and
iii) the gene of acyl-CoA: sterol-acyl transferase
(SAT1)
or
CA 02305780 2000-03-30
6
a-vi) first a plasmid is designed, into which the following
genes are inserted:
ii) the gene of squalene synthetase (ERGS),
and
iv) the gene of squalene epoxidase (ERG1),
or
a-vii) first a plasmid is designed, into which the following
genes are inserted:
iii) the gene of acyl-CoA: sterol-acyl transferase
(sATi),
and
iv) the gene of squalene epoxidase (ERG1),
or
b) first plasmids are designed, into which in each case
one of the genes that is mentioned under a-i) is
inserted,
and
c) microorganisms are transformed with the thus produced
plasmids, whereby the microorganisms are transformed
with a plasmid under a-i) to a-vii), or they are
transformed simultaneously or in succession with
several plasmids under b),
d) fermentation into ergosterol is performed with the thus
produced microorganisms,
e) after fermentation has ended, the ergosterol and its
intermediate products are extracted from the cells and
analyzed, and finally
CA 02305780 2000-03-30
7
f) the thus obtained ergosterol and its intermediate
products are purified using column chromatography and
isolated.
In addition, the gene of squalene epoxidase (ERGi) can be
inserted into the plasmids that are cited under a-ii), a-iii) and
a-v), and in addition, the gene of acyl-CoA: sterol-acyl
transferase (SATs) can be inserted into the plasmid that is cited
under a-ii). These plasmids are also subjects of this invention.
Intermediate products are defined as squalene, farnesol,
geraniol, lanosterol, zymosterol, 4,4-dimethylzymosterol, 4-
methylzymosterol, ergost-7-enol and ergosta-5,7-dienol,
especially sterols with 5,7-dime structure.
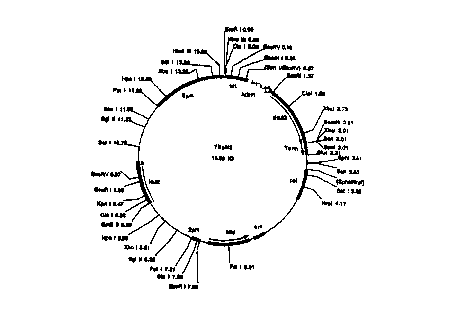
The plasmids that are used are preferably the plasmid YEpH2,
which contains the average ADH-promoter, t-HMG (altered variant
of HMG1) and the TRP-terminator (see Fig. 1), the plasmid
YDpUHK3, which contains the average ADH-promoter, t-HMG (altered
variant of HMG1) and the TRP-terminator, the gene for the
kanamycin resistance and the ura3 gene (see Fig. 2) and the
plasmid pADL-SAT1, which contains the sATi gene and the LEU2 gene
of YEpl3.
These plasmids and their use for the production of
ergosterol and its intermediate products, such as squalene,
farnesol, geraniol, lanosterol, zymosterol, 4,4-
dimethylzymosterol, 4-methylzymosterol, ergost-7-enol and
ergosta-5,7-dienol, especially sterols with 5,7-diene structure,
are also subjects of this invention.
CA 02305780 2000-03-30
8
As a host for the introduction of plasmids according to the
invention, in principle all microorganisms, especially yeasts,
are suitable.
The species S. cerevisiae, especially the strain 8.
cerevisiae AH22, is preferred.
The subject of this invention is also the yeast strain 8.
cerevisiae AH22, which contains one or more of the genes that are
mentioned in the process under a-i).
The subject of this invention is also the yeast strain 8.
cerevisiae AH22, which contains the plasmid pADL-SAT1.
In addition, the combined transformation of microorganisms
with the plasmids pADL-SAT1 and YDpUHK3, especially yeasts such
as 8. cerevisiae AH22, is preferred.
Viewed overall, the flow in the ergosterol metabolic process
is affected as follows:
The flow in the direction of ergosterol is maximized by the
activity of several bottle-neck enzymes being intensified
simultaneously. In this case, various enzymes play a decisive
role, whereby the combination of deregulation or over-expression
provides the decisive breakthrough for increasing the ergosterol
yield. As combinations, the enzymes or their genes HMG1 (Basson
et al., (1988)), ERGS (Fegueur et al., (1991)), acyl-CoA: sterol-
acyl transferase (sATi) (Yu et al. (1996)) and/or squalene
epoxidase (ERG1) (Jandrositz et al. (1991)) are introduced into a
yeast strain in altered form, whereby the genes are introduced
with one or more plasmids, whereby the DNA sequences are
CA 02305780 2000-03-30
9
contained either individually or in combination in the
plasmid(s).
In the case of gene HMG1, "altered" means that of the
corresponding genes, only the catalytic area is expressed without
the membrane-bound domains. This alteration was already
described (EP-0486 290). The purpose of the alteration of 8MG1
is to prevent the feedback regulation by intermediates of
ergosterol biosynthesis. Both HMG1 and the two other above-
mentioned genes are removed in the same way from the
transcriptional regulation. To this end, the promoter of the
genes is replaced by the "average" ADH1-promoter. This promoter
fragment of the ADH1-promoter shows an approximately constitutive
expression (Ruohonen et al., (1995)), so that the transcriptional
regulation no longer proceeds via intermediates of the ergosterol
biosynthesis.
The products that are produced in the over-expression can be
used in biotransformations or other chemical and therapeutic
purposes, e.g., obtaining vitamin D2 from ergosterol via W
irradiation, and obtaining steroid hormones via biotransformation
starting from ergosterol.
Subjects of this invention are also microorganisms,
especially yeast strains, which can produce an increased amount
of ergosterol and ergosterol in combination with increased
amounts of squalene by over-expression of the genes that are
mentioned in the process under a-i).
CA 02305780 2000-03-30
Preferred is an altered variant of the gene HMG1, in which
only the catalytic area is expressed without the membrane-bound
domain. This alteration is described (EP-0486 290).
A subject of this invention is also a process for the
production of ergosterol and its intermediate products, which is
characterized in that the genes that are mentioned in the process
under a), especially the genes that are mentioned in the
processes under a-i to a-vii) (two-, three-, and four-fold gene
combinations) in each case with the plasmids are first introduced
independently of one another into microorganisms of the same
species, and fermentation into ergosterol is performed with them
together, and the ergosterol that is thus obtained is extracted
from the cells, analyzed and purified using column chromatography
and isolated.
Subjects of this invention are also expression cassettes,
comprising the average ADH-promoter, the t-HMG gene, the TRP-
terminator, and the SAT1-gene with the average ADH-promoter and
the TRP-terminator and expression cassettes, comprising the
average ADH-promoter, the t-HMG gene, the TRP-terminator, the
8AT1 gene with the average ADH-promoter and the TRP-terminator,
and the ERG9-gene with the average ADH-promoter and the TRP-
terminator.
A subject of this invention is also a combination of
expression cassettes, whereby the combination consists of
a) a first expression cassette, in which the ADH-promoter,
the t-HMG gene and the TRP-terminator are located,
CA 02305780 2000-03-30
11
b) a second expression cassette, in which the ADH-
promoter, the SAT-1 gene and the TRP-terminator are
located,
and
c) a third expression cassette, in which the ADH-promoter
and the ERG9-gene with the TRP-terminator are located.
The subject of this invention is also the use of these
expression cassettes for the transformation of microorganisms,
which are used in the fermentation into ergosterol, whereby the
microorganisms are preferably yeasts.
Microorganisms such as yeasts, which contain these
expression cassettes, as well as their use in the fermentation
into ergosterol and ergosterol intermediate products, are also
subjects of the invention.
The following examples are used for the explanation with
respect to the implementation of the processes that are necessary
for the embodiments:
1. Restriction
The restriction of plasmids (1 to 10 fig) was performed in 30
~1 batches. To this end, the DNA was taken up in 24 ~1 of H20,
and mixed with 3 ~1 of the corresponding buffer, 1 ~1 of RSA
(bovine serum albumin) and 2 ~1 of enzyme. The enzyme
concentration was 1 unit/~1 or 5 units/~1 depending on the amount
of DNA. In some cases, 1 ~1 more of RNase was added to the batch
to degrade the tRNA. The restriction batch was incubated for two
hours at 37°C. The restriction was controlled with a minigel.
CA 02305780 2000-03-30
12
2. Gel Electrophoreses
The gel electrophoreses were performed in minigel or wide-
minigel equipment. The minigels (about 20 ml, 8 bags) and the
wide-minigels (50 ml, 15 or 30 bags) consisted of 1% agarose in
TAE. 1 x TAE was used as a mobile buffer. The samples (10 ~C1)
were mixed with 3 ~.1 of stopper solution and applied. I-DNA cut
with HindIII was used as a standard (bands at: 23.1 kb; 9.4 kb;
6.6 kb; 4.4 kb; 2.3 kb; 2.0 kb; 0.6 kb). For separation, a
voltage of 80 V for 45 to 60 minutes was prepared. Then, the gel
was stained in ethidium bromide solution and held under W light
with video-documentation system INTAS or photographed with an
orange filter.
3. Gel Elution
The desired fragments were isolated using gel elution. The
restriction preparation was applied in several bags of a minigel
and separated. Only 7l-HindIII and a "sacrifice trace" were
stained in ethidium bromide solution, viewed under W light, and
the desired fragment was labeled. As a result, DNA was prevented
from damaging the residual bags by the ethidium bromide and the
W light. By aligning the stained and unstained gel pieces, the
desired fragment from the unstained gel piece could be cut out
based on the labeling. The agarose piece with the fragment to be
isolated was added in a dialysis tube, sealed free of air bubbles
with a little TAE buffer and placed in the BioRad-minigel
apparatus. The mobile buffer consisted of 1 x TAE, and the
voltage was 100 V for 40 minutes. Then, the flow polarity was
CA 02305780 2000-03-30
13
varied for 2 minutes to loosen the DNA adhering to the dialysis
tube. The buffer that contains the DNA fragments of the dialysis
tube was moved into the reaction vessel and thus performed an
ethanol precipitation. To this end, 1/10 volume of 3 M sodium
acetate, tRNA (1 ul per 50 ~1 of solution) and 2.5 times the
volume of ice-cold 96% ethanol were added to the DNA solution.
The batch was incubated for 30 minutes at -20°C and then
centrifuged off at 12,000 rpm for 30 minutes at 4°C. The DNA
pellet was dried and taken up in 10 to 50 ~,1 of H20 (depending on
the amount of DNA).
4. Klenow Treatment
Projecting ends of DNA fragments are made up by the Klenow
treatment, so that "blunt ends" result. Per 1 ~Cg of DNA, the
following batch was pipetted together:
DNA-pellet +il ~,1 of H20
+ 1.5 ~1 of 10 x Klenow buffer
+ 1 ~,1 of O.1M DTT
+ 1 ~,1 of nucleotides (dNTP 2 mmol)
+ 1 ~,1 of Klenow-polymerase (1
unit/~,1)
In this case, the DNA should be derived from an ethanol
precipitation to prevent contaminants from inhibiting the Klenow-
polymerase. Incubation was carried out for 30 minutes at 37°C,
and then over another 5 minutes at 70°C the reaction was halted.
The DNA was obtained from the batch by an ethanol precipitation
and taken up in 10 ~1 of H20.
CA 02305780 2000-03-30
14
5. Ligation
The DNA fragments that were to be ligated were combined.
The end volume of 13.1 ~1 contained about 0.5 ~g of DNA with a
vector-insert ratio of 1:5. The sample was incubated for 45
seconds at 70°C, cooled to room temperature (about 3 minutes) and
then incubated on ice for 10 minutes. Then, the ligation buffers
were added: 2.6 ~1 of 500 mmol TrisHCl, pH 7.5, and 1.3 ~1 of
100 mmol MgClz, and they were incubated on ice for another 10
minutes. After 1 ~1 of 500 mmol DTT and 1 ~1 of 10 mmol ATP were
added, 1 ~1 of ligase (1 unit/~1) was added on ice for another 10
minutes. The entire treatment should be carried out with as
little shaking as possible so as to keep adjacent DNA ends from
reseparating. The ligation was carried out overnight at 14°C.
6. E. coli Transformation
Component Escherichia coli (E. coli) NM522 cells were
transformed with the DNA of the ligation preparation. As a
positive control, a batch was supplied with 50 ng of the pScL3
plasmid, and as a null control, a batch was supplied without DNA.
For each transformation preparation, 100 ~1 of 8% PEG solution,
~1 of DNA and 200 ~1 of competent cells (E. coli NM522) were
pipetted into a tabletop centrifuging tube. The batches were put
on ice for 30 minutes and shaken intermittently. Then, thermal
shock took place.: 1 minute at 42°C. For regeneration, 1 ml of
LB-medium was added to the cells and incubated on a shaker for 90
minutes at 37°C. 100 ~1 each of the undiluted batches, a 1:10
CA 02305780 2000-03-30
dilution and a 1:100 dilution were flattened out on LB +
ampicillin plates and incubated overnight at 37°C.
7. Plasmid Isolation from E. Coli (Miniprep)
E. coli colonies were cultured overnight in 1.5 ml of LB +
ampicillin medium in tabletop centrifuging tubes at 37°C and 120
rpm. The next day, the cells were centrifuged off for 5 minutes
at 5000 rpm and 4°C, and the pellet was taken up in 50 ~1 of TE-
buffer. Each batch was mixed with 100 ~1 of 0.2N NaoH, 1% SDS
solution, mixed and put on ice for 5 minutes (lysis of the
cells). Then, 400 ~1 of Na-acetate/NaCl solution (230 ~1 of H20,
130 ~1 of 3 M sodium acetate, and 40 ~1 of 5 M NaCl) was added,
the batch was mixed and put on ice for another 15 minutes
(protein precipitation). After 15 minutes of centrifuging at
11,000 rpm, the supernatant, which contains plasmid-DNA, was
moved into an Eppendorf vessel. If the supernatant was not
completely clear, it was centrifuged one more time. The
supernatant was mixed with 360 ~1 of ice-cooled isopropanol and
incubated for 30 minutes at -20°C (DNA precipitation). The DNA
was centrifuged off (15 minutes, 12,000 rpm, 4°C), the
supernatant was discarded, the pellet was washed in 100 ~1 of
ice-cooled 96% ethanol, incubated for 15 minutes at -20°C and
centrifuged off again (15 minutes, 12,000 rpm, 4°C). The pellet
was dried in a speed vacuum and then taken up in 100 ~1 of H20.
The plasmid-DNA was characterized by restriction analysis. To
this end, 10 ~1 of each batch was restricted and cleaved by gel
electrophoresis in a wide-minigel (see above).
CA 02305780 2000-03-30
16
8. Plasmid-Working-Up on E. coli (Maxiprep)
To isolate larger amounts of plasmid-DNA, the maxiprep
method was performed. Two plungers with 100 ml of LB +
ampicillin medium were inoculated with a colony or with 100 ~1 of
a frozen culture, which carries the plasmid that is to be
isolated, and it was incubated overnight at 37°C and 120 rpm.
The next day the culture (200 ml) was moved into a GSA beaker and
centrifuged for 10 minutes at 4000 rpm (2600 x g). The cell
pellet was taken up in 6 ml of TE-buffer. To digest the cell
wall, 1.2 ml of lysozyme solution (20 mg/ml of TE-buffer) was
added, and it was incubated for 10 minutes at room temperature.
Then, the lysis of the cells was carried out with 12 ml of 0.2N
NaOH, 1% SDS solution and for another 5 minutes of incubation at
room temperature. The proteins were precipitated by the addition
of 9 ml of cooled 3 M sodium acetate solution (pH 4.8) and a 15-
minute incubation on ice. After centrifuging (GSA: 13,000 rpm
(27,500 x g), 20 minutes, 4°C), the supernatant, which contained
the DNA, was moved into a new GSA beaker, and the DNA was
precipitated with 15 ml of ice-cold isopropanol and an incubation
of 30 minutes at -20°C. The DNA pellet was washed in 5 ml of
ice-cooled ethanol and dried in air (about 30-60 minutes). Then,
it was taken up in 1 ml of H20. An examination of the plasmid by
restriction analysis took place. The concentration was
determined by depositing dilutions on a minigel. To reduce the
salt content, a 30-60 minute microdialysis was carried out (pore
size 0.025 Vim).
CA 02305780 2000-03-30
17
9. Yeast Transformation
For the yeast transformation, a pre-culture of the strain
Saccharomyces cerevisiae (S. cerevisiae) AH22 was prepared. A
plunger with 20 ml of YE-medium was inoculated with 100 ~1 of the
frozen culture and incubated overnight at 28°C and 120 rpm. The
main cultivation was carried out under identical conditions in a
plunger with 100 ml of YE-medium, which was inoculated with 10
~1, 20 ~1 or 50 ~1 of the pre-culture.
9.1 Producing Competent Cells
The next day, the plungers were counted out using a Thoma
chamber, and the procedure was continued with the plunger, which
held 3-5 x 10~ cells/ml. The cells were harvested by
centrifuging (GSA: 5000 rpm (4000 x g), 10 minutes). The cell
pellet was taken up in 10 ml of TE-buffer and divided into two
tabletop centrifuging tubes (5 ml each). The cells were
centrifuged off for 3 minutes at 6000 rpm and washed twice more
with 5 ml of TE-buffer each. Then, the cell pellet was taken up
in 330 ~1 of lithium acetate buffer per 109 cells, moved into a
sterile 50 ml Erlenmeyer flask and shaken for one hour at 28°C.
As a result, the cells were competent for transformation.
9.2 Transformation
For each transformation preparation, 15 ~1 of herring sperm
DNA (10 mg/ml), 10 ~1 of DNA that is to be transformed (about 0.5
fig) and 330 ~1 of component cells were pipetted into a tabletop
centrifuging tube and incubated for 30 minutes at 28°C (without
CA 02305780 2000-03-30
18
shaking!). Then, 700 ~,1 of 50% PEG 6000 was added, and it was
incubated for one additional hour at 28°C, without shaking. A
thermal shock of 5 minutes at 42°C followed.
100 ~1 of the suspension was flattened out on the selection
medium (YNB, Difco) to select leukine prototrophy. In the case
of the selection on 6418 resistance, a regeneration of the cells
is carried out after the thermal shock (see under 9.3
Regeneration Phase).
9.3 Regeneration Phase
Since the selection marker is resistance to 6418, the cells
needed time for the expression of the resistance-gene. The
transformation preparations were mixed with 4 ml of YE-medium and
incubated overnight at 28°C in the shaker (120 rpm). The next
day, the cells were centrifuged off (6,000 rpm, 3 minutes), taken
up in 1 ml of YE-medium, and 100 ~1 or 200 ~1 was flattened out
on YE + 6418 plates. The plates were incubated for several days
at 28°C.
10. Reaction conditions for the PCR
The reaction conditions for the polymerase chain reaction
must be, optimized for the individual case and are not necessarily
valid for any batch. Thus, i.a., the amount of DNA used, the
salt concentrations and the melting temperature can be varied.
For our formulation of the problem, it has proven advantageous to
combine the following substances in an Eppendorf cap, which was
suitable for use in a thermocycler: 5 ~.1 of super buffer, 8 dal
CA 02305780 2000-03-30
19
of dNTP's (0.625 ~M each), 5'-primer, 3'-primer and 0.2 ~g of
matrix DNA, dissolved in enough water to yield a total volume of
50 ~1 for the PCR preparation, were added to 2 ~1 (-O.1 U) of
Super Taq polymerase. The batch was briefly centrifuged off and
covered with a drop of oil. Between 37 and 40 cycles were
selected for amplification.
CA 02305780 2000-03-30
The embodiments below describe the production of the
plasmids and yeast strains according to the invention as well as
their use, without, however, limiting the invention to these
examples.
Example 1
Expression of tHMG in 8. cerevisiae AH22
The DNA sequence for tHMG (Basson et al., (1988)) was
amplified by PCR from genomic DNA of saccharomyces cerevisiae
S288C (Mortimer and Johnston, (1986)) with use of standard
methods. The primers that are used in this case are the DNA
oligomer tHMG-5' and tHMG-3' (see Seq ID Nos. 1 and 2). The DNA-
fragment that was obtained was introduced in cloning vector pUCl9
(Yanisch-Perron et al., (1985)) after a Klenow treatment, and
yielded vector pUCl9-tHMG. After plasmid isolation and
restriction of pUC 19-tHMG with endonucleases EcoRl and BamHl,
the obtained fragment was introduced into yeast expression vector
pPT2b (Lang and Looman, (1995)), which also was treated with
EcoRl and BamHl. The plasmid pPT2b-tHMG that was produced
contains the ADH1-promoter (Bennetzen and Hall, (1982)) and the
TRP1-terminator (Tschumper and Carbon, (1980)), between which the
tHMG-DNA fragment is found. A DNA section was isolated from
vector pPT2b-tHMG via endonucleases EcoRV and Nrul, and said DNA
section contains the so-called average ADH1-promoter, the tHMG
and the TRP1-terminator. This DNA section was introduced into
yeast vector YEpl3 (Fischhoff et al., (1984)), which was treated
with endonuclease Sphl and a DNA polymerase. The vector that is
CA 02305780 2000-03-30
' 21
thus produced, the YEpH2 (Fig. 1), was treated with the
endonucleases EcoRV and Nrul. A DNA-fragment with the following
areas was thus produced: a transcription-activating area from
the tetracycline resistance gene (Sidhu and Bollon, (1990)), the
average ADH1-promoter, the tHMG and the TRP1-terminator
(expression cassette). This DNA-fragment was introduced into
vector YDpU (Berben et al., (1991)), which was treated with Stul.
Vector YDpUH2/12 that was thus produced was treated with
endonuclease Smal and ligated with a DNA-sequence that codes for
a kanamycin resistance (Webster and Dickson, (1983)). The
construct that is produced (YDpUHK3, Fig. 2) was treated with
EcoRV. The yeast strain Saccharomyces cerevisiae AH22 was
transformed with this construct. The transformation of the yeast
with a linearized vector, as it is in this example, results in a
chromosomal integration of the total vector at the URA3 gene
locus. To eliminate the areas from the integrated vector that
are not part of the expression cassette (E. coli origin, E. coli-
ampicillin resistance gene, TEF-promoter and kanamycin resistance
gene), transformed yeasts were subjected to a selection pressure
by FOA selection (Boeke et al., (1987)) that promotes uracil-
auxotrophic yeasts. The uracil-auxotrophic strain that is
described in the selection bears the name AH22/tH3ura8 and has
the tHMGi-expression cassette as chromosomal integration in the
URA3-gene.
The yeast strain AH22/tH3ura8 and the starting strain AH22
were cultivated for 48 hours in YE at 28°C and 160 rpm in a flow
spoiler plunger.
CA 02305780 2000-03-30
22
Cultivation conditions: Pre-culture WMVIII was prepared as
follows: 20 ml of WMVIII + histidine (20 ~,g/ml) + uracil (20
~g/ml) were inoculated with 100 ~,1 of frozen culture and
incubated for 2 days at 28°C and 120 rpm (reciprocal motion).
From the 20 ml of pre-culture, 100 ml of WMVIII + histidine (20
~,g/ml) + uracil (20 ~g/ml) were inoculated. For the main
culture, 50 ml of YE (in a 250 ml flow spoiler plunger) with 1 x
109 cells was inoculated. The plungers were incubated at 160 rpm
in a round shaker at 28°C for 48 hours. HMG-CoA-reductase
activities were determined (according to Qureshi et al., (1981)),
and produced the following values (see Table 1).
Table 1
Specific HMG-CoA-reductase
activity* (U/mg of protein)
AH22 3.99
AH22/tH3ura8 11.12
* A unit is defined as the reaction of 1 nmol of NADPH per minute
in a milliliter reaction mixture. The measurement was carried
out with total protein isolates.
The sterols were extracted (Parks et al., (1985)) and
analyzed using gas chromatography. The following values were
produced (see Table 2).
CA 02305780 2000-03-30
23
Table 2
Squalene (% w/w) Ergosterol (% w/w)
AH22 0.01794 1.639
AH22/tH3ura8 0.8361 1.7024
The values, in percent, relate to the dry weight of the
yeast.
Example 2
Expression of SAT1 in S. cerevisiae AH22
The sequence for the acyl-CoA: sterol transferase (SATs;
Yang et al., (1996)) was obtained by, as described above, PCR
from genomic DNA of Saccharomyces cerevisiae S288C. The primers
used in this case are the DNA-oligomers SAT1-5' and SAT1-3' (see
Seq ID Nos. 3 and 4). The DNA-fragment that was obtained was
cloned in cloning vector pGEM-T (Mezei and Storts, (1994)), which
resulted in vector pGEM-SAT1. By treatment of pGEM-SAT1 with
EcoRl, a fragment was obtained that was cloned in yeast
expression vector pADH1001, which also was treated with EcoRl.
Vector pADH-SAT1 that was thus produced was treated with the
endonuclease Nrul, and it was ligated with a fragment of YEpl3,
which contains the LEU2-gene.
Yeast expression vector pADL-SAT1 (Fig. 3), which was
introduced into yeast strain AH22, was thus produced. The thus
obtained strain AH22/pADL-SAT1 was incubated for 7 days in WMVIII
CA 02305780 2000-03-30
24
(Lang and Looman (1995)) in a minimal medium. Cultivation
conditions: (For pre-culture, see above) Main culture: 50 ml
of WMVIII + histidine (20 ~g/ml) + uracil (20 ~Cg/ml) of cultures
(in a 250 ml flow spoiler plunger) were inoculated with 1 x 109
cells: the plungers were incubated at 160 rpm on a round shaker
at 28°C for 7 days. The sterols formed were analyzed via gas
chromatography (see Table 3).
Table 3
Squalene (% w/w) Ergosterol (% w/w)
AH22 n.d. 1.254
AH22/pADL-SAT1 n.d. 1.831
The values, in percent, relate to the dry weight of the
yeast.
n.d.: indeterminate
Example 3
Combined Expression of the Shortened 3-Hydroxy-3-methylglutaryl-
CoA-Reductase (tHMG) and the Aoyl-CoA: Sterol-aayl Transferase
( SATl )
Example 3.1
Yeast strain AH22/tH3ura 8 was transformed with the 8AT1
expression vector pADL-SAT1, and yielded AH22/tH3ura8/pADL-SAT1.
CA 02305780 2000-03-30
This combined strain was cultivated for 7 days in WMVIII. The
sterols were extracted (see above) and analyzed via gas
chromatography. The following values were produced (see Table
4).
Table 4
Squalene (% w/w) Ergosterol (% w/w)
AH22/tH3ura8 1.602 3.798
AH22/tH3ura8/pADL- 1.049 5.540
SATl
The values, in percent, relate to the dry weight of the
yeast.
Example 3.2
Yeast cultures were cultivated for 7 days in WMVIII, but
different amounts of uracil were added to the cultures.
Concentrations of 10, 20, 40 and 100 ~,g/ml of uracil were set in
the medium. The ergosterol and the squalene amounts are at most
in a supplementation of 20 ~.g/ml of uracil. The results are
shown in Fig. 4.
It is shown that the ergosterol and squalene yield in strain
AH22tH3ura8/pADL-SAT1 depends greatly on the amount of uracil
that is added to cultivation medium WMVIII.
CA 02305780 2000-03-30
26
Example 3.3
Yeast cultures were cultivated for 7 days in WMVIII. Then,
the totality of the sterols was determined as described above.
The free sterols are determined by GC from yeasts that are
encapsulated with glass pearls and are extracted with n-hexane.
The results are shown in Table 5.
The results show that the enzyme sterol-acyl transferase
(Satl) esterifies with higher effectiveness in particular sterols
that are lacking the 4,4-dimethyl group. Thus, a technical
application for the separation of 4,-4-dimethylsterols from the
corresponding demethylated forms is also suitable.
Table 5
Proportion by percentage of free sterols. Each sterol was
determined as a free sterol (without solution) and was related to
the total amount of this sterol. The absolute total sterol
contents as area/g dry substance are indicated in parentheses.
Lanosterol and 4,4-dimethylzymosterol are sterols with a 4,4-
dimethyl group.
% of free sterols
Control AH22tH3ura8/pADL-SAT1
Lanosterol 54 (0.99) 59 (2.90)
4,4-dimethylzymosterol 58 (0.77) 84 (2.37)
4-methylzymosterol 7 (2.43) 10 (7.62)
zymosterol 10 (1.67 11 (5.85)
ergost-7-enol 24 (4.55) 12 (9.00)
ergosta-5,7-dienol
CA 02305780 2000-03-30
27
Description of the Figures
Fig. 1 shows plasmid YEpH2 with the corresponding interfaces.
Fig. 2 shows the plasmid YDpUHK3 with the corresponding
interf aces .
Fig. 3 shows the plasmid pADL-SATl with the corresponding
interfaces.
Fig. 4 shows the growth behavior and ergosterol and squalene
contents with different uracil supplementation. In the
figure, OD = optical density, Kultivierungszeit =
cultivation time, Hefe-Trockengewicht = yeast dry
weight, Uracilsupplementation = uracil supplementation.
CA 02305780 2000-03-30
28
WO 99/16886 PCT/EP98/06134
Bibliographic References
Basson, M. E.; Thorsness, M.; Finer-Moore, J.; Stroud, R. M.;
Rine, J. (1988) Structural and Functional Conservation between
Yeast and Human 3-Hydroxy-3-methylglutaryl Coenzyme A Reductases,
The Rate-limiting Enzyme of Sterol Biosynthesis. Mol. Cell.
Biol. 8: 3793-3808.
Bennetzen, J. L.; Hall, B. D. (1982) The Primary Structure of
the Baccharomyces cerevisiae Gene for Alcohol Dehydrogenase. J.
Biol. Chem. 257: 3018-3025.
Berben, G.; Dumont, J.; Gilliquet, V.; Bolle, P. A.; Hilger, F.
(1991) The YDp Plasmids: A Uniform Set of Vectors Bearing
Versatile Gene Disruption Cassettes for Saccharomyces cerevisiae.
Yeast 7: 475-477.
Boeke, J. D.; Trueheart, J.; Natsoulis, G.; Fink, G. (1987) 5-
Fluorootic Acid as a Selective Agent in Yeast Molecular Genetics.
Methods in Enzymology 154: 164-175.
Fegueur, M.; Richard, L.; Charles, A. D.; Karst, F. (1991)
Isolation and Primary Structure of the ERG9 Gene of Saccharomyces
cerevisiae Encoding Squalene Synthetase. Current Genetics 20:
365-372.
Fischhoff, D. A.; Waterston, R. H.; Olson, M. V. (1984) The Yeast
Cloning Vector YEpl3 Contains a tRNALeu3 Gene That Can Mutate to
an Amber Suppressor. Gene 27: 239-251.
Jandrositz, A.; Turnowsky, F.; Hogenauer, G. (1991) The Gene
Encoding Squalene Epoxidase from Saccharomyces cerevisiae:
Cloning and Characterization. Gene 107: 155-160.
Mezei, L. M.; Storts, D. R. (1994) in: PCR Technology: Current
Innovations, Griffin, H. G. and Griffin, A. M., eds. CRC Press,
Boca Raton, 21.
Mortimer, R. K.; Johnston, J. R. (1986) Genealogy of Principal
Strains of the Yeast Genetic Stock Center. Genetics 113: 35-43.
CA 02305780 2000-03-30
29
Lang, C.; Looman, A. C. (1995) Efficient Expression and
Secretion of Aspergillus niger RH5344 Polygalacturonase in
8accharomyoes cerevisiae. Appl. Microbiol. Biotechnol. 44: 147-
156.
Parks, L. W.; Bottema, C. D. K., Rodriguez, R. J.; Lewis, T. A.
(1985) Yeast Sterols: Yeast Mutants as Tools for the Study of
Sterol Metabolism. Meth. Enzymol. 111, 333-346.
Qureshi, N.; Nimmannit, S.; Porter, J. W. (1981) 3-Hydroxy-3-
methylglutaryl-CoA Reductase from Yeast. Meth. Enzymol. 71:
455-461.
Ruohonen, L.; Aalto, M. K.; Keranen, S. (1995) Modifications to
the ADH1-Promoter of Saccharomyces cerevisiae for Efficient
Production of Heterologous Proteins. Journal of Biotechnology 39:
193-203.
Siduh, R. S.; Bollon, A. P. (1990) Bacterial Plasmid pBR322
Sequences Serve as Upstream Activating Sequences in Saccharomyces
cerevisiae. Yeast 6: 221-229.
Tschumper, G.; Carbon, J. (1980) Sequence of a Yeast DNA Fragment
Containing a Chromosomal Replicator and the TRP1 Gene. Gene 10:
157-166.
Webster, T. D.; Dickson, R. C. (1983) Direct Selection of
Saccharomyces cerevisiae Resistant to the Antibiotic 6418
Following Transformation with a DNA Vector Carrying the
Kanamycin-Resistance Gene of Tn903. Gene 26: 243-252.
Yang, H.; Bard, M.; Bruner, D. A.; Gleeson, A.; Deckelbaum, R.
J.; Aljinovic, G.; Pohl, T. M.; Rothstein, R.; Sturley, S. L.
(1996) Sterol Esterification in Yeast: A Two-Gene Process.
Science 272: 1353-1356.
Yanisch-Perron, C.; Vieira, J.; Messing, J. Gene 33 (1985) 103-
119.
Yu, C.; Rothblatt, J. A. Cloning and Characterization of the
Saccharomyces cerevisiae Acyl-CoA: Sterol-Acyl Transferase
(1996). The Journal of Biological Chemistry, 271: 24157-24163.
CA 02305780 2000-03-30
WO 99/16886 PCT/EP98/06134
SEQUENCE LISTING
(1) GENERAL INFORMATION
(i) APPLICANT
(A) NAME: Schering AG
(B) STREET: Miillerstrasse 178
(C) CITY: Berlin
(E) COUNTRY: Germany
(F) POSTAL CODE (ZIP): D-13342
(G) TELEPHONE: (030)-4681 2085
(H) FAX: (030)-4681 2058
(ii) TITLE OF INVENTION: PROCESS FOR THE PRODUCTION OF
ERGOSTEROL AND ITS INTERMEDIATE PRODUCTS USING
RECOMBINANT YEASTS
(iii)NUMBER OF SEQUENCES: 4
(iv) COMPUTER READABLE FORM:
(A) MEDIUM TYPE: Floppy disk
(B) COMPUTER: IBM PC compatible
(C) OPERATING SYSTEM: PC-DOS/MS-DOS
(D) SOFTWARE: Patentin Release #1.0, Version #1.25
(EPO)
CA 02305780 2000-03-30
31
(2) INFORMATION FOR SEQ ID NO. 1:
(i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 25 bases
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(ii) HYPOTHETICAL: NO
(iii)SEQUENCE DESCRIPTION: SEQ ID NO: 1:
5'-ACTATGGACC AATTGGTGAA AACTG
(2) INFORMATION FOR SEQ ID NO. 2:
(i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 23 bases
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(ii) HYPOTHETICAL: NO
(iii)SEQUENCE DESCRIPTION: SEQ ID NO. 2:
5'-AGTCACATGG TGCTGTTGTG CTT
CA 02305780 2000-03-30
32
(2) INFORMATION FOR SEQ ID NO. 3:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 25 bases
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(ii) HYPOTHETICAL: NO
(iii)SEQUENCE DESCRIPTION: SEQ ID NO. 3:
5'-GAATTCAACC ATGGACAAGA AGAAG
(2) INFORMATION FOR SEQ ID NO. 4:
(i) SEQUENCE CHARACTERISTICS:
(A) LENGTH: 24 bases
(B) TYPE: nucleic acid
(C) STRANDEDNESS: single
(D) TOPOLOGY: linear
(ii) HYPOTHETICAL: NO
(iii)SEQUENCE DESCRIPTION: SEQ ID NO 4:
5'-AGAATTCCAC AGAACAGTTG CAGG