Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02338983 2001-O1-30
WO 00108043 PCT/US99/17487 ,
PREVENTION AND TREATMENT OF VIRAL DISEASE
The inventions consisting of monoclonal antibodies to the FRMS and
affinity tagged viral envelope protein, respectively, were made with
government support
under grant numbers RO1 AI44669-O1 and R21 AI44312-02 awarded by the National
Institutes of Health. The Government has certain rights in these inventions.
This application claims priority to U.S. Provisional Patent Application No.
60/095,105 filed August 3, 1998, and to U.S. Provisional Patent Application
No.
60/141,806 filed June 29, 1999, both of which are incorporated herein by
reference in their
entireties.
1. INTRODUCTION
The present invention relates to the surprising discovery that highly
effective
vaccines may be constructed which present transitional fusion-related
determinants to the
1 S immune system that engender unique antibodies capable of potently
neutralizing a broad
range of primary isolates, from worldwide locations and from different
phylogenetic viral
Glades. The invention relates to the generation of Fusion-Related Molecular
Structures
(FRMS) which comprise one or more fusion-related determinants. The invention
further
relates to the discovery that the broad and uniform neutralization of diverse
primary isolates
indicates that the critical determinants presented by FRMS is highly conserved
and may be
intimately tied to the basic functioning of the envelope protein in binding
and fusion. The
present invention provides methods for formation, isolation and purification
of the FRMS
as well as the use of such FRMS in a variety of compositions and methods,
including, for
example, as vaccine immunogens, diagnostics, and therapeutics.
The present invention concerns FRMS capable of eliciting neutralizing
antibodies to viral pathogens and primary isolates of a virus. Antibodies
raised to the
FRMS can be used to study the molecular pathway toward fusion of the virus and
host cell
and to identify points of a possible antibody-mediated blockade to that
pathway. The
invention also relates to the use of antibodies of the invention for anti-
viral agents, blood
product additives, contraceptive additives passive immunization in post-
exposure treatments
or fetus immunization.
2. BACKGROUND OF THE INVENTION
Citation of a reference herein shall not be construed as an admission that
such reference is prior art to the present invention.
CA 02338983 2001-O1-30
WO 00/08043 PCT/US99/17487
Viruses are infectious agents responsible for many diseases in humans,
animals, bacteria and plants, and are therefore of great medical, and
commercial
importance. Broadly speaking, a free virus particle, called a virion, is made
up of a genome
(which can be RNA or DNA), associated proteins or polyamides, and a protein
coat called a
capsid. Some virions are further surrounded by a membranous envelope. Capsid
and
envelope structures serve to protect the genome from nucleases present in the
environment
and also serve to facilitate viral attachment and entry into the cell in which
it will infect and
replicate.
2.1: VIRAL ENTRY
A virus must utilize the biosynthetic machinery of the host cell to replicate.
Viruses use the synthetic pathways and substrates available in animal cells to
maintain and
propagate their own genetic information. To gain access to the cell's
biomachinery, the
virus must enter or infect the cell. Entrance is facilitated by the presence
of one or more
viral receptor proteins on host cell membranes. Viral proteins bind to these
receptors
initiating a complex series of events by which the viral nucleic acid or
nucleoproteins are
released into the cell.
In the case of enveloped viruses, infection is facilitated by fusion of the
viral
envelope with a cellular membrane such as a cell surface membrane or an
internal
membrane. A variety of enveloped viruses are implicated in human and animal
disease.
2.2. HIV
One clinically important example of an enveloped virus is HIV. The human
immunodeficiency virus (HIV) has been implicated as the primary cause of the
slowly
degenerative immune system disease termed acquired immune deficiency syndrome
(AIDS)
(Bane-Sinoussi, F., et al., 1983, Science 220:868-870; Gallo, R., et al.,
1984, Science
224:500-503). In humans, HIV replication occurs prominently in CD4+ T
lymphocyte
populations, and HIV infection leads to depletion of this cell type and
eventually to immune
incompetence, opportunistic infections, neurological dysfunctions, neoplastic
growth, and
ultimately death. At least two distinct types of HIV exist including: HIV-1
(Barre-
Sinoussi, F., et al.; 1983, Science 220:868-870; Gallo, R., et al., 1984,
Science 224:500-
503) and HIV-2 (Clavel, F., et al., 1986, Science 233:343-346; Guyader, M., et
al., 1987,
Nature 326:662-669). Additionally, significant genetic heterogeneity exists
within
populations of each types.
HIV is a member of the class of lentivirus in the family of retroviruses
(Teich, N., et al., 1984, RNA Tumor Viruses, Weiss, R., et al., eds., CSH-
Press, pp. 949-
956). Retroviruses are small enveloped viruses that contain a single-stranded
RNA genome,
2
CA 02338983 2001-O1-30
WO 00/08043 PCTNS99/17487
and replicate via a DNA intermediate produced by a virally-encoded reverse
transcriptase,
an RNA-dependent DNA polymerase (Vannus, H., 1988, Science 240:1427-1439).
The HIV viral particle comprises a viral core, composed in part of capsid
proteins, together with the viral RNA genome and those enzymes required for
early
S replicative events. Myristylated gag protein forms an outer shell around the
viral core,
which, in turn, is surrounded by a lipid membrane envelope derived from an
infected cell
membrane. The HIV envelope surface glycoproteins are synthesized as a single
160
kilodalton precursor protein which is cleaved by a cellular protease during
viral budding
into two glycoproteins, gp41 and gp120. gp41 is a transmembrane glycoprotein
and gp120
is an extracelluiar glycoprotein which remains non-covalently associated with
gp4l,
possibly in a trimeric or multimeric form (Hammarskjold, M., et al., 1989,
Biochem.
Biophys. Acta 989:269-280).
HIV is targeted to CD4+ cells because a CD4 cellular membrane protein
(CD4) acts as a cellular receptor for the HIV-1 virus (Dalgleish, A., et al.,
1984, Nature
312:763-767; Klatzmann et al., 1984, Nature 312:767-768; Maddon et al., 1986,
Cell
47:333-348). Viral entry into cells is dependent upon gp120 binding the
cellular CD4
receptor molecules (McDougal, J.S., et al., 1986, Science 231:382-385; Maddon,
P.J., et al.,
1986, Cell 47:333-348), explaining HIV's tropism for CD4+ cells.
Upon interaction of the viral envelope protein and cellular receptor, the
envelope protein-CD4 complex undergoes conformational changes facilitating
subsequent
interaction with host cellular co-receptors to form a trimolecular complex.
Further
conformational changes in the trimolecular complex allow fusion of the apposed
cell
membrane and virus envelope releasing viral genetic material into the cell.
2.3. VIRAL TREATMENT STRATEGIES
2.3.1. DRUG THERAPY
Although considerable effort is being put into the design of effective
Therapeutics, currently no curative anti-retroviral drugs against AIDS exist.
Attempts to
develop such drugs have focused on several stages of the HIV life cycle
(Mitsuya, H., et al.,
1991, FASEB J. 5:2369-2381). Many viral targets for intervention with HIV life
cycle have
been suggested, since the prevailing view is that interference with a host
cell protein would
have deleterious side effects. For example, virally encoded reverse
transcriptase has been
one focus of drug development. A number of reverse-transcriptase-targeted
drugs,
including 2',3 '-dideoxynucleoside analogs such as AZT, ddI, ddC, and d4T have
been
developed which have been shown to been active against HIV (Mitsuya, H., et
al., 1991,
Science 249:1533-1544).
3
CA 02338983 2001-O1-30
-- WO 00/08043 PCT/US99/17487
The new treatment regimens for HIV-1 demonstrate that a combination of
anti-HIV compounds, which target reverse transcriptase (RT), such as
azidothymidine
(AZT), lamivudine (3TC), dideoxyinosine (ddI), dideoxycytidine {ddC) used in
combination with an HIV-1 protease inhibitor have a far greater effect (2 to 3
logs
reduction) on viral load compared to AZT alone (about 1 log reduction). For
example,
improved results have recently been obtained with a combination of AZT, ddI,
3TC and
ritonavir (Perelson, A.S., et al., 1996, Science 15:1582-1586). However, it is
likely that
long-term use of combinations of these chemicals will lead to toxicity,
especially to the
bone marrow. Long-term cytotoxic therapy may also lead to suppression of CD8+
T cells,
which are essential to the control of HIV, via killer cell activity (Blazevic,
V., et al., 1995,
AIDS Res. Hum. Retroviruses 11:1335-1342) and by the release of suppressive
factors,
notably the chemokines Rantes, MIP-la and MIP-lei (Cocchi, F., et al., 1995,
Science
270:1811-1815). Another major concern in long-term chemical anti-retroviral
therapy is the
development of HIV mutations with partial or complete resistance (Lange, J.M.,
1995,
ADS Res. Hum. Retroviruses 10:S77-82). It is thought that such mutations may
be an
inevitable consequence of anti-viral therapy. The pattern of disappearance of
wild-type
virus and appearance of mutant virus due to treatment, combined with
coincidental decline
in CD4+ T cell numbers strongly suggests that, at least with some compounds,
the
appearance of viral mutants is an underlying factor in the failure of AIDS
therapy.
Additionally, the failure of conventional drugs may also be attributable to
reservoirs of HIV in slowly growing cell populations which are latent and do
not actively
produce virus and therefore escape many of the conventional treatments.
Attempts are also being made to develop drugs which can inhibit viral entry
into the cell. For these studies, the focus has thus far focused on CD4, the
cell surface
receptor for HIV. Recombinant soluble CD4, for example, has been shown to
inhibit
infection of CD4~ T cells by some HIV-1 strains (Smith, D.H., et al., 1987,
Science
238:1704-1707). Certain primary HIV-I isolates, however, are relatively less
sensitive to
inhibition by recombinant CD4 (Daar, E., et al., 1990, Proc. Natl. Acad. Sci.
USA 87:6574-
6579). In addition, recombinant soluble CD4 clinical trials have produced
inconclusive
results (Schooley, R., et al., 1990, Ann. Int. Med. 112:247-253; Kahn, J.O.,
et al., 1990,
Ann. Int. Med. 112:254-261; Yarchoan, R., et al., 1989, Proc. Vth Int. Conf.
on AIDS, p.
564, MCP 137).
The late stages of HIV replication, which involve crucial virus-specific
processing of certain viral encoded proteins, have also been suggested as
possible anti-HIV
dmg targets. Late stage processing is dependent on the activity of a viral
protease, and
drugs are being developed which inhibit this protease (Erickson, J., 1990,
Science 249:527-
533).
4
CA 02338983 2001-O1-30
-- WO 00/08043 PC'r/US99/17487
Recently, chemokines produced by CD8+ T cells have been implicated in
suppression of HIV infection (Paul, W.E., 1994, Cell 82:177; Bolognesi, D.P.,
1993, Semin.
Immunol. 5:203). The chemokines RANTES, MIP-la and MIP-1 (I, which are
secreted by
CD8+ T cells, were shown to suppress HIV-1 p24 antigen production in cells
infected with
HIV-1 or HIV-2 isolates in vitro (Cocchi, F, et al., 1995, Science 270:1811-
1815). Thus,
these and other chemokines may prove useful in therapies for HIV infection.
The clinical
outcome, however, of all these and other candidate drugs is still in question.
2.3.2. VACCINES
bother important strategy in anti-viral treatment and prevention is the
development of vaccines. Due to the potential pandemic nature of viral
infection, such as
HIV, there is a significant need for effective vaccines.
Generally, traditional methods for preparing vaccines include the use of
inactivated or attenuated pathogens. A suitable inactivation of the pathogenic
1 S microorganism renders it han~nless as a biological agent but does not
destroy its
immunogenicity. Injection of these "killed" particles into a host will then
elicit an immune
response capable of preventing a future infection with a live microorganism.
However, a
major concern in the use of killed vaccines (using inactivated pathogen) is
failure to
inactivate all the microorganism particles. Even when this is accomplished,
since killed
pathogens do not multiply in their host, or for other unknown reasons, the
immunity
achieved is often incomplete, short lived and requires multiple immunizations.
Finally, the
inactivation process may alter the microorganism's antigens, rendering them
less effective
as immunogens. .
Attenuation refers to the production of strains of pathogenic microorganisms
which have essentially lost their disease-producing ability. One way to
accomplish this is to
subject the microorganism to unusual growth conditions and/or frequent passage
in cell
culture. Mutants are then selected which have lost virulence but yet are
capable of eliciting
an immune response. Attenuated pathogens often make good immunogens as they
actually
replicate in the host cell and elicit long lasting immunity. However, several
problems are
encountered with the use of live vaccines, the most worrisome being
insufficient attenuation
and the risk of reversion to virulence.
An alternative to the above methods is the use of subunit vaccines. This
involves immunization only with those components which contain the relevant
immunological material.
In the case of HIV, for example, envelope proteins (gp160, gp120, gp41)
have been shown to be the major antigens for anti-HIV antibodies present in
AIDS patients
(Barin et al., 1985, Science 228:1094-1096). Thus far, therefore, these
proteins have been
5
CA 02338983 2001-O1-30
WO 00/08043 PCT/US99/17487
the most common candidates to act as immunogens for anti-HIV vaccine
development.
Several groups have begun to use various portions of gp160, gp120, and/or gp41
as
immunogenic targets for the host immune system. See for example, Ivanoff, L.,
et al., U.S.
Pat. No. 5,141,867; Saith, G., et al., W092/22,654; Shafferman, A.,
W091/09,872;
Formoso, C., et al., W090/07,119.
Because the HIV envelope protein mediates the early binding and entry steps
in infection, many vaccine strategies have focused on blocking the function of
the viral
envelope glycoprotein. In 1993, two recombinant forms of the surface gp120
subunit of the
HIV envelope protein (rgp 120) were advanced as candidate vaccines for a large
scale
efficacy study sponsored by the National Institutes of Health (NIH). In
previous clinical
studies, these rgp 120 vaccines had been shown to be safe and to elicit
antibodies capable of
potently neutralizing related laboratory-adapted isolates of HIV (Belshe,
R.B., et al., 1994,
Journal of the American Medical Association 272:475; Kahn, J.O., et al., 1994,
Journal of
Infectious Diseases 170:1288). Progress was stalled, however, by findings that
Primary
Isolate ("PI") viruses were largely refractory to neutralization by rgp120
vaccine sera
(Cohen, J., 1993, Science 262:980; Cohen, J., 1994, Science 264:1839).
2.4. THE NEED FOR NEUTRALIZATION OF PRIMARY ISOLATES
The expanding epidemic of HIV infection threatens to infect more than 40
million persons worldwide by the year 2000 (UNAIDS Report
(www.unaids.org/unaids/report). The need for an effective HIV vaccine is
urgent, but
progress towards this goal has been blocked by the inability of any vaccine
candidate to
elicit antibodies capable of neutralizing infectivity of a variety of primary
isolates (PIs)
from HIV infected individuals {Wrin, T., et al., 1995, Journal of Virology
69:39; Moore,
J-P., et al., 1995, AIDS 9 (suppl A), S 117; Mascola, J.R., et al., 1996,
Journal of Infectious
Diseases 173:340.
Unfortunately, to date, researchers have been unsuccessful in producing
vaccines that generate antibodies which block a wide variety of primary
isolates of HIV.
Primary isolates are taken directly from infected persons and subjected to
only limited
~°wth in the laboratory in primary peripheral blood lymphocytes (PB
ls). Thus, primary
HIV isolates are clinically more relevant than laboratory-adapted strains.
Laboratory
adapted strains are those which are persistently grown in established T cell
lines in the
laboratory (See Wrin et al., 1995, J. of Virology 69:39). Previous vaccine
candidates have
elicited neutralizing antibodies to laboratory-adapted strains. For example,
Wrin et al.,
1995, Journal of Virology 69:39, tested HIV envelope proteins produced by
recombinant
DNA technology derived from laboratory adapted isolates. Antibodies generated
by
immunization with these recombinant proteins neutralized only laboratory-
adapted isolates
6
CA 02338983 2001-O1-30
WO 00/08043 PCT/US99/17487
of the virus. Recombinant viral vectors and DNA used to produce native
oligomeric
envelope protein in situ also failed to elicit neutralizing antibodies to
primary isolates.
Additionally, immunogens produced by recombinant technology comprising the
gp120 and
soluble CD4 receptor elicited antibodies to gp120 (Gershoni, 1993, FASEB
Journal
7:1185), but these antibodies were unable to neutralize primary isolates.
Neutralizing antibodies are important to prevent binding and fusion of the
virus to the host cell. The action of antibodies that neutralize laboratory-
adapted isolates
have been analyzed. Neutralizing antibodies directed specifically to the CD4
binding
domain on gp 120 interfere with CD4 binding. Most neutralizing antibodies,
however,
10 interfere with steps subsequent to the binding of CD4. For example, some
neutralizing
antibodies inhibit interaction with the co-receptor (Trkola et al., 1998,
Journal of Virology
72:1876; Wu, et al., 1996, Nature 384:179-83).
The identification of immunogens that elicit neutralizing antibodies in a host
is necessary to the development of efficacious vaccines. Neutralizing
antibodies must act
15 upon not only laboratory-adapted isolates, but the clinically relevant
primary isolates as
well. As can be understood from the above, there remains a need for new
immunogen
molecules which generate antibodies that effectively neutralize a wide variety
of primary
isolates.
20 3. SUMMARY OF THE INVENTION
The present invention is based on the surprising discovery that highly
effective vaccines may be constructed which present transitional fusion-
related determinants
to the immune system that engender unique antibodies capable of potently
neutralizing a
broad range of primary isolates of enveloped viruses, from worldwide locations
and from
25 different phylogenetic viral Glades. The vaccine of the invention comprise
as immunogens
Fusion-Related Molecular Structures (FRMS) which comprise one or more fusion-
related
determinants. The invention is further based on the discovery that the broad
and uniform
neutralization of diverse primary isolates indicates that the critical
determinants presented
by FRMS are highly conserved and may be intimately tied to the basic
functioning of the
30 envelope protein in binding and fusion. The present invention provides the
use of such
FRMS in a variety of compositions and methods, including, for example, as
vaccine
immunogens, blood product additives, anti-viral agents, diagnostics and
therapeutics.
The present invention concerns FRMS capable of eliciting neutralizing
antibodies to enveloped viral pathogens. Epitopes which elicit these
neutralizing antibody
35 result from conformational changes during the process of binding and fusion
of the viral
envelope and host cell membranes.
7
CA 02338983 2001-O1-30
WO 00/08043 PCT/US99/17487
In one embodiment, the complexes result from the interaction of viral
proteins with at least one host cellular receptor or co-receptor and are
created by
co-culturing cells transformed with a nucleic acid expressing a viral protein
and cells
expressing host cellular receptor(s). Preferably, the cells recombinantly
express the host cell
receptor(s). In a further embodiment, cultures are fixed at the onset of cell-
cell interaction
to preserve fusion-competent immunogens. In a preferred embodiment, the fusion-
related
immunogens are formed as a result of the interaction of the major human
immunodeficiency
virus type 1 (HIV-1) envelope protein, the host cellular receptor CD4 and the
host cellular
co-receptor CCRS.
Vaccinating with the FRMS of the subject invention as immunogens raises
an immune response to the viral pathogen and elicits the production of
neutralizing
antibodies in the vaccinated host. In addition to using the FRMS as a vaccine,
antibodies
raised to the FRMS can be used to study the molecular pathway toward fusion of
the virus
and host cell and to identify points of a possible antibody-mediated blockade
to that
pathway. Additionally, specific high-affinity "tags" may be expressed as part
of the
envelope protein or host cellular receptors) that faclilitates isolation and
purification of the
FRMS of the invention.
The present invention further relates to the development of monoclonal
antibodies (mAb) elicited by these FRMS immunogens. Monoclonal antibodies
which are
capable of neutralization of primary isolates are provided. Neutralizing mAbs
to
functionally conserved fusion-dependent epitopes may also be useful for
passive
immunization in post-exposure treatments or fetus immunization.
The present invention provides an isolated molecular structure comprising an
epitope formed as a result of association of (a) an envelope protein of an
enveloped virus,
with (b) one or more cellular membrane proteins, which envelope protein and
cellular
membrane proteins are necessary and sufficient under suitable conditions for
fusion of said
envelope of the virus with a cell membrane containing said cellular membrane
proteins.
The present invention provides an isolated molecular structure comprising an
epitope formed as a result of association of (a) an HIV envelope protein, or a
mutant thereof
that assembles into the viral envelope; with (b) human CD4 and a co-receptor
for HIV
fusion. In one embodiment, co-receptor is the chemokine receptor CCRS or
CXCR4. In
another embodiment, the molecular structure is formed by association of a
mutant of HIV
gp41 that is fusion-defective. In another embodiment, the mutant contains one
or more
mutations selected from the group consisting of V2E, GiOV, V570R, and Y586E.
In yet
another embodiment the molecular structure is formed by association of wild-
type HIV
envelope protein.
8
CA 02338983 2001-O1-30
WO 00/08043 PCTNS99/17487
The present invention provides an isolated molecular structure comprising an
epitope formed as a result of association of (a) a mutant envelope protein of
an enveloped
virus, which envelope protein in wild-type form functions in fusion ofthe
viral envelope
with a host cell membrane, and which mutant envelope protein is fusion-
defective; and (b)
one or more host cellular membrane proteins which function as receptors for
said envelope
protein.
In one embodiment, said virus is from a viral family is selected from the
group consisting of Retroviridae, Rhabdoviridae, Caronaviridae, Filoviridae,
Poxviridae,
Bunyaviridae, Flaviviridae, Togaviridae, Orthomyxoviridae, Paramyxoviridae,
and
Herpesviridae. In another embodiment; the envelope protein is E 1 and E2 of
HCV and the
cellular membrane protein is CD81. In another embodiment the molecular
structure is a
cross-linked cellular molecular structure. In yet another embodiment, the
molecular
structure is isolated from a cell lysate. In one embodiment, the cellular
molecular structure
comprises cells recombinantly expressing the envelope protein. In yet another
embodiment,
1 S the cell lysate is from a plurality of cells comprising cells
recombinantly expressing the
envelope protein. In still another embodiment, the cellular molecular
structure further
comprises cells recombinantly expressing the one or more host cellular
membrane proteins.
The present invention provides a recombinant enveloped virus, wherein said
virus recombinantly expresses on its envelope a cell receptor for a native
envelope protein
of said virus. In one embodiment, said cell receptor is human CD4 or a co-
receptor for
HIV, or said virus recombinantly expresses both human CD4 and said co-
receptor. In
another embodiment, said suitable conditions comprise a lowering of pH. In
another
embodiment, the cross-linked cellular molecular structure further comprises a
cross-linked
viral particle of said virus, containing said envelope protein.
The present invention provides a vaccine formulation comprising an
immunogenic amount of the molecular structure and a pharmaceutically
acceptable carrier.
The present invention provides a monoclonal antibody to the molecular
structure. In one embodiment, the antibody is labeled.
The present invention provides a purified polyclonal antiserum specific to
the molecular structure.
The present invention provides a contraceptive jelly, foam, cream, or
ointment comprising an amount of the antibody effective to inhibit or decrease
infection by
the virus.
The present invention provides a contraceptive jelly, foam, cream, or
ointment comprising an amount of the antibody effective to inhibit or decrease
infection by
HIV.
9
CA 02338983 2001-O1-30
WO 00/08043 PCT/US99/17487
The present invention provides a contraceptive jelly, foam, cream, or
ointment comprising an amount of the antiserum effective to inhibit or
decrease infection by
HIV.
The present invention provides a sample of mammalian blood, to which an
amount of the antibody has been added effective to inhibit or decrease
infection by the
virus.
The present invention provides a sample of human blood, to which an
amount of the antibody has been added effective to inhibit or decrease
infection by HIV. In
one embodiment, said envelope protein or host cellular membrane proteins
further
comprises an affinity tag. In another embodiment, said envelope protein, CD4,
or co-
receptor further comprises an affinity tag. In yet another embodiment, the
antibody is
labeled.
The present invention provides a kit comprising in one or more containers a
labeled monoclonal antibody to the molecular structure.
The present invention provides a kit comprising in one or more containers a
labeled monoclonal antibody to the molecular structure. in one embodiment, the
kit
comprises the molecular structure in a separate container.
The present invention provides a cell line that recombinantly expresses an
envelope protein of an enveloped virus that functions in fusion of the viral
envelope with a
host cell membrane, or a mutant form of said envelope protein that is fusion-
defective,
which cell line expresses one or more celluiar membrane proteins that function
as receptors
for said envelope protein. In one embodiment, said one or more proteins that
function as
receptors are recombinantly expressed.
The present invention provides a cell line that recombinantly expresses HIV
X160, which cell line expresses CD4 and a co-receptor for HIV; said cell line
lacking a
functional protease that cleaves gp160 to produce gp120 and gp4l.
The present invention provides a method of treating or preventing infection
by a virus in a subject comprising administering to the subject an immunogenic
amount of
the molecular structure effective to treat or prevent infection by the virus.
In one
embodiment, the subject is a human. In another embodiment, the subject is a
domestic
animal.
The present invention provides a method of treating or preventing infection
by HIV in a human comprising administering to the human an immunogenic amount
of the
molecular structure effective to treat or prevent infection by HIV.
The present invention provides a method of treating or preventing infection
by a virus in a subject comprising administering to the subject an amount of
the monoclonal
antibody effective to treat or prevent infection by the virus.
CA 02338983 2001-O1-30
WO 00/08043 PCT/US99/17487
The present invention provides a method of treating or preventing infection
by HIV in a human comprising administering to the human an amount of the
monoclonal
antibody effective to treat or prevent infection by HN. In one embodiment,
said human has
a high risk of HIV infection. In one embodiment, the method is for treatment
of AIDS in
said human.
The present invention provides a method for treating or preventing infection
by HIV in a human fetus comprising administering to a pregnant human
containing said
fetus an amount of the monoclonal antibody effective to treat or prevent
infection by HIV in
said fetus.
The present invention provides a method of inhibiting infection by a virus in
a sample of blood comprising contacting said sample of blood with an amount of
the
monoclonal antibody effective to inhibit infection by said virus.
The present invention provides a method of inhibiting infection by HIV in a
sample of human blood comprising contacting said sample of human blood with an
amount
of the monoclonal antibody effective to inhibit infection by HIV.
The present invention provides a method of decontaminating surgical or
dental tools comprising contacting said tools with an amount of the monoclonal
antibody
effective to inhibit infection by said virus.
The present invention provides a method of decontaminating surgical or
dental tools comprising contacting said tools with an amount of the monoclonal
antibody
effective to inhibit infection by HIV.
The present invention provides a method for monitoring the production of
antibody to the molecular structure in a subject previously administered an
amount of the
molecular structure comprising isolating from said subject a sample comprising
serum; and
detecting the presence of any antibodies to the molecular structure in said
serum. In one
embodiment, said detecting is carried out by a method comprising performing a
competitive
immunoassay with labeled antibody to the molecular structure.
The present invention provides a method of producing an immunogen for use
in a vaccine for the treatment or prevention of infection by a virus
comprising the following
steps in the order stated: (a) contacting an envelope protein or chimeric form
thereof of an
enveloped virus, which envelope protein functions in fusion of the viral
envelope with a cell
membrane, or a mutant form or chimeric form thereof of said envelope protein
that is
fusion-defective, with one or more cell proteins or chimeric forms thereof
that function as
receptors for said envelope protein; (b) exposing said envelope protein or
chimeric form
thereof or mutant form or chimeric form thereof, and said one or more host
cell proteins or
chimeric forms thereof, to a cross-linking agent; and (c) isolating a cross-
linked structure
11
CA 02338983 2001-O1-30
WO 00/0$043 PCT/US99/17487
comprising said envelope protein or chimeric form thereof or mutant form or
chimeric form
thereof.
The present invention provides a method of producing an immunogen for use
in a vaccine for the treatment or prevention of infection by HIV comprising
the following
s steps in the order stated: (a) co-culturing a first cell recombinantly
expressing HIV envelope
protein or a chimeric form thereof, or a mutant form or chimeric form thereof
of said
envelope protein that is fusion-defective, with a second cell that expresses
(i) human CD4
or a chimeric form thereof, and (ii) a co-receptor for HN or a chimeric form
thereof; (b)
exposing said envelope protein or chimeric form thereof or mutant form or
chimeric form
thereof, and said CD4 or chimeric form thereof and co-receptor or chimeric
form thereof, to
a cross-linking agent; and (c) isolating a cross-linked structure comprising
said envelope
protein or chimeric form thereof or mutant form or chimeric form thereof. In
one
embodiment, said virus is HIV and said host cell proteins are human CD4 and a
co-receptor
for HIV. In one embodiment, said second cell recombinantly expresses CD4 or
said co-
t S receptor or both CD4 and said co-receptor or chimeric forms of any of the
foregoing.
In other embodiment, said first and second cell are the same cell type. In yet
another embodiment, said envelope protein or chimeric form thereof or mutant
form or
chimeric form thereof is present on a viral particle or virus-like particle.
In still another
embodiment, said cross-linked structure is a cross-linked cellular complex.
In another embodiment, the virus is selected from the group consisting of
Retroviridae, Rhabdoviridae, Coronaviridae, Filoviridae, Poxviridae,
Bunyaviridae,
Flaviviridae, Togaviridae, Orthomyxoviridae, Paramyxoviridae, and
Herpesviridae.
In another embodiment, said contacting step occurs by infecting cells
expressing said host cell proteins with said virus expressing said envelope
protein or
chimeric form thereof or mutant form or chimeric form thereof. In another
embodiment, a
chimeric form of said envelope protein or one of said host cell proteins is
contacted, said
chimeric form comprising an affinity tag. In another embodiment, a chimeric
form of said
envelope protein or CD4 or said co-receptor is contacted, said chimeric form
comprising an
affinity tag.
The present invention provides a method of producing an immunogen for use
in a vaccine for the treatment or prevention of infection by HIV comprising
the following
steps in the order stated: (a) co-culturing a first cell recombinantly
expressing HIV envelope
protein or a chimeric form thereof, or a mutant form or chimeric form thereof
of said
envelope protein that is fusion-defective, with a second cell that expresses
(i) human CD4
or a chimeric form thereof, and (ii) a co-receptor for HIV or a chimeric form
thereof
wherein at least one of said chimeric forms comprising an affinity tag is
expressed; (b)
lysing said co-cultured cells to form a cell lysate under non-denaturing
conditions; and (c)
12
CA 02338983 2001-O1-30
WO 00/08043 PCT/US99/17487
isolating from said cell lysate a molecular structure comprising said envelope
protein or
chimeric form thereof or mutant form or chimeric form thereof by a method
comprising
contacting said cell lysate with a binding partner to said affinity tag and
recovering a
molecular structure bound to said affinity tag.
The present invention provides a cross-linked structure that is the product of
the method.
The present invention provides a monoclonal antibody to the structure of that
neutralizes in vitro the following primary isolates of HTV: 92US657, 92US660,
92RW023,
93IN101, 92UG035, and 92TH023.
The present invention provides a contraceptive, jelly, foam, cream or
ointment comprising an amount of the antibody to inhibit or decrease infection
by HIV.
The present invention provides a method of treating or preventing infection
by a virus in a subject comprising administering to the subject an immunogenic
amount of
the structure effective to treat or prevent infection by the virus.
The present invention provides a method of treating or preventing infection
by HIV in a human comprising administering to the human an itnmunogenic amount
of the
molecular structure effective to treat or prevent infection by HIV.
The present invention provides a method of decontaminating surgical or
dental tools comprising contacting said tools with an amount of the monoclonal
antibody
effective to inhibit infection by HIV.
The present invention provides a method of decontaminating surgical or
dental tools comprising contacting said tools with an amount of the monoclonal
antibody
effective to inhibit infection by said virus.
The present invention provides a method of screening a molecular structure
for vaccine efficacy comprising immunizing a transgenic non-human mammal with
the
molecular structure, wherein said transgenic non-human mammal expresses from
one or
more transgenes both human CD4 and a co-receptor for HIV, and detecting any
neutralizing
antibodies to HIV that are produced by said mammal.
The present invention provides a method of screening a molecular structure
for vaccine efficacy comprising immunizing a transgenic non-human mammal with
the
molecular structure, wherein said transgenic non-human mammal expresses from
one or
more transgenes said one or more host cellular membrane proteins; and
detecting any
neutralizing antibodies to said virus that are produced by said mammal. In one
embodiment,
the mammal is a mouse. In another embodiment, said first cell recombinantly
expresses
HIV envelope protein or a chimeric form thereof.
13
CA 02338983 2001-O1-30
WO 00/08043 PCTNS99/17487
4. BRIEF DESCRIPTION OF THE FIGURES
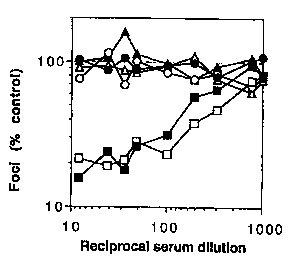
Figure 1. Neutralization of the homologous 168P PI virus by FC and FI vaccine
sera. Transgenic mice (hu CD4+, hu CCRS+, mouse CD4+) were immunized with FC
immunogen (COS-env with U87-CD4-CCRS; squares; n = 3 mice) or with cell
controls
(U87-CD4-CCRS cells alone or cocultured with mock-transfected COS cells;
circles n = 3
mice). Unimmunized mice were also used (triangles; n = 2 mice). Sera were
tested for
neutralization of 168P using U87-CD4 cells expressing either CXCR4 (black
symbols) or
CCRS (white symbols). Data represent averages of three to six neutralization
assays using
serum obtained 2 weeks following second and third immunization.
Figure 2A-B. Neutralization of P168 by FC but riot FI, vaccine sera. (A)
Transgenic
mice were immunized with FC immunogen (black squares; n = 4), FI immunogens
(COS-
env with U87 cells; gray circles n = 4; COS-env with U87-CD4 cells, gray
diamonds, n = 3;
COS-env with sCD4, white diamonds, n = 2; COS-env with U87-CD4-CCRS cells,
each
fixed separately prior to mixing for immunization, gray squares, n = 2), or
mock-transfected
cos cell immunogen (cocultured with U87-CD4-CCRS cells, white circles, n = 2).
Unimmunized mice (white triangles, n = 2) were also used. Neutralization was
independent
of specific co-receptor use and data here represent averages of three to six
neutralization
assays in U87-CD4-CXCR4 or -CCRS cells. In some cases, sera from all animals
within
each experimental group were pooled. (B) Neutralization of P168 by FC but not
FI, vaccine
sera. Neutralization of the homologous 168P PI virus in human PBL (lymphocyte)
culture.
PBLs were isolated, stimulated with phytohemagglutinin, and grown in the
presence of
interleukin-2; neutralization,was determined. HiV p24 antigen was determined
after 5 days
of culture by ELISA (Coulter Corporation) and values were normalized to the
virus control
(36 ng/ml). Asterisks indicates p24 antigen levels below the limit of
detection at the dilution
used in the ELISA.
Figure 3A-B. FC vaccine serum does not neutralize pseudotyped HIV virions
bearing
amphotropic MLV envelope protein (A) or primary SIVmac251 (B). For HIV bearing
an
amphotropic MLV envelope protein (ampho MLV pseudotype) neutralization
sensitivity
with pooled FC and FI antisera was determined in U87-CD4-CXCR4 cells. For
primary
isolate SIVmac251 neutralization was determined in U87-CD4-CCRS cells. Symbols
are:
FC Immunogen (black squares,) and FI immunogen (Cos-env + U87 cells, gray
circles).
Figure 4A-B Neutralization of TCLA 168C virus by FI vaccine sera.
Neutralization
sensitivity of the 168P PI virus (A) and its TCLA derivative 168C (B) were
tested in U87-
14
CA 02338983 2001-O1-30
-- WO 00/08043 PCT/US99/17487
CD4-CXCR4 cells with pooled sera: FC immunogen (black squares), FI immunogens
(COS-env + U87 cells, gray circles; COS-env + 1187-CD4 cells, gray diamonds;
COS-env
+sCD4, white diamonds), and mock-transfected cell controls (white circles).
Figure 5. Neutralization of diverse PI viruses from Glades A-E. Primary
isolates were
expanded in human PBLs, and neutralization was determined in permissive U87-
CD4-
CCRS (or -CXCR4) cells with pooled sera: FC immunogens (black squares), FI
immunogens (COS-env + U87 cells, gray circles; COS-env + U87-CD4, gray
diamonds;
COS-env + sCD4, white diamonds, and mock transfected cell controls (white
circles). Viral
biotype is indicated in lower right corner as SI or NSI where available. Viral
isotype is
indicated in lower corner of each graph.
Figure 6. Adsorption of PI virus neutralization activity by formaldehyde-fixed
COS-env
cells. FC vaccine serum was repeatedly incubated with formaldehyde-fixed COS-
env cells
(gray squares) or control COS cells (white squares). Serum obtained prior to
FC
immunization was similarly adsorbed (gray and white circles, respectively).
The starting
FC and preimmunization sera are indicated as black squares or black circles,
respectively.
Sear were tested for neutralization of 168P using U87-CD4-CXCR4 cells.
Figure 7. PI virus neutralization activity is not adsorbed by intact U87-CD4-
CCRS cells.
Pooled FC vaccine serum was incubated with U87-CD4 CCRS cells (while squares)
or in an
empty microculture well (mock, black squares). Pooled FI serum (COS-env + U98-
CD4)
was similarly treated (white and black circles, respectively). Sera were
tested for remaining
neutralization of 168P with U87-CD4-CCRS cells.
35
Figure 8. Neutralizing activity of two HIV Clade B primary isolates with
hybridoma
supernatants.
Figure 9. Serum titers of mice immunized with fusion-competent vaccine
immunogens.
~Ite square represents fusion-competent (FRMS) immunogen (env+with CD4/CCRS);
black circles represent immunogen from cells expressing envelope protein
immunogen and
cells expressing neither CD4 nor CCRS (env); white circles represent immunogen
from
cells expressing envelope protein and cells expressing CD4 immunogen
(env+CD4); white
triangle represent cell controls expressing CD4 and CCRS immunogen (CD4/CCRS).
Figure 10. Cross-neutralization of the 320SI primary virus isolate by
antibodies obtained
from mice immunized with a fusion-competent immunogen complex of the subject
CA 02338983 2001-O1-30
WO 00/08043 PCT/US99/17487
invention. Black circles represent fusion-competent (FRMS) immunogen (env with
CD4/CCRS); white squares represent immunogen from cells expressing envelope
protein
(env); white circles represent immunogen from cells expressing envelope
protein and CD4
(env + CD4).
Figure 11. Envelope protein co-purified with CD4-Spep and CCRS-Spep detected
by
Western blot analysis. a) 168P envelope protein isolated following co-culture
with cells
expressing CD4-Spep b) 168P envelope protein isolated following co-culture
with cells
expressing CD4 and CCRS-Spep c) 168P envelope protein was undetectable
following
co-culture with mock-transfected cells..
Figure 12. Co-culture of BSC40 cells infected with rV-168Penv and rV-CD4/CCRS
24 hr
post infection.
5. DETAILED DESCRIPTION OF THE INVENTION
The present invention concerns fusion-related molecular-structures (FRMS)
capable of eliciting neutralizing antibodies to enveloped viral pathogens. The
FRMS of the
invention comprise epitopes formed as a result of the association of one or
more cellular
molecules with one or more viral envelope molecules. The process of viral
envelope fusion
with a cell membrane is initiated following binding of a viral envelope
protein to specific
cell receptors and/or co-receptors on the host cell and involves fusion of the
viral envelope
with surface or internal cell membranes. Fusion is driven by conformational
changes which
occur within the envelope-receptor/co-receptor complex. As not intended to be
limited by a
particular mechanism, it is believed that the conformational changes that
occur during
Vision reveal new and unique immunogenic epitopes for neutralizing antibodies.
The
fusion-related molecular structures of the subject invention mimic the
conformational
intermediates formed during fusion. The FRMS of the invention may be used for
a variety
of purposes including but not limited to those presented in Sections S.S and
5.7, herein. For
example, in a preferred embodiment of the invention, the FRMS when used to
vaccinate
host animals elicits neutralizing antibodies to the infecting virus, and thus
provide novel
effective components of vaccine formulations of the invention, for treatment,
or prevention
of viral infection and its undesirable consequences.
Accordingly, the present invention provides an isolated molecular structure
comprising an epitope formed as a result of association of (a) an envelope
protein of an
enveloped virus, with (b) one or more cellular membrane proteins, which
envelope protein
and cellular membrane proteins are necessary and sufficient under suitable
conditions for
fusion of said envelope of the virus with a cell membrane containing said
cellular
16
CA 02338983 2001-O1-30
WO 00/08043 PCT/US99/17487
membrane proteins. As used herein a cellular membrane protein includes
proteins of the
cell surface membrane and internal membranes (including but not limited to the
endoplasmic reticulum membrane). Such cellular membrane protein may be an
integral
membrane protein (e.g., a trans-membrane protein), or may be attached to the
membrane by
an attached lipid (e.g., fatty acid chain or prenyl group) or by an
oligosaccharide (e.g., to a
phophoiipid, phosphatidylinositol), or the cellular membrane protein may also
be associated
with the membrane by non-covalent interactions (e.g., by association with an
integral
membrane protein).
The present invention also provides an isolated molecular structure
comprising an epitope formed as a result of association of (a) a mutant
envelope protein of
an enveloped virus, which envelope protein in wild-type form functions in
fusion of the
viral envelope with a host cell membrane, and which mutant envelope protein is
fusion-
defective; and (b) one or more host cellular membrane proteins which function
as receptors
for said envelope protein.
1 S In a specific embodiment, the present invention provides an isolated
molecular structure comprising an epitope formed as a result of association of
(a) an HIV
envelope protein, or a mutant thereof that assembles into the viral envelope;
with (b) human
CD4 and a co-receptor for HIV fusion. In one embodiment, the co-receptor is
the
chemokine receptor CCRS or CXCR4. In another specific embodiment, the
molecular
structure is formed by association of a mutant of HIV gp41 that is fusion-
defective. In yet
another embodiment, the mutant contains one or more mutations selected from
the group
consisting of V2E, G10V, V570R, and Y586E. In another embodiment the molecular
structure is formed by association of wild-type HIV envelope protein. In
another specific
embodiment, the envelope protein is E1 and E2 of Flavivirus HCV and the
cellular
membrane protein is CD81.
The present invention also provides a method of producing an immunogen
for use in a vaccine for the treatment or prevention of infection by a virus
comprising the
following steps in the order stated: (a) contacting an envelope protein or
chimeric form
thereof of an enveloped virus, which envelope protein functions in fusion of
the viral
envelope with a cell membrane, or a mutant form or chimeric form thereof of
said envelope
protein that is fusion-defective, with one or more cell proteins or chimeric
forms thereof
that function as receptors for said envelope protein; (b) exposing said
envelope protein or
chimeric form thereof or mutant form or chimeric form thereof, and said one or
more host
cell proteins or chimeric forms thereof, to a cross-linking agent; and (c)
isolating a cross-
linked structure comprising said envelope protein or chimeric form thereof or
mutant form
or chimeric form thereof. In one embodiment, said virus is HIV and said host
cell proteins
are human CD4 and a co-receptor for HIV.
17
CA 02338983 2001-O1-30
WO 00/08043 PCT/US99/17487
In a specific embodiment, the present invention also provides a method of
producing an immunogen for use in a vaccine for the treatment or prevention of
infection by
HIV comprising the following steps in the order stated: (a) co-culturing a
first cell
recombinantly expressing HIV envelope proteins) or a chimeric form thereof, or
a mutant
form or chimeric form thereof of said envelope protein that is fusion-
defective, with a
second cell that expresses (i) human CD4 or a chimeric form thereof, and (ii)
a co-receptor
for HIV or a chimeric form thereof; (b) exposing said envelope protein or
chimeric form
thereof or mutant form or chimeric form thereof, and said CD4 or chimeric form
thereof and
co-receptor or chimeric form thereof, to a cross-linking agent; and (c)
isolating a cross-
linked structure comprising said envelope protein or chimeric fonm thereof or
mutant form
or chimeric form thereof.
In one specific embodiment, said second cell recombinantly expresses CD4
or said co-receptor or both CD4 and said co-receptor or chimeric forms of any
of the
foregoing. In another specific embodiment, the present invention provides a
method of
producing an immunogen for use in a vaccine for the treatment or prevention of
infection by
HIV comprising the following steps in the order stated: (a) co-culturing a
first cell
recombinantly expressing HIV envelope protein or a chimeric form thereof, or a
mutant
form or chimeric form thereof of said envelope protein that is fusion-
defective, with a
second cell that expresses (i) human CD4 or a chimeric form thereof, and (ii)
a co-receptor
for HIV or a chimeric form thereof wherein at least one of said chimeric forms
comprising
an affinity tag is expressed; (b) lysing said co-cultured cells to form a cell
lysate under non-
denaturing conditions; and (c) isolating from said cell lysate a molecular
structure
comprising said envelope protein or chimeric form thereof or mutant form or
chimeric form
thereof by a method comprising contacting said cell lysate with a binding
partner to said
affinity tag and recovering a molecular structure bound to said affinity tag.
5.1. FORMATION OF THE FRMS
The present invention relates to FRMS which comprise an epitope formed as
a result of the association of one or more viral envelope molecules) and one
or more host
cell molecule(s). The FRMS of the invention encompass structures resulting
from the
association of at least one viral envelope protein and at least one host cell
protein. For
example, a FRMS of the invention may encompass fusion-competent complexes
(such as
complexes formed during fusion events of the viral envelope with a cellular
membrane),
fusion-defective complexes {such as those formed during pre-fusion events
between a
fusion-defective viral envelope and a cellular membrane) as well as complexes
formed by
the association of one or more viral envelope molecules with one or more
cellular receptor
18
CA 02338983 2001-O1-30
WO 00/08043 PCTNS99/17487
and/or co-receptor for the virus. In the case of HIV, the FRMS result from the
association
of HIV envelope protein with CD4 and a chemokine receptor.
The FRMS of the invention, thus comprise epitopes capable of eliciting
neutralizing antibodies to the virus.
The FRMS of the invention are formed by a variety of methods as described
herein. In a preferred embodiment, cells expressing one or more components
whose
association results in the FRMS are cultured to form the FRMS. All the
components may
be expressed on one cell, or a first component may be expressed on one cell,
and a second
component may be expressed on a different cell, and a third component (if any)
may be
expressed on one of the foregoing cells or a different cell. Alternatively,
one of the cells
may be replaced by a viral particle or pseudovirus or recombinant virus or
virus-like particle
containing the components) on its surface. The components (viral envelope
protein, host
cell receptor, and/or any necessary host cell co-receptor) may each be
endogenously or
recombinantly expressed by the cells. Thus, FRMS may be formed by the
interaction of
1 S endogenous molecules, exogenous molecules (e.g. recombinantly expressed)
or any
combination thereof.
In yet another embodiment of the invention, a soluble form of a viral
envelope protein of a envelope virus is used in the formation of a FRMS of the
invention.
For example, in one embodiment the soluble envelope protein may be added t o
host cells in
vitro which host cells express the receptors) for the virus. Thus the
association of the
soluble viral envelope protein and host cell receptors) results in the
formation of a FRMS.
In other embodiment of the invention one or more soluble cell recptor(s) are
used in formation of the FRMS. In one embodiment, for example, soluble
cellular
receptors) whose association with the viral envelope proteins) results in the
formation of a
FMS, may be used in the formation of a FRMS by contacting the viral envelope
proteins)
with said soluble cell receptor(s). In another specific embodiment, all of the
components
whose association result in the formation of the FRMS are in a soluble form
and the FRMS
is reconstitutional in vitro. In this embodiment, the reconstitution of a FRMS
may
optionally be aided by the addition of a mAb to the FRMS.
In yet other embodiments of the invention the FRMS is forced upon a
decrease in pH. For example, in some embodiments, such pH-medicated cell
fusion allows
for the formulation of the FRMS for enveloped viruses which cellular membrane
proteins
are located in internal cellular membranes.
19
CA 02338983 2001-O1-30
WO 00/08043 PCTNS99/17487
5.1.1. ENDOGENOUS EXPRESSION OF
COMPONENTS OF THE COMPLEX
The cellular components involved in the formation of a FRMS for a
particular virus include the cell receptors and/or co-receptors for the
particular virus. In a
S specific embodiment, at least some of these components are endogenously
expressed by the
cell. Thus, any cell known in the art which expresses a cellular component of
the FRMS
may be used in the formation of a FRMS.
Cells expressing native cellular receptors and/or co-receptors for viral
proteins can be used. For example, as described herein, human CD4 is known in
the art as a
cellular receptor for HIV. In one embodiment of the invention, cells
endogenously
expressing CD4 are used in the formation of a FRMS.
Cells endogenously expressing a component of the complexes of the
invention may comprise one or more of the cellular components used to form the
FRMS.
For example, cells may be used which express all of the required cellular
components of the
FMS or cells may be used which do not express all of the required cellular
components.
For example, in the case of an HIV FRMS, any cell which expresses both CD4 and
an HIV
co-receptor, such as CCRS may be used in the formation of an HIV FRMS.
Alternatively,
any cell which endogenously express CD4 but does not endogenously express an
HIV co-
receptor, in this embodiment, and as discussed below, components of the
complex which
~e not endogenously expressed may be introduced into the cell and
recombinantly
expressed. In this embodiment, additional cellular components of the complex
are
introduced to the cell such that the cell expresses both endogenous and
exogenous cellular
components. In one specific embodiment of the invention, the co-receptor CXCR4
is
predominantly located on T cell lines. In another specific embodiment of the
invention the
c°-receptor CCRS is predominantly located on macrophage cells. In one
specific
embodiment of the invention, the co-receptor CXCR4 is predominantly located on
T cell
lines. In another specific embodiment of the invention the co-receptor CCRS is
predominantly located on macrophage cells. Exogenous components may be
introduced
into the cell by any method known in the art, including those described in
Section 5.1.2.
below.
In yet another embodiment of the invention, a cell transformed by means of a
viral gene or genome integration, which expresses a viral component of the
FRMS may be
used in the formation of a fusion related complex. In this manner, the cell
may be said to
endogenously express a viral component important in the construction of an
FRMS. Such
virally-transformed cells may also endogenously express one or more cellular
components
of the FRMS. Alternatively, viral particles may be used as a source of
endogenous
expression of viral components of a FRMS.
CA 02338983 2001-O1-30
WO 00108043 PCTNS99/17487
5.1.2. RECOMBINANT EXPRESSION OF COMPONENTS
IMPORTANT IN THE FORMATION OF THE FRMS
Any of the components important in the construction of a FRMS or
fragments, analogs or derivatives thereof may be introduced into a cell such
that the
exogenous component is expressed in the cell.
The present invention encompasses expression systems, both eucaryotic and
procaryotic expression vectors, which may be used to express a component
important in the
formation of a FRMS or fragment, analog or derivative thereof of the present
invention.
In other embodiments of the invention, the viral envelope protein is
expressed in a host cell. In this embodiment, the exogenous viral envelope
protein may be
introduced into the cell by transfection or viral vectors as described herein,
or by infection
with the live or attenuated virus comprising the viral envelope protein. Thus,
viral infection
can lead to production of a viral envelope protein in preferred embodiments,
the expression
of a viral envelope protein on the cell membrane is enhanced by the
modification (e.g. by
recombinant means) of the viral envelope protein so as to increase its
expression on the cell
surface, or other cell membrane. For example, targeting signals are known in
the art that
enhance delivery of cellular proteins to particular subcellular compartments
or locations.
For example, an endoplasimic reticulum signal, KDEL, (one letter amino acid
codes) could
be engineered into the viral envelope protein to promote targetry of the
protein to the ER
when a cell is infected with the virus.
The invention also provides a cell expressing the viral and cellular
components important for the formation of a FRMS. In one embodiment, a cell is
constructed to express the cell receptor and/or co-receptor for a virus and to
recombinantly
express the viral envelope protein. Such cell may endogenously express the
host cell
receptors and/or co-receptors for the virus or recombinantly express one or
both of such
protein(s). In embodiments wherein viral and cellular proteins are
recombinantly
expressed, the nucleic acids encoding such proteins may be expressed from
different vectors
or from a single vector (e.g., wherein a vector comprises multiple genes
operably linked to a
single promoter). Thus, in this embodiment all of the components necessary for
the
formation of a FRMS are expressed on an individual cell. In a specific
embodiment, a cell
is constructed to recombinantly express CD4, a co-receptor CCRS or CXCR4 and
HIV
gp160. Alternatively, gp120 and gp41 may be used in place of gp160. In a
preferred
embodiment, when an individual cell is constructed to express gp 160, CD4 and
a co-
receptor for HIV-1 the viral envelope protein is also constructed such that it
is lacking the
native protease site responsible for cleavage of gp160 to gp120 and gp4l. In
this
embodiment an alternative protease site is constructed into the viral envelope
protein which
protease is not native to the host cell (such as a bacterial protease).
Formation of the FRMS
21
CA 02338983 2001-O1-30
WO 00/08043 PCT/L1S99/17487
is triggered on the cell expressing the three proteins, by contacting the cell
with the non-
native protease (such as by adding an amount effective to induce cleavage, to
the cell
media). A variety of proteases and protease sites are known in the art and may
be used in
accordance with the invention.
The nucleotide sequence coding for a viral envelope protein or cellular
receptor protein of the invention or a functionally active analog or fragment
or other
derivative thereof, can be inserted into an appropriate expression vector,
e.g., a vector which
contains the necessary elements for the transcription and translation of the
inserted protein-
coding sequence and/or for carrying out the methods of the invention.
The invention encompasses the use of DNA expression vectors and/or viral
vectors that contain coding sequences of a component of FRMS or analog,
derivative or
fragment thereof operatively associated with a regulatory element that directs
expression of
the coding sequences and genetically engineered host cells that contain any of
the foregoing
coding sequences operatively associated with a regulatory element that directs
the
expression of the coding sequences in the host cell.
The DNA expression vectors and viral vectors containing the nucleic acids
encoding a component important in the formation of a FRMS of the present
invention may
be produced by recombinant DNA technology using techniques well known in the
art.
Thus, methods for preparing the expression vectors and viral vectors of the
invention by
expressing nucleic acid containing sequences encoding a component important in
the
formation of a FRMS or analog, derivative or fragment thereof are described
herein.
Methods which are well known to those skilled in the art can be used to
construct
expression vectors containing gene product coding sequences and appropriate
transcriptional and translational control signals. These methods include, for
example, in
vitro recombinant DNA techniques, synthetic techniques, and genetic
recombination. See,
for example, the techniques described in Sambrook, et al., 1989, Molecular
Cloning, A
Laboratory Manual, Second Edition, Cold Spring Harbor Press, N.Y.; and
Ausubel, et al.,
1989-1999, Current Protocols in Molecular Biology, Green Publishing Associates
and
Wiley Interscience, N.Y., both of which are incorporated herein by reference
in their
entirety. Alternatively, gene product sequences may be chemically synthesized
using, for
example, oligonucleotide synthesizers. See, for example, the techniques
described in
"Oligonucleotide Synthesis", 1984, Gait, M.J. ed., IRL Press, Oxford, which is
incorporated
by reference herein in its entirety. Gene expression may be regulated by a
variety of
methods known in the art including but not limited to those presented in
Mizuno, T. et al.,
1984, Proc. Natl. Acad Sci USA. 81(7):1966-70.
The nucleic acids or DNA encoding one or more components which result in
the formation of the FRMS may be introduced into a cell may be accomplished by
a variety
22
CA 02338983 2001-O1-30
WO 00/08043 PCTNS99/17487
of methods, such as liposomes, electroporation, transfection, viral vectors,
bacteriophage,
etc.
In one embodiment, nucleic acids encoding one or more components
important in the formation of a FRMS is accomplished by packaging plasmid DNA
comprising the nucleic acids that code for the components) into liposomes or
by
complexing the plasmid DNA comprising nucleic acids that code for the
components) with
lipids or liposomes to form DNA-lipid or DNA-liposome complexes. The liposome
is
composed of cationic and neutral lipids commonly used to transfect cells in
vitro. The
cationic lipids complex with the plasmid DNA and form liposomes.
Cationic and neutral liposomes are contemplated by this invention. Cationic
liposomes can be complexed with the a negatively-charged biologically active
molecule
(e.g., DNA) by mixing these components and allowing them to charge-associate.
Cationic
liposomes are particularly useful with a nucleic acid because of the nucleic
acids negative
charge. Examples of cationic liposomes include lipofectin, lipofectamine,
lipofectace and
DOTAP (Hawley-Nelson et al., 1992, Focus 15(3):73-83; Felgner et al., 1992,
Proc. Natl.
Acad. Sci. U.S.A. 84:7413; Stewart et al., 1992, Human Gene Therapy 3:267-
275).
Commercially available cationic lipids are also available, e.g.,
dimethyldioctadeclammonium bromide (DDAB); a biodegradable lipid 1, 2-
bis(oleoyloxy)-
3-(trimethylammonio) propane (DOTAP); these liposomes may be mixed with a
neutral
lipid, e.g., L-a dioleoyl phosphatidylethanolamine (DOPE) or cholesterol
(Chol), two
commonly used neutral lipids for systemic delivery. DNA:liposome ratios may be
optimized using the methods used by those of skill in the art (see e.g.,
Nicolau et al., 198?,
Methods Enzymol. 149:157) and can readily be utilized herein by one of
ordinary skill in
the art to encase the complex of this invention.
In yet another embodiment of the present invention, the plasmid DNA
coding for the genes or nucleic acids encoding one or more components
resulting in the
formation of a FRMS of the invention may be delivered via polycations,
molecules which
carry multiple positive charges and are used to achieve gene transfer in
vitro, in vivo and ex
vivo. Polycations, such as polyethilenimine, may be used to achieve successful
gene
~~sfer (see e.g., Boletta et al., 1996, J. Am. Soc. Nephrol. 7:1728).
Recombinant viruses can also be used to deliver the components whose
association results in the FRMS. Cells infected by these viruses or
transformed by the viral
genome will thus express the desired components. Thus, in another embodiment
of the
present invention, either a live recombinant vector or an inactivated
recombinant viral
vector expressing one or more components) described herein can be engineered.
In this
regard, a variety of viruses may be genetically engineered to express a
component important
23
CA 02338983 2001-O1-30
WO 00/08043 PCT/US99/17487
in the formation of a FRMS. In some case, it may be desired that the
recombinant viruses
are either cold adapted, temperature sensitive, or attenuated.
In accordance with the present invention, a wide variety.of viruses and viral
vectors may be used to deliver the nucleotide sequences encoding one or more
S components) important to the formation of a FRMS of the present invention, a
few
examples of which are described below.
Retroviral vectors are commonly used to deliver genes to host cells.
Retroviral vectors are extremely efficient gene delivery vehicles that cause
no detectable
hanm as they enter the cells. The retroviral nucleic acid may integrate into
host
c~omosomal DNA allowing for long-term persistence and stable transmission to
future
progeny, such a vector would be useful for the delivery of one or more
components) which
result in the formation of a FRMS. An example of an appropriate retroviral
vector are
lentiviruses which have the advantage of infecting and transducing non-
dividing cells. In
such an embodiment, a lentiviral vector encoding a packagable RNA vector
genome and
°perably linked to a promoter in which all the functional retroviral
auxiliary genes are
absent, is used to transfer the DNA encoding a component important in the
formation of a
FRMS of the present invention. Examples of such vectors are described in WO
98/17815,
WO 98/17816 and WO 98/17817, each of which is incorporated herein by reference
in their
entirety.
In yet another embodiment, non-integrating viral vectors which infect and
transduce non-dividing cells, such as adenoviral vectors may be used to
deliver a
component important in the formation of a FRMS. Adenoviral vectors have
several
advantages since such vectors avoid risks associated with permanently altering
the host cell
genome or promoting insertional mutagenesis. Adenoviruses are one of the best
developed
non-integrating viral vectors and can be used to transfer expression cassettes
of up to 75 kb.
Recombinant adenoviruses can be produced at very high titers is highly
infectious and
efficiently transfer genes to a wide variety of non-replicating and
replicating cells and is
further ideal for in vivo mammalian gene transfer.
Adenovirus-based vectors are relatively safe and can be manipulated to
encode the desired components) and at the same time to be inactivated in terms
of its
ability to replicate in a normal iytic viral life cycle. Adenovirus has a
natural tropism for
airway epithelia. Therefore, adenovirus-based vectors are particularly
preferred for
epithelial delivery applications. In a particular embodiment, the adenovirus-
based gene
therapy vector comprises an adenovirus 2 serotype genome in which the Ela and
the Elb
3$ regions of the genome, which are involved in early stages of viral
replication have been
deleted and replaced by nucleotide sequences of interest. In a further
embodiment, the
adenovirus --based gene therapy vector contains only the essential open
reading frame
24
CA 02338983 2001-O1-30
WO 00/08043 PCT/US99/17487
(ORF3 or ORF6 of adenoviral early region 4 (E4) and is deleted of all other E4
open -
reading frames, or may additionally contain deletions in the E3 regions (see
e.g. U.S. Patent
No. 5,670,488, incorporated herein by reference in its entirety). In another
embodiment, the
adenovirus-based therapy vector used may be a pseudo-adenovirus (PAV), which
contain
no harmful viral genes and a theoretical capacity for foreign material of
nearly 36 kb.
In another embodiment, adeno-associated virus (AAV) systems may be used
to deliver the component important in the formation of a FRMS of the present
invention.
AAV has a wide host range and AAV vectors have currently have been designed
which do
not require helper virus. Examples of such AAV vectors are described in WO
97/17458.
Vaccinia viral vectors may be used in accordance with the present invention,
as large fragments of DNA are easily cloned into its genome and recombinant
attenuated
vaccinia variants have been described (Meyer, et al., 1991, J. Gen. Virol.
72:1031-1038). In
one embodiment of the invention, a vaccinia viral vector is used to deliver
the components
important to the formation of an FRMS such that the components are expressed
in the
1 S r~ombinant vaccinia infected cell. In a specific embodiment, the
components are human
CD4, a co-receptor (e.g., CCRS or CXCR4), and the HIV envelope protein. In
this specific
embodiment, infection of a cell with the recombinant vaccinia results in the
expression of
the components and formation of an HIV FRMS.
In another specific embodiment, two different recombinant vaccinia viruses
are constructed such that the first virus encodes the HIV env protein, and the
second virus
encodes human CD4 and a chemokine receptor. In this embodiment, the first
virus is used
to infect a first population of cells such that the infection results in the
expression of the
HIV env protein. The second virus is used to infect a second population of
cells such that
the infection results in the expression of CD4 and the chemokine receptor. Co-
culturing of
~e first infected population of cells with the second infected population of
cells results in
the formation of an HIV FRMS. In a further embodiment, the co-cultured cells
may be
cross-linked.
Orthomyxoviruses, including influenza; Paramyxoviruses, including
respiratory syncytial virus and Sendai virus; and Rhabdoviruses may be
engineered to
express mutations which result in attenuated phenotypes (see U.S. Patent
Serial No.
5,578,473, issued November 26, 1996 incorporated herein by reference in its
entirety).
These viral genomes may also be engineered to express foreign nucleotide
sequences, such
as a component important in the formation of a FRMS of the present invention
(see U.S.
Patent Serial No. 5,166,057, issued November 24, 1992 incorporated herein by
reference in
its entirety).
Reverse genetic techniques can be applied to manipulate negative and
positive strand RNA viral genomes to introduce mutations which result in
attenuated
CA 02338983 2001-O1-30
WO 00/08043 PGT/US99/17487
phenotypes, as demonstrated in influenza virus, measles virus, Sindbis virus
and poliovirus
(see Palese et al., 1996, Proc. Natl. Acad. Sci. USA 93:11354-11358). These
techniques
may also be utilized to introduce foreign DNA, i. e., encoding a component or
protein
important in the formation of a FRMS to create recombinant viral vectors to be
used in
accordance with the present invention. In addition, attenuated adenoviruses
and
retroviruses may be engineered to express a component important in the
formation of a
FRMS. Therefore, a wide variety of viruses may be engineered to deliver a
component
important in the formation of a FRMS of the present invention.
Bacteriophage may be used to specifically infects a bacterial cell (Soothill,
J~Sw 1992, J. Med. Microbiol. 37:358-261).
In the viral vectors of the present invention, the non-viral DNA (e.g.,
encoding a cell receptor) or non-native viral DNA (e.g., encoding a viral
envelope protein)
can encode any component important in the formation of a FRMS.
Thus, one skilled in the art would realize that viral envelope protein(s),
cell
1 S receptor(s), and/or co-receptor(s) used to construct the FRMS of the
subject invention can
be provided to cells by virions, virus-infected cells or
chemically/genetically inactivated
virions, viral vectors, viral replicons (i.e. non-replicating viral
constructs, e.g. VEE
replicons), virus-like particles produced by recombinant DNA methods or as
naked DNA.
As herein above, recombinant expression systems may employ regulatory
elements including not limited to, inducible and non-inducible promoters,
enhancers,
operators. Any of the methods previously described for the insertion of DNA or
nucleic
acid fragments into a vector may be used to construct expression vectors
containing a gene
or chimeric gene consisting of appropriate transcriptional/translational
control signals and
the protein coding sequences. These methods may include in vitro recombinant
DNA and
s~thetic techniques. In general, expression of a nucleic acid sequence
encoding a protein
or peptide fragment may be regulated by a second nucleic acid sequence so that
the protein
or peptide is expressed in a host cell transformed with the recombinant DNA
molecule. For
example, expression of a viral envelope protein may be controlled by any
promoter/enhancer element known in the art. A promoter/enhancer may be
homologous
(~~e~ native) or heterologous (i.e. not native). Promoters which may be used
to control the
expression of a protein include, but are not limited to, the SV40 early
promoter region
(Benoist et al., 1981, Nature 290:304-310), the promoter contained in the 3'
long terminal
repeat of Rous sarcoma virus (Yamamoto et al., i 980, Cell 22:787-797), the
herpes
thymidine kinase promoter (Wagner et al., 1981, Proc. Natl. Acad. Sci. U.S.A.
78:1441-
1445), the regulatory sequences of the metallothionein gene (Brinster et al.,
1982, Nature
296:39-42), plant expression vectors comprising the nopaline synthetase
promoter region
(Herrera-Estrella et al., Nature 303:209-213), the cauliflower mosaic virus
35S RNA
26
CA 02338983 2001-O1-30
WO 00/08043 PCTNS99/17487
promoter (Gardner et al., 1981, Nucl. Acids Res. 9:2871 ), and the promoter of
the
photosynthetic enzyme ribulose biphosphate carboxylase (Herrera-Estrella et
al., 1984,
Nature 310:115-120), promoter elements from yeast or other fungi such as the
Gal4-
responsive promoter, the ADC (alcohol dehydrogenase) promoter, PGK
(phosphoglycerol
S kinase) promoter, alkaline phosphatase promoter, and the following animal
transcriptional
control regions, which exhibit tissue specificity and have been utilized in
transgenic
animals: elastase I gene control region which is active in pancreatic acinar
cells (Swift et al.,
1984, Cell 38:639-646; Ornitz et al., 1986, Cold Spring Harbor Symp. Quart.
Biol. 50:399-
409; MacDonald, 1987, Hepatology 7:425-515); a gene control region which is
active in
p~creatic beta cells (Hanahan, 1985, Nature 315:115-122), an immunoglobulin
gene
control region which is active in lymphoid cells (Grosschedl et al., 1984,
Cell 38:647-658;
Adames et al., 1985, Nature 318:533-538; Alexander et al., 1987, Mol. Cell.
Biol. 7:1436-
1444), mouse mammary tumor virus control region which is active in testicular,
breast,
lymphoid and mast cells (Leder et al., 1986, Cell 45:485-495), albumin gene
control region
which is active in liver (Pinkert et al., 1987, Genes and Devel. 1:268-276),
alpha-fetoprotein
gene control region which is active in liver (Krumlauf et al., 1985, Mol.
Cell. Biol. 5: 1639-
1648; Hammer et al., 1987, Science 235:53-58), alpha 1-antitrypsin gene
control region
which is active in the liver (Kelsey et al., 1987, Genes and Devel. 1:161-
171), beta-giobin
gene control region which is active in erythroid cells (Mogram et al., 1985,
Nature 315:338-
340; Kollias et al., 1986, Cell 46:89-94), myelin basic protein gene control
region which is
active in oligodendrocyte cells in the brain (Readhead et al., 1987, Cell
48:703-712);
myosin light chain-2 gene control region which is active in skeletal muscle
(Sari, 1985,
Nature 314:283-286), and gonadotropic releasing hormone gene control region
which is
active in the hypothalamus (Mason et al., 1986, Science 234:1372-1378).
In a specific embodiment, a vector is used that comprises a promoter
operably linked to a nucleic acid encoding a viral or cellular protein or
chimeric protein and
one or more origins of replication, and, optionally, one or more selectable
markers (e.g., an
antibiotic resistance gene). In another embodiment, two or more promoters may
be used to
direct expression of two or more genes within a single plasmid.
In a specific embodiment the CMV immediate early (IE) promoter is used to
direct expression of a nucleic acid encoding a viral or cellulatprotein or
chimeric protein.
Promoters can be inducible, or constitutive. Expression from certain
promoters can be elevated in the presence of certain inducers; thus, for
example, expression
°f the genetically engineered protein chimeras may be controlled.
Inducible promoters may
be used to control expression of the proteins of the invention, such that the
protein is
produced only in the presence of the inducer.
27
CA 02338983 2001-O1-30
WO 00/08043 PCTNS99/17487
The present invention also provides methods for stable expression of the
components which result in the formation of an FRMS of the invention. For long
term,
high-yield production of recombinant proteins, stable expression is possible.
For example,
cell lines which stably express one or more cellular receptors and co-
receptors for the virus,
may be engineered. Rather than using expression vectors which contain viral
origins of
replication, host cells can be transformed with DNA controlled by appropriate
expression
control elements (e.g., promoter sequences, enhancer, sequences, transcription
terminators,
polyadenylation sites, etc.), and a selectable marker. For example, following
the
introduction of the foreign DNA, engineered cells may be allowed to grow for 1-
2 days in
~ e'mched media, and then are switched to a selective media. The selectable
marker in the
recombinant plasmid confers resistance to the selection foci (e.g., by stably
integrating the
plasmid into their chromosomes) and allows cells to and grow to form which in
turn can be
cloned and expanded into cell lines. This method may advantageously be used to
engineer
cell lines. This method may advantageously be used to engineer cell lines
which express
i5 the selected gene products. Such cell lines would be particularly useful in
screening and
evaluation of compounds that affect the endogenous activity of the selected
gene product.
A number of selection systems may be used in generating stably-expressing
cell lines, including but not limited to the herpes simplex virus thymidine
kinase (Wigler, et
al., 1977, Cell 11:223), hypoxanthine-guanine phosphoribosyltransferase
(Szybalska et al.,
1962, Proc. Natl. Acad. Sci. USA 48:2026), and adenine
phosphoribosyltransferase (Lowy,
et al., 1980, Cell 22:817) genes can be employed in tk', hgprt' or aprt'
cells, respectively.
Also, antimetabolite resistance can be used as the basis of selection for the
following genes:
DHFR, which confers resistance to methotrexate (Wigler, et al., 1980, Natl.
Acad. Sci. USA
77:3567; O'Hare, et al., 1981, Proc. Natl. Acad. Sci. USA 78:1527); gpt, which
confers
resistance to mycophenolic acid (Mulligan et al., 1981, Proc. Natl. Acad. Sci.
USA
78:2072); neo, which confers resistance to the aminoglycoside G-418 (Colberre-
Garapin, et
al., 1981, J. Mol. Biol. 150:1); and hygro, which confers resistance to
hygromycin
(Santerre, et al., 1984, Gene 30:147).
5.1.3. CELLS EXPRESSING COMPONENTS
WHOSE ASSOCIATION RESULTS IN FRMS
The present invention encompasses the expression of components) whose
association results in the formation of the FRMS. In several embodiments of
the invention,
expression may be in primary cells, animal cells, and insect cell lines. In
accordance with
~e present invention, a variety of primary or secondary cells or cell strains
may be used
including but not limited to cells isolated from skin, bone marrow, Liver,
spleen, pancreas,
kidney, adrenal and neurological tissue to name a few. Other cells types that
may be used
28
CA 02338983 2001-O1-30
WO 00/08043 PCT/US99/17487
in accordance with the present invention are immune cells (such as T-cells, B-
cells, natural
killer cells, etc.), macrophages/monocytes, adipocytes, pericytes,
fibroblasts, neuronal cells,
reticular cells etc. In a further embodiment, secondary cell lines may be
used, including,
but not limited to hepatic cell lines, such as CWSV, NR, Chang liver cells, or
other cell
lines such as CHO, VERO, BHK, Hela, COS, MDCK, 293, 373, CaSki and W138 cell
lines. In a preferred embodiment, host cells comprise one or more cell
receptors that
facilitate binding or viral infection.
In a preferred embodiment of the invention,~the cell is eukaryotic, preferably
a cell line, preferably mammalian, and most preferably human may be used for
the method
of the invention. Cells may be derived from human (e.g., HeLa cells), primate,
mouse,
rabbit, chicken, etc., although may also be from a transgenic non-human
animal.
Numerous eukaryotic cell lines may be purchased from ATCC (American Type
Culture
Collection, Rockville, MD). In a most preferred embodiment, eukaryotic cell is
derived
from a human. In other embodiments, the cell is derived from a mouse, monkey,
or rat.
Preferably non-tumor cell lines and autologous permissive cells are used in
preparing the
FRMS of the subject invention.
Thus, the present invention provides cells comprising one or more
components which result in the formation of an FRMS of the invention. The
present
invention also provides cells comprising all of the components required for
the formation of
the FRMS of the invention. In one embodiment, the present invention provides a
cell that
recombinantly expresses an envelope protein of an enveloped virus that
functions in fusion
of the viral envelope with a host cell membrane, or a mutant form of said
envelope protein
that is fusion-defective. In a fizrther embodiment the cell expresses one or
more cellular
membrane proteins that function as receptors for said envelope protein. In a
specific
embodiment, the invention provides a cell that recombinantly expresses HIV
gp160, human
CD4 and a co-receptor for HIV.
The present invention also provides a cell line that recombinantly expresses
an envelope protein of an enveloped virus that functions in fixsion of the
viral envelope with
a host cell membrane, or a mutant form of said envelope protein that is fusion-
defective. In
a ~~er embodiment the cell line expresses one or more cellular membrane
proteins that
function as receptors for said envelope protein. In a specific embodiment, the
invention
provides a cell line that recombinantly expresses HIV gp 160, which cell line
expresses CD4
and a co-receptor for HIV; said cell line lacking a functional protease that
cleaves gp160 to
produce gp 120 and gp41.
In other embodiments of the invention, cells comprising the FRMS of the
invention are administered as an immunogen(s) to a subject (see Section 5.5,
herein). In a
29
CA 02338983 2001-O1-30
WO 00/08043 PCT/US99/17487
preferred embodiment, when the subject is a human, the cells are human primary
cells. In a
further preferred embodiment, the cells are autologous.
5.1.4. USING FUSION DEFECTIVE MUTANTS
IN FORMATION OF THE COMPLEX
In one embodiment, any viral envelope protein containing wild type fusion
activity is used to make the FRMS. In an alternative embodiment, a fusion-
defective
envelope protein is used, as described in this section.
The present invention provides for the use of mutations to viral envelope
proteins or host cell receptor or co-receptor proteins which mutations inhibit
the completion
of process of viral envelope and cell membrane fusion. The invention provides
fusion-
defective mutations which do not allow completion of the process of viral
envelope-cell
membrane fusion, but which do result in the formation of a fusion-related
molecular
structure of the invention. The fusion-defective mutations are used to
construct epitopes
comprising an FRMS. In one embodiment of the invention, association of the
fusion
defective envelope protein with the cellular receptors) results in the
accumulation of the
FRMS. Thus the fusion-defective mutations of the invention may be used in the
formation
of an FRMS.
Any technique for mutagenesis known in the art can be used to modify
individual nucleotides in a DNA sequence, for purpose of making amino acid
substitutions)
in the expressed peptide sequence, for creating/deleting restriction sites, or
for adding
affinity tags. Such techniques include but are not limited to, chemical
mutagenesis, in vitro
site-directed mutagenesis (Hutchinson, C., et al., 1978, J. Biol. Chem
253:6551),
oligonucleotide-directed mutagenesis (Smith, 1985, Ann. Rev. Genet. 19:423-
463; Hill et
al., 1987, Methods Enzymol. 155:558-568), PCR-based overlap extension (Ho et
al., 1989,
Gene 77:51-59), PCR-based megaprimer mutagenesis {Sarkar et al., 1990,
Biotechniques,
8:404-407), etc. Modifications can be confirmed by DNA sequencing.
A fusion-defective mutation may be constructed for any enveloped virus. In
a specific embodiment, mutations are constructed that affect the ability to
complete fusion.
Selection of mutations may be based on published work, or based on molecular
modeling
from known or hypothetical structure predictions. Mutations known not to
detrimentally
affect envelope protein expression and transit to the cell surface will be
preferred.
In a further embodiment, a fusion-defective mutant is confirmed by
expressing the mutated gene in a cell (e.g., the methods of the invention)
such as COS,
293T and assayed for fusion competence. Cells may then be assayed for cell
surface
expression of the mutant component (by methods known in the art such as, e.g.,
by flow
cytometry or by live cell indirect immunofluorescence). Additionally,
proteolytic
CA 02338983 2001-O1-30
WO 00/08043 PCT/US99/17487
processing of a mutant protein such as a mutant of HIV gp 160 may be assayed
by Western
blot analysis or any other methods known in the art. Fusion competency of
mutant
components may be assayed as described in Section 5.7, herein, (e.g., by
syncytium
formation assay, e.g., in the case of HIV, U87-CD4-co-receptor cell fusion
assay).
In a further embodiment of the invention, binding-defective, fusion-defective
or fusion-arrested mutant envelope proteins are tested for the ability to
elicit primary isolate
virus neutralizing antibodies in the transgenic mouse vaccination assay. In
this
embodiment, immunogens may comprise cells expressing the test mutant envelope
protein
cocultured with e.g. U87-CD4-CCRS cells (when testing HIV related mutants).
In a preferred embodiment of the invention, mutations that elicit broad PI
virus neutralization are developed as subunit and recombinant virus-based
immunogens.
Table I provides a list of exemplary mutations which are provided for HIV. As
will be
apparent to one skilled in the art, similar mutations can be constructed for
other enveloped
viruses.
TABLE I:
Protein/site Mutationfs)
gp 120-gp41 cleavage site gp 120 C terminus
REKR to REKT
Vision peptide mutations gp41 V2E, G10V
gp41 coiled-coil mutations envelope V570R, Y586E, L568A,
W571R,
Q577R, N656L
enhanced gp120 cleavage alteration in furin protease cleavage
site of
gp120:C-terminus REKR to RSKR
Additional mutations within e.g. the bridging sheet of the gp120 core
structure include those
of Kwong et al. (Kwong PD, et al., 1998, Nature 393:648-59; Kwong PD, et al.,
1999, J
Biol Chem. 274:411 S-23) and regions of putative gp41/gp 120 interaction.
In another embodiment, the FRMS of the subject invention are prepared
using viral envelope proteins containing binding and fusion-related mutations.
In one
embodiment, the viral proteins bind host cell receptors, however, mutation
causes the fusion
process to be arrested before or during fusion of the viral envelope and host
cell membrane.
Thus, the FRMS of the subject invention include epitopes resulting from the
association of
one or more viral envelope proteins and one or more cell membrane proteins,
including
those proteins which are fusion-defective, fusion-an-ested or binding-
defective so long as
31
CA 02338983 2001-O1-30
WO 00/08043 PCT/US99/17487
the mutant proteins when used in the methods of the invention result in an
epitope elicits PI
neutralizing antibodies. Protein complexes arrested during the process of
fusion can be
isolated and used in the methods and compositions of the invention including
but not
limited to immunogens, diagnostics, kits, vaccines and compositions.
For example, in a specific embodiment, the FRMS comprise the major viral
envelope protein of HIV-1. There are a variety of classes of fusion-related
mutations which
may be used in constructing the complexes of the subject invention. Without
limitation as
to mechanism, during HIV-1 and host cell fusion, the oligomeric envelope
protein complex
gp160, comprising the gp120 surface and gp41 transmembrane portions, binds
initially to
the CD4 receptor on the host cell. Conformational changes in both the envelope
protein and
CD4 receptor facilitate interaction with a co-receptor molecule on the host
cell surface.
Further conformational changes in the viral protein-CD4-co-receptor
trimolecular complex
allow exposure of a viral gp41 pre-hairpin intermediate and the insertion of
the N-terminal
gp41 hydrophobic fusion domain into the host cell membrane. Helical heptad-
repeat
regions within the pre-hairpin intermediate subsequently collapse to form the
trimeric,
coiled-coil core of fusion active gp4l. The coiled-coil core drives the
ultimate fusion of the
opposed cell and virus membranes.
Viral envelope proteins with mutations that abrogate proteolytic cleavage of
the gp160 precursor protein preventing liberation of the gp41 hydrophobic
fusion domain
ZO °~ be used to construct the subject complexes. In particular, it has
been shown that certain
mutations that alter the highly conserved lys/arg-X-lys/arg-arg site at the C-
terminus of
gp 120 abrogates fusogenicity (see e.g., Lee CN, et al., 1994, AIDS Res. Hum.
Retroviruses.10:1065-9).
Additionally, mutations that affect the N-terminal gp41 fusion peptide can be
used in creating the complexes of the subject invention. The hydrophobic N-
terminal
region of gp41 mediates fusion by inserting into the cell membrane and
destabilizing the
lipid bilayer, perhaps by adopting a helical structure. Certain amino acid
changes within
this region render the envelope protein nonfusogenic. Synthetic peptides
bearing these
changes are either unable to bind to lipid bilayers unable to elicit fusion.
Two
Vision-defective mutations in gp41 have been well characterized: V2E and G10V
(Kliger Y,
et al., 1997, J Biol Chem.272:13496-505). The former involves a polar
substitution in the
hydrophobic peptide, whereas the later may affect the helical structure
assumed in the lipid
bilayer. These mutations can be introduced into the 168P envelope gene (gp41:
ala-val-gly-ile-gly-val-leu-phe-leu-glX-phe-leu-gly...) by site-directed
mutagenesis or any
method known in the art and the envelope proteins tested for expression and
fusogenicity.
Further, envelope proteins having mutations that alter the coiled-coil core
region of gp41 can be used to construct the structures of the subject
invention. The
32
CA 02338983 2001-O1-30
WO 00/08043 PCT/US99/17487
highly-conserved coiled-coil motif is found in proteins involved in fusion of
many virus
families, and similar structures have been identified in cellular proteins
that mediate
vesicular fusion. Synthetic peptides that mimic either the N- or C-helical
region (e.g.
DP107 and DP178) (Furuta et al., 1998, Nature Structural Biology 5:276-279.)
potently
neutralize virus infectivity, presumably by binding the cognate helical region
and interfering
with the collapses of the pre-hairpin intermediate. In addition to the
detailed structural
information available from crystallography, the N- and C-helical regions of
gp41 have also
been saturated by mutagenesis. Several mutations have been identified that
retain normal
envelope protein expression and assembly yet are non-fusogenic, presumably
though
disruption of the interface between N- and C-helical coils (e.g. V570R and
Y586E) (Weng
et al., 1998, Journal of Virology 72:9676-9682).
Competent or mutated viral envelope protein as well as cell receptors and/or
co-receptors can be incorporated into viral vectors or diploid cells by
methods known in the
art including those described herein. Viral genes encoding viral proteins are
routinely
cloned for expression in bacterial cells, in eucaryotic or procaryotic cells,
or in viral or DNA
vectors. Likewise, human cellular receptors can be cloned and expressed to
produce models
by which to study dangerous pathogenic diseases. Viral genes or host receptors
can be
incorporated into foreign DNA by transfection or through the use of viral
vectors.
5.1.5. OTHER METHODS TO FORM THE FRMS
Further, other methods by which to trigger formation of the subject
structures are contemplated including manipulation of pH conditions,
temperature and salt
concentration. Additionally, monoclonal antibodies that bind strongly enough
to envelope
protein may trigger the conformational changes necessary to form the subject
structures.
Thus, the FRMS of the subject invention can be formed as a result of the
interaction of viral
proteins and a variety of animal cellular components including but not limited
by those
listed above.
5.2. CAPTURING THE FRMS
Once a FRMS is formed a variety of methods may be used to preserve or
"capture" the FRMS in an immunogenic form that provides vaccine efficacy for
primary
viral isolates.
In one embodiment, the FRMS of the subject invention result from
interaction of one or more viral envelope proteins with one or more host
cellular receptors
~~°r co-receptors. In one embodiment, complexes are "captured" by
fixing or cross-
linking the FRMS. For example, a FRMS formed in co-cultured cells expressing
the viral
33
CA 02338983 2001-O1-30
WO 00/08043 PCT/US99/1748'7
and cellular components respectively, which result in a FRMA may be captured
by fixation
of the cells with a cross-linking agent.
In one specific embodiment, cells transformed with a nucleic acid expressing
a viral envelope protein and cells transformed with nucleic acids expressing
host cellular
receptors and co-receptors are cultured together. The viral protein expressed
on the surface
of a first cell binds to the receptors) expressed on the surface of a second
cell. The cells are
fixed at the initiation of this cell-cell interaction. Without limitation as
to mechanism,
crosslinking "captures" the intermediate fusion complex and provides fusion-
related
determinants which may be isolated and use as, for example, vaccine
components. Any
method known in the art may be used to express the viral or cellular
components of a fusion
complex in a cell (See, for example, Section 5.1.1, herein).
In capturing the FRMS, fusing cells or fusion-defective mutant cells can be
fixed or cross-linked by any method known in the art. Cross-linking reagents
that can be
used include but are not limited to formaldehyde gIutaraldehyde, formalin, p-
Azidobenzoyl
1 S hydrazide, N-(4-[p-Azidosalicyclamido]-butyl)-3'(2'-pyridyldithio)-
propionamide, Bis(beta-
[4-azidosaIicylamido]-ethyl)disulf de, 1,4-bismaleimidyl-2,3-dihydroxybutane,
1,6-
Bisrnaleimidohexane, 1,S-Difluoro-2,4-dinitrobenzene, Dimethyl adipimidate-
2HCl,
Dimethyl suberimidate-2HCl, Dimethyladipodimidate-2HC1, Dimethyl pimelimidate-
2HCl,
Disuccinimidyl glutarate, Disuccinimidyl tartrate, 1-Ethyl-3-[3-
Dimethylanonopropyl]
C~bodiimide Hydrochloride, (N-Hydroxy succinimidyl)-4-Azidosalicylic acid,
Sulfosuccinimdyl 2-[7-azido-4-methyl-coumarin-3-acetamidomethyl-1,3-
aminopropionate,
N-Succinimidyl-4-iodoacetylaminobenzoate, N-Succinimidyl-3-[2-
pyridylthio]propionate,
and Succinirnidyl 6-[3-(2-pyridylathio)-propionamide] hexanoate (Pierce
Chemical Co.,
Rockford, IL).
2S In a preferred embodiment of the invention, the cross-linking reagent is
formaldehyde. In one specific embodiment low levels of formaldehyde (0.2%) are
used in
order to conserve antigenicity. In other embodiments, higher concentrations
however can
be used to optimize stability. Formaldehyde in concentrations of from about
0.01% to
about 8% can be used in preparing the complexes of the subject invention.
Other agents
fat can be used to cross-link fusing cells include but are not limited to
glutaraldehyde
(O.OS-O.S%). One skilled in the art having the benefit of the disclosure
contained herein,
will recognize that fixing or cross-linking conditions (i.e., reagent used,
concentration of
reagents, time and temperature) can be varied to determine optimal conditions
for
immunogenicity of the FRMS.
3S In a further embodiment, the complexes of the subject invention can be
prepared by infecting human diploid cells in cell culture with, for example,
recombinant
vaccinia virus expressing viral envelope proteins and CD4/co-receptor (if not
endogenously
34
CA 02338983 2001-O1-30
WO 00/08043 PCT/US99/17487
present). During fusion, the cells are cross-linked to capture the fusion-
related structures
and determinants.
In yet another embodiment of the invention, when cells expressing the cell
receptors) and/or co-receptors for a virus are infected directly with the live
or attenuated
virus, the cells undergoing infection may be cross-linked in order to capture
the FRMS.
In other embodiments of the invention, a single cell may be engineered to
express all of the components important for the fonmation of a FRMS. In this
embodiment,
such a cells or cells may be cross-linked or fixed in order to capture the
FRMS. In this
embodiment, it is not necessary that a cell-cell interaction occur at the time
of fixation,
since all components are expressed on an individual cell.
The invention provides viral particles which are engineered to express in the
viral envelope the cellular components or proteins important for FRMS
formation. In this
embodiment, the recombinant viral particles may be cross-linked to capture the
FRMS
formed withing the viral particle envelope.
1 S In an alternate embodiment of the invention, the complexes need not be
fixed
or cross-linked. It has been found that the complexes of the subject invention
form in cell
lysates, in solution. Therefore, creation of the subject complexes by, for
example, the
simple lysis of cells transfected or infected with HIV-1 envelope protein as
well as CD4/co-
receptor where the FRMS form in solution. Cell lysis protocols are well known
in the art
(see, e.g., Sambrook et al., supra). Cell lysis protocols may involve
detergent lysis (e.g.,
1 % NP40) freeze thaw lysis, sonication lysis, or any method known in the art.
In a specific
embodiment, cell cultures are fixed at the onset of cell-cell interactions by
fixation in ice
cold 0.2% formaldehyde in phosphate buffered saline. In another embodiment of
the
invention detergent lysis of cells involves the use of the detergent Brij 97.
Accordingly, the invention provides a molecular structure which is a cross-
linked cellular molecular structure or is isolated from a cell lysate. In
other embodiments of
the invention provides fusion-defective mutations which allow accumulation of
the FRMS
and allows capturing of the FRMS.
5.3. ISOLATING THE FRMS
In one embodiment, cells (e.g. cross-linked cells) expressing the components
whose association results in the FRMS, can be used as immunogen in the
vaccines of the
invention. Alternatively, molecular structures comprising the FRMS may be
isolated for
use as immunogen.
Once the FRMS is formed, it may be isolated and purified by standard
methods including chromatography (e.g., ion exchange, affinity, and sizing
column
chromatography), centrifugation, differential solubility, or by any other
standard technique
CA 02338983 2001-O1-30
WO 00/0$043 PCT1IJS99/17487
for the purification of proteins. In addition, the components important in the
formation of a
FRMS can be synthesized comprising an affinity tag which facilitates recovery
and
purification. The peptide tag can be associated with any portion of the
protein, so long as
such association does not alter the epitope formation generated by association
of the
modified component with other members of the complex. In various embodiments,
such a
chimeric protein comprising an affinity tag can be made by ligating a gene
sequence coding
for the component to the sequence encoding the peptide tag in the proper
reading frame and
recombinantly expressing the protein. Care should be taken to ensure that the
modified
gene remains within the same translational reading frame, uninterrupted by
translational
stop signals.
A variety of peptide tags known in the art may be used in the modification of
a protein, such as but not limited to the polyhistidine sequence (Petty, 1996,
Metal-chelate
affinity chromatography, in Cun ent Protocols in Molecular Biology, Vol. 2,
Ed. Ausubel et
al., Greene Publish. Assoc. & Wiley Interscience), glutathione S-transferase
{GST; Smith,
1993, Methods Mol. Cell Bio. 4:220-229), the E. coli maltose binding protein
(Guan et al.,
1987, Gene 67:21-30), and various cellulose binding domains (U.S. Patent Nos.
5,496,934;
5,202,247; 5,137,819; Tomme et al., 1994, Protein Eng. 7:117-123), S TAGTM
System
(Novagen, Inc.), etc. Other possible peptide tags are short amino acid
sequences to which
monoclonal antibodies are available, such as but not limited to the following
well known
examples, the FLAG epitope, the myc epitope at amino acids 408-439, the
influenza virus
hemagglutinin (HA) epitope. Other peptide tags are recognized by specific
binding partners
and thus facilitate isolation by affinity binding to the binding partner,
which is preferably
immobilized and/or on a solid phase surface. As will be appreciated by those
skilled in the
art, many methods can be used to obtain the coding region of the above-
mentioned peptide
tags, including but not limited to, DNA cloning, DNA amplification, and
synthetic methods.
Some of the peptide tags and reagents for their detection and isolation are
available
commercially.
DNA sequences encoding desired peptide tags which are known or readily
available from libraries or commercial suppliers are suitable in the practice
of this
invention. These and other peptide tags are recognized by specific binding
partners and
thus facilitate isolation by affinity binding to the binding partner which can
be immobilized
onto a solid phase surface. The chimeric protein gene product can be prepared
using
recombinant DNA techniques. For example, gene sequence encoding a component
important in the formation of a FRMS can be introduced into a vector
containing the
sequence of a peptide tag, such that the component gene is expressed as a
peptide-tagged
chimeric protein. Peptide tags, which may be recognized by specific binding
partners, may
be used for affinity binding to the binding partner immobilized on a solid
phase surface.
36
CA 02338983 2001-O1-30
WO 00/08043 PCT/US99/17487
In one preferred embodiment, a poly-histidine tagged fusion-related protein
is constructed by insertion of a fusion-related protein gene or gene fragment
into an
expression vector such as one of the pET 15 series of vectors (Novagen), that
express
inserted sequences as chimeric proteins with N-terminal poly-histidine tags.
Proteins which
have a succession of six or more histidine residues at their amino or carboxyl
terminus have
a strong binding affinity to nickel. Poly-histidine-tagged chimeric proteins
will bind
specifically to the surface of a solid phase coated with chelated nickel. In a
specific
preferred embodiment, microtiter plates coated with metal' chelates are used
to isolate the
poly-histidine-tagged chirneric proteins (Pierce).
Alternatively, for example, a system described by Janknecht, et al. allows for
the ready purification of non-denatured chimeric proteins expressed in human
cell lines
(Janknecht, et al., 1991, Proc. Natl. Acad. Sci. USA 88, 8972-8976). In this
system, the
gene of interest is subcloned into a vaccinia recombination plasmid such that
the gene's
open reading frame is translationally fused to an amino-terminal tag
consisting of six
histidine residues. Extracts from cells infected with recombinant vaccinia
virus are loaded
onto Ni~+ nitriloacetic acid-agarose columns and histidine-tagged proteins are
selectively
eluted with imidazole-containing buffers.
In another specific embodiment, an expression construct can be made by
subcloning a fusion-related protein into the EcoRI restriction site of each of
the three pGEX
vectors (glutathione S-transferase expression vectors; Smith et al., 1988,
Gene 7:31-40).
This allows for the expression of a chimeric protein comprising the fusion-
related protein
linked to the GST binding domain in all three reading frames, such that, in
one frame, the
GST binding activity is maintained in the resulting chimeric protein. A GST
chimeric
peptide has a strong binding affinity for its substrate, glutathione. In a
specific
embodiment, FRMS comprising a GST tag is separated from a binding mixture
comprising
a cellular lysate or fixed cells by contacting such mixture with a glutathione-
linked solid
phase surface, such as glutathione sepharose beads. For example, the GST-
chimeric protein
can be anchored to glutathione-agarose beads or a glutathione-sepharose
column. The
mixture is then added to the column or beads in a manner that allows
interaction and
binding to occur. At the end of the reaction period, unbound material can be
washed away.
The interaction between the chimeric protein and the glutathione-agarose beads
allows
isolation of the complex.
In another embodiment, a chimeric protein may be readily purified by
utilizing an antibody specific for the chimeric protein being expressed.
In yet another embodiment, an affinity-tagged FRMS can be constructed by
conjugation of an affinity compound to the fusion-related protein. Affinity
compounds can
be used, such as, but not limited to, biotin, photobiotin, or other compounds
known in the
37
CA 02338983 2001-O1-30
WO 00/08043 PCT/US99/17487
art. In one embodiment, affinity compounds or affinity tags can be conjugated
to the
fusion-related protein through a polyfunctional crossIinker, and preferably a
bifunctional
molecule. As used herein, the term polyfunctional crosslinker encompasses
molecules
having more than one functional group that reacts with a functional group on
the fusion-
related protein. Typically, such crosslinker forms covalent bonds with an
amino or
sulfhydryl group on a polypeptide. For example, biotin N-hydroxysuccinimide
esters may
be used.
In a preferred embodiment of the invention, a highly-specific method to
isolate the FRMS via association with CD4, of a chemok co-receptor, or with
envelope
protein is the use of S-peptide affinity tagged molecules that takes advantage
of the specific
and high affinity interaction {Ka 10-' M) of the 15 amino acid S- peptide of
RNase A and
the larger subtilisin-digest fragment designated RNase S (S-protein; amino
acids 21-124 of
RNase A) (Potts et al., 1963; Dorai et al., 1994). This S~TAGT"' system was
developed
commercially by Novagen, Inc. for use in the detection and isolation of
recombinant
1 S chimeric proteins expressed in bacterial and baculovirus systems, and has
recently been
extended to mammalian chimeric proteins. The fusion-related proteins of the
FRMS is
tagged with S-peptides. The invention provides examples of components of
complexes
which were tagged with the S-peptide at the C-terminal ends of the CD4, CCRS
or envelope
protein molecules. Tagged complexes were readily isolated and purified by S-
protein
ag~ose affinity chromatography.
In one embodiment membrane-impermeable cross-linking reagents can be
used to fix cells containing tagged complexes. In one specific embodiment,
fixing cells
containing tagged complexes with a membrane impermeable cross-linking agent
facilitates
avoiding inactivation of the cytoplasmic S-peptide tag. Alternatively, the S-
peptide
s~uence may be altered (ala-glu-thr-ala-ala-ala-ala- phe-glu-arg-gln-his-met-
asp-ser) so
that it no longer contains vulnerable lysine residues and thus can be fixed
with
formaldehyde without resulting in inactivation of the cytoplasmic tag. Fusing
cells can be
fixed with a wide range of membrane impermeable cross-linking reagents
including, but not
limited to BS' and DTSSP (homobifunctional N-hydroxysuccinimidyl (NHS) esters
that
react through amines to form either non-cleavable or reversible linkages,
respectively);
Sulfo-SMPB (heterobifunctional NHS ester and maleimide to irreversibly cross-
link amine
and sulfhydryl groups); Sulfo-SANPAH and SASD (heterobifunctional NHS ester
and
photoreactive phenylazide cross-linkers which form either non-cleavable or
reversible
linkages, respectively) (Pierce Chemical Company).
Tagged complexes are readily purified providing an isolated preparation of
the fusion-related components which may be used for example, for inclusion in
vaccine
formulations. Purified or isolated tagged complexes can also be used as
diagnostic
38
CA 02338983 2001-O1-30
WO 00/08043 PCTNS99/17487
standards for in vitro assays. Further, tagged complexes permit the functional
analysis of
envelope-mediated fusion.
Accordingly, the invention provides a method of purifying a protein complex
comprising an envelope protein of human immunodeficiency virus type 1
functionally
interacting with human CD4 and human CCRS including the steps of a) tagging
the
complex with a peptide sequence to facilitate subsequent purification; and b)
isolating the
tagged complex.
In other specific embodiments, the invention provides an isolated protein
complex comprising an envelope protein of human immunodeficiency virus type 1
fictionally interacting with human CD4 and human CCRS.
5.4. TYPES OF FRMS
As will be apparent to one skilled in the art, any enveloped virus may be
used in the methods of the invention in order to construct or use the FRMS.
For example,
~ important use of the FRMS of the invention is as vaccine immunogens. One
advantage
of the vaccines comprising FRMS is that these immunogens provide significantly
higher
neutralization rates of primary isolates. The enveloped viruses that can be
used include but
are not limited to the families of Retroviridae, Rhabdoviridae, Coronaviridae,
Filoviridae,
Arenaviridae, Poxviridae, Bunyaviridae, Flaviviridae, Togaviridae,
Orthomyxoviridae,
P~~~°viridae, Herpesviridae, and Iridoviridae.
In one embodiment of the invention, a retroviral FRMS is formed by the
methods of the invention. For example, in a preferred embodiment the FRMS of
the
invention comprises the major viral envelope immunogen of human
immunodeficiency
virus type 1 (HIV-1 ) and arises from interaction with the host cellular
receptor for HIV-1
and a host cellular co-receptor. The host cellular co-receptor can be CCRS,
CXCR4, CCR3,
CCR2b or any other known in the art. An exemplified FRMS of the present
invention
results from interaction of the HIV-1 isolate 168P viral envelope protein
arising from
interaction with the host cellular receptor CD4, and the host cellular co-
receptor CCRS. It
should be apparent to those skilled in the art that other recombinant HIV
envelope proteins,
including those that act independently of CD4, could be used to prepare FRMS
within the
scope of the subject invention.
In a specific embodiment, by way of example but not limitation, for an HIV
vaccine, the immunogens of the subject invention are prepared by co-culturing
envelope-protein expressing COS-7 cells (LaCasse et al. 1998, Science, 283:357-
360) and
cells expressing the human CD4 receptor and CCRS co- receptor (LT87-CD4-CCRS).
In this
embodiment, the envelope-expressing cells are harvested using 0.5 mM EDTA 24
hr after
transfection (30-60% transfection efficiency) and co-cultured with an equal
number of
39
CA 02338983 2001-O1-30
WO 00/08043
PCTNS99/17487
F
U87-CD4-CCRS target cells. Progress towards fusion is assessed by staining
envelope-expressing cells and monitoring their incorporation into
multinucleated syncytia
microscopically. Fusion is complete within 12-24 hr. Complexes are formed
immediately.
Cells fixed after 1-5 hrs of co-culture are at an estimated 10-30% of maximal
syncytium
formation. This early time point in fusion was chosen initially in order to
capture the
transitional intermediates that lead to fusion. Other time points (e.g. 10 hr,
24 hr or greater)
can also be used to obtain the fusion-related complexes of the subject
invention.
Complexes from later points in the fusion process may reveal still further
neutralizing
epitopes and can be used to map appearance and course in time of critical
immunogens.
In other embodiments, Flavivirus FRMS is generated by the methods of the
invention. Viruses within the Flavivirus family include, for example,
Hepatitis C Virus
(HCV), Dengue virus, Yellow Fever virus, Tick-borne Encephalitis virus, and
Bovine Viral
Diarrhea virus. As a family, the Flaviviruses are similar to the Togaviridae
which include
the alphaviruses (e.g., Venezuelan Equine Encephalitis virus, Sindbis virus,
Semliki Forest
virus) and Rubella virus. By contrast to HIV, the Flaviviruses enter target
cells via
receptor-mediated endocytosis followed by pH-induced fusion within the
endosome.
Viruses within the Flavivirus family encode an envelope (E) protein which is
proteolytically cleaved in many family members to mature E 1 and E2 proteins.
In the
specific case of HCV (Hepatitis C Virus), the E2 protein is the receptor
binding moiety
(binding the cellular membrane protein CD81 ) and E 1 is the putative viral
fusion protein.
In another embodiment of the invention, a FIavivirus or Togavirus FRMS is
formed by the methods of the invention. For example, an HCV FRMS of the
invention
results from the association of E1, E2 and CD81. In a specific embodiment, the
HCV E1
and E2 proteins can be co-expressed in an acceptable cell substrate and will
become
localized in the cell endoplasmic reticulum (ER) and not significantly on the
cell surface. In
one embodiment, these cell cultures are incubated in medium buffered to pH 5
to 6.8 in
order to trigger E 1 E2 fusion of the intracellular membrane. The cultures are
treated with a
cross-linking agent to capture the intermediate fusion-competent structures,
and used
directly (or upon isolation of intracellular ER membranes) as vaccine
immunogen.
In another specific embodiment, the disrupted intracellular ER membranes
containing E 1 E2 protein are isolated as everted micelles and then incubated
with an
appropriate permissive cell expressing the E2 receptor, CD81. ElE2-containing
micelles are
internalized and E1-mediated fusion occurs in the endosome. Cells are then
cross-linked
and used for example, as vaccine immunogens. Alternatively, ElE2 proteins are
engineered
to alter ER retention signals and thus allow expression of E 1 E2 proteins on
the cell surface
thus obviating the need to isolate ER membranes.
CA 02338983 2001-O1-30
WO 00/08043 PCT/US99/17487
In another specific embodiment, recombinant DNA derived HCV virus-like
particles or HCV ElE2 containing pseudotyped virions (e.g. an alphavirus or
VSV) can be
produced and triggered for fusion by incubation with receptor-expressing cells
in medium
adjusted to pH S-6.8. The cultures are treated with a cross-linking agent such
as
formaldehyde and the FRMS is used as a vaccine immunogen. Alternatively,
virions can be
incubated with CD81-expressing cells; internalized virions will be induced to
fuse within
the endosome and can be captured by cross-linking treatment for use as a
vaccine
immunogen of the invention.
In yet another embodiment of the invention, an orthomyxovirus FRMS is
generated by the methods of the invention. The orthomyxoviridae family
includes all
influenza viruses, including but not limited to, influenza A, influenza B and
influenza C.
Thus, in a preferred embodiment, an influenza FRMS elicits neutralizing
antibodies against
a wide variety of flu strains and would allow for a drift-independent flu
vaccine.
Like the Flaviviruses and Togaviruses, the Orthomyxoviruses enter target
1 S cells via receptor-mediated endocytosis and fusion within the endosome. In
this case, the
viral neuraminidase (NA) protein functions as the sialic acid receptor binding
protein (a
ubiquitously expressed cell surface carbohydrate), and the hemagglutinin (HA)
protein
functions in pH-induced fusion. In contrast to the Flaviviruses, Influenza
virus buds from
the cell surface and thus recombinant NA and HA protein expression occurs on
the cell
surface.
In other embodiments influenza FRMS is generated by the methods of the
invention. Influenza FRMS is generated by the methods similar to those
described above
for the Flaviviruses and include pH-triggered HA-mediated fusion between
appropriate cells
or between cells and isolated cell membranes or virion particles, capturing
the complex may
be performed, for example, with a cross-linking agent allowing NA-mediated
endocytosis
and subsequent endosomal fusion to be captured.
In another embodiment of the invention, a paramyxovirus FRMS is
generated by the methods of the invention. The Paramyxovirus family
encompasses several
subgroups of significant medical and veterinary importance, including but not
limited to,
Morbilliviruses (measles virus, Canine Distemper virus, Rinderpest virus),
Rubulaviruses
(Mumps virus, Newcastle Disease), paramyxoviruses (human and bovine
parainfluenza),
and Pneumoviruses (Respiratory Syncytium virus). These viruses are similar to
the
Orthomyxoviruses, except that Paramyxovirus fusion occurs at the cell surface
rather than
in the endosome, and is pH independent. For the Paramyxoviruses, the
neuraminidase-
hemagglutinin (HN) (or simply hemagglutinin (H)) protein mediates cell
attachment
whereas the fusion (F) protein mediates virus-cell or cell-cell fusion. For
example, in the
case of measles virus, the H protein binds the cellular CD46 receptor and the
F protein
41
CA 02338983 2001-O1-30
WO 00/08043 PCT/US99/17487
mediates membrane fusion. In a specific embodiment, cells expressing H and F
proteins
from MV are co-cultured with cells expressing CD46 (and currently unidentified
co-
receptors) and result in cell-cell fusion that can be captured by cross-
linking or other
methods described herein. Alternatively, in another embodiment the co-receptor
may be
expressed on fusion-permissive cells expressing exogenus CD46. Specific
embodiments (as
described for HIV and other viruses above) may also be used.
In one embodiment of the invention, a herpesvirus FRMS is generated by the
methods of the invention. The Herpesviruses comprise a large family that
include members
of significant medical and veterinary importance. The human herpesviruses
include, but are
not limited to, HSV 1 and HSV 2, Varicella-zoster virus, Epstein-Barr virus,
Cytomegalovirus, and Kaposi's Herpesvirus HHV 8.
For example, in the case of HSV 1, binding and entry is a complex process
involving several viral glycoproteins (gD, gC, and gH/gL) and several cell
receptors and
adhesion molecules (glycosaminoglycans and Herpesvirus entry mediator (Hve)
proteins A-
C)' Binding and fusion occur on the cell surface in a pH-independent manner.
In a specific embodiment, HSV FRMS is constructed using a cell expressing
the cellular receptors and adhesion molecules (e.g. human MRCS cells) and co-
culturing
such cells with cells expressing the HSV glycoproteins gD, gC, gH, and gL.
Cell-cell fusion
is preferably arrested by fixation with a cross-linking agent, to allow
capturing of the
FMS.
As described herein, viral proteins and host cellular receptors can be
combined to form the subject FRMS for viruses including Herpes virus, pox
viruses,
paramyxovirus, measles, mumps, rubella, respiratory syncytial virus,
influenza, Hepatitis C,
ebola and flaviviruses such as Dengue and Yellow Fever. Additionally, the FRMS
of the
subject invention can include viral proteins from viruses of veterinary
importance including
rabies, feline leukemia, feline immunodeficiency virus and rinderpest.
Table II presents an exemplary list of the cellular receptors) for particular
enveloped viruses, which when expressed along with the viral envelope protein
and allowed
to associate with the envelope protein results in FRMS formation.
TABLE II:
virus Cellular Receptors)
Bovine coronavirus N-acetyl-9-O-acetylneuraminic acid
receptor
Choriomeningitis virus CD4+
42
CA 02338983 2001-O1-30
WO 00/08043 PCT/US99/17487
Virus Cellular Receptors)
Dengue virus Highly sulphated type heparin sulphate
pb5
Ebola CD 16b
Feline leukemia virus Extraceilular envelope glycoprotein
(Env-SU)
receptor
Gibbon ape leukemia virusGALV receptor
(GALV)
Herpes Simplex Virus Heparin sulphate glycosaminoglycan
receptor
Fibroblast growth factor receptor
HIV-1 CD4
CC-Chemokine receptor CCRS
Chemokine receptor CXCR4
Human cytomegalovirus Heparin sulphate proteoglycan
Annexin II
CD13 (aminopeptidase N)
Human coronovirus Human aminopeptidase N receptor
Influenza A, B & C Hemagglutinin receptor
Measles virus CD46 receptor
Morbilliviruses CD46 receptor
Mouse hepatitis virus Carcinoembryonic antigen family receptors
Carcinoembryonic antigen family Bgla
receptor
Murine leukemia virus Envelope glycoproteins
Murine gamma herpes virusgamma interferon receptor
Murine retrovirus Glycoprotein gp70
Rmc-1 receptor
Murine coronavirus Carcinoembryonic antigen family receptors
mouse hepatitis virus
Newcastle disease virus Hemagglutinin-neuraminidase protein
Fusion protein
Pox Virus Interferon gamma receptor
T-cell lymphotropic virusgp4b surface glycoprotein
1
43
CA 02338983 2001-O1-30
WO 00/08043 PCT/US99/17487
Virus Cellular Receptors)
Vaccinia virus TNFRp55 receptor
TNFRp75 receptor
Soluble Interleukin-1 beta receptor
Other viral diseases that can be treated or prevented by the methods of the
present invention include, but are not limited to, those caused by hepatitis
type C, influenza,
varicella, herpes simplex type I (HSV-I), herpes simplex type II (HSV-II),
rinderpest,
respiratory syncytial virus, cytomegalovirus, echinovirus, arbovirus,
hantavirus, mumps
virus, measles virus, rubella virus, human immunodeficiency virus type I (HIV-
1), human
immunodeficiency virus type II (HIV-2), any togaviruses (such as Dengue
virus),
alphaviruses, flaviviruses, coronaviruses, rabies virus, Marburg viruses,
ebola viruses,
parainfluenza virus, orthomyxoviruses, bunyaviruses, arenaviruses, human T
cell leukemia
virus type I, human T cell leukemia virus type II, simian immunodeficiency
virus,
Ientiviruses, Epstein-Barr virus, human herpesvirus-, cercopithecine herpes
virus 1 (B
virus), and poxviruses.
Table III presents an exemplary list of envelope proteins for particular
families of enveloped viruses, which when expressed along with the cellular
receptor
proteins) and allowed to associate with the cellular receptor proteins)
results in FRMS
formation.
TABLE III:
FAMILY EXAMPLES OF
ENVELOPE PROTEINS
Togaviridae E2
Flaviridae E
Coronaviridae E2 (S)
Rhabdoviridae G
P~amyxoviridae HN, H, F
Orthomyxoviridae HA
Bunyaviridae G1, G2
Arenaviridae Gl, G2
Retroviridae gp120, gp4l, gp160
Herpesviridae - gB, gD and gH
HSV
44
CA 02338983 2001-O1-30
WO 00/08043 PCT/US99/17487
- EBV gp350/220 and gp85
Poxviridae 56-Kd and 14-Kd
In other embodiments of the invention, a triple-stranded coiled-coil structure
of Orthomyxoviridae {such as Influenza), Filoviridae (such as Ebola), or
Retroviridae (such
as HIV) facilitates the formation of FRMS.
In another embodiment, synthetic peptides may be used to inhibit viral
infection. For example, by way of illustration, synthetic peptides that
comprise either of the
gp41 helical coils are able to bind the cognate helical region and broadly
inhibit viral
infectivity (see e.g., Wild, C., et al., 1992, Proceedings of the National
Academy of
Sciences USA 89:10537; Furuta, R.A., et al., 1998, Nature Structural Biology
5:276).
In other embodiments, neutralizing antibodies to FRMS immunogens may
likewise target structures involved in the activation of fusion.
In another embodiment of the invention, viral strains may be used which do
not require a host cell receptor, but require only a host cell co-receptor for
association with
the virus. For example, in the case of HIV, an HIV strain which does not
require CD4 for
viral binding may be used in the methods of the invention (see e.g., Hoxie, et
al., 1998,
J. Reprod. Immunol. 41:197-211.). Therefore, when using such a strain,
expression of CD4
is not necessary.
5.5. VACCINE FORMULATIONS AND ADMIhTISTRATION
The subject invention also concerns methods for vaccinating an individual or
animal to raise an immune response to and prevent infection by a virus. The
FRMS of the
subject invention can be presented to the vaccinee by several means. The FRMS
described
above can be administered to a vaccinee as fixed cells combined with an
adjuvant. Further,
isolated and purified FRMS can be administered to a vaccinee.
FRMS can be prepared in situ by the simultaneous immunization of a
vaccinee with transfected cells expressing viral protein and transfected cells
expressing host
cellular receptors and/or co-receptors. Likewise, vectors containing DNA
encoding viral
proteins and vectors containing DNA encoding receptor proteins can be used in
this
immunization strategy. Vectors can include viral vectors such as vaccirua and
vectors used
in DNA immunization. After simultaneous immunization, interaction between the
viral
protein and host cellular receptor protein occurs in vivo forming the FRMS of
the subject
invention and thereby exposing the vaccinee to the unique epitopes capable of
eliciting
neutralizing antibodies.
CA 02338983 2001-O1-30
WO 00/08043 PCTNS99/17487
Another method by which the FRMS can be formed in situ includes utilizing
the receptors and co-receptors on host cells. For example, in preparing the
exemplified HIV
envelope FRMS arising from interaction with CD4 and CCRS, transfected cells
expressing
the HIV glycoprotein or vectors containing DNA encoding the protein are
injected into or
targeted (e.g. by VEE or other vectors) the lymph node of a host. The viral
protein binds
and initiates fusion in vivo to host cells having viral receptors and co-
receptors to form the
FRMS whereby the vaccinee is exposed to the newly formed neutralizing
epitopes.
In a specific embodiment, a vaccine formulation comprises an isolated
protein complex comprising an envelope protein of human immunodeficiency virus
type 1
functionally interacting with human CD4 and human CCRS, and a pharmaceutically
acceptable carrier. In another embodiment, the invention provides a method of
immunizing
an animal to a virus comprising the steps of administering to the animal a
vaccine
formulation comprising a protein complex comprising one or more viral proteins
functionally interacting with one or more host cellular receptors or co-
receptors to mediate
1 S viral binding, entry and/or infection; whereby neutralizing antibodies to
the virus is
generated.
In one embodiment, the invention provides a vaccine formulation comprising
(a) a first nucleic acid encoding an envelope protein of an enveloped virus;
and (b) a second
nucleic acid encoding one or more cellular membrane proteins, which envelope
protein and
cellular membrane proteins are necessary and sufficient under suitable
conditions for fusion
of said envelope of the virus with a cell membrane containing said cellular
membrane
proteins, such that the envelope protein and cellular membrane proteins are
expressed in the
subject and neutralizing antibodies to the virus are produced; and (c) a
pharmaceutically
acceptable Garner.
In one embodiment, the invention provides a method of treating a host that
has been exposed to a virus, or preventing infection of a host by said virus,
the method
comprising the steps of administering to the host antibodies generated by
immunizing an
animal with an isolated protein complex comprising an envelope protein of
human
immunodeficiency virus type 1 functionally interacting with human CD4 and
human CCRS.
in an amount effective to treat or prevent infection of said host.
In another embodiment, the invention provides a method of preparing a
protein complex comprising one or more viral proteins functionally interacting
with one or
more host cellular receptors or co-receptors to mediate viral binding, entry
and/or infection,
including the steps of: a) culturing a first cell expressing one or more viral
proteins; b)
culturing a second cell expressing one or more host cellular receptors or co-
receptors for
said one or more viral proteins' c) co-culturing the first and second cells;
d) fixing said co-
culture during cell-cell fusion; and e) isolating the fixed cells.
46
CA 02338983 2001-O1-30
WO 00/08043 PCTNS99/17487
Many methods may be used to introduce the vaccine formulations of the
invention; these include but are not limited to oral, intradermal,
intramuscular,
intraperitoneal, intravenous, subcutaneous, intranasal routes, and via
scarification
(scratching through the top layers of skin, e.g., using a bifurcated needle).
The patient or subject to which the vaccine is administered is preferably a
mammal, most preferably a human, but can also be a non-human animal including
but not
limited to cows, horses, sheep, pigs, fowl (e.g., chickens), goats, cats,
dogs, hamsters, mice
and rats.
The patient or subject to which the vaccine is administered can also be a non-
human wild animal including but not limited to lion, cheetah, elephants,
giraffe, wildebeest,
leopards, panthers, etc. Thus, in this embodiment, the invention provides a
means of
wildlife management, and provides a method of preventing or treating a wild
animal or
animal population which has a high degree of inbreeding such that said
populations are
highly susceptible to viral disease.
In several embodiments of the methods herein, the subject is a human. In
other embodiments, said human has a high risk of HIV infection. In other
embodiments,
the subject is a domestic animal.
The present invention thus provides a method of immunizing an animal, or
treating or preventing viral diseases or disorders in an animal, comprising
administering to
the animal an effective immunizing dose of a vaccine of the present invention.
The vaccine formulations of the present invention can also be used to
produce antibodies for use in passive immunotherapy, in which short-term
protection of a
host is achieved by the administration of pre-formed antibody directed against
a
heterologous organism.
Vaccination of mice with immunogens comprising FRMS of the subject
invention elicits the production of neutralizing antibodies by immunized
animals. In an
exemplified embodiment, the complexes are administered as a vaccine comprising
fixed
whole cells formulated with an adjuvant. It would be apparent to one skilled
in the art
however that other vaccine strategies can be used in practicing the subj ect
invention. For
example, isolated and purified FRMS and/or epitopes thereof can be
administered as
subunit vaccines. Additionally, as mentioned previously, genes encoding the
functional
complexes can be constructed and placed in vectors or plasmids for use in live
vector or
DNA plasmid vaccines.
The virus vaccine formulations of the invention comprise an effective
l~~izing amount of one or more FRMS as a vaccine immunogens of the invention
and a
pharmaceutically acceptable carrier or excipient. Boosting is also
contemplated. In one
embodiment, vaccine compositions of the invention can comprise
"pharmaceutically
47
CA 02338983 2001-O1-30
WO 00/08043 PCT/US99/17487
acceptable Garners". As used in this description, pharmaceutically acceptable
carriers are
substances which do not interfere with the operation of the immunogen and are
non-toxic to
the animal. Pharmaceutically acceptable carriers are well known in the art and
include but
are not limited to saline, buffered saline, dextrose, water, glycerol, sterile
isotonic aqueous
buffer, oil-on-water or water-in-oil emulsions, aqueous compositions,
liposomes,
microbeads, microsomes or adjuvant compounds and combinations thereof. One
example
of such an acceptable carrier is a physiologically balanced culture medium
containing one
or more stabilizing agents such as stabilized, hydrolyzed proteins, lactose,
etc. The carrier
is preferably sterile. The formulation should suit the mode of administration.
The composition, if desired, can also contain minor amounts of wetting or
emulsifying agents, or pH buffering agents. The composition can be a liquid
solution,
suspension, emulsion, tablet, pill, capsule, sustained release formulation, or
powder. Oral
formulation can include standard carriers such as pharmaceutical grades of
mannitol,
lactose, starch, magnesium stearate, sodium saccharine, cellulose, magnesium
carbonate,
1 S etc.
Generally, the ingredients are supplied either separately or mixed together in
unit dosage form, for example, as a dry lyophilized powder or water free
concentrate in a
hermetically sealed container such as an ampoule or sachette indicating the
quantity of
active agent. Where the composition is administered by injection, an ampoule
of sterile
diluent can be provided so that the ingredients may be mixed prior to
administration.
In a specific embodiment, a lyophilized fusion-competent complex of the
invention is provided in a first container; a second container comprises
diluent consisting of
an aqueous solution of 50% glycerin, 0.25% phenol, and an antiseptic (e.g.,
0.005% brilliant
green).
The precise dose of immunogen or FRMS, to be employed in the
formulation will also depend on the route of administration, and the nature of
the patient,
and should be decided according to the judgment of the practitioner and each
patient's
circumstances according to standard clinical techniques. An effective
immunizing amount
is that amount sufficient to produce an immune response to the complex in the
host to
which the fusion-dependent complex, or vaccine immunogen, is administered.
In a specific embodiment, an effective immunizing amount of a vaccine
immunogen for a human subject of the present invention is within the range of
10 to100 mg
per kilogram body weight, more preferably 0.1 tolOmg per kilogram body weight.
Boosting is possible but not preferred.
The exact amount of vaccine immunogen utilized in a given preparation is
not critical, provided that the minimum amount necessary to provoke an immune
response
is given. A dosage range of as little as about 10 ug, up to amount a milligram
or more, is
48
CA 02338983 2001-O1-30
WO 00/08043 PCT/US99/17487
contemplated. As one example, in a specific embodiment, individual dosages may
range
from about 50-650 pg per immunization. In other embodiments, dosing is
dependent upon
the formulation of the vaccine immunogen (e.g. purified mimitope, vs. purified
FRMS, vs.
whole cell preparations).
in a preferred embodiment, the vaccine formulation comprises an effective
immunizing amount of the FRMS immunogen, preferably in combination with an
immunostimulant; and a pharmaceutically acceptable carrier. As used in the
present
context, "immunostimulant" is intended to encompass any compound or
composition which
has the ability to enhance the activity of the immune system, whether it be a
specific
p°tentiating effect in combination with a specific immunogen, or simply
an independent
effect upon the activity of one or more elements of the immune response. Some
of the more
commonly utilized immunostimulant compounds in vaccine compositions are the
adjuvants
alum or muramyl dipeptide (MDP) and its analogues. Methods of utilizing these
materials
are known in the art, and it is well within the ability of the skilled artisan
to determine an
°ptimum amount of stimulant for a given viral vaccine. It may also be
desired to use more
than one immunostimulant in a given formulation.
Use of purified immunogens or complexes vaccines can be carried out by
standard methods. For example, the purified proteins) should be adjusted to an
appropriate
concentration, formulated with any suitable vaccine adjuvant and packaged for
use.
Suitable adjuvants may include, but are not limited to: mineral gels, e.g.,
aluminum
hydroxide; surface active substances such as lysolecithin, pluronic polyols;
polyanions;
peptides; oil emulsions; alum, and MDP. The immunogen may also be incorporated
into
liposomes, or conjugated to polysaccharides and/or other polymers for use in a
vaccine
formulation. In instances where the complex is a hapten, i.e., a molecule that
is antigenic in
that it can react selectively with cognate antibodies, but not immunogenic in
that it cannot
elicit an immune response, the hapten may be covalently bound to a carrier or
immunogenic
molecule; for instance, a large protein such as serum albumin will confer
immunogenicity
to the hapten coupled to it. The hapten-carrier may be formulated for use as a
vaccine.
Effective doses (immunizing amounts) of the vaccines of the invention may
30. also be extrapolated from dose-response curves derived from animal model
test systems.
Accordingly, the present invention provides a method of treating or
preventing infection by a virus in a subject comprising administering to the
subject an
immunogenic amount of the FRMS effective to treat or prevent infection by the
virus. In a
specific embodiment, the present invention provides a method of treating or
preventing
infection by HN in a human comprising administering to the human an
immunogenic
amount of the FRMS effective to treat or prevent infection by HIV.
49
CA 02338983 2001-O1-30
WO 00/08043 PCT/US99/17487
The present invention also provides a method of treating or preventing
infection by a virus in a subject comprising administering to the subject an
amount of the
FRMS effective to treat or prevent infection by the virus. In a specific
embodiment, the
invention provides a method of treating or preventing infection by HIV in a
human
comprising administering to the human an amount of the monoclonal antibody
effective to
treat or prevent infection by HIV.
5.6. ANTIBODIES PRODUCED
The present invention relates to the formation of polyclonal and monoclonal
~tibodies reactive to a FRMS of the invention. According to the invention,
FRMS, its
fragments or other derivatives, or analogs thereof, may be used as an
immunogen to
generate antibodies which immunospecifically bind such an immunogen. Such
antibodies
include but are not limited to polyclonal, monoclonal, chimeric, single chain,
Fab
fragments, and an Fab expression library.
The subject invention also concerns methods for generating an immune
response, such as antibody production, in an animal. In one embodiment, an
animal is
administered a FRMS of the invention. Preferably, the FRMS comprises the HIV
envelope
protein and arises from interaction with CD4 and CCRS. The FRMS is
administered in a
manner such that an immune response is produced in the animal to the
immunogen.
Preferably, antibodies to epitopes on the FRMS are produced. More preferably,
the
antibodies produced include neutralizing antibodies. In a highly preferred
embodiment, the
antibodies produced are capable of neutralizing a wide variety of primary
isolates of the
VirllS.
Various procedures known in the art may be used for the production of
polyclonal antibodies to a FRMS or derivative or analog. In a particular
embodiment,
rabbit polyclonal antibodies to an epitope of FRMS, a subsequence thereof, can
be obtained.
For the production of antibody, various host animals can be immunized by
injection with
the native FRMS, or a synthetic version, or derivative (e.g., fragment or
chimera) thereof,
including but not limited to rabbits, mice, rats, etc. Various adjuvants may
be used to
increase the immunological response, depending on the host species, and
including but not
limited to Freund's (complete and incomplete), mineral gels such as aluminum
hydroxide,
surface active substances such as lysolecithin, pluronic polyols, polyanions,
peptides, oil
emulsions, keyhole limpet hemocyanins, dinitrophenol, and potentially useful
human
adjuvants such as BCG (bacille Calmette-Guerin) and corynebacterium parvum.
For preparation of monoclonal antibodies directed to a FRMS, derivative or
analog thereof, any technique which provides for the production of antibody
molecules by
continuous cell lines in culture may be used. For example, the hybridoma
technique
CA 02338983 2001-O1-30
WO 00/08043 PCT/US99/17487
originally developed by Kohler and Milstein, (Kohler et al., 1975, Nature
256:495-497), as
well as the trioma technique, the human B-cell hybridoma technique (Kozbor et
al., 1983,
Immunology Today 4:72), and the EBV-hybridoma technique to produce human
monoclonal antibodies (Cole et al., 1985, in Monoclonal Antibodies and Cancer
Therapy,
Alan R. Liss, Inc., pp. 77-96). In an additional embodiment of the invention,
monoclonal
antibodies can be produced in germ-free animals (see e.g., PCT/L1S90/022548).
According
to the invention, human antibodies may be used and can be obtained by using
human
hybridomas (Cole et al., 1983, Proc. Natl. Acad. Sci. U.S.A. 80:2026-2030) or
by
transforming human B cells with EBV virus in vitro (Cole et al., 1985, in
Monoclonal
~tibodies and Cancer Therapy, Alan R. Liss, pp:-77-96).
In a specific embodiment, spleens from mice immunized with FRMS are
harvested and splenocytes are isolated by teasing the spleen apart in culture
medium.
Splenocytes are fused with HPRT-negative mouse myeloma cells using
polyethylene glycol
{PEG). Cell pellets containing a 4:1 ratio of splenocytes to myeloma cells are
resuspended
~d treated in serum-free medium containing 50% pretested PEG-4000 (2.5 min
contact).
The cell suspension is then slowly diluted in serum-free medium, and cells are
plated in
96-well culture dishes in HAT selection medium: DMEM culture medium containing
20%
pretested fetal bovine serum, 10% NCTC-109 (Gibco), 1% nonessential amino
acids, 1X
glutamine and 1X Pen-Strep, 1X OPI (15 mg/ml oxaloacetate, 5 mg/ml sodium
pyruvate,
and 20 unitslml insulin), 100 pM hypoxanthine, 0.4 pM aminopterin, and 16 pM
thymidine. Wells containing hybridomas are monitored microscopically and
supernatants
harvested as appropriate/necessary for assay of antibody production.
In fact, according to the invention, techniques developed for the production
of "chimeric antibodies" (Mornson et al., 1984, Proc. Natl. Acad. Sci. U.S.A.
81:6851-
6855; Neuberger et al., 1984, Nature 312:604-608; Takeda et al., 1985, Nature
314:452-
454) by splicing the genes from a mouse antibody molecule specific for a FRMS
together
with genes from a human antibody molecule of appropriate biological activity
can be used;
such antibodies are within the scope of this invention. In another embodiment,
"humanized" antibodies are also provided by the invention (U.S. Patent No.
5,225,539).
According to the invention, techniques described for the production of single
chain antibodies (U.S. Patent No. 4,946,778) can be adapted to produce FRMS-
specific
single chain antibodies. An additional embodiment of the invention utilizes
the techniques
described for the construction of Fab' expression libraries (Huse et al.,
1989, Science
246:1275-1281) to allow rapid and easy identification of monoclonal Fab
fragments with
the desired specificity for tumor suppressor proteins, derivatives, or
analogs.
Antibody fragments which contain the idiotype of the molecule can be
generated by known techniques. For example, such fragments include but are not
limited
51
CA 02338983 2001-O1-30
WO 00/08043 PCT/US99/17487
to, the F(ab')2 fragment which can be produced by pepsin digestion of the
antibody
molecule, the Fab' fragments which can be generated by reducing the disulfide
bridges of
the F(ab')z fragment, the Fab fragments which can be generated by treating the
antibody
molecule with papain and a reducing agent, and Fv fragments.
Fab fragments from combinatorial libraries are also contemplated in the
invention (see e.g., Chanock et al., 1993, Infect Agents Dis. 2:118-31).
In the production of antibodies, screening for the desired antibody can be
accomplished by techniques known in the art (e.g., enzyme-linked immunosorbent
assay or
ELISA). For example, to select antibodies which specifically recognize FRMS,
one may
essay generated hybridomas for a product which binds to a FRMS epitope and
which does
not bind to the epitopes generated by other proteins. For selection of an
antibody that
specifically binds a first FRMS homolog but which does not specifically bind a
different
FRMS homolog, one can select on the basis of positive binding to the first
FRMS homolog
and a lack of binding to the second FRMS homolog. Screening of antibodies may
also be
perfonmed by functional assays such as virus neutralization assay, such as
those known in
the art or described herein.
Antibodies specific to a domain of a FRMS is also provided. Antibodies
specific to an epitope of a FRMS protein are also provided.
The generated antibodies may be isolated by standard techniques known in
the art (e.g., immunoaffinity chromatography, centrifugation, precipitation,
etc.) and used in
diagnostic immunoassays. The antibodies may also be used to monitor treatment
and/or
disease progression. Any immunoassay system known in the art, such as those
listed supra,
may be used for this purpose including but not limited to competitive and
noncompetitive
assay systems using techniques such as radioimmunoassays, ELISA (enzyme-linked
~unosorbent assays), "sandwich" immunoassays, precipitin reactions, gel
diffusion
precipitin reactions, immunodiffusion assays, agglutination assays, complement-
fixation
assays, immunoradiometric assays, fluorescent immunoassays, protein A
immunoassays
and immunoelectrophoresis assays, to name but a few.
The FRMS of the subject invention, when used as immunogens are capable
of eliciting neutralizing antibody to viral pathogens. In a preferred
embodiment, the
FRMS elicit neutralizing antibody to a wide variety of primary isolates of the
virus. In a
specific embodiment, the FRMS elicit neutralizing antibody to primary isolates
of HIV-1.
Neutralizing antibodies, while not elicited by static HIV envelope protein,
are able to bind
at least weakly to static envelope and are carried with the envelope protein
through the
binding and fusion process, during which the antibodies complete their high-
affinity
binding to critical intermediate structures. Such neutralizing antibodies can
be, in effect,
transported to the critical site to be available at the critical moment
regardless of the cellular
52
CA 02338983 2001-O1-30
WO 00/08043 PCTNS99/17487
site of action, in particular regardless of the cellular site of membrane
fusion (either at the
cell surface or at internal membranes such as the endosomal membrane). Thus,
viral
envelope proteins can serve as a vector to bring antibodies (e.g. linked to
e.g. toxin, other
protein, vaccine immunogens) into the cell, into the cytoplasm or into the
histocompatibility
complex presentation pathway.
Hybridoma cell supernatants of the invention can be assayed for the ability
to neutralize the homologous virus as well as for the ability to neutralize a
range of
genetically diverse viruses. The ability to neutralize genetically diverse
viruses suggests
recognition by the mAb of a conserved determinant. Further, mAb can be tested
for
inhibition of envelope-mediated cell-cell fusion. MAb can also be assayed for
the ability to
bind and/or immunoprecipitate envelope protein and envelope protein-containing
receptor-associated complexes. Of particular interest are those mAb that are
specific for
fusion-dependent epitopes and bind only weakly, if at all, to static envelope
protein.
In one embodiment, hybridomas are identified using high-throughput
screening assay to detect PI virus neutralization, and mAbs are characterized
to determine
the breadth and molecular targets of PI virus neutralization.
In various embodiments of the invention, the antibodies produced by the
methods of the invention are used individually (e.g. a single mAb) or in
combination (e.g.,
one or more mAbs directed to the same or different epitopes). Combinatorial
use of
~tibodies may include multiple mAbs directed to the same virus or to different
viruses. In
some embodiments of the invention it may be advantageous to use multiple mAbs
directed
to the same virus or multiple mAbs directed too the same FRMS of a virus.
5.7. ASSAYS FOR ANTIBODY BINDING
AND INHIBITION OF VIRAL INFECTII~N
The ability of antibodies of the invention or the derivatives or analogues
thereof to bind FRMS of the invention and thereby interfere with viral
infection can be
assayed by various methods.
Binding can be assayed by means well-known in the art. For example,
bioassays may be performed in which cells known to be expressing a chemokine
receptor
are exposed to the mAb derivative or analogue to be tested and assayed for a
known effect
(e.g., signal transduction). Alternatively, mAb, derivatives or analogues can
be tested for
the ability to bind chemokine receptors, host cell receptors or viral envelope
proteins by
procedures, including but not limited to, protein affinity chromatography,
affinity blotting,
l~unoprecipitation, cross-linking, and library based methods such as protein
pmbing,
phage display, and the two-hybrid system (see, generally, Phizicky et al.,
1995, Microbiol.
Rev. 59:94-123).
53
CA 02338983 2001-O1-30
WO 00/08043 PCTNS99/17487
High throughput screening for mAb, derivative or analogue binding may be
performed by methods known in the art, including but not limited to flow
cytometry. In a
specific embodiment, cells that express human CD4 and one of the HIV co-
receptors (e.g.,
CC CKR-5, CxC CKR4, CCRS, CXCR4 etc.) are treated with biotinylated mAb,
derivative,
or analogue and cell surface binding to the FRMS is detected with an avidin
FITC
conjugate. Alternatively, flow cytometry system may be used in a competitive
binding
assays using the monoclonal antibodies of the invention in the following
manner or by any
method known in the art. In a specific embodiment, a mAb of the invention is
labeled and
examined for the ability to bind an FRMS of the invention compared with the
ability of a
test antibody to bind to the same FRMS. Test antibodies of the invention may
be derived
from samples such as serum from a subject, or manufacturing samples such as
hybridoma
supernatants.
In another embodiment, the anti-viral activity exhibited by the mAb, mAb
derivative and/or analogue of the invention may be measured, for example, by
easily
performed in vitro assays, which can test the compound's ability to inhibit
syncytia
formation or to inhibit infection by cell-free virus and assess the effects of
the compound on
cell proliferation and viability. Applying these assays, the relative anti-
viral activity that a
mAb, derivative and/or analogue exhibits against a given virus or strain of
immunodeficiency virus formulation best suited for viral and strain specific
inhibitory
activity can be determined.
In one embodiment, a cell fusion assay is used to test the ability of mAb,
derivative or analogue, to inhibit virus-induced syncytia formation in vitro.
In a specific
embodiment, a cell fusion assay is used to test the ability of a mAb derivate
or analog of the
invention to inhibit HIV-induced syncytia formation in vitro. In this
embodiment, such an
Say involves culturing uninfected CD4+ cells in the presence of chronically
HIV-infected
cells and the composition containing the mAb, derivative or analogue to be
assayed. For
each, a range of concentrations may be tested. This range should include a
control culture
wherein no mAb, derivative and/or analogue has been added. Standard conditions
for
culturing, well known to those of ordinary skill in the art, are used. After
incubation for an
appropriate period, such as, for example, 24 hours at 37°C, the culture
is examined
microscopically for the presence of multinucleated giant cells, which are
indicative of cell
fusion and syncytia formation.
In another embodiment, an in vitro infectivity assay is performed using
primary macrophages and the macrophage-tropic isolate HIV-laaL, the first
described
macrophage-tropic HIV-1 isolate (see, Gartner et al., 1986, Science 233:215).
According to
this assay, primary macrophage cells isolated according to methods lmown in
the art are
infected with HN-IBaL that has been propagated and maintained only in primary
54
CA 02338983 2001-O1-30
WO 00/08043 PCT/US99/17487
macrophages. The input immunodeficiency virus is incubated with primary
macrophages in
the presence of concentrations of the mAb, derivative, or analogue to be
tested. After a
defined period of infection, unbound virus is removed by washing, and-the
cells are placed
in culture. The level of virus replication in this assay may be assessed by
techniques known
in the art, including but not limited to, measuring reverse transcriptase (RT)
levels, or the
release of extracellular p24 core antigen at different days post-infection. A
constant level of
inhibition of viral infection or replication is determined by measuring output
HIV p24 levels
(or another indicator of viral infection or replication, such as for example,
RT) relative to
control assays performed in the absence of the mAb, derivative or analogue.
Preferably, the
~b derivative or analogue reduces levels of virus, as measured by, for
example, p24, by Z
50% relative to control assays carried out in the absence of test compound.
The presence of
p24 may be determined using methods known in the art, such as commercially
available
enzyme-linked immunosorbent assays (Coulter, Hialeah, Florida; Abbott
Laboratories,
Hvalstad, Norway). Alternatively, RT activity may be tested by monitoring cell-
free
supernatant using standard techniques such as those described by, for example,
Goff et al.
(Goff et al., 1981, J. Virol. 38:239-248) and Willey et al. (Willey et al.,
1988, J. Virol.
62:139-147).
In other embodiments of the invention, a mAb of the invention is assayed in
a primary isolate neutralization assay. A variety of neutralization assays are
known in the
a~~ and are within the scope of the invention. In one embodiment, mAbs of the
invention
are assayed in a focus assay. In one embodiment of the focus assay, test
antibody is
incubated with a measured amount of virus prior to the addition of the virus
to host cells.
Infection of the host cells is measured by staining said cells by
immunohistochemical
methods known in the art using an antibody specific for the viral protein(s).
Generally, a
control assay is performed in parallel in which no test antibody is used. Test
antibody
which inhibits (e.g., prevents or decreases) infection compared to the control
infection is
indicative of a neutralizing antibody. In another embodiment of the invention,
a PBL assay
can be used to assay neutralization by a test antibody. In one embodiment of
the PBL
assay, test antibody is incubated with a measured amount of virus prior to the
addition of
~e virus to host cells. Host cell cultures are allowed to incubate for a
period of days, and
infection is assayed by measuring the virions present in a sample of host cell
supernatant.
Generally, a control assay is performed in parallel in which no test antibody
is used. Test
antibody which inhibits (e.g., prevents or decreases) infection compared to
the control
infection is indicative of a neutralizing antibody.
55
CA 02338983 2001-O1-30
WO 00/08043 PCT/US99/17487
5.7.1. TRANSGENIC MOUSE MODEL
The present invention encompasses the use of a non-human transgenic animal
which
expresses one or more components whose association results in the formation of
the FRMS
of the invention. In specific embodiments the components whose association
results in the
formation of the FRMS may include those listed in Table II and Table III,
herein. The
transgenic animal models of the invention may be used as Tolerance models
and/or
Infection models. In the case of Tolerance models, it is preferred that all of
the cellular
components important to the formation of the FRMS are expressed in the
transgenic animal.
In the case of Infection models, one or more of the components important for
the formation
of the FRMS may be expressed such that the transgenic animal is capable of
infection by
the virus to which the FRMS is derived.
Animals of any species, including, but not limited to, mice, rats, rabbits,
guinea pigs, pigs, micro-pigs, goats, sheep, and non-human primates, e.g.,
baboons,
monkeys, and chimpanzees may be used to generate transgenic animals expressing
a
component which results in the formation of a FRMS. The term "transgenic," as
used
herein, refers to animals expressing one or more components whose association
results in
the formation of the FRMS gene sequences from a different species (e.g., mice
expressing
human sequences encoding one or more component whose association results in
the
formation of a FRMS), as well as animals that have been genetically engineered
to over
express endogenous (i.e., same species) sequences encoding one or more
component whose
association results in the formation of a FRMS or animals that have been
genetically
engineered to no longer express endogenous gene sequences encoding one or more
component whose association results in the formation of a FRMS (i.e., "knock-
out"
animals), and their progeny.
~Y technique known in the art may be used to introduce an gene encoding
one or more component whose association results in the formation of a FRMS
transgene
into animals to produce the founder lines of transgenic animals. Such
techniques include,
but are not limited to pronuclear microinjection (Hoppe and Wagner, 1989, U.S.
Pat. No.
4,873,191); retrovirus mediated gene transfer into germ lines (Van der Putten,
et al., 1985,
Proc. Natl. Acad. Sci., USA 82, 6148-6152); gene targeting in embryonic stem
cells
(Thompson, et al., 1989, Cell 56, 313-321 ); electroporation of embryos (Lo,
1983, Mol.
Cell. Biol. 3, 1803-1814); and sperm-mediated gene transfer (Lavitrano et al.,
1989, Cell
57, 717-723). (For a review of such techniques, see Gordon, 1989, Transgenic
Animals,
Intl. Rev. Cytol. 115, 171-229.)
~Y technique known in the art may be used to produce transgenic animal
clones containing a one or more component whose association results in the
formation of a
FRMS transgene, for example, nuclear transfer into enucleated oocytes of
nuclei from
56
CA 02338983 2001-O1-30
WO 00/08043 PCT/US99/17487
cultured embryonic, fetal or adult cells induced to quiescence (Campbell, et
al., 1996,
Nature 380, 64-66; Wilmut, et al., Nature 385, 810-813).
The present invention provides for transgenic animals that carry a transgene
of one or more component whose association results in the formation of a FRMS
in all their
cells, as well as animals that carry the transgene in some, but not all their
cells, i.e., mosaic
animals. The transgene may be integrated as a single transgene or in
concatamers, e.g.,
head-to-head tandems or head-to-tail tandems. The transgene may also be
selectively
introduced into and activated in a particular cell type by following, for
example, the
teaching of Lasko et al. (Lasko, et al., 1992, Proc. Natl. Acad. Sci. USA 89,
6232-6236).
The regulatory sequences required for such a cell-type specific activation
will depend upon
the particular cell type of interest, and will be apparent to those of skill
in the art. When it
is desired that the gene transgene be integrated into the chromosomal site of
the endogenous
gene, gene targeting is preferred. Briefly, when such a technique is to be
utilized, vectors
containing some nucleotide sequences homologous to the endogenous gene are
designed for
I S the purpose of integrating, via homologous recombination with chromosomal
sequences,
into and disrupting the function of the nucleotide sequence of the endogenous
gene. The
transgene may also be selectively introduced into a particular cell type, thus
inactivating the
endogenous gene encoding one or more component whose association results in
the
formation of a FRMS in only that cell type, by following, for example, the
teaching of Gu,
et al. (Gu, et al., 1994, Science 265, 103-106). The regulatory sequences
required for such
a cell-type specific inactivation will depend upon the particular cell type of
interest, and will
be apparent to those of skill in the art.
Once transgenic animals have been generated, the expression of the
recombinant component whose association results in the formation of a FRMS
gene may be
essayed utilizing standard techniques. Initial screening may be accomplished
by Southern
blot analysis or PCR techniques to analyze animal tissues to assay whether
integration of
the transgene has taken place. The level of mRNA expression of the transgene
in the tissues
of the transgenic animals may also be assessed using techniques that include
but are not
limited to Northern blot analysis of tissue samples obtained from the animal,
in situ
hybridization analysis, and RT-PCR (reverse transcriptase PCR). Samples of the
component gene-expressing tissue, may also be evaluated immunocytochemically
using
antibodies specific for the component transgene product.
In a specific embodiment, the invention encompasses the use of a transgenic
mouse for the development of model system which allows screening of a test
vaccines
intended as vaccines for a species other than mouse. In a preferred
embodiment, the test
vaccine is a vaccine which is intended for a human.
57
CA 02338983 2001-O1-30
WO 00/08043 PCT/US99/17487
The transgenic mouse model of the invention encompasses a mouse which is
constructed to express the host cell receptors and/or co-receptors of another
species for a
virus which is not native to a mouse. Such mice are advantageous for the
analysis of virus
neutralization, since the effect of neutralizing antibodies directed to the
host cell receptors is
eliminated. These mice provide a model system for viral tolerance. For
example, a
transgenic mouse may be constructed or used which expresses human host cell
receptors)
for a virus capable of infecting a human {such as HIV).
In one embodiment of the invention, vaccine studies utilize transgenic mice
expressing human CD4 and CCRS co-receptor under the control of CD4 regulatory
elements which restrict co-expression to thymocytes and T helper lymphocytes
(Killeen et
al., 1993, EMBO J., 12:1547-53). These mice are used in order to allow for the
analysis of
virus neutralization without the confounding effect of neutralizing antibodies
directed to
CD4 or CCRS. In this regard, the immune response mimics that in humans;
immunogenic
epitopes are restricted to envelope and to HIV-dependent conformations of CD4
and co-
receptor. Although post-entry restrictions to HIV replication have to-date
limited the utility
of these mice for infectivity studies, the present invention provides a novel
use as a system
for the analysis of HIV vaccines. In this model, the transgenes function only
to provide
tolerance to the human CD4 and CCRS components of the vaccine.
In order to conduct the assay, transgenic mice are immunized one or more
times with the FRMS of the invention. Optionally, control immunogens may be
used to
vaccinate other mice which serve as a control. The FRMS immunogen may be in
any of
the forms described herein including in cell lysate, in solution, in a cross-
linked structure,
isolated, purified, etc. The FRMS immunogen may also be introduced to the
mouse in a
vaccine formulation.
Next, sera or antibodies produced from mice immunized the FRMS or
control immunogens are collected and assayed for the ability to neutralized PI
virus. Serum
is collected at an appropriate time following immunization to allow for the
production of an
immune response in the mouse. Such times are well known in the art. In one
embodiment,
sera is obtained 2 weeks following each immunization.
Preferably, sera or antibodies are tested for the ability to neutralize two,
three
or four primary isolates of the virus. More preferably, sera or antibodies are
tested for the
ability to neutralize four, six, eight primary isolates of the virus. Most
preferably, sera or
antibodies are tested for the ability to neutralize 10-15, 1 S-25, or more
primary isolates of
the virus.
Neutralization of a primary isolate may be assayed by methods known in the
art including those presented herein. In one embodiment, neutralization is
determined by
the inhibition of infectivity associated with incubating the test antibody
with a sample of the
58
CA 02338983 2001-O1-30
WO 00/08043 PCT/US99/17487
virus to be used in infection. An antibody which inhibits (e.g. decreases or
blocks) viral
infection, is indicative of a neutralizing antibody. In other embodiments,
sera or antibodies
are tested for the ability to inhibit syncytia formation or to inhibit
infection by cell-free
virus and assess the effects of the compound on cell proliferation and
viability. In a
preferred embodiment of the invention, the test antibody is a monoclonal
antibody. In a
more preferred embodiment of the invention, the test antibody is a human
monoclonal
antibody produced in response to a FRMS immunogen. As will be apparent to one
skilled
in the art, little or no inhibition of infectivity or neutralization activity
will be expected to be
observed in sera or antibodies obtained from mice immunized with control
immunogens.
In other embodiments of the invention, a virus neutralization assay is
performed on more than one primary isolates. In preferred embodiments, virus
neutralization assay are performed on 2-5, 5-10, 10-15 primary isolates. In a
more prefen ed
embodiment, neutralization assay are performed on 1 S-20, 20-30, or 30 or more
primary
isolates. In another preferred embodiment, the primary isolates assayed are
from more than
one viral Glade.
Neutralization may characterized in terms of percent neutralization of a
number of primary isolates. Generally, in this embodiment, a single assay is
used to assess
antibody neutralization. In one embodiment, an antibody of the invention
neutralizes 30-
40%, 40-SO%, or SO-60% of the primary isolates of the virus. In a prefer ed
embodiment,
~ ~tibody of the invention neutralizes 60-75%, 70-85%, or 80-90% of the
primary isolates
of the virus. In a most preferred embodiment, an antibody of the invention
neutralizes 90-
95%, 95-98%, or 99-100% of the primary isolates of the virus. In preferred
embodiments,
when mAb are tested for neutralization of primary isolates, neutralization is
expressed as
the number of foci in the presence of mAb (or hybridoma supernatant) relative
to the
number in the presence of medium alone.
In a specific embodiment, Transgenic mice (hu CD4+, hu CCRS+, mouse
CD4+) are immunized with an HIV FRMS immunogen (cross-linked COS-env co-
cultured
with U87-CD4-CCRS cells) or with cell controls (U87-CD4-CCRS cells alone or
cocultured
with mock-transfected COS cells). Sensitivity of the homologous 168P virus to
neutralization by vaccine sera is determined with U87-CD4 cells expressing
either CCRS or
CXCR4 co-receptor (see, LaCasse, et al., 1998, Science 72:2491; Follis, K.E.,
et a1.,1998,
Journal of Virology 72:7603).
In another specific embodiment of the invention, wherein HIV primary
isolates are used in a neutralization assay such primary isolates may include
but are not
limited to 92US657, 92US660, 92TH014, 89.6, 320NSI, 320SI, SHIV89.6P, 168P,
92RW023, 92UG031, 92UG037, 92RW008, 93IN101, 93IN999, 93IN905, 93IN904,
92UG035, 92UG021, 92UG024, 92UG046, 92TH023, 92TH024, 93TH051, and 93TH053.
59
CA 02338983 2001-O1-30
WO 00/08043 PCT/US99/17487
In one embodiment of the invention, an antibody produced to an HIV FRMS
by the methods of the invention (see Section 5.6) is capable of neutralization
of 60-70%,
70-80%, 80-85% of such primary isolates of HIV. In a preferred embodiment, an
antibody
produced to an HIV FRMS is capable of neutralization of 80-90%, 90-95%, or 95-
99% of
such primary isolates of HIV. In a most preferred embodiment, an antibody
produced to an
HIV FRMS is capable of neutralization of 100% of such primary isolates of HIV.
In another specific embodiment, primary isolates of HIV may include one or
more isolates of Glade A, B, C, D, E, F, G, H, or I. In other embodiments,
primary isolates
of HIV may include one or more isolates an M-group, O-group, or N-group.
In another embodiment, neutralization activity can be demonstrated as
antibody-mediated by methods known in the art. For example, sera can be
adsorbed to a
solid support containing Protein-A and Protein-G. In the case that an antibody
is
responsible for the virus neutralization activity of the sera, sera depleted
of Protein-A and
Protein-G binding-proteins will be expected to contain little or no virus
neutralization
activity, while eluate from the Protein-A and Protein-G solid support will be
expected to
contain virus neutralization activity.
Accordingly, the present invention provides a method of screening a
molecular structure for vaccine efficacy comprising immunizing a transgenic
non-human
mammal with the molecular structure, wherein said transgenic non-human mammal
expresses from one or more transgenes both human CD4 and a co-receptor for
HIV, and
detecting any neutralizing antibodies to HIV that are produced by said mammal.
The invention also provides a method of screening a molecular structure for
vaccine efficacy comprising immunizing a transgenic non-human mammal with the
molecular structure, wherein said transgenic non-human mammal expresses from
one or
more transgenes said one or more host cellular membrane proteins; and
detecting any
neutralizing antibodies to said virus that are produced by said mammal.
5.7.2. DETERMINATION QF VACCINE EFFICACY
Immunopotency of the one or more immunogens of the invention can be
determined by monitoring the immune response of test animals following
immunization
with the FRMS by use of any immunoassay known in the art. Generation of a
humoral
(antibody) response and/or cell-mediated immunity, may be taken as an
indication of an
immune response. Test animals may include mice, hamsters, dogs, cats, monkeys,
rabbits,
chimpanzees, etc., and eventually human subjects.
~ described in Section 5.10 herein, methods of introduction of the vaccine
may include oral, intracerebral, intradermal, intramuscular, intraperitoneal,
intravenous,
subcutaneous, intranasal or any other standard routes of immunization. The
immune
CA 02338983 2001-O1-30
wo ooiosoa3 Prrnrs~n ~as~
response of the test subjects can be analyzed by various approaches such as:
the reactivity of
the resultant immune serum to the immunogen of the invention, as assayed by
known
techniques, e.g., enzyme linked immunosorbent assay (ELISA), immunoblots,
radioimmune
assays (RIA) radioimmunoprecipitations, etc. Alternatively, protection of
immunized hosts
from infection by the pathogen and/or attenuation of symptoms due to infection
by the
pathogen in immunized hosts can serve as evidence of vaccine efficacy.
As one example of suitable animal testing of viral vaccine, the vaccine of the
invention may be tested in rabbits for the ability to induce 'an antibody
response to the
FRMS immunogen. Male specific-pathogen-free (SPF) young adult New Zealand
White
rabbits may be used. The test group each receives a fixed concentration of the
vaccine. A
control group receives an injection of 1 mM Tris-HCl pH 9.0 without the FRMS
immunogen. Blood samples may be drawn from the rabbits every one or two weeks,
and
serum analyzed for antibodies to FRMS. The presence of antibodies specific for
the
immunogen may be assayed, ~, using an ELISA.
In a preferred embodiment of the invention, animal testing of viral vaccine
effacy is by use of a transgenic mouse model as described herein in Section
5.7.1.
The invention also provides methods for optimizing and quantitating
vaccines comprising the subject FRMS. Preferably, the subject complexes are
prepared
using autologous cells negating reactivity toward host cellular components.
Vaccine
compositions comprising complexes prepared using non-autologous cells,
however, can be
assessed using transgenic animals genetically engineered to be tolerant to the
host cellular
components of the complexes. Using these transgenic animals it is possible to
determine
whether the success achieved by vaccinating with the fusion-complexes of the
subject
invention is due to novel fusion-competent epitopes presented by the
complexes. For
example, in one preferred embodiment, the FRMS result from the interaction of
HIV(168P)
viral envelope protein and the human proteins CD4 and CCRS. As described in
Section 6,
vaccine compositions have been tested in transgenic mice genetically
engineered to be
tolerant to the human components of the vaccine. Mice expressing both the
human CD4
receptor and the human CCRS co-receptor were used to evaluate the complex and
demonstrate that the neutralizing antibody generated was to immunologic
epitopes
restricted to envelope protein or HIV-dependent conformations of CD4 and co-
receptor. It
is noted that these transgenic mice were originally used as a mouse model for
HIV
infection. Utilizing transgenic animals to screen vaccine immunogens is a
novel use of
these mice. Transgenic mice exhibiting tolerance to host portions of vaccines
can be used
to optimize vaccine components, test variants and evaluate adjuvants.
Further, it should be apparent to one skilled in the art that this principal
can
be applied to any viral model to provide rapid and safe methods for screening
viral
61
CA 02338983 2001-O1-30
WO 00/08043 PCT/US99/17487
vaccines. Additionally, it should be apparent that transgenic animals other
than transgenic
mice can be used to screen candidate vaccines.
In one embodiment, the invention provides a method of screening a
molecular structure for vaccine efficacy comprising immunizing a transgenic
non-human
mammal with a molecular structure comprising an epitope formed as a result of
association
of (a) an envelope protein of an enveloped virus, with (b) one or more
cellular membrane
proteins, which envelope protein and cellular membrane proteins are necessary
and
sufficient under suitable conditions for fusion of said envelope of the virus
with a cell
membrane containing said cellular membrane proteins, wherein said transgenic
non-human
mammal expresses from one or more transgenes said one or more host cellular
membrane
proteins; and detecting any neutralizing antibodies to said virus that are
produced by said
mammal. In a preferred embodiment, the molecular structure is isolated.
In a specific embodiment, the invention provides a method of screening a
molecular structure for vaccine efficacy comprising immunizing a transgenic
non-human
1 S m~mal with a molecular structure comprising an epitope formed as a result
of association
of (a) an HIV envelope protein, or a mutant thereof that assembles into the
viral envelope;
with (b) human CD4 and a co-receptor for HIV fusion, wherein said transgenic
non-human
mammal expresses from one or more transgenes both human CD4 and a co-receptor
for
HIV, and detecting any neutralizing antibodies to HIV that are produced by
said mammal.
In a preferred embodiment, the mammal is a mouse. In another preferred
embodiment, the
molecular structure is isolated.
5.8. USES FOR ANTIBODIES
The antibodies generated by the vaccine formulations of the present
invention antibodies can be used in methods known in the art. For example, the
antibodies
generated against the FRMS or vaccine immunogen by immunization with the FRMS
of
the present invention also have potential uses such as diagnostic
immunoassays, passive
immunotherapy, viral decontamination, anti-viral treatment and prevention and
generation
of antiidiotypic antibodies.
The subject invention also encompasses antibodies that are elicited by the
novel epitopes of the FRMS of the subject invention contain antibodies useful
as reference
standards and components in diagnostic assays and kits. Additionally, mAb to
the FRMS of
the subject invention can be used as reference standards, diagnostic agents
for in vitro and
in vivo bioassays and as markers for critical determinants in binding and
fusion. For
example, during large scale production of the FRMS of the invention, the mAb
of the
invention are useful in determining the amount of FRMS produced in each batch
or lot. In
this embodiment, the mAb may be used to determine the amount to lot-to-lot
variation (e.g.,
62
CA 02338983 2001-O1-30
WO 00/08043 PCT/US99/17487
by ELISA, or RIA using the mAb). Alternatively, labeled FRMS may be used, by
way of a
competition assay with the test FRMS for binding a mAb specific for the FRMS.
Similar assays may be used to determine the antibody titer of a sample of
serum from a subject. In such an assay, a sample of serum from a subject is
taken at a time
following administration of the vaccine of the invention. The sample serum is
then tested
for ability to neutralize virus. The serum may also be tested for the ability
to bind to the
immunogen FRMS, or to compete with a neutralizing mAb for binding to the
immunogen
FRMS. The inability or weak ability of the serum sample to neutralize virus or
bind FRMS
provides an indication that booster vaccinations with the FRMS immunogen are
desired for
~e subject. Accordingly, the present invention provides a method for
monitoring the
production of antibody to the molecular structure in a subject previously
administered an
amount of the molecular structure comprising isolating from said subject a
sample
comprising serum; and detecting the presence of any antibodies to the
molecular structure in
said serum. In one embodiment, detecting is carned out by a method comprising
performing a competitive immunoassay with labeled antibody to the molecular
structure.
Further, polyclonal sera or monoclonal antibodies can be administered to
individuals for purposes of treatment or prevention of viral infection and its
sequences. In
particular embodiments, such antibody can be administered for purposes post-
exposure
prophylaxis or to prevent maternal transfer of virus to the neonate by way of
passive
2~ immunization. Humanized mAb of the invention can be used in protection
against
maternal-infant virus transmission or in post-exposure prophylaxis or
treatment. MAb of
the present invention are also useful in passive immunization studies in a hu-
PBL-scid
mouse model of HIV infection and in SHIV studies in rhesus macaques.
Accordingly, the
present invention provides a method for treating or preventing infection by
HIV in a human
fetus comprising administering to a pregnant human containing said fetus an
amount of the
monoclonal antibody effective to treat or prevent infection by HIV in said
fetus.
Production of mAb to the FRMS of the subject invention can be used to
further delineate the biochemical and immunochemical pathway to infection, and
define
critical epitopes for PI virus neutralization. Monoclonal antibodies (mAb)
generated to the
FMS of the subject invention can be used to dissect the biochemical basis for
binding and
entry, and the immunochemical basis for primary isolate virus neutralization.
In this
embodiment, immunogens should be chosen for MAb development that excel in
immunogenicity studies in vaccination models, are potent and elicit a broad
range of
primary isolate virus neutralizing antibodies.
The antibodies of the present invention can also be used in the production of
antiidiotypic antibody. The antiidiotypic antibody can then in turn be used
for
immunization, in order to produce a subpopulation of antibodies that bind the
initial antigen
63
CA 02338983 2001-O1-30
WO 00/08043 PCTNS99/17487
of the pathogenic microorganism (Jerne, 1974, Ann. Immunol. (Paris) 125c:373;
Jerne, et
al., 1982, EMBO J. 1:234).
Further, mAb can also be used as an anti-viral agent in blood and blood
products. The present invention provides neutralizing antibodies that are
useful as an
additive for the decontamination of blood or blood products which are
contaminated or
suspected of being contaminated with a virus. For example, the mAb of the
invention is
useful as an additive to donor blood (e.g., blood within a blood bank) which
is to be used in
the treatment of a subject. The mAb of the invention is useful as an additive
to blood
collection bags, gloves, blood sample collection containers, etc. In a
specific embodiment
of the embodiment, one or more mAb(s) of the invention are used to cleanse the
birth canal
prior to delivery of a child in order to prevent perinatal infection.
Effective amounts of the
mAb of the invention for blood and blood-product additives include 1 pg/ml to
l Omg/ml. As
will be apparent to one skilled in the art, effective amounts will be
influences by antibody
affinity, additives, and formulations. Any amount may be used such that the
effective
amount is capable of inhibiting (e.g. reducing or preventing) viral
infection.. In a preferred
embodiment, a mixture of neutralizing mAb against a variety of viruses is used
in
decontamination. In a specific embodiment, an effective amount of a
neutralizing mAb of
the invention which is immunospecific for HIV is added to a sample of human
blood before
such blood is transfused into a recipient subject. In this embodiment, the HN
is neutralized
by the mAb. Accordingly, the present invention provides a sample of mammalian
blood, to
which an amount of the antibody has been added effective to inhibit or
decrease infection
by the virus. In a specific embodiment, the present invention provides a
sample of human
blood, to which an amount of the antibody has been added effective to inhibit
or decrease
infection by HIV. The present invention also provides a method of inhibiting
infection by a
virus in a sample of blood comprising contacting said sample of blood with an
amount of
the monoclonal antibody effective to inhibit infection by said virus. In a
specific
embodiment, the present invention provides a method of inhibiting infection by
HIV in a
sample of human blood comprising contacting said sample of human blood with an
amount
of the monoclonal antibody effective to inhibit infection by HIV.
The neutralizing antibodies of the invention are also useful in
decontaminating any object exposed to the virus to which the antibody is
directed. Objects
which may be decontaminated with the mAb of the invention include but are not
limited to
surgical and dental tools (such as drills, picks, scalpels, etc). In a
preferred embodiment, a
mixture of neutralizing mAb against a variety of viruses is used in
decontamination. In a
preferred embodiment, objects which are not readily disposable (e.g., computer
aided tools,
robotic aided tools, specialized tools, etc.) are decontaminated with one or
more mAbs of
the invention. Effective amounts of the mAb of the invention for
decontamination include
64
CA 02338983 2001-O1-30
WO 00/08043 PCT/US99/17487
1 ug/ml to l Omg/ml. As will be apparent to one skilled in the art, effective
amounts will be
influences by antibody affinity, additives and formulations. Any amount may be
used such
that the effective amount is capable inhibiting viral infection- following
decontamination. In
a specific embodiment, HIV is neutralized in the decontamination. Accordingly,
the present
invention provides a method of decontaminating surgical or dental tools
comprising
contacting said tools with an amount of the monoclonal antibody effective to
inhibit
infection by said virus. In a specific embodiment, the present invention
provides a method
of decontaminating surgical or dental tools comprising contacting said tools
with an amount
of the monoclonal antibody effective to inhibit infection by HIV.
In another embodiment of the invention antibodies directed to an FRMS may
be used to screen molecules which comprise an epitope recognized by the
antibody. For
example, a mAb of t he invention can be used to screen small molecules for the
presence of
an antibody-recognized epitope. In one binding of the mAb to the small
molecules
indicates the presence of such epitope on the small molecule. Such small
molecules in turn
1 S may be tested as potential immunogens or therapeutics. In another
embodiment, the mAbs
of the invention may be used to identify peptides which bind to the antibody
(e.g., from a
phage display library). Such peptides may be tested, for example, for
immunogenicity (i.e.
as mimetopes).
The mAb of the invention are also useful as an anti-viral agent in
contraceptive or microbicide products. The addition of one or more mAb of the
invention
to a contraceptive or microbicide product is particularly preferred for
viruses which are
known to be sexually transmitted. Thus, the invention provides a contraceptive
product
comprising an effective amount of one or more neutralizing mAb(s) of the
invention.
Contraceptive or microbicide products to which the mAb of the invention may be
added
include but are not limited to creams, foams, jellies, ointment, condoms,
diaphragms, etc.
The contraceptive products may further comprise of spermicidal agent. In a
preferred
embodiment, a mixture of neutralizing mAbs against a variety of viruses is
used in the
contraceptive product. Effective amounts of the mAb of the invention for
contraceptive or
microbicide products include 1 ug/ml to l Omg/ml. As will be apparent to one
skilled in the
~~ elective amounts will be influences by antibody affinity, additives and
formulations.
Any amount may be used such that the effective amount is capable of inhibiting
(e.g.
reducing or preventing) viral infection. For example, for the use in an anti-
viral
contraceptive, an effective amount is that which is capable of inhibiting
infection upon
exposure by sexual transmission of a virus. In a specific embodiment, the
contraceptive or
microbicide products comprises mAb of the invention immunospecific to HIV or
other
sexually transmitted virus. Accordingly, the present invention provides a
contraceptive or
microbicide in the form of a jelly, foam, cream, or ointment comprising an
amount of the
CA 02338983 2001-O1-30
WO 00/08043 PCT/US99/17487
antibody of the invention effective to inhibit or decrease infection by the
virus. In a specific
embodiment, the present invention provides a contraceptive or microbicide in
the form of a
jelly, foam, cream, or ointment comprising an amount of the antibody of the
ineffective to
inhibit or decrease infection by HIV.
5.9. THERAPEUTIC COMPOSITIONS
The present invention provides methods of eliciting production of anti-
FRMS antibodies in a subject by the administration of a FRMS. Any of the FRMS
of the
invention, and functionally active fragments, analogs, and derivatives
thereof; nucleic acids
encoding the FRMS of the invention, and functionally active fragments and
derivatives
thereof; as well as antibodies which immunospecifically bind to a FRMS may be
used as
therapeutic, (termed herein "Therapeutic").
The present invention also provides pharmaceutical compositions. Such
compositions comprise a therapeutically effective amount of a Therapeutic, and
a
1 S ph~aceutically acceptable carrier. In a specific embodiment, the term
"phanmaceutically
acceptable" means approved by a regulatory agency of the Federal or a state
government or
listed in the U.S. Pharmacopeia or other generally recognized pharmacopeia for
use in
animals, and more particularly in humans. The term "carrier" refers to a
diluent, adjuvant,
excipient, or vehicle with which the Therapeutic is administered. Such
pharmaceutical
carvers can be sterile liquids, such as water and oils, including those of
petroleum, animal,
vegetable or synthetic origin, such as peanut oil, soybean oil, mineral oil,
sesame oil and the
like. Water is a preferred carrier when the pharmaceutical composition is
administered
intravenously. Saline solutions and aqueous dextrose and glycerol solutions
can also be
employed as liquid carriers, particularly for injectable solutions. Suitable
pharmaceutical
excipients include starch, glucose, lactose, sucrose, gelatin, malt, rice,
flour, chalk, silica
gel, sodium stearate, glycerol monostearate, talc, sodium chloride, dried skim
milk,
glycerol, propylene glycol, water, ethanol and the like. The composition, if
desired, can
also contain minor amounts of wetting or emulsifying agents, or pH buffering
agents.
These compositions can take the form of solutions, suspensions, emulsion,
tablets, pills,
capsules, powders, sustained-release formulations and the like. The
composition can be
formulated as a suppository, with traditional binders and carriers such as
triglycerides. Oral
formulation can include standard Garners such as pharmaceutical grades of
mannitol,
lactose, starch, magnesium stearate, sodium saccharine, cellulose, magnesium
carbonate,
etc. Examples of suitable pharmaceutical carriers are described in
"Remington's
Pharmaceutical Sciences" by E.W. Martin. Such compositions will contain a
therapeutically effective amount of'the Therapeutic, preferably in purified
form, together
66
CA 02338983 2001-O1-30
WO 00/08043 PCT/US99/17487
with a suitable amount of carrier so as to provide the form for proper
administration to the
patient. The formulation should suit the mode of administration.
In a preferred embodiment, the composition is formulated in accordance with
routine procedures as a pharmaceutical composition adapted for intravenous
administration
S to human beings. Typically, compositions for intravenous administration are
solutions in
sterile isotonic aqueous buffer. Where necessary, the composition may also
include a
solubilizing agent and a local anesthetic such as lignocaine to ease pain at
the site of the
injection. Generally, the ingredients are supplied either separately or mixed
together in unit
dosage form, for example, as a dry lyophilized powder or water free
concentrate in a
hermetically sealed container such as an ampoule or sachette indicating the
quantity of
active agent. Where the composition is to be administered by infusion, it can
be dispensed
with an infusion bottle containing sterile pharmaceutical grade water or
saline. Where the
composition is administered by injection, an ampoule of sterile water for
injection or saline
can be provided so that the ingredients may be mixed prior to administration.
1 S The Therapeutics of the invention can be formulated as neutral or salt
forms.
Pharmaceutically acceptable salts include those formed with free amino groups
such as
those derived from hydrochloric, phosphoric, acetic, oxalic, tartaric acids,
etc., and those
formed with free carboxyl groups such as those derived from sodium, potassium,
ammonium, calcium, fernc hydroxides, isopropylamine, triethylamine, 2-
ethylamino
ethanol, histidine, procaine, etc.
5.10. GENE THERAPY
Gene therapy refers to therapy performed by the administration of a nucleic
acid molecule to a subject. In this embodiment of the invention, the nucleic
acid
2S molecules) produces its encoded proteins) and mediates a therapeutic effect
by forming a
FRMS in vivo.
In a specific embodiment, a nucleic acid molecule comprising a sequence
encoding one or more components whose association results in the FRMS of the
invention
or a fimctional derivative thereof, are administered to produce an
immunological response
to the FRMS, by way of gene therapy. In more specific embodiments, a nucleic
acid or
nucleic acids encoding a viral envelope protein of an envelope virus, a host
cell receptor,
and a host cell co-receptor or fimctional derivatives thereof, are
administered by way of
gene therapy. In other embodiment, a viral envelope protein of an envelope
virus
administered by way of gene therapy.
3S ~Y of the methods for gene therapy available in the art can be used
according to the present invention. Exemplary methods are described below.
67
CA 02338983 2001-O1-30
WO 00/0$043 PCT/US99/17487
For general reviews of the methods of gene therapy, see Goldspiel et al.,
1993, Clinical Pharmacy 12:488-SOS; Wu and Wu, 1991, Biotherapy 3:87-95;
Tolstoshev,
1993, Ann. Rev. Pharmacol. Toxicol. 32:573-596; Mulligan, 1993, Science
260:926-932;
Morgan and Anderson, 1993, Ann. Rev. Biochem. 62:191-217; and May, 1993,
TIBTECH
11:155-215). Methods commonly known in the art for recombinant DNA technology
which
can be used are described in Ausubel et al. (eds.), 1993, Current Protocols in
Molecular
Biology, John Wiley & Sons, NY) and Kriegler, 1990, Gene Transfer and
Expression, A
Laboratory Manual, Stockton Press, NY.
In a preferred specific embodiment, the Therapeutic comprises an HIV
envelope protein and a human CD4 receptor, and a co-receptor (such as a
chemokine
receptor CCRS or CXCR4) in a suitable host. In another particular embodiment,
a nucleic
acid molecule is used in which the HIV envelope protein coding sequence is
delivered to a
subject, which subject expresses native CD4 and a native co-receptor.
Delivery of the nucleic acid into a patient may be either direct, in which
case
1 S the patient is directly exposed to the nucleic acid or nucleic acid-
carrying vector, or indirect,
in which case, cells are first transformed with the nucleic acid in vitro,
then transplanted
into the patient. These two approaches are known, respectively, as in vivo and
ex vivo gene
therapy.
In a specific embodiment, the nucleic acid is directly administered in vivo,
where it is expressed to produce the encoded product. This can be accomplished
by any of
numerous methods known in the art, e.g., by constructing it as part of an
appropriate nucleic
acid expression vector and administering it so that it becomes intracellular,
e.g., by infection
using a defective or attenuated retroviral or other viral vector (see, U.S.
Patent No.
4,980,286), or by direct injection of naked DNA, or by use of microparticle
bombardment
(e'g'~ a gene gun; Biolistic, Dupont), or coating with lipids or cell-surface
receptors or
transfecting agents, or by encapsulation in liposomes, microparticles, or
microcapsules, or
by administering it in linkage to a peptide which is known to enter the
nucleus, or by
administering it in linkage to a ligand subject to receptor-mediated
endocytosis (see, e.g.,
Wu and Wu, 1987, J. Biol. Chem. 262:4429-4432), which can be used to target
cell types
specifically expressing the receptors, etc: In another embodiment, a nucleic
acid-ligand
complex can be formed in which the ligand comprises a fusogenic viral peptide
that disrupts
endosomes, preventing lysosomal degradation of the nucleic acid. In yet
another
embodiment, the nucleic acid can be targeted in vivo for cell specific uptake
and expression
by targeting a specific receptor (see, e.g., International Patent Publications
WO 92/06180 by
Wu et al., WO 92/22635 by Wilson et al., WO 92/20316 by Findeis et al., WO
93/14188 by
Clarke et al., and WO 93/20221 by Young). Alternatively, the nucleic acid can
be
introduced intracellularly and incorporated within host cell DNA for
expression, by
68
CA 02338983 2001-O1-30
WO 00/08043 PCT/US99117487
homologous recombination (Koller and Smithies, 1989, Proc. Natl. Acad. Sci.
USA
86:8932-8935, Zijlstra et al., 1989, Nature 342:435-438).
In a specific embodiment, a viral vector that contains a nucleic acid encoding
a viral envelope protein, and host cellular membrane proteins) (e.g., host
cell receptor(s))
are used. Any of the viral vectors described in Section 5.1.2, may be used for
gene therapy
purposes. For example, a retroviral vector can be used (see Miller et al.,
1993, Meth.
Enzymol. 217:581-599}. These retroviral vectors have been modified to delete
retroviral
sequences that are not necessary for packaging of the viral genome and
integration into host
cell DNA. Other references illustrating the use of retroviral vectors in gene
therapy include:
Clowes et al., 1994, J. Clin. Invest. 93:644-651, Kiem et al., 1994, Blood
83:1467-1473,
Salmons and Gunzberg, 1993, Human Gene Therapy 4:129-141, and Grossman and
Wilson,
1993, Curr. Opin. in Genetics and Devel. 3:110-114.
As described in Section 5.1.2, Adenoviruses are other viral vectors that can
be used in gene therapy. Adenoviruses are especially attractive vehicles for
delivering
genes to respiratory epithelia. Adenoviruses naturally infect respiratory
epithelia where
they cause a mild disease. Other targets for adenovirus-based delivery systems
are liver, the
central nervous system, endothelial cells, and muscle. Adenoviruses have the
advantage of
being capable of infecting non-dividing cells. Kozarsky and Wilson, 1993,
Current Opinion
in Genetics and Development 3:499-503 present a review of adenovirus-based
gene therapy.
B°ut et al., 1994, Human Gene Therapy 5:3-10, demonstrated the use of
adenovirus vectors
to transfer genes to the respiratory epithelia of rhesus monkeys. Other
instances of the use
of adenoviruses in gene therapy can be found in Rosenfeld et al., 1991,
Science 252:431-
434, Rosenfeld et al., 1992, Cell 68:143-155, and Mastrangeli et al., 1993, J.
Clin. Invest.
91:225-234.
Adeno-associated virus (AAV) has also been proposed for use in gene
therapy (Walsh et al., 1993, Proc. Soc. Exp. Biol. Med. 204:289-300).
Another approach to gene therapy involves transferring a gene into cells in
tissue culture by such methods as electroporation, lipofection, calcium
phosphate mediated
transfection, or viral infection. Any method known in the art to transfer a
gene into a cell
may be used in connection with the gene therapy aspect of the invention,
including but not
limited to those described in Section 5.1.2. Usually, the method of transfer
includes the
transfer of a selectable marker to the cells. The cells are then placed under
selection to
isolate those cells that have taken up and are expressing the transferred
gene. Those cells
are then delivered to a patient.
In this embodiment, the nucleic acid is introduced into a cell prior to
administration in vivo of the resulting recombinant cell. Such introduction
can be carried
out by any method known in the art, including but not limited to transfection,
69
CA 02338983 2001-O1-30
WO 00/08043 PCT/US99/17487
electroporation, microinjection, infection with a viral or bacteriophage
vector containing the
nucleic acid sequences, cell fusion, chromosome-mediated gene transfer,
microcell-
mediated gene transfer, spheroplast fusion, etc. Numerous techniques are known
in the art
for the introduction of foreign genes into cells (see, e.g., Loeffler and
Behr, 1993, Meth.
Enzymol. 217:599-618, Cohen et al., 1993, Meth. Enzymol. 217:618-644, Cline,
1985,
Pharmac. Ther. 29:69-92) and may be used in accordance with the present
invention,
provided that the necessary developmental and physiological functions of the
recipient cells
are not disrupted. The technique should provide for the stable transfer of the
nucleic acid to
the cell, so that the nucleic acid is expressible by the cell, and is
heritable and expressible by
its cell progeny.
The resulting recombinant cells can be delivered to a patient by various
methods known in the art. In a preferred embodiment, epithelial cells are
injected, e.g.,
subcutaneousiy. In another embodiment, recombinant skin cells may be applied
as a skin
graft onto the patient. Recombinant blood cells (e.g., hematopoietic stem or
progenitor
cells) are preferably administered intravenously. The amount of cells
envisioned for use
depends on the desired effect, patient state, etc., and can be determined by
one skilled in the
art.
Cells into which a nucleic acid can be introduced for purposes of gene
therapy encompass any desired, available cell type, and include but are not
limited to
epithelial cells, endothelial cells, keratinocytes, fibroblasts, muscle cells,
hepatocytes, and
blood cells, such as T lymphocytes, B lymphocytes, monocytes, macrophages,
neutrophils,
eosinophils, megakaryocytes, granulocytes; various stem or progenitor cells,
in particular
hematopoietic stem or progenitor cells, e.g., as obtained from bone marrow,
umbilical cord
blood, peripheral blood, fetal liver, etc.
In a preferred embodiment, the cell used for gene therapy is autologous to
the patient.
In an embodiment in which recombinant cells are used in gene therapy, a
nucleic acid molecule encoding the components whose association results in the
formation
of a FRMS is used. In a specific embodiment, for example, the nucleic acid
encodes an
HIV viral envelope protein, human CD4 and a co-receptor (such as a chemokine
receptor
CCRS or CXCR4). The encoding nucleic acid molecule, is/are introduced into the
cells
such that the gene or genes are expressible by the cells or their progeny, and
the
recombinant cells are then administered in vivo for therapeutic effect. In a
specific
embodiment, stem or progenitor cells are used. Any stem and/or progenitor
cells which can
be isolated and maintained in vitro can potentially be used in accordance with
this
embodiment of the present invention. Such stem cells include but are not
limited to
hematopoietic stem cells (HSC), stem cells of epithelial tissues such as the
skin and the
CA 02338983 2001-O1-30
WO 00/08043 PCTNS99/17487
lining of the gut, embryonic heart muscle cells, liver stem cells
(International Patent
Publication WO 94/08598), and neural stem cells (Stemple and Anderson, 1992,
Celi
71:973-985).
In a specific embodiment, the nucleic acid to be introduced for purposes of
gene therapy comprises an inducible promoter operably linked to the coding
region (see
Section 5.1.2), such that expression of the nucleic acid is controllable by
controlling the
presence or absence of the appropriate inducer of transcription.
Additional methods can be adapted for use to deliver a nucleic acid molecule
encoding the components which result in the formation of the FRMS of the
invention will
be apparent to one skilled in the art, and, are within the scope of the
invention.
Accordingly, the invention provides a method of treating or preventing
infection by a virus in a subject comprising administering to the subject (a)
a first nucleic
acid encoding an envelope protein of an enveloped virus; and (b) a second
nucleic acid
encoding one or more cellular membrane proteins, which envelope protein and
cellular
membrane proteins are necessary and sufficient under suitable conditions for
fusion of said
envelope of the virus with a cell membrane containing said cellular membrane
proteins,
such that the envelope protein and cellular membrane proteins are expressed in
the subject
and neutralizing antibodies to the virus are produced. In one embodiment, the
method
wherein the first and second nucleic acids are the same. In another
embodiment, the first
and second nucleic acid are different nucleic acid vectors. In a specific
embodiment, the
envelope protein is en envelope protein of HIV, and the cellular membrane
proteins are
CD4 and an HIV co-receptor.
5.11. METHODS OF ADMIhiISTRA.TION
~e invention provides methods of treatment and prevention of viral
infection and disease by administration to a subject in need of such treatment
of a
therapeutically or prophylactically effective amount of a Therapeutic of the
invention. The
subject is preferably an arumai, including, but not limited to, animals such
as monkeys,
cows, pigs, horses, chickens, cats, dogs, etc., and is preferably a mammal,
and most
preferably human. In a specific embodiment, the subject is a human not
afflicted with HIV
infection.
Various delivery systems are known and can be used to administer a
Therapeutic of the invention, e.g., encapsulation in liposomes,
microparticies,
microcapsules, recombinant cells capable of expressing the Therapeutic,
receptor-mediated
endocytosis (see, e.g., Wu et al., 1987, J. Biol. Chem. 262:4429-4432),
construction of a
Therapeutic nucleic acid as part of a retroviral or other vector, etc. Methods
of introduction
include but are not limited to intradermal, intramuscular, intraperitoneal,
intravenous,
71
CA 02338983 2001-O1-30
WO 00/08043 PC'TNS99/17487
subcutaneous, intranasal, epidural, and oral routes. The compounds may be
administered by -
any convenient route, for example by infusion or bolus injection, by
absorption through
epithelial or mucocutaneous linings (e.g., oral mucosa, rectal and intestinal
mucosa, etc.)
and may be administered together with other biologically active agents.
Administration can
be systemic or local. In addition, it may be desirable to introduce the
pharmaceutical
compositions of the invention comprising antibody into the central nervous
system by any
suitable route, including intraventricular and intrathecal injection;
intraventricular injection
may be facilitated by an intraventricular catheter, for example, attached to a
reservoir, such
as an Ommaya reservoir. Pulmonary administration can also be employed, e.g.,
by use of
~ baler or nebulizer, and formulation with an aerosolizing agent.
In a specific embodiment, it may be desirable to administer the
pharmaceutical compositions of the invention comprising antibody locally to
the area in
need of treatment; this may be achieved, for example and not by way of
limitation, by
topical application, by injection, by means of a catheter, by means of a
suppository, or by
means of an implant, said implant being of a porous, non-porous, or gelatinous
material,
including membranes, such as sialastic membranes, or fibers. In a highly
specific
embodiment, the pharmaceutical compositions of the invention is administered
to an open
wound of a human, which wound is suspected of being exposed to HIV.
In another embodiment, the Therapeutic can be delivered in a vesicle, in
p~lcular a liposome (see, ~Langer, 1990, Science 249:1527-1533; Treat et al.,
1989 in
Liposomes in the Therapy of Infectious Disease and Cancer, Lopez-Berestein and
Fidler
(eds.), Liss, New York, pp. 353-365; Lopez-Berestein, Science., pp. 317-327;
see generally
Science. )
In yet another embodiment, the Therapeutic can be delivered in a controlled
release system. In one embodiment, a pump may be used (see Larger, supra;
Sefton, 1987,
CRC Crit. Ref. Biomed. Eng. 14:201; Buchwald et al., 1980, Surgery 88:507;
Saudek et al.,
1989, N. Engl. J. Med. 321:574). In another embodiment, polymeric materials
can be used
(see Medical Applications of Controlled Release, Larger and Wise (eds.), CRC
Pres., Boca
Raton, Florida (1974); Controlled Drug Bioavailability, Drug Product Design
and
Performance, Smolen and Ball (eds.), Wiley, New York (1984); Ranger et al.,
1983, J.
Macromol. Sci. Rev. Macromol. Chem. 23:61; see also Levy et al., 1985, Science
228:190;
During et al., 1989, Ann. Neurol. 25:351; Howard et al., 1989, J. Neurosurg.
71:105). In
yet another embodiment, a controlled release system can be placed in proximity
of the
therapeutic target, thus requiring only a fraction of the systemic dose (see,
e.g., Goodson, in
Medical Applications of Controlled Release, 1984, supra, vol. 2, pp. 115-138).
Other controlled release systems are discussed in the review by Larger
(Larger, 1990, Science 249:1527-1533).
72
CA 02338983 2001-O1-30
WO 00/08043 PCTNS99/17487
The compositions may, if desired, be presented in a pack or dispenser device
which may contain one or more unit dosage forms containing the active
ingredient. The
pack may for example comprise metal or plastic foil, such as a blister pack.
The pack or
dispenser device may be accompanied by instructions far administration.
Compositions
S comprising a compound of the invention formulated in a compatible
pharmaceutical carrier
may also be prepared, placed in an appropriate container, and labelled for
treatment of an
indicated condition.
5.12. DEMONSTRATION OF UTILITY AGAINST HIV
~e present invention provides assays to test the anti-HIV efficacy of
antibodies directed to an HIV-FRMS. The invention further provides methods to
monitor
FRMS vaccine efficacy in patients or subjects by assaying antibodies in a
sample of sera
derived from the patient or subject following vaccination The Therapeutics of
the
invention are preferably tested in vitro, and then in vivo for the desired
therapeutic or
prophylactic activity, prior to use in humans. Any in vitro or in vivo assay
known in the art
to measure viral infection or production can be used to test the efficacy of a
Therapeutic of
the invention.
In an embodiment of the invention, a method of screening a preparation
comprising a mAb for anti-HIV activity is provided, which assay comprises
assaying said
pr~~ation or fraction for the ability to inhibit HIV infection.
By way of example, to assay a Therapeutic in vitro, one can examine the
effect of the Therapeutic on HIV infection in cultured cells. Briefly,
cultured
hematopoietic cells (e.g., primary PBMCs, isolated macrophages, isolated CD4+
T cells or
cultured H9 human T cells) are acutely infected with HIV-1 using titers known
in the art to
acutely infect cells in vitro, such as 105 TCID~/ml. Then, appropriate amounts
of the
Therapeutic are added to the cell culture media. Cultures are assayed 3 and 10
days after
infection for HIV-1 production by measuring levels of HIV antigen using an
ELISA assay.
Reduction in HIV antigen levels over levels observed in untreated controls
indicates the
Therapeutic is effective for treatment of HIV infection. Alternatively, any
commercially
available HIV ELISA may be used as a comparative assay for pre- and post-
treatment with
the Therapeutic.
Additionally, assays for HIV-1 LTR driven transcription are useful for
testing the efficacy of Therapeutics of the invention. Specifically, a
reporter gene, i.e., a
gene the protein or RNA product of which is readily detected, such as, but not
limited to,
~e gene for chloramphenicol acetyltransferase (CAT), is cloned into a DNA
plasmid
construct such that the transcription of the reporter gene is driven by the
HIV-1 LTR
promoter. The resulting construct is then introduced by transfection, or any
other method
73
CA 02338983 2001-O1-30
WO 00/08043 PCT/US99/17487
- - . s
known in the art, into a cultured cell line, such as, but not limited to, the
human CD4+ T cell
line HUT 78. After exposure of the transformed cells to the Therapeutic,
transcription from
the HIV-1 LTR is determined by measurement of CAT activity using techniques
which are
routine in the art. Reduction in HIV-1 LTR driven transcription demonstrates
utility of the
Therapeutic for treatment and/or prevention of HN infection.
Exemplary tests in animal models are described in Section 5.7.1. herein.
The efficacy of Therapeutics of the invention can also be determined in SIV
infected rhesus monkeys (see, Letrin, N.L., et al., 1990, J. AIDS 3:1023-
1040), particularly
rhesus monkeys infected with SIV,",~s,, which SIV strain induces a syndrome in
experimentally infected monkeys which is very similar to human AIDS (Kestler,
H., et al.,
1990, Science 248:1109-1112). Specifically, monkeys can be infected with cell
free
SIV~ZS,, for example, with virus at a titer of 104'5 TCIDs~/ml. Infection is
monitored by the
appearance of SIV p27 antigen in PBMCs. Utility of the Therapeutic is
characterized by
normal weight gain, decrease in SIV titer in PBMCs and an increase in CD4+ T
cells.
Alternatively, a SHIV model is used in which an HIV env protein is constructed
into the
backbone of SIV. In one embodiment, monkeys are immunized using a FRMS vaccine
comprising cells expressing HIV env and cells expressing rhesus CD4 and CCRS
coreceptor. If adequate in vitro neutralization is obtained, then animals will
be challenged
with an infectious SHTV virus bearing a neutralization-sensitive HIV env.
Specifically,
monkeys are vaccinated with an immunogen comprising one or more FRMS and
challenged
with SHIV 89.6P primary isolate (see e.g., Montefiori et al., 1998, J Virol.
72:3427-31).
Once the Therapeutic has been tested in vitro, and also preferably in a non-
human animal model, the utility of the Therapeutic can be determined in human
subjects.
The efficacy of treatment with a Therapeutic can be assessed by measurement of
various
peters of HIV infection and HIV associated disease. Specifically, the change
in viral
load can be determined by quantitative assays for plasma HIV-1 RNA using
quantitative
RT-PCR (Van Gemen, B., et al., 1994, J. Virol. Methods 49:157-168; Chen, Y.H.,
et al.,
1992, AIDS 6:533-539) or by assays for viral production from isolated PBMCs.
Viral
production from PBMCs is determined by co-culturing PBMCs from the subject
with non-
infected PBL cells and subsequent measurement of HIV-1 titers using an ELISA
assay for
p24 antigen levels (see e.g.,Wrin, T., et al., 1995, Science 69:39; Popovic,
M., et al., 1984,
Science 204:497-500). Administration of the Therapeutic can also be evaluated
by
assessing changes in CD4+ T cell levels, body weight, or any other physical
condition
associated with HIV infection or AIDS or AmS Related Complex (ARC). Reduction
in
~ ~ml load or production, increase in CD4'" T cell or amelioration of HIV-
associated
symptoms demonstrates utility of a Therapeutic for administration in
treatment/prevention
of HIV infection.
74
CA 02338983 2001-O1-30
WO 00/08043 PCT/US99/17487
5.13. 1
The invention also concerns kits comprising the fusion-competent protein
complexes or antibodies generated by those complexes. Kits including.the
subject
complexes or antibodies can be used to analyze vaccine lots, conduct stability
studies or
develop vaccine release qualifications. Antibodies and the complexes can also
be used in
diagnostic kits to detect the presence of antibody or virus in the host
system. For example,
antibodies or the subject complexes can be used as substrate or reagent in a
kit providing a
standard diagnostic enzyme-linked immunosorbent assay (ELISA) or competitive
ELISA.
The invention also provides a pharmaceutical pack or kit comprising one or
more containers comprising one or more of the ingredients of the vaccine
formulations of
the invention. Associated with such containers) can be a notice in the form
prescribed by a
governmental agency regulating the manufacture, use or sale of pharmaceuticals
or
biological products, which notice reflects approval by the agency of
manufacture, use or
sale for human administration.
In one embodiment, the invention provides a kit comprising in one or more
containers a labeled monoclonal antibody to a molecular structure comprising
an epitope
formed as a result of association of (a) an envelope protein of an enveloped
virus, with (b)
one or more cellular membrane proteins, which envelope protein and cellular
membrane
proteins are necessary and sufficient under suitable conditions for fusion of
said envelope of
the virus with a cell membrane containing said cellular membrane proteins. In
a further
embodiment, the invention provides a kit which further comprises in a separate
container a
molecular structure comprising an epitope formed as a result of association of
(a) an
envelope protein of an enveloped virus, with {b) one or more cellular membrane
proteins,
which envelope protein and cellular membrane proteins are necessary and
sufficient under
suitable conditions for fusion of said envelope of the virus with a cell
membrane containing
said cellular membrane proteins.
In a specific embodiment, the invention provides a kit comprising in one or
more containers a labeled monoclonal antibody to a molecular structure
comprising an
epitope formed as a result of association of (a) an HIV envelope protein, or a
mutant thereof
fat assembles into the viral envelope; with (b) human CD4 and a co-receptor
for HN
fusion. In a further embodiment, the invention provides a kit which further
comprises in a
separate container an epitope formed as a result of association of (a) an HN
envelope
protein, or a mutant thereof that assembles into the viral envelope; with (b)
human CD4 and
a co-receptor for HIV fusion.
~ one embodiment, the invention provides a kit comprising in one or more
containers, a molecular structure comprising an epitope formed as a result of
association of
(a) an envelope protein of an enveloped virus, with (b) one or more cellular
membrane
CA 02338983 2001-O1-30
WO 00/08043 PCT/US99/17487
proteins, which envelope protein and cellular membrane proteins are necessary
and
sufficient under suitable conditions for fusion of said envelope of the virus
with a cell
membrane containing said cellular membrane proteins. In a preferred
embodiment, the
molecular structure is isolated. In a further embodiment, the invention
provides a kit which
further comprises a pharmaceutically acceptable carrier, and wherein said
molecular
structure is present in an immunological amount.
In another embodiment, the invention provides a kit comprising in one or
more containers (a) a first nucleic acid encoding an envelope protein of an
enveloped virus;
and (b) a second nucleic acid encoding one or more cellular membrane proteins,
which
I O envelope protein and cellular membrane proteins are necessary and
sufficient under suitable
conditions for fusion of said envelope of the virus with a cell membrane
containing said
cellular membrane proteins, such that the envelope protein and cellular
membrane proteins
are expressed in the subject and neutralizing antibodies to the virus are
produced.
6. EXAMPLES
The inventors of the present invention have made the surprising discovery
that the ability to neutralize PI viruses is related to the presentation of
functioning envelope
protein in active infection, as compared to the static, non-functioning
presentation of the
envelope protein in rgp120 vaccines.
As described herein, the HIV envelope protein orchestrates a complex series
of protein-protein interactions and structural changes that ultimately results
in fusion of the
virus and cell membranes, and infection of the cell. Upon binding to CD4, the
envelope
protein undergoes conformational change that facilitates subsequent
interaction with one of
several co-receptor molecules, predominantly the CC chemokine receptor 5
(CCRS) or the
CXC chemokine receptor 4 (CXCR4) (Berger, E.A., 1997, AIDS 11 (suppl A), S3;
Moore,
J.P., et al., 1997, Current Opinions in Immunology 9:551; Doranz, B.J., et
al., 1997,
Immunology Research 16:15). Without limitation as to mechanism, interaction
with either
co-receptor induces further conformational change in the envelope protein and
exposure of
the hydrophobic fusion domain of the transmembrane gp41 subunit, which then
mediates
Vision of the apposed cell and virus membranes. On the basis of this dynamic
model of HIV
binding and entry, HiV vaccine immunogens were developed by the methods of the
invention that explicitly incorporate these functional intermediate
structures.
The examples described herein serve to illustrate the methods of and
compositions of the invention.
76
CA 02338983 2001-O1-30
WO 00!08043 PCTNS99/17487
Example 1:Broad Primary Isolate Neutralization from HIV-FRMS Immunog_en
As one example of the invention, HIV vaccine immunogens were generated
that capture the transient structures that arise during HIV binding and
fusion. In a transgenic
mouse immunization model, formaldehyde-fixed whole cell vaccines elicited
antibodies
capable of neutralizing infectivity of 23 of 24 primary HIV isolates from
diverse geographic
locations and genetic Glades A-E. Development of these fusion-dependent
immunogens
provide clinically important and broadly effective HIV vaccines.
As described herein, the HIV envelope protein orchestrates a complex series
of protein-protein interactions and structural changes that ultimately results
in fusion of the
~~s ~d cell membranes, and infection, of the cell. Without limitation as to
mechanism,
upon binding to CD4, the envelope protein undergoes conformational change that
facilitates
subsequent interaction with one of several co-receptor molecules,
predominantly the CC
chemokine receptor 5 (CCRS) or the CXC chemokine receptor 4 (CXCR4) (Reviewed
in
Berger, E.A., 1997, AIDS 11(suppl A), S3; Moore, J.P., et al., 1997, Current
Opinions in
I~unology 9:551; Doranz, B.J., et al., 1997, Immunology Research 16:15).
Interaction
with either co-receptor induces further conformational change in the envelope
protein and
exposure of the hydrophobic fusion domain of the transmembrane gp41 subunit,
which then
mediates fusion of the apposed cell and virus membranes. The present example
illustrate
methods to develop HIV vaccine immunogens that explicitly incorporate these
functional
intermediate structures.
One measure of envelope protein function is the ability to mediate cell-cell
fusion. When cells expressing envelope protein are cocultured with cells
expressing CD4
and co-receptor, multinucleated syncytia form over the course of 6-24 hr. In
the instant
example, the process of binding and fusion was captured in progress by
formaldehyde-
crosslinking prior to extensive syncytium-formation.
Preparation of the FRMS
In these studies, the functioning envelope protein was derived from a T-
lymphocytropic PI virus obtained from the Amsterdam Cohort (ACH168.10; 168P).
The
molecularly cloned envelope gene of ACH168.10 was isolated by PCR using the
pCR3.1-
Uni plasmid (Invitrogen) {Tersmette, M., et al., 1989, Journal of Virology
63:2118; Wrin,
T., et al., 1995, Science 69:39; LaCasse, R.A., et al., 1998, Journal of
Virology 72:2491).
The molecularly cloned envelope protein, as well as the parental syncytium-
inducing(SI)
virus, utilizes both CCRS and CXCR4 co-receptors.
Cos-7 cells were transfected to express the envelope protein (COS-env) and
subsequently cocultured with human U87 glioma cells that express CD4 and CCRS
co-
receptor (LJ87-CD4-CCRS). The functional 168P (168P23) envelope gene was
transfected
77
CA 02338983 2001-O1-30
WO 00/08043 PCTNS99/17487
into COS-7 cells (American Type Culture Collection, Manassus, VA) by calcium
phosphate
precipitation (20 ~cg DNA/106 cells/10 cm culture dish) by methods known in
the art (see
e.g., Jordan, M., et al., 1996, Nucleic Acids Research 24:596). Transiently-
expressing COS-
7 cells were harvested 2 days later using 0.5 mM EDTA in phosphate-buffered
saline
(PBS), and U87-CD4-CCRS fusion partners (Hill, C.M., et al., 1997, Journal of
Virology
71:6296) were prepared using 0.1 mM EDTA in PBS. Cocultures were initiated by
mixing
the two cell types (1.5x106 cells each) in 10 cm culture dishes. The time
course of cell-cell
fusion was monitored microscopically and by immunocherriical staining (HMG) in
parallel
cocultures (LaCasse, R.A., et al., 1998, Science 72:2491). Cocultures were
typically
harvested by formaldehyde fixation at 4-5 hrs, when little or no overt
syncytium formation
was evident.
Capturing the FRMS
To capture transitional intermediates during the process of binding and
1 S Vision, cocultures were fixed in 0.2% formaldehyde after S hrs when few if
any
multinucleate cells were evident. Specifically, cultures were fixed in situ
using 0.2%
formaldehyde in PBS at 4° overnight by methods known in the art {see
e.g., Yamamoto,
J.K., et al., 1991, AIDS Research and Human Retroviruses 7:911; Verschoor,
E.J., et al.,
1995, Veterinary Immunology and Immunopathology 46:139). Cells were
subsequently
scraped, washed twice with PBS, resuspended at a nominal density of 3x106
cells/0.1 ml in
PBS containing 10% DMSO, and frozen at -80° for storage or used
immediately as
immunogens, as described herein below.
Immunization with a FRMS Immuno .den
The formaldehyde treated whole cell preparation was used as a fusion-
competent (FC) or FRMS immunogen. To test the ability of these immunogens to
elicit
neutralizing antibodies, it was necessary to restrict the immune response to
viral and virus-
induced epitopes. Otherwise, antibodies to CD4 and CCRS would be generated
that would
themselves block infectivity. Therefore, an animal model was used that was
l~~ologically tolerant to the human {hu) CD4 and CCRS components of the
vaccine.
Thus, immunogenicity studies were performed with transgenic mice that express
hu CD4
and hu CCRS co-receptor.
Construction of a CD4 targeted-deletion and hu CD4 transgenic mouse has
been described (Killeen, N., et al., 1993, EMBO Journal 12:1547). The design
of a hu
3$ CCRS transgenic mouse is summarized as follows: a 1.15 kb hu CCRS cDNA was
molecularly cloned into an engineered SaII site in exon 2 of a murine CD4
expression
cassette (construct c in (Sawada, et al., 1994, Cell 77:917). This minigene
contained the
78
CA 02338983 2001-O1-30
WO 00108043 PCT/US99/17487
marine CD4 enhancer, the CD4 promoter, the first (noncoding) exon, and intron
1 with an
internal deletion that eliminated the CD4 silencer. Transgenic founders were
identified by
flow cytometry using a mAb by methods known in the art. These animals were
bred to hu
CD4 expressing transgenic mice to yield progeny expressing hu CD4, hu CCRS,
and mouse
CD4. Pups were screened for expression of hu CD4, hu CCRS, and mouse CD4 by
flow
cytometry using a Coulter EPICS ELITE flow cytometer. The following antibody
reagents
were used: mouse a-human CD4/CyChrome (Pharmingen), mouse a-human CCRS MAB
180 (R&D Systems) with goat a-mouse Ig/FITC (Caltag), and rat a-mouse CD4
L3T4/PE.
Transgenic mice expressing hu CD4, hu CCRS, and mouse CD4 were
l~~zed with either FRMS immunogen or with cell controls (U87-CD4-CCRS cells,
alone or cocultured with mock-transfected COS cells). Vaccines comprised
formaldehyde-
fixed whole cells (3x106 cells/0.1 ml) formulated with an equal volume of Ribi
adjuvant (R-
700; reconstituted in half the recommended volume of PBS); in some
experiments, the
initial immunization was with adjuvant containing cell wall material (R-730).
Mice received
0'05 ml vaccine in four subcutaneous sites. Booster immunizations were at
three week
intervals, and mice were bled at 10-28 days post-immunizations from the tail.
Serum
antibodies directed to gp120 were quantitated by gp120 ELISA as per methods
known in
the art (see, Moore, J., et al., 1989, AIDS 3:155).
Virus Neutralization
Sensitivity of the homologous 168P virus to neutralization by vaccine sera
was determined with U87-CD4 cells expressing either CCRS or CXCR4 co-receptor.
This
PI virus neutralization assay has been validated relative to standard
neutralization assay in
PBL culture (LaCasse, R.A., et al., 1998, Science 72:2491; Follis, K.E., et
al., 1998, Journal
of Virology 72:7603) and was determined to perform well in the presence of
mouse serum.
All sera were heat-inactivated prior to use in neutralization assays.
Neutralization assay of the homologous 168P PI virus by fusion-competent
(FC) and fusion-incompetent (FI) vaccine sera was perfonmed as follows.
Transgenic mice
(hu CD4+, hu CCRS+, mouse CD4+) were immunized with FC immunogen (COS-env with
U87-CD4-CCRS; squares; n = 3 mice) or with cell controls (LJ87-CD4-CCRS cells
alone or
cocultured with mock-transfected COS cells; circles n = 3 mice). Unimmunized
mice were
also used (triangles; n = 2 mice). Sera were tested for neutralization of 168P
using U87-
CD4 cells expressing either CXCR4 (black symbols) or CCRS (white symbols).
Data
represent averages of three to six neutralization assays using serum obtained
2 weeks
following second and third immunization.
As indicated in Fig 1, results of virus neutralization assays indicated that
no
inhibition of infectivity was observed in sera from mice immunized with cell
controls,
79
CA 02338983 2001-O1-30
WO 00/08043 PCTNS99/17487
indicating that the transgenic mice were in fact tolerant to hu CD4 and CCRS
and that other
adventitious cellular reactivities did not interfere with the virus
infectivity assay (Fig 1).
Sera from mice immunized with FRMS immunogens neutralized the homologous 168P
PI
virus. Neutralization activity was further demonstrated to be antibody-
mediated and as such
activity could be adsorbed to, and subsequently eluted from, a solid support
containing
Protein-A and Protein-G . Specifically, serum was adsorbed sequentially to
Protein-A
Sepharose (Sigma) and Protein-G agarose (Sigma) at 4°. Adsorption of
antibody was
confirmed by gp120 ELISA. The solid supports were combined and antibodies were
eluted
using 100 mM glycine pH 2.5. The eluate was neutralized and dialyzed by
centrifugal
~10 ultrafiltration (Microcon-100; Amicon). Neutralization by FC sera in PBL
assay was
determined to be >99%, while neutralization by FC sera in U87-CD4-coR cell
assay was
determined to be 80-90%.
It is interesting to note that in the sensitive PBL neutralization assays
'fusion-incompetent' vaccine sera did demonstrate some inhibition of PI virus
replication,
albeit far less than 'fusion-competent' vaccine sera (see, LaCasse, RA, et
al.,1999, Science
283:357-62.) This minimal effect may account for intermittent claims in the
literature of PI
virus-neutralization by rgp120 vaccine sera (Devico, A, A Silver, et al.,
1996, Virology
218: 258-63;Berman, PW, et a1.,1998, AIDS Res & Hum Retrovirus 14 {suppl 3)
S277-
S89). The molecular basis for differences in neutralization sensitivity among
PI viruses is
u~own at this time, and may or may not correlate with phylogenetic Glade
groupings per
se. Serotypically-distinct determinants may arise independently in multiple
Glades. Thus,
'fusion-competent' envs derived from Glades other than B may be used to
determine
empirically the number of prototypic envs that define 'fusion-competent'
neutralization
serotypes. Regardless of the total number of HIV serotypes, it is significant
that 80% of PI
viruses tested could be strongly neutralized (>70%) using a single
representative but
appropriately presented Glade B env.
~XCR4 co-receptor may substitute for CCRS
Neutralization of the 168P virus by FC serum was observed regardless of the
co-receptor used in the U87-CD4 cell infection assay (Fig 1 ). Several reports
have
demonstrated that in general neutralization sensitivity is independent of
specific co-receptor
use (LaCasse, R.A., et al., 1998, Science 72:2491); Trkola, A. et al., 1998,
Journal of
Virology 72:1876; Mvntefiori, D.C., et al., 1998, Journal of Virology 72:3427;
Follis, K.E.,
et al., 1998, Science 72:7603).
~e fact that neutralization was observed herein with CXCR4, a co-receptor
to which the animal had not been exposed, argues that neutralization may not
directly target
CA 02338983 2001-O1-30
WO 00/08043 PCTNS99/17487
the CCRS component of the vaccine. Further, the CXCR4 co-receptor may
substitute for
CCRS.
Fusion-Incompetent Immunog_ens
The role of fusion-dependent determinants in the induction of PI virus
neutralization was examined. Fusion-incompetent (FI) immunogens - cocultures
that
express env but do not undergo cell-cell fusion. These include COS-env
cocultured with
U87 cells (no CD4 or CCRS co-receptor), COS-env cocultured with U87-CD4 cells,
and
COS-env cells to which soluble CD4 (sCD4) was complexed. Specifically,
envelope-
expressing cultures were incubated with, sCD4 (Berger, E.A., et al., 1988,
Proceedings of
the National Academy of Sciences USA 85:2357) (5 ~g/ml; 1 hr at 37°)
and subsequently
washed to remove unbound sCD4. All FI immunogens were fixed with formaldehyde
as
above. An additional FI immunogen comprised COS-env and U87-CD4-CCRS cells
that
were separately fixed with formaldehyde prior to mixing during the formulation
of the
vaccine.
In marked contrast to FC or FRMS immunogens, all FI immunogens were
unable to elicit significant neutralization of the homologous PI virus (Fig
2A). Specifically,
P168 was neutralized by FC, but not FI immunogens. As shown in Fig 2A,
transgenic mice
were immunized with FC immunogen (black squares; n = 4), FI immunogens (COS-
env
with U87 cells; gray circles n = 4; COS-env with U87-CD4 cells, gray diamonds,
n = 3;
COS-env with sCD4, white diamonds, n = 2; COS-env with U87-CD4-CCRS cells,
each
fixed separately prior to mixing for immunization, gray squares, n = 2), or
mock-transfected
cos cell immunogen (cocultured with U87-CD4-CCRS cells, white circles, n = 2).
Unimmunized mice (white triangles, n = 2) were also used. Neutralization was
independent
of specific co-receptor use and data here represent averages of three to six
neutralization
assays in U87-CD4-CXCR4 or -CCRS cells. In some cases, sera from all animals
within
each experimental group were pooled. These results are consistent with the
well-
documented failure of rgp120 vaccines to elicit PI virus neutralization. The
difference in
neutralization by FC and FI vaccine sera was also observed in assays utilizing
human
pnmary blood lymphocytes (PBLs) (Fig 2B).
Neutralization of the homologous 168P PI virus in human PBL culture by
FC, but not FI immunogens was also demonstrated. As shown in Fig 2B,
lymphocytes were
isolated, stimulated with phytohemagglutinin, and grown in the presence of
interleukin-2;
neutralization was determined as described herein. HIV p24 antigen was
determined after 5
days of culture by ELISA (Coulter Corporation) and values were normalized to
the virus
control (36 ng/ml). * indicates p24 antigen levels below the limit of
detection at the dilution
81
CA 02338983 2001-O1-30
WO 00/08043 PCT/US99/17487
used in the ELISA. Vaccine groups were as defned in Fig 2A, and sera were
pooled for this
assay.
Specificity of the FRMS immunogen
In order to exclude the possibility that FC vaccine sera inhibited viral
infectivity in a nonspecific manner, FC vaccine sera were shown not to inhibit
infectivity of
pseudotyped HIV virions bearing an amphotropic marine leukemia virus (MLV)
envelope
protein (Envelope-defective HIV NL4-3-Luc-R'E- provirus was pseudotyped using
amphotropic MLV envelope protein (Deng, H., et al., 1996, Nature 381:661).
Further, FC
vaccine sera were shown not to neutralize a primary isolate of the simian
immunodeficiency
virus SIVmac251. A primary isolate of SIVmac251 {Langlois, A.L., et al., 1998,
Journal of
Virology 72:6950) produced in rhesus PBLs was used {Fig 3).
As shown in Fig. 3, FC vaccine serum did not neutralize pseudotyped HIV
virions bearing amphotropic MLV envelope protein or primary SIVmac25l . HIV
bearing
an amphotropic MLV envelope protein {ampho MLV pseudotype) was used in
neutralization sensitivity using pooled FC and FI antisera determined in U87-
CD4-CXCR4
cells. Primary isolate SIVmac251 neutralization was determined in U87-CD4-CCRS
cells.
Symbols are as defined in Fig 2 FC immunogen (black squares) and FI immunogen
(Cos-
env + U87 cells, black circles).
Laboratory-adapted Isolate Neutralization
As with conventional rgp 120 vaccines, FI vaccines were able to elicit
neutralization of a related laboratory-adapted isolate of HIV, the T-cell line
adapted
derivative of 168P, 168C {described in Wrin, T., et al., 1995, Science 69:39;
LaCasse, R.A.,
et al., 1998, Science 72:2491). Neutralization sensitivity of the 168P PI
virus and its TCLA
derivative 168C were tested in U87-CD4-CXCR4 cells. Vaccine groups and symbols
are as
defined in Fig 2; sera were pooled for this assay. Results demonstrated in Fig
4, indicate
neutralization of TCLA 168C virus by FI vaccine sera. Neutralization titers of
the TCLA
168C virus were comparable among FC or FI vaccine sera, as were titers of anti-
gp120
antibodies, suggesting a similar degree of inherent immunogenicity among the
vaccines.
The failure of FI vaccines to elicit PI virus neutralization in the transgenic
mouse model highlights the specificity of the neutralization elicited by FC
vaccines. A
statistical comparison was performed on the data set comprising all
experimental animals
~d all virus neutralization assays. A simple model for virus-antibody binding
was used to
calculate a 'binding constant' K for each assay, and a mean K value was
determined for
each mouse. Analysis of variance with follow-up tests demonstrated a
significant difference
82
CA 02338983 2001-O1-30
WO 00/08043 PCT/US99/17487
in mean neutralization between FC and FI immunogens (p < 0,01 ). In all
experiments,
responses within experimental groups were consistent and unifonm.
Furthermore, the consistent failure of FI vaccine sera to inhibit PI virus
infectivity argues strongly that the immune response may not be directed to
adventitious
human cellular targets, such as those that confounded early studies of
inactivated SIV
vaccines (see, Cranage, M.P., et al., 1993, AIDS Research and Human
Retroviruses 9:13;
Putkonen, P., et al., 1993, Journal of Medical Primatology 22:100; Arthur,
L.O., et al.,
1992, Science 258:1935). Rather, the present invention recognizes that FC or
FRMS
immunogens present unique determinants that mediate neutralization of PI
viruses.
Primary Isolate Neutralization by FRMS Vaccinated Sera
A critical issue in HIV vaccine development centers on the ability of vaccine
antisera to neutralize a broad range of diverse PI viruses. In order to
demonstrate the
breadth of PI virus neutralization elicited by FRMS immunogens such as an FC
l~~ogen, we examined the sensitivity of a panel of representative PI viruses
from five
prevalent and geographically-diverse phylogenetic Glades. Infectious
proviruses
ACH320.2A.1.2 (320SI) and ACH320.2A.2.1 (320NSI) (Groenink, M., et al., 1991,
Journal
of Virology 65:1968; Guillon, C., et al., 1995, AIDS Research and Human
Retroviruses
11:1537) were obtained through the NIBSC (UK) AIDS Reagent Program). HIV89.6
(Collman, R., et al., 1992, Journal of Virology 66:7517), SHIV89.6, and
SHIV89.6P
(Reimann, K.A., et al., 1996, Journal of Virology 70:3198) were also used. Ali
other
primary isolates were obtained through the NIH AIDS Research and Reference
Reagent
Program and the UNAIDS Network for HIV -1 Isolation and Characterization. PI
viruses
were subjected to limited expansion in PHA-activated PBLs (Wrin, T., et al.,
1995, Science
69:39).
As depicted in Fig 5, FC sera elicited by a functioning Glade B envelope
protein were able to neutralize 23 of 24 PI viruses tested - monocytropic/NSI
and T-
lymphocytropic/SI viruses from North AmericalEurope (Glade B), Africa (Glades
A and D),
Thailand (Glades B and E), and India (Glade C). Vaccine groups and symbols are
as defined
in Fig 2; sera were pooled for this assay.
Despite the sequence diversity among these isolates, most were similarly
sensitive to neutralization by FC vaccine sera. One isolate (92RW008) failed
to attain >50%
neutralization and two others (93IN904 and 92UG024) showed limited
neutralization above
50%; these exceptions to the otherwise broad pattern of neutralization further
argue that the
FC immunogens of the invention may target primarily viral determinants.
FI sera were uniformly unable to neutralize these hetemlogous PI viruses.
The broad and uniform neutralization of diverse PI viruses by the FRMS
immunogens
83
CA 02338983 2001-O1-30
WO 00/08043 PCT/US99/17487
suggests that the critical determinants presented by FRMS immunogens (such as
FC
immunogens) are highly conserved, and may be intimately tied to the basic
functioning of
the envelope protein in binding and fusion.
Neutralizing Antibodies
In order to more precisely define the molecular target for PI virus
neutralization by FC vaccines, neutralizing antibodies were attempted to be
removed from
FC vaccine sera on incubation with envelope protein expressed on the surface
of transfected
COS cells.
Accordingly, formaldehyde-fixed COS cells expressing 168P envelope
protein were incubated with FC serum and the recovered serum was then tested
for PI virus
neutralization. FC vaccine serum was sequentially adsorbed four times with
approximately
l Ob formaldehyde-fixed COS cells expressing 168P envelope. Incubations were
for 1 hr at
4° with rocking. Controls included prebleed serum and formaldehyde-
fixed mock-
transfected COS cells. Adsorption of bulk anti-gp 120 antibody was monitored
by gp 120
ELISA. Final sera were tested for neutralization of HIV 168P using U87-CD4-
CXCR4
cells. Neutralization activity in FC vaccine serum was removed by incubation
with
envelope-expressing cells, but only minimally reduced by incubation with COS
cell
controls (Fig 6 and Fig 7). Specifically, FC vaccine serum was repeatedly
incubated with
formaldehyde-fixed COS-env or control COS cells and tested for residual
neutralization of
168P using U87-CD4-CXCR4 cells. Serum obtained prior to FC immunization was
similarly adsorbed.
Production of Neutralizing Antibodies
2$ In order to demonstrate that the methods of the invention are useful in
generating neutralizing antibodies, hybridoma supernatants were made by the
methods of
the invention (using cross-linked COS-env + U87-CD4-CCRS as an immunogen) were
capable of neutralizing primary isolates. Briefly, hybridomas assayed for
virus
neutralization in the U87-CD4-CXCR4 cell assay as described above. Hybridoma
supernatants were tested for the ability to neutralize two representative HIV
Glade B isolates
(ACH168.10 (168P) and 92US657). Neutralization is expressed as the number of
foci in
the presence of hybridoma supernatant relative to the number in the presence
of medium
alone. Each point represents a selected hybridoma. As shown in Fig. 8, a
majority of
hybridomas were found to neutralize both PI viruses to 60-80% (0.4 - 0.2
fraction
l~ectivity remaining, respectively).
Thus, the FRMS immunogens of the invention are capable of eliciting
neutralizing monoclonal antibodies against primary isolate virus. Taken
together, this
84
CA 02338983 2001-O1-30
WO 00/08043 PCTNS99/17487
example illustrates that an appropriately-presented Glade B envelope protein
can elicit
potent neutralization against most PI viruses from multiple HIV Glades and
suggest that
broad vaccine protection may not require an unlimited number HIV serotypes.
Further, although the static forms of the envelope protein does not function
as an effective immunogen, the present invention recognizes that the critical
fusion-related
epitopes are sufficiently represented on the static protein to allow binding.
When used in
vaccine formulations, the FRMS immunogens of the invention provide broad
protection
against a wide variety of viral serotypes and Glades.
Example 2: Fusion-Defective Mutations .
A variety of mutations have been examined for use in the methods of the
invention.
In the case of HIV, three classes of mutations of the envelope protein
include, but are not limited to: (1) mutations that abrogate proteolytic
cleavage of the
1S X160 precursor protein; (2) mutations that affect the N-terminal gp41
fusion peptide
domain; and (3) mutations that alter the coiled-coil domain.
Mutations that abrogated proteolytic cleavage of the gp160 precursor
proteins include certain mutations that altered the highly conserved K/R-X-K/R-
R site at the
C-terminus of gp 120 prevent proteolytic cleavage of the gp 160 polyprotein
and also
abrogate fusogenicity (Freed, E.O., et al., 1989, J. Virol. 63:4670-S; Guo
H.G., et al., 1990,
Virology, 174:217-24; Bosch V., et al., 1990, J. Virol. 1990:2337-44; Dubay,
J.W., et al.,
1995, J. Virol., 69:4675-82).
Site-directed mutagenesis (QuikChange, Stratagene) was used to introduce a
documented cleavage site mutation into the 168P envelope protein (REKR to
REKT). The
2S cleavage-defective 168P envelope protein gp 160 was expressed on the cell
surface but was
unable to mediate cell-cell fusion or infection by pseudotyped HIV virions.
Mutations that affect the N-terminal gp41 fusion peptide domain include
mutation of the hydrophobic N-terminal region of gp41 which region is believed
to mediate
fusion by inserting into the cell membrane and destablizing the lipid bilayer.
Certain amino
acids changes within this region render the envelope protein nonfusogenics
synthetic
peptides.
Mutations in the N-terminal fusion peptide of gp41 have been well
characterized: V2E (Freed, E.O., et al., 1992, Proc. Natl. Acad. Sci. USA
1992:70-4;
Pereira, F.B., et al., 1997, AIDS Res. & Hum Retrovirus 13:1203-11 ) and G10V
3S ~elahunty, M.D., et al., 1996, Virology 218-94-102). The former involves a
polar
substitution in the hydrophobic peptide, whereas the latter may affect the
helical structure
assumed in the lipid bilayer. These mutations are introduced into the 168P
envelope gene
8S
CA 02338983 2001-O1-30
WO 00/08043 PCT/US99/17487
(gp41: AVGIGVLFLC'~rFLG..) by site-directed mutagenesis and the envelope
proteins is
tested for expression are fusogenicity. Incorporation of these mutations into
pAbT4587-
168Penv allows facile generation of the cognate recombination vaccinia viruses
of the
invention.
Mutations that alter the coiled-coil regions include mutations that are within
the highly conserved coiled-coil motif is found in fusion proteins of many
virus families
(Weissenhorn, W., et al., 1998, Proc. Natl. Acad. Sci. USA 95:6032-6), for
example,
V570R and Y586E of HIV. In one embodiment, changes are introduced into the
168P
envelope gene and tested for expression and fusogenicity in cell assays before
being
~sferred to pAbT4587-168Penv for generation of the cognate recombinant
vaccinia virus.
Example 3 -Pre~,aration c~f FRMS
Fusion-competent immunogens were prepared by co-cultivating calcium
phosphate-transfected COS-7 (American Type Culture Collection, Rockville, MD)
cells
expressing the molecularly cloned HIV I68P envelope protein (from ACH168.10 a
syncytium-inducing primary isolate that utilized both CCRS and CXCR4)
{LaCasse, R.A.,
et al., 1998, Science 72:2491) with an equal number of cell fusion partners.
The cell fusion
partners were U87-CD4 cells expressing CCRS (U87-CD4-CCRS) (Dan Littman,
Skirball
Institute of Biomolecular Medicine, NYU Medical Center). Cultures were
terminated at the
onset of cell-cell interaction (3-5 hr) by fixation with ice-cold 0.2%
formaldehyde in
phosphate-buffered saline. The harvest was timed towards capturing
transitional
intermediates leading to cell-cell fusion. Preliminary and contemporaneous
immunostaining studies indicated 10-30% of maximal syncytium formation at the
time of
harvest. Cells were collected by scraping following overnight fixation.
Example 4-Immunization of mice with the FRMS
Transgenic mice expressing both human CD4 and CCRS co-receptor (Dan
Littman, Skirball Institute of Biomolecular Medicine, NYU Medical Center) were
immunized three times at 21 day intervals with cells prepared and collected as
in Example
I ~ Cells were formulated as vaccine using Ribi Adjuvant (Ribi ImmunoChem
Research,
Inc.) {2X106 cells/ 0.2 ml/mouse). Blood was collected from the tail vein.
Sera obtained
were used in neutralization assays in U87-CD4 cell lines expressing either.
CCRS or
CXCR4. This rapid PI virus neutralization assay has been validated relative to
a standard
neutralization assay in peripheral blood lymphocyte (PBL) culture (LaCasse,
R.A., et al.,
1998, Science 72:2491 ), and performs well in the presence of mouse serum. In
three
separate experiments, fusion-competent vaccines were administered to three
mice. Virus
neutralization assays using the homologous PI virus 168P were performed using
both
86
CA 02338983 2001-O1-30
WO 00/08043 PCT/US99/17487
U87-CD4-CCRS and -CXCR4 cells, and in some cases sera were analyzed after the
second
as well as the third immunization. In all cases, results were concordant and
are averaged in
the data presented in Fig 9. Potent neutralization of the homologous 168P PI
virus was
consistently observed; neutralization levels of Z 80% were typically obtained
at a 1:100
dilution of mouse serum. Neutralization was observed using CXCR4, a co-
receptor to
which the animal had not been exposed.
Several critical control immunizations were performed. One fundamental
control was to confirm that non-functioning envelope protein immunogens did
not elicit PI
virus neutralizing antibodies in the transgenic mouse vaccination model. In
one study, one
mouse received vaccine comprising 168P envelope/COS cells that had been
cocultured with
U87 cells expressing neither CD4 or co-receptor. Although some neutralization
of 168P PI
virus was observed in a total of three assays from two bleeds, the titer and
extent were quite
distinct from that observed using fusion-competent vaccines.
Conformational changes induced upon CD4 binding are believed to be
critical for subsequent interaction with co-receptor. Therefore, antibodies
directed to these
novel envelope proteins are advantageous for PI virus neutralization. In
parallel with the
envelope protein control vaccine, another mouse received control vaccine
comprising COS
cells expressing 168P envelope that had been cocultured with U87-CD4 cells. No
syncytium formation was observed using this fusion-incompetent system. It
appears that
~e addition of CD4 contributed little to the very minimal neutralization
obtained using
envelope protein alone.
Residual neutralization might arise from antibodies directed to CD4 and
CCRS were tolerance to the transgenes to be incomplete. Thus, control vaccines
comprising U87-CD4-CCRS cells, alone or cocultured with mock-transfected COS
cell,
were tested. In three studies, no inhibition of viral infectivity was
observed. These data
confirm tolerance in these transgenic mice and validate the vaccination model.
Specific
immune responses appear limited to viral and perhaps virus-induced epitopes.
Not only do
these mice appear tolerant to human CD4 and CCRS, as would human vaccinees,
but
antibodies directed to other cellular proteins do not appear to interfere with
viral infectivity
in this assay.
The possibility that the antibody response might be in part mediated by DNA
immunization was explored. During transfection, envelope-expressing cells were
exposed
to microgram quantities of plasmid DNA. To test whether animals were
responding to DNA
immunization, two mice were injected subcutaneously with 20 ug 168P23 plasmid
(in the
pCW 1-Uni expression vector). Although this route of administration is not
preferred for
DNA immunization, it does mirror that used with transfected cellular vaccines.
The DNA
87
CA 02338983 2001-O1-30
WO 00/08043 PCT/US99/17487
was not formalin-inactivated in this experiment. In a total of 8 virus
neutralization assays,
no PI virus neutralization was observed upon DNA immunization.
Example 5-Neutralization of Additional PI Using ~tibodies to the FRMS
ACH320.2A.1.2 (320SI) (Amsterdam Cohort) is a molecularly cloned
T-lymphocytropic PI virus which is particularly refractory to neutralization
by HIV-IG,
CD4-Ig, and IgG,-b12. Residual sera remaining in Example 3 were used to test
the
sensitivity of this PI virus to fusion-competent and fusion-incompetent
immunogens.
Although limited to a starting 1:100 dilution of fusion-competent serum, this
heterologous
clue B isolate was clearly sensitive to neutralization. Of interest also was
the failure of the
fusion-incompetent sera (env and env + CD4) to neutralize the heterologous
320SI virus, in
contrast to the partial neutralization seen against the homologous 168P virus.
(Fig. 10)
A critical issue in HIV vaccine development centers on the ability of vaccine
antisera to neutralize a broad range of diverse PI viruses. To determine the
breadth of PI
virus neutralization elicited by fusion-competent immunogens, the sensitivity
of a panel of
representative PI viruses was examined from five prevalent and geographically-
diverse
phylogenetic Glades. Neutralization assays in U87-CD4-co-receptor cells are as
described
above and in Example 1. Fusion-competent sera elicited by a functioning Glade
B envelope
protein were able to neutralize 23 of 24 PI viruses tested-monocytropic/NSI
and
T'l~phocytropic/SI viruses from North America/Europe (Glade B), Africa (Glades
A and
D), Thailand (Glades B and E), and India (Glade C). Despite the sequence
diversity among
these isolates, most were similarly sensitive to neutralization by fusion-
competent vaccine
sera. One isolate (92RW008) failed to attain >50% neutralization and two
others (931N904
and 92UG024) showed limited neutralization above 50%. Control sera were
uniformly
unable to neutralize these heterologous PI viruses, in keeping with the
historic failure of
rgp 120 immunogens. The broad and uniform neutralization of diverse PI viruses
elicited by
a single representative, but appropriately presented, Glade B envelope protein
suggests that
the critical determinants presented by fusion-competent immunogens are highly
conserved,
and may be intimately tied to the basic functioning of the envelope protein in
binding and
Vision. These findings suggest that the number of different HIV neutralization
serotypes
needed for worldwide protection against HIV infection may be limited.
Example 6-Preparation and isolation of tag end FRMS
The FRMS of the subject invention were tagged using a commercially
available system to facilitate their isolation and purification. The molecular
engineering
methods used to construct S-peptide tagged CD4, CCRS, CXCR4 and HIV envelope
are
parallel and will be described in detail only for CD4-Spep. Molecules were
tagged with an
88
CA 02338983 2001-O1-30
WO 00/08043 PCT/US99/17487
S-peptide at the C-terminal end of the CD4 molecule. The S peptide-encoding
sequence
(lys-glu-thr-ala-ala-ala-lys- phe-glu-arg-gln-his-met-asp-ser) was inserted
between the
cytoplasmic C-terminal isoleucine and termination codon (TGA) of the CD4 cDNA
expression plasmid (CD4-Spep) using a synthetic oligonucleotide and high
fidelity XL PCR
(PE Applied Biosystems). Functional expression of the modified CD4 was
confirmed by
infection of COS-7 cells expressing CD4-Spep and the CXCR4 co-receptor. The
C-terminally tagged CD4 was able to support infection by 168P virus comparable
to native
CD4. S-peptide tagged CD4 was also readily isolated using S-protein affinity
chromatography (Novagen, Inc.). Using similar methods, we have constructed
homologous
S-peptide tagged CCRS and CXCR4 co-receptor and HIV envelope molecules. All
constructs were shown to be functional in binding and fusion. COS-7 cells were
transfected
with CXCR4 and either CD4-Spep or native CD4 (in pcDNA3.1 expression vectors),
and 2
days subsequently labeled at the cell surface with biotin (EZ-Link Sulfo-NHS-
LC-Biotin;
Pierce Chemical Co.). Lysates were prepared in 0.5% Triton X-100 with protease
l~ibitors. CD4 molecules were purified either by 1 ) immunoprecipitation using
MAb T4
(Coulter Corp., Hialeah, FL) and rabbit-a-mouse Ig antibody-loaded Protein A
agarose, or
2) affinity purification using S-pmtein agarose. Proteins were released by
boiling in sodium
dodecyl sulfate (SDS) sample buffer, resolved by 10% polyacrylamide-SDS gel
electrophoresis, and transferred to nitrocellulose. Biotinylated proteins were
detected by
avidin-horseradish peroxidase (Pierce) and nickel-enhanced DAB substrate. S-
protein
agarose resulted in the specific purification of the 62kD CD4-Spep with yield
and purity
which surpassed that of a-CD4-antibody mediated immunoprecipitation.
The time course of S-peptide isolation following formalin crosslinking was
examined. COS-7 cells were transfected with CD4-Spep (f CXCR4) and were
subsequently harvested in 0.5% Triton X-100 lysis buffer (control), or
subjected to ice-cold
formaldehyde fixation prior to lysis. CD4-Spep pmtein was purified by S-
protein agarose,
and analyzed by Western blot using S-protein HRP (Novagen, Inc.). Minimal loss
of
S-peptide binding (and/or recovery) was seen with 0.2% formaldehyde fixation
for 1 hr;
some recovery was lost with 2% formaldehyde in 1 hr and more at 3 hr.
Intermediate
reaction conditions (eg. 0.2% formaldehyde for 1-2 hr) will allow for
efficient isolation of
effectively cross-linked complexes.
To identify specific CD4-Spep complexes, experiments were performed
using formaldehyde fixation as well as the specific cross-linking agent DTSSP
(Pierce
Chemical Company). DTSSP is a homobifimctional N-hydroxysuccinimidyl ester
that
reacts, as formaldehyde, with primary amines. Because of its negative charge,
DTSSP
cannot penetrate or cross the cell membrane and thus, in contrast to
formaldehyde, is
expected to spare the cytoplasmic S-peptide tag. Also unlike formaldehyde,
DTSSP
89
CA 02338983 2001-O1-30
WO 00/08043 PCT/US99/17487
cross-linking is reversible; an internal disulfide linkage can be cleaved to
release
cross-linked components.
COS-7 cells were transfected with 168P envelope or CD4-Spep plasmids;
envelope expressing cells as well as mock transfected cells were metabolically
labeled in a
cm dish for 8 hr using 1 mCi total 35S-methionine and cysteine. Envelope (or
mock) and
CD4-Spep expressing cells were then co-cultured for 4 hr to allow cell-cell
interaction, and
subjected to formaldehyde (0.2%, 1 hr on ice) or DTSSP (2 mM, 2X10 min on ice)
cross-linking. Lysates were prepared and S-peptide containing complexes were
isolated by
S-protein agarose affinity chromatography.
10 Formaldehyde cross-linked complexes were analyzed by 6% polyacrylamide
SDS gel electrophoresis. A high molecular weight complex that is not found in
the mock +
CD4-Spep control was identified. Viral proteins gp120 and gp160 were also
visible,
presumably isolated through non-covalent association with CD4-Spep. The
detection of
S-protein affinity-purified (i.e., CD4-Spep) and metabolically-labeled (i.e.,
envelope) high
molecular weight complexes suggests that cross-linked complexes can be
isolated for
further study.
To continue the analysis of these high molecular weight complexes, DTSSP
cross-linked complexes were examined. S-protein affinity purified complexes
resolved by
6% polyacrylamide/SDS gel electrophoresis with or without prior treatment with
50 mM
D~ were analyzed. Envelope-specific complexes were unable to be clearly
resolved above
those isolated from the mock + CD4-Spep control, with DTSSP cross-links
intact. Little
gp120/gp160 was detected. However, reversal of DTSSP cross-linking results in
the
specific release of gp120 and gp160 proteins. These data confirm that the
formation of
envelope-CD4 complexes observed using formaldehyde fixation, and highlight the
power in
the use of reversible cross-linking agents.
These data support the utility of the CD4-Spep affinity tag and the chemical
cross-linking methodologies in the isolation of fusion-active CD4-associated
complexes.
S-peptide tagged co-receptors CCRS (CCRS-Spep), CXCR4 (CXCR4-Spep), viral
envelope
(Env-Spep) have been produced and can also be used to isolate the fusion-
active complexes.
Isolated complexes can be used to define the molecular structure associated
with the
progression of envelope-mediated membrane fusion.
Studies were performed to determine whether envelope protein can be
co-purified with either CD4-Spep or CCRS-Spep. Human 293T cells were
separately
transfected with 168P envelope protein, and a) CD4-Spep, or b) CCRS-Spep and
CD4, or c)
mock. envelope-expressing cells were co-cultured with fusion partners (a-c)
for 5 hr and
complexes were solublized using 1 % Brij-97 detergent (Lapham et aL, 1996) and
isolated
using S-protein affinity chromatography. Co-purified envelope protein isolated
using
CA 02338983 2001-O1-30
WO 00/0$043 PCT/US99/17487
Brij-97 was detected by Western blot analysis using the HIV V3-directed mAb
50.1 (Fig
11). As anticipated, a large amount of 168P envelope protein was isolable
following
co-culture with cells expressing CD4-Spep. A lesser, but significant, amount
of envelope
protein was also isolable following co-culture with cells expressing CD4 and
CCRS-Spep.
Contaminating envelope protein was not detectable from co-cultures with mock-
transfected
cells. Complexes were isolated from formaldehyde cross-linked fusion-competent
co-cultures in similar experiments.
These results strongly suggest that complexes of envelope protein, CD4, and
CCRS arise early during fusion and can be isolated by S-protein affinity
chromatography.
These S-protein purified molecules and complexes can be used as "split virus"
or subunit
vaccine immunogens.
Example 7: Recombinant Vaccinia Viruses That Generate Fusion-Competent FRMS
in Cell Culture or at the Site of Vaccination.
1 S Other acceptable vaccine approaches to translate our invention into a
practical formulation include the use of viral vectors. For instance,
coadministration of
recombinant vaccinia viruses that express env and CD4/CCRS, respectively,
might drive
cell-cell fusion in situ. In one embodiment, two recombinant vaccinia viruses-
one
expressing HIV 168P envelope protein (rV-168Penv) and the second expressing
CD4 and
CCRS co-receptor (rV-CD4/CCRS) were constructed. Co-cultures of cells infected
with
rV-168Penv and cells infected with rV-CD4/CCRS were fusion-competent and yield
multinucleate syncytia (Fig. 12). rV-168Penv cells, rV-CD4/CCRS cells, or
cross-linked
cocultured rV-168Penv and rV-CD4/CCRS cells were used as FRMS to vaccinate
transgenic mice, as described above.
Sera obtained from mice are vaccinated with cross-linked cocultured
rV-168Penv and rV-CD4/CCRS cells are capable of virus neutralization across a
variety of
Glades of HN indicating that the viral vectors may successfully be used in the
methods of
the invention in the formation of the FRMS.
It should be understood that the examples and embodiments described herein
are for illustrative purposes only and that various modification or changes in
light thereof
will be suggested to persons skilled in the art and are to be included within
the spirit and
purview of this application and the scope of the appended claims.
The present invention is not to be limited in scope by the specific
embodiments described herein. Indeed, various modifications of the invention
in addition
to those described herein will become apparent to those skilled in the art
from the foregoing
91
CA 02338983 2001-O1-30
WO 00/08043 PCT/IJS99/17487
description and accompanying drawings. Such modifications are intended to fall
within the
scope of the appended claims.
Various references are cited herein above, including patent applications,
patents, and publications, the disclosures of which are hereby incorporated by
reference in
their entireties.
1S
25
35
92