Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02345923 2001-03-22
WO 99/16898 PCT/CA98/00923
TITLE OF THE INVENTION
MOLECULAR DIAGNOSTIC OF GLAUCOMAS
ASSOCIATED WITH CHROMOSOME 1, AND METHOD OF TREATMENT
THEREOF
FIELD OF THE INVENTION
The present invention relates to the identification of mutations
in the TIGRlMYOC gene in the GLCIA locus and the detection of these
mutations in individuals. The invention also relates to the identification of
individuals who are genotypically homozygote mutants of an autosomal
dominant inherited disease and yet display a normal phenotype.
BACKGROUND OF THE INVENTION
Glaucoma encompasses a complex of ocular-disease entities
characterized by an optic neuropathy in which degeneration of retinal ganglion
cells leads to a characteristic excavation of the head of the optic nerve
(Shields
et al., 1996, The Glaucomas, 2:717-725). Such damage causes progressive
narrowing of the visual fields and, when uncontrolled, blindness. Affected
people
often have ocular hypertension defined as intraocular pressures consistently
>21
mm Hg in both eyes. Although ocular hypertension is no longer an obligatory
diagnostic criterion for glaucoma, it is still recognized as one of the most
important risk factors (Wilson et al., 1996, The Glaucomas, 2_:753-763). Until
now, a diagnosis of glaucoma is made after observation of the characteristic
atrophy of the optic nerve, which is associated with typical visual field
defects.
In 1992, the World Health Organization estimated that, in the
global population, 5.2 million people were blind as a result of glaucoma
(Thylefors et al., 1994, World Health Organ. Bull., 72:323-326), making it the
third leading cause of blindness worldwide. The most common form is
adult-onset primary open-angle glaucoma (MIM 137760; McKusick, 1994. Johns
Hopkins University Press, p. 272), which represents ~50% of all cases of
CA 02345923 2001-03-22
WO 99/16898 PCT/CA98/00923
2
glaucoma. Among Caucasians, this form of the disorder affects ~2% of the
population >45 years old (Leske, 1983, Am. Epidemiol., 118:166-191; Thylefors
et al., 1994, supra; Wilson et al., 1996, supra). In African Americans,
prevalence
of adult-onset open-angle glaucoma is three to four times higher than that
5 observed in White Americans. More than 5 million North Americans may have
some form of glaucoma, but at least half of them may not be aware of it.
The glaucomas traditionally have been grouped into three
categories: open angle, closed angle (also termed "angle closure"), and
congenital. Each subtype has been further arbitrarily subdivided into primary,
10 when the anterior chamber of the eye appears normal and no cause for
glaucoma can be identified, or secondary, when glaucomas are caused by
underlying ocular or systemic conditions (Shields et al., 1996, supra).
Whereas
the division between open and closed angles refers to the configuration of the
irido-corneal angle in the anterior chamber of the eye, congenital glaucoma is
15 used to define one of the many types of developmental glaucoma that usually
occurs within the 1 st year of life. The majority (60%-70%) of primary
glaucomas
are of the open-angle type. Primary open-angle glaucomas have been further
subdivided into two groups according to age at onset, severity, and mode of
inheritance: the more prevalent is middle- to late-age-onset chronic open-
angle
20 glaucoma (COAL), by convention diagnosed after age 35 years and
characterized by its slow, insidious course (Shields et al., 1996, supra;
Wilson
et al., 1996, supra). The less common form, juvenile open-angle glaucoma
(JOAG), occurs between 3 years of age and early adulthood and generally
manifests highly elevated intraocular pressures with no angle abnormalities
25 (Goldwyn et al., 1970, Arch. Ophtalmol., 84:579-582; Fran~ois, 1980, Am. J.
Ophtalmol., 3:429-449; Johnson et al., 1996a, The Glaucomas, 1_:39-54).
Although the precise molecular defects leading to open-angle
glaucomas remain partly unknown, numerous advances in basic and clinical
sciences have begun to identify the molecular basis of glaucomas by mapping
30 the gene loci involved in the disease process. Due to recent mapping
CA 02345923 2001-03-22
WO 99/16898 PCT/CA98/00923
3
successes, the different forms of glaucoma will be further identified by the
names of the loci to which they have been localized. According to the Human
Genome Organization/Genome Database nomenclature, "GLC" is the general
symbol for the glaucoma genes; "1", "2", and "3" are, respectively, the
symbols
5 for the open-angle, angle-closure, and congenital subtypes of glaucoma; and
"A", "B", and "C" refer, respectively, to the first, second, or third gene
mapped
in each subgroup.
JOAG is a rare but aggressive form of glaucoma that usually
segregates in an autosomal dominant fashion with high penetrance (Stokes,
10 1940, Arch. Ophthalmol., 24:885-909; Crombie et al., 1964, Br. J.
Ophtalmol.,
48:143-147; Lee et al., 1985, Ann. Ophtalmol., 17:739-741; Johnson et al.,
1993, Ophthalmology, 100:524-529). In a single large American pedigree
affected by an autosomal dominant form of JOAG, Sheffield et al. (1993, Nat.
Genet., 4:47-50) located a gene responsible for this condition, at 1q21-q31.
This
15 locus, being the first open-angle glaucoma locus to be mapped, was named
"GLC1A." The TIGRlMYOC glaucoma disease gene consistently was first
associated with onset of the glaucoma phenotype before the age of 45 years,
highly elevated intraocular pressures, and typical excavation of the head of
the
optic nerve. Gonioscopy showed open angles with no anterior-chamber
20 abnormalities. The GLC9A locus has subsequently been reported by Nguyen et
al. in US Patent 5,606,043 to encode the trabecular meshwork induced
gfucocorticoid response (TIGR) gene. The gene sequence was first submitted
{13-JAN-1997) by Nguyen et al. to the GeneBank accession # 085257. The
TIGR sequence was modified on 19 April 1997 in GeneBank following
25 modifications by Nguyen submitted on 02-APR-1997. The accession number
stayed the same # 085257.
Genetic maps of the human genome can be exploited to
rapidly locate human monogenic disorders The final version of the Genethon
linkage map, which spans close to 100 % of the human genome, was published
30 in March 1996 (Dib et al., 1996, Nature 380 152-154). This map consists of
CA 02345923 2001-03-22
WO 99/16$98 PCT/CA98/00923
4
5,264 short tandem (ACITG)n repeat polymorphisms with a mean
heterozygosity of 70%.
The nomenclature system for the markers is well known in the
field. The nomenclature used is decided by the Human Genome Organization
5 (HUGO) nomenclature committee. It is as follows: for anonymous DNA
sequences, the convention is to use D which is equivalent to DNA followed by
1-22, X or Y to denote the chromosomal number and location, then S stands for
a unique segment and finally a serial number. For example, marker D2S2161
is a DNA marker located on chromosome 2 representing a unique segment. Its
10 serial number is 2161.
The nomenclature for the glaucoma genes is the following:
"GLC" is the general symbol for the glaucoma genes; "1 ", "2", and "3" are,
respectively, the symbols for the open-angle, angle-closure, and congenital
subtypes of glaucoma; and, "A", "B" and "C" refer, respectively, to the first,
15 second, or third gene mapped in each subgroup. For example, the GLC1A locus
was the first open-angle glaucoma locus to be mapped, in this case to
chromosome 1 q23-q25 in 1993. It was later identified as the trabecular
meshwork inducible giucocorticoid response gene product (TIGR) (Stone et al,
1997, Science, 275: 668-670). The TIGR gene is also known as MYOC.
20 These markers are accessible to all individuals. The central
data resource for the human gene mapping effort is the Genome Data Base
(GDB). It was established at Johns Hopkins University, School of Medicine. GDB
is updated regularly. It collects, organizes, stores and distributes human
genome
mapping information. GDB ~s accessible electronically at WWW-URL:
25 http://gdbwww.gdb.org/.
Alternatively, all the markers disclosed herein, except
D6S967, are short (CA)n repeat markers that have been developed in the
Genethon laboratory near Paris, France. These markers are also accessible
electronically at WWW-URL http://www.genethon.frl.
CA 02345923 2001-03-22
WO 99/16898 PCT/CA98/00923
5
Therefore markers are accessible either at GDB or at
Genethon.
The first mutations identified in the TIGR gene that have been
shown to give rise to glaucoma were first reported by Stone et. al (Science,
5 1997, 275:668-670). Twenty-eight mutations have now been reported. The
methodology used to identify these mutations was by amplifying overlapping
regions by polymerase chain reaction (PCR), performing single-strand
conformational polymorphism (SSCP) on the amplification products and
sequencing those DNA products that produced aberrant band pattern on the
10 SSCP. No quick method for mutational analyses for the TIGR has been
proposed.
The prior art as a whole teaches that a homozygote mutant
for an autosomal dominant disease should display a higher penetrance and
severity for a human disorder than a heterozygote mutant. Heterozygotes for an
15 autosomal dominant disease often exhibit variable penetrance.
The present description refers to a number of documents the
content of which is herein incorporated by reference.
SUMMARY OF THE INVENTfON
20 The invention concerns the mutational analyses in the GLC1A
gene locus encoding the TIGR gene (GeneBank accession no. U85257; SEQ
ID N0:1).
The present invention provides means to identify at least two
nucleotide changes in the DNA sequence coding for TIGR that result in an
25 amino acid change in the TIGR gene.
The invention further demonstrates that these amino acid
changes result in mutations producing a disease state in individuals, the
disease
being glaucoma.
The early detection of individuals at risk for developing
30 glaucoma is an important aspect of this invention. Early detection allows
for
CA 02345923 2001-03-22
WO 99/16898 PCT/CA98/00923
6
intervention prior to the genesis of the disease process and disease
progression
and may obviate the symptoms and the onset of the disease.
A method for mutation analyses called amplification refractory
mutation system (ARMS), that is simple, quick and adapted to glaucoma is
disclosed herein. The proposed invention relates to the inclusion of primers
and
probes for the amplification and detection of all mutations in the TIGR gene.
Although the invention teaches the use of the method of ARMS for glaucoma,
the invention is not so limited. Other methods of mutation analyses known in
the
field, such as allele specific oligonucleotide (ASO), denaturing gradient gel
electrophoresis (DGGE) and artificially created restriction site (ACRS), can
also
be used.
The mutation detection and analyses thereof can be
performed on either genomic DNA or cDNA by any method known to a person
skilled in the art.
In addition the applicant has demonstrated for the first time
a new type of dominance in mammals in which heterozygotes have a much
higher penetrance rate for a disease gene mutation than their homozygotic
counterparts.
Further, the present invention provides for the first time the
identification in an autosomal dominant disease, of a homozygote mutant which
is phenotypically normal, even though such an individual may give rise to an
affected heterozygote offspring. This homoallelic complementation
phenomenon has now been observed with 4 different patients.
The invention provides applications and uses for such a
discovery. These include but are not limited to:
a) treatment of a heterozygote mutant (affected individuals
with overexpressed mutant protein to induce protein complementation such that
normal protein function can be restored, this application will apply to any
autosomal dominant disease exhibiting the same mode of action as described
herein (i.e. homoallelic complementation).
CA 02345923 2001-03-22
WO 99/16898 PCT/CA98/00923
7
b) similarly an individual being a heterozygote for an
autosomal dominant disorder exhibiting the same mode of action as described
herein can be treated by gene therapy, such that a mutant allele is inserted
into
a vector and delivered to an individual thereby negating the effect of the
heterozygote mutation by either allelic or protein complementation.
c) with the new knowledge of the present invention, a
transgenic animal designed to carry a deleterious autosomal dominant mutation
can be used to assess the requirement to produce a phenotypically normal
animal, by either allelic complementation or protein complementation.
d) a diagnostic means to identify phenotypically normal
genotypically mutant individuals that can transmit the mutant allele to their
offsprings.
e) the teachings of the present invention can be used for
showing dimezisation of TIGR peptides.
The present invention therefore also provides the means to
identify novel mutations in the TIGR gene, wherein these mutations give rise
to
glaucoma. These mutations can also be identified by any other means known
to a person skilled in the art. As well, the diagnostic methods of the present
invention can be adapted in a kit format comprising probes, primers,
oligonucleotides and reagents commonly known in the art to provide the means
for detecting mutations that may cause glaucoma. The present invention further
provides methods to treat diseases or conditions associated with homoallelic
complementation.
The mutation analysis according to the present invention is
useful for screening individuals at risk for glaucoma. Such individuals may
have
a family history of glaucoma, and for identifying individuals carrying a
mutation
in the glaucoma gene enabling early treatment which may obviate or minimise
the progression of the disease.
In another embodiment of the present invention, there is
provided a kit comprising all the necessary reagents to carry out the herein
CA 02345923 2001-03-22
WO 99/16898 PCT/CA98/00923
described methods of detection. As known to the person of ordinary skill, the
kit comprises container means comprising oligonucleotide sequences or
antibodies (or binding proteins) and reagents such as washing reagents,
reagents for detection purposes and the like. It will also be readily
recognized
that the nucleic acid sequences or antibodies of the present invention could
be
incorporated into established kit formats.
The present invention thus discloses, for the first time, a
mechanism termed "homoallelic complementation" wherein for example the
K423E mutation is acting in a dominant negative fashion, thereby resulting in
a
defective TIGRwtlK423E protein heteromaltemers but functional TIGR
K23ElK23E homopolymers. This form of interaction may be interpreted as being
somewhat similar to the "metabolic interference model" that suggested a
deleterious defect due to interference between the protein products of the two
different alleles (Johnson, 1980, Am. J. Hum. Genet. 32:374-86). The
homoallelic complementation model of the present invention, however, further
proposes that functional TIGRwtlwt and/or TIGR K23ElK23E homopolymers are
generated by an admixture of normal and mutant subunits in TIGRwtlK432E
heterozygotes, thereby explaining, at least in part, the phenotypic
variability
observed in effected carriers as well as the unaffected mutant homozygotes
described herein. It should be noted that Crick et al. (1964, J. Molec. Biol.
8:161-165) had previously suggested the theory of inter-allelic
complementation.
White homoallelic complementation has been identified and
validated with glaucoma, it is suspected to be a phenomenon which is not
limited
thereto. For example, a form of epilepsy and mental retardation linked to
chromosome X, which only affects women, has been reported (Ryan et al.,
1997, Nature Genetics 17:92-95).
In accordance with the present invention, there is thus
provided an isolated DNA comprising the nucleotide sequence defined in SEQ.
ID. NO.: 1, wherein a mutation is identified therein
CA 02345923 2001-03-22
WO 99/16898 PCT/CA98/00923
9
There is also provided a method for detecting a mutant allele
of the TIGRlMYOC gene which comprises the steps of contacting a DNA sample
taken from an individual with an oiigonucleotide as defined in claims 3, 5 or
7
and with an oligonucleotide primer of the present invention; obtaining an
5 amplified product in an amplification reaction; and detecting the
amplification
product as an indication of the presence of said mutant allele.
In accordance with the present invention, there is also
provided a method for detecting a non-mutant allele of the TIGWMYOC gene
which comprises the steps of contacting a DNA sample taken from an individual,
10 with an oligonucleotide primer (or a probe enabling a distinction between
wild
type and mutant); obtaining an amplified product in an amplification reaction;
and detecting the amplification product as an indication of the presence of
the
non-mutant allele.
The application further relates to a kit for the detection of
15 mutations in the TIGR gene comprising an oligonucleotide of the present
invention; and suitable reagents required for obtaining amplified products in
an
amplification reaction.
In addition, in accordance with the present invention, there
is provided a method to counteract glaucoma in a heterozygotic carrier of TIGR
20 mutations, an overexpression of mutated TIGR protein in a patient, thereby
rendering the phenotype of said patient normal by homoallelic
complementation.
Further, in accordance with the present invention, there is
provided a method to counteract and/or treat heterozygotic carriers of an
25 autosomal dominant inherited disorder caused by a protein that forms
homomultimers, comprising at least one of an overexpression of the mutated
protein and an inhibition of the normal protein in a patient, thereby
rendering
said phenotype normal by homoallelic complementation.
Further, in accordance with the present invention, there is
30 provided a method to counteract a disease phenotype in a patient, wherein
the
CA 02345923 2001-03-22
WO 99/16898 PCT/CA98/00923
disease phenotype is associated with the presence of a heterozygotic mutation
in a protein in the patient, the method comprising at least one of an
elevation of
the expression of a mutated protein and an inhibition of expression and/or
activity of the normal protein, to counteract the affected phenotype, thereby
5 rendering the phenotype normal by one of homoallelic complementation and
haploinsufficiency.
DEFINITIONS AND TECHNOLOGICAL BACKGROUND
Nucleotide sequences are presented herein by single strand,
10 in the 5' to 3' direction, from left to right, using the one letter
nucleotide symbols
as commonly used in the art and in accordance with the recommendations of
the IUPAC-IUB Biochemical Nomenclature Commission.
Unless defined otherwise, the scientific and technological
terms and nomenclature used herein have the same meaning as commonly
understood by a person of ordinary skill to which this invention pertains.
Generally, the procedures for cell cultures, infection, molecular biology
methods
and the like are common methods used in the art. Such standard techniques
can be found in reference manuals such as for example Sambrook et al. (1989,
Molecular Cloning - A Laboratory Manual, Cold Spring Harbor Laboratories) and
Ausubel et al. (1994, Current Protocols in Molecular Biology, Wiley, New
York).
The present description refers to a number of routinely used
recombinant DNA (rDNA) technology terms. Nevertheless, definitions of
selected examples of such rDNA terms are provided for clarity and consistency.
As used herein, "TIGR" refers to the trabecular meshwork
inducible glucocorticoid response gene product, also known as MYOC, present
at the GLC1A locus. Herein, "TIGR" therefore refers to TIGRlMYOC. It should
be noted that the TIGRlMYOC gene is sometimes referred to as the GLC1A
gene.
As used herein. the terminology "homoallelic
complementation" refers to the novel description of mutant homozygotes being
CA 02345923 2001-03-22
WO 99/16898 PCT/CA98/00923
11
phenotypically normal with respect to an autosomal dominant inherited disease.
It should be noted that although the present invention focuses on autosomal
dominant inherited diseases, the present invention also applies to
haploinsufficiency. Broadly stated, the mechanism would be similar to a
5 suggested locus with a wild-type allele A and a mutant allele A', such that
homozygosity for either allele, has no phenotypic consequences, but the
heterozygous state AA' leads to a deleterious defect due to interference
between the protein products of the two different alleles.
As used herein, "nucleic acid molecule", refers to a polymer
of nucleotides. Non-limiting examples thereof include DNA (i.e. genomic DNA,
cDNA) and RNA molecules (i.e. mRNA). The nucleic acid molecule can be
obtained by cloning techniques or synthesized. DNA can be double-stranded or
single stranded (coding strand or non-coding strand [antisense]).
The term "recombinant DNA" as known in the art refers to a
DNA molecule resulting from the joining of DNA segments. This is often
referred
to as genetic engineering.
The term "DNA segment", is used herein, to refer to a DNA
molecule comprising a linear stretch or sequence of nucleotides. This sequence
when read in accordance with the genetic code, can encode a linear stretch or
sequence of amino acids which can be referred to as a polypeptide, protein,
protein fragment and the like.
The terminology "amplification pair" or "primer pair" refers
herein to a pair of oligonucleotides (oligos) of the present invention, which
are
selected to be used together in amplifying a selected nucleic acid sequence by
one of a number of types of amplification processes, preferably a polymerase
chain reaction. Other types of amplification processes include ligase chain
reaction, strand displacement amplification, or nucleic acid sequence-based
amplification. as explained in greater detail below. As commonly known in the
art, the oligos are designed to bind to a complementary sequence under
selected conditions.
CA 02345923 2001-03-22
WO 99/16898 PCT/CA98/00923
12
it will be clear to the person of ordinary skill that the present
invention can be adapted to the detection of numerous mutations in
TIGRlMYOC. As exemplified hereinbelow, using a particular embodiment of the
present invention using ARMS with oligos having the sequences of SEQ ID
5 N0:5 and SEQ ID N0:7, mutations His366G1n and LYS424GLn were identified.
The nucleic acid (i.e. DNA or RNA) for practising the present
invention may be obtained according to well known methods.
Oligonucleotide probes or primers of the present invention
may be of any suitable length, depending on the particular assay format and
the
10 particular needs and targeted genomes employed. In general, the
oligonucleotide probes or primers are at least 10 nucleotides in length,
preferably between 15 and 24 nucleotides, and they may be adapted to be
especially suited to a chosen nucleic acid amplification system. As commonly
known in the art, the oligonucleotide probes and primers can be designed by
15 taking into consideration the melting point of hydrizidation thereof with
its
targeted sequence (in Sambrook et al., 1989, Molecular Cloning - A Laboratory
Manual, 2nd Edition, CSH Laboratories; Ausubel et al., 1989, in Current
Protocols in Molecular Biology, John Wiley & Sons Inc., N.Y.).
"Nucleic acid hybridization" refers generally to the
20 hybridization of two single-stranded nucleic acid molecules having
complementary base sequences, which under appropriate conditions will form
a thermodynamically favored double-stranded structure. Examples of
hybridization conditions can be found in the two laboratory manuals referred
above (Sambrook et al., 1989, supra and Ausubel et al., 1989 supra) and are
25 commonly known in the art. In the case of a hybridization to a
nitrocellulose
filter, as for example in the well known Southern blotting procedure, a
nitrocellulose filter can be incubated overnight at 65°C with a labeled
probe in
a solution containing 50% formamide, high salt ( 5 x SSC or 5 x SSPE), 5 x
Denhardt's solution, 1 % SDS, and 100 Ng/ml denatured carried DNA ( i.e.
30 salmon sperm DNA). The non-specifically binding probe can then be washed
off
CA 02345923 2001-03-22
WO 99/16898 PCT/CA98/00923
13
the filter by several washes in 0.2 x SSC/0.1 % SDS at a temperature which is
selected in view of the desired stringency: room temperature (low stringency),
42°C (moderate stringency) or 65°C (high stringency). The
selected temperature
is based on the melting temperature (Tm) of the DNA hybrid. Of course,
5 RNA-DNA hybrids can also be formed and detected. In such cases, the
conditions of hybridization and washing can be adapted according to well known
methods by the person of ordinary skill. High stringency conditions will be
preferably used (Sambrook et a1.,1989, supra).
Probes or nucleic acid molecules of the invention can be
10 utilized with naturally occurring sugar-phosphate backbones as well as
modified
backbones including phosphorothioates, dithionates, alkyl phosphonates and
a-nucleotides and the like. Modified sugar-phosphate backbones are generally
taught by Miller, 1988, Ann. Reports Med. Chem. 23:295 and Moran et al., 1987,
Nucleic acid molecule. Acids Res., 14:5019. Probes of the invention can be
15 constructed of either ribonucleic acid {RNA) or deoxyribonucleic acid
(DNA), and
preferably of DNA.
The types of detection methods in which probes can be used
include Southern blots (DNA detection), dot or' slot blots (DNA, RNA), and
Northern blots (RNA detection). Although less preferred, labelled proteins
could
20 also be used to detect a particular nucleic acid sequence to which it
binds. Other
detection methods include kits containing probes on a dipstick setup and the
like.
Although the present invention is not specifically dependent
on the use of a label for the detection of a particular nucleic acid sequence,
such
25 a label might be beneficial, by increasing the sensitivity of the
detection.
Furthermore, it enables automation. Probes can be labelled according to
numerous well known methods (Sambrook et al., 1989, supra). Non-limiting
examples of labels include 3H, "C, 3zP, and 35S. Non-limiting examples of
detectable markers include iigands, fluorophores, chemiluminescent agents,
30 enzymes, and antibodies. Other detectable markers for use with probes,
which
CA 02345923 2001-03-22
WO 99/16898 PCT/CA98/00923
14
can enable an increase in sensitivity of the method of the invention, include
biotin and radionucleotides. It will become evident to the person of ordinary
skill
that the choice of a particular label dictates the manner in which it is bound
to
the probe.
5 As commonly known, radioactive nucleotides can be
incorporated into probes of the invention by several methods. Non-limiting
examples thereof include kinasing the 5' ends of the probes using gamma 32P
ATP and polynucleotide kinase, using the Kienow fragment of Pol I of E. coli
in
the presence of radioactive dNTP (i.e. uniformly labelled DNA probe using
10 random oligonucleotide primers in low-melt gels;), using the SP6/T7 system
to
transcribe a DNA segment in the presence of one or more radioactive NTP, and
the like.
As used herein, "oligonucleotides" or "oligos" define a
molecule having two or more nucleotides (ribo or deoxyribonucleotides). The
15 size of the oligo will be dictated by the particular situation and
ultimately by the
particular use thereof, and adapted accordingly by the person of ordinary
skill.
An oligonucleotide can be synthetised chemically or derived by cloning
according to well known methods.
As used herein, a "primer" defines an oligonucleotide which
20 is capable of annealing to a target sequence, thereby creating a double
stranded
region which can serve as an initiation point for DNA synthesis under suitable
conditions.
Amplification of a selected, or target, nucleic acid sequence
may be carried out by a number of suitable methods. See generally Kwoh et al.,
25 1990, (Am. Biotechnol. Lab. 8:14-25). Numerous amplification techniques
have
been described and can be readily adapted to suit the particular needs of a
person of ordinary skill. Non-limiting examples of amplification techniques
include polymerase chain reaction (PCR), ligase chain reaction (LCR), strand
displacement amplification (SDA), transcription-based amplification, the Q~
30 replicase system and NASBA (Kwoh et al 1989, Proc. Natl. Acad. Sci. USA 86,
CA 02345923 2001-03-22
WO 99/16898 PCT/CA98/00923
1173-1177; Lizardi et al., 1988, BioTechnology 6:1197-1202; Malek et al.,
1994,
Methods Mol. Biol., 28:253-260; and Sambrook et al., 1989, supra). Preferably,
amplification will be carried out using PCR.
Polymerase chain reaction (PCR) is carried out in accordance
5 with known techniques. See, e.g., U.S. Pat. Nos. 4,683,195; 4,683,202;
4,800,159; and 4,965,188 (the disclosures of all three U.S. Patent are
incorporated herein by reference). In general, PCR involves, a treatment of a
nucleic acid sample (e.g., in the presence of a heat stable DNA polymerase)
under hybridizing conditions, with one oligonucieotide primer for each strand
of
10 the specific sequence to be detected. An extension product of each primer
which
is synthesized is complementary to each of the two nucleic acid strands, with
the
primers sufficiently complementary to each strand of the specific sequence to
hybridize therewith. The extension product synthesized from each primer can
also serve as a template for further synthesis of extension products using the
15 same primers. Following a sufficient number of rounds of synthesis of
extension
products, the sample is analysed to assess whether the sequence or sequences
to be detected are present. Detection of the amplified sequence may be carried
out by visualization following EtBr staining of the DNA following gel
electrophoresis, or using a detectable label in accordance with known
techniques, and the like. For a review on PCR techniques (see PCR Protocols,
A Guide to Methods and Amplifications, Michael et al., Eds, Acad. Press,
1990).
Ligase chain reaction (LCR) is carried out in accordance with
known techniques (Weiss, 1991, Science 254:1292). Adaptation of the protocol
to meet the desired needs can be carried out by a person of ordinary skill.
Strand displacement amplification (SDA) is also carried out in accordance with
known techniques or adaptations thereof to meet the particular needs (Walker
et al., 1992, Proc. Natl. Acad. Sci. USA 89:392-396; and ibid., 1992, Nucleic
Acids Res. 20:1691-1696).
As used herein, the term "gene" is well known in the art and
relates to a nucleic acid sequence defining a single protein or polypeptide. A
CA 02345923 2001-03-22
WO 99/16898 PCT/CA98/00923
16
"structural gene" defines a DNA sequence which is transcribed into RNA and
translated into a protein having a specific amino acid sequence thereby giving
rise the a specific polypeptide or protein. It will be readily recognized by
the
person of ordinary skill, that the nucleic acid sequences of the present
invention
5 can be incorporated into anyone of numerous established kit formats which
are
well known in the art.
The term "vector" is commonly known in the art and defines
a plasmid DNA, phage DNA, viral DNA and the like, which can serve as a DNA
vehicle into which DNA of the present invention can be cloned. Numerous types
10 of vectors exist and are well known in the art.
The term "expression" defines the process by which a
structural gene is transcribed into mRNA (transcription), the mRNA is then
being
translated (translation) into one polypeptide (or protein) or more.
The terminology "expression vector" defines a vector or
15 vehicle, as described above, but designed to enable the expression of an
inserted sequence following transformation into a host. The cloned gene
(inserted sequence) is usually placed under the control of control element
sequences such as promoter sequences. The placing of a cloned gene under
such control sequences is often referred to as being "operably linked" to
control
20 elements or sequences.
Expression control sequences will vary depending on whether
the vector is designed to express the operably linked gene in a prokaryotic or
eukaryotic host or both (shuttle vectors) and can additionally contain
transcriptional elements such as enhancer elements, termination sequences,
25 tissue-specificity elements, and/or translational initiation and
termination sites.
As used herein, the designation "functional derivative"
denotes, in the context of a functional derivative of a sequence, whether
nucleic
acid or amino acid sequence. a molecule that retains a biological activity
(either
functional or structural) that is substantially similar to that of the
original
30 sequence. This functional derivative or equivalent may be a natural
derivative
CA 02345923 2001-03-22
WO 99/16898 PCT/CA98/00923
. 17
or may be prepared synthetically. Such derivatives include amino acid
sequences having substitutions, deletions, or additions of one or more amino
acids, provided that the biological activity of the protein is conserved. The
same
applies to derivatives of nucleic acid sequences which can have substitutions,
deletions, or additions of one or more nucleotides, provided that the
biological
activity of the sequence is generally maintained. When relating to a protein
sequence, the substituting amino acid has chemico-physical properties which
are similar to that of the substituted amino acid. The similar chemico-
physical
properties include, similarities in charge, bulkiness, hydrophobicity,
hydrophylicity and the like. The term "functional derivatives" is intended to
include "fragments", °segments", "variants", "analogs" or "chemical
derivatives"
of the subject matter of the present invention.
Thus, the term "variant" refers herein to a protein or nucleic
acid molecule which is substantially similar in structure and biological
activity to
the protein or nucleic acid of the present invention.
The functional derivatives of the present invention can be
synthesized chemically or produced through recombinant DNA technology. All
these methods are well known in the art.
As used herein, "chemical derivatives" is meant to cover
additional chemical moieties not normally part of the subject matter of the
invention. Such moieties could affect the physica-chemical characteristic of
the
derivative (i.e. solubility, absorption, half life and the like, decrease of
toxicity).
Such moieties are exemplified in Remington's Pharmaceutical Sciences (1980).
Methods of coupling these chemical-physical moieties to a polypeptide are well
known in the art.
The term "allele" defines an alternative form of a gene which
occupies a given locus on a chromosome.
As commonly known, a "mutation" is a detectable change in
the genetic material which can be transmitted to a daughter cell. As well
known,
a mutation can be, for example, a detectable change in one or more
CA 02345923 2001-03-22
WO 99/16898 PCT/CA98/00923
18
deoxyribonucleotide. For example, nucleotides can be added, deleted,
substituted for, inverted, or transposed to a new position. Spontaneous
mutations and experimentally induced mutations exist. The result of a
mutations
of nucleic acid molecule is a mutant nucleic acid molecule. A mutant
polypeptide
5 can be encoded from this mutant nucleic acid molecule.
As used herein, the term "purified" refers to a molecule
having been separated from a cellular component. Thus, for example, a
"purified
protein" has been purified to a level not found in nature. A "substantially
pure"
molecule is a molecule that is lacking in all other cellular components.
10 The term "autosome" defines any chromosome other that the
sex chromosomes, X and Y.
The term "dominant" refers to an allele that determines the
phenotype displayed in a heterozygote with another normal or mutant allele.
The terminology "transgenic animal" defines an animal that
15 has had its germ line genetically modified to give rise to a progeny animal
that
is different from the parental type and carrying the modification in its germ
line.
Non-human transgenic animals of the invention comprise
animals having transgenic alteration of an endogenous gene which shows
homoallelic complementation, in accordance with the present invention. Such
20 non-human animals are commonly known in the art and include vertebrates,
and
more especially mammalians, and especially rodents such as rats and more
particularly mice. These transgenic animals have had introduced into their
genomes, by non-natural means (i.e. by manipulation), one or more gene which
does not occur naturally in the animal These non-naturally occuring genes in
25 the animal {known as transgenes) may be from the same species as the
animal,
although in such a case, the gene or genes are in a different configuration
and/or chromosomal location. A transgenic non-human animal of the invention
can be produced in accordance with well-known methods in the art. Non-limiting
examples of such methods include viral integration, microinjection of zygotes
30 and the like.
CA 02345923 2001-03-22
WO 99/16898 PCT/CA98/00923
19
"Single Strand Conformational Polymorphism (SSCP)" refers
to a method for detecting the presence of a base pair change in an amplified
DNA fragment. The method involves denaturing the double stranded amplified
DNA and comparing the band pattern in a known non-mutant fragment to that
5 of an unknown fragment. A shift in the band pattern is indicative of a base
pair
change.
The designation "gene therapy" defines an attempt to treat
disease by genetic modification of the cells of a patient.
"Allele Specific Oligonucleotide (ASO)" are designed to detect
10 known and identified base pair change by designing oligonucleotides that
are
specific to the DNA fragment with and without the base change. These
oligonucleotides are used as probes in hybridisation protocols under stringent
conditions. Differences in the hybridization patterns is indicative of the
presence
or absence of the base change.
15 "Artificially Created Restrictian Site (ACRS)" refers to a
method for detection a known base change in a DNA sequence. It involves the
designing of a primer that may either create or obviate a restriction site in
the
vicinity of known base change, such that the restriction endonuclease used can
have a different digestion pattern for the changed and unchanged base.
20 For certainty, the sequences and poiypeptides useful to
practice the invention include without being limited thereto mutants,
homologs,
subtypes, alleles and the like. It shall be understood that generally, the
sequences of the present invention should encode a functional (albeit
defective)
interaction domain. it will be clear to the person of ordinary skill that
whether an
25 interaction domain of the present invention, variant, derivative, or
fragment
thereof retains its function in binding to its partner can be readily
determined by
using the teachings and assays of the present invention and the general
teachings of the art.
Also contemplated within the scope of the present invention
30 are fusion proteins comprising one of the interacting domains of the
present
CA 02345923 2001-03-22
WO 99/16898 PCT/CA98/00923
20
invention. As exemplified herein below in one embodiment, at feast one of an
interaction domain of the present invention may be provided as a fusion
protein.
The design of constructs therefor and the expression and production of fusion
proteins and are well known in the art (Sambrook et al., 1989, supra; and
5 Ausubel et al., 1994, supra). Non-limiting examples of fusion proteins
include
LexA-fusions, B42-fusions, hemaglutinin fusions and Gluthione-S-transferase
(GST) fusions and Maltose binding protein (MBP) fusions. In certain
embodiments, it might be beneficial to introduce a protease cleavage site
between the two polypeptide sequences which have been fused. Such protease
10 cleavage sites between two heterologously fused polypeptides are well known
in the art.
In certain embodiments, it might also be beneficial to fuse the
interaction domains of the present invention to signal peptide sequences
enabling a secretion of the fusion protein from the host cell. Signal peptides
from
15 diverse organisms are well known in the art. Bacterial OmpA and yeast Suc2
are
two non limiting examples of proteins containing signal sequences. In certain
embodiments, it might also be beneficial to introduce a linker {commonly
known)
between the interaction domain and the heterologous polypeptide portion. Such
fusion protein find utility in the assays of the present invention as well as
for
20 purification purposes, detection purposes and the like.
The interaction domains of the present invention can be
modified, for example by in vitro mutagenesis, to dissect the structure-
function
relationship thereof and permit a better design and identification of
modulating
compounds (i.e. to deactivate or inhibit the normal protein). However, some
25 derivative or analogs having lost their biological function of interacting
with their
respective interaction partner may provide further advantages as compared to
known mutant protein, as well they may also find utility, for example for
raising
antibodies. Such analogs or derivatives could be used for example to raise
antibodies to the interaction domains of the present invention. These
antibodies
30 could be used for detection or purification purposes. In addition, these
antibodies
CA 02345923 2001-03-22
WO 99/16898 PCT/CA98/00923
- 21
could also act as competitive or non-competitive inhibitor and be found to be
modulators of the protein-protein interactions of the present invention.
In general, techniques for preparing antibodies (including
monoclonal antibodies and hybridomas) and for detecting antigens using
antibodies are well known in the art (Campbell, 1984, In "Monoclonal Antibody
Technology: Laboratory Techniques in Biochemistry and Molecular Biology",
Elsevier Science Publisher, Amsterdam, The Netherlands) and in Harlow et al.,
1988 (in: Antibody - A Laboratory Manual, CSH Laboratories). The present
invention also provides polyclonal, monoclonal antibodies, or humanized
versions thereof, chimeric antibodies and the like which inhibit or neutralize
their
respective interaction domains and/or are specific thereto.
The present invention also provides antisense nucleic acid
molecules which can be used for example to decrease or abrogate the
expression of the normal allele. An antisense nucleic acid molecule according
to the present invention refers to a molecule capable of forming a stable
duplex
or triplex with a portion of its targeted nucleic acid sequence (DNA or RNA).
The
use of antisense nucleic acid molecules and the design and modification of
such
molecules is well known in the art as described for example in WO 96/32966,
WO 96/11266, WO 94/15646, WO 93/08845, and USP 5,593,974. Antisense
nucleic acid molecules according to the present invention can be derived from
the nucleic acid sequences and modified in accordance to well known methods.
For example, some antisense molecules can be designed to be more resistant
to degradation to increase their affinity to their targeted sequence, to
affect their
transport to chosen cell types or cell compartments, and/or to enhance their
lipid
solubility by using nucleotide analogs and/or substituting chosen chemical
fragments thereof, as commonly known in the art
From the specification and appended claims, the term
therapeutic agent should be taken in a broad sense so as to also include a
combination of at least two such therapeutic agents Further, the DNA segments
or proteins according to the present invention can be introduced into
individuals
CA 02345923 2001-03-22
WO 99/16898 PCT/CA98/00923
22
in a number of ways. For example, erythropoietic cells can be isolated from
the
afflicted individual, transformed with a DNA construct according to the
invention
and reintroduced to the afflicted individual in a number of ways, including
intravenous injection. Alternatively, the DNA construct can be administered
5 directly to the afflicted individual, for example, by injection in the bone
marrow.
The DNA construct can also be delivered through a vehicle such as a liposome,
which can be designed to be targeted to a specific cell type, and engineered
to
be administered through different routes.
For administration to humans, the prescribing medical
10 professional will ultimately determine the appropriate form and dosage for
a
given patient, and this can be expected to vary according to the chosen
therapeutic regimen (i.e DNA construct, protein, cells), the response and
condition of the patient as well as the severity of the disease.
Composition within the scope of the present invention should
15 contain the active agent (i.e. fusion protein, nucleic acid, and molecule)
in an
amount effective to achieve the desired therapeutic effect while avoiding
adverse side effects. Non-limiting examples of active agents include the
mutant
protein according to the present invention a nucleic acid molecule encoding
such
a mutant protein, an antisense molecule to a normal allele according to the
20 present invention, etc. Typically, the nucleic acids in accordance with the
present invention can be administered to mammals (i.e. humans) in doses
ranging from 0.005 to 1 mg per kg of body weight per day of the mammal which
is treated. Pharmaceutically acceptable preparations and salts of the active
agent are within the scope of the present invention and are well known in the
art
25 (Remington's Pharmaceutical Science, 16th Ed., Mack Ed.). For the
administration of polypeptides, antagonists, agonists and the like, the amount
administered should be chosen so as to avoid adverse side effects. The dosage
will be adapted by the clinician in accordance with conventional factors such
as
the extent of the disease and different parameters from the patient.
Typically,
30 0.001 to 50 mg/kg/day will be administered to the mammal.
CA 02345923 2001-03-22
r _~
r~: . . _ . . , ' r. ~~ on
., n
1 , . n ~ A f .~ ~, f1 C i/
fn . _ _ _. _ . O O
~ C C O 0
23
BRIEF DESCRIPTION OF THE DRAWINGS
Having thus generally described the invention, reference will
now be made to the accompanying drawings, showing by way of illustration a
preferred embodiment thereof, and in which:
Figure 1 a-Figure 1 a show the sequence that encodes the
wild-type TIGR~IVIYOC cDNA sequence (SEQ ID N0:1) localized at the GLC9A
locus on chromosome 1 q23-q25.
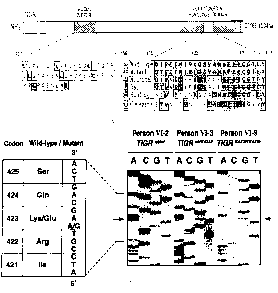
Figure 2 shows the characterization of a carrier homozygous
for the Lys423G1u TIGR mutation. a, Structure of the TIGR encoded protein. The
leucine zipper domain (amino acids 117-166) is shown within the N-terminal
half
of the protein. The Lys423G1u mutation is depicted by an open circle in the
olfactomedin homology domain represented by a striped box within the
C-terminal half of the protein. Amino acids comparison between human
TIGRlMYOC protein (amino acids 415-437), human neuronal olfactomedin, rat
and bullfrog neuronal olfactomedin-related proteins (GeneBank accession
079299, 003417, L13595, respectively) and C. elegans F11c3.2 protein
(GeneBank accession 281499) is represented. Identical amino acids are shaded
in black, conserved amino acids are further boxed by white squares. The codon
numbers correspond to those of the TIGWMYOC protein, b, Identification of an
homozygous carrier of the Lys423G1u TIGR mutation. Direct sequencing of
genomic DNA revealed that persons VI-3 and VI-9 were, respectively,
heterozygotic and homozygotic carriers of the Lys423G1u TIGR mutation. The
arrows indicate the A to G transition. Person VI-2 carried two wild-type TIGR
alleles.
Figure 3 shows the amplification refractory mutation system
(ARMS) as a method to type specific alleles at a polymorphic locus. In the
present invention, this method, ARMS, was used for detecting a specific
pathogenic mutation. The allele-specific oligonucleotide primers were designed
to discriminate between two target DNA sequences {wild-type (normal) versus
pathogenic) that differed by a single nucleotide in the region of interest
(either
AMENOEO SHEET
CA 02345923 2001-03-22
WO 99/16898 PCT/CA98/00923
24
one of the two mutations). Designed primers that differed at the extreme 3'
terminus were synthetised. This was done because the DNA synthesis step in
the PCR reaction is crucially dependent on correct base pairing at the 3' end.
The primers that were designed are differing in their 3' ends and can
therefore
5 specifically amplify the DNA fragment of interest, either normal or mutated.
This
figure is a pictorial representation of ARMS for the adenine to guanine
transition
at nucleotide 1267. The amplification strategy is demonstrated for the wild-
type;
or non-mutant allele, and the mutant aiiele.
Figure 4 shows the phenotypic status and segregation
10 analyses of the GLC1A disease haplotype and Lys423Gfu in family GV-510. All
living individuals were investigated for glaucoma, genotyped with
microsatelfite
markers spanning the GLC1A locus and tested for the presence of the
Lys423G1u TIGR mutation using ARMS. Selected AFM markers with their
corresponding GDB number, number of alleles observed for each marker in
15 pedigree GV-001 and sizes of the allele associated with the GLC1A disease
haplotype are represented on top. The position of the TIGR gene is indicated
relative to genetic markers. Sex-averaged recombination distances, depicted
between marker loci in centiMorgans, were not drawn to scale. Glaucoma
patients are depicted by solid black symbols, unaffected individuals by open
20 symbols, and deceased subjects reported as blind by at least two
independent
family members by a black quadrant in the upper left corner of their
respective
symbols. OHT persons are represented by open symbols containing a central
solid dot. Present ages of normal and OHT patients as well as ages of affected
carriers at time of diagnosis are depicted above their respective symbols. A
solid
25 black box indicates the common GLC1A disease hapiotype. The right side of
each phased haplotype indicates the haplotype inherited from the father; the
left
side indicates the haplotype inherited from the mother. An asterisk in the
genotype of person VII-5 represents a microsatellite mutation at locus
D152790.
Person VII-5 also inherited a paternal recombination between loci D152815 and
30 D1S2790. Results of the ARMS tests are depicted below each subject's
CA 02345923 2001-03-22
WO 99/16898 PCT/CA98/00923
genotype; W, ARMS test performed using the wild-type primers; M, ARMS test
performed using the Lys423G1u mutant primers. The internal control PCR
product is shown. Persons VI-2, VI-5, VI-6, VI-10 and VI-12 carried the wild-
type
allele on both chromosomes 1. Persons VI-1, Vl-7, Vl-9 and VI-11 are wild-type
5 negative and mutant positive, therefore, homozygous for the Lys423G1u
mutation. All other individuals are both wild-type positive and mutant
positive,
therefore, heterozygotes for the mutation.
Figure 5 shows the characterization of carriers for the
HIS366GIn and GIn368Stop TIGR mutations. a, Structure of the TIGR encoded
10 protein. The leucine zipper domain (amino acids 117-166) is shown within
the
N-terminal half of the protein. The His366G1n mutation is depicted by a black
circle in the olfactomedin homology domain represented by a striped box within
the C-terminal half of the protein. The GIn368Stop mutation is depicted by a
stop
codon in the olfactomedin homology domain. The codon numbers correspond
15 to those of the TIGR protein. b, Identification of carriers for the
His366G1n and
GIn368Stop TIGR mutations. Direct sequencing of genomic DNA revealed that
persons CT-003 and LA-002 were, respectively, heterozygotic carriers of the
GIn368Stop and His366G1n TIGR mutations. The arrows indicate the C to T
transition or C to G transversion.
20 Figure 6 shows that the TIGR wild-type protein and
Lys423G1u mutated TIGR protein form high molecular weight complexes in vivo
and in vitro. a, Western analyses of TIGR wild-type and TIGR Lys423G1u
polypeptides in transfected cells. COS-7 cells were transiently transfected
with
expression vectors containing wild-type TIGR, pRc/CMV or TIGRK423E
25 (pRcTIG432E) cDNA (20 mg per sample), alone or in combination. Twenty-four
hours after transfection, protein extracts from total cell lysates were
resolved by
SDS-PAGE and TIGR polypeptides were detected by immunoblotting. The two
fines above the immunoblot depict the relative amount of each cDNA tested per
sample on a scale of 0 (0 mg) to 100 (20 mg). For instance, TIGR wild-type
(TIGRwt) 50- TIGR Lys423G1u (TIGRK423E) 50 indicates that 10 mg of each
CA 02345923 2001-03-22
WO 99/16898 PCT/CA98/00923
26
cDNA were added in this transfection. Two high molecular weight complexes are
generated by TIGR polypeptides in COS-7 cells b, Western analyses of extracts
obtained from dissected human trabecular meshwork (HTM) tissues. This
analysis shows that TIGR polypeptides also form high molecular weight
5 complexes in vivo. COS-7 cell extracts from cultures transfected with TIGRwt
or TIGRK423E cDNA (20 mg) are depicted for comparison. Three distinct
subtypes of T1GR monomers are observed after treating HTM tissue extracts
with DTT, A brief exposure time period was performed to clearly visualize
these
monomers. c, Western analyses of in vitro synthesized TIGR wild-type and
10 TIGR Lys423G1u polypeptides. pRcTIG or pRcTIG423E plasmids were added
to a reticulocyte lysate transcription/translation coupled system containing
pancreatic microsomal membranes. 0.5 microgram of each plasmid were tested
per reaction. This analysis shows that both TIGR wild-type and TiGR Lys423Giu
polypeptides can form homodimers in vitro. a-c, All samples were separated on
15 6% denaturing polyacrylamide gels and analyzed using a specific polyclonal
antibody made against the non-glycosylated form of the protein. DTT indicates
samples that were treated with dithiothreitol (0.1 mM) prior to
electrophoresis.
Position of molecular weight markers and of the expected monomers, dimers
and tetramers are illustrated adjacent to the immunoblots.
20 Other objects, advantages and features of the present
invention will become more apparent upon reading of the following
non-restrictive description of preferred embodiments with reference to the
accompanying drawing which is exemplary and should not be interpreted as
limiting the scope of the present invention.
25
DESCRIPTION OF THE PREFERRED EMBODIMENT
The invention thus concerns novel mutations in the TIGR
gene, a quick method for an easy detection of identified mutations and the
teachings for the first time of mutant homozygotes being phenotypically normal
30 in an autosomally dominant inherited disease.
CA 02345923 2001-03-22
WO 99/16898 PCT/CA98/00923
27
The surprising and unexpected showing that homoallelic
complementation restores a normal phenotype opens the way to new and
powerful therapeutic approaches for diseases or conditions in which such a
type
of heterozygous state (AA') leads to a deleterious defect due to interference
5 between the protein products of the two different alleles A and A', and in
particular to an autosomal dominance state.
The present invention is illustrated in further detail by the
following non-limiting examples.
10 EXAMPLE 1
Pediarees and ophthalmologic assessments
1.1 Pediaree reconstitution
The pedigree genealogy was reconstituted using the registers compiled from the
Catholic parish records, which systematically list births, marriages, and
deaths
15 of 98% of the Quebec population. Validation of the family tree and new data
on
recent births were obtained through interviews with key family members. The
Archives Nationales du Quebec, the Quebec Civil register, and the Institut de
recherche sur /'etude des populations (/REP) data base (Bouchard et al., 1991,
Histoire d'un genome. Population et genetique dans Pest du Quebec, Presses
20 de 1'Universite Laval, Sillery, Quebec, pp 607) were also consulted.
1.2 Ophthalmologic investigations
All subjects, affected or not, gave informed consent before entering the
study.
Clinical assessments comprised complete ophthalmologic evaluation, including
best corrected visual acuity; optic disk examination; slit-lamp biomicroscopy;
25 applanation tonometry; gonioscopy; and visual-field evaluation. Three
criteria
were required for primary open-angle glaucoma (POAG) diagnosis: a)
intraocular pressures above 22 mm Hg in both eyes, b) characteristic optic
disk
damage and/or visual field impairment, and c) grade III or IV (open-angle)
gonioscopy. In the absence of optic disk damage or visual-field alteration,
30 subjects with intraocular pressures above 22 mm Hg in both eyes and grade
III
CA 02345923 2001-03-22
WO 99/16898 PCT/CA98/00923
28
or IV gonioscopy were diagnosed with ocular hypertension (OHT). Members of
the families were considered normal when they presented normal optic disks
and showed highest intraocular pressures ever recorded at 22 mm Hg or less.
Persons with other forms of giaucomas, including grade 0 (closed angle); grade
5 I or II (narrow-angle); congenital; and secondary glaucomas, or with other
nonglaucomatous ocular disorders were considered unaffected. Blindness in
deceased ancestors was confirmed by at least two independent sources.
EXAMPLE 2
10 Genetic analyses
2.1 Source of DNA
Blood samples were obtained from direct descendants of the founder as well as
spouses of affected patients with children; from each, 20 ml of blood was
drawn
by venipuncture in heparinized tubes. One additional 10 ml blood sample was
15 drawn from each subject to establish fymphoblastoid cell lines using the
method
of Anderson et al. (1984, In Vitro, 20:856-858).
2.2 Isolation of DNA
DNA was extracted from whole blood using the guanidine
hydrochloride-proteinase K method developed by Jeanpierre (1987, Nucl. Acids.
20 Res.15:9611-9611).
2.3 Genotyping procedures
To accelerate genotyping, a protocol similar to the procedure of Vignal et al.
(1993, Methods in molecular genetics, Academic Press, 1_:211-221 ) which was
derived from the multiplex sequencing technique of Church and Kieffer-Higgins
25 (1988 Science 240:185-188) was used. Briefly, polymerase chain reactions
(PCR) were performed in a total volume of 50 NI containing 100 ng of genomic
DNA, 50 pmol of each primer, 125 mM dNTPs, 50 mM KCI, 10 mM Tris (pH 9),
1.5 mM MgCl2, 0.01 % gelatin, 0.1 % Triton X-100, and 1 U Taq polymerase
(Perkin-Elmer-Cetus). Amplifications were carried out using a "hot-start"
30 procedure. Taq polymerase was added after a 5-min denaturation step at
96°C.
CA 02345923 2001-03-22
WO 99/16898 PCT/CA98/00923
29
Samples were then processed through 35 cycles of denaturation (94° C
for 40
s) and annealing (55°C for 30 s), followed by one last step of
elongation (2 min
at 72°C). Usually, three amplification products synthesized with
separate primer
sets on identical DNA samples were coprecipitated and comigrated in a single
5 lane of 6% polyacrylamide denaturing gels. Separated products were then
transferred onto Hybond N; nylon membranes (Arnersham), hybridized with a
(CA)Z° oligomer 3' labeled with Digoxigenin-11-ddUTP, and detected by
chemiluminescence using the DIG system (Boehringer-Mannheim) with Kodak
XAR-5 films. Genotypes were scored relative to reference alleles of the mother
10 of the CEPH family 1347 (individual 134702). Genotyping was repeated upon
detection of incompatibilities or recombination events.
2.4 Selection of microsatellite markers
In Figure 4, the markers used for haplotype analyses are shown. Wth the
exception of two markers (AFMGLC21 and AFMGLC22), all AFM (Genethon)
15 markers reported above were described in Dib et al. (1996, supra). For
AFMGLC21, the sequences were primer a: GATCTCTTATCAGTCAGGCA
(SEQ ID N0:7), and primer m: TTTCTAAGGCTGAATAATATTCG SEQ ID
N0:8). For AFMGLC22, the sequences were primer a:
TTAACTCACCACTCCCTGCC (SEQ ID N0:9), and primer m:
20 AATTATGGCCTTCGCCC (SEQ ID N0:10). Assignment of the genetic location
of these markers was established according to the method of Weissenbach et
al. (1992, Nature, 359:795-801) and has been validated by construction of a
10-cM physical map (Clepet et al., 1996, Eur. J. Hum.Genet., 4:250-259).
2.5 Haplotype analysis
25 Haplotypes were analysed to phase the marker genotypes with the disease
gene The haplotype inherited by an affected child constituted the "disease"
haplotype and was compared with the common disease hapiotype inherited from
the founder. The remaining three haplotypes were considered the "normal"
haplotypes.
CA 02345923 2001-03-22
WO 99/16898 PCT/CA98/00923
EXAMPLE 3
Discovery of the phenotyrpic normal-homozy~gote mutant
3.1 Phenotvc~ic normal-homozygote mutant (Figure 4~
Phenotypic status and segregation analyses of the GLC1A disease haplotype
5 and Lys423G1u TIGR mutation in family GV-510. All living individuals were
investigated for glaucoma, genotyped with microsatellite markers spanning the
GLC1A locus and tested for the presence of the Lys423G1u TIGR mutation using
ARMS. Selected AFM markers with their corresponding GDB number, number
of alleles observed for each marker in pedigree GV-001 and sizes of the allele
10 associated with the GLC1A disease haplotype are represented on top. The
position of the TIGRlMYOC gene is indicated relative to genetic markers.
Sex-averaged recombination distances, depicted between marker loci in
centiMorgans, were not drawn to scale. Glaucoma patients are depicted by solid
black symbols, unaffected individuals by open symbols, and deceased subjects
15 reported as blind by at least two independent family members by a black
quadrant in the upper left corner of their respective symbols. OHT persons are
represented by open symbols containing a central solid dot. Present ages of
normal and OHT patients as well as ages of affected carriers at time of
diagnosis are depicted above their respective symbols. A solid black box
20 indicates the common GLC9A disease haplotype. The right side of each phased
haplotype indicates the haplotype inherited from the father; the left side
indicates
the haplotype inherited from the mother. An asterisk in the genotype of person
VII-5 represents a microsatellite mutation at locus D152790. Person VII-5 also
inherited a paternal recombination between loci D152815 and D1S2790. Results
25 of the ARMS tests are depicted below each subject's genotype; W, ARMS test
performed using the wild-type primers; M, ARMS test performed using the
Lys423G1u mutant primers. The internal control PCR product is shown. Persons
VI-2, VI-5, VI-6, VI-10 and VI-12 carried the wild-type allele on both
chromosomes 1. Persons VI-1, VI-7, VI-9 and VI-11 are wild-type negative and
30 mutant positive, therefore, homozygous for the Lys423G1u mutation. All
other
CA 02345923 2001-03-22
WO 99/16898 PCT/CA98/00923
31
individuals are both wild-type positive and mutant positive, therefore,
heterozygotes for the mutation.
3.2 Initial screening for mutations (Figure 21
To obtain a wild-type TIGR cDNA, RT-PCR was performed using the Superscript
5 RT protocol (GibcoIBRL), 500 ng of oligo-dT and 10 Ng of total RNA isolated
from a pool of trabecular meshwork tissue dissected from 10 pairs of human
eyes. To obtain the mutated TIGR cDNA, the same protocol was followed using
10 pg of total RNA isolated from homozygote VI-9 immortalized lymphoblasts.
One to 3 NI of first strand cDNA synthesis was amplified with primers 41 F:
10 AGAGCTTTCCAGAGGAAGCC (SEQ ID N0:11 ), and 1731 R:
GGTCTACGCCCTCAGACTAC (SEQ 1D N0:12), before a second round of PCR
with internal primers 31 F: AGAGACAGCAGCACCCAACG (SEQ ID N0:13), and
21 R: TCTGCCATTGCCTGTACAGC (SEQ ID N0:14). PCR products were
directly cloned into the pCRll vector using the TA cloning kit (InV'~trogen)
15 according to the manufacturer's protocol. Cloned products were sequenced
using the T7 sequencing kit (Pharmacia).
3.3 Sequencing
To confirm mutations, genomic DNA sequencing was also performed on
selected individuals by direct asymmetric PCR sequencing using modifications
20 of the protocol described by Gyllensten et al. (1988, Proc. Natl. Acad.
Sci.,
$_5:7652-7656}. The mutation was recognized by the approximately equal peak
intensity of the bands on the autoradiogram. All sequencing was performed
bidirectionally.
25 EXAMPLE 4
Two mutations including ARMS
4.1 ARMS test for the Lyrs423G1u mutation (Figure ~
To test for the presence of the Lys423G1u mutation, we developed an
amplification refractory mutation system (ARMS) exploiting procedures
30 described by ~itt~e (1997, Current Protocols in human genetics, Eds.
Dracopoli,
CA 02345923 2001-03-22
WO 99/16898 PCT/CA98/00923
32
N.C. et al., 9.8.1. - 9.8.12). Two complementary PCR reactions were conducted
with.the same substrate. The first reaction contained a forward primer
specific
for the wild-type allele, SEQ. NO. 3, (GLC1A1313AA}:
TCGAACAAACCTGGGAGACAAACATCCGAA. The second reaction contained
5 a fonrvard primer specific for the Lys423G1u TIGR allele, SEQ. NO. 2,
(GLC1A1313GG): TCGAACAAACC T GGGAGACAAACATCCGGG. In each
reaction, a common reverse primer, GLC1A1479R, was used; its sequence was:
SEQ. NO. 4 CAAAGAGCTTCTTCTCCAGGGGGTTGTAGT. Both reactions
gave a 225 by amplified fragment. To serve as internal control, a second pair
of
10 primers that co-amplified a 438 by fragment within TIGR exon 1 was added to
the ARMS reaction. The forward TIGR exon 1 primer was:
AGAGCTTTCCAGAGGAAGCC (SEQ ID N0:11), the reverse TIGR exon 1
primer was TTGGGTTTCCAGCTGGTC (SEQ ID N0:15). PCR was performed
using standard protocols, annealing temperature was at 60°C.
Amplification
15 products were electrophoresed in 1,5% agarose gels before ethidium staining
and scored by two independent observers.
4.2 ARMS test for the His366G1n mutation
To test for the presence of the His366G1n mutation, we developed an
amplification refractory mutation system (ARMS) exploiting procedures
20 described by Little (1997). Two complementary PCR reactions were conducted
with the same substrate. The first reaction contained a forward primer
specific
for the wild-type allele, SEQ. NO. 6 (GLC1A1098C~:
GAGAAGGAAATCCCTGGAGCTGGCTACCTC. The second reaction contained
a forward primer specific for the His366G1n TIGR allele, SEQ. NO. 5,
25 (GLC1A1098GT): GAGAAGGAAATCCCTGGAGCTGGCTACCTG. In each
reaction, SEQ N0:4 was used as a common reverse primer. Both reactions
gave a 393 by amplified fragment. To serve as internal control, a second pair
of
primers that co-amplified a 438 by fragment within TIGR exon 1 was added to
the ARMS reaction. The forward TIGR exon 1 primer was:
30 AGAGCTTTCCAGAGGAAGCC (SEQ ID N0:11), the reverse TIGR exon 1
CA 02345923 2001-03-22
WO 99/16898 PCT/CA98/00923
33
primer was TTGGGTTTCCAGCTGGTC (SEQ ID NO:15). PCR was performed
using standard protocols, annealing temperature was at 60°C.
Amplification
products were electrophoresed in 1.5% agarose gels before ethidium staining
and scored by two independent observers.
5
EXAAAPLE 5
The TIGR wild-tvae protein and Lys423G1u mutated TIGR protein form hi4h
molecular weight complexes in vivo and in vitro
Methods
10 Construction of cDNA expression vectors. To create an
eukaryotic expression vector encoding the wild-type TIGR cDNA, a 1831 by
HindIII/Notl fragment encompassing 36 by of the 5' untranslated region, the
full-length 1512 by ORF and 188 by of the 3' untranslated region of TIGRwt
cDNA was purified from pTIGwt and subcloned directionnally into the
15 HindIII/Notl sites of plasmid pRc/CMV (Invitrogen). This new construct was
named pRcTIG and produced high constitutive levels of mRNA due to the
presence of CMV enhancer-promoter sequences. An expression vector
encoding a TIGR cDNA carrying the Lys423G1u mutation was then generated
by site-directed mutagenesis of pRcTIG using the QuikChange mutagenesis kit
20 (Stratagene). The primers used to generate the A to G transition in pRcTIG
were: 5'-GGGAGACAAACATCCGTGAGCAGTCAGTCGCC-3' (SEQ ID N0:16)
and 5'-GGCGACTGACTGCTCACGGATGTTTGTCTCCC-3' (SEQ ID N0:17).
This second expression vector was named pRcTIG423E. All constructions were
verified by sequencing the inserts bidirectionnally between the Hindlll and
Notl
25 sites.
Transfections
COS-7 cells were grown in Dulbecco's modified Eagle's
medium (DMEM) (Gibco BRL) supplemented with 10% fetal bovine serum
(Gibco BRL) and antibiotics (100 U/ml penicillin, 100 mg/ml streptomycin
(Gibco
30 BRL)). Cells were plated at a density of 2x106 cells per 75 cm2 tissue
culture
CA 02345923 2001-03-22
WO 99/16898 PCT/CA98/00923
34
flask. Twenty-four h later, transient transfection was performed using the
DNA/DEAE-dextran transfection method coupled with a chloroquine treatment
and followed by a dimethylsulfoxide shock. Each DNAIDEAE-dextran
transfection sample contained a total of 20 mg/flask of plasmid pRcTIG or
5 pRcTIG423E, alone or in combination. Briefly, COS-7 cells were exposed to
the
DNA/DEAE-dextran solution (0.5 mg/ml of DEAF in PBS; 5ml/flask) for 30 min
before addition of 20 ml of DMEM (w/o serum)/flask containing chforoquine to
reach a final concentration of 0.02 mg/ml for a further 2.5 h incubation
period.
The solutions were then aspirated and the cells exposed to DMSO (10% DMSO
10 in 5 ml DMEM with serum/flask) for exactly 90 seconds. The solutions were
again aspirated and the cells incubated in 25 ml DMEM supplemented with 10%
fetal bovine serum and antibiotics for 2 days before protein extraction. The
culture medium was replaced between the second and third day.
Reticulocyte lysate transcription-translation coupled system.
15 In vitro transcription/translation was performed using the TNT Coupled
Reticulocyte Lysate System (Promega) according to the manufacturer's protocol.
As both pRcTIG and pRcTIG423E constructs carried the T7 RNA promoter, no
additional DNA modifications were done before testing. Reactions contained 0.5
microgram of each plasmid and a complete amino acid mixture instead of
20 35S-methionine. Canine Pancreatic Microsomal Membranes (Promega) (2.5 ml
(2EQ/ml)/ 25 ml reaction) were added to the samples to optimize
cotranslational
processing. The samples were incubated for 90 min at 30°C before
protein
analysis.
Protein extraction
25 SDS-PAGE and Western-blot analysis Confluent cells from
tissue culture flasks were washed twice with cold PBS, scraped in 2 ml of ice
cold lysis buffer (10 mM tris-HCI pH 7.5) containing the Complete Protease
Inhibitor Cocktail (Boehringer Mannheim) (1 tablet/40 ml Tris-HCI). Human
trabeculum meshwork (HTM) tissues were dissected from donor eyes obtained
30 from the CHUL Eye Bank and lysed in 1 ml of cold lysis buffer. Protein
CA 02345923 2001-03-22
WO 99/16898 PCT/CA98/00923
concentration was estimated and, dithiothreitol (DDT; 0.1 mM) was added to
selected samples for 10 minutes, prior to electrophoresis, to reduce disulfide
links that may be involved in multimerization. SDS-PAGE was performed on 6%
resolving acrylamide gels using 5% stacking gels. Protein were transferred
onto
5 nitrocellulose, using a Trans-Blot SD Semi-Dry Transfer Cell apparatus
(Bio-Rad) for 1 h at 150 mA. Nitrocellulose membranes were saturated for 4 h
at 4°C in TBS containing 0.1 % Tween-20 (TBS-T) and 5% (w/v) skimmed
milk
powder (blotto), rinsed once (15 min) in TBS-T and incubated O/N at 4°C
with
purified TIGR antibodies diluted (1/2000) in blotto (1%). After 1-15 min and 3
-5
10 min washes with TBS-T, the nitrocellulose membranes were incubated for 1 h
at room temperature with goat-anti rabbit antibodies labeled with horseradish
peroxidase (1:5000, Amersham) in blotto (1%), washed four times (1-15 min and
3-5 min) with TBS-T, revealed 2 min with Renaissance Western blot
Chemiluminescence Reagent (Dupont NEN), and exposed to Reflection
15 autoradiography films (Dupont NEN) during increasing periods of time
ranging
from 30 s to 5 min.
According to homoallelic complementation, the Lys423G1u
mutation acts in a dominant negative fashion resulting in defective TIGR
wildtype/Lys423G1u protein heteromultimers but functional TIGR
20 Lys423G1u/Lys423G1u homopolymers. This model also demonstrates that a
locus with a wild-type allele A and a mutant allele A', such that homozygosity
for
either allele has no phenotypic consequence, but the heterozygous state AA'
leads to a deleterious defect due to interference between the protein products
of the two different alleles. Homoallelic complementation further refers to
25 affected heterozygotes in which some functional TIGR wild-type/wild-type
and/or
TIGR Lys423G1u/Lys423G1u homopolymers are also generated by an admixture
of normal and mutant subunits, thereby explaining part of the phenotypic
variability observed at this locus.
Examination of the amino acid sequence of the TIGR gene
30 product revealed two features potentially involved in protein-protein
interactions
CA 02345923 2001-03-22
WO 99!16898 PCT/CA98/00923
36
1 ) a leucine zipper domain containing seven motifs within the N-terminal half
of
the polypeptide and 2) a conserved cysteine at position 433 in a region highly
homologous to bullfrog olfactomedin in which the equivalent cysteine residue
may promote the formation of disulfide-linked homopolymers (Figure 2).
5 The discovery that the TIGR wild-type protein and Lys423G1u
TIGR mutated protein form high molecular weight complexes in vivo and in vitro
(Figure 6) provides evidence for homoallelic complementation as the mechanism
accounting for the unaffected status of TIGR Lys423G1u/Lys423G1u mutant
homozygotes. As shown in Figure 5, the TIGR wild-type protein and Lys423G1u
10 TIGR mutated protein form high molecular weight complexes in vivo and in
vitro.
COS-7 cells were transiently transfected with expression vectors encoding the
TIGR wild-type or TIGR Lys423G1u gene products, alone or in combination, and
newly synthesized proteins were analyzed by immunoblotting. In cells
transfected with the TIGR wild-type cDNA construct, pRcTIG, bands of
15 immunoreactivity were detected as two major complexes composed of TIGR
polypeptides migrating at approximately 120 kD and 240 kD (Figure 6 a and b).
When the denaturing agent dithiothreitol (DDT) was added to the extracts prior
to electrophoresis, both complexes were resolved into two lower molecular
weight forms migrating at approximately 57 kD and 62 kD (Figure 6 a and b). As
20 deduced from the nucleotide sequence, the 57 kD form corresponded to TIGR
monomers originating at the first translation initiation site of the TIGR mRNA
while the 62 kD form represented partially glycosylated monomers. In cells
transfected with increasing concentrations of TIGR Lys423G1u cDNA in the
presence of decreasing amount of the wild-type construct, the pattern of
25 immunoreactivity was identical to that detected when transfections were
performed with pRcTIG alone indicating that TIGR Lys423G1u gene products did
not disrupt the formation of the two major complexes (Figure 6 a and b).
Immunoblotting of protein extracts obtained from human trabecular meshwork
(HTM) tissues dissected from donor eyes revealed a similar pattern of
migration
30 establishing that TIGR polypeptides also formed high molecular weight
CA 02345923 2001-03-22
WO 99/16898 PCT/CA98/00923
37
complexes in vivo (Figure 6b). Under denaturing condition, HTM TIGR
polypeptides were resolved into three distinct monomers: the 57 and 62 kD
products and a smaller, approximately 55 kD, TIGR polypeptide resulting from
internal cleavage or, utilization of a second initiation site for translation
5 (Figure 6b).
To show that TIGR polypeptides did not required extraneous
proteins to undergo oligomerization, we used an in vitro reticulocyte lysate
system in which transcription of TIGR wild-type and TIGR Lys423G1u cDNAs
was coupled to translation of their respective mRNAs. Following protein
10 extraction and immunoblotting, we observed that both TIGR wild-type and
TIGR
Lys423G1u polypeptides formed homodimers migrating at the expected 120 kD
position (Figure 6c). These data showed that the high molecular weight
complexes observed in COS-7 cells were produced by multimerization of TIGR
monomers, generating homodimers and homotetramers.
15 Although the present invention has been described
hereinabove by way of preferred embodiments thereof, it can be modified,
without departing from the spirit and nature of the subject invention as
defined
in the appended claims.