Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02350829 2001-05-14
WO 00/29602 PCT/US99/08277
TRANSFECZ10N OF MALE GERM CELLS FOR GENERATION OF SELECTABLE TRANSGENIC STEM
CELLS
BACKGROUND OF THE INVENTION
Throughout this application various publications are referenced within
parentheses.
The disclosures of these publications in their entireties are hereby
incorporated by reference
in this application in order to more fully describe the state of the art to
which this invention
pertains.
1. THE FIELD OF THE INVENTION
This invention relates to the medical arts, particularly to the field of
transgenics and
gene therapy. The invention is particularly directed to the field of
transgenic vertebrate stem
cells.
2. DISCUSSION OF THE RELATED ART
The field of transgenics was initially developed to understand the action of a
single
gene in the context of the whole animal and phenomena of gene activation,
expression, and
interaction. This technology has been used to produce models for various
diseases in humans
and other animals. Transgenic technology is among the most powerful tools
available for the
study of genetics, and the understanding of genetic mechanisms and function.
It is also used
to study the relationship between genes and diseases. About 5,000 diseases are
caused by a
single genetic defect. More commonly, other diseases are the result of complex
interactions
between one or more genes and environmental agents, such as viruses or
carcinogens. The
understanding of such interactions is of prime importance for the development
of therapies,
such as gene therapy and drug therapies, and also treatments such as organ
transplantation.
Such treatments compensate for functional deficiencies and/or may eliminate
undesirable
CA 02350829 2001-05-14
WO 00/29602 PCT/US99/08277
functions expressed in an organism. Transgenesis has also been used for the
improvement of
livestock, and for the large scale production of biologically active
pharmaceuticals.
Historically, transgenic animals have been produced almost exclusively by
micro
injection of the fertilized egg. The pronuclei of fertilized eggs are micro-
injected in vitro with
foreign, i.e., xenogeneic or allogeneic DNA or hybrid DNA molecules. The micro-
injected
fertilized eggs are then transferred to the genital tract of a pseudopregnant
female. (E.g.,
P.J.A. Krimpenfort et al., Transgenic mice depleted in mature T-cells and
methods for making
transgenic mice, U.S. Pat. Nos. 5,175,384 and 5,434,340; P.J.A. Krimpenfort et
al.,
Transgenic mice depleted in mature lymphocytic cell-type, U.S. Pat. No.
5,591,669).
The generation of transgenic animals by this technique is generally
reproducible, and
for this reason little has been done to improve on it. This technique,
however, requires large
numbers of fertilized eggs. This is partly because there is a high rate of egg
loss due to lysis
during micro-injection. Moreover manipulated embryos are less likely to
implant and survive
in utero. These factors contribute to the technique's extremely Low
efficiency. For example,
300-500 fertilized eggs may need to be micro injected to produce perhaps three
transgenic
animals. Partly because of the need to micro-inject large numbers of embryos,
transgenic
technology has largely been exploited in mice because of their high fecundity.
While small
animals such as mice have proved to be suitable models for certain diseases,
their value in this
respect is Limited. Larger animals would be much more suitable to study the
effects and
treatment of most human diseases because of their greater similarity to humans
in many
aspects, and also the size of their organs. Now that transgenic animals with
the potential for
human xenotransplantation are being developed, larger animals, of a size
comparable to man
will be required. Transgenic technology will allow that such donor animals
will be
immunocompatible with the human recipient. Historical transgenic techniques,
however,
require that there be an ample supply of fertilized female germ cells or eggs.
Most large
mammals, such as primates, cows, horses and pigs produce only 10-20 or less
eggs per animal
per cycle even after hormonal stimulation. Consequently, generating large
animals with these
techniques is prohibitively expensive.
This invention relies on the fact that spermatogenesis in male vertebrates
produces vast
numbers of male germ cells that are more readily available than female germ
cells. Most male
mammals generally produce at least 10g spermatozoa (male germ cells) in each
ejaculate. This
2
CA 02350829 2001-05-14
w0 00/29602 PC'T/US99/08277
is in contrast to only 10-20 eggs in a mouse even after treatment with
superovulatory drugs.
A similar situation is true for ovulation in nearly all larger animals. For
this reason alone,
male germ cells will be a better target for introducing foreign DNA into the
germ line, leading
to the generation of transgenic animals with increased efficiency and after
simple, natural
mating.
Spermatogenesis is the process by which a diploid spermatogonial stem cell
provides
daughter cells which undergo dramatic and distinct morphological changes to
become self
propelling haploid cells (male gametes) capable, when fully mature, of
fertilizing an ovum.
Primordial germ cells are first seen in the endodermal yolk sac epithelium at
E8 and
are thought to arise from the embryonic ectoderm (A. McLaren and Buehr, Cell
Diff. Dev.
31:185 [1992]; Y. Matsui et al., Nature 353:750 [1991]). They migrate from the
yolk sac
epithelium through the hindgut endoderm to the genital ridges and proliferate
through mitotic
division to populate the testis.
At sexual maturity the spermatogonium goes through 5 or 6 mitotic divisions
before
it enters meiosis. The primitive spermatogonial stem cells (Ao/As) proliferate
and form a
population of intermediate spermatogonia types Apr, Aal, A1-4 after which they
differentiate
into type B spermatogonia. The type B spermatogonia differentiate to form
primary
spermatocytes which enter a prolonged meiotic prophase during which homologous
chromosomes pair and recombine. The states of meiosis that are morphologically
distinguishable are; preleptotene, leptotene, zygotene, pachytene, secondary
spermatocytes
and the haploid spermatids. Spermatids undergo great morphological changes
during
spermatogenesis, such as reshaping the nucleus, formation of the acrosome and
assembly of
the tail (A.R. Bellve et al., Recovery, capacitation, acrosome reaction, and
fractionation of
sperm, Methods Enzymol. 225:113-36 [1993]). The spermatocytes and spermatids
establish
vital contacts with the Sertoli cells through unique hemi functional
attachments with the
Sertoli cell membrane. The final changes in the maturing spermatozoan take
place in the
genital tract of the female prior to fertilization.
Initially, attempts were made to produce transgenic animals by adding DNA to
spermatozoa which were then used to fertilize mouse eggs in vitro. The
fertilized eggs were
then transferred to pseudopregnant foster females, and of the pups born, 30%
were reported
to be transgenic and express the transgene. Despite repeated efforts by
others, however, this
3
CA 02350829 2001-05-14
WO OOI29602 PCTIUS99/08277
experiment could not be reproduced and no transgenic pups were obtained.
Indeed, there
remains little doubt that the transgenic animals reputed to have been obtained
by this method
were not transgenic at all and the DNA incorporation reported was mere
experimental artifact.
Data collected from laboratories around the world engaged in testing this
method showed that
no transgenics were obtained from a total of 890 pups generated.
In summary, it is currently possible to produce live transgenic progeny but
the
previously available methods are costly and extremely inefficient. Therefore,
there is a
definite need for a simple, less costly and less invasive method of producing
transgenic
animals.
There has also been a need for a way of selecting or isolating stem cells from
non-stem
cells, for study or therapeutic uses, that does not require the use of
embryonic material,
because the use of embryonic material may present ethical problems. In
addition, the study
of stem cells specifically in the physiologic milieu of non-embryonic (e.g.;
adult) vertebrates
has been hampered by the difficulty of selecting, identifying, or isolating
stem cells from non-
stem cells in the tissues of these organisms.
A stem cell is an undifferentiated mother cell that is self renewable over the
life of the
organism and is multipotent, i.e., capable of generating various committed
progenitor cells
that can develop into fully mature differentiated cell lines. (T. Zigova and
P.R. Sanberg, The
rising star of neural stem cell research, Nature Biotechnol. 16(11):1007-08
[1998]). All
vertebrate tissues arise from stem cells, including hematopoietic stem cells,
from which
various types of blood cells derive; neural stem cells, from which brain and
nerve tissues
derive; and germ cells, from which male or female gametes derive.
Recently, there has been a great deal of interest in transgenic stem cells as
a potential
therapeutic tool for patients suffering from genetic diseases, metabolic
defects, varying kinds
of trauma, diseases of the nervous system, or cancers of the blood. In
manipulating stem cells
in vitro or in vivo it is important to be able to identify and select stem
cells of interest from
non-stem cells.
Tsukamoto et al. disclosed a method for identifying human hematopoietic stem
cells
based on specific antibody binding to Thy-1 and CD34 surface epitopes.
(A.Tsukamoto et al.,
Identification and isolation of human hematopoietic stem cells, U.S. Pat. No.
5,643,741).
Tsukamoto et al. taught embodiments of their method in which the antibodies
are labeled with
4
CA 02350829 2001-05-14
WO 00/29602 PC'T/US99/08277
a fluorochrome and detection of stem cells is by fluorescence activated cell
sorter (FACS).
Murray et al. taught a method of purifying a population of hematopoietic stem
cells
expressing a CDw109 marker that used binding of monoclonal antibodies specific
for
Cdw109. (L. Murray et al., Method of purling a population of cells enriched
for
S hematopoietic stem cells, populations of cells obtained thereby and methods
of use thereof,
U.S. Pat. No. 5,665,557).
Transgenic neural stem cells (NSCs) have also been identified and selected
using
immunofluorescence or other immunostaining techniques. (J.D. Flax et al.,
Engraftable
human neural stem cells respond to developmental cues, replace neurons, and
express foreign
genes, Nature Biotechnol. 16(11):1033-39 [1998]; O. Bruestle et al., Chimeric
brains
generated by intraventricular transplantation of fetal human brain cells into
embryonic rats,
Nature Biotechnol. 16(11):1040-44 [1998]).
However, such immunologically based methods as these have limited usefulness
in
identifying or selecting stem cells, because they rely on tissue- or lineage-
specific epitopes and
do not consistently leave the cells in a viable condition. Others have
addressed the latter
problem using non-lethal methods for labeling transgenic cells, particularly
using genes
encoding fluorescent or bioluminescent markers. For example, Chalfie et al.
disclosed a
recombinant DNA molecule comprising the green fluorescent protein gene
operatively linked
to any exogenous regulatory element. (M. Chalfie et al., Uses of green
fluorescent protein,
U.S. Pat. No. 5,491,084). Cormier et al. taught a recombinant DNA vector
comprising the
gene for apoaequorin, a bioluminescent protein. (M.J. Cormier et al.,
Recombinant DNA
vectors capable of expressing apoaequorin, USPN 5,422,266).
Contag et al. disclosed a method for detecting a transformed cell of interest
expressing
a light-generating moiety in vivo. (C.H. Contag, Non-invasive localization of
a light-emitting
conjugate in a mammal, U.S. Pat. No. 5,650,135). Similarly, Horan et al.
disclosed a method
for tracking cells in vivo related to labeling cells with a fluorecent cyanine
dye. (P.K. Horan
et al., In vivo cellular tracking, U.S. Pat. No. 4,762,701). And Patterson et
al. taught a
method of detecting cells expressing a specific nucleotide target sequence by
using
fluorescently labeled complementary nucleic acid probes and fluorescence-
activated flow
cytomety {FACS). (Patterson et aL, Method of detecting amplified nucleic
sequences in cells
by,flow cytometry, U.S. Pat. No. 5,840,478).
5
CA 02350829 2001-05-14
w0 00/29602 PCT/US99t08277
Lineage specific stem cell promoters and other regulatory elements are
available that
could be linked to the expression of a marker gene. For example, Burn et al.
taught the use
of a CD34 promoter, specific to hematopoietic stem cells. (T.C. Burn et al.,
Hematopoietic
stem cell specif c gene expression, U.S. Pat. No. 5,556,954}.
Gay disclosed a method of isolating a lineage specific stem cell in vitro.
(D.A. Gay,
Method of isolating a lineage specific stem cell in vitro, U.S. Patent No.
5,639,618). The
method involved in vitro transfection of pluripotent embryonic stem cells with
a construct
comprising a lineage specific promoter sequence operably linked to a DNA
encoding a
fluorescent or other reporter protein. But this method was not applicable in a
generalized way
to selecting stem cells in vitro or in vivo in transgenic animals. For this
purpose, there has
been a definite need for a promoter sequence that operates in a wide variety
of stem cells,
rather than regulating transcription in a lineage specific manner.
The differentiation of stem cells into somatic cells as well as normal cell
growth
depend on the regulation of the cell cycle. Dysfunction of this regulation can
lead to
uncontrolled cell growth and cancer (L.H. Hartwell and M.B. Kastan, Cell cycle
control and
cancer, Science 266:1821-28 [1994]). Important in the regulation of growth and
differentiation are the cyclins. Cyclins are positive regulators of cyclin-
dependent kinases
(CDKs), with which they can form activated complexes that play a central role
in driving the
cell through the cell cycle. The activities of these CDK's are regulated by
sequential
activating and inactivating phosphorylation and de-phosphorylation events.
(D.O. Morgan,
Principles of CDK regulation, Nature (Lond.) 374:131-34 [1995]; C.J. Sherr,
Phase
progression: cycling on cue, Cell 79:551-555 [1994]; P. Nurse, Ordering S
phase and M
phase in the cell cycle, Cell 79:547-50 [1994]). Negative regulators called
CDK inhibitors
can bind to and inhibit CDK's, adding another layer of regulation (T. Hirama
and H.P.
Koeffler, Role of the cyclin-dependent kinase inhibitors in the development of
cancer, Blood
86:841-54 [1995]; C.J. Sherr and J.M. Roberts, Inhibitors of mammalian Gl
cyclin-dependent
kinases, Genes Dev. 9:1149-1163 [1995]).
The kinase activity of the cyclin A/CDK2 complex, which rises at the G, to S
transition, is required for entry into S phase (K.A. Heichman and J.M.
Roberts, Rules to
replicate by, Cell 79:557-62 [1994]; M. Pagano et al., Cyclin A is required at
two points in
the human cell cycle, EMBO J. 11:961-71 [1992]; J. Pines and T. Hunter, Human
cyclin A is
6
CA 02350829 2001-05-14
WO 00/29602 PCTNS99/08277
adenovirus EIA-associated protein p60 and behaves differently from cyclin B,
Nature
346:760-63 [1990]; C. Desdouets et al., Cyclin A: function and expression
during cell
proliferation, Prog. Cell Cycle Res. 1:15-23 [1995]). Cyclin A also forms a
complex with
CDC2, the activity of which peaks at the GZ to M transition, and the kinase
activity of cyclin
A/CDC2 is also required for M-phase entry (M. Pagano et al. [1992]}.
Two kinds of cyclin A were first found in Xenopus; early embryos contained
both
cyclin A1 and cyclin A2. Later in development, cyclin A2, which shares
considerable
homology to mammalian cyclin A2, was found throughout the embryo, whereas
cyclin A1
was found only in the testis and ovary. ( J.A. Howe et al., Identification of
a developmental
timer regulating the stability of embryonic cyclin A and a new somatic A-type
cyclin at
gastrulation, Genes Dev. 9(10):164-76 [1995]). In the mouse, cyclin A2 was
found in a
number of tissues during development, but cyclin A1 expression was highly
restricted, with
high levels measured in late pachytene spermatocytes. (C. Sweeney et al., A
distinct cyclin
A is expressed in germ cells in the mouse, Development 122(1):53-64 [1996]).
1 S Cyclin A1 is not expressed in fully differentiated cells of non-embryonic
tissues, but
can be expressed in a wide variety of stem cells, including male and female
germ cells, brain
stem cells, hematopoietic progenitor cells, as well as in a majority of
myeloid leukemic cells
and undifferentiated hematological malignancies. (R. Yang et al.,
Characterization of a
second human cyclin A that is highly expressed in testis and in several
leukemic cell lines,
Cancer Res. 57(5):913-20 [1997]; A. Kramer et al., Cyclin AI is predominantly
expressed in
hematological malignancies with myeloid differentiation, Leukemia 12(6):893-98
[1998]; C.
Sweeney et al. [1996]; J.A. Howe et al. [1995]). The pattern of cyclin A1
expression
indicates that its regulation differs from that of cyclin A2, and this may be
related to
differential binding by cyclin A1 and cyclin A2 promoters of transcriptional
initiation factors,
such as the Spl family of initiation factors.
The Spl family of initiation factors is related to the regulation of
differentiation in
stem cells. (K.L. Block et al., Blood 88:2071-80 [1996]; H.M. Chen et al., J.
Biol. Chem.
268:8230-39 [1993]; R.K. Margana et al., J. Biol. Chem. 272:3083-90 [1997]).
Spl is
expressed at high levels in tissues where cyclin A1 expression is found. (C.
Sweeney et al.
[1996]). Also, induction of Spl was found to be associated with
differentiation of embryonal
carcinoma cells and Sp 1 was causally linked to expression of the fibronectin
gene, providing
7
CA 02350829 2001-05-14
WO 00/29602 PCTNS99/08277
evidence for a role of Spl in differentiation. (M. Suzuki et al., Molecular &
Cellular Biology
18: 3010-3020 [1998]). In adult tissue, high levels of Spl have been reported
in
hematopoietic progenitors and in the later stages of spermatogenesis. (J.D.
Safer et al.,
Molecular & Cellular Biology 11: 2189-2199 [1991]).
Levels of Spl vary up to 10-fold in different tissues. (J.D.Safer et al.
[1991]). This
could provide a basis for directing tissue specific expression in stem cells,
especially if the
affinity of the cis-acting Spl family binding sites of various promoters
differ. Another
mechanism of tissue-directed expression depends on the molar ratios of Spl
family members
to each other resulting in either activation or repression of transcription.
(A.P. Kumar et al.,
Nucleic Acids Res. 25:2012-19 [1997]; M.J. Birnbaum et al., Biochem.. 34:16503-
08
[1995]).
Spl has been shown to serve distinct roles in transcriptional activation: it
can directly
interact with the basal transcription complex. (A. Emili et al., Molec. Cell.
Biol. 14:1582
93 [1994]) and it can determine the transcription start site in TATA-less
promoters (J. Lu et
al., J. Biol. Chem. 269:5391-5402 [1994]). However, Spl can also function as a
more
general transcriptional activator, and an Spl family member, Sp3 protein, is
known to
function either as transcriptional activator or repressor depending on the
context of the
binding site in a promoter. (D. Apt et al., Virol. 224:281-91 [1996]; B.
Majello et al., J. Biol.
Chem. 272:4021-26 [1997]). When Sp3 binds to a single site, it can activate
transcription but
binding to multiple sites can lead to strong transcriptional repression ( M.J.
Birnbaum et al.,
Biochem.. 34:16503-08 [1995]).
Also, since myb was shown to be expressed in male germs cells, myb probably
acts
as an important transcriptional factor for expression from the cyclin A1
promoter during
spermatogenesis as well as hematopoiesis. (J. Sitzmann et al., Expression of B-
Myb during
mouse embryogenesis, Oncogene 12:1889-94 [1996]; K. Latham et al., Oncogene
13:1161-68
[1998]). The structure of myb protein includes a helix-turn-helix motif
involved with DNA
recognition. (M.D. Can et al., Eur. J. Biochem. 235:721-735 [1996]). The myb
proteins bind
DNA as monomers, with cooperative binding of the R2 and R3 regions within the
major
groove to the consensus myb binding site, MBS (c/TAAcNG). (K.M. Howe and R.J.
Watson,
EMBO J. 9:161-69 [1990]; K. Ogata et al., Nature Struct. Biol. 2:309-20
[1995]). The precise
role of myb transcription factors in cell cycle regulation is unknown but as a
transcriptional
8
CA 02350829 2001-05-14
WD OOI29602 PCTNS99/08277
activator they may be important for the activation of cell cycle genes such as
cyclin A1.
(Reviews: S.A. Ness, Biochim Biophys. Acta 1288:F123-F139 [1996]; M.K. Saville
and R.J.
Watson, Adv. Cancer Res. 72:109-40 [1998]).
The present invention addresses the need for spermatogenic transfection,
either in vitro
or in vivo, that is highly effective in transferring allogeneic as well as
xenogeneic genes into
the animal's germ cells and in producing transgenic vertebrate animals. The
present
technology addresses the requirements of germ line and stem cell line gene
therapies in
humans and other vertebrate species. Further, the method of the present
invention particularly
addresses the problem of identifying and selecting stem cells from non-stem
cells including
differentiated somatic cells, especially from non-embryonic biological
sources.
These and other benefits and features of the present invention are described
herein.
SUMMARY OF THE INVENTION
The present invention relates to the in vivo and ex vivo (in vitro)
transfection of
eukaryotic animal germ cells with a desired genetic material. Briefly, the in
vivo method
involves injection of genetic material together with a suitable vector
directly into the testicle
of the animal. In this method, all or some of the male germ cells within the
testicle are
transfected in situ, under effective conditions. The ex vivo method involves
extracting germ
cells from the gonad of a suitable donor or from the animal's own gonad, using
a novel
isolation method, transfecting them in vitro, and then returning them to the
testis under
suitable conditions where they will spontaneously repopulate it. The ex vivo
method has the
advantage that the transfected germ cells may be screened by various means
before being
returned to the testis to ensure that the transgene is incorporated into the
genome in a stable
state. Moreover, after screening and cell sorting only enriched populations of
germ cells may
be returned. This approach provides a greater chance of transgenic progeny
after mating.
This invention also relates to a novel method for the isolation of
spermatogonia,
comprising obtaining spermatogonia from a mixed population of testicular cells
by extruding
the cells from the seminiferous tubules and gentle enzymatic disaggregation.
The
spermatogonia or stem cells which are to be genetically modified, may be
isolated from a
mixed cell population by a novel method including the utilization of a
promoter sequence,
which is only active in stem cells, for example the cyclin A1 promoter, or in
cycling
9
CA 02350829 2001-05-14
WO 00/29602 PCTNS99/08277
spermatogonial stem cell populations, for example, B-Myb promoter or a
spermotogonia
specific promoter, such as the c-kit promoter region, c-raf 1 promoter, ATM
(ataxia-
telangiectasia) promoter, RBM (ribosome binding motif) promoter, DAZ (deleted
in
azoospermia) promoter, XRCC-1 promoter, HSP 90 (heat shock gene) promoter, or
FRMI
S (from fragile X site) promoter, optionally linked to a reporter construct,
for example, a
construct encoding Green Fluorescent Protein (EGFP), Yellow Fluorescent
Protein, Blue
Fluorescent Protein, a phycobiliprotein, such as phycoerythrin or phycocyanin,
or any other
protein which fluoresces under suitable wave-lengths of ultraviolet light.
These unique
promoter sequences drive the expression of the reporter construct only in the
cycling
spermatogonia or stem cells in which they operate. The spermatogonia or stem
cells, thus,
are the only cells in the mixed population which will express the reporter
construct and they,
thus, may be isolated on this basis. Transgenic cells expressing a fluorescent
reporter
construct can be sorted with the aid of, for example, a flow activated cell
sorter (FACS) set
at the appropriate wavelength or they may be selected by chemical methods.
The present invention also relates to a method of obtaining selectable
transgenic stem
cells by transfecting a male germ cell with a DNA construct comprising a stem
cell-specific
promoter, for example, a cyclin A1 promoter, operatively linked to a gene
encoding a
fluorescent or light-emitting reporter protein. The present invention also
relates to selectable
transgenic stem cells that have stably integrated the DNA and non-human
transgenic
vertebrates comprising them. In stem cells other than germ cells, expression
of the reporter
gene from a cyclin A1 promoter in vivo is facilitated by preventing the
methylation of
promoter DNA by the use of flanking insulator elements. Alternatively, when
transgenic stem
cells are grown in vitro, inhibitors of DNA methylation can be added to the
culture medium.
For transfection, the method of the invention comprises administering to the
animal,
or to germ cells in vitro, a composition comprising amounts of nucleic acid
comprising
polynucleotides encoding a desired trait. In addition, the composition
comprises, for example,
a relevant controlling promoter region made up of nucleotide sequences. This
is combined
with, for example, a gene delivery system comprising a cell transfection
promotion agent such
as retro viral vectors, adenoviral and adenoviral related vectors, or
liposamal reagents or other
agents used for gene therapy. These introduced under conditions effective to
deliver the
nucleic acid segments to the animal's germ cells optionally with the
polynucleotide inserted
CA 02350829 2001-05-14
WO OOI29602 PCT/US99/08277
into the genome of the germ cells. Following incorporation of the DNA, the
treated animal
is either allowed to breed naturally, or reproduced with the aid of assisted
reproductive
technologies, and the progeny selected for the desired trait.
This technology is applicable to the production of transgenic animals for use
as
animal models, and to the modification of the genome of an animal, including a
human, by
addition, modification, or subtraction of genetic material, often resulting in
phenotypic
changes. The present methods are also applicable to altering the carrier
status of an animal,
including a human, where that individual is carrying a gene for a recessive or
dominant gene
disorder, or where the individual is prone to pass a multigenic disorder to
his offspring.
A preparation suitable for use with the present methods comprises a
polynucleotide
segment encoding a desired trait and a transfection promotion agent, and
optionally an uptake
promotion agent which is sometime equipped with agents protective against DNA
breakdown.
The different components of the transfection composition (mixture) are also
provided in the
form of a kit, with the components described above in measured form in two or
more separate
containers. The kit generally contains the different components in separate
containers and
instructions for effective use. Other components may also be provided in the
kit as well as a
carrier.
Thus the present technology is of great value in the study of stem cells and
cellular
development, and in producing transgenic vertebrate animals as well as for
repairing genetic
defects. The present technology is also suitable for germ line and stem cell
line gene therapy
in humans and other vertebrate animal species. The present invention is also
valuable in
identifying cell lineages before full differentiation to facilitate
modification and/or
engineering of specific tissues in vitro for their subsequent transplantation
in the treatment of
disease or trauma.
BRIEF DESCRIPTION OF THE DRAWINGS
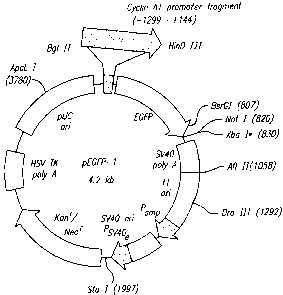
Figure 1 represents a map of DNA construct pCyclinAl-EGFP-1.
Figure 2 represents transcriptional start sites in the human cyclin A1 gene.
Figure 3 represents 5' upstream region of the human cyclin A1 gene.
Figure 4 represents transactivation activity of cyclin A1 promoter fragments
in Hela
cells.
11
CA 02350829 2001-05-14
W.O OO/Z9602 PCT/US99/08277
Figure 5 shows activity of the cyclin A1 promoter fragments in the Drosophila
cell
line S2.
Figure 6 shows effects of GC box (Spl site) mutations on promoter activity.
Figure 7 shows cell cycle regulated activity of the cyclin A1 promoter in Hela
cells.
Figure 8 shows germ line-specific expression of EGFP from a human cyclin A1
promoter in marine testicular tissue.
Figure 9 shows the positive association of cyclin A 1 promoter methylation
with
silencing of a cyclin A1 promoter - EGFP transgene in MG63 cells and the
repression of
cyclin Ai promoter activity by methylation and MeCP2 in S2 Drosophila cells.
I O Figure 10 shows a comparison of reporter gene expression from different
promoters,
including the cyclin A1 promoter, in cell lines from various tissues.
Figure 11 shows transactivation of the cyclin A1 promoter by c-myb.
DETAILED DESCRIPTION OF THE PREFERRED EMBODIMENTS
The present invention arose from a desire by the present inventors to improve
on
15 existing methods for the genetic modification of an animal's germ cells and
for producing
transgenic animals. The pre-existing art methods rely on direct injection of
DNA into zygotes
produced in vitro or in vivo, or by the production of chimeric embryos using
embryonal stem
cells incorporated into a recipient blastocyst. Following this, such treated
embryos are
transferred to the primed uterus or oviduct. The available methods are
extremely slow and
20 costly, rely on several invasive steps, and only produce transgenic progeny
sporadically and
unpredictably.
In their search for a less costly, faster, and more efficient approach for
producing
transgenics, the present inventors devised the present method which relies on
the in vivo or
ex vivo (in vitro) transfection of male animal germ cells with a nucleic acid
segment encoding
25 a desired trait. The present method relies on at least one of the following
strategies. A first
method delivers the nucleic acid segment using known gene delivery systems in
situ to the
gonad of the animal (in vivo transfection), allows the transfected germ cells
to differentiate
in their own milieu, and then selects for animals exhibiting the nucleic
acid's integration into
its germ cells (transgenic animals). The thus selected animals may be mated,
or their sperm
30 utilized for insemination or in vitro fertilization to produce transgenic
progeny. The selection
12
CA 02350829 2001-05-14
WO 00/29602 PCT/US99/08277
may take place after biopsy of one or both gonads, or after examination of the
animal's
ejaculate amplified by the polymerise chain reaction to confirm the
incorporation of the
desired nucleic acid sequence. In order to simplify the confirmation of the
actual
incorporation of the desired nucleic acid, the initial transfection may
include a co-transfected
reporter gene, such as a gene encoding for Green Fluorescent Protein (or
encoding enhanced
Green Fluorescent Protein [EGFP]), Yellow Fluorescent Protein, Blue
Fluorescent Protein,
a phycobiliprotein, such as phycoerythrin or phycocyanin, or any other protein
which
fluoresces under a suitable wave-length of ultraviolet light.
Alternatively, male germ cells may be isolated from a donor animal and
transfected,
IO or genetically altered in vitro to impart the desired trait. Following this
genetic manipulation,
germ cells which exhibit any evidence that the DNA has been modified in the
desired manner
are selected, and transferred to the testis of a suitable recipient animal.
Further selection may
be attempted after biopsy of one or both gonads, or after examination of the
animal's ejaculate
amplified by the polymerise chain reaction to confirm whether the desired
nucleic acid
sequence was actually incorporated. As described above, the initial
transfection may have
included a co-transfected reporter gene, such as a gene encoding the Green
Fluorescent
Protein (or enhanced Green Fluorescent Protein [EGFP]), Yellow Fluorescent
Protein, Blue
Fluorescent Protein, a phycobiliprotein, such as phycoerythrin or phycocyanin,
or any other
protein which fluoresces under light of suitable wave-lengths. Before transfer
of the germ
cells, the recipient testis are generally treated in one, or a combination, of
a number of ways
to inactivate or destroy endogenous germ cells, including by gamma
irradiation, by chemical
treatment, by means of infectious agents such as viruses, or by autoimmune
depletion or by
combinations thereof. This treatment facilitates the colonization of the
recipient testis by the
altered donor cells.
Animals that were shown to carry suitably modified sperm cells then may be
either
allowed to mate naturally, or alternatively their spermatozoa are used for
insemination or in
vitro fertilization. The thus obtained transgenic progeny may be bred, whether
by natural
mating or artificial insemination, to obtain further transgenic progeny. The
method of this
invention has a lesser number of invasive procedures than other available
methods, and a high
rate of success in producing incorporation into the progeny's genome of the
nucleic acid
sequence encoding a desired trait.
13
CA 02350829 2001-05-14
WO 00/29602 PCTNS99/08277
Primordial germ cells are thought to arise from the embryonic ectoderm, and
are first
seen in the epithelium of the endodermal yolk sac at the E8 stage. From there
they migrate
through the hindgut endoderm to the genital ridges. The primitive
spermatogonial stem cells,
known as AO/As, differentiate into type B spermatogonia. The latter further
differentiate to
form primary spermatocytes, and enter a prolonged meiotic prophase during
which
homologous chromosomes pair and recombine. Several morphological stages of
meiosis are
distinguishable : preleptotene, leptotene, zygotene, pachytene, secondary
spermatocytes, and
the haploid spermatids. The latter undergo further morphological changes
during
spermatogenesis, including the reshaping of their nucleus, the formation of
acrosome, and
assembly of the tail. The final changes in the spermatozoon take place in the
genital tract of
the female, prior to fertilization. The uptake of the nucleic acid segment
administered by the
present in vivo method to the gonads will reach germ cells that are at one or
more of these
stages, and be taken up by those that are at a more receptive stage. In the ex
vivo (in vitro)
method of genetic modification, generally only diploid spermatogonia are used
for nucleic
I S acid modification. The cells may be modified in vivo using gene therapy
techniques, or in
vitro using a number of different transfection strategies.
The inventors are, thus, providing in this patent a novel and unobvious method
for
isolation of male germ cells, and for the in vivo and ex vivo (in vitro)
transfection of
allogeneic as well as xenogeneic DNA into an animal's germ cells. This
comprises the
administration to an animal of a composition comprising a gene delivery system
and at least
one nucleic acid segment, in amounts and under conditions effective to modify
the animal's
germ cells, and allowing the nucleic acid segment to enter, and be released
into, the germ
cells, and to integrate into their genome.
The in vivo introduction of the gene delivery mixture to the germ cells may be
accomplished by direct delivery into the animal's testis (es}, where it is
distributed to male
germ cells at various stages of development. The in vivo method utilizes novel
technology,
such as injecting the gene delivery mixture either into the vasa efferentia,
directly into the
seminiferous tubules, or into the rete testis using, for example, a
micropipette. To ensure a
steady infusion of the gene delivery mixture, under pressures which will not
damage the
delicate tubule system in the testis, the injection may be made through the
micropipette with
the aid of a picopump delivering a precise measured volume under controlled
amounts of
14
CA 02350829 2001-05-14
WO 00/29602 PCT/US99/08277
pressure. The micropipette may be made of a suitable material, such as metal
or glass, and
is usually made from glass tubing which has been drawn to a fine bore at its
working tip, e.g.
using a pipette puller. The tip may be angulated in a convenient manner to
facilitate its entry
into the testicular tubule system. The micropipette may be also provided with
a beveled
working end to allow a better and less damaging penetration of the fine
tubules at the injection
site. This bevel may be produced by means of a specially manufactured grinding
apparatus.
The diameter of the tip of the pipette for the in vivo method of injection may
be about 15 to
45 microns, although other sizes may be utilized as needed, depending on the
animal's size.
The tip of the pipette may be introduced into the rete testis or the tubule
system of the testicle,
with the aid of a binocular microscope with coaxial illumination, with care
taken not to
damage the wall of the tubule opposite the injection point, and keeping trauma
to a minimum.
On average, a magnification of about x25 to x80 is suitable, and bench mounted
micromanipulators are not severally required as the procedure may be carried
out by a skilled
artisan without additional aids. A small amount of a suitable, non-toxic dye,
may be added
1 S to the gene delivery fluid to confirm delivery and dissemination to the
tubules of the testis.
It may include a dilute solution of a suitable, non-toxic dye, which may be
visualized and
tracked under the microscope.
In this manner, the gene delivery mixture is brought into intimate contact
with the
germ cells. The gene delivery mixture typically comprises the modified nucleic
acid encoding
the desired trait, together with a suitable promoter sequence, and optionally
agents which
increase the uptake of the nucleic acid sequence, such as liposomes,
retroviral vectors,
adenoviral vectors, adenovirus enhanced gene delivery systems, or combinations
thereof. A
reporter construct such as the gene encoding for Green Fluorescent Protein may
further be
added to the gene delivery mixture. Targeting molecules such as c-kit ligand
may be added
to the gene delivery mixture to enhance the transfer of the male germ cell.
For the ex vivo (in vitro) method of genetic alteration, the introduction of
the modified
germ cells into the recipient testis may be accomplished by direct injection
using a suitable
micropipette. Support cells, such as Leydig or Sertoli cells that provide
hormonal stimulus
to spermatogonial differentiation, may be transferred to a recipient testis
along with the
modif ed germ cells. These transferred support cells may be unmodified, or,
alternatively,
may themselves have been transfected, together with- or separately from the
germ cells.
CA 02350829 2001-05-14
WO 00/29602 PCTIUS99/08277
These transferred support cells may be autologous or heterologous to either
the donor or
recipient testis. A preferred concentration of cells in the transfer fluid may
easily be
established by simple experimentation, but will likely be within the range of
about 1 x 105 -
x 105 cells per 10 pl of fluid. This micropipette may be introduced into the
vasa efferentia,
5 the rete testis or the seminiferous tubules, optionally with the aid of a
picopump to control
pressure and/or volume, or this delivery may be done manually. The
micropipette employed
is in most respects similar to that used for the in vivo injection, except
that its tip diameter
generally will be about 70 microns. The microsurgical method of introduction
is similar in
all respects to that used for the in vivo method described above. A suitable
dyestuff may also
10 be incorporated into the carrier fluid for easy identification of
satisfactory delivery of the
transfected germ cells.
Once in contact with germ cells, whether they are in situ in the animal or
vitro, the
gene delivery mixture facilitates the uptake and transport of the xenogeneic
genetic material
into the appropriate cell location for integration into the genome and
expression. A number
of known gene delivery methods may be used for the uptake of nucleic acid
sequences into
the cell.
"Gene delivery (or transfection) mixture", in the context of this patent,
means selected
genetic material together with an appropriate vector mixed, for example, with
an effective
amount of lipid transfecting agent. The amount of each component of the
mixture is chosen
so that the transfection of a specific species of germ cell is optimized. Such
optimization
requires no more than routine experimentation. The ratio of DNA to lipid is
broad, preferably
about 1: 1, although other proportions may also be utilized depending on the
type of lipid
agent and the DNA utilized. This proportion is not crucial.
"Transfecting agent", as utilized herein, means a composition of matter added
to the
genetic material for enhancing the uptake of exogenous DNA segments) into a
eukaryotic
cell, preferably a mammalian cell, and more preferably a mammalian germ cell.
The
enhancement is measured relative to the uptake in the absence of the
transfecting agent.
Examples of transfecting agents include adenovirus-transferrin-polylysine-DNA
complexes.
These complexes generally augment the uptake of DNA into the cell and reduce
its
breakdown during its passage through the cytoplasm to the nucleus of the cell.
These
complexes may be targeted to the male germ cells using specific Iigands which
are recognized
16
CA 02350829 2001-05-14
WO 00/29602 PCT/US99/08277
by receptors on the cell swface of the germ cell, such as the c-kit ligand or
modifications
thereof.
Other preferred transfecting agents include lipofectin, lipfectamine, DIMRIE
C,
Superfect, and Effectin (Qiagen). Although these are not as efficient
transfecting agents as
viral transfecting agents, they have the advantage that they facilitate stable
integration of
xenogeneic DNA sequence into the vertebrate genome, without size restrictions
commonly
associated with virus-derived transfecting agents.
"Virus", as used herein, means any virus, or transfecting fragment thereof,
which may
facilitate the delivery of the genetic material into male germ cells. Examples
of viruses which
are suitable for use herein are adenoviruses, adeno-associated viruses,
retroviruses such as
human immune-deficiency virus, lentiviruses, such as Moloney marine leukemia
virus and
the retrovirus vector derived from Moloney virus called vesicular-stomatitis-
virus-
glycoprotein (VSV-G)-Moloney marine leukemia virus, mumps virus, and
transfecting
fragments of any of these viruses, and other viral DNA segments that
facilitate the uptake of
the desired DNA segment by, and release into, the cytoplasm of germ cells and
mixtwes
thereof. The mumps virus is particularly suited because of its affinity for
immatwe sperm
cells including spermatogonia. All of the above viruses may require
modification to render
them non-pathogenic or less antigenic. Other known vector systems, however,
may also be
utilized within the confines of the invention.
"Genetic material", as used herein, means DNA sequences capable of imparting
novel
genetic modification(s), or biologically functional characteristics) to the
recipient animal.
The novel genetic modifications) or characteristics) may be encoded by one or
more genes
or gene segments, or may be caused by removal or mutation of one or more
genes, and may
additionally contain regulatory sequences. The transfected genetic material is
preferably
functional, that is it expresses a desired trait by means of a product or by
suppressing the
production of another. Examples of other mechanisms by which a gene's function
may be
expressed are genomic imprinting, i.e. inactivation of one of a pair of genes
(alleles) during
very early embryonic development, or inactivation of genetic material by
mutation or deletion
of gene sequences, or by repression of a dominant negative gene product, among
others.
In addition, novel genetic modifications) may be artificially induced
mutations or
variations, or natural allelic mutations or variations of a gene(s). Mutations
or variations may
17
CA 02350829 2001-05-14
wo oon96o2 Pc~riusmosz~~
be induced artificially by a number of techniques, all of which are well known
in the art,
including chemical treatment, gamma irradiation treatment, ultraviolet
radiation treatment,
ultraviolet radiation, and the like. Chemicals useful for the induction of
mutations or
variations include carcinogens such as ethidium bromide and others known in
the art.
DNA segments of specific sequences may also be constructed to thereby
incorporate
any desired mutation or variation or to disrupt a gene or to alter genomic
DNA. Those skilled
in the art will readily appreciate that the genetic material is inheritable
and is, therefore,
present in almost every cell of future generations of the progeny, including
the germ cells.
Among novel characteristics are the expression of a previously unexpressed
trait,
I 0 augmentation or reduction of an expressed trait, over expression or under
expression of a trait,
ectopic expression, that is expression of a trait in tissues where it normally
would not be
expressed, or the attenuation or elimination of a previously expressed trait.
Other novel
characteristics include the qualitative change of an expressed trait, for
example, to palliate or
alleviate, or otherwise prevent expression of an inheritable disorder with a
multigenic basis.
For the expression of transfected genetic material to obtain a desired trait,
a promoter
sequence is operably linked to a polynucleotide sequence encoding the desired
trait or
product. A promoter sequence is chosen that operates in the cell type of
interest.
A promoter sequence, which is only active in cycling spermatogonial stem cell
populations can be used for differential expression in male germ cells, for
example, B-Myb
or a spermotogonia specific promoter, such as the c-kit promoter region, c-raf
I promoter,
ATM (ataxia-telangiectasia) promoter, RBM (ribosome binding motif) promoter,
DAZ
(deleted in azoospermia) promoter, XRCC-1 promoter, HSP 90 (heat shock gene)
promoter,
or FRMI (from fragile X site) promoter.
The human cyclin A1 promoter region is a most preferred promoter sequence for
driving the expression of a reporter construct or for driving the expression
of another desired
xenogeneic gene sequence, when expression is desired in germ cells,
hematopoietic cells,
other stem cells of a vertebrate.
The following nucleotide sequence represents the 5' end of the human cyclin A1
gene.
An untranscribed region extends from nucleotide -1299 to -1; a transcribed but
untranslated
region extends from +1 to +127, where the first ATG sequence begins; also
represented are
cyclin Al exon 1 (+1 to +234), intron 1 (+235 to +537), and part of exon 2
(beginning at
I8
CA 02350829 2001-05-14
WO 00/29602 PCT/US99/08277
+538), with transcribed regions being underlined and the translational start
site at nt. +127 to
+129 being bolded:
-1299 TCGATCTGAT TTAGAGATTT AGGGATGGAT GTTTTAAAAA AAGCAAAAGT
-1249 AGTAACAGAC TATAGCATTG GTAATGTGTG TGTGCATATA TACATATTAT
-1199 TTTTAAAAAA ATAAAGTTCG ATTATTTCAC CTGGCTTGTC AGTCACCTAT
-1149 GCAGGCGTCT GAGCCCCCGG GTTTCCAGGA GCCCCCCGTA TAAGGACCCC
-1099 AGGGACTCCT CTCCCCACGC GGCCGGGCCG CCCGCCCGGC CCCCAGCCCG
-1049 GAGAGCTGCC ACCGACCCCC TCAACGTCCC AAGCCCCAGC TCTGTCGCCC
-0999 GCGTTCCTTC CTCTTCCTGG GCCACAATCT TGGCTTTCCC GGGCCGGCTT
-0949 CACGCAGTTG CGCAGGAGCC CGCGGGGGAA GACCTCTCGTGGGGACCTCG
-0899 AGCACGACGT GCGACCCTAA ATCCCCACAT CTCCTCTGCC GCCTCGCAGG
-0849 CCACATGCAC CGGGAGCCGG GCGGGGCAGG CGCGGCCCGC AAGGACCCCC
-0799 GCGATGGAGA CGCAACACTG CCGCGACTGC ACTTGGGGCA GCCCCGCCGC
-0749 GTCCCAGCCG CCTCCCGGCA GGAAGCGTAG GTGTGTGAGC CGACCCGGAG
1 S -0699 CGAGCCGCGC CCTCGGGCCA GCGTGGGCAG GGCGCCGCAG CCTGCGCAGC
-0649 CCCGAGGACC CCGCGTCGCT CTCCCGAGCC AGGGTTCTCA GGAGCGGGCC
-0599 GCGCAGGAGA CGTTAGAGGG GGTTGTTAGC GGCTGTTGGG AGAACGGGTC
-0549 ACGGAAACAG TCCCTTCCAA AGCCGGGGCC ATCGTGGGGT GGGCGAGTCC
-0499 GCCCTCCCAG GCCGGGGGCG CGGACCAGAG GGGACGTGTG CAGACGGCCG
-0449 CGGTCAGCCC CACCTCGCCC GGGCGGAGAC GCACAGCTGG AGCTGGAGGG
-0399 CCGTCGCCCG TTGGGCCCTC AGGGGCCTGA ACGCCCAGGG GTCGCGGCGA
-0349 GTCCACCCGG AGCGAGTCAG GTGAGCAGGT CGCCATGGCG ATGCGGCCCC
-0299 GGAGAGCGCA CGCCTGCCGC GGTCGGCATG GAAACGCTCC CGCTAGGTCC
-0249 GGGGGCGCCG CTGATTGGCC GATTCAACAG ACGCGGGTGG GCAGCTCAGC
-0199 CGCATCGCTA AGCCCGGCCG CCTCCCAGGC TGGAATCCCT CGACACTTGG
-0149 TCCTTCCCGC CCCGCCCTTC CGTGCCCTGC CCTTCCCTGC CCTTCCCCGC
-0099 CCTGCCCCGC CCGGCCCGGC CCGGCCCTGC CCAACCCTGC CCCGCCCTGC
-0049 CCCGCCCAGC CGGCCACCTC TTAACCGCGA TCCTCCAGTG CACTTGCCA~
+0003 TTGTTCCGGA CACATAGAAA GATAACGACG GGAAGACGGG GCCCCGTTTG
+0053 GGGTCCAGGC AGGTTTTGGG GCCTCCTGTC TGGTGGGAGG AGGCCGCAGC
+0103 ~CAGCA~~'rT GCTCGTCACT TGGGAT GAG ACCGGCTTTC CCGCAATCAT
+0153 GTAC~''~'T~rA TS~AT TG G~~GCTGGGG AGAAGAGTAT CTCAGCTGGG
+0203 ~AGGACCC~G GCTCCCAGAT TTCGTCTTCC AGGTAACGTG GGTTTAGTAT
19
CA 02350829 2001-05-14
WO 00/29602 PCT/US99/08Z77
+0253 CCCGACTTGG AGGCTTGTCA GAATGTTTCT CTCCTTCCAG CCCAACACGA
+0303 AGTCTTGGGA TAAAAAGCCT CCCTCAGGGA TTCAAATAAC TGZTITGATT
+0353 CAGAGCAACT TTGATCGCCT GTGCGGTCGC ACCTGCCCTT TCAGCCCCAA
+0403 TAATTACTGG GAAGATCAGC AATTGGTGTT AGTCCCATTG CTTGGTGCTC
+0453 TCCCTCCTAG AGGTTCGCTG TGTCCTTGGA GCCCGGGGTG GACGGAATCG
+0503 ACTAAACAGC TTGTCTGTTT CTCTTTCCCT GGTAGCAGCA GCCCGTGGAG
+0553 TCTGA_A_GCAA TGCACTGCAG CA_ACCCCAAG AGTGGAGTTG TGCTGGCTAC
+0603 AGTGGCCCGA GGTCCCGATG CTTGTCAGAT ACTCACCAGA GCCCCGCTGG
+0653 GCC~1GGAT (SEQ. ID. NO.:1).
A most preferred embodiment of the cyclin A 1 promoter of the present
invention is
a DNA fragment with the sequence of nt. -1299 to +144, inclusive, having the
first
translational start site (the ATG in bold at nt. +127 to +129 of the human
sequence above)
changed to ATT (SEQ. ID. N0.2). Other preferred embodiments of a cyclin A1
promoter
include any operative fragment of SEQ. ID. N0.:2 or non-human homologue
thereof, or an
operative derivative of any of these. Preferred examples of an operative
fragment include the
-1151 to +144 fragment (SEQ. ID. N0.:3), the -454 to +144 fragment (SEQ. ID.
N0.:4), the
-326 to +144 fragment (SEQ. ID. NO.:S), the -190 to +144 fragment (SEQ. ID.
N0.:6), the
-160 to +144 fragment (SEQ. ID. N0.:7), the -120 to +144 fragment (SEQ. ID.
N0.:8), the
-I 12 to +144 fragment (SEQ. ID. N0.:9), all with ATG at +127 to +129 changed
as described
above. But any cyclin Al promoter fragment that includes the nucleotide
sequence extending
from nt. -112 downstream to at least nt. +5 or beyond, up to and including nt.
+144, is also
operative and useful, as long as the translational start site at +127 to +129
is no longer intact
and the essential Spl binding sites between -112 and -37 (GC Box Nos. 1, 2,
and 3 and/or 4)
are intact, as described below. Other preferred fragments, in accordance with
the present
invention, include those extending from -190 to +20 (SEQ. ID. NO.:10), or from
+190 to any
nucleotide between nt. +20 up to nt. +144 (without the translational start
site). But shorter
fragments such as -190 to +13 (SEQ. ID. NO.:11), -190 to +6 (SEQ. LD. N0.:12),
or -190 to
+5 (SEQ. ID. N0.:13) are also operative and useful. Non-human homologues
include any
cyclin A1 promoter sequence of non-human origin that functions in a vertebrate
stem cell type
of interest.
Another preferred embodiment of a cyclin A 1 promoter is an operative
derivative of
CA 02350829 2001-05-14
WO 00129602 PCT/US99/08277
SEQ. ID. N0:2, or of any operative fragment of SEQ. ID. N0.:2 or non-human
homologue
thereof, in which the codon of the first translational start site is changed
to another codon
sequence, other than ATT, that is also not recognized as a translational start
site; another
preferred cyclin A1 promoter is a derivative of SEQ. ID. N0.:2 with the codon
of the first
translational start site deleted altogether. Other operative derivatives
include cyclin Al promoter
sequences containing a mutation, polymorphism, or variant allele with respect
to any nucleotide
position of SEQ. ID. N0.:2 that does not eliminate promoter activity.
Similar to promoters in other cell cycle regulatory genes (B. Henglein et al.,
Proc.
Natl. Acad. Sci. (USA) 91:5490-94 [1994]; A. Hwang et al., J. Biol. Chem.
270:28419-24
[1995]; E.W. Lam et al., Oncogene 7:1885-90 [1992]), the cyclin A1 promoter
does not
possess a TATA-box motif. The nucleotides surrounding the transcriptional
start site are
likely to function as an initiator. The cyclin A1 promoter region contains
multiple binding
sites for transcription factor including GC boxes, Myb, and E2F sites.
The upstream region contains a GC rich region with multiple Sp 1 binding sites
that
are essential for transcription from the cyclin A 1 promoter. In contrast,
predicted GC boxes
in the cyclin A2 promoter are located more than 120 by upstream of the
transcriptional start
site and these have not been shown to be essential for gene expression. GC
boxes and the Spl
family transcription factors are important in the regulation of expression
from the cyclin A1
promoter. Six GC boxes are found in the first 200 by upstream of the
transcription start site.
Omitting the four GC boxes between -112 and -37 almost completely abrogates
promoter
activity. Among GC boxes Nos. 1-4, the two closest to the transcriptional
start sites are most
critical. Of GC boxes Nos. 3 and 4, only one of these is necessary for a basal
level of
transcriptional activity of the promoter.
Spl, the main activating factor of the Spl family, and Sp3 can bind to GC
boxes Nos.
1 + 2 and 3 + 4. Analysis of these fragments in insect cells demonstrates that
Spl
reconstitutes cyclin A1 promoter activity in all fi~agments that involve the
GC boxes Nos. 1-4.
Spl (or at least a member of the Spl family) is required for cyclin A1
promoter activity
through interaction with elements located between -112 and -37. Repression is
likely to be
accomplished by Sp3 and an as yet unidentified repressor mechanism that does
not depend
on E2F, CDE or CHR elements.
The DNA of animal cells is subject to methylation at the 5' carbon position of
the
21
CA 02350829 2001-05-14
WO 00/29602 PGTNS99/08Z77
cytidine bases of CpG dinucleotides. Unmethylated CpGs are found
preferentially in
transcriptionally active chromatin. (T. Naveh-Many et al., Active gene
seguences are
undermethylated, Proc. Natl. Acad. Sci. USA 78:4246-50 [1981]}.
Hypermethylation is
associated with transcriptional repression. (R. Holliday, The inheritance of
epigenetic defects,
Science 238:163-70 [1987]).
Tissue-specific expression from the cyclin A1 promoter in male germ cells is
seen
irrespective of promoter methylation status. Even high levels of methylation
do not inhibit
cyclin A1 promoter expression during spermatogenesis. In contrast, expression
from the
cyclin A 1 promoter in somatic tissues has been observed only in a transgenic
mouse line that
does not methylate the cyclin A1 promoter. This is evidence that the effects
of methylation
on gene expression are tissue-specific and can differ between somatic and germ
cells.
High in vivo expression levels of cyclin Al in mice and healthy humans are
restricted
to germ cells. (R. Yang et al. [1997]; Sweeney et al. [1996]). For an unknown
reason, cyclin
A1 is also frequently expressed at high levels in acute myeloid leukemia (R.
Yang et al.
[1997]; R. Yang et al., Cyclin A1 expression in leukemia and normal
hematopoietic cells.
Blood 93:2067-74 [1999]). Chromatin structure and probably changes in the
methylation
pattern contribute to tissue-specific expression. The cyclin A1 promoter is
highly GC rich and
bears a CpG island that extends over several hundred base pairs and ends about
50 base pairs
upstream of the main transcriptional start site. When the methylation pattern
of the CpG
dinucleotides in the critical parts of the promoter was analyzed using
bisulfate sequencing, as
described in Example 22 below, a high degree of CpG methylation was observed
in somatic,
adherent cell lines but not in cyclin A1-expressing leukemia cell lines.
Hypomethylation in
leukemic cell lines is clearly restricted to the CpG island since a CpG at
+114 outside of the
CpG island was found to be completely methylated in all cell lines tested.
Therefore, for the purposes of obtaining selectable transgenic stem cells in
accordance
with the present method, silencing of expression from the cyclin A1 promoter
in stem cell
types other than germ cells is preferably prevented by flanking the promoter
sequence and the
reporter gene with insulator elements. For example, by including double copies
of the 1.2
kb chicken (i-globin insulator element 5' to the cyclin A1 promoter sequence
and 3' to the
reporter protein gene in the present DNA construct, methylation will be
substantially
prevented at CG dinucleotide sites within the CpG island of the cyclin AI
promoter sequence
22
CA 02350829 2001-05-14
WO 00/29602 PGT/US99/08277
and thus expression of the reporter gene occurs within stem cell types other
than germ cells.
(M.J. Pikaart et al., Loss of transcriptional activity of a transgene is
accompanied by DNA
methylation and histone deacetylation and is prevented by insulators, Genes
Dev. 12:2852-62
[1998]; Chung et al., DNA sequence which acts as a chromatin insulator element
to protect
expressed genes from cis-acting regulatory sequences in mammalian cells, U.S.
Patent No.
5,610,053).
Alternatively, when the method of obtaining selectable transgenic stem cells
is
practiced to select stem cells grown in vitro, inhibitors of histone
deacetylation and DNA
methylation, such as trichostatin A or sodium butyrate, can be included in the
culture medium
to prevent silencing of reporter expression from the cyclin A1 promoter in a
wide variety of
cultured stem cells. (M.J. Pikaart et al. [1998]).
Suppression of methylation of the cyclin A 1 promoter sequence can sometimes
cause
expression from a cyclin A1 promoter in kidney podocytes or in B-cells.
Consequently, in
applications in which selectable kidney stem cells are of interest, in
accordance with the
present method of obtaining selectable transgenic stem cells, fluorescent or
luminescent
podocytes that express a reporter gene from a cyclin A 1 promoter are easily
distinguished
from fluorescing or light-emitting transgenic kidney stem cells by the
distinct podocyte
morphology (including protruding pedicels). In applications in which
hematopoietic stem
cells are of interest, fluorescent or luminescent B-cells are distinguished
from transgenic
hematopoietic stem cells by additionally using a B-cell-specific antibody
conjugated to a
fluorescent label that fluoresces or emits at a different wavelength from that
of the reporter
protein expressed as a result of cyclin A1-promoted transcription. For
example,
phycoerythricin-conjugated monoclonal antibodies against B-cell-specific
surface epitopes
can be applied to a cell population sample from bone marrow to distinguish B-
cells from
transgenic hematopoietic stem cells.
Three potential binding sites for Myb proteins are present within 100 by of
the
transcription start sites of the cyclin A1 gene, located starting at -66, -27,
and +2. (Fig. 3).
Binding of c-myb protein occurs at the sites starting at -27 and +2, and c-myb
protein
transactivates expression from the human cyclin A 1 promoter, as described in
Example 23.
In contrast, no consensus myb sites have been found for either the marine or
human cyclin A2
promoter ( X. Huet et al., Molecular & Cellular Biology 16:3789-98 [1996]).
23
CA 02350829 2001-05-14
WO 00/29602 PGT/US99/08277
Similar to the cyclin A2 gene, two potential binding sites for transcription
factor E2F
are downstream of the transcriptional start site of cyclin A 1. These E2F
sites are not required
for repression of cyclin A2 transcription in the G1 phase. (J. Zwicker et al.
(1995) EMBO
Journal 14, 4514-4522; X. Huet et al. [1996]). Likewise, the introduction of
mutations in
these sites in the cyclin A1 promoter does not alter the regulation of
expression. Further
evidence that these E2F sites are not relevant for regulation was shown using
a 3 deletion (-
190 to +13) that showed cell cycle regulation in vivo similar to the
constructs containing both
E2F sites (data not shown). Likewise, a 6-by sequence that resembles the CDE
of the human
cyclin A2 gene was found in an antisense direction at position -19 to -24
(TCGCGG; SEQ.
ID. N0.:32) of the cyclin A1 promoter. No significant differences in cell
cycle regulation
were found when these nucleotides were mutated (Fig. 9). This is consistent
with the finding
that these elements need to be in a 5' to 3' orientation to be functional
(J.Zwicker et al. [1995];
N. Liu et al., Oncogene 16:2957-63 [1998]; N. Liu et al., Nucleic Acids Res.
25: 491 S-20
[1997]).
The present invention relates to a method of obtaining a selectable transgenic
stem cell
from a vertebrate. The method involves transfecting a male germ cell or germ
cell precursor
with a transfection mixture, as described herein, that includes a
polynucleotide, comprising
a stem cell-specific promoter sequence, for example, a human or other
homologous vertebrate
cyclin A1 promoter sequence, or an operative fragment thereof, operatively
linked to a gene
encoding a fluorescent or light-emitting reporter protein, oriented so as to
comprise a
transcriptional unit. A polynucleotide containing the operatively linked stem
cell-specific
promoter and reporter gene, is incorporated in to the genome of a transfected
male germ cell,
or precursor, and can be transmitted to progeny after breeding, where it
operates in stem cells
of the progeny in vivo, such that in a cell population, taken from a progeny
vertebrate's tissue
or viewed in situ, stem cells differentially express the reporter gene
compared to non-stem
cells. Thus, these stem cells are readily selectable from the population of
non-stem cells
present in the tissue. Types of stem cells for which the method is useful
include pluripotent,
multipotent, bipotent, or monopotent stem cells, which includes male or female
germ cells or
stem cells related to any tissue of the vertebrate including, but not limited
to, spermatogonial,
embryonic, osteogenic, hematopoietic, granulopoietic, sympathoadrenal,
mesenchymal,
epidermal, neuronal, neural crest, O-2A progenitor, brain, kidney, pancreatic,
liver or cardiac
24
CA 02350829 2001-05-14
WO 00/29602 PCTNS99/08277
stem cells. And the present invention is also directed to a selectable
transgenic stem cell, of
any type, obtained by the method.
Preferred reporter genes encode fluorescent proteins including Green
Fluorescent
Protein (or EGFP), Yellow Fluorescent Protein, Blue Fluorescent Protein, a
phycobiliprotein,
such as phycoerythrin or phycocyanin, or any other protein which fluoresces
under suitable
wave-lengths of ultra-violet or other light. Another reporter gene suitable
for some
applications is a gene encoding a protein that can enzymatically lead to the
emission of light
from a substrate(s); for purposes of the present invention, such a protein is
a "light-emitting
protein." For example, a light-emitting protein includes proteins such as
luciferase or
apoaequorin.
In particular applications involving a transfected cell that expresses
additional
xenogeneic genes from any promoter, this expression may be linked to a
reporter gene that
encodes a different fluorescent or light-emitting protein from the reporter
gene linked to the
cyclin A1 promoter. Thus, multiple reporters fluorescing or emitting at
different wavelengths
can be chosen and cell selections based on the expression of multiple traits
can be made: The
selectable transgenic stem cells may be sorted, isolated or selected from non-
stem cells with
the aid of, for example, a FACS scanner set at the appropriate wavelength(s).
Alternatively,
they are isolated or selected manually from non-stem cells using conventional
microscopic
technology. It is an advantage of the present method of obtaining selectable
transgenic stem
cells that it allows stem cells to be selected or isolated from non-embryonic
tissue.
The invention also relates to a nucleic acid construct comprising a human
cyclin A 1
promoter sequence in accordance with the present invention, or an operative
fragment thereof.
In a preferred embodiment for use in the method of obtaining a selectable
transgenic stem cell,
the cyclin A1 promoter is operatively linked to a DNA having a nucleotide
sequence encoding
a fluorescent protein or a light emitting protein. Other preferred embodiments
employ a
xenogeneic nucleic acid encoding any desired product or trait. For purposes of
the present
invention, "operatively linked" means that the promoter sequence, is located
directly upstream
from the coding sequence and that both sequences are oriented in a 5' to 3'
manner, such that
transcription could take place in vitro in the presence of all essential
enzymes, transcription
factors, co-factors, activators, and reactants, under favorable physical
conditions, e.g., suitable
pH and temperature. This does not mean that, in any particular cell,
conditions will favor
CA 02350829 2001-05-14
WO 00/29602 PCT/US99/08277
transcription. For example, transcription from a cyclin AI promoter is not
favored in most
differentiated cell types in transgenic animals.
The present invention also relates to a transgenic vertebrate cell containing
the nucleic
acid construct of the present invention, regardless of the method by which the
construct was
introduced into the cell. The present invention also relates to transgenic non-
human
vertebrates comprising such cells.
The present invention also relates to a kit for transfecting a male
vertebrate's germ
cells, which is useful for obtaining selectable transgenic stem cells. The kit
is a ready
assemblage of materials for facilitating the transfection of a vertebrate male
germ cell. A kit
of the present invention contains a transfecting agent, as described above,
and a
polynucleotide that includes a stem cell-specific promoter sequence
operatively linked to a
DNA sequence encoding a fluorescent or light-emitting protein, together with
instructions for
using the components effectively. Preferably, the kit includes a nucleic acid
construct of the
present invention. Optionally, the kit can include an immunosuppressing agent,
such as
cyclosporin or a corticosteroid, and/or an additional nucleotide sequence
encoding for the
expression of a desired trait. The materials or components assembled in the
kit are provided
to the practitioner stored in any convenient and suitable way that preserves
their operability
and utility. For example the components can be in dissolved, dehydrated, or
lyophilized
form; they can be provided at room, refrigerated or frozen temperatures.
This invention also relates to a method for the isolation of spermatogonia,
comprising
obtaining spermatogonia from a mixed population of testicular cells by
extruding the cells
from the seminiferous tubules and gentle enzymatic disaggregation. The
spermatogonia or
stem cells which are to be genetically modified, may be isolated from a mixed
cell population
by a novel method including the utilization of a promoter sequence, which is
only active in
stem cells, such as human cyclin A1 promoter, or in cycling spermatogonia stem
cell
populations, for example, B-Myb or a spermotogonia specific promoter, such as
the c-kit
promoter region, c-raf I promoter, ATM (ataxia-telangiectasia) promoter, RBM
(ribosome
binding motif) promoter, DAZ (deleted in azoospermia) promoter, XRCC-1
promoter, HSP
90 (heat shock gene) promoter, or FRMI (from fragile X site) promoter, linked
to a reporter
construct, for example, a construct comprising a gene encoding Green
Fluorescent Protein (or
EGFP), Yellow Fluorescent Protein, Blue Fluorescent Protein, a
phycobiliprotein, such as
26
CA 02350829 2001-05-14
WO 00/29602 PC'T/US99/08277
phycoerythrin or phycocyanin, or any other protein which fluoresces under
suitable wave-
lengths of light. These unique promoter sequences drive the expression of the
reporter
construct only in the cycling spermatogonia. The spermatogonia, thus, are the
only cells in
the mixed population which will express the reporter constructs) and they,
thus, may be
isolated on this basis. In the case of a fluorescent reporter construct, the
cells may be sorted
with the aid of, for example, a FACS set at the appropriate wavelengths) or
they may be
selected by chemical methods.
The method of the invention is suitable for application to a variety of
vertebrate
animals, all of which are capable of producing gametes, i.e. sperm or ova.
Thus, in
accordance with the invention novel genetic modifications) and/or
characteristics) may be
imparted to animals, including mammals, such as humans, non-human primates,
for example
simians, marmosets, domestic agricultural (farm) animals such as sheep, cows,
pigs, horses,
particularly race horses, marine mammals, feral animals, felines, canines,
pachyderms, rodents
such as mice and rats, gerbils, hamsters, rabbits, and the like. Other animals
include fowl such
as chickens, turkeys, ducks, ostriches, emus, geese, guinea fowl, doves,
quail, rare and
ornamental birds, and the like. Of particular interest are endangered species
of wild animal,
such rhinoceros, tigers, cheetahs, certain species of condor, and the like.
The present invention is also related to a transgenic non-human vertebrate
comprising
a selectable transgenic stem cell in accordance with the present invention.
Broadly speaking,
a "transgenic" vertebrate animal is one that has had foreign DNA permanently
introduced into
its cells. The foreign genes) which (have) been introduced into the animal's
cells is (are)
called a "transgene(s)". The present invention is applicable to the production
of transgenic
animals containing xenogeneic, i.e., exogenous, transgenic genetic material,
or material from
a different species, including biologically functional genetic material, in
its native,
undisturbed form in which it is present in the animal's germ cells. In other
instances, the
genetic material is "allogeneic" genetic material, obtained from different
strains of the same
species, for example, from animals having a "normal" form of a gene, or a
desirable allele
thereof. Also the gene may be a hybrid construct consisting of promoter DNA
sequences and
DNA coding sequences linked together. These sequences may be obtained from
different
species or DNA sequences from the same species that are not normally
juxtaposed. The DNA
27
CA 02350829 2001-05-14
WO OOIZ9602 PCT/US99/08277
construct may also contain DNA sequences from prokaryotic organisms, such as
bacteria, or
viruses.
In one preferred embodiment, the transfected germ cells of the transgenic
animal have
the non-endogenous (exogenous) genetic material integrated into their
chromosomes. This is
what is referred to as a "stable transfection". This is applicable to all
vertebrate animals,
including humans. Those skilled in the art will readily appreciate that any
desired traits
generated as a result of changes to the genetic material of any transgenic
animal produced by
this invention are inheritable. Although the genetic material was originally
inserted solely
into the germ cells of a parent animal, it will ultimately be present in the
germ cells of future
progeny and subsequent generations thereof. The genetic material is also
present in all other
cells of the progeny, including somatic cells and all non-stem cells, of the
progeny. This
invention also encompasses progeny resulting from breeding of the present
transgenic
animals. The transgenic animals bred with other transgenic or non-transgenic
animals of the
same species will produce some transgenic progeny, which should be fertile.
This invention,
thus, provides animal lines) which result from breeding of the transgenic
animals) provided
herein, as well as from breeding their fertile progeny.
"Breeding", in the context of this patent, means the union of male and female
gametes
so that fertilization occurs. Such a union may be brought about by natural
mating, i.e.
copulation, or by in vitro or in vivo artificial means. Artificial means
include, but are not
limited to, artificial insemination, in vitro fertilization, cloning and
embryo transfer,
intracytoplasmic spermatozoa) microinjection, cloning and embryo splitting,
and the like.
However, others may also be employed.
The transfection of mature male germ cells may be also attained utilizing the
present
technology upon isolation of the cells from a vertebrate, as is known in the
art, and
exemplified in Example 10. The thus isolated cells may then be transfected ex
vivo (in vitro),
or prepared for cryostorage, as described in Example 11. The actual
transsection of the
isolated testicular cells may be accomplished, for example, by isolation of a
vertebrate's
testes, decapsulation and teasing apart and mincing of the seminiferous
tubules. The
separated cells may then be incubated in an enzyme mixture comprising enzymes
known for
gently breaking up the tissue matrix and releasing undamaged cells such as,
for example,
pancreatic trypsin, collagenase type I, pancreatic DNAse type I, as well as
bovine serum
28
CA 02350829 2001-05-14
WO OO/Z9602 PCTNS99/08277
albumin and a modified DMEM medium. The cells may be incubated in the enzyme
mixture
for a period of about 5 min to about 30 min, more preferably about 15 to about
20 min, at a
temperature of about 33°C to about 37°C, more preferably about
36 to 37°C. After washing
the cells free of the enzyme mixture, they may be placed in an incubation
medium such as
DMEM, and the like, and plated on a culture dish. Any of a number of
commercially
available transfection mixtures may be admixed with the polynucleotide
encoding a desire
trait or product for transfection of the cells. The transfection mixture may
then be admixed
with the cells and allowed to interact for a period of about 2 hrs to about 16
hrs, preferably
about 3 to 4 hrs, at a temperature of about 33°C to about 37°C,
preferably about 36°C to
37°C, and more preferably in a constant and/or controlled atmosphere.
After this period, the
cells are preferably placed at a lower temperature of about 33 °C to
about 34°C, preferably
about 30-35°C for a period of about 4 hrs to about 20 hrs, preferably
about 16 to 18 hrs.
Other conditions which do not deviate radically from the ones described may
also be utilized
as an artisan would know.
I S The present method is applicable to the field of gene therapy, since it
permits the
introduction of genetic material encoding and regulating specific genetic
traits. Thus, in the
human, for example, by treating parents it is possible to correct many single
gene disorders
which otherwise might affect their children. It is similarly possible to alter
the expression of
fully inheritable disorders or those disorders having at least a partially
inherited basis, which
are caused by interaction of more than one gene, or those which are more
prevalent because
of the contribution of multiple genes. This technology may also be applied in
a similar way
to correct disorders in animals other than human primates. In some instances,
it may be
necessary to introduce one or more "gene(s}" into the germ cells of the animal
to attain a
desired therapeutic effect, as in the case where multiple genes are involved
in the expression
or suppression of a defined trait. In the human, examples of multigenic
disorders include
diabetes mellitus caused by deficient production of, or response to, insulin,
inflammatory
bowel disease, certain forms of atheromatus cardiovascular disease and
hypertension,
schizophrenia and some forms of chronic depressive disorders, among others. In
some cases,
one gene may encode an expressible product, whereas another gene encodes a
regulatory
function, as is known in the art. Other examples are those where homologous
recombinant
methods are applied to repair point mutations or deletions in the genome,
inactivation of a
29
CA 02350829 2001-05-14
WO 00129602 PCT/US99/08277
gene causing pathogenesis or disease, or insertion of a gene that is expressed
in a dominant
negative manner, or alterations of regulating elements such as gene promoters,
enhancers, the
untranslated tail region of a gene, or regulation of expansion of repeated
sequences of DNA
which cause such diseases as Huntingdon's chorea, Fragile-X syndrome and the
like.
A specific reproductive application of the present method is to the treatment
of
animals, particularly humans, with disorders of spermatogenesis. Defective
spermatogenesis
or spermiogenesis frequently has a genetic basis, that is, one or several
mutations in the
genome may result in failure of production of normal sperm cells. This may
happen at
various stages of the development of germ cells, and may result in male
infertility or sterility.
The present invention is applicable, for example, to the insertion or
incorporation of nucleic
acid sequences into a recipient's genome and, thereby, establish
spermatogenesis in the
correction of oligozoospermia or azoospermia in the treatment of infertility.
Similarly, the
present methods are also applicable to males whose subfertility or sterility
is due to a motility
disorder with a genetic basis.
The present method is additionally applicable to the generation of transgenic
animals
expressing agents which are of therapeutic benefit for use in human and
veterinary medicine
or well being. Examples include the production of pharmaceuticals in domestic
cows' milk,
such as factors which enhance blood clotting for patients with types of
haemophilia, or
hormonal agents such as insulin and other peptide hormones.
The present method is further applicable to the generation of transgenic
animals of a
suitable anatomical and physiological phenotype for human xenograft
transplantation.
Transgenic technology permits the generation of animals which are immune-
compatible with
a human recipient. Appropriate organs, for example, may be removed from such
animals to
allow the transplantation of, for example, the heart, lung and kidney.
In addition, germ cells transfected in accordance with this invention may be
extracted
from the transgenic animal, and stored under conditions effective for later
use, as is known
in the art. Storage conditions include the use of cryopreservation using
programmed freezing
methods and/or the use of cryoprotectants, and the use of storage in
substances such as liquid
nitrogen. The germ cells may be obtained in the form of a male animal's semen,
or separated
spermatozoa, or immature spermatocytes, or whole biopsies of testicular tissue
containing the
primitive germ cells. Such storage techniques are particularly beneficial to
young adult
CA 02350829 2001-05-14
WO 00/29602 PCT/US99/08277
humans or children, undergoing oncological treatments for such diseases such
as leukemia or
Hodgkin's lymphoma. These treatments frequently irreversibly damage the
testicle and, thus,
render it unable to recommence spermatogenesis after therapy by, for example,
irradiation or
chemotherapy. The storage of germ cells and subsequent testicular transfer
allows the
restoration of fertility. In such circumstances, the transfer and manipulation
of germ cells as
taught in this invention are accomplished, but transfection is generally not
relevant or needed.
In species other than humans, the present techniques are valuable for
transport of
gametes as frozen germ cells. Such transport will facilitate the establishment
of various
valued livestock or fowl, at a remote distance from the donor animal. This
approach is also
applicable to the preservation of endangered species across the globe.
The method of obtaining selectable transgenic stem cells, the selectable
transgenic
stem cells, the transgenic non-human vertebrates and vertebrate semen, and the
nucleic acid
contructs and kits, in accordance with the present invention, are valuable
tools in the study
of cellular differentiation and development and in developing new therapies
for diseases
related to cell differentiation, such as cancer, or for the regeneration of
tissues after traumatic
injuries. The present invention is valuable in identifying cell lineages
before full
differentiation to facilitate modification and/or engineering of specific
tissues in vitro for their
subsequent transplantation in the treatment of disease or trauma. It is an
advantage of the
present method of obtaining selectable transgenic stem cells that it allows
stem cells to be
selected or isolated from non-embryonic tissue, thus avoiding potential
ethical and legal
problems associated with the use of embryonic tissue. It is a further
advantage that in
accordance with the present invention, selectable transgenic stem cells can be
selected and
analyzed whether grown in vivo (i.e., in the whole organism) or in vitro.
The invention will now be described in greater detail by reference to the
following
non-limiting examples. The pertinent portions of the contents of all
references, and published
patent applications cited throughout this patent necessary for enablement
purposes are hereby
incorporated by reference.
EXAMPLES
In Vivo and In Vitro Adenovirus-enhanced Transferrin-polylysine-mediated
Delivery
31
CA 02350829 2001-05-14
WO 00/29602 PCT/US99/08277
of Green Fluorescent Protein Reporter Gene to Testicular Cells and Expression
The adenovirus enhanced transferrin-polylysine-mediated gene delivery system
has
been described and patented by Curiel et al. (D.T. Curiel et al. Adenovirus
enhancement of
transferrin-polylysine-mediated gene delivery, PNAS USA 88: 8850-8854 (1991).
The
S delivery of DNA depends upon endocytosis mediated by the transferrin
receptor (Wagner et
al., Transferrin polycation conjugates as carriers for DNA uptake into cells,
Proc. Natl. Acad.
Sci. (USA) 87:3410-3414 (1990). In addition this method relies on the capacity
of
adenoviruses to disrupt cell vesicles, such as endosomes and release the
contents entrapped
therein. This system can enhance the gene delivery to mammalian cells by as
much as 2,000
fold over other methods.
The gene delivery system employed for the in vivo and in vitro experiments was
prepared as shown in examples below.
Example 1: Preparation of Transferrin-poly-L-Lysine Complexes
Human transferrin was conjugated to poly (L-lysine) using EDC (1-ethyl-3-(3-
dimethyl aminopropyl carbodiimide hydrochloride) (Pierce), according to the
method of
Gabarek and Gergely (Gabarek & Gergely, Zero-length cross-linking procedure
with the use
of active esters, Analyt. Biochem 185 : 131 (1990)). In this reaction, EDC
reacts with a
carboxyl group of human transferrin to form an amine-reactive intermediate.
The activated
protein was allowed to react with the poly (L-lysine) moiety for 2 hrs at room
temperature,
and the reaction was quenched by adding hydroxylamine to a final concentration
of 10 mM.
The conjugate was purified by gel filtration, and stored at -20°C.
Example 2: Preparation of DNA for In Vivo Trasfection
The Green Lantern-1 vector (Life Technologies, Gibco BRL, Gaithersberg, MD) is
a reporter construct used for monitoring gene transfection in mammalian cells.
It consists of
the gene encoding the Green Fluorescent Protein (GFP) driven by the
cytomegalovirus (CMV)
immediate early promoter. Downstream of the gene is a SV40 polyadenylation
signal. Cells
transfected with Green Lantern-1 fluoresce with a bright green light when
illuminated with
blue light. The excitation peak is 490 nm.
32
CA 02350829 2001-05-14
WO 00/29602 PCT/U599/08277
Example 3: Preparation of Adenoviral Particles
Adenovirus dI312, a replication-incompetent strain deleted in the Ela region,
was
propagated in the Ela traps-complementing cell line 293 as described by Jones
and Shenk
(Jones and Shenk, PNAS USA (1979) 79: 3665-3669). A large scale preparation of
the virus
was made using the method of Mittereder and Trapnell (Mittereder et al.,
"Evaluation of the
concentration and bioactivity of adenovirus vectors for gene therapy", J.
Urology, 70: 7498-
7509 (1996)). The virion concentration was determined by UV spectroscopy, 1
absorbance
unit being equivalent to 10 viral particles /ml. The purified virus was stored
at -70°C.
Example 4: Formation of Transferrin-poly-L Lysine-DNA-Viral Complexes
6 leg transferrin-polylysine complex from Example 1 were mixed in 7.3 x 10'
adenovirus d1312 particles prepared as in Example 3, and then mixed with 5 ug
of the Green
Lantern DNA construct of Example 2, and allowed to stand at room temperature
for 1 hour.
About 100 ul of the mixture were drawn up into a micropipette, drawn on a
pipette pullet, and
slightly bent on a microforge. The filled micropipette was then attached to a
picopump
(Eppendorf), and the DNA complexes were delivered under continuous pressure,
in vivo to
mice as described in Example 6.
Controls were run following the same procedure, but omitting the
transferrin-poly-lysine-DNA-viral complexes from the administered mixture.
Example 5: Comparison of Adenovirus-enhanced Transferrin-polylysine
& Lipofectin Mediated Transfection Efficiency
The conjugated adenovirus particle complexed with DNA were tested on CHO cells
in vitro prior to in vivo testing. For these experiments a luciferase reporter
gene was used due
to the ease of quantifying luciferase activity. The expression construct
consists of a reporter
gene encoding luciferase, is driven by the CMV promoter (Invitrogen, Carlsbad,
CA 92008).
CHO cells were grown in Dulbecco's modified Eagle's medium (DMEM) with 10%
fetal calf
serum. For gene transfer experiments CHO cells were seeded into 6 cm tissue
culture plates
and grown to about 50% confluency (5x105 cells). Prior to transfection the
medium was
aspirated and replaced with serum free DMEM. Cells were either transfected
with transferrin-
polylysine-DNA complexes or with lipofectin DNA aggregates. For the
transferrin-
33
CA 02350829 2001-05-14
WO 00/29602 PCT/US99/08277
polylysine mediated DNA transfer, the DNA-adenovirus complexes were added to
the cells
at a concentration of 0.05-3.2 x 104 adenovirus particles per cell. Plates
were returned to the
5% COZ incubator for 1 hour at 37°C. After 1 hour 3 ml of complete
media was added to the
wells and the cells were allowed to incubate for 48 hours before harvesting.
The cells were
removed from the plate, counted and then lysed for measurement of luciferase
activity.
For cells transfected by lipofectin, l ltg of CMV-luciferase DNA was incubated
with
17p,1 of Lipofectin (Life Technologies). The DNA-lipofectin aggregates were
added to the
CHO cells and allowed to incubate at 37°C at 5% COZ for 4 hours. Three
mls of complete
medium was added then to the cells and they were allowed to incubate for 48
hours. The cells
were harvested, counted and Iysed for luciferase activity. The luciferase
activity was
measured by a luminometer. The results obtained are shown in Table 1.
The data included in Table 1 below show that the adenovirus-enhanced
transferrin-
polylysine gene delivery system is 1,808 fold more efficient than lipofection
for transfection
of CHO cells.
Table 1: Comparison of Lipofection & Adenovirus Enhanced
Transferrin-polylysine Transfection of CHO Cells
Sample Treatment L a c i f a r a s a
Activity (RLU)
1 1x10'particles + hug CMV-Luc 486
2 2.5 x 10' particles + hug CMV-Luc 1,201
3 5.0 x 10' particles + hug CMV-luc 11,119
4 1 x 109 particles + dug CMV-Luc 2,003,503
5 Lipofection 1,108
6 Unmanipulated cells 155
Example 6: In Vivo Delivery of DNA to Animal's Germ Cells
via Tranfernn-L-lysine-DNA-Viral Complexes
The CMV-EGFP (Gibco-BRL, Life Technologies, Gaithersburg, MD 20884)
DNA-transferrin-polylysine viral complexes, prepared as described in Example 4
above, were
delivered into the seminiferous tubules of three (3)-week-old B6D2F 1 male
mice. The DNA
delivery by transferrin receptor-mediated endocytosis is described by Schmidt
et al. and
34
CA 02350829 2001-05-14
WO 00/29602 PCT/US99/08277
Wagner et al. (Schmidt et al., Cell 4: 41-51 (1986); Wagner, E., et al. PNAS
(1990), (IJSA)
81: 3410-3414 (1990)). In addition, this delivery system relies on the
capacity of
adenoviruses to disrupt cell vesicles, such as endosomes and release the
contents entrapped
therein. The transfection efficiency of this system is almost 2,000 fold
higher than
lipofection.
The male mice were anesthetized with 2% Avertin ( 100% Avertin comprises 10 g
2,2,2-tribromoethanol (Aldrich) and 10 ml t-amyl alcohol (Sigma), and a small
incision made
in their skin and body wall, on the ventral side of the body at the level of
the hind leg. The
animal's testis was pulled out through the opening by grasping at the testis
fat pad with
forceps, and the vas efferens tubules exposed and supported by a glass
syringe. The EGFP
DNA-transferrin-polylysine viral complexes were injected into a single vasa
efferentia using
a glass micropipette attached to a hand held glass syringe or a pressurized
automatic pipettor
(Eppendorf), and Trypan blue added to visualize the entry of the mixture into
the seminiferous
tubules. The testes were then placed back in the body cavity, the body wall
was sutured, the
skin closed with wound clips, and the animal allowed to recover on a warm pad.
Example 7: Detection of DNA and Transcribed Message
Nine (9) days after delivery of the genetic material to the animals' testis,
two of the
animals were sacrificed, their testes removed, cut in half, and frozen in
liquid nitrogen. The
DNA from one half of the tissues, and the RNA from the other half of the
tissues were
extracted and analyzed.
(a) Detection of DNA
The presence of DNA encoding enhanced green fluorescent protein (EGFP DNA) in
the extracts was tested 9 days after administration of the transfection
mixture using the
polymerase chain reaction, and EGFP specific oligonucleotides. EGFP DNA was
present in
the testes of the animals that had received the DNA complexes, but was absent
from sham
operated animals.
(b) Detection of RNA
CA 02350829 2001-05-14
WO 00/29602 PCT/US99/08Z77
The presence of CMV-EGFP mRNA was assayed in the testes of experimental
animals
as follows. RNA was extracted from injected, and non-injected testes, and the
presence of the
EGFP messages was detected using reverse transcriptase PCR (RTPCR) with EGFP
specific
primers. The EGFP message was present in the injected testes, but not in the
control testes.
Thus, the DNA detected above by PCR analysis is, in fact, episomal EGFP DNA,
or EGFP
DNA which has integrated into the chromosomes of the animal. The transfected
gene was
being expressed.
Northern blot analysis was also done to confirm transcription regulated by the
human
cyclin Al promoter. Total RNA was prepared from tissues using a RNA tissue
preparation
kit (Qiagen). Polyadenylated RNA was prepared by passage over an oligo(dT)-
cellulose
column. The RNA is polyoxylated and applied to 1.5% Agarose gel. After
electrophoresis
the RNA is transferred to nitrocelullose paper and hybridized with a cyclin A1
cDNA
riboprobe. After hybridization the membrane was washed twice with lx SSC at
60°C for 1
hour. The washed membrane was exposed to X-ray film.
Example 8: Expression of Non-endogenous DNA
Two males, one having received an injection with the EGFP transfection mixture
and
a control to whom only surgery was administered, were sacrificed 4 days after
injection, and
their testes excised, and fixed in 4% paraformaldehyde for 18 hours at 4
° C. The fixed testis
was then placed in 30% sucrose in PBS with 2 mM MgCl2 for 18 hours at
4°C, embedded in
OCT frozen on dry ice, and sectioned. When the testes of both animals were
examined with
a confocal microscope with fluorescent light at a wavelength of 488 nM, bright
fluorescence
was detected in the tubules of the EGFP-injected mice, but nat in the testes
of the controls.
Many cells within the seminferous tubules of the EGFP-injected mouse showed
bright
fluorescence, which evidences that they were expressing Green Fluorescent
Protein.
Example 9: Generation of Offspring from Normal Matings
EGFP-transfected males were mated with normal females. The females were
allowed
to complete gestation, and the pups to be born. The pups (F 1 offspring or
progeny) were
screened for the presence of the novel genetic material(s).
36
CA 02350829 2001-05-14
WO 00/29602 PCT/US99/08277
Example 10: In Vitro Transfection of Testicular Cells
Cells were isolated from the testes of three 10-day-old mice. The testes were
decapsulated and the seminiferous tubules were teased apart and minced with
sterile needles.
The cells were incubated in enzyme mixture for 20 minutes at 37°C. The
enzyme mixture was
made up of 10 mg bovine serum albumin (embryo tested), 50 mg bovine pancreatic
trypsin
type III, Clostridium collagenase type I, 1 mg bovine pancreatic DNAse type I
in 10 mls of
modified HTF medium (Irvine Scientific, Irvine, CA). The enzymes were obtained
from
Sigma Company (St. Louis, Missouri 63178). After digestion, the cells were
washed twice
by centrifugation at 500 x g with HTF medium and resuspended in 250~c1 HTF
medium. The
cells were counted, and 0.5 x 106 cells were plated in a 60mm culture dish in
a total volume
of Sml DMEM (Gibco-BRL, Life Technologies, Gaithesburg, MD 20884). A
transfection
mixture was prepared by mixing S,ug Green Lantern DNA (Gibco-BRL, Life
Technologies,
Gaithesburg, MD 20884) with 20~c1 Superfect (Qiagen, Santa Clarita, CA 91355)
and 1501
DMEM. The transfection mix was added to the cells and they were allowed to
incubate for
3 hours at 37°C, 5% COZ The cells were transferred to a 33 C incubator
and incubated
overnight.
The following morning the cells were assessed for transfection efficiency by
counting
the number of fluorescent cells. In this experiment the transfection
efficiency was 90%
(Figure not shown). The testicular cells transfected with Green Lantern viewed
with Nomaski
optics x20 show the same cells viewed with FITC. Nearly all the cells were
fluorescent, which
is confirmation of their successful transfection.
The cells were injected into the testis via the vasa efferentia using a
micropipette.
3 x 105 cells in a total volume of SO~cI were used for the injection. The
cells were mixed with
Trypan blue prior to the injection. Three adult mice were injected with
transfected cells. The
Balb/cByJ recipient mice had been irradiated 6 weeks prior to the injection
with 800 Rads of
gamma irradiation. One mouse became sick and was sacrificed 48 hours after the
injection.
The testes from this mouse were dissected, fixed and processed for histology.
The two remaining males were bred with normal females as shown. After 4 months
pups were born. Litters are currently being screened for the integration of
the transgene.
Example 11: Preparation of a Cell Suspension from Testicular Tissue for
Cryopreservation
37
CA 02350829 2001-05-14
WO 00/29602 PCT/US99/08277
A cell suspension was prepared from mice of different ages as described below.
Group I: 7-10 day olds
Group II: I S-17 day olds
Group III: 24-26 day olds
The mice's testes were dissected, placed in phosphate buffered saline (PBS)
decapsulated, and the seminiferous tubules were teased apart. Seminiferous
tubules from
groups I and II were transferred to HEPES buffered culture medium (D-MEM)
(Gibco-BRL,
Life Technologies, Gaithesburg, MD 20884} containing Img/ml Bovine serum
albumin
(BSA) (Sigma, St. Louis, MO 63178) and Collagenase Type I (Sigma) for the
removal of
interstitial cells. After a 10 minute incubation at 33'C, the tubules were
lifted into fresh
culture medium. This enzymatic digestion was not carried out on the testes
from group I
because of their fragility.
The tubules from group II and III mice or the whole tissue from group I mice
were
transferred to a Petri dish with culture medium and were cut into 0.1-Imm
pieces using a
sterile scalpel and needle. The minced tissue was centrifuged at 500 x g for S
minutes and the
pellet was resuspended in 1 ml of enzyme mix. The enzyme mix was made up in D-
DMEM
with HEPES (Gibco-BRL) and consisted of lmg/ml bovine serum albumin (BSA)
(Sigma,
embryo tested), lmg/ml collagenase I (Sigma) and 5 mg/ml bovine pancreatic
trypsin (Sigma)
and O.Img/ml deoxyribonuclease I (DN-EP, Sigma). The tubules were incubated in
enzyme
mix for 30 minutes at 33'C. After the incubation, ImI of medium was added to
the mix and
the cells were centrifuged at 500 x g for 5 min. The cells were washed twice
in medium by
centrifugation and resuspension. After the final wash the cell pellet was
resuspended in 250,u1
of culture medium and counted.
Example 12: Cloning of the cyclin A1 gene and construction of DNA constructs
containing
cyclin AI-luciferase
.lo ing of t_he genomic fraement of t_he human cyclin Al . The cyclin A1 gene
was cloned by
screening a genomic Fix II lambda library made from placenta (Stratagene)
using the cyclin
A 1 cDNA as a probe. (R. Yang et al. [ 1997]). Of the several phage clones
obtained, one
contained all the exons and included a 1.3 kb region upstream of the 5' end of
the cDNA. A
2.2 kb NotI-Bam HI fragment from the 5' end of the gene was subcloned into the
pRS316
38
CA 02350829 2001-05-14
WO 00/19602 PCT/US99/08277
cloning vector. The construct was further digested using Sma I; and three
fragments were
subcloned into PUC 19. The fragments were sequenced in both directions using
cycle
sequencing and an automated sequencer (ABI373) or Sequenase 2.0 (Amersham).
The
positions and lengths of the introns were determined by PCR amplification of
the entire cyclin
A1 coding region with different primers. Subsequently, PCR products were
either subcloned
using pGEM-T-Easy (Promega) or directly sequenced using cycle sequencing.
Boundaries
of the ~4.5 kb intron 2 were determined by direct sequencing of the lambda
phage clone.
Gennratinn of r~clin At-luciferase DNA constructs. The initial luciferase
constructs were
generated by PCR amplification of the pRS316 plasmid containing the 2.2 kb
cyclin A1
fragment. A BgIII site at the 5' end and a Bam HI site at the 3' end were
introduced and the Pfu
amplified fragment was cloned into the BgIII site of PGL3-Basic. The +144
fragment was
generated to include the potential E2F site starting at +I39. (Figure 3). The
ATG in the
primer (the initiating codon for cyclin A1 at nt. +127 to +129) was mutated to
ATT to avoid
the initiation of translation. All constructs were confirmed to have the
correct sequence by
DNA sequencing. The 5' deletions were generated by exonuclease III treatment
using Kpn
1/Sac I digested PGL3-Basic containing the -1299 to +144 fragment and the
Erase-a-base kit
(Promega). The endpoints of the deletions were determined by sequencing. The -
37 fragment
was constructed by digesting the -190 to +144 containing PGL3-Basic with NaeI
and Hind III
and subsequent cloning of the 200 by fragment into PGL3-Basic digested with
Sma I and Hind
III.
('ell cul_tu_re a_nd tr~n~fe-cton. Hela cells were cultured in DMEM medium
supplemented with
10% fetal calf serum (FCS) containing 100 U/ml Penicillin and 100 ~cg/mL
Streptomycin. For
transfection, 5 x lOs cells were seeded into 60 mm plates 16 hours before
transfection.
Transfection was carried out using IipofectAMINE (Gibco, Life Technology)
according to the
manufacturer's protocol. Two ,ug of luciferase reporter plasmid was
transfected together with
300 ng of a CMV-(3-gal expression vector used for standardization. Cells were
harvested and
assayed for luciferase and ~i-galactosidase activity after 48 hours. All
experiments were
carried out in duplicate and were independently performed at least 3 times.
Data of luciferase
assays are shown as mean t SEM of three independent experiments unless stated
otherwise.
39
CA 02350829 2001-05-14
WO 00/29602 PCT/US99/08277
The Drosophila cell line S2 was obtained from ATCC and grown at room
temperature in
Schneider's insect cell medium (Gibco) supplemented with 10% FCS. Insect cells
were
transfected using Superfect (Qiagen). Briefly, Sx105 cells were seeded into 6
well plates and
the superfect-DNA mixture was added dropwise. One ~cg of the luciferase
reporter was
transfected with or without 100 ng of the Spl expression vector pAC-Spl which
was a kind
gift from Dr. E. Stanbridge (UC Irvine). Luciferase activity was analyzed
after 48 hours.
Luciferase values could not be standardized using (3-galactosidase activity
because the viral
promoters in the available plasmids also depended strongly on Spl for adequate
expression.
All experiments were carried out in duplicate and independently performed at
least 3 times.
Cell cXcle a~gndent promoter activity. Hela cells were transfected using
lipofectAMINE as
described above. After transfection, cells were cultured in 0.1% FCS
containing medium.
After 16 h, medium was exchanged and cells were synchronized essentially as
described. (D.
Carbonaro-Hall et al., Oncogene 8:1649-59 [1993]). Cells were arrested in G,
by serum
starvation (0.1 % FCS), in early S phase by aphidicolin (2,u/ml), and in S
phase by aphidicolin
treatment and release into fresh medium 6 hours before harvest. Cells were
arrested in GZ/M
phase by nocodazole (0.1 ~cg/ml). Appropriate synchronization was confirmed by
DNA
quantitation using flow cytometry and the experiments were performed at least
three times.
For the cell cycle release experiments, Hela cells were arrested using
aphidicolin as described
above but cells were harvested at the different time points. The time course
experiments were
independently performed two times.
RACE anal primer extension. The rapid amplification of 5' cDNA ends (RACE) was
performed using a 5' RACE system (Gibco). The procedure was performed as
suggested in
the manufacturer's protocol using RNA of the myeloid leukemia cell lines ML1
and U937.
RNA was reversed transcribed using the primer 5'-CCC TCT CAG AAC AGA CAT ACA
(SEQ. ID. N0.:14; positions +981 to +961 of the cDNA) and Superscript II
reverse
transcriptase (Gibco). Gene-specific cDNA was PCR-amplified using the gene-
specific
primer 5'-CTG ATC CAG AAT AAC ACC TGA (SEQ. ID. NO.:15; positions +460 to +440
of the cDNA) and the universal 5' RACE Abridged Anchor Primer 5'-GGC CAC GCG
TCG
ACT AGT ACG GGI IGG GII GGG IIG (SEQ. ID. N0.:16; I=inosine). PCR-
amplifications
from both RNA samples yielded a single band of about 450 bp. The entire PCR
product was
CA 02350829 2001-05-14
WO 00/29602 PCT/US99/08277
phenol-chloroform extracted, precipitated using NH4' acetate and finally
cloned into pGEM-T-
Easy and sequenced.
The primer extension assay was carned out by reverse transcription of l0~cg
RNA
(U937) using a 32P-labeled primer 5'-CTC CTC CCA CCA GAC AGG A (SEQ. ID.
N0.:17)
corresponding to +95 to +76 on the cDNA. Hybridization was carried out
overnight at 58°C.
Superscript II was used for reverse transcription at 42°C for 50
minutes. Extension products
were resolved on a 8% sequencing gel with a sequencing reaction being run in
parallel. As
negative controls, we used l0,ug of t-RNA and a sample without RNA.
F,lect_ro~oretic Mobility Shift Assavs. Nuclear extracts from Hela cells were
prepared as
described (A.M. Chumakov et al., Oncogene 8:3005-lI [1993]). For gel
retardation
experiments, 1 ng of 32P- labeled double stranded oligonucleotides containing
either GC boxes
1+2 (5'-CCT GCC CCG CCC TGC CCC GCC CAG CC; SEQ. ID. NO.:18) or GC boxes 3+4
(5'-CCT TCC CCG CCC TGC CCC GCC CGG CCC; SEQ. ID. NO.:19) were incubated for
min at room temperature with 5 ~cg of Hela nuclear extract. The final reaction
contained:
15 10 mM Tris-HCL, pH 7.5, 5% glycerol, 1 mM MgCl2, 0.5 mM EDTA, 0.5 mM DTT,
100
mM NaCI and l,ug poly(dI-dC)-poly(dI-dC). For competition experiments, 100 ng
of double
stranded oligonucleotide containing either a Spl consensus site (5'-ATT CGA
TCG GGG
CGG GGC GAG C; SEQ. ID. N0.:20), the oligonucleotide used for gel retardation
(see
above) or a non-specific oligonucleotide (5'-GAG ACC GGC TCG AAC GCA ATC ATG
T;
20 SEQ. ID. N0.:21 ) were preincubated for 15 min at room temperature with the
nuclear extracts
before the addition of the labeled oligonucleotide. For supershift
experiments, 2-3 ,ug of
polyclonal antibody against Spl (Pep2, Santa Cruz) or Sp3 (D20, Santa Cruz)
were
preincubated with the nuclear extracts. Reactions were loaded on a O.Sx TBE
/4% non-
denaturing polyacrylamide gel and run for 2-3 h at 10 V/cm. Gels were dried
and
autoradiographed.
Site directed mutag neci . Site directed mutagenesis was performed according
to the method
from Deng and Nickoloff (W.P. Deng and J.A. Nickoloff, Analyt. Biochem.200:81-
88 [1992])
using the Transformer site directed mutagenesis kit (Clontech). In brief,
phosphorylated
oligonucleotides containing the desired mutation were annealed on the single-
stranded PGL3-
41
CA 02350829 2001-05-14
WO 00/29602 PCT/US99/08277
Basic plasmid (containing the fragment -190 to +144) together with the
oligonucleotide 5'-
AAT CGA TAA GAA TTC GTC GAC CGA (SEQ. ID. N0.:22) that changes the unique Bam
HI site to an Eco RI site. The complementary strand was extended and completed
from the
annealed oligos using T4 Polymerase and T4 Ligase. Selection for the mutant
plasmid was
performed by two rounds of digestion with Bam HI and subsequent
transformations, first into
the repair deficient strain BMH 71-18 mutS and finally into DHSa. The entire
promoter
fragment was sequenced to verify desired mutations and to exclude second site
mutations.
Because of the short distances between GC boxes 1+2 and 3+4, oligonucleotides
were
designed to mutate both GC boxes simultaneously. Mutations in all 4 GC boxes
were
introduced by simultaneously adding oligos 1+2 and 3+4. All oligonucleotides
used in these
experiments were 5'-phosphorylated. The following oligonucleotides were used
(mutated
bases underlined):
GC box 1: CCC CGC CCT GCC CCT TAC AGC CGG CCA CC (SEQ. ID. N0.:23),
GC box 2: CCA ACC CTG CCC T~A.CCT GCC CCG (SEQ. ID. N0.:24),
GC box 3: CCC TGC CCC FTC CGG CCC GGC C (SEQ. ID. N0.:25),
GC box 4: CTG CCC TTC CCT_TCC CTG CCC C (SEQ. ID. N0.:26),
GC boxes 1+2: GCC CAA CCC TGC CSC CTG CCC COCA GCC GGC CAC CTC
(SEQ. ID. N0::27),
GC boxes 3+4: CTT CCC TGC CCT TCC C~CC TGC CCC ~ CGG CCC GGG CCG
GCC (SEQ. ID. N0.:28).
The potential CDE element in the cylin A 1 promoter was mutated using the
following
oligonucleotide: CCA CCT CTT AAC AAG CTT CCT CCA GTG CA (SEQ. ID. N0.:29).
The cyclin Al-EGFP construct was finally constructed by cloning a Bglll -
HindIII
fi~ag<nent from the PGL3-Basic-Cyclin A1 Promoter construct into the
promoterless EGFP-1
(Clontech} plasmid.
Example 13 : Genomic cloning and gene structure of the human cyclin A 1 gene.
A human genomic lambda phage library was screened using the cDNA of cyclin A 1
as a probe. Several clones containing pieces of the gene were obtained and one
clone with a
42
CA 02350829 2001-05-14
WO 00/29602 PCT/US99/08277
14.5 kb insert contained the entire gene. A 2.2 kb fragment at the 5' end of
the gene was
subcloned and sequenced. The 2.2 kb fragment contained the first intron and
parts of exon 2.
The other exon-intron boundaries were analyzed by PCR-amplification and
sequencing using
sets of primers that span the entire coding region. The human cyclin A1 gene
consists of 9
exons and 8 introns which extend over ~ 13 kb.
Example 14: Analysis of transcription start sites.
Transcription start sites were determined using primer extension analysis and
5'
RACE. Primer extension was carried out as outlined in Example 12. A sample
without RNA
and a sample of t-RNA (10 ~cg) were used as negative controls. The primer
extension
products shown in Fig. 2 are indicated by an asterisk above the appropriate
nucleotide of the
indicated sequence. Starting points of the RACE products are indicated by an
arrow
underneath the sequence. The number of RACE clones (total 25) starting at a
particular base
is indicated by the number shown below the arrows. The site where 44% (11/25)
RACE
clones started was assigned +1.
Both methods demonstrated the existence of several transcription start sites.
The PCR
product from the RACE reaction consisted of a single band of ~-450 bp.
Sequencing of the
inserts after cloning revealed that 80% of the RACE products (20/25) started
from a 4 base
pair stretch, and thus the predominant start site was assigned +1. This site
is 130 by upstream
of the translation initiating ATG codon. Primer extension analysis identified
the same start
sites, but minor products were also seen further upstream (Fig. 2). The major
start site
coincides with the RACE results.of the 5' end of the cDNA clone described by
Yang et al.
(1997). Neither RACE clones nor primer extension assays showed evidence for a
second
transcript in myeloid leukemia cells that could indicate a transcriptional
start site upstream of
the second ATG in intron 1 (data not shown).
Example 15: Potential transcription factor binding sites in the 5' upstream
region.
Genomic sequences 1299 by upstream of the transcription start site were cloned
and
sequenced. No TATA box was found in proximity to the putative transcriptional
start site.
The main transcriptional start site is likely to function as an initiator
region (Inr) since the
43
CA 02350829 2001-05-14
WO 00129602 PCT/US99/08277
sequence "CCAGTT" is very similar to the consensus Inr sequence "TCA G/T T
T/C" (T.W.
Burke and J.T. Kadonaga, Genes & Development 1:3020-31 [I997]). No DPE element
was
found downstream of the main transcriptional start site. (See id.). Several
potential binding
sites for transcription factors occur within the sequence.
Figure 3 represents the 5' upstream region of the human cyclin A1 gene. The
first
bases of the different fiagments are indicated, as well as potential
transcription factor binding
sites between -190 to +144. The transcriptional start site is marked with ari
arrow and the
translational initiation codon is boldfaced. An E2F site is located at nt.
+139 to +144 and
another possible site starting at +67. A site that resembles the cycle
dependent element
(CDE) of the cyclin A2 promoter was found at -28. (J. Zwicker et al., EMBO J.
14:4514-22
[ 1995]). However, this element was located on the antisense strand. No cell
cycle genes
homology region (CHR) was found. Potential Myb sites were predicted starting
at positions
+2, -27 and -66. However, c-myb protein bound only at the first two of these
sites. (See Fig.
3 and Example 23). The nucleotide sequences contain two CpG islands of up to
90% GC
content reaching from -1000 to -700 and from -550 to -50. Multiple GC boxes
are found in
this region, and six GC boxes grouped as three double sites are located
between nt -150 and
-45.
Example 16: Functional analysis of the basal activity of the cyclin A1
promoter.
Portions of the cyclin A1 promoter were Pfu-PCR amplified and cloned into the
promoterless PGL3-Basic Luciferase vector. Promoter activity was analyzed
after transient
transfection into Hela cells. Figure 4 represents transactivation activity of
cyclin A1 promoter
fragments in Hela cells. Activity of 5' deletion constructs was analyzed in
luciferase assays.
Values are expressed as fold activation (PGL3-Basic=1); means and SEM of three
independent experiments are shown. The construct containing nucleotides from -
1299 to
+144 from the 5' cyclin A1 upstream region showed significant promoter
activity when cloned
in the sense direction. The same fragment cloned in the opposite direction or
a construct
containing solely exon 1 and intron 1 did not show promoter activity (data not
shown).
Deletions from the 5' end were made for the -1299 to +144 firagment using
exonuclease
III treatment. Transient transfection and subsequent luciferase assays
revealed the strongest
activity occurred in the construct containing the fragment from -190 to +144
bp. (Fig. 4).
44
CA 02350829 2001-05-14
WO 00/Z9602 PCT/US99/08277
Both the -1299 to +144 and the -190 to +144 constructs exhibited promoter
activity in a
variety of cell lines including Cos-7(monkey kidney cell), MCF-7 (breast
cancer cell), U937
(myeloid leukemia cell), KCL22 (myeloid leukemia cell), PC3 (prostatic cancer
cell}, Hela
(cervical cancer cell) and Jurkat (T-cell lymphoma). (Data not shown). In all
of these
mammalian cell lines, luciferase activities generated by the -190 to +144
construct were higher
than those by the -1299 to +144 construct. Constructs with a 5' end containing
less than 190
by upstream of the transcription start site showed a progressive loss of
promoter activity. A
construct containing by -37 to +144 showed only two-fold higher activity than
the
promoterless vector PGL3-Basic.
Example 17: Role of Sp 1 and GC boxes for transcriptional activity of the
cyclin A 1 promoter.
TATA-less promoters frequently depend on GC boxes to activate transcription.
(J. Lu
et al., J. Biol. Chem. 269:5391-5402 [1994]; M.C. Blake et al., Molec. Cell.
Biol. 10:6632-41
[ 1990]). One of the main classes of transcription factors binding to these
sites are Sp 1 family
proteins (A.J. Courey and R. Tjian, Cell 55:887-98 [1988]; A.P. Kumar and A.P.
Butler,
Nucleic Acids Res. 25:2012-19 [1997]; G. Hagen et al., J. Biol. Chem.
270:24989-94 [1995]).
The cyclin A1 promoter contains at least six potential GC boxes between 190
and 37 by
upstream of the transcription start site. The importance of Sp 1 for the
activity of the cyclin
A 1 promoter, was demonstrated by the use of various promoter constructs that
were
transfected into the Drosophila cell line S2, which lacks endogenous Spl and
Sp3.
Figure S shows activity of the cyclin A 1 promoter fragments in the Drosophila
cell line
S2. Activity is indicated as fold activation of PGL3-Basic as compared to
reporter gene
activity without addition of Spl expression plasmid. The punctated and solid
bars represent
activities without and with Spl co-expression, respectively. When transfected
alone, the
activity of all cyclin A1 promoter fragments was not significantly different
from the empty
vector control. (Fig. 5, dotted bars).
The addition of a Sp 1 expression plasmid strongly activated transcription by
15- to 25-
fold from the cyclin A1 promoter. (Fig. 5, solid bars). Increased
transcriptional activity was
observed only for constructs containing sequences starting between -1299 and -
112 by
upstream of the transcription start site. The construct containing the
nucleotide sequences
between -37 and +I44 did not show any increase in activity, implying that Spl
binding sites
CA 02350829 2001-05-14
WO 00/29602 PCT/US99/08277
between -112 and -37 are essential for Spl mediated transcriptional activity
of the cyclin A1
promoter in Drosophila cells. This region contains four GC boxes which are
grouped in two
pairs. (Fig.3).
The ability of Spl and other Spl family members to bind to these sites was
shown by
gel-shift experiments performed using 5 ~cg Hela nuclear extract. Complexes
bound to a 32p-
end labeled oligonucleotide containing GC box Nos. 1 and 2, and the labeled
oligonucleotide
containing GC boxes Nos. 3+4. Binding was competed away with a 100-fold excess
of cold
Spl consensus oligonucleotide and by a 100-fold excess of cold
oligonucleotides using either
GC box Nos. 1 + 2 or GC box Nos. 3+4. A 100-fold excess of a non-specific
oligonucleotide
did not alter specific complex binding. Antibodies against Sp 1 were added to
some samples,
and antibodies against Sp3 were present in reactions in others. These
supershift experiments
with antibody against either Sp 1 or Sp3 demonstrated the presence of Sp 1 in
one complex, and
the presence of Sp3 in two other complexes. (Data not shown).
The relevance of the GC boxes for promoter activity was further studied by
mutational
analysis. Point mutations were made in each GC box. Each mutant was tested
either alone
with the remaining sites unaltered or in combination with the other mutant
sites. Luciferase
analyses demonstrated that a mutation in either GC box No. 1 or 2 reduced
promoter activity
by about 40 and 75%, respectively, whereas a single mutation of either GC box
No. 3 or 4 did
not have a major effect on promoter activity.
Figure 6 shows effects of GC box mutations on promoter activity. Individual GC
boxes or their combinations were mutated and transiently transfected into Hela
cells. Activity
of the wild type construct containing nt -190 to +144 was set as 100%. Wild
type GC boxes
are indicated in white and mutated GC boxes are shown in black. Mutation of GC
Box Nos.
1 and 2 together, decreased promoter activity by 85%. The presence of at least
one of the two
upstream GC boxes (GC Box Nos. 3 or 4) being intact was essential for cyclin
A1 promoter
activity, as mutations in both reduced promoter activity by about 80%.
Mutations of all four
GC boxes reduced activity of the cyclin A1 promoter by 95%.
Example 18: Cell cycle regulation of promoter activity.
The concentration of cyclins vary during the cell cycle, and one mechanism of
their
regulation occurs at the transcriptional level. (R. Muller, Trends in Genetics
11:173-78
46
CA 02350829 2001-05-14
WO 00/29602 PCTNS99/08277
[1995]). To analyze cell cycle regulation of promoter activity, transiently
transfected cells
were arrested in different phases of the cell cycle and subsequently analyzed
for luciferase
activity. Cell cycle regulated activity was found for the full length promoter
as well as for the
construct containing the -190 to +144 fragment.
Figure 7 shows Cell cycle regulated activity of the cyclin A1 promoter in Hela
cells.
In Figure 7(A), Hela cells were cell cycle arrested after transfection with a
luciferase construct
containing nt -190 to +144 of the cyclin A1 promoter. Cells were subsequently
analyzed for
luciferase activity. Cell cycle synchronization was confirmed by flow
cytometry (data not
shown). The bars represent means and SEM of at least three independent
experiments.
Promoter activity at 0 h was set as 1. The cyclin A1 promoter activity was
relatively low
during the G°/G, phase. It increased after the cell cycle progressed
beyond the G,/S boundary.
In Figure 7(B), Hela cells were synchronized at the G,/S boundary using
aphidicolin,
following transient transfection and serum starvation. Cells were released
from the block and
harvested at the indicated time points for luciferase and cell cycle analyses.
The graph depicts
data from a representative experiment. When transiently transfected Hela cells
were released
from an aphidicolin block, luciferase values started to increase after 6 hours
and reached a
maximum after 12-16 h.
Figure 7(C) shows cell cycle distribution at the different time points of the
time-release
experiment. The hatched, open and solid bars represent G,, S and Gz/M phases,
respectively.
The highest levels of activity were observed in the S and GZ/M phases. The
maximum
promoter activity corresponded to the percentage of cells present in the S and
Gz/M phases.
This is consistent with data showing that levels of cyclin A1 mRNA accumulate
during S
phase, with the highest levels present at the S and Gz/M phases. (Yang et al.,
Mol. Cell. Biol.
[in press 1999]).
Fragments containing nucleotides -1299 to +144, -190 to +144, or -190 to +13
performed similarly in all these experiments (data not shown).
Various point mutations and deletions were generated in the presumed E2F sites
and
the suspected CDE element in order to define the regions that are relevant for
cell cycle
regulation of the cyclin A1 promoter. Activity of the wild type construct
(containing the -1299
to +144 fragment} in aphidicolin arrested cells was set as 1.0 and compared to
the other
constructs. Only a 40%~decrease was detected for the construct containing the
four mutated
47
CA 02350829 2001-05-14
WO 00/29602 PCT/US99/08277
GC boxes. Nucleotides in the suspected CDE in antisense direction were mutated
in the
construct called mutation -19 to -24. There was a strong increase in promoter
activity after
release from a G,/S block by aphidicolin. Constructs lacking the four GC boxes
either due to
mutation or 5' deletion were not induced upon entering the S phase. No
significant difference
was observed between wild type and the mutation -19 to -24 construct.
These findings are consistent with repression of Spl mediated activity in the
G1 phase
of the cell cycle. Selective repression of Spl mediated activity by Sp3 has
been demonstrated
to be relevant in cell cycle regulated promoters containing several Spl sites.
The dihydrofolate
reductase (DHFR) promoter contains four Spl sites and is specifically
repressed by Sp3. (M.J.
Birnbaum et al., Biochemistry 34:16503-08 [1995]). Besides repression by Sp3,
other
mechanisms probably contribute to repression of the cyclin A1 promoter in G1.
Studies have
shown that repression of glutamine rich activators such as Spl and NF-Y is the
predominant
mechanism of cell cycle regulation for several promoters (J. Zwicker et al.,
(1995) Nucleic
Acids Research 23:3822-30 [1995]; J. Zwicker et al., (I998) Nucleic Acids
Research 26:4926-
4932 [1998]). However, none of the known repressor elements (CDE, CHR, E2F)
appears
to be relevant for the cyclin A1 promoter.
A 3' deletion construct (-190 to + 13) was generated by PCR that deleted the
two
presumed E2F sites downstream of the transcriptional start site. Mutations in
these two
presumed E2F sites, the mutation in the inverted presumed CDE element, and the
3' deletion
showed an indistinguishable pattern of cell cycle regulation when compared to
the wildtype.
(Data not shown).
Hence, these E2F sites and the inverted CDE element are unlikely to play a
role in cell
cycle regulation of the promoter. Analysis of 5' deletions and the constructs
containing the
mutated GC boxes revealed that the four GC boxes are essential for cell cycle
regulation. The
activity of the construct containing the mutated GC boxes showed 60% of the
activity of the
wild type reporter construct in G, phase. However, the activity of the
construct failed to
increase when cells entered S phase and showed only 4% of the wild type cyclin
A1 promoter
activity. Similar data were obtained for the 5' deletion lacking the four GC
boxes.
Example 19: Screening transgenic vertebrates for the presence of cyclin A1-
EGFP DNA
Transgenic mice were screened by PCR-amplification of DNA sampled from their
48
CA 02350829 2001-05-14
WO 00/29602 PCT/US99/08277
tails. The mice were anesthetized with metafane, and a I -cm piece of tail tip
was cut using
a sterile scalpel. The tail biopsy was incubated with 100~cg of Proteinase K
in 700,uL lysis
buffer (10 mM Tris, pH7.5, 1mM EDTA, and 10% SDS) overnight at 50°C.
The lysate was
extracted once with SOO,uI phenol, twice with phenol/chloroform (1:1) and was
precipitated
with ice cold isopropanol. The precipitate was centrifuged and the pellet was
washed once
with 70% ethanol. The pellet was allowed to air dry for 30 minutes at room
temperature and
was then resuspended in 200,uL 10 mM Tris, pH 7.5, 0.1 mM EDTA. The tail DNA
was
allowed to incubate at 65 ° C for 10 min, and it was then stored at 4
° C.
For each sample, 100 ng of tail DNA was added to a PCR cocktail mix in a total
volume of SO~cL. For each sample tube, the PCR cocktail contained l O~cL of
Qiagen Q buffer,
S~cL of PCR buffer (Qiagen), dNTPs and a pair of EGFP-specific primers, 5'-TTG
TCG GGC
AGC AGC ACG GGG CCG-3' (SEQ. ID. N0.:30) and 5'-TCA CCG GGG TGG TGC CAT
CCT TGG-3' (SEQ. ID. N0.:31). A 600 by fragment was amplified. A positive
control
contained the cyclin A1-EGFP plasmid DNA, and a negative control contained no
DNA.
Example 20: Selectable fluorescent vertebrate germ cells expressing EGFP by
the cyclin A1
promoter
Five lines of transgenic mice were generated that contain DNA construct
pCyclinAl-
EGFP-1 and express the flourescent green reporter gene (EGFP) under the
control of the cylin
A1 promoter (cyclin A1-EGFP mice). Flourescent green protein is seen in male
germ cells
with FITC filter. The mice were transfected with a construct containing a 1.4
kb 5' flanking
region DNA of human cyclin A1 including, nt. -1299 to +144, inserted into the
BgIIIlHindIII
site of the promoterless fluorescent green protein (EGFP) expression vector
pEGFP-1
(Clontech; Figure 1). The vector also contained a SV40 splice and
polyadenylation signal 3'
to the EGFP gene, as well as kanamycin and neomycin resistance genes for
selection purposes.
The pCyclinAl-EGFP-I construct was expressed in Cos-7, MCF-7, and U937 cells
in vitro.
For the generation of transgenic mice, the vector sequences were removed from
the construct,
and the DNA fragment which comprised the cyclin A1 promoter, the EGFP gene,
and the
SV40 splice and polyadenylation signal was purified on a 10%-40% sucrose
gradient. One-
milliliter fractions were collected from the gradient, and the fraction
containing the construct
49
CA 02350829 2001-05-14
WO 00/29602 PCT/US99/08277
was dialized in a slide cassette dialysis membrane (Pierce) against 4 liters
of 10 mM Tris, pH
7.5, 0.1 mM EDTA for 48 hours with 3 changes.
The purified pCyclinAl-EGFP construct was used to generate transgenic mice by
microinjection of DNA into the pronucleus of fertilized eggs. (Gordon and
Ruddle, 1980;
S Hogan, Costantini and Lacy, 1996). The surrogate mothers delivered 3 8 pups,
8 (21 %) of
which had integrated the transgene as was shown by PCR and Southern Blot
analysis. Two
of the founder animals failed to breed and one did not show expression of the
transgene in the
testis. The remaining 5 animals expressed EGFP in male germ cells.
Example 21: FACS Analysis of Testicular Cells from Transgenic Mice
Testicular cell suspensions from cyclin A1-EGFP transgenic mice were made
using
an enzymatic digestion method modified from Dym et al. (M. Dym et al., Biol.
Reprod. 52:8-
9 [1995]). Testes were dissected from euthanized transgenic mice and
decapsulated. The
seminiferous tubules were spread apart in Enzyme Mix I: Collagenase I (1
mg/mL; bovine
pancreatic, Sigma) in modified HTF medium (Irvine Scientific) containing 1%
BSA (1
mg/mL; Sigma, embryo tested) and incubated for 10 min at 37°C. This
first enzymatic step
is aimed at eliminating cells external to the seminiferous tubules, such as
Leydig cells.
The tubules were then lifted into 1 mL of Enzyme Mix II: Collagenase I (1
mg/mL;
bovine pancreatic), trypsin type III (50 mg/mL; Sigma), DNAase I (1 mg/mL;
Sigma) in
modified HTF medium, which contained BSA (1 mg/mL), and the tubules were cut
into small
pieces using sterile needles attached to 1 mL syringes. The cut tubules were
incubated in
Enzyme Mix II for 15 min at 37°C. After this incubation, the cells were
washed 3 times in
10 mL of the modified HTF by centrifugation at 2,000 rpm and were resuspended.
The cells
were resuspended in 2 mL of modified HTF and were filtered through 70 um mesh
(Corning)
to be analyzed by FACS. Typically, about 3 x 10' testicular cells were
harvested from a
mature male mouse. The cells were tested for viability with trypan blue.
Kidney cells were
prepared in the same way except that the first collagenase incubation was
omitted.
The transgenic testicular cells were analyzed for fluorescence and for
sideward scatter
using a Becton-Dickenson cell sorter on channel 1 (FITC for Green Fluorescent
Protein).
Based on these properties, four populations were distinguishable: 1 ) a EGFP-
negative
CA 02350829 2001-05-14
WO 00/29602 PGTNS99/08277
population; and populations 2 through 4, which had increasing fluorescence and
scatter
properties reflecting different cell types.
The cyclin Al-EGFP cells were also tested with PE conjugated PE anti-c-kit
antibodies
and analyzed with FACS. The FACS analysis showed that there is a population of
fluorescent
cells which expresses EGFP under the cyclin A1 promoter and that these cells
are positive for
c-kit. Some of the c-kit cells were not EGFP positive.
Figure 8 shows frozen sections from testis of adult mice that were cut, rinsed
in
phosphate buffered saline (PBS) for 10 min and analyzed by confocal laser
scanning
microscopy. Whereas no fluorescence could be observed in testicular tubuli of
control mice
(Fig. 8a), strong and highly specific expression of EGFP (Fig. 8b and c) was
detected in testis
of transgenic mice. Maximal EGFP expression was observed during and after the
first meiotic
division and a weaker staining was present in spermatogonia. Magnifications
are 400x (Fig.
8a and b) and 100x (Fig. 8c).
Example 22: The effect of CpG methylation of the cyclin A1 promoter.
Bisulfate sequencing was carried out according to the method described by
Clark et al.
with minor modifications. (S.J. Clark et al., High sensitivity mapping of
methylated cytosines.
Nucleic Acids Res 22: 2990-2997 [1994]). Ten mg of DNA was incubated with the
bisulfiteJhydroquinone solution for six hours. A nested PCR was performed
(detailed Primer
information will be provided on request) and the final PCR product (ca. 400
bp) was gel
purified. The PCR products were either blunt end cloned and at least 10 clones
were
sequenced, or the purified PCR product was directly sequenced using '3P-cycle
sequencing of
nucleotides.
The cyclin A1 promoter - luciferase reporter
construct was in vitro methylated by SssI following the recommendations of the
manufacturer
(New Enigland Biolabs). (S. Kudo, Methyl-CpG binding protein MeCP2 represses
Spl -activated
transcription of the human leukosialin gene when the promoter is methylated,
Mol. Cell.
Bio1.18:5492-99 (1998]). S2 Drosophila cells were transfected as described
previously using 1 wg
of methylated or mock-methylated luciferase - reporter plasmid, 100 ng of Spl
expression
plasmid and 1 ~,g of a CMV-[3-galactosidase expression plasmid used for
standardization
purposes. One ~,g of human MeCP2 expression vector or empty vector control
were co-
51
CA 02350829 2001-05-14
WO OO/Z9602 PCT/US99/08277
transfected. (S. Kudo [1998]). Luciferase experiments were performed in
duplicate and
independently repeated three times. The human MeCP2 expression vector was a
kind gift from
Dr. S. Kudo, Hokkaido Institute, Sapporo, Japan.
As described above, the cyclin A1 promoter is highly GC rich and bears a CpG
island that extends over several hundred base pairs and ends 50 base pairs
upstream of the
main transcriptional start site. When the methylation pattern of the CpG
dinucleotides in
the critical parts of the promoter was analyzed using bisulfate sequencing
(S.J. Clark et al.
[1994]), a high degree of CpG methylation was observed in somatic, adherent
cell lines but
not in cyclin A 1 expressing leukemia cell lines. Hypomethylation in the
leukemic cell
lines was clearly restricted to the CpG island since a CpG at nt. +114 outside
of the CpG
island was found to be completely methylated in all cell lines tested.
To analyze whether methylation of the cyclin A1 promoter was associated with
gene
silencing, MG63 osteosarcoma cells were stably transfected with a Cyclin A 1
promoter - EGFP
construct. After prolonged culture of cells (2 months), there were two
populations of neomycin-
resistant cells, i.e., that showed stable integration of the transgene. One
part of the population
maintained relatively high expression of EGFP (Fig. 9a, left hand peak), while
a fraction of the
population of neomycin-resistant cells lost EGFP expression over time. (Fig.
9a, right hand peak).
Both EGFP-expressing and non-expressing cell populations were sorted by flow
cytometry and
analyzed for CpG methylation of the cyclin A1 promoter transgene. Specific
primers were
designed for the promoter-EGFP construct to avoid analysis of the endogenous
cyclin Al locus.
The transgenic cyclin A1 promoter was substantially non-methylated in the
expressing cells (Fig.
9a, left hand sequencing ladder), but cells that had lost EGFP expression
showed strong
methylation of the cyclin A1 promoter. (Fig. 9a, right hand sequencing
ladder).
Studies recently have shown that the methyl CpG binding protein 2 (MeCP2) is
an
important mediator of methylation-induced gene silencing. (P.L. Jones et al.,
Methylated
DNA and MeCP2 recruit histone deacetylase to repress transcription, Nature
Genetics 19:
187-91 [1998]); X. Nan et al., Transcriptional repression by the methyl CpG-
binding
protein MeCP2 involves a histone deacetylase complex, Nature 393:386-89
[1998]; X. Nan
et al., MeCP2 is a transcriptional repressor with abundant binding sites in
genomic
chromatin, Cell 88:471-78 [1997]). MeCP2 binds specifically to methylated DNA
and
recruits co-repressors, such as mSin3A, leading to the deacetylation of
histones and
52
CA 02350829 2001-05-14
WO 00/29602 PCT/US99/08277
repression of transcriptional activity. (X. Nan et al. [1997] and [1998]). The
human
leukosialin gene is one of the genes shown to be negatively regulated by MeCP2
when its
promoter is methylated. (S. Kudo [1998]). Leukosialin (similar to cyclin Al)
is tissue-
specifically expressed in hematopoietic cells and its transcriptional activity
depends on the
Spl transcription factor. Using S2 Drosophila cells that do not express
endogenous
MeCP2, it was analyzed whether co-transfected MeCP2 would suppress activity of
the
methylated (+) or unmethylated (-) cyclin A1 promoter (Fig. 9b). Upon
transfection of in
vitro methylated cyclin A 1 promoter constructs into Drosophila cells, we
noticed 3-fold
repression without MeCP2. When MeCP2 was co-expressed with the methylated
cyclin
A1 promoter constructs, promoter activity was inhibited by 12-fold, indicating
that MeCP2
can suppress transcriptional activation of the methylated cyclin A1 promoter.
(Fig. 9b).
Since methylation appeared to be involved in regulation of the cyclin A1 gene
in the
mammalian cell lines, it was investigated whether the site of chromosomal
integration would
determine the patterns of methylation and expression of the transgenic cyclin
A 1 promoter. Four
lines of transgenic mice carried the cyclin A 1 promoter - EGFP reporter
construct, as described
above; this was the same nucleic acid construct used to generate the stable
MG63 cell line. All
lines of transgenic mice showed highly specific expression in the testis
resembling the expression
pattern previously determined by in-situ hybridization techniques. (Fig. 8; C.
Sweeney et al.
[1996]). The EGFP expression pattern in testis was indistinguishable among the
different lines.
The cyclin A1 promoter was able to direct tissue specific expression in the
testis independent of
the chromosomal integration site. The methylation status of a transgene is
thought to be largely
determined by either the chromatin structure at the site of integration, the
cis-acting sequences in
the transgene, and/or the influence of a locus control region. (J.R. Chaillet
et al., Parental-speck
methylation of imprinted transgene is established duringgametogenesis and
progressively changes
during embryogenesis, Cell 66:77-83 [1991]; K. Matsuo et al., An embryonic
demethylation
mechanism imrolving binding of transcription factors to replicating DNA, EMBO
J. 17:1446-53
[1998]; M. Brandeis et al., Nature 371:435-38 [1994]). Transgene activity has
also been reported
to be associated with hypomethylation. (E.g., Pilcaart et al. [1998]).
Analysis of the methylation
status of the human cyclin A1 promoter in the testis of four transgenic mouse
lines showed that
the promoter and the transgene were not methylated in the testis of two lines.
However, the
promoter and the transgene were heavily methylated in testis of the two other
lines. No difference
53
CA 02350829 2001-05-14
WO 00/29602 PCTNS99/08277
in the EGFP expression pattern in testis could be found between the marine
lines either with or
without CpG methylation. To confine that EGFP was highly expressed despite
methylation in
these male germ cells, testis cells were disaggregated and sorted by flow
cytometry as described
above.
Bisulfate sequencing confirmed that methylation of the cyclin A 1 promoter in
germ
cells did not inhibit expression of the transgene in testis. One of the marine
lines without
methylation in testis showed promoter methylation in the kidney and bone
marrow, but not
in the liver and did not express the transgene in any organ besides the
testis. The silencing
of a gene in the absence of methylation has been described for other genes as
well. (E.g.,
P.M. Warnecke and S.J. Clark, DNA methylation profile of the mouse skeletal
alpha-actin
promoter during development and differentiation, Mol. Cell Biol. 19:164-72
[1999]). One
transgenic marine line did not show a significant degree of methylation of the
transgenic
cyclin Al promoter anywhere and expressed EGFP in a subset of cells in the
kidney
(25%), spleen (10%) and bone marrow (16%). Taken together, transcriptional
activity of
the cyclin A 1 promoter transgene outside of the testis was only seen when the
promoter
was not methylated. This finding might supports a linkage of methylation of
the cyclin A1
promoter to transcriptional repression in somatic cells. In contrast,
methylation of the
cyclin A 1 promoter - EGFP transgene did not lead to silencing in marine male
germ cells.
Example 23: Transactivation of cyclin A1 promoter by c-myb.
Analysis of the cyclin A1 promoter sequence showed potential binding sites for
c-
myb within the -190 to +144 fragment. (Fig. 3). To analyze fizrther the
differences in
expression, four human cell lines were chosen that differed in the degree of
cyclin A1
expression. Two were derived from myeloid cells (IJ937, KCL22) and two others
from
solid carcinomas (PC3 prostate cancer, Hela cervical carcinoma). Expression of
cyclin A1
was analyzed by RT-PCR followed by Southern blotting. The RT-PCR results
confirmed
that cyclin A 1 expression differed between the myeloid and the non-myeloid
cell lines.
The highest RNA levels were found in U937 and the lowest occurred in Hela
cells.
To analyze whether differences in RNA levels could be related to promoter
activity, the cyclin A1 promoter was transiently transfected into several
myeloid and
adherent cells lines (Fig. 10). Both cyclin A1 promoter luciferase constructs
ranging finm
54
CA 02350829 2001-05-14
WO 00/29602 PCT/US99/08277
-1299 to +144 and from -190 to +144 showed activity in all four cell lines
(Fig. 10). The
reporter activity of the shorter promoter fragment was always higher than the
activity of
the longer fragment. In addition, the activity of the cyclin Al promoter was
higher than
that of the SV40 promoter (without enhancer) in all four cell lines.
The cyclin A2 promoter is tightly cell cycle regulated and is assumed to be
transactivated in all cycling mammalian cells. Activity of the cyclin A2
promoter was
detectable in all four cell lines, but the degree of activity was inversely
correlated with the
cyclin A1 promoter activity. Cyclin A2 promoter activity was higher in PC3 and
Hela cells
and it was lower in the myeloid cell lines as compared to the cyclin A 1
promoter activity.
(Fig. 10}. Preferential activity of the cyclin A1 promoter in myeloid cells
(compared to the
cyclin A2 promoter) was evident for both promoter constructs tested. The
inverse
relationship between cyclin A2 and cyclin A 1 was also present at the RNA
level in
samples from patients with acute myeloid leukemia. (R. Yang et al. [1999]).
However,
activity of the cyclin A1 promoter by transient transfection was not limited
to the myeloid
cell lines but was also present in PC3 and Hela cells. The tissues from which
these cell
lines derived express very low levels of cyclin A1. An explanation could be
that
transcription factors expressed in the cell lines, but not expressed in the
normal tissue, lead
to aberrant promoter activity. One transcription factor expressed in a wide
variety of cell
lines is c-myb. Western blot analysis demonstrated expression of c-myb in all
four cell
lines as well as in ML,-l, another myeloid cell Line that expresses high
levels of cyclin A1.
The non-myeloid cell lines appeared to have only a high molecular weight form
while the
myeloid lines had both a high and a low molecular weight form. This may
reflect a
phosphorylated and a non-phosphorylated myb protein.
CA 02350829 2001-05-14
WO 00/29602 PCTNS99/08277
To analyze promoter transactivation by c-myb, a c-myb expression vector was
transfected (0 to 5 ~g of co-transfected plasmid DNA encoding c-myb) along
with the -190
to +144 cyclin A1 promoter construct into CV-1 cells that do not express c-
myb. A dose-
dependent increase in cyclin A1 promoter activity occurred (Fig. l la), and no
increase in
activity was observed when c-myb was co-transfected with the empty reporter
plasmid (data
not shown). The same experiments were repeated using U937 myeloid cells, which
express
rather Sow levels of c-myb. As in CV-1 cells, in U937 c-myb clearly
transactivated the
promoter with maximal activity occurring when 3 p,g of c-myb-encoding DNAs
were co-
transfected. (Fig. 11 b). These findings indicate that the cyclin A 1 promoter
can be
transactivated by c-myb in adherent as well as in myeloid cell lines.
To analyze whether c-myb directly affected the cyclin A1 promoter, binding of
c-myb
protein to the predicted myb binding sites in the promoter region was
examined. Gel-shift
experiments were performed with c-myb protein expressed in Cos-1 cells and 32P-
labelled
oligonucleotides constituting the myb-binding sites of the cyclin A 1
promoter. Experiments
1 S showed that c-myb was able to bind to the cyclin A1 promoter at the +2 to
+5 binding site.
Weak binding was seen at the potential myb site at -27 to -24 and no specific
binding at the
site at position -66 could be detected. Nuclear extracts from Cos-1 cells
expressing c-myb
led to the appearance of two new bands compared to nuclear extract prepared
from Cos-1
cells transfected with empty expression vector, only. Specificity of the
binding to the +2 site
was confirmed using competitor oligonucleotides and c-myb specific antibody.
Addition of
c-myb specific antibody led to extinction of both bands. The faster migrating
band appeared
at the same position as the c-myb band produced on a myb consensus binding
site (data not
shown). Therefore the slower migrating band might be a complex of proteins
with one of them
being c-myb. No c-myb binding could be detected using a potential binding site
at -66. The
binding site at -27 showed a rather weak band after incubation with the c-myb
expressing
nuclear extract. (Data not shown). Also, the band did not disappear after
addition of c-myb
56
CA 02350829 2001-05-14
WO 00/29602 PCT/US99/08277
antibody implying that c-myb either did not or only weakly bound this site. To
test
whether c-myb activation of the promoter was affected by alteration of the myb
binding sites,
different sites were mutated and the resulting constructs were transfected in
KCL22 cells.
These cells showed the highest c-myb expression of all the cell lines.
Abrogation of the myb
site at +2 clearly diminished promoter activity by 50% whereas a mutation at
either -27 or
mutation of the ets site at -15 did not lead to a decrease in promoter
activity. The myb site
at +2 to +S is close to the transcriptional start site and the base pairs
surrounding the
transcriptional start site could function as an Initiator (Inr). To rule out
that the observed
effects of the mutation at +2 depended on the loss of binding of the basal
transcriptional
machinery, we transfected the mutated reporter construct together with the c-
myb expression
plasmid or an empty vector control into CV-1 cells and compared the results
with
transfections using the wildtype promoter plasmid. The mutation at +2 led to a
minor
reduction in promoter activity when transfected with the empty vector control.
However,
transactivation of the mutated reporter plasmid by c-myb was reduced by more
than 50%,
indicating that c-myb can transactivate the cyclin A1 promoter through this
site. Other sites
or indirect effects may contribute to the cyclin A1 promoter activation,
because the mutation
at +2 did not abolish the increase in promoter activity entirely. Different
amounts of c-myb
were co-expressed with a cyclin Al promoter construct (-190 to +144 fragment).
Empty
vector was used to reach the same total amount of DNA in all experiments. Mean
and
standard error for three independent experiments are shown.
The foregoing examples being illustrative but not an exhaustive description of
the embodiments of the present invention, the following claims are presented.
57
CA 02350829 2001-05-14
WO 00/29602 PCT/US99/08277
SEQUENCE LISTING
<110> Cedars-Sinai Medical Center
Readhead, Carol W.
Winston, Robert
Koeffler, H. Phillip
Miiller, Carsten
<120> Transfection, Storage and Transfer of
Male Germ Cells for Generation of Selectable Transgenic Stem
Cells
<130> P07 41795
<140> Unassigned
<141> 1999-04-15
<150> US 09/191,920
<151> 1998-11-13
<150> US 60/065,825
<151> 1997-11-14
<150> US 09/272,443
<151> 1999-03-19
<150> PCT/US98/24238
<151> 1998-11-13
<160> 32
<170> FastSEQ for Windows Version 3.0
<210> 1
<211> 1958
<212> DNA
<213> HUMAN
<400> 1
tcgatctgatttagagattt agggatggatgttttaaaaa aagcaaaagtagtaacagac 60
tatagcattggtaatgtgtg tgtgcatatatacatattat ttttaaaaaaataaagttcg 120
attatttcacctggcttgtc agtcacctatgcaggcgtct gagcccccgggtttccagga 180
240
gccccccgtataaggacccc agggactcctctccccacgc ggccgggccgcccgcccggc 300
ccccagcccggagagctgcc accgaccccctcaacgtccc aagccccagctctgtcgccc 360
gcgttccttcctcttcctgg gccacaatcttggctttccc gggccggcttcacgcagttg 420
cgcaggagcccgcgggggaa gacctctcgtggggacctcg agcacgacgtgcgaccctaa 480
atccccacatctcctctgcc gcctcgcaggccacatgcac cgggagccgggcggggcagg 540
cgcggcccgcaaggaccccc gcgatggagacgcaacactg ccgcgactgcacttggggca 600
gccccgccgcgtcccagccg cctcccggcaggaagcgtag gtgtgtgagccgacccggag 660
cgagccgcgccctcgggcca gcgtgggcagggcgccgcag cctgcgcagccccgaggacc 720
ccgcgtcgctctcccgagcc agggttctcaggagcgggcc gcgcaggagacgttagaggg 780
ggttgttagcggctgttggg agaacgggtcacggaaacag tcccttccaaagccggggcc
1
CA 02350829 2001-05-14
WO 00/29602 PCT/US99/08277
atcgtggggtgggcgagtcc gccctcccaggccgggggcg cggaccagaggggacgtgtg 840
cagacggccgcggtcagccc cacctcgcccgggcggagac gcacagctggagctggaggg 900
ccgtcgcccgttgggccctc aggggcctgaacgcccaggg gtcgcggcgagtccacccgg 960
agcgagtcaggtgagcaggt cgccatggcgatgcggcccc ggagagcgcacgcctgccgc 1020
ggtcggcatggaaacgctcc cgctaggtccgggggcgccg ctgattggccgattcaacag 1080
acgcgggtgggcagctcagc cgcatcgctaagcccggccg cctcccaggctggaatccct 1140
cgacacttggtccttcccgc cccgcccttccgtgccctgc ccttccctgcccttccccgc 1200
cctgccccgcccggcccggc ccggccctgcccaaccctgc cccgccctgccccgcccagc 1260
cggccacctcttaaccgcga tcctccagtgcacttgccag ttgttccggacacatagaaa 1320
gataacgacgggaagacggg gccccgtttggggtccaggc aggttttggggcctcctgtc 1380
tggtgggaggaggccgcagc gcagcaccctgctcgtcact tgggatggagaccggctttc 1440
ccgcaatcatgtaccctgga tcttttattgggggctgggg agaagagtatctcagctggg 1500
aaggaccggggctcccagat ttcgtcttccaggtaacgtg ggtttagtatcccgacttgg 1560
aggcttgtcagaatgtttct ctccttccagcccaacacga agtcttgggataaaaagcct 1620
ccctcagggattcaaataac tgttttgattcagagcaact ttgatcgcctgtgcggtcgc 1680
acctgccctttcagccccaa taattactgggaagatcagc aattggtgttagtcccattg 1740
cttggtgctctccctcctag aggttcgctgtgtccttgga gcccggggtggacggaatcg 1800
actaaacagcttgtctgttt ctctttccctggtagcagca gcccgtggagtctgaagcaa 1860
tgcactgcagcaaccccaag agtggagttgtgctggctac agtggcccgaggtcccgatg 1920
cttgtcagatactcaccaga gccccgctgggccaggat 1958
<210> 2
<211> 1442
<212> DNA
<213> HUMAN
<220>
<221> promoter
<222> (1)...(1442)
<221> mutation
<222> (1427)...(1427)
<400> 2
tcgatctgatttagagatttagggatggat gttttaaaaaaagcaaaagt agtaacagac 60
tatagcattggtaatgtgtgtgtgcatata tacatattatttttaaaaaa ataaagttcg 120
attatttcacctggcttgtcagtcacctat gcaggcgtctgagcccccgg gtttccagga 180
gccccccgtataaggaccccagggactcct ctccccacgcggccgggccg cccgcccggc 240
ccccagcccggagagctgccaccgaccccc tcaacgtcccaagccccagc tctgtcgccc 300
gcgttccttcctcttcctgggccacaatct tggctttcccgggccggctt cacgcagttg 360
cgcaggagcccgcgggggaagacctctcgt ggggacctcgagcacgacgt gcgaccctaa 420
atccccacatctcctctgccgcctcgcagg ccacatgcaccgggagccgg gcggggcagg 480
cgcggcccgcaaggacccccgcgatggaga cgcaacactgccgcgactgc acttggggca 540
gccccgccgcgtcccagccgcctcccggca ggaagcgtaggtgtgtgagc cgacccggag 600
cgagccgcgccctcgggccagcgtgggcag ggcgccgcagcctgcgcagc cccgaggacc 660
ccgcgtcgctctcccgagccagggttctca ggagcgggccgcgcaggaga cgttagaggg 720
ggttgttagcggctgttgggagaacgggtc acggaaacagtcccttccaa agccggggcc 780
atcgtggggtgggcgagtccgccctcccag gccgggggcgcggaccagag gggacgtgtg 840
cagacggccgcggtcagccccacctcgccc gggcggagacgcacagctgg agctggaggg 900
ccgtcgcccgttgggccctcaggggcctga acgcccaggggtcgcggcga gtccacccgg 960
agcgagtcaggtgagcaggtcgccatggcg atgcggccccggagagcgca cgcctgccgc 1020
ggtcggcatggaaacgctcccgctaggtcc gggggcgccgctgattggcc gattcaacag 1080
acgcgggtgggcagctcagccgcatcgcta agcccggccgcctcccaggc tggaatccct 1140
2
CA 02350829 2001-05-14
WO 00/29602 PCT/US99/08Z77
cgacacttggtccttcccgc cccgcccttccgtgccctgc ccttccctgcccttccccgc 1200
cctgccccgcccggcccggc ccggccctgcccaaccctgc cccgccctgccccgcccagc 1260
cggccacctcttaaccgcga tcctccagtgcacttgccag ttgttccggacacatagaaa 1320
gataacgacgggaagacggg gccccgtttggggtccaggc aggttttggggcctcctgtc 1380
tggtgggaggaggccgcagc gcagcaccctgctcgtcact tgggattgagaccggctttc 1440
cc 1442
<210> 3
<211> 1294
<212 > DNA
<213> HUMAN
<220>
<221> promoter
<222> (1)...(1294)
<221> mutation
<222> (1279)...(1279)
<400> 3
atgcaggcgtctgagccccc gggtttccaggagccccccg tataaggaccccagggactc 60
ctctccccacgcggccgggc cgcccgcccggcccccagcc cggagagctgccaccgaccc 120
cctcaacgtcccaagcccca gctctgtcgcccgcgttcct tcctcttcctgggccacaat 180
cttggctttcccgggccggc ttcacgcagttgcgcaggag cccgcgggggaagacctctc 240
gtggggacctcgagcacgac gtgcgaccctaaatccccac atctcctctgccgcctcgca 300
ggccacatgcaccgggagcc gggcggggcaggcgcggccc gcaaggacccccgcgatgga 360
gacgcaacactgccgcgact gcacttggggcagccccgcc gcgtcccagccgcctcccgg 420
caggaagcgtaggtgtgtga gccgacccggagcgagccgc gccctcgggccagcgtgggc 480
agggcgccgcagcctgcgca gccccgaggaccccgcgtcg ctctcccgagccagggttct 540
caggagcgggccgcgcagga gacgttagagggggttgtta gcggctgttgggagaacggg 600
tcacggaaacagtcccttcc aaagccggggccatcgtggg gtgggcgagtccgccctccc 660
aggccgggggcgcggaccag aggggacgtgtgcagacggc cgcggtcagccccacctcgc 720
ccgggcggagacgcacagct ggagctggagggccgtcgcc cgttgggccctcaggggcct 780
gaacgcccaggggtcgcggc gagtccacccggagcgagtc aggtgagcaggtcgccatgg 840
cgatgcggccccggagagcg cacgcctgccgcggtcggca tggaaacgctcccgctaggt 900
ccgggggcgccgctgattgg ccgattcaacagacgcgggt gggcagctcagccgcatcgc 960
taagcccggccgcctcccag gctggaatccctcgacactt ggtccttcccgccccgccct 1020
tccgtgccctgcccttccct gcccttccccgccctgcccc gcccggcccggcccggccct 1080
gcccaaccctgccccgccct gccccgcccagccggccacc tcttaaccgcgatcctccag 1140
tgcacttgccagttgttccg gacacatagaaagataacga cgggaagacggggccccgtt 1200
tggggtccaggcaggttttg gggcctcctgtctggtggga ggaggccgcagcgcagcacc 1260
ctgctcgtcacttgggattg agaccggctttccc 1294
<210> 4
<211> 597
<212> DNA
<213> HUMAN
<220>
<221> promoter
<222> (1)...(597)
<221> mutation
CA 02350829 2001-05-14
WO 00/29602 PCT/US99/08277
<222> (582)...(582)
<400> 4
ggccgcggtcagccccacct cgcccgggcggagacgcaca gctggagctggagggccgtc 60
gcccgttgggccctcagggg cctgaacgcccaggggtcgc ggcgagtccacccggagcga 120
gtcaggtgagcaggtcgcca tggcgatgcggccccggaga gcgcacgcctgccgcggtcg 180
gcatggaaacgctcccgcta ggtccgggggcgccgctgat tggccgattcaacagacgcg 240
ggtgggcagctcagccgcat cgctaagcccggccgcctcc caggctggaatccctcgaca 300
cttggtccttcccgccccgc ccttccgtgccctgcccttc cctgcccttccccgccctgc 360
cccgcccggcccggcccggc cctgcccaaccctgccccgc cctgccccgcccagccggcc 420
acctcttaaccgcgatcctc cagtgcacttgccagttgtt ccggacacatagaaagataa 480
cgacgggaagacggggcccc gtttggggtccaggcaggtt ttggggcctcctgtctggtg 540
ggaggaggccgcagcgcagc accctgctcgtcacttggga ttgagaccggctttccc 597
<210> 5
<211> 469
<212> DNA
<213> HUMAN
<220>
<221> promoter
<222> (1) . . . (469)
<221> mutation
<222> (454) . . . (454)
<400> 5
agcaggtcgccatggcgatgcggccccgga gagcgcacgcctgccgcggt cggcatggaa 60
acgctcccgctaggtccgggggcgccgctg attggccgattcaacagacg cgggtgggca 120
gctcagccgcatcgctaagcccggccgcct cccaggctggaatccctcga cacttggtcc 180
ttcccgccccgcccttccgtgccctgccct tccctgcccttccccgccct gccccgcccg 240
gcccggcccggccctgcccaaccctgcccc gccctgccccgcccagccgg ccacctctta 300
accgcgatcctccagtgcacttgccagttg ttccggacacatagaaagat aacgacggga 360
agacggggccccgtttggggtccaggcagg ttttggggcctcctgtctgg tgggaggagg 420
ccgcagcgcagcaccctgctcgtcacttgg gattgagaccggctttccc 469
<210> 6
<211> 333
<212 > DNA
<213> HUMAN
<220>
<22I> promoter
<222> (1)...(333)
<221> mutation
<222> (318) . . . (318)
<400> 6
aagcccggcc gcctcccagg ctggaatccc tcgacacttg gtccttcccg ccccgccctt 60
ccgtgccctg cccttccctg cccttccccg ccctgccccg cccggcccgg cccggccctg 120
cccaaccctg ccccgccctg ccccgcccag ccggccacct cttaaccgcg atcctccagt 180
gcacttgcca gttgttccgg acacatagaa agataacgac gggaagacgg ggccccgttt 240
4
CA 02350829 2001-05-14
WO 00/29602 PCT/US99/08277
ggggtccagg caggttttgg ggcctcctgt ctggtgggag gaggccgcag cgcagcaccc 300
tgctcgtcac ttgggattga gaccggcttt ccc 333
<210> 7
<211> 303
<212> DNA
<213> HUMAN
<220>
<221> promoter
<222> (1) . . . (303)
<221> mutation
<222> (288)...(288)
<400> 7
tcgacacttg gtccttcccgccccgcccttccgtgccctg cccttccctg cccttccccg 60
ccctgccccg cccggcccggcccggccctgcccaaccctg ccccgccctg ccccgcccag 120
ccggccacct cttaaccgcgatcctccagtgcacttgcca gttgttccgg acacatagaa 180
agataacgac gggaagacggggccccgtttggggtccagg caggttttgg ggcctcctgt 240
ctggtgggag gaggccgcagcgcagcaccctgctcgtcac ttgggattga gaccggcttt 300
ccc 303
<210> 8
<211> 263
<212> DNA
<213> HUMAN
<220>
<221> promoter
<222> (1) . . . (263)
<221> mutation
<222> (248) . . . (248)
<400> 8
cccttccctg cccttccccgccctgccccgcccggcccgg cccggccctgcccaaccctg 60
ccccgccctg ccccgcccagccggccacctcttaaccgcg atcctccagtgcacttgcca 120
gttgttccgg acacatagaaagataacgacgggaagacgg ggccccgtttggggtccagg 280
caggttttgg ggcctcctgtctggtgggaggaggccgcag cgcagcaccctgctcgtcac 240
ttgggattga gaccggctttccc 263
<210> 9
<211> 255
<212> DNA
<213> HUMAN
<220>
<221> promoter
<222> (1) . . . (255)
<221> mutation
<222> (240)...(240)
CA 02350829 2001-05-14
WO 00/29602 PCT/US99/08277
<400> 9
tgcccttccccgccctgccc cgcccggcccggcccggccc tgcccaaccctgccccgccc 60
tgccccgcccagccggccac ctcttaaccgcgatcctcca gtgcacttgccagttgttcc 120
ggacacatagaaagataacg acgggaagacggggccccgt ttggggtccaggcaggtttt 180
ggggcctcctgtctggtggg aggaggccgcagcgcagcac cctgctcgtcacttgggatt 240
gagaccggctttccc 255
<210> 10
<211> 209
<212> DNA
<213> HUMAN
<220>
<221> promoter
<222> (1) . . . (209)
<400> 10
aagcccggcc gcctcccagg ctggaatccc tcgacacttg gtccttcccg ccccgccctt 60
ccgtgccctg cccttccctg cccttccccg ccctgccccg cccggcccgg cccggccctg 120
cccaaccctg ccccgccctg ccccgcccag ccggccacct cttaaccgcg atcctccagt 180
gcacttgcca gttgttccgg acacataga 209
<210> 11
<211> 202
<2I2> DNA
<213> HUMAN
<220>
<221> promoter
<222> (1) . . . (202)
<400> 11
aagcccggcc gcctcccagg ctggaatccc tcgacacttg gtccttcccg ccccgccctt 60
ccgtgccctg cccttccctg cccttccccg ccctgccccg cccggcccgg cccggccctg 120
cccaaccctg ccccgccctg ccccgcccag ccggccacct cttaaccgcg atcctccagt 180
gcacttgcca gttgttccgg ac 202
<210> 12
<211> 195
<212> DNA
<213> HUMAN
<220>
<221> promoter
<222> (1)...(195)
<400> 12
aagcccggcc gcctcccagg ctggaatccc tcgacacttg gtccttcccg ccccgccctt 60
ccgtgccctg cccttccctg cccttccccg ccctgccccg cccggcccgg cccggccctg 120
cccaaccctg ccccgccctg ccccgcccag ccggccacct cttaaccgcg atcctccagt 180
gcacttgcca gttgt 195
6
CA 02350829 2001-05-14
WO 00/29602 PCT/US99/08277
<210> 13
<211> 194
<212> DNA
<213> HUMAN
<220>
<221> promoter
<222> (1)...(194)
<400> 13
aagcccggcc gcctcccagg ctggaatccc tcgacacttg gtccttcccg ccccgccctt 60
ccgtgccctg cccttccctg cccttccccg ccctgccccg cccggcccgg cccggccctg 120
cccaaccctg ccccgccctg ccccgcccag ccggccacct cttaaccgcg atcctccagt 180
gcacttgcca gttg 194
<210> 14
<211> 21
<212> DNA
<213> Artificial Sequence
<220>
<223> Single-stranded oligonucleotide primer sequence
<400> 14
ccctctcaga acagacatac a 21
<210> 15
<211> 21
<212> DNA
<213> Artificial Sequence
<220>
<223> Single-stranded oligonucleotide primer sequence
<400> 15
ctgatccaga ataacacctg a 21
<210> 16
<211> 36
<212> DNA
<213> Artificial Sequence
<220>
<223> n equals inosine; Universal 5' RACE abridged
anchor primer
<400> 16
ggccacgcgt cgactagtac gggnngggnn gggnng 36
<210> 17
<211> 19
<212> DNA
<213> Artificial Sequence
7
CA 02350829 2001-05-14
WO 00/29602 PCT/US99/08277
<220>
<223> Single-stranded oligonucleotide primer sequence
<400> 17
ctcctcccac cagacagga 19
<210> 18
<211> 26
<212> DNA
<213> Artificial Sequence
<220>
<223> Double-stranded oligonucleotide
<400> 18
cctgccccgc cctgccccgc ccagcc 26
<210> 19
<211> 27
<212> DNA
<213> Artificial Sequence
<220>
<223> Double-stranded oligonucleotide
<400> 19
ccttccccgc cctgccccgc ccggccc 27
<210> 20
<211> 22
<212 > DNA
<213> Artificial Sequence
<220>
<223> Double-stranded oligonucleotide
<400> 20
attcgatcgg ggcggggcga gc 22
<210> 21
<211> 25
<212> DNA
<213> Artificial Sequence
<220>
<223> Double-stranded oligonucleotide
<400> 21
gagaccggct cgaacgcaat catgt 25
<210> 22
<211> 24
8
CA 02350829 2001-05-14
WO 00/29602 PCT/US99/08277
<212> DNA
<213> Artificial Sequence.
<220>
<223> Single-stranded oligonucleotide
<400> 22
aatcgataag aattcgtcga ccga 24
<210> 23
<211> 29
<212> DNA
<213> Artificial Sequence
<220>
<223> Single-stranded oligonucleotide
<400> 23
ccccgccctg ccccttacag ccggccacc 29
<210> 24
<211> 29
<212> DNA
<213> Artificial Sequence
<220>
<223> Single-stranded oligonucleotide
<400> 24
ccccgccctg ccccttacag ccggccacc 29
<210> 25
<211> 22
<212> DNA
<213> Artificial Sequence
<220>
<223> Single-stranded oligonucleotide
<400> 25
ccctgcccct tccggcccgg cc 22
<210> 26
<211> 22
<212> DNA
<213> Artificial Sequence
<220>
<223> Single-stranded oligonucleotide
<400> 26
ctgcccttcc cttccctgcc cc 22
9
CA 02350829 2001-05-14
WO 00/29602 PCT/US99/08277
<210> 27
<211> 42
<212> DNA
<213> Artificial Sequence
<220>
<223> Single-stranded oligonucleotide
<400> 27
gcccaaccct gcccttacct gccccttaca gccggccacc tc 42
<210> 28
<211> 45
<212> DNA
<213> Artificial Sequence
<220>
<223> Single-stranded oligonucleotide
<400> 28
cttccctgcc cttcccttac ctgcccctta cggcccggcc cggcc 45
<210> 29
<211> 29
<212> DNA
<213> Artificial Sequence
<220>
<223> Single-stranded oligonucleotide
<400> 29
ccacctctta acaagcttcc tccagtgca 29
<210> 30
<211> 24
<212> DNA
<213> Artificial Sequence
<220>
<223> EGFP-specific single-stranded oligonucleotide
<400> 30
ttgtcgggca gcagcacggg gccg 24
<210> 31
<211> 24
<212> DNA
<213> Artificial Sequence
<220>
<223> EGFP-specific single-stranded oligonucleotide
<400> 31
CA 02350829 2001-05-14
WO 00/29602 PCT/US99/08277
tcaccggggt ggtgccatcc tttg , 24
<210> 32
<211> 6
<212> DNA
<213> HUMAN
<220>
<221> misc_feature
<222> (0) . . (0)
<400> 32
6
tcgcgg
11