Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02369002 2001-09-26
WO 00/50644 PCT/US00/04939
METHODS FOR PURIFYING DNA USING
IMMOBILIZED CAPTURE PROBES
BACKGROUND OF THE INVENTION
Nucleic acid sequence information plays a vital role in both basic and
applied biomedical research. The nucleotide sequence of a particular portion
of
DNA can be instructive as to the molecular basis for a given disease, such as
Huntington's Disease. Once a segment of genome has been identified as being
potentially responsible for a particular affliction, elucidating the
nucleotide sequence
becomes very important. The sequence, once lmown, can play a part in the
therapeutic regime to be provided, such as in the case of gene therapy. This
is most
evident when the basis of the disease is a genetic mutation of the normal
gene. One
methodology employed for treating genetic mutation-based diseases is the
introduction of the wild-type nucleotide sequence. But first it must be
established
that in fact a gene, or an aberrant form of a gene, is the etiologic agent for
a
particular disease or syndrome. This information is most often provided
through the
isolation and characterization of the putative aberrant gene. Characterization
often
involves the sequence analysis of the nucleotide sequence itself that defines
the gene
of interest. This will often involve understanding both the wild-type, or
physiologically normal, and mutant genes.
In practice, the quality of the sequence analysis is, in part, a reflection of
the
quality of the starting material. It is vital that the preparation that is to
be subjected
to sequence analysis be of high quality, that is, relatively pure and free of
CA 02369002 2001-09-26
WO 00/50644 PCT/US00/04939
-2-
contaminating species like proteins and small molecules, such as salts, that
can
interfere with obtaining a high quality result from sequence analysis. Current
protocols involve ethanol precipitation in order to remove unincorporated
nucleotides and salt from, for example, extension products prior to sequence
analysis. Also, with regard to the extension products, it is often desirable
to remove
any template DNA and excess primers from the preparation prior to sequence
analysis. Precipitation is time consuming and requires care to achieve
consistent
product yields. Nevertheless, it is critical to the success of performing an
informative sequence analysis on a target nucleotide sequence that the target
sequence preparation be as free of contaminating molecules as possible.
Additionally, it would be advantageous to have a purification system that
could sort
the products of multiplexed sequencing reactions.
SUMMARY OF THE INVENTION
The present invention pertains to methods of purifmg a target molecule
contained within a test sample. Typically, the target molecule in a test
sample will
be a nucleic acid molecule, in particular, single-stranded DNA primer
extension
sequencing reaction products of a dideoxy sequencing reaction, for example,
the
Sanger method, Sanger, F., et al., Proc. Natl. Acad. Sci. USA, 74:5463-5467
(1977),
or a cycle sequencing method, Carothers, Biotechniques" 7:494-499 (1989), the
entire teachings of which are herein incorporated by reference in their
entirety.
Once purified, these purified nucleic acid molecules can be used in a variety
of ways
including being subjected to capillary or slab gel electrophoresis for DNA
sequence
analysis. Nucleic acid molecule capture probes modified with S'-acrylamide
groups
are copolymerized within an electrophoresis gel, such as a polyacrylamide gel.
Single-stranded target nucleic acid molecules can bind to their complementary
sequence contained within a capture probe, if there is sufficient
complementarity
between the two molecules. Double-stranded target nucleic acid molecules can
bind
to a complementary sequence forming a triple helical arrangement.
The target molecule, or molecules, present in a test sample can be placed in
an electrophoresis gel containing immobilized capture probes and undergo
electrophoresis. The target molecule will migrate through the gel medium until
it
CA 02369002 2001-09-26
WO 00/50644 PCT/US00/04939
-3-
comes in contact with its complementary immobilized capture probe. Once the
target and capture probe are in contact with one another, they can hybridize
forming
a complex. The non-target molecules contained in the test sample can continue
electrophoresis and are effectively removed from the target molecule.
In one embodiment of the present invention, the target molecules are DNA
extension products formed during a primer extension sequencing reaction. A
reaction mixture from a primer extension sequencing reaction is loaded into a
purification device, for example, a microtiter plate containing multiple wells
(having, e.g., 6, 12, 96 or 384 wells). The purification device comprises an
electrophoresis gel containing immobilized capture probes that are
complementary
to at least one nucleotide sequence region contained within the target
molecules.
Preferably, an electric field is applied such that all negatively charged
molecules
migrate through the electrophoresis gel toward the a positively charged
electrode.
The positively charged electrode can be housed in a positively charged
electrode
buffer chamber. This chamber can be used to collect molecules that exit the
electrophoretic medium as a result of their electrophoretic migration. The.
target
molecules will be captured by complementary, immobilized capture probes that
are
within the gel. The non-target molecules contained within the test sample will
pass
through the gel and into the positively charged electrode buffer (also
referred to
herein as the collecting chamber). The collecting chamber can then be replaced
with
fresh positively charged electrode buffer. A sufficient voltage can be applied
so as
to denature the hybridization complex formed between the target molecule and
capture probe thereby releasing the target molecule. The electric field can be
applied using the same polarity as originally applied, thereby allowing for
the
continued migration of the released target molecule into the collecting
chamber
containing fresh positively charged electrode buffer. Alternatively, the
electric field
can be reversed drawing the released target molecule back into the test sample
well
of the purification device. The purified target molecule can now be accessed
and
subjected to further analysis, such as capillary or slab gel electrophoresis
for
sequence analysis.
Another embodiment of the invention is a kit for purifying a primer
extension sequencing reaction. The kit contains an electrophoretic medium
which
CA 02369002 2001-09-26
WO 00/50644 PCT/IJS00/04939
-4-
has a capture probe, or a set of capture probes. At least one capture probe
has a
sequence of at least 5 nucleotides in length which is substantially
complementary to
a portion of at least one primer extension sequencing reaction product.
In a further embodiment, the kit contains multiple electrophoretic media for
purifying multiple primer extension sequencing reactions. Each electrophoretic
medium in the kit contains a capture probe, or set of capture probes. At least
one
capture probe in each medium has a sequence of at least 5 nucleotides in
length
which is substantially complementary to a portion of at least one primer
extension
sequencing reaction product.
Using the method of the invention primer extension sequencing reaction
products can be purified directly without time consuming concentration or
precipitation steps typically required in current protocols for DNA
sequencing. In
addition, the method of the invention can be used to sort out primer extension
sequencing reaction products from multiple primer extension sequencing
reactions
carned out in the same reaction mixture, by selecting the appropriate capture
probes.
Thus, the method of the invention allows many primer extension sequencing
reactions to be carned out together saving preparation time required to set up
multiple separate sequencing reactions.
BRIEF DESCRIPTION OF THE DRAWINGS
Fig. 1 is a schematic representation of purifying a target nucleic acid
molecule from a extension sequencing reaction using an electrophoresis gel
with
capture probes immobilized within a region of the gel.
Fig. 2 is a schematic representation of the steps involved in purifying
extension products using a microtiter well comprising an electrophoretic
medium
containing capture probes immobilized within the medium. In Fig. 2, "ddNTPs"
represents dideoxynucleotide triphosphates, "pol" represents a DNA polymerase,
and "ex. products" represents primer extension sequencing reaction products.
Fig. 3 is the organization of the forward primer (SEQ ID NO 1 ), the template
(SEQ ID NO 2) and the capture probe (SEQ ID NO 3) used in Example 1.
Fig. 4 is a schematic drawing illustrating the experimental design for DNA
isolation using an electrophoretic medium.
CA 02369002 2001-09-26
WO 00/50644 PCT/US00/04939
-5-
Fig. 5 is shows the effects of varying the elution voltage.
Fig. 6 is shows results obtained from subjecting extension sequencing
reaction products to electrophoresis in which the electrophoretic medium
contained
immobilized capture probes; Fig. 6A shows the results of the experiment after
running the gel for thirty minutes; Fig. 6B shows the results of the
experiment after
sixty minutes.
Fig. 7A is a schematic representation of a device and a method used to purify
the primer extension sequencing reaction products of Example 2.
Fig. 7B shows the distribution of the primer extension sequencing reaction
products in the lower gel-tip which contained the capture probe.
Fig. 8 is a photograph of an electrophoresis gel showing the distribution of
component of a primer extension sequencing reaction taken prior to
purification
(Lane 1 ), after purification using with the upper gel-tip alone (Lane 2), and
after
purification with the lower gel-tip containing a capture probe (Lane 3). Lanes
4-6
contain purified M13 DNA of varying concentrations.
Fig. 9A is a schematic representation of forward and reverse primer
extension sequencing reactions carried out simultaneously in Example 3.
Fig. 9B is an image of the forward and reverse primer extension sequencing
reaction product separated using two gel-tips. The upper gel-tip contains a
capture
probe complementary to the forward primer extension sequencing reaction
products.
The lower gel-tip contanins a capture probe complementary to the reverse
primer
extension sequencing reaction products.
Fig. 9C is an image of the separation of the products of a forward and reverse
primer extension sequencing reaction product on a slab gel which contained two
capture probes (Lane 1). The purity of the reverse primer extension sequencing
reaction products after separation from the forward primer extension
sequencing
reaction products is shown in Lane 2. One capture probe was complementary to
the
reaction products of the forward sequencing reaction and the other capture
probe
was complementary to the reaction products from the reverse sequencing
reaction.
Fig. 9D is an analysis of the reverse sequencing reaction products after
purification by the method of the invention.
CA 02369002 2001-09-26
WO 00/50644 PCT/US00/04939
-6-
Fig. 10 is a temperature gradient gel used to determine the temperature at
which a target is released from a capture probe.
Fig. 11 is an image of a gel used to determine the temperature at which a
target will be released from capture probes of varying lengths.
DETAILED DESCRIPTION OF THE INVENTION
The present invention provides both methods and devices employed to purify
a test sample containing a target molecule. It should be understood that the
use of
the singular term "target molecule" is only used for simplicity throughout
this
application, and the plural form "target molecules" is implied therein. The
methods
described herein employ nucleic acid molecule (e.g., oligonucleotide) capture
probes
immobilized within an electrophoretic medium. The capture probe can be
dispersed
throughout the electrophoretic medium, or immobilized within discrete layers
of the
medium as described in U. S. Serial No. 08/971,845, the entire teaching of
which is
incorporated herein by reference in its entirety. This electrophoretic medium
is
contained within a purification device, such as a microtiter plate. The
capture
probes can be designed to specifically interact with, and hybridize to, a
target
molecule contained within a test sample. The test sample comprising target and
non-target molecules can be introduced into the device, for example, a
microtiter
plate comprising an electrophoretic medium containing immobilized capture
probes.
An electric field can be applied to the purification device so that charged
molecules
in the test sample will migrate within the electrophoretic medium toward the
appropriate pole. For example, for the device used in Example l, voltages for
capture would fall in the range of 0.1 to 200 V, more preferably, between 50
and 150
V. Typically, the molecules of interest will possess a negative charge and
therefore
migrate toward the positively charged electrode. The target molecule will
continue
to migrate until it has come into contact with an immobilized capture probe
which is
specific for that particular target molecule. A hybridization complex can then
form
between the immobilized capture probe and target molecule. (See Fig. 1). This
hybridization complex prevents further migration of the target molecule and
allows
for the continued migration of non-target molecules, thereby effectuating
purification of the target molecule contained within the test sample. Non-
target
CA 02369002 2001-09-26
WO 00/50644 PCT/IJS00/04939
_'7_
molecules can include proteins, such as enzymes, small molecules like salts,
non-
targeted nucleotides as well as other non-target molecules, that is, those
molecules
not targeted for further processing. (See Fig. 2). The target molecule can
subsequently be released from the capture probe by applying a sufficient
voltage or
temperature and exit the gel for further analysis. For example, using the
device used
in Example 1, the target molecule can be eluted from the capture layer (i.e.,
that
layer in the electrophoresis medium containing the immobilized capture probes)
at
voltages of 250V or higher. Preferably, voltages for elution would be in the
range of
250 to 1000 V, more preferably, 250 to 300 V. Suitable voltages for capture
and
elution using the purification devices described herein can be easily
determined by
one of skill in the art. Examples 4 and 5 provide methods for determining the
temperature at which the target molecules are released from capture probes of
varying lengths.
The methods of the present invention use a purification device which
comprises three regions. The first region comprises a test sample receptacle
which
receives a given test sample. The test sample receptacle can be positioned in
such a
manner as to be proximal to at least one orifice that allows for the delivery
of a test
sample (e.g., a reaction mixture from a primer extension sequencing reaction).
In
one embodiment, this orifice is the opening at the top of a microtiter well.
The
second region of the purification device comprises an electrophoretic medium.
Preferably, the electrophoretic medium comprises capture probes immobilized
within the medium. Preferably, this second region is physically positioned
adjacent
to the first region. In one embodiment, the second region is positioned
basally to the
first position and is also adjacent to the first region. In a preferred
embodiment, this
second region is formed within one, or more, microtiter wells, though still
allowing
for the first region to receive and store test sample. The third region of the
purification device can be physically contiguous with, or attached to the
second
region of the purification device. This third region can house a chamber that
can
collect molecules that exit the second region, in this instance the chamber is
referred
to as a collecting chamber. The chamber can also perform other functions such
as to
house buffer. The purification device can also be attached to, or have the
capacity to
connect with, a power source which generates DC voltage (e.g., a battery).
CA 02369002 2001-09-26
WO 00/50644 PCT/US00/04939
_g_
In one embodiment, the purification device is a microtiter plate containing a
set of multiple wells, for example 6, 12, 48, 96 or 384 wells. The well, or
wells, of
the microtiter plate comprises the three regions elucidated above for the
purification
device. The first region comprises a test sample receptacle for receiving test
sample.
The second region comprises an electrophoretic medium that contains
immobilized
capture probes. The third region can be formed by excising the bottom support
of
the well creating an orifice, in some instances the microtiter well has a
pointed tip
which can be trimmed to provide an opening. This bottom orifice can optionally
be
covered using a porous membrane to provide support for the gel layer (second
region). Preferably, this porous membrane has a molecular weight cutoff
greater
than 15,000 daltons. More preferably, the molecular weight cutoff is
approximately
3,000 daltons. Preferably, the porous membrane should demonstrate negligible
binding of nucleic acid molecules. This bottom orifice can be used to gain
access to
the third region which can be physically attached or detached from the well
itself.
The third region can comprise a collecting chamber which can contain buffer.
Specifically encompassed by the present invention is a method for purifying
primer extension sequencing reaction products from a primer extension sequencW
g
reaction using a purification device described herein. The target molecule is
one, or
more, of the primer extension sequencing reaction products formed during a
particular stage of a DNA sequencing protocol. Typically, the primer extension
sequencing reaction product can have a size from about 20 to about 2000
nucleotides
in length. For example, a DNA molecule that is destined for nucleotide
sequencing
can be placed into an appropriate sequencing vector, such as the M13 phage
vector.
Under suitable conditions well known to those skilled in the art, extension
nucleic
acid products can be produced using this vector, preferably using the cycle
sequencing method. (See Fig. 1; Carothers, Biotechniques, 7:494-499 (1989),
and
Murray, Nucleic Acid Res., 17:8889 (1989)). Some of the reactants employed in
this
primer extension sequencing reaction are DNA Polymerase, primers,
deoxynucleotides, and appropriate salts. Those skilled in the art will be
familiar
with standard DNA sequencing protocols. (See, Ausbel, F.M., et al. (eds);
Current
Protocols in Molecular Biology, vol.l, ch.7, (1995)). The target molecule
(primer
CA 02369002 2001-09-26
WO 00/50644 PCT/LTS00/04939
-9-
extension sequencing reaction product) can subsequently undergo purification
using
a purification device.
Another embodiment of the invention is a kit for purifying a primer
extension sequencing reaction. The kit contains a electrophoretic medium which
has
a capture probe, or a set of capture probes. At least one capture probe has a
sequence of at least 5 nucleotides in length which is substantially
complementary to
a portion of at least one primer extension sequencing reaction product. The
kit can,
optionally, include a sample receptacle and a collecting chamber. In one
embodiment, the sample receptacle and the collecting chamber are located at
opposite ends of the electrophoretic medium.
In a further embodiment, the kit contains multiple electrophoretic media for
purifying multiple primer extension sequencing reactions. The electrophoretic
media can be segregated from each other. Each electrophoretic medium in the
kit
contains a capture probe, or set of capture probes. At least one capture probe
in each
medium has a sequence of at least 5 nucleotides in length which is
substantially
complementary to a portion of at least one primer extension sequencing
reaction
product. Each electrophoretic medium can contain the same capture probe, or
set of
capture probes. This type of kit could be used to purify the reaction products
from
the same primer extension sequencing reaction which has been earned out on
multiple samples in separate reaction vessels. Alternatively, the each
electrophoretic
medium in the kit can have a different capture probe or set of capture probes.
This
type of kit can be used to purify multiple primer extension sequencing
reactions
which are carried out in the same reaction vessel.
When the kit contains multiple electrophoretic media, each media can be
segregated from other electrophoretic media in wells of a microtiter plate.
Each
electrophoretic medium can be attached to a separate sample receptacle and a
separate collection chamber.
In one embodiment of the present invention, a method for purifying multiple
primer extension sequencing reaction products which are formed by synthesizing
target molecules (i.e., primer extension sequencing reaction products) using
both a
first-end (e.g., near or at the 5' end) and a second-end (e.g., near or at the
3' end) of
the DNA template simultaneously is described. Primer extension sequencing can
CA 02369002 2001-09-26
WO 00/50644 PCT/US00/04939
-10-
occur in both directions of the template simultaneously. In this embodiment, a
first-
end primer and a second-end primer would be concurrently annealed to the
template
DNA allowing for extension in both direction using the one template DNA. The
primer extension sequencing reaction would then occur and produce primer
extension sequencing reaction products arising from both the first-end and the
second-end of the DNA template. The primer extension sequencing reaction
products can then be added to a purification device that can purify the target
molecule based upon whether it was synthesized using the first-end primer or
second-end primer.
In this embodiment, the purification device comprises at least two
electrophoretic gel cartridges that can be brought together to form a
continuity
between the two cartridges. A gel cartridge is a device that can house and
support
an electrophoretic medium. The gel cartridges comprise electrophoretic medium
containing capture probes immobilized within the medium. However, each
cartridge
comprises an electrophoretic medium containing different immobilized capture
probes. For example, one cartridge can comprise an electrophoretic medium
containing an immobilized capture probe that contains a nucleotide sequence
which
is substantially identical to a nucleotide sequence that lies adjacent, or
close to, the
first-end of the template (capture probe "A"), whereas, the second cartridge
can
comprise an electrophoretic medium which contains an immobilized capture probe
that contains a nucleotide sequence which is substantially identical to a
nucleotide
sequence that lies adjacent, or close to, the second-end of the template
(capture
probe "B"). By "substantially identical to," it is meant a nucleotide sequence
with
greater than 70% sequence identity and/or similarity (e.g., 75%, 80%, 85%,
90%, or
95% or greater homology). Initial search for substantially identical
nucleotide
sequences can be performed at NCBI against the GenBank (release 87.0), EMBL
(release 39.0), and SwissProt (release 30.0) databases using the BLAST network
service. Altshul, S.F., et al., Basic Local Alignment Search Tool, J. Mol.
Biol.,
215:403 (1990), the entire teachings of which are incorporated herein by
reference in
its entirety. Computer analysis of nucleotide sequences can be performed using
MOTIFS and the FindPatterns subroutines of the Genetics Computing Group (GCG,
version 8.0) software. Nucleotide comparisons can also be performed according
to
CA 02369002 2001-09-26
WO 00/50644 PCT/US00/04939
-11-
Higgins and Sharp (Higgins, D.G. and P.M. Sharp, Description of the Method
used
in CLUSTAL, Gene, 73:237-244 (1998)).
The two cartridges can be positioned in such a way as to allow for the
migration of target molecules through one cartridge into the next cartridge.
For example, the cartridge that has the "A" capture probe is positioned such
that it first receives the sample, and the second cartridge, which has the "B"
capture
probe, is positioned to receive the migrating sample from the first cartridge.
If a test
sample containing a heterogeneous mixture of target molecules (those that were
synthesized using the first-end primer together with those that employed the
second-
end primer) is added to this purification device, then the target molecules
can be
purified or separated based upon the primer used to synthesize the target
molecule.
When an electric field is applied to the purification device, the target
molecules in
the test sample can undergo electrophoretic migration. Those target molecules
that
used the first-end primer for synthesis will be captured in the first
cartridge
containing "A" as capture probes (its appropriate capture probe), while those
target
molecules that used the second-end primer will migrate through the first
cartridge
and will subsequently be captured in the second cartridge containing "B"
capture
probes (its appropriate capture probe). Following electrophoretic migration,
the
cartridges can be separated and placed into separate collecting chambers,
thereby
allowing for the collecting of the first-end primer target molecules and the
second-
end primer target molecules separately.
Any electrophoretic matrix suitable for electrophoresis can be used for the
methods of the present invention. Suitable matrices include acrylamide and
agarose,
both commonly used for nucleic acid electrophoresis. However, other materials
may
be used as well. Examples include chemically modified acrylamides, starch,
dextrans, cellulose-based polymers. Additional examples include modified
acrylamides and acrylate esters (for examples see Polysciences, Inc., Polymer
&
Monomer catalog, 1996-1997, Warrington, PA), starch (Smithies, Biochem. J.
71:585 (1959); product number S5651, Sigma Chemical Co., St. Louis, MO),
dextrans (for examples see Polysciences, Inc., Polymer & Monomer Catalog, 1996-
1997, Warrington, PA), and cellulose-based polymers (for examples see Quesada,
Current Opin. in Biotechnology, 8: 82-93 (1997)). Any of these polymers listed
CA 02369002 2001-09-26
WO 00/50644 PCT/US00/04939
-12-
above can be chemically modified to allow specific attachment of capture
probes for
use in the present invention.
The capture probes of the instant invention are typically nucleic acids,
modified nucleic acids, or nucleic acid analogs. The capture probes are
complementary to the primer extension sequencing reaction products, but not to
the
primer extension sequencing primer. Methods of coupling nucleic acids to
create
nucleic acid-containing gels are known to those of skill in the art. Nucleic
acids,
modified nucleic acids and nucleic acid analogs can be coupled to agarose,
dextrans,
cellulose, and starch polymers using cyanogen bromide or cyanuric chloride
activation. Polymers containing carboxyl groups can be coupled to synthetic
capture
probes having primary amine groups using carbodiimide coupling. Polymers
carrying primary amines can be coupled to amine-containing probes with
glutaraldehyde or cyanuric chloride. Many polymers can be modified with thiol-
reactive groups which can be coupled to thiol-containing synthetic probes.
Many
other suitable methods can be found in the literature. (For review see Wong,
"Chemistry of Protein Conjugation and Cross-linking", CRC Press, Boca Raton,
FL,
1993).
A variety of capture probes can be used in the methods of the present
invention. Typically, the capture probes of the present invention comprise a
nucleic
acid (e.g., oligonucleotide) with a nucleotide sequence substantially
complementary
to a nucleotide sequence region contained within the target molecule wherein
the
target molecule hybridizes to the capture probe. It is important to note that
the
capture probe is not complementary to the primer used in the primer extension
sequencing reaction. The complementarity of the capture probe to the target
molecule need only be sufficient so as to specifically bind the target
molecule and
effectuate the purification of the target molecule in the reaction mixture.
Probes
suitable for use in the present invention comprise RNA, DNA, nucleic acid
analogs
(such as PNA), modified nucleic acids and chimeric probes of a mixed class
comprising a nucleic acid with another organic component, e.g., peptide
nucleic
acids (PNA). Capture probes can be single-stranded or double-stranded nucleic
acids. Typically, the length of a capture probe will be at least 5 nucleotides
in
CA 02369002 2001-09-26
WO 00/50644 PCT/US00/04939
-13-
length, more typically between 5 and 50 nucleotides, and can be as long as
several
thousand bases in length.
Methods for covalently attaching the capture probes described herein to
polymerizable chemical groups have also been developed. When copolymerized
with suitable mixtures of polymerizable monomer compounds, matrices containing
high concentrations of immobilized nucleic acids can be produced. Examples of
methods for covalently attaching nucleic acids to polymerizable chemical
groups are
found in U.S. Serial No. 08/812,105; U.S. Serial No. 08/971,845, and Rehman,
F.N.,
et al., Nucleic Acid Res., 27:649-655 (1999), the teachings of which are
herein
incorporated by reference in their entirety.
For some methods, it may be useful to use composite matrices containing a
mixture of two or more matrix forming materials, an example is the composite
acrylamide-agarose gel. These gels typically contain from 2-5% acrylamide and
0.5%-1% agarose. In these gels the acrylamide provides the chief sieving
function,
but without the agarose, such low concentration acrylamide gels lack
mechanical
strength for convenient handling. The agarose provides mechanical support
without
significantly altering the sieving properties of the acrylamide. In such
cases, it is
preferred that the nucleic acid can be attached to the component that confers
the
sieving function of the gel, since that component makes the most intimate
contacts
with the solution phase nucleic acid target.
For many applications gel-forming matrices such as agarose and cross-linked
polyacrylamide will be preferred. However, for capillary electrophoresis (CE)
applications it is convenient and reproducible to use soluble polymers as
electrophoretic matrices. Examples of soluble polymers that have proven to be
useful for CE analysis are linear polymers of polyacrylamide, poly(N,N-
dimethylacrylamide), poly(hydroxyethylcellulose), poly(ethyleneoxide) and
poly(vinylalcohol) as described in Quesada (Current Opinion in Biotechnology,
8:82-93 (1997)). These soluble matrices can also be used to practice the
methods of
the present invention. It is particularly convenient to use the methods found
in the
application U.S. Serial No. 08/812,105, entitled "Nucleic Acid-Containing
Polymerizable Complex" for preparation of soluble polymer matrices containing
immobilized capture probes. Another approach for attaching nucleic acid
molecule
CA 02369002 2001-09-26
WO 00/50644 PCT/CTS00/04939
-14-
probes to preformed polyacrylamide gels found in Timofeev, et al., Nucleic
Acids
Res., 24:3142-3148 (1996), can also be used to attach capture probes to
prepolymerized soluble linear polyacrylamide.
Nucleic acids may be attached to particles which themselves can be
incorporated into electrophoretic matrices. The particles can be macroscopic,
microscopic, or colloidal in nature. (See Polyciences, Inc., 1995-1996
particle
Catalog, Warrington, PA). Cantor, et al., U.S. Patent No. 5,482,863 describes
methods for casting electrophoresis gels containing suspensions or particles.
The
particles are linked to nucleic acids using methods similar to those described
above
mixed with gel forming compounds and cast as a suspension into the desired
matrix
form.
As defined herein, the term "nucleic acid" includes DNA (deoxyribonucleic
acid) or RNA (ribonucleic acid). Nucleic acids referred to herein as
"isolated" are
nucleic acids separated away from the components of their source of origin
(e.g., as
1 S it exists in cells, or a mixture of nucleic acids such as a library) and
may have
undergone further processing. Isolated nucleic acids include nucleic acids
obtained
by methods known to those of skill in the art. These isolated nucleic acids
include
substantially pure nucleic acids, nucleic acids produced by chemical
synthesis, by
combinations of biological and chemical methods and recombinant nucleic acids
which are isolated.
"Nucleic acid analogs", as used herein, include nucleic acids containing
modified sugar groups, phosphate groups or modified bases. Examples of nucleic
acids having modified bases, include, for example, acetylated, carboxylated or
methylated bases (e.g., 4-acetylcytidine, 5-carboxymethylaminomethyluridine, 1-
methylinosine, norvaline or alto-isoleucine). Such nucleic acid analogs are
known
to those of skill in the art. One example of a useful nucleic acid analog is
peptide
nucleic acid (PNA), in which standard nucleotide bases are attached to a
modified
peptide backbone comprised of repeating N-(2-aminoethyl)glycine units (Nielsen
et
al., Science, 254:1497-1500, (1991)). The peptide backbone is capable of
holding
the bases at the proper distance to base pair with standard DNA and RNA single
strands. PNA-DNA hybrid duplexes are much stronger than equivalent DNA-DNA
duplexes, probably due to the fact that there are no negatively charged
CA 02369002 2001-09-26
WO 00/50644 PCT/US00/04939
-15-
phosphodiester linkages in the PNA strand. In addition, because of their
unusual
structure PNAs are very resistant to nuclease degradation. For these reasons,
PNA
nucleic acid analogs are useful for immobilized probe assays. It will be
apparent to
those skilled in the art that similar design strategies can be used to
construct other
nucleic acid analogs that will have useful properties for immobilized probe
assays.
Probes containing modified nucleic acid molecules may also be useful. For
instance, nucleic acid molecules containing deazaguanine and uracil bases can
be
used in place of guanine and thymine-containing nucleic acid molecules to
decrease
the thermal stability of hybridized probes (Wetmur, Critical reviews in
Biochemistry
and Molecular Biology, 26:227-259 (1991)). Similarly, 5-methylcytosine can be
substituted for cytosine if hybrids of increased thermal stability are desired
(Wetmur, Critical reviews in Biochemistry and Molecular Biology, 26:227-259
(1991)). Modifications to the ribose sugar group, such as the addition of
2'-O-methyl groups can reduce the nuclease susceptibility of immobilized RNA
probes (Wagner, Nature, 372:333-335 (1994)). Modifications that remove
negative
charge from the phosphodiester backbone can increase the thermal stability of
hybrids (Moody et al. Nucleic Acids Res., 17:4769-4782 (1989); Iyer et al. J.
Biol.
Chem., 270:14712-14717 (1995)).
As defined herein, "substantially complementary" means that the nucleic
acid molecule sequence of the capture probe need not reflect the exact nucleic
acid
molecule sequence of the microbial target molecule, but must be sufficiently
similar
in identity of sequence to hybridize with the target molecule under specified
conditions. For example, non-complementary bases, or additional nucleic acid
molecules can be interspersed in sequences provided that the sequences have
sufficient complementary bases to hybridize therewith. Generally, the degree
of
complementarity using short capture probes (approximately 20 nucleotides in
length) is approximately greater than 95%. For longer probes significantly
less
complementarity is required if there are contiguous segments of from about 15
to
about 20 nucleotides in length being complementary to each other.
Specified conditions of hybridization can be determined empirically by those
of skill in the art. For example, conditions of stringency should be chosen
that
significantly decrease non-specific hybridization reactions. Stringency
conditions
CA 02369002 2001-09-26
WO 00/50644 PCT/US00/04939
-16-
for nucleic acid hybridizations are explained in e.g., Current Protocols in
Molecular
Biology, Ausubel, F.M., et al., eds., Vol. 1, Suppl, 26, 1991; the teachings
of which
are herein incorporated by reference in their entirety. Factors such as probe
length,
base composition, percent mismatch between the hybridizing sequences,
temperature and ionic strength influence the stability of nucleic acid
hybrids.
Stringent conditions, e.g., moderate, or high stringency, can be determined
empirically, depending on part of the characteristics of the probe and
microbial
target molecule.
The features and other details of the invention will now be more particularly
described and pointed out in the exemplification. It will be understood that
the
particular embodiments of the invention are shown by way of illustration and
not as
limitations of the invention. The principle features of this invention can be
employed in various embodiments without departing from the scope of the
invention.
EXAMPLES
The following examples are not intended to limit the scope of the invention.
They are provided merely to teach concrete and practical means for carrying
out
the invention.
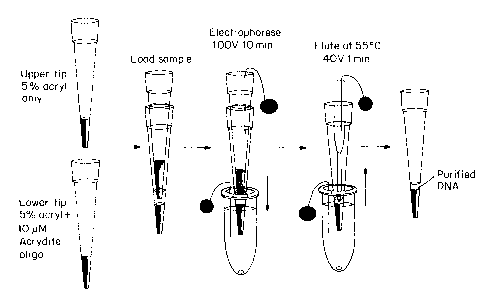
EXAMPLE 1: Gel-based DNA Isolation and Elution
Sequencing reaction products were prepared using the Thermo SequenaseTm
DYEnamic direct cycle sequencing kit with -21 M13 forward primer (5'-dyel-
spacer-TGT*AAAACGACGGCCAGT-3' [SEQ ID No. 1]), where * indicates the
position of base modification with one of four fluorescence energy transfer
dyes
according to the manufacturer's instructions (Amersham Pharmacia Biotech,
Piscataway, NJ). Each of four reactions was prepared by mixing 1 mL of M13mp18
single-stranded DNA (0.25 mg/mL, New England BioLabs catalog #404-C), 14 mL
of distilled H20, and 2 mL of the manufacturer's reaction mixture. Each of the
four
reactions used a different dye-labeled primer, designated "dyel", "dye2",
"dye3" and
"dye4", and a different ddNTP nucleotide mixture. These four tubes were then
placed in a thermal cycler (PTC100, MJ Research, Watertown, MA) and subjected
CA 02369002 2001-09-26
WO 00/50644 PCT/US00/04939
-17-
to 30 cycles of 95° C for 30 seconds, 45° C for 15 seconds, and
70° C for 30 seconds.
The four reactions were then pooled (100 mL total volume) and 11 mL of loading
buffer was added (2.5% wt/vol Xylene Cyanol, 2.5% wt/vol Bromophenol Blue, 20
mM EDTA, pH 8.0, 15% (wt/vol) Ficoll 400,000 average molecular weight in a
total of 10 mL distilled HZO).
Polyacrylamide gels for electrophoretic hybridization purification were cast
in
standard micropipette tips for 1-200 ~,L micropipettes (Fisher Brand yellow
tips for
Gilson P200, Fisher Scientific, Pittsburgh, PA). For the purification step,
two gel
tips were stacked so that the sequencing reaction could be subjected to
electrophoresis through each tip sequentially in one step. (See Fig. 4A). The
gel in
the upper tip (10) comprised a 20 ~,L 5% polyacrylamide gel (29:1 monomer:bis
wt/wt) cast in 1 x TBE buffer (89 mM Tris-Borate pH 8.3, 2 mM EDTA (Bio-Rad).
This upper gel is designed to trap the high molecular weight M13 template DNA
which has negligible electrophoretic mobility under the conditions used for
capture
of the extension sequencing reaction products. Removal of the high molecular
weight template improves quality of sequencing results on capillary
electrophoresis
instruments such as the Megabase from Molecular Dynamics (Sunnyvale, CA).
The gel in the lower tip (20) is the same as that of the upper tip, except
that it
contains an immobilized nucleic acid molecule capture probe (5'-acrylamide-GGG
ATC CTC TAG AGT CGA CCT 3' [SEQ ID NO 3]) at a concentration of 10 ~.M
(refernng to nucleic acid molecule strands). The capture probe is
complementary to
a sequence within the extension products that is located immediately 3' of the
sequencing primer, as shown in Fig. 3.
As shown if Fig. 3, the cloned insert to be sequenced is located on the 5'
side
of the template region shown. Thus, as shown in the diagram, the capture probe
is
complementary to the extended sequencing reaction products, but not to the
sequencing primer. In this way, electrophoresis of the extension sequencing
reaction
products through the gel of the lower tip will allow hybridization capture of
the
extension products without impeding electrophoresis of the excess primers
through
the tip.
The capture probe was modified with a 5'-acrylamide group using an
acrylamide phosphoramidite (AcryditeTM, Mosaic Technologies, Boston, MA). The
CA 02369002 2001-09-26
WO 00/50644 PCT/US00/04939
-18-
probe was immobilized on the gel matrix by adding it to the unpolymerized
acrylamide mixture and allowing it to copolymerize directly in the gel tip.
The tips were stacked as shown in Fig. 4A, where the probe-containing tip
(20) is on the bottom. The region between the two tips (30) is filled with
electrophoresis buffer (1 x TBE which is 89 mM Tris-borate, pH 8.3, 2 mM
EDTA),
as well as the region above the upper gel tip (40). The lower tip (10) is
immersed in
a buffer-filled 1.5 mL microcentrifuge tube (50). Separate platinum electrodes
(60)
are placed in the buffer above the gel in the higher tube and in the buffer in
the
microcentrifuge tube (50). The upper electrode is connected to a negative lead
of
the power supply, while the lower electrode is attached to a positive lead.
The upper tip of the device shown in Fig. 4A was loaded with 75 ~L of the
pooled sequencing reaction in 15 ~,L aliquots every 10 minutes for one hour,
while
subjecting the tips to electrophoresis at an applied field of 100 V throughout
the
loading process. The field was applied for an additional 3 hours to ensure
that all of
the sequencing reaction products become trapped on the gel in the lower tip.
The
primers, which are not complementary to the immobilized capture nucleic acid
molecule probes in the lower gel, nucleotides, and excess salts pass through
the gel
into the lower tube.
Following electrophoresis, the upper gel tip containing the slow-moving
template was discarded and the lower gel tip (20) was then placed into a
second
apparatus, depicted in Fig. 4B. The lower end of the tip is placed into an
electrophoresis buffer held in an ultrafiltration device (70) with a 3000
Dalton
molecular weight cutoff membrane (75) on its bottom surface (Microcon 3,
Amicon/Millipore, Bedford, MA). The ultrafiltration unit was partially
immersed in
a 1 x TBE-filled microcentrifuge tube (80) containing a positively charge
platinum
electrode (60). A negatively-charged electrode (60) was immersed the buffer (1
x
TBE) above the gel in the tip. The ultrafiltration membrane was used to
prevent the
migration of the eluted sequencing reaction products onto the electrode, where
they
would be damaged by electrochemical reactions. To elute the sequencing
reaction
products, a field of 300 V was applied to the device for 3 minutes. This
voltage was
sufficient to elute the sequencing reaction products from the gel capture
probes and
cause it to collect in the ultrafiltration unit.
CA 02369002 2001-09-26
WO 00/50644 PCT/US00/04939
-19-
Fig. 5 shows the effects of varying the elution voltage. Sequencing reaction
products were captured and purified by electrophoretic hybridization capture
as
described above. The tip was then subjected to the indicated electrophoresis
conditions, and then scanned in a fluorescence imaging device (Fluoroimager
595,
Molecular Dynamics, Sunnyvale, CA) to visualize the fluorescent sequencing
reaction products. As seen, voltages above 250 V cause complete elution of the
fluorescent sequencing reaction products.
To characterize the eluted products, samples of purified and crude sequencing
reaction products were subjected to electrophoresis in a polyacrylamide gel
containing a discrete layer of gel immobilized capture probe arranged as a
horizontal
band across the width of the gel (see "Capture layer" in Fig. 6). The gel was
composed of 5% polyacrylamide ( 29:1 monomer:bis wt/wt), 1 x TBE. The capture
layer contained the same polyacrylamide and buffer with 10 ~,M of the 5'-
acrylamide capture probe (5'-acrylamide- GGG ATC CTC TAG AGT CGA CCT 3'
[SEQ ID NO 3]). The samples were subjected to electrophoresis run at 150 Volts
for 30 minutes (Fig. 6A) and 60 minutes (Fig. 6B). Lane 1 contains 15 ~.L of
the
sample that had been purified by electrophoretic capture and elution, and lane
2
contains S ~,L of the unpurified sequencing reaction product. Fig. 6A shows
that the
hybridization-purified product (lane 1) has been purified away from the excess
primers, which are seen in the unpurified sample at the bottom of lane 2.
EXAMPLE 2: Purification of a Single DNA Product Complementary to
M13mp18 Sequence
Fig. 7A provides a schematic representation of the method and devices)
described below for purifying, and optionally concentrating, products of DNA
sequencing reactions. Fig. 7B provides a photograph of a capture probe-gel-tip
after
electrophoresis and prior to elution of the captured nucleotide sequence.
Fig. 8 provides a photograph of a gel showing the results of purification of
a desired oligonucleotide sequence from a DNA sequencing reaction that
included
primers, salts, DNA template, unincorporated nucleotides, and dye terminators.
First the DNA sequencing reaction was purified by gel-loading tip to provide a
crude
sample, then the crude sample was purified by capture probe-gel-tip. Lane 1
shows
CA 02369002 2001-09-26
WO 00/50644 PCT/US00/04939
-20-
the pattern provided prior to purification. Lane 2 provides the pattern seen
after
purification using a gel-loading tip. Removal of DNA template is demonstrated.
Lane 3 shows the pattern seen after purification using a capture probe-gel-
tip.
Localization of the desired sequence in the absence of DNA template is
demonstrated.
A. Preparation of a Separation Device With and Without an
Immobilized Capture Probe
Preparation of the separation device containing AcryditeTM oligonucleotide
gel was performed as follows. A Hybrigel solution was prepared from stock
solutions of 40% acrylamide (29:1 acrylamide monomer:bis-acrylamide) and l Ox
TBE (90mM Tris-Borate-EDTA buffer pH 8.3; reagents were purchased from
Biorad Laboratories, Inc.; Hercules, CA). The AcryditeTM oligonucleotide for
the
capture probe was synthesized using oligonucleotides (obtained from Operon
Technologies, Inc.; Alameda, CA) and acrylamide phosphoramidte (Acrydite
polymer available from Mosaic Technologies, Inc.; Boston, MA) according the
method disclosed in U.S. Patent No. 5,641,658 and Kenney, Ray, and Boles,
BioTechniques 25, 516-521 (1998), the disclosure of each of which is
incorporated
herein by reference in its entirety). The Hybrigel contained 5% acrylamide
(29:1), 1
x TBE and 10 qM AcryditeTM oligonucleotide capture probe (SEQ ID NO 4)
having the following sequence:
5'-acrylamide GCT GAG ATC TCC TAG GG 3' (SEQ ID NO 4)
The selected capture probe for this example has a sequence that is
complementary to
a portion of the polylinker of vector Ml3mpl8 which provides the target
molecule, a
product of a DNA sequencing reaction, but the capture probe does not include
any of
the primer sequence. When 10% ammonium persulfate and N,N,N',N'-tetramethyl
ethylenediamine (TEMED; BioRad, Hercules, CA) were added to the Hybrigel
solution, polymerization was rapid (within 2 minutes).
To prepare the probe-gel-tip, 1~1 of 10% ammonium persulfate and O.SqI
of TEMED were added to 2001 of Hybrigel solution. 101 of the polymerizing
CA 02369002 2001-09-26
WO 00/50644 PCT/LJS00/04939
-21-
Hybrigel oligonucleotide containing solution were quickly pipetted into a 2001
tip
and allowed to polymerize. The probe-gel-tips were made 8 or 12 at a time
using a
multipipeting device. For storage, probe-gel-tips were ejected into eppendorf
tubes
containing approximately 0.3 ml of lx TBE. Care must be taken not to dislodge
the
S gel from the tip. Then the probe-gel-tips were overlaid with 1501 of lx TBE.
Gel-loading tips were used to remove template DNA. Gel-loading tips
were prepared containing acrylamide (29:1) only (i.e. no Acrydite capture
probe).
The gel-loading-tip was prepared as described above using a solution
containing 5%
acrylamide (29:1), 1 x TBE, made from stock solutions of 40% acrylamide (29:1
monomer:bis) and l Ox TBE. 10% ammonium persulfate and TEMED were added to
the solution and 200 ~,1 of the final solution was pipetted into each tip.
Gel-loading-tips may also be stored as described above.
B. Preparation of a DNA Sequencing Reaction Product and Capture
Probes
PE-Applied Biosystems sequencing reaction products were prepared
following the protocol of PE Applied Biosystems BigDye Primer Cycle Sequencing
Kit (available from PE-Applied BioSystems) with the -21 M13 forward primer in
a
GeneAmp 2400 using the cycling conditions recommended by PE Applied
Biosystems. Vector M13mp18 was used. A DNA segment having a known
sequence was inserted after the primer site. Extension products were prepared.
Capture probes were made to the region between the primer and the inserted
DNA.
The capture probes capable of hybridizing to extension products of the forward
primer were selected. The capture probes comprise an oligonucleotide
synthesized
with Acrydite TM at the 5' end.
Alternatively, the DYEnamic ET Terminator Cycle Sequencing Kit from
Amersham-Pharmacia may be used.
C. Capture of DNA Sequencing Extension Product
Electrophoretic capture and separation of a chosen extension product
(target in test sample) were performed as follows: 101 of sequencing reaction
solution (i.e. 1/2 of one reaction) that contained primers, salt,
unincorporated
CA 02369002 2001-09-26
WO 00/50644 PCT/US00/04939
-22-
nucleotides, dye terminators, template DNA, and the target-DNA extension
products
synthesized from the template DNA were added to 1pl of lOx ficoll loading
buffer
(35% ficoll 400, 0.1% bromophenol blue, 0.1% xylene cyanol, 100mM EDTA) to
provide a sample solution. The sample solution was layered onto the surface of
the
gel in the gel-loading-tip, (i.e., no Acrydite capture probe), to remove
template
DNA. This tip was stacked above a probe-gel-tip, a Hybrigel containing tip, as
shown in Fig. 7A. A small volume of electrophoresis buffer was layered onto
the
surface of the gel in the probe-gel-tip and the gel-loading-tip was placed in
contact
with the electrophoresis buffer. Thus, a liquid connection was formed between
the
two gels allowing for an electrical connection when the electrodes and power
source
were in place.
After the sample solution was loaded onto the surface of the gel of the
gel-loading-tip, both tips were placed into an ependorf tube containing lx
TBE,
electrophoresis buffer. Platinum electrodes were placed in the electrophoresis
buffer
above and below the stacked gel tips and a voltage of 100V was applied for 10
minutes. The upper electrode was connected to the negative lead of the power
supply, while the lower electrode was attached to the power supply's positive
electrode. The gel-loading tip thus provided a partially purified sample
solution for
introduction into the probe-gel-tip. The smaller DNA fragments pass into the
buffer
and then into the probe-gel-tip more rapidly than the DNA template and dye
which
are retained in the gel-loading-tip
The electrophoresis buffer remaining above the probe-gel-tip gel surface
was removed. The electrophoresis buffer was replaced with 4~1 of formamide
loading dye (5:1 deionized formamide, 25mg/ml blue dextran, 25mM EDTA). The
temperature in the gel in the probe-gel-tip was raised to 55 °C to
facilitate
detachment of the target oligonucleotide sequence from the capture probe by
placing
the eppendorf tube containing the lower buffer reservoir into a drybath.
(VWR). A
clean second gel-loading tip with a 30% acrylamide gel containing 5 % acrylic
acid
was lowered into the formamide loading dye. A platinum electrode was placed in
the
top gel-loading tip. The direction of the current was reversed to drive the
oligonucleotide released from the hybridization complex upwards. One minute at
40V was sufficient to drive the oligonucleotide out of the probe-gel-tip and
into the
CA 02369002 2001-09-26
WO 00/50644 PCT/US00/04939
-23-
formamide loading dye. A 4 ~l sample was removed by pipet and retained for
sequence analysis. After electrophoresis, the gel in the probe-gel-tip was
visualized
using a Molecular Dynamics Fluorimager 595. The results shown in Fig. 7B
demonstrate that capture occurs at the upper surface of the gel in the probe-
gel-tip.
D. Analysis of Sequencing Purification by Hybrigel Assay
Glass plates for a vertical polyacrylamide minigel (10 x 10 cm, 0.75 mm
spacers) were assembled and the sandwich was filled approximately half way
with
20% acrylamide (29:1; Bio-Rad), lx TBE (90 mM Tris-borate buffer, pH 8.3, 2 mM
EDTA). Polymerization was initiated by inclusion of 10% aqueous ammonium
persulfate (APS) and TEMED at 1/100th and 1/1000th gel volume, respectively.
For
gels containing one capture layer, 600 ~1 of gel solution (20% polyacrylamide,
lx
TBE, 4 ~l 10% APS and 4 ~1 10% TEMED) containing Acrydite-labeled
oligonucleotide at a final concentration of 10 ~M were polymerized. After
polymerization of the capture layer, the remaining space in the plate sandwich
was
filled with a 5% gel. This composite gel was then assembled in a minigel
apparatus
containing lx TBE and subjected to electrophoresis at 100-150 V for ~45 min.
After
electrophoresis, the gel was visualized using a Molecular Dynamics Fluorimager
595. The results confirm that the sequence captured by the Hybrigel probe is
the
complement of the template DNA.
E. Automated Sequencing of the Captured Oligonucleotide
Following the procedures described above and analyzing the
oligonucleotide sequence purified by the inventive device with an automated
sequencer, repeated experiments with standard vectors have demonstrated that
the
accuracy of sequencing of the first 500 nucleotides is always >99%. The
readable
sequence extends to at least 750 nucleotides.
EXAMPLE 3: Simultaneous Separation of Multiple DNA Sequencing reaction
products
This example demonstrates that the method of the invention is useful for
sequencing an oligonucleotide insert replicated in a plasmid. Both forward and
CA 02369002 2001-09-26
WO 00/50644 PCT/US00/04939
-24-
reverse primers are used as illustrated in Fig. 9A. A plasmid with a large
insert was
sequenced simultaneously using two primers in one reaction vessel. Two
probe-gel-tips each containing a different capture probe were arranged in
tandem.
The probe sequences were designed based upon the forward and reverse primers
used. From the slab gel illustrated in Fig. 9C, purification of the two
oligonucleotide
targets in the test sample as compared to the crude product is demonstrated.
Note
that the sequence of the reverse product (right) shows no contamination with
the
forward sequence. Note also that DNA template contamination is removed.
A. Preparation of Separation Device With and Without Immobilized
Capture Probe
Gel-loading tips were prepared as described in Example 2. Probe-gel-tips
were prepared as described in Example 2 except that one was provided with a
forward primer probe and the other was provided with a reverse primer probe as
shown in Fig. 9A. Thus, two separate Hybrigel probe-gel-tips were made, one
with
acrydite oligonucleotide (SEQ ID NO 5) and the other with acrydite
olgonucleotide
(SEQ ID NO 6). Plasmid p698 which is plasmid vector pGEM3Zf(-) (Promega
Biotech, WI) with a 3.8kb insert in the Xba I site of the polylinker was used.
The
two probes derive from sequence on either side of the Xba I site within the
polylinker. SEQ ID NO 5 is complementary to, and therefore capable of,
capturing
sequencing reaction products made using the reverse primer. Similarly, SEQ ID
NO
6 can capture sequence from the forward primer.
SEQ ID NO 5 5'acrylamide -TGCAGGCATGCAAGCTT
SEQ ID NO 6 5'acrylamide -GGGTACCGAGCTCGAATTC
Thus, each oligonucleotide primer probe is synthesized with Acrydite at
the 5' end and is capable of hybridizing to an extension product. Each capture
probe
sequence is both complementary to an extension product sequence and to the
primer.
Each capture probe sequence is specific for a particular vector derived from
the
region between the primer site and the insert DNA.
CA 02369002 2001-09-26
WO 00/50644 PCT/US00/04939
-25-
B. Purification of Products of Multiple Reactions
Probe2-gel-tip, containing SEQ ID NO 6 was placed in tandem (stacked)
with a probe3-gel-tip, containing SEQ ID NO 5 as is illustrated in Fig. 9A.
Running
electrophoresis buffer was placed on the upper surface of probe3-gel-tip to
provide
electrical contact and to prevent drying. The test sample from a multiplex
sequencing reaction was electrophoresed through both probe2-gel-tip and
probe3-gel-tip. Fig. 9B illustrates capture in the two separate tips by the
two distinct
probes.
EXAMPLE 4: Elution of Target from Capture Probe using a Temperature
Gradient
The stability of the hybridization complex is dependent on temperature. A
vertical slab gel containing a layer of AcryditeTM capture probe sandwiched
between
layers of gel without capture probe was made. The gel for the upper and lower
layers
was that used for the gel-loading-tip. The Acrydite probe layer was made as
described in Example 2 for the probe-gel-tip using the capture probe sequence
SEQ
ID NO 5. The sample in this case was a fluorescent oligonucleotide with a
complementary sequence to SEQ ID NO 5. The same sample was loaded in each
well. The whole gel was subjected to a temperature gradient using an aluminum
backplate and two water baths. The gel temperature on the left is 23 °
C. The
temperature increased across the gel up to 53 °C on the right. At low
temperature the
target was efficiently captured at the top of the capture layer. As the
temperature was
increased, target capture was inhibited until the sample runs right through
the layer
(see gel image in Fig. 10). The transition temperature, i.e. the temperature
at which
the target stops ceases to be captured is related to the Tm but that was not
the only
factor found to be involved.
EXAMPLE 5: Temperature and Capture Probe Size Dependence of Sequence
Elution
To define temperature conditions for capture and elution of sequencing
reaction products the experiment shown in Fig. 11 was performed. This
experiment
demonstrates that the temperature of elution is affected by the size of the
capture
CA 02369002 2001-09-26
WO 00/50644 PCT/US00/04939
-26-
probe. The temperature of elution was shown to be affected~by the size of the
capture probe. The five panels shown in Fig. 11 show the same vertical slab
gel run
at five different temperatures starting with 23 °C wherein five
different capture
probes were used. The number of bases in each probe sequence was 13, 15, 17,
19,
and 21 nucleotides as indicated in Fig. 11 (from left to right). An M13
sequencing
reaction (DynamicTM ET) was loaded in each lane. The l amer capture probe did
not
effectively captured the sequencing reaction products at 23 °C. All the
others
capture probe (i.e., having lengths 15, 17, 17, and 21) captured the
sequencing
reaction products at 23 °C. At 30 °C, the 1 Smer released the
sequence while the
l7mer started to release at 35 °C and so on until at 50 °C all
target sequences came
off. These results illustrate temperature conditions for capture and elution
of
sequencing reaction products.
The l7mer Acrydite probe was chosen as the capture probe in the standard
procedure with elution occurring at 45°C.
EQUIVALENTS
While this invention has been particularly shown and described with
references to preferred embodiments thereof, it will be understood by those
skilled
in the art that various changes in form and details may be made therein
without
departing from the spirit and scope of the invention as defined by the
appended
claims.