Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02387185 2002-04-10
WO 01/29234 PCT/EP00/10423
Self-cleaving RNA sequences and their use for the control
of protein synthesis
Background of the invention
This invention relates to the control of protein synthesis from a mRNA
sequence
(activation or repression) by inserting a sequence into an untranslated region
(UTR) of a
gene which confers conditional self cleavage to the transcribed RNA.
Synthesis of a protein in vivo is controlled at several levels. The first
level is the
transcription of the gene to generate a pre-messengerRNA (pre-mRNA). The rate
of
transcription initiation as well as the transcript elongation are under tight
physiological
to control by the basal transcription machinery. Another level is pre-mRNA
splicing into a
mature mRNA. Alternative splicing events can occur in tissue-specific manner
or under
hormonal control, hence the active protein is synthesized in a restricted set
of tissue or
physiological environment. Other levels of control are the mRNA turn-over and
the
ability of the mRNA to be translated into protein. Hence drug-control of
protein synthesis
can theoretically be addressed at each of the aforementioned levels.
A drug can control the expression of a target gene within a genome by altering
the rate of synthesis of the RNA. It can be achieved by drugs which bind DNA
with high
affinity and sequence-specificity and hence compete with the transcription
factors
necessary for the transcription of the target gene. Such drugs may be triple
helix-forming
oligonucleotides (C. Helene et al., Ciba Found Symp. (1997) 209: 94-106),
peptide
nucleic acids (Pooga, M. et al.; Nat Biotechnol. (1998) 16(9):857-61) or
pyrrole-
imidazole polyamides (Dickinson, L.A. et al.; Proc Natl Acad Sci U S A.
(1998); 95(22):
12890-5). It can also achieved by xenogenic transcription factors which bind
specifically
to the promoter region of the gene and which are activated upon binding of the
inducer
molecule (Allgood, V.E. et al.; Annual Reports in Medicinal Chemistry (1997)
32: 231-
239). The drugs which activate the xenogenic transcription factors are
antibiotics,
estrogen analogs or immuno-suppressors.
CA 02387185 2002-04-10
WO 01/29234 PCT/EP00/10423
2
Decreased protein production can be achieved by rising the turnover of the
mRNA. Antisense oligonucleotides anneal with the target RNA which can induce
its
cleavage by RNaseH, or prevent its translation or induce faster degradation by
disturbing
the secondary structure of the RNA (Kumar, M. et al.; Microbiol Mol Biol Rev.
(1998)
62(4):1415-34). In contrast ribozymes anneal with a target RNA and cleave it
(Forster,
A.C. & Altman, S.; Science (1990) 249: 783-6). Ribozymes are typically RNA
molecules
which have enzyme-like catalytic activities usually associated with cleavage,
splicing or
ligation of nucleic acid sequence. The typical substrates for catalytically
active ribozymes
are RNA molecules, although ribozymes can catalyze reactions in which DNA
molecules
1o serve as substrates. Naturally occuring ribozymes which are active
intracellularly work in
cis, catalyzing only a single turnover reaction, and are usually self modified
during the
reaction (Cech, T.R.; Biosci Rep. 1990;10(3):239-61). However, ribozymes can
be
engineered to act in traps, in a truly catalytic manner, with a turnover
greater than one
and without being self modified. Two distinct regions can be identified in a
ribozyme: the
binding region which gives the ribozyme its specificity through hybridization
to a
specific nucleic acid sequence, and a catalytic region which gives the
ribozyme the
activity of cleavage, ligation or splicing.
Drug-control of the protein production from a specific target gene within a
genome can be addressed by controlling the translation of the mRNA into
protein.
2o Control of gene expression which takes advantage of the interaction of a
specific ligand-
binding RNA (aptamer) and its cognate ligand has been published (Werstuck, G.
&
Green, M.R.; Science ( 1998) 282, 296-298). Translation of the mRNA is
inhibited by a
drug which binds in the vicinity of the 5' cap structure of the mRNA. In this
setting, the
intracellular presence of the drug is repressing protein production.
The control of gene expression and protein production is advantageous in the
field of gene therapy and DNA vaccines. It is intended to prevent the
occurrence of
adverse side effects and to tune the level of expression within an efficient
therapeutic
window. The use of the above-mentioned systems for the control of gene
expression in
gene therapy, however, faces major hurdles. The DNA binding drugs, as well as
anti-
sense and ribozyme oligonucleotides have a poor bio-availability and are
readily
CA 02387185 2002-04-10
WO 01/29234 PCT/EP00/10423
3
degraded by nucleases and proteases within tissues or body fluids. Xenogenic
transcription factors may elicit a cytotoxic immune response which will
destroy the cells
expressing the transgenes and hence undermine the therapy or the vaccination
(S. K.
Tripathy et al., Nature Medicine (1996) 2, 545-550). Furthermore the drugs
which
activate the xenogenic transcription factors are antibiotics, estrogen analogs
or immuno-
suppressors. They are not therapeutics per se for the gene therapy and may
have
undesired side effects. Therefore it would be advantageous to choose an
inducer drug
which is at least innocuous, for the therapy considered. An additional
drawback is the
large cloning space needed in the vectors, in order to accommodate the control
sequences
to and the genes encoding the transcription factors. Therefore it is highly
advantageous i) to
avoid the use of xenogenic proteins for the control of gene expression and ii)
to choose
the inducer drug according to the therapeutic objectives.
Control of gene expression and protein production is advantageous in the field
of
functional genomics, transgenic plants and transgenic animals. The conditional
expression of a gene into cell cultures or whole plants or animals and the
comparison of
the phenotypes with and without gene expression enables one to decipher the
function of
the gene. The drawbacks of the xenogenic transcription factor-based gene
switches are
two-fold: i) the expression of the target gene and the translation of the mRNA
are no
more under control of the endogenous physiological stimuli and ii) the effect
of the
2o inducer drug on the physiology of the cells. Therefore it is highly
advantageous i) to keep
the transgene under the control of its endogenous transcription promoter, as
well as
maintain the same translation control sequences on the mRNA and ii) to choose
an
inducer drug which does not alter cell physiology.
The invention described hereafter features the advantageous characteristics
aforementioned.
Description of the invention
Definitions
CA 02387185 2002-04-10
WO 01/29234 PCT/EP00/10423
4
Gene:
Any nucleic acid molecule encoding a biological product, i.e., a RNA, protein,
polypeptide or peptide. A gene, within the context of the instant invention,
therefore
includes gDNA, cDNA or synthetic or semi-synthetic DNAs. In particular, genes
according to the instant invention can be any nucleic acid encoding a
biological product,
comprising one or more naturally present or artifically produced untranslated
region(s).
Untranslated region (LTTR) of a gene:
A DNA sequence of a gene which is transcribed into RNA but which is not
translated into a protein. The UTR may be located before the 5'-end of the
coding region
(5'-UTR), after the 3'-end of the coding (3'-UTR), or may be an intron
inserted within
the coding region.
Untranslated region of a pre-mRNA:
A sequence of the pre-mRNA which is not translated into protein. The UTR may
be located before the 5'-end of the coding region (S'-UTR), after the 3'-end
of the coding
(3'-UTR), or may be an intron inserted within the coding region.
Ligand:
The ligand can be a nucleic acid molecule, a protein, polysaccharide or sugar,
or
an organic or inorganic molecule which interacts with the self cleaving RNA
sequence
and either inhibits or stimulates self cleavage.
2o Description of the invention
The instant invention provides methods and compositions for the control of
protein production in a cell. More particularly, the invention provides
methods and
compositions for the control of protein production using particular nucleic
acid
sequences, inserted within at least one UTR of a gene, which confer ligand-
dependant
self cleavage to RNAs transcribed from said gene. Hence, the pre-mRNA or the
mRNA
can undergo self cleavage that can be regulated through the presence, or the
absence of a
ligand. The ligand-controlled cleavage enables one to control protein
production with
this ligand (figure 1 ).
CA 02387185 2002-04-10
WO 01/29234 PCT/EP00/10423
This method is useful in situations where a gene is transfected into cultured
cells
or into live animals and one needs to control the expression of this gene. The
amount of a
protein synthesized in a cell is proportional to the amount of its mRNA, and
depends on
efficient translation of its mRNA into protein. Two features of mRNAs are
essential for
5 their efficient translation into proteins (Gallie, R.; Genes & Dev. ( 1991 )
5: 2108-2116)
and for their stability (Beelman, C.A. & Parker, R.; Cell (1995) 81: 179-183):
the 5'-cap
structure and the 3'-polyA tail. Figure 2A shows that the expression of the
luciferase gene
can be controlled by removing either 3'-polyA or S'-cap from the mRNA. The
insertion
of an active nucleic acid sequence of this invention in the 5'- or 3'-UTR of
the reporter
gene entails the cleavage of the transcribed RNA and hence the removal of,
respectively,
the 5'-cap and 3'-polyA. The active nucleic acid sequence; in either location;
decreases
the luciferase protein production whereas an inactive nucleic acid sequence in
the same
location does not affect gene expression.
This invention can be used to control production of essentially all types of
proteins, in essentially all types of cells, in vitro ex vivo or in vivo.
A first object of this invention resides more particularly in a modified gene
encoding a protein, polypeptide or peptide, wherein said modified gene
contains, inserted
in an untranslated region thereof (UTR), a nucleic acid sequence conferring
ligand-
dependant self cleavage to a RNA molecule transcribed from the gene.
2o As indicated above, the modified gene can be a genomic DNA, a cDNA or a
synthetic DNA. The modified gene comprises at least one UTR region, which can
be
naturally present in said gene or artificially inserted therein. For instance,
where the gene
is a gDNA molecule, the gene comprises naturally-ocurring UTRs such as 5'-UTR,
introns and 3'-UTRs. Where the gene is a cDNA or a synthetic (or semi-
synthetic)
molecule, UTR regions may be introduced, such as introns. Furthermore, even
where
natural UTRs are present within the gene, additional UTR can be inserted, in
addition
thereto or in replacement thereof. The modified gene may further comprise a
transcriptional promoter, to direct transcription of the coding region into
pre-mRNA. The
gene may, in addition, be contained in a vector, such as a plasmid, a virus, a
cosmid, a
3o phage, an artificial chromosome, etc. The modified gene may encode any type
of protein,
CA 02387185 2002-04-10
WO 01/29234 PCT/EP00/10423
6
polypeptide or peptide, including protein, polypeptide or peptide of human,
other
mammalian, plant, viral or bacterial origin, or derivatives thereof for
instance. The
encoded biological product may exhibit therapeutic or prophylactic activity,
and may also
represent a marker molecule, for instance.
As indicated above, the modified gene of this invention comprises at least one
particular nucleic acid sequence inserted in a UTR of said gene. The UTR can
be a 5'-
UTR, a 3'-UTR or an intron. Although the insertion of one single copy of a
nucleic acid
sequence in a gene of the invention is sufficient to provide control over
protein
production, in particular embodiments, the modified gene may comprise several
copies of
to such a nucleic acid sequence, inserted in various UTRs or in various
locations of a UTR,
or different nucleic acid sequences inserted in various UTRs or in various
locations of a
UTR.
In one embodiment the self cleavage of the RNA sequence inserted into a UTR of
the pre-mRNA is inhibited by a ligand. Hence the pre-mRNA or mRNA is cleaved
in the
absence of ligand and protein production is decreased ; the pre-mRNA or mRNA
is not
cleaved in the presence of ligand and protein production is restored.
A particular modified gene of this invention thus comprises a nucleic acid
sequence which confers ligand-inhibited self cleavage to a RNA transcribed
from the
gene.
In another embodiment the self cleavage of the RNA sequence inserted into a
UTR of the pre-mRNA is activated by a ligand. Hence the pre-mRNA or mRNA is
cleaved in the presence of ligand and protein production is repressed; the pre-
mRNA or
mRNA is not cleaved in the absence of ligand and protein production is
restored.
Another particular modified gene of this invention thus comprises a nucleic
acid
sequence which confers ligand-activated self cleavage to a RNA transcribed
from the
gene.
The invention also relates to a method of modifying a gene, comprising
inserting,
within at least one untranslated region of the gene, a nucleic acid sequence
conferring
ligand-dependent self cleavage to a RNA transcribed from said gene.
CA 02387185 2002-04-10
WO 01/29234 PCT/EP00/10423
7
The nucleic acid can be inserted in various sites within a UTR sequence. Where
5'-UTR or 3'-UTR are concerned, insertion can be performed in different sites
which
essentially do not affect transcription of the DNA into pre-mRNA. Such sites
may be
determined by analyzing the sequence of a UTR, and by using restriction sites
available
or artificially created. Insertion may also be accomplished by recombination,
mutagenesis, etc. Where introns are concerned, insertion can take place in any
region
which do not affect transcription. Since the efficiency of self cleavage of
the nucleic acid
sequence within a UTR may depend on the surrounding nucleotide sequence, it is
preferred to use computer programs like RNAfold (Zuker, M.; Methods Mol Biol (
1994)
25:267-94) which predict the most stable conformations, and assess whether the
nucleic
acid sequence folds in a productive conformation when inserted in a particular
location
within a UTR. Indeed the surrounding sequence may provide the nucleic acid
sequence
with alternative folding pathways which lead to inactive or ligand-independent
conformations (Stage-Zimmermann, T.K. RNA (1998) 4:875-889). Various sites of
insertion of the nucleic acid sequence within a UTR can be selected according
to the
above methodologies.
The particular nucleic acid sequence used in the instant invention to provide
production control can be any nucleic acid sequence encoding a ligand-
dependant self
cleavable RNA. The size of this nucleic acid sequence can vary, depending on
its nature
and/or origin. Generally, the nucleic acid sequence comprises between 10 and
500 base
pairs, preferably below 300 base pairs. Typical such nucleic acids comprise
between 20
and 200 bp.
The nucleic acid can be of different origin. In particular, the nucleic acid
can be
derived from naturally occuring self cleavable RNA sequences. In this regard,
one can
choose DNA sequences whose RNA transcripts have a self cleavage activity which
is
known to be affected by various ligands such as hammerhead ribozymes,
hepatitis delta
virus ribozymes, Neurospora VS ribozyme, hairpin ribozymes, group I intron,
group II
intron and Rnase P, for instance (Clouet-d'Orval, B.et al.; Biochemistry
(1995) 34:
11186-90; Olive, J.E. et al.; EMBO J. (1995) 14: 3247-51; Rogers, J. et al.;
J. Mol. Biol.
CA 02387185 2002-04-10
WO 01/29234 PCT/EP00/10423
8
( 1996) 259: 916-25). These sequences can be prepared artificially and used
according to
the instant invention. In particular, DNAs encoding such RNAs (or functional
derivatives
thereof) can be prepared by conventional techniques and used to modify a gene
in
accordance with the instant invention.
Alternatively, any DNA sequences whose RNA transcripts have self cleaving
activity may be chosen and inhibitors of self cleavage may be identified from
a library of
compounds.
In a preferred embodiment, however, an advantageous ligand is chosen first and
DNA sequences whose RNA transcripts feature ligand-dependant self cleavage are
1 o produced in vitro. In this regard, the invention also describes a
particular method that can
be used to produce artificial, non-naturally ocurnng ligand-dependant self
cleavable RNA
and DNA encoding them. Said artificial, non-naturally ocurring ligand-
dependant self
cleavable RNA are called aptazymes, and represent another object of the
instant
invention.
Accordingly, in a particular embodiment of this invention, the nucleic acid
sequence is an artificial DNA encoding an aptazyme. Said aptazyme can be
obtained by
an in vitro evolution and selection method, as described below.
Aptazyme production
In the method disclosed in this invention, the ligand can be selected first,
and then
corresponding activated or inhibited aptazymes are produced.
Selection of the ligand
The ligand can be a nucleic acid molecule, a protein, polysaccharide or sugar,
or
an organic or inorganic molecule. The nature of the ligand can be chosen to be
exogenously supplied, such as some non-toxic molecule or drug which readily
enters at
least the cells transfected with the transgene. Alternatively, an entirely
endogenous
system can be designed in which the controlling ligand is a molecule (e.g.,
some small
3o metabolite or macromolecule) present within the target cell. In this
regard, the ligand can
CA 02387185 2002-04-10
WO 01/29234 PCT/EP00/10423
9
be a molecule present within the target cell population and essentially absent
(or present
at a lower concentration) within other cell population, thereby conferring
tissue
selectivity to the expression. More particularly, the ligand may be a molecule
present
within the target cell population, which is directly or indirectly related to
a pathology or
condition to be corrected, and essentially absent from cells or tissues not
affected by the
pathology. The activity of the ligand-dependant self cleavage nucleic acid is
then
dependent on its binding to the metabolite or macromolecule. If the ligand-
dependant
self cleaving nucleic acid is inactivated by (i.e., not cleaved in the
presence of) the
metabolite or macromolecule (e.g., a pathogenic protein), the expression of
the gene is
to then restricted essentially to the cells which express sufficient amounts
of the metabolite
or macromolecule, i.e., the diseased cell population (examples of such ligands
include
mutated p53 molecules, activated oncogenes, etc.). Other examples of ligands
include
antibiotics (e.g., doxycycline, pefloxacine, etc.), molecules which are used
in humans
(such as drugs, adjuvents, substitutes, etc.) and any molecule which would be
innocuous
in a human subject, for instance. This process requires a very high degree of
specificity in
the molecular recognition between the aptazyme and the ligand. Such a
specificity can in
principle is achieved by a nucleic acid aptamers (Famulok, M. ; Curr. Opin.
Struct. Biol.
(1999); 9: 324-329).
2o Selection of the aptazyme
Aptazymes that undergo ligand-dependent self cleavage can be prepared and
isolated by an in vitro evolution and selection method. This method is related
to the
SELEX technology (US Patent 5270163, incorporated therein by reference) which
is a
technique that allows the simultaneous screening of highly diverse
combinatorial libraries
of different RNA or DNA (single stranded or double stranded DNA) molecules for
a
particular feature. These features may be i) catalytic activity of a nucleic
acid, ii) the
ability of a nucleic acid to specifically complex a desired target molecule
with high
affinity and selectivity.
CA 02387185 2002-04-10
WO 01/29234 PCT/EP00/10423
The present method differs from methods which have been previously described
for the selection of allosteric ribozymes (W094/13791; W098/08974). More
specifically,
a production method of this invention (as depicted in Figure 3A and 3B),
comprises:
1. preparing a pool of different double-stranded deoxyribonucleic acid (DNA)
5 molecules, at least part of the sequence in the molecules of the pool is a
random or
partially random sequence, such that said part has a different sequence in
different
molecules of the pool. Generally a random sequence may be prepared, for
example, by
utilizing a nucleic acid synthesizer,
2. transcribing the pool of DNA molecules into a pool of ribonucleic acid
(RNA)
1o molecules under conditions where no-self cleavage should occur: in the
presence of
ligand for the selection of ligand-inhibited self cleaving nucleic acid
sequences, or in the
absence of ligand for the selection of ligand-activated self cleaving nucleic
acid
sequences,
3. immobilizing all members of the said RNA pool on a solid support under
conditions where no self cleavage should occur (presence or absence of
ligand),
4. incubating said RNA pool, in the presence or absence of said ligand, under
conditions where no self cleavage should occur (presence or absence of
ligand),
5. separating between the RNA molecules bound to the solid support, and the
RNA molecules present in the liquid phase which result from self cleavage.
Denaturing
2o and washing the RNA molecules bound to the solid support,
6. repeating steps 3-5 over a plurality of cycles, e.g. about 1-50 cycles, to
eliminate the RNA sequences which are capable of self cleavage under non-
permissive
conditions, albeit with low efficiency, due to multiple, non-productive
conformational
states,
7. adding or removing said ligand to said pool and incubating under conditions
permitting self cleavage, and
8. separating between the RNA molecules of said pool which feature self
cleavage
(present in the liquid phase) and such which do not (bound.to solid support),
to obtain a
first selected pool of RNA molecules which feature ligand-dependent self
cleavage.
CA 02387185 2002-04-10
WO 01/29234 PCT/EP00/10423
11
In a preferred embodiment, the method of this invention further comprises:
9. reverse-transcribing the said first selected pool of RNA molecules into a
pool of
single-stranded complementary DNA (cDNA) molecules and amplifying (for
instance by
PCR) the single-stranded cDNA pool into a double-stranded DNA pool, and
10. repeating steps 2-9 over a plurality of cycles, e.g. about 10-100 cycles
to
obtain said aptazymes.
This method can also be used to select aptamers, i.e. ligand-binding RNA
sequences.
In one embodiment, the molecules in the pool are comprised of an entirely
random sequence with the exception of two short flanking sequences which
encompass
the RNA polymerase promoter and the attachment sites of the amplification
primers.
In another embodiment of the invention the molecules in the pool are
constructed
based on a known ribozyme sequence. For example, the molecule may be comprised
of a
constant, ribozyme-derived sequence attached to a random or semi-random
sequence.
There is no set number of random nucleotides. However, in general the random
sequence contains between 10 and 300 nucleotides, preferably between 20 and
200, more
preferably between 20 and 180 nucleotides. Although referred to herein as a
»random»
sequence, it is understood that the sequence is random only as originally used
in the
selection process, that the product of the selection process is not random but
a set of
specific sequences displaying ligand-dependent self cleaving ability.
The initial pool may comprise a various number of DNA molecules. In
particular,
the initial pool may comprise up to 102° molecules or more. Typical
pools comprise
between 104 and 10'6 molecules.
It should be understood that the above production method may be applied to
different pools of DNA, including genomic, cDNA or RNA libraries, for
instance.
In the steps of the selection, it is desired to apply conditions which mimic
the
cellular environment in which the aptazyme will eventually be used. For
example where
the aptazyme is intended to control protein production in mammalian cells, the
ionic
CA 02387185 2002-04-10
WO 01/29234 PCT/EP00/10423
12
composition of the selection medium will be set accordingly and the
temperature will be
set to 37 degrees Celsius.
It is useful to progressively increase the threshold criteria for selection in
each
round of selection and amplification. For example, as the steps of the in
vitro evolution
proceeds, lower concentration of ligand may be used, resulting in the
selection of species
having progressively higher affinity to the desired ligand. Number and time of
incubation
at steps 3-5 (non permissive conditions) may be progressively extended, thus
increasing
the control of the ligand on the self cleavage nucleic acid sequences. Time of
incubation
during step 7, under conditions permitting self cleavage, may be progressively
shortened,
1o thus species having faster cleavage rate are selected.
In addition, mutations can be introduced in the selected DNA pool obtained
during step 9 by performing the amplification (e.g., the PCR) under mutagenic
conditions. This mutagenic step broadens the sequence diversity of the pool
which is
subjected to the selection.
Selection of ligand-controlled gene expression cassette
The in vitro selection of the aptazyme generally yields a set of several
sequences,
which exhibit the desired property of ligand-dependent self cleavage. It is
then possible
to clone the selected aptazymes and determine their exact sequence. This can
be achieved
2o by ligation of the double-stranded DNA pool encoding the aptazymes to an
appropriate
linearized plasmid vector and transforming bacteria with the resulting
circular plasmid
vectors (Sambrook et al (1989). Molecular cloning, a laboratory manual, Second
Edition,
N. Ford, ed., Cold Spring Harbor: Cold Spring Harbor Laboratory Press). The
cloned
plasmids containing the aptazymes can be sequenced by the method of Sanger et
al.
(Proc. Natl. Acad. Sci. USA, 74 (1977) 5463-5467) with an Applied Biosystems
kit
according to the manufacturer's instructions. Generally, between 10 and 1000
clones are
sequenced, among which a subset of different families exhibiting the desired
property can
be identified.
It is then desired to select for aptazymes which confer ligand-dependent
expression of a reporter gene into the target cells. Reporter genes expression
cassettes and
CA 02387185 2002-04-10
WO 01/29234 PCT/EP00/10423
13
vectors carrying them can be constructed by standard molecular biology
techniques
(Sambrook et al (1989). Molecular cloning, a laboratory manual, Second
Edition, N.
Ford, ed., Cold Spring Harbor: Cold Spring Harbor Laboratory Press). The
target cells are
transfected with two reporter genes; reporter gene ( 1 ) contains the sequence
of an in
vitro-selected aptazyme within at least one UTR, and reporter gene (2) does
not contain
any aptazyme. Two sets of cells are cultured in the absence and in the
presence of the
ligand. The ratio of reporter gene ( 1 ) expression over reporter gene (2)
expression is
assessed in the presence and in the absence of ligand. The aptazyme clone
which yields
the lowest ratio where protein production is to be repressed and the highest
ratio where
to protein production is to be induced is selected.
The invention can be used to control protein production in vitro, in vivo or
ex
vivo in various cell types.
In this regard, the invention also resides in a method for controlling
production of
a protein, polypeptide or peptide from a gene, comprising inserting, within at
least one
untranslated region of the gene, a nucleic acid sequence conferring ligand-
dependent self
cleavage to a RNA transcribed from said gene.
In a particular embodiment, the invention provides a method for removing, in a
ligand-dependant manner, the 5'-cap structure and/or the 3'-polyA tail from a
pre-mRNA
2o or a mRNA of a gene whose expression is to be controlled. This can be
achieved by
inserting a sequence in at least one UTR of the gene which entails ligand-
dependent self
cleavage of the transcribed RNA.
The invention thus also resides in a method of removing a 5'-cap structure
and/or
a 3'-polyA tail from a pre-mRNA or a mRNA transcribed from a gene, comprising
inserting, within at least one untranslated region of the gene, a nucleic acid
sequence
conferring ligand-dependent self cleavage to a RNA transcribed from said gene.
In a particular embodiment, the method is for removing a 5'-cap structure from
a
pre-mRNA or a mRNA transcribed from a gene, and the nucleic acid sequence is
inserted
within at least a 5'-untranslated region of the gene.
CA 02387185 2002-04-10
WO 01/29234 PCT/EP00/10423
14
In another particular embodiment, the method is for removing a 3'-polyA
structure from a pre-mRNA or a mRNA transcribed from a gene, and the nucleic
acid
sequence is inserted within at least a 3'-untranslated region of the gene.
The invention also relates to a method of controlled production of a protein,
polypeptide or peptide within a cell, comprising (i) introducing into a cell a
modified
gene encoding the protein, polypeptide or peptide, wherein said modified gene
contains,
inserted in an untranslated region thereof (UTR), a nucleic acid sequence
confernng
ligand-dependant self cleavage to a RNA molecule transcribed from the gene,
and (ii)
1o contacting the cell in the presence or absence of the ligand.
In a particular embodiment, the modified gene contains, inserted in an
untranslated region thereof (UTR), a nucleic acid sequence confernng ligand-
activated
self cleavage to a RNA molecule transcribed from the gene, and production is
repressed
by contacting the cell with the ligand, while production is restored (or
increased) by
removing (or stopping contacting or in the absence of) the ligand.
In another particular embodiment, the modified gene contains, inserted in an
untranslated region thereof (UTR), a nucleic acid sequence conferring ligand-
inhibited
self cleavage to a RNA molecule transcribed from the gene, and production is
increased
by contacting the cell with the ligand, while production is depressed (or
inhibited) by
removing (or stopping contacting or in the absence of) the ligand.
In the above method, the ligand can be either exogenously supplied to the cell
or
be an endogenous component of the cell, as described above. In this regard,
the ligand
can be a molecule which is preferentially present in cells where production of
the protein,
polypeptide or peptide is sought (and essentially absent or express at a lower
concentration in other cells or tissues).
The above method can be used for production of a protein, polypeptide or
peptide
in a cell (or in a cell population or culture) in vitro or ex vivo.
The above method can also be applied to the production of a protein,
polypeptide
or peptide in a cell, tissue or organ in vivo.
CA 02387185 2002-04-10
WO 01/29234 PCT/EP00/10423
The above cell can be selected from prokaryotic cells, eukaryotic cells,
mammalian cells and plant cells, for instance. It may be a tissue, organ, an
isolated cell
culture, established cell lines or primary cultures. Preferred cells are
mammalian cells, in
particular marine or human cells, more preferably ex vivo or in vivo. For in
vivo uses, the
5 gene can be introduced into the cells by administration to an organism, and
the ligand can
be supplied exogenously or contained within the cell. Furthermore, the gene
may be
contained in a vector selected from plasmids, viruses, cosmids, artificial
chromosomes,
etc., or combinations thereof. Preferred vectors include plasmids and viruses,
such as
adenoviruses, retroviruses, AAV, HSV, HIV, etc.
10 The instant invention also discloses and claims combinations of a ligand
and a
modified gene (or vector containing the same) as described above, for
simultaneous,
separate or sequential use.
Preferred combinations comprise:
- a ligand and a modified gene, wherein the self cleavage of the RNA
transcribed
15 from the said sequence is inhibited by said ligand;
- a ligand and a modified gene, wherein the self cleavage of the RNA
transcribed
from the said gene is activated by said ligand.
Preferably, the ligand is selected from the group of nucleic acid molecules,
proteins,
polysaccharides, sugars and organic and inorganic molecules.
Other aspects and advantages of the instant invention will be disclosed in the
following experimental section, which should be considered as merely
illustrative and not
limiting the scope of the invention.
Legend to the Figures
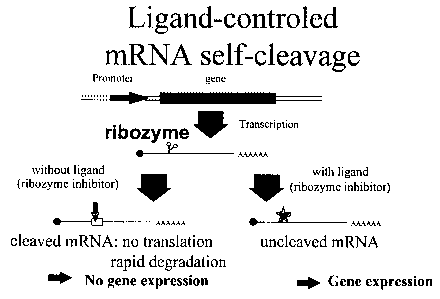
Figure 1:
Description of the ligand-dependant control of protein production according to
the
invention
CA 02387185 2002-04-10
WO 01/29234 PCT/EP00/10423
16
Figures 2A, 2B and 2C:
Demonstration that a self cleaving RNA sequence (e.g., a ribozyme hammerhead
sequence) inserted within a UTR of a pre-mRNA can control gene expression.
Figure 2A:
reporter protein production without ribozyme sequence, with inactive ribozyme
sequence
s or with active ribozyme sequence in one UTR of the gene. Figure 2B: site of
insertion of
the ribozyme sequences (restriction enzymes sites) within the pre-mRNA of the
reporter
gene. Figure 2C: sequence of the active and the inactive ribozyme.
Figures 3A and 3B:
Figure 3A: outline of the selection procedure for ligand-inhibited self
cleaving
l0 RNA sequences.
Figure 3B: outline of the selection procedure for ligand-activated self
cleaving
RNA sequences.
Figure 4:
Description of the plasmids used for the cloning of the aptazyme pool.
is Figure 5:
Design of the initial pool. The complete sequence of the double stranded DNA
molecules used for the first selection cycle is shown, as well as the relative
positions and
sequences of the different primers synthesized.
Figure 6:
20 Time course of the two selections. The percentage of RNA eluted after each
selection cycle is shown for the two selections, using doxycycline or
pefloxacine as a
ligand. An empty space between two cycles indicates an increase in the
selection pressure
(either by changing incubation times or/and by decreasing ligand concentration
(see
Table 1 ).
CA 02387185 2002-04-10
WO 01/29234 PCT/EP00/10423
17
Ficture 7:
Kinetics of self cleavage of the selected RNA pool in the presence of
increasing
amounts of doxycycline. Left panel: pool after selection cycle #10; Right
panel pool after
selection cycle #13.
s Figure 8:
Self cleavage-activity of a doxycycline-sensitive aptazyme and conversion of
the
aptazyme into aptamer. Aptazyme RNA was incubated in SOmM Tris-HCl pH7.5 at
37°C, self cleavage was initiated by addition of IOmM MgClz and
assessed during 3
minutes. When indicated 200nM of doxycycline (Dox) or tetracycline (Tet) were
added in
the cleavage reaction. The self cleavage reaction was performed in the absence
of added
RNA (left panel: Control), in the presence of 1 ~M and 2pM aptazyme-derived
aptamer
(2°d and 3'~ panels from the left: 1 pM aptamer, 2uM aptamer), in the
presence of 1 pM
and 2pM non-cognate RNA (4~' and 5'" panels from the left: 1 pM non-cognate
RNA,
2pM non-cognate RNA).
~s Examples
Example 1: Selection of aptazymes inhibited by doxycycline and
pefloxacine
An in vitro selection was carried out to select for aptazymes inhibited by
(i.e., not
cleaved in the presence of) either doxycycline and pefloxacine (designed as
the ligand in
2o sections i to iv). The conditions used during the two selections were
identical, except for
the ligand used.
i. Preparation of initial degenerated pool
The initial degenerated pool was produced using standard DNA synthesis
chemistry on an Expedite nucleic acids synthesis system (Millipore).
Oligonucleotides
25 were purified on denaturating polyacrylamide gels. The following primers
were
synthesized:
CA 02387185 2002-04-10
WO 01/29234 PCT/EP00/10423
18
Pool-Primer:
5'-CGCGTTGTGTTTACGCGTCTGATGAGT-N(40)-ACGAAACTACCTCGA
GACGT (SEQ ID NO: 1 )
Primer 1:
5'-CGCGTTGTGTTTACGCGTCTGATG (SEQ ID N0:2)
Primer 2:
5'-AGCTGGTACCTAATACGACTCACTATAGGAGCTCGGTAGTCACGCG
TTGTGTTTACGCGTCTGATG (SEQ ID N0:3)
Primer 3:
l0 S'-ACGTCTCGAGGTAGTTTCGT (SEQ ID N0:4)
After purification, 6 nmol of Pool-Primer were obtained (4.10'5 molecules).
The
total DNA was amplified by PCR using Primer 1 and 3 as previously described
(Bartel
D.P. & Szostak J.W.; Science (1993) 261: 1411-1418). A further amplification
step was
performed under the same conditions using 6 nmol of pre-amplified double
stranded
product as template, primer 2 and 3. The final DNA molecules contained the
following
features (from 5' to 3'): KpnI restriction site, T, RNA polymerise promoter,
SacI
restriction site, Hammerhead ribozyme sequence containing 40 random
nucleotides
located in the Helix II domain, XhoI restriction (see Figure 5).
This DNA library was used as a template for T, RNA polymerise during the
2o initial in vitro selection cycle.
ii. General selection scheme
The selection scheme used is outlined in Figures 3A and 3B. Several
modifications of the selection scheme were done during the selection and are
detailed
later (section iii).
a) Transcription and immobilization on a column
Standard transcription reactions contained the following components: 250 units
T,
RNA polymerise (Stratagene), T, Buffer (Stratagene), 2,5 mM of each dNTP, 20
mM of
GMPS, 20 ~Ci a32P GTP, RNasin (50 U), ligand (1mM). The transcription reaction
was
incubated over night at 37°C. The uncleaved RNA transcripts were then
purified on a 8%
CA 02387185 2002-04-10
WO 01/29234 PCT/EP00/10423
19
denaturating polyacrylamide gel. After purification, the RNA was treated with
Iodo-
Acetyl-LC-Biotin (Pierce - 200 fold excess) for 90 min. at room temperature.
Primed
RNA was gel purified under the same conditions as described above and
incubated with
streptavidine agarose (Pierce) for 30 min. at room temperature. The column
material was
then washed 6 times alternatively in lml washing buffer A (WA ; 25 mM HEPES pH
7.4,
1M NaCI, SmM EDTA), washing buffer B (WB ; 3M urea, SmM EDTA) and water.
Finally, it was rinsed five times with water.
b) Negative selection
The immobilized RNA was incubated at 37°C for 2 to 4 hours in selection
buffer
(SB ; 40 mM Tris HCl pH 8, 50 mM NaCI, 10 mM Spermidine, 8 mM MgCl2) in the
presence of the ligand ( 1 mM to 1 pM). The incubation was started upon the
addition of
magnesium. After round 5, this incubation was interrupted 10 times during the
first 2h30
of incubation and the column was washed twice with 1 ml WB (denaturating
conditions)
and rinsed 3 times with water. The column material was then washed again
thoroughly (6
times 1 ml WA - WB - HzO; 5 times 1 ml H20).
c) Positive selection
The positive selection was performed under similar conditions as the negative
selection, without the ligand. Incubation time was considerably shorter (10 to
1 min.).
Cleaved nucleic acid sequences were eluted with 1 ml of WB. Eluted RNA was
purified
by 2 phenol-chloroform extractions and ethanol precipitation.
d) Reverse transcription and amplification
The RNA was finally reverse transcribed with Tth DNA polymerase (Boehringer)
and amplified by PCR using primers 3 and 4 under standard conditions. This DNA
template was used as a template for T, RNA polymerase for the next round of
selection.
iii. Modifications of the selection scheme during selection
During selection, several modifications of the selection scheme were made (see
Table 1). During the first cycle, 16 nmol of transcribed RNA were used in
order to
preserve the diversity of the pool. Column material and washing volumes were
scaled up
CA 02387185 2002-04-10
WO 01/29234 PCT/EP00/10423
accordingly . During later cycles, RNA quantity was reduced. The ligand
concentration,
and the different incubation times were also adjusted to increase the
stringency. After
selection cycles 10, and 13, the pool was subjected to a mutagenic PCR under
previously
described conditions (Cadwell R.C. & Joyce G.F.; in PCR Methods and
Applications
5 (1992): 28-33). The mutagenic PCR creates a new diversity of RNA molecules
closely
related to selected sequences.
iv. Enrichment during selection (Figure 6)
Elution profiles during selections for doxycycline inhibited aptazymes and
1 o pefloxacine inhibited aptazymes were extremely similar, suggesting that
this type of
selection scheme is extremely robust. First hints for enrichment could be
observed during
cycle 4 and were confirmed during cycle S. However a time course experiment of
the
selected pools showed no significant difference between cleavage in the
presence or
absence of the ligand. Furthermore, cleavage efficiency of this pool was
severely reduced
15 compared to the unselected pool (data not shown). It was hypothesized that
this result
was due to co-selection of RNA species able of self cleavage under non-
permissive
conditions, albeit with low efficiency, due to multiple non productive
conformational
states.
To counter-select against these parasites, the selection scheme was modified
by
2o introduction of several denaturation steps during the negative selection
(see ii-b).
During cycle 7 a new enrichment could be observed. Thus, selection stringency
was increased by reducing ligand concentration to 100 ~,M and elution time to
1 min.
After 3 cycles a new enrichment could be observed. The selected pool at this
stage was
subjected to mutagenic PCR and the selection stringency was increased by a 10
fold
decrease in ligand concentration. After 3 cycles, a new enrichment could be
observed.
This procedure (mutagenic PCR - increase in selection stringency - 3 selection
cycles)
was reproduced on the selected pool and yielded a new enrichment of the pool.
After this
stage, a last cycle was performed with a highly reduced elution time ( 10
sec.), to select
for the fastest cleaving nucleic acid sequences.
CA 02387185 2002-04-10
WO 01/29234 PCT/EP00/10423
21
v. Pool kinetics (figure 7)
During and after selection, pools obtained after different cycles were
transcribed
under standard conditions (2s0 units T, RNA polymerase (Stratagene), T, Buffer
(Stratagene), 2,s mM of each dNTP, 20 ~Ci a32P GTP, s0 U RNasin , 1mM ligand).
s Uncleaved transcripts were gel purified, dephosphorylated using Calf
Intestinal Alkaline
Phosphatase (Stratagene) and 5' radio-labeled using T4 polynucleotide kinase
(Stratagene) and y3zP-ATP. Radio-labeled RNA molecules were gel purified and
incubated at a 10 nM concentration in SB with various amounts of ligand at
37°C.
Reaction was initiated upon addition of magnesium. Aliquots were taken at
different time
l0 points and mixed with an equal volume of loading buffer (9M urea, s mM
EDTA) on ice
to quench the reaction. The samples were then loaded on a 12 % sequencing gel,
and the
bands corresponding to intact ( 103 nt) or cleaved ( 13 nt) nucleic acid
sequences were
quantified using a phosphor imager (Molecular Dynamics).
is Example 2: Selection of doxycycline- and pefloxacine-induced expression
cassettes in mammalian cells
The DNA pools obtained after selection cycles 10, 13 and 16 are cut by the
restriction enzymes XhoI and KpnI. The plasmids pNPGI and pNPG2 (figure 4) are
cut
with the same enzymes and the three DNA pools are ligated to each of the two
plasmids.
2o E. Coli strain DHSa is transfected by each one of the six ligation mixture
according to
standard procedures (Sambrook et al (1989). Molecular cloning, a laboratory
manual,
Second Edition, N. Ford, ed., Cold Spring Harbor: Cold Spring Harbor
Laboratory Press).
One hundred colonies are isolated for each transfection. Plasmid DNA is
extracted and
purified from each of these colonies. The aptazymes cloned into the plasmids
are
2s sequenced by the method of Sanger et al. (Proc. Natl. Acad. Sci. USA, 74
(1977) s463-
s467) with an Applied Biosystems kit according to the manufacturer's
instructions.
Human cultured cell lines HeLa and HEK293 are transfected by plasmid DNA
complexed to lipofectamine (Gibco-Life Sciences, Bethesda, MD USA) according
to the
manufacturer's instructions (6 p1 lipofectamine per ~g DNA). In one set of
experiments
CA 02387185 2002-04-10
WO 01/29234 PCT/EP00/10423
22
the cells are incubated with 10 pM doycycline or 10 ~M pefloxacine, in another
set of
experiment the cells are incubated without ligand. 48 hours after
transfection, the cells are
lysed and the amount of Renilla luciferase and firefly lucifease are recorded
according to
the manufacturer's instructions (Promega, Madison, WI USA). The ratio of
firefly
luciferase activity (aptazme-controlled) over Renilla luciferase activity
(Ratio R) is
computed for each experimental condition (i.e. plasmid, ligand). The plasmids
which
yield production of high amount of firefly luciferase in the presence of
ligand
(doxycycline or pefloxacine) and low amount of firefly luciferase in the
absence of ligand
are identified by the highest ratio R. These plasmids are selected for further
use in gene
to transfer experiments where production of a protein is to be controlled by
doxycycline or
pefloxacine.
Example 3: Doxycycline- induced protein production in mice
pg of aptazyme-containing plasmids pNPG 1 or pNPG2 (selected as in example
2) are injected into tibialis cranialis muscles (posterior limbs) of eight
weeks-old Balb/C
mice, the limbs are subsequently subjected to electroporation in order to
enhance gene
transfer (Mir, L.M. et al.; P.N.A.S. USA (1999); 96(8):4262-7). One group of
five mice is
given doxycycline in the drinking water (0.2 mg/ml), the other group is not. 7
days after
gene transfer the animals are sacrificed and the tibialis cranialis muscles
are collected.
The muscles are resuspended in lml of lysis buffer (Dual-Luciferase reporter
Assay
2o System, Promega, Madison, WI USA) homogenized by glass and ceramic beads
into a
Fast-Prep homogenizer (Bio 101, Vista, CA USA). The amount of firefly
luciferase and
Renilla luciferase are assessed according to Promega's instructions. The
amount of firefly
luciferase synthesized by the mice of each group (treated or not by
doxycycline) is
compared. Inter-individual variations in gene transfer efficiency can be
compensated by
comparing the ratio firefly luciferase over renilla luciferase between the 2
groups of mice.
Hence one can verify that luciferase production in mice is induced by
doxycycline.
Example 4: selection of RNA aptamers binding to doxycycline (Figure 8)
Aptazymes were selected according to the procedure outlined in example 1. The
CA 02387185 2002-04-10
WO 01/29234 PCT/EP00/10423
23
DNA pool was inserted into pNPG2 plasmid according to example 2. Several
clones were
sequenced by the method of Sanger et al. (Proc. Natl. Acad. Sci. USA, 74
(1977) 5463-
5467) with an Applied Biosystems kit according to the manufacturer's
instructions.
Clone 6 was further investigated. The clone 6 DNA sequence was amplified by
PCR using primer 2 and primer 3 (example 1 ). The amplified sequence (upper
strand) is
described below:
AGCTGGTACCTAATACGACTCACTATAGGAGCTCGGTAGTCACGCGTT
GTGTTTACGCGTCTGATGAGTGGTACAGTCCAGGGTGAAGTTCCAATTTTGAA
CACCTCCACGAAACTACCTCGAGACGT (SEQ ID NO:S)
l0 Twice-underlined: T7 RNA polymerase transcription promoter
Underlined: random sequence in the original DNA pool (example 1 )
Bold: cleavage site
The amplified DNA sequence (SEQ ID NO: 5) was transcribed by T7 RNA
polymerase and the resulting RNA was purified as described in example 1. The
RNA
sequence is (hereafter referred to as the aptazyme):
GGAGCUCGGUAGUCACGCGUUGUGUUUACGCGUCUGAUGAGUGGUA
CAGUCCAGGGUGAAGUUCCAAUUUUGAACACCUCCACGAAACUACCUCGAG
ACGU (SEQ IDN0:8)
The clone 6 DNA sequence was also amplified by PCR using primer 4 and primer
3. Primer 4 and the amplified sequence (upper strand) are described below:
Primer 4:
5'-AGCTGGTACCTAATACGACTCACTATAGGAGCTCGGTAGTCACGCG
TTGTGTTTACGCGTCTGATG (SEQ ID N0:8)
Amplified sequence:
AGCTGGTACCTAATACGACTCACTATAGGAGCTCGGTAGTGACGCGTT
GTGTTTACGCGTCTGATGAGTGGTACAGTCCAGGGTGAAGTTCCAATTTTGAA
CACCTCCACGAAACTACCTCGAGACGT (SEQ ID N0:7)
Twice-underlined: T7 RNA polymerase transcription promoter
Underlined: random sequence in the original DNA pool (example 1 )
3o Bold: mutated base
CA 02387185 2002-04-10
WO 01/29234 PCT/EP00/10423
24
Primer 4 introduces a C to G mutation which inactivates the self cleavage
activity
of hammerhead ribozymes but minimally affects their tertiary structure
(Baidya, N. and
Uhlenbeck, O.C.; Biochemistry (1997) 36: 1108-1114).
The amplified DNA sequence (SEQ ID NO: 7) was transcribed by T7 RNA
polymerase and the resulting RNA was purified as described in example 1. The
RNA
sequence is (hereafter referred to as the aptamer):
GGAGCUCGGUAGUGACGCGUUGUGUUUACGCGUCUGAUGAGUGGUA
CAGUCCAGGGUGAAGUUCCAAUUUUGAACACCUCCACGAAACUACCUCGAG
ACGU
1o The experiment described in figure 8 shows 1) that the aptazyme, features
self
cleavage activity which is inhibited specifically by doxycycline
(tetracycline, an isomer
of doxycycline, inhibits cleavage to a much lesser extent) and 2) that the
aptamer is able
to relieve inhibition of self cleavage by doxycycline but not inhibition of
self cleavage by
tetracycline. The interpretation is that the aptamer competes out the aptazyme
for the
binding of doxycycline. This is a demonstration that mutation of the
nucleotide C 5' to
the cleavage site into nucleotide G, converts the aptazyme into a RNA ligand
which binds
doxycycline specifically.
CA 02387185 2002-04-10
WO 01/29234 PCT/EP00/10423
0
~
"' .
o ,...,
0
M
O
O
O
.-, O
N
*
O
O
O~
O
O
00
O
i
N
d.
M
I
N
I
i
w C N O
'~ o .~ '.~ U
~ ~
ao ~ w
'~ '~
U ~ ~ N ~ '~'
b O O
N
~
z...~.
CA 02387185 2002-04-10
WO 01/29234 PCT/EP00/10423
1
LISTE DE SEQUENCES
<110> AVENTIS PHARMA S.A.
<120> SELF CLEAVING RNA SEQUENCES AND THEIR USE FOR THE
CONTROL OF PROTEIN SYNTHESIS
<130> Ribo
<140> 99402552.6
<141> 2000-10-13
<150> 99402552.6
<151> 1999-10-15
<160> 9
<170> PatentIn Ver. 2.1
<210> 1
<211> 87
<212> ADN
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence:PRIMER
<400> 1
cgcgttgtgt ttacgcgtct gatgagtnnn nnnnnnnnnn nnnnnnnnnn nnnnnnnnnn 60
nnnnnnnacg aaactacctc gagacgt 87
<210> 2
<211> 24
<212> ADN
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence:PRIMER
<400> 2
cgcgttgtgt ttacgcgtct gatg 24
<210> 3
<211> 66
<212> ADN
<213> Artificial Sequence
<220>
<223> Description of Artificial Sequence:PRIMER
<400> 3
agctggtacc taatacgact cactatagga gctcggtagt cacgcgttgt gtttacgcgt 60
ctgatg 66
<210> 4
<211> 20
<212> ADN
<213> Artificial Sequence
CA 02387185 2002-04-10
WO 01/29234 PCT/EP00/10423
2
<220>
<223> Description of Artificial Sequence: PRIMER
<400> 4
acgtctcgag gtagtttcgt 20
<210> 5
<211> 128
<212> ADN
<213> Sequence artificielle
<220>
<223> Description de la sequence artificielle:
oligonucleotide
<400> 5
agctggtacc taatacgact cactatagga gctcggtagt cacgcgttgt gtttacgcgt 60
ctgatgagtg gtacagtcca gggtgaagtt ccaattttga acacctccac gaaactacct 120
cgagacgt 128
<210> 6
<211> 101
<212> ARN
<213> Sequence artificielle
<220>
<223> Description de la sequence artificielle:
oligonucleotide
<400> 6
ggagcucggu agucacgcgu uguguuuacg cgucugauga gugguacagu ccagggugaa 60
guuccaauuu ugaacaccuc cacgaaacua ccucgagacg a 101
<210> 7
<211> 128
<212> ADN
<213> Sequence artificielle
<220>
<223> Description de la sequence
artificielle:oligonucleotide
<400> 7
agctggtacc taatacgact cactatagga gctcggtagt cacgcgttgt gtttacgcgt 60
ctgatgagtg gtacagtcca gggtgaagtt ccaattttga acacctccac gaaactacct 120
cgagacgt 128
<210> 8
<211> 66
<212> ADN
<213> Sequence artificielle
<220>
<223> Description de la sequence artificielle: PRIMER
<400> 8
CA 02387185 2002-04-10
WO 01/29234 PCT/EP00/10423
3
agctggtacc taatacgact cactatagga gctcggtagt cacgcgttgt gtttacgcgt 60
66
ctgatg
<210> 9
<211> 101
<212> ARN
<213> Sequence artificielle
<220>
<223> Description de la sequence
artificielle:oligonucleotide
<400> 9
ggagcucggu agugacgcgu uguguuuacg cgucugauga gugguacagu ccagggugaa 60
guuccaauuu ugaacaccuc cacgaaacua ccucgagacg a 101
3/3
3/3