Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
NUCLEIC ACID MOLECULES DERIVED FROM
RAT BRAIN AND PROGRAMMED CELL DEATH MODELS
FIELD OF THE INVENTION
The invention relates to nucleic acid molecules derived from rat brain and
programmed cell death expression libraries. Also provided are vectors, host
cells, and
methods for making and using the novel molecules of the invention.
BACKGROUND OF THE INVENTION
A great deal of effort has been expended by the modern scientific research
community to identify and sequence genes, particularly human genes. The
identification of genes and knowledge of their nucleic acid sequences pave the
way
for many scientific and commercial advancements, both in research applications
and
in diagnostic and therapeutic applications. For example, advances in gene
identification and sequencing allow the production of the products encoded by
these
genes, such as by recombinant and synthetic means. Furthermore, identification
of
I S genes and the products they encode provide important information about the
mechanism of disease and can provide new diagnostic tests and therapeutic
treatments
for the diagnosis and treatment of disease. Thus, identification and
sequencing of
genes provide valuable information and compositions for use in the
biotechnology
and pharmaceutical industries.
In multicellular organisms, homeostasis is maintained by balancing the rate of
cell proliferation against the rate of cell death. Cell proliferation is
influenced by
numerous growth factors and the expression of proto-oncogenes, which typically
encourage progression through the cell cycle. In contrast, numerous events,
including
the expression of tumor suppressor genes, can lead to an arrest of cellular
proliferation.
In differentiated cells, a particular type of cell death called apoptosis
occurs
when an internal suicide program is activated. This program can be initiated
by a
variety of external signals as well as signals that are generated within the
cell in
response to, for example, genetic damage. Dying cells are eliminated by
phagocytes,
without an inflammatory response.
-1-
CA 02388617 2002-05-07
WO 01/31007 PCT/USUO/29132
Programmed cell death (PCD) is a highly regulated process (Wilson (1998)
Biochem. Cell. Biol. 76:573-582). The death signal is then transduced through
various signaling pathways that converge on caspase-mediated degradative
cascades
resulting in the activation of late effectors of morphological and
physiological aspects
of apoptosis, including DNA fragmentation and cytoplasmic condensation. In
addition, regulation of programmed cell death may be integrated with
regulation of
energy, redox- and ion homeostasis in the mitochondria (reviewed by Kroemer (
1998)
Cell Death and Differentiation 5:547), and/or cell-cycle control in the
nucleus and
cytoplasm (reviewed by Choisy-Rossi and Yonish-Rouach (1998) Cell Death and
Differentiation 5:129-131; Dang (1999) Molecular and Cellular Biology 19:1-11;
and
Kasten and Giordano (1998) Cell Death and Differentiation 5:132-140). Many
mammalian genes regulating apoptosis have been identified as homologs of genes
originally identified genetically in Caenorhabditis elegans or Drosophila
melanogaster, or as human oncogenes. Other programmed cell death genes have
been
found by domain homology to known motifs, such as death domains, that mediate
protein-protein interactions within the programmed cell death pathway.
The mechanisms that mediate apoptosis include, but are not limited to, the
activation of endogenous proteases, loss of mitochondrial function, and
structural
changes such as disruption of the cytoskeleton, cell shrinkage, membrane
blebbing,
and nuclear condensation due to degradation of DNA. The various signals that
trigger
apoptosis may bring about these events by converging on a common cell death
pathway that is regulated by the expression of genes that are highly
conserved.
Caspases (cysteine proteases having specificity for aspartate at the substrate
cleavage site) are central to the apoptotic program, are. These proteases are
responsible for degradation of cellular proteins that lead to the
morphological changes
seen in cells undergoing apoptosis. One of the human caspases was previously
known
as the interleukin-113 (IL-113) converting enzyme (ICE), a cysteine protease
responsible for the processing of pro-IL-113 to the active cytokine.
Overexpression of
ICE in Rat-1 fibroblasts induces apoptosis (Miura et al. (1993) Cell 75:653).
Many caspases and proteins that interact with caspases possess domains of
about 60 amino acids called a caspase recruitment domain (CARD). Apoptotic
proteins may bind to each other via their CARDs. Different subtypes of CARDs
may
-2-
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
confer binding specificity, regulating the activity of various caspases.
(Hofmann et al.
(1997) TIBS 22:155).
The functional significance of CARDs have been demonstrated in two recent
publications. Duan et al. ( 1997) Nature 385:86 showed that deleting the CARD
at
the N-terminus of RAIDD, a newly identified protein involved in apoptosis,
abolished
the ability of RAIDD to bind to caspases. In addition, Li et al. (1997) Cell
91:479
showed that the N-terminal 97 amino acids of apoptotic protease activating
factor-1
(Apaf 1 ) was sufficient to confer caspase-9-binding ability.
Thus, programmed cell death (apoptosis) is a normal physiological activity
necessary to proper and differentiation in all vertebrates. Defects in
apoptosis
programs result in disorders including, but not limited to, neurodegenerative
disorders, cancer, immunodeficiency, heart disease and autoimmune diseases
(Thompson et al. (1995) Science 267:1456).
In vertebrate species, neuronal programmed cell death mechanisms have been
associated with a variety of developmental roles, including the removal of
neuronal
precursors which fail to establish appropriate synaptic connections (Oppenheim
et al.
(1991) Annual Rev. Neuroscience 14:453-501 ), the quantiative matching of pre-
and
post-synaptic population sizes (Herrup et al. (1987) J. Neurosci. 7:829-836),
and
sculpting of neuronal circuits, both during development and in the adult
(Bottjer et al.
(1992) J. Neurobiol. 23:1172-1191).
Inappropriate apoptosis has been suggested to be involved in neuronal loss in
various neurodegenerative diseases such as Alzheimer's disease (Loo et al. (
1993)
Proc. Natl. Acad. Sci. 90:7951-7955), Huntington's disease (Portera-Cailliau
et al.
(1995) J. Neurosc. 15:3775-3787), amyotrophic lateral sclerosis (Rabizadeh et
al.
(1995) Proc. Natl. Acad. Sci. 92:3024-3028), and spinal muscular atrophy (Roy
et al.
(1995) Cell 80:167-178).
In addition, improper expression of genes involved in apoptosis has been
implicated in carcinogenesis. Thus, it has been shown that several "oncogenes"
are in
fact involved in apoptosis, such as in the Bcl family.
Accordingly, genes involved in apoptosis are important targets for therapeutic
intervention. It is important, therefore, to identify novel genes involved in
apoptosis
or to discover whether known genes function in this process.
-3-
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
Nucleic acid probes have long been used to detect complementary nucleic acid
sequences in a nucleic acid of interest (the "target" nucleic acid). In some
assay
formats, the nucleic acid is tethered, i.e., by covalent attachment, to a
solid support.
Arrays of nucleic acid sequences immobilized on solid supports have been used
to
detect specific nucleic acid sequences in a target nucleic acid. See, e.g.,
PCT patent
publication Nos. WO 89/10977 and 89/11548. Others have proposed the use of
large
numbers of nucleic acid sequences to provide the complete nucleic acid
sequence of a
target nucleic with methods for using arrays of immobilized nucleic acid
sequences
for this purpose. See U.S. Pat. Nos. 5,202,231 and 5,002,867 and PCT patent
publication No. WO 93/17126.
The development of specific microarray technology has provided methods for
making very large arrays of nucleic acid sequences in very small physical
arrays. See
U.S. Pat. No. 5,143,854 and PCT patent publication Nos. WO 90/15070 and
92/10092, each of which is incorporated herein by reference. U.S. patent
application
No. 082,937, filed Jun. 25, 1993, describes methods for making arrays of
sequences
that can be used to provide the complete sequence of a target nucleic acid and
to
detect the presence of a nucleic acid containing a specific nucleotide
sequence. Thus,
microfabricated arrays of large numbers of nucleic acid sequences, called "DNA
chips" offer great promise for a wide variety of applications.
SUMMARY OF THE INVENTION
The present invention is based on the identification of novel nucleic acid
molecules derived from rat brain and programmed cell death cDNA libraries.
Thus, in one aspect, the invention provides an isolated nucleic acid molecule
that comprises a nucleotide sequence selected from the group consisting of the
sequences shown in SEQ ID NOS: I-6, 8, and 10 and the complements of the
sequences shown in SEQ ID NOS: 1-6, 8, and 10.
The invention also provides an isolated fragment or portion of any of the
sequences shown in SEQ ID NOS: 1-6, 8, and 10 and the complement of the
sequences shown in SEQ ID NOS: 1-6, 8, and 10. In some embodiments, the
fragment is useful as a probe or primer, and/or is at least 15, at least 18,
or at least 20,
22,25, 30, 35, 50, 100, 200 or more nucleotides in length.
-4-
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
In another embodiment, the invention provides an isolated nucleic acid
molecule that comprises a nucleotide sequence that is at least about 60%
identical,
about 65% identical, about 70% identical, about 80% identical, about 90%
identical,
about 95% identical, about 96% identical, about 97% identical, about 98%
identical,
or about 99% or more identical to a nucleotide sequence selected from the
group
consisting of the sequences shown in SEQ ID NOS: 1-6, 8, and 10, and the
complements of the sequences shown in SEQ ID NOS: 1-6, 8, and 10.
In another embodiment, the invention provides an isolated nucleic acid
molecule that hybridizes under highly stringent conditions to a nucleotide
sequence
selected from the group consisting of the sequences shown in SEQ ID NOS: I-6,
8,
and 10, and the complements of the sequences shown in SEQ ID NOS: 1-6, 8, and
10.
The invention further provides nucleic acid vectors comprising the nucleic
acid molecules described above. In one embodiment, the nucleic acid molecules
of
the invention are operatively linked to at least one expression control
element.
The invention further includes host cells, such as bacterial cells, fungal
cells,
plant cells, insect cells and mammalian cells, comprising the nucleic acid
vectors
described above.
In another aspect, the invention provides isolated gene products, proteins and
polypeptides encoded by nucleic acid molecules of the invention.
The invention further provides antibodies, including monoclonal antibodies, or
antigen-binding fragments thereof, which selectively bind to the isolated
proteins and
polypeptides of the invention.
The invention also provides methods for preparing proteins and polypeptides
encoded by isolated nucleic acid molecules described herein by culturing a
host cell
containing a vector molecule of the invention.
Additionally, the invention provides a method for assaying for the presence of
a nucleic acid sequence, protein or polypeptide of the present invention, in a
biological sample, e.g., in a tissue sample, by contacting said sample with an
agent
(e.g., an antibody or a nucleic acid molecule) suitable for specific detection
of the
nucleic acid sequence, protein or polypeptide.
The invention also provides a kit comprising a nucleic acid probe which
hybridizes to a nucleotide sequence of claim 1 and instructions for use, and a
kit
comprising an agent which binds to a polypeptide of claim I 0 and instructions
for use.
-5-
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
BRIEF DESCRIPTION OF THE FIGURES
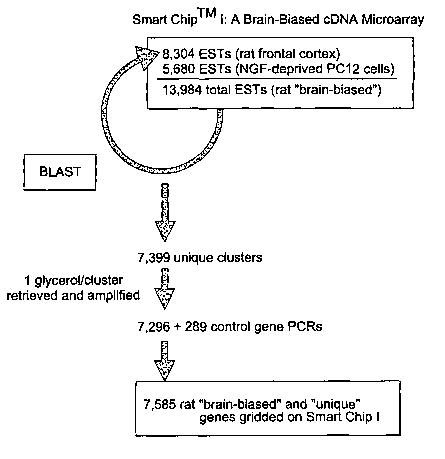
Figure 1 illustrates the construction of the "Smart ChipTM I". cDNAs were
cloned from rat frontal cortex and from differentiated PC 12 cells deprived of
nerve
growth factor, a model of programmed cell death as described in detail in the
experimental section. PC 12 cells are an adrenal gland cell line from rat that
provides
a pre-neuron set that can be differentiated in vitro. The application of nerve
growth
factor induces the formation of axons and dendritic structures. This serves as
a model
for neuronal differentiation. When the nerve growth factor is withdrawn, the
cells
undergo programmed cell death (apoptosis). Approximately 300 control nucleic
acid
sequences (of known function) were added as an internal control and for
transcriptional profiling of the cloned cDNA sequences. These sequences were
then
subjected to BLASTX analysis to determine the correspondence between the cDNA
and a known cDNA and to determine to which protein family, if any, the
proteins
encoded by each cDNA belong. Computer analysis was used to assemble the cDNA
sequences into unique clusters. The majority of the clusters as well as
control genes
were gridded on Smart ChipTM I.
Figure 2 shows the coefficient of variation (standard deviation/mean for
triplicate hybridizations) after normalization for each array element plotted
against the
mean intensity for the gene (gene expression intensity). The figure shows the
moving
average (with a window of 200) for three different mRNA probes, 3 hour KC1-
withdrawn, 3 hour control, and 6 hour control (See the examples and figures 3
and 4).
As is typical for all probes, past a threshold of 30 to 40, the coefficient of
variation
averages below 0.2. The inset compares one triplicate hybridization (Filter Y)
to
another (Filter Z). Each point represents a different gene graphed on log-log
axis
comparing the intensity measured on one filter versus the other.
Figure 3 shows temporal expression clusters observed following KC1 and
serum withdrawal. A hierarchical clustering algorithm was used to cluster
genes
based on expression patterns across 10 time points (from left to right), 1, 3,
6, 12, and
24 hours post-KCl/serum-replacement (sham), and 1, 3, 6, 12, and 24 hours post-
KCl/serum-withdrawal (treatment) (See Examples). Expression values for each
gene
were scaled based on the number of standard deviations from the mean intensity
of
each gene across all 10 time points. Scaled expression values are color-coded
such
-6-
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
that red, yellow, and blue indicated above, at, and below mean intensity,
respectively.
The correlation between expression patterns of neighboring genes is depicted
by the
dendrogram on the right. Genes regulated by programmed cell death (KC 1 /serum-
withdrawal alone) are enlarged in B. Representative non-scaled gene expression
bar
graphs with standard deviation error bars are aligned next to the four major
clusters
for Late Effector, Middle, Early, and Immediate Early gene expression classes.
Regulated genes within each temporal expression class are listed in order of
hierarchical clustering in SEQ ID NOS: 1-6, 8, and 106.
Figure 4 shows expression clusters for all of the CGN programmed cell death
models (KCl and serum withdrawal, KCl withdrawal alone, and kainate
treatment).
Figure 4A shows a self organizing map (SOM) algorithm (See, e.g. Kohonen, Self
Organizing Maps: Springer, Berlin ( 1997)) that was used to cluster genes
based on
expression in 26 experiments (in order: serum added back, I, 3, 6, 12, 24
hours;
KCl/serum withdrawal, 1, 3, 6, 12, 24 hours; controls for KCl withdrawal, 1,
3, 6, 12
hours; KCI withdrawal alone, 1, 3, 6, 12 hours; controls for kainate
treatment, 2, 4, 8,
12 hours; kainate treatment, 2, 4, 8, I2 hours; see examples for experimental
details).
As shown, a S x 4 geometry was used to organize the genes into 20 groups. A
cluster
(3,3) of 17 programmed cell death-induced genes is highlighted. The inset
shows a
tiled depiction of all the genes in the (3,3) cluster; red = above mean
expression, white
= mean expression, blue = below mean expression; the tiles are ordered in
columns as
indicated above for experimental order; each row represents a different array
element
gene in the order indicated by distance from the cluster centroid. Caspase 3,
a gene
involved in apoptosis, is part of the array and depicted in the raw values
graph (i.e.
relative expression in the 26 experiments); each experiment is represented in
order on
the x-axis; the y-axis indicates gene expression intensity.
Figure 4 B, C, D, and E show the raw gene expression intensity plotted for a
representative gene from programmed cell death-regulated, regulated by KC1
withdrawal only, immediate early genes, and serum-repressed constitutive
expression
classes, respectively. Each panel shows the data for a representative member
of the
cluster (indicated in the gene list by *), along with a list of genes included
in the
expression cluster.
_7_
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
Figure 4B shows the raw gene expression intensity for a gene representative
from the list on the right. The graph shows increased expression with KCl and
serum
withdrawal, and kainate treatment. Accordingly, genes with these
characteristics are
designated "programmed cell death regulated." The list of genes with this
pattern (on
the chip) is shown on the right. Known genes include genes regulated in
apoptosis.
Figure 4C lists genes which show increased expression after withdrawal of
KCL or KCL and serum, but following kainate treatment. The list includes genes
known to be involved in apoptosis.
Figure 4D shows genes that demonstrate constitutive immediate early
expression.
Figure 4E shows genes that demonstrate constitutive expression in the absence
of serum. The list on the right shows that this class contains mediators of
programmed cell death.
Figure 5 shows information relating to various NARC genes. Accordingly the
first column gives the NARC (neuronal apoptosis regulated candidate)
designation.
The second column provides specific information, such as the number of
nucleotides
sequenced, the region sequenced, for example, the 3' untranslated region,
information
regarding open reading frames, information regarding human orthologs (whose
sequences may also be found in SEQ ID NOS: 1-6, 8, and 10), information
regarding
homology to known amino acid or nucleotide sequences, information regarding
function, and other information related to specific physical or functional
characteristics. The third column shows the gene expression class as described
and
designated in Figure 4. The fourth column shows the results of Northern blot
hybridization, for example whether expression is restricted to specific organs
or
ubiquitous, and transcript size.
Figure 6 shows a tabulation of expression data of genes known to be related to
programmed cell death, the data being obtained from experiments disclosed
herein
wherein nucleic acid sequences on the microarray were hybridized to mRNA
derived
from the two programmed cell death models {see examples). The first column
indicates the clone designation. Where the clone is a previously known gene
(for
example, c-fos and c-jun), the gene name is given rather than the cDNA clone
designation. The second column indicates the gene designation for each clone
based
on a BLASTX search. The third column indicates the expression pattern for each
of
_g_
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
the clones. This tabulation can serve as an internal control to assess the
fidelity of the
experimental conditions and thus can serve as a background to compare the
expression pattern of uncharacterized clones in the array. Accordingly, this
figure
shows a subarray that can serve as an internal control for discovering genes
related to
apoptosis and cell proliferation.
Figure 7 shows all genes (i.e., that are represented by nucleic acid sequences
on the chip) that are regulated in specific experimental conditions described
in the
examples and shown in Figure 4. Specific genes are clustered (in an underlined
category). Each cluster represents clones having a specific expression
characteristic.
For example, the first cluster is transiently down-regulated by serum and down-
regulated by KCl withdrawal. The second column identifies cDNA clones whose
function is previously known. The third column indicates the cluster number.
See
Figure 4A. In addition, an analysis of the functions of the genes in each
cluster
showed that within a cluster, certain functional classes of genes may be over-
represented. Thus, the material in parentheses indicates the biological
functions that
are associated with a disproportionate number of genes in the cluster. This
includes
secretion and synaptic vesicle release (cluster 0,0), cell proliferation
(cluster 0,3),
secretion/synaptic vesicle release/cytoskeletal reorganization (cluster 1,0),
stress
response/hormone response (cluster 1,3), stress response/hormone response
(cluster
1,4), calcium signal transduction (cluster 2,0), and cytoskeleton/synapse
cytoskeleton
(cluster 2,4).
Figure 8 summarizes tissue expression data for the Smart Chip IT"" microarray
elements. The data were obtained by membrane blotting of the microarray
against
mRNA from testes, brain, heart, smooth muscle, spleen, kidney, skeletal
muscle, lung,
liver, and pancreatic tissue. Following hybridization with labeled cDNA
synthesized
from RNA from the indicated tissue type, the signal from each sequence on the
array
filters was quantitated by phosphorimaging.
Figure 9 provides a list of genes that were shown to be regulated by KCl and
serum withdrawal in the microarray experiments described herein.
-9-
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
DETAILED DESCRIPTION OF THE INVENTION
Isolated Nucleic Acid Molecules
The invention encompasses the discovery and isolation of nucleic acid
molecules that are expressed in rat brain and in programmed cell death in
vitro models
(neuronal apoptosis regulated candidates or NARCs) and their human homologs.
The
sequences of these human homologs are specifically disclosed in SEQ ID NOS:1
(human NARC 9B), 2 (human NARC 8B), 3 (human NARC 2A), 4 (human NARC
16B), 5 (human NARC 1 OC), 6 (human NARC 1 C), 8 (human NARC 1 A), and 10
(human NARC 25).
As appropriate, the isolated nucleic acid molecules of the present invention
can be RNA, for example, mRNA, or DNA, such as cDNA and genomic DNA. DNA
molecules can be double-stranded or single-stranded; single stranded RNA or
DNA
can be either the coding, or sense, strand or the non-coding, or antisense,
strand. The
nucleic acid molecule can include all or a portion of the coding sequence of
the genes
of the invention. Additionally, the nucleic acid molecule can be fused to a
marker
sequence, for example, a sequence that encodes a polypeptide to assist in
isolation or
purification of the polypeptide. Such sequences include, but are not limited
to, those
which encode a glutathione-S-transferase (GST) fusion protein and those which
encode a hemaglutin A (HA) polypeptide marker from influenza.
An "isolated" nucleic acid molecule, as used herein, is one that is separated
from nucleic acid which normally flanks the nucleic acid molecule in nature.
With
regard to genomic DNA, the term "isolated" refers to nucleic acid molecules
which
are separated from the chromosome with which the genomic DNA is naturally
associated. For example, the isolated nucleic acid molecule can contain less
than
about 5 kb, 4 kb, 3 kb, 2 kb, 1 kb, 0.5 kb or 0.1 kb of nucleotides which
flank the
nucleic acid molecule in the genomic DNA of the cell from which the nucleic
acid is
derived.
Moreover, an isolated nucleic acid of the invention, such as a cDNA or RNA
molecule, can be substantially free of other cellular material, or culture
medium when
produced by recombinant techniques, or chemical precursors or other chemicals
when
chemically synthesized. However, the nucleic acid molecule can be fused to
other
-10-
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/21132
coding or regulatory sequences and still be considered isolated. In some
instances,
the isolated material will form part of a composition (for example, a crude
extract
containing other substances), buffer system or reagent mix. In other
circumstances,
the material may be purified to essential homogeneity, for example as
determined by
PAGE or column chromatography such as HPLC. Preferably, an isolated nucleic
acid
comprises at least about 50, 80 or 90% (on a molar basis) of all
macromolecular
species present.
Further, recombinant DNA contained in a vector is included in the definition
of "isolated" as used herein. Also, isolated nucleic acid molecules include
recombinant DNA molecules in heterologous host cells, as well as partially or
substantially purified DNA molecules in solution. "Isolated" nucleic acid
molecules
also encompass in vivo and ~n vitro RNA transcripts of the DNA molecules of
the
present invention.
The invention further provides variants of the isolated nucleic acid molecules
of the invention. Such variants can be naturally occurring, such as allelic
variants
(same locus), homologs (different locus), and orthologs (different organism),
or may
be constructed by recombinant DNA methods or by chemical synthesis. Such
non-naturally occurring variants can be made using well-known mutagenesis
techniques, including those applied to polynucleotides, cells, or organisms.
Accordingly, variants can contain nucleotide substitutions, deletions,
inversions
and/or insertions in either or both the coding and non-coding region of the
nucleic
acid molecule. Further, the variations can produce both conservative and
non-conservative amino acid substitutions.
Typically, variants have a substantial identity with a nucleic acid molecule
selected from the group consisting of the sequences shown in SEQ ID NOS:1-6,
8,
and 10 and the complements thereof. Particularly preferred are nucleic acid
molecules and fragments which have at least about 60%, at least about 70%, at
least
about 80%, at least about 85%, at least about 90%, at least about 95%, at
least about
96%, at least about 97%, at least about 98%, or at least about 99% or more
identity
with nucleic acid molecules described herein.
Such nucleic acid molecules can be readily identified as being able to
hybridize under stringent conditions to a nucleotide sequence selected from
the group
consisting of the sequences shown in SEQ ID NOS:l-6, 8, and 10 and the
-11-
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
complements thereof. In one embodiment, the variants hybridize under high
stringency hybridization conditions (e.g., for selective hybridization) to a
nucleotide
sequence selected from the sequences shown in SEQ ID NOS:1-6, 8, and 10.
As used herein, the term "hybridizes under stringent conditions" describes
conditions for hybridization and washing. Stringent conditions are known to
those
skilled in the art and can be found in Current Protocols in Molecular Biology,
John
Wiley & Sons, N.Y. (1989), 6.3.1-6.3.6. Aqueous and nonaqueous methods are
described in that reference and either can be used. A preferred, example of
stringent
hybridization conditions are hybridization in 6X sodium chloride/sodium
citrate
(SSC) at about 45°C, followed by one or more washes in 0.2X SSC, 0.1%
SDS at
50°C. Another example of stringent hybridization conditions are
hybridization in 6X
sodium chloride/sodium citrate (SSC) at about 45°C, followed by one or
more washes
in 0.2X SSC, 0.1% SDS at 55°C. A further example of stringent
hybridization
conditions is hybridization in 6X sodium chloride/sodium citrate (SSC) at
about 45°C,
followed by one or more washes in 0.2X SSC, 0.1 % SDS at 60°C.
Preferably,
stringent hybridization conditions are hybridization in 6X sodium
chloride/sodium
citrate (SSC) at about 45°C, followed by one or more washes in 0.2X
SSC, 0.1% SDS
at 65°C. Particularly preferred stringency conditions (and the
conditions that should
be used if the practitioner is uncertain about what conditions should be
applied to
determine if a molecule is within a hybridization limitation of the invention)
are O.SM
Sodium Phosphate, 7% SDS at 65°C, followed by one or more washes at
0.2X SSC,
1 % SDS at 65°C. The hybridization step may be performed for 4, 8, 12,
or 16 hours,
and the wash steps are generally 15 minutes or 30 minutes in length.
The percent identity of two nucleotide or amino acid sequences can be
determined by aligning the sequences for optimal comparison purposes (e.g.,
gaps can
be introduced in the sequence of a first sequence). The nucleotides or amino
acids at
corresponding positions are then compared, and the percent identity between
the two
sequences is a function of the number of identical positions shared by the
sequences.
In certain embodiments, the length of a sequence aligned for comparison
purposes is
at least 30%, preferably at least 40%, more preferably at least 60%, and even
more
preferably at least 70%, 80% or 90% of the length of the reference sequence.
The
actual comparison of the two sequences can be accomplished by well-known
methods, for example, using a mathematical algorithm. A preferred, non-
limiting
-12-
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
example of such a mathematical algorithm is described in Karlin et al. (1993)
Proc.
Natl. Acad. Sci. USA, 90:5873-5877. Such an algorithm is incorporated into the
NBLAST and XBLAST programs (version 2.0) as described in Altschul et al. (
1997)
Nucleic Acids Res., 25:389-3402. When utilizing BLAST and Gapped BLAST
programs, the default parameters of the respective programs (e.g., NBLAST) can
be
used. See http://www.ncbi.nlm.nih.gov. In one embodiment, parameters for
sequence comparison can be set at score=100, wordlength=12, or can be varied
(e.g.,
W=5 or W=20).
Another preferred, non-limiting example of a mathematical algorithm utilized
for the comparison of sequences is the algorithm of Myers and Miller, CABIOS
(1989). Such an algorithm is incorporated into the ALIGN program (version 2.0)
which is part of the CGC sequence alignment software package. When utilizing
the
ALIGN program for comparing amino acid sequences, a PAM120 weight residue
table, a gap length penalty of 12, and a gap penalty of 4 can be used.
Additional
algorithms for sequence analysis are known in the art and include ADVANCE and
ADAM as described in Torellis and Robotti (1994) Comput. Appl. Biosci. 10:3-5;
and
FASTA described in Pearson and Lipman (1988) PNAS, 85:2444-8.
In another embodiment, the percent identity between two amino acid
sequences can be accomplished using the GAP program in the CGC software
package
(available at http://www.cgc.com) using either a BLOSUM 63 matrix or a PAM250
matrix, and a gap weight of 12, 10, 8, 6, or 4 and a length weight of 2, 3, or
4. In yet
another embodiment, the percent identity between two nucleic acid sequences
can be
accomplished using the GAP program in the CGC software package (available at
http://www.cgc.com), using a gap weight of 50 and a length weight of 3.
The present invention also provides isolated nucleic acids that contain a
fragment or portion that hybridizes under highly stringent conditions to a
nucleotide
sequence selected from the group consisting of the sequences shown in SEQ ID
NOS:1-6, 8, and 10 and the complements of the sequences shown in SEQ ID NOS:1-
6, 8, and 10. In one embodiment, the nucleic acid consists of a fragment of a
nucleotide sequence selected from the group consisting of the sequences shown
in
SEQ ID NOS: 1-6, 8, and l0and the complements of the sequences shown in SEQ ID
NOS:1-6, 8, and 10. The nucleic acid fragments of the invention are at least
about 15,
preferably at least about 18, 20, 23 or 25 nucleotides, and can be 30, 40, 50,
100, 200
-13-
CA 02388617 2002-05-07
WO 01/31007 PCT/US110/29132
or more nucleotides in length. Longer fragments, for example, 30 or more
nucleotides
in length, which encode antigenic proteins or polypeptides described herein
are
useful. Additionally, nucleotide sequences described herein can also be
contigged
(e.g., overlapped or joined) to produce longer sequences (see, for example,
http://bozeman.mbt.washington.edu/phrap.docs/phrap.html).
In a related aspect, the nucleic acid fragments of the invention are used as
probes or primers in assays such as those described herein. "Probes" are
oligonucleotides that hybridize in a base-specific manner to a complementary
strand
of nucleic acid. Such probes include polypeptide nucleic acids, as described
in
Nielsen et al. ( 1991 ) Science, 254, 1497-1500. Typically, a probe comprises
a region
of nucleotide sequence that hybridizes under highly stringent conditions to at
least
about 15, typically about 20-25, and more typically about 40, 50 or 75
consecutive
nucleotides of a nucleic acid selected from the group consisting of the
sequences
shown in SEQ ID NOS:1-6, 8, and 10 and the complements thereof. More
typically,
the probe further comprises a label, e.g., radioisotope, fluorescent compound,
enzyme,
or enzyme co-factor.
As used herein, the term "primer" refers to a single-stranded oligonucleotide
which acts as a point of initiation of template-directed DNA synthesis using
well-known methods (e.g., PCR, LCR) including, but not limited to those
described
herein. The appropriate length of the primer depends on the particular use,
but
typically ranges from about 15 to 30 nucleotides. The term "primer site"
refers to the
area of the target DNA to which a primer hybridizes. The term "primer pair"
refers to
a set of primers including a 5' (upstream) primer that hybridizes with the 5'
end of the
nucleic acid sequence to be amplified and a 3' (downstream) primer that
hybridizes
with the complement of the sequence to be amplified.
The nucleic acid molecules of the invention such as those described above can
be identified and isolated using standard molecular biology techniques and the
sequence information provided in the sequences shown in SEQ ID NOS: l-6, 8,
and
10. For example, nucleic acid molecules can be amplified and isolated by the
polymerase chain reaction using synthetic oligonucleotide primers designed
based on
one or more of the sequences provided in the sequences shown in SEQ ID NOS: 1-
6,
8, and l0and the complements thereof. See generally PCR Technology: Principles
and Applications for DNA Amplification (ed. H.A. Erlich, Freeman Press, NY,
NY,
-14-
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
1992); PCR Protocols: A Guide to Methods and Applications (Eds. Innis, et al.
Academic Press, San Diego, CA, 1990); Manila et al. (1991) Nucleic Acids Res.
19:4967; Eckert et al. ( 1991 ) PCR Methods and Applications, 1:17; PCR (eds.
MePherson et al. IRL Press, Oxford); and U.S. Patent 4,683,202. The nucleic
acid
molecules can be amplified using cDNA, mRNA or genomic DNA as a template,
cloned into an appropriate vector and characterized by DNA sequence analysis.
Other suitable amplification methods include the ligase chain reaction (LCR)
{see Wu and Wallace (I989) Genomics, 4:560, Landegren et al. (1988) Science,
241:1077, transcription amplification (Kwoh et al. ( 1989) Proc. Natl. Acad.
Sci. USA,
86:1173), and self sustained sequence replication (Guatelli et al. (1990)
Proc. Nat.
Acad. Sci_ USA, 87:1874) and nucleic acid based sequence amplification
(NASBA).
The latter two amplification methods involve isothermal reactions based on
isothermal transcription, which produce both single stranded RNA (ssRNA) and
double stranded DNA (dsDNA) as the amplification products in a ratio of about
30 or
100 to 1, respectively.
The amplified DNA can be radiolabelled and used as a probe for screening a
eDNA library, mRNA in zap express, ZIPLOX or other suitable vector.
Corresponding clones can be isolated, DNA can obtained following in vivo
excision,
and the cloned insert can be sequenced in either or both orientations by art
recognized
methods to identify the correct reading frame encoding a protein of the
appropriate
molecular weight. For example, the direct analysis of the nucleotide sequence
of
nucleic acid molecules of the present invention can be accomplished using
well-known methods that are commercially available. See, for example, Sambrook
et
al. Molecular Cloning, A Laboratory Manual (2nd Ed., CSHP, New York 1989);
Zyskind et al. Recombinant DNA Laboratory Manual, (Acad. Press, 1988)). Using
these or similar methods, the proteins) and the DNA encoding the protein can
be
isolated, sequenced and further characterized.
Antisense nucleic acids of the invention can be designed using the nucleotide
sequences of the sequences shown in SEQ ID NOS: 1-6, 8, and 10, and
constructed
using chemical synthesis and enzymatic ligation reactions using procedures
known in
the art. For example, an antisense nucleic acid (e.g., an antisense
oligonucleotide) can
be chemically synthesized using naturally occurring nucleotides or variously
modified
nucleotides designed to increase the biological stability of the molecules or
to increase
-15-
CA 02388617 2002-05-07
WO 01/31007 PCT/1IS00/29132
the physical stability of the duplex formed between the antisense and sense
nucleic
acids, e.g., phosphorothioate derivatives and acridine substituted nucleotides
can be
used. Examples of modified nucleotides which can be used to generate the
antisense
nucleic acid include 5-fluorouracil, 5-bromouracil, 5-chlorouracil, 5-
iodouracil,
hypoxanthine, xanthine, 4-acetylcytosine, 5-(carboxyhydroxylmethyl) uracil,
5-carboxymethylaminomethyl-2-thiouridine, 5-carboxymethylaminomethyluracil,
dihydrouracil, beta-D-galactosylqueosine, inosine, N6-isopentenyladenine,
1-methylguanine, I-methylinosine, 2,2-dimethylguanine, 2-methyladenine,
2-methylguanine, 3-methylcytosine, 5-methylcytosine, N6-adenine, 7-
methylguanine,
5-methylaminomethyluracil, 5-methoxyaminomethyl-2-thiouracil,
beta-D-mannosylqueosine, 5'-methoxycarboxymethyluracil, 5-methoxyuracil,
2-methylthio-N6-isopentenyladenine, uracil-5-oxyacetic acid (v), wybutoxosine,
pseudouracil, queosine, 2-thiocytosine, 5-methyl-2-thiouracil, 2-thiouracil,
4-thiouracil, 5-rnethyluracil, uracil-5-oxyacetic acid methylester, uracil-5-
oxyacetic
acid (v), 5-methyl-2-thiouracil, 3-(3-amino-3-N-2-carboxypropyl) uracil,
(acp3)w,
and 2,6-diaminopurine. Alternatively, the antisense nucleic acid can be
produced
biologically using an expression vector into which a nucleic acid has been
subcloned
in an antisense orientation (i.e., RNA transcribed from the inserted nucleic
acid will
be of an antisense orientation to a target nucleic acid of interest).
Additionally, the nucleic acid molecules of the invention can be modified at
the base moiety, sugar moiety or phosphate backbone to improve, e.g., the
stability,
hybridization, or solubility of the molecule. For example, the deoxyribose
phosphate
backbone of the nucleic acids can be modified to generate peptide nucleic
acids (see
Hyrup et al. (1996) Bioorganic & Medicinal Chemistry, 4:5). As used herein,
the
terms "peptide nucleic acids" or "PNAs" refer to nucleic acid mimics, e.g.,
DNA
mimics, in which the deoxyribose phosphate backbone is replaced by a
pseudopeptide
backbone and only the four natural nucleobases are retained. The neutral
backbone of
PNAs has been shown to allow for specific hybridization to DNA and RNA under
conditions of low ionic strength. The synthesis of PNA oligomers can be
performed
using standard solid phase peptide synthesis protocols as described in Hyrup
et al.
( 1996), supra; Perry-O'Keefe et al. ( 1996) Proc. Natl. Acad. Sci. USA,
93:14670.
PNAs can be further modified, e.g., to enhance their stability, specificity or
cellular
uptake, by attaching lipophilic or other helper groups to PNA, by the
formation of
-16-
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
PNA-DNA chimeras, or by the use of liposomes or other techniques of drug
delivery
known in the art. The synthesis of PNA-DNA chimeras can be performed as
described in Hyrup (1996), supra, Finn et al. (1996) Nucleic Acids Res.
29(17):3357-63, Mag et al. (1989) Nucleic Acids Res. 17:5973, and Peterser et
al.
(1975) Bioorganic Med. Chem. Lett. 5:1119.
The nucleic acid molecules and fragments of the invention can also include
other appended groups such as peptides (e.g., for targeting host cell
receptors in vivo),
or agents facilitating transport across the cell membrane (see, e.g.,
Letsinger et al.
(1989) Proc. Natl. Acad. Sci. USA, 86:6553-6556; Lemaitre et al. (1987) Proc.
Natl.
Acad. Sci. USA, 84:648-652; PCT Publication No. W088/0918) or the blood brain
barrier (see, e.g., PCT Publication No. W089/10134). In addition,
oligonucleotides
can be modified with hybridization-triggered cleavage agents (see, e.g., Krol
et al.
(1988) Bio-Techniques, 6:958-976) or intercalating agents (see, e.g., Zon
(1988)
Pharm Res. 5:539-549).
Uses of the nucleic acids of the invention are described in detail in below.
In
general, the isolated nucleic acid sequences can be used as molecular weight
markers
on Southern gels, and as chromosome markers which are labeled to map related
gene
positions. The nucleic acid sequences can also be used to compare with
endogenous
DNA sequences in patients to identify genetic disorders, and as probes, such
as to
hybridize and discover related DNA sequences or to subtract out known
sequences
from a sample. The nucleic acid sequences can further be used to derive
primers for
genetic fingerprinting, to raise anti-protein antibodies using DNA
immunization
techniques, and as an antigen to raise anti-DNA antibodies or elicit immune
responses. Additionally, the nucleotide sequences of the invention can be used
identify and express recombinant proteins for analysis, characterization or
therapeutic
use, or as markers for tissues in which the corresponding protein is
expressed, either
constitutively, during tissue differentiation, or in disease states.
II. Vectors and Host Cells
Another aspect of the invention pertains to nucleic acid vectors containing a
nucleic acid selected from the group consisting of the sequences shown in SEQ
ID
NOS: 1-6, 8, and 10. These vectors comprise a sequence of the invention that
has
been inserted in a sense or antisense orientation. As used herein, the term
"vector"
-17-
CA 02388617 2002-05-07
WO (11/31007 PCT/USOU/29132
refers to a nucleic acid molecule capable of transporting another nucleic acid
to which
it has been linked. One type of vector is a "plasmid", which refers to a
circular double
stranded DNA loop into which additional DNA segments can be ligated. Another
type of vector is a viral vector, wherein additional DNA segments can be
ligated into
the viral genome. Certain vectors are capable of autonomous replication in a
host cell
into which they are introduced (e.g., bacterial vectors having a bacterial
origin of
replication and episomal mammalian vectors). Other vectors (e.g., non-episomal
mammalian vectors) are integrated into the genome of a host cell upon
introduction
into the host cell, and thereby are replicated along with the host genome.
Moreover,
certain vectors, expression vectors, are capable of directing the expression
of genes to
which they are operably linked. In general, expression vectors of utility in
recombinant DNA techniques are often in the form of plasmids (vectors).
However,
the invention is intended to include such other forms of expression vectors,
such as
viral vectors {e.g., replication defective retroviruses, adenoviruses and
adeno-associated viruses) that serve equivalent functions.
Preferred recombinant expression vectors of the invention comprise a nucleic
acid of the invention in a form suitable for expression of the nucleic acid in
a host
cell. This means that the recombinant expression vectors include one or more
regulatory sequences, selected on the basis of the host cells to be used for
expression,
which is operably linked to the nucleic acid sequence to be expressed. Within
a
recombinant expression vector, "operably linked" is intended to mean that the
nucleotide sequence of interest is linked to the regulatory sequences) in a
manner
which allows for expression of the nucleotide sequence (e.g., in an in vitro
transcription/translation system or in a host cell when the vector is
introduced into the
host cell). The term "regulatory sequence" is intended to include promoters,
enhancers and other expression control elements {e.g., polyadenylation
signals). Such
regulatory sequences are described, for example, in Goeddel, Gene Expression
Technology: Methods in Enzymology 185, Academic Press, San Diego, CA (1990).
Regulatory sequences include those which direct constitutive expression of a
nucleotide sequence in many types of host cell and those which direct
expression of
the nucleotide sequence only in certain host cells (e. g., tissue-specific
regulatory
sequences). It will be appreciated by those skilled in the art that the design
of the
expression vector can depend on such factors as the choice of the host cell to
be
-18-
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
transformed, the level of expression of protein desired, etc. The expression
vectors of
the invention can be introduced into host cells to thereby produce proteins or
peptides,
including fusion proteins or peptides, encoded by nucleic acids as described
herein .
The recombinant expression vectors of the invention can be designed for
expression of a polypeptide of the invention in prokaryotic or eukaryotic
cells, e.g.,
bacterial cells such as E. coli, insect cells (using baculovirus expression
vectors),
yeast cells or mammalian cells. Suitable host cells are discussed further in
Goeddel,
supra. Alternatively, the recombinant expression vector can be transcribed and
translated in vitro, for example using T7 promoter regulatory sequences and T7
polymerase.
Expression of proteins in prokaryotes is most often carried out in E. coli
with
vectors containing constitutive or inducible promoters directing the
expression of
either fusion or non-fusion proteins. Fusion vectors add a number of amino
acids to a
protein encoded therein, usually to the amino terminus of the recombinant
protein.
Such fusion vectors typically serve three purposes: 1) to increase expression
of
recombinant protein; 2) to increase the solubility of the recombinant protein;
and 3) to
aid in the purification of the recombinant protein by acting as a ligand in
affinity
purification. Often, in fusion expression vectors, a proteolytic cleavage site
is
introduced at the junction of the fusion moiety and the recombinant protein to
enable
separation of the recombinant protein from the fusion moiety subsequent to
purification of the fusion protein. Such enzymes, and their cognate
recognition
sequences, include Factor Xa, thrombin and enterokinase. Typical fusion
expression
vectors include pGEX (Pharmacia Biotech Inc; Smith and Johnson (1988) Gene,
67:31-40), pMAL (New England Biolabs, Beverly, MA) and pRITS (Pharmacia,
Piscataway, NJ) which fuse glutathione S-transferase (GST), maltose E binding
protein, or protein A, respectively, to the target recombinant protein.
Examples of suitable inducible non-fusion E. coli expression vectors include
pTrc (Amann et al. (1988) Gene, 69:301-315) and pET 1 1d (Studier et al. Gene
Expression Technology: Methods in Enzymology, 185, Academic Press, San Diego,
California (1990) 60-89). Target gene expression from the pTrc vector relies
on host
RNA polymerase transcription from a hybrid trp-lac fusion promoter. Target
gene
expression from the pET 1 1d vector relies on transcription from a T7 gnl0-lac
fusion
promoter mediated by a coexpressed viral RNA polymerase (T7 gnl). This viral
-19-
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
polymerase is supplied by host strains BL21 (DE3) or HMS 174(DE3) from a
resident
prophage harboring a T7 gnl gene under the transcriptional control of the
lacUV 5
promoter.
One strategy to maximize recombinant protein expression in E. coli is to
express the protein in a host bacteria with an impaired capacity to
proteolytically
cleave the recombinant protein (Gottesman, Gene Expression Technology: Methods
in
Enzymology, 185, Academic Press, San Diego, California (1990) 119-128).
Another
strategy is to alter the nucleic acid sequence of the nucleic acid to be
inserted into an
expression vector so that the individual codons for each amino acid are those
preferentially utilized in E. coli (Wada et al. (1992) Nucleic Acids Res.
20:2111-2118). Such alteration of nucleic acid sequences of the invention can
be
carried out by standard DNA synthesis techniques.
In another embodiment, the expression vector is a yeast expression vector.
Examples of vectors for expression in yeast S. cerivisae include pYepSecl
(Baldari
et al. (1987) EMBD J. 6:229-234), pMFa (Kurjan and Herskowitz (1982) Cell
30:933-943), pJRY88 (Schultz et al. (1987) Gene, 54:113-123), pYES2
(Invitrogen
Corporation, San Diego, CA), and pPicZ (InVitrogen Corp, San Diego, CA).
Alternatively, a nucleic acid of the invention can be expressed in insect
cells
using baculovirus expression vectors. Baculovirus vectors available for
expression of
proteins in cultured insect cells (e.g., Sf 9 cells) include the pAc series
(Smith et al.
(1983) Mol. Cell Biol. 3:2156-2165) and the pVL series (Lucklow and Summers
(1989) Virology, 170:31-39).
In yet another embodiment, a nucleic acid of the invention is expressed in
mammalian cells using a mammalian expression vector. Examples of mammalian
expression vectors include pCDM8 (Seed (i987) Nature, 329:840) and pMT2PC
(Kaufman et al. ( 1987) EMBO J. 6:187-195). When used in mammalian cells, the
expression vector's control functions are often provided by viral regulatory
elements.
For example, commonly used promoters are derived from polyoma, Adenovirus 2,
cytomegalovirus and Simian Virus 40. For other suitable expression systems for
both
prokaryotic and eukaryotic cells see chapters 16 and 17 of Sambrook et al.
supra.
In another embodiment, the recombinant mammalian expression vector is
capable of directing expression of the nucleic acid preferentially in a
particular cell
type (e.g., tissue-specific regulatory elements are used to express the
nucleic acid).
-20-
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
Tissue-specific regulatory elements are known in the art. Non-limiting
examples of
suitable tissue-specific promoters include the albumin promoter (liver-
specific;
Pinkert et al. (1987) Genes Dev. 1:268-277), lymphoid-specific promoters
(Calame
and Eaton (1988) Adv. Immunol. 43:235-275), in particular promoters of T cell
receptors (Winoto and Baltimore (1989) EMBO J. 8:729-733) and immunoglobulins
(Banerji et al. (1983) Cell, 33:729-740; Queen and Baltimore (1983) Cell,
33:741-748), neuron-specific promoters (e.g., the neurofilament promoter;
Byrne and
Ruddle ( 1989) Proc. Natl. Acad. Sci. , USA, 86:5473-5477), pancreas-specific
promoters (Edlund et al. (1985) Science, 230:912-916), and mammary gland-
specific
promoters (e.g., milk whey promoter; U.S. Patent No. 4,873,316 and European
Application Publication No. 264,166). Developmentally regulated promoters are
also
encompassed, for example the murine hox promoters (Kessel and Gruss ( 1990)
Science, 249:374-379) and the alpha-fetoprotein promoter (Campes and Tilghman
(1989) Genes Dev. 3:537-546).
I 5 The invention further provides a recombinant expression vector comprising
a
DNA molecule of the invention cloned into the expression vector in an
antisense
orientation. That is, the DNA molecule is operably linked to at least one
expression
control element in a manner which allows for expression (by transcription of
the DNA
molecule) of an RNA molecule which is antisense to an mRNA of the invention.
Regulatory sequences operably linked to a nucleic acid cloned in the antisense
orientation can be chosen which direct the continuous expression of the
antisense
RNA molecule in a variety of cell types, for instance viral promoters and/or
enhancers, or regulatory sequences can be chosen which direct constitutive,
tissue
specific or cell type specific expression of antisense RNA. The antisense
expression
vector can be in the form of a recombinant plasmid, phagemid or attenuated
virus in
which antisense nucleic acids are produced under the control of a high
efficiency
regulatory region, the activity of which can be determined by the cell type
into which
the vector is introduced. For a discussion of the regulation of gene
expression using
antisense genes see Weintraub et al. (Reviews - Trends in Genetics, Vol. I ( 1
) 1986).
Another aspect of the invention pertains to host cells into which a
recombinant
expression vector of the invention has been introduced. The terms "host cell"
and
"recombinant host cell" are used interchangeably herein. It is understood that
such
terms refer not only to the particular subject cell but also to the progeny or
potential
-21-
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
progeny of such a cell. Because certain modifications may occur in succeeding
generations due to either mutation or environmental influences, such progeny
may
not, in fact, be identical to the parent cell, but are still included within
the scope of the
term as used herein.
A host cell can be any prokaryotic or eukaryotic cell. For example, a nucleic
acid of the invention can be expressed in bacterial cells (e.g., E. coli),
insect cells,
yeast or mammalian cells (such as Chinese hamster ovary cells (CHO) or COS
cells).
Other suitable host cells are known to those skilled in the art.
Vector DNA can be introduced into prokaryotic or eukaryotic cells via
conventional transformation or transfection techniques. As used herein, the
terms
"transformation" and "transfection" are intended to refer to a variety of art-
recognized
techniques for introducing foreign nucleic acid (e.g., DNA) into a host cell,
including
calcium phosphate or calcium chloride co-precipitation, DEAE-dextran-mediated
transfection, lipofection, or electroporation. Suitable methods for
transforming or
transfecting host cells can be found in Sambrook, et al. (supra), and other
laboratory
manuals.
For stable transfection of mammalian cells, it is known that, depending upon
the expression vector and transfection technique used, only a small fraction
of cells
may integrate the foreign DNA into their genome. In order to identify and
select
these integrants, a gene that encodes a selectable marker (e.g., for
resistance to
antibiotics) is generally introduced into the host cells along with the gene
of interest.
Preferred selectable markers include those that confer resistance to drugs,
such as
6418, hygromycin and methotrexate. Nucleic acid encoding a selectable marker
can
be introduced into a host cell on the same vector as that nucleic acid of the
invention
or can be introduced on a separate vector. Cells stably transfected with the
introduced
nucleic acid can be identified by drug selection (e.g., cells that have
incorporated the
selectable marker gene will survive, while the other cells die).
A host cell of the invention, such as a prokaryotic or eukaryotic host cell in
culture, can be used to produce (i. e., express) a polypeptide of the
invention.
Accordingly, the invention further provides methods for producing a
polypeptide
using the host cells of the invention. In one embodiment, the method comprises
culturing the host cell of invention (into which a recombinant expression
vector
encoding a polypeptide of the invention has been introduced) in a suitable
medium
-22-
CA 02388617 2002-05-07
WO 01/31007 PCT/USOU/29132
such that the polypeptide is produced. In another embodiment, the method
further
comprises isolating the polypeptide from the medium or the host cell.
The host cells of the invention can also be used to produce nonhuman
transgenic animals. For example, in one embodiment, a host cell of the
invention is a
fertilized oocyte or an embryonic stem cell into which a nucleic acid of the
invention
have been introduced. Such host cells can then be used to create non-human
transgenic animals in which exogenous nucleotide sequences have been
introduced
into their genome or homologous recombinant animals in which endogenous
nucleotide sequences have been altered. Such animals are useful for studying
the
function and/or activity of the nucleotide sequence and polypeptide encoded by
the
sequence and for identifying and/or evaluating modulators of their activity.
As used
herein, a "transgenic animal" is a non-human animal, preferably a mammal, more
preferably a rodent such as a rat or mouse, in which one or more of the cells
of the
animal includes a transgene. Other examples of transgenic animals include
non-human primates, sheep, dogs, cows, goats, chickens, amphibians, etc. A
transgene is exogenous DNA which is integrated into the genome of a cell from
which
a transgenic animal develops and which remains in the genome of the mature
animal,
thereby directing the expression of an encoded gene product in one or more
cell types
or tissues of the transgenic animal. As used herein, an "homologous
recombinant
animal" is a non-human animal, preferably a mammal, more preferably a mouse,
in
which an endogenous gene has been altered by homologous recombination between
the endogenous gene and an exogenous DNA molecule introduced into a cell of
the
animal, e.g., an embryonic cell of the animal, prior to development of the
animal.
A transgenic animal of the invention can be created by introducing a nucleic
acid of the invention into the male pronuclei of a fertilized oocyte, e.g., by
microinjection, retroviral infection, and allowing the oocyte to develop in a
pseudopregnant female foster animal. The sequence can be introduced as a
transgene
into the genome of a non-human animal. Intronic sequences and polyadenylation
signals can also be included in the transgene to increase the efficiency of
expression
of the transgene. A tissue-specific regulatory sequences) can be operably
linked to
the transgene to direct expression of a polypeptide in particular cells.
Methods for
generating transgenic animals via embryo manipulation and microinjection,
particularly animals such as mice, have become conventional in the art and are
-23-
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/21132
described, for example, in U.S. Patent Nos. 4,736,866 and 4,870,009, U.S.
Patent No.
4,873,191 and in Hogan, Manipulating the Mouse Embryo (Cold Spring Harbor
Laboratory Press, Cold Spring Harbor, N.Y., 1986). Similar methods are used
for
production of other transgenic animals. A transgenic founder animal can be
identified
based upon the presence of the transgene in its genome and/or expression of
mRNA in
tissues or cells of the animals. A transgenic founder animal can then be used
to breed
additional animals carrying the transgene. Moreover, transgenic animals
carrying a
transgene encoding the transgene can further be bred to other transgenic
animals
carrying other transgenes.
Homologously recombinant host cells can also be produced that allow the in
situ
alteration of endogenous polynucleotide sequences of the invention in a host
cell
genome. The host cell includes, but is not limited to, a stable cell line,
cell in vivo, or
cloned microorganism. This technology is more fully described in WO 93/09222,
WO
91/12650, WO 91/06667, U.S. 5,272,071, and U.S. 5,641,670. Briefly, specific
polynucleotide sequences corresponding to the polynucleotides or sequences
proximal or
distal to a gene are allowed to integrate into a host cell genome by
homologous
recombination where expression of the gene can be affected. In one embodiment,
regulatory sequences are introduced that either increase or decrease
expression of an
endogenous sequence. Accordingly, a protein can be produced in a cell not
normally
producing it. Alternatively, increased expression of a protein can be effected
in a cell
normally producing the protein at a specific level. Further, expression can be
decreased
or eliminated by introducing a specific regulatory sequence. The regulatory
sequence
can be heterologous to the protein sequence or can be a homologous sequence
with a
desired mutation that affects expression. Alternatively, the entire gene can
be deleted.
The regulatory sequence can be specific to the host cell or capable of
functioning in
more than one cell type. Still further, specific mutations can be introduced
into any
desired region of the gene to produce mutant proteins of the invention. Such
mutations
could be introduced, for example, into the specific functional regions.
To create an homologous recombinant animal, a vector is prepared which
contains at least a portion of a nucleic acid of the invention into which a
deletion,
addition or substitution has been introduced to thereby alter, e.g.,
functionally disrupt,
the endogenous gene. In one embodiment, the vector is designed such that, upon
homologous recombination, the endogenous gene is functionally disrupted (l.
e., no
-24-
CA 02388617 2002-05-07
WO 01/31007 PCTlUS00/29132
longer encodes a functional protein; also referred to as a "knock out"
vector).
Alternatively, the vector can be designed such that, upon homologous
recombination,
the endogenous gene is mutated or otherwise altered but still encodes
functional
protein (e.g., the upstream regulatory region can be altered to thereby alter
the
expression of the endogenous protein). In the homologous recombination vector,
the
altered portion of the gene is flanked at its 5' and 3' ends by additional
nucleic acid of
the gene to allow for homologous recombination to occur between the exogenous
gene carried by the vector and an endogenous gene in an embryonic stem cell.
The
additional flanking nucleic acid is of sufficient length for successful
homologous
recombination with the endogenous gene. Typically, several kilobases of
flanking
DNA (both at the 5' and 3' ends) are included in the vector (see, e.g., Thomas
and
Capecchi (1987) Cell 51:503 for a description of homologous recombination
vectors).
The vector is introduced into an embryonic stem cell line (e.g., by
electroporation)
and cells in which the introduced nucleic acid has homologously recombined
with the
endogenous gene are selected (see, e.g., Li et al. (1992) Cell 69:915). The
selected
cells are then injected into a blastocyst of an animal (e.g., a mouse) to form
aggregation chimeras (see, e.g., Bradley in Teratocarcinomas and Embryonic
Stem
Cells: A Practical Approach, Robertson, ed. (IRL, Oxford, 1987) pp. 113-152).
A
ehimeric embryo can then be implanted into a suitable pseudopregnant female
foster
animal and the embryo brought to term. Progeny harboring the homologously
recombined DNA in their germ cells can be used to breed animals in which all
cells of
the animal contain the homologously recombined DNA by germline transmission of
the transgene. Methods for constructing homologous recombination vectors and
homologous recombinant animals are described further in Bradley ( 1991 )
Current
Opinion in BiolTechnology 2:823-829 and in PCT Publication Nos. WO 90/11354,
WO 91/01140, WO 92/0968, and WO 93/04169.
In another embodiment, transgenic non-human animals can be produced which
contain selected systems that allow for regulated expression of the transgene.
One
example of such a system is the crelloxP recombinase system of bacteriophage P
1.
For a description of the crelloxP recombinase system, see, e.g., Lakso et al.
(1992)
Proc. Natl. Acad Sci. USA 89:6232-6236. Another example of a recombinase
system
is the FLP recombinase system of Saccharomyces cerevisiae (O'Gorman et al.
(1991)
Science 251:1351-1355. If a crelloxP recombinase system is used to regulate
-25-
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
expression of the transgene, animals containing transgenes encoding both the
Cre
recombinase and a selected protein are required. Such animals can be provided
through the construction of "double" transgenic animals, e.g., by mating two
transgenic animals, one containing a transgene encoding a selected protein and
the
other containing a transgene encoding a recombinase.
Clones of the non-human transgenic animals described herein can also be
produced according to the methods described in Wilmut et al. (1997) Nature
385:810-813 and PCT Publication Nos. WO 97/07668 and WO 97/07669.
III. Polypeptides
The present invention also provides isolated polypeptides and variants and
fragments thereof that are encoded by the nucleic acid molecules of the
invention,
especially as shown in SEQ ID NOS: 1-6, 8, and 10. For example, as described
above, the nucleotide sequences can be used to design primers to clone and
express
cDNAs encoding the polypeptides of the invention. Further, the nucleotide
sequences
of the invention, e.g., the sequences shown in SEQ ID NOS: 1-6, 8, and 10, can
be
analyzed using routine search algorithms (e.g., BLAST, Altschul et al. (1990)
J. Mol.
Biol. 215:403-410; BLAZE, Brutlag et al. (1993) Comp. Chem. 17:203-207) to
identify open reading frames (ORFs).
As used herein, a polypeptide is said to be "isolated" or "purified" when it
is
substantially free of cellular material when it is isolated from recombinant
and
non-recombinant cells, or free of chemical precursors or other chemicals when
it is
chemically synthesized. A polypeptide, however, can be joined to another
polypeptide with which it is not normally associated in a cell and still be
"isolated" or
"purified."
The polypeptides of the invention can be purified to homogeneity. It is
understood, however, that preparations in which the polypeptide is not
purified to
homogeneity are useful and considered to contain an isolated form of the
polypeptide.
The critical feature is that the preparation allows for the desired function
of the
polypeptide, even in the presence of considerable amounts of other components.
Thus, the invention encompasses various degrees of purity. In one embodiment,
the
language "substantially free of cellular material" includes preparations of
the
polypeptide having less than about 30% (by dry weight) other proteins (i.e.,
-26-
CA 02388617 2002-05-07
WO 01/31007 PCT/US(10/29132
contaminating protein), less than about 20% other proteins, less than about
10% other
proteins, or less than about 5% other proteins.
When a polypeptide is recombinantly produced, it can also be substantially
free of culture medium, i.e., culture medium represents less than about 20%,
less than
about 10%, or less than about 5% of the volume of the protein preparation. The
language "substantially free of chemical precursors or other chemicals"
includes
preparations of the polypeptide in which it is separated from chemical
precursors or
other chemicals that are involved in its synthesis. In one embodiment, the
language
"substantially free of chemical precursors or other chemicals" includes
preparations of
I 0 the polypeptide having less than about 30% (by dry weight) chemical
precursors or
other chemicals, less than about 20% chemical precursors or other chemicals,
less
than about 10% chemical precursors or other chemicals, or less than about 5%
chemical precursors or other chemicals.
In one embodiment, a polypeptide comprises an amino acid sequence encoded
by a nucleic acid comprising a nucleotide sequence selected from the group
consisting
of the sequences shown in SEQ ID NOS: 1-6, 8, and 10 and the complements
thereof.
However, the invention also encompasses sequence variants. Variants include a
substantially homologous protein encoded by the same genetic locus in an
organism,
i.e., an allelic variant. Variants also encompass proteins derived from other
genetic
loci in an organism, but having substantial homology to a polypeptide encoded
by a
nucleic acid comprising a nucleotide sequence selected from the group
consisting of
the sequences shown in SEQ ID NOS: 1-6, 8, and 10 and the complements thereof.
Variants also include proteins substantially homologous to these polypeptides
but
derived from another organism, i.e., an ortholog. Variants also include
proteins that
are substantially homologous to these polypeptides that are produced by
chemical
synthesis. Variants also include proteins that are substantially homologous or
identical to these polypeptides that are produced by recombinant methods.
As used herein, two proteins (or a region of the proteins) are substantially
homologous or identical when the amino acid sequences are at least about 45-
55%,
typically at least about 70-75%, more typically at least about 80-85%, and
most
typically at least about 90, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99% or
more identical. A substantially homologous amino acid sequence, according to
the
present invention, will be encoded by a nucleic acid hybridizing to a nucleic
acid
-27-
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
sequence selected from the group consisting of the sequences shown in SEQ ID
NOS:
1-6, 8, and 10, or fragment thereof under stringent conditions as more
described
above.
To determine the percent similarity or identity of two amino acid sequences,
or of two nucleic acids, the sequences are aligned for optimal comparison
purposes
(e.g., gaps can be introduced in the sequence of one protein or nucleic acid
for optimal
alignment with the other protein or nucleic acid). The amino acid residues or
nucleotides at corresponding amino acid positions or nucleotide positions are
then
compared. When a position in one sequence is occupied by the same amino acid
residue or nucleotide as the corresponding position in the other sequence,
then the
molecules are homologous at that position. As used herein, amino acid or
nucleic
acid "homology" is equivalent to amino acid or nucleic acid "identity". The
percent
homology between the two sequences is a function of the number of identical
positions shared by the sequences (i.e., per cent homology equals the number
of
identical positions/total number of positions times 100).
The invention also encompasses polypeptides having a lower degree of
identity but having sufficient similarity so as to perform one or more of the
same
functions performed by a polypeptide encoded by a nucleic acid of the
invention.
Similarity is determined by conserved amino acid substitution. Such
substitutions are
those that substitute a given amino acid in a polypeptide by another amino
acid of like
characteristics. Conservative substitutions are likely to be phenotypically
silent.
Typically seen as conservative substitutions are the replacements, one for
another,
among the aliphatic amino acids Ala, Val, Leu, and Ile; interchange of the
hydroxyl
residues Ser and Thr, exchange of the acidic residues Asp and Glu,
substitution
between the amide residues Asn and Gln, exchange of the basic residues Lys and
Arg
and replacements among the aromatic residues Phe, Tyr. Guidance concerning
which
amino acid changes are likely to be phenotypically silent are found in Bowie
et al.
{1990) Science 247:1306-1310.
-28-
CA 02388617 2002-05-07
WO 01/31007 PCT/US(1(I/29132
TABLE 1. Conservative Amino Acid Substitutions.
Aromatic Phenylalani
Tryptophan
Tyrosine
Hydrophobic Leucine
Isoleucine
Valine
Polar Glutamine
Asparagine
Basic Arginine
Lysine
Histidine
Acidic Aspartic Acid
Glutamic Acid
Small Alanine
Serine
Threonine
Methionine
Glycine
Both identity and similarity can be readily calculated (Computational
Molecular Biology, Lesk, A.M., ed., Oxford University Press, New York, 1988;
Biocomputing: Informatics and Genome Projects, Smith, D.W., ed., Academic
Press,
New York, 1993; Computer Analysis of Sequence Data, Part 1, Griffin, A.M., and
Griffin, H.G., eds., Humana Press, New Jersey, 1994; Sequence Analysis in
Molecular
Biology, von Heinje, G., Academic Press, 1987; and Sequence Analysis Primer,
Gribskov, M. and Devereux, J., eds., M Stockton Press, New York, 1991 ).
Preferred computer program methods to determine identify and similarity
between two sequences include, but are not limited to, GCG program package
(Devereux, J., et al. (1984) Nucleic Acids Res. 12(1):387), BLASTP, BLASTN,
FASTA (Atschul, S.F. et al. (1990) J. Molec. Biol. 21:403).
-29-
CA 02388617 2002-05-07
WO 01/31007 PCT/USUO/29132
A variant polypeptide can differ in amino acid sequence by one or more
substitutions, deletions, insertions, inversions, fusions, and truncations or
a
combination of any of these. Further, variant polypeptides can be fully
functional or
can lack function in one or more activities. Fully functional variants
typically contain
only conservative variation or variation in non-critical residues or in non-
critical
regions. Functional variants can also contain substitution of similar amino
acids that
result in no change or an insignificant change in function. Alternatively,
such
substitutions may positively or negatively affect function to some degree.
Non-functional variants typically contain one or more non-conservative amino
acid substitutions, deletions, insertions, inversions, or truncation or a
substitution,
insertion, inversion, or deletion in a critical residue or critical region.
As indicated, variants can be naturally-occurring or can be made by
recombinant means or chemical synthesis to provide useful and novel
characteristics
for the polypeptide. This includes preventing immunogenicity from
pharmaceutical
formulations by preventing protein aggregation.
Amino acids that are essential for function can be identified by methods
known in the art, such as site-directed mutagenesis or alanine-scanning
mutagenesis
(Cunningham et al. (1989) Science 244:1081-1085). The latter procedure
introduces
single alanine mutations at every residue in the molecule. The resulting
mutant
molecules are then tested for biological activity in vitro, or in vitro
proliferative
activity. Sites that are critical for polypeptide activity can also be
determined by
structural analysis such as crystallization, nuclear magnetic resonance or
photoaffinity
labeling (Smith et al. (1992) J. Mol. Biol. 224:899-904; de Vos et al. (1992)
Science
255:306-312).
The invention also includes polypeptide fragments of the polypeptides of the
invention. Fragments can be derived from a polypeptide encoded by a nucleic
acid
comprising a nucleotide sequence selected from the group consisting of the
sequences
shown in SEQ ID NOS: 1-6, 8, and 10, and the complements thereof. However, the
invention also encompasses fragments of the variants of the polypeptides
described
herein.
As used herein, a fragment comprises at least 6 contiguous amino acids.
Useful fragments include those that retain one or more of the biological
activities of
-3 0-
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
the polypeptide as well as fragments that can be used as an immunogen to
generate
polypeptide specific antibodies.
Biologically active fragments (peptides which are, for example, 6, 9, 12, 15,
20, 30, 35, 36, 37, 38, 39, 40, 50, 100 or more amino acids in length) can
comprise a
domain, segment, or motif that has been identified by analysis of the
polypeptide
sequence using well-known methods, e.g., signal peptides, extracellular
domains, one
or more transmembrane segments or loops, ligand binding regions, zinc finger
domains, DNA binding domains, acylation sites, glycosylation sites, or
phosphorylation sites.
The invention also provides fragments with immunogenic properties. These
contain an epitope-bearing portion of the polypeptides and variants of the
invention.
These epitope-bearing peptides are useful to raise antibodies that bind
specifically to a
polypeptide or region or fragment. These peptides can contain at least 6, 7,
8, 9, 12,
at least 14, or between at least about 1 S to about 30 amino acids. The
epitope-bearing
peptide and polypeptides may be produced by any conventional means (Houghten
(1985) Proc. Natl. Acad. Sci. LISA 82:5131-5135). Simultaneous multiple
peptide
synthesis is described in U.S. Patent No. 4,631,211.
Fragments can be discrete (not fused to other amino acids or polypeptides) or
can be within a larger polypeptide. Further, several fragments can be
comprised
within a single larger polypeptide. In one embodiment a fragment designed for
expression in a host can have heterologous pre- and pro-polypeptide regions
fused to
the amino terminus of the polypeptide fragment and an additional region fused
to the
carboxyl terminus of the fragment.
The invention thus provides chimeric or fusion proteins. These comprise a
polypeptide of the invention operatively linked to a heterologous protein
having an
amino acid sequence not substantially homologous to the polypeptide.
"Operatively
linked" indicates that the polypeptide protein and the heterologous protein
are fused
in-frame. The heterologous protein can be fused to the N-terminus or C-
terminus of
the polypeptide. In one embodiment the fusion protein does not affect function
of the
polypeptide per se. For example, the fusion protein can be a GST-fusion
protein in
which the polypeptide sequences are fused to the C-terminus of the GST
sequences.
Other types of fusion proteins include, but are not limited to, enzymatic
fusion
proteins, for example beta-galactosidase fusions, yeast two-hybrid GAL
fusions,
-31-
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
poly-His fusions and Ig fusions. Such fusion proteins, particularly poly-His
fusions,
can facilitate the purification of recombinant polypeptide. In certain host
cells (e.g.,
mammalian host cells), expression and/or secretion of a protein can be
increased by
using a heterologous signal sequence. Therefore, in another embodiment, the
fusion
protein contains a heterologous signal sequence at its N-terminus.
EP-A-O 464 533 discloses fusion proteins comprising various portions of
immunoglobulin constant regions. The Fc is useful in therapy and diagnosis and
thus
results, for example, in improved pharmacokinetic properties (EP-A 0232 262).
In
drug discovery, for example, human proteins have been fused with Fc portions
for the
purpose of high-throughput screening assays to identify antagonists. Bennett
et al.
(1995) Journal of Molecular Recognition 8:52-58 and Johanson et al. (1995) The
Journal of Biological Chemistry 270,16:9459-9471. Thus, this invention also
encompasses soluble fusion proteins containing a polypeptide of the invention
and
various portions of the constant regions of heavy or light chains of
immunoglobulins
1 S of various subclass (IgG, IgM, IgA, IgE). Preferred as immunoglobulin is
the
constant part of the heavy chain of human IgG, particularly IgGI, where fusion
takes
place at the hinge region. For some uses it is desirable to remove the Fc
after the
fusion protein has been used for its intended purpose, for example when the
fusion
protein is to be used as antigen for immunizations. In a particular
embodiment, the Fc
part can be removed in a simple way by a cleavage sequence that is also
incorporated
and can be cleaved with factor Xa.
A chimeric or fusion protein can be produced by standard recombinant DNA
techniques. For example, DNA fragments coding for the different protein
sequences
are ligated together in-frame in accordance with conventional techniques. In
another
embodiment, the fusion gene can be synthesized by conventional techniques
including
automated DNA synthesizers. Alternatively, PCR amplification of nucleic acid
fragments can be carried out using anchor primers which give rise to
complementary
overhangs between two consecutive nucleic acid fragments which can
subsequently
be annealed and re-amplified to generate a chimeric nucleic acid sequence (see
Ausubel et al., Current Protocols in Molecular Biology, 1992). Moreover, many
expression vectors are commercially available that already encode a fusion
moiety
(e.g., a GST protein). A nucleic acid encoding a polypeptide of the invention
can be
-32-
CA 02388617 2002-05-07
W O l11/31007 PCT/US00/29132
cloned into such an expression vector such that the fusion moiety is linked in-
frame to
the polypeptide protein.
The isolated polypeptide can be purified from cells that naturally express it,
purified from cells that have been altered to express it (recombinant), or
synthesized
using known protein synthesis methods.
In one embodiment, the protein is produced by recombinant DNA techniques.
For example, a nucleic acid molecule encoding the polypeptide is cloned into
an
expression vector, the expression vector introduced into a host cell and the
protein
expressed in the host cell. The protein can then be isolated from the cells by
an
appropriate purification scheme using standard protein purification
techniques.
Polypeptides often contain amino acids other than the 20 amino acids
commonly referred to as the 20 naturally-occurring amino acids. Further, many
amino acids, including the terminal amino acids, may be modified by natural
processes, such as processing and other post-translational modifications, or
by
chemical modification techniques well known in the art. Common modifications
that
occur naturally in polypeptides are described in basic texts, detailed
monographs, and
the research literature, and they are well known to those of skill in the art.
Accordingly, the polypeptides also encompass derivatives or analogs in which
a substituted amino acid residue is not one encoded by the genetic code, in
which a
substituent group is included, in which the mature polypeptide is fused with
another
compound, such as a compound to increase the half life of the polypeptide (for
example, polyethylene glycol), or in which the additional amino acids are
fused to the
mature polypeptide, such as a leader or secretory sequence or a sequence for
purification of the mature polypeptide or a pro-protein sequence.
Known modifications include, but are not limited to, acetylation, acylation,
ADP-ribosylation, amidation, covalent attachment of flavin, covalent
attachment of a
heme moiety, covalent attachment of a nucleotide or nucleotide derivative,
covalent
attachment of a lipid or lipid derivative, covalent attachment of
phosphotidylinositol,
cross-linking, cyclization, disulfide bond formation, demethylation, formation
of
covalent crosslinks, formation of cystine, formation of pyroglutamate,
formylation,
gamma carboxylation, glycosylation, GPI anchor formation, hydroxylation,
iodination, methylation, myristoylation, oxidation, proteolytic processing,
-33-
CA 02388617 2002-05-07
WO 01/31007 PCT/USOU/29132
phosphorylation, prenylation, racemization, selenoylation, sulfation, transfer-
RNA
mediated addition of amino acids to proteins such as arginylation, and
ubiquitination.
Such modifications are well-known to those of skill in the art and have been
described in great detail in the scientific literature. Several particularly
common
modifications, glycosylation, lipid attachment, sulfation, gamma-carboxylation
of
glutamic acid residues, hydroxylation and ADP-ribosylation, for instance, are
described in most basic texts, such as Proteins - Structure and Molecular
Properties,
2nd Ed., T.E. Creighton, W. H. Freeman and Company, New York (1993). Many
detailed reviews are available on this subject, such as by Wold, F.,
Posttranslational
Covalent Modifrcation of Proteins, B.C. Johnson, Ed., Academic Press, New York
1-12 (1983); Seifter et al., Meth. Enzymol. 182: 626-646 (1990) and Rattan et
al.
(1992)Ann. N. Y. Acad Sci. 663:48-62.
As is also well known, polypeptides are not always entirely linear. For
instance, polypeptides may be branched as a result of ubiquitination, and they
may be
circular, with or without branching, generally as a result of post-translation
events,
including natural processing event and events brought about by human
manipulation
which do not occur naturally. Circular, branched and branched circular
polypeptides
may be synthesized by non-translational natural processes and by synthetic
methods.
Modifications can occur anywhere in a polypeptide, including the peptide
backbone, the amino acid side-chains and the amino or carboxyl termini.
Blockage of
the amino or carboxyl group in a polypeptide, or both, by a covalent
modification, is
common in naturally-occurring and synthetic polypeptides. For instance, the
amino
terminal residue of polypeptides made in E coli, prior to proteolytic
processing,
almost invariably will be N-formylmethionine.
The modifications can be a function of how the protein is made. For
recombinant polypeptides, for example, the modifications will be determined by
the
host cell posttranslational modification capacity and the modification signals
in the
polypeptide amino acid sequence. Accordingly, when glycosylation is desired, a
polypeptide should be expressed in a glycosylating host, generally a
eukaryotic cell.
Insect cells often carry out the same posttranslational glycosylations as
mammalian
cells and; for this reason, insect cell expression systems have been developed
to
efficiently express mammalian proteins having native patterns of
glycosylation.
Similar considerations apply to other modifications.
-34-
CA 02388617 2002-05-07
WO 01/311107 PCT/US00/29132
The same type of modification may be present in the same or varying degree
at several sites in a given polypeptide. Also, a given polypeptide may contain
more
than one type of modification.
Uses of the polypeptides of the invention are described in detail below. In
general, polypeptides or proteins of the present invention can be used as a
molecular
weight marker on SDS-PAGE gels or on molecular sieve gel filtration columns
using
art-recognized methods. The polypeptides of the present invention can be used
to
raise antibodies or to elicit an immune response. The polypeptides can also be
used as
a reagent, e.g., a labeled reagent, in assays to quantitatively determine
levels of the
protein or a molecule to which it binds (e.g., a receptor or a ligand) in
biological
fluids. The polypeptides can also be used as markers for tissues in which the
corresponding protein is preferentially expressed, either constitutively,
during tissue
differentiation, or in a diseased state. The polypeptides can be used to
isolate a
corresponding binding partner, e.g., receptor or ligand, such as, for example,
in an
interaction trap assay, and to screen for peptide or small molecule
antagonists or
agonists of the binding interaction.
IV. Antibodies
In another aspect, the invention provides antibodies to the polypeptides and
polypeptide fragments of the invention, e.g., having an amino acid encoded by
a
nucleic acid comprising all or a portion of a nucleotide sequence selected
from the
group consisting of the sequences shown in SEQ ID NOS: 1-6, 8, and 10. The
term
"antibody" as used herein refers to immunoglobulin molecules and
immunologically
active portions of immunoglobulin molecules, i.e., molecules that contain an
antigen
binding site that specifically binds an antigen. A molecule that specifically
binds to a
polypeptide of the invention is a molecule that binds to that polypeptide or a
fragment
thereof, but does not substantially bind other molecules in a sample, e. g., a
biological
sample, which naturally contains the polypeptide. Examples of immunologically
active portions of immunoglobulin molecules include Flab) and F(ab')2
fragments
which can be generated by treating the antibody with an enzyme such as pepsin.
The
invention provides polyclonal and monoclonal antibodies that bind to a
polypeptide of
the invention. The term "monoclonal antibody" or "monoclonal antibody
composition", as used herein, refers to a population of antibody molecules
that
-35-
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
contain only one species of an antigen binding site capable of immunoreacting
with a
particular epitope of a polypeptide of the invention. A monoclonal antibody
composition thus typically displays a single binding affinity for a particular
polypeptide of the invention with which it immunoreacts.
Polyclonal antibodies can be prepared as described above by immunizing a
suitable subject with a desired immunogen, e.g., polypeptide of the invention
or
fragment thereof. The antibody titer in the immunized subject can be monitored
over
time by standard techniques, such as with an enzyme linked immunosorbent assay
(ELISA) using immobilized polypeptide. If desired, the antibody molecules
directed
against the polypeptide can be isolated from the mammal (e.g., from the blood)
and
further purified by well-known techniques, such as protein A chromatography to
obtain the IgG fraction. At an appropriate time after immunization, e.g., when
the
antibody titers are highest, antibody-producing cells can be obtained from the
subject
and used to prepare monoclonal antibodies by standard techniques, such as the
hybridoma technique originally described by Kohler and Milstein (1975) Nature
256:495-497, the human B cell hybridoma technique (Kozbor et al. (I 983)
Immunol.
Today 4:72), the EBV-hybridoma technique (Cole et al. (1985), Monoclonal
Antibodies and Cancer Therapy, Alan R. Liss, Ine., pp. 77-96) or trioma
techniques.
The technology for producing hybridomas is well known (see generally Current
Protocols in Immunology (1994) Coligan et al. (eds.) John Wiley & Sons, Inc.,
New
York, NY). Briefly, an immortal cell line (typically a myeloma) is fused to
lymphocytes (typically splenocytes) from a mammal immunized with an immunogen
as described above, and the culture supernatants of the resulting hybridoma
cells are
screened to identify a hybridoma producing a monoclonal antibody that binds a
polypeptide of the invention.
Any of the many well known protocols used for fusing lymphocytes and
immortalized cell lines can be applied for the purpose of generating a
monoclonal
antibody to a polypeptide of the invention (see, e.g., Current Protocols in
Immunology, supra; Galfre et al. (1977) Nature 266:55052; R.H. Kenneth, in
Monoclonal Antibodies: A New Dimension In Biological Analyses, Plenum
Publishing
Corp., New York, New York (1980); and Lerner (1981) Yale J. Biol. Med.
54:387-402. Moreover, the ordinarily skilled worker will appreciate that there
are
many variations of such methods that also would be useful. Typically, the
immortal
-36-
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
cell line (e.g., a myeloma cell line) is derived from the same mammalian
species as
the lymphocytes. For example, murine hybridomas can be made by fusing
lymphocytes from a mouse immunized with an immunogenic preparation of the
present invention with an immortalized mouse cell line, e.g., a myeloma cell
line that
is sensitive to culture medium containing hypoxanthine, aminopterin and
thymidine
("HAT medium"). Any of a number of myeloma cell lines can be used as a fusion
partner according to standard techniques, e.g., the P3-NS1/1-Ag4-1, P3-x63-
Ag8.653
or Sp2/O-Agl4 myeloma lines. These myeloma lines are available from ATCC.
Typically, HAT-sensitive mouse myeloma cells are fused to mouse splenocytes
using
polyethylene glycol ("PEG"). Hybridoma cells resulting from the fusion are
then
selected using HAT medium, which kills unfused and unproductively fused
myeloma
cells (unfused splenocytes die after several days because they are not
transformed).
Hybridoma cells producing a monoclonal antibody of the invention are detected
by
screening the hybridoma culture supernatants for antibodies that bind a
polypeptide of
the invention, e.g., using a standard ELISA assay.
Alternative to preparing monoclonal antibody-secreting hybridomas, a
monoclonal antibody to a polypeptide of the invention can be identified and
isolated
by screening a recombinant combinatorial immunoglobulin library (e.g., an
antibody
phage display library) with the polypeptide to thereby isolate immunoglobulin
library
members that bind the polypeptide. Kits for generating and screening phage
display
libraries are commercially available (e.g., the Pharmacia Recombinant Phage
Antibody System, Catalog No. 27-9400-01; and the Stratagene SurfZAPTM Phage
Display Kit, Catalog No. 240612). Additionally, examples of methods and
reagents
particularly amenable for use in generating and screening antibody display
library can
be found in, for example, U.S. Patent No. 5,223,409; PCT Publication No. WO
92/18619; PCT Publication No. WO 91/17271; PCT Publication No. WO 92/20791;
PCT Publication No. WO 92/15679; PCT Publication No. WO 93/01288; PCT
Publication No. WO 92/01047; PCT Publication No. WO 92/09690; PCT Publication
No. WO 90/02809; Fuchs et al. (1991) BiolTechnology 9:1370-1372; Hay et al.
( 1992) Hum. Antibod. Hybridomas 3:81-85; Huse et al. ( 1989) Science
246:1275-1281; Griffiths et al. (1993) EMBO J. 12:725-734.
Additionally, recombinant antibodies, such as chimeric and humanized
monoclonal antibodies, comprising both human and non-human portions, which can
-3 7-
CA 02388617 2002-05-07
WO 01/31007 PCT/USO(1/29132
be made using standard recombinant DNA techniques, are within the scope of the
invention. Such chimeric and humanized monoclonal antibodies can be produced
by
recombinant DNA techniques known in the art, for example using methods
described
in PCT Publication No. WO 87/02671; European Patent Application 184,187;
S European Patent Application 171,496; European Patent Application 173,494;
PCT
Publication No. WO 86/01533; U.S. Patent No. 4,816,567; European Patent
Application 125,023; Better et al. (1988) Science 240:1041-1043; Liu et al.
(1987)
Proc. Natl. Acad. Sci. USA 84:3439-3443; Liu et al. ( 1987) J. Immunol.
139:3521-3526; Sun et al. (1987) Proc. Natl. Acad. Sci. TJSA 84:214-218;
Nishimura
et al. (1987) Canc. Res. 47:999-1005; Wood et al. (1985) Nature 314:446-449;
and
Shaw et al. (1988) J. Natl. Cancer Inst. 80:1553-1559); Morrison (1985)
Science
229:1202-1207; Oi et al. {1986) BiolTechniques 4:214; U.S. Patent 5,225,539;
Jones
et al. (1986) Nature 321:552-525; Verhoeyan et al. (1988) Science 239:1534;
and
Beidler et al. (1988) .l. Immunol. 141:4053-4060.
Completely human antibodies are particularly desirable for therapeutic
treatment of human patients. Such antibodies can be produced using transgenic
mice
that are incapable of expressing endogenous immunoglobulin heavy and light
chains
genes, but which can express human heavy and light chain genes. The transgenic
mice are immunized in the normal fashion with a selected antigen, e.g., all or
a
portion of a polypeptide of the invention. Monoclonal antibodies directed
against the
antigen can be obtained using conventional hybridoma technology. The human
immunoglobulin transgenes harbored by the transgenic mice rearrange during B
cell
differentiation, and subsequently undergo class switching and somatic
mutation.
Thus, using such a technique, it is possible to produce therapeutically useful
lgG, IgA
and IgE antibodies. For an overview of this technology for producing human
antibodies, see Lonberg and Huszar (1995) Int. Rev. Immunol. 13:65-93. For a
detailed discussion of this technology for producing human antibodies and
human
monoclonal antibodies and protocols for producing such antibodies, see, e.g.,
U.S.
Patent 5,625,126; U.S. Patent 5,633,425; U.S. Patent 5,569,825; U.S. Patent
5,661,016; and U.S. Patent 5,545,806. In addition, companies such as Abgenix,
Inc.
(Freemont, CA), can be engaged to provide human antibodies directed against a
selected antigen using technology similar to that described above.
-3 8-
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
Completely human antibodies that recognize a selected epitope can be
generated using a technique referred to as "guided selection." This technology
is
described, for example, in Jespers et al. (1994) Bioltechnology 12:899-903).
Uses of the antibodies of the invention are described in detail below. In
general, antibodies of the invention (e.g., a monoclonal antibody) can be used
to
isolate a polypeptide of the invention by standard techniques, such as
affinity
chromatography or immunoprecipitation. A polypeptide specific antibody can
facilitate the purification of natural polypeptide from cells and of
recombinantly
produced polypeptide expressed in host cells. Moreover, an antibody specific
for a
polypeptide of the invention can be used to detect the polypeptide (e.g., in a
cellular
lysate, cell supernatant, or tissue sample) in order to evaluate the abundance
and
pattern of expression of the polypeptide. Antibodies can be used
diagnostically to
monitor protein levels in tissue as part of a clinical testing procedure,
e.g., to, for
example, determine the efficacy of a given treatment regimen. Detection can be
1 S facilitated by coupling the antibody to a detectable substance. Examples
of detectable
substances include various enzymes, prosthetic groups, fluorescent materials,
luminescent materials, bioluminescent materials, and radioactive materials.
Examples
of suitable enzymes include horseradish peroxidase, alkaline phosphatase,
(-galactosidase, or acetylcholinesterase; examples of suitable prosthetic
group
complexes include streptavidin/biotin and avidin/biotin; examples of suitable
fluorescent materials include umbelliferone, fluorescein, fluorescein
isothiocyanate,
rhodamine, dichlorotriazinylamine fluorescein, dansyl chloride or
phycoerythrin; an
example of a luminescent material includes luminol; examples of bioluminescent
materials include luciferase, luciferin, and aequorin, and examples of
suitable
radioactive material include ~ZSI, ~3~I, 3sS or 3H.
V. Computer Readable Means
The nucleotide or amino acid sequences of the invention are also provided in a
variety of mediums to facilitate use thereof. As used herein, "provided"
refers to a
manufacture, other than an isolated nucleic acid or amino acid molecule, which
contains a nucleotide or amino acid sequence of the present invention. Such a
manufacture provides the nucleotide or amino acid sequences, or a subset
thereof
(e.g., a subset of open reading frames (ORFs)) in a form which allows a
skilled artisan
-3 9-
CA 02388617 2002-05-07
W O l11 /3100 7 PCT/US00/29132
to examine the manufacture using means not directly applicable to examining
the
nucleotide or amino acid sequences, or a subset thereof, as they exists in
nature or in
purified form.
In one application of this embodiment, a nucleotide or amino acid sequence of
the present invention can be recorded on computer readable media. As used
herein,
"computer readable media" refers to any medium that can be read and accessed
directly by a computer. Such media include, but are not limited to: magnetic
storage
media, such as floppy discs, hard disc storage medium, and magnetic tape;
optical
storage media such as CD-ROM; electrical storage media such as RAM and ROM;
and hybrids of these categories such as magnetic/optical storage media. The
skilled
artisan will readily appreciate how any of the presently known computer
readable
mediums can be used to create a manufacture comprising computer readable
medium
having recorded thereon a nucleotide or amino acid sequence of the present
invention.
As used herein, "recorded" refers to a process for storing information on
computer readable medium. The skilled artisan can readily adopt any of the
presently
known methods for recording information on computer readable medium to
generate
manufactures comprising the nucleotide or amino acid sequence information of
the
present invention.
A variety of data storage structures are available to a skilled artisan for
creating a computer readable medium having recorded thereon a nucleotide or
amino
acid sequence of the present invention. The choice of the data storage
structure will
generally be based on the means chosen to access the stored information. In
addition,
a variety of data processor programs and formats can be used to store the
nucleotide
sequence information of the present invention on computer readable medium. The
sequence information can be represented in a word processing text file,
formatted in
commercially-available software such as WordPerfect and Microsoft Word, or
represented in the form of an ASCII file, stored in a database application,
such as
DB2, Sybase, Oracle, or the like. The skilled artisan can readily adapt any
number of
dataprocessor structuring formats (e.g_, text file or database) in order to
obtain
computer readable medium having recorded thereon the nucleotide sequence
information of the present invention.
By providing the nucleotide or amino acid sequences of the invention in
computer readable form, the skilled artisan can routinely access the sequence
-40-
CA 02388617 2002-05-07
WO O1/31U07 PCT/US00/29132
information for a variety of purposes. For example, one skilled in the art can
use the
nucleotide or amino acid sequences of the invention in computer readable form
to
compare a target sequence or target structural motif with the sequence
information
stored within the data storage means. Search means are used to identify
fragments or
regions of the sequences of the invention which match a particular target
sequence or
target motif.
As used herein, a "target sequence" can be any DNA or amino acid sequence
of six or more nucleotides or two or more amino acids. A skilled artisan can
readily
recognize that the longer a target sequence is, the less likely a target
sequence will be
present as a random occurrence in the database. The most preferred sequence
length
of a target sequence is from about 10 to 100 amino acids or from about 30 to
300
nucleotide residues. However, it is well recognized that commercially
important
fragments, such as sequence fragments involved in gene expression and protein
processing, may be of shorter length.
As used herein, "a target structural motif," or "target motif," refers to any
rationally selected sequence or combination of sequences in which the
sequences) are
chosen based on a three-dimensional configuration which is formed upon the
folding
of the target motif. There are a variety of target motifs known in the art.
Protein
target motifs include, but are not limited to, enzyme active sites and signal
sequences.
Nucleic acid target motifs include, but are not limited to, promoter
sequences, hairpin
structures and inducible expression elements (protein binding sequences).
Computer software is publicly available which allows a skilled artisan to
access sequence information provided in a computer readable medium for
analysis
and comparison to other sequences. A variety of known algorithms are disclosed
publicly and a variety of commercially available software for conducting
search
means are and can be used in the computer-based systems of the present
invention.
Examples of such software includes, but is not limited to, MacPattern (EMBL),
BLASTN and BLASTX (NCBIA).
For example, software which implements the BLAST (Altschul et al. ( 1990) J.
Mol. Biol. 215:403-410) and BLAZE (Brutlag et al. (1993) Comp. Chem. 17:203-
20?)
search algorithms on a Sybase system can be used to identify open reading
frames
(ORFs) of the sequences of the invention which contain homology to ORFs or
proteins from other libraries. Such ORFs are protein encoding fragments and
are
-41-
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
useful in producing commercially important proteins such as enzymes used in
various
reactions and in the production of commercially useful metabolites.
VI. Detection Assays
S Portions or fragments of the nucleotide sequences identified herein (and the
corresponding complete gene sequences) can be used in numerous ways as
polynucleotide reagents. For example, these sequences can be used to: (i) map
their
respective genes on a chromosome; and, thus, locate gene regions associated
with
genetic disease; (ii) identify an individual from a minute biological sample
(tissue
typing); and (iii) aid in forensic identification of a biological sample.
These
applications are described in the subsections below.
Chromosome Mapping
Once the nucleic acid (or a portion of the sequence) has been isolated, it can
be used to map the location of the gene on a chromosome. The mapping of the
sequences to chromosomes is an important first step in correlating these
sequences
with genes associated with disease. Briefly, genes can be mapped to
chromosomes by
preparing PCR primers (preferably 15-25 by in length) from the nucleic acid
molecules described herein. Computer analysis of the sequences can be used to
predict primers that do not span more than one exon in the genomic DNA, thus
complicating the amplification process. These primers can then be used for PCR
screening of somatic cell hybrids containing individual human chromosomes.
Only
those hybrids containing the human gene corresponding to the appropriate
nucleotide
sequences will yield an amplified fragment.
Somatic cell hybrids are prepared by fusing somatic cells from different
mammals (e.g., human and mouse cells). As hybrids of human and mouse cells
grow
and divide, they gradually lose human chromosomes in random order, but retain
the
mouse chromosomes. By using media in which mouse cells cannot grow, because
they lack a particular enzyme, but human cells can, the one human chromosome
that
contains the gene encoding the needed enzyme, will be retained. By using
various
media, panels of hybrid cell lines can be established. Each cell line in a
panel
contains either a single human chromosome or a small number of human
chromosomes, and a full set of mouse chromosomes, allowing easy mapping of
-42-
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
individual genes to specific human chromosomes. (D'Eustachio et al. (1983)
Science
220:919-924). Somatic cell hybrids containing only fragments of human
chromosomes can also be produced by using human chromosomes with
translocations
and deletions.
PCR mapping of somatic cell hybrids is a rapid procedure for assigning a
particular sequence to a particular chromosome. Three or more sequences can be
assigned per day using a single thermal cycle. Using the nucleic acid
molecules of the
invention to design oligonucleotide primers, sublocalization can be achieved
with
panels of fragments from specific chromosomes. Other mapping strategies which
can
similarly be used to map a specified sequence to its chromosome include in
situ
hybridization (described in Fan et al. (1990) PNAS 97:6223-2?), pre-screening
with
labeled flow-sorted chromosomes, and pre-selection by hybridization to
chromosome
specific cDNA libraries.
Fluorescence in situ hybridization (FISH) of a nucleotide sequence to a
metaphase chromosomal spread can further be used to provide a precise
chromosomal
location in one step. Chromosome spreads can be made using cells whose
division
has been blocked in metaphase by a chemical such as colcemid that disrupts the
mitotic spindle. The chromosomes can be treated briefly with trypsin, and then
stained with Giemsa. A pattern of light and dark bands develops on each
chromosome, so that the chromosomes can be identified individually. The FISH
technique can be used with a nucleotide sequence as short as 500 or 600 bases.
However, clones larger than 1,000 bases have a higher likelihood of binding to
a
unique chromosomal location with sufficient signal intensity for simple
detection.
Preferably 1,000 bases, and more preferably 2,000 bases will suffice to get
good
results at a reasonable amount of time. for a review of this technique, see
Verma et
al., Human Chromosomes: A Manual of Basic Techniques (Pergamon Press, New
York 1988).
Reagents for chromosome mapping can be used individually to mark a single
chromosome or a single site on that chromosome, or panels of reagents can be
used
for marking multiple sites and/or multiple chromosomes. Reagents corresponding
to
noncoding regions of the genes actually are preferred for mapping purposes.
Coding
sequences are more likely to be conserved within gene families, thus
increasing the
chance of cross hybridizations during chromosomal mapping.
-43-
CA 02388617 2002-05-07
WO 01/31007 PCT/USOU/29132
Once a sequence has been mapped to a precise chromosomal location, the
physical position of the sequence on the chromosome can be correlated with
genetic
map data. (Such data are found, for example, in V. McKusick, Medelian
Inheritance
in Man, available on-line through Johns Hopkins University Welch Medical
Library).
The relationship between a gene and a disease, mapped to the same chromosomal
region, can then be identified through linkage analysis (co-inheritance of
physically
adjacent genes), described in, for example, Egeland et al. (1987) Nature
325:783-787.
Moreover, differences in the DNA sequences between individuals affected and
unaffected with a disease associated with a specified gene, can be determined.
If a
mutation is observed in some or all of the affected individuals but not in any
unaffected individuals, then the mutation is likely to be the causative agent
of the
particular disease. Comparison of affected and unaffected individuals
generally
involves first looking for structural alterations in the chromosomes, such as
deletions
or translocations that are visible form chromosome spreads or detectable using
PCR
based on that DNA sequence. Ultimately, complete sequencing of genes from
several
individuals can be performed to confirm the presence of a mutation and to
distinguish
mutations from polymorphisms.
2. Tissue Typing
The nucleotide sequences of the present invention can also be used to identify
individuals from minute biological samples. The United States military, for
example,
is considering the use of restriction fragment length polymorphism (RFLP) for
identification of its personnel. In this technique, an individual's genomic
DNA is
digested with one or more restriction enzymes, and probed on a Southern blot
to yield
unique bands for identification. This method does not suffer from the current
limitations of "Dog Tags" which can be lost, switched, or stolen, making
positive
identification difficult. The sequences of the present invention are useful as
additional DNA markers for RFLP (described in U.S. Patent 5,272,057).
Furthermore, the sequences of the present invention can be used to provide an
alternative technique that determines the actual base-by-base DNA sequence of
selected portions of an individual's genome. Thus, the nucleic acid molecules
described herein can be used to prepare two PCR primers from the 5' and 3'
ends of
-44-
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
the sequences. These primers can then be used to amplify an individual's DNA
and
subsequently sequence it.
Panels of corresponding DNA sequences from individuals, prepared in this
manner, can provide unique individual identifications, as each individual will
have a
unique set of such DNA sequences due to allelic differences. The sequences of
the
present invention can be used to obtain such identification sequences from
individuals
and from tissue. The nucleic acid molecules of the invention uniquely
represent
portions of the human genome. Allelic variation occurs to some degree in the
coding
regions of these sequences, and to a greater degree in the noncoding regions.
It is
estimated that allelic variation between individual humans occurs with a
frequency of
about once per each 500 bases. Each of the sequences described herein can, to
some
degree, be used as a standard against which DNA from an individual can be
compared
for identification purposes. Because greater numbers of polymorphisms occur in
the
noncoding regions, fewer sequences are necessary to differentiate individuals.
The
noncoding sequences of these sequences can comfortably provide positive
individual
identification with a panel of perhaps 10 to 1,000 primers which each yield a
noncoding amplified sequence of 100 bases. If predicted coding sequences are
used,
a more appropriate number of primers for positive individual identification
would be
500-2,000.
If a panel of reagents from nucleic acid molecules described herein is used to
generate a unique identification database for an individual, those same
reagents can
later be used to identify tissue from that individual. Using the unique
identification
database, positive identification of the individual, living or dead, can be
made from
extremely small tissue samples.
3. Use of Partial Sequences in Forensic Biology
DNA-based identification techniques can also be used in forensic biology
Forensic biology is a scientific field employing genetic typing of biological
evidence
found at a crime scene as a means of positively identifying, for example, a
perpetrator
of a crime. To make such an identification, PCR technology can be used to
amplify
DNA sequences taken from very small biological samples such as tissues, e.g.,
hair or
skin, or body fluids, e.g., blood, saliva, or semen found at a crime scene.
The
-45-
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
amplified sequence can then be compared to a standard, thereby allowing
identification of the origin of the biological sample.
The sequences of the present invention can be used to provide polynucleotide
reagents, e.g., PCR primers, targeted to specific loci in the human genome,
which can
enhance the reliability of DNA-based forensic identifications by, for example,
providing another "identification~marker" (i.e. another DNA sequence that is
unique
to a particular individual). As mentioned above, actual base sequence
information can
be used for identification as an accurate alternative to patterns formed by
restriction
enzyme generated fragments. Sequences targeted to noncoding regions of
sequences
described herein are particularly appropriate for this use, as greater numbers
of
polymorphisms occur in the noncoding regions, making it easier to
differentiate
individuals using this technique. Examples of polynucleotide reagents include
the
nucleic acid molecules or the invention, or portions thereof, e.g., fragments
having a
length of at least 20 bases, preferably at least 30 bases.
The nucleic acid molecules described herein can further be used to provide
polynucleotide reagents, e.g., labeled or labelable probes which can be used
in, or
example, an in situ hybridization technique, to identify a specific tissue.
This can be
very useful in cases where a forensic pathologist is presented with a tissue
of
unknown origin. Panels of such probes can be used to identify tissue by
species
and/or by organ type.
In a similar fashion, these reagents, primers or probes can be used to screen
tissue culture for contamination (i.e., screen for the presence of a mixture
of different
types of cells in a culture).
VII. Predictive Medicine:
The present invention also pertains to the field of predictive medicine in
which
diagnostic assays, prognostic assays, and monitoring clinical trials are used
for
prognostic (predictive) purposes to thereby treat an individual
prophylactically.
Accordingly, one aspect of the present invention relates to diagnostic assays
for
determining protein and/or nucleic acid expression as well as activity of
proteins of
the invention, in the context of a biological sample (e.g., blood, serum,
cells, tissue) to
thereby determine whether an individual is afflicted with a disease or
disorder, or is at
risk of developing a disorder, associated with aberrant expression or
activity. The
-46-
CA 02388617 2002-05-07
WO 01/31007 PCTlUSO(1/29132
invention also provides for prognostic (or predictive) assays for determining
whether
an individual is at risk of developing a disorder associated with activity ox
expression
of proteins or nucleic acids of the invention.
Disorders relating to programmed cell death are particularly relevant as
S discussed in detail herein below.
For example, mutations in a specified gene can be assayed in a biological
sample. Such assays can be used for prognostic or predictive purpose to
thereby
prophylactically treat an individual prior to the onset of a disorder
characterized by or
associated with expression or activity of nucleic acid molecules or proteins
of the
invention.
Another aspect of the invention pertains to monitoring the influence of agents
(e.g., drugs, compounds) on the expression or activity of proteins of the
invention in
clinical trials.
These and other agents are described in further detail in the following
sections.
1. Diagnostic Assays
An exemplary method for detecting the presence or absence of proteins or
nucleic acids of the invention in a biological sample involves obtaining a
biological
sample from a test subject and contacting the biological sample with a
compound or
an agent capable of detecting the protein, or nucleic acid (e.g., mRNA,
genomic
DNA) that encodes the protein, such that the presence of the protein or
nucleic acid is
detected in the biological sample. A preferred agent for detecting mRNA or
genomic
DNA is a labeled nucleic acid probe capable of hybridizing to mRNA or genomic
DNA sequences described herein. The nucleic acid probe can be, for example, a
full-length nucleic acid, or a portion thereof, such as an oligonucleotide of
at least 15,
30, 50, 100, 250 or 500 nucleotides in length and sufficient to specifically
hybridize
under stringent conditions to appropriate mRNA or genomic DNA. For example,
the
nucleic acid probe can be all or a portion of the sequences shown in SEQ ID
NOS: 1-
6, 8, and 10, or the complement of the sequences shown in SEQ ID NOS: 1-6, 8,
and
10, or a portion thereof. Other suitable probes for use in the diagnostic
assays of the
invention are described herein.
In one embodiment, the agent for detecting proteins of the invention is an
antibody capable of binding to the protein, preferably an antibody with a
detectable
-47-
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
label. Antibodies can be polyclonal, or more preferably, monoclonal. An intact
antibody, or a fragment thereof (e.g., Fab or F(ah')2) can be used. The term
"labeled",
with regard to the probe or antibody, is intended to encompass direct labeling
of the
probe or antibody by coupling (i. e., physically linking) a detectable
substance to the
probe or antibody, as well as indirect labeling of the probe or antibody by
reactivity
with another reagent that is directly labeled. Examples of indirect labeling
include
detection of a primary antibody using a fluorescently labeled secondary
antibody and
end-labeling of a DNA probe with biotin such that it can be detected with
fluorescently labeled streptavidin. The term "biological sample" is intended
to
include tissues, calls and biological fluids isolated from a subject, as well
as tissues,
cells and fluids present within a subject. That is, the detection method of
the
invention can be used to detect mRNA, protein, or genomic DNA of the invention
in a
biological sample in vitro as well as in vivo. For example, in vitro
techniques for
detection of mRNA include Northern hybridizations and in situ hybridizations.
In
vitro techniques for detection of protein include enzyme linked immunosorbent
assays
(ELISAs), Western blots, immunoprecipitations and immunofluorescence. In vitro
techniques for detection of genomic DNA include Southern hybridizations.
Furthermore, in vivo techniques for detection of protein include introducing
into a
subject a labeled anti-protein antibody. For example, the antibody can be
labeled with
a radioactive marker whose presence and location in a subject can be detected
by
standard imaging techniques.
In one embodiment, the biological sample contains protein molecules from the
test subject. Alternatively, the biological sample can contain mRNA molecules
from
the test subject or genomic DNA molecules from the test subject. A preferred
biological sample is a serum sample or biopsy isolated by conventional means
from a
subj ect.
In another embodiment, the methods further involve obtaining a control
biological sample from a control subject, contacting the control sample with a
compound or agent capable of detecting protein, mRNA, or genomic DNA of the
invention, such that the presence of protein, mRNA or genomic DNA is detected
in
the biological sample, and comparing the presence of protein, mRNA or genomic
DNA in the control sample with the presence of protein, mRNA or genomic DNA in
the test sample.
-48-
CA 02388617 2002-05-07
WO 01/31007 PCT/US110/29132
The invention also encompasses kits for detecting the presence of proteins or
nucleic acid molecules of the invention in a biological sample. For example,
the kit
can comprise a labeled compound or agent capable of detecting protein or mRNA
in a
biological sample; means for determining the amount of in the sample; and
means for
comparing the amount of in the sample with a standard. The compound or agent
can
be packaged in a suitable container. The kit can further comprise instructions
for
using the kit to detect protein or nucleic acid.
2. Prognostic Assays
The diagnostic methods described herein can furthermore be utilized to
identify subjects having or at risk of developing a disease or disorder
associated with
aberrant expression or activity of proteins and nucleic acid molecules of the
invention.
Accordingly, the term "diagnostic" refers not only to ascertaining whether a
subject
has an active disease but also relates to ascertaining whether a subject is
predisposed
to developing active disease as well as ascertaining the probability that
treatment of
active disease will be effective. For example, the assays described herein,
such as the
preceding diagnostic assays or the following assays can be utilized to
identify a
subject having or at risk of developing a disorder associated with protein or
nucleic
acid expression or activity such as a proliferative disorder, a
differentiative or
developmental disorder, or a hematopoietic disorder. Alternatively, the
prognostic
assays can be utilized to identify a subject having or at risk for developing
a
differentiative or proliferative disease (e.g., cancer). Thus, the present
invention
provides a method for identifying a disease or disorder associated with
aberrant
expression or activity of proteins or nucleic acid molecules of the invention,
in which
a test sample is obtained from a subject and protein or nucleic acid (e.g.,
mRNA,
genomic DNA) is detected, wherein the presence of protein or nucleic acid is
diagnostic for a subject having or at risk of developing a disease or disorder
associated with aberrant expression or activity of the protein or nucleic acid
sequence
of the invention. As used herein, a "test sample" refers to a biological
sample
obtained from a subject of interest. For example, a test sample can be a
biological
fluid (e.g., serum), cell or tissue sample.
Disorders relating to programmed cell death are particularly relevant as
discussed in detail herein below.
-49-
CA 02388617 2002-05-07
WO 01/31007 PCT/USUO/29132
Furthermore, the prognostic assays described herein can be used to determine
whether a subject can be administered an agent (e.g., an agonist, antagonist,
peptidomimetic, protein, polypeptide, nucleic acid, small molecule, or other
drug
candidate) to treat a disease or disorder associated with aberrant expression
or activity
of a protein or nucleic acid molecule of the invention. For example, such
methods
can be used to determine whether a subject can be effectively treated with an
agent for
a disorder, such as a proliferative disorder, a differentiative or a
developmental
disorder. Alternatively, such methods can be used to determine whether a
subject can
be effectively treated with an agent for a differentiative or proliferative
disease (e.g.,
cancer). Thus, the present invention provides methods for determining whether
a
subject can be effectively treated with an agent for a disorder associated
with aberrant
expression or activity of a protein or nucleic acid of the present invention,
in which a
test sample is obtained and protein or nucleic acid expression or activity is
detected
(e.g., wherein the abundance of particular protein or nucleic acid expression
or
activity is diagnostic for a subject that can be administered the agent to
treat a
disorder associated with aberrant expression or activity.)
Disorders relating to programmed cell death are particularly relevant as
discussed in detail herein below.
The methods of the invention can also be used to detect genetic alterations in
genes or nucleic acid molecules of the present invention, thereby determining
if a
subject with the altered gene is at risk for a disorder characterized by
aberrant
development, aberrant cellular differentiation, aberrant cellular
proliferation or an
aberrant hematopoietic response. In certain embodiments, the methods include
detecting, in a sample of cells from the subject, the presence or absence of a
genetic
alteration characterized by at least one of an alteration affecting the
integrity of a gene
encoding a particular protein, or the mis-expression of the gene. For example,
such
genetic alterations can be detected by ascertaining the existence of at least
one of (I) a
deletion of one or more nucleotides; (2) an addition of one or more
nucleotides; (3) a
substitution of one or more nucleotides, (4) a chromosomal rearrangement; (5)
an
alteration in the level of a messenger RNA transcript; (6) aberrant
modification, such
as of the methylation pattern of the genomic DNA; (7) the presence of a non-
wild
type splicing pattern of a messenger RNA transcript; (8) a non-wild type
level; (9)
allelic loss; and (10) inappropriate post-translational modification. As
described
-50-
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
herein, there are a large number of assay techniques known in the art that can
be used
for detecting alterations in a particular gene. A preferred biological sample
is a tissue
or serum sample isolated by conventional means from a subject.
In certain embodiments, detection of the alteration involves the use of a
probe/primer in a polymerase chain reaction (PCR) (see, e.g., U.S. Patent Nos.
4,683,195 and 4,683,202), such an anchor PCR or RACE PCR, or, alternatively,
in a
ligation chain reaction (LCR) (see, e.g., Landegran et al. (1988) Science
2:17:1077-1080; and Nakazawa et al. (1994) PNAS 91:360-364), the latter of
which
can be particularly useful for detecting point mutations (see Abravaya et al.
(1995)
Nucleic Acids Res. 23:675-682). This method can include the steps of
collecting a
sample of cells from a patient, isolating nucleic acid (e.g., genomic, mRNA or
both)
from the cells of the sample, contacting the nucleic acid sample with one or
more
primers which specifically hybridize to the gene under conditions such that
hybridization and amplification of the gene (if present) occurs, and detecting
the
l 5 presence or absence of an amplification product, or detecting the size of
the
amplification product and comparing the length to a control sample. It is
anticipated
that PCR and/or LCR may be desirable to use as a preliminary amplification
step in
conjunction with any of the techniques used for detecting mutations described
herein.
Alternative amplification methods include: self sustained sequence replication
(Guatelli, J.C. et al. (1990) Proc. Natl. Acad Sci. USA 87:1874-1878),
transcriptional
amplification system (Kwoh et al., (1989) Proc. Natl. Acad. Sci. USA 86:1173-
1177),
Q-Beta Replicase (Lizardi et al. (1988) BiolTechnology 6:1197), or any other
nucleic
acid amplification method, followed by the detection of the amplified
molecules using
techniques well known to those of skill in the art. These detection schemes
are
especially useful for the detection of nucleic acid molecules if such
molecules are
present in very low numbers.
In an alternative embodiment, mutations in a given gene from a sample cell
can be identified by alterations in restriction enzyme cleavage patterns. For
example,
sample and control DNA is isolated, amplified (optionally), digested with one
or more
restriction endonucleases, and fragment length sizes are determined by gel
electrophoresis and compared. Differences in fragment length sizes between
sample
and control DNA indicate mutations in the sample DNA. Moreover, the use of
sequence specific ribozymes {see, for sample, U.S. Patent No. 5,498,531 ) can
be used
-51-
CA 02388617 2002-05-07
WO 01/31(107 PCT/USO(1/29132
to score for the presence of specific mutations by development or loss of a
ribozyme
cleavage site.
In other embodiments, genetic mutations can be identified by hybridizing a
sample and control nucleic acids, e.g., DNA or RNA, to high density arrays
containing hundreds or thousands of oligonucleotide probes (Cronin et al.
(1996)
Human Mutation 7:244-2~5; Kozal et a1.(1996) Nature Medicine 2:753-759). For
example, genetic mutations can be identified in two dimensional arrays
containing
light-generated DNA probes as described in Cronin, M.T. et al. supra. Briefly,
a first
hybridization array of probes can be used to scan through long stretches of
DNA in a
sample and control to identify base changes between the sequences by making
linear
arrays of sequential overlapping probes. This step allows the identification
of point
mutations. This step is followed by a second hybridization array that allows
the
characterization of specific mutations by using smaller, specialized probe
arrays
complementary to all variants or mutations detected. Each mutation array is
composed of parallel probe sets, one complementary to the wild-type gene and
the
other complementary to the mutant gene.
In yet another embodiment, any of a variety of sequencing reactions known in
the art can be used to directly sequence the gene and detect mutations by
comparing
the sequence of the gene from the sample with the corresponding wild-type
(control)
gene sequence. Examples of sequencing reactions include those based on
techniques
developed by Maxim and Gilbert (( 1997) PNAS 74:560) or Sanger (( 1977) PNAS
7=1:5463). It is also contemplated that any of a variety of automated
sequencing
procedures can be utilized when performing the diagnostic assays ((1995)
Biotechnigues 19:448), including sequencing by mass spectrometry (see, e.g.,
PCT
International Publication No. WO 94116101; Cohen et al. (1996) Adv.
Chromatogr.
36:127-162; and Griffin et al. (1993) Appl. Biochem. Biotechnol. 38:147-159).
Other methods for detecting mutations include methods in which protection
from cleavage agents is used to detect mismatched bases in RNA/RNA or RNA/DNA
heteroduplexes (Myers et al. (1985) Science 230:1242). In general, the art
technique
of "mismatch cleavage" starts by providing heteroduplexes of formed by
hybridizing
(labeled) RNA or DNA containing the wild-type sequence with potentially mutant
RNA or DNA obtained from a tissue sample. The double-standard duplexes are
treated with an agent that cleaves single-stranded regions of the duplex such
as which
-52-
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
will exist due to base pair mismatches between the control and sample strands.
For
instance, RNA/DNA duplexes can be treated with Rnase and DNA/DNA hybrids
treated with S1 nuclease to enzymatically digest the mismatched regions. After
digestion of the mismatched regions, the resulting material is then separated
by size
on denaturing polyacrylamide gels to determine the site of mutation. See, for
example Cotton et al. (1988) Proc. Natl. Acad. Sci. USA 8:4397; Saleeba et al.
(1992) Methods Enzymol. 217:286-295. In certain embodiments, the control DNA
or
RNA can be labeled for detection.
In still another embodiment, the mismatch cleavage reaction employs one or
more proteins that recognize mismatched base pairs in double-stranded DNA (so
called "DNA mismatch repair" enzymes) in defined systems for detecting and
mapping point mutations in cDNAs obtained from samples of cells. For example,
the
mutt enzyme of E. coli cleaves A at G/A mismatches and the thymidine DNA
glycosylase from HeLa cells cleaves T at G/T mismatches (Hsu et al. (1994)
Carcinogenesis 15:1657-1662). According to an exemplary embodiment, a probe
based on an nucleotide sequence of the invention is hybridized to a cDNA or
other
DNA product from a test cell(s). The duplex is treated with a DNA mismatch
repair
enzyme, and the cleavage products, if any, can be detected from
electrophoresis
protocols or the like. See, for example, U.S. Patent No. 5,459,039.
In other embodiments, alterations in electrophoretic mobility will be used to
identify mutations in genes. For example, single strand conformation
polymorphism
(SSCP) may be used to detect differences in electrophoretic mobility between
mutant
and wild type nucleic acids (Orita et al. (1989) Proc. Natl. Acad Sci. USA
86:2766,
see also Cotton (1993) Mutat Res 285:125-144; and Hayashi (1992) Genet Anal.
Tech.
Appl. 9:73-79). Single-stranded DNA fragments of sample and control nucleic
acids
will be denatured and allowed to renature. The secondary structure of single-
stranded
nucleic acids varies according to sequence, the resulting alteration in
electrophoretic
mobility enables the detection of even a single base change. The DNA fragments
may be labeled or detected with labeled probes. The sensitivity of the assay
may be
enhanced by using RNA (rather than DNA), in which the secondary structure is
more
sensitive to a change in sequence. In one embodiment, the subject method
utilizes
heteroduplex analysis to separate double stranded heteroduplex molecules on
the basis
of changes in electrophoretic mobility (Keen et al. (1991 ) Trends Genet.
7:5).
-53-
CA 02388617 2002-05-07
WO U1/311)U7 PCT/USIIU/29132
In yet another embodiment the movement of mutant or wild-type fragments in
polyacrylamide gels containing a gradient of denaturant is assayed using
denaturing
gradient gel electrophoresis (DGGE) (Myers et al. (1985) Nature 313:495). When
DGGE is used as the method of analysis, DNA will be modified to insure that it
does
not completely denature, for example by adding a GC clamp of approximately 40
by
of high-melting GC-rich DNA by PCR. In a further embodiment, a temperature
gradient is used in place of a denaturing gradient to identify differences in
the
mobility of control and sample DNA (Rosenbaum and Reissner (1987) Biophys.
Chem. 265:12753).
Examples of other techniques for detecting point mutations include, but are
not limited to, selective oligonucleotide hybridization, selective
amplification, or
selective primer extension. For example, oligonucleotide primers may be
prepared in
which the known mutation is placed centrally and then hybridized to target DNA
under conditions which permit hybridization only if a perfect match is found
(Saiki et
al. (1986) Nature 324:163); Saiki et al. (1989) Proc. Natl. Acid. Sci. USA
86:6320).
Such allele-specific oligonucleotides are hybridized to PCR amplified target
DNA or
a number of different mutations when the oligonucleotides are attached to the
hybridizing membrane and hybridized with labeled target DNA.
Alternatively, allele specific amplification technology that depends on
selective PCR amplification may be used in conjunction with the instant
invention.
Oligonucleotides used as primers for specific amplification may carry the
mutation of
interest in the center of the molecule (so that amplification depends on
differential
hybridization) (Gibbs et al. (1989) Nucleic Acids Res. 17:2437-2448) or at the
extreme 3' end of one primer where, under appropriate conditions, mismatch can
prevent, or reduce polymerise extension (Prossner (1993) Tibtech 11:238). In
addition it may be desirable to introduce a novel restriction site in the
region of the
mutation to create cleavage-based detection (Gasparini et al. (1992) Mol. Cell
Probes
6:1). It is anticipated that in certain embodiments amplification may also be
performed using Taq ligase for amplification (Barany (1991) Proc. Natl. Acid.
Sci.
USA 88:189). In such cases, ligation will occur only if there is a perfect
match at the
3' end of the 5' sequence making it possible to detect the presence of a known
mutation at a specific site by looking for the presence or absence of
amplification.
-54-
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
The methods described herein may be performed, for example, by utilizing
pre-packaged diagnostic kits comprising at least one probe nucleic acid or
antibody
reagent described herein, which may be conveniently used, e.g., in clinical
settings to
diagnose patients exhibiting symptoms or family history of a disease or
illness
involving a gene of the present invention. Any cell type or tissue in which
the gene is
expressed may be utilized in the prognostic assays described herein.
3. Monitoring of Effects During Clinical Trials
Monitoring the influence of agents (e.g., drugs, compounds) on the expression
or activity of nucleic acid molecules or proteins of the present invention
(e.g.,
modulation of cellular signal transduction, regulation of gene transcription
in a cell
involved in development or differentiation, regulation of cellular
proliferation) can be
applied not only in basic drug screening, but also in clinical trials. For
example, the
effectiveness of an agent determined by a screening assay as described herein
to
increase gene expression, protein levels, or upregulate protein activity, can
be
monitored in clinical trials of subjects exhibiting decreased gene expression,
protein
levels, or downregulated protein activity. Alternatively, the effectiveness of
an agent
determined by a screening assay to decrease gene expression, protein levels,
or
downregulate protein activity, can be monitored in clinical trials of subjects
exhibiting
increased gene expression, protein levels, or upregulated protein activity. In
such
clinical trials, the expression or activity of the specified gene and,
preferably, other
genes that have been implicated in, for example, a proliferative disorder can
be used
as a "read out" or markers of the phenotype of a particular cell.
For example, and not by way of limitation, genes that are modulated in cells
by treatment with an agent (e.g., compound, drug or small molecule) which
modulates
protein activity (e.g., identified in a screening assay as described herein)
can be
identified. Thus, to study the effect of agents on proliferative disorders,
developmental or differentiative disorder, or hematopoietic disorder, for
example, in a
clinical trial, cells can be isolated and RNA prepared and analyzed for the
levels of
expression of the specified gene and other genes implicated in the
proliferative
disorder, developmental or differentiative disorder, or hematopoietic
disorder,
respectively. The levels of gene expression (i.e., a gene expression pattern)
can be
quantified by Northern blot analysis or RT-PCR, as described herein, or
alternatively
-55-
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
by measuring the amount of protein produced, by one of the methods as
described
herein, or by measuring the levels of activity of the specified gene or other
genes. In
this way, the gene expression pattern can serve as a marker, indicative of the
physiological response of the cells to the agent. Accordingly, this response
state may
be determined before, and at various points during, treatment of the
individual with
the agent.
Disorders relating to programmed cell death are particularly relevant as
discussed in detail herein below.
In one embodiment, the present invention provides a method for monitoring
the effectiveness of treatment of a subject with an agent (e.g., an agonist,
antagonist,
peptidomimetic, protein, polypeptide, nucleic acid, small molecule, or other
drug
candidate identified by the screening assays described herein) comprising the
steps of
(i) obtaining a pre-administration sample from a subject prior to
administration of the
agent; (ii) detecting the level of expression of a specified protein, mRNA, or
genomic
DNA of the invention in the pre-administration sample; (iii) obtaining one or
more
post-administration samples from the subject; (iv) detecting the level of
expression or
activity of the protein, mRNA, or genomic DNA in the post-administration
samples;
(v) comparing the level of expression or activity of the protein, mRNA, or
genomic
DNA in the pre-administration sample with the protein, mRNA, or genomic DNA in
the post-administration sample or samples; and (vi) altering the
administration of the
agent to the subject accordingly. For example, increased administration of the
agent
may be desirable to increase the expression or activity of the protein or
nucleic acid
molecule to higher levels than detected, i.e., to increase effectiveness of
the agent.
Alternatively, decreased administration of the agent may be desirable to
decrease
effectiveness of the agent. According to such an embodiment, protein or
nucleic acid
expression or activity may be used as an indicator of the effectiveness of an
agent,
even in the absence of an observable phenotypic response.
VIII. Screening Assays
The invention provides a method (also referred to herein as a "screening
assay") for identifying modulators, i.e., candidate or test compounds or
agents (e.g.,
antisense, polypeptides, peptidomimetics, small molecules or other drugs)
which bind
to nucleic acid molecules, polypeptides or proteins described herein or have a
-56-
CA 02388617 2002-05-07
WO 01/311107 PCT/USUU/29132
stimulatory or inhibitory effect on, for example, expression or activity of
the nucleic
acid molecules, polypeptides or proteins of the invention.
As an example, apoptosis-specific assays may be used to identify modulators
of any of the target nucleic acids or proteins of the present invention, which
proteins
and/or nucleic acids are related to apoptosis. Accordingly, an agent that
modulates
the level or activity of any of these nucleic acids or proteins can be
identified by
means of apoptosis-specific assays. For example, high throughput screens exist
to
identify apoptotic cells by the use of chromatin or cytoplasmic-specific dyes.
Thus,
hallmarks of apoptosis, cytoplasmic condensation and chromosome fragmentation,
can be used as a marker to identify modulators of any of the genes related to
programmed-cell death described herein. Other assays include, but are not
limited to,
the activation of specific endogenous proteases, loss of mitochondrial
function,
cytoskeleial disruption, cell shrinkage, membrane blebbing, and nuclear
condensation
due to degradation of DNA.
1 ~ In one embodiment, the invention provides assays for screening candidate
or
test compounds that bind to or modulate the activity of protein or polypeptide
described herein or biologically active portion thereof. The test compounds of
the
present invention can be obtained using any of the numerous approaches in
combinatorial library methods known in the art, including: biological
libraries;
spatially addressable parallel solid phase or solution phase libraries;
synthetic library
methods requiring deconvolution; the 'one-bead one-compound' library method;
and
synthetic library methods using affinity chromatography selection. The
biological
library approach is limited to polypeptide libraries, while the other four
approaches
are applicable to polypeptide, non-peptide oligomer or small molecule
libraries of
compounds (Lam, K.S. (1997) Anticancer Drug Des. 12:145).
Examples of methods for the synthesis of molecular libraries can be found in
the art, for example in DeWitt et al. ( 1993) Proc. Natl. Acad. Sci. U.S.A.
90:6909; Erb
et al. (1994) Proc. Natl. Acad. Sci. U.S_A. 91:11422; Zuckermann et al.
(1994). J.
Med Chem. 37:2678; Cho et a1.(1993) Science 261:1303; Carell et al. (1994)
Angew.
Chem. Int. Ed. Engl. 33:2059; Carell et al. ( 1994) Angew. Chem. Int. Ed.
Engl.
33:2061; and in Gallop et al. ( 1994) J. Med. Chem. 3 7:123 3.
Libraries of compounds may be presented in solution (e.g., Houghten (1992)
Biotechnigues 13:412-421 ), or on beads (Lam ( 1991 ) Nature 35-1:82-84),
chips (Fodor
-57-
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
(1993) Nature 364:555-556), bacteria (Ladner U.S. Patent No. 5,223,409),
spores
(Ladner USP '409), plasrnids (Cull et a1.(1992) Proc. Natl. Acad. Sci. U.S.A.
89:1865-1869) or on phage (Scott and Smith (1990) Science 249:386-390);
(Devlin
(1990) Science 2=19:404-406); (Cwirla et al. (1990) Proc. Natl. Acad. Sci.
S 97:6378-6382); (Felici (1991) J. Mol. Biol. 222:301-310); (Ladner supra).
In one embodiment, an assay is a cell-based assay in which a cell that
expresses an encoded polypeptide (e.g., cell surface protein such as a
receptor) is
contacted with a test compound and the ability of the test compound to bind to
the
polypeptide is determined. The cell, for example, can be of mammalian origin,
such
as a keratinocyte. Determining the ability of the test compound to bind to the
polypeptide can be accomplished, for example, by coupling the test compound
with a
radioisotope or enzymatic label such that binding of the test compound to the
polypeptide can be determined by detecting the labeled with ~ZSI, 355, ~4C, or
3H,
either directly or indirectly, and the radioisotope detected by direct
counting of
I 5 radioemmission or by scintillation counting. Alternatively, test compounds
can be
enzymatically labeled with, for example, horseradish peroxidase, alkaline
phosphatase, or luciferase, and the enzymatic label detected by determination
of
conversion of an appropriate substrate to product.
It is also within the scope of this invention to determine the ability of a
test
compound to interact with the polypeptide without the labeling of any of the
interactants. For example, a microphysiometer can be used to detect the
interaction of
a test compound with the polypeptide without the labeling of either the test
compound
or the polypeptide. McConnell et al. (1992) Science 257:1906-1912. As used
herein,
a "microphysiometer" (e.g., CytosensorTM) is an analytical instrument that
measures
the rate at which a cell acidifies its environment using a light-addressable
potentiometric sensor (LAPS). Changes in this acidification rate can be used
as an
indicator of the interaction between ligand and polypeptide.
In one embodiment, the assay comprises contacting a cell which expresses an
encoded protein described herein on the cell surface (e.g., a receptor) with a
polypeptide ligand or biologically-active portion thereof, to form an assay
mixture,
contacting the assay mixture with a test compound, and determining the ability
of the
test compound to interact with the polypeptide, wherein determining the
ability of the
test compound to interact with the polypeptide comprises determining the
ability of
-58-
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
the test compound to preferentially bind to the polypeptide as compared to the
ability
of the ligand, or a biologically active portion thereof, to bind to the
polypeptide.
In another embodiment, an assay is a cell-based assay comprising contacting a
cell expressing a particular target molecule described herein with a test
compound and
determining the ability of the test compound to modulate or alter (e.g.
stimulate or
inhibit) the activity of the target molecule. Determining the ability of the
test
compound to modulate the activity of the target molecule can be accomplished,
for
example, by determining the ability of a known ligand to bind to or interact
with the
target molecule. Determining the ability of the known ligand to bind to or
interact
with the target molecule can be accomplished by one of the methods described
above
for determining direct binding. In one embodiment, determining the ability of
the
known ligand to bind to or interact with the target molecule can be
accomplished by
determining the activity of the target molecule. For example, the activity of
the target
molecule can be determined by detecting induction of a cellular second
messenger of
the target (e.g., intracellular Ca2+, diacylglycerol, IP3, etc.), detecting
catalytic/enzymatic activity of the target an appropriate substrate, detecting
the
induction of a reporter gene (comprising a target-responsive regulatory
element
operatively linked to a nucleic acid encoding a detectable marker, e.g.,
luciferase), or
detecting a cellular response, for example, development, differentiation or
rate of
proliferation.
In yet another embodiment, an assay of the present invention is a cell-free
assay in which protein of the invention or biologically active portion thereof
is
contacted with a test compound and the ability of the test compound to bind to
the
protein or biologically active portion thereof is determined. Binding of the
test
compound to the protein can be determined either directly or indirectly as
described
above. In one embodiment, the assay includes contacting the protein or
biologically
active portion thereof with a known compound which binds the protein to form
an
assay mixture, contacting the assay mixture with a test compound, and
determining
the ability of the test compound to interact with the protein. Determining the
ability of
the test compound to interact with the protein comprises determining the
ability of the
test compound to preferentially bind to the protein or biologically active
portion
thereof as compared to the known compound.
-59-
CA 02388617 2002-05-07
WO 01131007 PCT/USUO/29132
In another embodiment, the assay is a cell-free assay in which a protein of
the
invention or biologically active portion thereof is contacted with a test
compound and
the ability of the test compound to modulate or alter (e.g., stimulate or
inhibit) the
activity of the protein or biologically active portion thereof is determined.
Determining the ability of the test compound to modulate the activity of the
protein
can be accomplished, for example, by determining the ability of the protein to
bind to
a known target molecule by one of the methods described above for determining
direct binding. Determining the ability of the protein to bind to a target
molecule can
also be accomplished using a technology such as real-time Bimolecular
Interaction
Analysis (BIA). Sjolander and Urbaniczky (1991 ) Anal. Chem. 63:2338-2345 and
Szabo et al. (1995) Curr. Opin. Struct. Biol. 5:699-705. As used herein, "BIA"
is a
technology for studying biospecific interactions in real time, without
labeling any of
the interactants (e.g., BIAcoreTM). Changes in the optical phenomenon surface
plasmon resonance (SPR) can be used as an indication of real-time reactions
between
biological molecules.
In an alternative embodiment, determining the ability of the test compound to
modulate the activity of a protein of the invention can be accomplished by
determining the ability of the protein to further modulate the activity of a
target
molecule. For example, the catalytic/enzymatic activity of the target molecule
on an
appropriate substrate can be determined as previously described.
In yet another embodiment, the cell-free assay involves contacting a protein
of
the invention or biologically active portion thereof with a known compound
which
binds the protein to form an assay mixture, contacting the assay mixture with
a test
compound, and determining the ability of the test compound to interact with
the
protein, wherein determining the ability of the test compound to interact with
the
protein comprises determining the ability of the protein to preferentially
bind to or
modulate the activity of a target molecule.
The cell-free assays of the present invention are amenable to use of both
soluble and/or membrane-bound forms of isolated proteins. In the case of cell-
free
assays in which a membrane-bound form an isolated protein is used it may be
desirable to utilize a solubilizing agent such that the membrane-bound form of
the
isolated protein is maintained in solution. Examples of such solubilizing
agents
include non-ionic detergents such as n-octylglucoside, n-dodecylglucoside,
-60-
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
n-dodecylmaltoside, octanoyl-N-methylglucamide, decanoyl-N-methylglucamide,
Triton~X-100, Triton~ X-114, Thesit~, Isotridecypoly(ethylene glycol
ether)n,3-[(3-cholamidopropyl)dimethylamminio]-1-propane sulfonate (CHAPS),
3-[(3-cholamidopropyl)dimethylamminio]-2-hydroxy-1-propane sulfonate
(CHAPSO), or N-dodecyl-N,N-dimethyl-3-ammonio-1-propane sulfonate.
In more than one embodiment of the above assay methods of the present
invention, it may be desirable to immobilize either the protein or its target
molecule to
facilitate separation of complexed from uncomplexed forms of one or both of
the
proteins, as well as to accommodate automation of the assay. Binding of a test
compound to the protein, or interaction of the protein with a target molecule
in the
presence and absence of a candidate compound, can be accomplished in any
vessel
suitable for containing the reactants. Examples of such vessels include
microtitre
plates, test tubes, and micro-centrifuge tubes. In one embodiment, a fusion
protein
can be provided which adds a domain that allows one or both of the proteins to
be
bound to a matrix. For example, glutathione-S-transferase fusion proteins can
be
adsorbed onto glutathione sepharose beads (Sigma Chemical, St. Louis, MO) or
glutathione derivatized microtitre plates, which are then combined with the
test
compound or the test compound and either the non-adsorbed target protein or
protein
of the invention, and the mixture incubated under conditions conducive to
complex
formation (e.g., at physiological conditions for salt and pH). Following
incubation,
the beads or microtitre plate wells are washed to remove any unbound
components,
the matrix immobilized in the case of beads, complex determined either
directly or
indirectly, for example, as described above. Alternatively, the complexes can
be
dissociated from the matrix, and the level of binding or activity determined
using
standard techniques.
Other techniques for immobilizing proteins on matrices can also be used in the
screening assays of the invention. For example, either a protein of the
invention or a
target molecule can be immobilized utilizing conjugation of biotin and
streptavidin.
Biotinylated protein of the invention or target molecules can be prepared from
biotin-NHS(N-hydroxy-succinimide) using techniques well known in the art
(e.g.,
biotinylation kit, Pierce Chemicals, Rockford, IL), and immobilized in the
wells of
streptavidin-coated 96 well plates (Pierce Chemical). Alternatively,
antibodies
reactive with a protein of the invention or target molecules, but which do not
interfere
-61-
CA 02388617 2002-05-07
WO Ol/310U7 PCT/US00/29132
with binding of the protein to its target molecule, can be derivatized to the
wells of the
plate, and unbound target or protein trapped in the wells by antibody
conjugation.
Methods for detecting such complexes, in addition to those described above for
the
GST-immobilized complexes, include immunodetection of complexes using
antibodies reactive with the protein or target molecule, as well as enzyme-
linked
assays which rely on detecting an enzymatic activity associated with the
protein or
target molecule.
In another embodiment, modulators of expression of nucleic acid molecules of
the invention are identified in a method wherein a cell is contacted with a
candidate
compound and the expression of appropriate mRNA or protein in the cell is
determined. The level of expression of appropriate mRNA or protein in the
presence
of the candidate compound is compared to the level of expression of mRNA or
protein in the absence of the candidate compound. The candidate compound can
then
be identified as a modulator of expression based on this comparison. For
example,
I 5 when expression of mRNA or protein is greater (statistically significantly
greater) in
the presence of the candidate compound than in its absence, the candidate
compound
is identified as a stimulator or enhancer of the mRNA or protein expression.
Alternatively, when expression of the mRNA or protein is less (statistically
significantly less) in the presence of the candidate compound than in its
absence, the
candidate compound is identified as an inhibitor of the mRNA or protein
expression.
The level of mRNA or protein expression in the cells can be determined by
methods
described herein for detecting mRNA or protein.
In yet another aspect of the invention, the proteins of the invention can be
used
as "bait proteins" in a two-hybrid assay or three-hybrid assay (see, e.g.,
U.S. Patent
No. 5,283,317; Zervos et al. (1993) Cell 72:223-232; Madura et al. (1993) J.
Biol.
Chem. 268:12046-12054; Bartel et al. (1993) Biotechnigues X4:920-924; Iwabuchi
et
al. (1993) Oncogene 8:1693-1696; and Brent W094/10300), to identify other
proteins
(captured proteins) which bind to or interact with the proteins of the
invention and
modulate their activity. Such captured proteins are also likely to be involved
in the
propagation of signals by the proteins of the invention as, for example,
downstream
elements of a protein-mediated signaling pathway. Alternatively, such captured
proteins are likely to be cell-surface molecules associated with non-protein-
expressing
cells, wherein such captured proteins are involved in signal transduction.
-62-
CA 02388617 2002-05-07
WO Ill/31U07 PCT/OSU(1/29132
This invention further pertains to novel agents identified by the
above-described screening assays. Accordingly, it is within the scope of this
invention to further use an agent identified as described herein in an
appropriate
animal model. For example, an agent identified as described herein (e.g., a
modulating agent, an antisense nucleic acid molecule, a specific antibody, or
a
protein-binding partner) can be used in an animal model to determine the
efficacy,
toxicity, or side effects of treatment with such an agent. Alternatively, an
agent
identified as described herein can be used in an animal model to determine the
mechanism of action of such an agent. Furthermore, this invention pertains to
uses of
novel agents identified by the above-described screening assays for treatments
as
described herein.
IX. Methods of Treatment
The present invention provides for both prophylactic and therapeutic methods
of treating a subject at risk of (or susceptible to) a disorder or having a
disorder
associated with aberrant expression or activity of or related to proteins or
nucleic
acids of the invention. Methods of treatment involve modulating nucleic acid
or
polypeptide level or activity in a subject having a disorder that can be
treated by such
modulation. Accordingly, modulation can cause up regulation or down regulation
of
the levels of expression or up regulation or down regulation of the activity
of the
nucleic acid or protein. Disorders relating to programmed cell death are
particularly
relevant as discussed in detail herein below.
Expression of the nucleic acids of the invention has been shown for the
following tissues: testes, brain, heart, kidney, skeletal muscle, spleen,
lung, smooth
muscle, pancreas, and liver as shown in Figure 8. Accordingly, disorders to
which the
methods disclosed herein are particularly relevant include those involving
these
tissues.
Disorders involving the spleen include, but are not limited to, splenomegaly,
including nonspecific acute splenitis, congestive spenomegaly, and spenic
infarcts;
neoplasms, congenital anomalies, and rupture. Disorders associated with
splenomegaly include infections, such as nonspecific splenitis, infectious
mononucleosis, tuberculosis, typhoid fever, brucellosis, cytomegalovirus,
syphilis,
malaria, histoplasmosis, toxoplasmosis, kala-czar, trypanosomiasis,
schistosomiasis,
-63-
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/2'1132
leishmaniasis, and echinococcosis; congestive states related to partial
hypertension,
such as cirrhosis of the liver, portal or splenic vein thrombosis, and cardiac
failure;
lymphohematogenous disorders, such as Hodgkin disease, non-Hodgkin
iymphomas/leukemia, multiple myeloma, myeloproliferative disorders, hemolytic
ancmias, and thrombocytopenic purpura; immunologic-inflammatory conditions,
such
as rheumatoid arthritis and systemic lupus erythematosus; storage diseases
such as
Gaucher disease, Niemann-Pick disease, and mucopolysaccharidoses; and other
conditions, such as amyloidosis, primary neoplasms and cysts, and secondary
neoplasms.
Disorders involving the lung include, but are not limited to, congenital
anomalies; atelectasis; diseases of vascular origin, such as pulmonary
congestion and
edema, including hemodynamic pulmonary edema and edema caused by
microvascular injury, adult respiratory distress syndrome (diffuse alveolar
damage),
pulmonary embolism, hemorrhage, and infarction, and pulmonary hypertension and
vascular sclerosis; chronic obstructive pulmonary disease, such as emphysema,
chronic bronchitis, bronchial asthma, and bronchiectasis; diffuse interstitial
(infiltrative, restrictive) diseases, such as pneumoconioses, sarcoidosis,
idiopathic
pulmonary fibrosis, desquamative interstitial pneumonitis, hypersensitivity
pneumonitis, pulmonary eosinophilia (pulmonary infiltration with
eosinophilia),
Bronchiolitis obliterans-organizing pneumonia, diffuse pulmonary hemorrhage
syndromes, including Goodpasture syndrome, idiopathic pulmonary hemosiderosis
and other hemorrhagic syndromes, pulmonary involvement in collagen vascular
disorders, and pulmonary alveolar proteinosis; complications of therapies,
such as
drug-induced lung disease, radiation-induced lung disease, and lung
transplantation;
tumors, such as bronchogenic carcinoma, including paraneoplastic syndromes,
bronchioloalveolar carcinoma, neuroendocrine tumors, such as bronchial
carcinoid,
miscellaneous tumors, and metastatic tumors; pathologies of the pleura,
including
inflammatory pleural effusions, noninflammatory pleural effusions,
pneumothorax,
and pleural tumors, including solitary fibrous tumors (pleural fibroma) and
malignant
mesothelioma.
Disorders involving the liver include, but are not limited to, hepatic injury;
jaundice and cholestasis, such as bilirubin and bile formation; hepatic
failure and
cirrhosis, such as cirrhosis, portal hypertension, including ascites,
portosystemic
-64-
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
shunts, and splenomegaly; infectious disorders, such as viral hepatitis,
including
hepatitis A-E infection and infection by other hepatitis viruses,
clinicopathologic
syndromes, such as the carrier state, asymptomatic infection, acute viral
hepatitis,
chronic viral hepatitis, and fulminant hepatitis; autoimmune hepatitis; drug-
and
toxin-induced liver disease, such as alcoholic liver disease; inborn errors of
metabolism and pediatric liver disease, such as hemochromatosis, Wilson
disease, a,-
antitrypsin deficiency, and neonatal hepatitis; intrahepatic biliary tract
disease, such as
secondary biliary cirrhosis, primary biliary cirrhosis, primary sclerosing
cholangitis,
and anomalies of the biliary tree; circulatory disorders, such as impaired
blood flow
into the liver, including hepatic artery compromise and portal vein
obstruction and
thrombosis, impaired blood flow through the liver, including passive
congestion and
centrilobular necrosis and peliosis hepatis, hepatic vein outflow obstruction,
including
hepatic vein thrombosis (Budd-Chiari syndrome) and veno-occlusive disease;
hepatic
disease associated with pregnancy, such as preeclampsia and eclampsia, acute
fatty
liver of pregnancy, and intrehepatic cholestasis of pregnancy; hepatic
complications
of organ or bone marrow transplantation, such as drug toxicity after bone
marrow
transplantation, graft-versus-host disease and liver rejection, and
nonimmunologic
damage to liver allografts; tumors and tumorous conditions, such as nodular
hyperplasias, adenomas, and malignant tumors, including primary carcinoma of
the
liver metastatic tumors, and liver fibrosis.
Disorders involving the brain include, but are not limited to, disorders
involving neurons, and disorders involving glia, such as astrocytes,
oligodendrocytes,
ependymal cells, and microglia; cerebral edema, raised intracranial pressure
and
herniation, and hydrocephalus; malformations and developmental diseases, such
as
neural tube defects, forebrain anomalies, posterior fossa anomalies, and
syringomyelia
and hydromyelia; perinatal brain injury; cerebrovascular diseases, such as
those
related to hypoxia, ischemia, and infarction, including hypotension,
hypoperfusion,
and low-flow states--global cerebral ischemia and focal cerebral ischemia--
infarction
from obstruction of local blood supply, intracranial hemorrhage, including
intracerebral (intraparenchymal) hemorrhage, subarachnoid hemorrhage and
ruptured
berry aneurysms, and vascular malformations, hypertensive cerebrovascular
disease,
including lacunar infarcts, slit hemorrhages, and hypertensive encephalopathy;
infections, such as acute meningitis, including acute pyogenic (bacterial)
meningitis
-65-
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
and acute aseptic (viral) meningitis, acute focal suppurative infections,
including brain
abscess, subdural empyema, and extradural abscess, chronic bacterial
meningoencephalitis, including tuberculosis and mycobacterioses,
neurosyphilis, and
neuroborreliosis (Lyme disease), viral meningoencephalitis, including
arthropod-
borne (Arbo) viral encephalitis, Herpes simplex virus Type 1, Herpes simplex
virus
Type 2, Vcrricalla-zoster virus (Herpes zoster), cytomegalovirus,
poliomyelitis, rabies,
and human immunodeficiency virus 1, including HIV-1 meningoencephalitis
(subacute encephalitis), vacuolar myelopathy, AIDS-associated myopathy,
peripheral
neuropathy, and AIDS in children, progressive multifocal leukoencephalopathy,
subacute sclerosing panencephalitis, fungal meningoencephalitis, other
infectious
diseases of the nervous system; transmissible spongiform encephalopathies
(prion
diseases); demyelinating diseases, including multiple sclerosis, multiple
sclerosis
variants, acute disseminated encephalomyelitis and acute necrotizing
hemorrhagic
encephalomyelitis, and other diseases with demyelination; degenerative
diseases, such
as degenerative diseases affecting the cerebral cortex, including Alzheimer
disease
and Pick disease, degenerative diseases of basal ganglia and brain stem,
including
Parkinsonism, idiopathic Parkinson disease (paralysis agitans), progressive
supranuclear palsy, corticobasal degenration, multiple system atrophy,
including
striatonigral degenration, Shy-Drager syndrome, and olivopontocerebellar
atrophy,
and Huntington disease; spinocerebellar degenerations, including
spinocerebellar
ataxias, including Friedreich ataxia, and ataxia-telanglectasia, degenerative
diseases
affecting motor neurons, including amyotrophie lateral sclerosis (motor neuron
disease), bulbospinal atrophy (Kennedy syndrome), and spinal muscular atrophy;
inborn errors of metabolism, such as leukodystrophies, including Krabbe
disease,
metachromatic leukodystrophy, adrenoleukodystrophy, Pelizaeus-Merzbacher
disease, and Canavan disease, mitochondria) encephalomyopathies, including
Leigh
disease and other mitochondria) encephalomyopathies; toxic and acquired
metabolic
diseases, including vitamin deficiencies such as thiamine (vitamin B~)
deficiency and
vitamin B~2 deficiency, neurologic sequelae of metabolic disturbances,
including
hypoglycemia, hyperglycemia, and hepatic encephatopathy, toxic disorders,
including
carbon monoxide, methanol, ethanol, and radiation, including combined
methotrexate
and radiation-induced injury; tumors, such as gliomas, including astrocytoma,
including fibrillary (diffuse) astrocytoma and glioblastoma multiforme,
pilocytie
-66-
CA 02388617 2002-05-07
WO 01/31007 PCT/iIS00/29132
astrocytoma, pleomorphic xanthoastrocytoma, and brain stem glioma,
oligodendroglioma, and ependymoma and related paraventricular mass lesions,
neuronal tumors, poorly differentiated neoplasms, including medulloblastoma,
other
parenchymal tumors, including primary brain lymphoma, germ cell tumors, and
pineal parenchymal tumors, meningiomas, metastatic tumors, paraneoplastic
syndromes, peripheral nerve sheath tumors, including schwannoma, neurofibroma,
and malignant peripheral nerve sheath tumor (malignant schwannoma), and
neurocutaneous syndromes (phakomatoses), including neurofibromotosis,
including
Type 1 neurofibromatosis (NF1) and TYPE 2 neurofibromatosis (NF2), tuberous
sclerosis, and Von Hippel-Lindau disease.
Disorders involving the heart, include but are not limited to, heart failure,
including but not limited to, cardiac hypertrophy, left-sided heart failure,
and right-
sided heart failure; ischemic heart disease, including but not limited to
angina
pectoris, myocardial infarction, chronic ischemic heart disease, and sudden
cardiac
death; hypertensive heart disease, including but not limited to, systemic
(left-sided)
hypertensive heart disease and pulmonary (right-sided) hypertensive heart
disease;
valvular heart disease, including but not limited to, valvular degeneration
caused by
calcification, such as calcific aortic stenosis, calcification of a
congenitally bicuspid
aortic valve, and mural annular calcification, and myxomatous degeneration of
the
mitral valve (mural valve prolapse), rheumatic fever and rheumatic heart
disease,
infective endocarditis, and noninfected vegetations, such as nonbacterial
thrombotic
endocarditis and endocarditis of systemic lupus erythematosus (Libman-Sacks
disease), carcinoid heart disease, and complications of artificial valves;
myocardial
disease, including but not limited to dilated cardiomyopathy, hypertrophic
cardiomyopathy, restrictive cardiomyopathy, and myocarditis; pericardial
disease,
including but not limited to, pericardial effusion and hemopericardium and
pericarditis, including acute pericarditis and healed pericarditis, and
rheumatoid heart
disease; neoplastic heart disease, including but not limited to, primary
cardiac tumors,
such as myxoma, lipoma, papillary fibroelastoma, rhabdomyoma, and sarcoma, and
cardiac effects of noncardiac neoplasms; congenital heart disease, including
but not
limited to, left-to-right shunts--late cyanosis, such as atrial septal defect,
ventricular
septal defect, patent ductus arteriosus, and atrioventricular septal defect,
right-to-left
shunts--early cyanosis, such as tetralogy of fallot, transposition of great
arteries,
-67-
CA 02388617 2002-05-07
WO 01131007 PCT/US00/29132
truncus arteriosus, tricuspid atresia, and total anomalous pulmonary venous
connection, obstructive congenital anomalies, such as coarctation of aorta,
pulmonary
stenosis and atresia, and aortic stenosis and atresia, and disorders involving
cardiac
transplantation.
S Disorders involving the kidney include, but are not limited to, congenital
anomalies including, but not limited to, cystic diseases of the kidney, that
include but are
not limited to, cystic renal dysplasia, autosomal dominant (adult) polycystic
kidney
disease, autosomal recessive (childhood) polycystic kidney disease, and cystic
diseases
of renal medulla, which include, but are not limited to, medullary sponge
kidney, and
nephronophthisis-uremic medullary cystic disease complex, acduired (dialysis-
associated) cystic disease, such as simple cysts; glomerular diseases
including
pathologies of glomerular injury that include, but are not limited to, in situ
immune
complex deposition, that includes, but is not limited to, anti-GBM nephritis,
Heymann
nephritis, and antibodies against planted antigens, circulating immune complex
nephritis,
antibodies to glomerular cells, cell-mediated immunity in glomerulonephritis,
activation
of alternative complement pathway, epithelial cell injury, and pathologies
involving
mediators of glomerular injury including cellular and soluble mediators, acute
glomerulonephritis, such as acute proliferative (poststreptococcal,
postinfectious)
glomerulonephritis, including but not limited to, poststreptococcal
glomerulonephritis
and nonstreptococcal acute glomerulonephritis, rapidly progressive
(crescentic)
glomerulonephritis, nephrotic syndrome, membranous glomerulonephritis
(membranous
nephropathy), minimal change disease (lipoid nephrosis), focal segmental
glomerulosclerosis, membranoproliferative glomerulonephritis, IgA nephropathy
{Berger disease), focal proliferative and necrotizing glomerulonephritis
(focal
glomerulonephritis), hereditary nephritis, including but not limited to,
Alport syndrome
and thin membrane disease (benign familial hematuria), chronic
glomerulonephritis,
glomerular lesions associated with systemic disease, including but not limited
to,
systemic lupus erythematosus, Henoch-Schonlein purpura, bacterial
endocarditis,
diabetic glomerulosclerosis, amyloidosis, fibrillary and immunotactoid
glomerulonephritis, and other systemic disorders; diseases affecting tubules
and
interstitium, including acute tubular necrosis and tubulointerstitial
nephritis, including
but not limited to, pyelonephritis and urinary tract infection, acute
pyelonephritis,
chronic pyelonephritis and reflux nephropathy, and tubulointerstitial
nephritis induced
-68-
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
by drugs and toxins, including but not limited to, acute drug-induced
interstitial
nephritis, analgesic abuse nephropathy, nephropathy associated with
nonsteroidal anti-
inflammatory drugs, and other tubulointerstitial diseases including, but not
limited to,
orate nephropathy, hypercalcemia and nephrocalcinosis, and multiple myeloma;
diseases
of blood vessels including benign nephrosclerosis, malignant hypertension and
accelerated nephrosclerosis, renal artery stenosis, and thrombotic
microangiopathies
including, but not limited to, classic (childhood) hemolytic-uremic syndrome,
adult
hemolytic-uremic syndrome/thrombotic thrombocytopenic purpura, idiopathic
HUS/TTP, and other vascular disorders including, but not limited to,
atherosclerotic
ischemic renal disease, atheroembolic renal disease, sickle cell disease
nephropathy,
diffuse cortical necrosis, and renal infarcts; urinary tract obstruction
(obstructive
uropathy); urolithiasis (renal calculi, stones); and tumors of the kidney
including, but not
limited to, benign tumors, such as renal papillary adenoma, renal fibroma or
hamartoma
(renomedullary interstitial cell tumor), angiomyolipoma, and oncocytoma, and
malignant
tumors, including renal cell carcinoma (hypernephroma, adenocarcinoma of
kidney),
which includes urothelial carcinomas of renal pelvis.
Disorders involving the testis and epididymis include, but are not limited to,
congenital anomalies such as cryptorchidism, regressive changes such as
atrophy,
inflammations such as nonspecific epididymitis and orchitis, granulomatous
(autoimmune) orchids, and specific inflammations including, but not limited
to,
gonorrhea, mumps, tuberculosis, and syphilis, vascular disturbances including
torsion,
testicular tumors including germ cell tumors that include, but are not limited
to,
seminoma, spermatocytic seminoma, embryonal carcinoma, yolk sac tumor
choriocarcinoma, teratoma, and mixed tumors, tumore of sex cord-gonadal stroma
including, but not limited to, leydig (interstitial) cell tumors and sertoli
cell tumors
(androblastoma), and testicular lymphoma, and miscellaneous lesions of tunica
vaginalis.
Disorders involving the skeletal muscle include tumors such as
rhabdomyosarcoma.
Disorders involving the pancreas include those of the exocrine pancreas such
as
congenital anomalies, including but not limited to, ectopic pancreas;
pancreatitis,
including but not limited to, acute pancreatitis; cysts, including but not
limited to,
pseudocysts; tumors, including but not limited to, cystic tumors and carcinoma
of the
pancreas; and disorders of the endocrine pancreas such as, diabetes mellitus;
islet cell
-69-
CA 02388617 2002-05-07
WO fll/310f17 PCT/USOII/29132
tumors, including but not limited to, insulinomas, gastrinomas, and other rare
islet cell
tumors.
Preferred disorders include those involving the central nervous system and
particularly the brain.
With regard to both prophylactic and therapeutic methods of treatment, such
treatments may be specifically tailored or modified, based on knowledge
obtained
from the field of pharmacogenomics. "Pharmacogenomics", as used herein, refers
to
the application of genomics technologies such as gene sequencing, statistical
genetics,
and gene expression analysis to drugs in clinical development and on the
market.
More specifically, the term refers the study of how a patient's genes
determine his or
her response to a drug (e.g., a patient's "drug response phenotype", or "drug
response
genotype".) Thus, another aspect of the invention provides methods for
tailoring an
individual's prophylactic or therapeutic treatment with the molecules of the
present
invention or modulators according to that individual's drug response genotype.
Pharmacogenomies allows a clinician or physician to target prophylactic or
therapeutic treatments to patients who will most benefit from the treatment
and to
avoid treatment of patients who will experience toxic drug related side
effects.
Prophylactic Methods
In one aspect, the invention provides a method for preventing in a subject, a
disease or condition associated with aberrant expression or activity of genes
or
proteins of the present invention, by administering to the subject an agent
which
modulates expression or at least one activity of a gene or protein of the
invention.
Subjects at risk for a disease that is caused or contributed to by aberrant
gene
expression or protein activity can be identified by, for example, any or a
combination
of diagnostic or prognostic assays as described herein. Administration of a
prophylactic agent can occur prior to the manifestation of symptoms
characteristic of
the aberrancy, such that a disease or disorder is prevented or, alternatively,
delayed in
its progression. Depending on the type of aberrancy, for example, an agonist
or
antagonist agent can be used for treating the subject. The appropriate agent
can be
determined based on screening assays described herein.
-70-
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
2. Therapeutic Methods
Another aspect of the invention pertains to methods of modulating expression
or activity of genes or proteins of the invention for therapeutic purposes.
The
modulatory method of the invention involves contacting a cell with an agent
that
modulates one or more of the activities of the specified protein associated
with the
cell. An agent that modulates protein activity can be an agent as described
herein,
such as a nucleic acid or a protein, a naturally-occurring target molecule of
a protein
described herein, a polypeptide, a peptidomimetic, or other small molecule. In
one
embodiment, the agent stimulates one or more protein activities. Examples of
such
stimulatory agents include active protein as well as a nucleic acid molecule
encoding
the protein that has been introduced into the cell. In another embodiment, the
agent
inhibits one or more protein activities. Examples of such inhibitory agents
include
antisense nucleic acid molecules and anti-protein antibodies. These modulatory
methods can be performed in vitro (e.g., by culturing the cell with the agent)
or,
alternatively, irc vivo (e.g., by administering the agent to a subject). As
such, the
present invention provides methods of treating an individual afflicted with a
disease
or disorder characterized by aberrant expression or activity of a protein or
nucleic acid
molecule of the invention. In one embodiment, the method involves
administering an
agent (e.g., an agent identified by a screening assay described herein), or
combination
of agents that modulates (e.g., upregulates or downregulates) expression or
activity of
a gene or protein of the invention. In another embodiment, the method involves
administering a protein or nucleic acid molecule of the invention as therapy
to
compensate for reduced or aberrant expression or activity of the protein or
nucleic
acid molecule.
Stimulation of protein activity is desirable in situations in which the
protein is
abnormally downregulated and/or in which increased protein activity is likely
to have
a beneficial effect. Likewise, inhibition of protein activity is desirable in
situations in
which the protein is abnormally upregulated and/or in which decreased protein
activity is likely to have a beneficial effect. One example of such a
situation is where
a subject has a disorder characterized by aberrant development or cellular
differentiation. Another example of such a situation is where the subject has
a
proliferative disease (e.g., cancer) or a disorder characterized by an
aberrant
-71-
CA 02388617 2002-05-07
WO 01/31007 PCT/US00J29132
hematopoietic response. Yet another example of such a situation is where it is
desirable to achieve tissue regeneration in a subject (e.g., where a subject
has
undergone brain or spinal cord injury and it is desirable to regenerate
neuronal tissue
in a regulated manner).
Pharmaceutical Compositions
The nucleic acid molecules, protein modulators of the protein, and antibodies
(also referred to herein as "active compounds") can be incorporated into
pharmaceutical
compositions suitable for administration to a subject, e.g., a human. Such
compositions
typically comprise the nucleic acid molecule, protein, modulator, or antibody
and a
pharmaceutically acceptable carrier.
The term "administer" is used in its broadest sense and includes any method of
introducing the compositions of the present invention into a subject. This
includes
producing polypeptides or polynucleotides in vivo as by transcription or
translation, in
vivo, of polynucleotides that have been exogenously introduced into a subject.
Thus,
polypeptides or nucleic acids produced in the subject from the exogenous
compositions
are encompassed in the term "administer."
As used herein the language "pharmaceutically acceptable carrier" is intended
to
include any and all solvents, dispersion media, coatings, antibacterial and
antifungal
agents, isotonic and absorption delaying agents, and the like, compatible with
pharmaceutical administration. The use of such media and agents for
pharmaceutically
active substances is well known in the art. Except insofar as any conventional
media or
agent is incompatible with the active compound, such media can be used in the
compositions of the invention. Supplementary active compounds can also be
incorporated into the compositions. A pharmaceutical composition of the
invention is
formulated to be compatible with its intended route of administration.
Examples of
routes of administration include parenteral, e.g., intravenous, intradermal,
subcutaneous,
oral (e.g., inhalation), transdermal (topical), transmucosal, and rectal
administration.
Solutions or suspensions used for parenteral, intradermal, or subcutaneous
application
can include the following components: a sterile diluent such as water for
injection, saline
solution, fixed oils, polyethylene glycols, glycerine, propylene glycol or
other synthetic
solvents; antibacterial agents such as benzyl alcohol or methyl parabens;
antioxidants
such as ascorbic acid or sodium bisulfate; chelating agents such as
-72-
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
ethylenediaminetetraacetic acid; buffers such as acetates, citrates or
phosphates and
agents for the adjustment of tonicity such as sodium chloride or dextrose. pH
can be
adjusted with acids or bases, such as hydrochloric acid or sodium hydroxide.
The
parenteral preparation can be enclosed in ampules, disposable syringes or
multiple dose
vials made of glass or plastic.
Pharmaceutical compositions suitable for injectable use include sterile
aqueous
solutions (where water soluble) or dispersions and sterile powders for the
extemporaneous preparation of sterile injectable solutions or dispersion. For
intravenous
administration, suitable carriers include physiological saline, bacteriostatic
water,
Cremophor ELT"" (BASF, Parsippany, NJ) or phosphate buffered saline (PBS). In
all
cases, the composition must be sterile and should be fluid to the extent that
easy
syringability exists. It must be stable under the conditions of manufacture
and storage
and must be preserved against the contaminating action of microorganisms such
as
bacteria and fungi. The carrier can be a solvent or dispersion medium
containing, for
example, water, ethanol, polyol (for example, glycerol, propylene glycol, and
liquid
polyethylene glycol, and the like), and suitable mixtures thereof. The proper
fluidity can
be maintained, for example, by the use of a coating such as lecithin, by the
maintenance
of the required particle size in the case of dispersion and by the use of
surfactants.
Prevention of the action of microorganisms can be achieved by various
antibacterial and
antifungal agents, for example, parabens, chlorobutanol, phenol, ascorbic
acid,
thimerosal, and the like. In many cases, it will be preferable to include
isotonic agents,
for example, sugars, polyalcohols such as mannitol, sorbitol, sodium chloride
in the
composition. Prolonged absorption of the injectable compositions can be
brought about
by including in the composition an agent which delays absorption, for example,
aluminum monostearate and gelatin.
Sterile injectable solutions can be prepared by incorporating the active
compound (e.g., a ubiquitin protease protein or anti- ubiquitin protease
antibody) in the
required amount in an appropriate solvent with one or a combination of
ingredients
enumerated above, as required, followed by filtered sterilization. Generally,
dispersions
are prepared by incorporating the active compound into a sterile vehicle which
contains
a basic dispersion medium and the required other ingredients from those
enumerated
above. In the case of sterile powders for the preparation of sterile
injectable solutions,
the preferred methods of preparation are vacuum drying and freeze-drying which
yields
-73-
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
a powder of the active ingredient plus any additional desired ingredient from
a
previously sterile-filtered solution thereof.
Oral compositions generally include an inert diluent or an edible carrier.
They
can be enclosed in gelatin capsules or compressed into tablets. For oral
administration,
the agent can be contained in enteric forms to survive the stomach or further
coated or
mixed to be released in a particular region of the GI tract by known methods.
For the
purpose of oral therapeutic administration, the active compound can be
incorporated
with excipients and used in the form of tablets, troches, or capsules. Oral
compositions
can also be prepared using a fluid carrier for use as a mouthwash, wherein the
compound
in the fluid Garner is applied orally and swished and expectorated or
swallowed.
Pharmaceutically compatible binding agents, and/or adjuvant materials can be
included
as part of the composition. The tablets, pills, capsules, troches and the like
can contain
any of the following ingredients, or compounds of a similar nature: a binder
such as
microcrystalline cellulose, gum tragacanth or gelatin; an excipient such as
starch or
lactose, a disintegrating agent such as alginic acid, Primogel, or corn
starch; a lubricant
such as magnesium stearate or Sterotes; a glidant such as colloidal silicon
dioxide; a
sweetening agent such as sucrose or saccharin; or a flavoring agent such as
peppermint,
methyl salicylate, or orange flavoring.
For administration by inhalation, the compounds are delivered in the form of
an
aerosol spray from pressured container or dispenser, which contains a suitable
propellant, e.g., a gas such as carbon dioxide, or a nebulizer.
Systemic administration can also be by transmueosal or transdermal means. For
transmucosal or transdermal administration, penetrants appropriate to the
barrier to be
permeated are used in the formulation. Such penetrants are generally known in
the art,
and include, for example, for transmucosal administration, detergents, bile
salts, and
fusidic acid derivatives. Transmucosal administration can be accomplished
through the
use of nasal sprays or suppositories. For transdermal administration, the
active
compounds are formulated into ointments, salves, gels, or creams as generally
known in
the art.
The compounds can also be prepared in the form of suppositories (e.g., with
conventional suppository bases such as cocoa butter and other glycerides) or
retention
enemas for rectal delivery.
-74-
CA 02388617 2002-05-07
WO 01/31007 PCT/US110/29132
In one embodiment, the active compounds are prepared with earners that will
protect the compound against rapid elimination from the body, such as a
controlled
release formulation, including implants and microencapsulated delivery
systems.
Biodegradable, biocompatible polymers can be used, such as ethylene vinyl
acetate,
polyanhydrides, polyglycolic acid, collagen, polyorthoesters, and polylactic
acid.
Methods for preparation of such formulations will be apparent to those skilled
in the art.
The materials can also be obtained commercially from Alza Corporation and Nova
Pharmaceuticals, Inc. Liposomal suspensions (including liposomes targeted to
infected
cells with monoclonal antibodies to viral antigens) can also be used as
pharmaceutically
acceptable carriers. These can be prepared according to methods known to those
skilled
in the art, for example, as described in U.S. Patent No. 4,522,811.
It is especially advantageous to formulate oral or parenteral compositions in
dosage unit form for ease of administration and uniformity of dosage. "Dosage
unit
form" as used herein refers to physically discrete units suited as unitary
dosages for the
subject to be treated; each unit containing a predetermined quantity of active
compound
calculated to produce the desired therapeutic effect in association with the
required
pharmaceutical carrier. The specification for the dosage unit forms of the
invention are
dictated by and directly dependent on the unique characteristics of the active
compound
and the particular therapeutic effect to be achieved, and the limitations
inherent in the art
of compounding such an active compound for the treatment of individuals.
The nucleic acid molecules of the invention can be inserted into vectors and
used
as gene therapy vectors. Gene therapy vectors can be delivered to a subject
by, for
example, intravenous injection, local administration (U.S. 5,328,470) or by
stereotactic
injection (see e.g., Chen et al. ( 1994) PNAS 91:3054-3057). The
pharmaceutical
preparation of the gene therapy vector can include the gene therapy vector in
an
acceptable diluent, or can comprise a slow release matrix in which the gene
delivery
vehicle is imbedded. Alternatively, where the complete gene delivery vector
can be
produced intact from recombinant cells, e.g. retroviral vectors, the
pharmaceutical
preparation can include one or more cells which produce the gene delivery
system.
The pharmaceutical compositions can be included in a container, pack, or
dispenser together with instructions for administration.
As defined herein, a therapeutically effective amount of protein or
polypeptide
(i.e., an effective dosage) ranges from about 0.001 to 30 mg/kg body weight,
-75-
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
preferably about 0.01 to 25 mg/kg body weight, more preferably about 0.1 to 20
mg/kg body weight, and even more preferably about 1 to 10 mg/kg, 2 to 9 mg/kg,
3 to
8 mg/kg, 4 to 7 mg/kg, or 5 to 6 mg/kg body weight.
The skilled artisan will appreciate that certain factors may influence the
dosage required to effectively treat a subject, including but not limited to
the severity
of the disease or disorder, previous treatments, the general health and/or age
of the
subject, and other diseases present. Moreover, treatment of a subject with a
therapeutically effective amount of a protein, polypeptide, or antibody can
include a
single treatment or, preferably, can include a series of treatments. In a
preferred
example, a subject is treated with antibody, protein, or polypeptide in the
range of
between about 0.1 to 20 mg/kg body weight, one time per week for between about
1
to 10 weeks, preferably between 2 to 8 weeks, more preferably between about 3
to 7
weeks, and even more preferably for about 4, 5, or 6 weeks. It will also be
appreciated that the effective dosage of antibody, protein, or polypeptide
used for
treatment may increase or decrease over the course of a particular treatment.
Changes
in dosage may result and become apparent from the results of diagnostic assays
as
described herein.
The present invention encompasses agents which modulate expression or
activity. An agent may, for example, be a small molecule. For example, such
small
molecules include, but are not limited to, peptides, peptidomimetics, amino
acids,
amino acid analogs, polynucleotides, polynucleotide analogs, nucleotides,
nucleotide
analogs, organic or inorganic compounds (i.e., including heteroorganic and
organometallic compounds) having a molecular weight less than about 10,000
grams
per mole, organic or inorganic compounds having a molecular weight less than
about
5,000 grams per mole, organic or inorganic compounds having a molecular weight
less than about 1,000 grams per mole, organic or inorganic compounds having a
molecular weight less than about 500 grams per mole, and salts, esters, and
other
pharmaceutically acceptable forms of such compounds.
It is understood that appropriate doses of small molecule agents depends upon
a number of factors within the ken of the ordinarily skilled physician,
veterinarian, or
researcher. The doses) of the small molecule will vary, for example, depending
upon
the identity, size, and condition of the subject or sample being treated,
further
depending upon the route by which the composition is to be administered, if
-76-
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
applicable, and the effect which the practitioner desires the small molecule
to have
upon the nucleic acid or polypeptide of the invention. Exemplary doses include
milligram or microgram amounts of the small molecule per kilogram of subject
or
sample weight (e.g., about 1 microgram per kilogram to about 500 milligrams
per
kilogram, about 100 micrograms per kilogram to about S milligrams per
kilogram, or
about 1 microgram per kilogram to about 50 micrograms per kilogram. It is
furthermore understood that appropriate doses of a small molecule depend upon
the
potency of the small molecule with respect to the expression or activity to be
modulated. Such appropriate doses may be determined using the assays described
herein. When one or more of these small molecules is to be administered to an
animal
(e.g., a human) in order to modulate expression or activity of a polypeptide
or nucleic
acid of the invention, a physician, veterinarian, or researcher may, for
example,
prescribe a relatively low dose at first, subsequently increasing the dose
until an
appropriate response is obtained. In addition, it is understood that the
specific dose
level for any particular animal subject will depend upon a variety of factors
including
the activity of the specific compound employed, the age, body weight, general
health,
gender, and diet of the subject, the time of administration, the route of
administration,
the rate of excretion, any drug combination, and the degree of expression or
activity to
be modulated.
3. Pharmacogenomics
The molecules of the present invention, as well as agents, or modulators which
have a stimulatory or inhibitory effect on the protein activity (e.g., gene
expression) as
identified by a screening assay described herein can be administered to
individuals to
treat (prophylactically or therapeutically) disorders (e.g., proliferative or
developmental disorders) associated with aberrant protein activity. In
conjunction
with such treatment, pharmacogenomics (i. e., the study of the relationship
between an
individual's genotype and that individual's response to a foreign compound or
drug)
may be considered. Differences in metabolism of therapeutics can lead to
severe
toxicity or therapeutic failure by altering the relation between dose and
blood
concentration of the pharmacologically active drug. Thus, a physician or
clinician
may consider applying knowledge obtained in relevant pharmacogenomics studies
in
determining whether to administer a molecule of the invention or modulator
thereof,
_77_
CA 02388617 2002-05-07
WO 01/31007 PCT/IJS00/29132
as well as tailoring the dosage and/or therapeutic regimen of treatment with
such a
molecule or modulator.
Pharmacogenomics deals with clinically significant hereditary variations in
the
response to drugs due to altered drug disposition and abnormal action in
affected
persons. See e.g., Eichelbaum (1996) Clin Exp. Pharmacol. Physiol.
23(10-11):983-985 and Linder (1997) Clin. Chem. =13(2):254-266. In general,
two
types of pharmacogenetic conditions can be differentiated. Genetic conditions
transmitted as a single factor altering the way drugs act on the body (altered
drug
action) or genetic conditions transmitted as single factors altering the way
the body
acts on drugs (altered drug metabolism). These pharmacogenetic conditions can
occur either as rare genetic defects or as naturally-occurring polymorphisms.
For
example, glucose-6-phosphate dehydrogenase deficiency (G6PD) is a common
inherited enzymopathy in which the main clinical complication is haemolysis
after
ingestion of oxidant drugs (anti-malarials, sulfonamides, analgesics,
nitrofurans) and
consumption of fava beans.
One pharmacogenomics approach to identifying genes that predict drug
response, known as "a genome-wide association", relies primarily on a
high-resolution map of the human genome consisting of already known gene-
related
markers (e.g., a "bi-allelic" gene marker map which consists of 60,000-100,000
polymorphic or variable sites on the human genome, each of which has two
variants).
Such a high-resolution genetic map can be compared to a map of the genome of
each
of a statistically significant number of patients taking part in a Phase
II/III drug trial to
identify markers associated with a particular observed drug response or side
effect.
Alternatively, such a high resolution map can be generated from a combination
of
some ten-million known single nucleotide polymorphisms (SNPs) in the human
genome. As used herein, a "SNP" is a common alteration that occurs in a single
nucleotide base in a stretch of DNA. For example, a SNP may occur once per
every
1,000 bases of DNA. A SNP may be involved in a disease process, however, the
vast
majority may not be disease-associated. Given a genetic map based on the
occurrence
of such SNPs, individuals can be grouped into genetic categories depending on
a
particular pattern of SNPs in their individual genome. In such a manner,
treatment
regimens can be tailored to groups of genetically similar individuals, taking
into
account traits that may be common among such genetically similar individuals.
_78_
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
Alternatively, a method termed the "candidate gene approach", can be utilized
to identify genes that predict drug response. According to this method, if a
gene that
encodes a drug's target is known (e.g., a protein or a polypeptide of the
present
invention), all common variants of that gene can be fairly easily identified
in the
population and it can be determined if having one version of the gene versus
another
is associated with a particular drug response.
As an illustrative embodiment, the activity of drug metabolizing enzymes is a
major determinant of both the intensity and duration of drug action. The
discovery of
genetic polymorphisms of drug metabolizing enzymes (e. g., N-acetyltransferase
2(NAT 2) and cytochrome P450 enzymes CYP2D6 and CYP2C 19) has provided an
explanation as to why some patients do not obtain the expected drug effects or
show
exaggerated drug response and serious toxicity after taking the standard and
safe dose
of a drug. These polymorphisms are expressed in two phenotypes in the
population,
the extensive metabolizes (EM) and poor metabolizes (PM). The prevalence of PM
is
different among different populations. For example, the gene coding for CYP2D6
is
highly polymorphic and several mutations have been identified in PM, which all
lead
to the absence of functional CYP2D6. Poor metabolizers of CYP2D6 and CYP2C 19
quite frequently experience exaggerated drug response and side effects when
they
receive standard doses. If a metabolite is the active therapeutic moiety, PM
show no
therapeutic response, as demonstrated for the analgesic effect of codeine
mediated by
its CYP2D6-formed metabolite morphine. The other extreme is the so called
ultra-rapid metabolizers who do not respond to standard doses. Recently, the
molecular basis of ultra-rapid metabolism has been identified to be due to
CYP2D6
gene amplification.
Alternatively, a method termed the "gene expression profiling", can be
utilized
to identify genes that predict drug response. For example, the gene expression
of an
animal dosed with a drug (e.g., a molecule or modulator of the present
invention) can
given an indication whether gene pathways related to toxicity have been turned
on.
Information generated from more than one of the above pharmacogenomics
approaches can be used to determine appropriate dosage and treatment regimens
for
prophylactic or therapeutic treatment an individual. This knowledge, when
applied to
dosing or drug selection, can avoid adverse reactions or therapeutic failure
and thus
enhance therapeutic or prophylactic efficiency when treating a subject with a
-79-
CA 02388617 2002-05-07
WO 01/31007 PCT/lJS()0/29132
molecule or modulator of the invention, such as a modulator identified by one
of the
exemplary screening assays described herein.
Disorders which may be treated or diagnosed by methods described herein
include, but are not limited to disorders involving apoptosis. Certain
disorders are
associated with an increased number of surviving cells, which are produced and
continue to survive or proliferate when apoptosis is inhibited.
As used herein, "programmed cell death" refers to a genetically regulated
process involved in the normal development of multicellular organisms. This
process
occurs in cells destined for removal in a variety of normal situations,
including larval
development of the nematode C. elegans, insect metamorphosis, development in
mammalian embryos, including the nephrogenic zone in the developing kidney,
and
regression or atrophy (e.g., in the prostate after castration). Programmed
cell death
can occur following the withdrawal of growth and trophic factors in many
cells,
nutritional deprivation, hormone treatment, ultraviolet irradiation, and
exposure to
toxic and infectious agents including reactive oxygen species and phosphatase
inhibitors, e.g., okadaic acid, calcium ionophores, and a number of cancer
chemotherapeutic agents. See Wilson (1998) Biochem. Cell Biol. 76:573-582 and
Hetts (1998) JAMA 279:300-307, the contents of which are incorporated herein
by
reference. Thus, the proteins of the invention, by being differentially
expressed
during programmed cell death, e.g., neuronal programmed cell death, can
modulate a
programmed cell death pathway activity and provide novel diagnostic targets
and
therapeutic agents for disorders characterized by deregulated programmed cell
death,
particularly in cells that express the protein.
As used herein, a "disorder characterized by deregulated programmed cell
death" refers to a disorder, disease or condition which is characterized by a
deregulation, e.g., an upregulation or a downregulation, of programmed cell
death.
Programmed cell death deregulation can lead to deregulation of cellular
proliferation
and/or cell cycle progression. Examples of disorders characterized by
deregulated
programmed cell death include, but are not limited to, neurodegenerative
disorders,
e.g., Alzheimer's disease, demential related to Alzheimer's disease (such as
Pick's
disease), Parkinson's and other Lewy diffuse body diseases, multiple
sclerosis,
amyotrophic lateral sclerosis, progressive supranuclear palsy, epilepsy, Jakob-
Creutzfieldt disease, or AIDS related dementias; myelodysplastic syndromes,
e.g.,
-80-
CA 02388617 2002-05-07
WO 01/31007 PCT/USOII/29132
aplastic anemia; ischemic injury, e.g., myocardial infarction, stroke, or
reperfusion
injury; autoimmune disorders, e.g., systemic lupus erythematosus, or immune-
mediated glomerulonephritis; or profilerative disorders, e.g., cancer, such as
follicular
lymphomas, carcinomas with p53 mutations, or hormone-dependent tumors, e.g.,
breast cancer, prostate cancer, or ovarian cancer). Clinical manifestations of
faulty
apoptosis are also seen in stroke and in rheumatoid arthritis. Wilson (1998)
Biochem.
Cell. Biol. 76:573-582.
Failure to remove autoimmune cells that arise during development or that
develop as a result of somatic mutation during an immune response can result
in
autoimmune disease. One of the molecules that plays a critical role in
regulating cell
death in lymphocytes is the cell surface receptor for Fas.
Viral infections, such as those caused by herpesviruses, poxviruses, and
adenoviruses, may result in aberrant apoptosis. Populations of cells are often
depleted
in the event of viral infection, with perhaps the most dramatic example being
the cell
depletion caused by the human immunodeficiency virus (HIV). Most T cells that
die
during HIV infections do not appear to be infected with HIV. Stimulation of
the CD4
receptor may result in the enhanced susceptibility of uninfected T cells to
undergo
apoptosis.
Many disorders can be classified based on whether they are associated with
abnormally high or abnormally low apoptosis. Thompson (1995) Science 267:1456-
1462. Apoptosis may be involved in acute trauma, myocardial infarction,
stroke, and
infectious diseases, such as viral hepatitis and acquired immunodeficiency
syndrome.
Primary apoptosis deficiencies include graft rejection. Accordingly, the
invention is relevant to the identification of genes useful in inhibiting
graft rejection.
Primary apoptosis deficiencies also include autoimmune diabetes.
Accordingly, the invention is relevant to the identification of genes involved
in
autoimmune diabetes and accordingly, to the identification of agents that act
on these
targets to modulate the expression of these genes and hence, to treat or
diagnose this
disorder. Further, it has been suggested that all autoimmune disorders can be
viewed
as primary deficiencies of apoptosis (Hens, above). Accordingly, the invention
is
relevant for screening for gene expression and transcriptional profiling in
any
autoimmune disorder and for screening for agents that affect the expression or
transcriptional profile of these genes.
-81-
CA 02388617 2002-05-07
WO 01/31007 PCT/USOO129132
Primary apoptosis deficiencies also include local self reactive disorder. This
includes Hashimoto thyroiditis.
Primary apoptosis deficiencies also include lymphoproliferation and
autoimmunity. This includes, but is not limited to, Canale-Smith syndrome.
Primary apoptosis deficiencies also include cancer. For example, p53 induces
apoptosis by acting as a transcription factor that activates expression of
various
apoptosis-mediating genes or by upregulating apoptosis-mediating genes such as
Bax.
Primary apoptosis excesses are associated with neurodegenerative disorders
including Alzheimer's disease, Parkinson's disease, spinal muscular atrophy,
and
amyotrophic lateral sclerosis.
Primary apoptosis excesses are also associated with heart disease including
idiopathic dilated cardiomyopathy, ischemic cardiomyopathy, and valvular heart
disease. Evidence has also been shown of apoptosis in heart failure resulting
from
arrhythmogenic right ventricular dysplasia. For all these disorders, see
Hetts, above.
1 S Death receptors also include the TNF receptor-1 and hence, TNF acts as a
death ligand.
A wide variety of neurological diseases are characterized by the gradual loss
of specific sets of neurons. Such disorders include Alzheimer's disease,
Parkinson's
disease, amyotrophic lateral sclerosis (ALS) retinitis pigmentosa, spinal
muscular
atrophy, and various forms of cerebellar degeneration. The cell loss in these
diseases
does not induce an inflammatory response, and apoptosis appears to be the
mechanism of cell death.
In addition, a number of hematologic diseases are associated with a decreased
production of blood cells. These disorders include anemia associated with
chronic
disease, aplastic anemia, chronic neutropenia, and the myelodysplastic
syndromes.
Disorders of blood cell production, such as myelodysplastic syndrome and some
forms of aplastic anemia, are associated with increased apoptotic cell death
within the
bone marrow.
These disorders could result from the activation of genes that promote
apoptosis, acquired deficiencies in stromal cells or hematopoietic survival
factors, or
the direct effects of toxins and mediators of immune responses.
Two common disorders associated with cell death are myocardial infarctions
and stroke. In both disorders, cells within the central area of ischemia,
which is
-82-
CA 02388617 2002-05-07
WO 01/31007 PCT/USUO/29132
produced in the event of acute loss of blood flow, appear to die rapidly as a
result of
necrosis. However, outside the central ischemic zone, cells die over a more
protracted
time period and morphologically appear to die by apoptosis.
The invention also pertains to disorders of the central nervous system (CNS).
These disorders include, but are not limited to cognitive and
neurodegenerative
disorders such as Alzheimer's disease, senile dementia, Huntington's disease,
amyotrophic lateral sclerosis, and Parkinson's disease, as well as Gilles de
la
Tourette's syndrome, autonomic function disorders such as hypertension and
sleep
disorders, and neuropsychiatric disorders that include, but are not limited to
schizophrenia, schizoaffective disorder, attention deficit disorder, dysthymic
disorder,
major depressive disorder, mania, obsessive-compulsive disorder, psychoactive
substance use disorders, anxiety, panic disorder, as well as bipolar affective
disorder,
e.g., severe bipolar affective (mood) disorder (BP-I), bipolar affective
(mood)
disorder with hypomania and major depression (BP-II). Further CNS-related
1 S disorders include, for example, those listed in the American Psychiatric
Association's
Diagnostic and Statistical manual of Mental Disorders (DSM), the most current
version of which is incorporated herein by reference in its entirety.
As used herein, "differential expression" or differentially expressed"
includes
both quantative and qualitative differences in the temporal and/or cellular
expression
pattern of a gene, e.g., the programmed cell death genes disclosed herein,
among, for
example, normal cells and cells undergoing programmed cell death. Genes which
are
differentially expressed can be used as part of a prognostic or diagnostic
marker for
the evaluation of subjects at risk for developing a disorder characterized by
deregulated programmed cell death. Depending on the expression level of the
gene,
the progression state of the disorder can also be evaluated.
X. Arrays and Microarrays
The term "array" refers to a set of nucleic acid sequences that comprise at
least
one of SEQ ID NOS: 1-6, 8, and 10. Preferred arrays contain numerous genes.
The
term can refer to all of the sequences in SEQ ID NOS: 1-6, 8, and 10 but could
also
include additional sequences, for example, sequences included as controls for
specific
biological processes. A "subarray" is also an array but is obtained by
creating an
array of less than all of the sequences in a starting array.
-83-
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
In one embodiment of the invention, the functional subarray comprises nucleic
acid sequences expressed in programmed cell death as disclosed herein.
The array comprises not only the specific designated sequences but also
variants of these sequences, as described herein. As described, variants
include,
allelic variants, homologs from other loci in the same animal, orthologs, and
sequences sufficiently similar such that they fulfill the requisites for
sequence
similarity/homology as described herein.
Further, the array not only comprises the specific designated sequences, but
also comprises fragments thereof. As described herein, the range of fragments
will
vary depending upon the specific sequence involved. Accordingly, the range of
fragments is considerable, for example, 10, 15, 20, 25, 30, 35, 40, 45, 50,
55, 60, 65,
70, 75, 80, 85, 90, 95, 100, 125, 150, 200, 250, 300, 350, 400, 450, 500, 550,
600,
650, 700, 750, 800, 850, 900, 950, 1000 etc. In no way, however, is a fragment
to be
construed as having a sequence identical to that which may be found in the
prior art.
The array can be used to assay expression of one or more genes in the array.
In one embodiment, the array can be used to assay gene expression in a tissue
to ascertain tissue specificity of genes in the array.
In addition to such qualitative determination, the invention allows the
quantitation of gene expression. Thus, not only tissue specificity, but also
the level of
expression of a battery of genes in the tissue is ascertainable. Thus, genes
can be
grouped on the basis of their tissue expression per se and level of expression
in that
tissue. This is useful, for example, in ascertaining the relationship of gene
expression
between or among tissues. Thus, one tissue can be perturbed and the effect on
gene
expression in a second tissue can be determined. In this context, the effect
of one cell
type on another cell type in response to a biological stimulus can be
determined. Such
a determination is useful, for example, to know the effect of cell-cell
interaction at the
level of gene expression. If an agent is administered therapeutically to treat
one cell
type but has an undesirable effect on another cell type, the invention
provides an
assay to determine the molecular basis of the undesirable effect and thus
provides the
opportunity to co-administer a counteracting agent or otherwise treat the
undesired
effect. Similarly, even within a single cell type, undesirable biological
effects can be
deterniined at the molecular level. Thus, the effects of an agent on
expression of
other than the target gene can be ascertained and counteracted.
-84-
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
In another embodiment, the array can be used to monitor the time course of
expression of one or more genes in the array. This can occur in various
biological
contexts, as disclosed herein, for example development and differentiation,
tumor
progression, progression of other diseases, in vitro processes, such as
cellular
transformation and senescence, autonomic neural and neurological processes,
such as,
for example, pain and appetite, and cognitive functions, such as learning or
memory.
The array is also useful for ascertaining the effect of the expression of a
gene
on the expression of other genes in the same cell or in different cells. This
provides,
for example, for a selection of alternate molecular targets for therapeutic
intervention
if the ultimate or downstream target cannot be regulated.
The array is also useful for ascertaining differential expression patterns of
one
or more genes in normal and abnormal cells. This provides a battery of genes
that
could serve as a molecular target for diagnosis or therapeutic intervention.
In one embodiment, the array, and particularly subarrays containing one or
I 5 more of the nucleic acid sequences related to programmed cell death, are
useful for
diagnosing disease or predisposition to disease involving apoptosis. These
disorders
include, but are not limited to, those discussed in detail herein. In
addition, the array
or subarrays created therefrom are useful for diagnosing active disorders of
the central
nervous system or for predicting the tenancy to develop such disorders.
Disorders of
the central nervous system include, but are not limited to, those disclosed in
detail
herein. Furthermore, the array and subarrays thereof are useful for diagnosing
an
active disorder or predicting the tendency to develop a disorder including,
but not
limited to, disorders involving secretion/synaptic vesicle release, cell
proliferation,
cytoskeletal reorganization, stress response/hormone response; and calcium
signal
transduction.
The array is also useful for ascertaining expression of one or more genes in
model systems in vitro or in vivo. Various model systems have been developed
to
study normal and abnormal processes, including, but not limited to, apoptosis.
Apoptosis can be actively induced in animal cells by a diverse array of
triggers
that range from ionizing radiation to hypothermia to viral infections to
immune
reactions. Majno et al. (1995) Amer. J. Pathol. I 16:3-15; Hockenberry et al.
(1995)
Bio Essays 17:631-638; Thompson et al. Science 267:1456-1462 (1995).
-85-
CA 02388617 2002-05-07
WO 01131007 PCT/US00/29132
Transgenic mouse models have been developed for familial amyotrophic
lateral sclerosis, familial Alzheimer's disease and Huntington's disease,
reviewed in
Price et crl. (1998) Science 282:1079-1083. Amyotrophic lateral sclerosis is
the most
common adult onset motor neuron disease. Alzheimer's disease is the most
common
cause of dementia in adult life. It is associated with the damage of regions
and
neurocircuits critical for cognition and memory, including neurons in the
neocortex,
hippocampus, amygdala, basal forebrain cholinergic system, and brain stem
monoaminergic nuclei. Neurological diseases that are associated with autosomal
dominant trinucleotide repeat mutations include Huntington's disease, several
spinal
cerebellar ataxias and dentatorubral pallidoluysian atrophy. SCA-1 and SCA-3
or
Machado-Joseph disease are characterized by ataxia and lack of coordination.
In
Huntington's disease, symptoms are related to degeneration of subsets of
striatal and
cortical neurons. Apoptosis is thought to play a role in the degeneration of
these cells.
In SCA-1, SCA-3, and in dentatorubral pallidoluysian atrophy, a variety of
cell
populations, and particularly cells in the cerebellum, have been shown to
degenerate.
See Price et al. above, which is incorporated by reference in its entirety for
the
teachings of model systems related to neurodegenerative diseases.
Mouse models have been developed for non-obese diabetic mice, to study
disease progression for the treatment of autoimmune diabetes mellitus.
Bellgrau et al.
(1995) Nature 377:630-632. Models have also been developed in mice wherein the
mice lack one or two copies of the p53 gene. Study of these mice has shown
that
apoptosis is involved in suppressing tumor development in vivo. Lozano et al.
( 1998)
Semin. Canc. Biol. 8:337-344. Another animal model relevant to the study of
apoptosis involves the targeted gene disruption of caspase genes creating
caspase
gene knockout mice. Colussi et a1.(1999) J. Immure. Cell. Biol. 77:58-63. A
further
mouse model pertains to cold injury in mice, such injury inducing neuronal
apoptosis.
Murakami et al. ( 1999) Prog. Neurobiol. ~ 7:289-299.
Knockout mice have been created for Apafl . In these mice, defects are found
in essentially all tissues whose development depends on cell death, including
loss of
interdigital webs, formation of the palate, control of neuron cell number, and
development of the lens and retina. Cecconi et al. (1998) Cell 94:727-737.
Caspase knockout mice have also been achieved for caspase 1, 2, 3, and 9.
Green (1998) Cell 94:695-698.
-86-
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
The array allows the simultaneous determination of a battery of genes
involved in these processes and thus provides multiple candidates for in vivo
verification and clinical testing. Because the array allows the determination
of
expression of multiple genes; it provides a powerful tool to ascertain
coordinate gene
expression, that is co-expression of two or more genes in a time and/or tissue-
specific
manner, both qualitatively and quantitatively. Thus, genes can be grouped on
the
basis of their expression per se and/or level of expression. This allows the
classification of genes into functional categories even when the gene is
completely
uncharacterized with respect to function. Accordingly, if a first gene is
expressed
coordinately with a second gene whose function is known, a putative function
can be
assigned to that first gene. This first gene thus provides a new target for
affecting that
function in a diagnostic or therapeutic context. The larger the number of
genes in an
array, the greater is the probability that numerous known genes having the
same or
similar function will be expressed. In this case, the coordinate expression of
one or
more novel genes (with respect to function and/or structure) strongly allows
discovery
of genes in the same functional category as the known genes.
Accordingly, the array of the invention provides for "internal control" groups
of genes whose functions are known and can thus be used to identify genes as
being in
the same functional category of the control group if they are coordinated
expressed.
As an alternative to relying on such internal control groups, external control
groups can be added to the array. The genes in such a group would have a known
function. Genes coordinately expressed with these genes would thus be prima
facie
involved in the same function.
Therefore, the array provides a method not only for discovering novel genes
having a specific function but also for assigning function to genes whose
function is
unknown or assigning to a known gene an additional function, previously
unknown
for that gene.
Accordingly, as disclosed and exemplified herein, previously characterized
genes were grouped into new functional categories (i.e., previously the
function was
not known to be possessed by that gene). Furthermore, several uncharacterized
genes
could be functionally classified on the basis of coordinate expression with
the
"internal control group of genes". In a specific embodiment, disclosed and
exemplified herein, genes related to programmed cell death in brain were
selected.
_87_
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
The array could, accordingly be used to select for genes related to other
important
biological processes, such as those disclosed herein. Nucleic acid from any
tissue in
any biological process is hybridized to nucleic acid sequences in an array.
The
expression pattern of genes in the array allows for their classification into
functional
groups based on specific expression patterns. Internal or external control
genes (i.e.
genes known to be expressed in the specific tissue/biological process) provide
verification to classify other genes in the specific category.
Just as the array was useful for identifying programmed cell death genes,
other
relevant normal biological models include differentiation programs and
disorders such
as those disclosed herein.
The array is also useful for drug discovery. Candidate compounds can be used
to screen cells and tissues in any of the biological contexts disclosed
herein, such as
pathology, development, differentiation, etc. Thus the expression of one or
more
genes in the array can be monitored by using the array to screen for RNA
expression
in a cell or tissue exposed to a candidate compound. Compounds can be selected
on
the basis of the overall effect on gene expression, not necessarily on the
basis of its
effect on a single gene. Thus, for example, where a compound is desired that
affects a
particular first gene or genes but has no effect on a second gene or genes,
the array
provides a way to globally monitor the effect on gene expression of a
compound.
Alternatively, it may be desirable to target more than one gene, i.e. to
modulate the expression of more than one gene. The array provides a way to
discover
compounds that will modulate a set of genes. All genes of the set can be
upregulated
or downregulated. Alternatively, some of the genes may be upregulated and
others
downregulated by the same compound. Moreover, compounds are discoverable that
modulate desired genes to desired degrees.
In the context of drug discovery, functional subarrays of genes are especially
useful. Thus, using the methods disclosed herein and those routinely
available,
groups of genes can be assembled based on their relationships to a specific
biological
function. The expression of this group of genes can be used for diagnostic
purposes
and to discover compounds relevant to the biological function. Thus, the
subarray can
provide the basis for discovering drugs relevant to treatment and diagnosis of
disease,
for example those disclosed herein.
_88_
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
In the present case, the group of genes whose expression is correlated with ,
programmed cell death can be used to discover compounds that affect programmed
cell death, and especially disorders in which programmed cell death is
involved.
These include but are not limited to those disclosed herein.
Apoptosis can be triggered by the addition of apoptosis-promoting ligands to a
cell in culture or in vivo. In one embodiment of the invention, therefore, the
arrays
and subarrays described herein are useful to identify genes that respond to
apoptosis-
promoting ligands and conversely to identify ligands that act on genes
involved in
apoptosis. Apoptosis can also be triggered by decreasing or removing an
apoptosis-
inhibiting or survival-promoting ligand. Accordingly, apoptosis is triggered
in view
of the fact that the cell lacks a signal from a cell surface survival factor
receptor.
Ligands include, but are not limited to, Fast. Death-inhibiting ligands
include, but
are not limited to, IL-2. See Hetts et al. (1998) JAMA 279:300-307
(incorporated by
reference in its entirety for teaching of ligands involved in active and
passive
apoptosis pathways.) Central in the pathway, and also serving as potential
molecules
for inducing (or releasing from inhibition) apoptosis pathways include FADD,
caspases, human CED4 homolog (also called apoptotic protease activating factor
1 ),
the Bcl-2 family of genes including, but not limited to, apoptosis promoting
(for
example, Bax and Bad) and apoptosis inhibiting (for example, Bcl-2 and Bcl-xi)
molecules. See Hetts et al., above.
Multiple caspases upstream of caspase-3 can be inhibited by viral proteins
such as cowpox, CrmA, and baculovirus, p35, synthetic tripeptides and
tetrapeptides
inhibit casepase-3 specifically (Hens, above). Accordingly, the arrays and
subarrays
are useful for determining the modulation of gene expression in response to
these
agents.
The array is also useful for obtaining a set of human (or other animal)
orthologs that can be used for drug discovery, treatment, diagnosis, and the
other uses
disclosed herein. The subarrays can be used to specifically create a
corresponding
human (or other animal) subarray that is relevant to a specific biological
function.
Accordingly, a method is provided for obtaining sets of genes from other
organisms,
which sets are correlated with, for example, disease or developmental
disorders.
In a preferred embodiment of the invention, the arrays and subarrays disclosed
herein are in a "microarray". The term "microarray" is intended to designate
an array
-89-
CA 02388617 2002-05-07
WO 01/31007 PCT/USUO/29132
of nucleic acid sequences on a chip. This includes in situ synthesis of
desired nucleic
acid sequences directly on the chip material, or affixing previously
chemically
synthesized nucleic acid sequences or nucleic acid sequences produced by
recombinant DNA methodology onto the chip material. In the case of recombinant
DNA methodology, nucleic acids can include whole vectors containing desired
inserts, such as phages and plasmids, the desired inserts removed from the
vector as
by, PCR cloning, cDNA synthesized from mRNA, mRNA modified to avoid
degradation, and the like.
A series of state-of the-art reviews of the technology for production of
nucleic
acid microarrays in various formats and examples of their utilization to
address
biological problems is provided in Nata~re Genetics, 21 Supplement, January
1999.
These topics include molecular interactions on microarrays, expression
profiling using
eDNA microarrays, making and reading microarrays, high density synthetic
oligonueleotide arrays, sequencing and mutation analysis using oligonucleotide
microarrays, the use of microarrays in drug discovery and development, gene
expression informatics, and use of arrays in population genetics. Various
microarray
substrates, methods for processing the substrates to affix the nucleic acids
onto the
substrates, processes for hybridization of the nucleic acid on the substrate
to an
external nucleic acid sample, methods for detection, and methods for analyzing
expression data using specific algorithms have been widely disclosed in the
art.
References disclosing various microarray technologies are listed below.
Lashkari et al. ( 1997) "Yeast Microarrays for Genome Wide Parallel Genetic
and Gene Expression Analysis", Proc. Natl. Acad. Sci. 9=1:13057-13062; Ramsay
(1998) "DNA Chips: State-of the-Art", Nature Biotechnology 16:40-44; Marshall
et
al. (1998) " DNA Chips: An Array of Possibilities", Nature Biotechnology 16:27-
31;
Wodicka et al. (1997) "Genome-Wide Expression Monitoring In Saccharomyces
Cerevi.siae", Nature Biotechnology 15:1359-1367; Southern et al. (1999)
"Molecular
Interactions On Microarrays", Nature Genetics 21(1):5-9; Duggan, e1 al. (1999)
Nature Genetics 21(1):10-14; Cheung et al. (1999) "Making and Reading
Microarrays", Nature Genetics 21(1):15-19; Lipshutz et al. (1999) "High
Density
Synthetic Oligonucleotide Arrays", Nature Genetics 21 (1):20-24; Bowtell
(1999)
Nature Genetics 21:25-32; Brown et al. (1999) "Exploring the New World of the
Genome with DNA Microarrays" Nature Genetics 21(1):33-37; Cole et al. (1999)
-90-
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
"The Genetics of Cancer--A 3D Model" Nature Genetics 21(1):38-41; Hacia (1999)
"Resequencing and Mutational Analysis Using Oligonucleotide Microarrays",
Nature
Genetics 21(1):42-47; Debouck et al. (1999) "DNA Microarrays in Drug Discovery
and Development", Nature Genetics 21(1):48-50; Bassett, Jr. et al. (1999)
"Gene
Expression Informatics--It's All In Your Mine", Nature Genetics 21 (I):51-55;
Chakravarti (1999) "Population Genetic--Making Sense Out of Sequence", Nature
Genetics 21(1):56-60; Chee et al. (1996) "Accessing Genetic Information with
High-
Density DNA Arrays", Science 274:610-614; Lockhart et al. (1996) "Expression
Monitoring by Hybridization to High-Density Oligonucleotide Arrays", Nature
Biotechnology 14:1675-1680; Tamayo et al. (1999) "Interpreting Patterns of
Gene
Expression with Self Organizing Maps: Methods and Application to Hematopoietic
Differentiation", Proc. Nat!. Acad. Sci. 96:2907-2912; Eisen et al. ( 1998)
"Cluster
Analysis and Display of Genome-Wide Expression Patterns", Proc. Nat!. Acad.
Sci.
95:14863-14868; Wen et al. (1998) "Large-Scale Temporal Gene Expression
Mapping of Central Nervous System Development", Proc. Nat!. Acad. Sci. 9.5:334-
339; Ermolaeva et al. (1998) "Data Management and Analysis for Gene Expression
Arrays", Nature Genetics 20:19-23; Wang et al. (1998) "A Strategy for Genome-
Wide Gene Analysis: Integrated Procedure for Gene Identification", Proc. Nat!.
Acad. Sci. 95:11909-11914; U.S. Patent No. 5,837,832; U.S. Patent No.
5,861,242;
WO 97110363.
In the instant case, the microarray contains nucleic acid sequences on a
Biodyne B filter. However, any medium, including those that are well-known and
available to the person of ordinary skill in the art, to which nucleic acids
can be
affixed in a manner suitable to allow hybridization, are encompassed by the
invention.
This includes, but is not limited to, any of the membranes disclosed in the
references
above, which are incorporated herein for reference to those membranes, and
other
membranes that are commercially available, including but not limited to,
nitrocellulose-1, supported nitrocellulose-1, and Biodyne A, which is a
neutrally-
charged nylon membrane suitable for Southern transfer and dot blotting
procedures.
(All are available from Life Technologies.)
-91-
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
EXAMPLE
Summar
Programmed cell death (PCD) in rat cerebellar granule neurons (CGNs)
induced by potassium (K+) withdrawal has been shown to depend on de novo RNA
synthesis. The inventors characterized this transcriptional component of CGN
programmed cell death using a custom-built brain-biased cDNA array
representing
over 7000 different rat genes. Consistent with carefully orchestrated mRNA
regulation, the profiles of 234 differentially expressed genes segregated into
distinct
temporal groups (immediate early, early, middle, and late) encompassing genes
involved in distinct physiological responses including cell-cell signaling,
nuclear
reorganization, apoptosis, and differentiation. A set of 64 genes, including
22 novel
genes, were regulated by both K+ withdrawal and kainate treatment. Human
homologs were isolated for 8 of these novel regulated genes: The sequences of
these
human homologs are shown in SEQ ID NOS:1 (human NARC 9B), 2 (human NARC
8B), 3 (human NARC 2A), 4 (human NARC 16B), 5 (human NARC l OC), 6 (human
NARC 1 C), 8 (human NARC 1 A), and 10 (human NARC 25).
Thus, array technology was used to broadly characterize physiological
responses at
the transcriptional level and identify novel genes induced by multiple models
of
programmed cell death.
Background
In neurons, programmed cell death is an essential component of neuronal
development (Jacobson et al. 1997; Pettmann and Henderson (1998); Pettmann and
Henderson (1998) Neuron 20:633-747) and has been associated with many forms of
neurodegeneration (Hefts (1998) Journal of the American Medical Association
279:300-307). In the cerebellum, granule cell development occurs postnatally.
The
final number of neurons represents the combined effects of additive processes
such as
cell division and subtractive processes such as target-related programmed cell
death.
Depolarization due to high concentrations (25 mM) of extracellular potassium
(K+)
promotes the survival of cerebellar granule neurons (CGNs) in vitro. CGNs
maintained in serum containing medium with high K+ will undergo programmed
cell
death when switched to serum-free medium with low K+ (5 mM) (D'Mello et al.
-92-
CA 02388617 2002-05-07
WO 01/3100'7 PCT/US00/29132
(1993) Proc. Natl. Acad. Sci. USA 90:10989-10993; Miller and Johnson (1996)
Journal of Neueroscience 16:7487-7495). The resulting programmed cell death
has a
transcriptional component that can be blocked by inhibitors of new RNA
synthesis
{Galli et al. (1995) Journal of Neuroscience l~:l 172-1179; and Schulz and
Klockgether ( I 996) Journal of Neuroscience 16:4696-4706). Traditionally, the
regulation of limited numbers of specific genes were characterized during CGN
programmed cell death using Northern nucleic acid hybridization (e.g. PTZ-17,
Roschier et al. (1998) Biochemical and Biophysical Research Communications
252:10-13), reverse transcription polymerase chain reaction (RT-PCR; e.g. c-
jun,
eyclophilin, cyclin D1, c fo.s and caspase (Miller et al. (1997) Journal of
Cell Biology
139:205-217), and in situ hybridization (e.g. RP-8; Owens et al. (1995)
Developmental Brain Research 86:35-47).
High-density eDNA arrays have been successfully used to characterize
genome-wide mRNA expression in yeast (Lashkari et al. ( 1997) Proc. Natl. Acad
Sci. USA 9=1:13057-13062; Wodicka et al. (1997) Nature Biotechnology 15:1997).
In
higher eukaryotes, the strategy has been to array as many sequences as
possible from
known genes, from expressed sequence tags (ESTs), or from uncharacterized cDNA
clones from a library (Bowtell (1999) Nature Genetics 21:25-32; Duggan et al.
(1999)
Nature Genetics 21:10-14; Marshall and Hodgson (1998) Nature Biotechnology
16:27-31; and Ramsay (1998) Nature Biotechnology 16:40-44). Global RNA
regulation during cellular processes including cell-cycle regulation (Cho et
al. (1998)
Molecular Cell 2:65-73, and Spellman et al. (1998) Mol. Biol. Cell. 95:14863-
14868),
fibroblast growth control (Iyer et al. (1999) Science 283:83-87), metabolic
responses
to growth medium (Derisi and Brown ( 1997) Science 278: 680-686), and germ
cell
development (Chu et al. (1998) Science 282:699-705) have been temporally
monitored using arrays. The program of gene expression delineated in these
studies
demonstrated a correlation between common function and coordinate expression,
and
also provided a comprehensive, dynamic picture of the processes involved
(Brown
and Botstein (1999) Nature Genetics 21:33-37). For the cellular process of
programmed cell death, a DNA chip has been used to identify twelve known genes
as
differentially expressed between two conditions, etoposide-treated and
untreated cells
(Wang et al. (1999) FEBS Letters 44:269-273).
-93-
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
A genome-wide approach for the comprehensive characterization of the
transcriptional component of rat CGN programmed cell death and for
identification of
novel neuronal apoptosis genes requires an array consisting of both known and
novel
rat cDNAs. The inventors constructed a brain-biased and programmed cell death-
enriched clone set by arraying 7300 consolidated ESTs from two cDNA libraries
cloned from rat frontal cortex and differentiated PC12 cells deprived of nerve
growth
factor (NGF), and >300 genes that are known markers for the central nervous
system
and/or programmed cell death. They reproducibly and simultaneously monitored
the
expression of the genes at 1, 3, 6, 12, and 24 hours after K+ withdrawal. They
then
categorized the regulated genes by time course expression pattern to identify
cellular
processes mobilized by CGN programmed cell death at the RNA level. In
particular
they focused on the expression profiles of many known pro- and anti-apoptotic
regulatory proteins, including transcription factors, Bcl-2 family members,
caspases,
cyclins, heat shock proteins (HSPs), inhibitors of apoptosis (IAPs), growth
factors and
receptors, other signal transduction molecules, p53, superoxide dismutases
(SODs);
and other stress response genes. Finally, they compared the time courses of
regulated
genes induced by K+ withdrawal in the presence or absence of serum to those
induced
by glutamate toxicity. Thus, they identified a restricted set of relevant
genes
regulated by multiple models of programmed cell death in CGNs.
Results
Construction and validation of a brain-biased cDNA microarray
In order to characterize the transcriptional component of neuronal apoptosis
in
rat cerebellar granule neurons, the inventors constructed a cDNA array, called
Smart
ChipTM I, that contains primarily rat brain genes. Figure 1 shows a schematic
representation of the construction of the microarray. Two cDNA libraries were
cloned from rat frontal cortex and nerve growth factor-deprived rat PC12 cells
to
enrich for cDNAs expressed in the central nervous system and in one in vitro
model
of neuronal apoptosis. Expressed sequence tags (ESTs) from the 5'-end were
identified for 8,304 clones in the cortical library and 5,680 in the PC 12
library. These
13,984 ESTs were condensed into 7,399 unique sequence clusters by using the
Basic
Local Alignment Search Tool (BLAST) sequence comparison analysis (Altschul et
al.
1990) to identify ESTs with overlapping sequence. One representative clone was
-94-
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
chosen from each of 7,296 of the unique sequence clusters and prepared for PCR
amplification using a robotic sample processor. In addition to the ESTs, PCR
templates were prepared for 289 known DNA sequences, including negative
controls,
genes with known function in the CNS and/or during programmed cell death, and
genes previously identified as regulated by CGN programmed cell death using
differential display (data not shown). To check the fidelity of the set of
array
elements, a robotic sample processor was used to randomly choose 212 clones
for
sequencing. Ten clones produced poor sequence. The remaining 202 matched their
seed sequence (data not shown), implicating 100% fidelity in sample tracking.
A sample volume of 20 n1 from each of the 7584 PCR products was arrayed
onto nylon filters at a density of ~64/cm2 using a pin robot. The arrayed DNA
elements were denatured and covalently attached to the nylon filters for use
in reverse
Northern nucleic acid hybridization experiments. In a typical experiment,
"radiolabeled RNA", 1 pg polyA RNA radiolabeled by 33P-dCTP incorporation
during eDNA synthesis, was hybridized to triplicate arrays following RNA
hydrolysis. Subsequently, the filters were washed and exposed to phosphoimage
screens. Gene expression was quantified for each array element by digitizing
the
phosphoimage-captured hybridization signal intensity. Figure 2 illustrates
that the
coefficient of variation between triplicate hybridizations averaged less than
0.2 for
genes whose intensities were above a threshold of 30-40 units. From control
experiments when in vitro transcribed RNAs were deliberately spiked into
samples,
this threshold amounted to a copy number of less than 1 in 100,000 (data not
shown).
Tissue distribution of brain-biased Smart Chip ESTs
To characterize the brain-biased eDNA array and possibly identify brain-
specific genes, radiolabeled RNA from ten different normal rat tissues was
hybridized
to Smart Chip. Compared to heart, kidney, liver, lung, pancreas, skeletal
muscle,
smooth muscle, spleen, and testes, radiolabeled rat brain RNA produced more
hybridization signal intensity against most of the brain-biased array
elements. After
data normalization and averaging between replicates, the threshold of
detection was
determined for each experiment and the number of genes detected for each
tissue was
tabulated (Figure 6). Most (6127 out of 7296) but not all of the ESTs were
detected in
at least one of the tissues profiled. The number of genes detected in brain
was the
-95-
CA 02388617 2002-05-07
WO 01/31007 PCT/USUO/29132
highest. 582 genes appeared to be brain-specific, as defined by detection
above
threshold for brain but below threshold for any of the other nine tissues.
The physiology of CGN KCUserum-withdrawal as characterized by transcription
profiling on Smart Chip
Using the brain-biased, programmed cell death nucleic acid-enriched Smart
Chip, global mRNA expression was profiled throughout a time course of
KCl/serum-
withdrawal-induced cell death in primary cultures of CGNs. The transcription-
dependent CGN programmed cell death was coordinated, resulting in less than
30%
survival at 24 hours post-withdrawal as quantified by cell counting (data not
shown).
RNA samples, designated "treated", were isolated at 1, 3, 6, 12, and 24 hours
after
switching post-natal day eight CGNs from medium containing 5% serum and 25 mM
KCl to serum-free medium with 5 mM KCI. For controls, the 5% serum/25 mM KC1
medium was replaced, and "sham" RNA at 1, 3, 6, 12, and 24 hours was isolated.
Since the average coefficient of variation for gene expression intensities
between triplicate hybridizations was less than 0.2, genes regulated at least
three-fold
during the time course (790 out of 6818 detected; data not shown) were further
addressed. Using hierarchical clustering algorithms {see Experimental
Procedures),
the regulated genes were ordered based on their gene expression pattern across
the ten
experimental points (five time points, sham and treated (Figure 3)). The
dendrogram
in Figure 3 depicts the hierarchy of relatedness between gene expression
profiles. The
first major branch point segregated those genes regulated by sham treatment
(first five
columns), and those regulated by KCl/serum-withdrawal treatment only (last
five
columns). A majority of genes (556) were regulated by sham treatment. These
genes
included trkA, PSD-95, SV 2A, and VAMP 1, and were most likely induced by
serum-add-back in the sham since the medium was exchanged at t=0 with
unconditioned medium.
Figure 3 shows the expression pattern of 234 programmed cell death-induced
genes that were regulated by KCI/serum-withdrawal only, and were not regulated
by
serum-add-back in the sham experiments. Their coefficient of variation in
expression
level throughout the five serum-add-back experiments was less than 20%. Since
the
serum-add-back experiments were non-discriminating for these genes, the serum-
add-
back data were averaged to generate a single control data set for clustering
with the
-96-
CA 02388617 2002-05-07
WO 01/310(17 PCT/US00/29132
KCl/serum withdrawal time course. Four apparent temporal regulation classes
were
designated immediate early (peaking at 1 hour followed by rapid decay), early
(peaking at 3-6 hours), middle (peaking at 6-12 hours), and late (up-regulated
at 24
hours). Almost all of the immediate early genes encoded proteins with known
roles in
regulating secretion and synaptic vesicle release including synaptotagmin,
synaphin,
NSG-1, calcium calmodulin-dependent kinase II, synapsin, complexin, LDL
receptor,
and fodrin (Figure 7). Histones l, 2A, and 3 fell in the early class. Middle
genes
comprised several known genes induced by programmed cell death or stress,
including caspase 3, the mammalian oxy R homolog, cytochrome c oxidase and
protein phosphatase Wip-1. Functions encoded for by late genes could be
effectors of
survival mechanisms including inhibitory neurotransmission (GAD, GABA-A
receptor, GABA transporter), cell adhesion (nexin, basement membrane protein
40,
phosphacan, rat GRASP), down-regulation of excitatory neurotransmission
(glutamate transporter, sodium-dependent glutamate/aspartate transporter),
leukotriene metabolism (dithiolethione-induced NADP-dependent leukotriene B4
12-
hydroxydegydrogenase, leukotriene A-4 hydrolase), protein stabilization
(cysteine
proteinase inhibitor cystatin C, N-alpha-acetyl transferase, CaBP2, elongation
factor
1-gamma, APG-1), and ionic balance and cell volume (SLC12A integral membrane
protein transporter). Based on four distinct waves of gene expression, the
major
transcriptional reponses observed for KCI/serum-withdrawal included initial up-
regulation of synaptic vesicle release/recycling, then, of histone
biosynthesis,
followed by various constituents of programmed cell death regulation and
stress-
response signaling, and finally, of multiple survival mechanisms. The apparent
changes in transcription most likely also reflect changes in the relative cell
populations, since late mRNAs may be markers of neurons and non-neuronal cells
which have survived KCI/serum-withdrawal at 24 hours. Another contributing
factor
may be the presence of two populations of dying neurons that respond with
different
kinetics to serum versus KCl withdrawal, as has been described by other
groups.
Neuronal apoptosis regulated candidates (NARCs) regulated by multiple models
of programmed cell death
112 novel ESTs were significantly regulated by KC1/serum-withdrawal in rat
CGNs (data not shown). Some exhibited similar expression profiles throughout
-97-
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
KCl/serum-withdrawal and serum-add-back to genes with known function during
programmed cell death, such as caspase 3. The temporally-coupled expression of
these novel genes may reflect related functionality with caspase 3, since they
probably
share common RNA regulatory elements, including those regulating initiation,
elongation, processing, and/or stability. Apparent coordinate transcriptional
up-
regulation of synaptic vesicle release/recycling possibly reflects a
physiological
response to near cessation of synaptic transmission that may or may not
contribute to
the programmed cell death pathway. To help further distinguish genes that are
specifically regulated in response to programmed cell death, CGN programmed
cell
death induced by glutamate (excitatory neurotransmitter) toxicity was studied.
In
addition, the effect of KCl-withdrawal alone on gene expression was examined.
This
was done under defined medium conditions to minimize the effect of serum on
the
sham and treated samples.
Rat CGNs from post-natal day seven pups were isolated as before and plated
into basal medium Eagle containing "high", 10% dialysed fetal bovine serum,
and
"high", 25 mM KCI. After two days in culture, the medium was replaced with
neurobasal medium supplemented with "low", 0.5% serum, and high KCI. To
initiate
KCl-withdrawal on day eight, the KCl concentration was switched to 5 mM for
the
treated samples. The same low serum, high KCI, neurobasal medium was replaced
in
the controls to minimize gene induction by high serum. For the glutamate
toxicity
experiment, the cells were treated for 30 rnin in sodium-free Locke's medium
withvor
without 100 gM kainate for treated samples and controls, respectively.
After isolation from treated and control samples at 1, 3, 6, and 12 hours
after
KCl-withdrawal and 2, 4, 6, 12 hours after kainate treatment, mRNA was
subjected to
expression profiling analysis on Smart Chip I. Figure 4 illustrates the
changes in gene
expression that occur over time when CGNs are induced to undergo programmed
cell
death by KCl/serum-withdrawal, KCl-withdrawal alone, or kainate treatment. In
the
scatter plots, due to differential expression, large numbers of regulated
genes migrated
away from a line of slope one when withdrawn (W) or treated (T) samples were
compared to control (C). The sham treated cells for the KCl/serum-withdrawal
clearly responded to basal medium serum-add-back, whereas shams for KCl-
withdrawal alone and kainate treatment did not respond to conditioned
neurobasal
-98-
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
medium add-back. Profiling across the mRNA levels of thousands of genes
provided
a clear index of changes in overall cell physiology.
In general, apparent changes in gene expression were less robust in the cells
cultured on neurobasal medium. The number of genes detected above threshold
was
similar for all three paradigms, 6634, 7017, and 6818, respectively, for KC1-
withdrawal, kainate treatment, and KCl/serum withdrawal (data not shown). Yet
the
number of genes regulated by at least three-fold during KCl-withdrawal and
kainate
treatment was only 156 and 167, respectively (data not shown), compared to the
790
discussed above for KCl/serum withdrawal.
A hierarchical clustering algorithm was used to order the regulated genes
based on their gene expression pattern across all CGN programmed cell death
paradigms investigated. Twenty-six individual profiling experiments in
duplicate or
triplicate were performed across the 7584 rat genes on Smart Chip I using mRNA
isolated from 5 serum-add-back time points, 5 KCl/serum-withdrawal time
points, 4
time points each for sham and KCl-withdrawal, and 4 time points each for sham
and
kainate treatment.
Figure 4 shows expression clusters generated by one hierarchical clustering
algorithm. The inset shows a specific group of genes having similar expression
patterns. This group includes genes known to be regulated in programmed cell
death,
for example caspase 3 and Wip 1, as well as other nucleic acid sequences on
the array
not previously known to be regulated. Those sequences meeting specific
criteria were
designated "neuronal apoptosis regulated candidate" (NARC). Criteria for
designating such genes were based on specific expression criteria as shown in
Figure
4. Nucleic acid sequences having an expression pattern similar to genes known
to be
involved in apoptosis were designated as NARC sequences.
Gene expression validation by RT-PCR
Although the reproducibility in transcription profiling experiments was quite
high (average CV<0.2), the gene expression regulation of known and novel genes
was
validated by semi-quantitative RT-PCR. The rat CGN model system was used to
independently validate the expression of several NARC genes that had shown
expression (when hybridized with sequences on the chip) related to programmed
cell
death. Reverse transcriptase-assisted PCR was performed to assess expression
of
-99-
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
NARC I -7, 9, 12, 13, 15, and 16. Experimental samples received KCI withdrawal
treatment. Control samples show cells receiving no treatment. The PCR
reactions
contained 10, 5, 2.5, 1.3, and 0.7 ng of total RNA each. The RT-PCR protocol
is
disclosed in the exemplary material herein. NARC l, 2, 4, 5, 7, 9, 12, 13, ,15
and 16
all showed significantly increased expression 3-6 hours after KCl withdrawal.
The
designation "N" above is an abbreviation of the acronym "NARC" which is an
abbreviation of "neuronal apoptosis regulated candidate" as described in the
Examples section.
NARC1 and NARC2 regulation in vivo during cerebellar development
Two novel neuronal apoptosis regulated candidates, NARC 1 and NARC2,
were validated by in situ hybridization and shown to be coordinately up-
regulated
with caspase 3 during postnatal development when increased apoptosis is
associated
I S with synapse consolidation in the cerebellum (not shown).
Experimental Procedures
BLAST sequence comparison analysis
ESTs determined for the 5'-end of cDNA clones picked from two cDNA
libraries, rat frontal cortex (8,304 clones) and NGF-deprived differentiated
PC 12 cells
(5,680 clones), ranged from 100-1000 nt in sequence length and averaged 500 nt
(data
not shown). Sequence comparisons were done using BLAST (Altschul et al. 1990).
Contiguous matches defined a sequence cluster. Large clusters were checked by
hand
to eliminate apparent chimeras. From 13,984 sequences inputted, the analysis
identified 5,779 singletons and 1,620 larger clusters (data not shown). The 5'-
most
clone was selected from the larger clusters. Because two 96-well microtiter
plates of
clones were missing, a total of 7,296 out of the 7,399 identified were
selected for
Smart ChipTM I.
cDNA microarray construction
Using a Genesis RSP 150 robotic sample processor (Tecan AG, Switzerland),
bacterial cultures of individual EST clones from the two libraries were
consolidated
-100-
CA 02388617 2002-05-07
WO 01/31007 PCT/US(10/29132
from 13,792 clones spanning 144 96-well microtiter plates to 7296 Smart Chip I
clones spanning 76 plates. To prepare templates for array elements,
oligonucleotide
primers specific for vector sequences up- and downstream of the cloning site
were
used to amplify the cDNA insert by PCR. Following ethanol precipitation and
concentration (to 1-10 mg/ml), the array element templates were resuspended in
3X
SSC (1X SSC: 150 mM sodium chloride, 15 mM sodium citrate, pH 7.0). A sample
volume of 20 n1 frorn each template was arrayed onto nylon filters (Biodyne B,
Gibco
BRL Life Technologies, Gaithersburg, MD) at a density of ~64/cm2 using a 96-
well
format pin robot (THOR). After the filters were dry, the arrayed DNA was
denatured
in 0.4 M sodium hydroxide, neutralized in 0.1 M Tris-HCI, pH 7.5, rinsed in 2X
SSC,
and dried to completion.
Array hybridization
Rat poly A+ RNA was purchased from Clontech (Palo Alto, CA) for the organ
recital (Figure 8) or was isolated as total RNA from cultured CGNs using RNA
STAT-60TM (Tel-Test, Inc., Friendswood, TX) and then prepared using OligotexTM
(Qiagen, Inc., Chatsworth, CA). Re-annealed 1 ~g mRNA and 1 p.g oligo(dT)3o
was
incubated at 50°C for 30 min with SuperScriptTM II as recommended by
Gibco in the
presence of 0.5 mM each deoxynucleotide dATP, dGTP, and dTTP, and 100pCi a33P-
dCTP (2000-4000 Ci/mmol; NENTM Life Science Products, Boston, MA). After
purification over Chroma SpinTM +TE-30 columns (Clontech), the labeled cDNA
was
annealed with 10 pg poly(dA)>ZOo and 10 pg rat Cot-1 DNA (prepared as
described in
Britten et al. (1974) Methods in Enzymology 29:263-418). At 2 x 106 cpm/m1,
the
annealed cDNA mixture was added to array filters in pre-annealing solution
containing 100 mg/ml sheared salmon sperm DNA in 7% SDS (sodium dodecyl
sulfate), 0.25 M sodium phosphate, 1 mM ethylenediaminetetraacetic acid, and
10%
formamide. Following over night hybridization at 65°C in a rotisserie-
style incubator
(Robbins Scientific, Sunnyvale, CA), the array filters were washed twice for
15 min at
22°C in 2X SSC, 1% SDS, twice for 30 min at 65°C in 0.2X SSC,
0.5% SDS, and
twice for 15 min at 22°C in 2X SSC. The array filters were then dried
and exposed to
phosphoimage screens for 48 h. The radioactive hybridization signals were
captured
with a Fuji BAS 2500 phosphoimager and quantified using Array VisionTM
software
(Imaging Research Inc., Canada). Array hybridizations for the organ recital,
the CGN
-101-
CA 02388617 2002-05-07
WO 01/31007 PCT/US110/29132
KCl only-withdrawal, and the CGN kainate treatment experiments were performed
in
triplicate; for the CGN KCl/serum-withdrawal, they were performed in
duplicate.
Transcription profiling data analysis
For replicate array hybridizations, the distribution of signal intensities
across
all rat genes was normalized to a median of 100. Replicate measurements were
averaged and a coefficient of variation (CV; standard deviation/mean for
triplicates or
the absolute value of the difference/mean for duplicates) was determined for
each
gene. The detection threshold was chosen for each hybridization experiment by
graphing the moving average (with a window of 200) for CV versus mean gene
expression intensity (Figure 2). The threshold was defined as the intensity at
which
lower intensities exhibited an average CV that was greater than 0.3. For most
experiments, this threshold ranged from 10 to 40, and the number of genes
detected
above threshold ranged from 70% to 95%.
IS
CGN cell culture
CGNs were prepared from seven day old rat pups as previously described
(Johnson and Miller ( 1996) Journal of Neuroscience 16:74877-7495). Briefly,
cerebella were isolated, and meningeal layers and blood vessels were removed
under
a dissecting scope. Dissociated cells were plated at a density of 2.3 x 105
cells/cm2 in
basal medium Eagle (BME; Gibco) supplemented with 25 mM KCI, 10% dialyzed
fetal bovine serum (Summit Biotechnology lot #04D35, Ft. Collins, CO), 100
U/ml
penicillin, and 100 pg/ml streptomycin. Aphidicolin (Sigma, St. Louis, MO) was
added to the cultures at 3.3 pg/ml, 24 hours after initial plating to reduce
the number
of non-neuronal cells to less than I-5%.
For KCl/serum-withdrawal experiments, after seven days in culture, the
treated cells were switched to S mM KCI, BME, no serum, while the shams
received a
medium replacement. By 24 hours post-withdrawal, less than 30% of the cells
were
surviving as assayed by Hoechts cell counts (data not shown). This apparent
cell
death could be rescued by actinomycin D at 2 ~g/ml (data not shown).
For the KCI-withdrawal alone and kainate treatment experiments, on day two
in culture, the medium was replaced with neurobasal medium (Gibco)
supplemented
with 25 mM KCI, 0.5% dialyzed fetal bovine serum, B27 supplement (Gibco), 0.5
-102-
CA 02388617 2002-05-07
WO 01/31007 PCT/US00129132
mM L-glutamine (Gibco), 0.1 mg/ml AIbuMAX I (Gibco), 100 U/ml penicillin, 100
~tg/ml streptomycin, and 3.3 pg/ml aphidicolin. On day seven, KCl-withdrawal
was
initiated by replacing the medium with 5 mM KCl while the shams received 25
mM.
By 24 hours post-withdrawal, 40% of the cells were surviving as assayed by
Hoechts
cell counts (data not shown). As previously described, glutamate toxicity was
induced by replacing the medium for 30 min with 5 mM KCI, 100 pM kainic acid
(Sigma) in sodium free Locke's buffer, while the shams received no kainic acid
(Coyle et al. (1996) Neuroscience 7=1:675-683). After 30 min, the supplemented
neurobasal medium was replaced. By 12 hours post-withdrawal, 30% of the cells
were surviving as assayed by Hoechts cell counts (data not shown). The KCl-
withdrawal induced cell death was rescued by actinomycin D, whereas the
kainate-
induced was not.
Expression data clustering algorithms
After normalization and averaging of the KCl/serum-withdrawal data, 790
genes passed the following criteria over the 10 time points (5 treated, 5
sham) for
input into heirarchical clustering analysis: 1. detection, maximum intensity
greater
than 30; 2. noise filter, the difference between maximum and minimum intensity
greater than 30; and 3. regulation, fold induction between maximum and minimum
intensity of at least 3 (data not shown). Hierarchical clusters were ordered
based on
Euclidian distances. 234 out of 790 genes that passed the significance filter
described
above were not regulated in the controls based on CV less than 0.2 for all
five control
time points (data not shown).
RT-PCR
Oligonucleotide primer sequences specific for each EST validated by RT-PCR
were selected from quality sequence regions and designed to obtain a melting
temperature of 55-60°C as predicted by PrimerSelect software (DNASTAR,
Inc.,
Madison, WI) based on DNA stability measurements by ( Breslauer et al. ( 1986)
Proc. Natl. Acad. Sci. USA 83:3746-3750). The Stratagene Opti-PrimeTM Kit (La
Jolla, CA) was used to determine optimal RT-PCR amplification conditions for
each
primer pair. RT-PCR reactions on 2-fold serially diluted CGN programmed cell
death
cDNA were set up using the Genesis RSP 150 robotic sample processor and
-103-
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
incorporating the optimal buffer conditions for each primer pair. Every robot
run
included primers specific for housekeeping genes to control for day to day
differences
in cDNA template dilutions. The number of cycles was adjusted to obtain a
linear
range of amplification by comparing the amount of product made from the
serially
diluted templates as assessed by agarose gel electrophoresis.
Preparation of Array on Nylon
I. Procedure for Generating Labeled First Strand cDNA Using
Superscript II Reverse Transcriptase
1. 10 mL (100 mCi) 33P a-dCTP was dried down by SpeedVac.
2. In a separate tube, the following components were mixed:
1.0 ug Poly A+ RNA or 10 ug Total RNA
1 uL 1 ug/uL oligo-dT(30)
x uL DEPC-H20, to 10 uL
The above sample was heated at 70°C for 4 minutes and then placed
on ice.
3. 8uL from the oligo/RNA mixture (#2) was removed and used to
resuspend the dried 3P3. The following components were added to the reaction:
4 uL 5X First Strand Buffer (comes with Superscript II RT)
2 uL 100 mM DTT
1 uL 10 mM dAGT-TPs
1 uL 0.1 mM cold dCTP
1 uL Rnase Inhibitor
1 uL Superscript II RT
The reaction was incubated for 30 minutes at 50°C.
4. After incubation, 2 uL 0.5 M NaOH, and 2 uL 10 mM EDTA
were added. The reaction was heated at 65°C, for 10 minutes to degrade
RNA
template.
5. The volume was brought to 50 uL (i.e., add 26 uL H20).
6. One Choma-Spin +TE 30 column (Clontech, #K1321) was
prepared for every probe made.
a. Air bubbles were removed from the column.
-104-
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
b. The break-away end of the column was removed and
the column placed in an empty 2 mL tube and spun for 5 minutes at 700g (in
Eppendorf 541 SC "3.5").
c. The column was removed and the flow-through
discarded. The column was placed in clean tube. The probe was added slowly to
the
center of the column bed without disturbing the matrix so that the liquid did
not touch
the side of the column and flow down the edge of the column wall.
d. The probe was eluted by spinning the column as above.
II. Hybridization
1. The hybridization chamber was preheated to 65°C.
2. 10 mL of 10% Formamide Church Buffer was added. This was
placed in the hybridization chamber for around 15 minutes.
3. Sheared salmon sperm DNA was denatured at 95°C for 5
minutes, placed on ice, and then added to the hybridization mixture at a final
concentration of 100 ug/mL. Prehybridization was for 1.5 hours.
4. The amount of probe was calculated necessary to achieve 2 x
106 cpm/mL for 10 mL.
5. The Cot Annealing Reactions (per bottle) were as follows:
Rat probe with Rat Filters:
l0ug Poly dA (>200nt)
l0ug Rat Cot 10 DNA
25uL 20 x SSC
probe + water to 1 OOuL
Mouse probe with Rat Filters:
l0ug Poly dA (>200nt)
l0ug Mouse Cot 1 DNA
25uL 20 x SSC
probe + water to 1 OOuL
Also added Sug Rat Cot 10 DNA to the
prehybridization.
Human probe with Human Filters:
l0ug Poly dA (>200nt)
-105-
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
I Oug Human Cot 1 DNA
25uL 20 x SSC
probe + water to 100uL
The probe was heated to 95°C, and then probe was allowed to preanneal
at 65°C, for
1.5 hours.
6. The probe was added to prehybridizing filters (directly to the
solution and not onto the filters) and hybridization was for approximately 20
hours.
III. Washing
1. Probe was removed.
2. Three quick washes were performed with preheated 2 x
SSC/1% SDS, 65°C (washes could be done in roller bottles).
3. Two washes were performed for 15 minutes each with
preheated high stringency wash buffer:
0.5 x SSC, 0.1% SDS for cross species washes
0.5 x SSC, 0.1% SDS for normal washes
0.1 x SSC, 0.1 % SDS for very high stringency washes
4. After the high stringency washes, the filters were rinsed in a
large square petri dish in 2 x SSC, no SDS. For experiments in which many
filters are
used, the 2 x SSC is frequently changed so there is no residual SDS left on
the filters.
S. The filters were removed from the 2 x SSC and placed on
Whatman filter paper. Filters were baked at 85°C for 1 hour or longer.
Screens were
protected against any moisture. Filters were placed on a blank phosphorimager
screen. No yellowed phosphoimager screens were used since they may not respond
to
exposure linearly. Screens had been erased on a light box for no less than 20
minutes.
6. Blots were exposed to the screen at least 48 hours or as
necessary.
IV. Scanning Filters on Fuji Phosphorimager
Gradation 16 bit, Resolution SOm, Dynamic Range S4000, select Read
and Launch Image Gauge. Image was saved on the hard drive.
APPENDIX I:
-106-
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
10% Formamide-Church Buffer:
59.6mL water
70mL 20% SDS
50mL 2M NaP04 pH 7.2
20mL Ultrapure Formamide
0.4mL O.SM EDTA pH 8.0
The above components were added to water, mixed, and filtered through a 0.2 um
filter.
RT-PCR Protocol
I. For one PCR reaction mix, the following components were used:
28u1 SX First Strand Buffer
14u1 0.1 M DTT
4u1 dNTPs (20 mM)
7u1 Rnase Inhibitor
7u1 Superscript II
This buffer can be stored at -80°C for 3 months.
II. Total RNA was reversed transcribed as follows:
l.4ug Total RNA (DNAsed)
14u1 Random Primers (50ng/ul--Gibco)
Water was added to 60u1. The mixture was incubated at 70°C for 10
minutes and then
placed on ice for 2 minutes. 60u1 of the RT Reaction Mix was added. Incubation
was
at room temperature for 10 minutes, then 50°C for 30 minutes, then
90°C for 10
minutes. The sample was diluted with 480u1 water to result in long per Sul.
III. The PCR reaction was performed with the following ingredients:
Sul 4x PCR Buffer
Sul cDNA (at lOng/Sul)
Sul luM Primer Pair
Sul Enzyme Cocktail (0.2u1 Hot Start
Taq, lul 2mM
dNTPs, 3.8u1 water
-107-
CA 02388617 2002-05-07
WO 01131007 PCT/LTS00129132
IV. Cycling was as follows:
95°C 15 minutes
94°C 30 seconds
52°C 30 seconds
72°C 1 minute
Cycle 26-30 times
72°C 10 minutes
4°C Hold
Cerebellar granule cell isolation was performed according to the method
disclosed in Johnson et al. (1996) J. Neurosci. 16:74877-7495.
The induction of apoptosis in neurites induced by kainate is described in
Neurosci. 75:675-683 (1996). The procedure shown in this reference was
followed.
The following parameters were checked:
(1) Cerebellum granule neuron viability following potassium and serum
withdrawal at time points corresponding to PCR-based methods for differential
gene
expression (Hoechst stain).
{2) Effects of 2 ug/ml actinomycin D on potassium and serum withdrawal
at 24 hours on cerebellar granule neurons; viability by Hoeschst stained cell
counts.
(3) Time course of kainate-induced cell death for parallel analysis of PCR-
based method for differential gene expression of CGN Poly A mRNA.
(4) Time course of kainate-induced (30 minute exposure) apoptosis in
CGNs; analysis by Hoechst cell counts.
(5) Time course of potassium withdrawal apoptosis in CGNs in defined
media for PCR-based method for differential gene expression of analysis by
Hoechst
counts.
While this invention has been particularly shown and described with reference
to preferred embodiments thereof, it will be understood by those skilled in
the art that
various changes in form and details may be made therein without departing from
the
spirit and scope of the invention as defined by the appended claims.
-108-
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
SEQUENCE LISTING
<110> Chiang, Lillian Wei-Ming
<120> Nucleic Acid Molecules Derived from
Rat Brain and Programmed Cell Death Models
<130> 35800/205244 (5800-37-1)
<160> 11
<170> FastSEQ for Windows Version 3.0
<210> 1
<211> 2738
<212> DNA
<213> Homo Sapiens
<400>
1
gtcgacccacgcgtccggagatatccttaataagcgacaatgagttcaagtgcaggcatt60
cacagccggagtgtggttatggcttgcagcctgatcgttggacagagtacagcatacaga120
cgatggaaccagataacctggaactaatctttgattttttcgaagaagatctcagtgagc180
acgtagttcagggtgatgcccttcctggacatgtgggtacagcttgtctcttatcatcca240
ccattgctgagagtggaaagagtgctggaattcttactcttcccatcatgagcagaaatt300
cccggaaaacaataggcaaagtgagagttgactatataattattaagccattaccaggat360
acagttgtgacatgaaatcttcattttccaagtattggaagccaagaataccattggatg420
ttggccatcgaggtgcaggaaactctacaacaactgcccagctggctaaagttcaagaaa480
atactattgcttctttaagaaatgctgctagtcatggtgcagcctttgtagaatttgacg540
tacacctttcaaaggactttgtgcccgtggtatatcatgatcttacctgttgtttgacta600
tgaaaaagaaatttgatgctgatccagttgaattatttgaaattccagtaaaagaattaa660
catttgaccaactccagttgttaaagctcactcatgtgactgcactgaaatctaaggatc720
ggaaagaatctgtggttcaggaggaaaattccttttcagaaaatcagccatttccttctc780
ttaagatggttttagagtctttgccagaagatgtagggtttaacattgaaataaaatgga840
tctgccagcaaagggatggaatgtgggatggtaacttatcaacatattttgacatgaatc900
tgtttttggatataattttaaaaactgttttagaaaattctgggaagaggagaatagtgt960
tttcttcatttgatgcagatatttgcacaatggttcggcaaaagcagaacaaatatccga1020
tactatttttaactcaaggaaaatctgagatttatcctgaactcatggacctcagatctc1080
ggacaacccccattgcaatgagctttgcacagtttgaaaatctactggggataaatgtac1140
atactgaagacttgctcagaaacccatcctatattcaagaggcaaaagctaagggactag1200
tcatattctgctggggtgatgataccaatgatcctgaaaacagaaggaaattgaaggaac1260
ttggagttaatggtctaatttatgataggatatatgattggatgcctgaacaaccaaata1320
tattccaagtggagcaattggaacgcctgaagcaggaattgccagagcttaagagctgtt1380
tgtgtcccactgttagccgctttgttccctcatctttgtgtggggagtctgatatccatg1440
tggatgccaacggcattgataacgtggagaatgcttagtttttattgcacagaggtcatt1500
ttgggggcgtgcaccgctgttctgggtattcatttttcatcactgagcattgttgatcta1560
tgccttttgggcttctcagttcaatgaagcaataatgaagtatttaactctttcactaca1620
gttcttgcaagtatgctatttaaattacttggccaggtataattgccagtcagtctcttt1680
atagtgagaaaatttattggttagtaatataaatattttaaactaaatatataaatctat1740
aatgttaaacatatgttcattaaaagcatagcactttgaaattaactatataaatagctc1800
atatttacacttacagcttttcatttgatcaggtctgaaatctttagcacttaaggaaaa1860
tgactatgcataattatacctgaccatgaaaaaaataagtacctcaaatgcatgcatttg1920
cactggtgattccaactgcacaaatctttgtgccatcttgtatataggtattttttacat1980
gggttgacatgcacacaacaccattttcattcagtatgaaccttgaggctgctgccattt2040
ttccacttaaccaaaccagcctgaaggtgaacctcgaaacttgtttcataaatctttcaa2100
aagttgttttacatcaatgttaaaatttcaaaatgctgcagggtaatttaatgtataaaa2160
tattagtaagaaaaagtatgtattgcatacttagtagaatagatcacaacatacaaattc2220
aattcagtgcatgctttaggtgttaagcatgagattgtacatgtttactgttaggtcctt2280
gcatctgtggtgctaggtgagtatgagaagatgtcaaggactggacgtattttgttgcct2340
aaaaaaaaaaggctgtttgtaggcgttttaaatatgcttattttgtgtgtctctcactac2400
ctattacacactgttgctttgtgggtttgttttgtatgtgcgtgtgttatacagtagtta2960
aatttccatgcagaaaaataaatgtcctgaattctcatattagtattctttattgtatat2520
catgcatgtaatttatttagaaatgtaggtcttactaaatgtatatgcatgtatttcaga2580
ttatactaggatttcttggattagaagcagattgtgttaactgtaacttaaagaatgaat2640
gttaaataaaatgatacagatttattttcttcattacaaaaaaaaaaaaaaaaaaaaaaa2700
aaaaaaaaaaaaaaaaaaaaaaaaaaaagggcggccgc 2738
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
<210> 2
<211> 1907
<212> DNA
<213> Homo Sapiens
<400> 2
gtcgacccacgcgtccggttggagcgagcatgtgggtctgcagtaccctgtggcgggtgc60
gaacccccgcccggcagtggcgggggctgctcccagcttctggctgtcacggacctgccg120
cctcctcctactccgcatccgccgagcctgcccgggtccgggcgcttgtctatgggcacc180
acggggatccagccaaggtcgtcgaactcaagaacctggagctagctgctgtgagaggat240
cagatgtccgtgtgaagatgctggcggcccctatcaatccatctgacataaatatgatcc300
aaggaaactacggactccttcctgaactgcctgctgttggagggaacgaaggtgttgcac360
aggtggtagcggtgggcagcaatgtgaccgggctgaagccaggagactgggtgattccag920
caaatgctggtttaggaacctggcggaccgaggctgtgttcagcgaggaagcactgatcc480
aagttccgagtgacatccctcttcagagcgctgccaccctgggtgtcaatccctgcacag540
cctacaggatgttgatggatttcgagcaactgcagccaggggattctgtcatccagaatg600
catccaacagcggagtggggcaagcggtcatccagatcgccgcagccctgggcctaagaa660
ccatcaatgtggtccgagacagacctgatatccagaagctgagtgacagactgaagagtc720
tgggggctgagcatgtcatcacagaagaggagctaagaaggcccgaaatgaaaaacttct780
ttaaggacatgccccagccacggcttgctctcaactgtgttggtgggaaaagctccacag840
agctgctgcggcagttagcgcgtggaggaaccatggtaacctatggggggatggccaagc900
agcccgtcgtagcctctgtgagcctgctcatttttaaggatctcaaacttcgaggctttt960
ggttgtcccagtggaagaaggatcacagtccagaccagttcaaggagctgatcctcacac1020
tgtgcgatctcatccgccgaggccagctcacagcccctgcctgctcccaggtcccgctgc1080
aggactaccagtctgccttggaagcctccatgaagcccttcatatcttcaaagcagattc1140
tcaccatgtgatcatcccaaaagagctggagtgacatgggaggggaggcggatctgaggg1200
gctgggtgcaggcccctcagttggggctcccaccttccccagactactgttctcctcact1260
gcctcttcctattaggaggatggtgaagccagccacggttttccccagggccagccttaa1320
ggtatctaataaagtctgaactctcccttccaaaaaaaaaaaaaaaaaaaaaaaaaaaaa1380
aaaaaaaaaaaaaaaaagggcggccgc 1407
<210> 3
<211> 1664
<212> DNA
<213> Homo Sapiens
<400> 3
gcggccgcagccccggccgagcaggcgccgcgggccaagggccgcccgagacggtcccca60
gagagccaccggaggagcagctcacctgagagacggagccccggctcgcccgtgtgcaga120
gcggacaaggcaaaatctcagcaagttcggacctctagtacaataaggcgaacctcctct180
ttggatacaataacaggaccttacctcacaggacagtggccacgggatcctcatgttcac240
tacccttcatgcatgaaagacaaagctactcagacacctagctgttgggcagaagagggt300
gcagaaaagaggtcacatcagcgttctgcgtcatgggggagtgctgatcaactaaaagag360
atcgccaaactgaggcagcaactacaacgcagtaaacagagtagtcgtcacagtaaggag420
aaagatcgccagtcacctcttcatggcaaccatataacaatcagtcacactcaggctact480
ggatcaaggtcagttcctatgccactgtcaaatatatcagtgccaaaatcatctgtttcg590
cgtgtgccctgcaatgtagaaggaataagtcctgaattagaaaaggtattcattaaagaa600
aataatgggaaggaagaagtatccaagccgttggacataccagatggtcgaagagctcca660
cttcctgctcattaccggagcagtagtactcgcagcattgacactcagactccttctgtc720
caggagcgcagcagtagctgcagcagtcattcaccctgtgtctcccctttttgtcccccg780
gaatcccaggatggtagcccttgctcaacagaagatttgctctatgatcgtgataaaggt840
ctcgtcagcctatctcggcccctctcttttcatgtcctgacaaaaacaaggttaatttca900
tcccaaccggatcagctttctgtcctgtaaaacttctaggccccctcttacctgcttctg960
accttatgctcaagaactctcctaactctggccagagctcagctttggcaactctgaccg1020
ttgagcagctctcatcccgggtttcctttacgtctctttctgatgacaccagcacagcgg1080
gctccatggaggcctctgtccagcagccatcccagcagcagcagctcctgcaggaactgc1140
agggtgaggaccacatctctgctcagaactatgtgatcatctaaaaaagggggagctggc1200
ctccaccctgtgttccatggattcggaacaagatttcagacatctgcatgagtgacaaac1260
tttctgaacaccaccaccaccaataatacttatcagcatcataaagtatctcttaaacac1320
tgatcttggcagggacggaactcctattcagcagtttttgtggaaagcagtaatgcttgc1380
aaaacgtgtgtgtcattcagcattttaagtggagactatgcatttcatagtatatttgac1490
agattagtactgtgtcctgtgttttgttccagattcttcagtataaataagctctatatc1500
aaaaagttgcctgtctaaatagaaaatgtcttgctgtgttttgtcctatggaaaatactg1560
taattcaggattatgtttacaattgatccaggtgtttgtttctaacttctgtaatacata1620
caatgcaaaaaaaaaaaaaaaaaacggacgcgtgggtcgactcc 1664
2
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
<210> 9
<211> 3206
<212> DNA
<213> Homo Sapiens
<400> 4
gtcgacccacgcgtccgggcgaggcacggacggcgggcgcccggtacctctgcccgcggt60
cctcgctctcgggcggggcggcggcgacgcggacctgcggactagcgaacccggagcacg120
acatcataaaataaatccatcagaatgacaccttctcaggttgcctttgaaataagagga180
actcttttaccaggagaagtttttgcgatatgtggaagctgtgatgctttgggaaactgg240
aatcctcaaaatgctgtggctcttcttccagagaatgacacaggtgaaagcatgctatgg300
aaagcaaccattgtactcagtagaggagtatcagttcagtatcgctacttcaaagggtac360
tttttagaaccaaagactatcggtggtccatgtcaagtgatagttcacaagtgggagact420
catctacaaccacgatcaataacccctttagaaagcgaaattattattgacgatggacaa980
tttggaatccacaatggtgttgaaactctggattctggatggctgacatgtcagactgaa590
ataagattacgtttgcattattctgaaaaacctcctgtgtcaataaccaagaaaaaatta600
aaaaaatctagatttagggtgaagctgacactagaaggcctggaggaagatgacgatgat660
agggtatctcccactgtactccacaaaatgtccaatagcttggagatatccttaataagc720
gacaatgagttcaagtgcaggcattcacagccggagtgtggttatggcttgcagcctgat780
cgttggacagagtacagcatacagacgatggaaccagataacctggaactaatctttgat840
tttttcgaagaagatctcagtgagcacgtagttcagggtgatgcccttcctggacatgtg900
ggtacagcttgtctcttatcatccaccattgctgagagtggaaagagtgctggaattctt960
actcttcccatcatgagcagaaattcccggaaaacaataggcaaagtgagagttgactat1020
ataattattaagccattaccaggatacagttgtgacatgaaatcttcattttccaagtat1080
tggaagccaagaataccattggatgttggccatcgaggtgcaggaaactctacaacaact1140
gcccagctggctaaagttcaagaaaatactattgcttctttaagaaatgctgctagtcat1200
ggtgcagcctttgtagaatttgacgtacacctttcaaaggactttgtgcccgtggtatat1260
catgatcttacctgttgtttgactatgaaaaagaaatttgatgctgatccagttgaatta1320
tttgaaattccagtaaaagaattaacatttgaccaactccagttgttaaagctcactcat1380
gtgactgcactgaaatctaaggatcggaaagaatctgtggttcaggaggaaaattccttt1990
tcagaaaatcagccatttccttctcttaagatggttttagagtctttgccagaagatgta1500
gggtttaacattgaaataaaatggatctgccagcaaagggatggaatgtgggatggtaac1560
ttatcaacatattttgacatgaatctgtttttggatataattttaaaaactgttttagaa1620
aattctgggaagaggagaatagtgttttcttcatttgatgcagatatttgcacaatggtt1680
cggcaaaagcagaacaaatatccgatactatttttaactcaaggaaaatctgagatttat1740
cctgaactcatggacctcagatctcggacaacccccattgcaatgagctttgcacagttt1800
gaaaatctactggggataaatgtacatactgaagacttgctcagaaacccatcctatatt1860
caagaggcaaaagctaagggactagtcatattctgctggggtgatgataccaatgatcct1920
gaaaacagaaggaaattgaaggaacttggagttaatggtctaatttatgataggatatat1980
gattggatgcctgaacaaccaaatatattccaagtggagcaattggaacgcctgaagcag2040
gaattgccagagcttaagagctgtttgtgtcccactgttagccgctttgttccctcatct2100
ttgtgtggggagtctgatatccatgtggatgccaacggcattgataacgtggagaatgct2160
tagtttttattgcacagaggtcattttgggggcgtgcaccgctgttctgggtattcattt2220
ttcatcactgagcattgttgatctatgccttttgggcttctcagttcaatgaagcaataa2280
tgaagtatttaactctttcactacagttcttgcaagtatgctatttaaattacttggcca2340
ggtataattgccagtcagtctctttatagtgagaaaatttattggttagtaatataaata2400
ttttaaactaaatatataaatctataatgttaaacatatgttcattaaaagcatagcact2460
ttgaaattaactatataaatagctcatatttacacttacagcttttcatttgatcaggtc2520
tgaaatctttagcacttaaggaaaatgactatgcataattatacctgaccatgaaaaaaa2580
taagtacctcaaatgcatgcatttgcactggtgattccaactgcacaaatctttgtgcca2640
tcttgtatataggtattttttacatgggttgacatgcacacaacaccattttcattcagt2700
atgaaccttgaggctgctgccatttttccacttaaccaaaccagcctgaaggtgaacctc2760
gaaacttgtttcataaatctttcaaaagttgttttacatcaatgttaaaatttcaaaatg2820
ctgcagggtaatttaatgtataaaatattagtaagaaaaagtatgtattgcatacttagt2880
agaatagatcacaacatacaaattcaattcagtgcatgctttaggtgttaagcatgagat2940
tgtacatgtttactgttaggtccttgcatctgtggtgctaggtgagtatgagaagatgtc3000
aaggactggacgtattttgttgcctaaaaaaaaaaggctgtttgtaggcgttttaaatat3060
gcttattttgtgtgtctctcactacctattacacactgttgctttgtgggtttgttttgt3120
atgtgcgtgtgttatacagtagttaaatttccatgcagaaaaataaatgtcctgaattct3180
caaaaaaaaaaaaaaagggcggccgc 3206
<210> 5
<211> 2034
<212> DNA
<213> Homo Sapiens
<400> 5
3
CA 02388617 2002-05-07
WO (11/310U7 PCT/USUO/29132
gtcgacccacgcgtccggcaagatctctctggaccagctcgggtgcagggcctctgcggg60
agccctcctagacctctgcggcttctcctctaacatggccgactcggaaaaccaggggcc120
tgcggagcctagccaggcggcggcagcggcggaggcagcggcagaggaggtaatggcgga180
aggcggtgcgcagggtggagactgtgacagcgcggctggtgaccctgacagcgcggctgg290
tcagatggctgaggagccccagacccctgcagagaatgccccaaagccgaaaaatgactt300
tatcgagagcctgcctaattcggtgaaatgccgagtcctggccctcaaaaagctgcagaa360
gcgatgcgataagatagaagccaaatttgataaggaatttcaggctctggaaaaaaagta920
taatgacatctataagcccctactcgccaagatccaagagctcaccggcgagatggaggg480
gtgtgcatggaccttggagggggaggaggaggaggaagaggagtacgaggatgacgagga540
ggagggggaagacgaggaggaggaggaggctgcggcagaggctgccgcgggggccaaaca600
tgacgatgcccacgccgagatgcctgatgacgccaagaagtaaggggggcagagatggat660
gaagagaaagcccacgaagaaaaaagcctggttttgtttttcccagaatatcgatggact720
taaaaaggctcaggtttttgaccaaaatacaatgtgaatttattctgacattcctaaaat780
agattaaattaaagcaattagatcctggccagctcgattcaaatttgactttcattttga840
acataataaatatatcaaaaggtgttaaagaaaactgaattaaacccaaaattatgtttt900
catggtctcttctctgaggattgaggtttacaaagggtgttagcagatgcgaagtaaaga960
acgtcactttgaaacccattcatcacacagcatacgctacacatggaacacccaagccat1020
gactgaacacgttctcagtgcttaattcttaaatttctttactcatgacatttcgcagtg1080
cagagaaggcagaacccaagaaaaacgtcatctttgagactttgcttttgtaacgcagac1140
atcagctttacacttcacaggagattgatggcattgaggaagattgcaatggagatcatg1200
acactactgttaataaggccaggaaaactgccatttcaagttctgaaaaatgttttgagt1260
atttgaatttagagaaacaacatggttccaagaaggagggtgtaaaacctgtaaaatact1320
gtcaacatatgtattcattagttacaatctcatgtttgtgttttcttagtactgtctatt1380
tacaaacacgtaaaaaataccccaaatatgtttaagtattaaatcactttacctagcgtt1440
ttagaaatattaatttacttgaagagatgtagaatgtagcaaattatgtaaagcatgtgt1500
atccagcgttatgtactttgcgccttgtgacgtctttctgtcatgtagcttttagggtgt1560
agctgtgaaaatcatcagaactcttcactgaagctaatgtttggaaaaaatatatacttg1620
aagaaccaatccaagtgtgtgcccctacccccagctcagaagtagaaagggtttaagttt1680
gcttgtattagctgtgccttcattattttgctatgtaaatgtgacatattaattataaaa1740
tggtgcataatcaaattttactgcttgaggacagatgcatacagtaaggatttttaggaa1800
gaatatatttaatgtaaagactcttagcttctgtgtgggttttgaattatgtgtgagcca1860
gtgatctataaagaaacataagcttaaagttgtttatcactgtggtgttaataaaacagt1920
attttcaaaaaataaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaa1980
aaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaagggcggccgc 2039
<210> 6
<211> 3605
<212> DNA
<213> Homo sapiens
<220>
<221> CDS
<222> (233)...(2308)
<400> 6
gcctggagga gtgagccagg ggctcgggcg ggccgggacg 60
cagtgagact cgtcgttgca
gcagcggctc ccagctccca cgcgcgcccc ttcacgcgcc 120
gccaggattc ctgctcctga
acttcagctc ctgcacagtc caaggctcaa ggcgccgccg 180
ctccccaccg gcgtggaccg
cgcacggcct ctaggtctcc agcaacctct cccctggccc 238
tcgccaggac tc
atg
ggc
Met Gly
1
acc gtc agc tcc agg cgg tgg ctgccactg ctg ctg 286
tcc tgg ccg ctg
Thr Val Ser Ser Arg Arg Trp LeuProLeu Leu Leu
Ser Trp Pro Leu
10 15
ctg ctg ctg ctc ctg ggt ggc cgtgcgcag gag gac 334
ccc gcg gcc gag
Leu Leu Leu Leu Leu Gly Gly ArgAlaGln Glu Asp
Pro Ala Ala Glu
20 25 30
gac ggc gac tac gag gag cta ttgcgttcc gag gag 382
ctg gtg gcc gac
Asp Gly Asp Tyr G1u Glu Leu LeuArgSer Glu Glu
Leu Val Ala Asp
35 40 45 50
ggc ctg gcc gaa gca ccc gga acagccacc ttc cac 430
gag cac acc cgc
Gly Leu Ala G1u Ala Pro Gly ThrAlaThr Phe His
Glu His Thr Arg
55 60 65
4
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
tgc gccaaggatccgtggaggttgcctggcacctacgtggtggtgctg 478
Cys AlaLysAspProTrpArgLeuProGlyThrTyrValValValLeu
70 75 80
aag gaggagacccacctctcgcagtcagagcgcactgcccgccgcctg 526
Lys GluGluThrHisLeuSerGlnSerGluArgThrAlaArgArgLeu
85 90 95
cag gcccaggetgcccgccggggatacctcaccaagatcctgcatgtc 574
Gln AlaGlnAlaAlaArgArgGlyTyrLeuThrLysIleLeuHisVal
100 105 110
ttc catggccttcttcctggcttcctggtgaagatgagtggcgacctg 622
Phe HisGlyLeuLeuProGlyPheLeuValLysMetSerGlyAspLeu
115 120 125 130
ctg gagctggccttgaagttgccccatgtcgactacatcgaggaggac 670
Leu GluLeuAlaLeuLysLeuProHisValAspTyrIleGluGluAsp
135 140 145
tcc tctgtctttgcccagagcatcccgtggaacctggagcggattacc 718
Ser SerValPheAlaGlnSerIleProTrpAsnLeuGluArgIleThr
150 155 160
cct ccacggtaccgggcggatgaataccagccccccgacggaggcagc 766
Pro ProArgTyrArgAlaAspGluTyrGlnProProAspGlyGlySer
165 170 175
ctg gtggaggtgtatctcctagacaccagcatacagagtgaccaccgg 814
Leu ValGluValTyrLeuLeuAspThrSerIleGlnSerAspHisArg
180 185 190
gaa atcgagggcagggtcatggtcaccgacttcgagaatgtgcccgag 862
Glu IleGluGlyArgValMetValThrAspPheGluAsnValProGlu
195 200 205 210
gag gacgggacccgcttccacagacaggccagcaagtgtgacagtcat 910
Glu AspGlyThrArgPheHisArgGlnAlaSerLysCysAspSerHis
215 220 225
ggc acccacctggcaggggtggtcagcggccgggatgccggcgtggcc 958
Gly ThrHisLeuAlaGlyValValSerGlyArgAspAlaGlyValA1a
230 235 240
aag ggtgccagcatgcgcagcctgcgcgtgctcaactgccaagggaag 1006
Lys GlyAlaSerMetArgSerLeuArgValLeuAsnCysGlnGlyLys
295 250 255
ggc acggttagcggcaccctcataggcctggagtttattcggaaaagc 1054
Gly ThrValSerGlyThrLeuIleGlyLeuGluPheIleArgLysSer
260 265 270
cag ctggtccagcctgtggggccactggtggtgctgctgcccctggcg 1102
Gln LeuValGlnProValGlyProLeuValValLeuLeuProLeuAla
275 280 285 290
ggt gggtacagccgcgtcctcaacgccgcctgccagcgcctggcgagg 1150
Gly GlyTyrSerArgValLeuAsnAlaAlaCysGlnArgLeuAlaArg
295 300 305
get ggggtcgtgctggtcaccgetgccggcaacttccgggacgatgcc 1198
Ala GlyValValLeuValThrAlaAlaGlyAsnPheArgAspAspAla
310 315 320
tgc ctctactccccagcctcagetcccgaggtcatcacagttggggcc 1246
Cys LeuTyrSerProAlaSerAlaProGluValIleThrValGlyAla
5
CA 02388617 2002-05-07
WO 01/31007 PCT/iTS00/29132
325 330 335
accaatgcccaggaccagccggtgaccctggggactttggggaccaac 1294
ThrAsnAlaGlnAspGlnProValThrLeuGlyThrLeuGlyThrAsn
340 345 350
tttggccgctgtgtggacctctttgccccaggggaggacatcattggt 1342
PheGlyArgCysValAspLeuPheAlaProGlyGluAspIleIleGly
355 360 365 370
gcctccagcgactgcagcacctgctttgtgtcacagagtgggacatca 1390
AlaSerSerAspCysSerThrCysPheValSerGlnSerGlyThrSer
375 380 385
caggetgetgcccacgtggetggcattgcagccatgatgctgtctgcc 1938
GlnAlaAlaAlaHisValAlaGlyIleAlaAlaMetMetLeuSerAla
390 395 400
gagccggagctcaccctggccgagttgaggcagagactgatccacttc 1986
GluProGluLeuThrLeuAlaGluLeuArgGlnArgLeuIleHisPhe
405 910 415
tctgccaaagatgtcatcaatgaggcctggttccctgaggaccagcgg 1534
SerAlaLysAspValIleAsnGluAlaTrpPheProGluAspGlnArg
420 425 430
gtactgacccccaacctggtggccgccctgcccccc.agcacccatggg 1582
ValLeuThrProAsnLeuValAlaAlaLeuProProSerThrHisGly
435 440 995 450
gcaggttggcagctgttttgcaggactgtgtggtcagcacactcgggg 1630
AlaGlyTrpGlnLeuPheCysArgThrValTrpSerAlaHisSerGly
455 460 465
cctacacggatggccacagccatcgcccgctgcgccccagatgaggag 1678
ProThrArgMetAlaThrAlaIleAlaArgCysAlaProAspGluGlu
470 475 480
ctgctgagctgctccagtttctccaggagtgggaagcggcggggcgag 1726
LeuLeuSerCysSerSerPheSerArgSerGlyLysArgArgGlyGlu
985 490 495
cgcatggaggcccaagggggcaagctggtctgccgggcccacaacget 1779
ArgMetGluAlaGlnGlyGlyLysLeuValCysArgAlaHisAsnAla
500 505 510
tttgggggtgagggtgtctacgccattgccaggtgctgcctgctaccc 1822
PheGlyGlyGluGlyValTyrAlaIleAlaArgCysCysLeuLeuPro
515 520 525 530
caggccaactgcagcgtccacacagetccaccagetgaggccagcatg 1870
GlnAlaAsnCysSerValHisThrAlaProProAlaGluAlaSerMet
535 540 545
gggacccgtgtccactgccaccaacagggccacgtcctcacaggctgc 1918
GlyThrArgValHisCysHisGlnGlnGlyHisValLeuThrGlyCys
550 555 560
agctcccactgggaggtggaggaccttggcacccacaagccgcctgtg 1966
SerSerHisTrpGluValGluAspLeuGlyThrHisLysProProVal
565 570 575
ctgaggccacgaggtcagcccaaccagtgcgtgggccacagggaggcc 2014
LeuArgProArgGlyGlnProAsnGlnCysValGlyHisArgGluAla
580 585 590
agcatccacgettcctgctgccatgccccaggtctggaatgcaaagtc 2062
6
CA 02388617 2002-05-07
WO 01/31007 PCT/US(10/29132
Ser Ile His Ala Ser Cys Cys His Ala Pro Gly Leu Glu Cys Lys Val
595 600 605 610
aag gag cat gga atc ccg gcc cct cag gag cag gtg acc gtg gcc tgc 2110
Lys Glu His Gly Ile Pro Ala Pro Gln Glu Gln Val Thr Val Ala Cys
615 620 625
gag gag ggc tgg acc ctg act ggc tgc agt gcc ctc cct ggg acc tcc 2158
Glu Glu Gly Trp Thr Leu Thr Gly Cys Ser Ala Leu Pro Gly Thr Ser
630 635 640
cac gtc ctg ggg gcc tac gcc gta gac aac acg tgt gta gtc agg agc 2206
His Val Leu Gly Ala Tyr Ala Val Asp Asn Thr Cys Val Val Arg Ser
695 650 655
cgg gac gtc agc act aca ggc agc acc agc gaa gag gcc gtg aca gcc 2254
Arg Asp Val Ser Thr Thr Gly Ser Thr Ser Glu Glu Ala Val Thr Ala
660 665 670
gtt gcc atc tgc tgc cgg agc cgg cac ctg gcg cag gcc tcc cag gag 2302
Val Ala Ile Cys Cys Arg Ser Arg His Leu Ala Gln Ala Ser Gln Glu
675 680 6B5 690
ctc cag tgacagcccc atcccaggat gggtgtctgg ggagggtcaa gggctggggc 2358
Leu Gln
tgagctttaa aatggttccg acttgtccct ctctcagccc tccatggcct ggcacgaggg 2918
gatggggatg cttccgcctt tccggggctg ctggcctggc ccttgagtgg ggcagcctcc 2478
ttgcctggaa ctcactcact ctgggtgcct cctccccagg tggaggtgcc aggaagctcc 2538
ctccctcact gtggggcatt tcaccattca aacaggtcga gctgtgctcg ggtgctgcca 2598
gctgctccca atgtgccgat gtccgtgggc agaatgactt ttattgagct cttgttccgt 2658
gccaggcatt caatcctcag gtctccacca aggaggcagg attcttccca tggatagggg 2718
agggggcggt aggggctgca gggacaaaca tcgttggggg gtgagtgtga aaggtgctga 2778
tggccctcat ctccagctaa ctgtggagaa gcccctgggg gctccctgat taatggaggc 2838
ttagctttct ggatggcatc tagccagagg ctggagacag gtgtgcccct ggtggtcaca 2898
ggctgtgcct tggtttcctg agccaccttt actctgctct atgccaggct gtgctagcaa 2958
cacccaaagg tggcctgcgg ggagccatca cctaggactg actcggcagt gtgcagtggt 3018
gcatgcactg tctcagccaa cccgctccac tacccggcag ggtacacatt cgcaccccta 3078
cttcacagag gaagaaacct ggaaccagag ggggcgtgcc tgccaagctc acacagcagg 3138
aactgagcca gaaacgcaga ttgggctggc tctgaagcca agcctcttct tacttcaccc 3198
ggctgggctc ctcattttta cgggtaacag tgaggctggg aaggggaaca cagaccagga 3258
agctcggtga gtgatggcag aacgatgcct gcaggcatgg aactttttcc gttatcaccc 3318
aggcctgatt cactggcctg gcggagatgc ttctaaggca tggtcggggg agagggccaa 3378
caactgtccc tccttgagca ccagccccac ccaagcaagc agacatttat cttttgggtc 3438
tgtcctctct gttgcctttt tacagccaac ttttctagac ctgttttgct tttgtaactt 3498
gaagatattt attctgggtt ttgtagcatt tttattaata tggtgacttt ttaaaataaa 3558
aacaaacaaa cgttgtccta aaaaaaaaaa aaaaaawaaa aaaaaaa 3605
<210> 7
<211> 692
<212> PRT
<213> Homo sapiens
<400> 7
Met Gly Thr Val Ser Ser Arg Arg Ser Trp Trp Pro Leu Pro Leu Leu
1 5 10 15
Leu Leu Leu Leu Leu Leu Leu Gly Pro Ala Gly Ala Arg Ala Gln Glu
20 25 30
Asp Glu Asp Gly Asp Tyr Glu Glu Leu Val Leu Ala Leu Arg Ser Glu
35 40 45
Glu Asp Gly Leu Ala Glu Ala Pro Glu His Gly Thr Thr Ala Thr Phe
50 55 60
His Arg Cys Ala Lys Asp Pro Trp Arg Leu Pro Gly Thr Tyr Val Val
65 70 75 80
Val Leu Lys Glu Glu Thr His Leu Ser Gln Ser Glu Arg Thr Ala Arg
85 90 95
7
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
Arg Leu Gln Ala Gln Ala Ala Arg Arg Gly Tyr Leu Thr Lys Ile Leu
100 105 110
His Val Phe His Gly Leu Leu Pro Gly Phe Leu Va1 Lys Met Ser Gly
115 120 125
Asp Leu Leu Glu Leu Ala Leu Lys Leu Pro His Val Asp Tyr Ile Glu
130 135 140
Glu Asp Ser Ser Val Phe Ala Gln Ser Ile Pro Trp Asn Leu Glu Arg
145 150 155 160
Ile Thr Pro Pro Arg Tyr Arg Ala Asp Glu Tyr Gln Pro Pro Asp Gly
165 170 175
Gly Ser Leu Val Glu Val Tyr Leu Leu Asp Thr Ser Ile Gln Ser Asp
180 185 190
His Arg Glu Ile Glu Gly Arg Val Met Val Thr Asp Phe Glu Asn Val
195 200 205
Pro Glu Glu Asp Gly Thr Arg Phe His Arg Gln Ala Ser Lys Cys Asp
210 215 220
Ser His Gly Thr His Leu Ala Gly Val Val Ser Gly Arg Rsp Ala Gly
225 230 235 240
Val Ala Lys Gly Ala Ser Met Arg Ser Leu Arg Val Leu Asn Cys Gln
245 250 255
Gly Lys Gly Thr Val Ser Gly Thr Leu Ile Gly Leu Glu Phe Ile Arg
260 265 270
Lys Ser Gln Leu Val Gln Pro Val Gly Pro Leu Val Val Leu Leu Pro
275 280 285
Leu Ala Gly Gly Tyr Ser Arg Val Leu Asn Ala Ala Cys Gln Arg Leu
290 295 300
Ala Arg Ala Gly Val Val Leu Val Thr Ala Ala Gly Asn Phe Arg Asp
305 310 315 320
Asp Ala Cys Leu Tyr Ser Pro Ala Ser Ala Pro Glu Val Ile Thr Val
325 330 335
Gly Ala Thr Asn Ala Gln Asp Gln Pro Val Thr Leu Gly Thr Leu Gly
340 345 350
Thr Asn Phe Gly Arg Cys Val Asp Leu Phe Ala Pro Gly Glu Asp Ile
355 360 365
Ile Gly Ala Ser Ser Asp Cys Ser Thr Cys Phe Val Ser Gln Ser Gly
370 375 380
Thr Ser Gln Ala Ala Ala His Val Ala Gly Ile Ala Ala Met Met Leu
385 390 395 400
Ser Ala Glu Pro Glu Leu Thr Leu Ala Glu Leu Arg Gln Arg Leu Ile
405 410 915
His Phe Ser Ala Lys Asp Val Ile Asn Glu Ala Trp Phe Pro Glu Asp
420 425 430
Gln Arg Val Leu Thr Pro Asn Leu Val Ala Ala Leu Pro Pro Ser Thr
935 440 945
His Gly Ala Gly Trp Gln Leu Phe Cys Arg Thr Val Trp Ser Ala His
450 955 460
Ser Gly Pro Thr Arg Met Ala Thr Ala Ile Ala Arg Cys Ala Pro Asp
965 470 975 980
Glu Glu Leu Leu Ser Cys Ser Ser Phe Ser Arg Ser Gly Lys Arg Arg
485 490 495
Gly Glu Arg Met Glu Ala Gln Gly Gly Lys Leu Val Cys Arg Ala His
500 505 510
Asn Ala Phe Gly Gly Glu Gly Val Tyr Ala Ile Ala Arg Cys Cys Leu
515 520 525
Leu Pro Gln Ala Asn Cys Ser Val His Thr Ala Pro Pro Ala Glu Ala
530 535 540
Ser Met Gly Thr Arg Val His Cys His Gln Gln Gly His Val Leu Thr
545 550 555 560
Gly Cys Ser Ser His Trp Glu Val Glu Asp Leu Gly Thr His Lys Pro
565 570 575
Pro Val Leu Arg Pro Arg Gly Gln Pro Asn Gln Cys Val Gly His Arg
580 585 590
Glu Ala Ser Ile His Ala Ser Cys Cys His Ala Pro Gly Leu Glu Cys
595 600 605
Lys Val Lys Glu His Gly Ile Pro Ala Pro Gln Glu Gln Val Thr Val
610 615 620
Ala Cys Glu Glu Gly Trp Thr Leu Thr Gly Cys Ser Ala Leu Pro Gly
g
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
625 630 635 640
Thr Ser His Val Leu Gly Ala Tyr Ala Val Asp Asn Thr Cys Val Val
645 650 655
Arg
Ser
Arg
Asp
Val
Ser
Thr
Thr
Gly
Ser
Thr
Ser
Glu
Glu
Ala
Val
660 665 670
Thr Ile eu
Ala Cys Ala
Val Cys Gln
Ala Arg Ala
Ser Ser
Arg
His
L
675 680 685
Gln Gln
Glu
Leu
690
<210>8
<211>3583
<212>DNA
<213>HomoSapiens
<220>
<221>CDS
<222>(97)...(1863 )
<400>8
cggacgcgtggcgcaaggc aaggcgcc gccggcg tggaccgcgcacg cctctaggt60
g tc g
ctcctcgccagacagcaac ctcccctggccctc atgggcaccgtcagctcc 114
g ct
MetGlyThrValSer5er
1 5
agg cgg tggtgg ccgctg ccactgctgctgctgctgctgctgctc 162
tcc
Arg Arg TrpTrp ProLeu ProLeuLeuLeuLeuLeuLeuLeuLeu
Ser
10 15 20
ctg ggt gcgggc gcccgt gcgcaggaggacgaggacggcgactac 210
ccc
Leu Gly AlaGly AlaArg AlaGlnGluAspGluAspGlyAspTyr
Pro
25 30 35
gag gag gtgcta gccttg cgttccgaggaggacggcctggccgaa 258
ctg
Glu Glu ValLeu AlaLeu ArgSerGluGluAspGlyLeuAlaGlu
Leu
40 95 50
gca ccc cacgga accaca gccaccttccaccgctgcgccaaggat 306
gag
Ala Pro HisGly ThrThr AlaThrPheHisArgCysAlaLysAsp
Glu
55 60 65 70
ccg tgg ttgcct ggcacc tacgtggtggtgctgaaggaggagacc 354
agg
Pro Trp LeuPro GlyThr TyrValValValLeuLysGluGluThr
Arg
75 80 85
cac ctc cagtca gagcgc actgcccgccgcctgcaggcccagget 402
tcg
His Leu GlnSer GluArg ThrAlaArgArgLeuGlnAlaGlnAla
Ser
gp 95 100
gcc cgc ggatac ctcacc aagatcctgcatgtcttccatggcctt 450
cgg
Ala Arg GlyTyr LeuThr LysIleLeuHisValPheHisGlyLeu
Arg
105 110 115
ctt cct ttcctg gtgaag atgagtggcgacctgctggagctggcc 498
ggc
Leu Pro PheLeu ValLys MetSerGlyAspLeuLeuGluLeuAla
Gly
120 125 130
ttg aag ccccat gtcgac tacatcgaggaggactcctctgtcttt 546
ttg
Leu Lys ProHis ValAsp TyrIleGluGluAspSerSerValPhe
Leu
135 140 145 150
gcc cag atcccg tggaac ctggagcggattacccctccacggtac 594
agc
Ala Gln IlePro TrpAsn LeuGluArgIleThrProProArgTyr
Ser
155 160 165
cgg gcg gaatac cagccc cccgacggaggcagcctggtggaggtg 642
gat
Arg Ala GluTyr GlnPro ProAspGlyGlySerLeuValGluVal
Asp
9
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
170 175 180
tatctcctagac accagcatacagagtgaccaccgg gaaatcgagggc 690
TyrLeuLeuAsp SerIleGlnSerAspHisArg GluIleGluGly
Thr
185 190 195
agggtcatggtc accgacttcgagaatgtgcccgag gaggacgggacc 738
ArgValMetVal ThrAspPheGluAsnValProGlu GluAspGlyThr
200 205 210
cgcttccacaga caggccagcaagtgtgacagtcat ggcacccacctg 786
ArgPheHisArg GlnAlaSerLysCysAspSerHis GlyThrHisLeu
215 220 225 230
gcaggggtggtc agcggccgggatgccggcgtggcc aagggtgccagc 839
AlaGlyValVal SerGlyArgAspAlaGlyValAla LysGlyAlaSer
235 240 245
atgcgcagcctg cgcgtgctcaactgccaagggaag ggcacggttagc 882
MetArgSerLeu ArgValLeuAsnCysGlnGlyLys GlyThrValSer
250 255 260
ggcaccctcata ggcctggagtttattcggaaaagc cagctggtccag 930
GlyThrLeuIle GlyLeuGluPheIleArgLysSer GlnLeuValGln
265 270 275
cctgtggggcca ctggtggtgctgctgcccctggcg ggtgggtacagc 978
ProValGlyPro LeuValValLeuLeuProLeuAla GlyGlyTyrSer
280 285 290
cgcgtcctcaac gccgcctgccagcgcctggcgagg gttggggtcgtg 1026
ArgValLeuAsn AlaAlaCysGlnArgLeuAlaArg ValGlyValVal
295 300 305 310
ctggtcaccget gccggcaacttccgggacgatgcc tgcctctactcc 1074
LeuValThrAla AlaGlyAsnPheArgAspAspAla CysLeuTyrSer
315 320 325
ccagcctcaget cccgaggtcatcacagttggggcc accaatgcccag 1122
ProAlaSerAla ProGluValI1eThrValGlyAla ThrAsnAlaGln
330 335 340
gaccagccggtg accctggggactttggggaccaac tttggccgctgt 1170
AspGlnProVal ThrLeuGlyThrLeuGlyThrAsn PheGlyArgCys
345 350 355
gtggacctcttt gccccaggggaggacatcattggt gcctccagcgac 1218
ValAspLeuPhe AlaProGlyGluAspIleIleGly AlaSerSerAsp
360 365 370
tgcagcacctgc tttgtgtcacagagtgggacatca caggetgetgcc 1266
CysSerThrCys PheValSerGlnSerGlyThrSer GlnAlaAlaAla
375 380 385 390
cacgtggetggc attgcagccatgatgctgtctgcc gagccggagctc 1314
HisValAlaGly IleAlaAlaMetMetLeuSerAla GluProGluLeu
395 400 405
accctggccgag ttgaggcagagactgatccacttc tctgccaaagat 1362
ThrLeuAlaGlu LeuArgGlnArgLeuIleHisPhe SerAlaLysAsp
410 415 420
gtcatcaatgag gcctggttccctgaggaccagcgg gtactgaccccc 1410
ValIleAsnGlu AlaTrpPheProGluAspGln ValLeuThrPro
Arg
425 430 435
aacctggtggcc gcc ccccccagcacccatggg gcaggttggcag 1458
ctg
1~
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
AsnLeuValAlaAla LeuProProSerThrHisGly AlaGlyTrpGln
q40 445 950
ctgttttgcaggact gtgtggtcagcacactcgggg cctacacggatg 1506
LeuPheCysArgThr ValTrpSerAlaHisSerGly ProThrArgMet
955 460 965 470
gccacagccatcgcc cgctgcgccccagatgaggag ctgctgagctgc 1554
AlaThrAlaIleAla ArgCysAlaProAspGluGlu LeuLeuSerCys
475 480 485
tccagtttctccagg agtgggaagcggcggggcgag cgcatggaggcc 1602
SerSerPheSerArg SerGlyLysArgArgGlyGlu ArgMetGluAla
990 495 500
caagggggcaagctg gtctgccgggcccacaacget tttgggggtgag 1650
GlnGlyGlyLysLeu ValCysArgAlaHisAsnAla PheGlyGlyGlu
505 510 515
ggtgtctacgccatt gccaggtgctgcctgctaccc caggccaactgc 1698
GlyValTyrAlaIle AlaArgCysCysLeuLeuPro GlnAlaAsnCys
520 525 530
agcgtccacacaget ccaccagetgaggccagcatg gggacccgtgtc 1746
SerValHisThrAla ProProAlaGluAlaSerMet GlyThrArgVal
535 540 545 550
cactgccaccaacag ggccacgtcctcacaggtttc ctagetcttgcc 1794
HisCysHisGlnGln GlyHisValLeuThrGlyPhe LeuAlaLeuAla
555 560 565
tcagaccttaaagag agagggtctgatggggatggg cactggagacgg 1842
SerAspLeuLysGlu ArgGlySerAspGlyAspGly HisTrpArgArg
570 575 580
agcatcccagcattt cacatctgagctggctttcctctgcc caggctgca 1893
c
SerIleProAlaPhe HisIle
585
gctcccactg ggaggtggag gaccttggca cccacaagcc gcctgtgctg aggccacgag 1953
gtcagcccaa ccagtgcgtg ggccacaggg aggccagcat ccacgcttcc tgctgccatg 2013
ccccaggtct ggaatgcaag tcaaggagca tggaatcccg gcccctcagg agcaggtgac 2073
cgtggcctgc gaggagggct ggaccctgac tggctgcagt gccctccctg ggacctccca 2133
cgtcctgggg gcctacgccg tagacaacac gtgtgtagtc aggagccggg acgtcagcac 2193
tacaggcagc accagcgaag aggccgtgac agccgttgcc atctgctgcc ggagccggca 2253
cctggcgcag gcctcccagg agctccagtg acagccccat cccaggatgg gtgtctgggg 2313
agggtcaagg gctggggctg agctttaaaa tggttccgac ttgtccctct ctcagccctc 2373
catggcctgg cacgagggga tggggatgct tccgcctttc cggggctgct ggcctggccc 2433
ttgagtgggg cagcctcctt gcctggaact cactcactct gggtgcctcc tccccaggtg 2493
gaggtgccag gaagctccct ccctcactgt ggggcatttc accattcaaa caggtcgagc 2553
tgtgctcggg tgctgccagc tgctcccaat gtgccgatgt ccgtgggcag aatgactttt 2613
attgagctct tgttccgtgc caggcattca atcctcaggt ctccaccaag gaggcaggat 2673
tcttcccatg gataggggag ggggcggtag gggctgcagg gacaaacatc gttggggggt 2733
gagtgtgaaa ggtgctgatg gccctcatct ccagctaact gtggagaagc ccctgggggc 2793
tccctgatta atggaggctt agctttctgg atggcatcta gccagaggct ggagacaggt 2853
gtgcccctgg tggtcacagg ctgtgccttg gtttcctgag ccacctttac tctgctctat 2913
gccaggctgt gctagcaaca cccaaaggtg gcctgcgggg agccatcacc taggactgac 2973
tcggcagtgt gcagtggtgc atgcactgtc tcagccaacc cgctccacta cccggcaggg 3033
tacacattcg cacccctact tcacagagga agaaacctgg aaccagaggg ggcgtgcctg 3093
ccaagctcac acagcaggaa ctgagccaga aacgcagatt gggctggctc tgaagccaag 3153
cctcttctta cttcacccgg ctgggctcct catttttacg ggtaacagtg aggctgggaa 3213
ggggaacaca gaccaggaag ctcggtgagt gatggcagaa cgatgcctgc aggcatggaa 3273
ctttttccgt tatcacccag gcctgattca ctggcctggc ggagatgctt ctaaggcatg 3333
gtcgggggag agggccaaca actgtccctc cttgagcacc agccccaccc aagcaagcag 3393
acatttatct tttgggtctg tcctctctgt tgccttttta cagccaactt ttctagacct 3453
gttttgcttt tgtaacttga agatatttat tctgggtttt gtagcatttt tattaatatg 3513
gtgacttttt aaaataaaaa caaacaaacg ttgtcctaaa aaaaaaaaaa aaaaaaaaaa 3573
11
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
gggcggccgc
<210> 9
<211> 589
<212> PRT
<213> Homo Sapiens
<400> 9
Met Gly Thr Val Ser Ser Arg Arg Ser Trp Trp Pro Leu Pro Leu Leu
1 5 10 15
Leu Leu Leu Leu Leu Leu Leu Gly Pro Ala Gly Ala Arg Ala Gln Glu
20 25 30
Asp Glu Asp Gly Asp Tyr Glu Glu Leu Val Leu Ala Leu Arg Ser Glu
35 40 45
Glu Asp Gly Leu Ala Glu Ala Pro Glu His Gly Thr Thr Ala Thr Phe
50 55 60
His Arg Cys Ala Lys Asp Pro Trp Arg Leu Pro Gly Thr Tyr Val Val
65 70 75 80
Val Leu Lys Glu Glu Thr His Leu Ser Gln Ser Glu Arg Thr Ala Arg
85 90 95
Arg Leu Gln Ala Gln Ala Ala Arg Arg Gly Tyr Leu Thr Lys Ile Leu
100 105 110
His Val Phe His Gly Leu Leu Pro Gly Phe Leu Val Lys Met Ser Gly
115 120 125
Asp Leu Leu Glu Leu Ala Leu Lys Leu Pro His Val Asp Tyr Ile Glu
130 135 140
Glu Asp Ser Ser Val Phe Ala Gln Ser Ile Pro Trp Asn Leu Glu Arg
145 150 155 160
Ile Thr Pro Pro Arg Tyr Arg Ala Asp Glu Tyr Gln Pro Pro Asp Gly
165 170 175
Gly Ser Leu Val Glu Val Tyr Leu Leu Asp Thr Ser Ile Gln Ser Asp
180 185 190
His Arg Glu Ile Glu Gly Arg Val Met Val Thr Asp Phe Glu Asn Val
195 200 205
Pro Glu Glu Asp Gly Thr Arg Phe His Arg Gln Ala Ser Lys Cys Asp
210 215 220
Ser His Gly Thr His Leu Ala Gly Val Val Ser Gly Arg Asp Ala Gly
225 230 235 240
Val Ala Lys Gly Ala Ser Met Arg Ser Leu Arg Val Leu Asn Cys Gln
245 250 255
Gly Lys Gly Thr Val Ser Gly Thr Leu Ile Gly Leu Glu Phe Ile Arg
260 265 270
Lys Ser Gln Leu Val Gln Pro Val Gly Pro Leu Val Val Leu Leu Pro
275 280 285
Leu Ala Gly Gly Tyr Ser Arg Va1 Leu Asn Ala Ala Cys Gln Arg Leu
290 295 300
Ala Arg Val Gly Val Val Leu Val Thr Ala Ala Gly Asn Phe Arg Asp
305 310 315 320
Asp Ala Cys Leu Tyr Ser Pro Ala Ser Ala Pro Glu Val Ile Thr Val
325 330 335
Gly Ala Thr Asn Ala Gln Asp Gln Pro Val Thr Leu Gly Thr Leu G1y
340 345 350
Thr Asn Phe Gly Arg Cys Val Asp Leu Phe Ala Pro Gly Glu Asp Ile
355 360 365
Ile Gly Ala Ser Ser Asp Cys Ser Thr Cys Phe Val Ser Gln Ser Gly
370 375 380
Thr Ser Gln Ala Ala Ala His Val Ala Gly Ile Ala Ala Met Met Leu
385 390 395 400
Ser Ala Glu Pro Glu Leu Thr Leu Ala Glu Leu Arg Gln Arg Leu Ile
405 410 415
His Phe Ser Ala Lys Asp Val Ile Asn Glu Ala Trp Phe Pro Glu Asp
420 425 430
Gln Arg Val Leu Thr Pro Asn Leu Val Ala Ala Leu Pro Pro Ser Thr
435 940 445
His Gly Ala Gly Trp Gln Leu Phe Cys Arg Thr Val Trp Ser Ala His
450 455 460
Ser Gly Pro Thr Arg Met Ala Thr Ala Ile Ala Arg Cys Ala Pro Asp
12
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
465 470 475 480
Glu Glu Leu Leu Ser Cys Ser Ser Phe Ser Arg Ser Gly Lys Arg Arg
485 990 995
Gly GluArgMetGluAlaGlnGlyGly LysLeu Cys Ala
Val Arg His
500 505 510
Asn AlaPheGlyGlyGluGlyValTyr AlaIleAla CysCysLeu
Arg
515 520 525
Leu ProGlnAlaAsnCysSerValHis ThrAlaProProAlaGlu
Ala
530 535 540
Ser MetGlyThrArgValHisCysHis GlnGlnGlyHisValLeuThr
545 550 555 560
Gly PheLeuAlaLeuAlaSerAspLeu LysGluArgGlySerAspGly
565 570 575
Asp GlyHisTrpArgArgSerIlePro AlaPheHisIle
580 585
<2 10>10
<2 11>5145
<2 12>DNA
<213> Homo
Sapiens
<220>
<221> CDS
<222> 1113)... (1390)
<400> 10
ggcggcggga agctgcctggagg 60
gagctgctgg cgggcccggc
ctcgcccgga
tcccggg
ccggggaagg ggagcgggca 118
tgagcggctg cc
cgggacccag atg
cccctcgccg gtg
Met
Val
1
ctg tcggtgcctgtgatcgcgctgggc gccacgctgggcacagccacc 166
Leu SerValProValIleAlaLeuGly AlaThrLeuGlyThrAlaThr
5 10 15
agc atcctcgcgttgtgcggggtcacc tgcctgtgtcggcacatgcac 214
Ser IleLeuAlaLeuCysGlyValThr CysLeuCysArgHisMetHis
20 25 30
ccc aagaaggggctgctgccgcgggac caggaccccgacctggagaag 262
Pro LysLysGlyLeuLeuProArgAsp GlnAspProAspLeuGluLys
35 40 45 50
gcg aagcccagcttgctcgggtctgca caacagttcaatgttaaaaag 310
Ala LysProSerLeuLeuGlySerAla GlnGlnPheAsnValLysLys
55 60 65
tcc acggaacctgttcagccccgtgcc ctcctcaagttcccagacatc 358
Ser ThrGluProValGlnProArgAla LeuLeuLysPheProAspIle
70 75 80
tat ggacccaggccagetgtgacgget ccagaggtcatcaactatgca 406
Tyr GlyProArgProAlaValThrAla ProGluValIleAsnTyrAla
85 90 95
gac tattcactgaggtctacggaggag cccactgcacctgccagcccc 454
Asp TyrSerLeuArgSerThrGluGlu ProThrAlaProAlaSerPro
100 105 110
caa cccccgaatgacagtcgcctcaag aggcaggtcacagaggagctg 502
Gln ProProAsnAspSerArgLeuLys ArgGlnValThrGlu Leu
Glu
115 120 125 130
ttc atcctccctcagaatggtgtggtg gaggatgtctgtgtc gag 550
atg
Phe IleLeuProGlnAsnGlyValVal GluAspValCysValMetGlu
135 I40 145
13
CA 02388617 2002-05-07
WO 01/31007 PCT/US00/29132
acctggaac gag agt ccc ctc 598
cca aag tgg aaa
get aac
gcc cag
gcc
ThrTrpAsn Glu Ser Pro Leu
Pro Lys Trp Lys
Ala Asn
Ala Gln
A1a
150 155 160
cactactgc ctggac tat cagaaggcagaattgttt act 646
gac gtg
tgt
HisTyrCys LeuAsp Tyr GlnLys GluLeuPhe Thr
Asp Ala Val
Cys
165 170 175
cgcctggaa getgtg accagcaaccacgacggaggctgtgac tac 694
tgc
ArgLeuGlu AlaVal ThrSer HisAspGlyGlyCysAsp Tyr
Asn Cys
180 185 190
gtccaaggg agtgtg gccaataggaccggctctgtggaggetcagaca 792
ValGlnGly SerVal AlaAsnArgThrGlySerValGluAla Thr
Gln
195 200 205 210
gccctaaag aagcgg cagctgcacaccacctgggaggagggcctggtg 790
AlaLeuLys LysArg GlnLeuHisThrThrTrpGluGluGlyLeuVal
215 220 225
ctccccctg gcggag gaggagctccccacagccaccctgacgctgacc 838
LeuProLeu AlaGlu GluGluLeuProThrAlaThrLeuThrLeuThr
230 235 240
ttgaggacc tgcgac cgcttctcccgtcacagcgtggccggggagctc 886
LeuArgThr CysAsp ArgPheSerArgHisSerValAlaGlyGluLeu
245 250 255
cgcctgggc ctggac gggacatctgtgcctctaggggetgcccagtgg 939
ArgLeuGly LeuAsp GlyThrSerValProLeuGlyAlaAlaGlnTrp
260 265 270
ggcgagctg aagact tcagcgaaggagccatctgcaggagetggagag 982
GlyGluLeu LysThr SerAlaLysGluProSerAlaGlyAlaGlyGlu
275 280 285 290
gtcctacta tccatc agctacctcccggetgccaaccgcctcctggtg 1030
ValLeuLeu SerIle SerTyrLeuProAlaAlaAsnArgLeuLeuVal
295 300 305
gtgctgatt aaagcc aagaacctccactctaaccagtccaaggagctc 1078
ValLeuIle LysAla LysAsnLeuHisSerAsnGlnSerLysGluLeu
310 315 320
ctggggaag gatgtc tctgtcaaggtgaccttgaagcaccaggetcgg 1126
LeuGlyLys AspVal SerValLysValThrLeuLysHisGlnAlaArg
325 330 335
aagctgaag aagaag cagactaaacgagetaagcacaagatcaacccc 1174
LysLeuLys LysLys GlnThrLysArgAlaLysHisLysIleAsnPro
340 345 350
gtgtggaac gagatg atcatgtttgagctgcctgacgacctgctgcag 1222
ValTrpAsn GluMet IleMetPheGluLeuProAspAspLeuLeuGln
355 360 365 370
gcctccagt gtggag ctggaagtgctgggccaggacgattcagggcag 1270
AlaSerSer ValGlu LeuGluValLeuGlyGlnAspAspSerGlyGln
375 380 385
agctgtgcg cttggc cactgcagcctgggcctgcacacc ggctct 1318
tcg
SerCysAla Leu HisCysSerLeu Thr Gly
Gly Gly Ser Ser
Leu
His
390 395 400
gag cac gaggagatg aac cgc cag 1366
cgc tgg ctc cct cgg att
agc aaa
Glu His GluGluMet Asn Gln
Arg Trp Leu Pro Ile
Ser Lys Arg
Arg
405 910 415
14
CA 02388617 2002-05-07
WO 01/31007 PCT/US011/29132
gcc atg tgg cac cag ctg cac ctg taaccagctg cccagctgcc tcccttcttg 1420
Ala Met Trp His Gln Leu His Leu
920 425
gacagccctg acccgtcctc tgcaacctcc tttctgtgcc cctttcctca ttctgacacc 1480
cagaagacag tgacagatgt gtttgcaagg ctgggatggc tctctcatca tactcttgtt 1540
tcttagaaat aagcaagaca gagcaggaaa tggaatatgc gggtcacact gaggaatgca 1600
ttttgctcat ctgtgttatt gaaggaggtg cttattaaat acagttccta tgcctgtttt 1660
ataggtgggg ttaggccaga tgcagagaaa gctaaatgtg ggaatcatgg atgcaaagaa 1720
gaatttggct ttttgaaaaa caagcatttc aaaaatgatg aaggaagtga aagtatcctg 1780
gatcaactcc tagagttaga gattgcccag gtggaaagaa accttagcca gcgttcaatc 1840
aagctcacca tgcagggcag tcacccggca gttctcaaac tttagcatgt gaagagtcac 1900
cagcagattc ctgggctcgc ctggagacat tcctagtcgg tattcctggt cgaagcccag 1960
gagccttcct ttttaacaag ctgatgtaga gggtggagca ctgtatgtgg agaaattcct 2020
tctacaatat tccacacagg tttttggcca cagtccttga tggagtccca aaaccatggt 2080
gcagccagtt ccaatgctgg acacctcaac catcagggtg aaatctgggg cctcagcttt 2140
ttaatttaat tattttaatt cttaatactt taatttgtgc atttcataag ccccctgctc 2200
ttggactgaa ttttgtgctt tttattgaag aattttattg tttttatctt aaaatcagtt 2260
tctattatcc ttggggagac catccctaac aaagtacagg tgggatctcc tgtgagtcat 2320
tggctgggtt ctgattgcta gatgtcacac ccaccagcat caccaaagtg actctgagat 2380
agaccggtcc cttctcagcg ttccagtcac ttcaggagga atttagttat tgacttagtc 2490
tatgacatct ggctacatgt aggtagagaa gaaagacaat tttaaaaagg aaatcaggtc 2500
ttttgcaact gtgcctccct ctgtctgttt tcacttgaat gggtaaataa ccagcagcta 2560
ggttttgaat tcctaccttg ttattctaaa cagatgtcca cattgttaat taaatctaaa 2620
ttatgagcct tgctgagtgg atacggtact tacacctgaa ccaggattcc tgggttctgt 2680
tgttgacatt gcccttcagc acctgtttgg ccagctgtat aagataggac taatgactag 2740
gaagcctacc ccaatgaatg atatactaga tgaaatagtg ttcaaaacct gtaggcactc 2800
tctggctaaa aacaaactct gaggccacca gcagatcatc tttaagctaa gttactattt 2860
ttcacctttt tttttagacg gagttttgct ctttgttgcc caggctggag tgcagtggca 2920
cgatctcggc tcactgcaac ctccgcctcc caagttcaag cgattctcct gtctcagcct 2980
cctgggtagc tgggattaca ggtgcccacc aacatgcctg gctaattttt gtacttttag 3040
tagagatggg gtttcaccat gttggccagg ctggtcttca actccagatc tcaggtgatc 3100
taccctcctc ggcctcccaa agtactggga ttacaggcct gagccaccgc gcccggccta 3160
tttttcactt taatttggca gctgagaatg cccaaaaagt gccagaagca tcgtggcatt 3220
tccagaacca tggattctgc ctttggaccc ctctctatta atattaaaac tctgggcctt 3280
cagatgtcac cctaatccac tgccctaaga cagaatttct ggacaagatg ggtaagggct 3390
tcattccttc aacaagtcaa gtcatacttg gcctctccct gagaatctga gcaggagcct 3400
tataacctgt ggtcattatt ttttctttct gtacagaaat agaaaagcat tagaaataac 3460
ttctaaccat cctctgaaaa aacagaaaaa atatcgaatc cctctttcat gagaagtctt 3520
ttggataatt ggaaaccttc atcactgagg ttggccagcc cctgccaagt gttgtgtagg 3580
caaagcactt gttagtggct tcctatgaaa tgttttagag atctcttcac catactggtt 3640
tcttctcttt ggttggtgtg ggtaaaagaa aacaaaacat ttcctataag ctgaaagctg 3700
accagcattc tcttcttggt aacatctact actccaacct agaaaatttg gattctagac 3760
caaaaatcag gaaacatggc tccttataaa tctgtgcagc tgccttatag taccatcaaa 3820
ggaatttcag gtgggctggg cggggccccg atcccagaat tatcaactcc acccatcatc 3880
atttggtcat gaagcatcct ttcattcttc ttcttctttt ttttgggggg ggcggggcgg 3940
gggagggatc tcaaagtttt agtcttccag aatccaaatt aaaggttgcc cctgatgggg 4000
gccaggttcc gccacagaac atcttagatg tcagccttga cctcacttag cagggattac 4060
agaaatgaga tacattttga aggagagttg tctgttatgt tcactgtatt ctaagtgcct 4120
gggataaagc tgtctcatgg gtgctccata tatattcata tatatttgtt gagtgaatta 4180
atgaattaag agtggctggc agagtaggca gaaaaagaca ctgcaaatgg cataaaaatt 4240
aaagtcctag ctgagttctc aatggtaaag gcatcagatg tcttagcagt caagctagaa 4300
attcatgaca atgagtatta ctatttgcct aatgacaact cattgctctc catgtaaatg 4360
taatcaacag atgaagagaa tataattgct ctgcttttcc actaaaactc catcttagtg 4920
aattttaaat tatccagaga tgtcaaactg ccaaataaaa atatttcagt agtctttgca 4480
tcagcttacc ttgtaccaga aacatttcca atttactatc aaattatagt aactgagcct 4540
gtgtgaagta tctcatcatt ttcgaaagga acaccttgtg tgatgccagt gagcatttct 9600
aaaaagggtg tgaggtagag gtaaaaataa ggtgagagac catttcagaa tgcactgttg 4660
ctcaaaaagg tgatctggtt ctttcttcag agatttctac ggggatagaa aatcgggagt 9720
ctgccctcat taatctgtga ctccacctct tgcatcaaat caatatctat ttgttgagca 9780
cttattgatt aagaccttgc atatgtctgt ccattttgat ttgagataca actttttgtg 4840
tgggttgaat gacaaatcac tccaaacaaa actgggcaca gagaatcagc taggagacca 4900
gttattcagg gtccatttct cttggatgta aaggagtcct gggtaaaatg tggctgtaac 4960
ctaaaccaac tagtccttgt gatttgtttc tgccctctgt gtttcctgtt gtcaaatgct 5020
aagtgtgtgt tttgcagtca tgaactaaag cacaaaaaga tgcatgagac attgtagtca 5080
tatgtctggt gtgacacttt ggagcaaaaa ccttgcagtg gtaaataaaa aatttccaac 5140
1$
CA 02388617 2002-05-07
WO 01/31007 PCT/US(10/29132
agggt
<210> 11
<211> 426
<212> PRT
<223> Homo Sapiens
<400> 11
Met Val Leu Ser Val Pro Val Ile Ala Leu Gly Ala Thr Leu Gly Thr
1 5 10 15
Ala Thr Ser Ile Leu Ala Leu Cys Gly Val Thr Cys Leu Cys Arg His
20 25 30
Met His Pro Lys Lys Gly Leu Leu Pro Arg Asp Gln Asp Pro Asp Leu
35 40 45
Glu Lys Ala Lys Pro Ser Leu Leu Gly Ser Ala Gln Gln Phe Asn Val
50 55 60
Lys Lys Ser Thr Glu Pro Val Gln Pro Arg Ala Leu Leu Lys Phe Pro
65 70 75 80
Asp Ile Tyr Gly Pro Arg Pro Ala Val Thr Ala Pro Glu Val I1e Asn
85 90 95
Tyr A1a Asp Tyr Ser Leu Arg Ser Thr Glu Glu Pro Thr Ala Pro Ala
100 105 110
Ser Pro Gln Pro Pro Asn Asp Ser Arg Leu Lys Arg Gln Val Thr Glu
115 120 125
Glu Leu Phe Ile Leu Pro Gln Asn Gly Val Val Glu Asp Val Cys Val
130 135 140
Met Glu Thr Trp Asn Pro Glu Lys Ala Ala Ser Trp Asn Gln Ala Pro
145 150 155 160
Lys Leu His Tyr Cys Leu Asp Tyr Asp Cys Gln Lys Ala Glu Leu Phe
165 170 175
Val Thr Arg Leu Glu Ala Val Thr Ser Asn His Asp Gly Gly Cys Asp
180 185 190
Cys Tyr Val Gln Gly Ser Val Ala Asn Arg Thr Gly Ser Val Glu Ala
195 200 205
Gln Thr Ala Leu Lys Lys Arg Gln Leu His Thr Thr Trp Glu Glu G1y
210 215 220
Leu Val Leu Pro Leu Ala Glu Glu Glu Leu Pro Thr Ala Thr Leu Thr
225 230 235 240
Leu Thr Leu Arg Thr Cys Asp Arg Phe Ser Arg His Ser Val Ala Gly
245 250 255
Glu Leu Arg Leu Gly Leu Asp Gly Thr Ser Val Pro Leu Gly Ala Ala
260 265 270
Gln Trp Gly Glu Leu Lys Thr Ser Ala Lys Glu Pro Ser Ala Gly Ala
275 280 285
Gly Glu Val Leu Leu Ser Ile Ser Tyr Leu Pro Ala Ala Asn Arg Leu
290 295 300
Leu Val Val Leu Ile Lys Ala Lys Asn Leu His Ser Asn Gln Ser Lys
305 310 315 320
Glu Leu Leu Gly Lys Asp Val Ser Val Lys Val Thr Leu Lys His Gln
325 330 335
Ala Arg Lys Leu Lys Lys Lys Gln Thr Lys Arg Ala Lys His Lys Ile
340 345 350
Asn Pro Val Trp Asn Glu Met Ile Met Phe Glu Leu Pro Asp Asp Leu
355 360 365
Leu Gln Ala Ser Ser Val Glu Leu G1u Val Leu Gly Gln Asp Asp Ser
370 375 380
Gly Gln Ser Cys Ala Leu Gly His Cys Ser Leu Gly Leu His Thr Ser
385 390 395 400
Gly Ser Glu Arg Ser His Trp G1u Glu Met Leu Lys Asn Pro Arg Arg
405 910 415
Gln Ile Ala Met Trp His Gln Leu His Leu
420 425
16