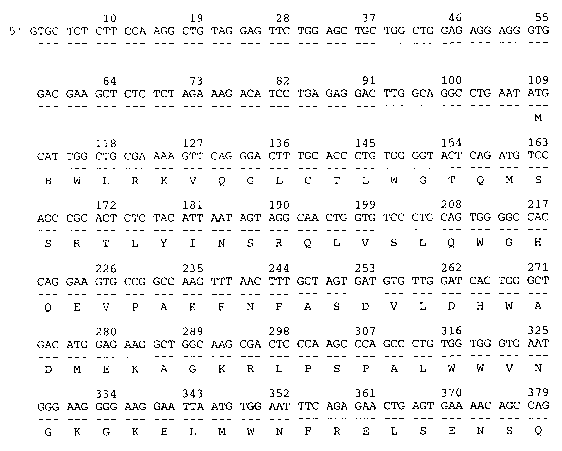Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02420101 2003-02-19
WO 02/16595 PCT/USO1/26317
KIDNEY-SPECIFIC PROTEIN
TECHNICAL FIELD
This invention relates to a mammalian cDNA which encodes a kidney-specific
protein
which is diagnostic of renal cell carcinoma (KSRCC) and to the use of the cDNA
and the encoded
protein in the diagnosis and treatment of kidney disorders.
BACKGROUND OF THE INVENTION
Phylogenetic relationships among organisms have been demonstrated many times,
and
studies from a diversity of prokaryotic and eukaryotic organisms suggest a
more or less gradual
evolution of molecules, biochemical and physiological mechanisms, and
metabolic pathways.
Despite different evolutionary pressures, the proteins of nematode, fly, rat,
and man have common
chemical and structural features and generally perform the same cellular
function. Comparisons of
the nucleic acid and protein sequences from organisms where structure and/or
function are known
accelerate the investigation of human sequences and allow the development of
model systems for
testing diagnostic and therapeutic agents for human conditions, diseases, and
disorders.
The human kidneys are two bean-shaped organs on either side of the backbone.
The
kidneys filter waste from the blood, form urine, and regulate the water and
electrolyte content of the
body. They reabsorb and retain proteins, glucose, amino acids, and bicarbonate
and inorganic
phosphate, and release hormones which interact to maintain intravascular
volume. They contribute
to maintenance of the body's acid/base balance and of homeostasis by
regulating blood pH
electrolyte levels and blood pressure.
Renal cell carcinoma (RCC), originating within the proximal renal tubular
epithelium, is the
most common type of kidney cancer accounting for approximately 3% of all adult
cancers. There
are four main types of renal cell carcinoma, with the majority being clear
cell type, and mixed
granular and clear cell type. RCC is curable only in its very early stages
through surgery. It is
resistant to chemotherapy and relatively resistant to radiotherapy. Risk
factors for development of
RCC include cigarette smoking and development of acquired cystic kidney
disease.
The discovery of a mammalian cDNA encoding KSRCC satisfies a need in the art
by
providing compositions which are useful in the diagnosis and treatment of
kidney disorders,
particularly, renal cell carcinoma.
SUMMARY OF THE INVENTION
The invention is based on the discovery of a mammalian cDNA which encodes a
mammalian kidney specific-protein which is useful in the diagnosis and
treatment of kidney
disorders including acquired cystic kidney disease and, particularly, renal
cell carcinoma.
The invention provides an isolated mammalian cDNA or a fragment thereof
encoding a
CA 02420101 2003-02-19
WO 02/16595 PCT/USO1/26317
mammalian protein or a portion thereof selected from the group consisting of
an amino acid
sequence of SEQ )D NO:1, a variant having 80% identity to the amino acid
sequence of SEQ >D
NO:1, an antigenic epitope of SEQ ID NO:1, an oligopeptide of SEQ U7 NO:1, and
a biologically
active portion of SEQ )D N0:1. The invention also provides an isolated
mammalian cDNA or the
complement thereof selected from the group consisting of a nucleic acid
sequence of SEQ m N0:2,
a variant having 83% identity to the nucleic acid sequence of SEQ >D N0:2, a
fragment of SEQ m
NOs:3-14, an oligonucleotide of SEQ m NOs:2-14. The invention additionally
provides a
composition, a substrate, and a probe comprising the cDNA ,or the complement
of the cDNA,
encoding KSRCC. The invention further provides a vector containing the cDNA, a
host cell
containing the vector and a method for using the cDNA to make KSRCC. The
invention still
further provides a transgenic cell line or organism comprising the vector
containing the cDNA
encoding KSRCC. The invention additionally provides a mammalian fragment or
the complement
thereof selected from the group consisting of SEQ m NOs: l l-14. In one
aspect, the invention
provides a substrate containing at least one of these fragments. In a second
aspect, the invention
provides a probe comprising the fragment which can be used in methods of
detection, screening,
and purification. In a further aspect, the probe is a single stranded
complementary RNA or DNA
molecule.
The invention provides a method for using a cDNA to detect the differential
expression of a
nucleic acid in a sample comprising hybridizing a probe to the nucleic acids,
thereby forming
hybridization complexes and comparing hybridization complex formation with a
standard, wherein
the comparison indicates the differential expression of the cDNA in the
sample. In one aspect, the
method of detection further comprises amplifying the nucleic acids of the
sample prior to
hybridization. In another aspect, the method showing differential expression
of the cDNA is used
to diagnose renal cell carcinoma. In another aspect, the cDNA or a fragment or
a complement
thereof may comprise an element on an array.
The invention additionally provides a method for using a cDNA or a fragment or
a
complement thereof to screen a library or plurality of molecules or compounds
to identify at least
one ligand which specifically binds the cDNA, the method comprising combining
the cDNA with
the molecules or compounds under conditions allowing specific binding, and
detecting specific
binding to the cDNA , thereby identifying a ligand which specifically binds
the cDNA. In one
aspect, the molecules or compounds are selected from aptamers, DNA molecules,
RNA molecules,
peptide nucleic acids, artificial chromosome constructions, peptides,
transcription factors,
repressors, and regulatory molecules.
The invention provides a purified mammalian protein or a portion thereof
selected from the
group consisting of an amino acid sequence of SEQ m N0:1, a variant having 80%
identity to the
2
CA 02420101 2003-02-19
WO 02/16595 PCT/USO1/26317
amino acid sequence of SEQ m NO:1, an antigenic epitope of SEQ m NO:1, an
oligopeptide of
SEQ ~ N0:1, and a biologically active portion of SEQ ID NO:1. The invention
also provides a
composition comprising the purified protein or a portion thereof in
conjunction with a
pharmaceutical carrier. The invention further provides a method of using the
KSRCC to treat a
subject with renal cell carcinoma comprising administering to a patient in
need of such treatment
the composition containing the purified protein. The invention still further
provides a method for
using a protein to screen a library or a plurality of molecules or compounds
to identify at least one
ligand , the method comprising combining the protein with the molecules or
compounds under
conditions to allow specific binding and detecting specific binding, thereby
identifying a ligand
which specifically binds the protein. In one aspect, the molecules or
compounds are selected from
DNA molecules, RNA molecules, peptide nucleic acids, peptides, proteins,
mimetics, agonists,
antagonists, antibodies, immunoglobulins, inhibitors, and drugs. In another
aspect, the ligand is
used to treat a subject with renal cell carcinoma.
The invention provides a method of using a mammalian protein to screen a
subject sample
for antibodies which specifically bind the protein comprising isolating
antibodies from the subject
sample, contacting the isolated antibodies with the protein under conditions
that allow specific
binding, dissociating the antibody from the bound-protein, and comparing the
quantity of antibody
with known standards, wherein the presence or quantity of antibody is
diagnostic of renal cell
carcinoma.
The invention also provides a method of using a mammalian protein to prepare
and purify
antibodies comprising immunizing a animal with the protein under conditions to
elicit an antibody
response, isolating animal antibodies, attaching the protein to a substrate,
contacting the substrate
with isolated antibodies under conditions to allow specific binding to the
protein, dissociating the
antibodies from the protein, thereby obtaining purified antibodies.
The invention provides a purified antibody which binds specifically to a
protein which is
expressed in renal cell carcinoma. The invention also provides a method of
using an antibody to
diagnose renal cell carcinoma comprising combining the antibody comparing the
quantity of bound
antibody to known standards, thereby establishing the presence of renal cell
carcinoma. The
invention further provides a method of using an antibody to treat renal cell
carcinoma comprising
administering to a patient in need of such treatment a pharmaceutical
composition comprising the
purified antibody.
The invention provides a method for inserting a marker gene into the genomic
DNA of a
mammal to disrupt the expression of the endogenous polynucleotide. The
invention also provides a
method for using a cDNA to produce a mammalian model system, the method
comprising
constructing a vector containing the cDNA selected from SEQ ID NOs:2-14,
transforming the
CA 02420101 2003-02-19
WO 02/16595 PCT/USO1/26317
vector into an embryonic stem cell, selecting a transformed embryonic stem,
microinjecting the
transformed embryonic stem cell into a mammalian blastocyst, thereby forming a
chimeric
blastocyst, transferring the chimeric blastocyst into a pseudopregnant dam,
wherein the dam gives
birth to a chimeric offspring containing the cDNA in its germ line, and
breeding the chimeric
mammal to produce a homozygous, mammalian model system.
BRIEF DESCRIPTION OF THE FIGURES AND TABLE
Figures 1A, 1B, 1C, 1D, 1E, and 1F show the mammalian KSRCC (SEQ ID NO:1)
encoded
by the cDNA (SEQ ID N0:2). The alignment was produced using MACDNASIS PRO
software
(Hitachi Software Engineering, South San Francisco CA).
Figures 2A and 2B demonstrate the conserved chemical and structural
similarities among
the domains of KSRCC (SEQ ID NO:1), Rattus norve ig cus KS (g3127193), and
Homo Sapiens KS
(g3219339), SEQ ID NOs:l, and 15-16, respectively. The aligmnent was produced
using the
MEGALIGN program of LASERGENE software (DNASTAR, Madison WI).
Figure 3 shows the northern analysis for I~SRCC produced using the LIFESEQ
Gold
database (Incyte Genomics, Palo Alto CA). The first column presents the tissue
categories; the
second column, the number of clones in the tissue category; the third column,
the number of
libraries in which at least one transcript was found; the fourth column,
absolute abundance of the
transcript; and the fifth column, percent abundance of the trancript.
Figure 4 shows the hydrophilicity plots and antigenic indices for KSRCC, rat
g3127193 and
human g3219339.
DESCRIPTION OF THE INVENTION
It is understood that this invention is not limited to the particular
machines, materials and
methods described. It is also to be understood that the terminology used
herein is for the purpose of
describing particular embodiments and is not intended to limit the scope of
the present invention
which will be limited only by the appended claims. As used herein, the
singular forms "a", "an",
and "the" include plural reference unless the context clearly dictates
otherwise. For example, a
reference to "a host cell" includes a plurality of such host cells known to
those skilled in the art.
Unless defined otherwise, all technical and scientific terms used herein have
the same
meanings as commonly understood by one of ordinary skill in the art to which
this invention
belongs. All publications mentioned herein are cited for the purpose of
describing and disclosing
the cell lines, protocols, reagents and vectors which are reported in the
publications and which
might be used in connection with the invention. Nothing herein is to be
construed as an admission
that the invention is not entitled to antedate such disclosure by virtue of
prior invention.
Definitions
"KSRCC" refers to a substantially purified protein obtained from any mammalian
species,
4
CA 02420101 2003-02-19
WO 02/16595 PCT/USO1/26317
including bovine, canine, marine, ovine, porcine, rodent, simian, and
preferably the human species,
and from any source, whether natural, synthetic, semi-synthetic, or
recombinant.
"Array" refers to an ordered arrangement of at least two cDNAs on a substrate.
At least one
of the cDNAs represents a control or standard sequence, and the other, a cDNA
of diagnostic
interest. The arrangement of from about two to about 40,000 cDNAs on the
substrate assures that
the size and signal intensity of each labeled hybridization complex formed
between a cDNA and a
sample nucleic acid is individually distinguishable.
The "complement" of a cDNA of the Sequence Listing refers to a nucleic acid
molecule
which is completely complementary over its full length and which will
hybridize to the cDNA or an
mRNA under conditions of high stringency.
"cDNA" refers to an isolated polynucleotide, nucleic acid molecule, or any
fragment or
complement thereof. It may have originated recombinantly or synthetically, be
double-stranded or
single-stranded, represent coding and/or noncoding sequence, an exon with or
without an intron
from a genomic DNA molecule.
The phrase "cDNA encoding a protein" refers to a nucleic acid sequence that
closely aligns
with sequences which encode conserved regions, motifs or domains that were
identified by
employing analyses well known in the art. These analyses include BLAST (Basic
Local Alignment
Search Tool; Altschul (1993) J Mol Evol 36: 290-300; Altschul et al. (1990) J
Mol Biol
215:403-410) which provides identity within the conserved region.
"Derivative" refers to a cDNA or a protein that has been subjected to a
chemical
modification. Derivatization of a cDNA can involve substitution of a
nontraditional base such as
queosine or of an analog such as hypoxanthine. These substitutions are well
known in the art.
Derivatization of a protein involves the replacement of a hydrogen by an
acetyl, acyl, alkyl, amino,
formyl, or marpholino group. Derivative molecules retain the biological
activities of the naturally
occurnng molecules but may confer advantages such as longer lifespan or
enhanced activity.
"Differential expression" refers to an increased, upregulated or present, or
decreased,
downregulated or absent, gene expression as detected by the absence, presence,
or at least two-fold
changes in the amount of transcribed messenger RNA or translated protein in a
sample.
"Disorder" refers to conditions, diseases or syndromes in which the cDNAs and
KSRCC are
differentially expressed.
"Fragment" refers to a chain of consecutive nucleotides from about 200 to
about 700 base
pairs in length. Fragments may be used in PCR or hybridization technologies to
identify related
nucleic acid molecules and in binding assays to screen for a ligand. Nucleic
acids and their ligands
identified in this manner are useful as therapeutics to regulate replication,
transcription or
translation.
CA 02420101 2003-02-19
WO 02/16595 PCT/USO1/26317
"Guilt by association" (GBA) is a method for identifying cDNAs or proteins
that are
associated with a specific disease, regulatory pathway, subcellular
compartment, cell type, tissue
type, or species. In particular, the method identifies cDNAs useful in
diagnosis, prognosis,
treatment, and evaluation of therapies for kidney disorders.
A "hybridization complex" is formed between a cDNA and a nucleic acid of a
sample when
the purines of one molecule hydrogen bond with the pyrimidines of the
complementary molecule,
e.g., 5'-A-G-T-C-3' base pairs with 3'-T-C-A-G-5'. The degree of
complementarity and the use of
nucleotide analogs affect the efficiency and stringency of hybridization
reactions.
"Ligand" refers to any agent, molecule, or compound which will bind
specifically to a
complementary site on a cDNA molecule or polynucleotide, or to an epitope or a
protein. Such
ligands stabilize or modulate the activity of polynucleotides or proteins and
may be composed of
inorganic or organic substances including nucleic acids, proteins,
carbohydrates, fats, and lipids.
"Oligonucleotide" refers a single stranded molecule from about 18 to about 60
nucleotides
in length which may be used in hybridization or amplification technologies or
in regulation of
replication, transcription or translation. Substantially equivalent terms are
amplimer, primer, and
oligomer.
"Portion" refers to any part of a protein used for any purpose; but
especially, to an epitope
for the screening of ligands or for the production of antibodies.
"Post-translational modification" of a protein can involve lipidation,
glycosylation,
phosphorylation, acetylation, racemization, proteolytic cleavage, and the
like. These processes may
occur synthetically or biochemically. Biochemical modifications will vary by
cellular location, cell
type, pH, enzymatic milieu, and the like.
"Probe" refers to a cDNA that hybridizes to at least one nucleic acid in a
sample. Where
targets are single stranded, probes are complementary single strands. Probes
can be labeled with
reporter molecules for use in hybridization reactions including Southern,
northern, in situ, dot blot,
array, and like technologies or in screening assays.
"Protein" refers to a polypeptide or any portion thereof. A "portion" of a
protein refers to
that length of amino acid sequence which would retain at least one biological
activity, a domain
identified by PFAM or PRINTS analysis or an antigenic epitope of the protein
identified using
Kyte-Doolittle algorithms of the PROTEAN program (DNASTAR, Madison Wl]. An
"oligopeptide" is an amino acid sequence from about five residues to about 15
residues that is used
as part of a fusion protein to produce an antibody.
"Purified" refers to any molecule or compound that is separated from its
natural
environment and is from about 60% free to about 90% free from other components
with which it is
naturally associated.
CA 02420101 2003-02-19
WO 02/16595 PCT/USO1/26317
"Sample" is used in its broadest sense as containing nucleic acids, proteins,
antibodies, and
the like. A sample may comprise a bodily fluid; the soluble fraction of a cell
preparation, or an
aliquot of media in which cells were grown; a chromosome, an organelle, or
membrane isolated or
extracted from a cell; genomic DNA, RNA, or cDNA in solution or bound to a
substrate; a cell; a
tissue; a tissue print; a fingerprint, buccal cells, skin, or hair; and the
like.
"Specific binding" refers to a special and precise interaction between two
molecules which
is dependent upon their structure, particularly their molecular side groups.
For example, the
intercalation of a regulatory protein into the major groove of a DNA molecule,
the hydrogen
bonding along the backbone between two single stranded nucleic acids, or the
binding between an
epitope of a protein and an agonist, antagonist, or antibody.
"Similarity" as applied to sequences, refers to the quantification (usually
percentage) of
nucleotide or residue matches between at least two sequences aligned using a
standardized
algorithm such as Smith-Waterman alignment (Smith and Waterman (1981) J Mol
Biol 147:195-
197) or BLAST2 (Altschul et al. (1997) Nucleic Acids Res 25:3389-3402). BLAST2
may be used
in a standardized and reproducible way to insert gaps in one of the sequences
in order to optimize
alignment and to achieve a more meaningful comparison between them.
"Substrate" refers to any rigid or semi-rigid support to which cDNAs or
proteins are bound
and includes membranes, filters, chips, slides, wafers, fibers, magnetic or
nonmagnetic beads, gels,
capillaries or other tubing, plates, polymers, and microparticles with a
variety of surface forms
including wells, trenches, pins, channels and pores.
"Variant" refers to molecules that are recognized variations of a cDNA or a
protein encoded
by the cDNA. Splice variants may be determined by BLAST score, wherein the
score is at least
100, and most preferably at least 400. Allelic variants have a high percent
identity to the cDNAs
and may differ by about three bases per hundfed bases. "Single nucleotide
polymorphism" (SNP)
refers to a change in a single base as a result of a substitution, insertion
or deletion. The change
may be conservative (purine for purine) or non-conservative (purine to
pyrimidine) and may or may
not result in a change in an encoded amino acid or its secondary, tertiary, or
quaternary structure.
THE INVENTION
The invention is based on the discovery of a cDNA which encodes KSRCC and on
the use
of the cDNA, or fragments thereof, and protein, or portions thereof, directly
or as compositions in
the characterization, diagnosis, and treatment of kidney disorders.
Nucleic acids encoding the KSRCC of the present invention were first
identified (in Incyte
Clone 3481942CB 1 from kidney tissue cDNA library, KJDNNOT31), as kidney-
specific through
GBA analysis of sequences co-regulated in kidney pathways. Eleven known kidney
disease-associated genes were selected to identify novel genes that are
closely associated with
CA 02420101 2003-02-19
WO 02/16595 PCT/USO1/26317
diseases of the kidney. These known genes were uromodulin, NKCC2, NCCT,
aldolase B,
ROMKl, ATP1G1, PDZKl, NPT-l, calbindin, kininogen, and CIC-Kb. The degree of
association
was measured by probability values using a cutoff p value less than 0.00001.
The sequences were
further examined to ensure that the genes that passed the probability test had
strong association with
known kidney disease-associated genes. Nine novel genes showed strong
association with known
kidney disease-associated genes from a total of 41,419. Each of the nine novel
genes was
coexpressed with at least one of the 11 known genes with a p-value of less
than 10e °5. KSRCC
cDNA is 2054 nucleic acids in length. SEQ ID N0:3 (Incyte Clone 3481942CB 1)
is 521
nucleotides in length and has 82% identity using BLAST2 to the nucleic acid
sequence of KS, a rat
kidney-specific protein (g3127192).
The cDNA far SEQ ID N0:2 was derived from the following overlapping and/or
extended
nucleic acid sequences (SEQ ID N0:3-10): Incyte Clones 3481942CB1, 5519427H1
(LIVRDIR01),
5390984F8 (KIDNNOT32), 433223079 (KIDNNOT32), 76083581 (BRATTLJT02), 76254081
(BRAITU°T02), 209942086 (BRATTUT02), and 7695905J1
(LGcompseqsJLJN2000new).
Figure 3 shows expression of the transcript in kidney tissue, particularly in
tissues from
patients with renal cell carcinoma. Therefore, the cDNA is useful in assays to
diagnose renal cell
carcinoma. A fragment of SEQ lD N0:2 from nucleotide 1 to nucleotide 106 is
useful as a
hybridization probe. An oligonucleotide from 54 to 69 or 80 to 95 is useful as
a diagnostic to
distinguish the transcript encoding KSRCC from other kidney-specific proteins.
In one embodiment, the invention encompasses a palypeptide comprising the
amino acid
sequence of SEQ ID N0:1 as shown in Figures 1A through 1F. KSRCC is 577 amino
acids in
length and has 77% identity to the amino acid sequence of the rat kidney-
specific protein
(g3127193) using BLAST2 analysis. BLIMPS analysis shows that SEQ ID NO:1 has a
16 amino
acid segment that matches the AMP-binding domains of g3127193. BLOCKS analysis
indicates
that the regions of KSRCC from Q213 to 7224 and 5225 to H233 are similar to
AMP-binding sites.
Pfam analysis indicates that the region of KSRCC from N82 to V493 is sinular
to an AMP-binding
site.
As shown in Figures 2A and 2B, KSRCC has chemical and structural homology with
kidney-specific protein from rat (g3127193:SEQ ID NO:15) and a human homolog
of the rat protein
(g3219339:SEQ ~ N0:16). In particular, KSRCC and the rat kidney-specific
protein share 77%
sequence identity over proteins of nearly identical length (577 AA residues
and 572 AA residues,
respectively). The human homolog of the rat protein (g3219339) is a shorter
protein (207 AA
residues), but is 97% identical to KSRCC over this entire length.
As shown in Figures 4A and 4B, the three proteins have similar hydrophilicity
plats and
antigenic indices.
CA 02420101 2003-02-19
WO 02/16595 PCT/USO1/26317
Mammalian variants of the cDNA encoding KSRCC were identified using BLAST2
with
default parameters and the ZOOSEQ databases (Incyte Genomics). Mammalian
variants of the
cDNA encoding the KSRCC include 701648693H1 (RALITXT40), 212451 Rn.l
(template),
202264_Rn.l (template), and 70183634671 (RAKITXT11); SEQ m NOs:l1-14 of the
Sequence
Listing, respectively. These cDNAs are particularly useful for producing
transgenic cell lines or
organisms which model human kidney disorders and upon which potential
therapeutic treatments
for such kidney disorders may be tested Table 1 below shows the percent
identity between the
cDNA encoding KSRCC and its mammalian variants, SEQ ID NOs: l l-14. The first
column shows
the SEQ ID for the human cDNA; the second column, the SEQ IDvar for variant
cDNAs; the third
column, the clone number for the variant cDNAs; the fourth column, the percent
identity to the
human cDNA; and the fifth column, the alignment of the variant cDNA to the
human cDNA.
SEQ )DH SEQ ID~n~ Clone~"~ Identity NtH Alignment
2 11 701648693H1 83% 407-513
2 12 212451_Rn.l 84% 709-986
2 13 202264_Rn.l 83% 1067-1764
2 14 70183634671 84% 1655-1812
These cDNAs are particularly useful for producing transgenic cell lines or
organisms which model
human kidney disorders and upon which potential therapeutic treatments for
such kidney disorders
may be tested.
It will be appreciated by those skilled in the art that as a result of the
degeneracy of the
genetic code, a multitude of cDNA encoding KSRCC, some bearing minimal
similarity to the
cDNAs of any known and naturally occurring gene, may be produced. Thus, the
invention
contemplates each and every possible variation of cDNA that could be made by
selecting
combinations based on possible codon choices. These combinations are made in
accordance with
the standard triplet genetic code as applied to the polynucleotide encoding
naturally occurring
HSTK, and all such variations are to be considered as being specifically
disclosed.
The cDNA and fragments thereof (SEQ ID NOs:2-14) may be used in hybridization,
amplification, and screening technologies to identify and distinguish among
SEQ ID N0:2 and
related molecules in a sample. The mammalian cDNAs may be used to produce
transgenic cell
lines or organisms which are model systems for human renal cell carcinoma and
upon which the
toxicity and efficacy of potential therapeutic treatments may be tested.
Toxicology studies, clinical
trials, and subject/patient treatment profiles may be performed and monitored
using the cDNAs,
proteins, antibodies and molecules and compounds identified using the cDNAs
and proteins of the
present invention. '
Characterization and Use of the Invention
GBA Analysis
GBA identifies cDNAs that are expressed in a plurality of cDNA libraries. The
cDNAs
9
CA 02420101 2003-02-19
WO 02/16595 PCT/USO1/26317
include genes of known or unknown function which are expressed in a specific
disease process,
subcellular compartment, cell type, tissue type, or species. The expression
patterns of genes with
known function are compared with those of cDNAs with unknown function to
determine whether a
specified co-expression probability threshold is met. Through this comparison,
a subset of the
cDNAs having a high co-expression probability with the known genes can be
identified. The high
co-expression probability correlates with a particular coexpression
probability threshold which is
preferably less than 0.001 and more preferably less than 0.00001.
The cDNAs originate from cDNA libraries derived from a variety of sources
including, but
not limited to, eukaryotes such as human, mouse, rat, dog, monkey, plant, and
yeast; prokaryotes
such as bacteria; and viruses. These cDNAs can also be selected from a variety
of sequence types
including, but not limited to, expressed sequence tags (ESTs), assembled
polynucleotides, full
length gene coding regions, promoters, introns, enhancers, 5' untranslated
regions, and 3'
untranslated regions. To have statistically significant analytical results,
the cDNAs need to be
expressed in at least five cDNA libraries.
The cDNA libraries used in the co-expression analysis of the present invention
can be
obtained from any cell or cell line, tissue, or organ and may be from adrenal
gland, biliary tract,
bladder, blood cells, blood vessels, bone marrow, brain, bronchus, cartilage,
chromaffm system,
colon, connective tissue, cultured cells, embryonic stem cells, endocrine
glands, epithelium,
esophagus, fetus, ganglia, heart, hypothalamus, immune system, intestine,
islets of Langerhans,
kidney, larynx, liver, lung, lymph, muscles, neurons, ovary, pancreas, penis,
peripheral nervous
system, phagocytes, pituitary, placenta, pleurus, prostate, salivary glands,
seminal vesicles,
skeleton, spleen, stomach, testis, thymus, tongue, ureter, uterus, and the
like. The number of cDNA
libraries selected can range from as few as 500 to greater than 10,000.
Known kidney specific genes can be selected based on the use of the genes as
diagnostic or
prognostic markers or as therapeutic targets for diseases associated with
kidney disorders.
The method for identifying cDNAs that exhibit a statistically significant co-
expression
pattern with known kidney specific genes is as follows. First, the presence or
absence of a gene
sequence in a cDNA library is defined: a gene is present in a cDNA library
when at least one cDNA
fragment corresponding to that gene is detected in a cDNA sample taken from
the library, and a
gene is absent from a library when no corresponding cDNA fragment is detected
in the sample.
Second, the significance of gene co-expression is evaluated using a
probability method to
measure a due-to-chance probability of the co-expression. The probability
method can be the Fisher
exact test, the chi-squared test, or the kappa test. These tests and examples
of their applications are
well known in the art and can be found in standard statistics texts (Agresti
(1990) Categorical Data
Analysis, John Wiley & Sons, New York NY; Rice (1988) Mathematical Statistics
and Data
CA 02420101 2003-02-19
WO 02/16595 PCT/USO1/26317
Analysis, Duxbury Press, Pacific Grove CA). A Bonferroni correction (Rice,
supra, p. 384) can
also be applied in combination with one of the probability methods for
correcting statistical results
of one gene versus multiple other genes. In a preferred embodiment, the due-to-
chance probability
is measured by a Fisher exact test, and the threshold of the due-to-chance
probability is set
preferably to less than 0.001, more preferably to less than 0.00001.
To determine whether two genes, A and B, have similar co-expression patterns,
occurrence
data vectors can be generated as illustrated in the table below. The presence
of a gene occurring at
least once in a library is indicated by a one, and its absence from the
library, by a zero.
Library Library Library ... Library
1 2 3 N
Gene 1 1 0 ... 0
A
Gene 1 0 1 ... 0
B
For a given pair of genes, the co-occurrence data can be summarized in a 2 x 2
contingency table.
Gene A Present Gene A Absent Total
Gene B Present 8 2 10
Gene B Absent 2 18 20
Total 10 20 30
The contingency table shows the co-occurrence data for gene A and gene B in a
total of 30
libraries. Both gene A and gene B occur 10 times in the libraries, and the
table summarizes and
presents: 1) the number of times gene A and B are both present in a library;
2) the number of times
gene A and B are both absent in a library; 3) the number of times gene A is
present, and gene B is
absent; and 4) the number of times gene B is present, and gene A is absent.
The upper left entry is
the number of times the two genes co-occur in a library, and the middle right
entry is the number of
times neither gene occurs in a library. The off diagonal entries are the
number of times one gene
occurs, and the other does not. Both A and B are present eight times and
absent 18 times. Gene A
is present, and gene B is absent, two times; and gene B is present, and gene A
is absent, two times.
The probability ("p-value") that the above association occurs due to chance as
calculated using a
Fisher exact test is 0.0003. Associations are generally considered significant
if a p-value is less
than 0.01 (Agresti, supra; Rice, supra).
This method of estimating the probability for co-expression of two genes makes
several
assumptions. The method assumes that the libraries are independent and are
identically sampled.
However, in practical situations, the selected cDNA libraries are not entirely
independent, because
more than one library may be obtained from a single subject or tissue. Nor are
they entirely
11
CA 02420101 2003-02-19
WO 02/16595 PCT/USO1/26317
identically sampled, because different numbers of cDNAs may be sequenced from
each library.
The number of cDNAs sequenced typically ranges from 5,000 to 10,000 cDNAs per
library. In
addition, because a Fisher exact co-expression probability is calculated for
each gene versus every
other assembled gene that occur in at least five libraries, a Bonferroni
correction for multiple
statistical tests is used.
cDNA libraries
In a particular embodiment disclosed herein, mRNA was isolated from mammalian
cells
and tissues using methods which are well known to those skilled in the art and
used to prepare the
cDNA libraries. The Incyte clones listed above were isolated from mammalian
cDNA libraries.
Three library preparations representative of the invention are described in
the EXAMPLES below.
The consensus sequences were chemically andlor electronically assembled from
fragments
including Incyte clones and extension and/or shotgun sequences using computer
programs such as
PHRtIP (P Green, University of Washington, Seattle WA), and AUTOASSEMBLER
application
(Applied Biosystems, Foster City CA). Clones, extension and/or shotgun
sequences are
electronically assembled into clusters and/or master clusters.
Sequencing
Methods for sequencing nucleic acids are well known in the art and may be used
to practice
any of the embodiments of the invention. These methods employ enzymes such as
the Klenow
fragment of DNA polymerise I, SEQUENASE, Taq DNA polymerise and thermostable
T7 DNA
polymerise (Amersham Pharmacia Biotech (APB), Piscataway NJ), or combinations
of
polymerises and proofreading exonucleases such as those found in the ELONGASE
amplification
system (Life Technologies, Gaithersburg MD). Preferably, sequence preparation
is automated with
machines such MICROLAB 2200 system (Hamilton, Reno NV) and the DNA ENGINE
thermal
cycler (MJ Research, Watertown MA). Machines commonly used for sequencing
include the ABI
PRISM 3700, 377 or 373 DNA sequencing systems (Applied Biosystems), the
MEGABACE 1000
DNA sequencing system (APB), and the like. The sequences may be analyzed using
a variety of
algorithms well known in the art and described in Ausubel et al. (1997; Short
Protocols in
Molecular Biolo~y, John Wiley & Sons, New York NY, unit 7.7) and in Meyers
(1995; Molecular
Biology and Biotechnolo~y, Wiley VCH, New York NY, pp. S56-g53).
Shotgun sequencing may also be used to complete the sequence of a particular
cloned insert
of interest. Shotgun strategy involves randomly breaking the original insert
into segments of
various sizes and cloning these fragments into vectors. The fragments are
sequenced and
reassembled using overlapping ends until the entire sequence of the original
insert is known.
Shotgun sequencing methods are well known in the art and use thermostable DNA
polymerises,
heat-labile DNA polymerises, and primers chosen from representative regions
flanking the cDNAs
12
CA 02420101 2003-02-19
WO 02/16595 PCT/USO1/26317
of interest. Incomplete assembled sequences are inspected for identity using
various algorithms or
programs such as CONSED (Gordon (1998) Genome Res 8:195-202) which are well
known in the
art. Contaminating sequences including vector or chimeric sequences or deleted
sequences can be
removed or restored, respectively, organizing the incomplete assembled
sequences into finished
sequences.
Extension of a Nucleic Acid Sequence
The sequences of the invention may be extended using various PCR-based methods
known
in the art. For example, the XL-PCR kit (Applied Biosystems), nested primers,
and commercially
available cDNA or genomic DNA libraries may be used to extend the nucleic acid
sequence. For
all PCR-based methods, primers may be designed using commercially available
software, such as
OLIGO primer analysis software (Molecular Biology Insights, Cascade CO) to be
about 22 to 30
nucleotides in length, to have a GC content of about 50% or more, and to
anneal to a target
molecule at temperatures from about 55C to about 68C. When extending a
sequence to recover
regulatory elements, it is preferable to use genomic, rather than cDNA
libraries.
Hybridization
The cDNA and fragments thereof can be used in hybridization technologies for
various
purposes. A probe may be designed or derived from unique regions such as the
5'regulatory region
or from a nonconserved region (i.e., 5' or 3' of the nucleotides encoding the
conserved catalytic
domain of the protein) and used in protocols to identify naturally occurring
molecules encoding the
KSRCC, allelic variants, or related molecules. The probe may be DNA or RNA,
may be single
stranded and should have at least 50% sequence identity to any of the nucleic
acid sequences, SEQ
ID NOs:2-14. Hybridization probes may be produced using oligolabeling, nick
translation,
end-labeling, or PCR amplification in the presence of a reporter molecule. A
vector containing the
cDNA or a fragment thereof may be used to produce an mRNA probe in vitro by
addition of an
RNA polymerase and labeled nucleotides. These procedures may be conducted
using commercially
available kits such as those provided by APB.
The stringency of hybridization is determined by G+C content of the probe,
salt
concentration, and temperature. In particular, stringency can be increased by
reducing the
concentration of salt or raising the hybridization temperature. In solutions
used for some membrane
based hybridizations, addition of an organic solvent such as formamide allows
the reaction to occur
at a lower temperature. Hybridization can be performed at low stringency with
buffers, such as
SxSSC with 1 % sodium dodecyl sulfate (SDS) at 60C, which permits the
formation of a
hybridization complex between nucleic acid sequences that contain some
mismatches. Subsequent
washes are performed at higher stringency with buffers such as 0.2xSSC with
0.1% SDS at either
45C (medium stringency) or 68C (high stringency). At high stringency,
hybridization complexes
13
CA 02420101 2003-02-19
WO 02/16595 PCT/USO1/26317
will remain stable only where the nucleic acids are completely complementary.
In some membrane-
based hybridizations, preferably 35% or most preferably 50%, formamide can be
added to the
hybridization solution to reduce the temperature at which hybridization is
performed, and
background signals can be reduced by the use of other detergents such as
Sarkosyl or TRITON X-
100 (Sigma-Aldrich, St. Louis MO) and a blocking agent such as denatured
salmon sperm DNA.
Selection of components and conditions for hybridization are well known to
those skilled in the art
and are reviewed in Ausubel (supra) and Sambrook et al. (1989) Molecular
Cloning, A Laboratory
Manual, Cold Spring Harbor Press, Plainview NY.
Arrays may be prepared and analyzed using methods known in the art.
Oligonucleotides
may be used as either probes or targets in an array. The array can be used to
monitor the expression
level of large numbers of genes simultaneously and to identify genetic
variants, mutations, and
single nucleotide polymorphisms. Such information may be used to determine
gene function; to
understand the genetic basis of a condition, disease, or disorder; to diagnose
a condition, disease, or
disorder; and to develop and monitor the activities of therapeutic agents.
(See, e.g., Brennan et al.
(1995) USPN 5,474,796; Schena et al. (1996) Proc Natl Acad Sci 93:10614-10619;
Baldeschweiler
et al. (1995) PCT application WO95/251116; Shalon et al. (1995) PCT
application W095/35505;
Heller et al. (1997) Proc Natl Acad Sci 94:2150-2155; and Heller et al. (1997)
USPN 5,605,662.)
Hybridization probes are also useful in mapping the naturally occurring
genomic sequence.
The probes may be hybridized to: 1) a particular chromosome, 2) a specific
region of a
chromosome, or 3) an artificial chromosome construction such as human
artificial chromosome
(HAC), yeast artificial chromosome (YAC), bacterial artificial chromosome
(BAC), bacterial P1
construction, or single chromosome cDNA libraries.
Ex ression
Any one of a multitude of cDNAs encoding KSRCC may be cloned into a vector and
used
to express the protein, or portions thereof, in host cells. The nucleic acid
sequence can be
engineered by such methods as DNA shuffling (USPN 5,830,721) and site-directed
mutagenesis to
create new restriction sites, alter glycosylation patterns, change codon
preference to increase
expression in a particular host, produce splice variants, extend half life,
and the like. The
expression vector may contain transcriptional and translational control
elements (promoters,
enhancers, specific initiation signals, and polyadenylated 3' sequence) from
various sources which
have been selected for their efficiency in a particular host. The vector,
cDNA, and regulatory
elements are combined using in vitro recombinant DNA techniques, synthetic
techniques, and/or in
vivo genetic recombination techniques well known in the art and described in
Sambrook su ra, ch.
4, 8, 16 and 17).
A variety of host systems may be transformed with an expression vector. These
include,
14
CA 02420101 2003-02-19
WO 02/16595 PCT/USO1/26317
but are not limited to, bacteria transformed with recombinant bacteriophage,
plasmid, or cosmid
DNA expression vectors; yeast transformed with yeast expression vectors;
insect cell systems
transformed with baculovirus expression vectors; plant cell systems
transformed with expression
vectors containing viral and/or bacterial elements, or animal cell systems
(Ausubel supra, unit 16).
For example, an adenovirus transcriptionltranslation complex may be utilized
in mammalian cells.
After sequences are ligated into the E1 or E3 region of the viral genome, the
infective virus is used
to transform and express the protein in host cells. The Rous sarcoma virus
enhancer or SV40 or
EBV-based vectors may also be used for high-level protein expression.
Routine cloning, subcloning, and propagation of nucleic acid sequences can be
achieved
using the multifunctional PBLUESCRIfT vector (Stratagene, La Jolla CA) or
PSPORT1 plasmid
(Life Technologies). Introduction of a nucleic acid sequence into the multiple
cloning site of these
vectors disrupts the lacZ gene and allows colorimetric screening for
transformed bacteria. In
addition, these vectors may be useful for in vitro transcription, dideoxy
sequencing, single strand
rescue with helper phage, and creation of nested deletions in the cloned
sequence.
For long term production of recombinant proteins, the vector can be stably
transformed into
cell lines along with a selectable or visible marker gene on the same or on a
separate vector. After
transformation, cells are allowed to grow fox about 1 to 2 days in enriched
media and then are
transferred to selective media. Selectable markers, antimetabolite,
antibiotic, or herbicide
resistance genes, confer resistance to the relevant selective agent and allow
growth and recovery of
cells which successfully express the introduced sequences. Resistant clones
identified either by
survival on selective media or by the expression of visible markers, such as
anthocyanins, green
fluorescent protein (GFP),13 glucuronidase, luciferase and the like, may be
propagated using culture
techniques. Visible markers are also used to quantify the amount of protein
expressed by the
introduced genes. Verification that the host cell contains the desired
mammalian cDNA is based on
DNA-DNA or DNA-RNA hybridizations or PCR amplification techniques.
The host cell may be chosen for its ability to modify a recombinant protein in
a desired
fashion. Such modifications include acetylation, carboxylation, glycosylation,
phosphorylation,
lipidation, acylation and the like. Post-translational processing which
cleaves a "prepro" form may
also be used to specify protein targeting, folding, and/or activity. Different
host cells available from
the ATCC (Manassas VA) which have specific cellular machinery and
characteristic mechanisms
for post-translational activities may be chosen to ensure the correct
modification and processing of
the recombinant protein.
Recovery of Proteins from Cell Culture
Heterologous moieties engineered into a vector for ease of purification
include glutathione
S-transferase (GST), 6xHis, FLAG, MYC, and the like. GST and 6-His are
purified using
CA 02420101 2003-02-19
WO 02/16595 PCT/USO1/26317
commercially available affinity matrices such as immobilized glutathione and
metal-chelate resins,
respectively. FLAG and MYC are purified using commercially available
monoclonal and
polyclonal antibodies. For ease of separation following purification, a
sequence encoding a
proteolytic cleavage site may be part of the vector located between the
protein and the heterologous
moiety. Methods for recombinant protein expression and purification are
discussed in Ausubel
(supra, unit 16) and are commercially available.
Chemical Synthesis of Peptides
Proteins or portions thereof may be produced not only by recombinant methods,
but also by
using chemical methods well known in the art. Solid phase peptide synthesis
may be carried out in
a batchwise or continuous flow process which sequentially adds a-amino- and
side chain-protected
amino acid residues to an insoluble polymeric support via a linker group. A
linker group such as
methylamine-derivatized polyethylene glycol is attached to polystyrene-co-
divinylbenzene) to form
the support resin. The amino acid residues are N-a-protected by acid labile
Boc
(t-butyloxycarbonyl) or base-labile Fmoc (9-fluorenylmethoxycarbonyl). The
carboxyl group of the
protected amino acid is coupled to the amine of the linker group to anchor the
residue to the solid
phase support resin. Trifluoroacetic acid or piperidine are used to remove the
protecting group in
the case of Boc or Fmoc, respectively. Each additional amino acid is added to
the anchored residue
using a coupling agent or pre-activated amino acid derivative, and the resin
is washed. The full
length peptide is synthesized by sequential deprotection, coupling of
derivitized amino acids, and
washing with dichloromethane and/or N, N-dimethylformamide. The peptide is
cleaved between
the peptide carboxy terminus and the linker group to yield a peptide acid or
amide. (Novabiochem
1997/98 Catalog and Peptide Synthesis Handbook, San Diego CA pp. S 1-S20).
Automated
synthesis may also be carried out on machines such as the ABI 431A peptide
synthesizer (Applied
Biosystems). A protein or portion thereof may be substantially purified by
preparative high
performance liquid chromatography and its composition confirmed by amino acid
analysis or by
sequencing (Creighton (1984) Proteins, Structures and Molecular Properties, WH
Freeman, New
York NY).
Preparation and Screening of Antibodies
Various hosts including goats, rabbits, rats, mice, humans, and others may be
immunized by
injection with KSRCC or any portion thereof. Adjuvants such as Freund's,
mineral gels, and
surface active substances such as lysolecithin, pluronic polyols, polyanions,
peptides, oil emulsions,
keyhole limpet hemacyanin (KLH), and dinitrophenol may be used to increase
immunological
response. The oligopeptide, peptide, or portion of protein used to induce
antibodies should consist
of at least about five amino acids, more preferably ten amino acids, which are
identical to a portion
of the natural protein. Oligopeptides may be fused with proteins such as KLH
in order to produce
16
CA 02420101 2003-02-19
WO 02/16595 PCT/USO1/26317
antibodies to the chimeric molecule.
Monoclonal antibodies may be prepared using any technique which provides for
the
production of antibodies by continuous cell lines in culture. These include,
but are not limited to,
the hybridoma technique, the human B-cell hybridoma technique, and the EBV-
hybridoma
technique. (See, e.g., Kohler et al. (1975) Nature 256:495-497; Kozbor et al.
(1985) J. Itnmunol
Methods 81:31-42; Cote et al. (1983) Proc Natl Acad Sci 80:2026-2030; and Cole
et al. (1984) Mol
Cell Biol 62:109-120.)
Alternatively, techniques described for the production of single chain
antibodies may be
adapted, using methods known in the art, to produce epitope specific single
chain antibodies.
Antibody fragments which contain specific binding sites for epitopes of the
protein may also be
generated. For example, such fragments include, but are not limited to, F(ab~2
fragments produced
by pepsin digestion of the antibody molecule and Fab fragments generated by
reducing the disulfide
bridges of the F(ab~2 fragments. Alternatively, Fab expression libraries may
be constructed to
allow rapid and easy identification of monoclonal Fab fragments with the
desired specificity. (See,
e.g., Huse et al. (1989) Science 246:1275-1281.)
The KSRCC or a portion thereof may be used in screening assays of phagemid or
B-
lymphocyte immunoglobulin libraries to identify antibodies having the desired
specificity.
Numerous protocols for competitive binding or immunoassays using either
polyclonal or
monoclonal antibodies with established specificities are well known in the
art. Such immunoassays
typically involve the measurement of complex formation between the protein and
its specific
antibody. A two-site, monoclonal-based immunoassay utilizing monoclonal
antibodies reactive to
two non-interfering epitopes is preferred, but a competitive binding assay may
also be employed
(Pound (1998) Immunochemical Protocols, Humana Press, Totowa NJ).
Labeling of Molecules for Assay
A wide variety of reporter molecules and conjugation techniques are known by
those skilled
in the art and may be used in various nucleic acid, amino acid, and antibody
assays. Synthesis of
labeled molecules may be achieved using commercially available kits (Promega,
Madison WI) for
incorporation of a labeled nucleotide such as 3zP-dCTP (APB), Cy3-dCTP or Cy5-
dCTP (Operon
Technologies, Alameda CA), or amino acid such as 35S-methionine (APB).
Nucleotides and amino
acids may be directly labeled with a variety of substances including
fluorescent, chemiluminescent,
or chromogenic agents, and the like, by chemical conjugation to amines, thiols
and other groups
present in the molecules using reagents such as BIODIPY or FITC (Molecular
Probes, Eugene OR).
DIAGNOSTICS
The cDNAs, fragments, oligonucleotides, complementary RNA and DNA molecules,
and
PNAs and may be used to detect and quantify differential gene expression,
absence/presence vs.
17
CA 02420101 2003-02-19
WO 02/16595 PCT/USO1/26317
excess, expression of mRNAs or to monitor mRNA levels during therapeutic
intervention.
Similarly antibodies which specifically bind KSRCC may be used to quantitate
the protein. Kidney
disorders associated with differential expression include acquired cystic
kidney disease and, in
particular, renal cell carcinoma. The diagnostic assay may use hybridization
or amplification
technology to compare gene expression in a biological sample from a patient to
standard samples in
order to detect differential gene expression. Qualitative or quantitative
methods for this comparison
are well known in the art.
For example, the cDNA or probe may be labeled by standard methods and added to
a
biological sample from a patient under conditions for the formation of
hybridization complexes.
After an incubation period, the sample is washed and the amount of label (or
signal) associated with
hybridization complexes, is quantified and compared with a standard value. If
complex formation
in the patient sample is significantly altered (higher or lower) in comparison
to either a normal or
disease standard, then differential expression indicates the presence of a
disorder.
In order to provide standards for establishing differential expression, normal
and disease
expression profiles are established. This is accomplished by combining a
sample taken from normal
subjects, either animal or human, with a cDNA under conditions for
hybridization to occur.
Standard hybridization complexes may be quantified by comparing the values
obtained using
normal subjects with values from an experiment in which a known amount of a
substantially
purified sequence is used. Standard values obtained in this manner may be
compared with values
obtained from samples from patients who were diagnosed with a particular
condition, disease, or
disorder. Deviation from standard values toward those associated with a
particular disorder is used
to diagnose that disorder.
Such assays may also be used to evaluate the efficacy of a particular
therapeutic treatment
regimen in animal studies and in clinical trial or to monitor the treatment of
an individual patient.
Once the presence of a condition is established and a treatment protocol is
initiated, diagnostic
assays may be repeated on a regular basis to determine if the level of
expression in the patient
begins to approximate that which is observed in a normal subject. The results
obtained from
successive assays may be used to show the efficacy of treatment over a period
ranging from several
days to months.
Immunological Methods
Detection and quantification of a protein using either specific polyclonal or
monoclonal
antibodies are known in the art. Examples of such techniques include enzyme-
linked
immunosorbent assays (ELISAs), radioimmunoassays (RIAs), and fluorescence
activated cell
sorting (FAGS). A two-site, monoclonal-based immunoassay utilizing monoclonal
antibodies
reactive to two non-interfering epitopes is preferred, but a competitive
binding assay may be
18
CA 02420101 2003-02-19
WO 02/16595 PCT/USO1/26317
employed. (See, e.g., Coligan et al. (1997) Current Protocols in Immunolo~y,
Wiley-Interscience,
New York NY; and Pound, supra.)
THERAPEUTICS
Chemical and structural similarity, in the context of the AMP-binding domain,
exists
between regions of KSRCC (SEQ ll~ NO:1) and the kidney-specific proteins from
rat and human
(g312719, and g3219339, respectively) as shown in Figures 2A and 2B. In
addition, differential
expression is highly associated with tissues and with renal cell carcinoma as
shown in Figure 3.
KSRCC clearly plays a role in acquired cystic kidney disease and, in
particular, renal cell
carcinoma.
In the treatment of conditions associated with increased expression of the
protein such as
renal cell carcinoma, it is desirable to decrease expression or protein
activity. In one embodiment,
the an inhibitor, antagonist or antibody of the protein may be administered to
a subject to treat a
condition associated with increased expression or activity. In another
embodiment, a
pharmaceutical composition comprising an inhibitor, antagonist or antibody in
conjunction with a
pharmaceutical carrier may be administered to a subject to treat a condition
associated with the
increased expression or activity of the endogenous protein. In an additional
embodiment, a vector
expressing the complement of the cDNA or fragments thereof may be administered
to a subject to
treat the disorder.
Any of the cDNAs, complementary' molecules, or fragments thereof, proteins or
portions
thereof, vectors delivering these nucleic acid molecules or expressing the
proteins, and their ligands
may be administered in combination with other therapeutic agents. Selection of
the agents for use
in combination therapy may be made by one of ordinary skill in the art
according to conventional
pharmaceutical principles. A combination of therapeutic agents may act
synergistically to affect
treatment of a particular disorder at a lower dosage of each agent.
Modification of Gene Expression Using Nucleic Acids
Gene expression may be modified by designing complementary or antisense
molecules
(DNA, RNA, or PNA) to the control, 5', 3', or other regulatory regions of the
gene encoding
KSRCC. Oligonucleotides designed with reference to the transcription
initiation site are preferred.
Similarly, inhibition can be achieved using triple helix base-pairing which
inhibits the binding of
polymerases, transcription factors, or regulatory molecules (Gee et al. In:
Huber and Carr (1994)
Molecular and Immunologic Approaches, Futura Publishing, Mt. Kisco NY, pp. 163-
177). A
complementary molecule may also be designed to block translation by preventing
binding between
ribosomes and mRNA. In one alternative, a library or plurality of cDNAs or
fragments thereof may
be screened to identify those which specifically bind a regulatory,
nontranslated sequence .
Ribozymes, enzymatic RNA molecules, may also be used to catalyze the specific
cleavage
19
CA 02420101 2003-02-19
WO 02/16595 PCT/USO1/26317
of RNA. The mechanism of ribozyme action involves sequence-specific
hybridization of the
ribozyme molecule to complementary target RNA followed by endonucleolytic
cleavage at sites
such as GUA, GUU, and GUC. Once such sites are identified, an oligonucleotide
with the same
sequence may be evaluated for secondary structural features which would render
the
oligonucleotide inoperable. The suitability of candidate targets may also be
evaluated by testing
their hybridization with complementary oligonucleotides using ribonuclease
protection assays.
Complementary nucleic acids and ribozymes of the invention may be prepared via
recombinant expression, in vitro or in vivo, or using solid phase
phosphoramidite chemical
synthesis. In addition, RNA molecules may be modified to increase
intracellular stability and
half-life by addition of flanking sequences at the 5' and/or 3' ends of the
molecule or by the use of
phosphorothioate or 2' O-methyl rather than phosphodiesterase linkages within
the backbone of the
molecule. Modification is inherent in the production of PNAs and can be
extended to other nucleic
acid molecules. Either the inclusion of nontraditional bases such as inosine,
queosine, and
wybutosine, and or the modification of adenine, cytidine, guanine, thymine,
and uridine with acetyl-
, methyl-, thio- groups renders the molecule less available to endogenous
endonucleases.
Screening and Purification Assay
The cDNA encoding KSRCC may be used to screen a library of molecules or
compounds
for specific binding affinity. The libraries may be aptamers, DNA molecules,
RNA molecules,
PNAs, peptides, proteins such as transcription factors, enhancers, repressors,
and other ligands
which regulate the activity, replication, transcription, or translation of the
cDNA in the biological
system. The assay involves combining the cDNA or a fragment thereof with the
library of
molecules under conditions allowing specific binding, and detecting specific
binding to identify at
least one molecule which specifically binds the single stranded or, if
appropriate, double stranded
molecule.
In one embodiment, the cDNA of the invention may be incubated with a plurality
of
purified molecules or compounds and binding activity determined by methods
well known in the
art, e.g., a gel-retardation assay (USPN 6,010,849) or a reticulocyte lysate
transcriptional assay. In
another embodiment, the cDNA may be incubated with nuclear extracts from
biopsied and/or
cultured cells and tissues. Specific binding between the cDNA and a molecule
or compound in the
nuclear extract is initially determined by gel shift assay and may be later
confirmed by recovering
and raising antibodies against that molecule or compound. When these
antibodies are added into the
assay, they cause a supershift in the gel-retardation assay.
In another embodiment, the cDNA may be used to purify a molecule or compound
using
affinity chromatography methods well known in the art. In one embodiment, the
cDNA is
chemically reacted with cyanogen bromide groups on a polymeric resin or gel.
Then a sample is
CA 02420101 2003-02-19
WO 02/16595 PCT/USO1/26317
passed over and reacts with or binds to the cDNA. The molecule or compound
which is bound to
the cDNA may be released from the cDNA by increasing the salt concentration of
the flow-through
medium and collected.
In a further embodiment" the protein or a portion thereof may be used to
purify a ligand
from a sample. A method for using a mammalian protein or a portion thereof to
purify a ligand
would involve combining the protein or a portion thereof with a sample under
conditions to allow
specific binding, detecting specific binding between the protein and ligand,
recovering the bound
protein, and using an appropriate chaotropic agent to separate the protein
from the purified ligand.
In a preferred embodiment, KSRCC or a portion thereof may be used to screen a
plurality
of molecules or compounds in any of a variety of screening assays. The portion
of the protein
employed in such screening may be free in solution, affixed to an abiotic or
biotic substrate (e.g.
borne on a cell surface), or located intracellularly. For example, in one
method, viable or fixed
prokaryotic host cells that are stably transformed with recombinant nucleic
acids that have
expressed and positioned a peptide on their cell surface can be used in
screening assays. The cells
are screened against a plurality or libraries of ligands and the specificity
of binding or formation of
complexes between the expressed protein and the ligand may be measured.
Specific binding
between the protein and molecule may be measured. Depending on the kind of
library being
screened, the assay may be used to identify DNA molecules, RNA molecules,
peptide nucleic acids,
peptides, proteins, mimetics, agonists, antagonists, antibodies,
immunoglobulins, inhibitors, and
drugs or any other ligand, which specifically binds the protein.
In one aspect, this invention comtemplates a method for high throughput
screening using
very small assay volumes and very small amounts of test compound as described
in USPN
5,876,946, incorporated herein by reference. This method is used to screen
large numbers of
molecules and compounds via specific binding. In another aspect, this
invention also contemplates
the use of competitive drug screening assays in which neutralizing antibodies
capable of binding the
protein specifically compete with a test compound capable of binding to the
protein or oligopeptide
or portion thereof. Molecules or compounds identified by screening may be used
in a mammalian
model system to evaluate their toxicity, diagnostic, or therapeutic potential.
Pharmacoloey
Pharmaceutical compositions are those substances wherein the active
ingredients are
contained in an effective amount to achieve a desired and intended purpose.
The determination of
an effective dose is well within the capability of those skilled in the art.
For any compound, the
therapeutically effective dose may be estimated initially either in cell
culture assays or in animal
models. The animal model is also used to achieve a desirable concentration
range and route of
administration. Such information may then be used to determine useful doses
and routes for
21
CA 02420101 2003-02-19
WO 02/16595 PCT/USO1/26317
administration in humans.
A therapeutically effective dose refers to that amount of protein or inhibitor
which
ameliorates the symptoms or condition. Therapeutic efficacy and toxicity of
such agents may be
determined by standard pharmaceutical procedures in cell cultures or
experimental animals, e.g.,
EDSO (the dose therapeutically effective in 50% of the population) and LDso
(the dose lethal to 50%
of the population). The dose ratio between toxic and therapeutic effects is
the therapeutic index,
and it may be expressed as the ratio, LDSO/EDSO. Pharmaceutical compositions
which exhibit large
therapeutic indexes are preferred. The data obtained from cell culture assays
and animal studies are
used in formulating a range of dosage for human use.
I0 Model Systems
Animal models may be used as bioassays where they exhibit a phenotypic
response similar
to that of humans and where exposure conditions are relevant to human
exposures. Mammals are
the most common models, and most infectious agent, cancer, drug, and toxicity
studies are
performed on rodents such as rats or mice because of low cost, availability,
lifespan, reproductive
potential, and abundant reference literature. Inbred and outbred rodent
strains provide a convenient
model for investigation of the physiological consequences of under- or over-
expression of genes of
interest and for the development of methods for diagnosis and treatment of
diseases. A mammal
inbred to over-express a particular gene (for example, secreted in milk) may
also serve as a
convenient source of the protein expressed by that gene.
Toxicolo~y
Toxicology is the study of the effects of agents on living systems. The
majority of toxicity
studies are performed on rats or mice. Observation of qualitative and
quantitative changes in
physiology, behavior, homeostatic processes, and lethality in the rats or mice
are used to generate a
toxicity profile and to assess potential consequences on human health
following exposure to the
agent.
Genetic toxicology identifies and analyzes the effect of an agent on the rate
of endogenous,
spontaneous, and induced genetic mutations. Genotoxic agents usually have
common chemical or
physical properties that facilitate interaction with nucleic acids and are
most harmful when
chromosomal aberrations are transmitted to progeny. Toxicological studies may
identify agents that
increase the frequency of structural or functional abnormalities in the
tissues of the progeny if
administered to either parent before conception, to the mother during
pregnancy, or to the
developing organism. Mice and rats are most frequently used in these tests
because their short
reproductive cycle allows the production of the numbers of organisms needed to
satisfy statistical
requirements.
Acute toxicity tests are based on a single administration of an agent to the
subject to
22
CA 02420101 2003-02-19
WO 02/16595 PCT/USO1/26317
determine the symptomology or lethality of the agent. Three experiments are
conducted: 1) an
initial dose-range-finding experiment, 2) an experiment to narrow the range of
effective doses, and
3) a final experiment for establishing the dose-response curve.
Subchronic toxicity tests are based on the repeated administration of an
agent. Rat and dog
are commonly used in these studies to provide data from species in different
families. With the
exception of carcinogenesis, there is considerable evidence that daily
administration of an agent at
high-dose concentrations for periods of three to four months will reveal most
forms of toxicity in
adult animals.
Chronic toxicity tests, with a duration of a year or more, are used to
demonstrate either the
absence of toxicity or the carcinogenic potential of an agent. When studies
axe conducted on rats, a
minimum of three test groups plus one control group are used, and animals are
examined and
monitored at the outset and at intervals throughout the experiment.
Trans~enic Animal Models
Transgenic rodents that over-express or under-express a gene of interest may
be inbred and
used to model human diseases or to test therapeutic or toxic agents. (See,
e.g., USPN 5,175,383 and
USPN 5,767,337.) In some cases, the introduced gene may be activated at a
specific time in a
specific tissue type during fetal or postnatal development. Expression of the
transgene is monitored
by analysis of phenotype, of tissue-specific mRNA expression, or of serum and
tissue protein levels
in transgenic animals before, during, and after challenge with experimental
drug therapies.
Embryonic Stem Cells
Embryonic (ES) stem cells isolated from rodent embryos retain the potential to
form
embryonic tissues. When ES cells are placed inside a carrier embryo, they
resume normal
development and contribute to tissues of the live-born animal. ES cells are
the preferred cells used
in the creation of experimental knockout and knockin rodent strains. Mouse ES
cells, such as the
mouse 129/SvJ cell line, are derived from the early mouse embryo and are grown
under culture
conditions well known in the art. Vectors used to produce a transgenic strain
contain a disease gene
candidate and a marker gen, the latter serves to identify the presence of the
introduced disease gene.
The vector is transformed into ES cells by methods well known in the art, and
transformed ES cells
are identified and microinjected into mouse cell blastocysts such as those
from the C57BL/6 mouse
strain. The blastocysts are surgically transferred to pseudopregnant dams, and
the resulting
chimeric progeny are genotyped and bred to produce heterozygous or homozygous
strains.
ES cells derived from human blastocysts may be manipulated in vitro to
differentiate into at
least eight separate cell lineages. These lineages are used to study the
differentiation of various cell
types and tissues in vitro, and they include endoderm, mesoderm, and
ectodermal cell types which
differentiate into, for example, neural cells, hematopoietic lineages, and
cardiomyocytes.
23
CA 02420101 2003-02-19
WO 02/16595 PCT/USO1/26317
Knockout Analysis
In gene knockout analysis, a region of a mammalian gene is enzymatically
modified to
include a non-mammalian gene such as the neomycin phosphotransferase gene
(neo; Capecchi
(1989) Science 244:1288-1292). The modified gene is transformed into cultuxed
ES cells and
integrates into the endogenous genome by homologous recombination. The
inserted sequence
disrupts transcription and translation of the endogenous gene. Transformed
calls are injected into
rodent blastulae, and the blastulae are implanted into pseudopregnant dams.
Transgenic progeny
are crossbred to obtain homozygous inbred lines which lack a functional copy
of the mammalian
gene. In one example, the mammalian gene is a human gene.
Knockin AnalXsis
ES cells can be used to create knockin humanized animals (pigs) or transgenic
animal
models (mice or rats) of human diseases. With knockin technology, a region of
a human gene is
injected into animal ES cells, and the human sequence integrates into the
animal cell genome.
Transformed cells are injected into blastulae and the blastulae are implanted
as described above.
Transgenic progeny or inbred lines are studied and treated with potential
pharmaceutical agents to
obtain information on treatment of the analogous human condition. These
methods have been used
to model several human diseases.
Non-Human Primate Model
The field of animal testing deals with data and methodology from basic
sciences such as
physiology, genetics, chemistry, pharmacology and statistics. These data are
paramount in
evaluating the effects of therapeutic agents on non-human primates as they can
be related to human
health. Monkeys are used as human surrogates in vaccine and drug evaluations,
and their responses
are relevant to human exposures under similar conditions. Cynomolgus and
Rhesus monkeys
(Macaca fascicularis and Macaca mulatta, respectively) and Common Marmosets
Callithrix
'act thus) are the most common non-human primates (NHPs) used in these
investigations. Since
great cost is associated with developing and maintaining a colony of NHPs,
early research and
toxicological studies are usually carried out in rodent models. In studies
using behavioral measures
such as drug addiction, NHPs are the first choice test animal. In addition,
NHPs and individual
humans exhibit differential sensitivities to many drugs and toxins and can be
classified as a range of
phenotypes from "extensive metabolizers" to "poor metabolizers" of these
agents.
In additional embodiments, the cDNAs which encode the mammalian protein may be
used
in any molecular biology techniques that have yet to be developed, provided
the new techniques
rely on properties of cDNAs that are currently known, including, but not
limited to, such properties
as the triplet genetic code and specific base pair interactions.
EXAMPLES
24
CA 02420101 2003-02-19
WO 02/16595 PCT/USO1/26317
The examples below are provided to illustrate the subject invention and are
not included for
the purpose of limiting the invention. For purposes of example, preparation of
the human kidney
(KIDNNOT32) library will be described.
I cDNA Library Construction
The tissue used for kidney library construction was obtained from a a 49-year-
old
Caucasian male who died from an intracranial hemorrhage and cerebrovascular
accident. The
frozen tissue was-homogenized and lysed in TRIZOL reagent (1 g tissue/10 ml
TRIZOL; Life
Technologies) using a POLYTRON homogenizer (Brinkmann Instruments, Westbury
NY). After
brief incubation on ice, chloroform was added (1:5 v/v), and the mixture was
centrifuged to
separate the phases. The upper aqueous phase was removed to a fresh tube, and
isopropanol was
added to precipitate RNA. The RNA was resuspended in RNase-free water and
treated with DNase.
The RNA was re-extracted with acid phenol-chloroform and reprecipitated with
sodium acetate and
ethanol. Poly(A+) RNA was isolated using the OLIGOTEX mRNA purification kit
(QIAGEN,
Valencia CA) and used to-construct the cDNA library. .
II Construction of pINCY Plasmid
The plasmid was constructed by digesting the pSPORTl plasmid (Life
Technologies) with
EcoRI restriction enzyme (New England Biolabs, Beverly MA) and filling the
overhanging ends
using Klenow enzyme (New England Biolabs) and 2'-deoxynucleotide 5'-
triphosphates (dNTPs).
The plasmid was self ligated and transformed into the bacterial host, E. coli
strain JM109.
An intermediate plasmid produced by the bacteria (pSPORT 1-SRI) showed no
digestion
with EcoRI and was digested with Hind III (New England Biolabs) and the
overhanging ends were
again filled in with Klenow and dNTPs. A linker sequence was phosphorylated,
ligated onto the 5'
blunt end, digested with EcoRI, and self ligated. Following transformation
into JM109 host cells,
plasmids were isolated and tested for preferential digestibility with EcoRI,
but not with Hind III. A
single colony that met this criteria was designated plNCY plasmid.
After testing the plasmid for its ability to incorporate cDNAs from a library
prepared using
NotI and EcoRI restriction enzymes, several clones were sequenced; and a
single clone containing
an insert of approximately 0.8 kb was selected from which to prepare a large
quantity of the
plasmid. After digestion with NotI and EcoRI, the plasmid was isolated on an
agarose gel and
purified using a QIAQUICK column (Qiagen) for use in library construction.
III Isolation and Sequencing of cDNA Clones
Plasmid DNA was released from the cells and purified using either the MIN1PREP
kit
(Edge Biosystems, Gaithersburg MD) or the REAL PREP 96 plasmid kit (Qiagen).
This kit
consists of a 96-well block with reagents for 960 purifications. The
recommended protocol was
employed except for the following changes: 1) the bacteria were cultured in 1
ml of sterile
CA 02420101 2003-02-19
WO 02/16595 PCT/USO1/26317
TERRIFIC BROTH (BD Biosciences, Sparks MD) with carbenicillin at 25 mg/1 and
glycerol at
0.4%; 2) after inoculation, the cells were cultured for 19 hours and then
lysed with 0.3 ml of lysis
buffer; and 3) following isopropanol precipitation, the plasmid DNA pellet was
resuspended in 0.1
ml of distilled water. After the last step in the protocol, samples were
transferred to a 96-well block
for storage at 4C.
The cDNAs were prepared for sequencing using the MICROLAB 2200 system
(Hamilton)
in combination with the DNA ENGINE thermal cyclers (MJ Research). The cDNAs
were
sequenced by the method of Sanger and Coulson (1975; J Mol Biol 94:441-448)
using an ABI
PRISM 377 sequencing system (Applied Biosystems) or the MEGABACE 1000 DNA
sequencing
system (APB). Most of the isolates were sequenced according to standard ABI
protocols and kits
(Applied Biosystems) with solution volumes of 0.25x-l.Ox concentrations. In
the alternative,
cDNAs were sequenced using solutions and dyes from APB.
IV Extension of cDNA Sequences
The cDNAs were extended using the cDNA clone and oligonucleotide primers. One
primer
was synthesized to initiate 5' extension of the known fragment, and the other,
to initiate 3' extension
of the known fragment. The initial primers were designed using OLIGO software
(Molecular
Biology Insights), to be about 22 to 30 nucleotides in length, to have a GC
content of about 50% or
more, and to anneal to the target sequence at temperatures of about 68C to
about 72C. Any stretch
of nucleotides that would result in hairpin structures and primer-primer
dimerizations was avoided.
Selected cDNA libraries were used as templates to extend the sequence. If more
than one
extension was necessary, additional or nested sets of primers were designed.
Preferred libraries
have been size-selected to include larger cDNAs and random primed to contain
more sequences
with 5' or upstream regions of genes. Genomic libraries are used to obtain
regulatory elements,
especially extension into the 5' promoter binding region.
High fidelity amplification was obtained by PCR using methods such as that
taught in
USPN 5,932,451. PCR was performed in 96-well plates using the DNA ENGINE
thermal cycler
(MJ Research). The reaction mix contained DNA template, 200 nmol of each
primer, reaction
buffer containing Mg2+, (NH4)ZSO4, and (3-mercaptoethanol, Taq DNA polymerase
(APB),
ELONGASE enzyme (Life Technologies), and Pfu DNA polymerase (Stratagene), with
the
following parameters for primer pair PCI A and PCI B (Incyte Genomics): Step
1: 94C, three min;
Step 2: 94C, 15 sec; Step 3: 60C, one min; Step 4: 68C, two min; Step 5: Steps
2, 3, and 4 repeated
20 times; Step 6: 68C, five min; Step 7: storage at 4C. In the alternative,
the parameters for primer
pair T7 and SK+ (Stratagene) were as follows: Step l: 94C, three min; Step 2:
94C, 15 sec; Step 3:
57C, one min; Step 4: 68C, two min; Step 5: Steps 2, 3, and 4 repeated 20
times; Step 6: 68C, five
min; Step 7: storage at 4C.
26
CA 02420101 2003-02-19
WO 02/16595 PCT/USO1/26317
The concentration of DNA in each well was determined by dispensing 100 ~.l
PICOGREEN
quantitation reagent (0.25% reagent in lx TE, v/v; Molecular Probes) and 0.5
~,1 of undiluted PCR
product into each well of an opaque fluorimeter plate (Corning, Acton MA) and
allowing the DNA
to bind to the reagent. The plate was scanned in a Fluoroskan II (Labsystems
Oy) to measure the
fluorescence of the sample and to quantify the concentration of DNA. A 5 ~.1
to 10 ~,1 aliquot of the
reaction mixture was analyzed by electrophoresis on a 1 % agarose mini-gel to
determine which
reactions were successful in extending the sequence.
The extended clones were desalted, concentrated, transferred to 384-well
plates, digested
with CviJI cholera virus endonuclease (Molecular Biology Research, Madison
WI), and sonicated
or sheared prior to religation into pUCl8 vector (APB). For shotgun sequences,
the digested
nucleotide sequences were separated on low concentration (0.6 to 0.8%) agarose
gels, fragments
were excised, and the agar was digested with AGARACE enzyme (Promega).
Extended clones
were religated using T4 DNA ligase (New England Biolabs) into pUCl8 vector
(APB), treated with
Pfu DNA polymerise (Stratagene) to fill-in restriction site overhangs, and
transfected into E. coli
competent cells. Transformed cells were selected on antibiotic-containing
media, and individual
colonies were picked and cultured overnight at 37C in 384-well plates in LB/2x
carbenicillin liquid
media.
The cells were lysed, and DNA was amplified using primers, Taq DNA polymerise
(APB)
and Pfu DNA polymerise (Stratagene) with the following parameters: Step 1:
94C, three min; Step
2: 94C, 15 sec; Step 3: 60C, one min; Step 4: 72C, two min; Step 5: steps 2,
3, and 4 repeated 29
times; Step 6: 72C, five min; Step 7: storage at 4C. DNA was quantified using
PICOGREEN
quantitative reagent (Molecular Probes) as described above. Samples with low
DNA recoveries
were reamplified using the conditions described above. Samples were diluted
with 20%
dimethylsulfoxide (DMSO; 1:2, v/v), and sequenced using DYENAMIC energy
transfer sequencing
primers and the DYENAMIC DIRECT cycle sequencing kit (APB) or the ABI PRISM
BIGDYE
terminator cycle sequencing kit (Applied Biosystems).
V Homology Searching of cDNA Clones and Their Deduced Proteins
The cDNAs of the Sequence Listing or their deduced amino acid sequences were
used to
query databases such as GenBank, SwissProt, BLOCKS, and the like. These
databases that contain
previously identified and annotated sequences or domains were searched using
BLAST or BLAST
2 (Altschul et al. supra; Altschul, supra) to produce alignments and to
determine which sequences
were exact matches or homologs. The alignments were to sequences of
prokaryotic (bacterial) or
eukaryotic (animal, fungal, or plant) origin. Alternatively, algorithms such
as the one described in
Smith and Smith (1992, Protein Engineering 5:35-51) could have been used to
deal with primary
sequence patterns and secondary structure gap penalties. All of the sequences
disclosed in this
27
CA 02420101 2003-02-19
WO 02/16595 PCT/USO1/26317
application have lengths of at least 49 nucleotides, and no more than 12%
uncalled bases (where N
is recorded rather than A, C, G, or T).
As detailed in Karlin su re), BLAST matches between a query sequence and a
database
sequence were evaluated statistically and only reported when they satisfied
the threshold of 10-25 for
nucleotides and 10-'4 for peptides. Homology was also evaluated by product
score calculated as
follows: the % nucleotide or amino acid identity [between the query and
reference sequences] in
BLAST is multiplied by the % maximum possible BLAST score [based on the
lengths of query and
reference sequences] and then divided by 100. In comparison with hybridization
procedures used in
the laboratory, the electronic stringency for an exact match was set at 70,
and the conservative
lower limit for an exact match was set at approximately 40 (with 1-2% error
due to uncalled bases).
The BLAST software suite, freely available sequence comparison algorithms
(NCBI,
Bethesda MD; http://www.ncbi.nlm.nih.gov/gorf/bl2.htm1), includes various
sequence analysis
programs including "blastn" that is used to align nucleic acid molecules and
BLAST 2 that is used
for direct pairwise comparison of either nucleic or amino acid molecules.
BLAST programs are
commonly used with gap and other parameters set to default settings, e.g.:
Matrix: BLOSLTM62;
Reward for match: 1; Penalty for mismatch: -2; Open Gap-.. 5 and Extension
Gap: 2 penalties; Gap x
drop-off: 50; Expect: 10; Word Size: 11; and Filter: on. Identity is measured
over the entire length
of a sequence or some smaller portion thereof. Brenner et al. (1998; Proc Natl
Acad Sci 95:6073-
6078, incorporated herein by reference) analyzed the BLAST for its ability to
identify structural
homologs by sequence identity and found 30% identity is a reliable threshold
for sequence
alignments of at least I50 residues and 40%, for alignments of at least 70
residues.
The mammalian cDNAs of this application were compared with assembled consensus
sequences or templates found in the LIFESEQ GOLD database. Component sequences
from
cDNA, extension, full length, and shotgun sequencing projects were subjected
to PHRED analysis
and assigned a quality score. All sequences with an acceptable quality score
were subjected to
various pre-processing and editing pathways to remove low quality 3' ends,
vector and linker
sequences, polyA tails, Alu repeats, mitochondria) and ribosomal sequences,
and bacterial
contamination sequences. Edited sequences had to be at least 50 by in length,
and low-information
sequences and repetitive elements such as dinucleotide repeats, Alu repeats,
and the like, were
replaced by "Ns" or masked.
Edited sequences were subjected to assembly procedures in which the sequences
were
assigned to gene bins. Each sequence could only belong to one bin, and
sequences in each bin were
assembled to produce a template. Newly sequenced components were added to
existing bins using
BLAST and CROSSMATCH. To be added to a bin, the component sequences had to
have a
BLAST quality score greater than or equal to 150 and an alignment of at least
82% local identity.
28
CA 02420101 2003-02-19
WO 02/16595 PCT/USO1/26317
The sequences in each bin were assembled using PHRAP. Bins with several
overlapping
component sequences were assembled using DEEP PHRAP. The orientation of each
template was
determined based on the number and orientation of its component sequences.
Bins were compared to one another and those having local similarity of at
least 82% were
combined and reassembled. Bins having templates with less than 95% local
identity were split.
Templates were subjected to analysis by STITCHER/BXON MAPPER algorithms that
analyze the
probabilities of the presence of splice variants, alternatively spliced exons,
splice junctions,
differential expression of alternative spliced genes across tissue types or
disease states, and the like.
Assembly procedures were repeated periodically, and templates were annotated
using BLAST
against GenBank databases such as GBpri. An exact match was defined as having
from 95% local
identity over 200 base pairs through 100% local identity over 100 base pairs
and a homolog match
as having an E-value (or probability score) of <1 x 10-$. The templates were
also subjected to
frameshift FASTx against GENPEPT, and homolog match was defined as having an E-
value of <1
x 10-8. Template analysis and assembly was described in USSN 09/276,534, filed
March 25, 1999.
Following assembly, templates were subjected to BLAST, motif, and other
functional
analyses and categorized in protein hierarchies using methods described in
USSN 08/812,290 and
USSN 08/811,758, both filed March 6, 1997; in USSN 08/947,845, filed October
9, 1997; and in
USSN 09/034,807, filed March 4, 1998. Then templates were analyzed by
translating each template
in all three forward reading frames and searching each translation against the
PFAM database of
hidden Markov model-based protein families and domains using the HMMER
software package
(Washington University School of Medicine, St. Louis MO;
http://pfam.wustl.edu/). Using
BLAST2 analysis against the Incyte LifeGold human template database, Clone
3481942CB 1 was
found to be the 5' end of the 279978.9 template. Further analysis showed that
the 3' end was Clone
4331016CT1 within a related template. These two templates were spliced
together to form SEQ ~
N0:2. The cDNA was further analyzed using MACDNASIS PRO software (Hitachi
Software
Engineering), and LASERGENE software (DNASTAR) and queried against public
databases such
as the GenBank rodent, mammalian, vertebrate, prokaryote, and eukaryote
databases, SwissProt,
BLOCKS, PRINTS, PFAM, and Prosite.
VT Chromosome Mapping
Radiation hybrid and genetic mapping data available from public resources such
as the
Stanford Human Genome Center (SHGC), Whitehead Institute for Genome Research
(WIGR), and
Genethon are used to determine if any of the cDNAs presented in the Sequence
Listing have been
mapped. Any of the fragments of the cDNA encoding KSRCC that have been mapped
result in the
assignment of all related regulatory and coding sequences mapping to the same
location. The
genetic map locations are described as ranges, or intervals, of human
chromosomes. The map
29
CA 02420101 2003-02-19
WO 02/16595 PCT/USO1/26317
position of an interval, in cM (which is roughly equivalent to 1 megabase of
human DNA), is
measured relative to the terminus of the chromosomal p-arm.
VII Hybridization Technologies and Analyses
Immobilization of cDNAs on a Substrate
The cDNAs are applied to a substrate by one of the following methods. A
mixture of
cDNAs is fractionated by gel electrophoresis and transferred to a nylon
membrane by capillary
transfer. Alternatively, the cDNAs are individually ligated to a vector and
inserted into bacterial
host cells to form a library. The cDNAs are then arranged on a substrate by
one of the following
methods. In the first method, bacterial cells containing individual clones are
robotically picked and
arranged on a nylon membrane. The membrane is placed on LB agar containing
selective agent
(carbenicillin, kanamycin, ampicillin, or chloramphenicol depending on the
vector used) and
incubated at 37C for 16 hr. The membrane is removed from the agar and
consecutively placed
colony side up in 10% SDS, denaturing solution (I.5 M NaCI, 0.5 M NaOH ),
neutralizing solution
(1.5 M NaCI, 1 M Tris, pH 8.0), and twice in 2xSSC for 10 min each. The
membrane is then UV
irradiated in a STRATALINI~ER UV-crosslinker (Stratagene).
In the second method, cDNAs are amplified from bacterial vectors by thirty
cycles of PCR
using primers complementary to vector sequences flanking the insert. PCR
amplification increases
a starting concentration of 1-2 ng nucleic acid to a final quantity greater
than 5 ~,g. Amplified
nucleic acids from about 400 by to about 5000 by in length are purified using
SEPHACRYL-400
beads (APB). Purified nucleic acids are arranged on a nylon membrane manually
or using a dot/slot
blotting manifold and suction device and are immobilized by denaturation,
neutralization, and UV
irradiation as described above. Purified nucleic acids are robotically
arranged and immobilized on
polymer-coated glass slides using the procedure described in USPN 5,807,522.
Polymer-coated
slides are prepared by cleaning glass microscope slides (Corning, Acton MA) by
ultrasound in 0.1 %
SDS and acetone, etching in 4% hydrofluoric acid (VWR Scientific Products,
West Chester PA),
coating with 0.05% aminopropyl silane (Sigma Aldrich) in 95% ethanol, and
curing in a 110C oven.
The slides are washed extensively with distilled water between and after
treatments. The nucleic
acids are arranged on the slide and then immobilized by exposing the array to
UV irradiation using
a STRATALINKER W-crosslinker (Stratagene). Arrays are then washed at room
temperature in
0.2% SDS and rinsed three times in distilled water. Non-specific binding sites
are blocked by
incubation of arrays in 0.2% casein in phosphate buffered saline (PBS; Tropix,
Bedford MA) for 30
min at 60C; then the arrays are washed in 0.2% SDS and rinsed in distilled
water as before .
Probe Preparation for Membrane Hybridization
Hybridization probes derived from the cDNAs of the Sequence Listing are
employed for
screening cDNAs, mRNAs, or genomic DNA in membrane-based hybridizations.
Probes are
CA 02420101 2003-02-19
WO 02/16595 PCT/USO1/26317
prepared by diluting the cDNAs to a concentration of 40-50 ng in 45 ~,I TE
buffer, denaturing by
heating to 100C for five min, and briefly centrifuging. The denatured cDNA is
then added to a
REDIPRnVxE tube (APB), gently mixed until blue color is evenly distributed,
and briefly
centrifuged. Five ~,1 of [32P]dCTP is added to the tube, and the contents are
incubated at 37C for 10
min. The labeling reaction is stopped by adding 5 ~l of 0.2M EDTA, and probe
is purified from
unincorporated nucleotides using a PROBEQUANT G-50 microcolurnn (APB). The
purified probe
is heated to 100C for five min, snap cooled for two min on ice, and used in
membrane-based
hybridizations as described below.
Probe Preparation for Polymer Coated Slide Hybridization
Hybridization probes derived from mRNA isolated from samples are employed for
screening cDNAs of the Sequence Listing in array-based hybridizations. Probe
is prepared using
the GEMbright kit (Incyte Genomics) by diluting mRNA to a concentration of 200
ng in 9 ~.1 TE
buffer and adding 5 ~.15x buffer, 1 x,10.1 M DTT, 3 ~.l Cy3 or Cy5 labeling
mix, 1 ~,1 RNase
inhibitor, 1 ~,1 reverse transcriptase, and 5 ~.1 lx yeast control mRNAs.
Yeast control mRNAs are
synthesized by in vitro transcription from noncoding yeast genomic DNA (W.
Lei, unpublished).
As quantitative controls, one set of control mRNAs at 0.002 ng, 0.02 ng, 0.2
ng, and 2 ng are
diluted into reverse transcription reaction mixture at ratios of 1:100,000,
1:10,000, 1:1000, and
1:100 (w/w) to sample mRNA respectively. To examine mRNA differential
expression patterns, a
second set of control mRNAs are diluted into reverse transcription reaction
mixture at ratios of 1:3,
3:1, 1:10, 10:1, 1:25, and 25:1 (w/w). The reaction mixture is mixed and
incubated at 37C for two
hr. The reaction mixture is then incubated for 20 min at 85C, and probes are
purified using two
successive CHROMA SPIN+TE 30 columns (Clontech, Palo Alto CA). Purified probe
is ethanol
precipitated by diluting probe to 90 ,u1 in DEPC-treated water, adding 2 ,u1
lmg/ml glycogen, 60 ~1
5 M sodium acetate, and 300 ~,1 100% ethanol. The probe is centrifuged for 20
min at 20,800xg,
and the pellet is resuspended in 12 ~,1 resuspension buffer, heated to 65C for
five min, and mixed
thoroughly. The probe is heated and mixed as before and then stored on ice.
Probe is used in high
density array-based hybridizations as described below.
Membrane-based Hybridization
Membranes are pre-hybridized in hybridization solution containing 1% Sarkosyl
and lx
high phosphate buffer (0.5 M NaCI, 0.1 M Na2HP04, 5 mM EDTA, pH 7) at 55C for
two hr. The
probe, diluted in 15 ml fresh hybridization solution, is then added to the
membrane. The membrane
is hybridized with the probe at 55C for 16 hr. Following hybridization, the
membrane is washed for
15 min at 25C in 1mM Tris (pH 8.0), 1% Sarkosyl, and four times for 15 min
each at 25C in 1mM
Tris (pH 8.0). To detect hybridization complexes, XOMAT-AR film (Eastman
Kodak, Rochester
NY) is exposed to the membrane overnight at -70C, developed, and examined
visually.
31
CA 02420101 2003-02-19
WO 02/16595 PCT/USO1/26317
Polymer Coated Slide-based Hybridization
Probe is heated to 65C for five min, centrifuged five min at 9400 rpm in a
5415C
microcentrifuge (Eppendorf Scientific, Westbury NY), and then 18 ~,1 is
aliquoted onto the array
surface and covered with a coverslip. The arrays are transferred to a
waterproof chamber having a
cavity just slightly larger than a microscope slide. The chamber is kept at
100% humidity internally
by the addition of 140 ~,1 of SxSSC in a corner of the chamber. The chamber
containing the arrays
is incubated for about 6.5 hr at 60C. The arrays are washed for 10 min at 45C
in lxSSC, 0.1%
SDS, and three times for 10 min each at 45C in O.IxSSC, and dried.
Hybridization reactions are performed in absolute or differential
hybridization formats. In
the absolute hybridization format, probe from one sample is hybridized to
array elements, and
signals are detected after hybridization complexes form. Signal strength
correlates with probe
mRNA levels in the sample. In the differential hybridization format,
differential expression of a set
of genes in two biological samples is analyzed. Probes from the two samples
are prepared and
labeled with different labeling moieties. A mixture of the two labeled probes
is hybridized to the
array elements, and signals are examined under conditions in which the
emissions from the two
different labels are individually detectable. Elements on the array that are
hybridized to
substantially equal numbers of probes derived from both biological samples
give a distinct
combined fluorescence (Shalon W095/35505).
Hybridization complexes are detected with a microscope equipped with an Innova
70 mixed
gas 10 W laser (Coherent, Santa Clara CA) capable of generating spectral lines
at 488 nm for
excitation of Cy3 and at 632 nm for excitation of CyS. The excitation laser
light is focused on the
array using a 20X microscope objective (Nikon, Melville NY). The slide
containing the array is
placed on a computer-controlled X-Y stage on the microscope and raster-scanned
past the objective
with a resolution of 20 micrometers. In the differential hybridization format,
the two fluorophores
are sequentially excited by the laser. Emitted light is split, based on
wavelength, into two
photomultiplier tube detectors (PMT 81477, Hamamatsu Photonics Systems,
Bridgewater NJ)
corresponding to the two fluorophores. Appropriate filters positioned between
the array and the
photomultiplier tubes are used to filter the signals. The emission maxima of
the fluorophores used
are 565 nm for Cy3 and 650 nm for CyS. The sensitivity of the scans is
calibrated using the signal
intensity generated by the yeast control mRNAs added to the probe mix. A
specific location on the
array contains a complementary DNA sequence, allowing the intensity of the
signal at that location
to be correlated with a weight ratio of hybridizing species of 1:100,000.
The output of the photomultiplier tube is digitized using a 12-bit RTI-835H
analog-to-
digital (A/D) conversion board (Analog Devices, Norwood MA) installed in an
IBM-compatible PC
computer. The digitized data are displayed as an image where the signal
intensity is mapped using
32
CA 02420101 2003-02-19
WO 02/16595 PCT/USO1/26317
a linear 20-color transformation to a pseudocolor scale ranging from blue (low
signal) to red (high
signal). The data is also analyzed quantitatively. Where two different
fluorophores are excited and
measured simultaneously, the data are first corrected for optical crosstalk
(due to overlapping
emission spectra) between the fluorophores using the emission spectrum for
each fluorophore. A
grid is superimposed over the fluorescence signal image such that the signal
from each spot is
centered in each element of the grid. The fluorescence signal within each
element is then integrated
to obtain a numerical value corresponding to the average intensity of the
signal. The software used
for signal analysis is the GEMTOOLS program (fiicyte Genomics).
VIII Electronic Analysis
BLAST was used to search for identical or related molecules in the GenBank or
LIFESEQ
databases (Incyte Genomics). The product score for human and rat sequences was
calculated as
follows: the BLAST score is multiplied by the % nucleotide identity and the
product is divided by
(5 times the length of the shorter of the two sequences), such that a 100%
alignment over the length
of the shorter sequence gives a product score of 100. The product score takes
into account both the
degree of similarity between two sequences and the length of the sequence
match. For example,
with a product score of 40, the match will be exact within a 1% to 2% error,
and with a product
score of at least 70, the match will be exact. Similar or related molecules
are usually identified by
selecting those which show product scores between 8 and 40.
Electronic northern analysis was performed at a product score of 70 as shown
in Figure 3.
All sequences and cDNA libraries in the LIFESEQ database were categorized by
system,
organ/tissue and cell type. The categories included cardiovascular system,
connective tissue,
digestive system, embryonic structures, endocrine system, exocrine glands,
female and male
genitalia, germ cells, hemic/immune system, liver, musculoskeletal system,
nervous system,
pancreas, respiratory system, sense organs, skin, stomatognathic system,
unclassified/mixed, and
the urinary tract. For each category, the number of libraries in which the
sequence was expressed
were counted and shown over the total number of libraries in that category. In
a non-normalized
library, expression levels of two or more are significant.
IX Complementary Molecules
Molecules complementary to the cDNA, from about 5 (PNA) to about 5000 by
(complement of a cDNA insert), are used to detect or inhibit gene expression.
These molecules are
selected using OLIGO software (Molecular Biology Insights). Detection is
described in Example
VII. To inhibit transcription by preventing promoter binding, the
complementary molecule is
designed to bind to the most unique 5' sequence and includes nucleotides of
the 5' UTR upstream of
the initiation codon of the open reading frame. Complementary molecules
include genomic
sequences (such as enhancers or introns) and are used in "triple helix" base
pairing to compromise
33
CA 02420101 2003-02-19
WO 02/16595 PCT/USO1/26317
the ability of the double helix to open sufficiently for the binding of
polymerases, transcription
factors, or regulatory molecules. To inhibit translation, a complementary
molecule is designed to
prevent ribosomal binding to the mRNA encoding the mammalian protein.
Complementary molecules are placed in expression vectors and used to transform
a cell line
to test efficacy; into an organ, tumor, synovial cavity, or the vascular
system for transient or short
term therapy; or into a stem cell, zygote, or other reproducing lineage for
long term or stable gene
therapy. Transient expression lasts for a month or more with a non-replicating
vector and for three
months or more if appropriate elements for inducing vector replication are
used in the
transformation/expression system.
Stable transformation of appropriate dividing cells with a vector encoding the
complementary molecule produces a transgenic cell line, tissue, or organism
(USPN 4,736,866).
Those cells that assimilate and replicate sufficient quantities of the vector
to allow stable integration
also produce enough complementary molecules to compromise or entirely
eliminate activity of the
cDNA encoding the mammalian protein.
X Expression of I~SRCC
Expression and purification of the mammalian protein are achieved using either
a
mammalian cell expression system or an insect cell expression system. The
pUB6/V5-His vector
system (Invitrogen, Carlsbad CA) is used to express KSRCC in CHO cells. The
vector contains the
selectable bsd gene, multiple cloning sites, the promoter/enhancer sequence
from the human
ubiquitin C gene, a C-terminal V5 epitope for antibody detection with anti-V5
antibodies, and a C-
terminal polyhistidine (6xHis) sequence for rapid purification on PROBOND
resin (Invitrogen).
Transformed cells are selected on media containing blasticidin.
Spodoptera f~iperda (Sf9) insect cells are infected with recombinant
Auto~raphica
californica nuclear polyhedrosis virus (baculovirus). The polyhedrin gene is
replaced with the
mammalian cDNA by homologous recombination and the polyhedrin promoter drives
cDNA
transcription. The protein is synthesized as a fusion protein with 6xhis which
enables purification
as described above. Purified protein is used in the following activity and to
make antibodies
XI Production of Antibodies
KSRCC is purified using polyacrylamide gel electrophoresis and used to
immunize mice or
rabbits. Antibodies are produced using the protocols below. Alternatively, the
amino acid sequence
of KSRCC is analyzed using LASERGENE software (DNASTAR) to determine regions
of high
antigenicity. An antigenic epitope, usually found near the C-terminus or in a
hydrophilic region is
selected, synthesized, and used to raise antibodies. Typically, epitopes of
about 15 residues in
length are produced using an ABI 431A peptide synthesizer (Applied Biosystems)
using Fmoc-
chemistry and coupled to KLH (Sigma-Aldrich) by reaction with N-
maleimidobenzoyl-N-
34
CA 02420101 2003-02-19
WO 02/16595 PCT/USO1/26317
hydroxysuccinimide ester to increase antigenicity.
Rabbits are immunized with the epitope-KLH complex in complete Freund's
adjuvant.
Immunizations are repeated at intervals thereafter in incomplete Freund's
adjuvant. After a
minimum of seven weeks for mouse or twelve weeks for rabbit, antisera are
drawn and tested for
antipeptide activity. Testing involves binding the peptide to plastic,
blocking with 1 % bovine
serum albumin, reacting with rabbit antisera, washing, and reacting with radio-
iodinated goat anti-
rabbit IgG. Methods well known in the art are used to determine antibody titer
and the amount of
complex formation.
XII Purification of Naturally Occurring Protein Using Specific Antibodies
Naturally occurring or recombinant protein is purified by immunoaffmity
chromatography
using antibodies which specifically bind the protein. An immunoaffinity column
is constructed by
covalently coupling the antibody to CNBr-activated SEPHAROSE resin (APB).
Media containing
the protein is passed over the immunoaffmity column, and the column is washed
using high ionic
strength buffers in the presence of detergent to allow preferential absorbance
of the protein. After
coupling, the protein is eluted from the column using a buffer of pH 2-3 or a
high concentration of
urea or thiocyanate ion to disrupt antibody/protein binding, and the protein
is collected.
XIII Screening Molecules for Specific Binding with the cDNA or Protein
The cDNA, or fragments thereof, or the protein, or portions thereof, are
labeled with 32P-
dCTP, Cy3-dCTP, or Cy5-dCTP (APB), or with BIODIPY or FITC (Molecular Probes,
Eugene
OR), respectively. Libraries of candidate molecules or compounds previously
arranged on a
substrate are incubated in the presence of labeled cDNA or protein. After
incubation under
conditions for either a nucleic acid or amino acid sequence, the substrate is
washed, and any
position on the substrate retaining label, which indicates specific binding or
complex formation, is
assayed, and the ligand is identified. Data obtained using different
concentrations of the nucleic
acid or protein are used to calculate affinity between the labeled nucleic
acid or protein and the
bound molecule.
XIV Two-Hybrid Screen
A yeast two-hybrid system, MATCHMAKER LexA Two-Hybrid system (Clontech ,
Laboratories, Palo Alto CA), is used to screen for peptides that bind the
mammalian protein of the
invention. A cDNA encoding the protein is inserted into the multiple cloning
site of a pLexA
vector, ligated, and transformed into E. coli. cDNA, prepared from mRNA, is
inserted into the
multiple cloning site of a pB42AD vector, ligated, and transformed into E.
coli to construct a cDNA
library. The pLexA plasmid and pB42AD-cDNA library constructs are isolated
from E. coli and
used in a 2:1 ratio to co-transform competent yeast EGY48[p8op-lacZ] cells
using a polyethylene
glycol/lithium acetate protocol. Transformed yeast cells are plated on
synthetic dropout (SD) media
CA 02420101 2003-02-19
WO 02/16595 PCT/USO1/26317
lacking histidine (-His), tryptophan (-Trp), and uracil (-Ura), and incubated
at 30C until the colonies
have grown up and are counted. The colonies are pooled in a minimal volume of
lx TE (pH 7.5),
replated on SD/-His/-Leu/-Trp/-Ura media supplemented with 2% galactose (Gal),
1% raffinose
(Raf), and 80 mg/ml 5-bromo-4-chloro-3-indolyl (3-d-galactopyranoside (X-Gal),
and subsequently
examined for growth of blue colonies. Interaction between expressed protein
and cDNA fusion
proteins activates expression of a LEU2 reporter gene in EGY48 and produces
colony growth on
media lacking leucine (-Leu). Interaction also activates expression of 13-
galactosidase from the
p8op-lacZ reporter construct that produces blue color in colonies grown on X-
Gal.
Positive interactions between expressed protein and cDNA fusion proteins are
verified by
isolating individual positive colonies and growing them in SD/-Trp/-Ura liquid
medium for 1 to 2
days at 30C. A sample of the culture is plated on SD/-Trp/-Ura media and
incubated at 30C until
colonies appear. The sample is replica-plated on SD/-Trp/-Ura and SD/-His/-
Trpl-Ura plates.
Colonies that grow on SD containing histidine but not on media lacking
histidine have lost the
pLexA plasmid. Histidine-requiring colonies are grown on SD/Gal/Raf/X-Gal/-
Trp/-Ura, and white
colonies are isolated and propagated. The pB42AD-cDNA plasmid, which contains
a cDNA
encoding a protein that physically interacts with the mammalian protein, is
isolated from the yeast
cells and characterized.
XV KSRCC Assay
AMP-binding activity of KSRCC is determined in a ligand-binding assay using
candidate
ligand molecules in the presence of'ZSI-labeled KSRCC. KSRCC is labeled
with'ZSI Bolton-Hunter
reagent. (See, e.g., Bolton, A.E. and W.M. Hunter (1973) Biochem. J. 133:529-
539.) Candidate
AMP molecules, previously arrayed in the wells of a multi-well plate, are
incubated with the
labeled KSRCC, washed, and any wells with labeled KSRCC complex are assayed.
Data obtained
using different concentrations of KSRCC are used to calculate values for the
number, affinity, and
association of KSRCC with the candidate molecules.
All patents and publications mentioned in the specification are incorporated
by reference
herein. Various modifications and variations of the described method and
system of the invention
will be apparent to those skilled in the art without departing from the scope
and spirit of the
invention. Although the invention has been described in connection with
specific preferred
embodiments, it should be understood that the invention as claimed should not
be unduly limited to
such specific embodiments. Indeed, various modifications of the described
modes for carrying out
the invention that are obvious to those skilled in the field of molecular
biology or related fields are
intended to be within the scope of the following claims.
36
CA 02420101 2003-02-19
WO 02/16595 PCT/USO1/26317
<110> TNCYTE GENOMICS, INC.
WALKER, Michael G.
KRASNOW, Randi E.
<120> KIDNEY-SPECIFIC PROTEIN
<130> PC-0020 OCT
<140> To Be Assigned
<141> Herewith
<150> 09/645,961
<151> 2000-08-24
<160> 16
<170> PERL Program
<210> 1
<211> 577
<212> PRT
<213> Homo Sapiens
<220>
<221> misc_feature
<223> Incyte ID No: 3481942
<400> 1
Met His Trp Leu Arg Lys Val Gln Gly Leu Cys Thr Leu Trp Gly
1 5 10 15
Thr Gln Met Ser Ser Arg Thr Leu Tyr Ile Asn Ser Arg Gln Leu
20 25 30
Val Sex Leu Gln Trp Gly His Gln Glu Val Pro Ala Lys Phe Asn
35 40 45
Phe Ala Ser Asp Val Leu Asp His Trp Ala Asp Met Glu Lys Ala
50 55 60
Gly Lys Arg Leu Pro Ser Pro Ala Leu Trp Trp Val Asn Gly Lys
65 70 75
G1y Lys Glu Leu Met Trp Asn Phe Arg Glu Leu Ser Glu Asn Ser
80 85 90
Gln G1n Ala Ala Asn Val Leu Ser Gly Ala Cys Gly Leu Gln Arg
95 100 105
Gly Asp Arg Val Ala Val Val Leu Pro Arg Val Pro Glu Trp Trp
110 115 120
Leu Val Ile Leu Gly Cys Ile Arg Ala Gly Leu Ile Phe Met Pro
125 130 135
Gly Thr Ile Gln Met Lys Ser Thr Asp Ile Leu Tyr Arg Leu Gln
140 145 150
Met Ser Lys Ala Lys Ala Ile Val Ala Gly Asp Glu Val Ile Gln
155 160 165
Glu Val Asp Thr Val Ala Ser Glu Cys Pro Ser Leu Arg Ile Lys
170 175 180
Leu Leu Val Ser Glu Lys Ser Cys Asp Gly Trp Leu Asn Phe Lys
185 190 195
Lys Leu Leu Asn Glu Ala Ser Thr Thr His His Cys Val Glu Thr
200 205 210
Gly Ser Gln Glu Ala Ser Ala Ile Tyr Phe Thr Ser Gly Thr Ser
215 220 225
Gly Leu Pro Lys Met Ala Glu His Ser Tyr Ser Ser Leu Gly Leu
230 235 240
Lys Ala Lys Met Asp Ala G1y Trp Thr Gly Leu Gln Ala Ser Asp
245 250 255
Ile Met Trp Thr Ile Ser Asp Thr Gly Trp Ile Leu Asn Ile Leu
1/9
CA 02420101 2003-02-19
WO 02/16595 PCT/USO1/26317
260 265 270
Gly Ser Leu Leu Glu Ser Trp Thr Leu Gly Ala Cys Thr Phe Val
275 280 285
His Leu Leu Pro Lys Phe Asp Pro Leu Val Ile Leu Lys Thr Leu
290 295 300
Ser Ser Tyr Pro Ile Lys Ser Met Met Gly Ala Pro Ile Val Tyr
305 310 315
Arg Met Leu Leu Gln Gln Asp Leu Ser Ser Tyr Lys Phe Pro His
320 325 330
Leu Gln Asn Cys Leu Ala Gly Gly Glu Ser Leu Leu Pro Glu Thr
335 340 345
Leu Glu Asn Trp Arg Ala Gln Thr G1y Leu Asp Ile Arg Glu Phe
350 355 360
Tyr Gly Gln Thr Glu Thr Gly Leu Thr Cys Met Val Ser Lys Thr
365 370 375
Met Lys Ile Lys Pro Gly Tyr Met Gly Thr A1a Ala Ser Cys Tyr
380 385 390
Asp Val Gln Val Ile Asp Asp Lys Gly Asn Val Leu Pro Pro Gly
395 400 405
Thr Glu G1y Asp Ile Gly Ile Arg Val Lys Pro Ile Arg Pro Ile
410 415 420
Gly Ile Phe Ser Gly Tyr Val Glu Asn Pro Asp Lys Thr Ala Ala
425 430 435
Asn Ile Arg Gly Asp Phe Trp Leu Leu Gly Asp Arg G1y Ile Lys
440 445 450
Asp Glu Asp Gly Tyr Phe Gln Phe'Met Gly Arg Ala Asp Asp Ile
455 460 465
Tle Asn Ser Ser Gly Tyr Arg Ile Gly Pro Ser Glu Val Glu Asn
470 475 480
Ala Leu Met Lys His Pro Ala Val Val Glu Thr Ala Val Ile Ser
485 490 495
Ser Pro Asp Pro Val Arg Gly Glu Val Val Lys A1a Phe Val Ile
500 505 510
Leu Ala Ser Gln Phe Leu Ser His Asp Pro Glu Gln Leu Thr Lys
515 520 525
Glu Leu Gln Gln His Val Lys Ser Val Thr Ala Pro Tyr Lys Tyr
530 535 540
Pro Arg Lys Ile Glu Phe Val Leu Asn Leu Pro Lys Thr Val Thr
545 550 555
Gly Lys I1e Gln Arg Thr Lys Leu Arg Asp Lys Glu Trp Lys Met
560 565 570
Ser Gly Lys Ala Arg Ala Gln
575
<210> 2
<211> 2054
<212> DNA
<213> Homo Sapiens
<220>
<221> misc_feature
<223> Incyte ID No: 3481942
<400> 2
gtgctctctt ccaaggctgt aggagttctg gagctgctgg ctggagagga gggtggacga 60
agctctctct agaaagacat cctgagagga cttggcaggc ctgaatatgc attggctgcg 120
aaaagttcag ggactttgca ccctgtgggg tactcagatg tccagccgca ctctctacat 180
taatagtagg caactggtgt ccctgcagtg gggccaccag gaagtgccgg ccaagtttaa 240
ctttgctagt gatgtgttgg atcactgggc tgacatggag aaggctggca agcgactccc 300
aagcccagcc ctgtggtggg tgaatgggaa ggggaaggaa ttaatgtgga atttcagaga 360
actgagtgaa aacagccagc aggcagccaa cgtcctctcg ggagcctgtg gcctgcagcg 420
tggggatcgt gtggcagtgg tgctgccccg agtgcctgag tggtggctgg tgatcctggg 480
ctgcattcga gcaggtctca tctttatgcc tggaaccatc cagatgaaat ccactgacat 540
actgtatagg ttgcagatgt ctaaggccaa ggctattgtt gctggggatg aagtcatcca 600
agaagtggac acagtggcat ctgaatgtcc ttctctgaga attaagctac tggtgtctga 660
2/9
CA 02420101 2003-02-19
WO 02/16595 PCT/USO1/26317
gaaaagctgc gatgggtggc tgaacttcaa gaaactacta aatgaggcat ccaccactca 720
tcactgtgtg gagactggaa gccaggaagc atctgccatc tacttcacta gtgggaccag 780
tggtcttccc aagatggcag aacattccta ctcgagcctg ggcctcaagg ccaagatgga 840
tgctggttgg acaggcctgc aagcctctga tataatgtgg accatatcag acacaggttg 900
gatactgaac atcttgggct cacttttgga atcttggaca ttaggagcat gcacatttgt 960
tcatctcttg ccaaagtttg acccactggt tattctaaag acactctcca gttatccaat 1020
caagagtatg atgggtgccc ctattgttta ccggatgttg ctacagcagg atctttccag 1080
ttacaagttc ccccatctac agaactgcct cgctggaggg gagtcccttc ttccagaaac 1140
tctggagaac tggagggccc agacaggact ggacatccga gaattctatg gccagacaga 1200
aacgggatta acttgcatgg tttccaagac aatgaaaatc aaaccaggat acatgggaac 1260
ggctgcttcc tgttatgatg tacaggttat agatgataag ggcaacgtcc tgccccccgg 1320
cacagaagga gacattggca tcagggtcaa acccatcagg cctataggca tcttctctgg 1380
ctatgtggaa aatcccgaca agacagcagc caacattcga ggagactttt ggctccttgg 1440
agaccgggga atcaaagatg aagatgggta tttccagttt atgggacggg cagatgatat 1500
cattaactcc agcgggtacc ggattggacc ctcggaggta gagaatgcac tgatgaagca 1560
ccctgctgtg gttgagacgg ctgtgatcag cagcccagac cccgtccgag gagaggtggt 1620
gaaggcattt gtgatactgg cctcgcagtt cctatcccat gacccagaac agctcaccaa 1680
ggagctgcag cagcatgtga agtcagtgac agccccatac aagtacccaa gaaagataga 1740
gtttgtcttg aacctgccca agactgtcac agggaaaatt caacgaaaca aacttcgaga 1800
caaggagtgg aagatgtccg gaaaagcccg tgcgcagtga ggcgtctagg agacattcat 1860
ttggattccc ctcttctttc tctttctttt ccctttgggc ccttggcctt actatgatga 1920
tatgagattc tttatgaaag aacatgaatg taagttttgt cttgccctgg ttattagcac 1980
aaaacattac tatgttagat attgaaataa gaaagggaag gaatgagaga gagtgaaaag 2040
gagagggtaa saga 2054
<210> 3
<211> 521
<212> DNA
<213> Homo Sapiens
<220>
<221> misc_feature
<223> Incyte ID No: 3481942CB1
<400> 3
tctggagctg ctggctggag aggagggtgg acgaagctct ctctagaaag acatcctgag 60
aggacttggc aggcctgaac atgcattggc tgcgaaaagt tcagggactt tgcaccctgt 120
ggggtactca gatgtccagc cgcactctct acattaatag taggcaactg gtgtccctgc 180
agtggggcca ccaggaagtg ccggccaagt ttaactttgc tagtgatgtg ttggatcact 240
gggctgacat ggagaaggct ggcaagcgac tcccaagccc agccctgtgg tgggtgaatg 300
ggaaggggaa ggaattaatg tggaatttca gagaactgag tgaaaacagc cagcaggcag 360
ccaacgtcct ctcgggagcc tgtggcctgc agcgtgggga tcgtgtggca gtggtgctgc 420
cccgagtgcc tgagtggtgg ctggtgatcc tgggctgcat tcgagcaggg ctcatcttta 480
tgcctggaac catcagatga gatccactga catactgtat a 521
<210> 4
<211> 251
<212> DNA
<213> Homo Sapiens
<220>
<221> misc_feature
<223> Incyte ID No: 5519427H1
<220>
<221> unsure
<222> 25, 55
<223> a, t, c, g, or other
<400> 4
gtgctctctt ccaaggctgt aggangttct ggagctgctg gctggagagg agggnggacg 60
aagctctctc cagaaagaca tcctgagagg acttggcagg cctgaacatg cattggctgc 120
gaaaagttca gggactttgc accctgtggg gtactcagat gtccagccgc actctctaca 180
ttaatagtag gcaactggtg tccctgcagt ggggccacca ggaattccgg ccaagtttaa 240
3/9
CA 02420101 2003-02-19
WO 02/16595 PCT/USO1/26317
252
ctttgctagt g
<210> 5
<211> 594
<212> DNA
<213> Homo sapiens
<220>
<221> misc_feature
<223> Incyte ID No: 5390984F8
<400> 5
ggctgtagga gttctggagc tgctggctgg agaggagggt ggatgaagct ctctctagaa 60
agacatcctg agaggacttg gcaggcctga atatgcattg gctgcgaaaa gttcagggac 120
tttgcaccct gtggggtact cagatgtcca gccgcactct ctacattaat agtaggcaac 180
tggtgtccct gcagtggggc caccaggaag tgccggccaa gtttaacttt gctagtgatg 240
tgttggatca ctgggctgac atggagaagg ctggcaagcg actcccaagc ccagccctgt 300
ggtgggtgaa tgggaagggg aaggaattaa tgtggaattt cagagaactg agtgaaaaca 360
gccagcaggc agccaacgtc ctctcgggag cctgtggcct gcagcgtggg gatcgtgtgg 420
cagtggtgct gccccgagtg cctgagtggt ggctggtgat cctgggctgc attcgagcag 480
gtctcatctt tatgcctgga accatccaga tgaaatccac tgacatactg tataggttgc 540
agatgtctaa ggccaaggct attgttgctg gggatgaagt catccaagaa gtgg 594
<210> 6
<211> 444
<212> DNA
<213> Homo Sapiens
<220>
<221> misc_feature
<223> Incyte ID No: 433223079
<400> 6
gttgatctaa agacctttag aataaccagt gggtcaaact ttggcaagag atgaacaaat 60
gtgcatgctc ctaatgtcca agattccaaa agtgagccca agatgttcag tatccaacct 120
gtgtctgata tggtccacat tatatcagag gcttgcaggc ctgtccaacc agcatccatc 180
ttggccttga ggcccaggct cgagtaggaa tgttctgcca tcttgggaag accactggtc 240
ccactagtga agtagatggc agatgcttcc tggcttccag tctccacaca gtgatgagtg 300
gtggatgcct catttagtag tttcttgaag ttcagccacc catcgcagct tttctcagac 360
accagtagct taattctcag agaaggacat tcagatgcca ctgtgtccac ttcttggatg 420
acttcatccc cagcaacaat agcc 444
<210> 7
<211> 535
<212> DNA
<213> Homo Sapiens
<220>
<221> misc_feature
<223> Incyte ID No: 76083581
<400> 7
atgtggacca tatcagacac aggttggata ctgaacatct tgggctcact tttggaatct 60
tggacattag gagcatgcac atttgttcat ctcttgccaa agtttgaccc actggttatt 120
ctaaagacac tctccagtta tccaatcaag agtatgatgg gtgcccctat tgtttaccgg 180
atgttgctac agcaggatct ttccagttac aagttccccc atctacagaa ctgcctcgct 240
ggaggggagt cccttcttcc agaaactctg gagaactgga gggcccagac aggactggac 300
atccgagaat tctatggcca gacagaaacg ggattaactt gcatggtttc caagacaatg 360
aaaatcaaac caggatacat gggaacggct gcttcctgtt atgatgtaca ggttatagat 420
gataagggca acgtcctgcc ccccggcaca gaaggagaca ttggcatcag ggtcaaaccc 480
atcaggccta taggcatctt ctctggctat gtggtatcat ccccatgatc tatgc 535
<210> 8
<211> 487
4/9
CA 02420101 2003-02-19
WO 02/16595 PCT/USO1/26317
<212> DNA
<213> Homo sapiens
<220>
<221> misc_feature
<223> Incyte ID No: 76254081
<400> 8
agttccccca tctacagaac tgcctcgctg gaggggagtc ccttcttcca gaaactctgg 60
agaactggag ggcccagaca ggactggaca tccgagaatt ctatggccag acagaaacgg 120
gattaacttg catggtttcc aagacaatga aaatcaaacc aggatacatg ggaacggctg 180
cttcctgtta tgatgtacag gttatagatg ataagggcaa cgtcctgccc cccggcacag 240
aaggagacat tggcatcagg gtcaaaccca tcaggcctat aggcatcttc tctggctatg 300
tggaaaatcc cgacaagaca gcagccaaca ttcgaggaga cttttggctc cttggagacc 360
ggggaatcaa agatgaagat gggtatttcc agtttatggg acgggcagat gatatcatta 420
actccagcgg gtacggattg gaccctcgga ggtagagaat gcactgatga agcaccctgc 480
tgtggtt
487
<210> 9
<211> 488
<212> DNA
<213> Homo sapiens
<220>
<221> misc_feature
<223> Incyte ID No: 209942086
<400> 9
caacattcga ggagactttt ggctccttgg agaccgggga atcaaagatg aagatgggta 60
tttccagttt atgggacggg cagatgatat cattaactcc agcgggtacc ggattggacc 120
ctcggaggta gagaatgcac tgatgaagca ccctgctgtg gttgagacgg ctgtgatcag 180
cagcccagac cccgtccgag gagaggtggt gaaggcattt gtgatactgg cctcgcagtt 240
cctatcccat gacccagaac agctcaccaa ggagctgcag cagcatgtga agtcagtgac 300
agccccatac aagtacccaa gaaagataga gtttgtcttg aacctgccca agactgtcac 360
agggaaaatt caacgaacca aacttcgaga caaggagtgg aagatgtccg gaaaagcccg 420
tgcgcagtga ggcgtctagg agacattcat ttggattccc ctcttctttc tctttctttt 480
ccccttgg 488
<210> 10
<211> 528
<212> DNA
<213> Homo Sapiens
<220>
<221> misc_feature
<223> Incyte ID No: 7695905J1
<400> 10
gaggtggtga aggcatttgt gatactggcc tcgcagttcc tatcccatga cccagaacag 60
ctcaccaagg agctgcagca gcatgtgaag tcagtgacag ccccatacaa gtacccaaga 120
aagatagagt ttgtcttgaa cctgcccaag actgtcacag ggaaaattca acgaaccaaa 180
cttcgagaca aggagtggaa gatgtccgga aaagcccgtg cgcagtgagg cgtctaggag 240
acattcattt ggattcccct cttctttctc tttcttttcc ctttgggccc ttggccttac 300
tatgatgata tgagattctt tatgaaagaa catgaatgta agttttgtct tgccctggtt 360
attagcacaa aacattacta tgttagatat tgaaataaga aagggaagga atgagagaga 420
gtgaaaagga gagggtaaca gaaaaagagg aaagaaaagt aagtcaggga aatattaaaa 480
actgcaagag aaagcaattg aaaaagaaat aaagtaggga gggaaggc 528
<210> 11
<211> 310
<212> DNA
<213> Rattus norvegicus
<220>
5/9
CA 02420101 2003-02-19
WO 02/16595 PCT/USO1/26317
<221> misc_feature
<223> Incyte ID No: 701648693H1
<400> 11
gtttcaggga gctgagggac ctcagccgcc gtgctgccaa cgtctttgag cagacttgtg 60
gcctgcagca tggagatcgc ctggccttga ttctgcctcg agtgccggag tggtggctgg 120
tgacagtggg ctgcattcga acaggagtca tCttCattCC tgggactgcc caaatgaaag 180
ccaaggatat tctctaccga atacaaatgt ctcaagccaa agccattgtg accacagaca 240
gccttgttcc agaggtggaa tctgtggctt ctgagtgtcc tggtctgaaa accaagatag 300
tggtgtctga 310
<210> 12
<211> 358
<212> DNA
<213> Rattus norvegicus
<220>
<221> misc_feature
<223> Incyte ID No: 212451 Rn.1
<400> 12
atcccccatt catcagtgtg tggagactgt aagccaagaa tcagctgcca tctatttcac 60
tagcgggacc agtggccctc ccaagatggc agagcactcc cactgcagcc tgggcctcaa 120
ggccaagatg gatgctggct ggacaggact gggaccttct gacacaatgt ggaccatctc 180
agacacaggc tggatattaa acattttagg gtcatttctg gaaccttggg tattgggaac 240
gtgcatattt gtccatcttt tgccaaagtt tgatccacaa actgttctaa aggtgctttc 300
cagctacccc atcaataccc tgctgggtgc ccccctcatt taccggatgt tgctacaa 358
<210> 13
<211> 1428
<212> DNA
<213> Rattus norvegicus
<220>
<221> misc_feature
<223> Incyte TD No: 202264 Rn.1
<400> 13
attcgaaacg aggaacagga tctttccagt tacaagttcc cacatctgca tagctgcttc 60
agtggaggag agaccctcct cccggagact ctggagagtt ggcaagccaa gacaggactg 120
ggaatccgag aaatctatgg ccagacagaa acgggaatta cctgcagagt ttctaggaca 180
atgaaagtca aaccaggcta cctgggaaca gccattgtcc cttatgatgt ccaggtcata 240
gatgagcagg gcaatgtcct gccccctggc aaggaaggag acatggctct cagggtgaag 300
cccatcaggc ctataggcat gttctctgga tatgtggaca atcccaagaa gacacaggct 360
aatattcgag gagacttttg gcttctggga gaccggggaa ttaaggatac agaagggtat 420
ttccacttca tgggacggac agacgatatc attaattcca gtgggtaccg gattgggacc 480
ttccgaggtg gagaatgcac tgatggaaca tcctgccgtg gttgaaacag ctgtgatcag 540
cagcccagac cctatcagaa gagaggtggt gaaggcattt gtggttctag cccctgagtt 600
cctgtcccat gaccaagacc agctcaccaa ggttcttcag gaacacgtga agtcagtgac 660
agcaccctac aagtacccca ggaaggtgga gtttgtctta gacctgccca agaccatcat 720
caggaaaaat tgagcgagct aaaccttcga gccaaggaat ggaaaacatc aggataagcc 780
caggcccaag tgagactccc aaggctcttg ctctgtctgt tcccaatcct tttcttatga 840
ctccttagtc ttcctatagc aatatgaaat tattctatgt agaggcatgt atgtgattta 900
ggtctttgct tggtttattg actactagaa cagatgacac gtttctatgc taaaagatag 960
tacagcatag agggaataag aggtttgaag tctagaaaag gctaaaacag caaaggggaa 1020
agaaattgcc aattcatgtg gtcaggaatg gggtagagag aaggggatat agcagaatga 1080
ggagaagaag agaagaatga gaatggaatc tggagagaaa tagtataaaa gggattgatg 1140
tagaaggaca ctagtgtcat tcgaccaaga ttgtgtcaat ccttagggac atcatgaccc 1200
aaaggagaat ggaaatacca cttacagagc atgacccagg gacaactgat tatacacttg 1260
tcatgagtcc atataccccg cgtacatgag ccatcttaca ttcaataatg ggtgacatct 1320
gcatggctca tcattcatgc accccataaa tgtatcacca ttcctgtagc tcacattttt 1380
tgttctagct ttatttcctt gaattaataa taaaatcact tcaaaatc 1428
<210> 14
6/9
CA 02420101 2003-02-19
WO 02/16595 PCT/USO1/26317
<211> 464
<212> DNA
<213> Rattus norvegicus
<220>
<221> misc_feature
<223> Incyte ID No: 70183634671
<400> 14
tcttggcacc gtccgctttc ctcttctctc taccccttcc tgaccacatg aattggcaat 60
ttctttccct tgctgtttta gccttttctg acttcaaacc tcttattccc tctttctttc 120
ttcttttagc atgaaacgtg tcattgttct gtgtcaataa ccaagcaaag acctaaatca 180
catacatgcc tctacatgaa taatttcata ttgctatagg aagactaagg agtcataaga 240
aaaggattgg gaaagacaga gcaagagcct tgggagtctc actgggcctg ggcttatcct 300
gatgttttcc attccttggc tcgaagttta gctcgctcaa tttttcctgt gatggtcttg 360
ggcaggtcta agacaaactc caccttcctg gggtacttgt agggtgctgt cactgacttc 420
acgtgttcct gaagaacctt ggtgagctgg tcttggtcat ggga 464
<210> 15
<211> 572
<212> PRT
<213> Rattus norvegicus
<220>
<221> misc_feature
<223> Incyte ID No: g3127193
<400> 15
Met His Trp Leu Trp Lys I1e Pro Arg Leu Cys Thr Phe Trp Gly
1 5 10 15
Thr Glu Met Phe His Arg Ser Phe His Met Asn Ile Lys Lys Leu
20 25 30
Met Pro Ile Gln Trp Gly His Gln Glu Val Pro Ala Lys Phe Asn
35 40 45
Phe Ala Ser Asp Val Ile Asp His Trp Ala Ser Leu Glu Lys A1a
50 55 60
Gly Lys Arg Ser Pro Gly Pro Ala Leu Trp Trp Met Asn Gly Ser
65 70 75
Gly Glu G1u Leu Lys Trp Asn Phe Arg Glu Leu Ser Glu Ile Ser
80 85 90
Lys Gln Thr Ala Asn Val Leu Thr Gly Ala Cys Gly Leu Gln Arg
95 100 105
Gly Asp Arg Val Ala Val Val Leu Pro Arg Val Pro Glu Trp Trp
110 115 120
Leu Val Thr Leu Gly Cys Met Arg Ser Gly Leu Val Phe Met Pro
125 230 135
Gly Thr Thr Gln Met Lys Ser Thr Asp Ile Leu Tyr Arg Leu Gln
140 145 150
Ser Ser Lys Ala Arg Ala Ile Val Ala Gly Asp Glu Val Val Gln
155 160 165
Glu Val Asp Ala Val Ala Pro Asp Cys Ser Phe Leu Lys Ile Lys
170 175 180
Leu Leu Va1 Ser Glu Lys Asn Arg Glu Gly Trp Leu Asn Phe Lys
185 190 195
Ala Leu Leu Lys Asp Ala Ser Pro Ile His Gln Cys Val G1u Thr
200 205 210
Val Ser Gln Glu Ser Ala Ala Ile Tyr Phe Thr Ser Gly Thr Ser
215 220 225
Gly Pro Pro Lys Met Ala Glu His Ser His Cys Ser Leu Gly Leu
230 235 240
Lys Ala Lys Met Asp Ala Gly Trp Thr Gly Leu G1y Pro Ser Asp
245 250 255
Thr Met Trp Thr I1e Ser Asp Thr Gly Trp Ile Leu Asn Ile Leu
260 265 270
7/9
CA 02420101 2003-02-19
WO 02/16595 PCT/USO1/26317
Gly Ser Phe Leu Glu Pro Trp Val Leu Gly Thr Cys Ile Phe Val
275 280 285
His Leu Leu Pro Lys Phe Asp Pro Gln Thr Val Leu Lys Val Leu
290 295 300
Ser Ser Tyr Pro Ile Asn Thr Leu Leu Gly Ala Pro Leu Ile Tyr
305 310 315
Arg Met Leu Leu Gln Gln Asp Leu Ser Ser Tyr Lys Phe Pro His
320 325 330
Leu His Ser Cys Phe Ser Gly Gly Glu Thr Leu Leu Pro Glu Thr
335 340 345
Leu Glu Ser Trp Lys Ala Lys Thr Gly Leu Glu Ile Arg Glu Ile
350 355 360
Tyr Gly G1n Thr Glu Thr Gly Ile Thr Cys Arg Val Ser Arg Thr
365 370 375
Met Lys Val Lys Pro Gly Tyr Leu Gly Thr Ala Ile Val Pro Tyr
380 385 390
Asp Val Gln Val Ile Asp Glu Gln Gly Asn Val Leu Pro Pro Gly
395 400 405
Lys Glu Gly Asp Met Ala Leu Arg Val Lys Pro Ile Arg Pro Ile
410 415 420
Gly Met Phe Ser Gly Tyr Val Asp Asn Pro Lys Lys Thr Gln Ala
425 430 435
Asn I1e Arg Gly Asp Phe Trp Leu Leu Gly Asp Arg Gly Ile Lys
440 445 450
Asp Thr Glu Gly Tyr Phe His Phe Met Gly Arg Thr Asp Asp Ile
455 460 465
Ile Asn Ser Ser Gly Tyr Arg Ile Gly Pro Ser Glu Val Glu Asn
470 475 480
Ala Leu Met Glu His Pro Ala Val Val Glu Thr Ala Val Ile Ser
485 490 495
Ser Pro Asp Pro Ile Arg Arg Glu Val Val Lys Ala Phe Val Val
500 505 510
Leu Ala Pro Glu Phe Leu Ser His Asp Gln Asp Gln Leu Thr Lys
515 520 525
Val Leu Gln Glu His Val Lys Ser Val Thr Ala Pro Tyr Lys Tyr
530 535 540
Pro Arg Lys Val Glu Phe Val Leu Asp Leu Pro Lys Thr I1e Thr
545 550 555
Gly Lys Ile Glu Arg Ala Lys Leu Arg Ala Lys Glu Trp Lys Thr
560 565 570
Ser Gly
<210> 16
<211> 207
<212> PRT
<213> Homo sapiens
<220>
<221> misc_feature
<223> Incyte ID No: 83219339
<400> 16
Met Val Ser Lys Thr Met Lys Ile Lys Pro Gly Tyr Met Gly Thr
1 5 10 15
Ala Ala Ser Cys Tyr Asp Val Gln Ile Ile Asp Asp Lys Gly Asn
20 25 30
Val Leu Pro Pro Gly Thr Glu Gly Asp Ile G1y Ile Arg Val Lys
35 40 45
Pro Ile Arg Pro Ile Gly Ile Phe Ser Gly Tyr Val Asp Asn Pro
50 55 60
Asp Lys Thr Ala Ala Asn Ile Arg Gly Asp Phe Trp Leu Leu Gly
65 70 75
Asp Arg Gly Ile Lys Asp Glu Asp Gly Tyr Phe Gln Phe Met Gly
80 85 90
8/9
CA 02420101 2003-02-19
WO 02/16595 PCT/USO1/26317
Arg Ala Asp Asp Ile I1e Asn Ser Ser Gly Tyr Arg Ile Gly Pro
95 100 105
Ser Glu Val Glu Asn Ala Leu Met Glu His Pro Ala Val Val Glu
110 115 120
Thr Ala Val Ile Ser Ser Pro Asp Pro Val Arg Gly Glu Val Val
125 130 135
Lys A1a Phe Val Val Leu Ala Leu Gln Phe Leu Ser His Asp Pro
140 145 150
Glu Gln Leu Thr Lys Glu Leu Gln Gln His Val Lys Ser Val Thr
155 160 165
Ala Pro Tyr Lys Tyr Pro Arg Lys Ile Glu Phe Val Leu Asn Leu
170 175 180
Pro Lys Thr-Val Thr Gly Lys Ile Gln Arg Ala Lys Leu Arg Asp
185 190 195
Lys Glu Trp Lys Met Ser Gly Lys A1a Arg Ala Gln
200 205
9/9