Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
Treatment of Inflammatory Bowel Disease Using Growth Factors
FIELD OF THE INVENTION
The present invention generally relates to methods of treatment of
inflammatory
conditions in the intestinal tract of mammals using growth factor related
polypeptides. More
specifically, the polypeptides employed in the methods of the invention are
related to a
member of the fibroblast growth factor family and to a member of the platelet
derived growth
factor family.
BACKGROUND OF THE INVENTION
Inflammatory bowel disease comprises two distinct subsets: ulcerative colitis
and
Crohn's disease. In 1999, approximately 1.7 million people were diagnosed with
this
debilitating disease. Satisfactory treatment of IBD is an unmet medical need,
as existing
therapeutics have not been successful in curtailing the disease and preventing
surgeries. Up to
forty percent of all ulcerative colitis patients undergo surgery, which
typically includes the
removal of part of the large intestine or a full colostomy. Such surgery is
not curative for
Crohn's disease, as 75% of all patients undergo at least one surgery in their
lifetime, and up to
90% of these patients require additional surgeries. Consequently a therapeutic
that can
successfully treat inflammatory bowel disease will have the beneficial effects
of improving a
patient's quality of life, while potentially saving the healthcare system
millions of dollars in
costs associated with invasive surgical procedures.
SUMMARY OF THE INVENTION
The present invention is based upon methods of treating inflammatory
conditions in
the intestinal tract of mammals using growth factor related polypeptides.
Methods of using
fibroblast growth factor-CX (FGF-CX) polynucleotide sequences and the FGF-CX
polypeptides encoded by such nucleic acid sequences, or variants, fragments
and homologs
thereof, are claimed in the invention. Similarly, methods of using FCTRX
polynucleotide
sequences and the FCTRX polypeptides encoded by such nucleic acid sequences,
or variants,
fragments and homologs thereof, alone or in combination, are also claimed in
the invention.
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
FCTRX collectively refers to any of six variant FCTRX seduences, designated
FCTRl,
FCTR2, FCTR3, FCTR4, FCTRS and FCTR6.
In one aspect, the invention provides a method of promoting the growth of a
population
of cells whereby the cells are placed into contact with a composition
including a FGF-CX or
FCTRX polypeptide, or a composition including FGF-CX and FCTRX polypeptides.
In
another aspect, the invention provides a method of treating an inflammatory
pathology in a
subject, whereby an FGF-CX or an FCTRX polypeptide composition is administered
to the
subject. In yet another aspect, the invention provides a method of delaying
the onset of an
inflammatory pathology in a subject, whereby a composition including a FGF-CX
or FCTRX
polypeptide, or a composition including FGF-CX and FCTRX polypeptides, is
administered to
the subject. In a further aspect, the invention provides a method of
ameliorating an
inflammatory pathology in a subject, whereby a composition including a FGF-CX
or FCTRX
polypeptide, or a composition including FGF-CX and FCTRX polypeptides, is
administered to
the subj ect.
, In one embodiment, the subject is a mammal. In another embodiment, the
subject is
human. In yet another embodiment, the inflammatory pathology is inflammatory
bowel
disease, an inflammatory condition occurring in the colon, an inflammatory
condition
occurring in the small intestine, or Crohn's disease. In yet another
embodiment, the FGF-CX
polypeptide is given by SEQ ID N0:2, or a variant, deletion mutant, or a
variant of the
deletion mutant thereof, wherein up to 15% of the residues of either variant
are changed
according to a conservative amino acid substitution. In still yet another
embodiment, the
FCTRX polypeptide is given by any one of SEQ ID NOS:4, 6, 8, 10, 12, and 14,
or a variant,
deletion mutant, variant of the deletion mutant, p35 form, or a variant of the
p35 form thereof,
wherein up to 15% of the residues of any variant are changed according to a
conservative
amino acid substitution. In yet a further embodiment, the polypeptide
composition is
administered intravenously or subcutaneously.
The invention further provides a method of preparing a pharmaceutical
composition,
whereby a polypeptide effective in treating an inflammatory pathology is
combined with a
pharmaceutically acceptable Garner.
In one embodiment, the pharmaceutical composition is suitable for intravenous,
or
subcutaneous administration to the subject. In another embodiment, the
polypeptide is FGF-
CX. In yet another embodiment, the FGF-CX polypeptide is given by SEQ ID N0:2,
or a
2
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
variant, deletion mutant, or a variant of the deletion mutant thereof, wherein
up to 15% of the
residues of either variant are changed according to a conservative amino acid
substitution. In
a further embodiment, the polypeptide is FCTRX. In yet a further embodiment,
the FCTRX
polypeptide is given by any one of SEQ m NOS: 4, 6, 8, 10, 12, and 14, or a
variant, deletion
mutant, variant of the deletion mutant, p35 form, or a variant of the p35 form
thereof, wherein
up to 15% of the residues of any variant are changed according to a
conservative amino acid
substitution. In yet a further embodiment, the inflammatory pathology is
inflammatory bowel
disease, an inflammatory condition occurring in the colon, an inflammatory
condition
occurring in the small intestine, or Crohn's disease.
Contemplated disorders within the invention include pathology such as
inflammatory
conditions in the gastrointestinal tract, including but not limited to
inflammatory bowel disease
such as ulcerative colitis and Crohn's disease, growth and proliferative
diseases such as
cancer, angiogenesis, atherosclerotic plaques, collagen formation, cartilage
and bone
formation, cardiovascular and fibrotic diseases and diabetic ulcers. In
addition, FCTRX
nucleic acids and their encoded polypeptides will be therapeutically useful
for the prevention
of aneurysms and the acceleration of wound closure through gene therapy.
Furthermore,
FCTRX nucleic acids and their encoded polypeptides can be utilized to
stimulate cellular
growth. wound healing, neovascularization and tissue growth, and similar
tissue regeneration
needs. More specifically, a FCTRX nucleic acid or polypeptide may be useful in
treatment of
anemia and leukopenia, intestinal tract sensitivity and baldness. Treatment of
such conditions
may be indicated, e. g., in patients having undergone radiation or
chemotherapy, wherein
treatment would minimize any hyperproliferative side effects.
Unless otherwise defined, all technical and scientific terms used herein have
the
same meaning as commonly understood by one of ordinary skill in the art to
which this
invention belongs. Although methods and materials similar or equivalent to
those described
herein can be used in the practice or testing of the present invention,
suitable methods and
materials are described below. All publications, patent applications, patents,
and other
references mentioned herein are incorporated by reference in their entirety.
In the case of
conflict, the present specification, including definitions, will control. In
addition, the
materials, methods, and examples are illustrative only and not intended to be
limiting. Other
features and advantages of the invention will be apparent from the following
detailed
description and claims.
3
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
Other features and advantages of the invention will be apparent from the
following
detailed description and claims.
BRIEF DESCRIPTION OF THE DRAWINGS
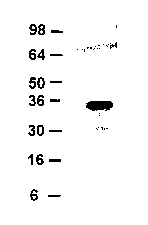
FIG. 1 shows a Western analysis of FGF-CX protein secreted by 293 cells.
FIG. 2 shows a Western analysis of FGF-CX protein expressed in E. coli cells.
FIG. 3 shows a Western analysis of FGF-CX. Samples from 293 cells (Panel A) or
NIH 3T3 cells (Panel B) transiently transfected with the indicated construct
were examined by
Western analysis using anti-VS antibody. CM=conditioned media, SE=suramin-
extracted
conditioned media. Molecular mass markers are indicated on the left.
FIG. 4 presents an image of a Coomassie Blue stained SDS-PAGE gel of purified
samples of FGF-CX prepared under reducing and nonreducing conditions.
FIG. 5 provides the results of a dose titration growth experiment carried out
using 786-
0 human renal carcinoma cells. In this experiment incorporation of
bromodeoxyuridine
induced by varying amounts of FGF-CX (designated in FIGS as 20858) was
determined.
FIG. 6 shows the results of experiments assessing the receptor binding
specificity of
FGF-CX. NIH 3T3 cells were serum-starved, incubated with the indicated growth
factor
(square=PDGF-BB; triangle=aFGF; circle=FGF-CX) either alone or together with
the
indicated soluble FGFR, and analyzed by a BrdU incorporation assay.
Experiments were
performed in triplicate and are represented as the percent BrdU increase in
incorporation of
BrdU relative to cells receiving the growth factor alone.
FIG. 7 shows an image of a Coomassie Blue stained SDS-PAGE gel of the arginine
supernatant obtained when plasmid pET24a- FGF20X-de154-codon was expressed in
E.coli
strain BL21 (DE3).
FIG. 8 displays the biological activity of a truncated form of recombinant FGF-
CX
(denoted by (dl-23)FGF20 in the FIG.) as represented by its effects on DNA
synthesis,
compared to that of full length FGF-CX (denoted FGF20 in the FIG.). NIH 3T3
mouse
fibroblasts were serum-starved, incubated with the indicated factor for 18 hr,
and analyzed by
a BrdU incorporation assay.
FIG. 9 is a representation of a Western blot of a 30664188.m99 protein
expressed in E.
coli cells.
4
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
FIG. 10 is a representation of a Western blot of a 30664188.m99 protein
secreted by
human 293 cells.
FIG. 11. Panel A is a schematic representation of a scheme for the recombinant
production, purification and apparent molecular weight of a mature form of the
protein of
clone 30664188Ø99. Panel B includes representations of two Western blot
analyses showing
expression of a 30664188Øm99 polypeptide.
FIG. 12 is a graph showing incorporation ofBrdU into NIH 3T3 cells and CCD-
1070
cells in response to various treatments.
FIG. 13 is a graph showing proliferation of NIH 3T3 5-24 cells in response to
various
treatments.
FIG. 14 is a graph showing cell number in NIH 3T3 cells exposed to a mock
treatment
or 30664188.
FIG. 15 is a depiction of a photomicrograph showing cell density and cell
morphology
of NIH 3T3 cells in response to treatment with pCEP4sec CM or 30664188
protein.
FIG. 16 is a depiction of a photomicrograph showing changes in cell number in
NHost
osteoblast cells in response to various treatments.
FIG. 17. Panel A is a representation of a western blot of 30664188.m99
expressed by
HEK 293 cells cultured in the absence of serum. Panel B is a representation of
SDS-PAGE
30664188.m99 protein expressed by HEK 293 cells cultured in the presence of
serum.
FIG. 18 is a representation of dose titration of BrdU incorporation into NIH
3T3 cells
stimulated by p85 (bars 4-10) and by the p35 fragment of 30664188.m99 protein
(bars 11-17).
FIG. 19 is a representation of a Western blot and SDS PAGE analysis of PDGF D.
In
Panel A, samples from the conditioned medium of HEK 293 cells transiently
transfected with
pCEP4/Sec (lane 1) or pCEP4/Sec-PDGF D (lanes 2 & 3) and cultured in the
presence (lane 3)
or absence (lanes 1 & 2) of FBS were examined by SDS-PAGE under reducing
conditions,
followed by immunoblot analysis using anti-VS antibody. In Panel B, purified
PDGF-D from
pCEP4/Sec-PDGF D transfected HEK 293 cells cultured in the presence (lanes 3 &
4) or
absence (lanes 1 & 2) of FBS was resolved by SDS-PAGE and stained with
Coomassie Blue.
Samples were treated with (+) and without (-) DTT. Molecular weight markers
are indicated
on the left.
FIG. 20 is a representation of fragments obtained from p35 and identified by N-
terminal sequencing. In each panel, the upper sequence in black is the
predicted sequence
5
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
from the clone, and the lower sequence in gray is the sequence provided by N-
terminal
sequencing. The diagonal shadings represent two fragments of p35. Horizontal
shading
represents the VS epitope and vertical shading represents the 6His tag, both
of which originate
from vector pCEP4/Sec-30664188 (Example 3). In Panel A, two sequences were
identified,
one beginning with GlyArg (shown with these two residues underlined), and the
second
beginning with the third residue, Ser.
FIG. 21 is a depiction of the SDS-PAGE of the 30664188 gene product in the
presence
of fetal bovine serum (Panel B) and Calf Serum (Panel A). Lanes 1 and 2 in
each panel show
authentic 30664188 p35 alone or in the presence of serum, respectively. Lane 3
in each panel
shows p85 in the absence of serum, and lanes 4-6 show p85 in the presence of
increasing
concentrations of the respective serum.
FIG. 22 is a depiction of the stimulation of the growth of pulmonary artery
smooth
muscle cells by growth factors. Smooth muscle cells were treated with purified
p35 PDGF
DD, PDGF AA or PDGF BB at the concentrations indicated, and the amount of BrdU
incorporated into DNA was determined.
FIG. 23 is a diagram showing the proliferation of pulmonary artery smooth
muscle
cells in response to various treatments.
FIG. 24 presents bar graphs representing mean body weights of mice on day 0,
and on
day 6 after various treatments.
FIG. 25 presents bar graphs representing changes in mean body weights of mice
between day 0 and day 6 after various treatments.
FIG. 26 presents bar graphs representing percent changes in mean body weights
of
mice between day 0 and day 6 after various treatments.
FIG. 27 presents bar graphs representing changes in mean weights of the
spleens of
mice between day 0 and day 6 after various treatments.
FIG. 28 presents bar graphs representing changes in mean spleen weights of
mice
between day 0 and day 6 after various treatments.
FIG. 29 presents bar graphs representing changes in mean colon weights of mice
between day 0 and day 6 after various treatments.
FIG. 30 presents bar graphs representing changes in mean colon weights of mice
between day 0 and day 6 after various treatments.
6
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
FIG. 31 presents bar graphs representing percent changes in mean colon weights
of
mice between day 0 and day 6 after various treatments.
FIG. 32 presents bar graphs representing changes in mean colon lengths of mice
between day 0 and day 6 after various treatments.
FIG. 33 presents bar graphs representing percent changes in mean colon lengths
of
mice between day 0 and day 6 after various treatments.
FIG. 34 presents bar graphs representing mean colon blood content scores in
mice after
various treatments.
FIG. 35 presents bar graphs representing mean colon edema scores in mice after
various treatments.
FIG. 36 presents bar graphs representing mean colon inflammation scores in
mice after
various treatments.
FIG. 37 presents bar graphs representing mean colon epithelial loss scores in
mice after
various treatments.
FIG. 38 presents bar graphs representing mean colon erosion content scores in
mice
after various treatments.
FIG. 39 presents bar graphs representing sum of histopathology scores in mice
after
various treatments.
FIG. 40 presents bar graphs representing histopathology score differencess in
mice
after various treatments.
FIG. 41 presents bar graphs representing mean splenic lynphoid atrophy scores
in
mice after various treatments.
FIG. 42 presents photomicrographs at 400x in the original image of mouse colon
cross
sections. Panel A, DSS plus Vehicle; Panel B, DSS+AB020858; Panel C, Normal
mouse.
FIG. 43 presents photomicrographs at SOx in the original image of mouse colon
crossections. Panel A, DSS plus Vehicle; Panel B, DSS+AB020858; Panel C,
Normal mouse.
FIG. 44 presents the change in mean body weight from day 0 upon treating mice
with
varying doses of AB020258.
FIG. 45 presents the percent change in mean body weight from day 0 upon
treating
mice with varying doses of AB020258.
FIG. 46 presents mean colon blood content score upon treating mice with
varying
doses of AB020258.
7
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
FIG. 47 presents mean colon lengths upon treating mice with varying doses of
AB020258.
FIG. 48 presents mean colon lengths as a percent of normal, upon treating mice
with
varying doses of AB020258.
FIG. 49 presents mean colon weights upon treating mice with varying doses of
AB020258.
FIG. 50 presents mean colon colon weights as a percent of normal, upon
treating mice
with varying doses of AB020258.
FIG. 51 presents mean spleen weights upon treating mice with varying doses of
AB020258.
FIG. 52 presents mean distal colon inflammation score upon treating mice with
varying
doses of AB020258.
FIG. 53 presents mean distal colon gland loss score upon treating mice with
varying
doses of AB020258.
FIG. 54 presents mean distal colon erosion score upon treating mice with
varying
doses of AB020258.
FIG. 55 presents mean sums of histopathology scores upon treating mice with
varying
doses of AB020258.
FIG. 56 presents mean splenic lymphoid atrophy score upon treating mice with
varying
doses of AB020258.
FIG. 57 presents mean splenic extramedullary hematopoiesis score upon treating
mice
with varying doses of AB020258.
FIG. 58 presents the effect of CG53135 Treatment on Weight Loss in
Indomethacin-
treated rats. Body weight change from Day 0 to Day 5 is shown in grams.
FIG. 59 presents the effect of CG53135 Treatment on Small Intestine Weight in
Indomethacin-treated rats.
FIG. 60 presents effect of CG53135 Treatment on absolute neutrophil and
lymphocyte
counts in indomethacin-treated rats. Blood was collected on Day 5 at necropsy
and the cell
counts were determined.
FIG. 61 presents effect of CG53135 Treatment on Histopathology Scores in
Indomethacin-treated rats. Five sections of affected intestine were evaluated
and scored for
necrosis and inflammation as described in the methods.
8
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
FIG. 62 presents images showing the protective effect of CG53135 on intestinal
architecture. Panel A: Small intestine from normal control animal treated iv
with vehicle
(BSA). Panel. B: Small intestine from indomethacin- treated rat, further
treated with vehicle
(BSA) iv. Panel C: Small intestine from indomethacin-treated rat further
treated with
CG53135, 0.2 mg/kg iv. Sections were stained with H&E and visualized at a
magnification of
25). FIG. 62 shows the protective Effect of CG53135 on Intestinal
Architecturein
indomethacin treated rats. Panel A, normal control; Panel B, disease control
(indomethacin
treated); Panel C, disease model animal treated with 0.2 mg/kg iv CG53135.
Photomicrographs were obtained on sections stained with hemotoxylin and eosin,
at 25X
magnification.
FIG. 63 shows the effect of CG53135 treatment on BrdU Labeling in the
Intestine.
BrdU incorporation was detected by Immunoperoxidase staining. Panel A: Small
intestine
from normal control animal (100X). Panel B: Small intestine from indomethacin
+ vehicle
(BSA) treated animal (50X). Panel C: Small intestine from indomethacin +
CG53135 0.2
mg/kg iv treated rat (50X).
DETAILED DESCRIPTION OF THE INVENTION
This invention is related in part to the discovery of novel FGF-CX nucleic
acid
sequences that encode polypeptides that are members of the fibroblast growth
factor ("FGF")
family. As used herein the designation "FGF-CX" relates to nucleic acids,
polynucleotides,
proteins, polypeptides, and variants, derivatives and fragments of any of
them, as well as to
antibodies that bind irnmunospecifically to any of these classes of compounds.
In the present
disclosure, FGF-CX polypeptides are alternatively identified by the internal
accession
numbers AB020858, CG53135-O1 and CG53135-02.
The invention further is based on the discovery of nucleic acids that encode
polypeptides related to bone-morphogen protein-1 ("BMP-1"), to vascular
endothelial growth
factor ("VEGF-E"), and to platelet derived growth factors ("PDGF"). These
sequences are
collectively referred to as "FCTRX nucleic acids" or FCTRX polynucleotides"
and the
corresponding encoded polypeptide is referred to as a "FCTRX polypeptide" or
"FCTRX
protein." Unless indicated otherwise, "FCTRX" is meant to refer to any of the
novel
sequences disclosed herein. In addition, the polypeptides and nucleic acids of
the invention
are alternately referred to herein collectively as "PDGF D", since they are
considered to
9
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
represent a heretofore unknown PDGF, i. e., one that differs from PDGF A, PDGF
B and
PDGF C. Furthermore, when reference is made to "PDGFXX" or "PDGF XX" wherein
"X"
is either the A, B, C or D, this is meant to refer to homodimers of the
particular PDGF.
Alternately, when reference is made to "PDGFXY" wherein X and Y are either the
A, B, C or
D, and "X" is different from "Y" this is meant to refer to PDGF heterodimers.
It is shown herein that PDGF D has a high molecular weight latent form,
designated
p85, and a low molecular weight active form, designated p35. In the present
disclosure, the
FCTRX or PDGF D polypeptides are alternatively designated by the identifiers
30664188 and
variations thereof such as 30664188Ø99 or 30664188Ø331, and CG52053 and
variations
thereof such as CG52053-01 and CG52053-02.
Inflammatory bowel disease
Inflammatory bowel disease ("IBD") refers to a group of chronic inflammatory
disorders involving the gastrointestinal tract. Although IBD is diagnosed
largely by exclusion,
there are characteristic features associated with it that allows accurate
diagnosis.
Chronic IBD is sub-divided into two major groups, namely, ulcerative colitis
("UC")
and Crohn's disease ("CD"). Clinically IBD is characterized by recurrent
inflammatory
involvement of intestinal segments with diverse clinical manifestations.
Typically UC affects
the rectum and extends proximally to involve part or all of the colon. Lesions
are restricted to
the mucosal or submucosal layers of the colon with deeper layers unaffected
except in
fulminant disease. Symptoms include rectal bleeding, mucus containing
diarrhea, abdominal
pain and weight loss. CD affects the full thickness of the gut wall in both
the small and large
intestines in contrast to UC. The clinical symptoms of UC vary according to
the region
affected. In general, fever, malaise, weight loss, abdominal pain and cramps
are the common
symptoms of CD. Full thickness bowel lesions can progress to bowel
perforations and local
abscesses, fistulas in the adjoining abdominal and pelvic organs, and fibrosis
of the bowel wall
with obstruction.
The etiology of UC or CD remains unknown. However, a combination of factors
including abnormalities in the immune system, genetic predisposition,
environmental and
psychological factors, may be of importance in determining the outcome of the
disease.
In Europe and the United States, incidence and prevalence of CD is
approximately 1-6
and 10-100 cases per 100, 000 population respectively. For UC the incidence
and prevalence
rates are respectively 2-10 and 35-100 per 100, 000. There is a slight
preponderence in
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
females over males for contracting the disease. UC and CD affect primarily
individuals
between the ages of 15 and 35 years.
Therapeutic options for inflammatory bowel disease
Choice of therapy for IBD is dependent on pharmacodynamic considerations that
govern drug and patient characteristics. Clinical remission (relief of
inflammatory symptoms)
and mucosal healing are two vital aspects that need to be treated. Many of the
current drugs of
choice have a poor correlation between symptomatic relief and rnucosal
healing. Thus agents
that can maintain remission as well as accomplish healing will be of
particular interest in the
management of IBD. In the past decade several drugs have been used in the
treatment of
IBD. These include conventional salicylates, antibiotics and corticosteroids
as well as
immunomodulators and biological response modifiers.
5-Aminosalicylates (5-ASAs)
The 5-ASAs (sulfasalazine and the sulfa-free agents) are known to alter the
immune
response by down-regulationg antibody secretion and lymphocyte function,
inhibit neutrophil
and macrophage chemotaxis and protect intestinal epithelium by enhancing
expression of heat
shock proteins. In addition, they also inhibit the cyclooxygenase and 5-
lipoxygenase
pathways of arachidonic acid metabolism that may inhibit the release of
chemotactic
substances (Grisham, M. B. Lancet, 1994, 344:59-X61). 5-ASAs are effective
therapeutic
agents for mild to moderate conditions of UC. However, 5-ASAs are not the
drugs of choice
for IBD due to their side effects that may include nausea, allergic reactions
and reversible
oligospermia.
Antibiotics
Historically, antibiotics like metronidazole and quinolones have been used to
treat CD,
although their effectiveness in ameliorating the condition has not been well
documented. The
presumed effect of these agents may be in the alteration of the bacterial
flora associated with
IBD. Antibiotics are not only less effective for IBD but also have associated
side effects
(anorexia, nausea, rash) and thus may not be the treatment of choice for IBD.
Corticosteroids
Corticosteroids have been the oldest of the nonspecific but effective
therapeutic
regimen used for IBD. Corticosteroids modulate both immunologic and
inflammatory
responses and inhibit an array of leukocyte functions such as adherence,
chemotaxis,
11
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
phagocytosis arachidocic acid metabolism and eicosanoids production. Although
their use in
short-term treatment of CD and UC have been shown, their efficacy in
maintenance therapy is
far from satisfactory (Munkholin et al.. Gut, 1994, 35:360-362). The failure
of corticosteroids
in maintenance therapy coupled with the known detrimental side effects of this
agent limit
their use in the treatment for IBD.
Immunomodulators
The thiopurine agents 6-mercaptoputine ("6-MP") and azathioprine ("AZA") have
been used in the treatment of CD and UC as steroid sparing agents (Pearson et
al.. Annals of
Internal Medicine, 1995, 123:1320142). Side effects such as leukopenia,
thrombocytopenia
associated with these drugs are further complicated by the genetic
predisposition of the
patient. (Yates et al Ann. Intern. Med. 1997, 126:608-614). Additional side
effects such as
pancreatitis, hepatitis, nausea and rash are also reported.
Methotrexate has been shown to be effective in steroid-dependent CD but not in
UC
The side effects of methotrexate include bone marrow suppression, interstitial
pneumonitis
and neuropathy.
Cyclosporine has been effective in the treatment of both CD and UC.
Cyclosporine
has been particularly shown to be effective in patients with active CD or UC
that are resistant
or intolerant to corticosteriods (Lichtiger et al.. New England Journal of
Medicine, 1994,
330:1841-1845). The side effects of cyclosporin include reversible or
irreversible decrease in
renal function, hypertension, tremor, and seizure.
Biological Response Modifiers
The agent Infliximab, a chimeric monoclonal IgGl antibody directed against TNF-
a,
has been effectively used in the treatment of CD. Although it is effective in
maintenance
therapy and healing fistulas (Present et al.. New England Journal of Medicine,
1999,
340:1398-1405), side effects include delayed hypersensitivity reactions and
lymphoproliferative disorders.
Fibroblast Growth Factors
The fibroblast growth factor (FGF) group of cytokines includes at least 21
members
that regulate diverse cellular functions such as growth, survival, apoptosis,
motility and
differentiation. These molecules transduce signals via high affinity
interactions with cell
surface tyrosine kinase FGF receptors (FGFRs). FGF receptors are expressed on
most types of
12
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
cells in tissue culture. Dimerization of FGF receptor monomers upon ligand
binding has been
reported to be a requisite for activation of the kinase domains, leading to
receptor trans
phosphorylation. FGF receptor-1 (FGFR-1), which shows the broadest expression
pattern of
the four FGF receptors, contains at least seven tyrosine phosphorylation
sites. A number of
signal transduction molecules are affected by binding with different
affinities to these
phosphorylation sites.
In addition to participating in normal growth and development, known FGFs have
also
been implicated in the generation of pathological states, including cancer.
FGFs may
contribute to malignancy by directly enhancing the growth of tumor cells. For
example,
autocrine growth stimulation through the co-expression of FGF and FGFR in the
same cell has
been reported to lead to cellular transformation.
Previously described members of the FGF family regulate diverse cellular
functions
such as growth, survival, apoptosis, motility and differentiation (Szebenyi &
Fallon (1999) W t.
Rev. Cytol. 185, 45-106). These molecules transduce signals intracellularly
via high affinity
interactions with cell surface tyrosine kinase FGF receptors (FGFRs), four of
which have been
identified to date (Xu et al. (1999) Cell Tissue Res. 296, 33-43; Flint &
Claesson-Welsh
(1999) Front. Biosci. 4, 165-177). These FGF receptors are expressed on most
types of cells
in tissue culture. Dimerization of FGF receptor monomers upon ligand binding
has been
reported to be a requisite for activation of the kinase domains, leading to
receptor trans
phosphorylation. FGF receptor-1 (FGFR-1), which shows the broadest expression
pattern of
the four FGF receptors, contains at least seven tyrosine phosphorylation
sites. A number of
signal transduction molecules axe affected by binding with different
affinities to these
phosphorylation sites.
FGFs also bind, albeit with low affinity, to heparin sulfate proteoglycans
(HSPGs)
present on most cell surfaces and extracellular matrices (ECM). Interactions
between FGFs
and HSPGs serve to stabilize FGF/FGFR interactions, and to sequester FGFs and
protect them
from degradation (Szebenyi. & Fallon (1999)). Due to its growth-promoting
capabilities, one
member of the FGF family, FGF-7, is currently in clinical trials for the
treatment of
chemotherapy-induced mucositis (Danilenko (1999) Toxicol. Pathol. 27, 64-71).
In addition to participating in normal growth and development, known FGFs have
also
been implicated in the generation of pathological states, including cancer
(Basilico &
Moscatelli (1992) Adv. Cancer Res. 59, 115-165). FGFs may contribute to
malignancy by
13
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
directly enhancing the growth of tumor cells. For example, autocrine growth
stimulation
through the co-expression of FGF and FGFR in the same cell leads to cellular
transformation
(Matsumoto-Yoshitomi, et al., (1997) Int. J. Cancer 71, 442-450). Likewise,
the constitutive
activation of FGFR via mutation or rearrangement leads to uncontrolled
proliferation (Lorenzi,
et al., (1996) Proc. Natl. Acad. Sci. USA. 93, 8956-8961; Li, et al., (1997)
Oncogene 14,
1397-1406). Furthermore, some FGFs are angiogenic (Gerwins, et al., (2000)
Crit. Rev.
Oncol. Hematol. 34, 185-194). Such FGFs may contribute to the tumorigenic
process by
facilitating the development of the blood supply needed to sustain tumor
growth. Not
surprisingly, at least one FGF is currently under investigation as a potential
target for cancer
therapy (Gasparini (1999) Drugs 58, 17-38).
Expression of FGFs and their receptors in the brains of perinatal and adult
mice has
been examined. Messenger RNA all FGF genes, with the exception of FGF-4, is
detected in
these tissues. FGF-3, FGF-6, FGF-7 and FGF-8 genes demonstrate higher
expression in the
late embryonic stages than in postnatal stages, suggesting that these members
are involved in
the late stages of brain development. In contrast, expression of FGF-1 and FGF-
5 increased
after birth. In particular, FGF-6 expression in perinatal mice has been
reported to be restricted
to the central nervous system and skeletal muscles, with intense signals in
the developing
cerebrum in embryos but in cerebellum in 5-day-old neonates. FGF-receptor
(FGFR)-4, a
cognate receptor for FGF-6, demonstrate similar spatiotemporal expression,
suggesting that
FGF-6 and FGFR-4 plays significant roles in the maturation of nervous system
as a ligand-
receptor system. According to Ozawa et al., these results strongly suggest
that the various
FGFs and their receptors are involved in the regulation of a variety of
developmental
processes of brain, such as proliferation and migration of neuronal progenitor
cells, neuronal
and glial differentiation, neurite extensions, and synapse formation.
Glia-activating factor ("GAF"), another FGF family member, is a heparin-
binding
growth factor that was purified from the culture supernatant of a human glioma
cell line. See,
Miyamoto et al., 1993, Mol Cell Biol 13(7): 4251-4259. GAF shows a spectrum of
activity
slightly different from those of other known growth factors, and is designated
as FGF-9. The
human FGF-9 cDNA encodes a polypeptide of 208 amino acids. Sequence similarity
to other
members of the FGF family was estimated to be around 30%. Two cysteine
residues and other
consensus sequences found in other family members were also well conserved in
the FGF-9
14
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
sequence. FGF-9 was found to have no typical signal sequence in its N terminus
like those in
acidic FGF and basic FGF.
Acidic FGF and basic FGF are known not to be secreted from cells in a
conventional
manner. However, FGF-9 was found to be secreted efficiently from cDNA-
transfected COS
cells despite its lack of a typical signal sequence. It could be detected
exclusively in the culture
medium of cells. The secreted protein lacked no amino acid residues at the N
terminus with
respect to those predicted by the cDNA sequence, except the initiation
methionine. The rat
FGF-9 cDNA was also cloned, and the structural analysis indicated that the FGF-
9 gene is
highly conserved.
Platelet Derived Growth Factors
Polypeptide growth factors exerting effects in a variety of tissues have been
described.
Among these growth factors are bone morphogenetic protein-1 ("BMP-1"),
vascular
endothelial growth factor (VEGF), and platelet-derived growth factor ("PDGF").
Multiple effects have been attributed to BMP-1. For example, BMP-1 is capable
of
inducing formation of cartilage in vivo. BMP1 is also identical to purified
procollagen C
proteinase ("PCP"), a secreted calcium-dependent metalloprotease that has been
reported to be
required for cartilage and bone formation. BMP-1 cleaves the C-terminal
propeptides of
procollagen I, II, and III and its activity is increased by the procollagen C-
endopeptidase
enhancer protein.
Vascular endothelial growth factor ("VEGF") polypeptides have been reported to
act
as mitogens primarily for vascular endothelial cells. The specificity for
vascular endothelial
cells contrasts VEGF polypeptides from other polypeptide mitogens, such as
basic fibroblast
growth factor and platelet-derived growth factors, which are active on a wider
range of cell
types.
VEGF has also been reported to affect tumor angiogenesis. For example, VEGF
has
been shown to stimulate the elongation, network formation, and branching of
nonproliferating
endothelial cells in culture that are deprived of oxygen and nutrients.
The platelet derived growth factor ("PDGF") family currently consists of at
least 3
distinct genes, PDGF A, PDGF B, and PDGF C whose gene products selectively
signal
through two PDGFRs to regulate diverse cellular functions. PDGF A, PDGF B, and
PDGF C
dimerize in solution to form homodimers, as well as the heterodimer.
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
Expression of RNA encoding the PDGF A and PDGF B subunits of has been reported
in vascular tissues involved in atherosclerosis. PDGF A and PDGF B mRNA have
been
reported to be present in mesenchymal-appearing intimal cells and endothelial
cells,
respectively, of atherosclerotic plaques. In addition, PDGF receptor mRNA has
also been
localized predominantly in plaque intimal cells.
The PDGF B is related to the transforming gene (v-sis) of simian sarcoma
virus. The
PDGF B has also been reported to be mitogen for cells of mesenchymal origin.
The PDGF B
has in addition been implicated in autocrine growth stimulation in the
pathologic proliferation
of endothelial cells characteristically found in glioblastomas. PDGF has also
been reported to
promote cellular proliferation and inhibits apoptosis.
FGF-CX
The present invention is related to a novel human FGF as well as its
corresponding
cDNA. The protein product of this gene has been shown to exhibit growth
stimulatory and
growth promoting properties.
The nucleotide sequence and translated polypeptide sequence of Fibroblast
Growth
Factor-CX ("FGF-CX," also referred to as AB020858) is presented in Table 1
(see Example
1; see also disclosure in U. S. Ser. No. 60/145,899, filed July 27, 1999, U.
S. Ser. No.
09/494585, filed January 31, 2000 and U. S. Ser. No. 09/609543, filed July 3,
2000, all of
which are incorporated herein by reference in their entireties). The start and
stop codons are
shown in bold.
Table 1. Nucleotide (SEQ ID NO:1) and Protein (SEQ ID N0:2)
Sequence of Fibroblast Growth Factor-CX (FGF-CX )
1 ATGGCTCCCTTAGCCGAAGTCGGGGGCTTTCTGGGCGGCCTGGAG
MetAlaProLeuAlaGluValGlyGlyPheLeuGlyGlyLeuGlu
46 GGCTTGGGCCAGCAGGTGGGTTCGCATTTCCTGTTGCCTCCTGCC
GlyLeuGlyGlnGlnValGlySerHisPheLeuLeuProProAla
91 GGGGAGCGGCCGCCGCTGCTGGGCGAGCGCAGGAGCGCGGCGGAG
GlyGluArgProProL,euLeuGlyGluArgArgSerAlaAlaGlu
136 CGGAGCGCGCGCGGCGGGCCGGGGGCTGCGCAGCTGGCGCACCTG
ArgSerAlaArgGlyGlyProGlyAlaAlaGlnLeuAlaHisLeu
181 CACGGCATCCTGCGCCGCCGGCAGCTCTATTGCCGCACCGGCTTC
16
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
HisGlyIleLeuArgArgArgGlnLeuTyrCysArgThrGlyPhe
226 CACCTGCAGATCCTGCCCGACGGCAGCGTGCAGGGCACCCGGCAG
HisLeuGlnIleLeuProAspGlySerValGlnGlyThrArgGln
271 GACCACAGCCTCTTCGGTATCTTGGAATTCATCAGTGTGGCAGTG
AspHisSerLeuPheGlyIleLeuGluPheIleSerValAlaVa1
316 GGACTGGTCAGTATTAGAGGTGTGGACAGTGGTCTCTATCTTGGA
GlyLeuValSerIleArgGlyValAspSerGlyLeuTyrLeuGly
361 ATGAATGACAAAGGAGAACTCTATGGATCAGAGAAACTTACTTCC
MetAsnAspLysGlyGluLeuTyrGlySerGluLysLeuThrSer
406 GAATGCATCTTTAGGGAGCAGTTTGAAGAGAACTGGTATAACACC
GluCysIlePheArgGluGlnPheGluGluAsnTrpTyrAsnThr
451 TATTCATCTAACATATATAAACATGGAGACACTGGCCGCAGGTAT
TyrSerSerAsnIleTyrLysHisGlyAspThrGlyArgArgTyr
496 TTTGTGGCACTTAACAAAGACGGAACTCCAAGAGATGGCGCCAGG
PheValAlaLeuAsnLysAspGlyThrProArgAspGlyAlaArg
541 TCCAAGAGGCATCAGAAATTTACACATTTCTTACCTAGACCAGTG
SerLysArgHisGlnLysPheThrHisPheLeuProArgProVa1
586 GATCCAGAAAGAGTTCCAGAATTGTACAAGGACCTACTGATGTAC
AspProGluArgValProGluLeuTyrLysAspLeuLeuMetTyr
631 ACT* (SEQ ID N0:1)
Thr (SEQ TD N0:2)
Included in the invention is a nucleotide sequence (SEQ m NO:l) encoding a
novel
fibroblast growth factor designated fibroblast growth factor-20X (FGF-CX )
(see Table 1;
SEQ ID N0:1). This coding sequence was identified in human genomic DNA
sequences. The
disclosed DNA sequence has 633 bases that encode a polypeptide predicted to
have 211 amino
acid residues (Table 1; SEQ ID NO:2). The predicted molecular weight of FGF-
CX, based on
the sequence shown in Table 1 and SEQ ID N0:2, is 23498.4 Da.
The FGF-CX nucleic acid sequence was used as a query nucleotide sequence in a
BLASTN search to identify related nucleic acid sequences. The FGF-CX
nucleotide sequence
has a high similarity to marine fibroblast growth factor 9 ("FGF-9") (392 of
543 bases
identical, or 72%; GenBank Accession Number 582023) and to human DNA encoding
glia
activating factor (GAP) (385 of 554 bases identical, or 69%; GenBank Accession
Number
E05822, also termed FGF-9). In addition, FGF-CX was found to have a comparable
degree of
identity (311 of 424 bases identical, or 73%) to a GAF sequence (SEQ ID NO:S)
disclosed by
17
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
Naruo et al. in Japanese Patent: JP 1993301893 entitled "Glia-Activating
Factor And Its
Production".
To verify that the open reading frame (ORF) identified by genomic mining was
correct, PCR amplification was used to obtain a cDNA corresponding to the
predicted
genomic clone. The nucleotide sequence of the obtained product precisely
matches that of the
predicted gene (see Example 2).
The protein encoded by the cDNA is most closely related to Xenopus FGF-20X
(designated XFGF-CX or XFGF-20X herein), as well as to human FGF-9 and human
FGF-16
(80%, 70% and 64% amino acid identity, respectively). Based on the strong
homology with
XFGF-CX, the gene identified in the present disclosure is believed to
represent its human
ortholog, and is named FGF-CX herein.
A BLASTP analyses of the polypeptide of SEQ ID N0:2 shows that the first 208
amino acids of the FGF-CX polypeptide sequence (SEQ ID N0:2) aligns with a
human FGF-
9. See, e.g., SWISSPROT Accession Number P31371 for Glia-Activating Factor
Precursor
(GAF) (Fibroblast Growth Factor-9); Miyamoto et al. 1993 Mol. Cell. Biol.
13:4251-4259;
and Naruo et al. 1993 J. Biol. Chem. 268:2857-2864. BLASTX analysis shows that
the first
208 amino acids of the FGF-CX polypeptide (SEQ ID N0:2 aligns with the mouse
FGF-9 and
rat FGF-9 sequences. See, e.g., SWISSPROT Accession Number P54130 for Glia-
Activating
Factor Precursor (GAF) (Fibroblast Growth Factor-9), Santos-Ocampo et al.,
1996 J. Biol.
Chem. 271:1726-1731, for mouse FGF-9; and SWISSPROT Accession Number P36364
Glia-
Activating Factor Precursor (GAF) (Fibroblast Growth Factor-9) (FGF-9),
Miyamoto, 1993
Mol. Cell. Biol. 13:4251-4259, for rat FGF-9.
The full length FGF-CX polypeptide (SEQ ID NO:2) was also aligned by BLASTX
with Xenopus XFGF-CX (See, I~oga et al., 1999 Bioclaem Biophys Res Commute
261(3):756-
765). It was found that FGF-CX has 170 of 211 (80%) identical residues, and
189 of 211
(89%) positive residues compared with Xenopus XFGF-CX. The deduced 208 amino
acid
sequence of the XFGF-CX open reading frame contains a motif characteristic of
the FGF
family. XFGF-CX has a 73.1 % overall similarity to XFGF-9 but differs from
XFGF-9 in its
amino-terminal region (33.3% similarity). This resembles the similarity seen
for the presently
disclosed SEQ ID N0:2 with respect to various mammalian FGF-9 and FGF-16
sequences,
including human (see above).
18
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
FGF-CX lacks a classical amino-terminal signal sequence as predicted by PSORT
(Nakai & Kanehisa (1992) Genomics 14, 897-911) and SIGNALP (Nielsen et al.
(1997)
Protein Ehg. 10, 1-6) computer algorithms, just as found for some of its
closest human family
members (e. g. FGF-9 and FGF-16). Nonetheless, both FGF-9 and FGF-16 are
secreted
(Matsumoto-Yoshitomi et al. (1997) Int. J. Cahce~ 71, 442-450; Miyake et al.
(1998)
Biochem. Biophys. Res. Comm. 243, 148-152; Miyakawa et al. (1999) J. Biol.
Chem. 274,
29352-29357; Revest et al. (2000) JBiol. Chem. 275, 8083-8090). To determine
whether
FGF-CX is also secreted, the cDNA encoding the full length FGF-CX protein was
subcloned
into a mammalian expression vector designated pFGF-CX. The protein expressed
when
human embryonic kidney 293 cells are transfected with this vector is found in
the conditioned
medium, and exhibits a band detected by an antibody to a C-terminal VS
epitope, with an
appaxent molecular weight in a Western blot of ~27 kDa (FIG. 4). An additional
portion of
the expressed protein is released from sequestration on the 293 cells by
treatment with a
substance that inhibits interaction with heparin sulfate proteoglycan (HSPG).
The protein
released in this way also exhibits a similar Western blot pattern (FIG. 4).
Similarly when the
protein is expressed in HEK293 cells from a recombinant plasmid incorporating
an Ig Kappa
signal sequence, a band is detected by Western blot with an apparent molecular
weight of
approximately 34 kDa (FIG. 1, Example 4).
FCTRl
A polynucleotide of the invention includes the nucleic acid of FCTRl (also
referred to
as clone 30664188Ø99). FCTRl is 1828 nucleotides in length. The nucleotide
sequence of
FCTRl (also referred to as 30664188Ø99 or PDGFD) is reported in Table 2 (SEQ
ID N0:3).
The clone was originally obtained from RNA from pituitary gland tissues is
also present in
RNA from human uterine microvascular endothelial cells (Clonetics, San Diego,
CA), human
erythroleukemia cells (ATCC, Manassas, VA), thyroid, small intestine,
lymphocytes, adrenal
gland and salivary gland. The untranslated regions upstream of the start site
and downstream
of the stop codon are underlined, and the start and stop codons are shown in
bold.
TABLE 2. Nucleotide (SEQ ID N0:3) and Protein (SEQ ID N0:4)
Sequence of FCTR1
Translated Protein - Frame: 2 - Nucleotide 182 to 1291
1 CTAAAAAATATGTTCTCTACAACACCAAGGCTCATTAAAATATTT
19
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
46 TAAATATTAATATACATTTCTTCTGTCAGAAATACATAAAACTTT
91 ATTATATCAGCGCAGGGCGGCGCGGCGTCGGTCCCGGGAGCAGAA
136 CCCGGCTTTTTCTTGGAGCGACGCTGTCTCTAGTCGCTGATCCCA
181 _AATGCACCGGCTCATCTTTGTCTACACTCTAATCTGCGCAAACTT
MetHisArgLeuIlePheValTyrThrLeuIleCysAlaAsnPh
226 TTGCAGCTGTCGGGACACTTCTGCAACCCCGCAGAGCGCATCCAT
eCysSerCysArgAspThrSerAlaThrProGlnSerAlaSerll
271 CAAAGCTTTGCGCAACGCCAACCTCAGGCGAGATGAGAGCAATCA
eLysAlaLeuArgAsnAlaAsnLeuArgArgAspGluSerAsnHi
316 CCTCACAGACTTGTACCGAAGAGATGAGACCATCCAGGTGAAAGG
sLeuThrAspLeuTyrArgArgAspGluThrIleGlnValLysG1
361 AAACGGCTACGTGCAGAGTCCTAGATTCCCGAACAGCTACCCCAG
yAsnGlyTyrValGlnSerProArgPheProAsnSerTyrProAr
406 GAACCTGCTCCTGACATGGCGGCTTCACTCTCAGGAGAATACACG
gAsnLeuLeuLeuThrTrpArgLeuHisSerGlnGluAsnThrAr
451 GATACAGCTAGTGTTTGACAATCAGTTTGGATTAGAGGAAGCAGA
glleGlnLeuValPheAspAsnGlnPheGlyLeuGluGluAlaG1
496 AAATGATATCTGTAGGTATGATTTTGTGGAAGTTGAAGATATATC
uAsnAspIleCysArgTyrAspPheValGluValGluAspIleSe
541 CGAAACCAGTACCATTATTAGAGGACGATGGTGTGGACACAAGGA
rGluThrSerThrIleIleArgGlyArgTrpCysGlyHisLysG1
586 AGTTCCTCCAAGGATAAAATCAAGAACGAACCAAATTAAAATCAC
uValProProArgIleLysSerArgThrAsnGlnIleLysIleTh
631 ATTCAAGTCCGATGACTACTTTGTGGCTAAACCTGGATTCAAGAT
rPheLysSerAspAspTyrPheValAlaLysProGlyPheLysIl
676 TTATTATTCTTTGCTGGAAGATTTCCAACCCGCAGCAGCTTCAGA
eTyrTyrSerLeuLeuGluAspPheGlnProAlaAlaAlaSerG1
72l GACCAACTGGGAATCTGTCACAAGCTCTATTTCAGGGGTATCCTA
uThrAsnTrpGluSerValThrSerSerIleSerGlyValSerTy
766 TAACTCTCCATCAGTAACGGATCCCACTCTGATTGCGGATGCTCT
rAsnSerProSerValThrAspProThrLeuTleAlaAspAlaLe
811 GGACAAAAAAATTGCAGAATTTGATACAGTGGAAGATCTGCTCAA
uAspLysLysIleAlaGluPheAspThrValGluAspLeuLeuLy
856 GTACTTCAATCCAGAGTCATGGCAAGAAGATCTTGAGAATATGTA
sTyrPheAsnProGluSerTrpGlnGluAspLeuGluAsnMetTy
901 TCTGGACACCCCTCGGTATCGAGGCAGGTCATACCATGACCGGAA
rLeuAspThrProArgTyrArgGlyArgSerTyrHisAspArgLy
946 GTCAAAAGTTGACCTGGATAGGCTCAATGATGATGCCAAGCGTTA
sSerLysValAspLeuAspArgLeuAsnAspAspAlaLysArgTy
99l CAGTTGCACTCCCAGGAATTACTCGGTCAATATAAGAGAAGAGCT
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
rSerCysThrProArgAsnTyrSerValAsnIleArgGluGluLe
1036 GAAGTTGGCCAATGTGGTCTTCTTTCCACGTTGCCTCCTCGTGCA
uLysLeuAlaAsnValValPhePheProArgCysLeuLeuValGl
1081 GCGCTGTGGAGGAAATTGTGGCTGTGGAACTGTCAACTGGAGGTC
nArgCysGlyGlyAsnCysGlyCysGlyThrValAsnTrpArgSe
1126 CTGCACATGCAATTCAGGGAAAACCGTGAAAAAGTATCATGAGGT
rCysThrCysAsnSerGlyLysThrValLysLysTyrHisGluVa
1171 ATTACAGTTTGAGCCTGGCCACATCAAGAGGAGGGGTAGAGCTAA
lLeuGlnPheGluProGlyHisIleLysArgArgGlyArgAlaLy
1216 GACCATGGCTCTAGTTGACATCCAGTTGGATCACCATGAACGATG
sThrMetAlaLeuValAspIleGlnLeuAspHisHisGluArgCy
1261 TGATTGTATCTGCAGCTCAAGACCACCTCGATAAGAGAATGTGCA
SAspCysTleCysSerSerArgProProArg
1306 CATCCTTACATTAAGCCTGAAAGAACCTTTAGTTTAAGGAGGGTG
1351 AGATAAGAGACCCTTTTCCTACCAGCAACCAAACTTACTACTAGC
1396 CTGCAATGCAATGAACACAAGTGGTTGCTGAGTCTCAGCCTTGCT
1441 TTGTTAATGCCATGGCAAGTAGAAAGGTATATCATCAACTTCTAT
1486 ACCTAAGAATATAGGATTGCATTTAATAATAGTGTTTGAGGTTAT
1531 ATATGCACAAACACACACAGAAATATATTC~-1TGTCTATGTGTATA
1576 TAGATCAAATGTTTTTTTTGGTATATATAACCAGGTACACCAGAG
1621 CTTACATATGTTTGAGTTAGACTCTTAAAATCCTTTGCCAAAATA
1666 AGGGATGGTCAAATATATGAAACATGTCTTTAGAAAATTTAGGAG
1711 ATAAATTTATTTTTAAATTTTGAAACACAAAACAATTTTGAATCT
1756 TGCTCTCTTAAAGAAAGCATCTTGTATATTAAAAATCAAAAGATG
1801 AGGCTTTCTTACATATACATCTTAGTTG
Nucleotides 182 to 1292 of SEQ ID N0:3 encode a 370 amino acid protein (SEQ ID
N0:4) that includes sequences characteristic of secreted proteins. The
sequence of the
encoded protein, which is also referred to herein as "FCTRl protein,"
"30664188Ø99
protein," "30664188Ø99," "PDGFD," or "human PDGFD" is presented in Table 2.
The
predicted molecular weight of the 30664188Ø99 protein is 42847.8 daltons
with a pI of 7.88.
BLASTN and BLASTP analyses indicate the 30664188Ø99 polypeptide has a
similarity to human vascular endothelial growth factor E (VEGF-E), as well as
to VEGF-E
from other vertebrate species. For example, there is a 44% identity to human
secretory growth
factor-like protein (VEGF-E, or fallotein; Acc. No.: AAF00049 which references
GenBank-
ID: AF091434 for the nucleotide sequence). An aligmnent of the amino acid
sequence of the
30664188Ø99 polypeptide with that of VEGF-E is shown in FIG. 1. BLASTP
analyses also
indicate that FCTRl is related to human PDGF C, PDGF B, and PDGF A (42%, 27%,
and
25% overall amino acid identity, respectively)
21
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
PFAM and PROSITE analyses indicte that 30664188Ø99 polypeptide amino acid
sequence conatains a PDGF domain (aa 272-362) and a N-linked glycosylation
site (residue
276).
The 30664188Ø99 polypeptide amino acid sequence shows similarity to the
sequence
of human procollagen C-endopeptidase (bone morphogenetic protein-1; BMP-1; PIR-
m:A58788), which is a polypeptide of 823 residues. Residues 54 to 169 of the
30664188Ø99
polypeptide show 30-41% identity over three segments of the BMP-1 polypeptide.
The
30664188Ø99 polypeptide also shows a similar degree of identity is to BMP-1
from Xenopus
laevis (ACC NO:P98070), which is a 707 residue protein. The latter protein may
act as a zinc
protease in promoting cartilage and bone formation (Wozney et al., Science
242: 1528-34,
1988).
The 30664188Ø99 polypeptide is also related to other growth factors. For
example, it
shows 42% identity and 59% similarity to chicken spinal cord-derived growth
factor
(TREMBLNEW-ACC:BAB03265), 42% identity and 59% identity to human secretory
growth
factor-like protein fallotein (SPTREMBL-ACC:Q9UL22), 42% identity and 39%
similarity to
human platelet-derived growth factor C (TREMBLNEW-ACC:AAF80597), and 39%
identity
and 59% similarity to mouse fallotein (SPTREMBL-ACC:Q9QY71).
The homologies discussed above identify the 30664188Ø99 polypeptide as a
member
of the BMP-1/VEGF-E/PDGF protein family. BMP-1 proteins include an EGF-like
domain,
three CUB domains, and PDGF/VEGF domains. BMP-1 proteins are also members of
the
astacin subfamily.
SignalP and PSORT analyses predict that the amino acid sequence for
30664188Ø99
includes a cleavable amino terminal signal peptide with a cleavage site
between positions 23
and 24 (TSA-TP). The protein is most likely secreted and localized outside of
the cell. The
InterPro software program predicts the presence of a CUB domain in
30664188Ø99 from
residue 53 to residue 167, a PDGF domain spanning residues 272-306 and 350-
362, and a
metallothionein domain from residue 302 to residue 365. A FCTRl polypeptide of
the
invention includes a polypeptide having one, two, three, or four of these
domains, or a
combination thereof.
A FCTR1 polypeptide of the invention includes a mature form of a FCTRl
polypeptide
that includes amino acids 24-370 of SEQ m N0:4. These sequences are also
encoded in a
construct encoded by clone 30664188Øm99, which is described in more detail
below. Also
22
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
within the invention are nucleic acids encoding FCTRX polypeptide fragments
that include
amino acid sequences 247-370, 247-338, or 339-370, or their variant forms. In
some
embodiments, the fragments stimulate proliferation of cells. Also within the
invention are the
FCTRX polypeptide fragments, or their variants, homologs or analogs encoded by
these
nucleic acids.
FCTR2 Nucleic Acids and Polypeptides
A polynucleotide of the invention includes the nucleic acid sequence of FCTR2
(also
referred to as clone 30664188Ø331). FCTR2 is 1587 nucleotides in length and
was originally
isolated from RNA from pituitary gland tissues. The nucleotide sequence of
FCTR2 is shown
in Table 3 (SEQ ID NO:S). The untranslated regions upstream of the start site
and
downstream of the stop codon are underlined, and the start and stop codons are
shown in bold.
TABLE 3. Nucleotide (SEQ ID NO:S) and Protein (SEQ ID N0:6)
Sequence of FCTR2
Translated Protein - Frame: 3 - Nucleotide 540 to 935
1 AGAGGCTCTCAAATTAGATCAAGAAATGCCTTTAACAGAAGTGAA
46 GAGTGAACCTGCTCCTGACATGGCGGCTTCACTCTCAGGAGAATA
91 CACGGATACAGCTAGTGTTTGACAATCAGTTTGGATTAGAGGAAG
136 CAGAAAATGATATCTGTAGGTATGATTTTGTGGAAGTTGAAGATA
181 TATCCGAAACCAGTACCATTATTAGAGGACGATGGTGTGGACACA
226 AGGAAGTTCCTCCAAGGATAAAATCAAGAACGAACCAAATTAAAA
271 TCACATTCAAGTCCGATGACTACTTTGTGGCTAAACCTGGATTCA
316 AGATTTATTATTCTTTGCTGGAAGATTTCCAACCCGCAGCAGCTT
361 CAGAGACCAACTGGGAATCTGTCACAAGCTCTATTTCAGGGGTAT
406 CCTATAACTCTCCATCAGTAACGGATCCCACTCTGATTGCGGATG
451 CTCTGGACAAAAAAATTGCAGAATTTGATACAGTGGAAGATCTGC
496 TCAAGTACTTCAATCCAGAGTCATGGCAAGAAGATCTTGAGAATA
M
541 TGTATCTGGACACCCCTCGGTATCGAGGCAGGTCATACCATGACC
etTyrLeuAspThrProArgTyrArgGlyArgSexTyrHisAspA
586 GGAAGTCAAAAGTTGACCTGGATAGGCTCAATGATGATGCCAAGC
rgLysSerLysValAspLeuAspArgLeuAsnAspAspAlaLysA
631 GTTACAGTTGCACTCCCAGGAATTACTCGGTCAATATAAGAGAAG
rgTyrSerCysThrProArgAsnTyrSerValAsnIleArgGluG
676 AGCTGAAGTTGGCCAATGTGGTCTTCTTTCCACGTTGCCTCCTCG
luLeuLysLeuAlaAsnValValPhePheProArgCysLeuLeuV
721 TGCAGCGCTGTGGAGGAAATTGTGGCTGTGGAACTGTCAACTGGA
alGlnArgCysGlyGlyAsnCysGlyCysGlyThrValAsnTrpA
766 GGTCCTGCACATGCAATTCAGGGAAA.ACCGTGAAAAAGTATCATG
rgSerCysThrCysAsnSerGlyLysThrValLysLysTyrHisG
811 AGGTATTACAGTTTGAGCCTGGCCACATCAAGAGGAGGGGTAGAG
luValLeuGlnPheGluProGlyHisIleLysArgArgGlyArgA
856 CTAAGACCATGGCTCTAGTTGACATCCAGTTGGATCACCATGAAC
laLysThrMetAlaLeuValAspIleGlnLeuAspHisHisGluA
901 GATGTGATTGTATCTGCAGCTCAAGACCACCTCGATAAGAGAATG
RgCysAspCysIleCysSerSerArgProProArg
946 TGCACATCCTTACATTAAGCCTGAAAGAACCTTTAGTTTAAGGAG
23
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
991 GGTGAGATAAGAGACCCTTTTCCTACCAGCAACCAAACTTACTAC
1036 TAGCCTGCAATGCAATGAACACAAGTGGTTGCTGAGTCTCAGCCT
1081 TGCTTTGTTAATGCCATGGCAAGTAGAAAGGTATATCATCAACTT
1126 CTATACCTAAGAATATAGGATTGCATTTAATAATAGTGTTTGAGG
1171 TTATATATGCACAAACACACACAGAAATATATTCATGTCTATGTG
1216 TATATAGATCAAATGTTTTTTTTGGTATATATAACCAGGTACACC
1261 AGAGCTTACATATGTTTGAGTTAGACTCTTAAAATCCTTTGCCAA
1306 AATAAGGGATGGTCAAATATATGAAACATGTCTTTAGAAAATTTA
1351 GGAGATAAATTTATTTTTAAATTTTGAAACACAAAACAATTTTGA
1396 ATCTTGCTCTCTTAAAGAAAGCATCTTGTATATTAAAAATCAAAA
1441 GATGAGGCTTTCTTACATATACATCTTAGTTGATTATTAAAAAAG
1486 GAAAAATATGGTTTCCAGAGAAAAGGCCAATACCTAAGCATTTTT
1531 TCCATGAGAAGCACTGCATACTTACCTATGTGGACTATAATAACC
1576 TGTCTCCAAAAC
Clone 30664188Ø331 includes an open reading frame from nucleotides 540 to
936.
The open reading frame encodes a polypeptide of 132 amino acids (SEQ ID N0:6).
The
encoded polypeptide is referred to herein as the "30664188Ø331 protein" or
the
"30664188Ø331 polypeptide". The predicted amino acid sequence of the
30664188Ø331
nucleic acid sequence is shown in Table 3 (SEQ ID N0:6).
Nucleotides 50 to 1472 of clone 30664188Ø331 are 100% identical to
nucleotides
406-1828 of clone 30664188Ø99. The 132 amino acids of the clone
30664188Ø331 protein
are 100% identical to the carboxy-terminal region of the protein sequence of
30664188Ø99.
Thus, the nucleic acids of clones 30664188Ø99 and 30664188Ø331 are
therefore related as
splice variants of a common gene.
The 30664188Ø331 protein shows similarity to human growth factor FIGF (c-fos-
induced growth factor; ptnr:SPTREMBL-ACC:043915), a member of the platelet-
derived
growth factor/vascular endothelial growth factor (PDGF/VEGF) family, and to
rat vascular
endothelial growth factor D (ptnr:SPTREMBL-ACC:035251).
FCTR3 Nucleic Acids and Polypeptides
A FCTR3 (also refered to within the specification as PDGFD or marine PDGFD or
mPDGFD) nucleic acid and polypeptide according to the invention includes the
nucleic acid
and encoded polypeptide sequence shown in Table 4 (SEQ ID N0:7 and 8). The
start and stop
codons are shown in bold. The FCTR3 nucleic acid sequence was identified from
a marine
brain library. The predicted open reading frame codes for a 370 amino acid
long secreted
protein. The FCTR3 has a predicted molecular weight of 42,808 daltons and a pI
of 7.53.
Protein structure analysis using PFAM and PROSITE identified the core PDGF
domain within
the FCTR3 polypeptide sequence.
24
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
TABLE 4. Nucleotide (SEQ ID N0:7) and Protein (SEQ ID N0:8) Sequence of FCTR3
1
ATGCAACGGCTCGTTTTAGTCTCCATTCTCCTGTGCGCGAACTTTAGCTGCTATCCGGACACTTTTGCGACTCCGCAGA
G
M Q R L V L V S I L L C A N F S C Y P D T F A T P Q R
81
AGCATCCATCAAAGCTTTGCGCAATGCCAACCTCAGGAGAGATGAGAGCAATCACCTCACAGACTTGTACCAGAGAGAG
G
A S I K A L R N A N L R R D E S N H L T D L Y Q R E E
161
AGAACATTCAGGTGACAAGCAATGGCCATGTGCAGAGTCCTCGCTTCCCGAACAGCTACCCAAGGAACCTGCTTCTGAC
A
N I Q V T S N G H V Q S P R F P N S Y P R N L L L T
241
TGGTGGCTCCGTTCCCAGGAGAAAACACGGATACAACTGTCCTTTGACCATCAATTCGGACTAGAGGAAGCAGAAAATG
A
W W L R S Q E K T R 2 Q L S F D H Q F G L E E A E N D
321
CATTTGTAGGTATGACTTTGTGGAAGTTGAAGAAGTCTCAGAGAGCAGCACTGTTGTCAGAGGAAGATGGTGTGGCCAC
A
I C R Y D F V E V E E V S E S S T V V R G R W C G H K
401
AGGAGATCCCTCCAAGGATAACGTCAAGAACAAACCAGATTAAAATCACATTTAAGTCTGATGACTACTTTGTGGCAAA
A
E I P P R I T S R T N Q I K I T F K S D D Y F V A K
481
CCTGGATTCAAGATTTATTATTCATTTGTGGAAGATTTCCAACCGGAAGCAGCCTCAGAGACCAACTGGGAATCAGTCA
C
P G F K I Y Y S F V E D F Q P E A A S E T N W E S V T
561
AAGCTCTTTCTCTGGGGTGTCCTATCACTCTCCATCAATAACGGACCCCACTCTCACTGCTGATGCCCTGGACAAAACT
G
S S F S G V S Y H S P S I T D P T L T A D A L D K T V
641
TCGCAGAATTCGATACCGTGGAAGATCTACTTAAGCACTTCAATCCAGTGTCTTGGCAAGATGATCTGGAGAATTTGTA
T
A E F D T V E D L L K H F N P V S . W Q D D L E N L Y
721
CTGGACACCCCTCATTATAGAGGCAGGTCATACCATGATCGGAAGTCCAAAGTGGACCTGGACAGGCTCAATGATGATG
T
L D T P H Y R G R S Y H D R K S K V D L D R L N D D V
801
CAAGCGTTACAGTTGCACTCCCAGGAATCACTCTGTGAACCTCAGGGAGGAGCTGAAGCTGACCAATGCAGTCTTCTTC
C
K R Y S C T P R N H S V N L R E E L K L T N A V F F P
881
CACGATGCCTCCTCGTGCAGCGCTGTGGTGGCAACTGTGGTTGCGGAACTGTCAACTGGAAGTCCTGCACATGCAGCTC
A
R C L L V Q R C G G N C G C G T V N W K S C T C S S
961
GGGAAGACAGTGAAGAAGTATCATGAGGTATTGAAGTTTGAGCCTGGACATTTCAAGAGAAGGGGCAAAGCTAAGAATA
T
G K T V K K Y H E V L K F E P G H F K R R G K A K N M
1041 GGCTCTTGTTGATATCCAGCTGGATCATCATGAGCGATGTGACTGTATCTGCAGCTCAAGACCACCTCGATAA
A L V D I Q L D H H E R C D C I C S S R P P R
FCTR4 Nucleic Acids and Polypeptides
A FCTR4 (also refered to within the specification as PDGFD or marine PDGFD or
mPDGFD) nucleic acid and polypeptide according to the invention includes the
nucleic acid
and encoded polypeptide sequence shown in Table 5 (SEQ m N0:9 and 10). The
start and
stop codons are shown in bold. The FCTR4 nucleic acid sequence was identified
from a
marine brain library and is a splice variant of FCTR3. FCTR4 has an internal
stop codon in
comparison with FCTR3. See Table 8. Unlike FCTR3, however, FCTR4 lacks a
significant
portion of the PDGF-like domain. See Table 9.
TABLE 5. Nucleotide (SEQ ID N0:9) and Protein (SEQ ID NO:10) Sequence of FCTR4
ATGCAACGGCTCGTTTTAGTCTCCATTCTCCTGTGCGCGAACTTTAGCTGCTATCCGGACACTTTTGCGACTCCGCAGA
GAGCA
TCCATCAAAGCTTTGCGCAATGCCAACCTCAGGAGAGATGAGAGCAATCACCTCACAGACTTGTACCAGAGAGAGGAGA
ACATT
CAGGTGACAAGCAATGGCCATGTGCAGAGTCCTCGCTTCCCGAACAGCTACCCAAGGAACCTGCTTCTGACATGGTGGC
TCCGT
TCCCAGGAGAAAACACGGATACAACTGTCCTTTGACCATCAATTCGGACTAGAGGAAGCAGAAAATGACATTTGTAGGT
ATGAC
TTTGTGGAAGTTGAAGAAGTCTCAGAGAGCAGCACTGTTGTCAGAGGAAGATGGTGTGGCCACAAGGAGATCCCTCCAA
GGATA
ACGTCAAGAACAAACCAGATTAAAATCACATTTAAGTCTGATGACTACTTTGTGGCAAAACCTGGATTCAAGATTTATT
ATTCA
TTTGTGGAAGATTTCCAACCGGAAGCAGCCTCAGAGACCAACTGGGAATCAGTCACAAGCTCTTTCTCTGGGGTGTCCT
ATCAC
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
TCTCCATCAATAACGGACCCCACTCTCACTGCTGATGCCCTGGACAAAACTGTCGCAGAATTCGATACCGTGGAAGATC
TACTT
AAGCACTTCAATCCAGTGTCTTGGCAAGATGATCTGGAGAATTTGTATCTGGACACCCCTCATTATAGAGGCAGGTCAT
ACCAT
GATCGGAAGTCCAAAGGTATTGAAGTTTGAGCCTGGACATTTCAAGAGAAGGGGCAAAGCTAAGAATATGGCTCTTGTT
GATAT
CCAGCTGGATCATCATGAGCGATGTGACTGTATCTGCAGCTCAAGACCACCTCGATAA
MQRLVLVSILLCANFSCYPDTFATPQRASIKALRNANLRRDESNHLTDLYQREENIQVTSNGHVQSPRFPNSYPRNLLL
TWWLR
SQEKTRIQLSFDHQFGLEEAENDICRYDFVEVEEVSESSTWRGRWCGHKEIPPRITSRTNQIKITFKSDDYFVAKPGFK
IYYS
FVEDFQPEAASETNWESVTSSFSGVSYHSPSITDPTLTADALDKTVAEFDTVEDLLKHFNPVSWQDDLENLYLDTPHYR
GRSYH
DRKSKGIEV
FCTRS Nucleic Acids and Polypeptides
A FCTRS (also refered to within the specification as PDGFD or human PDGFD or
hPDGFD or clone pCR2.1-552 2B) nucleic acid and polypeptide according to the
invention
includes the nucleic acid and encoded polypeptide sequence of FCTRS and is
shown in Table
6 (SEQ ID NO:11 and SEQ ID N0:12). The FCTRS nucleic acid sequence was
identified as a
splice variant of FCTR1.
Similar to FCTR1, protein structure analysis programs PSORT , PFAM and PROSITE
predicted that FCTRS contains a characteristic signal peptide (aa 1-23), PDGF
domain (aa
272-362) and a N-linked glycosylation site (residue 276). BLASTP analysis
revealed that the
human FGTRS is most closely related to human PDGF C, PDGF B, and PDGF A (42%,
27%,
and 25% overall amino acid identity, respectively).
TABLE 6. Nucleotide (SEQ ID NO:11) and Protein (SEQ ID N0:12) Sequence of
FCTRS
ATGCACCGGCTCATCTTGTTCTACACTCTAATCTGCGCAAACTTTTGCAGCTGTCGGGACACTTCTGCAACCCCGCAGA
GCGCATC
CATCAAAGCTTTGCGCAACGCCAACCTCAGGCGAGATGTTGACCTGGATAGGCTCAATGATGATGCCAAGCGTTACAGT
TGCACTC
CCAGGAATTACTCGGTCAATATAAGAGAAGAGCTGAAGTTGGCCAATGTGGTCTTCTTTCCACGTTGCCTCCTCGTGCA
GCGCTGT
GGAGGAAATTGTGGCTGTGGAACTGTCAACTGGAGGTCCTGCACATGCAATTCAGGGAAAACCGTGAAAAAGTATCATG
AGGTATT
ACAGTTTGAGCCTGGCCACATCAAGAGGAGGGGTAGAGCTAAGACCATGGCTCTAGTTGACATCCAGTTGGATCACCAT
GAACGAT
GCGATTGTATCTGCAGCTCAAGACCACCTCGA
MHRLILFYTLICANFCSCRDTSATPQSASIKALRNANLRRDVDLDRLNDDAKRYSCTPRNYSVNIREELKLANVVFFPR
CLLVQRC
GGNCGCGTVNWRSCTCNSGKTVKKYHEVLQFEPGHIKRRGRAKTMALVDIQLDHHERCDCICSSRPPR
FCTR6 Nucleic Acids and Polypeptides
A FCTR6 (also refered to within the specification as PDGFD or human PDGFD or
hPDGFD) nucleic acid and polypeptide according to the invention includes the
nucleic acid
and encoded polypeptide sequence of FCTR6 and is shown in Table 7 (SEQ m N0:13
and
SEQ ID N0:14). The FCTR6 sequence (also referred to as clone pCR2.1-569 4B)
was
identified as a splice variant of FCTRl .
26
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
FCTR6 contains much of the 5' end of the full length gene (FCTRl), but it is
spliced to
a cryptic, non-consensus splice site at the extreme 3' end of the coding
sequence. This
splicing introduces a STOP codon immediately downstream to the splice site.
This splice
variant contains the intact CUB domain of 30664188Ø99, but deletes the PDGF
domains,
indicating a possible regulatory function of the molecule.
Similar to FCTRl, however, protein structure analysis programs PSORT , PFAM
and
PROSITE predicted that FCTR6 contains a characteristic signal peptide (aa 1-
23), a CUB
domain (aa 53-167) and an N-linked glycosylation site (residue 276). BLASTP
analysis
revealed that the human FGTRS is most closely related to human PDGF C, PDGF B,
and
PDGF A (42%, 27%, and 25% overall amino acid identity, respectively).
TABLE 7. Nucleotide (SEQ ID N0:13) and Protein (SEQ ID N0:14) Sequence of
FCTR6
ATGCACCGGCTCATCTTTGTCTACACTCTAATCTGCGCAAACTTTTGCAGCTGTCGGGACACTTCTGCAACCCCGCA
GAGCGCATCCATCAAAGCTTTGCGCAACGCCAACCTCAGGCGAGATGAGAGCAATCACCTCACAGACTTGTACCGAA
GAGATGAGACCATCCAGGTGAAAGGAAACGGCTACGTGCAGAGTCCTAGATTCCCGAACAGCTACCCCAGGAACCTG
CTCCTGACATGGCGGCTTCACTCTCAGGAGAATACACGGATACAGCTAGTGTTTGACAATCAGTTTGGATTAGAGGA
AGCAGAAAATGATATCTGTAGGTAGAGCTAAGACCATGGCTCTAGTTGACATCCAGTTGGATCACCATGAACGATGC
GATTGTATCTGCAGCTCAAGACCACCTCGA
MHRLIFVYTLICANFCSCRDTSATPQSASIKALRNANLRRDESNHLTDLYRRDETIQVKGNGYVQSPRFPNSYPRNL
LLTWRLHSQENTRIQLVFDNQFGLEEAENDICR
FCTRX sequences
The various FCTRX nucleic acids and polypeptides are disclosed in related
applications USSN 60/158,083, filed October 7, 1999; USSN 60/159, 231, filed
October 13,
1999; USSN 60/174,485 filed January 4, 2000; USSN 60/186,707 filed March 3,
2000; USSN
60/188,250, filed March 10, 2000; USSN 60/223,879, filed August 8, 2000; USSN
60/234,082, filed on September 20, 2000; USSN 09/685,330, filed on October 5,
2000; PCT
Application US00/27671, filed October 6, 2000; USSN 09/688,312, filed October
13, 2000;
USSN 09/715,332 filed November 16, 2000; and USSN 09/775,482 filed February 2,
2001.
Each of these applications is incorporated by reference in its entirety.
FCTRX amino acid sequence variants were analyzed with ClustalW software. The
resulting sequence alignment is shown in Table 8.
Table 8: Alignment of FCTRX Polypeptide Sequences.
27
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
20 30 40 50
.I....I....I....~. ..I....~.. .I. .I. .I. ..I
FCTR1 ~H~F~YT~T~CSC'~ .~ ~~S~ ~. ~ '~ n 50
FCTR2 ----------L~YLBRPRY'G THDKS---------------------- 18
FCTR3 Q' VL SI L ~ SCYP~ F. ~. .. r~ ~ ~~ ~ 50
FCTR4 Q' VL~SI L ' SCYP~ F' ~ ' ~~ ~ '~ ~ 50
FCTR5 H' I'LFYT I ~ CSC' ~ ~~S~ ' ' ~ ~~--------- 41
FCTR6 H' LFDYT I ~ CSC ~ ~ ~~S~ ' ~ ~ '~ ~ 50
60
70
80
90
100
...I....~....I.
FCTR1.I. 100
.~....I....~....I....I
~D~T~KG~Y
~
'
R~H~N~VmN~
FCTR2-_________________________________________________
18
FCTR3Q~E~N 100
TS
H
~
'
5
~
FCTR4~ 100
~
~
~
~
Q
E
N
TS
H
~
'~D~
ii
S
1Hi
FCTR5
_____________
_____________________________________ 1
FCTR6~D~T~KG~Y 10
0
~R~H~N
'~~
VmN~
110
120
130
140
150
I
.
FCTR1.. 15
.I.. 0
.I.
..1....I.
I
I....I.
.I..
.I
.
,
~
'
~
.
'
DI~T~I
I~~V~IC~
FCTR2-_________________________________________________
18
FCTR3 ~ ' 1 Et/~ V ~ T 15
0
FCTR4a a ~ ~ -. ~ ~ t~ I~ T' ~ 15
~ ;~ E~~ 0
5
FCTR5-__~_____ 41
______==_______________________________
FCTR6~ 110
_______________________________________
160
170
180
190
200
I
I
.
FCTR1.... 2
. 0
..~ 0
.I..
.I....~.
.I.
.I...
.
~
v
'
,I
.
.
.
L3L
.
.
.
.
,
I~~~'
FCTR2__________________________________________________
18
FCTR3W 2
0
Frc' 0
~
E
'
F
H
'
1
FCTR4~~
200
~~
F
~
~
E
F~~~~
FCTR5_
_ 41
________
______________
-__________
------
FCTR6__________________________________________________
110
210
220
230
240
250
~.~.. 250
FCTR1I
.
..
I
.
.
.
I
~I~.
.
I
.~...~~~
v
I
'
~
n
K~
n'
v
Y
E
NE
~
~
FCTR2__________________________________________
18
FCTR3~T 2
' 5
~ 0
'
~
T;
~
~
U
Q
H
FCTR4H~ 250
~
p~~,~
~
~
T~'
~
T
~
~
H
V
Q'D~
L
~
H
FCTR5. 41
__________________________________________________
FCTR6__________________________________________________
110
260 270 280 290 300
....1....I. .I. .I. .I. .1_.__1___m i
FCTR1 m ~ ~~ m Y I 299
FCTR2 _____ ~ ~~ ~1 ~~ Y I' ~ 61
FCTR3 1~ ~ 1~ 1~ H L' T ~' 299
FCTR4 ~~ GIEV-_______________-______________________ 261
FCTR5 _______ m ~y~T~~ 83
FCTR6 ___________________~i_i_~_i_~_.__~__C_1_i_~_____~____
°__________ 110
310 320 330 340 350
.1....I. .~. .I....I....I....I....I....I....I
FCTRl T.~ Q I ' ~ T ' 349
FCTR2 N Q I ' ~ T ' 111
28
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
FCTR3 ~T S ' F ~ ~ N ~ 349
FCTR4 ______________.___________________________________ 261
FCTR5 i~ i 1 T~R~N ~y~Q~I~RmT~ 133
~~FCTR6 ________________________________._________________ 110
360 370
FCTR1 .~~ ~ ~ ~~ 370
FCTR2 ~ i ~ ~ ~~' 132
FCTR3 ~ i ~ ~ "~ 370
FCTR4 ------------_-------- 261
FCTR5 ~ i ~ ~~ ~ R~ 154
FCTR6 _____________________ 110
Nucleic acids of FCTR1, FCTR3, FCTR4 and FCTR6 are aligned with each other
over
the nucleotide residues shown in Tahle 9_
Table 9: Alignment of SEQ ID NOS:3, 7, 9 and 13, respectively.
160 170 180 190 200
....~....~....~....~....p..
FCTR1 GAGCGACGCTGTCTCTAGTCGCTGATCCC ~. ~C. C..T 200
FCTR3 _______________________________. . G T l9
FCTR4 ________.______________________. . G T 19
FCTR6 _______________________________. .C C T 19
210 220 ~ 230 240 250
FCTR1 250
FCTR3 69
FCTR4 69
FCTR6
69
260 270 280 290 300
FCTR1
300
FCTR3
119
FCTR4
119
FCTR6 119
310 320 330 340 350
FCTR1 350
FCTR3 169
FCTR4
169
FCTR6
169
360 370 380 390 400
FCTR1 400
FCTR3 219
FCTR4 219
FCTR6 219
410 420 430 440 450
29
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
FCTRl 450
FCTR3 269
FCTR4 269
FCTR6 269
460 470 480 490 500
FCTR1 500
FCTR3 319
FCTR4 319
FCTR6 319
5l0 520 530 540 550
.I. .1~~..I. .I. .I. ~I~ ~I.~ .I. .1....I
FCTR1 ~~,T~ C ~~~ ~ ~T. ~-~. '~ ~T-. .C ' 'C ~ 548
FCTR3 f,~'1,C ' T ~ ~ ~ ~ - ~ ~ ~~~ ~ ~, -G C ' ~ G ~' ~, 3 67
FCTR4 ilC' T ~ ~ ~ ~-~ ~~ ~ -G C 'G~ ~ 367
FCTR6 ~T~C ~ 'GAG CCiTG C CT' TTGAC CAGTT Gi 369
560 570 580 590 600
FCTR1 5gg
FCTR3 417
FCTR4 417
FCTR6 415
610 620 630 640 650
FCTR1 648
FCTR3 467
FCTR4 467
FCTR6 __________________________________________________ 415
660 670 680 690 700
FCTR1 698
FCTR3 517
FCTR4 517
FCTR6 __________________________________________________ 415
710 720 730 740 750
FCTR1 748
FCTR3 567
FCTR4 567
FCTR6 __________________________________________________ 415
760 770 780 790 800
FCTR1 798
FCTR3 617
FCTR4 617
FCTR6 __________________________________________________ 415
810 820 830 840 850
FCTRl .G ~ T . ~ ~~~~ T .~ ~~ .T ~ ~ ~~ ~ g4g
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
FCTR3 T ~ C ~ ~~~~CTG C ~ ~~ C ~ ~ C ~~ ~ 667
FCTR4 ~T~C ~ " ~~CTGBC ~ '~ C ~ ~ C ~~ ~ 667
FCTR6 __________________________________________________ 415
FCTR1 ggg
FCTR3 717
FCTR4 717
FCTR6 _______________________________-__________________ 415
FCTR1 g4g
FCTR3 767
FCTR4 767
FCTR6 __________________________________________________ 415
960 980 990 1000
970
FCTR1 .' TTGACCTG T GCT . TG .C TTAC TTGCA 998
GC
FCTR3 C TGGACCTG C GCT TG 817
GT C TTAC TTGCA
FCTR4 C GTATTG TTT GC - AC ' GGG CAAAG 815
G TT
FCTR6 ___________________________ _______________________415
1010 1020 1030 1040 1050
FCTR1 ~CCC~,~'1,GG~ TTACTCG C ~T GAG ~G TG~ GT GC ~ 1048
FCTR3 CCC~G~TCACTCT G ~ CC CAGGGAG ~ G TG~GC C ~ 8 67
FCTR4 G~~~'n'TiTGGCTCTT TG~T CCAGCTG ~T - ~iTC G G~ 863
FCTR6 _________-____________-_________________-_________ 415
1060 1070 1080
1090
1100
FCTR1 TGGTCT.C TTC GTTG T. TGC~IGCGCTGTGGAGGAAA1095
--- '
FCTR3 CAGTCT C TCC GATG TGCGCGCTGTGGTGGCAA914
--- T
FCTR4 TGACTG CTG G T ---------------ggg
GCT
FCTR6 __________________ ________________________________
415
1110 1120 1130 1140 1150
FCTR1 TTGTGGCTGTGGAACTGTCAACTGGAGGTCCTGCACATGCAATTCAGGGA 1145
FCTR3 CTGTGGTTGCGGAACTGTCAACTGGAAGTCCTGCACATGCAGCTCAGGGA 964
FCTR4 __________________________________________________ 898
FCTR6 __________________________________________________ 415
1160 1170 1180 1190 1200
FCTR1 AAACCGTGAAAAAGTATCATGAGGTATTACAGTTTGAGCCTGGCCACATC 1195
FCTR3 AGACAGTGAAGAAGTATCATGAGGTATTGAAGTTTGAGCCTGGACATTTC 1014
FCTR4 __________________________________________________ g98
FCTR6 __________________________________________________ 415
1210 1220 1230 1240 1250
..
FCTR1 AAGAGGAGGGGTAGAGCTAAGACCATGGCTCTAGTTGACATCCAGTTGGA 1245
FCTR3 AAGAGAAGGGGCAAAGCTAAGAATATGGCTCTTGTTGATATCCAGCTGGA 1064
FCTR4 __________________________________________________ 898
31
860 870 880 890 900
910 920 930 940 950
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
FCTR6 --_________________________________________________ 415
1260 1270 1280 1290 1300
....~....~....~....~....~....~....~....~....~....~
FCTR1 TCACCATGAACGATGTGATTGTATCTGCAGCTCAAGACCACCTCGATAAG 1295
FCTR3 TCATCATGAGCGATGTGACTGTATCTGCAGCTCAAGACCACCTCGATAA- 1113
FCTR4 __________________________________________________ g98
FCTR6 __________________________________________________ 415
Amino acids of FCTR1, FCTR3, FCTR4 and FCTR6 are aligned with each other as
shown in Table 10.
Table 10: Alignment of SEQ ID NOS:4, 8,10 and 14, respectively.
20 30 40 50
FCTR1 50
FCTR3 50
FCTR4 50
FCTR6 50
60 70 80 90 100
FCTR1 100
FCTR3 100
FCTR4
100
FCTR6 100
110 l20 130 140 150
FCTR1 .~ ~' ~. ~p1 T' ~~~' ~~ ~.~ 150
FCTR3 ~ ~ ~ ~V S tT- ~ 1~T15 0
FCTR4 ~ 1 ~ ~~7 3 I ~ ~ T 150
FCTR6 ~ ~ _______________________________________ 110
1.60 170 180 190 200
FCTR1 200
FCTR3 200
FCTR4
200
FCTR6 __________________________________________________ 110
210 220 230 240 250
FCTR1
250
FCTR3 250
FCTR4
250
FCTR6 _______________________________,__________________ 110
260 270 280 290 300
.~....~.. .~.. .~....~....~.. .~. .~. .~. .)
FCTR1 ' 1 ~ tj'p1, ~ ~ y ~
FCTR3 ~ ~T~1, ~ ~ ~ ~ . ~ 2 9 9
H~L~a5 ~~T 299
FCTR4 ~~ G~~~-______________________________________ 261
FCTR6 __________________________________________________ 110
32
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
310 320 330 340 350
.I. .~. .~. .~....~. .~. .I.
FCTR1 . ~ F~y, N1. 349
FCTR3 1'
FCTR4 __________________________________________________ 261
FCTR6 __________________________________________________ 110
360 370
FCTR1 1 1 ~ ~~' 370
FCTR3 ~ 1 ~ R " 3 7 0
FCTR4 _____________________ 361
FCTR6 --------------_------ 110
Nucleic acids of FCTR2 and FCTRS are aligned with each other over the
nucleotide
residues shown in Table 11.
Table 11: Alignment of SEQ ID NO:S and SEQ ID NO:11, respectively.
460 470 480 490 500
....I....I....1....1....1....1....1....1. .I. ..I
FCTR2 CTCTGGACAAAAAAATTGCAGAATTTG~ ~GT~G~GC~CAAG 500
FCTR5 ' G ~CC CTC~ TG~TCTA 23
510 520 530 540 550
.I....I....I....1....1....1....1....1....1....1
FCTR2 T~TC~~ CA~TC'TGGC~C~T---~T~T T 547
FCTR5 C ~~~~,, CT ~ ~ T-,~~, C~C ~ CTTTT C ~ C G,~~, C~G~,,r~~ CACT Ciili~~C ~
C 7 2
560 570 580 590 600
.I....I....I....1....1....1....1....1....1....1
FCTR2 GGA~CCC~T~GT-~ G~~1G GGT~T~~'IC T-G' GG~TC~ ~ 595
FCTR5 CCG~~GAG G TCC~ ~ TTTG GC~ GCC ~ TC~G CG~G~ 122
610 620 630 640 650
.1....1....I.. .I.. .I.. .I.. .1....I. .I. .I
FCTR2 ~ ~ ~ '~ ~~ '. ~ ~~ . . ~~ 645
FCTR5 T ~ ~ ~ ~~ ~ ~ Ii ~~ ~ ~ ~ ~ 172
660 670 680 690 700
..I.. .I. .1....I. ..1....I.. .I.. .I. .I. .I
FCTR2 ~ . .. . ~. . . . . ~.I . . . ~.. . . ~ ~ 695
FCTR5 . .. . ~.. . .. ~ .. ~ .. .. 222
710 720 730 740 750
.I. .1....1....1....1....1.. .I.. .1....1....I
FCTR2 ~I ~'v .
i ., ... 745
FCTR5 ~~
., ... i 272
760 770 780 790 800
.1....1....1....1.. .I.. .I.. .I.. .1....1....I
FCTR2 .. .. . . . ~ .. . .i... 795
FCTR5 I, . . . . . . . . . .. .~ . 322
810 820 830 840 850
.1....1....1....1.. .I.. .I.. .I. .1....1....I
FCTR2 ~..... . . . . . . . . . .. . 845
FCTR5 ..... . , I. . . . . ~ . .. . 372
33
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
860 870 880 890 900
FCTR2 ~.. ~ . .. . . . . . . ~. .~ ~ 895
FCTR5 ~ ~ . .. . . . . . . . . . 422
910 920 930 940 950
FCTR2 ~~~ ~~~T '. ~. ~ ~~ ~~ i~ ~TAAGAGAATG 945
FCTR5 ~ ' ~ C ' ~ ~ ~ ~ ~ I~ ~ ~ 4 62
Amino acids of FCTR2 and FCTRS are aligned with each other as shown in Table
12.
Table 12: Alignment of SEQ ID N0:6 and SEQ ID N0:12, respectively.
20 30 40 50
..
FCTR2 ------- L~YLDRPRY G .THD~t~------------KS n ~ ~~w 28
FCTR5 MHRLILFYTL;CANFCSCBDTBATPQSASIKALRNANL~RD ~ ~~ m 50
60 70 80 90 100
~I~ .I. .I. .I. .I. .I. .I. .I. .I. .I
FCTR2 ~. ~ ~ ~ ~~ 1~ '~'~~ ,,, 78
FCTR5 ~ .. I~ . . ~,1I . ~,~a~~ 1~ . ~T~~ 100
110 120 130 140 150
~I. .I. .I. .I. .I. .I. .I. .I. .1....I
FCTR2 1~5 ~~ ~ 1 ~ ~ 128
FCTR5 ~ ~ ~. ~ ~ ~ ~ m v ~ ~ ;~ ~ ~ m ~ . 15 0
FCTR2 ~ 132
FCTR5 ~~ 154
The similarities of the disclosed FCTRX polypeptides to previously described
BMP-1
5 VEGF-E and PDGF polypeptides indicate a similarity of functions by the FCTRX
nucleic
acids and polypeptides of the invention. These utilities are described in more
detail below.
FCTRX nucleic acids and polypeptides may be use to induce formation of
cartilage, as
BMP-1 is also capable of inducing formation of cartilage in vivo (Wozney et
al., Science 242:
1528-1534 (1988)).
10 An additional use for the FCTRX nucleic acids and polypeptides is in the
modulation
of collagen formation. Recombinantly expressed BMP 1 and purified procollagen
C proteinase
(PCP), a secreted metalloprotease requiring calcium and needed for cartilage
and bone
formation, are, in fact, identical. See, Kessler et al., Science 271:360-62
(1996). BMP-1
cleaves the C-terminal propeptides of procollagen I, II, and III and its
activity is increased by
the procollagen C-endopeptidase enhancer protein. FCTRX nucleic acids and
polypeptides
may play similar roles in collagen modulation pathways.
34
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
It is shown in the Examples below that FCTRX polypeptides have the ability to
reduce
or ameliorate the extent of inflammatory response in two animal models of
inflammatory
bowel disease.
The similarity between FCTRX polypeptides and PDGF polypeptides suggests that
FCTRX nucleic acids and their encoded polypeptides can be used in various
therapeutic and
diagnostic applications. For example, FCTRX nucleic acids and their encoded
polypeptides
can be used to treat cancer, cardiovascular and fibrotic diseases and diabetic
ulcers. In
addition, FCTRX nucleic acids and their encoded polypeptides will be
therapeutically useful
for the prevention of aneurysms and the the acceleration of wound closure
through gene
therapy. Furthermore, FCTRX nucleic acids and their encoded polypeptides can
be utilized
to stimulate cellular growth.
A FCTRX nucleic acid or gene product, e. g., a nucleic acid encoding SEQ ID
N0:4 or
SEQ ID N0:6, is useful as a therapeutic agent in promoting wound healing,
neovascularization
and tissue growth, and similar tissue regeneration needs. More specifically, a
FCTRX nucleic
acid or polypeptide may be useful in treatment of anemia and leukopenia,
intestinal tract
sensitivity and baldness. Treatment of such conditions may be indicated in, e.
g., patients
having undergone radiation or chemotherapy. It is intended in such cases that
administration
of a FCTX nucleic acid or polypeptide, e. g., a polypeptide including the
amino acid sequence
of SEQ ID N0:4 or SEQ ID N0:6, or a nucleic acid sequence encoding these
polypeptides (e.
g., SEQ ID N0:3 or SEQ ID NO:S) will be controlled in dose such that any
hyperproliferative
side effects are minimized.
The invention also includes mature FGF-CX and/or FCTRX polypeptides, variants
of
mature FGF-CX and/or FCTRX polypeptides, fragments of mature and mature
variant FGF-
CX and/or FCTRX polypeptides, and nucleic acids encoding these polypeptides
and
fragments. As used herein, a "mature" form of a FGF-CX and/or FCTRX
polypeptide or
protein disclosed in the present invention is the product of a naturally
occurnng polypeptide or
precursor form or proprotein. The naturally occurring polypeptide, precursor
or proprotein
includes, by way of nonlimiting example, the full length gene product, encoded
by the
corresponding gene. In some embodiments, the mature form include an FGF-CX
and/or
FCTRX polypeptide, precursor or proprotein encoded by an open reading frame
described
herein. The product "mature" form can arise, e. g., as a result of one or more
naturally
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
occurring processing steps as they may take place within the cell, or host
cell, in which the
gene product arises.
Examples of such processing steps leading to a "mature" form of a polypeptide
or
protein include the cleavage of the N-terminal methionine residue encoded by
the initiation
codon of an open reading frame, or the proteolytic cleavage of a signal
peptide or leader
sequence. Thus a mature form arising from a FGF-CX or a FCTRX precursor
polypeptide or
protein that has residues 1 to N, where residue 1 is the N-terminal
methionine, would have
residues 2 through N remaining after removal of the N-terminal methionine.
Alternatively, a
mature form arising from a precursor polypeptide or protein having residues 1
to N, in which
an N-terminal signal sequence from residue 1 to residue M is cleaved, would
have the residues
from residue M+1 to residue N remaining. Additionally, a "mature" protein or
fragment may
arise from a cleavage event other than removal of an initiating methionine or
removal of a
signal peptide. Further as used herein, a "mature" form of a FGF-CX and/or a
FCTRX
polypeptide or protein may arise from a step of post-translational
modification other than a
proteolytic cleavage event. Such additional processes include, by way of non-
limiting
example, glycosylation, myristoylation or phosphorylation. In general, a
mature polypeptide
or protein may result from the operation of only one of these processes, or a
combination of
any of them.
As used herein, "identical" residues correspond to those residues in a
comparison
between two sequences where the equivalent nucleotide base or amino acid
residue in an
alignment of two sequences is the same residue. Residues are alternatively
described as
"similar" or "positive" when the comparisons between two sequences in an
alignment show
that residues in an equivalent position in a comparison are either the same
amino acid or a
conserved amino acid as defined below.
Included within the invention are FGF-CX and FCTRX nucleic acids, isolated
nucleic
acids that encode FGF-CX and FCTRX polypeptides or a portion thereof, FGF-CX
and
FCTRX polypeptides, vectors containing these nucleic acids, host cells
transformed with the
FGF-CX and/or FCTRX nucleic acids, anti-FGF-CX and/or FCTRX antibodies, and
pharmaceutical compositions. Also disclosed are methods of making FGF-CX
andlor FCTRX
polypeptides, as well as methods of screening, diagnosing, treating conditions
using these
compounds, and methods of screening compounds that modulate FGF-CX and/or
FCTRX
polypeptide activity. The FGF-CX and/or FCTRX nucleic acids and polypeptides,
as well as
36
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
FGF-CX and/or FCTRX antibodies, therapeutic agents and pharmaceutical
compositions
discussed herein, are useful, inter alia, in treating inflammatory conditions,
as well as tissue
proliferation-associated disorders.
FGF-CX and/or FCTRX Nucleic Acids and Polypeptides
A summary of the FGF-CX and/or FCTRX nucleic acids and proteins of the
invention
is provided in Table 13.
TABLE 13: Summary Of Nucleic Acids And Proteins Of The Invention
Clone Table Clone alias Nucleic Amino
Acid SEQ Acid SEQ
ID NO ID NO
FGF-CX 1 AB020858; CG53135-O1; CG53135-02;1 2
TA-AB02085-5274-F19; 20858
FCTRl 2 PDGFD; 30664188; 30664188Ø99;3 4
CG52053; CG52053-02; 30664188Øm99;
30664188-S311a;30664188-Slla
FCTR2 3 PDGFD; 30664188Ø331; CG52053-O15 6
FCTR3 4 PDGFD; marine PDGFD; mPDGFD 7 g
FCTR4 5 PDGFD; marine PDGFD; mPDGFD 9 10
FCTRS 6 PDGFD; human PDGFD; hPDGFD; 11 12
clone
CR2.1-5852 2B
FCTR6 7 PDGFD; human PDGFD; hPDGFD; 13 14
clone
pCR2.1- 5869 4B
One aspect of the invention pertains to isolated nucleic acid molecules that
encode
FGF-CX and/or FCTRX polypeptides or biologically active portions thereof. Also
included in
the invention are nucleic acid fragments sufficient for use as hybridization
probes to identify
FGF-CX and/or FCTRX-encoding nucleic acids (e.g., FGF-CX and/or FCTRX mRNAs)
and
fragments for use as PCR primers for the amplification and/or mutation of FGF-
CX and/or
FCTRX nucleic acid molecules. As used herein, the term "nucleic acid molecule"
is intended
to include DNA molecules (e.g., cDNA or genomic DNA), RNA molecules (e.g.,
mRNA),
analogs of the DNA or RNA generated using nucleotide analogs, and derivatives,
fragments
and homologs thereof. The nucleic acid molecule may be single-stranded or
double-stranded,
but preferably is comprised double-stranded DNA.
An FGF-CX and/or FCTRX nucleic acid can encode a mature FGF-CX and/or FCTRX
polypeptide. As used herein, a "mature" form of a polypeptide or protein
disclosed in the
37
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
present invention is the product of a naturally occurring polypeptide or
precursor form or
proprotein. The naturally occurring polypeptide, precursor or proprotein
includes, by way of
nonlimiting example, the full-length gene product, encoded by the
corresponding gene.
Alternatively, it may be defined as the polypeptide, precursor or proprotein
encoded by an
ORF described herein. The product "mature" form arises, again by way of
nonlimiting
example, as a result of one or more naturally occurring processing steps as
they may take place
within the cell, or host cell, in which the gene product arises. Examples of
such processing
steps leading to a "mature" form of a polypeptide or protein include the
cleavage of the N-
terminal methionine residue encoded by the initiation codon of an ORF, or the
proteolytic
cleavage of a signal peptide or leader sequence. Thus a mature form arising
from a precursor
polypeptide or protein that has residues 1 to N, where residue 1 is the N-
terminal rriethionine,
would have residues 2 through N remaining after removal of the N-terminal
methioiune.
Alternatively, a mature form arising from a precursor polypeptide or protein
having residues 1
to N, in which an N-terminal signal sequence from residue 1 to residue M is
cleaved, would
have the residues from residue M+1 to residue N remaining. Further as used
herein, a
"mature" form of a polypeptide or protein may arise from a step of post-
translational
modification other than a proteolytic cleavage event. Such additional
processes include, by
way of non-limiting example, glycosylation, myristoylation or phosphorylation.
In general, a
mature polypeptide or protein may result from the operation of only one of
these processes, or
a combination of any of them.
The term "probes", as utilized herein, refers to nucleic acid sequences of
variable
length, preferably between at least about 10 nucleotides (nt), 100 nt, or as
many as
approximately, e.g., 6,000 nt, depending upon the specific use. Probes are
used in the
detection of identical, similar, or complementary nucleic acid sequences.
Longer length
probes are generally obtained from a natural or recombinant source, are highly
specific, and
much slower to hybridize than shorter-length oligomer probes. Probes may be
single- or
double-stranded and designed to have specificity in PCR, membrane-based
hybridization
technologies, or ELISA-like technologies.
The term "isolated" nucleic acid molecule, as utilized herein, is one, which
is separated
from other nucleic acid molecules which are present in the natural source of
the nucleic acid.
Preferably, an "isolated" nucleic acid is free of sequences which naturally
flank the nucleic
acid (i.e., sequences located at the 5'- and 3'-termini of the nucleic acid)
in the genomic DNA
3S
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
of the organism from which the nucleic acid is derived. For example, in
various embodiments,
the isolated FGF-CX and/or FCTRX nucleic acid molecules can contain less than
about 5 kb,
4 kb, 3 kb, 2 kb, 1 kb, 0.5 kb or 0.1 kb of nucleotide sequences which
naturally flank the
nucleic acid molecule in genomic DNA of the cell/tissue from which the nucleic
acid is
derived (e.g., brain, heart, liver, spleen, etc.). Moreover, an "isolated"
nucleic acid molecule,
such as a cDNA molecule, can be substantially free of other cellular material
or culture
medium when produced by recombinant techniques, or of chemical precursors or
other
chemicals when chemically synthesized.
A nucleic acid molecule of the invention, e.g., a nucleic acid molecule having
the
nucleotide sequence of SEQ m NOS:1, 3, 5, 7, 9, 11 and 13, or a complement of
this
aforementioned nucleotide sequence, can be isolated using standard molecular
biology
techniques and the sequence information provided herein. Using all or a
portion of the nucleic
acid sequence of SEQ )D NOS:1, 3, 5, 7, 9, 11 and 13 as a hybridization probe,
FGF-CX
and/or FCTRX molecules can be isolated using standard hybridization and
cloning techniques
(e.g., as described in Sambrook, et al., (eds.), MOLECULAR CLONING: A
LABORATORY
MANUAL 2"d Ed., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY,
1989; and
Ausubel, et al., (eds.), CURRENT PROTOCOLS IN MOLECULAR BIOLOGY, John Wiley &
Sons,
New York, NY, 1993.)
A nucleic acid of the invention can be amplified using cDNA, mRNA or
alternatively,
genomic DNA, as a template and appropriate oligonucleotide primers according
to standard
PCR amplification techniques. The nucleic acid so amplified can be cloned into
an
appropriate vector and characterized by DNA sequence analysis. Furthermore,
oligonucleotides corresponding to FGF-CX and/or FCTRX nucleotide sequences can
be
prepared by standard synthetic techniques, e.g., using an automated DNA
synthesizer.
As used herein, the term "oligonucleotide" refers to a series of linked
nucleotide
residues, which oligonucleotide has a sufficient number of nucleotide bases to
be used in a
PCR reaction. A short oligonucleotide sequence may be based on, or designed
from, a
genomic or cDNA sequence and is used to amplify, confirm, or reveal the
presence of an
identical, similar or complementary DNA or RNA in a particular cell or tissue.
Oligonucleotides comprise portions of a nucleic acid sequence having about 10
nt, 50 nt, or
100 nt in length, preferably about 15 nt to 30 nt in length. In ones
embodiment of the
invention, an oligonucleotide comprising a nucleic acid molecule less than 100
nt in length
39
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
would fiuther comprise at least 6 contiguous nucleotides of SEQ m NOS:1, 3, 5,
7, 9, 11 and
13, or a complement thereof. Oligonucleotides may be chemically synthesized
and may also
be used as probes.
In another embodiment, an isolated nucleic acid molecule of the invention
comprises a
nucleic acid molecule that is a complement of the nucleotide sequence shown in
SEQ m
NOS:1, 3, 5, 7, 9, 11 and 13, or a portion of this nucleotide sequence (e.g.,
a fragment that can
be used as a probe or primer or a fragment encoding a biologically-active
portion of an FGF-
CX and/or FCTRX polypeptide). A nucleic acid molecule that is complementary to
the
nucleotide sequence shown in SEQ ID NOS:1, 3, 5, 7, 9, 11 and 13 is one that
is sufficiently
complementary to the nucleotide sequence shown in SEQ m NOS:1, 3, 5, 7, 9, 11
and 13 that
it can hydrogen bond with little or no mismatches to the nucleotide sequence
shown SEQ ID
NOS:1, 3, 5, 7, 9, 11 and 13, thereby forming a stable duplex.
As used herein, the term "complementary" refers to Watson-Crick or Hoogsteen
base
pairing between nucleotides units of a nucleic acid molecule, and the term
"binding" means
the physical or chemical interaction between two polypeptides or compounds or
associated
polypeptides or compounds or combinations thereof. Binding includes ionic, non-
ionic, van
der Waals, hydrophobic interactions, and the like. A physical interaction can
be either direct
or indirect. Indirect interactions may be through or due to the effects of
another polypeptide or
compound. Direct binding refers to interactions that do not take place
through, or due to, the
effect of another polypeptide or compound, but instead are without other
substantial chemical
intermediates.
Fragments provided herein are defined as sequences of at least 6 (contiguous)
nucleic
acids or at least 4 (contiguous) amino acids, a length sufficient to allow for
specific
hybridization in the case of nucleic acids or for specific recognition of an
epitope in the case of
amino acids, respectively, and are at most some portion less than a full
length sequence.
Fragments may be derived from any contiguous portion of a nucleic acid or
amino acid
sequence of choice. Derivatives are nucleic acid sequences or amino acid
sequences formed
from the native compounds either directly or by modification or partial
substitution. Analogs
are nucleic acid sequences or amino acid sequences that have a structure
similar to, but not
identical to, the native compound but differs from it in respect to certain
components or side
chains. Analogs may be synthetic or from a different evolutionary origin and
may have a
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
similar or opposite metabolic activity compared to wild type. Homologs are
nucleic acid
sequences or amino acid sequences of a particular gene that are derived from
different species.
Derivatives and analogs may be full length or other than full length, if the
derivative or
analog contains a modified nucleic acid or amino acid, as described below.
Derivatives or
analogs of the nucleic acids or proteins of the invention include, but are not
limited to,
molecules comprising regions that are substantially homologous to the nucleic
acids or
proteins of the invention, in various embodiments, by at least about 70%, 80%,
or 95%
identity (with a preferred identity of 80-95%) over a nucleic acid or amino
acid sequence of
identical size or when compared to an aligned sequence in which the alignment
is done by a
computer homology program known in the art, or whose encoding nucleic acid is
capable of
hybridizing to the complement of a sequence encoding the aforementioned
proteins under
stringent, moderately stringent, or low stringent conditions. See e.g.
Ausubel, et al., CURRENT
PROTOCOLS irr MOLECULAR BIOLOGY, John Wiley & Sons, New York, NY, 1993, and
below.
A "homologous nucleic acid sequence" or "homologous amino acid sequence," or
variations thereof, refer to sequences characterized by a homology at the
nucleotide level or
amino acid level as discussed above. Homologous nucleotide sequences encode
those
sequences coding for isoforms of FGF-CX and/or FCTRX polypeptides. Isoforms
can be
expressed in different tissues of the same organism as a result of, for
example, alternative
splicing of RNA. Alternatively, isoforms can be encoded by different genes. In
the invention,
homologous nucleotide sequences include nucleotide sequences encoding for an
FGF-CX
and/or FCTRX polypeptide of species other than humans, including, but not
limited to:
vertebrates, and thus can include, e.g., frog, mouse, rat, rabbit, dog, cat
cow, horse, and other
organisms. Homologous nucleotide sequences also include, but are not limited
to, naturally
occurring allelic variations and mutations of the nucleotide sequences set
forth herein. A
homologous nucleotide sequence does not, however, include the exact nucleotide
sequence
encoding human FGF-CX and/or FCTRX protein. Homologous nucleic acid sequences
include those nucleic acid sequences that encode conservative amino acid
substitutions (see
below) in SEQ ID NOS:1, 3, 5, 7, 9, 11 and 13, as well as a polypeptide
possessing FGF-CX
and/or FCTRX biological activity. Various biological activities of the FGF-CX
and/or
FCTRX proteins are described below.
As used herein, "identical" residues correspond to those residues in a
comparison
between two sequences where the equivalent nucleotide base or amino acid
residue in an
41
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
alignment of two sequences is the same residue. Residues are alternatively
described as
"similar" or "positive" when the comparisons between two sequences in an
alignment show
that residues in an equivalent position in a comparison are either the same
amino acid or a
conserved amino acid as defined below.
An FGF-CX and/or FCTRX polypeptide is encoded by the open reading frame
("ORF") of an FGF-CX and/or FCTRX nucleic acid. An ORF corresponds to a
nucleotide
sequence that could potentially be translated into a polypeptide. A stretch of
nucleic acids
comprising an ORF is uninterrupted by a stop codon. An ORF that represents the
coding
sequence for a full protein begins with an ATG "start" codon and terminates
with one of the
three "stop" codons, namely, TAA, TAG, or TGA. For the purposes of this
invention, an ORF
may be any part of a coding sequence, with or without a start codon, a stop
codon, or both.
For an ORF to be considered as a good candidate for coding for a bona fide
cellular protein, a
minimum size requirement is often set, e.g., a stretch of DNA that would
encode a protein of
50 amino acids or more.
The nucleotide sequences determined from the cloning of the human FGF-CX
and/or
FCTRX genes allows for the generation of probes and primers designed for use
in identifying
and/or cloning FGF-CX and/or FCTRX homologues in other cell types, e.g. from
other tissues,
as well as FGF-CX and/or FCTRX homologues from other vertebrates. The
probe/primer
typically comprises substantially purified oligonucleotide. The
oligonucleotide typically
comprises a region of nucleotide sequence that hybridizes under stringent
conditions to at least
about 12, 25, 50, 100, 150, 200, 250, 300, 350 or 400 consecutive sense strand
nucleotide
sequence of SEQ m NOS:l, 3, 5, 7, 9, 11 and 13; or an anti-sense strand
nucleotide sequence
of SEQ m NOS:1, 3, 5, 7, 9, 11 and 13; or of a naturally occurring mutant of
SEQ m NOS:1,
3, 5, 7, 9, 11 and 13.
Probes based on the human FGF-CX and/or FCTRX nucleotide sequences can be used
to detect transcripts or genomic sequences encoding the same or homologous
proteins. In
various embodiments, the probe further comprises a label group attached
thereto, e.g. the label
group can be a radioisotope, a fluorescent compound, an enzyme, or an enzyme
co-factor.
Such probes can be used as a part of a diagnostic test kit for identifying
cells or tissues which
mis-express an FGF-CX and/or FCTRX protein, such as by measuring a level of an
FGF-CX
and/or FCTRX-encoding nucleic acid in a sample of cells from a subject e.g.,
detecting FGF-
42
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
CX and/or FCTRX mRNA levels or determining whether a genomic FGF-CX and/or
FCTIRX
gene has been mutated or deleted.
"A polypeptide having a biologically-active portion of an FGF-CX and/or FCTIZX
polypeptide" refers to polypeptides exhibiting activity similar, but not
necessarily identical to,
an activity of a polypeptide of the invention, including mature forms, as
measured in a
particular biological assay, with or without dose dependency. A nucleic acid
fragment
encoding a "biologically-active portion of FGF-CX and/or FCTRX" can be
prepared by
isolating a portion SEQ ID NOS:1, 3, 5, 7, 9, 11 and 13 that encodes a
polypeptide having an
FGF-CX and/or FCTRX biological activity (the biological activities of the FGF-
CX and/or
FCTRX proteins are described below), expressing the encoded portion of FGF-
CX,and/or
FCTRX protein (e.g., by recombinant expression ih vitro) and assessing the
activity of the
encoded portion of FGF-CX and/or FCTRX.
FGF-CX and/or FCTRX Nucleic Acid and Polypeptide Variants
The invention further encompasses nucleic acid molecules that differ from the
nucleotide sequences shown SEQ ID NOS:1, 3, 5, 7, 9, 11 and 13 due to
degeneracy of the
genetic code and thus encode the same FGF-CX and/or FCTRX proteins as that
encoded by
the nucleotide sequences shown in SEQ ID NOS:1, 3, 5, 7, 9, 11 and 13. In
another
embodiment, an isolated nucleic acid molecule of the invention has a
nucleotide sequence
encoding a protein having an amino acid sequence shown in SEQ ID NOS:2, 4, 6,
8, 10, 12
and 14.
In addition to the human FGF-CX and/or FCTRX nucleotide sequences shown in SEQ
ID NOS:1, 3, 5, 7, 9, 11 and 13 it will be appreciated by those skilled in the
art that DNA
sequence polymorphisms that lead to changes in the amino acid sequences of the
FGF-CX
and/or FCTRX polypeptides may exist within a population (e.g., the human
population). Such
genetic polymorphism in the FGF-CX and/or FCTRX genes may exist among
individuals
within a population due to natural allelic variation. As used herein, the
terms "gene" and
"recombinant gene" refer to nucleic acid molecules comprising an open reading
frame (OI2F)
encoding an FGF-CX and/or FCTItX protein, preferably a vertebrate FGF-CX
and/or FCTRX
protein. Such natural allelic variations can typically result in 1-5% variance
in the nucleotide
sequence of the FGF-CX and/or FCTRX genes. Any and all such nucleotide
variations and
resulting amino acid polymorphisms in the FGF-CX and/or FCTRX polypeptides,
which are
43
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
the result of natural allelic variation and that do not alter the functional
activity of the FGF-CX
and/or FCTRX polypeptides, are intended to be within the scope of the
invention.
Moreover, nucleic acid molecules encoding FGF-CX and/or FCTRX proteins from
other species, and thus that have a nucleotide sequence that differs from the
human sequence
SEQ m NOS:1, 3, 5, 7, 9, 11 and 13 are intended to be within the scope of the
invention.
Nucleic acid molecules corresponding to natural allelic variants and
homologues of the FGF-
CX and/or FCTRX cDNAs of the invention can be isolated based on their homology
to the
human FGF-CX and/or FCTRX nucleic acids disclosed herein using the human
cDNAs, or a
portion thereof, as a hybridization probe according to standard hybridization
techniques under
stringent hybridization conditions.
Accordingly, in another embodiment, an isolated nucleic acid molecule of the
invention is at least 6 nucleotides in length and hybridizes under stringent
conditions to the
nucleic acid molecule, comprising the nucleotide sequence of SEQ m NOS:1, 3,
5, 7, 9, 11 and
13. In another embodiment, the nucleic acid is at least 10, 25, 50, 100, 250,
500, 750, 1000,
1500, or 2000 or more nucleotides in length. In yet another embodiment, an
isolated nucleic
acid molecule of the invention hybridizes to the coding region. As used
herein, the term
"hybridizes under stringent conditions" is intended to describe conditions for
hybridization and
washing under which nucleotide sequences at least 60% homologous to each other
typically
remain hybridized to each other.
Homologs (i. e., nucleic acids encoding FGF-CX and/or FCTRX proteins derived
from
species other than human) or other related sequences (e.g., paralogs) can be
obtained by low,
moderate or high stringency hybridization with all or a portion of the
particular human
sequence as a probe using methods well known in the art for nucleic acid
hybridization and
cloning.
As used herein, the phrase "stringent hybridization conditions" refers to
conditions
under which a probe, primer or oligonucleotide will hybridize to its target
sequence, but to no
other sequences. Stringent conditions are sequence-dependent and will be
different in
different circumstances. Longer sequences hybridize specifically at higher
temperatures than
shorter sequences. Generally, stringent conditions are selected to be about 5
°C lower than the
thermal melting point (Tm) for the specific sequence at a defined ionic
strength and pH. The
Tm is the temperature (under defined ionic strength, pH and nucleic acid
concentration) at
which 50% of the probes complementary to the target sequence hybridize to the
target
44
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
sequence at equilibrium. Since the target sequences are generally present at
excess, at Tm,
50% of the probes are occupied at equilibrium. Typically, stringent conditions
will be those in
which the salt concentration is less than about 1.0 M sodium ion, typically
about 0.01 to 1.0 M
sodium ion (or other salts) at pH 7.0 to 8.3 and the temperature is at least
about 30°C for short
probes, primers or oligonucleotides (e.g., 10 nt to 50 nt) and at least about
60°C for longer
probes, primers and oligonucleotides. Stringent conditions may also be
achieved with the
addition of destabilizing agents, such as formamide.
Stringent conditions are known to those skilled in the art and can be found in
Ausubel,
et al., (eds.), CURRENT PROTOCOLS IN MOLECULAR BIOLOGY, John Wiley & Sons,
N.Y.
(1989), 6.3.1-6.3.6. Preferably, the conditions are such that sequences at
least about 65%,
70%, 75%, 85%, 90%, 95%, 98%, or 99% homologous to each other typically remain
hybridized to each other. A non-limiting example of stringent hybridization
conditions are
hybridization in a high salt buffer comprising 6X SSC, 50 mM Tris-HCl (pH
7.5), 1 mM
EDTA, 0.02% PVP, 0.02% Ficoll, 0.02% BSA, and 500 mg/ml denatured salmon sperm
DNA
at 65°C, followed by one or more washes in 0.2X SSC, 0.01% BSA at
50°C. An isolated
nucleic acid molecule of the invention that hybridizes under stringent
conditions to the
sequences of SEQ )D NOS:1, 3, 5, 7, 9, 11 and 13 corresponds to a naturally-
occurring nucleic
acid molecule. As used herein, a "naturally-occurnng" nucleic acid molecule
refers to an
RNA or DNA molecule having a nucleotide sequence that occurs in nature (e.g.,
encodes a
natural protein).
In a second embodiment, a nucleic acid sequence that is hybridizable to the
nucleic
acid molecule comprising the nucleotide sequence of SEQ )D NOS:1, 3, 5, 7, 9,
1 l and 13 or
fragments, analogs or derivatives thereof, under conditions of moderate
stringency is provided.
A non-limiting example of moderate stringency hybridization conditions are
hybridization in
6X SSC, SX Denhardt's solution, 0.5% SDS and 100 mg/ml denatured salmon sperm
DNA at
55°C, followed by one or more washes in 1X SSC, 0.1% SDS at
37°C. Other conditions of
moderate stringency that may be used are well-known within the art. See, e.g.,
Ausubel, et al.
(eds.), 1993, CURRENT PROTOCOLS IN MOLECULAR BIOLOGY, John Wiley & Sons, NY,
and
Kriegler, 1990; GENE TRANSFER AND EXPRESSION, A LABORATORY MANUAL, Stockton
Press,
NY.
In a third embodiment, a nucleic acid that is hybridizable to the nucleic acid
molecule
comprising the nucleotide sequences of SEQ )D NOS:1, 3, 5, 7, 9, 11 and 13 or
fragments,
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
analogs or derivatives thereof, under conditions of low stringency, is
provided. A non-limiting
example of low stringency hybridization conditions are hybridization in 35%
formamide, SX
SSC, 50 mM Tris-HCl (pH 7.5), 5 mM EDTA, 0.02% PVP, 0.02% Ficoll, 0.2% BSA,
100
mg/ml denatured salmon sperm DNA, 10% (wt/vol) dextran sulfate at 40°C,
followed by one
or more washes in 2X SSC, 25 mM Tris-HCl (pH 7.4), 5 mM EDTA, and 0.1% SDS at
50°C.
Other conditions of low stringency that may be used are well known in the art
(e.g., as
employed for cross-species hybridizations). See, e.g., Ausubel, et al. (eds.),
1993, CURRENT
PROTOCOLS IN MOLECULAR BIOLOGY, John Wiley & Sons, NY, and Kriegler, 1990,
GENE
TRANSFER AND EXPRESSION, A LABORATORY MANUAL, Stockton Press, NY; Shilo and
Weinberg, 1981. Proc Natl Acad Sci USA 78: 6789-6792.
Conservative Mutations
In addition to naturally-occurnng allelic variants of FGF-CX andlor FCTRX
sequences
that may exist in the population, the skilled artisan will further appreciate
that changes can be
introduced by mutation into the nucleotide sequences of SEQ 1D NOS:1, 3, 5, 7,
9, 1 l and 13
thereby leading to changes in the amino acid sequences of the encoded FGF-CX
and/or
FCT12X proteins, without altering the functional ability of said FGF-CX and/or
FCT12X
proteins. For example, nucleotide substitutions leading to amino acid
substitutions at
"non-essential" amino acid residues can be made in the sequence of SEQ ID
NOS:2, 4, 6, 8,
10, 12 and 14. A "non-essential" amino acid residue is a residue that can be
altered from the
wild-type sequences of the FGF-CX and/or FCTRX proteins without altering their
biological
activity, whereas an "essential" amino acid residue is required for such
biological activity. For
example, amino acid residues that are conserved among the FGF-CX and/or FCTRX
proteins
of the invention are predicted to be particularly non-amenable to alteration.
Amino acids for
which conservative substitutions can be made are well-known within the art.
Another aspect of the invention pertains to nucleic acid molecules encoding
FGF-CX
and/or FCTRX proteins that contain changes in amino acid residues that are not
essential for
activity. Such FGF-CX and/or FCTRX proteins differ in amino acid sequence from
SEQ m
NOS:2, 4, 6, 8, 10, 12 and 14 yet retain biological activity. In one
embodiment, the isolated
nucleic acid molecule comprises a nucleotide sequence encoding a protein,
wherein the protein
comprises an amino acid sequence at least about 45% homologous to the amino
acid
sequences of SEQ ID NOS:2, 4, 6, 8, 10, 12 and 14. Preferably, the protein
encoded by the
nucleic acid molecule is at least about 60% homologous to SEQ m NOS:2, 4, 6,
8, 10, 12 and
46
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
14; more preferably at least about 70% homologous to SEQ ID NOS:2, 4, 6, 8,
10, 12 and 14;
still more preferably at least about 80% homologous to SEQ ID NOS:2, 4, 6, 8,
10, 12 and 14;
even more preferably at least about 90% homologous to SEQ ID NOS:2, 4, 6, 8,
10, 12 and
14; and most preferably at least about 95% homologous to SEQ ID NOS:2, 4, 6,
8, 10, 12 and
14.
An isolated nucleic acid molecule encoding an FGF-CX and/or FCTRX protein
homologous to the protein of SEQ m NOS:2, 4, 6, 8, I0, 12 and 14 can be
created by
introducing one or more nucleotide substitutions, additions or deletions into
the nucleotide
sequence of SEQ ID NOS:1, 3, 5, 7, 9, 11 and 13 such that one or more amino
acid
substitutions, additions or deletions are introduced into the encoded protein.
Mutations can be introduced into SEQ ID NOS:2, 4, 6, 8, 10, 12 and 14 by
standard
techniques, such as site-directed mutagenesis and PCR-mediated mutagenesis.
Preferably,
conservative amino acid substitutions are made at one or more predicted, non-
essential amino
acid residues. A "conservative amino acid substitution" is one in which the
amino acid residue
is replaced with an amino acid residue having a similar side chain. Families
of amino acid
residues having similar side chains have been defined within the art. These
families include
amino acids with basic side chains (e.g., lysine, arginine, histidine), acidic
side chains (e.g.,
aspartic acid, glutamic acid), uncharged polar side chains (e.g., glycine,
asparagine, glutamine,
serine, threonine, tyrosine, cysteine), nonpolar side chains (e.g., alanine,
valine, leucine,
isoleucine, proline, phenylalanine, methionine, tryptophan), beta-branched
side chains (e.g.,
threonine, valine, isoleucine) and aromatic side chains (e.g., tyrosine,
phenylalanine,
tryptophan, histidine). Thus, a predicted non-essential amino acid residue in
the FGF-CX
and/or FCTRX protein is replaced with another amino acid residue from the same
side chain
family. Alternatively, in another embodiment, mutations can be introduced
randomly along all
or part of an FGF-CX and/or FCTRX coding sequence, such as by saturation
mutagenesis, and
the resultant mutants can be screened for FGF-CX and/or FCTRX biological
activity to
identify mutants that retain activity. Following mutagenesis of SEQ ID NOS:1,
3, 5, 7, 9, 11
and 13, the encoded protein can be expressed by any recombinant technology
known in the art
and the activity of the protein can be determined.
The relatedness of amino acid families may also be determined based on side
chain
interactions. Substituted amino acids may be fully conserved "strong" residues
or fully
conserved "weak" residues. The "strong" group of conserved amino acid residues
may be any
47
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
one of the following groups: STA, NEQK, NHQK, NDEQ, QHRK, MILV, MILF, HY, FYW,
wherein the single letter amino acid codes are grouped by those amino acids
that may be
substituted for each other. Likewise, the "weak" group of conserved residues
may be any one
of the following: CSA, ATV, SAG, STNK, STPA, SGND, SNDEQK, NDEQHK, NEQ13RK,
VLIM, HFY, wherein the letters within each group represent the single letter
amino acid code.
In one embodiment, a mutant FGF-CX and/or FCTRX protein can be assayed for (i)
the ability to form protein:protein interactions with other FGF-CX and/or
FCTRX proteins,
other cell-surface proteins, or biologically-active portions thereof, (ii)
complex formation
between a mutant FGF-CX and/or FCTRX protein and an FGF-CX and/or FCTRX
ligand; or
(iii) the ability of a mutant FGF-CX andlor FCTRX protein to bind to an
intracellular target
protein or biologically-active portion thereof; (e.g. avidin proteins).
In yet another embodiment, a mutant FGF-CX and/or FCTRX protein can be assayed
for the ability to regulate a specific biological function (e.g., regulation
of insulin release).
Antisense Nucleic Acids
Another aspect of the invention pertains to isolated antisense nucleic acid
molecules
that are hybridizable to or complementary to the nucleic acid molecule
comprising the
nucleotide sequence of SEQ m NOS:l, 3, 5, 7, 9, 11 and 13, or fragments,
analogs or
derivatives thereof. An "antisense" nucleic acid comprises a nucleotide
sequence that is
complementary to a "sense" nucleic acid encoding a protein (e.g.,
complementary to the
coding strand of a double-stranded cDNA molecule or complementary to an ml2NA
sequence). In specific aspects, antisense nucleic acid molecules are provided
that comprise a
sequence complementary to at least about 10, 25, 50, 100, 250 or S00
nucleotides or an entire
FGF-CX and/or FCTRX coding strand, or to only a portion thereof. Nucleic acid
molecules
encoding fragments, homologs, derivatives and analogs of an FGF-CX and/or
FCTRX protein
of SEQ ID NOS:2, 4, 6, 8, 10, 12 and 14, or antisense nucleic acids
complementary to an
FGF-CX and/or FCTRX nucleic acid sequence of SEQ ID NOS:1, 3, 5, 7, 9, 11 and
13, are
additionally provided.
In one embodiment, an antisense nucleic acid molecule is antisense to a
"coding
region" of the coding strand of a nucleotide sequence encoding an FGF-CX
and/or FCTRX
protein. The term "coding region" refers to the region of the nucleotide
sequence comprising
codons which are translated into amino acid residues. In another embodiment,
the antisense
nucleic acid molecule is antisense to a "noncoding region" of the coding
strand of a nucleotide
48
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
sequence encoding the FGF-CX and/or FCTRX protein. The term "noncoding region"
refers
to 5' and 3' sequences which flank the coding region that are not translated
into amino acids
(i.e., also referred to as 5' and 3' untranslated regions).
Given the coding strand sequences encoding the FGF-CX and/or FCTRX protein
disclosed herein, antisense nucleic acids of the invention can be designed
according to the
rules of Watson and Crick or Hoogsteen base pairing. The antisense nucleic
acid molecule
can be complementary to the entire coding region of FGF-CX and/or FCTRX mRNA,
but
more preferably is an oligonucleotide that is antisense to only a portion of
the coding or
noncoding region of FGF-CX and/or FCTRX mRNA. For example, the antisense
oligonucleotide can be complementary to the region surrounding the translation
start site of
FGF-CX and/or FCTRX mRNA. An antisense oligonucleotide can be, for example,
about 5,
10, 15, 20, 25, 30, 35, 40, 45 or 50 nucleotides in length. An antisense
nucleic acid of the
invention can be constructed using chemical synthesis or enzymatic ligation
reactions using
procedures known in the art. ' For example, an antisense nucleic acid (e.g.,
an antisense
oligonucleotide) can be chemically synthesized using naturally-occurring
nucleotides or
variously modified nucleotides designed to increase the biological stability
of the molecules or
to increase the physical stability of the duplex formed between the antisense
and sense nucleic
acids (e.g., phosphorothioate derivatives and acridine substituted nucleotides
can be used).
Examples of modified nucleotides that can be used to generate the antisense
nucleic
acid include: 5-fluorouracil, 5-bromouracil, 5-chlorouracil, 5-iodouracil,
hypoxanthine,
xanthine, 4-acetylcytosine, 5-(carboxyhydroxylmethyl) uracil, 5-
carboxymethylaminomethyl-
2-thiouridine, 5-carboxymethylaminomethyluracil, dihydrouracil, beta-D-
galactosylqueosine,
inosine, N6-isopentenyladenine, 1-methylguanine, 1-methylinosine, 2,2-
dimethylguanine,
2-methyladenine, 2-methylguanine, 3-methylcytosine, 5-methylcytosine, N6-
adenine,
7-methylguanine, 5-methylaminomethyluracil, 5-methoxyaminomethyl-2-thiouracil,
beta-D-mannosylqueosine, 5'-methoxycarboxymethyluracil, 5-methoxyuracil, 5-
methyluracil,
2-methylthio-N6-isopentenyladenine, uracil-5-oxyacetic acid (v), wybutoxosine,
pseudouracil,
queosine, 2-thiocytosine, 5-methyl-2-thiouracil, 2-thiouracil, 4-thiouracil,
uracil-5-oxyacetic
acid methylester, uracil-5-oxyacetic acid (v), 5-methyl-2-thiouracil, 2,6-
diaminopurine,
(acp3)w, and 3-(3-amino-3-N-2-carboxypropyl) uracil. Alternatively, the
antisense nucleic
acid can be produced biologically using an expression vector into which a
nucleic acid has
been subcloned in an antisense orientation (i.e., RNA transcribed from the
inserted nucleic
49
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
acid will be of an antisense orientation to a target nucleic acid of interest,
described further in
the following subsection).
The antisense nucleic acid molecules of the invention are typically
administered to a
subject or generated in situ such that they hybridize with or bind to cellular
mRNA andlor
genomic DNA encoding an FGF-CX and/or FCTRX protein to thereby inhibit
expression of
the protein (e.g., by inhibiting transcription andlor translation). The
hybridization can be by
conventional nucleotide complementarity to form a stable duplex, or, for
example, in the case
of an antisense nucleic acid molecule that binds to DNA duplexes, through
specific
interactions in the major groove of the double helix. An example of a route of
administration
of antisense nucleic acid molecules of the invention includes direct inj
ection at a tissue site.
Alternatively, antisense nucleic acid molecules can be modified to target
selected cells and
then administered systemically. For example, for systemic administration,
antisense
molecules can be modified such that they specifically bind to receptors or
antigens expressed
on a selected cell surface (e.g., by linking the antisense nucleic acid
molecules to peptides or
antibodies that bind to cell surface receptors or antigens). The antisense
nucleic acid
molecules can also be delivered to cells using the vectors described herein.
To achieve
sufficient nucleic acid molecules, vector constructs in which the antisense
nucleic acid
molecule is placed under the control of a strong pol II or pol III promoter
are preferred.
In yet another embodiment, the antisense nucleic acid molecule of the
invention is an
a-anomeric nucleic acid molecule. An a-anomeric nucleic acid molecule forms
specific
double-stranded hybrids with complementary RNA in which, contrary to the usual
(3-units, the
strands run parallel to each other. See, e.g., Gaultier, et al., 1987. Nucl.
Acids Res. 15:
6625-6641. The antisense nucleic acid molecule can also comprise a
2'-o-methylribonucleotide (see, e.g., moue, et al. 1987. Nucl. Acids Res. 15:
6131-6148) or a
chimeric RNA-DNA analogue (see, e.g., moue, et al., 1987. FEBSLett. 215: 327-
330.
Ribozymes and PNA Moieties
Nucleic acid modifications include, by way of non-limiting example, modified
bases,
and nucleic acids whose sugar phosphate backbones are modified or derivatized.
These
modifications are carried out at least in part to enhance the chemical
stability of the modified
nucleic acid, such that they may be used, for example, as antisense binding
nucleic acids in
therapeutic applications in a subject.
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
In one embodiment, an antisense nucleic acid of the invention is a ribozyme.
Ribozymes are catalytic RNA molecules with ribonuclease activity that are
capable of
cleaving a single-stranded nucleic acid, such as an mRNA, to which they have a
complementary region. Thus, ribozyrnes (e.g., hammerhead ribozymes as
described in
Haselhoff and Gerlach 1988. Nature 334: 585-591) can be used to catalytically
cleave FGF-
CX and/or FCTRX mRNA transcripts to thereby inhibit translation of FGF-CX
andlor FCTRX
mRNA. A ribozyme having specificity for an FGF-CX and/or FCTRX-encoding
nucleic acid
can be designed based upon the nucleotide sequence of an FGF-CX and/or FCTRX
cDNA
disclosed herein (i.e., SEQ )D NOS:1, 3, 5, 7, 9, 11 and 13). For example, a
derivative of a
Tetrahymeha L-19 IVS RNA can be constructed in which the nucleotide sequence
of the
active site is complementary to the nucleotide sequence to be cleaved in an
FGF-CX andlor
FCTRX-encoding mRNA. See, e.g., U.S. Patent 4,987,071 to Cech, et al. and U.S.
Patent
5,116,742 to Cech, et al. FGF-CX and/or FCTRX mRNA can also be used to select
a catalytic
RNA having a specific ribonuclease activity from a pool of RNA molecules. See,
e.g., Bartel
et al., (1993) Science 261:1411-1418.
Alternatively, FGF-CX andlor FCTRX gene expression can be inhibited by
targeting
nucleotide sequences complementary to the regulatory region of the FGF-CX
and/or FCTRX
nucleic acid (e.g., the FGF-CX and/or FCTRX promoter and/or enhancers) to form
triple
helical structures that prevent transcription of the FGF-CX and/or FCTRX gene
in target cells.
See, e.g., Helene, 1991. Ahticahcer Drug Des. 6: 569-84; Helene, et al. 1992.
Ann. N. Y. Acad.
Sci. 660: 27-36; Maher, 1992. Bioassays 14: 807-15.
In various embodiments, the FGF-CX andlor FCTRX nucleic acids can be modified
at
the base moiety, sugar moiety or phosphate backbone to improve, e.g., the
stability,
hybridization, or solubility of the molecule. For example, the deoxyribose
phosphate
backbone of the nucleic acids can be modified to generate peptide nucleic
acids. See, e.g.,
Hyrup, et al., 1996. Bioorg Med Che~ra 4: 5-23. As used herein, the terms
"peptide nucleic
acids" or "PNAs" refer to nucleic acid mimics (e.g., DNA mimics) in which the
deoxyribose
phosphate backbone is replaced by a pseudopeptide backbone and only the four
natural
nucleobases are retained. The neutral backbone of PNAs has been shown to allow
for specific
hybridization to DNA and RNA under conditions of low ionic strength. The
synthesis of PNA
oligomers can be performed using standard solid phase peptide synthesis
protocols as
51
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
described in Hyrup, et al., 1996. supra; Perry-O'Keefe, et al., 1996. PYOC.
Natl. Acad. Sci. USA
93: 14670-14675.
PNAs of FGF-CX and/or FCTRX can be used in therapeutic and diagnostic
applications. For example, PNAs can be used as antisense or antigene agents
for
S sequence-specific modulation of gene expression by, e.g., inducing
transcription or translation
arrest or inhibiting replication. PNAs of FGF-CX and/or FCTRX can also be
used, for
example, in the analysis of single base pair mutations in a gene (e.g., PNA
directed PCR
clamping; as artificial restriction enzymes when used in combination with
other enzymes, e.g.,
S1 nucleases (see, Hyrup, et al., 1996.sup~a); or as probes or primers for DNA
sequence and
hybridization (see, Hyrup, et al., 1996, supra; Perry-O'Keefe, et al., 1996.
supra).
In another embodiment, PNAs of FGF-CX and/or FCTRX can be modified, e.g., to
enhance their stability or cellular uptake, by attaching lipophilic or other
helper groups to
PNA, by the formation of PNA-DNA chimeras, or by the use of liposomes or other
techniques
of drug delivery known in the art. For example, PNA-DNA chimeras of FGF-CX
and/or
1S FCTRX can be generated that may combine the advantageous properties of PNA
and DNA.
Such chimeras allow DNA recognition enzymes (e.g., RNase H and DNA
polyrnerases) to
interact with the DNA portion while the PNA portion would provide high binding
affinity and
specificity. PNA-DNA chimeras can be linked using linkers of appropriate
lengths selected in
terms of base stacking, number of bonds between the nucleobases, and
orientation (see, Hyrup,
et al., 1996. supra). The synthesis of PNA-DNA chimeras can be performed as
described in
Hyrup, et al., 1996. supra and Finn, et al., 1996. Nucl Acids Res 24: 3357-
3363. For example,
a DNA chain can be synthesized on a solid support using standard
phosphoramidite coupling
chemistry, and modified nucleoside analogs, e.g.,
5'-(4-methoxytrityl)amino-S'-deoxy-thymidine phosphoramidite, can be used
between the
2S PNA and the S' end of DNA. See, e.g., Mag, et al., 1989. Nucl Acid Res 17:
5973-5988. PNA
monomers are then coupled in a stepwise manner to produce a chimeric molecule
with a S'
PNA segment and a 3' DNA segment. See, e.g., Finn, et al., 1996. supra.
Alternatively,
chimeric molecules can be synthesized with a S' DNA segment and a 3' PNA
segment. See,
e.g., Petersen, et al., 1975. Bioorg. Med. Chem. Lett. S: 1119-11124.
In other embodiments, the oligonucleotide may include other appended groups
such as
peptides (e.g., for targeting host cell receptors in vivo), or agents
facilitating transport across
the cell membrane (see, e.g., Letsinger, et al., 1989. P~oc. Natl. Acad. Sci.
U.S.A. 86:
S2
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
6553-6556; Lemaitre, et al., 1987. P~oc. Natl. Acad. Sci. 84: 648-652; PCT
Publication No.
W088/09810) or the blood-brain barner (see, e.g., PCT Publication No. WO
89/10134). In
addition, oligonucleotides can be modified with hybridization triggered
cleavage agents (see,
e.g., Krol, et al., 1988. BioTechfziques 6:958-976) or intercalating agents
(see, e.g., Zon, 1988.
Phar~z. Res. 5: 539-549). To this end, the oligonucleotide may be conjugated
to another
molecule, e.g., a peptide, a hybridization triggered cross-linking agent, a
transport agent, a
hybridization-triggered cleavage agent, and the like.
FGF-CX and/or FCTRX Polypeptides
A polypeptide according to the invention includes a polypeptide including the
amino
acid sequence of FGF-CX and/or FCTRX polypeptides whose sequences are provided
in SEQ
m NOS:2, 4, 6, 8, 10, 12 and 14. The invention also includes a mutant or
variant protein any
of whose residues may be changed from the corresponding residues shown in SEQ
m NOS:2,
4, 6, 8, 10, 12 and 14 while still encoding a protein that maintains its FGF-
CX and/or FCTRX
activities and physiological functions, or a functional fragment thereof.
In general, an FGF-CX and/or FCTRX variant that preserves FGF-CX and/or FCTRX-
like function includes any variant in which residues at a particular position
in the sequence
have been substituted by other amino acids, and further include the
possibility of inserting an
additional residue or residues between two residues of the parent protein as
well as the
possibility of deleting one or more residues from the parent sequence. Any
amino acid
substitution, insertion, or deletion is encompassed by the invention. In
favorable
circumstances, the substitution is a conservative substitution as defined
above.
One aspect of the invention pertains to isolated FGF-CX and/or FCTRX proteins,
and
biologically-active portions thereof, or derivatives, fragments, analogs or
homologs thereof.
Also provided are polypeptide fragments suitable for use as immunogens to
raise anti-FGF-
CX and/or FCTRX antibodies. In one embodiment, native FGF-CX and/or FCTRX
proteins
can be isolated from cells or tissue sources by an appropriate purification
scheme using
standard protein purification techniques. In another embodiment, FGF-CX and/or
FCTRX
proteins are produced by recombinant DNA techniques. Alternative to
recombinant
expression, an FGF-CX and/or FCTRX protein or polypeptide can be synthesized
chemically
using standard peptide synthesis techniques.
An "isolated" or "purified" polypeptide or protein or biologically-active
portion thereof
is substantially free of cellular material or other contaminating proteins
from the cell or tissue
53
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
source from which the FGF-CX and/or FCTRX protein is derived, or substantially
free from
chemical precursors or other chemicals when chemically synthesized. The
language
"substantially free of cellular material" includes preparations of FGF-CX
and/or FCTRX
proteins in which the protein is separated from cellular components of the
cells from which it
is isolated or recombinantly-produced. In one embodiment, the language
"substantially free of
cellular material" includes preparations of FGF-CX and/or FCTRX proteins
having less than
about 30% (by dry weight) of non-FGF-CX and/or FCTRX proteins (also referred
to herein as
a "contaminating protein"), more preferably less than about 20% of non-FGF-CX
and/or
FCTRX proteins, still more preferably less than about 10% of non-FGF-CX and/or
FCTRX
proteins, and most preferably less than about 5% of non-FGF-CX and/or FCTRX
proteins.
When the FGF-CX and/or FCTRX protein or biologically-active portion thereof is
recombinantly-produced, it is also preferably substantially free of culture
medium, i.e., culture
medium represents less than about 20%, more preferably less than about 10%,
and most
preferably less than about 5% of the volume of the FGF-CX and/or FCTRX protein
preparation.
The language "substantially free of chemical precursors or other chemicals"
includes
preparations of FGF-CX and/or FCTRX proteins in which the protein is separated
from
chemical precursors or other chemicals that are involved in the synthesis of
the protein. In one
embodiment, the language "substantially free of chemical precursors or other
chemicals"
includes preparations of FGF-CX and/or FCTRX proteins having less than about
30% (by dry
weight) of chemical precursors or non-FGF-CX and/or FCTRX chemicals, more
preferably
less than about 20% chemical precursors or non-FGF-CX and/or FCTRX chemicals,
still more
preferably less than about 10% chemical precursors or non-FGF-CX and/or FCTRX
chemicals, and most preferably less than about 5% chemical precursors or non-
FGF-CX
and/or FCTRX chemicals.
Biologically-active portions of FGF-CX and/or FCTRX proteins include peptides
comprising amino acid sequences sufficiently homologous to or derived from the
amino acid
sequences of the FGF-CX andlor FCTRX proteins (e.g., the amino acid sequence
shown in
SEQ ID NOS:2, 4, 6, ~, 10, 12 and 14) that include fewer amino acids than the
full-length
FGF-CX and/or FCTRX proteins, and exhibit at least one activity of an FGF-CX
and/or
FCTRX protein. Typically, biologically-active portions comprise a domain or
motif with at
least one activity of the FGF-CX and/or FCTRX protein. A biologically-active
portion of an
54
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
FGF-CX and/or FCTRX protein can be a polypeptide which is, for example, 10,
25, 50, 100 or
more amino acid residues in length.
Moreover, other biologically-active portions, in which other regions of the
protein are
deleted, can be prepared by recombinant techniques and evaluated for one or
more of the
functional activities of a native FGF-CX and/or FCTRX protein.
In an embodiment, the FGF-CX and/or FCTRX protein has an amino acid sequence
shown in SEQ ID NOS:2, 4, 6, 8, 10, I2 and 14. In other embodiments, the FGF-
CX and/or
FCTRX protein is substantially homologous to SEQ ID NOS:2, 4, 6, 8, 10, 12 and
14, and
retains the functional activity of the protein of SEQ ID NOS:2, 4, 6, 8, 10,
12 and 14, yet
differs in amino acid sequence due to natural allelic variation or
mutagenesis, as described in
detail, below. Accordingly, in another embodiment, the FGF-CX and/or FCTRX
protein is a
protein that comprises an amino acid sequence at least about 45% homologous to
the amino
acid sequence SEQ ID NOS:2, 4, 6, 8, 10, 12 and 14, and retains the functional
activity of the
FGF-CX and/or FCTRX proteins of SEQ ID NOS:2, 4, 6, 8, 10, 12 and 14.
Determining Homology Between Two or More Sequences
To determine the percent homology of two amino acid sequences or of two
nucleic
acids, the sequences are aligned for optimal comparison purposes (e.g., gaps
can be introduced
in the sequence of a first amino acid or nucleic acid sequence for optimal
alignment with a
second amino or nucleic acid sequence). The amino acid residues or nucleotides
at
corresponding amino acid positions or nucleotide positions are then compared.
When a
position in the first sequence is occupied by the same amino acid residue or
nucleotide as the
corresponding position in the second sequence, then the molecules are
homologous at that
position (i.e., as used herein amino acid or nucleic acid "homology" is
equivalent to amino
acid or nucleic acid "identity").
The nucleic acid sequence homology may be determined as the degree of identity
between two sequences. The homology may be determined using computer programs
known
in the art, such as GAP software provided in the GCG program package. See,
Needleman and
Wunsch, 1970. JMoI Biol 48: 443-453. Using GCG GAP software with the following
settings
for nucleic acid sequence comparison: GAP creation penalty of 5.0 and GAP
extension
penalty of 0.3, the coding region of the analogous nucleic acid sequences
referred to above
exhibits a degree of identity preferably of at least 70%, 75%, 80%, 85%, 90%,
95%, 98%, or
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
99%, with the CDS (encoding) part of the DNA sequence shown in SEQ ID NOS:1,
3, 5, 7, 9,
11 and 13.
The term "sequence identity" refers to the degree to which two polynucleotide
or
polypeptide sequences are identical on a residue-by-residue basis over a
particular region of
comparison. The term "percentage of sequence identity" is calculated by
comparing two
optimally aligned sequences over that region of comparison, determining the
number of
positions at which the identical nucleic acid base (e.g., A, T, C, G, U, or I,
in the case of
nucleic acids) occurs in both sequences to yield the number of matched
positions, dividing the
number of matched positions by the total number of positions in the region of
comparison (i. e.,
the window size), and multiplying the result by 100 to yield the percentage of
sequence
identity. The term "substantial identity" as used herein denotes a
characteristic of a
polynucleotide sequence, wherein the polynucleotide comprises a sequence that
has at least 80
percent sequence identity, preferably at least 85 percent identity and often
90 to 95 percent
sequence identity, more usually at least 99 percent sequence identity as
compared to a
reference sequence over a comparison region.
Chimeric and Fusion Proteins
The invention also provides FGF-CX and/or FCTRX chimeric or fusion proteins.
As
used herein, an FGF-CX and/or FCTRX "chimeric protein" or "fusion protein"
comprises an
FGF-CX and/or FCTRX polypeptide operatively-linked to a non-FGF-CX and/or
FCTR.X
polypeptide. An "FGF-CX and/or FCTRX polypeptide" refers to a polypeptide
having an
amino acid sequence corresponding to an FGF-CX and/or FCTRX protein (SEQ ID
NOS:2, 4,
6, 8, 10, 12 and 14), whereas a "non-FGF-CX and/or FCTRX polypeptide" refers
to a
polypeptide having an amino acid sequence corresponding to a protein that is
not substantially
homologous to the FGF-CX and/or FCTRX protein, e.g., a protein that is
different from the
FGF-CX and/or FCTItX protein and that is derived from the same or a different
organism.
Within an FGF-CX and/or FCTRX fusion protein the FGF-CX and/or FCTRX
polypeptide
can correspond to all or a portion of an FGF-CX and/or FCTRX protein. In one
embodiment,
an FGF-CX and/or FCTRX fusion protein comprises at least one biologically-
active portion of
an FGF-CX and/or FCTRX protein. In another embodiment, an FGF-CX and/or FCTRX
fusion protein comprises at least two biologically-active portions of an FGF-
CX and/or
FCTRX protein. In yet another embodiment, an FGF-CX and/or FCTRX fusion
protein
comprises at least three biologically-active portions of an FGF-CX and/or
FCTRX protein.
56
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
Within the fusion protein, the term "operatively-linked" is intended to
indicate that the FGF-
CX and/or FCTRX polypeptide and the non-FGF-CX and/or FCTRX polypeptide are
fused
in-frame with one another. The non-FGF-CX and/or FCTRX polypeptide can be
fused to the
N-terminus or C-terminus of the FGF-CX and/or FCTRX polypeptide.
In one embodiment, the fusion protein is a GST-FGF-CX and/or FCTRX fusion
protein in which the FGF-CX and/or FCTRX sequences are fused to the C-terminus
of the
GST (glutathione S-transferase) sequences. Such fusion proteins can facilitate
the purification
of recombinant FGF-CX and/or FCTRX polypeptides.
In another embodiment, the fusion protein is an FGF-CX and/or FCTRX protein
containing a heterologous signal sequence at its N-terminus. In certain host
cells (e.g.,
mammalian host cells), expression and/or secretion of FGF-CX and/or FCTRX can
be
increased through use of a heterologous signal sequence.
In yet another embodiment, the fusion protein is an FGF-CX and/or
FCTRX-immunoglobulin fusion protein in which the FGF-CX and/or FCTRX sequences
are
fused to sequences derived from a member of the immunoglobulin protein family.
The FGF-
CX and/or FCTRX-immunoglobulin fusion proteins of the invention can be
incorporated into
pharmaceutical compositions and administered to a subject to inhibit an
interaction between an
FGF-CX and/or FCTRX ligand and an FGF-CX and/or FCTRX protein on the surface
of a
cell, to thereby suppress FGF-CX and/or FCTRX-mediated signal transduction in
vivo. The
FGF-CX and/or FCTRX-immunoglobulin fusion proteins can be used to affect the
bioavailability of an FGF-CX and/or FCTRX cognate ligand. Inhibition of the
FGF-CX
and/or FCTRX ligand/FGF-CX and/or FCTRX interaction may be useful
therapeutically for
both the treatment of proliferative and differentiative disorders, as well as
modulating (e.g.
promoting or inhibiting) cell survival. Moreover, the FGF-CX and/or
FCTRX-immunoglobulin fusion proteins of the invention can be used as
immunogens to
produce anti-FGF-CX and/or FCTRX antibodies in a subject, to purify FGF-CX
and/or
FCTRX ligands, and in screening assays to identify molecules that inhibit the
interaction of
FGF-CX and/or FCTRX with an FGF-CX and/or FCTR.X ligand.
An FGF-CX and/or FCTRX chimeric or fusion protein of the invention can be
produced by standard recombinant DNA techniques. For example, DNA fragments
coding for
the different polypeptide sequences are ligated together in-frame in
accordance with
conventional techniques, e.g., by employing blunt-ended or stagger-ended
termini for ligation,
57
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
restriction enzyme digestion to provide for appropriate termini, filling-in of
cohesive ends as
appropriate, alkaline phosphatase treatment to avoid undesirable joining, and
enzymatic
ligation. In another embodiment, the fusion gene can be synthesized by
conventional
techniques including automated DNA synthesizers. Alternatively, PCR
amplification of gene
fragments can be carried out using anchor primers that give rise to
complementary overhangs
between two consecutive gene fragments that can subsequently be annealed and
reamplified to
generate a chimeric gene sequence (see, e.g., Ausubel, et al. (eds.) CURRENT
PROTOCOLS IN
MOLECULAR BIOLOGY, John Wiley & Sons, 1992). Moreover, many expression vectors
are
commercially available that already encode a fusion moiety (e.g., a GST
polypeptide). An
FGF-CX andlor FCTRX-encoding nucleic acid can be cloned into such an
expression vector
such that the fusion moiety is linked in-frame to the FGF-CX and/or FCTRX
protein.
FGF-CX and/or FCTRX Agonists and Antagonists
The invention also pertains to variants of the FGF-CX and/or FCTRX proteins
that
function as either FGF-CX and/or FCTRX agonists (i. e., mimetics) or as FGF-CX
and/or
FCTRX antagonists. Variants of the FGF-CX and/or FCTRX protein can be
generated by
mutagenesis (e.g., discrete point mutation or tnmcation of the FGF-CX and/or
FCTRX
protein). An agonist of the FGF-CX and/or FCTRX protein can retain
substantially the same,
or a subset of, the biological activities of the naturally occurring form of
the FGF-CX and/or
FCTRX protein. An antagonist of the FGF-CX and/or FCTRX protein can inhibit
one or more
of the activities of the naturally occurring form of the FGF-CX and/or FCTRX
protein by, for
example, competitively binding to a downstream or upstream member of a
cellular signaling
cascade which includes the FGF-CX and/or FCTRX protein. Thus, specific
biological effects
can be elicited by treatment with a variant of limited function. In one
embodiment, treatment
of a subject with a variant having a subset of the biological activities of
the naturally occurring
form ofthe protein has fewer side effects in a subject relative to treatment
with the naturally
occurring form of the FGF-CX and/or FCTRX proteins.
Variants of the FGF-CX and/or FCTRX proteins that function as either FGF-CX
and/or FCTRX agonsts (i.e., mimetics) or as FGF-CX and/or FCTRX antagonists
can be
identified by screening combinatorial libraries of mutants (e.g., truncation
mutants) of the
FGF-CX and/or FCTRX proteins for FGF-CX and/or FCTRX protein agonist or
antagonist
activity. In one embodiment, a variegated library of FGF-CX and/or FCTRX
variants is
generated by combinatorial mutagenesis at the nucleic acid level and is
encoded by a
5~
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
variegated gene library. A variegated library of FGF-CX and/or FCTRX variants
can be
produced by, for example, enzymatically ligating a mixture of synthetic
oligonucleotides into
gene sequences such that a degenerate set of potential FGF-CX and/or FCTRX
sequences is
expressible as individual polypeptides, or alternatively, as a set of larger
fusion proteins (e.g.,
for phage display) containing the set of FGF-CX and/or FCTRX sequences
therein. There are
a variety of methods which can be used to produce libraries of potential FGF-
CX and/or
FCTRX variants from a degenerate oligonucleotide sequence. Chemical synthesis
of a
degenerate gene sequence can be performed in an automatic DNA synthesizer, and
the
synthetic gene then ligated into an appropriate expression vector. Use of a
degenerate set of
genes allows for the provision, in one mixture, of all of the sequences
encoding the desired set
of potential FGF-CX and/or FCTRX sequences. Methods for synthesizing
degenerate
oligonucleotides are well-known within the art. See, e.g., Narang, 1983.
Tetrahedron 39: 3;
Itakura, et al., 1984. Annu. Rev. Biochem. 53: 323; Itakura, et al., 1984.
Science 198: 1056;
Ike, et al., 1983. Nucl. Acids Res. 11: 477.
Polypeptide Libraries
In addition, libraries of fragments of the FGF-CX and/or FCTRX protein coding
sequences can be used to generate a variegated population of FGF-CX and/or
FCTRX
fragments for screening and subsequent selection of variants of an FGF-CX
and/or FCTRX
protein. In one embodiment, a library of coding sequence fragments can be
generated by
treating a double stranded PCR fragment of an FGF-CX and/or FCTRX coding
sequence with
a nuclease under conditions wherein nicking occurs only about once per
molecule, denaturing
the double stranded DNA, renaturing the DNA to form double-stranded DNA that
can include
sense/antisense pairs from different nicked products, removing single stranded
portions from
reformed duplexes by treatment with S1 nuclease, and ligating the resulting
fragment library
into an expression vector. By this method, expression libraries can be derived
which encodes
N-terminal and internal fragments of various sizes of the FGF-CX and/or FCTRX
proteins.
Various techniques are known in the art for screening gene products of
combinatorial
libraries made by point mutations or truncation, and for screening cDNA
libraries for gene
products having a selected property. Such techniques are adaptable for rapid
screening of the
gene libraries generated by the combinatorial mutagenesis of FGF-CX and/or
FCTRX
proteins. The most widely used techniques, which are amenable to high
throughput analysis,
for screening large gene libraries typically include cloning the gene libraxy
into replicable
59
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
expression vectors, transforming appropriate cells with the resulting library
of vectors, and
expressing the combinatorial genes under conditions in which detection of a
desired activity
facilitates isolation of the vector encoding the gene whose product was
detected. Recursive
ensemble mutagenesis (REM), a new technique that enhances the frequency of
functional
mutants in the libraries, can be used in combination with the screening assays
to identify FGF-
CX and/or FCTRX variants. See, e.g., Arkin and Yourvan, 1992. Proc. Natl.
Acad. Sci. USA
89: 7811-7815; Delgrave, et al., 1993. Protein E~gineerihg 6:327-331.
Anti-FGF-CX and/or FCTRX Antibodies
Also included in the invention are antibodies to FGF-CX and/or FCTRX proteins,
or
fragments of FGF-CX and/or FCTRX proteins. The term "antibody" as used herein
refers to
immunoglobulin molecules and immunologically active portions of immunoglobulin
(Ig)
molecules, i.e., molecules that contain an antigen binding site that
specifically binds
(immunoreacts with) an antigen. Such antibodies include, but are not limited
to, polyclonal,
monoclonal, chimeric, single chain, Fab, Fab' and F~ab~>2 fragments, and an
Fab expression
library. In general, an antibody molecule obtained from humans relates to any
of the classes
IgG, IgM, IgA, IgE and IgD, which differ from one another by the nature of the
heavy chain
present in the molecule. Certain classes have subclasses as well, such as
IgGI, IgG2, and
others. Furthermore, in humans, the light chain may be a kappa chain or a
lambda chain.
Reference herein to antibodies includes a reference to all such classes,
subclasses and types of
human antibody species.
An isolated FGF-CX and/or FCTRX-related protein of the invention may be
intended
to serve as an antigen, or a portion or fragment thereof, and additionally can
be used as an
immunogen to generate antibodies that immunospecifically bind the antigen,
using standard
techniques for polyclonal and monoclonal antibody preparation. The full-length
protein can
be used or, alternatively, the invention provides antigenic peptide fragments
of the antigen for
use as immunogens. An antigenic peptide fragment comprises at least 6 amino
acid residues
of the amino acid sequence of the full length protein and encompasses an
epitope thereof such
that an antibody raised against the peptide forms a specific immune complex
with the full
length protein or with any fragment that contains the epitope. Preferably,,
the antigenic peptide
comprises at least 10 amino acid residues, or at least 15 amino acid residues,
or at least 20
amino acid residues, or at least 30 amino acid residues. Preferred epitopes
encompassed by
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
the antigenic peptide are regions of the protein that are located on its
surface; commonly these
are hydrophilic regions.
In certain embodiments of the invention, at least one epitope encompassed by
the
antigenic peptide is a region of FGF-CX andlor FCTRX-related protein that is
located on the
surface of the protein, e.g., a hydrophilic region. A hydrophobicity analysis
of the human
FGF-CX and/or FCTRX-related protein sequence will indicate which regions of a
FGF-CX
and/or FCT12X-related protein are particularly hydrophilic and, therefore, are
likely to encode
surface residues useful for targeting antibody production. As a means for
targeting antibody
production, hydropathy plots showing regions of hydrophilicity and
hydrophobicity may be
generated by any method well known in the art, including, for example, the
Kyte Doolittle or
the Hopp Woods methods, either with or without Fourier transformation. See,
e.g., Hopp and
Woods, 1981, P~oc. Nat. Acad. Sci. USA 78: 3824-3828; Kyte and Doolittle 1982,
J. Mol.
Biol. 157: 105-142, each of which is incorporated herein by reference in its
entirety.
Antibodies that are specific for one or more domains within an antigenic
protein, or
derivatives, fragments, analogs or homologs thereof, are also provided herein.
A protein of the invention, or a derivative, fragment, analog, homolog or
ortholog
thereof, may be utilized as an immunogen in the generation of antibodies that
immunospecifically bind these protein components.
Various procedures known within the art may be used for the production of
polyclonal
or monoclonal antibodies directed against a protein of the invention, or
against derivatives,
fragments, analogs homologs or orthologs thereof (see, for example,
Antibodies: A Laboratory
Manual, Harlow and Lane, 1988, Cold Spring Harbor Laboratory Press, Cold
Spring Harbor,
NY, incorporated herein by reference). Some of these antibodies are discussed
below.
Polyclonal Antibodies
For the production of polyclonal antibodies, various suitable host animals
(e.g., rabbit,
goat, mouse or other mammal) may be immunized by one or more injections with
the native
protein, a synthetic variant thereof, or a derivative of the foregoing. An
appropriate
immunogenic preparation can contain, for example, the naturally occurring
immunogenic
protein, a chemically synthesized polypeptide representing the immunogenic
protein, or a
recombinantly expressed immunogenic protein. Furthermore, the protein may be
conjugated
to a second protein known to be immunogenic in the mammal being immunized.
Examples of
such immunogenic proteins include but are not limited to keyhole limpet
hemocyanin, serum
61
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
albumin, bovine thyroglobulin, and soybean trypsin inhibitor. The preparation
can further
include an adjuvant. Various adjuvants used to increase the immunological
response include,
but are not limited to, Freund's (complete and incomplete), mineral gels
(e.g., aluminum
hydroxide), surface active substances (e.g., lysolecithin, pluronic polyols,
polyanions,
peptides, oil emulsions, dinitrophenol, etc.), adjuvants usable in humans such
as Bacille
Calmette-Guerin and Corynebacterium parvum, or similar immunostimulatory
agents.
Additional examples of adjuvants which can be employed include MPL-TDM
adjuvant
(monophosphoryl Lipid A, synthetic trehalose dicorynomycolate).
The polyclonal antibody molecules directed against the immunogenic protein can
be
isolated from the mammal (e.g., from the blood) and further purified by well
known
techniques, such as affinity chromatography using protein A or protein G,
which provide
primarily the IgG fraction of immune serum. Subsequently, or alternatively,
the specific
antigen which is the target of the immunoglobulin sought, or an epitope
thereof, may be
immobilized on a column to purify the immune specific antibody by
immunoaffinity
chromatography. Purification of immunoglobulins is discussed, for example, by
D. Wilkinson
(The Scientist, published by The Scientist, Inc., Philadelphia PA, Vol. 14,
No. 8 (April 17,
2000), pp. 25-28).
Monoclonal Antibodies
The term "monoclonal antibody" (MAb) or "monoclonal antibody composition", as
used herein, refers to a population of antibody molecules that contain only
one molecular
species of antibody molecule consisting of a unique light chain gene product
and a unique
heavy chain gene product. In particular, the complementarity determining
regions (CDRs) of
the monoclonal antibody are identical in all the molecules of the population.
MAbs thus
contain an antigen binding site capable of immunoreacting with a particular
epitope of the
antigen characterized by a unique binding affinity for it.
Monoclonal antibodies can be prepared using hybridoma methods, such as those
described by Kohler and Milstein, Nature, 256:495 (1975). In a hybridoma
method, a mouse,
hamster, or other appropriate host animal, is typically immunized with an
immunizing agent to
elicit lymphocytes that produce or are capable of producing antibodies that
will specifically
bind to the immunizing agent. Alternatively, the lymphocytes can be immunized
in vitro.
The immunizing agent will typically include the protein antigen, a fragment
thereof or
a fusion protein thereof. Generally, either peripheral blood lymphocytes are
used if cells of
62
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
human origin are desired, or spleen cells or lymph node cells are used if non-
human
mammalian sources are desired. The lymphocytes are then fused with an
immortalized cell
line using a suitable fusing agent, such as polyethylene glycol, to form a
hybridoma cell
(Goding, MONOCLONAL ANTIBODIES: PRINCIPLES AND PRACTICE, Academic Press,
(1986) pp.
59-103). Immortalized cell lines are usually transformed mammalian cells,
particularly
myeloma cells of rodent, bovine and human origin. Usually, rat or mouse
myeloma cell lines
are employed. The hybridoma cells can be cultured in a suitable culture medium
that
preferably contains one or more substances that inhibit the growth or survival
of the unfused,
immortalized cells. For example, if the parental cells lack the enzyme
hypoxanthine guanine
phosphoribosyl transferase (HGPRT or HPRT), the culture medium for the
hybridomas
typically will include hypoxanthine, aminopterin, and thymidine ("HAT
medium"), which
substances prevent the growth of HGPRT-deficient cells.
Preferred immortalized cell lines are those that fuse efficiently, support
stable high
level expression of antibody by the selected antibody-producing cells, and are
sensitive to a
medium such as HAT medium. More preferred immortalized cell lines are marine
myeloma
lines, which can be obtained, for instance, from the Sallc Institute Cell
Distribution Center, San
Diego, California and the American Type Culture Collection, Mantissas,
Virginia. Human
myeloma and mouse-human heteromyeloma cell lines also have been described for
the
production of human monoclonal antibodies (I~ozbor, J. Immunol., 133:3001
(1984); Brodeur
et al., MONOCLONAL ANTIBODY PRODUCTION TECHNIQUES AND APPLICATIONS, Marcel
Dekker, Inc., New York, (1987) pp. 51-63).
The culture medium in which the hybridoma cells are cultured can then be
assayed for
the presence of monoclonal antibodies directed against the antigen.
Preferably, the binding
specificity of monoclonal antibodies produced by the hybridoma cells is
determined by
immunoprecipitation or by an in vitro binding assay, such as radioimmunoassay
(RIA) or
enzyme-linked immunoabsorbent assay (ELISA). Such techniques and assays are
known in
the art. The binding affinity of the monoclonal antibody can, for example, be
determined by
the Scatchard analysis of Munson and Pollard, Ahal. Biochem., 107:220 (1980).
Preferably,
antibodies having a high degree of specificity and a high binding affinity for
the target antigen
are isolated.
After the desired hybridoma cells are identified, the clones can be subcloned
by
limiting dilution procedures and grown by standard methods. Suitable culture
media for this
63
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
purpose include, for example, Dulbecco's Modified Eagle's Medium and RPMI-1640
medium.
Alternatively, the hybridoma cells can be grown in vivo as ascites in a
mammal.
The monoclonal antibodies secreted by the subclones can be isolated or
purified from
the culture medium or ascites fluid by conventional immunoglobulin
purification procedures
such as, for example, protein A-Sepharose, hydroxylapatite chromatography, gel
electrophoresis, dialysis, or affinity chromatography.
The monoclonal antibodies can also be made by recombinant DNA methods, such as
those described in U.S. Patent No. 4,816,567. DNA encoding the monoclonal
antibodies of
the invention can be readily isolated and sequenced using conventional
procedures (e.g., by
using oligonucleotide probes that are capable of binding specifically to genes
encoding the
heavy and light chains of marine antibodies). The hybridoma cells of the
invention serve as a
preferred source of such DNA. Once isolated, the DNA can be placed into
expression vectors,
which are then transfected into host cells such as simian COS cells, Chinese
hamster ovary
(CHO) cells, or myeloma cells that do not otherwise produce immunoglobulin
protein, to
obtain the synthesis of monoclonal antibodies in the recombinant host cells.
The DNA also
can be modified, for example, by substituting the coding sequence for human
heavy and light
chain constant domains in place of the homologous marine sequences (U.S.
Patent No.
4,816,567; Mornson, Nature 368, 812-13 (1994)) or by covalently joining to the
immunoglobulin coding sequence all or part of the coding sequence for a non-
imrnunoglobulin
polypeptide. Such a non-immunoglobulin polypeptide can be substituted for the
constant
domains of an antibody of the invention, or can be substituted for the
variable domains of one
antigen-combining site of an antibody of the invention to create a chimeric
bivalent antibody.
Humanized Antibodies
The antibodies directed against the protein antigens of the invention can
further
comprise humanized antibodies or human antibodies. These antibodies are
suitable for
administration to humans without engendering an immune response by the human
against the
administered immunoglobulin. Humanized forms of antibodies are chimeric
immunoglobulins,
immunoglobulin chains or fragments thereof (such as Fv, Fab, Fab', F(ab')Z or
other antigen-
binding subsequences of antibodies) that are principally comprised of the
sequence of a human
immunoglobulin, and contain minimal sequence derived from a non-human
immunoglobulin.
Humanization can be performed following the method of Winter and co-workers
(Jones et al.,
Nature, 321:522-525 (1986); Riechmann et al., Nature, 332:323-327 (1988);
Verhoeyen et al.,
64
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
Science, 239:1534-1536 (1988)), by substituting rodent CDRs or CDR sequences
for the
corresponding sequences of a human antibody. (See also U.S. Patent No.
5,225,539.) In some
instances, Fv framework residues of the human immunoglobulin are replaced by
corresponding non-human residues. Humanized antibodies can also comprise
residues which
are found neither in the recipient antibody nor in the imported CDR or
framework sequences.
In general, the humanized antibody will comprise substantially all of at least
one, and typically
two, variable domains, in which all or substantially all of the CDR regions
correspond to those
of a non-human immunoglobulin and all or substantially all of the framework
regions are
those of a human immunoglobulin consensus sequence. The humanized antibody
optimally
also will comprise at least a portion of an immunoglobulin constant region
(Fc), typically that
of a human immunoglobulin (Jones et al., 1986; Riechmann et al., 1988; and
Presta, Curr. Op.
St~uct. Biol., 2:593-596 (1992)).
Human Antibodies
Fully human antibodies relate to antibody molecules in which essentially the
entire
sequences of both the light chain and the heavy chain, including the CDRs,
arise from human
genes. Such antibodies are termed "human antibodies", or "fully human
antibodies" herein.
Human monoclonal antibodies can be prepared by the trioma technique; the human
B-cell
hybridoma technique (see Kozbor, et al., 1983 Immunol Today 4: 72) and the EBV
hybridoma
technique to produce human monoclonal antibodies (see Cole, et al., 1985 In:
MONOCLONAL
2O ANTIBODIES AND CANCER THERAPY, Alan R. Liss, Inc., pp. 77-96). Human
monoclonal
antibodies may be utilized in the practice of the present invention and may be
produced by
using human hybridomas (see Cote, et al., 1983. Proc Natl Acad Sci USA 80:
2026-2030) or
by transforming human B-cells with Epstein Barr Virus in vitro (see Cole, et
al., 1985 In:
MONOCLONAL ANTIBODIES AND CANCER THERAPY, Alan R. Liss, Inc., pp. 77-96).
In addition, human antibodies can also be produced using additional
techniques,
including phage display libraries (Hoogenboom and Winter, J. Mol. Biol.,
227:381 (1991);
Marks et al., .I. Mol. Biol., 222:581 (1991)). Similarly, human antibodies can
be made by
introducing human immunoglobulin loci into transgenic animals, e.g., mice in
which the
endogenous immunoglobulin genes have been partially or completely inactivated.
Upon
challenge, human antibody production is observed, which closely resembles that
seen in
humans in all respects, including gene rearrangement, assembly, and antibody
repertoire. This
approach is described, for example, in U.S. Patent Nos. 5,545,807; 5,545,806;
5,569,825;
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
5,625,126; 5,633,425; 5,661,016, and in Marks et al. (BiolTechnology 10, 779-
783 (1992));
Lonberg et al. (Nature 368 856-859 (1994)); Morrison ( Nature 368, 812-13
(1994)); Fishwild
et al,( Nature Biotechnology 14, 845-51 (1996)); Neuberger (Nature
Biotechnology 14, 826
(1996)); and Lonberg and Huszar (Intern. Rev. Imnaunol. 13 65-93 (1995)).
Human antibodies may additionally be produced using transgenic nonhuman
animals
which are modified so as to produce fully human antibodies rather than the
animal's
endogenous antibodies in response to challenge by an antigen. (See PCT
publication
W094/02602). The endogenous genes encoding the heavy and light immunoglobulin
chains in
the nonhuman host have been incapacitated, and active loci encoding human
heavy and light
chain immunoglobulins are inserted into the host's genome. The human genes are
incorporated, for example, using yeast artificial chromosomes containing the
requisite human
DNA segments. An animal which provides all the desired modifications is then
obtained as
progeny by crossbreeding intermediate transgenic animals containing fewer than
the full
complement of the modifications. The preferred embodiment of such a nonhuman
animal is a
mouse, and is termed the Xenomouse~ as disclosed in PCT publications WO
96/33735 and
WO 96/34096. This animal produces B cells which secrete fully human
immunoglobulins.
The antibodies can be obtained directly from the animal after immunization
with an
immunogen of interest, as, for example, a preparation of a polyclonal
antibody, or alternatively
from immortalized B cells derived from the animal, such as hybridomas
producing
monoclonal antibodies. Additionally, the genes encoding the immunoglobulins
with human
variable regions can be recovered and expressed to obtain the antibodies
directly, or can be
further modified to obtain analogs of antibodies such as, for example, single
chain Fv
molecules.
An example of a method of producing a nonhuman host, exemplified as a mouse,
lacking expression of an endogenous immunoglobulin heavy chain is disclosed in
U.S. Patent
No. 5,939,598. It can be obtained by a method including deleting the J segment
genes from at
least one endogenous heavy chain locus in an embryonic stem cell to prevent
rearrangement of
the locus and to prevent formation of a transcript of a rearranged
irnmunoglobulin heavy chain
locus, the deletion being effected by a targeting vector containing a gene
encoding a selectable
marker; and producing from the embryonic stem cell a transgenic mouse whose
somatic and
germ cells contain the gene encoding the selectable marker.
66
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
A method for producing an antibody of interest, such as a human antibody, is
disclosed
in U.S. Patent No. 5,916,771. It includes introducing an expression vector
.that contains a
nucleotide sequence encoding a heavy chain into one mammalian host cell in
culture,
introducing an expression vector containing a nucleotide sequence encoding a
light chain into
another mammalian host cell, and fusing the two cells to form a hybrid cell.
The hybrid cell
expresses an antibody containing the heavy chain and the light chain.
In a further improvement on this procedure, a method for identifying a
clinically
relevant epitope on an immunogen, and a correlative method for selecting an
antibody that
binds immunospecifically to the relevant epitope with high affinity, are
disclosed in PCT
publication WO 99/53049.
Fab Fragments and Single Chain Antibodies
According to the invention, techniques can be adapted for the production of
single-chain antibodies specific to an antigenic protein of the invention (see
e.g., U.S. Patent
No. 4,946,778). In addition, methods can be adapted for the construction of
Fab expression
libraries (see e.g., Huse, et al., 1989 Science 246: 1275-1281) to allow rapid
and effective
identification of monoclonal Fab fragments with the desired specificity for a
protein or
derivatives, fragments, analogs or homologs thereof. Antibody fragments that
contain the
idiotypes to a protein antigen may be produced by techniques known in the art
including, but
not limited to: (i) an F~ab')2 fragment produced by pepsin digestion of an
antibody molecule; (ii)
an Fab fragment generated by reducing the disulfide bridges of an F~ab')2
fragment; (iii) an Fab
fragment generated by the treatment of the antibody molecule with papain and a
reducing
agent and (iv) F~ fragments.
BispeciBc Antibodies
Bispecific antibodies are monoclonal, preferably human or humanized,
antibodies that
have binding specificities for at least two different antigens. In the present
case, one of the
binding specificities is for an antigenic protein of the invention. The second
binding target is
any other antigen, and advantageously is a cell-surface protein or receptor or
receptor subunit.
Methods for making bispecific antibodies are known in the art. Traditionally,
the
recombinant production of bispecific antibodies is based on the co-expression
of two
immunoglobulin heavy-chain/light-chain pairs, where the two heavy chains have
different
specificities (Milstein and Cuello, Nature, 305:537-539 (1983)). Because of
the random
assortment of immunoglobulin heavy and light chains, these hybridomas
(quadromas) produce
67
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
a potential mixture of ten different antibody molecules, of which only one has
the correct
bispecific structure. The purification of the correct molecule is usually
accomplished by
affinity chromatography steps. Similar procedures are disclosed in WO
93/08829, published
13 May 1993, and in Traunecker et al., 1991 EMBO J., 10:3655-3659.
Antibody variable domains with the desired binding specificities (antibody-
antigen
combining sites) can be fused to immunoglobulin constant domain sequences. The
fusion
preferably is with an immunoglobulin heavy-chain constant domain, comprising
at least part
of the hinge, CH2, and CH3 regions. It is preferred to have the first heavy-
chain constant
region (CH1) containing the site necessary for light-chain binding present in
at least one of the
fusions. DNAs encoding the immunoglobulin heavy-chain fusions and, if desired,
the
immunoglobulin light chain, are inserted into separate expression vectors, and
are co-
transfected into a suitable host organism. For further details of generating
bispecific
antibodies see, for example, Suresh et al., Methods in Enzymology, 121:210
(1986).
According to another approach described in WO 96/27011, the interface between
a pair
of antibody molecules can be engineered to maximize the percentage of
heterodimers which
are recovered from recombinant cell culture. The preferred interface comprises
at least a part
of the CH3 region of an antibody constant domain. In this method, one or more
small amino
acid side chains from the interface of the first antibody molecule are
replaced with larger side
chains (e.g. tyrosine or tryptophan). Compensatory "cavities" of identical or
similar size to the
large side chains) are created on the interface of the second antibody
molecule by replacing
large amino acid side chains with smaller ones (e.g. alanine or threonine).
This provides a
mechanism for increasing the yield of the heterodimer over other unwanted end-
products such
as homodimers.
Bispecific antibodies can be prepaxed as full length antibodies or antibody
fragments
(e.g. F(ab')2 bispecific antibodies). Techniques for generating bispecific
antibodies from
antibody fragments have been described in the literature. For example,
bispecific antibodies
can be prepared using chemical linkage. Brennan et al., ,Science 229:81 (1985)
describe a
procedure wherein intact antibodies are proteolytically cleaved to generate
F(ab')2 fragments.
These fragments are reduced in the presence of the dithiol complexing agent
sodium arsenite
to stabilize vicinal dithiols and prevent intermolecular disulfide formation.
The Fab'
fragments generated axe then converted to thionitrobenzoate (TNB) derivatives.
One of the
Fab'-TNB derivatives is then reconverted to the Fab'-thiol by reduction with
68
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
mercaptoethylamine and is mixed with an equimolar amount of the other Fab'-TNB
derivative
to form the bispecific antibody. The bispecific antibodies produced can be
used as agents for
the selective immobilization of enzymes.
Additionally, Fab' fragments can be directly recovered from E. coli and
chemically
coupled to form bispecific antibodies. Shalaby et al., J. Exp. Med. 175:217-
225 (1992)
describe the production of a fully humanized bispecific antibody F(ab')a
molecule. Each Fab'
fragment was separately secreted from E. coli and subjected to directed
chemical coupling in
vitro to form the bispecific antibody. The bispecific antibody thus formed was
able to bind to
cells overexpressing the ErbB2 receptor and normal human T cells, as well as
trigger the lytic
activity of human cytotoxic' lymphocytes against human breast tumor targets.
Various techniques for making and isolating bispecific antibody fragments
directly
from recombinant cell culture have also been described. For example,
bispecific antibodies
have been produced using leucine zippers. Kostelny et al., J. Immuhol.
148(5):1547-1553
(1992). The leucine zipper peptides from the Fos and Jun proteins were linked
to the Fab'
portions of two different antibodies by gene fusion. The antibody homodimers
were reduced
at the hinge region to form monomers and then re-oxidized to form the antibody
heterodimers.
This method can also be utilized for the production of antibody homodimers.
The "diabody"
technology described by Hollinger et al., Proc. Natl. Acad. Sci. USA 90:6444-
6448 (1993) has
provided an alternative mechanism for making bispecific antibody fragments.
The fragments
comprise a heavy-chain variable domain (VH) connected to a light-chain
variable domain (VL)
by a linker which is too short to allow pairing between the two domains on the
same chain.
Accordingly, the VH and VL domains of one fragment are forced to pair with the
complementary VL and VH domains of another fragment, thereby forming two
antigen-binding
sites. Another strategy for making bispecific antibody fragments by the use of
single-chain Fv
(sFv) dimers has also been reported. See, Gruber et al., J. Immuraol. 152:5368
(1994).
Antibodies with more than two valencies are contemplated. For example,
trispecific
antibodies can be prepared. Tutt et al., J. Immuhol. 147:60 (1991).
Exemplary bispecific antibodies can bind to two different epitopes, at least
one of
which originates in the protein antigen of the invention. Alternatively, an
anti-antigenic arm
of an immunoglobulin molecule can be combined with an arm which binds to a
triggering
molecule on a leukocyte such as a T-cell receptor molecule (e.g. CD2, CD3,
CD28, or B7), or
Fc receptors for IgG (FcyR), such as FcyRI (CD64), FcyRII (CD32) and FcyRIII
(CD16) so as
69
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
to focus cellular defense mechanisms to the cell expressing the particular
antigen. Bispecific
antibodies can also be used to direct cytotoxic agents to cells which express
a particular
antigen. These antibodies possess an antigen-binding arm and an arm which
binds a cytotoxic
agent or a radionuclide chelator, such as EOTUBE, DPTA, DOTA, or TETA. Another
bispecific antibody of interest binds the protein antigen described herein and
further binds
tissue factor (TF).
Heteroconjugate Antibodies
Heteroconjugate antibodies are also within the scope of the present invention.
Heteroconjugate antibodies are composed of two covalently joined antibodies.
Such
antibodies have, for example, been proposed to target immune system cells to
unwanted cells
(U.S. Patent No. 4,676,980), and for treatment of HIV infection (WO 91/00360;
WO
92/200373; EP 03089). It is contemplated that the antibodies can be prepared
in vitro using
known methods in synthetic protein chemistry, including those involving
crosslinking agents.
For example, immunotoxins can be constructed using a disulfide exchange
reaction or by
forming a thioether bond. Examples of suitable reagents for this purpose
include iminothiolate
and methyl-4-mercaptobutyrimidate and those disclosed, for example, in U.S.
Patent No.
4,676,980.
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
Effector Function Engineering
It can be desirable to modify the antibody of the invention with respect to
effector
function, so as to enhance, e.g., the effectiveness of the antibody in
treating cancer. For
example, cysteine residues) can be introduced into the Fc region, thereby
allowing interchain
disulfide bond formation in this region. The homodimeric antibody thus
generated can have
improved internalization capability and/or increased complement-mediated cell
killing and
antibody-dependent cellular cytotoxicity (ADCC). See Caron et al., J. Exp
Med., 176: 1191-
1195 (1992) and Shopes, J. Iminunol., 148: 2918-2922 (1992). Homodimeric
antibodies with
enhanced anti-tumor activity can also be prepared using heterobifunctional
cross-linkers as
described in Wolff et al. Cancer Research, 53: 2560-2565 (1993).
Alternatively, an antibody
can be engineered that has dual Fc regions and can thereby have enhanced
complement lysis
and ADCC capabilities. See Stevenson et al., Anti-Cancer Drug Design, 3: 219-
230 (1989).
Immunoconjugates
The invention also pertains to immunoconjugates comprising an antibody
conjugated
to a cytotoxic agent such as a chemotherapeutic agent, toxin (e.g., an
enzymatically active
toxin of bacterial, fungal, plant, or animal origin, or fragments thereof), or
a radioactive
isotope (i.e., a radioconjugate).
Chemotherapeutic agents useful in the generation of such immunoconjugates have
been described above. Enzymatically active toxins and fragments thereof that
can be used
include diphtheria A chain, nonbinding active fragments of diphtheria toxin,
exotoxin A chain
(from Pseudomonas aeruginosa), ricin A chain, abrin A chain, modeccin A chain,
alpha-sarcin,
Aleurites fordii proteins, dianthin proteins, Phytolaca americana proteins
(PAPI, PAPII, and
PAP-S), momordica charantia inhibitor, curcin, crotin, sapaonaria officinalis
inhibitor,
gelonin, mitogellin, restrictocin, phenomycin, enomycin, and the
tricothecenes. A variety of
radionuclides are available for the production of radioconjugated antibodies.
Examples
include alaBi, l3ih i3y~ 90~,~ ~d 186Re.
Conjugates of the antibody and cytotoxic agent are made using a variety of
bifunctional protein-coupling agents such as N-succinimidyl-3-(2-
pyridyldithiol) propionate
(SPDP), iminothiolane (IT), bifunctional derivatives of imidoesters (such as
dimethyl
adipimidate HCL), active esters (such as disuccinimidyl suberate), aldehydes
(such as
glutareldehyde), bis-azido compounds (such as bis (p-azidobenzoyl)
hexanediamine), bis-
diazonium derivatives (such as bis-(p-diazoniumbenzoyl)-ethylenediamine),
diisocyanates
71
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
(such as tolyene 2,6-diisocyanate), and bis-active fluorine compounds (such as
1,5-difluoro-
2,4-dinitrobenzene). For example, a ricin immunotoxin can be prepared as
described in
Vitetta et al., Science, 238: 1098 (1987). Carbon-14-labeled 1-
isothiocyanatobenzyl-3-
methyldiethylene triaminepentaacetic acid (MX-DTPA) is an exemplary chelating
agent for
conjugation of radionucleotide to the antibody. See W094/11026.
In another embodiment, the antibody can be conjugated to a "receptor" (such
streptavidin) for utilization in tumor pretargeting wherein the antibody-
receptor conjugate is
administered to the patient, followed by removal of unbound conjugate from the
circulation
using a clearing agent and then administration of a "ligand" (e.g., avidin)
that is in turn
conjugated to a cytotoxic agent.
In one embodiment, methods for the screening of antibodies that possess the
desired
specificity include, but are not limited to, enzyme-linked immunosorbent assay
(ELISA) and
other immunologically-mediated techniques known within the art. In a specific
embodiment,
selection of antibodies that are specific to a particular domain of an FGF-CX
and/or FCTRX
protein is facilitated by generation of hybridomas that bind to the fragment
of an FGF-CX
and/or FCTRX protein possessing such a domain. Thus, antibodies that are
specific for a
desired domain within an FGF-CX and/or FCTRX protein, or derivatives,
fragments, analogs
or homologs thereof, are also provided herein.
Anti-FGF-CX and/or FCTRX antibodies may be used in methods known within the
art
relating to the localization and/or quantitation of an FGF-CX and/or FCTRX
protein (e.g., for
use in measuring levels of the FGF-CX and/or FCTRX protein within appropriate
physiological samples, for use in diagnostic methods, for use in imaging the
protein, and the
like). In a given embodiment, antibodies for FGF-CX and/or FCTRX proteins, or
derivatives,
fragments, analogs or homologs thereof, that contain the antibody derived
binding domain, are
utilized as pharmacologically-active compounds (hereinafter "Therapeutics").
An anti-FGF-CX and/or FCTRX antibody (e.g., monoclonal antibody) can be used
to
isolate an FGF-CX and/or FCTRX polypeptide by standard techniques, such as
affinity
chromatography or immunoprecipitation. An anti-FGF-CX and/or FCTRX antibody
can
facilitate the purification of natural FGF-CX and/or FCTRX polypeptide from
cells and of
recombinantly-produced FGF-CX and/or FCTRX polypeptide expressed in host
cells.
Moreover, an anti-FGF-CX and/or FCTRX antibody can be used to detect FGF-CX
and/or
FCTRX protein (e.g., in a cellular lysate or cell supernatant) in order to
evaluate the
72
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
abundance and pattern of expression of the FGF-CX and/or FCTRX protein. Anti-
FGF-CX
and/or FCTRX antibodies can be used diagnostically to monitor protein levels
in tissue as part
of a clinical testing procedure, e.g., to, for example, determine the efficacy
of a given
treatment regimen. Detection can be facilitated by coupling (i. e., physically
linking) the
antibody to a detectable substance. Examples of detectable substances include
various
enzymes, prosthetic groups, fluorescent materials, luminescent materials,
bioluminescent
materials, and radioactive materials. Examples of suitable enzymes include
horseradish
peroxidase, alkaline phosphatase, (3-galactosidase, or acetylcholinesterase;
examples of
suitable prosthetic group complexes include streptavidin/biotin and
avidin/biotin; examples of
suitable fluorescent materials include umbelliferone, fluorescein, fluorescein
isothiocyanate,
rhodamine, dichlorotriazinylamine fluorescein, dansyl chloride or
phycoerythrin; an example
of a luminescent material includes luminol; examples of bioluminescent
materials include
luciferase, luciferin, and aequorin, and examples of suitable radioactive
material include lash
isih sss or 3H.
FGF-CX and/or FCTRX Recombinant Expression Vectors and Host Cells
Another aspect of the invention pertains to vectors, preferably expression
vectors,
containing a nucleic acid encoding an FGF-CX and/or FCTRX protein, or
derivatives,
fragments, analogs or homologs thereof. As used herein, the term "vector"
refers to a nucleic
acid molecule capable of transporting another nucleic acid to which it has
been linked. One
type of vector is a "plasmid", which refers to a circular double stranded DNA
loop into which
additional DNA segments can be ligated. Another type of vector is a viral
vector, wherein
additional DNA segments can be ligated into the viral genome. Certain vectors
are capable of
autonomous replication in a host cell into which they are introduced (e.g.,
bacterial vectors
having a bacterial origin of replication and episomal mammalian vectors).
Other vectors (e.g.,
non-episomal mammalian vectors) are integrated into the genome of a host cell
upon
introduction into the host cell, and thereby are replicated along with the
host genome.
Moreover, certain vectors are capable of directing the expression of genes to
which they are
operatively-linked. Such vectors are referred to herein as "expression
vectors". In general,
expression vectors of utility in recombinant DNA techniques are often in the
form of plasmids.
In the present specification, "plasmid" and "vector" can be used
interchangeably as the
plasmid is the most commonly used form of vector. However, the invention is
intended to
73
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
include such other forms of expression vectors, such as viral vectors (e.g.,
replication defective
retroviruses, adenoviruses and adeno-associated viruses), which serve
equivalent functions.
The recombinant expression vectors of the invention comprise a nucleic acid of
the
invention in a form suitable for expression of the nucleic acid in a host
cell, which means that
the recombinant expression vectors include one or more regulatory sequences,
selected on the
basis of the host cells to be used for expression, that is operatively-linked
to the nucleic acid
sequence to be expressed. Within a recombinant expression vector, "operably-
linked" is
intended to mean that the nucleotide sequence of interest is linked to the
regulatory
sequences) in a manner that allows for expression of the nucleotide sequence
(e.g., in an in
vitro transcriptionltranslation system or in a host cell when the vector is
introduced into the
host cell).
The term "regulatory sequence" is intended to includes promoters, enhancers
and other
expression control elements (e.g., polyadenylation signals). Such regulatory
sequences are
described, for example, in Goeddel, GENE EXPRESSION TECHNOLOGY: METHODS IN
ENZYMOLOGY 185, Academic Press, San Diego, Calif. (1990). Regulatory sequences
include
those that direct constitutive expression of a nucleotide sequence in many
types of host cell
and those that direct expression of the nucleotide sequence only in certain
host cells (e.g.,
tissue-specific regulatory sequences). It will be appreciated by those skilled
in the art that the
design of the expression vector can depend on such factors as the choice of
the host cell to be
transformed, the level of expression of protein desired, etc. The expression
vectors of the
invention can be introduced into host cells to thereby produce proteins or
peptides, including
fusion proteins or peptides, encoded by nucleic acids as described herein
(e.g., FGF-CX and/or
FCTRX proteins, mutant forms of FGF-CX and/or FCTRX proteins, fusion proteins,
etc.).
The recombinant expression vectors of the invention can be designed for
expression of
FGF-CX and/or FCTRX proteins in prokaryotic or eukaryotic cells. For example,
FGF-CX
and/or FCTRX proteins can be expressed in bacterial cells such as EsclZerichia
coli, insect
cells (using baculovirus expression vectors) yeast cells or mammalian cells.
Suitable host cells
are discussed further in Goeddel, GENE EXPRESSION TECHNOLOGY: METHODS IN
ENZYMOLOGY 185, Academic Press, San Diego, Calif. (1990). Alternatively, the
recombinant
expression vector can be transcribed and translated ih vitro, for example
using T7 promoter
regulatory sequences and T7 polymerase.
74
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
Expression of proteins in prokaryotes is most often carried out in EscheYichia
coli with
vectors containing constitutive or inducible promoters directing the
expression of either fusion
or non-fusion proteins. Fusion vectors add a number of amino acids to a
protein encoded
therein, usually to the amino terminus of the recombinant protein. Such fusion
vectors
typically serve three purposes: (i) to increase expression of recombinant
protein; (ii) to
increase the solubility of the recombinant protein; and (iii) to aid in the
purification of the
recombinant protein by acting as a ligand in affinity purification. Often, in
fusion expression
vectors, a proteolytic cleavage site is introduced at the junction of the
fusion moiety and the
recombinant protein to enable separation of the recombinant protein from the
fusion moiety
subsequent to purification of the fusion protein. Such enzymes, and their
cognate recognition
sequences, include Factor Xa, thrombin and enterokinase. Typical fusion
expression vectors
include pGEX (Pharmacia Biotech Inc; Smith and Johnson, 1988. Geue 67: 31-40),
pMAL
(New England Biolabs, Beverly, Mass.) and pRITS (Pharmacia, Piscataway, N.J.)
that fuse
glutathione S-transferase (GST), maltose E binding protein, or protein A,
respectively, to the
target recombinant protein.
Examples of suitable inducible non-fusion E. coli expression vectors include
pTrc
(Amrann et al., (1988) Geue 69:301-315) and pET l 1d (Studier et al., GENE
EXPRESSION
TECHNOLOGY: METHODS IN ENZYMOLOGY 185, Academic Press, San Diego, Calif.
(1990)
60-89).
One strategy to maximize recombinant protein expression in E. coli is to
express the
protein in a host bacteria with an impaired capacity to proteolytically cleave
the recombinant
protein. See, e.g., Gottesman, GENE EXPRESSION TECHNOLOGY: METHODS IN
ENZYMOLOGY
185, Academic Press, San Diego, Calif. (1990) 119-128. Another strategy is to
alter the
nucleic acid sequence of the nucleic acid to be inserted into an expression
vector so that the
individual codons for each amino acid are those preferentially utilized in E.
coli (see, e.g.,
Wada, et al., 1992. Nucl. Acids Res. 20: 2111-2118). Such alteration of
nucleic acid
sequences of the invention can be carried out by standard DNA synthesis
techniques.
In another embodiment, the FGF-CX and/or FCTRX expression vector is a yeast
expression vector. Examples of vectors for expression in yeast Saccha~o~rayces
ce~ivisae
include pYepSecl (Baldari, et al., 1987. EMBO J. 6: 229-234), pMFa (Kurjan and
Herskowitz, 1982. Cell 30: 933-943), pJRY88 (Schultz et al., 1987. Gehe 54:
113-123),
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
pYES2 (Invitrogen Corporation, San Diego, Calif.), and picZ (InVitrogen Corp,
San Diego,
Calif.).
Alternatively, FGF-CX and/or FCTRX can be expressed in insect cells using
baculovirus expression vectors. Baculovirus vectors available for expression
of proteins in
cultured insect cells (e.g., SF9 cells) include the pAc series (Smith, et al.,
1983. Mol. Cell.
Biol. 3: 2156-2165) and the pVL series (Lucklow and Summers, 1989. Virology
170: 31-39).
In yet another embodiment, a nucleic acid of the invention is expressed in
mammalian
cells using a mammalian expression vector. Examples of mammalian expression
vectors
include pCDM8 (Seed, 1987. Nature 329: 840) and pMT2PC (Kaufinan, et al.,
1987. EMBO
.I. 6: 187-195). When used in mammalian cells, the expression vector's control
functions are
often provided by viral regulatory elements. For example, commonly used
promoters are
derived from polyoma, adenovirus 2, cytomegalovirus, and simian virus 40. For
other suitable
expression systems for both prokaryotic and eukaryotic cells see, e.g.,
Chapters 16 and 17 of
Sambrook, et al., MOLECULAR CLONING: A LABORATORY MANUAL. 2nd ed., Cold Spring
Harbor Laboratory, Cold Spring Harbor Laboratory Press, Cold Spring Harbor,
N.Y., 1989.
In another embodiment, the recombinant mammalian expression vector is capable
of
directing expression of the nucleic acid preferentially in a particular cell
type (e.g.,
tissue-specific regulatory elements are used to express the nucleic acid).
Tissue-specific
regulatory elements axe known in the art. Non-limiting examples of suitable
tissue-specific
promoters include the albumin promoter (liver-specific; Pinkert, et al., 1987.
Genes Dev. 1:
268-277), lymphoid-specific promoters (Calame and Eaton, 1988. Adv. Immuyaol.
43:
235-275), in particular promoters of T cell receptors (Winoto and Baltimore,
1989. EMBO J.
8: 729-733) and immunoglobulins (Banerji, et al., 1983. Cell 33: 729-740;
Queen and
Baltimore, 1983. Cell 33: 741-748), neuron-specific promoters (e.g., the
neurofilament
promoter; Byrne and Ruddle, 1989. Proc. Natl. Acad. Sci. USA 86: 5473-5477),
pancreas-specific promoters (Edlund, et al., 1985. Scierace 230: 912-916), and
mammary
gland-specific promoters (e.g., milk whey promoter; LT.S. Pat. No. 4,873,316
and European
Application Publication No. 264,166). Developmentally-regulated promoters are
also
encompassed, e.g., the marine hox promoters (Kessel and Grass, 1990. Science
249: 374-379)
and the oc-fetoprotein promoter (Campes and Tilghman, 1989. Gehes Dev. 3: 537-
546).
The invention further provides a recombinant expression vector comprising a
DNA
molecule of the invention cloned into the expression vector in an antisense
orientation. That
76
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
is, the DNA molecule is operatively-linked to a regulatory sequence in a
manner that allows
for expression (by transcription of the DNA molecule) of an RNA molecule that
is antisense to
FGF-CX and/or FCTRX mRNA. Regulatory sequences operatively linked to a nucleic
acid
cloned in the antisense orientation can be chosen that direct the continuous
expression of the
antisense RNA molecule in a variety of cell types, for instance viral
promoters and/or
enhancers, or regulatory sequences can be chosen that direct constitutive,
tissue specific or cell
type specific expression of antisense RNA. The antisense expression vector can
be in the form
of a recombinant plasmid, phagemid or attenuated virus in which antisense
nucleic acids are
produced under the control of a high efficiency regulatory region, the
activity of which can be
determined by the cell type into which the vector is introduced. For a
discussion of the
regulation of gene expression using antisense genes see, e.g., Weintraub, et
al., "Antisense
RNA as a molecular tool for genetic analysis," Reviews-Trends ira Genetics,
Vol. 1(1) 1986.
Another aspect of the invention pertains to host cells into which a
recombinant
expression vector of the invention has been introduced. The terms "host cell"
and
"recombinant host cell" are used interchangeably herein. It is understood that
such terms refer
not only to the particular subj ect cell but also to the progeny or potential
progeny of such a
cell. Because certain modifications may occur in succeeding generations due to
either
mutation or environmental influences, such progeny may not, in fact, be
identical to the parent
cell, but are still included within the scope of the term as used herein.
A host cell can be any prokaryotic or eukaryotic cell. For example, FGF-CX
and/or
FCTRX protein can be expressed in bacterial cells such as E. coli, insect
cells, yeast or
mammalian cells (such as Chinese hamster ovary cells (CHO) or COS cells).
Other suitable
host cells are known to those skilled in the art.
Vector DNA can be introduced into prokaryotic or eukaryotic cells via
conventional
transformation or transfection techniques. As used herein, the terms
"transformation" and
"transfection" are intended to refer to a variety of art-recognized techniques
for introducing
foreign nucleic acid (e.g., DNA) into a host cell, including calcium phosphate
or calcium
chloride co-precipitation, DEAF-dextran-mediated transfection, lipofection, or
electroporation. Suitable methods for transforming or transfecting host cells
can be found in
Sambrook, et al. (MOLECULAR CLONING: A LABORATORY MANUAL. 2nd ed., Cold Spring
Harbor Laboratory, Cold Spring Harbor Laboratory Press, Cold Spring Harbor,
N.Y., 1989),
and other laboratory manuals.
77
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
For stable transfection of mammalian cells, it is known that, depending upon
the
expression vector and transfection technique used, only a small fraction of
cells may integrate
the foreign DNA into their genome. In order to identify and select these
integrants, a gene that
encodes a selectable marker (e.g., resistance to antibiotics) is generally
introduced into the
host cells along with the gene of interest. Various selectable markers include
those that confer
resistance to drugs, such as 6418, hygromycin and methotrexate. Nucleic acid
encoding a
selectable marker can be introduced into a host cell on the same vector as
that encoding FGF-
CX and/or FCTRX or can be introduced on a separate vector. Cells stably
transfected with the
introduced nucleic acid can be identified by drug selection (e.g., cells that
have incorporated
the selectable marker gene will survive, while the other cells die).
A host cell of the invention, such as a prokaryotic or eukaryotic host cell in
culture, can
be used to produce (i.e., express) FGF-CX and/or FCTIZX protein. Accordingly,
the invention
further provides methods for producing FGF-CX and/or FCTRX protein using the
host cells of
the invention. In one embodiment, the method comprises culturing the host cell
of invention
(into which a recombinant expression vector encoding FGF-CX and/or FCTRX
protein has
been introduced) in a suitable medium such that FGF-CX and/or FCT1RX protein
is produced.
In another embodiment, the method further comprises isolating FGF-CX and/or
FCTIRX
protein from the medium or the host cell.
Transgenic FGF-CX and/or FCTRX Animals
The host cells of the invention can also be used to produce non-human
transgenic
animals. For example, in one embodiment, a host cell of the invention is a
fertilized oocyte or
an embryonic stem cell into which FGF-CX and/or FCTRX protein-coding sequences
have
been introduced. Such host cells can then be used to create non-human
transgenic animals in
which exogenous FGF-CX and/or FCTIZX sequences have been introduced into their
genome
or homologous recombinant animals in which endogenous FGF-CX and/or FCTIZX
sequences
have been altered. Such animals are useful for studying the function and/or
activity of FGF-
CX and/or FCTRX protein and for identifying and/or evaluating modulators of
FGF-CX
and/or FCTRX protein activity. As used herein, a "transgenic animal" is a non-
human animal,
preferably a mammal, more preferably a rodent such as a rat or mouse, in which
one or more
of the cells of the animal includes a transgene. Other examples of transgenic
animals include
non-human primates, sheep, dogs, cows, goats, chickens, amphibians, etc. A
transgene is
exogenous DNA that is integrated into the genome of a cell from which a
transgenic animal
78
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
develops and that remains in the genome of the mature animal, thereby
directing the
expression of an encoded gene product in one or more cell types or tissues of
the transgenic
animal. As used herein, a "homologous recombinant animal" is a non-human
animal,
preferably a mammal, more preferably a mouse, in which an endogenous FGF-CX
and/or
FCTRX gene has been altered by homologous recombination between the endogenous
gene
and an exogenous DNA molecule introduced into a cell of the animal, e.g., an
embryonic cell
of the animal, prior to development of the animal.
A transgenic animal of the invention can be created by introducing FGF-CX
and/or
FCTRX-encoding nucleic acid into the male pronuclei of a fertilized oocyte
(e.g., by
microinjection, retroviral infection) and allowing the oocyte to develop in a
pseudopregnant
female foster animal. The human FGF-CX and/or FCTRX cDNA sequences of SEQ m
NOS:1, 3, 5, 7, 9, 11 and 13 can be introduced as a transgene into the genome
of a non-human
animal. Alternatively, a non-human homologue of the human FGF-CX and/or FCTRX
gene,
such as a mouse FGF-CX and/or FCTRX gene, can be isolated based on
hybridization to the
human FGF-CX and/or FCTRX cDNA (described further supra) and used as a
transgene.
Intronic sequences and polyadenylation signals can also be included in the
transgene to
increase the efficiency of expression of the transgene. A tissue-specific
regulatory sequences)
can be operably-linked to the FGF-CX and/or FCTRX transgene to direct
expression of FGF-
CX and/or FCTRX protein to particular cells. Methods for generating transgenic
animals via
embryo manipulation and microinjection, particularly animals such as mice,
have become
conventional in the art and are described, for example, in U.S. Patent Nos.
4,736,866;
4,870,009; and 4,873,191; and Hogan, 1986. In: MANIPULATING THE MousE EMBRYO,
Cold
Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y. Similar methods are
used for
production of other transgenic animals. A transgenic founder animal can be
identified based
upon the presence of the FGF-CX and/or FCTRX transgene in its genome and/or
expression of
FGF-CX and/or FCTRX mRNA in tissues or cells of the animas. A transgenic
founder
animal can then be used to breed additional animals carrying the transgene.
Moreover,
transgenic animals carrying a transgene-encoding FGF-CX and/or FCTRX protein
can further
be bred to other transgenic animals carrying other transgenes.
To create a homologous recombinant animal, a vector is prepared which contains
at
least a portion of an FGF-CX and/or FCTRX gene into which a deletion, addition
or
substitution has.been introduced to thereby alter, e.g., functionally disrupt,
the FGF-CX andJor
79
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
FCTRX gene. The FGF-CX and/or FCTRX gene can be a human gene (e.g., the cDNA
of
SEQ m NOS:l, 3, 5, 7, 9, 11 and 13), but more preferably, is a non-human
homologue of a
human FGF-CX and/or FCTRX gene. For example, a mouse homologue of human FGF-CX
and/or FCTRX gene of SEQ ID NOS:1, 3, 5, 7, 9, 11 and 13 can be used to
construct a
homologous recombination vector suitable for altering an endogenous FGF-CX
and/or
FCTRX gene in the mouse genome. In one embodiment, the vector is designed such
that,
upon homologous recombination, the endogenous FGF-CX and/or FCTRX gene is
functionally disrupted (i.e., no longer encodes a functional protein; also
referred to as a "knock
out" vector).
Alternatively, the vector can be designed such that, upon homologous
recombination,
the endogenous FGF-CX and/or FCTRX gene is mutated or otherwise altered but
still encodes
functional protein (e.g., the upstream regulatory region can be altered to
thereby alter the
expression of the endogenous FGF-CX and/or FCTRX protein). In the homologous
recombination vector, the altered portion of the FGF-CX and/or FCTRX gene is
flanked at its
5'- and 3'-termini by additional nucleic acid of the FGF-CX andlor FCTRX gene
to allow for
homologous recombination to occur between the exogenous FGF-CX and/or FCTRX
gene
carried by the vector and an endogenous FGF-CX and/or FCTRX gene in an
embryonic stem
cell. The additional flanking FGF-CX and/or FCTRX nucleic acid is of
sufficient length for
successful homologous recombination with the endogenous gene. Typically,
several kilobases
of flanking DNA (both at the 5'- and 3'-termini) are included in the vector.
See, e.g., Thomas,
et al., 1987. Cell 51: 503 for a description of homologous recombination
vectors. The vector
is ten introduced into an embryonic stem cell line (e.g., by electroporation)
and cells in which
the introduced FGF-CX and/or FCTRX gene has homologously-recombined with the
endogenous FGF-CX and/or FCTRX gene are selected. See, e.g., Li, et al., 1992.
Cell 69:
915.
The selected cells are then injected into a blastocyst of an animal (e.g., a
mouse) to
form aggregation chimeras. See, e.g., Bradley, 1987. In: TERATOCARCINOMAS AND
EMBRYONIC STEM CELLS: A PRACTICAL APPROACH, Robertson, ed. IRL, Oxford, pp.
113-152.
A chimeric embryo can then be implanted into a suitable pseudopregnant female
foster animal
and the embryo brought to term. Progeny harboring the homologously recombined
DNA in
their germ cells can be used to breed animals in which all cells of the animal
contain the
homologously-recombined DNA by germline transmission of the transgene. Methods
for
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
constructing homologous recombination vectors and homologous recombinant
animals are
described further in Bradley, 1991. Curr. Opin. Biotechraol. 2: 823-829; PCT
International
Publication Nos.: WO 90/11354; WO 91/01140; WO 92/0968; and WO 93/04169.
In another embodiment, transgenic non-humans animals can be produced that
contain
selected systems that allow for regulated expression of the transgene. One
example of such a
system is the cre/loxP recombinase system of bacteriophage P 1. For a
description of the
cre/loxP recombinase system, See, e.g., Lakso, et al., 1992. Proc. Natl. Acad.
Sci. USA 89:
6232-6236. Another example of a recombinase system is the FLP recombinase
system of
Saccharomyces cerevisiae. See, O'Gorman, et al., 1991. Science 251:1351-1355.
If a cre/loxP
recombinase system is used to regulate expression of the transgene, animals
containing
transgenes encoding both the Cre recombinase and a selected protein are
required. Such
animals can be provided through the construction of "double" transgenic
animals, e.g., by
mating two transgenic animals, one containing a transgene encoding a selected
protein and the
other containing a transgene encoding a recombinase.
Clones of the non-human transgenic animals described herein can also be
produced
according to the methods described in Wilmut, et al., 1997. Nature 385: 810-
813. In brief, a
cell (e.g., a somatic cell) from the transgenic animal can be isolated and
induced to exit the
growth cycle and enter Go phase. The quiescent cell can then be fused, e.g.,
through the use of
electrical pulses, to an enucleated oocyte from an animal of the same species
from which the
quiescent cell is isolated. The reconstructed oocyte is then cultured such
that it develops to
morula or blastocyte and then transferred to pseudopregnant female foster
animal. The
offspring borne of this female foster animal will be a clone of the animal
from which the cell
(e.g., the somatic cell) is isolated.
Pharmaceutical Compositions
The FGF-CX and/or FCTRX nucleic acid molecules, FGF-CX and/or FCTRX
proteins, and anti-FGF-CX and/or FCTRX antibodies (also referred to herein as
"active
compounds") of the invention, and derivatives, fragments, analogs and homologs
thereof, can
be incorporated into pharmaceutical compositions suitable for administration.
Such
compositions typically comprise the nucleic acid molecule, protein, or
antibody and a
pharmaceutically acceptable carrier. As used herein, "pharmaceutically
acceptable carrier" is
intended to include any and all solvents, dispersion media, coatings,
antibacterial and
antifungal agents, isotonic and absorption delaying agents, and the like,
compatible with
81
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
pharmaceutical administration. Suitable carriers are described in the most
recent edition of
Remington's Pharmaceutical Sciences, a standard reference text in the field,
which is
incorporated herein by reference. Preferred examples of such carriers or
diluents include, but
are not limited to, water, saline, finger's solutions, dextrose solution, and
5% human serum
albumin. Liposomes and non-aqueous vehicles such as fixed oils may also be
used. The use of
such media and agents for pharmaceutically active substances is well known in
the art. Except
insofar as any conventional media or agent is incompatible with the active
compound, use
thereof in the compositions is contemplated. Supplementary active compounds
can also be
incorporated into the compositions.
A pharmaceutical composition of the invention is formulated to be compatible
with its
intended route of administration. Examples of routes of administration include
parenteral,
e.g., intravenous, intradermal, subcutaneous, oral (e.g., inhalation),
transdermal (i.e., topical),
transmucosal, and rectal administration. Solutions or suspensions used for
parenteral,
intradermal, or subcutaneous application can include the following components:
a sterile
diluent such as water for injection, saline solution, fixed oils, polyethylene
glycols, glycerine,
propylene glycol or other synthetic solvents; antibacterial agents such as
benzyl alcohol or
methyl parabens; antioxidants such as ascorbic acid or sodium bisulfate;
chelating agents such
as ethylenediaminetetraacetic acid (EDTA); buffers such as acetates, citrates
or phosphates,
and agents for the adjustment of tonicity such as sodium chloride or dextrose.
The pH can be
adjusted with acids or bases, such as hydrochloric acid or sodium hydroxide.
The parenteral
preparation can be enclosed in ampoules, disposable syringes or multiple dose
vials made of
glass or plastic.
Pharmaceutical compositions suitable for injectable use include sterile
aqueous
solutions (where water soluble) or dispersions and sterile powders for the
extemporaneous
preparation of sterile injectable solutions or dispersion. Fox intravenous
administration,
suitable carriers include physiological saline, bacteriostatic water,
Cremophor ELTM (BASF,
Parsippany, N.J.) or phosphate buffered saline (PBS). In all cases, the
composition must be
sterile and should be fluid to the extent that easy syringeability exists. It
must be stable under
the conditions of manufacture and storage and must be preserved against the
contaminating
action of microorganisms such as bacteria and fungi. The carrier can be a
solvent or
dispersion medium containing, for example, water, ethanol, polyol (for
example, glycerol,
propylene glycol, and liquid polyethylene glycol, and the like), and suitable
mixtures thereof.
82
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
The proper fluidity can be maintained, for example, by the use of a coating
such as lecithin, by
the maintenance of the required particle size in the case of dispersion and by
the use of
surfactants. Prevention of the action of microorganisms can be achieved by
various
antibacterial and antifungal agents, for example, parabens, chlorobutanol,
phenol, ascorbic
acid, thimerosal, and the like. In many cases, it will be preferable to
include isotonic agents,
for example, sugars, polyalcohols such as manitol, sorbitol, sodium chloride
in the
composition. Prolonged absorption of the injectable compositions can be
brought about by
including in the composition an agent which delays absorption, for example,
aluminum
monostearate and gelatin.
Sterile injectable solutions can be prepared by incorporating the active
compound (e.g.,
an FGF-CX and/or FCTRX protein or anti-FGF-CX and/or FCTRX antibody) in the
required
amount in an appropriate solvent with one or a combination of ingredients
enumerated above,
as required, followed by filtered sterilization. Generally, dispersions are
prepared by
incorporating the active compound into a sterile vehicle that contains a basic
dispersion
medium and the required other ingredients from those enumerated above. In the
case of sterile
powders for the preparation of sterile injectable solutions, methods of
preparation are vacuum
drying and freeze-drying that yields a powder of the active ingredient plus
any additional
desired ingredient from a previously sterile-filtered solution thereof.
Oral compositions generally include an inert diluent or an edible carrier.
They can be
enclosed in gelatin capsules or compressed into tablets. For the purpose of
oral therapeutic
administration, the active compound can be incorporated with excipients and
used in the form
of tablets, troches, or capsules. Oral compositions can also be prepared using
a fluid carrier
for use as a mouthwash, wherein the compound in the fluid Garner is applied
orally and
swished and expectorated or swallowed. Pharmaceutically compatible binding
agents, and/or
~,5 adjuvant materials can be included as part of the composition. The
tablets, pills, capsules,
troches and the like can contain any of the following ingredients, or
compounds of a similar
nature: a binder such as microcrystalline cellulose, gum tragacanth or
gelatin; an excipient
such as starch or lactose, a disintegrating agent such as alginic acid,
Primogel, or corn starch; a
lubricant such as magnesium stearate or Sterotes; a glidant such as colloidal
silicon dioxide; a
sweetening agent such as sucrose or saccharin; or a flavoring agent such as
peppermint,
methyl salicylate, or orange flavoring.
~3
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
For administration by inhalation, the compounds are delivered in the form of
an
aerosol spray from pressured container or dispenser which contains a suitable
propellant, e.g.,
a gas such as carbon dioxide, or a nebulizer.
Systemic administration can also be by transmucosal or transdermal means. For
transmucosal or transdermal administration, penetrants appropriate to the
barrier to be
permeated are used in the formulation. Such penetrants are generally known in
the art, and
include, for example, for transmucosal administration, detergents, bile salts,
and fusidic acid
derivatives. Transmucosal administration can be accomplished through the use
of nasal sprays
or suppositories. For transdermal administration, the active compounds are
formulated into
ointments, salves, gels, or creams as generally known in the art.
The compounds can also be prepared in the form of suppositories (e.g., with
conventional suppository bases such as cocoa butter and other glycerides) or
retention enemas
for rectal delivery.
In one embodiment, the active compounds are prepared with carriers that will
protect
the compound against rapid elimination from the body, such as a controlled
release
formulation, including implants and microencapsulated delivery systems.
Biodegradable,
biocompatible polymers can be used, such as ethylene vinyl acetate,
polyanhydrides,
polyglycolic acid, collagen, polyorthoesters, and polylactic acid. Methods for
preparation of
such formulations will be apparent to those skilled in the art. The materials
can also be
obtained commercially from Alza Corporation and Nova Pharmaceuticals, Inc.
Liposomal
suspensions (including liposomes targeted to infected cells with monoclonal
antibodies to viral
antigens) can also be used as pharmaceutically acceptable carriers. These can
be prepared
according to methods known to those skilled in the art, for example, as
described in U.S.
Patent No. 4,522,811.
It is especially advantageous to formulate oral or parenteral compositions in
dosage
unit form for ease of administration and uniformity of dosage. Dosage unit
form as used
herein refers to physically discrete units suited as unitary dosages for the
subject to be treated;
each unit containing a predetermined quantity of active compound calculated to
produce the
desired therapeutic effect in association with the required pharmaceutical
Garner. The
specification for the dosage unit forms of the invention are dictated by and
directly dependent
on the unique characteristics of the active compound and the particular
therapeutic effect to be
84
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
achieved, and the limitations inherent in the art of compounding such an
active compound for
the treatment of individuals.
The nucleic acid molecules of the invention can be inserted into vectors and
used as
gene therapy vectors. Gene therapy vectors can be delivered to a subject by,
for example,
intravenous injection, local administration (see, e.g., U.S. Patent No.
5,328,470) or by
stereotactic injection (see, e.g., Chen, et al., 1994. Proc. Natl. Acad. Sci.
USA 91: 3054-3057).
The pharmaceutical preparation of the gene therapy vector can include the gene
therapy vector
in an acceptable diluent, or can comprise a slow release matrix in which the
gene delivery
vehicle is imbedded. Alternatively, where the complete gene delivery vector
can be produced
intact from recombinant cells, e.g., retroviral vectors, the pharmaceutical
preparation can
include one or more cells that produce the gene delivery system.
The pharmaceutical compositions can be included in a container, pack, or
dispenser
together with instructions for administration.
Screening and Detection Methods
The isolated nucleic acid molecules of the invention can be used to express
FGF-CX
and/or FCTRX protein (e.g., via a recombinant expression vector in a host cell
in gene therapy
applications), to detect FGF-CX and/or FCTRX mRNA (e.g., in a biological
sample) or a
genetic lesion in an FGF-CX and/or FCTRX gene, and to modulate FGF-CX and/or
FCTRX
activity, as described further, below. In addition, the FGF-CX and/or FCTRX
proteins can be
used to screen drugs or compounds that modulate the FGF-CX and/or FCTRX
protein activity
or expression as well as to treat disorders characterized by insufficient or
excessive production
of FGF-CX and/or FCTRX protein or production of FGF-CX andlor FCTRX protein
forms
that have decreased or aberrant activity compared to FGF-CX and/or FCTRX wild-
type
protein (e.g.; diabetes (regulates insulin release); obesity (binds and
transport lipids);
metabolic disturbances associated with obesity, the metabolic syndrome X as
well as anorexia
and wasting disorders associated with chronic diseases and various cancers,
and infectious
disease(possesses anti-microbial activity) and the various dyslipidemias. In
addition, the
anti-FGF-CX and/or FCTRX antibodies of the invention can be used to detect and
isolate
FGF-CX and/or FCTRX proteins and modulate FGF-CX and/or FCTRX activity. In yet
a
further aspect, the invention can be used in methods to influence appetite,
absorption of
nutrients and the disposition of metabolic substrates in both a positive and
negative fashion.
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
The invention further pertains to novel agents identified by the screening
assays
described herein and uses thereof for treatments as described, supra.
Screening Assays
The invention provides a method (also referred to herein as a "screening
assay") for
identifying modulators, i.e., candidate or test compounds or agents (e.g.,
peptides,
peptidomimetics, small molecules or other drugs) that bind to FGF-CX andlor
FCT12X
proteins or have a stimulatory or inhibitory effect on, e.g., FGF-CX and/or
FCTRX protein
expression or FGF-CX and/or FCTRX protein activity. The invention also
includes
compounds identified in the screening assays described herein.
In one embodiment, the invention provides assays for screening candidate or
test
compounds which bind to or modulate the activity of the membrane-bound form of
an FGF-
CX and/or FCT12X protein or polypeptide or biologically-active portion
thereof. The test
compounds of the invention can be obtained using any of the numerous
approaches in
combinatorial library methods known in the art, including: biological
libraries; spatially
addressable parallel solid phase or solution phase libraries; synthetic
library methods requiring
deconvolution; the "one-bead one-compound" library method; and synthetic
library methods
using affinity chromatography selection. The biological library approach is
limited to peptide
libraries, while the other four approaches are applicable to peptide, non-
peptide oligomer or
small molecule libraries of compounds. See, e.g., Lam, 1997. Ahticance~ Drug
Design 12:
145.
A "small molecule" as used herein, is meant to refer to a composition that has
a
molecular weight of less than about 5 kD and most preferably less than about 4
kD. Small
molecules can be, e.g., nucleic acids, peptides, polypeptides,
peptidomimetics, carbohydrates,
lipids or other organic or inorganic molecules. Libraries of chemical and/or
biological
mixtures, such as fungal, bacterial, or algal extracts, are known in the art
and can be screened
with any of the assays of the invention.
Examples of methods for the synthesis of molecular libraries can be found in
the art,
for example in: DeWitt, et al., 1993. Proc. Natl. Acad. Sci. U.S.A. 90: 6909;
Erb, et al., 1994.
Proc. Natl. Acad. Sci. U.S.A. 91: 11422; Zuckermann, et al., 1994. J. Med.
Chem. 37: 2678;
Cho, et al., 1993. Science 261: 1303; Carrell, et al., 1994. Ahgew. ChenZ.
hZt. Ed. Engl. 33:
2059; Carell, et al., 1994. Aragew. Chem. Int. Ed. Eragl. 33: 2061; and
Gallop, et al., 1994. J.
Med. Chem. 37: 1233.
86
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
Libraries of compounds may be presented in solution (e.g., Houghten, 1992.
Bioteclaniques 13: 412-421), or on beads (Lam, 1991. Nature 354: 82-84), on
chips (Fodor,
1993. Nature 364: 555-556), bacteria (Ladner, U.S. Patent No. 5,223,409),
spores (Ladner,
U.S. Patent 5,233,409), plasmids (Cull, et al., 1992. Proc. Natl. Acad. Sci.
USA 89:
1865-1869) or on phage (Scott and Smith, 1990. Science 249: 386-390; Devlin,
1990. Science
249: 404-406; Cwirla, et al., 1990. PYOG. Natl. Acad. Sci. U.S.A. 87: 6378-
6382; Felici, 1991.
J. Mol. Biol. 222: 301-310; Ladner, U.S. Patent No. 5,233,409.).
In one embodiment, an assay is a cell-based assay in which a cell which
expresses a
membrane-bound form of FGF-CX and/or FCTRX protein, or a biologically-active
portion
thereof, on the cell surface is contacted with a test compound and the ability
of the test
compound to bind to an FGF-CX and/or FCTRX protein determined. The cell, for
example,
can of mammalian origin or a yeast cell. Determining the ability of the test
compound to bind
to the FGF-CX and/or FCTRX protein can be accomplished, for example, by
coupling the test
compound with a radioisotope or enzymatic label such that binding of the test
compound to
the FGF-CX and/or FCTRX protein or biologically-active portion thereof can be
determined
by detecting the labeled compound in a complex. For example, test compounds
can be labeled
with lash 3sS~ i4C, or 3H, either directly or indirectly, and the radioisotope
detected by direct
counting of radioemission or by scintillation counting. Alternatively, test
compounds can be
enzymatically-labeled with, for example, horseradish peroxidase, alkaline
phosphatase, or
luciferase, and the enzymatic label detected by determination of conversion of
an appropriate
substrate to product. In one embodiment, the assay comprises contacting a cell
which
expresses a membrane-bound form of FGF-CX and/or FCTRX protein, or a
biologically-
active portion thereof, on the cell surface with a known compound which binds
FGF-CX .
and/or FCTRX to form an assay mixture, contacting the assay mixture with a
test compound,
and determining the ability of the test compound to interact with an FGF-CX
and/or FCTRX
protein, wherein determining the ability of the test compound to interact with
an FGF-CX
and/or FCTRX protein comprises determining the ability of the test compound to
preferentially bind to FGF-CX and/or FCTRX protein or a biologically-active
portion thereof
as compared to the known compound.
In another embodiment, an assay is a cell-based assay comprising contacting a
cell
expressing a membrane-bound form of FGF-CX and/or FCTRX protein, or a
biologically-
active portion thereof, on the cell surface with a test compound and
determining the ability of
87
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
the test compound to modulate (e.g., stimulate or inhibit) the activity of the
FGF-CX and/or
FCTRX protein or biologically-active portion thereof. Determining the ability
of the test
compound to modulate the activity of FGF-CX and/or FCTRX or a biologically-
active portion
thereof can be accomplished, for example, by determining the ability of the
FGF-CX and/or
FCTRX protein to bind to or interact with an FGF-CX and/or FCTRX target
molecule. As
used herein, a "target molecule" is a molecule with which an FGF-CX and/or
FCTRX protein
binds or interacts in nature, for example, a molecule on the surface of a cell
which expresses
an FGF-CX and/or FCTRX interacting protein, a molecule on the surface of a
second cell, a
molecule in the extracellular milieu, a molecule associated with the internal
surface of a cell
membrane or a cytoplasmic molecule. An FGF-CX and/or FCTRX target molecule can
be a
non-FGF-CX and/or FCTRX molecule or an FGF-CX and/or FCTRX protein or
polypeptide
of the invention. In one embodiment, an FGF-CX and/or FCTRX target molecule is
a
component of a signal transduction pathway that facilitates transduction of an
extracellular
signal (e.g. a signal generated by binding of a compound to a membrane-bound
FGF-CX
and/or FCTRX molecule) through the cell membrane and into the cell. The
target, for
example, can be a second intercellular protein that has catalytic activity or
a protein that
facilitates the association of downstream signaling molecules with FGF-CX
and/or FCTRX.
Determining the ability of the FGF-CX and/or FCTRX protein to bind to or
interact
with an FGF-CX and/or FCTRX target molecule can be accomplished by one of the
methods
described above for determining direct binding. In one embodiment, determining
the ability of
the FGF-CX and/or FCTRX protein to bind to or interact with an FGF-CX andlor
FCTRX
target molecule can be accomplished by determining the activity of the target
molecule. For
example, the activity of the target molecule can be determined by detecting
induction of a
cellular second messenger of the target (i. e. intracellular Ca2+,
diacylglycerol, IP3, etc.),
detecting catalytic/enzymatic activity of the target an appropriate substrate,
detecting the
induction of a reporter gene (comprising an FGF-CX and/or FCTRX-responsive
regulatory
element operatively linked to a nucleic acid encoding a detectable marker,
e.g., luciferase), or
detecting a cellular response, for example, cell survival, cellular
differentiation, or cell
proliferation.
In yet another embodiment, an assay of the invention is a cell-free assay
comprising
contacting an FGF-CX and/or FCTRX protein or biologically-active portion
thereof with a test
compound and determining the ability of the test compound to bind to the FGF-
CX and/or
88
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
FCTRX protein or biologically-active portion thereof. Binding of the test
compound to the
FGF-CX and/or FCTRX protein can be determined either directly or indirectly as
described
above. In one such embodiment, the assay comprises contacting the FGF-CX
and/or FCTRX
protein or biologically-active portion thereof with a known compound which
binds FGF-CX
and/or FCTRX to form an assay mixture, contacting the assay mixture with a
test compound,
and determining the ability of the test compound to interact with an FGF-CX
and/or FCTRX
protein, wherein determining the ability of the test compound to interact with
an FGF-CX
and/or FCTRX protein comprises determining the ability of the test compound to
preferentially bind to FGF-CX and/or FCTRX or biologically-active portion
thereof as
compared to the known compound.
In still another embodiment, an assay is a cell-free assay comprising
contacting FGF-
CX and/or FCTRX protein or biologically-active portion thereof with a test
compound and
determining the ability of the test compound to modulate (e.g. stimulate or
inhibit) the activity
of the FGF-CX and/or FCTRX protein or biologically-active portion thereof.
Determining the
ability of the test compound to modulate the activity of FGF-CX and/or FCTRX
can be
accomplished, for example, by determining the ability of the FGF-CX and/or
FCTRX protein
to bind to an FGF-CX and/or FCTRX target molecule by one of the methods
described above
for determining direct binding. In an alternative embodiment, determining the
ability of the
test compound to modulate the activity of FGF-CX and/or FCTRX protein can be
accomplished by determining the ability of the FGF-CX and/or FCTRX protein
further
modulate an FGF-CX and/or FCTRX target molecule. For example, the
catalytic/enzymatic
activity of the target molecule on an appropriate substrate can be determined
as described,
supra.
In yet another embodiment, the cell-free assay comprises contacting the FGF-CX
and/or FCTRX protein or biologically-active portion thereof with a known
compound which
binds FGF-CX and/or FCTRX protein to form an assay mixture, contacting the
assay mixture
with a test compound, and determining the ability of the test compound to
interact with an
FGF-CX and/or FCTRX protein, wherein determining the ability of the test
compound to
interact with an FGF-CX and/or FCTRX protein comprises determining the ability
of the FGF-
CX and/or FCTRX protein to preferentially bind to or modulate the activity of
an FGF-CX
and/or FCTRX target molecule.
~9
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
The cell-free assays of the invention are amenable to use of both the soluble
form or
the membrane-bound form of FGF-CX and/or FCTRX protein. In the case of cell-
free assays
comprising the membrane-bound form of FGF-CX andlor FCTRX protein, it may be
desirable
to utilize a solubilizing agent such that the membrane-bound form of FGF-CX
and/or FCTRX
protein is maintained in solution. Examples of such solubilizing agents
include non-ionic
detergents such as n-octylglucoside, n-dodecylglucoside, n-dodecylmaltoside,
octanoyl-N-methylglucamide, decanoyl-N-methylglucamide, Triton~ X-100, Triton~
X-114,
Thesit~, Isotridecypoly(ethylene glycol ether)", N-dodecyl--
N,N-dimethyl-3-ammonio-1-propane sulfonate, 3-(3-cholamidopropyl)
dimethylamminiol-
1-propane sulfonate (CHAPS), or 3-(3-cholamidopropyl)dimethylamminiol-2-
hydroxy-
1-propane sulfonate (CHAPSO).
In more than one embodiment of the above assay methods of the invention, it
may be
desirable to immobilize either FGF-CX and/or FCTR.X protein or its target
molecule to
facilitate separation of complexed from uncomplexed forms of one or both of
the proteins, as
well as to accommodate automation of the assay. Binding of a test compound to
FGF-CX
and/or FCTRX protein, or interaction of FGF-CX and/or FCTRX protein with a
target
molecule in the presence and absence of a candidate compound, can be
accomplished in any
vessel suitable for containing the reactants. Examples of such vessels include
microtiter
plates, test tubes, and micro-centrifuge tubes. In one embodiment, a fusion
protein can be
provided that adds a domain that allows one or both of the proteins to be
bound to a matrix.
For example, GST-FGF-CX and/or FCTRX fusion proteins or GST-target fusion
proteins can
be adsorbed onto glutathione sepharose beads (Sigma Chemical, St. Louis, MO)
or glutathione
derivatized microtiter plates, that are then combined with the test compound
or the test
compound and either the non-adsorbed target protein or FGF-CX and/or FCTRX
protein, and
the mixture is incubated under conditions conducive to complex formation
(e.g., at
physiological conditions for salt and pH). Following incubation, the beads or
microtiter plate
wells are washed to remove any unbound components, the matrix immobilized in
the case of
beads, complex determined either directly or indirectly, for example, as
described, supra.
Alternatively, the complexes can be dissociated from the matrix, and the level
of FGF-CX
and/or FCTRX protein binding or activity determined using standard techniques.
Other techniques for immobilizing proteins on matrices can also be used in the
screening assays of the invention. For example, either the FGF-CX and/or FCTRX
protein or
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
its target molecule can be immobilized utilizing conjugation of biotin and
streptavidin.
Biotinylated FGF-CX andlor FCTRX protein or target molecules can be prepared
from
biotin-NHS (N-hydroxy-succinimide) using techniques well-known within the art
(e.g.,
biotinylation kit, Pierce Chemicals, Rockford, Ill.), and immobilized in the
wells of
streptavidin-coated 96 well plates (Pierce Chemical). Alternatively,
antibodies reactive with
FGF-CX and/or FCTRX protein or target molecules, but which do not interfere
with binding
of the FGF-CX and/or FCTRX protein to its target molecule, can be derivatized
to the wells of
the plate, and unbound target or FGF-CX and/or FCTRX protein trapped in the
wells by
antibody conjugation. Methods for detecting such complexes, in addition to
those described
above for the GST-immobilized complexes, include immunodetection of complexes
using
antibodies reactive with the FGF-CX and/or FCTRX protein or target molecule,
as well as
enzyme-linked assays that rely on detecting an enzymatic activity associated
with the FGF-CX
and/or FCTRX protein or target molecule.
In another embodiment, modulators of FGF-CX and/or FCTRX protein expression
are
identified in a method wherein a cell is contacted with a candidate compound
and the
expression of FGF-CX and/or FCTRX mRNA or protein in the cell is determined.
The level
of expression of FGF-CX andlor FCTRX mRNA or protein in the presence of the
candidate
compound is compared to the level of expression of FGF-CX and/or FCTRX mRNA or
protein in the absence of the candidate compound. The candidate compound can
then be
identified as a modulator of FGF-CX and/or FCTRX mRNA or protein expression
based upon
this comparison. For example, when expression of FGF-CX and/or FCTRX mRNA or
protein
is greater (i.e., statistically significantly greater) in the presence of the
candidate compound
than in its absence, the candidate compound is identified as a stimulator of
FGF-CX and/or
FCTRX mRNA or protein expression. Alternatively, when expression of FGF-CX
and/or
FCTRX mRNA or protein is less (statistically significantly less) in the
presence of the
candidate compound than in its absence, the candidate compound is identified
as an inhibitor
of FGF-CX and/or FCTRX mRNA or protein expression. The level of FGF-CX and/or
FCTRX mRNA or protein expression in the cells can be determined by methods
described
herein for detecting FGF-CX and/or FCTRX mRNA or protein.
In yet another aspect of the invention, the FGF-CX and/or FCTRX proteins can
be
used as "bait proteins" in a two-hybrid assay or three hybrid assay (see,
e.g., U.S. Patent No.
5,283,317; Zervos, I et al., 1993. Cell 72: 223-232; Madura, et al., 1993. J.
Biol. Che~ra. 268:
91
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
12046-12054; Bartel, et al., 1993. Biotechhiques 14: 920-924; Iwabuchi, et
al., 1993.
Ohcogehe ~: 1693-1696; and Brent WO 94/10300), to identify other proteins that
bind to or
interact with FGF-CX and/or FCTRX ("FGF-CX and/or FCTRX-binding proteins" or
"FGF-
CX and/or FCTRX-by") and modulate FGF-CX and/or FCTRX activity. Such FGF-CX
and/or FCTRX-binding proteins are also likely to be involved in the
propagation of signals by
the FGF-CX and/or FCTRX proteins as, for example, upstream or downstream
elements of the
FGF-CX and/or FCTRX pathway.
The two-hybrid system is based on the modular nature of most transcription
factors,
which consist of separable DNA-binding and activation domains. Briefly, the
assay utilizes
two different DNA constructs. In one construct, the gene that codes for FGF-CX
and/or
FCTRX is fused to a gene encoding the DNA binding domain of a known
transcription factor
(e.g., GAL-4). In the other construct, a DNA sequence, from a library of DNA
sequences, that
encodes an unidentified protein ("prey" or "sample") is fused to a gene that
codes for the
activation domain of the known transcription factor. If the "bait" and the
"prey" proteins are
able to interact, in vivo, forming an FGF-CX and/or FCTRX-dependent complex,
the
DNA-binding and activation domains of the transcription factor are brought
into close
proximity. This proximity allows transcription of a reporter gene (e.g., LacZ)
that is operably
linked to a transcriptional regulatory site responsive to the transcription
factor. Expression of
the reporter gene can be detected and cell colonies containing the functional
transcription
factor can be isolated and used to obtain the cloned gene that encodes the
protein which
interacts with FGF-CX and/or FCTRX.
The invention further pertains to novel agents identified by the
aforementioned
screening assays and uses thereof for treatments as described herein.
Detection Assays
Portions or fragments of the cDNA sequences identified herein (and the
corresponding
complete gene sequences) can be used in numerous ways as polynucleotide
reagents. By way
of example, and not of limitation, these sequences can be.used to: (i) map
their respective
genes on a chromosome; and, thus, locate gene regions associated with genetic
disease; (ii)
identify an individual from a minute biological sample (tissue typing); and
(iii) aid in forensic
identification of a biological sample. Some of these applications are
described in the
subsections, below.
92
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
Chromosome Mapping
Once the sequence (or a portion of the sequence) of a gene has been isolated,
this
sequence can be used to map the location of the gene on a chromosome. This
process is called
chromosome mapping. Accordingly, portions or fragments of the FGF-CX and/or
FCTRX
sequences, SEQ m NOS:1, 3, 5, 7, 9, 11 and 13, or fragments or derivatives
thereof, can be
used to map the location of the FGF-CX and/or FCTRX genes, respectively, on a
chromosome. The mapping of the FGF-CX and/or FCTRX sequences to chromosomes is
an
important first step in correlating these sequences with genes associated with
disease.
Briefly, FGF-CX and/or FCTRX genes can be mapped to chromosomes by preparing
PCR primers (preferably 15-25 by in length) from the FGF-CX and/or FCTRX
sequences.
Computer analysis of the FGF-CX and/or FCTRX, sequences can be used to rapidly
select
primers that do not span more than one exon in the genomic DNA, thus
complicating the
amplification process. These primers can then be used for PCR screening of
somatic cell
hybrids containing individual human chromosomes. Only those hybrids containing
the human
gene corresponding to the FGF-CX and/or FCTRX sequences will yield an
amplified
fragment.
Somatic cell hybrids are prepared by fusing somatic cells from different
mammals
(e.g., human and mouse cells). As hybrids of human and mouse cells grow and
divide, they
gradually lose human chromosomes in random order, but retain the mouse
chromosomes. By
using media in which mouse cells cannot grow, because they lack a particular
enzyme, but in
which human cells can, the one human chromosome that contains the gene
encoding the
needed enzyme will be retained. By using various media, panels of hybrid cell
lines can be
established. Each cell line in a panel contains either a single human
chromosome or a small
number of human chromosomes, and a full set of mouse chromosomes, allowing
easy
mapping of individual genes to specific human chromosomes. See, e.g.,
D'Eustachio, et al.,
193. Science 220: 919-924. Somatic cell hybrids containing only fragments of
human
chromosomes can also be produced by using human chromosomes with
translocations and
deletions.
PCR mapping of somatic cell hybrids is a rapid procedure for assigning a
particular
sequence to a particular chromosome. Three or more sequences can be assigned
per day using
a single thermal cycler. Using the FGF-CX and/or FCTRX sequences to design
93
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
oligonucleotide primers, sub-localization can be achieved with panels of
fragments from
specific chromosomes.
Fluorescence ih situ hybridization (FISH) of a DNA sequence to a metaphase
chromosomal spread can further be used to provide a precise chromosomal
location in one
step. Chromosome spreads can be made using cells whose division has been
blocked in
metaphase by a chemical like colcemid that disrupts the mitotic spindle. The
chromosomes
can be treated briefly with trypsin, and then stained with Giemsa. A pattern
of light and dark
bands develops on each chromosome, so that the chromosomes can be identified
individually.
The FISH technique can be used with a DNA sequence as short as 500 or 600
bases.
However, clones larger than 1,000 bases have a higher likelihood of binding to
a unique
chromosomal location with sufficient signal intensity for simple detection.
Preferably 1,000
bases, and more preferably 2,000 bases, will suffice to get good results at a
reasonable amount
of time. For a review of this technique, see, Verma, et al., HUMAN
CHROMOSOMES: A
MANUAL OF BASIC TECHNIQUES (Pergamon Press, New York 1988).
Reagents for chromosome mapping can be used individually to mark a single
chromosome or a single site on that chromosome, or panels of reagents can be
used for
marking multiple sites and/or multiple chromosomes. Reagents corresponding to
noncoding
regions of the genes actually are preferred for mapping purposes. Coding
sequences are more
likely to be conserved within gene families, thus increasing the chance of
cross hybridizations
during chromosomal mapping.
Once a sequence has been mapped to a precise chromosomal location, the
physical
position of the sequence on the chromosome can be correlated with genetic map
data. Such
data are found, e.g., in McKusick, MENDELIAN INHERITANCE IN MAN, available on-
line
through Johns Hopkins University Welch Medical Library). The relationship
between genes
and disease, mapped to the same chromosomal region, can then be identified
through linkage
analysis (co-inheritance of physically adjacent genes), described in, e.g.,
Egeland, et al., 1987.
Nature, 325: 783-787.
Moreover, differences in the DNA sequences between individuals affected and
unaffected with a disease associated with the FGF-CX and/or FCTRX gene, can be
determined. If a mutation is observed in some or all of the affected
individuals but not in any
unaffected individuals, then the mutation is likely to be the causative agent
of the particular
disease. Comparison of affected and unaffected individuals generally involves
first looking
94
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
for structural alterations in the chromosomes, such as deletions or
translocations that are
visible from chromosome spreads or detectable using PCR based on that DNA
sequence.
Ultimately, complete sequencing of genes from several individuals can be
performed to
confirm the presence of a mutation and to distinguish mutations from
polymorphisms.
Tissue Typing
The FGF-CX and/or FCTRX sequences of the invention can also be used to
identify
individuals from minute biological samples. In this technique, an individual's
genomic DNA
is digested with one or more restriction enzymes, and probed on a Southern
blot to yield
unique bands for identification. The sequences of the invention are useful as
additional DNA
markers for 1RF'LP ("restriction fragment length polymorphisms," described in
U.S. Patent No.
5,272,057).
Furthermore, the sequences of the invention can be used to provide an
alternative
technique that determines the actual base-by-base DNA sequence of selected
portions of an
individual's genome. Thus, the FGF-CX andlor FCTRX sequences described herein
can be
used to prepare two PCR primers from the 5'- and 3'-termini of the sequences.
These primers
can then be used to amplify an individual's DNA and subsequently sequence it.
Panels of corresponding DNA sequences from individuals, prepared in this
manner,
can provide unique individual identifications, as each individual will have a
unique set of such
DNA sequences due to allelic differences. The sequences of the invention can
be used to
obtain such identification sequences from individuals and from tissue. The FGF-
CX andlor
FCTRX sequences of the invention uniquely represent portions of the human
genome. Allelic
variation occurs to some degree in the coding regions of these sequences, and
to a greater
degree in the noncoding regions. It is estimated that allelic variation
between individual
humans occurs with a frequency of about once per each 500 bases. Much of the
allelic
variation is due to single nucleotide polymorphisms (SNPs), which include
restriction
fragment length polymorphisms (RFLPs).
Each of the sequences described herein can, to some degree, be used as a
standaxd
against which DNA from an individual can be compared for identification
purposes. Because
greater numbers of polymorphisms occur in the noncoding regions, fewer
sequences are
necessary to differentiate individuals. The noncoding sequences can
comfortably provide
positive individual identification with a panel of perhaps 10 to 1,000 primers
that each yield a
noncoding amplified sequence of 100 bases. If predicted coding sequences, such
as those in
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
SEQ m NOS:1, 3, 5, 7, 9, 11 and 13 are used, a more appropriate number of
primers for
positive individual identification would be 500-2,000.
Predictive Medicine
The invention also pertains to the field of predictive medicine in which
diagnostic
assays, prognostic assays, pharmacogenomics, and monitoring clinical trials
are used for
prognostic (predictive) purposes to thereby treat an individual
prophylactically. Accordingly,
one aspect of the invention relates to diagnostic assays for determining FGF-
CX and/or
FCTRX protein and/or nucleic acid expression as well as FGF-CX and/or FCTRX
activity, in
the context of a biological sample (e.g., blood, serum, cells, tissue) to
thereby determine
whether an individual is afflicted with a disease or disorder, or is at risk
of developing a
disorder, associated with aberrant FGF-CX expression or activity, aberrant
FCTRX expression
or activity, or both. The disorders include pathology such as inflammatory
conditions in the
gastrointestinal tract, including but not limited to inflammatory bowel
disease such as
ulcerative colitis and Crohn's disease, growth and proliferative diseases such
as cancer,
angiogenesis, atherosclerotic plaques, collagen formation, cartilage and bone
formation,
cardiovascular and fibrotic diseases and diabetic ulcers. In addition, FCTRX
nucleic acids and
their encoded polypeptides will be therapeutically useful for the prevention
of aneurysms and
the acceleration of wound closure through gene therapy. Furthermore, FCTRX
nucleic acids
and their encoded polypeptides can be utilized to stimulate cellular growth.
wound healing,
neovascularization and tissue growth, and similar tissue regeneration needs.
More
specifically, a FCTRX nucleic acid or polypeptide may be useful in treatment
of anemia and
leukopenia, intestinal tract sensitivity and baldness. Treatment of such
conditions may be
indicated, e. g., in patients having undergone radiation or chemotherapy,
wherein treatment
would minimize any hyperproliferative side effects.
The invention also provides for prognostic (or predictive) assays for
determining
whether an individual is at risk of developing a disorder associated with FGF-
CX and/or
FCTRX protein, nucleic acid expression or activity. For example, mutations in
an FGF-CX
and/or FCTRX gene can be assayed in a biological sample. Such assays can be
used for
prognostic or predictive purpose to thereby prophylactically treat an
individual prior to the
onset of a disorder characterized by or associated with FGF-CX and/or FCTRX
protein,
nucleic acid expression, or biological activity.
96
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
Another aspect of the invention provides methods for determining FGF-CX and/or
FCTRX protein, nucleic acid expression or activity in an individual to thereby
select
appropriate therapeutic or prophylactic agents for that individual (referred
to herein as
"pharmacogenomics"). Pharmacogenomics allows for the selection of agents
(e.g., drugs) for
therapeutic or prophylactic treatment of an individual based on the genotype
of the individual
(e.g., the genotype of the individual examined to determine the ability of the
individual to
respond to a particular agent.)
Yet another aspect of the invention pertains to monitoring the influence of
agents (e.g.,
drugs, compounds) on the expression or activity of FGF-CX and/or FCTRX in
clinical trials.
These and other agents are described in further detail in the following
sections.
Diagnostic Assays
An exemplary method for detecting the presence or absence of FGF-CX and/or
FCTRX in a biological sample involves obtaining a biological sample from a
test subject and
contacting the biological sample with a compound or an agent capable of
detecting FGF-CX
and/or FCTRX protein or nucleic acid (e.g., mRNA, genomic DNA) that encodes
FGF-CX
and/or FCTRX protein such that the presence of FGF-CX and/or FCTRX is detected
in the
biological sample. An agent for detecting FGF-CX and/or FCTRX mRNA or genomic
DNA
is a labeled nucleic acid probe capable of hybridizing to FGF-CX and/or FCTRX
mRNA or
genomic DNA. The nucleic acid probe can be, for example, a full-length FGF-CX
and/or
FCTRX nucleic acid, such as the nucleic acid of SEQ m NOS:1, 3, 5, 7, 9, 11
and 13, or a
portion thereof, such as an oligonucleotide of at least 15, 30, 50, 100, 250
or 500 nucleotides
in length and sufficient to specifically hybridize under stringent conditions
to FGF-CX and/or
FCTRX mRNA or genomic DNA. Other suitable probes for use in the diagnostic
assays of
the invention are described herein.
An agent for detecting FGF-CX and/or FCTRX protein is an antibody capable of
binding to FGF-CX and/or FCTRX protein, preferably an antibody with a
detectable label.
Antibodies can be polyclonal, or more preferably, monoclonal. An intact
antibody, or a
fragment thereof (e.g., Fab or F(ab')a) can be used. The term "labeled", with
regard to the
probe or antibody, is intended to encompass direct labeling of the probe or
antibody by
coupling (i.e., physically linking) a detectable substance to the probe or
antibody, as well as
indirect labeling of the probe or antibody by reactivity with another reagent
that is directly
labeled. Examples of indirect labeling include detection of a primary antibody
using a
97
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
fluorescently-labeled secondary antibody and end-labeling of a DNA probe with
biotin such
that it can be detected with fluorescently-labeled streptavidin. The term
"biological sample" is
intended to include tissues, cells and biological fluids isolated from a
subject, as well as
tissues, cells and fluids present within a subject. That is, the detection
method of the invention
can be used to detect FGF-CX and/or FCTRX mRNA, protein, or genomic DNA in a
biological sample in vitro as well as i~r. vivo. For example, in vitro
techniques for detection of
FGF-CX and/or FCTRX mRNA include Northern hybridizations and iu situ
hybridizations. Ih
vitro techniques for detection of FGF-CX and/or FCTRX protein include enzyme
linked
imrnunosorbent assays (ELISAs), Western blots, immunoprecipitations, and
immunofluorescence. 1h vitro techniques for detection of FGF-CX and/or FCTR.X
genomic
DNA include Southern hybridizations. Furthermore, ih vivo techniques for
detection of FGF-
CX and/or FCTRX protein include introducing into a subject a labeled anti-FGF-
CX and/or
FCTRX antibody. For example, the antibody can be labeled with a radioactive
marker whose
presence and location in a subject can be detected by standard imaging
techniques.
In one embodiment, the biological sample contains protein molecules from the
test
subject. Alternatively, the biological sample can contain mRNA molecules from
the test
subject or genomic DNA molecules from the test subject. A preferred biological
sample is a
peripheral blood leukocyte sample isolated by conventional means from a
subject.
In another embodiment, the methods further involve obtaining a control
biological
sample from a control subj ect, contacting the control sample with a compound
or agent
capable of detecting FGF-CX and/or FCTRX protein, mRNA, or genomic DNA, such
that the
presence of FGF-CX and/or FCTRX protein, mRNA or genomic DNA is detected in
the
biological sample, and comparing the presence of FGF-CX and/or FCTRX protein,
mRNA or
genomic DNA in the control sample with the presence of FGF-CX and/or FCTRX
protein,
mRNA or. genomic DNA in the test sample.
The invention also encompasses kits for detecting the presence of FGF-CX
and/or
FCTRX in a biological sample. For example, the kit can comprise: a labeled
compound or
agent capable of detecting FGF-CX and/or FCTRX protein or mRNA in a biological
sample;
means for determining the amount of FGF-CX and/or FCTRX in the sample; and
means for
comparing the amount of FGF-CX and/or FCTRX in the sample with a standard. The
compound or agent can be packaged in a suitable container. The kit can further
comprise
instructions for using the kit to detect FGF-CX and/or FCTRX protein or
nucleic acid.
9~
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
Prognostic Assays
The diagnostic methods described herein can furthermore be utilized to
identify
subjects having or at risk of developing a disease or disorder associated with
aberrant FGF-CX
andlor FCTRX expression or activity. For example, the assays described herein,
such as the
preceding diagnostic assays or the following assays, can be utilized to
identify a subject
having or at risk of developing a disorder associated with FGF-CX and/or FCTRX
protein,
nucleic acid expression or activity. Alternatively, the prognostic assays can
be utilized to
identify a subj ect having or at risk for developing a disease or disorder.
Thus, the invention
provides a method for identifying a disease or disorder associated with
aberrant FGF-CX
and/or FCTRX expression or activity in which a test sample is obtained from a
subject and
FGF-CX and/or FCTRX protein or nucleic acid (e.g., mRNA, genomic DNA) is
detected, ,
wherein the presence of FGF-CX and/or FCTRX protein or nucleic acid is
diagnostic for a
subject having or at risk of developing a disease or disorder associated with
aberrant FGF-CX
and/or FCT1RX expression or activity. As used herein, a "test sample" refers
to a biological
sample obtained from a subject of interest. For example, a test sample can be
a biological
fluid (e.g., serum), cell sample, or tissue.
Furthermore, the prognostic assays described herein can be used to determine
whether
a subject can be administered an agent (e.g., an agonist, antagonist,
peptidomimetic, protein,
peptide, nucleic acid, small molecule, or other drug candidate) to treat a
disease or disorder
associated with aberrant FGF-CX and/or FCTRX expression or activity. For
example, such
methods can be used to determine whether a subject can be effectively treated
with an agent
for a disorder. Thus, the invention provides methods for determining whether a
subject can be
effectively treated with an agent for a disorder associated with aberrant FGF-
CX and/or
FCTRX expression or activity in which a test sample is obtained and FGF-CX
and/or FCTRX
protein or nucleic acid is detected (e.g., wherein the presence of FGF-CX
and/or FCTRX
protein or nucleic acid is diagnostic for a subject that can be administered
the agent to treat a
disorder associated with aberrant FGF-CX and/or FCTRX expression or activity).
The methods of the invention can also be used to detect genetic lesions in an
FGF-CX
and/or FCTRX gene, thereby determining if a subject with the lesioned gene is
at risk for a
disorder characterized by aberrant cell proliferation and/or differentiation.
In various
embodiments, the methods include detecting, in a sample of cells from the
subject, the
presence or absence of a genetic lesion characterized by at least one of an
alteration affecting
99
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
the integrity of a gene encoding an FGF-CX and/or FCTRX-protein, or the
misexpression of
the FGF-CX andlor FCTRX gene. For example, such genetic lesions can be
detected by
ascertaining the existence of at least one of (i) a deletion of one or more
nucleotides from an
FGF-CX and/or FCTRX gene; (ii) an addition of one or more nucleotides to an
FGF-CX
and/or FCTRX gene; (iii) a substitution of one or more nucleotides of an FGF-
CX and/or
FCTRX gene, (iv) a chromosomal rearrangement of an FGF-CX and/or FCTRX gene;
(v) an
alteration in the level of a messenger RNA transcript of an FGF-CX and/or
FCTRX gene, (vi)
aberrant modification of an FGF-CX and/or FCTRX gene, such as of the
methylation pattern
of the genomic DNA, (vii) the presence of a non-wild-type splicing pattern of
a messenger
RNA transcript of an FGF-CX and/or FCTRX gene, (viii) a non-wild-type level of
an FGF-CX
and/or FCTRX protein, (ix) allelic loss of an FGF-CX and/or FCTRX gene, and
(x)
inappropriate post-translational modification of an FGF-CX and/or FCTRX
protein. As
described herein, there are a large number of assay techniques known in the
art which can be
used for detecting lesions in an FGF-CX and/or FCTRX gene. A preferred
biological sample
is a peripheral blood leukocyte sample isolated by conventional means from a
subj ect.
However, any biological sample containing nucleated cells may be used,
including, for
example, buccal mucosal cells.
In certain embodiments, detection of the lesion involves the use of a
probe/primer in a
polymerase chain reaction (PCR) (see, e.g., U.S. Patent Nos. 4,683,195 and
4,683,202), such
as anchor PCR or RACE PCR, or, alternatively, in a ligation chain reaction
(LCR) (see, e.g.,
Landegran, et al., 1988. Science 241: 1077-1080; and Nakazawa, et al., 1994.
Proc. Natl.
Acad. Sci. USA 91: 360-364), the latter of which can be particularly useful
for detecting point
mutations in the FGF-CX and/or FCTRX-gene (see, Abravaya, et al., 1995. Nucl.
Acids Res.
23: 675-682). This method can include the steps of collecting a sample of
cells from a patient,
isolating nucleic acid (e.g., genomic, mRNA or both) from the cells of the
sample, contacting
the nucleic acid sample with one or more primers that specifically hybridize
to an FGF-CX
andlor FCTRX gene under conditions such that hybridization and amplification
of the FGF-
CX andlor FCTRX gene (if present) occurs, and detecting the presence or
absence of an
amplification product, or detecting the size of the amplification product and
comparing the
length to a control sample. It is anticipated that PCR and/or LCR may be
desirable to use as a
preliminary amplification step in conjunction with any of the techniques used
for detecting
mutations described herein.
100
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
Alternative amplification methods include: self sustained sequence replication
(see,
Guatelli, et al., 1990. Proc. Natl. Acad. Sci. USA 87: 1874-1878),
transcriptional amplification
system (see, Kwoh, et al., 1989. Proc. Natl. Acad. Sci. USA 86: 1173-1177);
Q(3 Replicase
(see, Lizardi, et al, 1988. BioTechnology 6: 1197), or any other nucleic acid
amplification
method, followed by the detection of the amplified molecules using techniques
well known to
those of skill in the art. These detection schemes are especially useful for
the detection of
nucleic acid molecules if such molecules are present in very low numbers.
In an alternative embodiment, mutations in an FGF-CX and/or FCTRX gene from a
sample cell can be identified by alterations in restriction enzyme cleavage
patterns. For
example, sample and control DNA is isolated, amplified (optionally), digested
with one or
more restriction endonucleases, and fragment length sizes are determined by
gel
electrophoresis and compared. Differences in fragment length sizes between
sample and
control DNA indicates mutations in the sample DNA. Moreover, the use of
sequence specific
ribozymes (see, e.g., U.S. Patent No. 5,493,531) can be used to score for the
presence of
specific mutations by development or loss of a ribozyme cleavage site.
In other embodiments, genetic mutations in FGF-CX and/or FCTRX can be
identified
by hybridizing a sample and control nucleic acids, e.g., DNA or RNA, to high-
density arrays
containing hundreds or thousands of oligonucleotides probes. See, e.g.,
Cronin, et al., 1996.
Human Mutation 7: 244-255; Kozal, et al., 1996. Nat. Med. 2: 753-759. For
example, genetic
mutations in FGF-CX and/or FCTRX can be identified in two dimensional arrays
containing
light-generated DNA probes as described in Cronin, et al., supra. Briefly, a
first hybridization
array of probes can be used to scan through long stretches of DNA in a sample
and control to
identify base changes between the sequences by making linear arrays of
sequential
overlapping probes. This step allows the identification of point mutations.
This is followed
by a second hybridization array that allows the characterization of specific
mutations by using
smaller, specialized probe arrays complementary to all variants or mutations
detected. Each
mutation array is composed of parallel probe sets, one complementary to the
wild-type gene
and the other complementary to the mutant gene.
In yet another embodiment, any of a variety of sequencing reactions known in
the art
can be used to directly sequence the FGF-CX andlor FCTRX gene and detect
mutations by
comparing the sequence of the sample FGF-CX and/or FCTRX with the
corresponding
wild-type (control) sequence. Examples of sequencing reactions include those
based on
101
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
techniques developed by Maxim and Gilbert, 1977. Proc. Natl. Acad. Sci. USA
74: 560 or
Sanger, 1977. Proc. Natl. Acad. Sci. USA 74: 5463. It is also contemplated
that any of a
variety of automated sequencing procedures can be utilized when performing the
diagnostic
assays (see, e.g., Naeve, et al., 1995. Biotechhiques 19: 448), including
sequencing by mass
spectrometry (see, e.g., PCT International Publication No. WO 94/16101; Cohen,
et al., 1996.
Adv. Chromatography 36: 127-162; and Griffin, et al., 1993. Appl. Biochem.
Biotechnol. 38:
147-159).
Other methods for detecting mutations in the FGF-CX and/or FCTRX gene include
methods in which protection from cleavage agents is used to detect mismatched
bases in
RNA/RNA or RNA/DNA heteroduplexes. See, e.g., Myers, et al., 1985. Science
230: 1242.
In general, the art technique of "mismatch cleavage" starts by providing
heteroduplexes of
formed by hybridizing (labeled) RNA or DNA containing the wild-type FGF-CX
and/or
FCTRX sequence with potentially mutant RNA or DNA obtained from a tissue
sample. The
double-stranded duplexes are treated with an agent that cleaves single-
stranded regions of the
duplex such as which will exist due to basepair mismatches between the control
and sample
strands. For instance, RNA/DNA duplexes can be treated with RNase and DNA/DNA
hybrids
treated with S1 nuclease to enzymatically digesting the mismatched regions. In
other
embodiments, either DNA/DNA or RNA/DNA duplexes can be treated with
hydroxylamine or
osmium tetroxide and with piperidine in order to digest mismatched regions.
After digestion
of the mismatched regions, the resulting material is then separated by size on
denaturing
polyacrylamide gels to determine the site of mutation. See, e.g., Cotton, et
al., 1988. Proc.
Natl. Acad. Sci. USA 85: 4397; Saleeba, et al., 1992. Methods Enzymol. 217:
286-295. In an
embodiment, the control DNA or RNA can be labeled for detection.
In still another embodiment, the mismatch cleavage reaction employs one or
more
proteins that recognize mismatched base pairs in double-stranded DNA (so
called "DNA
mismatch repair" enzymes) in defined systems for detecting and mapping point
mutations in
FGF-CX and/or FCTRX cDNAs obtained from samples of cells. For example, the
mutt
enzyme of E. coli cleaves A at G/A mismatches and the thymidine DNA
glycosylase from
HeLa cells cleaves T at G/T mismatches. See, e.g., Hsu, et al., 1994.
Carcinogenesis 15:
1657-1662. According to an exemplary embodiment, a probe based on an FGF-CX
and/or
FCTRX sequence, e.g., a wild-type FGF-CX and/or FCTRX sequence, is hybridized
=to a
cDNA or other DNA product from a test cell(s). The duplex is treated with a
DNA mismatch
102
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
repair enzyme, and the cleavage products, if any, can be detected from
electrophoresis
protocols or the like. See, e.g., U.S. Patent No. 5,459,039.
In other embodiments, alterations in electrophoretic mobility will be used to
identify
mutations in FGF-CX and/or FCTRX genes. For example, single strand
conformation
polymorphism (SSCP) may be used to detect differences in electrophoretic
mobility between
mutant and wild type nucleic acids. See, e.g., Orita, et al., 1989. Proc.
Natl. Acad. Sci. USA:
86: 2766; Cotton, 1993. Mutat. Res. 285: 125-144; Hayashi, 1992. Genet. Anal.
Tech. Appl. 9:
73-79. Single-stranded DNA fragments of sample and control FGF-CX and/or FCTRX
nucleic acids will be denatured and allowed to renature. The secondary
structure of
single-stranded nucleic acids varies according to sequence, the resulting
alteration in
electrophoretic mobility enables the detection of even a single base change.
The DNA
fragments may be labeled or detected with labeled probes. The sensitivity of
the assay may be
enhanced by using RNA (rather than DNA), in which the secondary structure is
more sensitive
to a change in sequence. In one embodiment, the subject method utilizes
heteroduplex
analysis to separate double stranded heteroduplex molecules on the basis of
changes in
electrophoretic mobility. See, e.g., Keen, et al., 1991. Trends Genet. 7: 5.
In yet another embodiment, the movement of mutant or wild-type fragments in
polyacrylamide gels containing a gradient of denaturant is assayed using
denaturing gradient
gel electrophoresis (DGGE). See, e.g., Myers, et al., 1985. Nature 313: 495.
When DGGE is
used as the method of analysis, DNA will be modified to insure that it does
not completely
denature, for example by adding a GC clamp of approximately 40 by of high-
melting GC-rich
DNA by PCR. In a further embodiment, a temperature gradient is used in place
of a
denaturing gradient to identify differences in the mobility of control and
sample DNA. See,
e.g., Rosenbaum and Reissner, 1987. Biophys. Ch.eTn. 265: 12753.
Examples of other techniques for detecting point mutations include, but are
not limited
to, selective oligonucleotide hybridization, selective amplification, or
selective primer
extension. For example, oligonucleotide primers may be prepared in which the
known
mutation is placed centrally and then hybridized to target DNA under
conditions that permit
hybridization only if a perfect match is found. See, e.g., Saiki, et al.,
1986. Nature 324: 163;
Saiki, et al., 1989. Proc. Natl. Acad. Sci. USA 86: 6230. Such allele specific
oligonucleotides
are hybridized to PCR amplified target DNA or a number of different mutations
when the
103
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
oligonucleotides are attached to the hybridizing membrane and hybridized with
labeled target
DNA.
Alternatively, allele specific amplification technology that depends on
selective PCR
amplification may be used in conjunction with the instant invention.
Oligonucleotides used as
primers for specific amplification may carry the mutation of interest in the
center of the
molecule (so that amplification depends on differential hybridization; see,
e.g., Gibbs, et al.,
1989. Nucl. Acids Res. 17: 2437-2448) or at the extreme 3'-terminus of one
primer where,
under appropriate conditions, mismatch can prevent, or reduce polymerase
extension (see, e.g.,
Prossner, 1993. Tibtech. 11: 238). In addition it may be desirable to
introduce a novel
restriction site in the region of the mutation to create cleavage-based
detection. See, e.g.,
Gasparini, et al., 1992. Mol. Cell Probes 6: 1. It is anticipated that in
certain embodiments
amplification may also be performed using Taq ligase for amplification. See,
e.g., Barany,
1991. P~oc. Natl. Acad. Sci. USA 88: 189. In such cases, ligation will occur
only if there is a
perfect match at the 3'-terminus of the 5' sequence, making it possible to
detect the presence of
a known mutation at a specific site by looking for the presence or absence of
amplification.
The methods described herein may be performed, for example, by utilizing
pre-packaged diagnostic kits comprising at least one probe nucleic acid or
antibody reagent
described herein, which may be conveniently used, e.g., in clinical settings
to diagnose
patients exhibiting symptoms or family history of a disease or illness
involving an FGF-CX
and/or FCTRX gene.
Furthermore, any cell type or tissue, preferably peripheral blood leukocytes,
in which
FGF-CX and/or FCTRX is expressed may be utilized in the prognostic assays
described
herein. However, any biological sample containing nucleated cells may be used,
including, for
example, buccal mucosal cells.
Pharmacogenomics
Agents, or modulators that have a stimulatory or inhibitory effect on FGF-CX
and/or
FCTRX activity (e.g., FGF-CX and/or FCTRX gene expression), as identified by a
screening
assay described herein can be administered to individuals to treat
(prophylactically or
therapeutically) disorders. The disorders include pathology such as
inflammatory conditions in
the gastrointestinal tract, including but not limited to inflammatory bowel
disease such as
ulcerative colitis and Crohn's disease, growth and proliferative diseases such
as cancer,
angiogenesis, atherosclerotic plaques, collagen formation, cartilage and bone
formation,
104
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
cardiovascular and fibrotic diseases and diabetic ulcers. In addition, FCTRX
nucleic acids and
their encoded polypeptides will be therapeutically useful for the prevention
of aneurysms and
the acceleration of wound closure through gene therapy. Furthermore, FCTRX
nucleic acids
and their encoded polypeptides can be utilized to stimulate cellular growth.
wound healing,
neovascularization and tissue growth, and similar tissue regeneration needs.
More
specifically, a FCTRX nucleic acid or polypeptide may be useful in treatment
of anemia and
leukopenia, intestinal tract sensitivity and baldness. Treatment of such
conditions may be
indicated, e. g., in patients having undergone radiation or chemotherapy,
wherein treatment
would minimize any hyperproliferative side effects.
In conjunction with such treatment, the pharmacogenomics (i.e., the study of
the
relationship between an individual's genotype and that individual's response
to a foreign
compound or drug) of the individual may be considered. Differences in
metabolism of
therapeutics can lead to severe toxicity or therapeutic failure by altering
the relation between
dose and blood concentration of the pharmacologically active drug. Thus, the
pharmacogenomics of the individual permits the selection of effective agents
(e.g., drugs) for
prophylactic or therapeutic treatments based on a consideration of the
individual's genotype.
Such pharmacogenomics can further be used to determine appropriate dosages and
therapeutic
regimens. Accordingly, the activity of FGF-CX and/or FCTRX protein, expression
of FGF-
CX and/or FCTRX nucleic acid, or mutation content of FGF-CX and/or FCTRX genes
in an
individual can be determined to thereby select appropriate agents) for
therapeutic or
prophylactic treatment of the individual.
Pharmacogenomics deals with clinically significant hereditary variations in
the
response to drugs due to altered drug disposition and abnormal action in
affected persons. See
e.g., Eichelbaum, 1996. Clin. Exp. Pharmacol. Physiol., 23: 983-985; Linden
1997. Clin.
Chem., 43: 254-266. In general, two types of pharmacogenetic conditions can be
differentiated. Genetic conditions transmitted as a single factor altering the
way drugs act on
the body (altered drug action) or genetic conditions transmitted as single
factors altering the
way the body acts on drugs (altered drug metabolism). These pharmacogenetic
conditions can
occur either as rare defects or as polymorphisms. For example, glucose-6-
phosphate
dehydrogenase (G6PD) deficiency is a common inherited enzymopathy in which the
main
clinical complication is hemolysis after ingestion of oxidant drugs (anti-
malarials,
sulfonamides, analgesics, nitrofurans) and consumption of fava beans.
105
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
As an illustrative embodiment, the activity of drug metabolizing enzymes is a
major
determinant of both the intensity and duration of drug action. The discovery
of genetic
polymorphisms of drug metabolizing enzymes (e.g., N-acetyltransferase 2 (NAT
2) and
cytochrome P450 enzymes CYP2D6 and CYP2C19) has provided an explanation as to
why
some patients do not obtain the expected drug effects or show exaggerated drug
response and
serious toxicity after taking the standard and safe dose of a drug. These
polymorphisms are
expressed in two phenotypes in the population, the extensive metabolizer (EM)
and poor
metabolizer (PM). The prevalence of PM is different among different
populations. For
example, the gene coding for CYP2D6 is highly polymorphic and several
mutations have been
identified in PM, which all lead to the absence of functional CYP2D6. Poor
metabolizers of
CYP2D6 and CYP2C19 quite frequently experience exaggerated drug response and
side
effects when they receive standard doses. If a metabolite is the active
therapeutic moiety, PM
show no therapeutic response, as demonstrated for the analgesic effect of
codeine mediated by
its CYP2D6-formed metabolite morphine. At the other extreme are the so called
ultra-rapid
metabolizers who do not respond to standard doses. Recently, the molecular
basis of
ultra-rapid metabolism has been identified to be due to CYP2D6 gene
amplification.
Thus, the activity of FGF-CX and/or FCTRX protein, expression of FGF-CX and/or
FCTRX nucleic acid, or mutation content of FGF-CX and/or FCTRX genes in an
individual
can be determined to thereby select appropriate agents) for therapeutic or
prophylactic
treatment of the individual. In addition, pharmacogenetic studies can be used
to apply
genotyping of polymorphic alleles encoding drug-metabolizing enzymes to the
identification
of an individual's drug responsiveness phenotype. This knowledge, when applied
to~dosing or
drug selection, can avoid adverse reactions or therapeutic failure and thus
enhance therapeutic
or prophylactic efficiency when treating a subj ect with an FGF-CX andlor
FCTRX modulator,
such as a modulator identified by one of the exemplary screening assays
described herein.
Monitoring of Effects During Clinical Trials
Monitoring the influence of agents (e.g., drugs, compounds) on the expression
or
activity of FGF-CX and/or FCTRX (e.g., the ability to modulate aberrant cell
proliferation
and/or differentiation) can be applied not only in basic drug screening, but
also in clinical
trials. For example, the effectiveness of an agent determined by a screening
assay as described
herein to increase FGF-CX andlor FCTRX gene expression, protein levels, or
upregulate FGF-
CX and/or FCTRX activity, can be monitored in clinical trails of subjects
exhibiting decreased
106
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
FGF-CX and/or FCTRX gene expression, protein levels, or downregulated FGF-CX
and/or
FCTRX activity. Alternatively, the effectiveness of an agent determined by a
screening assay
to decrease FGF-CX and/or FCTRX gene expression, protein levels, or
downregulate FGF-CX
and/or FCTRX activity, can be monitored in clinical trails of subjects
exhibiting increased
FGF-CX and/or FCTRX gene expression, protein levels, or upregulated FGF-CX
and/or
FCTRX activity. In such clinical trials, the expression or activity of FGF-CX
and/or FCTRX
and, preferably, other genes that have been implicated in, for example, a
cellular proliferation
or immune disorder can be used as a "read out" or markers of the immune
responsiveness of a
particular cell.
By way of example, and not of limitation, genes, including FGF-CX and/or
FCTRX,
that are modulated in cells by treatment with an agent (e.g., compound, drug
or small
molecule) that modulates FGF-CX and/or FCTRX activity (e.g., identified in a
screening assay
as described herein) can be identified. Thus, to study the effect of agents on
cellular
proliferation disorders, for example, in a clinical trial, cells can be
isolated and RNA prepared
and analyzed for the levels of expression of FGF-CX and/or FCTRX and other
genes
implicated in the disorder. The levels of gene expression (i.e., a gene
expression pattern) can
be quantified by Northern blot analysis or RT-PCR, as described herein, or
alternatively by
measuring the amount of protein produced, by one of the methods as described
herein, or by
measuring the levels of activity of FGF-CX and/or FCTRX or other genes. In
this manner, the
gene expression pattern can serve as a marker, indicative of the physiological
response of the
cells to the agent. Accordingly, this response state may be determined before,
and at various
points during, treatment of the individual with the agent.
In one embodiment, the invention provides a method for monitoring the
effectiveness
of treatment of a subject with an agent (e.g., an agonist, antagonist,
protein, peptide,
peptidomimetic, nucleic acid, small molecule, or other drug candidate
identified by the
screening assays described herein) comprising the steps of (i) obtaining a pre-
administration
sample from a subject prior to administration of the agent; (ii) detecting the
level of expression
of an FGF-CX and/or FCTRX protein, mRNA, or genomic DNA in the
preadministration
sample; (iii) obtaining one or more post-administration samples from the
subject; (iv)
detecting the level of expression or activity of the FGF-CX and/or FCTRX
protein, mRNA, or
genomic DNA in the post-administration samples; (v) comparing the level of
expression or
activity of the FGF-CX and/or FCTRX protein, mRNA, or genomic DNA in the
107
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
pre-administration sample with the FGF-CX and/or FCT1RX protein, mRNA, or
genomic DNA
in the post administration sample or samples; and (vi) altering the
administration of the agent
to the subject accordingly. For example, increased administration of the agent
may be
desirable to increase the expression or activity of FGF-CX and/or FCTRX to
higher levels
than detected, i.e., to increase the effectiveness of the agent.
Alternatively, decreased
administration of the agent may be desirable to decrease expression or
activity of FGF-CX
and/or FCTRX to lower levels than detected, i.e., to decrease the
effectiveness of the agent.
Methods of Treatment
The invention provides for both prophylactic and therapeutic methods of
treating a
subject at risk of (or susceptible to) a disorder or having a disorder
associated with aberrant
FGF-CX and/or FCTRX expression or activity. The disorders include
cardiorriyopathy,
atherosclerosis, hypertension, congenital heart defects, aortic stenosis,
atrial septal defect
(ASD), atrioventricular (A-V) canal defect, ductus arteriosus, pulmonary
stenosis, subaortic
stenosis, ventricular septal defect (VSD), valve diseases, tuberous sclerosis,
scleroderma,
obesity, transplantation, adrenoleukodystrophy, congenital adrenal
hyperplasia, prostate
cancer, neoplasm; adenocarcinoma, lymphoma, uterus cancer, fertility,
hemophilia,
hypercoagulation, idiopathic thrombocytopenic purpura, immunodeficiencies,
graft versus
host disease, AIDS, bronchial asthma, Crohn's disease; multiple sclerosis,
treatment of
Albright Hereditary Ostoeodystrophy, and other diseases, disorders and
conditions of the like.
These methods of treatment will be discussed more fully, below.
Disease and Disorders
Diseases and disorders that are characterized by increased (relative to a
subject not
suffering from the disease or disorder) levels or biological activity may be
treated with
Therapeutics that antagonize (i.e., reduce or inhibit) activity. Therapeutics
that antagonize
activity may be administered in a therapeutic or prophylactic manner.
Therapeutics that may
be utilized include, but are not limited to: (i) an aforementioned peptide, or
analogs,
derivatives, fragments or homologs thereof; (ii) antibodies to an
aforementioned peptide; (iii)
nucleic acids encoding an aforementioned peptide; (iv) administration of
antisense nucleic acid
and nucleic acids that axe "dysfunctional" (i.e., due to a heterologous
insertion within the
coding sequences of coding sequences to an aforementioned peptide) that are
utilized to
"knockout" endoggenous function of an aforementioned peptide by homologous
recombination (see, e.g., Capecchi, 1989. Science 244: 1288-1292); or (v)
modulators ( i.e.,
108
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
inhibitors, agonists and antagonists, including additional peptide mimetic of
the invention or
antibodies specific to a peptide of the invention) that alter the interaction
between an
aforementioned peptide and its binding partner.
Diseases and disorders that are characterized by decreased (relative to a
subject not
suffering from the disease or disorder) levels or biological activity may be
treated with
Therapeutics that increase (i.e., are agonists to) activity. Therapeutics that
upregulate activity
may be administered in a therapeutic or prophylactic manner. Therapeutics that
may be
utilized include, but are not limited to, an aforementioned peptide, or
analogs, derivatives,
fragments or homologs thereof; or an agonist that increases bioavailability.
Increased or decreased levels can be readily detected by quantifying peptide
andlor
RNA, by obtaining a patient tissue sample (e.g., from biopsy tissue) and
assaying it ih vitro for
RNA or peptide levels, structure and/or activity of the expressed peptides (or
mRNAs of an
aforementioned peptide). Methods that are well-known within the art include,
but are not
limited to, immunoassays (e.g., by Western blot analysis, immunoprecipitation
followed by
sodium dodecyl sulfate (SDS) polyacrylamide gel electrophoresis,
imrnunocytochemistry, etc.)
and/or hybridization assays to detect expression of mRNAs (e.g., Northern
assays, dot blots, in
situ hybridization, and the like).
Prophylactic Methods
In one aspect, the invention provides a method for preventing, in a subject, a
disease or
condition associated with an aberrant FGF-CX and/or FCTRX expression or
activity, by
administering to the subj ect an agent that modulates FGF-CX andlor FCTRX
expression or at
least one FGF-CX and/or FCTRX activity. Subjects at risk for a disease that is
caused or
contributed to by aberrant FGF-CX and/or FCTRX expression or activity can be
identified by,
for example, any or a combination of diagnostic or prognostic assays as
described herein.
Administration of a prophylactic agent can occur prior to the manifestation of
symptoms
characteristic of the FGF-CX and/or FCTRX aberrancy, such that a disease or
disorder is
prevented or, alternatively, delayed in its progression. Depending upon the
type of FGF-CX
and/or FCTRX aberrancy, for example, an FGF-CX and/or FCTRX agonist or FGF-CX
and/or
FCTRX antagonist agent can be used for treating the subject. The appropriate
agent can be
determined based on screening assays described herein. The prophylactic
methods of the
invention are further discussed in the following subsections.
109
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
Therapeutic Methods
Another aspect of the invention pertains to methods of modulating FGF-CX
and/or
FCTRX expression or activity for therapeutic purposes. The modulatory method
of the
invention involves contacting a cell with an agent that modulates one or more
of the activities
of FGF-CX and/or FCTRX protein activity associated with the cell. An agent
that modulates
FGF-CX and/or FCTRX protein activity can be an agent as described herein, such
as a nucleic
acid or a protein, a naturally-occurnng cognate ligand of an FGF-CX andlor
FCTRX protein, a
peptide, an FGF-CX and/or FCTRX peptidomimetic, or other small molecule. In
one
embodiment, the agent stimulates one or more FGF-CX and/or FCTRX protein
activity.
Examples of such stimulatory agents include active FGF-CX and/or FCTRX protein
and a
nucleic acid molecule encoding FGF-CX and/or FCTRX that has been introduced
into the cell.
In another embodiment, the agent inhibits one or more FGF-CX and/or FCTRX
protein
activity. Examples of such inhibitory agents include antisense FGF-CX and/or
FCTRX
nucleic acid molecules and anti-FGF-CX and/or FCTRX antibodies. These
modulatory
methods can be performed ih vitro (e.g., by culturing the cell with the agent)
or, alternatively,
ih vivo (e.g., by administering the agent to a subject). As such, the
invention provides methods
of treating an individual afflicted with a disease or disorder characterized
by aberrant
expression or activity of an FGF-CX and/or FCTRX protein or nucleic acid
molecule. In one
embodiment, the method involves administering an agent (e.g., an agent
identified by a
screening assay described herein), or combination of agents that modulates
(e.g., up-regulates
or down-regulates) FGF-CX and/or FCTRX expression or activity. In another
embodiment,
the method involves administering an FGF-CX and/or FCTRX protein or nucleic
acid
molecule as therapy to compensate for reduced or aberrant FGF-CX and/or FCTRX
expression or activity.
Stimulation of FGF-CX and/or FCTRX activity is desirable in situations in
which
FGF-CX and/or FCTRX is abnormally downregulated and/or in which increased FGF-
CX
and/or FCTRX activity is likely to have a beneficial effect. One example of
such a situation is
where a subject has a disorder characterized by aberrant cell proliferation
and/or
differentiation (e.g., cancer or immune associated disorders). Another example
of such a
situation is where the subject has a gestational disease (e.g., preclampsia).
110
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
Determination of the Biological Effect of the Therapeutic
In various embodiments of the invention, suitable ifa vitro or in vivo assays
are
performed to determine the effect of a specific Therapeutic and whether its
administration is
indicated for treatment of the affected tissue.
In various specific embodiments, ih vitro assays may be performed with
representative
cells of the types) involved in the patient's disorder, to determine if a
given Therapeutic exerts
the desired effect upon the cell type(s). Compounds for use in therapy may be
tested in
suitable animal model systems including, but not limited to rats, mice,
chicken, cows,
monkeys, rabbits, and the like, prior to testing in human subjects. Similarly,
for i~ vivo
testing, any of the animal model system known in the art may be used prior to
administration
to human subj ects.
Prophylactic and Therapeutic Uses of the Compositions of the Invention
The FGF-CX and/or FCTRX nucleic acids and proteins of the invention are useful
in
potential prophylactic and therapeutic applications implicated in a variety of
disorders
including, but not limited to: inflammatory bowel disease and disorders
associated with FGF-
CX, with FCTRX, or with both FGF-CX and/or FCTIZX.
As an example, a cDNA encoding the FGF-CX and/or FCTRX protein of the
invention
may be useful in gene therapy, and the protein may be useful when administered
to a subject
in need thereof. By way of non-limiting example, the compositions of the
invention will have
efficacy for treatment of patients suffering from: inflammatory conditions in
the
gastrointestinal tract, including but not limited to inflammatory bowel
disease such as
ulcerative colitis and Crohn's disease, growth and proliferative diseases such
as cancer,
angiogenesis, atherosclerotic plaques, collagen formation, cartilage and bone
formation,
cardiovascular and fibrotic diseases and diabetic ulcers. In addition, FCTRX
nucleic acids and
their encoded polypeptides will be therapeutically useful for the prevention
of aneurysms and
the acceleration of wound closure through gene therapy. Furthermore, FCTRX
nucleic acids
and their encoded polypeptides can be utilized to stimulate cellular growth.
wound healing,
neovascularization and tissue growth, and similar tissue regeneration needs.
More
specifically, a FCT1RX nucleic acid or polypeptide may be useful in treatment
of anemia and
leukopenia, intestinal tract sensitivity and baldness. Treatment of such
conditions may be
indicated, e. g., in patients having undergone radiation or chemotherapy,
wherein treatment
would minimize any hyperproliferative side effects.
111
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
Both the novel nucleic acid encoding the FGF-CX and/or FCTRX protein, and the
FGF-CX andlor FCTRX protein of the invention, or nucleic acid or protein
fragments,
analogs, homologs or derivative thereof, may also be useful in diagnostic
applications,
wherein the presence or amount of the nucleic acid or the protein are to be
assessed. A further
use could be as an anti-bacterial molecule (i.e., some peptides have been
found to possess anti-
bacterial properties). These materials are further useful in the generation of
antibodies which
immunospecifically-bind to the novel substances of the invention for use in
therapeutic or
diagnostic methods.
EXAMPLES
It is shown in several Examples below that both FGF-CX and FCTRX induce the
growth and proliferation of various mammalian cells in culture. It is further
demonstrated in
animal models of inflarmnatory bowel disease that these proteins have
beneficial effects in
treating, ameliorating and delaying the onset of inflammatory bowel disease.
By "treating" is
meant the administration of a protein used in the present invention to a
subject suffering from
a pathology such as inflammatory bowel disease with the obj ective of
providing a beneficial
therapeutic effect. By "ameliorating" a pathology such as inflammatory bowel
disease, it is
meant that a) in a subject in which the pathology is becoming more severe, one
or more
symptoms of the pathology cease becoming more severe and stabilize or improve;
or b) in a
subject in which the pathology is considered to be at a stable state, one or
more symptoms of
the pathology improve or become less severe. By "delaying the onset" of a
pathology such as
inflammatory bowel disease, it is meant that administering a prophylactic dose
or dosing
regimen of a therapeutic agent such as the FGF-CX and FCTRX proteins employed
in the
present invention results in the delay of appearance, or the delay of
worsening, of one or more
symptoms of a pathology such as inflammatory bowel disease. Such a delay may
be for an
indeterminate period, in which the symptoms essentially never appear or never
worsen, or it
may be for A more limited period, in which the symptoms appear or worsen at a
later time
than would be expected, based on the experience of patients not treated by the
compositions
envisioned in the present methods, in the absence of administering the
therapeutic agent.
The results of experiments reported below in three Examples indicate that, in
mice in
which inflammatory bowel disease is induced by oral administration of DSS for
7 days,
112
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
simultaneous treatment with the growth factors employed here during the course
of exposure
to DSS lead to significant therapeutic benefits compared to untreated DSS
controls.
An additional Example reports results on rats treated with indomethacin which
results
in gross and histopathologic intestinal alterations that are similar to those
occurring in Crohn's
Disease. Administration of CG53135 (0.2 mg/kg iv) to indomethacin-treated rats
results in
significant reductions in weight loss, small intestine weight, absolute
neutrophil counts, and
jejunal necrosis and inflammation scores.
Example 1. Identification of the FG>i-CX gene.
The FGF-CX gene was identified following a TBLASTN (Altschul, S. F., Gish, W.,
Miller, W., Myers, E. W. & Lipman, D. J. (1990) J. Mol. Biol. 215, 403-410)
search of
Genbank human genomic DNA sequences with Xenopus FGF-CX (Koga, C., Adati, N.,
Nakata, K., Mikoshiba, K., Furuhata, Y., Sata, S., Tei, H., Sakati, Y.,
Kurokawa, T.,
Shiokawa, K. ~ Yokoyama, K. K. (1999) Biochem. Biophys. Res. Comm. 261, 756-
765;
Accession No. AB012615) as query. This search identified a locus (Accession
No. AB020858)
of high homology on chromosome 8. Intron/exon boundaries were deduced using
standard
consensus splicing parameters (Mount, S. M. (1996) Science 271, 1690-1692),
together with
homologies derived from known FGFs. The FGF-CX initiation codon localizes to
by 16214 of
the sequence of AB020858, and the remaining 3' portion of this exon continues
to by 15930.
The 5' UTR of FGF-CX was extended upstream of the initiation codon by an
additional 606
by using public ESTs (Accession Nos. AA232729, AA236522, AI272876 and
AI272878). The
remaining structure of the FGF-CX gene as it relates to locus AB020858 is as
follows: intron 1
(bp 15929-9942); exon 2 (bp 9941-9838); intron 2 (bp 9837-7500); exon 3
(begins at by
7499).
The gene discovered by the procedure in the preceding paragraph includes 3
exons and
2 introns. The DNA sequence predicts an ORF of 211 amino acid residues (see
Table 1), with
an in-frame stop codon 117 by upstream of the initiator methionine. The DNA
segment from
which the gene was mined maps to chromosome 8p21.3-p22, a location that was
confirmed by
radiation hybrid analysis.
Example 2. Molecular Cloning of the Sequence Encoding a FGF-CX Protein.
Oligonucleotide primers were designed for the amplification by PCR of a DNA
segment, representing an open reading frame, coding for the full length FGF-
CX. The forward
primer includes a BgIII restriction site (AGATCT) and a consensus Kozak
sequence
113
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
(CCACC). The reverse primer contains an in-frame XhoI restriction site for
further
subcloning purposes. Both the forward and the reverse primers contain a 5'
clamp sequence
(CTCGTC). The sequences of the primers are the following:
FGF-CX-Forward: 5' - CTCGTC AGATCT CCACC ATG GCT CCC TTA GCC
GAA GTC - 3' (SEQ ID NO:15)
FGF-CX-Reverse: 5' - CTCGTC CTCGAG AGT GTA CAT CAG TAG GTC CTT G
- 3' (SEQ D7 N0:16)
PCR reactions were performed using a total of Sng human prostate cDNA
template, 1
p,M of each of the FGF-CX-Forward and FGF-CX-Reverse primers, 5 micromoles
dNTP
(Clontech Laboratories, Palo Alto CA) and 1 microliter of SOxAdvantage-HF 2
polymerase
(Clontech Laboratories) in 50 microliter volume. The following PCR reaction
conditions were
used:
a) 96°C 3 minutes
b) 96°C 30 seconds denaturation
c) 70°C 30 seconds, primer annealing. This temperature was gradually
decreased by 1 °C/cycle.
d) 72°C 1 minute extension.
Repeat steps (b)-(d) ten times
e) 96°C 30 seconds denaturation
f) 60°C 30 seconds annealing
g) 72°C 1 minute extension
Repeat steps (e)-(g) 25 times
h) 72°C 5 minutes final extension
A single PCR product, with the expected size of approximately 640 bp, was
isolated
after electrophoresis on agarose gel and ligated into a pCR2.1 vector
(Invitrogen, Carlsbad,
CA). The cloned insert was sequenced using vector specific M13 Forward(-40)
and M13
Reverse primers, which verified that the nucleotide sequence was 100%
identical to the
sequence in Table 1 (SEQ 117 NO:1) inserted directly between the upstream
BgIII cloning site
and the downstream XhoI cloning site. The cloned sequence constitutes an open
reading
frame coding for the predicted FGF-CX full length protein. The clone is called
TA-AB020~5-
5274-F 19.
114
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
Example 3. Preparation of Mammalian Expression Vector pCEP4/Sec.
The oligonucleotide primers pSec-V5-His Forward CTCGT CCTCG AGGGT
AAGCC TATCC CTAAC (SEQ ID N0:17) and pSec-V5-His Reverse CTCGT CGGGC
CCCTG ATCAG CGGGT TTAAA C (SEQ ID N0:18), were designed to amplify a fragment
from the pcDNA3.1-VSHis (Invitrogen, Carlsbad, CA) expression vector that
includes V5 and
His6. The PCR product was digested with XhoI and ApaI and ligated into the
XhoI/ApaI
digested pSecTag2 B vector harboring an Ig kappa leader sequence (Invitrogen,
Carlsbad CA).
The correct structure of the resulting vector, pSecVSHis, including an in-
frame Ig-kappa
leader and V5-His6 was verified by DNA sequence analysis. The vector pSecVSHis
was
digested with PmeI and NheI to provide a fragment retaining the above elements
in the correct
frame. The PmeI-NheI fragment was ligated into the BamHI/I~lenow and NheI
treated vector
pCEP4 (Invitrogen, Carlsbad, CA). The resulting vector was named pCEP4/Sec and
includes
an in-frame Ig kappa leader, a site for insertion of a clone of interest, and
theVS epitope and
6xHis under control of the PCMV and/or the PT7 promoter. pCEP4/Sec is an
expression
vector that allows heterologous protein expression and secretion by fusing any
protein into a
multiple cloning site following the Ig kappa chain signal peptide. Detection
and purification of
the expressed protein are aided by the presence of the V5 epitope tag and
6xHis tag at the
C-terminus (Invitrogen, Carlsbad, CA).
Example 4. Expression of FGF-CX in human embryonic kidney (HEIR 293 cells.
The BgIII-Xhol fragment containing the FGF-CX sequence was isolated from TA-
AB02085-5274-F19 (Example 2) and subcloned into the BamHI-XhoI digested
pCEP4lSec to
generate the expression vector pCEP4/Sec-FGF-CX. The pCEP4/Sec-FGF-CX vector
was
transfected into 293 cells using the LipofectaminePlus reagent following the
manufacturer's
instructions (GibcolBRL/Life Technologies, Rockville, MD). The cell pellet and
supernatant
were harvested 72 hours after transfection and examined for FGF-CX expression
by Western
blotting (reducing conditions) with an anti-V5 antibody. FIG. 1 shows that FGF-
CX is
expressed as a polypeptide having an apparent molecular weight (Mr) of
approximately 34
kDa proteins secreted by 293 cells. In addition a minor band is observed at
about 31 kDa.
Example 5. Expression of FGF-CX in E. coli.
~ The vector pRSETA (InVitrogen Inc., Carlsbad, CA) was digested with XhoI and
NcoI
restriction enzymes. Oligonucleotide linkers of the sequence
115
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
5' CATGGTCAGCCTAC 3' (SEQ ID N0:19) and
5' TCGAGTAGGCTGAC 3' (SEQ ID N0:20)
were annealed at 37 degree Celsius and ligated into the XhoI-NcoI treated
pRSETA.
The resulting vector was confirmed by restriction analysis and sequencing and
was named
pETMY. The BgllI-XhoI fragment of the sequence encoding FGF-CX (see Example 2)
was
ligated into vector pETMY that was digested with BamHI and XhoI restriction
enzymes. The
expression vector is named pETMY-FGF-CX. In this vector, hFGF-CX was fused to
the 6xHis
tag and T7 epitope at its N-terminus. The plasmid pETMY-FGF-CX was then
transfected into
the E. coli expression host BL21(DE3, pLys) (Novagen, Madison, WI) and
expression of
protein FGF-CX was induced according to the manufacturer's instructions. After
induction,
total cells were harvested, and proteins were analyzed by Western blotting
using anti-HisGly
antibody (Invitrogen, Carlsbad, CA). FIG. 2 shows that FGF-CX was expressed as
a protein
of apparent molecular weight Mr approximately 32 kDa.
Example 6. Comparison of Expression of Recombinant FGF-CX Protein With and
Without a Cloned Signal Peptide.
a) Expression Without a Signal Peptide
As noted in the Detailed Description of the Invention, FGF-CX apparently lacks
a
classical amino-terminal signal sequence. To determine whether FGF-CX is
secreted from
mammalian cells, cDNA obtained as the BgIII XhoI fragment, encoding the full
length FGF-
CX protein, was subcloned from TA-AB02085-5274-F19 (Example 2) into BamHIlXhoI-
digested pcDNA3.1 (Invitrogen). This provided a mammalian expression vector
designated
pFGF-CX. This construct incorporates the VS epitope tag and a polyhistidine
tag into the
carboxy-terminus of the protein to aid in its identification and purification,
respectively, and
should generate a polypeptide of about 27 kDa. Following transient
transfection into 293
human embryonic kidney cells, conditioned media was harvested 48 hr post
transfection.
In addition to secretion of FGF-CX into conditioned media, it also found to be
associated with the cell pellet/ECM (data not shown). Since FGFs are known to
bind to
heparin sulfate proteoglycan (HSPG) present on the surface of cells and in the
extracellular
matrix (ECM), the inventors investigated the possibility that FGF-CX was
sequestered in this
manner. To this end, FGF-CX-transfected cells were extracted by treatment with
0.5 ml
DMEM containing 100 p,M suramin, a compound known to disrupt low affinity
interactions
between growth factors and HSPGs (La Rocca, R.V., Stein, C.A. & Myers, C.E.
(1990)
116
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
Cancer Cells 2, 106-115), for 30 min at 4°C. The suramin-extracted
conditioned media was
then harvested and clarified by centrifigation (5 min; 2000 X g).
The conditioned media and the suramin extract were then mixed with equal
volumes of
2X gel-loading buffer. Samples were boiled for 10 min, resolved by SDS-PAGE on
4-20%
gradient polyacrylamide gels (Novex, Dan Diego, CA) under reducing conditions,
and
transferred to nitrocelluose filters (Novex). Western analysis was performed
according to
standard procedures using HRP-conjugated anti-VS antibody (Invitrogen) and the
ECL
detection system (Amersham Pharmacia Biotech, Piscataway, NJ).
One band having the expected Mr was identified in conditioned media from 293
cells
transfected with pFGF-CX (FIG. 3, Panel A, lane 1). Conditioned media from
cells transfected
with control vector did not react with the antibody (FIG. 3, Panel A, lane 5).
After suramin
treatment, it was found that a significant quantity of FGF-CX could in fact be
released from
the cell surface/ECM, indicating that HSPGs are likely to play a role in
sequestering this
protein (FIG. 3, Panel A, lane 2). These results indicate that FGF-CX can be
secreted without
a classical signal peptide.
Recombinant FGF-CX protein stimulates DNA synthesis and cell proliferation,
effects
that are likely to be mediated via high affinity binding of FGF-CX to a cell
surface receptor,
and modulated via low affinity interactions with HSPGs. The suramin extraction
data
suggests that FGF-CX binds to HSPGs present on the cell surface and/or the
ECM.
b) Expression With a Signal Peptide
With the goal of enhancing protein secretion, a construct (pCEP4/Sec-FGF-CX)
was
generated in which the FGF-CX cDNA was fused in frame with a cleavable amino-
terminal
secretory signal sequence derived from the Ig~ gene. The resulting protein
also contained
carboxy-terminal VS and polyhistidine tags as described above for pFGF-CX.
Following
transfection into 293 cells, a protein product having the expected Mr of about
31 kDa was
obtained, and suramin was again found to release a significant quantity of
sequestered FGF-
CX protein (FIG. 3, Panel A; lanes 3 and 4). As expected, pCEP4/Sec-FGF-CX
generated
more soluble FGF-CX protein than did pFGF-CX.
Results similar to those described above for 293 cells were also obtained with
N1H 3T3
cells (FIG. 3, Panel B).
117
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
Example 7. Expression of FGF-CX.
FGF-CX was expressed essentially as described in Example 5. The protein was
purified using Ni2+-affinity chromatography, subj ected to SDS-PAGE under both
reducing and
nonreducing conditions, and stained using Coomassie Blue. The results are
shown in FIG. 4.
It is seen that under both sets of conditions, the protein migrates with an
apparent molecular
weight of approximately 29-30 kDa.
Example 8. Stimulation of Sromodeoxyuridine Incorporation by Recombinant FGF-
CX.
A dose response experiment for incorporation of BrdU was carned out using
human
renal carcinoma cells (786-0; American Type Culture Collection, Manassas, VA).
293-EBNA
cells (Invitrogen) were transfected using Lipofectamine 2000 according to the
manufacturer's
protocol (Life Technologies, Gaithersburg, MD). Cells were supplemented with
10% fetal
bovine senun (FBS; Life Technologies) 5 hr post-transfection. To generate
protein for BrdU
and growth assays (Example 10), cells were washed and fed with Dulbecco's
modified Eagle
medium (DMEM; Life Technologies) 18 hr post-transfection. After 48 hr, the
media was
discarded and the cell monolayer was incubated with 100 pM suramin (Sigma, St.
Louis, MO)
in 0.5 ml DMEM for 30 min at 4°C. The suramin-extracted conditioned
media was then
removed, clarified by centrifugation (5 min; 2000 X g), and subjected to TALON
metal
affinity chromatography according to the manufacturer's instructions
(Clontech, Palo Alto,
CA) taking advantage of the carboxy-terminal polyhistidine tag. Retained
fusion protein was
released by washing the column with imidazole.
To generate control protein, 293-EBNA cells were transfected with pCEP4
plasmid
(Invitrogen) and subjected to the purification procedure outlined above.
Recombinant FGF-CX was tested for its ability to induce DNA synthesis in a
bromodeoxyuridine (BrdU) incorporation assay. 786-0 cells were cultured in 96-
well plates to
approximately 100% confluence, washed with DMEM, and serum-starved in DMEM for
24
hr. Recombinant FGF-CX or control protein was then added to the cells for 18
hr. The BrdU
assay was performed according to the manufacturer's specifications (Roche
Molecular
Biochemicals, Indianapolis, IN) using a 5 hr BrdU incorporation time.
The results are shown in FIG. 5, in which FGF-CX is designated "20858". It is
seen
that FGF-CX stimulates proliferation of renal carcinoma cells by more than 4-
fold over
controls, with a half effective dose being about 2.5 ng/mL.
118
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
Example 9. Receptor binding specificity of FGF-CX.
To determine the receptor binding specificity of FGF-CX, we examined the
effect of
soluble FGF receptors (FGFRs) on the induction of DNA synthesis in NIH 3T3
cells by
recombinant FGF-CX. Four receptors have been identified to date (Klint P and
Claesson-
Welsh L. Front. Biosci., 4: 166-177, 1999; Xu X, et al. Cell Tissue Res., 296:
33-43, 1999).
Soluble receptors for FGFRl(3(IIIc), FGFR2a(IIIb), FGFR2(3(IIIb),
FGFR2a(IIIc),
FGFR3a(IIIc) and FGFR4 were utilized. It was found that soluble forms of each
of these
FGFRs were able to specifically inhibit the biological activity of FGF-CX (see
FIG. 6).
Complete or nearly complete inhibition was obtained with soluble FGFR2a(IIIb),
FGFR2(3(IIIb), FGFR2a(IIIc), and FGFR3a(IIIc), whereas partial inhibition was
achieved
with soluble FGFR1 (3(IIIc) and FGFR4. None of the soluble receptor reagents
interfered with
the induction of DNA synthesis by PDGF-BB, thereby demonstrating their
specificity. The
integrity of each soluble receptor reagent was demonstrated by showing its
ability to inhibit
the induction of DNA synthesis by aFGF (acidic FGF), a factor known to
interact with all of
the FGFRs under analysis.
Example 10. Cloning and Expression of an N-terminal Deletion Form of FGF-CX.
E. coli strain BL21 (DE3) (Invitrogen) harboring the plasmid pET24a- FGF20X-
de154-
codon were grown in LB medium at 37°C. This plasmid encodes the C-
terminal portion of
FGF-CX beginning at position 55. When cell densities reached an OD of 0.6,
IPTG was added
to final concentration of lmM. Induced cultures were then incubated for an
additional 4 hours
at 37°C. Cells were harvested by centrifugation at 3000Xg for 15
minutes at 4°C, suspended
in PBS and then disrupted with two passes through a microfluidizer. To
separate soluble and
insoluble proteins, the lysate was subjected to centrifugation at 10,000Xg for
20 minutes at
4°C. The insoluble fraction (pellet) was extracted with PBS containing
1M L-arginine. The
remaining insoluble material was then removed by centrifugation and the
soluble fraction of
the arginine extract was filtered through 0.2 micron low-protein binding
membrane and
analyzed by SDS PAGE. The result is shown in FIG. 7, which indicates that the
product is a
polypeptide with an apparent molecular weight of approximately 20 kDa (see
arrow). N-
terminal sequencing of the expressed polypeptide provides the sequence
AQLAHLHGILRRRQL which is 100% identical to residues 54-64 of FGF-CX (Table 1,
SEQ
ID N0:2).
119
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
Example 11. Stimulation of Bromodeoxyuridine Incorporation into NIH 3T3 Cells
in
Response to a Truncated Form of FGF-CX.
A vector expressing residues 24-211 of FGF-CX, referred to as (dl-23)FGF-CX,
was
prepaxed. See Table 1 and SEQ ID N0:2. The incorporation of BrdU by NIH 3T3
cells
treated with conditioned medium obtained using the vector incorporating this
truncated form
was compared to the incorporation in response to treatment with conditioned
medium using a
vector encoding full length FGF-CX. This experiment was carried out as
described in
Example 8.
The results are shown in FIG. 8. It is seen that (dl-23)FGF-CX retains high
activity at
the lowest concentration tested, 10 ng/mL. At this concentration, the activity
of full length
FGF-CX has fallen considerably, approaching the level of the control. It is
estimated that
(dl-23)FGF-CX may be at least 5-fold more active than full length FGF-CX.
Example 12. Molecular cloning of a mature FCTR1 form (30664188Øm99)
polypeptide
from cline 30664188Ø99
A mature form of clone 30664188Ø99, coding for residues 24 to 370 of the
amino
acid sequence of Table 2 (SEQ ID N0:4) was cloned. This fragment was
designated
30664188Øm99 and corresponds to the polypeptide sequence remaining after a
signal peptide
predicted to be cleaved between residues 23 and 24 has been removed. The
following
oligonucleotide primers were designed to PCR amplify the predicted mature form
of
30664188Ø99: 30664188 Eco Forward - CTCGTC GAATTC ACC CCG CAG AGC GCA
TCC ATC AAA GC (SEQ 117 N0:21), and 3066418 Xho Reverse - CTCGTC CTC GAG
TCG AGG TGG TCT TGA GCT GCA GAT ACA (SEQ ID N0:22).
The forward primer included an in frame EcoRI restriction site, and the
reverse primer
included an XhoI restriction site. The EcoRI/XhoI fragment is compatible with
the pET28a
E.coli expression vector and with the pMelVSHis baculovirus expression vector.
PCR reactions were set up using 5 ng human spleen and fetal lung cDNA
templates.
The reaction mixtures contained 1 microM of each of the 30664188 Eco Forward
and
30664188 Xho Reverse primers, 5 micromoles dNTP (Clontech Laboratories, Palo
Alto CA)
and 1 microliter of SOxAdvantage-HF 2 polymerase (Clontech Laboratories, Palo
Alto CA) in
. 50 microliter volume. The following reaction conditions were used:
a) 96°C 3 minutes
b) 96°C 30 seconds denaturation
120
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
c) 70°C 30 seconds, primer annealing. This temperature was gradually
decreased by 1°C per cycle
d) 72°C 1 minute extension.
Repeat steps b-d 10 times
e) 96°C 30 seconds denaturation
f) 60°C 30 seconds annealing
g) 72°C 1 minute extension
Repeat steps e-g 25 times
h) 72°C 5 minutes final extension
The amplified product expected to have 1041 by was detected by agarose gel
electrophoresis in both samples. The fragments were purified from agarose gel
and ligated to
pCR2.1 vector (Invitrogen, Carlsbad, CA). The cloned inserts were sequenced
using M13
Forward, M13 Reverse and the following gene specific primers:
3066418 S 1: GGA CGA TGG TGT GGA CAC AAG (SEQ ID N0:23),
~ 3066418 S2: CTT GTG TCC ACA CCA TCG TCC (SEQ ID N0:24),
3066418 S3: TAT CGA GGC AGG TCA TAC CAT (SEQ ID N0:25) and
3066418 S4: ATG GTA TGA CCT GCC TCG ATA (SEQ ID NO:26).
The cloned inserts were verified as an open reading frame coding for the
predicted
mature form of 30664188Ø99. The construct derived from fetal lung, called
30664188-
5311 a, was used for further subcloning into expression vectors (see below).
The nucleotide
sequence of 30664188-S1 la within the restriction sites was found to be 100%
identical to the
corresponding fragment in the ORF of 30664188Ø99 (Table 2; SEQ ID N0:4).
Example 13. Expression of 30664188.m99 polypeptide in E. coli
The vector pRSETA (InVitrogen Inc., Carlsbad, CA) was digested with XhoI and
NcoI
restriction enzymes. Oligonucleotide linkers CATGGTCAGCCTAC (SEQ ID N0:27);
and
TCGAGTAGGCTGAC (SEQ ID N0:28) were annealed at 37 degrees Celsius and ligated
into
the XhoI-NcoI treated pRSETA. The resulting vector was confirmed by
restriction analysis
and sequencing and was named pETMY. The BamHI-XhoI fragment containing the
30664188
sequence ( Example 12) was ligated into BamHI-XhoI digested pETMY. The
resulting
expression vector was named pETMY-30664188. In this vector, 30664188 is fused
to the T7
epitope and a 6xHis tag at its N-terminus The plasmid pETMY-30664188 was then
transfected into the E. coli expression host BL21 (DE3, pLys) (Novagen,
Madison, WI) and
121
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
expression of the protein was induced according to the manufacturer's
instructions. After
induction, the E. coli cells were harvested, and proteins were analyzed by
Western blotting
using anti-His6Gly antibody (Invitrogen, Carlsbad, CA). FIG. 9 shows that the
resulting
polypeptide, termed 30664188.m99 herein, was expressed as a protein of
apparent molecular
weight 40 kDa. This approximates the molecular weight expected for the
30664188.m99
sequence.
Example 14. Expression of 30664188.m99 polypeptide in human embryonic kidney
293
cells.
The EcoRI-XhoI fragment containing the 30664188.m99 sequence was isolated from
30664188-5311 a (Example 12) and subcloned into the vector pE28a (Novagen,
Madison, WI)
to give the plasmid pET28a-30664188. Subsequently, pET28a-30664188 was
partially
digested with BamHI restriction enzyme, and then completely digested with
XhoI. A
fragment of 1.1 kb was isolated and ligated into BamHI-XhoI digested pCEP4/Sec
(Example
3) to generate expression vector pCEP4/Sec-30664188.m99. The pCEP4/Sec-
30664188.m99.
vector was transfected into human embryonic kidney 293 cells (ATCC No. CRL-
1573,
Manassas, VA) using the LipofectaminePlus reagent following the manufacturer's
instructions
(Gibco/BRL/Life Technologies, Rockville, MD). The cell pellet and supernatant
were
harvested 72 hours after transfection and examined for expression of the
30664188.m99
protein by Western blotting of an SDS-PAGE run under reducing conditions using
an anti-VS
antibody. FIG. 10 shows that 30664188.m99 is expressed as three discrete
protein bands of
apparent molecular weight 50, 60, and 98 kDa secreted by 293 cells. The 50 kDa
band
migrated at a sized expected for a monomer glycosylated form of 30664188.m99,
and the 98
kDa band migrated at a size consistent with a dimer of the monomer form.
Example 15. Expression and Purification of 30664188.m99 protein
HEIR 293 cells were grown in Dulbecco's modified eagle's medium (DMEM)/10%
fetal bovine serum medium to 90 % confluence. The cells were transfected with
pCEP4sec or
pCEP4sec/30664188.m99 using Lipofectamine 2000 according to the manufacturer's
specifications (GibcoBRLlLife Technologies, Rockville, MD). Transfected cells
were
incubated for 2 days with DMEM and conditioned medium was prepared by
collection of cell
supernatants. The conditioned medium was enriched by Talon metal affinity
chromatography
(Clontech, Palo Alto, CA). Briefly, 7 ml of conditioned medium was incubated
with 1 ml of
Talon metal affinity resin in spin columns. The spin columns were washed twice
with one ml
122
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
of PBS. The columns were then eluted twice with 0.65 ml of PBS/O.SM imidazole
pH 8.0 and
the eluates pooled. Imidazole was removed by buffer exchange dialysis into PBS
using
Microcon centrifugal filter devices (Millipore Corp., Bedford, MA). The
enriched gene
products were stored at 4°C.
The purified protein obtained was subjected to SDS-PAGE under reducing
conditions
and probed with an anti-VS antibody, which was detected with an enzyme label.
The results
of two separate transfection and purification runs are shown in the gels. They
show that the
product is a mixture of VS-containing polypeptides. The largest has an
apparent molecular
weight of about 50 kDa (FIG. 1 l, Panel B). The program ProSite predicts one N-
glycosylation site in the mature protein. Glycosylation may explain the
apparent molecular
weight found. Thus the SOkDa band is consistent with the length expected for
full length gene
product. Other bands, preponderantly having apparent molecular weights of
about 20-25 kDa
also arise. These are presumed to be the result of proteolysis occurring
either intracellularly
within the 293 cells or extracellularly after secretion from them. In another
run (not shown)
the broad band extending from about 6 kDa to about 14 kDa is reolved into two
bands of about
7-8 kDa and about 10 kDa.
Example 16. The clone 30664188Øm99 protein induces cellular DNA synthesis
Human CCD-1070 fibroblast cells (ATCC No. CRL-2091, Manassas, VA) or marine
NIH 3T3 (ATCC No. CRL-1658, Manassas, VA) fibroblast cells were cultured in
DMEM
supplemented with 10% fetal bovine serum or 10% calf serum respectively.
Fibroblasts were
grown to confluence at 37°C in 10% COa/air. Cells were then starved in
DMEM for 24 h.
pCEP4/Sec ( Example 3) or pCEP4/Sec/30664188.m99 ( Example 14) enriched
conditioned
medium was added (10 microL/100 microL of culture ) for 18 h. BrdU (10 uM) was
then
added and incubated with the cells for 5 h. BrdU incorporation was assayed by
colorimetric
immunoassay according to the manufacturer's specifications (Boehringer
Mannheim,
Indianapolis, IN).
FIG. 12 demonstrates that 30664188.m99 induced an approximate four- to five-
fold
increase in BrdU incorporation in either cell type compared to cells treated
with control
conditioned medium or untreated cells. The proliferative increase observed was
similar to the
increase in BrdU incorporation induced by platelet derived FCTRX (PDGF), basic
fibroblast
growth factor (bFGF), or serum treatment. Additionally, 30664188.m99 partially
purified
conditioned medium did not induce BrdU incorporation in human MG-63 epithelial
cells or
123
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
CCD1106 keratinocytes (data not shown). These results suggest that 30664188
selectively
induces DNA synthesis in human and mouse fibroblasts, but not in epithelial
cell lines.
In separate experiments, CCD-1070 cells and MG-63 osteosarcoma cells (ATCC
Cat.
No. CRL-1427) treated with pCEP4/Sec/30664188 each incorporated BrdU in a dose-
s dependent fashion, with 1 ug/mL providing the full effect (approximately 2.5-
to 3-fold
increase over control), 100 ng/mL providing slightly less than one-half the
effect, and 10 and 1
ng/mL providing approximately control levels of incorporation. Furthermore,
the dose
response of NIH 3T3 cells shows that a 50% response occurs between doses of 10
and 50
ng/mL of pCEP4/Sec/30664188.M99 (FIG. 13).
In additional dose titration experiments using both NIH/3T3 cells and CCD1070
cells,
the half maximal effect occurred at or below 25 ng/mL.
Example 17. Induction of Proliferation of NIH 3T3 cells by 30664188.m99
Murine NIH 3T3 fibroblasts were plated at 40% confluency and cultured in DMEM
supplemented with 10% fetal bovine serum or 10% calf serum for 24 hrs. The
culture medium
was removed and replaced with an equivalent volume of pCEP4/Sec (Example 3) or
pCEP4/Sec/30664188.m99.m99. (Example 14) conditioned medium. After 48 h, cells
were
photographed with a Zeiss Axiovert 100. Cell numbers were determined by
trypsinization
followed by counting using a Coulter Zl Particle Counter.
Treatment of N1H 3T3 fibroblasts with conditioned medium from 30664188
transfected HEI~293 kidney epithelial cells resulted in a 6 to 8 fold increase
in cell number
over a two day period (FIG. 14). Cells treated with control conditioned medium
from
HEK293 cells transfected with the pCEP4/Sec vector alone demonstrated little
or no growth
(FIG. 14).
To determine whether 30664188.m99 conditioned medium was able to induce
phenotypic changes characteristic of cellular transformation, cells treated
with either
30664188 conditioned medium or mock conditioned medium were examined by light
microscopy. FIG. 15 shows that NIH 3T3 cells treated with 30664188.m99, but
not control
treated NIH 3T3 cells, showed a marked increase in cell number, as well as
refractile
properties. Loss of contact inhibition of growth was evident. The cobblestone
appearance
characteristic of confluent NIH 3T3 cells was lost and density independent
growth was
evident. The latter was also suggested by the more rounded appearance of the
NIH 3T3 cells
due to subtle retraction. Transfection ofpCEP4/Sec/30664188.m99.m99 also
showed nearly
124
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
identical potency in transformation potential 2 to 5 days in culture. After 7
to 10 days in
culture, however, the morphologically transformed phenotype appeared to
revert.
Example 1~8. Induction of proliferation of human primary osteoblast cells by
the
30664188 protein
In an experiment similar to that described in Example 17, human primary
osteoblast
cells (NHost; Clonetics)also underwent a dose-dependent increase in cell
number by 3- to 4-
fold (FIG. 16). The dose required to elicit a 50% response in FIG. 16 is below
100 ng/mL of
pCEP4/Sec130664188.m99. In addition, Jurkat cells contacted with partially
purified
conditioned medium containing the 30664188 gene product exhibited a doubling
of BrdU
uptake compared to the medium from mock transfection, whereas the same cells
contacted
with 13 other CuraGen Corporation gene products thought to have growth
promoting activity
elicited no effect.
In summary, the observations that the 30664188 protein induces DNA synthesis
(Example 16), cell growth (Examples 16 and 17), and morphological
transformation (Example
17) indicate that the 30664188 protein possesses growth promoting and
stimulating properties.
Example 19. Purification of Intact and Cleaved Products of the 30664188.m99
Protein.
It was observed that in certain experiments treatment with the vector
pCEP4/Sec/
30664188.m99 did not result in DNA synthesis or cell proliferation. In
additional
experiments, medium conditioned with 30664188.m99 was obtained from HEK 293
cells
grown in the presence of serum (Examples 15-17). The 30664188.m99 gene product
was
purified by cation exchange chromatography, followed by nickel affinity
chromatography.
The protein product was run under nonreducing and reducing conditions on SDS-
PAGE, and
developed by Coomassie stain. The results are shown in FIG. 17, Panels A and
B. In the
presence of serum, the 30664188.m99 gene product appeared as a protein of
about 35 kDa
under nonreducing conditions (FIG. 17 Panel B). However, this polypeptide
appears as three
degraded bands when run under reducing conditions. The apparent molecular
weights of the
two bands were 22-25 kDa (band I), about 16 kDa (band II) and about 5-6 kDa
(band III). N-
terminal amino acid analysis of these fragments indicates that bands I and II
both appear to
result from cleavage between residues 247 and 248, such that the peptide
product begins at
residue 248 of the 30664188Ø99 (Table 2, SEQ ID N0:4) amino acid sequence,
and that band
III begins at residue 339. These results are consistent with cleavage of the
polypeptide
corresponding to band I to provide the fragments of bands II and III. It is
possible that the 35
125
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
kDa band observed under nonreducing conditions is a dimer composed of band I,
and/or the
bonded polypeptide composed of bands II and III, observed under reducing
conditions.
Aminoa terminal analysis indicates that the gene product from
pCEP4sec/30664188.m99-transfected 293 cells grown in the presence of serum,
isolated
according to the procedure described above, is a carboxyl-terminal fragment of
the full length
protein. The 35 kDa band found under nonreducing conditions is termed p35
below. These
results are expanded in Example 21.
When 293 cells were cultured in the absence of serum, and the same isolation
and
detection procedure described in the preceding paragraph is followed, a
different gene product
is observed. Under nonreducing conditions a band was found at about 85 kDa
(FIG. 17 Panel
A). This protein is termed p85 below. The corresponding gene product observed
under
reducing conditions a major band is found at about 53-54 kDa. N-terminal amino
acid
analysis of this gene product provides the amino acids at the multiple cloning
site used in
pCEP4sec/ 30664188.m99 (Example 14). The residues corresponding to the Ig
kappa leader
sequence, cloned upstream from the multiple cloning site, are absent. These
results indicate
that the gene product obtained in the absence of serum represents the full
amino acid sequence
encoded in pCEP4sec/30664188.m99. The p85 polypeptide is thought to be a dimer
of the 50
kDa species observed on reducing SDS-PAGE. These results are expanded in
Example 21.
Example 20. Activity of Intact and Cleaved Fragments of the 30664188.m99
Protein
Purified p85 and p35 FCTRX proteins were separately applied to NIH 3T3 cells
in a
range of concentrations. Incorporation of BrdU was evaluated as described in
Example 8.
The results are shown in FIG. 18. It is seen that p85 has growth-promoting
activity that does
not differ from control levels except at the highest concentration used (bars
4-10). p35, on the
other hand, was at least as active, if not more so, than unfractionated
pCEP4/Sec/30664188.m99 conditioned medium (bars 11-17). The concentration of
p35
giving 50% of the maximum DNA synthesis falls between 20 and 50 ng/mL.
These results suggest that the p35 fragment derived from intact 30664188.m99
has
growth-promoting activity but that the intact dimeric form of the .m99
protein, p85, does not.
Example 21. Purification of recombinant PDGF DD.
The gene product of PDGFD was expressed in HEK293 cells grown on porous
microcarriers (Cultisphere-GL, Hyclone; Logan, UT) in 1 L spinner flasks. As
noted in
Examples 2 and 4, the recombinant PDGF D gene includes a 6xHis fusion at the
3' end. Cells
126
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
were grown in DMEMlFI2 media containing 1% penicillin/ streptomycin in the
presence or
absence of 5% fetal bovine serum (FBS). The conditioned medium was harvested
by
centrifugation (4000 x g for 15 minutes at 4~C) and loaded onto a POROS HS50
column (PE
Biosystems; Foster City, CA), pre-equilibrated with 20 mM Tris-acetate (pH
7.0). After
washing with the equilibration buffer, bound proteins were eluted with a NaCI
step gradient
(0.25 M, 0.5 M, 1.0 M and 2.0 M). Fractions containing PDGF DD p35 (1.0 M NaCI
step
elution) or p85 (0.5 M NaCI step elution) (see Example 19) were pooled and
diluted with an
equal volume of phosphate-buffered saline (PBS), pH 8.0 containing 0.5 M NaCI,
then loaded
onto a POROS MC20 column pre-charged with nickel sulfate (PE Biosystems).
After washing
with PBS/0.5 M NaCI, bound proteins were eluted with a linear gradient of
imidazole (0 - 0.5
M). Fractions containing PDGF DD (homodimers of PDGFD) (100 - 150 mM
imidazole)
were pooled and dialyzed twice against 1000 volumes of 20 mM Tris-HCI, pH 7.5,
50 mM
NaCI. The protein purity was estimated to be > 95% by sodium dodecyl sulfate-
polyacrylamide gel electrophoresis (SDS-PAGE; 4-20% Tris-glycine gradient gel;
Invitrogen,
Carlsbad, CA) analysis (See, for example, the results in Example 19, including
FIG. 17).
Biochemical Properties of PDGF D. To examine the biochemical properties of the
gene product of PDGF D, the cDNA encoding PDGF D protein was subcloned into a
mammalian expression vector, pCEP4/Sec-30664188m99 (Example 14). This
construct
incorporates an epitope tag (VS) and a polyhistidine tag into the COOH
terminus of the protein
to aid in its identification and purification (expression vector pCEP4/Sec-
30664188m99;
Example 14).
Following transfection into 293 HEIR cells and growth in serum-free culture, a
secreted
polypeptide with an apparent molecular weight of ~49 kDa (p49 species) was
identified by
Western blot analysis under reducing conditions (FIG. 19 Panel A, lane 2). The
fact that the
apparent molecular weight of p49 is greater than the expected value of ~43-kDa
may be
attributable to glycosylation. In contrast, a 20-kD protein was secreted when
PDGF D-
transfected cells were grown in the presence of FBS (FIG. 19 Panel A, lane 3).
Conditioned
media from mock transfected cells did not react with the anti-VS antibody
(FIG. 19 Panel A,
lane 1).
In addition, PDGF D was expressed in the presence or absence of FBS and
purified to
>95% homogeneity. As shown in FIG. 19 Panel B (lane 2), expression of PDGF D
under
serum-free conditions resulted in the detection of the expected 49-kD gene
product under
127
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
reducing conditions, when the gel was stained using Coomassie Blue. A
polypeptide species
with an apparent molecular weight of about 84 kDa, corresponding to a dimeric
p85 species of
p49, was seen under non-reducing conditions (FIG. 19 Panel B, lane 1). When
PDGF DD was
purified from serum-containing conditioned medium and run under nonreducing
conditions, a
species with an apparent molecular weight of about 35 kDa (p35) was observed
(FIG. 19 Panel
B, lane 3). Under reducing conditions, p35 was found to yield three bands when
visualized
with Coomassie Blue, which migrate with apparent molecular weights of
approximately 20,
14, and 6 kDa (FIG. 19 Panel B, lane 4).
Amino terminal sequence analysis of p35 demonstrated proteolytic cleavage
after
Arg247 (R247) or Arg249 (R249) (FIG. 20). As indicated in Panel A of FIG. 20,
two peptides
were found, one beginning with GlyArg (GRSYHDR ...; shown with the GR residues
underlined), and the second beginning with the third residue, Ser (SYHDR ...).
The ratio of
these peptides was found to be SYHDR:GRSYHDR = 4:1. The additional sequencing
results
in FIG. 20 (Panels B and C) indicate that further processing produces the
remaining
polypeptides seen with Coomassie blue staining but not with anti-VS Westerns,
namely the 16
kDa and 6 kDa species shown. These are joined together to provide p35.
The results presented in this Example indicate that the PDGF D gene products
are
dimers in both the holoprotein form (p85) and the C-terminal fragment (p35).
The p85 form
appears to be processed in the presence of FBS to provide the p35 form. These
dimeric forms
are designated PDGF DD.
Example 22. Processing of the 30664188 Gene Product in the Presence of Fetal
Bovine
Serum and Calf Serum.
The 30664188 gene product was incubated in the presence of increasing
concentrations
of calf serum (FIG. 21, Panel A) or fetal bovine serum (Panel B). The results
demonstrate that
only fetal bovine serum (Panel B) but not calf serum (Panel A) processes the
p85 form of the
30664188 gene product to provide p35.
Example 23. Stimulation of Growth of Pulmonary Artery Smooth Muscle Cells by
Growth Factors.
This EXAMPLE demonstrates the ability of PDGF DD to stimulate growth of
pulmonary artery smooth muscle cells.
The p35 dimer of 30664188, PDGF AA or PDGF BB were added at various
concentrations to pulmonary artery smooth muscle cells (Clonetics) after being
cultured in
128
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
6-well plates to approximately 35% confluence, washed with DMEM, and starved
overnight..
After 18 hrs, BrdU was added, and 5 hrs later the cells were analyzed for BrdU
incorporation
using a BrdU-directed ELISA.
The results are shown in FIG. 22. It is seen that the maximal effect achieved
by
treatment with p35 dimer exceeds that given by both PDGF AA and PDGF BB. It is
seen that
the effects of p35 dimer and PDGF BB resemble each other more closely than the
effect
obtained with PDGF AA. Of all three growth factors tested, p35 dimer induced
the greatest
growth in smooth muscle cells, as determined by BrdU incorporation, with 50%
maximal
effect obtained at less than 12.5 ng/mL.
Example 24. Proliferation of Pulmonary Artery Smooth Muscle Cells in Response
to
Various Growth-Promoting Treatments.
This EXAMPLE demonstrates the ability of PDGF DD to stimulate proliferation of
pulmonary artery smooth' muscle cells.
Pulmonary artery smooth muscle cells were cultured in 6-well plates to
approximately
35% confluence, washed with DMEM, and starved overnight. Cells were then fed
with
DMEM supplemented with recombinant 30664188, a known PDGF (200 ng/ml) or 10%
FBS
for three days. Culture fluids were removed and replaced with same media for
an additional 2-
3 days. To quantitate the smooth muscle cell growth assay, cells were
trypsinized and counted
with a Beckman Coulter Z1 series counter (Beckman Coulter, Fullerton, CA).
The results are shown in FIG. 23. It is seen that PDGF produces a modest
increase in
cell number, whereas treatment with 30664188 provides an effect, compared with
control, that
is almost double that observed with PDGF. A positive control using treatment
with 10% FBS
gave a very pronounced effect. Treatment of smooth muscle cells with 30664188
and PDGF
BB led to elongated bipolar spindle shaped phenotype in contrast to the flat
club shaped
phenotype observed with serum.
30664188 is an effective stimulant of pulmonary artery smooth muscle cell
proliferation, and suggests that 30664188 has a therapeutic use in wound
healing, tissue repair
and cartilage repair. Furthermore, antibodies directed against 30664188 may
have therapeutic
use in inhibiting or preventing restenosis of patent vasculature.
129
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
Example 25. Proliferation of Saphenous Vein Cells in Response to Various
Growth-
Promoting Treatments.
This EXAMPLE illustrates the ability of PDGF DD to stimulate proliferation in
saphenous vein cells. Saphenous vein cells (Clonetics) were treated and
analyzed as described
in Example 24. The results (not shown) indicate that PDGF produces a slightly
lower increase
in cell number than does treatment with 30664188, which provides proliferation
to almost 5
times the cell number seen with the control. A positive control using
treatment with 10% FBS
gave a very pronounced effect. 30664188 is an effective stimulant of saphenous
vein cell
proliferation, and suggests that 30664188 and 30664188 antibodies has a
therapeutic use in
wound healing, tissue repair and cartilage repair., Furthermore, antibodies
directed against
30664188 may have therapeutic use in inhibiting or preventing restenosis of
patent
vasculature.
Example 26. Mouse model for inflammatory bowel disease
A widely recognized animal model for inflammatory bowel disease is the mouse
dosed
with the sodium form of dextran sulfate.
Materials and Methods
Colitis Study Design. Normal female Balb/c mice (Harlan Labs), 6 - 8 weeks old
weighing approximately 20 g, were housed 3-5 animals per cage in polycarbonate
cages with
filter tops and given food (Harlan Teklad mouse chow) and tap water ad
libitum. Mice were
acclimated for 6 days (Day -7 through Day -1) and then given water orally (po)
ad libitum
containing 5% dextran sulfate sodium (DSS) or control water ad libitum for 7
days (Day 0
through Day 6). DSS (Spectrum Chemicals, Gardena CA) was made as a 5% solution
in tap
water; DSS was made every other day and stored at 4°C. Mice were
divided into 4 treatment
groups (Table 14). On Day 0, daily intraperitoneal (ip) treatments with
vehicle (1M L-
arginine in phosphate buffered saline) or protein (CG53135 or CG52053, 5
mg/kg) were
initiated and continued each morning through Day 6. On Day 7, mice were
sacrificed by
exposure to C02.
Table 14: Treatment Groups
Group Na Treatment
1 5 Normal control: no DSS water + vehicle ip
2 10 Disease control: DSS water o + vehicle i
130
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
3 10 CG53135: DSS water po + 5 mg/kg CG 53135 ip
4 5 CG52053: DSS water po + 5 mg/kg CG52053 ip
a N =
number
of animals
per
group
Protein production. The cDNA for CG52053 was identified and cloned into the
pCEP4/Sec vector (Invitrogen, Carlsbad, CA) and transfected into human
embryonic kidney
' cells (HEIR 293). The transfected cells were selected using Hygromycin B and
then scaled up
in 10 L bioreactors using DMEM medium containing 10% FBS. The CG52053 protein
was
purified from the culture medium by ion-exchange and metal affinity
chromatography. The
final purified CG52053 was diluted in 20 mM Tris HCl (pH 7.4) and 50 mM NaCI.
The cDNA for CG53135 was identified and cloned into the pRSET vector
(Invitrogen)
to provide the vector pETMY-FGF-CX described in Example 5. The gene product of
this
construct provides a polypeptide incorporating (His)6-(enterokinase cleavage
site)-
(multicloning site) at the N-terminal end of the polypeptide; in addition, in
this construct, the
FGF-CX sequence begins with the Ala at position 2 of Table 1 (SEQ ID N0:2).
This vector
was transformed into Escherichia coli. The E. coli cells were grown up to 10 L
scale and
infected with CE6 phage to produce the recombinant CG53135. The recombinant
protein was
purified by disrupting the E. coli cells in a microfluidizer and extraction
with 1M L-arginine
solution, followed by multiple metal affinity chromatography steps. The final
purified protein
was dialyzed into phosphate buffered saline containing 1M L-arginine. Protein
purity was
determined by SDS-PAGE analysis and identities were confirmed by Western blot
analysis.
Activity of proteins was determined by BrdU incorporation assay (Roche
Molecular
Biochemicals) using a 5 hr incorporation time and NIH 3T3 cells.
Body weights were measured daily and at termination on day 7. Additional
parameters
measured at necropsy included colon length, colon weight and spleen weights.
Colon and
spleen were collected into formalin for histopathologic evaluation.
Colon content was scored at necropsy according to the following criteria:
0 = normal to semi-solid stool, no blood observed
1 = normal to semi-solid stool, blood tinged
2 = semi-solid to fluid stool with definite evidence of blood
3 = bloody fluid
131
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
Pathology Methods
Three sections approximately 1 cm apart from the distal end (area that is most
severely
affected in this model) and 3 sections approximately 1 cm apart from the
proximal end (less
severely affected area) were processed for paraffin embedding, sectioned and
stained with
hematoxylin and eosin for pathologic evaluation.
For each section, submucosal edema was quantitated by measuring the distance
from
the muscularis mucosa to the internal border of the outer muscle layer.
Inflammation (foamy
macrophage, lymphocyte and PMN infiltrate) was assigned severity scores
according to the
following:
Normal = 0
Minimal =1
Mild = 2
Moderate = 3
Marked = 4
S evere = 5
Splenic lymphoid atrophy was also scored by the above criteria.
The parameters reflecting epithelial cell loss/damage were scored individually
using a
area involved scoring method:
None = 0
1-10% of the mucosa affected ~ 1
11-25% of the mucosa affected = 2
26-50% of the mucosa affected = 3
51-75% of the mucosa affected = 4
76-100% of the mucosa affected = 5
Parameters that were scored using % involvement included:
Colon glandular epithelial loss-this includes crypt epithelial as well as
remaining gland
epithelial loss and would equate to crypt damage score.
Colon Erosion-this reflects loss of surface epithelium and generally was
associated
with mucosal hemorrhage (reflective of the bleeding seen clinically and at
necropsy).
For each.animal, 3 proximal (less severe lesions) and 3 distal (most severe
lesion) areas
were scored and the mean of the scores for each of the regions was determined.
Group means
and % inhibition from disease control were determined. By doing it this way
(rather than
132
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
summing the scores from various sections) one can look at the mean + SE for in
individual
parameter (represented by 3 sections) and equate it to a delineated severity.
As an example, if
the mean is 4 for gland epithelial loss one knows that 51-75% of the mucosa
was devoid of
epithelium.
The 3 important scored parameters (inflammation, glandular epithelial loss,
erosion)
were ultimately summed to arrive at a sum of histopathology score which
indicates the overall
damage and would have a maximum score of 15.
One final summation of proximal + distal summed scores was done to reflect the
overall total colonic severity score.
Statistics. The mean and standard error (SE) for each treatment group was
determined
for each parameter scored; the data were compared to the data for the disease
controls (Group
2) using a 2-tailed Student's t test with significance at p <_ 0.05.
Results
Live Phase, Necropsy and Organ Weight
All animals except DSS+vehicle control mouse 4 survived to study termination.
Mouse
4 was found dead the morning of necropsy on day 7.
DSS treatment-related changes in body weight were present by day 3 in all DSS
treated
mice. At study termination, DSS+vehicle controls had a 25% decrease in body
weight (FIGS.
24, 25 and 26). A significant beneficial effect on DSS induced weight loss was
seen in mice
given FGF-CX, referred to as AB020858 (FIGS. 25 and 26).
Clinical evidence of bloody diarrhea was evident in all DSS+vehicle animals
except
animal 1. At necropsy all DSS controls had blood or blood tinged fluid in the
colon. In
contrast, mice treated with AB020858 generally had semi-solid stool and little
evidence of
blood. Similar findings occurred in mice treated with 30664188.
Colon content scores reflecting colonic hemorrhage were dramatically decreased
(93%) in mice treated with AB020858 and (79%) in mice treated with 30664188
(FIG. 34).
Absolute spleen weights (FIGS. 27 and 28) were decreased approximately 30% in
mice
treated with vehicle. Treatment with AB020858 resulted in 55% reduction of the
DSS-induced
losses in spleen weights. Treatment with 30664188 reduced the splenic weight
losses by 62%.
Absolute colon weights (FIGS. 29 and 30) were decreased approximately 26% in
mice
treated with vehicle. Treatment with AB020858 resulted in slight but not
significant reduction
133
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
of the DSS-induced changes in colon weights. Treatment with 30664188 reversed
the colon
weight decreases (FIGS. 30 and 31).
Absolute colon lengths (FIGS. 32 and 33) were decreased approximately 40% in
mice
treated with DSS + vehicle. Treatment with AB020858 resulted in significant
(40%) reduction
of the DSS-induced changes in colon length. Treatment with 30664188 reduced
the colon
length loss 36%.
l3istopathology Findings
Histopathology was conducted on the full length of the colon. Lesions were
much
greater in the distal vs. proximal colon, as expected. Quantitation of
efficacy of treatment is
based primarily on inhibition of pathological changes in this location.
Colonic edema in the
distal colon was inhibited 76% by treatment with AB020858 whereas treatment
with
30664188 did not inhibit the edema (FIG. 35).
Colonic inflammation in the distal colon was inhibited 55% by treatment with
AB020858 and 41% by treatment with 30664188 (FIG. 36).
Protection of colonic epithelium (both crypts and remainder of the gland), as
determined by the epithelial loss score, was 57% in mice given AB020858 and
41% in those
treated with 30664188 (FIG. 37). Further evidence of mucosal epithelial
protection in the
distal colon was evident on evaluation of degree of surface epithelial loss
leading to
erosion/ulceration. As shown by the colon erosion scores, AB020858 treatment
gave 84%
inhibition of the erosive lesions and 30664188 treatment resulted in 74%
inhibition (FIG. 38).
Summing the important histologic scores for inflammation, glandular epithelial
damage and erosion (FIG. 39), it is seen that an overall protective effect
results from the
treatment with AB020858, which provides 66% inhibition of the pathology.
Treatment with
30664188 resulted in 53% inhibition of the overall score. Slight but not
significant (33-37%)
inhibition of the total histologic scores was evident for proximal colon.
Results for the colon
overall are shown in FIG. 40.
Splenic weight decreases were largely a result of splenic lymphoid atrophy.
Treatment
with both proteins inhibited this parameter as well (FIG. 41).
Discussion and Conclusions
In this model of inflammatory bowel disease, in which mice are exposed to 5%
DSS
for 7 days, most animals develop marked to severe distal colonic
inflammation/edema in
134
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
association with crypt and colonic glandular epithelial loss and
erosion/ulceration leading to
marked hemorrhage. Lesions in the proximal colon are much milder but similar
in character.
Cotemporaneous treatment with AB020858 (5 mg/kg, qd, d0-6) resulted in
clinical
benefit (reduced body weight loss) as well as protection against development
of hemorrhagic
diarrhea, a common feature of this model. Stressed unhealthy DSS treated mice
have splenic
lymphoid atrophy. This parameter (reflected by weight changes and histologic
alterations)
was also benefited by treatment with AB020858.
Colonic shortening (due to inflammation and mucosal tissue loss) was inhibited
40%
by treatment with AB020858. This gross observation was strongly supported by
the histologic
observations of mucosal epithelial preservation in the crypts, colonic glands
and surface
epithelium (see FIGS. 42 and 43). In FIG. 42, viewed at 400x in the original
images, the
normal colonic mucosa has uniform glandular architecture and no submucosal
edema (upper
left). The disease control has no mucosal glands and surface epithelium,
exposing blood
vessels of the severely inflamed lamina propria to the lumen and resulting in
hemorrhage
(upper right). Itreatment with CG53135 preserves mucosal integrity and results
in decreased
epithelial loss and reduced inflammation in the lamina propria (lower left).
Treatment with
CG52053 decreases epithelial loss and mucosal inflammation, although to a
lesser degree than
treatment with CG53135 (lower right). In FIG. 43, viewed at 50x in the
original images, the
normal control shows normal colonic mucosa with uniform glandular architecture
and no
submucosal edema (upper left). DSS-induced colitis results in loss of
glandular architecture
and edema that separates the mucosa from the outer muscle layers (upper
right). Treatment
with CG53135 inhibits the severe mucosal changes and submucosal edema induced
by DSS
(lower left). Treatment with CG52053 results in some inhibition of
inflammation and loss of
glandular architecture but no inhibition of submucosal edema (lower right).
This histologic
evidence of mucosal protection corroborates the dramatic necropsy observation
that very little
hemorrhagic diarrhea occurs.
The results of the experiments reported in this Example indicate that, in mice
in which
inflammatory bowel disease is induced by oral administration of DSS for 7
days, simultaneous
treatment with the growth factors employed here during the course of exposure
to DSS led to
significant therapeutic benefits compared to untreated DSS controls.
135
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
Example 27. Dose Responsive Effects of AB020858 Female Swiss Webster Mice with
Dextran Sulfate-induced Colitis
The experiments reported in this Example report the results of dose titration
experiments in an animal model of inflammatory bowel disease using a different
strain of
mouse than that used in Example 26.
Introduction and General Methods
Colitis Study Design. Normal female Swiss-Webster mice (Harlan Labs), 6 - 8
weeks
old weighing approximately 20 g, were acclimated for 4 days (Day -4 through
Day -1) and
then given water orally (po) ad libitum containing 5% dextran sulfate sodium
(DSS) or control
water ad libitum for 7 days (Day 0 through Day 6). DSS (Spectrum Chemicals,
Gardena CA)
was made as a 5% solution in tap water; DSS was made every other day and
stored at 4°C.
Mice were divided into 8 treatment groups including QD doses of 0.3, 1, 3 and
10 mg/kg, and
a BID dose regimen of 5 mg/kg per dose (Table 15). On Day 0, daily
intraperitoneal (ip)
treatments with vehicle (1M L-arginine in phosphate buffered saline) or
CG53135 protein in
vehicle were initiated and continued through Day 6. On Day 7, mice were
sacrificed with
COa.
Table 15: Treatment Groups
TreatmentNormal Disease Disease
b CG53135CG53135CG53135CG53135b CG53135
l
tr
Control
Group Controlso D QD QD QD BID
QD Q
BID
Group 1 2 3 4 5 6 7 8
#
CG 53135
0 0 10 3 1 0.3 0 5
~mg~b)
Number
of
Test 4 10 10 10 10 10 10 10
Animals
a normal
control
= vehicle
only;
b disease
control
= 5%
DSS
+ vehicle
Protein production. The CG53135 protein was produced in E. coli as described
in
Example 26. The recombinant protein was purified by disrupting the E. coli
cells
(resuspended in a 1 M L-arginine solution) in a microfluidizer, followed by
multiple metal
affinity chromatography steps. The final purified protein was dialyzed into
phosphate
buffered saline containing 1M L-arginine.
Colon content was scored as described in Example 1.
136
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
Pathology Methods
Three sections equidistant apart from the distal one third of the colon (area
that is most
severely affected in this model) were processed for paraffin embedding,
sectioned and stained
with hematoxylin and eosin for pathologic evaluation.
For each section, scoring was done as described in Example 26.
Splenic lymphoid atrophy was also scored by the above criteria.
Epithelial cell loss/damage was scored as described in Example 26.
Parameters that were scored using % involvement included:
Colon glandular epithelial loss-this includes crypt epithelial as well as
remaining gland
epithelial loss and would equate to crypt damage score.
Colon Erosion-this reflects loss of surface epithelium and generally was
associated
with mucosal hemorrhage (reflective of the bleeding seen clinically and at
necropsy).
For each animal, 3 distal (most severe lesion) areas were scored. Scoring and
analysis
was done as described in Example 26.
Live Phase, Necropsy and Organ Weight Results
Four animals died during the course of the study (#10 in vehicle control group
2 on day
7, #3 in group 6, 0.3 mg/kg on day 6, #5 in group 8 vehicle control BID on day
7, and #6 in
group 7 5 mg/kg BID on day 6).
DSS treatment-related changes in body weight were obvious by day 5 in all DSS
treated mice and ultimately were most severe in animals treated with vehicle
(FIG. 44). At
study termination, DSS+vehicle controls had a 28% decrease in body weight. A
significant
beneficial effect on DSS induced weight loss was seen in mice given AB020858
QD at all
doses (FIG. 45).
Clinical evidence of bloody diarrhea was evident in all DSS+vehicle animals.
At
necropsy all DSS controls had blood or blood tinged fluid in the colon. In
contrast, mice
treated QD with 10 mglkg AB020858 generally had semi-solid stool and less
blood (except
animals #5). Clinical benefit was also evident but less impressive in those
given doses of 3 or
1 mg/kg QD and absent in those treated with 0.3 mglkg (FIG. 46). Mice treated
BID with 5
mglkg had the most impressive clinical benefit (68% inhibition) and clinically
these mice had
the best overall improvement.
Absolute colon lengths (FIGS. 47 and 48) were decreased 41% in mice treated
with
vehicle. Treatment with AB020858 QD at 10 mg/kg resulted in significant (21%)
inhibition of
137
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
the DSS-induced changes in colon length. Treatment with AB020858 BID at 5
mg/kg reduced
the colon length decrease 36%.
Absolute colon weights (FIGS. 49 and 50) were decreased approximately 26% in
mice
treated with DSS in vehicle. Treatment with AB020858 at 10 mglkg QD or 5 mg/kg
BID
resulted in significant reduction of the DSS-induced changes in colon weights.
Absolute spleen weights (FIG. 51) were increased approximately 40% in mice
treated
with DSS+vehicle (due to extreme extramedullary hematopoiesis). Spleen weights
were
significantly greater in all DSS treated animals vs. normal.
Histopathology Findings
Significant reduction of colonic inflammation, gland loss, erosion and total
histopathology scores occurred in mice treated with AB020858 QD (10 mg/kg) and
BID (5
mg/kg) and was of approximately equal magnitude (FIGS. 52, 53, 54 and 55).
Splenic lymphoid atrophy (an indication of stress) was inhibited in these same
animals
47% and 46% respectively (FIG. 56). Inhibition of induction of splenic
extramedullary
hematopoiesis was greater in mice treated BID vs. QD and occurred in all
treatment groups
(FIG. 57).
Discussion and Conclusions
The experiments reported in this Example provide dose-response information for
the
administration of AB020258, using a different strain of mouse than those in
Example 26
(which used Balb/c mice). The results indicate that simultaneous
administration of AB020258
is effective in inhibiting the appearance of markers of DSS-induced
inflammatory bowel
disease, especially with the highest doses used.
Example 28. Administering CG53135 Subcutaneously
An additional experiment was carned out in which mice were also treated
subcutaneously with CG53135. Together with the results in Examples 26 and 27,
these
studies demonstrate that prophylactic administration of CG53135 at doses of 5
or 10 mg/kg ip
and 5 or 1 mg/kg sc significantly reduce the extent and severity of mucosal
damage induced
by dextran sulfate sodium in a marine model of colitis.
Example 29. Effects of Administering CG53135 to Indomethacin-treated rats
Treatment of rats with indomethacin results in gross and histopathologic
intestinal
alterations that are similar to those occurring in Crohn's Disease. The
experiments provided in
138
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
this Example report on the efficacy of CG53135 in treating the rat model of
indomethacin-
induced intestinal injury. The efficacy of this protein in an alternate model
of intestinal injury
adds support to the therapeutic potential of CG53135 in treatment of
inflammatory bowel
disease.
Materials & Methods
Protein production. Preparation of CG53135 protein was the same as described
in
Example 26.
Study Design. Female Lewis rats (Harlan, Indianapolis, INS weighing 175-200 g
were
acclimated for 8 days (Day -8 through Day -1). Rats were divided into 8
treatment groups:
four groups receiving CG53135 (three groups iv and one group sc), two iv
controls for normal
and the disease model, and two sc controls for normal and the disease model.
On Day -1,
treatments with CG53135 or vehicle were initiated and continued through Day 4.
CG53135
was injected iv (tail vein) at doses of 5, 1 or 0.2 mg/kg, or 1 mg/kg sc;
vehicle controls were
injected with BSA (5 mg/mL in PBS + 1M L-agrinine). On Days 0 and 1 rats were
treated
with indomethacin (Sigma Chemical Co., St. Louis, MO; 7.5 mg/kg doses) in
order to induce
gross and histopathologic intestinal alterations similar to those occurring in
Crohn's Disease.
Indomethacin was prepared in 5% sodium bicarbonate. On Day 5, rats were
injected with a
single ip dose of 50 mg/kg 5-bromo-2'deoxyuridine (BrdU, Calbiochem, LaJolla,
CA) 1 hour
prior to necropsy in order to pulse label proliferating cells in the intestine
and spleen.
Following termination, a 10 cm section of jejunum in the area at risk for
lesions was weighed,
given a gross pathology score, and then collected into formalin for
histopathologic evaluation
and scoring of necrosis and inflammation. Blood was collected for CBC
analysis.
Observations and Analysis of Markers of Pathology
Gross Observations. Body weight was measured daily beginning on Day 0. At
necropsy, liver and spleen weights were measured, and a 10 cm section of
jejunum in the area
at risk was weighed, scored for gross pathology, and collected into formalin
for
histopathologic evaluation and scoring of necrosis and inflammation. The area
at risk for
indomethacin-induced injury was scored at necropsy according to the following
criteria:
0 = normal
1 = minimal thickening of the mesentery/mesenteric border of the intestine
2 = mild to moderate thickening of the mesentery/mesenteric border of
intestine, but no
adhesions
139
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
3 = moderate thickening with 1 or more definite adhesions that are easily
separated
4 = marked thickening with 1 to numerous hard to separate adhesions
= severe intestinal lesions resulting in death.
Histopathology. Five sections (approximately equally spaced) taken from the
weighed
5 l Ocm area at risk of small intestine for indomethacin-induced lesions were
fixed in 10%
neutral buffered formalin, processed for paraffin embedding, sectioned at 5
~.m and stained
with hematoxylin and eosin for histopathologic evaluation. Necrosis was scored
according to
the percent area of the section affected in the same way as described in
Example 26 for scoring
epithelial cell loss.
Inflammation was scored according to the following criteria:
0 = none
1 = minimal inflammation in mesentery and muscle or lesion
2= mild inflammation in mesentery and muscle or lesion
3 = moderate inflammation in mesentery and muscle or lesion
4 = marked inflammation in lesion
5 = severe inflammation in lesion.
The means for inflammation and necrosis were determined for each animal, and
then
the means for each group were calculated.
Statistics. The mean and standard error (SE) for each treatment group were
determined
for each parameter scored; the data were compared to the data for the disease
controls using a
2-tailed Student's T test with significance at p < 0.05.
Results
Weight loss was observed in all animals treated with indomethacin. A slight,
but
significant reduction in weight loss was observed in animals treated with
CG53135 (0.2 mg/kg
iv) as compared with disease controls (iv). Other doses of CG53135 (both iv
and sc routes of
administration) provided diminished, but not statistically significant,
indomethacin-induced
weight loss (FIG. 58).
At necropsy, a 10 cm section of jejunum in the area at risk from each animal
was
weighed. Indomethacin treatment resulted in an elevation in small intestine
weight as
compared with normal iv and sc controls, consistent with edema and
inflammation associated
with this model of intestinal injury. Treatment with CG53135 (1 mg/kg or 0.2
mg/kg iv)
resulted in significant reductions in small intestine weight as compared with
disease controls
140
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
(FIG. 59). A slight reduction in the small intestine clinical score was
observed, with the
greatest benefit occurnng with the 1.0 mg/kg iv dose (38%) and the 0.2 mg/kg
iv dose (25%);
these benefits, however, were not statistically significant. Relative spleen
and liver weights
were increased in animals treated with indomethacin. Administration of CG53135
produced
moderate additional increases in these weights (data not shown).
Hematology. Administration of 2 doses of indomethacin to rats increased the
total
white blood cell count as a result of increased neutrophils and lymphocytes.
Reductions in red
blood cell count, hematocrit, and hemoglobin concentration were also observed.
Treatment
with CG53135 (5 mg/kg and 0.2 mg/kg iv) resulted in significant reductions in
absolute
neutrophil counts as compared with disease controls (FIG. 60). Hemoglobin
concentration
was diminished in the indomethacin controls compared to normal controls, and
slightly fizrther
diminished in rates treated with CG53135 (data not shown).
Histopathology. Evaluation and scoring of 5 sections of intestine were
conducted for
each animal. Histologic evidence of a protective effect on the intestine was
observed only in
animals treated with CG53135 (0.2 mglkg iv). A 53% reduction in jejunal
necrosis and 38%
reduction in inflammation score were observed for the 0.2 mg/kg iv CG53135
dose as
compared with disease controls iv (FIG. 61). Photomicrographs of affected
small intestine are
shown in FIG. 62 for a normal and disease control, and a rat treated with 0.2
mglkg CG53135.
Panel A shows the small intestine from a normal control animal treated iv with
vehicle (BSA).
Normal villous architecture and mesentery (arrow) are apparent. Panel B
presents a
photomicrograph of the small intestine from an indomethacin- treated rat, with
vehicle (BSA)
iv. Focal mucosal necrosis extending across most of the area associated with
attachment of
the mesentery is apparent (see, for example, the asterisks at upper right
intestinal wall and
lower right intestinal wall). Marked inflammatory cell infiltrate is present
in the mesentery
(arrow). Panel C shows the image of the small intestine from an indomethacin-
treated rat
further treated with CG53135, 0.2 mg/kg iv. There is no apparent necrosis, in
contrast to the
disease control shown in Panel B. There is a focal area of attenuated villi
and cellular
infiltration into muscle layer (see, for example, the three asterisks at the
upper right, right and
lower right of the intestinal wall). Mesentery (arrow) is infiltrated by
inflammatory cells. The
photomicrographs in FIG. 62 provide further support for the protective effect
of 0.2 mg/kg iv
CG53135
141
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
BrdU labeling was carried out by injecting 50 mg/kg lhr prior to necropsy. In
the
small intestine from a normal control animal, normal pattern of crypt labeling
is seen at 100X
(FIG. 63, Panel A). BrdU incorporation in the disease model was decreased or
absent in
eptithelial cells in mucosal areas of necrosis, but increased in subaj acent
inflammatory tissue
in which fibroblast labeling was prominent (FIG. 63, Panel B, visualized at
50X). Focal
mucosal necrosis (arrow) is delineated by an absence of BrdU immunostaining as
well as
severe infiltration of inflammatory cells and fibroblast proliferation. Small
intestine from a rat
treated with indomethacin + CG53135 0.2 mg/kg iv shows an absence of crypt
labeling, but
relatively intact mucosa (arrow in FIG. 63, Panel C, visualized at 50X).
Subadjacent smooth
muscle and mesentery is only mildly infiltrated with inflammatory cells,
compared with that
seen in the disease control (Panel B). In certain animals treated with
CG53135, in which
preservation of mucosal integrity occurred, increased crypt labeling was also
observed; this is
in the direction found in the normal control.
The results of the experiments in this Example may be summarized as follows.
Treatment of rats with indomethacin results in gross and histopathologic
intestinal alterations
that are similar to those occurring in Crohn's Disease. Administration of
CG53135 (0.2 mg/kg
iv) to indomethacin-treated rats resulted in significant reductions in weight
loss, small intestine
weight, absolute neutrophil counts, and jejunal necrosis and inflammation
scores. Higher
doses of CG53135 (5, 1 mg/kg iv and 1 mg/lcg sc) were less efficacious. The
morphological
appearance of tissues collected from animals injected with BrdU 1 hour prior
to necropsy
suggested that the beneficial effects of CG53135 in this model of intestinal
injury were the
result of mucosal protection rather than a proliferative effect on target
cells.
EQUIVALENTS
From the foregoing detailed description of the specific embodiments of the
invention,
it should be apparent that particular novel compositions and methods involving
nucleic acids,
polypeptides, antibodies, detection and treatment have been described.
Although these
particular embodiments have been disclosed herein in detail, this has been
done by way of
example for purposes of illustration only, and is not intended to be limiting
with respect to the
scope of the appended claims that follow. In particular, it is contemplated by
the inventors that
various substitutions, alterations, and modifications may be made as a matter
of routine for a
person of ordinary skill in the art to the invention without departing from
the spirit and scope
142
CA 02428084 2003-05-05
WO 02/058716 PCT/USO1/43846
of the invention as defined by the claims. Indeed, various modifications of
the invention in
addition to those described herein will become apparent to those skilled in
the art from the
foregoing description and accompanying figures. Such modifications are
intended to fall
within the scope of the appended claims.
143