Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02457318 2004-02-20
WO 03/016498 PCT/US02/26418
MOLECITLAR INTERACTION SITES OF RNase P RNA
AND METHODS OF MODULATING THE SAME
FIELD OF THE INVENTION
The present invention relates to identification of molecular interaction sites
of
RNase P RNA, virtual or actual screening of. compounds that bind thereto, and
to
modulating the activity of RNase P RNA with such compounds identified in the
actual
or virtual screening.
to BACKGROUND OF THE INVENTION
Ribonuclease P (RNase P) is the endoribonuclease responsible for removing
the leader sequence from the tRNA precursors during the maturation of the 5'
end of
tRNAs. Altman et al., FASEB J., 1993, 7, 7-14 and Pace et al., J. Bacteriol.,
1995,
177, 1919-1928. Rnase P is a ribonucleoprotein whose catalytic function, at
least in
bacteria, is carried out by its RNA component (RNase P RNA) rather than by the
protein. Guerrier-Takada et al., Cell, 1983, 35, 849-857. Another feature of
RNase P
RNA is its ability to recognize the tertiary structure of its pre-tRNA
substrates. Kahle
et al., EMBO J., 1990, 9, 1929-1937. The secondary structure of bacterial
RNase P
RNA was inferred from a first comparative analysis of sequences (James et al.,
Cell,
1988, 52, 19-26) and refined as further sequences became available (Haas et
al.,
Science, 1991, 254, 853-856; Haas et al., Froc. Natl. Acad. Sci. USA, 1994,
91, 2527
2531; Brown et al., Nucl. Acids Res., 1993, 21, 671-679). In addition,
derivation of the
three-dimensional architecture of bacterial RNase P RNAs from E. coli and
Bacillus
subtilzs are described in Massire et al., J. Mol. Biol., 1998, 279, 773-793,
which is
incorporated herein by reference in its entirety.
Recent advances in genomics, molecular biology, and structural biology have
highlighted how RNA molecules participate in or controls many of the events
required
to express proteins in cells. Rather than function as simple intermediaries,
RNA
molecules actively regulate their own transcription from DNA, splice and edit
mRNA
molecules and tRNA molecules, synthesize peptide bonds in the ribosome,
catalyze
CA 02457318 2004-02-20
WO 03/016498 PCT/US02/26418
the migration of nascent proteins to the cell membrane, and provide fine
control over
the rate of translation of messages. RNA molecules can adopt a variety of
unique
structural motifs that provide the framework required to perform these
functions.
"Small" molecule therapeutics, which bind specifically to structured RNA
molecules, are organic chemical molecules that are not polymers. "Small"
molecule
therapeutics include, for example, the most powerful naturally-occurring
antibiotics.
For example, the aminoglycoside and macrolide antibiotics are "small"
molecules that
bind to defined regions in ribosomal RNA (rRNA) structures and work, it is
believed,
by blocking conformational changes in the RNA required for protein synthesis.
In
1o addition, changes in the conformation of RNA molecules have been shown to
regulate
rates of transcription and translation of mRNA molecules. Small molecules are
generally less than 10 kDa.
RNA molecules or groups of related RNA molecules are believed by
Applicants to have regulatory regions that are used by the cell to control
synthesis of
proteins. The cell is believed to exercise control over both the timing and
the amount
of protein that is synthesized by direct, specific interactions with RNA. This
notion is
inconsistent with the impression obtained by reading the scientific literature
on gene
regulation, which is highly focused on transcription. The process of RNA
maturation,
transport, intracellular localization and translation are rich in RNA
recognition sites
that provide good opportunities for drug binding. Applicants' invention is
directed,
inter alia, to finding these regions of RNA molecules, in particular the RNase
P RNA,
in the microbial genome. Applicants' invention also makes use of combinatorial
chemistry to make and/or screen, actually or virtually, a large number of
chemical
entities for their ability to bind and/or modulate these drug binding sites.
The determination of potential three dimensional structures of nucleic acids
and their attendant structural motifs affords insights into areas such as the
study of
catalysis by RNA, RNA-RNA interactions, RNA-nucleic acid interactions, RNA-
protein interactions, and the recognition of small molecules by nucleic acids.
Four
general approaches to the generation of model three dimensional structures of
RNA
have been demonstrated in the literature. All of these employ sophisticated
molecular
modelling and computational algorithms for the simulation of folding and
tertiary
interactions within target nucleic acids, such as RNA. Westhof and Altman
(Proc.
Natl. Acad. Sci., 1994, 91, 5133, incorporated herein by reference in its
entirety) have
CA 02457318 2004-02-20
WO 03/016498 PCT/US02/26418
-3-
described the generation of a three-dimensional working model of Ml RNA, the
catalytic RNA subunit of RNase P from E. coli via an interactive computer
modelling
protocol. Leveraging the significant body of work in the area of cryo-electron
microscopy (cryo-EM) and biochemical studies on ribosomal RNAs, Mueller and
Brimacombe (J. Mol. Biol., 1997, 271, 524) have constructed a three
dimensional
model of E. coli 16S Ribosomal RNA. A method to model nucleic acid hairpin
motifs
has been developed based on a set of reduced coordinates for describing
nucleic acid
structures and a sampling algorithm that equilibriates structures using Monte
Carlo
(MC) simulations (Tung, Biophysical J., 1997, 72, 876, incorporated herein by
1o reference in its entirety). MC-SYM is yet another approach to predicting
the three
dimensional structure of RNAs using a constraint-satisfaction method. Major et
al.,
Proc. Natl. Acad. Sci., 1993, 90, 9408. The MC-SYM program is an algorithm
based
on constraint satisfaction that searches conformational space for all models
that satisfy
query input constraints, and is described in, for example, Cedergren et al.,
RNA
Structure And FunctiofZ, 1998, Cold Spring Harbor Lab. Press, p.37-75. Three
dimensional structures of RNA are produced by that method by the stepwise
addition
of nucleotide having one or several different conformations to a growing
oligonucleotide model.
Westhof and Altman (Proc. Natl. Acad. Sci., 1994, 91, 5133) have described
the generation of a three-dimensional working model of Ml RNA, the catalytic
RNA
subunit of RNase P from E. coli via an interactive computer modelling
protocol. This
modelling protocol incorporated data from chemical and enzymatic protection
experiments, phylogenetic analysis, studies of the activities of mutants and
the
kinetics of reactions catalyzed by the binding of substrate to Ml RNA.
Modelling was
performed for the most part as described in the literature. Westhof et. al.,
in
"Theoretical Biochemistry and Molecular Biophysics," Beveridge and Lavery
(Eds.),
Adenine, NY, 1990, 399. In general, starting with the primary sequence of Ml
RNA,
the stem-loop structures and other elements of secondary structure were
created.
Subsequent assembly of these elements into a three dimensional structure using
a
3o computer graphics station and FRO1~0 (Jones, J. Appl. Crystallogr., 1978,
11, 268)
followed by refinement using NUCLIN-NLTCLSQ afforded a RNA model that had
correct geometries, the absence of bad contacts, and appropriate
stereochemistry. The
model so generated was found to be consistent with a large body of empirical
data on
CA 02457318 2004-02-20
WO 03/016498 PCT/US02/26418
-4-
Ml RNA and opens the door for hypotheses about the mechanism of action of
RNase
P. The models generated by this method, however, are less well resolved that
the
structures determined via X-ray crystallography.
Mueller and Brimacombe (J. Mol. Biol., 1997, 271, 524, which is
incorporated herein by reference in its entirety) have constructed a three
dimensional
model of E. coli 16S ribosomal RNA using a modelling program called ERNA-3D.
This program generates three dimensional structures such as A-form RNA helices
and
single-strand regions via the dynamic docking of single strands to fit
electron density
obtained from low resolution diffraction data. After helical elements have
been
defined and positioned in the model, the configurations of the single strand
regions is
adjusted, so as to satisfy any known biochemical constraints such as RNA-
protein
cross-linking and foot-printing data.
A method to model nucleic acid hairpin motifs has been developed based on
a set of reduced coordinates for describing nucleic acid structures and a
sampling
algorithm that equilibrates structures using Monte Carlo (MC) simulations.
Tung,
Biophysical J., 1997, 72, 876, incorporated herein by reference in its
entirety. The
stem region of a nucleic acid can be adequately modelled by using a canonical
duplex
formation. Using a set of reduced coordinates, an algorithm that is capable of
generating structures of single stranded loops with a pair of fixed ends was
created.
This allows efficient structural sampling of the loop in conformational space.
Combining this algorithm with a modified Metropolis Monte Carlo algorithm
afforded
a structure simulation package that simplifies the study of nucleic acid
hairpin
structures by computational means. Once the RNA subdomains have been
identified,
they can, if desired, be stabilized by the methods disclosed in U.S. Patent
No.
5,712,096.
While X-ray crystallography is a very powerful technique that can allow for
the determination of some secondary and tertiary structure of biopolymeric
targets
(Erikson et al., Anfa. Rep. in Med. Cl2em., 1992, 27, 271-289), this technique
can be an
expensive procedure and very difficult to accomplish. Crystallization of
biopolymers
is extremely challenging, difficult to perform at adequate resolution, and is
often
considered to be as much an art as a science. Further confounding the utility
of X-ray
crystal structures in the drug discovery process is the inability of
crystallography to
reveal insights into the solution-phase, and therefore the biologically
relevant,
CA 02457318 2004-02-20
WO 03/016498 PCT/US02/26418
-S-
structures of the targets of interest. Some analysis of the nature and
strength of
interaction between a ligand (agonist, antagonist, or inhibitor) and its
target can be
performed by ELISA (Kemeny and Challacombe, in ELISA and other Solid Phase
Immunoassays: 1988), radioligand binding assays (Berson et al., CliyZ. 1968;
Chard, in
"An Introduction to Radioimmunoassay and Related Techniques," 1982), surface-
plasmon resonance (Karlsson et al., 1991, Jonsson et al., Biotechf2iques,
1991), or
scintillation proximity assays (Udenfriend et al., AsZal. Biochef~a., 1987),
all cited
previously. The radioligand binding assays are typically useful only when
assessing
the competitive binding of the unknown at the binding site for that of the
radioligand
and also require the use of radioactivity. The surface-plasmon resonance
technique is
more straightforward to use, but is also quite costly. Conventional
biochemical assays
of binding kinetics, and dissociation and association constants are also
helpful in
elucidating the nature of the target-ligand interactions.
Accordingly, one aspect of the invention identifies molecular interaction
sites
in RNase P RNA. These molecular interaction sites, which comprise secondary
structural elements, are highly likely to give rise to significant
therapeutic, regulatory,
or other interactions with "small" molecules and the like. Another aspect of
the
invention is to compare molecular interaction sites of RNase P RNA with
compounds
proposed for interaction therewith.
Yet another aspect of the present invention is the establishment of databases
of the numerical representations of three-dimensional structures of molecular
interaction sites of RNase P RNA. Such databases libraries provide powerful
tools for
the elucidation of structure and interactions of molecular interaction sites
with
potential ligands and predictions thereof. Another aspect of the present
invention is to
provide a general method for the screening of combinatorial libraries
comprising
individual compounds or mixtures of compounds against RNase P RNA, so as to
determine which components of the library bind to the target.
SUMMARY OF THE INVENTION
3o The present invention is directed to identification of molecular
interaction
sites of RNase P RNA that comprise particular secondary structure.
The present invention is also directed to nucleic acid molecules,
polynucleotides or oligonucleotides comprising the molecular interaction sites
that can
CA 02457318 2004-02-20
WO 03/016498 PCT/US02/26418
-6-
be used to screen, virtually or actually, combinatorial libraries of compounds
that bind
thereto.
The present invention is also directed to computer-readable medium
comprising three dimensional representations of the structures of the
molecular
interaction sites.
The present invention is also directed to modulating the activity of RNase P
RNA by contacting RNase P RNA or prokaryotic cells comprising the same with a
compound identified by such virtual or actual screening.
The present invention is also directed to modulating prokaryotic cell growth
l0 comprising contacting a prokaryotic cell with a compound identified by such
virtual or
actual screening.
BRIEF DESCRIPTION OF THE DRAWINGS
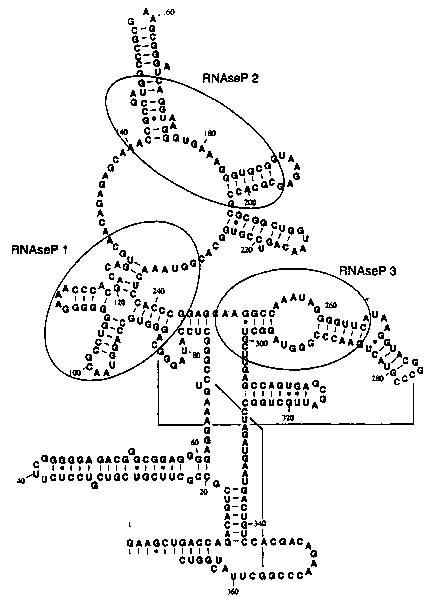
Figures l, 1A, 1B and 1C show representative structures of E. coli RNase P
RNA showing sites 1, 2, and 3.
Figures 2, 2A, 2B and 2C show representative structures of B. subtilis RNase
P RNA showing sites 4, 5, and 6.
DESCRIPTION OF PREFERRED EMBODIMENTS OF THE INVENTION
The present invention is directed to, inter alia, identification of molecular
interaction sites of RNase P RNA. Such molecular interaction sites comprise
secondary structure capable of interacting with cellular components, such as
factors
and proteins ~ required for translation and other cellular processes. Nucleic
acid
molecules or polynucleotides comprising the molecular interaction sites can be
used to
screen, virtually or actually, combinatorial libraries of compounds that bind
thereto.
The compounds identified by such screening are used to modulate the activity
of
RNase P RNA and, thus, can be used to modulate, either inhibit or stimulate,
prokaryotic cell growth. Thus, novel drugs, agricultural chemicals, industrial
chemicals and the like that operate through the modulation of RNase P RNA can
be
3o identified.
A number of procedures and protocols are preferably integrated to provide
powerful drug and other biologically useful compound identification.
Pharmaceuticals, veterinary drugs, agricultural chemicals, pesticides,
herbicides,
CA 02457318 2004-02-20
WO 03/016498 PCT/US02/26418
_7_
fungicides, industrial chemicals, research chemicals and many other beneficial
compounds useful in pollution control, industrial biochemistry, and
biocatalytic
systems can be identified in accordance with embodiments of this invention.
Novel
combinations of procedures provide extraordinary power and versatility to the
present
methods. While it is preferred in some embodiments to integrate a number of
processes developed by the assignee of the present application as will be set
forth
more fully herein, it should be recognized that other methodologies can be
integrated
herewith to good effect. Thus, while it is greatly advantageous to determine
molecular
binding sited on RNase P RNA in accordance with the teachings of this
invention, the
to interactions of ligands and libraries of ligands with other RNase P RNA
identified as
being of interest may ,greatly benefit from other aspects of this invention.
All such
combinations are within the spirit of the invention.
One aspect of Applicants' invention is directed to identifying secondary
structures in RNase P RNA termed "molecular interaction sites." As used
herein,
"molecular interaction sites" are regions of RNase P RNA that have secondary
structure. Molecular interaction sites can be conserved among a plurality of
different
taxonomic species of RNase P RNA. Molecular interaction sites are small,
preferably
less than 200 nucleotides, preferably less than 150 nucleotides, preferably
less than 70
nucleotides, preferably less than 50 nucleotides, alternatively less than 30
nucleotides,
2o independently folded, functional subdomains contained within a larger RNA
molecule. Molecular interaction sites can contain both single-stranded and
double-
stranded regions. Thus, molecular interaction sites are capable of undergoing
interaction with "small" molecules and otherwise, and are expected to serve as
sites
for interacting with "small" molecules, oligomers such as oligonucleotides,
and other
compounds in therapeutic and other applications. Molecular interaction sites
also
comprise a pocket for binding small molecules, drugs and the like.
The molecular interaction sites are present within at least RNase P RNA. In
accordance with some embodiments of this invention, it will be appreciated
that the
RNase P RNAs having a molecular interaction site or sites may be derived from
a
3o number of sources. Thus, such RNase P RNAs can be identified by any means,
rendered into three dimensional representations and employed for the
identification of
compounds that can interact with them to effect modulation of the RNase P RNA.
In
some embodiments, the molecular interaction sites that are identified in RNase
P RNA
CA 02457318 2004-02-20
WO 03/016498 PCT/US02/26418
_$_
are absent from eukaryotes, particularly humans, and, thus, can serve as sites
for
"small" molecule binding with concomitant modulation of the RNase P RNA of
prokaryotic organisms without effecting human toxicity.
The molecular interaction sites can be identified by any means known to the
skilled artisan. In some embodiments of the invention, the molecular
interaction sites
in RNase P RNA are identified according to the general methods described in
International Publication WO 99/58719, which is incorporated herein by
reference in
its entirety. Briefly, a target RNase P RNA nucleotide sequence is chosen from
among
known sequences. Any RNase P RNA nucleotide sequence can be chosen. The
l0 nucleotide sequence of the target RNase P RNA is compared to the nucleotide
sequences of a plurality of RNase P RNA from different taxonomic species. At
least
one sequence region that is effectively conserved among the plurality of RNase
P
RNAs and the target RNase P RNA is identified. Such conserved region is
examined
to determine whether there is any secondary structure, and, for conserved
regions
having secondary structure, such secondary structure is identified.
In accordance with some embodiments of the invention, the nucleotide
sequence of the target RNase P RNA is compared with the nucleotide sequences
of a
plurality of corresponding RNase P RNAs from different taxonomic species.
Initial
selection of a particular target nucleic acid can be based upon any functional
criteria.
RNase P RNA known to be involved in pathogenic genomes such as, for example,
bacterial and yeast, are exemplary targets. Pathogenic bacteria and yeast are
well
known to those skilled in the art. Additional RNase P RNA targets can be
determined
independently or can be selected from publicly available prokaryotic genetic
databases
known to those skilled in the art. Databases include, for example, Online
Mendelian
Inheritance in .Man (OMIM), the Cancer Genome Anatomy Project (CGAP),
GenBank, EMBL, PIR, SWISS-PROT, and the like. OMIM, which is a database of
genetic mutations associated with disease, was developed, in part, for the
National
Center for Biotechnology Information (NCBI). OM1M can be accessed through the
world wide web of the Internet at, for example, ncbi.nlm.nih.gov/Omim/. CGAP,
3o which is an interdisciplinary program to establish the information and
technological
tools required to decipher the molecular anatomy of a cancer cell, can be
accessed
through the world wide web of the Internet at, for example,
ncbi.nlm.nih.govlncicgap/.
Some of these databases may contain complete or partial nucleotide sequences.
In
CA 02457318 2004-02-20
WO 03/016498 PCT/US02/26418
-9-
addition, RNase P RNA targets can also be selected from private genetic
databases.
Alternatively, RNase P RNA targets can be selected from available publications
or
can be determined especially for use in connection with the present invention.
After a RNase P RNA target is selected or provided, the nucleotide sequence
of the RNase P RNA target is determined and then compared to the nucleotide
sequences of a plurality of RNase P RNAs from different taxonomic species. In
one
embodiment of the invention, the nucleotide sequence of the RNase P RNA target
is
determined by scanning at least one genetic database or is identified in
available
publications. Databases known and available to those skilled in the art
include, for
to example, GenBank, and the like. These databases can be used in connection
with
searching programs such as, for example, Entrez, which is known and available
to
those skilled in the art, and the like. Entrez can be accessed through the
world wide
web of the Internet at, for example, ncbi.nlm.nih.gov/Entrez/. Preferably, the
most
complete nucleic acid sequence representation available from various databases
is
used. The GenBank database, which is known and available to those skilled in
the art,
can also be used to obtain the most complete nucleotide sequence. GenBank is
the
NIH genetic sequence database and is an annotated collection of all publicly
available
DNA sequences. GenBank is described in, for example, Nuc. Acids Res., 1998,
26, 1-
7, which is incorporated herein by reference in its entirety, and can be
accessed by
those skilled in the art through the world wide web of the Internet at, for
example,
ncbi.nlm.nih.gov/Web/Genbank/index.html. Alternatively, partial nucleotide
sequences of RNase P RNA targets can be used when a complete nucleotide
sequence
is not available.
The nucleotide sequence of the RNase P RNA target is compared to the
nucleotide sequences of a plurality of RNase P RNAs from different taxonomic
species. A plurality of RNase P RNAs from different taxonomic species, and the
nucleotide sequences thereof, can be found in genetic databases, from
available
publications, or can be determined especially for use in connection with the
present
invention. In one embodiment of the invention, the RNase P RNA target is
compared
to the nucleotide sequences of a plurality of RNase P RNAs from different
taxonomic
species by performing a sequence similarity search, an ortholog search, or
both, such
searches being known to persons of ordinary skill in the art.
CA 02457318 2004-02-20
WO 03/016498 PCT/US02/26418
-10-
The result of a sequence similarity search is a plurality of RNase P RNAs
having at least a portion of their nucleotide sequences which are homologous
to at
least an 8 to 20 nucleotide region of the target RNase P RNA, referred to as
the
window region. Preferably, the plurality of RNase P RNAs comprise at least one
portion which is at least 60% homologous to any window region of the target
RNase P
RNA. More preferably, the homology is at least 70%. More preferably, the
homology
is at least 80%. Most preferably, the homology is at least 90% or 95%. For
example,
the window size, the portion of the target RNase P RNA to which the plurality
of
sequences are compared, can be from about 8 to about 20, preferably from about
10 to
to about 15, most preferably from about 11 to about 12, contiguous
nucleotides. The
window size can be adjusted accordingly. A plurality of RNase P RNAs from
different
taxonomic species is then preferably compared to each likely window in the
target
RNase P RNA until all portions of the plurality of sequences is compared to
the
windows of the target RNase P RNA. Sequences of the plurality of RNase P RNAs
from different taxonomic species which have portions which are at least 60%,
preferably at least 70%, more preferably at least 80%, or most preferably at
least 90%
homologous to any window sequence of the target RNase P RNA are considered as
likely homologous sequences.
Sequence similarity searches can be performed manually or by using several
available computer programs known to those skilled in the art. Preferably,
Blast and
Smith-Waterman algorithms, which are available and known to those skilled in
the art,
and the like can be used. Blast is NCBI's sequence similarity search tool
designed to
support analysis of nucleotide and protein sequence databases. Blast can be
accessed
through the world wide web of the Internet at, for example,
ncbi.nlm.nih.gov/BLAST/. The GCG Package provides a local version of Blast
that
can be used either with public domain databases or with any locally available
searchable database. GCG Package v.9.0 is a commercially available software
package that contains over 100 interrelated software programs that enables
analysis of
sequences by editing, mapping, comparing and aligning them. Other programs
included in the GCG Package include, for example, programs which facilitate
RNA
secondary structure predictions, nucleic acid fragment assembly, and
evolutionary
analysis. In addition, the most prominent genetic databases (GenBank, EMBL,
PIR,
and SWISS-PROT) are distributed along with the GCG Package and are fully
CA 02457318 2004-02-20
WO 03/016498 PCT/US02/26418
-11-
accessible with the database searching and manipulation programs. GCG can be
accessed through the world wide web of the Internet at, for example, gcg.com/.
Fetch
is a tool available in GCG that can get annotated GenBank records based on
accession
numbers and is similar to Entrez. Another sequence similarity search can be
performed with GeneWorld and GeneThesaurus from Pangea. GeneWorld 2.5 is an
automated, flexible, high-throughput application for analysis of
polynucleotide and
protein sequences. GeneWorld allows for automatic analysis and annotations of
sequences. Like GCG, GeneWorld incorporates several tools for homology
searching,
gene finding, multiple sequence alignment, secondary structure prediction, and
motif
1o identification. GeneThesaurus 1.OTM is a sequence and annotation data
subscription
service providing information from multiple sources, providing a relational
data
model for public and local data.
Another alternative sequence similarity search can be performed, for
example, by BlastParse. BlastParse is a PERL script running on a iJNE~
platform that
automates the strategy described above. BlastParse takes a list of target
accession
numbers of interest and parses all the GenBank fields into "tab-delimited"
text that
can then be saved in a "relational database" format for easier search and
analysis;
which provides flexibility. The end result is a series of completely parsed
GenBank
records that can be easily sorted, filtered, and queried against, as well as
an
2o annotations-relational database.
Another toolkit capable of doing sequence similarity searching and data
manipulation is SEALS, also from NCBI. This tool set is written in perl and C
and can
run on any computer platform that supports these languages. It is available
for
download, for example, at the world wide web of the Internet at
ncbi.nlm.nih.gov/Walker/SEALS/. This toolkit provides access to Blast2 or
gapped
blast. It also includes a tool called tax collector which, in conjunction with
a tool
called tax break, parses the output of Blast2 and returns the identifier of
the sequence
most homologous to the query sequence for each species present. Another useful
tool
is feature2fasta which extracts sequence fragments from an input sequence
based on
the annotation.
Preferably, the plurality of RNase P RNAs from different taxonomic species
which have homology to the target nucleic acid, as described above in the
sequence
similarity search, are further delineated so as to find orthologs of the
target RNase P
CA 02457318 2004-02-20
WO 03/016498 PCT/US02/26418
-12-
RNA therein. An ortholog is a term defined in gene classification to refer to
two genes
in widely divergent organisms that have sequence similarity, and perform
similar
functions within the context of the organism. In contrast, paralogs are genes
within a
species that occur due to gene duplication, but have evolved new functions,
and are
also referred to as isotypes. Optionally, paralog searches can also be
performed. By
performing an ortholog search, an exhaustive list of homologous sequences from
diverse organisms is obtained. Subsequently, these sequences are analyzed to
select
the best representative sequence that fits the criteria for being an ortholog.
An
ortholog search can be performed by programs available to those skilled in the
art
including, for example, Compare. Preferably, an ortholog search is performed
with
access to complete and parsed GenBank annotations for each of the sequences.
Currently, the records obtained from GenBank are "flat-files", and are not
ideally
suited for automated analysis. Preferably, the ortholog search is performed
using a Q-
Compare program. The Blast Results-Relation database and the Annotations-
Relational database are used in the Q-Compare protocol, which results in a
list of
ortholog sequences to compare in the interspecies sequence comparisons
programs
described below.
The above-described similarity searches provide results based on cttt-off
values, referred to as e-scores. E-scores represent the probability of a
random
sequence match within a given window of nucleotides. The lower the e-score,
the
better the match. One skilled in the art is familiar with e-scores. The user
defines the
e-value cut-off depending upon the stringency, or degree of homology desired,
as
described above. In some embodiments of the invention, it is preferred that
any
homologous nucleotide sequences of RNase P RNA that are identified not be
present
in the human genome.
In another embodiment of the invention, the sequences required are obtained
by searching ortholog databases. One such database is Hovergen, which is a
curated
database of vertebrate orthologs. Ortholog sets may be exported from this
database
and used as is, or used as seeds for further sequence similarity searches as
described
above. Further searches may be desired, for example, to find invertebrate
orthologs.
Hovergen can be downloaded as a file transfer program at, for example,
pbil.univ-
lyonl.fr/pub/hovergen/. A database of prokaryotic orthologs, COGS, is
available and
CA 02457318 2004-02-20
WO 03/016498 PCT/US02/26418
-13-
can be used interactively through the world wide web of the Internet at, for
example,
ncbi.nlm.nih.gov/COG/.
After the orthologs or virtual transcripts described above are obtained
through either the sequence similarity search or the ortholog search, at least
one
sequence region which is conserved among the plurality of RNase P RNAs from
different taxonomic species and the target RNase P RNA is identified.
Interspecies
sequence comparisons can be performed using numerous computer programs which
are available and known to those skilled in the art. Preferably, interspecies
sequence
comparison is performed using Compare, which is available and known to those
1o skilled in the art. Compare is a GCG tool that allows pair-wise comparisons
of
sequences using a window/stringency criterion. Compare produces an output file
containing points where matches of specified quality are found. These can be
plotted
with another GCG tool, DotPlot.
Alternatively, the identification of a conserved sequence region is performed
by interspecies sequence comparisons using the ortholog sequences generated
from Q-
Compare in combination with CompareOverWins. Preferably, the list of sequences
to
compare, i.e., the ortholog sequences, generated from Q-Compare is entered
into the
CompareOverWins algorithm. Preferably, interspecies sequence comparisons are
performed by a pair-wise sequence comparison in which a query sequence is slid
over
2o a window on the master target sequence. Preferably, the window is from
about 9 to
about 99 contiguous nucleotides.
Sequence homology between the window sequence of the target RNase P
RNA and the query sequence of any of the plurality of RNase P RNAs obtained as
described above, is preferably at least 60%, more preferably at least 70%,
more
preferably at least ~0%, and most preferably at least 90% or 95%. The most
preferable
method of choosing the threshold is to have the computer automatically try all
thresholds from 50% to 100% and choose a threshold based a metric provided by
the
user. One such metric is to pick the threshold such that exactly n hits are
returned,
where n is usually set to 3. This process is repeated until every base on the
query
nucleic acid, which is a member of the plurality of RNase P RNAs described
above,
has been compared to every base on the master target sequence. The resulting
scoring
matrix can be plotted as a scatter plot. Based on the match density at a given
location,
there may be no dots, isolated dots, or a set of dots so close together that
they appear
CA 02457318 2004-02-20
WO 03/016498 PCT/US02/26418
-14-
as a line. The presence of lines, however small, indicates primary sequence
homology.
Sequence conservation within RNase P RNA in divergent species is likely to be
an
indicator of conserved regulatory elements that are also likely to have a
secondary
structure. The results of the interspecies sequence comparison can be analyzed
using
MS Excel and visual basic tools in an entirely automated manner as known to
those
skilled in the art.
After at least one region that is conserved between the nucleotide sequence of
the RNase P RNA target and the plurality of RNase P RNAs from different
taxonomic
species, preferably via the orthologs, is identified, the conserved region is
analyzed to
l0 determine whether it contains secondary structure. Determining whether the
identified
conserved regions contain secondary structure can be performed by a number of
procedures known to those skilled in the art. Determination of secondary
structure is
preferably performed by self complementarity comparison, alignment and
covariance
analysis, secondary structure prediction, or a combination thereof.
In one embodiment of the invention, secondary structure analysis is
performed by alignment and covariance analysis. Numerous protocols for
alignment
and covariance analysis are known to those skilled in the art. Preferably,
alignment is
performed by ClustalW, which is available and known to those skilled in the
art.
ClustalW is a tool for multiple sequence alignment that, although not a part
of GCG,
2o can be added as an extension of the existing GCG tool set and used with
local
sequences. ClustalW can be accessed through the world wide web of the Internet
at,
for example, dot.imgen.bcm.tmc.edu:9331/multi-align/Options/clustalw.html.
ClustalW is also described in Thompson, et al., Nuc. Acids Res., 1994, 22,
4673-4680,
which is incorporated herein by reference in its entirety. These processes can
be
scripted to automatically use conserved UTR regions identified in earlier
steps. Seqed,
a UNIX command line interface available and known to those skilled in the art,
allows
extraction of selected local regions from a larger sequence. Multiple
sequences from
many different species can be clustered and aligned for further analysis.
In another embodiment of the invention, the output of all possible pair-wise
3o CompareOverWindows comparisons are compiled and aligned to a reference
sequence using a program called AlignHits, a program that can be reproduced by
one
skilled in the art. One purpose of this program is to map all hits made in
pair-wise
comparisons back to the position on a reference sequence. This method
combining
CA 02457318 2004-02-20
WO 03/016498 PCT/US02/26418
-15-
CompareOverWindows and AlignHits provides more local alignments (over 20-100
bases) than any other algorithm. This local alignment is required for the
structure
finding routines described later such as covariation or RevComp. This
algorithm
writes a fasta file of aligned sequences. It is important to differentiate
this from using
ClustalW by itself, without CompareOverWindows and AlignHits.
Covariation is a process of using phylogenetic analysis of primary sequence
information for consensus secondary structure prediction. Covariation is
described in
the following references, each of which is incorporated herein by reference in
their
entirety: Gutell et al., "Comparative Sequence Analysis Of Experiments
Performed
During Evolution" In Ribosomal RNA Group I Introns, Green, Ed., Austin:
Landes,
1996; Gautheret et al., Nuc. Acids Res., 1997, 25, 1559-1564; Gautheret et
al., RNA,
1995, l, 807-814; Lodmell et al., Proc. Natl. Acad. Sci. USA,1995, 92, 10555-
10559;
Gautheret et al., J. Mol. Biol., 1995, 248, 27-43; Gutell, Nuc. Acids Res.,
1994, 22,
3502-3517; Gutell, Nuc. Acids Res., 1993, 21, 3055-3074; Gutell, Nuc. Acids
Res.,
1993, 21, 3051-3054; Woese, Proc. Natl. Aead. Sci. USA, 1989, 86, 3119-3122;
and
Woese et al., Nuc. Acids Res., 1980, 8, 2275-2293, each of which is
incorporated
herein by reference in its entirety. Preferably, covariance software is used
for
covariance analysis. Preferably, Covariation, a set of programs for the
comparative
analysis of RNA structure from sequence alignments, is used. Covariation uses
phylogenetic analysis of primary sequence information for consensus secondary
structure prediction. Covariation can be obtained through the world wide web
of the
Internet at, for example, mbio.ncsu.edu/RNaseP/info/programs/programs.html. A
complete description of a version of the program has been published (Brown, J.
W.
1991, Phylogenetic analysis of RNA structure on the Macintosh computer. CABIOS
7:391-393). The current version is v4.1, which can perform various types of
covariation analysis from RNA sequence alignments, including standard
covariation
analysis, the identification of compensatory base-changes, and mutual
information
analysis. The program is well-documented and comes with extensive example
files. It
is compiled as a stand-alone program; it does not require Hypercard (although
a much
3o smaller 'stack' version is included). This program will run in any
Macintosh
environment running MacOS v7.1 or higher. Faster processor machines (68040 or
PowerPC) is suggested for mutual information analysis or the analysis of large
sequence alignments.
CA 02457318 2004-02-20
WO 03/016498 PCT/US02/26418
-16-
In another embodiment of the invention, secondary structure analysis is
performed by secondary structure prediction. There are a number of algorithms
that
predict RNA secondary structures based on thermodynamic parameters and energy
calculations. Preferably, secondary structure prediction is performed using
either M-
fold or RNA Structure 2.52. M-fold can be accessed through the world wide web
of
the Internet at, for example, ibc.wustl.edu/-zuker/ma/form2.cgi or can be
downloaded
for local use on UNIX platforms. M-fold is also available as a part of GCG
package.
RNA Structure 2.52 is a windows adaptation of the M-fold algorithm and can be
accessed through the world wide web of the Internet at, for example,
l0 12~.151.176.70/RNAstructure.html.
In another embodiment of the invention, secondary structure analysis is
performed by self complementarity comparison. Preferably, self complementarity
comparison is performed using Compare, described above. More preferably,
Compare
can be modified to expand the pairing matrix to account for G-U or U-G
basepairs in
addition to the conventional Watson-Crick G-C/C-G or A-U/LT-A pairs. Such a
modified Compare program (modified Compare) begins by predicting all possible
base-pairings within a given sequence. As described above, a small but
conserved
region is identified based on primary sequence comparison of a series of
orthologs. In
modified Compare, each of these sequences is compared to its own reverse
2o complement. Allowable base-pairings include Watson-Crick A-U, G-C pairing
and
non-canonical G-U pairing. An overlay of such self complementarity plots of
all
available orthologs, and selection for the most repetitive pattern in each,
results in a
minimal number of possible folded configurations. These overlays can then used
in
conjunction with additional constraints, including those imposed by energy
considerations described above, to deduce the most likely secondary structure.
In another embodiment of the invention, the output of AlignHits is read by a
program called RevComp. This program could be reproduced by one skilled in the
art.
One purpose of this program is to use base pairing rules and ortholog
evolution to
predict RNA secondary structure. RNA secondary structures are composed of
single
stranded regions and base paired regions, called stems. Since structure
conserved by
evolution is searched, the most probable stem for a given alignment of
ortholog
sequences is the one which could be formed by the most sequences. Possible
stem
formation or base pairing rules is determined by, for example, analyzing base
pairing
CA 02457318 2004-02-20
WO 03/016498 PCT/US02/26418
-17-
statistics of stems which have been determined by other techniques such as
NMR. The
output of RevComp is a sorted list of possible structures, ranked by the
percentage of
ortholog set member sequences which could form this structure. Because this
approach uses a percentage threshold approach, it is insensitive to noise
sequences.
Noise sequences are those that either not true orthologs, or sequences that
made it into
the output of AlignHits due to high sequence homology even though they do not
represent an example of the structure which is searched. A very similar
algorithm is
implemented using Visual basic for Applications (VBA) and Microsoft Excel to
be
run on PCs, to generate the reverse complement matrix view for the given set
of
to sequences.
A result of the secondary structure analysis described above, whether
performed by alignment and covariance, self complementarity analysis,
secondary
structure predictions, such as using M-fold or otherwise, is the
identification of
secondary structure in the conserved regions among the target RNase P RNA and
the
plurality of RNase P RNAs from different taxonomic species. Exemplary
secondary
structures that may be identified include, but are not limited to, bulges,
loops, stems,
hairpins, knots, triple interacts, cloverleafs, or helices, or a combination
thereof.
Alternatively, new secondary structures may be identified.
The present invention is also directed to nucleic acid molecules, such as
2o polynucleotides and oligonucleotides, comprising a molecular interaction
site present
in 16S rRNA. Nucleic acid molecules include the physical compounds themselves
as
well as iyz silico representations of the same. Thus, the nucleic acid
molecules are
derived from RNase P RNA. The molecular interaction site serves as a binding
site for
at least one molecule which, when bound to the molecular interaction site,
modulates
the expression of the RNase P RNA in a cell. The nucleotide sequence of the
polynucleotide is selected to provide the secondary structure of the molecular
interaction sites described in grater detail in the Examples. The nucleotide
sequence of
the polynucleotide is preferably the nucleotide sequence of the target RNase P
RNAs,
described above. Alternatively, the nucleotide sequence is preferably the
nucleotide
3o sequence of RNase P RNAs from a plurality of different taxonomic species
which also
contain the molecular interaction site.
The polynucleotides of the invention comprise the molecular interaction sites
of the RNase P RNAs. Thus, the polynucleotides of the invention comprise the
CA 02457318 2004-02-20
WO 03/016498 PCT/US02/26418
-18-
nucleotide sequences of the molecular interaction sites. In addition, the
polynucleotides can comprise up to 50, more preferably up to 40, more
preferably up
to 30, more preferably up to 20, and most preferably up to 10 additional
nucleotides at
either the 5' or 3', or combination thereof, ends of each polynucleotide.
Thus, for
example, if a molecular interaction site comprises 25 nucleotides, the
polynucleotide
can comprise up to 75 nucleotides. The nucleotides that are in addition to
those
present in the molecular interaction site are selected to preserve the
secondary
structure of the molecular interaction site. One skilled in the art can select
such
additional nucleotides so as to conserve the secondary structure. The
polynucleotides
l0 can comprise either RNA or DNA or can be chimeric RNA/DNA. The
polynucleotides can comprise modified bases, sugars and backbones that are
well
known to the skilled artisan. Further, a single polynucleotide can comprise a
plurality
of molecular interaction sites. In addition, a plurality of polynucleotides
can, together,
comprise a single molecular interaction site. Alternatively, when a plurality
of
polynucleotides together comprise a molecular interaction site, one skilled in
the art
can attach the polynucleotides to one another, thus, forming a single
polynucleotide.
The portion of the polynucleotide comprising the molecular interaction site
can comprise one or more deletions, insertions and substitutions. Stems, end
loops,
bulges, internal loops, and dangling regions can comprise one or more
deletions,
insertions and substitutions. Thus, for example, an end loop of a molecular
interaction
site that consists of 10 nucleotides can be modified to contain one or more
insertions,
deletions or substitutions, thus, resulting in a shortening or lengthening of
the stem
preceding the end loop. In addition, unpaired, dangling nucleotides that are
adjacent
to, for example, a double-stranded region can be deleted or can be basepaired
with the
addition of another nucleotide, thus, lengthening the stem. In addition,
nucleotide base
pairings within a stem can also be substituted, deleted, or inserted. Thus,
for example,
an A-U basepair within a stem portion of a molecular interaction site can be
replaced
with a G-C basepair. Further, non-canonical base pairing (e.g., G-A, C-T, G-U,
etc.)
can also be present within the polynucleotide. Thus, polynucleotides having at
least
70%, more preferably 80%, more preferably 90%, more preferably 95%, and most
preferably 99% homology with the molecular interaction sites, such as those
set forth
in the Examples below, are included within the scope of the invention. Percent
homology can be determined by, for example, the Gap program (Wisconsin
Sequence
CA 02457318 2004-02-20
WO 03/016498 PCT/US02/26418
-19-
Analysis Package, Version 8 for Unix, Genetics Computer Group, University
Research Park, Madison WI), using the default settings, which uses the
algorithm of
Smith and Waterman (Adv. Appl. Math., 1981, 2, 482-489, which is incorporated
herein by reference in its entirety).
The present invention is also directed to the purified and isolated nucleic
acid
molecules, or polynucleotides, described above, that are present within RNase
P RNA.
The polynucleotides comprising the molecular interaction site mimic the
portion of
the RNase P RNA comprising the molecular interaction site.
Polynucleotides, and modifications thereof, are well known to those skilled in
1o the art. The polynucleotides of the invention can be used, for example, as
research
reagents to detect, for example, naturally occurring molecules that bind the
molecular
interaction sites. Alternatively, the polynucleotides of the invention can be
used to
screen, either actually or virtually, small molecules that bind the molecular
interaction
sites, as described below in greater detail. Virtual generation of compounds
and
screening thereof for binding to molecular interaction sites is described in,
for
example, International Publication WO 99/58947, which is incorporated herein
by
reference in its entirety. The polynucleotides of the invention can also be
used as
decoys to compete with naturally-occurring molecular interaction sites within
a cell
for research, diagnostic and therapeutic applications. In particular, the
polynucleotides
2o can be used in, for example, therapeutic applications to inhibit bacterial
growth.
Molecules that bind to the molecular interaction site modulate, either by
augmenting
or diminishing, the function of RNase P RNA in translation. The
polynucleotides can
also be used in agricultural, industrial and other applications.
The present invention is also directed to compositions comprising at least one
polynucleotide described above. In some embodiments of the invention, two
polynucleotides are included within a composition. The compositions of the
invention
can optionally comprise a carrier. A "carrier" is an acceptable solvent,
diluent,
suspending agent or any other inert vehicle for delivering one or more nucleic
acids to
an animal, and are well known to those skilled in the art. The carrier can be
a
3o pharmaceutically acceptable carrier. The carrier can be liquid or solid and
is selected,
with the planned manner of administration in mind, so as to provide for the
desired
bulk, consistency, etc., when combined with the other components of the
composition.
Typical pharmaceutical carriers include, but are not limited to, binding
agents (e.g.,
CA 02457318 2004-02-20
WO 03/016498 PCT/US02/26418
-20-
pregelatinised maize starch, polyvinylpyrrolidone or hydroxypropyl
methylcellulose,
etc.); fillers (e.g., lactose and other sugars, microcrystalline cellulose,
pectin, gelatin,
calcium sulfate, ethyl cellulose, polyacrylates or calcium hydrogen phosphate,
etc.);
lubricants (e.g., magnesium stearate, talc, silica, colloidal silicon dioxide,
stearic acid,
metallic stearates, hydrogenated vegetable oils, corn starch, polyethylene
glycols,
sodium benzoate, sodium acetate, etc.); disintegrates (e.g., starch, sodium
starch
glycolate, etc.); or wetting agents (e.g., sodium lauryl sulphate, etc.).
The present invention is also directed to methods of identifying compounds
that bind to a molecular interaction site of RNase P RNA comprising providing
a
to numerical representation of the three-dimensional structure of the
molecular
interaction site and providing a compound data ~ set comprising numerical
representations of the three dimensional structures of a plurality of organic
compounds. The numerical representation of the molecular interaction site is
then
compared with members of the compound data set to generate a hierarchy of
organic
compounds ranked in accordance with the ability of the organic compounds to
form
physical interactions with the molecular interaction site.
The present invention is also directed to methods of identifying compounds
that bind to a molecular interaction site of RNase P RNA, or a polynucleotide
comprising the same. In some embodiments of the invention, compounds that bind
to
a molecular interaction site of RNase P RNA, or a polynucleotide comprising
the
same, are identified according to the general methods described in
International
Publication WO 99/58947, which is incorporated herein by reference in its
entirety.
Briefly, the methods comprise providing a numerical representation of the
three
dimensional structure of the molecular interaction site, or a polynucleotide
comprising
the same, providing a compound data set comprising numerical representations
of the
three dimensional structures of a plurality of organic compounds, comparing
the
numerical representation of the molecular interaction site with members of the
compound data set to generate a hierarchy of organic compounds which is ranked
in
accordance with the ability of the organic compounds to form physical
interactions
with the molecular interaction site.
While there are a number of ways to characterize binding between molecular
interaction sites and ligands, such as for example, organic compounds,
methodologies
are described in International Publications WO 99/58719, WO 99/59061, WO
CA 02457318 2004-02-20
WO 03/016498 PCT/US02/26418
-21-
99/58722, WO 99145150, WO 99/58474, and WO 99/58947, each of which is
assigned to the assignee of the present inventions, and each of which is
incorporated
by reference herein in their entirety.
In addition, the present invention is also directed to three dimensional
representations of the nucleic acid molecules, and compositions comprising the
same,
described above. The three dimensional structure of a molecular interaction
site of
RNase P RNA can be manipulated as a numerical representation. The three
dimensional representations, i.e., in silico (e.g. in computer-readable form)
representations can be generated by methods disclosed in, for example,
International
1o Publication WO 99/58947, which is incorporated herein by reference in its
entirety.
Briefly, the three dimensional structure of a molecular interaction site,
preferably of
an RNA, can be manipulated as a numerical representation. Computer software,
that
provides one skilled in the art with the ability to design molecules based on
the
chemistry being performed and on available reaction building blocks is
commercially
available. Software packages such as, for example, SybylBase (Tripos, St.
Louis,
MO), Insight II (Molecular Simulations, San Diego, CA), and Sculpt (MDL
Information Systems, San Leandro, CA) provide means for computational
generation
of structures. These software products also provide means for evaluating and
comparing computationally generated molecules and their structures. b2 silico
2o collections of molecular interaction sites can be generated using the
software from any
of the above-mentioned vendors and others which are or may become available.
The
three dimensional representations can be used, for example, to dock the
molecules) to
potential therapeutic compounds. Thus, the three dimensional representations
can be
used in drug screening procedures. Accordingly, the nucleic acid molecules and
compositions comprising the same of the present invention include the three
dimensional representations of the same.
A set of structural constraints for the molecular interaction site of the
RNase
P RNA can be generated from biochemical analyses such as, for example,
enzymatic
mapping and chemical probes, and from genomics information such as, for
example,
3o covariance and sequence conservation. Information such as this can be used
to pair
bases in the stem or other region of a particular secondary structure.
Additional
structural hypotheses can be generated for noncanonical base pairing schemes
in loop
and bulge regions. A Monte Carlo search procedure can sample the possible
CA 02457318 2004-02-20
WO 03/016498 PCT/US02/26418
-22-
conformations of the RNase P RNA consistent with the program constraints and
produce three dimensional structures.
Reports of the generation of three dimensional, in silico representations are
available from the standpoint of library design, generation, and screening
against
protein targets. Likewise, some efforts in the area of generating RNA models
have
been reported in the literature. However, there are no reports on the use of
structure-
based design approaches to query zf2 silico representations of organic
molecules,
"small" molecules, polynucleotides or other nucleic acids, with three
dimensional, in
silico, representations of RNase P RNA structures. The present invention
preferably
employs computer software that allows the construction of three dimensional
models
of RNase P RNA structure, the construction of three dimensional, in silico
representations of a plurality of organic compounds, "small" molecules,
polymeric
compounds, polynucleotides and other nucleic acids, screening of such in
silico
representations against RNase P RNA molecular interaction sites in silico,
scoring and
identifying the best potential binders from the plurality of compounds, and
finally,
synthesizing such compounds in a combinatorial fashion and testing them
experimentally to identify new ligands for such RNase P RNA targets.
The molecules that may be screened by using the methods of this invention
include, but are not limited to, organic or inorganic, small to large
molecular weight
2o individual compounds, and combinatorial mixture or libraries of ligands,
inhibitors,
agonists, antagonists, substrates, and biopolymers, such as peptides or
polynucleotides. Combinatorial mixtures include, but are not limited to,
collections of
compounds, and libraries of compounds. These mixtures may be generated via
combinatorial synthesis of mixtures or via admixture of individual compounds.
Collections of compounds include, but are not limited to, sets of individual
compounds or sets of mixtures or pools of compounds. These combinatorial
libraries
may be obtained from synthetic or from natural sources such as, for example
to,
microbial, plant, marine, viral and animal materials. Combinatorial libraries
include at
least about twenty compounds and as many as a thousands of individual
compounds
3o and potentially even more. When combinatorial libraries are mixtures of
compounds
these mixtures typically contain from 20 to 5000 compounds preferably from 50
to
1000, more preferably from 50 to 100. Combinations of from 100 to 500 are
useful as
are mixtures having from 500 to 1000 individual species. Typically, members of
CA 02457318 2004-02-20
WO 03/016498 PCT/US02/26418
-23-
combinatorial libraries have molecular weight less than about 10,000 Da, more
preferably less than 7,500 Da, and most preferably less than 5000 Da.
A significant advance in the area of virtual screening was the development of
a software program called DOCK that allows structure-based database searches
to find
and identify the interactions of known molecules to a receptor of interest
(Kuntz et al.,
Acc. Chem. Res., 1994, 27, 117; Geschwend and Kuntz, J. Compt.-Aided Mol.
Des.,
1996, 10, 123). DOCK allows the screening of molecules, whose 3D structures
have
been generated in silico, but for which no prior knowledge of interactions
with the
receptor is available. DOCK, therefore, provides a tool to assist in
discovering new
to ligands to a receptor of interest. DOCK can thus be used for docking the
compounds
prepared according to the methods of the present invention to desired target
molecules. Implementation of DOCK is described in, for example, International
Publication WO 99/58947, which is incorporated herein by reference in its
entirety.
In some embodiments of the invention, an automated computational search
algorithm, such as those described above, is used to predict all of the
allowed three
dimensional molecular interaction site structures from RNase P RNA, which are
consistent with the biochemical and genomic constraints specified by the user.
Based,
for example, on their root-mean-squared deviation values, these structures are
clustered into different families. A representative member or members of each
family
can be subjected to further structural refinement via molecular dynamics with
explicit
solvent and cations.
Structural enumeration and representation by these software programs is
typically done by drawing molecular scaffolds and substituents in two
dimensions.
Once drawn and stored in the computer, these molecules may be rendered into
three
dimensional structures using algorithms present within the commercially
available
software. Preferably, MC-SYM is used to create three dimensional
representations of
the molecular interaction site. The rendering of two dimensional structures of
molecular interaction sites into three dimensional models typically generates
a low
energy conformation or a collection of low energy conformers of each molecule.
The
3o end result of these commercially available programs is the conversion of a
RNase P
RNA sequence containing a molecular interaction site into families of similar
numerical representations of the three dimensional structures of the molecular
interaction site. These numerical representations form an ensemble data set.
CA 02457318 2004-02-20
WO 03/016498 PCT/US02/26418
-24-
The three dimensional structures of a plurality of compounds, preferably
"small" organic compounds, can be designated as a compound data set comprising
numerical representations of the three dimensional structures of the
compounds.
"Small" molecules in this context refers to non-oligomeric organic compounds.
Two
dimensional structures of compounds can be converted to three dimensional
structures, as described above for the molecular interaction sites, and used
for
querying against three dimensional structures of the molecular interaction
sites. The
two dimensional structures of compounds can be generated rapidly using
structure
rendering algorithms commercially available. The three dimensional
representation of
1o the compounds which are polymeric in nature, such as polynucleotides or
other
nucleic acids structures, may be generated using the literature methods
described
above. A three dimensional structure of "small" molecules or other compounds
can be
generated and a low energy conformation can be obtained from a short molecular
dynamics minimization. These three dimensional structures can be stored in a
relational database. The compounds upon which three dimensional structures are
constructed can be proprietary, commercially available, or virtual.
In some embodiments of the invention, a compound data set comprising
numerical representations of the three dimensional structure of a plurality of
organic
compounds is provided by, for example, Converter (MSI, San Diego) from two
dimensional compound libraries generated by, for example, a computer program
modified from a commercial program. Other suitable databases can be
constructed by
converting two dimensional structures of chemical compounds into three
dimensional
structures, as described above. The end result is the conversion of a two
dimensional
structure of organic compounds into numerical representations of the three
dimensional structures of a plurality of organic compounds. These numerical
representations are presented as a compound data set.
After both the numerical representations of the three-dimensional structure of
the polynucleotides comprising the molecular interaction sites and the
compound data
set comprising numerical representations of the three dimensional structures
of a
plurality of organic compounds are obtained, the numerical representations of
the
molecular interaction sites are compared with members of the compound data set
to
generate a hierarchy of the organic compounds. The hierarchy is ranked in
accordance
with the ability of the organic compounds to form physical interactions with
the
CA 02457318 2004-02-20
WO 03/016498 PCT/US02/26418
-2$-
molecular interaction site. Preferably, the comparing is carried out seriatim
upon the
members of the compound data set. In accordance with some embodiments, the
comparison can be performed with a plurality of polynucleotides comprising
molecular interaction sites at the same time.
A variety of theoretical and computational methods are known by those
skilled in the art to study and optimize the interactions of "small" molecules
or
organic compounds with biological targets such as nucleic acids. These
structure-
based drug design tools have been very useful in modelling the interactions of
proteins
with small molecule ligands and in optimizing these interactions. Typically
this type
of study has been performed when the structure of the protein receptor was
known by
querying individual small molecules, one at a time, against this receptor.
Usually these
small molecules had either been co-crystallized with the receptor, were
related to
other molecules that had been co-crystallized or were molecules for which some
body
of knowledge existed concerning their interactions with the receptor. DOCK, as
described above, can be used to find and identify molecules that are expected
to bind
to polynucleotides comprising the molecular interaction sites and, hence,
RNase P
RNA of interest. DOCK 4.0 is commercially available from the Regents of the
University of California. Equivalent programs are also comprehended in the
present
invention.
2o The DOCK program has been widely applied to protein targets and the
identification of ligands that bind to them. Typically, new classes of
molecules that
bind to known targets have been identified, and later verified by in vitro
experiments.
The DOCK software program consists of several modules, including SPHGEN (Kuntz
et al., T. Mol. Biol., 1982,161, 269) and CHEMGRm (Meng et al., J. Comput.
Chem.,
1992, 13, 505, each of which is incorporated herein by reference in its
entirety).
SPHGEN generates clusters of overlapping spheres that describe the solvent-
accessible surface of the binding pocket within the target receptor. Each
cluster
represents a possible binding site for small molecules. CHEMGRm precalculates
and
stores in a grid file the information necessary for force field scoring of the
interactions
3o between binding molecule and target RNase P RNA. The scoring function
approximates molecular mechanics interaction energies and consists of van der
Waals
and electrostatic components. DOCK uses the selected cluster of spheres to
orient
ligands molecules in the targeted site on RNase P RNA. Each molecule within a
CA 02457318 2004-02-20
WO 03/016498 PCT/US02/26418
-26-
previously generated three dimensional database is tested in thousands of
orientations
within the site, and each orientation is evaluated by the scoring function.
Only that
orientation with the best score for each compound so screened is stored in the
output
file. Finally, all compounds of the database are ranked in a hierarchy in
order of their
scores and a collection of the best candidates may then be screened
experimentally.
Using DOCK, numerous ligands have been identified for a variety of protein
targets. Recent efforts in this area have resulted in reports of the use of
DOCK to
identify and design small molecule ligands that exhibit binding specificity
for nucleic
acids such as RNA double helices. While RNA plays a significant role in many
diseases such as AIDS, viral and bacterial infections, few studies have been
made on
small molecules capable of specific RNA binding. Compounds possessing
specificity
for the RNA double helix, based on the unique geometry of its deep major
groove,
were identified using the DOCK methodology. Chen et al., Biochemistry, 1997,
36,
11402 and Kuntz et al., Acc. Chem. Res., 1994, 27, 117. Recently, the
application of
DOCK to the problem of ligand recognition in DNA quadruplexes has been
reported.
Chen et al., Proc. lVatl. Acad. Sci., 1996, 93, 2635.
Preferably, individual compounds are designated as mol files, for example,
and combined into a collection of in silieo representations using an
appropriate
chemical structure program or equivalent software. These two dimensional mol
files
are exported and converted into three dimensional structures using commercial
software such as Converter (Molecular Simulations Inc., San Diego) or
equivalent
software, as described above. Atom types suitable for use with a docking
program
such as DOCK or QXP are assigned to all atoms in the three dimensional mol
file
using software such as, for example, Babel, or with other equivalent software.
A low-energy conformation of each molecule is generated with software such
as Discover (MSI, San Diego). An orientation search is performed by bringing
each
compound of the plurality of compounds into proximity with the molecular
interaction
site in many orientations using DOCK or QXP. A contact score is determined for
each
orientation, and the optimum orientation of the compound is subsequently used.
3o Alternatively, the conformation of the compound can be determined from a
template
conformation of the scaffold determined previously.
The interaction of a plurality of compounds and molecular interaction sites is
examined by comparing the numerical representations of the molecular
interaction
CA 02457318 2004-02-20
WO 03/016498 PCT/US02/26418
-27-
sites with members of the compound data set. Preferably, a plurality of
compounds
such as those generated by a computer program or otherwise, is compared to the
molecular interaction site and undergoes random "motions" among the dihedral
bonds
of the compounds. Preferably about 20,000 to 100,000 compounds are compared to
at
least one molecular interaction site. Typically, 20,000 compounds are compared
to
about five molecular interaction sites and scored. Individual conformations of
the
three dimensional structures are placed at the target site in many
orientations.
Moreover, during execution of the DOCK program, the compounds and molecular
interaction sites are allowed to be "flexible" such that the optimum hydrogen
bonding,
electrostatic, and van der Waals contacts can be realized. The energy of the
interaction
is calculated and stored for 10-15 possible orientations of the compounds and
molecular interaction sites. QXP methodology allows true flexibility in both
the ligand
and target and is presently preferred.
The relative weights of each energy contribution are updated constantly to
insure that the calculated binding scores for all compounds reflect the
experimental
binding data. The binding energy for each orientation is scored on the basis
of
hydrogen bonding, van der Waals contacts, electrostatics,
solvationldesolvation, and
the quality of the fit. The lowest-energy van der Waals, dipolar, and hydrogen
bonding
interactions between the compound and the molecular interaction site are
determined,
and summed. In some embodiments, these parameters can be adjusted according to
the
results obtained empirically. The binding energies for each molecule against
the target
are output to a relational database. The relational database contains a
hierarchy of the
compounds ranked in accordance with the ability of the compounds to form
physical
interactions with the molecular interaction site. The higher ranked compounds
are
better able to form physical interactions with the molecular interaction site.
In another embodiment, the highest ranking, i.e., the best fitting compounds,
are selected for synthesis. In some embodiments of the invention, those
compounds
which are likely to have desired binding characteristics based on binding data
are
selected for synthesis. Preferably the highest ranking 5% are selected for
synthesis.
3o More preferably, the highest ranking 10% are selected for syntheses. Even
more
preferably, the highest ranking 20% are selected for synthesis. The synthesis
of the
selected compounds can be automated using a parallel array synthesizer or
prepared
using solution-phase or other solid-phase methods and instruments. In
addition, the
CA 02457318 2004-02-20
WO 03/016498 PCT/US02/26418
_~$_
interaction of the highly ranked compounds with the nucleic acid containing
the
molecular interaction site is assessed as described below.
The interaction of the highly ranked organic compounds with the
polynucleotide comprising the RNase P RNA molecular interaction site can be
assessed by numerous methods known to those skilled in the art. For example,
the
highest ranking compounds can be tested for activity in high-throughput (HTS)
functional and cellular screens. HTS assays can be determined by scintillation
proximity, precipitation, luminescence-based formats, filtration based assays,
colorometric assays, and the like. Lead compounds can then be scaled up and
tested in
to animal models for activity and toxicity. The assessment preferably
comprises mass
spectrometry of a mixture of the RNase P RNA polynucleotide and at least one
of the
compounds or a functional bioassay.
Certain evaluation techniques employing mass spectroscopy are disclosed in
International Publication WO 99145150, which is incorporated herein by
reference in
its entirety, as exemplary of certain useful and mass spectrometric techniques
for use
herewith. It is to be specifically understood, however, that it is not
essential that these
particular mass spectrometric techniques be employed in order to perform the
present
invention. Rather, any evaluative technique may be undertaken so long as the
objectives of the present invention are maintained.
2o In some embodiments of the invention, the highest ranking 20% of
compounds from the hierarchy generated using the DOCK program or QXP are used
to generate a further data set of three dimensional representations of organic
compounds comprising compounds which are chemically related to the compounds
ranking high in the hierarchy. Although the best fitting compounds are likely
to be in
the highest ranking 1%, additional compounds, up to about 20%, are selected
for a
second comparison so as to provide diversity (ring size, chain length,
functional
groups). This process insures that small errors in the molecular interaction
sites are not
propagated into the compound identification process. The resulting
structure/score
data from the highest ranking 20%, for example, is studied mathematically
(clustered)
to find trends or features within the compounds which enhance binding. The
compounds are clustered into different groups. Chemical synthesis and
screening of
the compounds, described above, allows the computed DOCK or QXP scores to be
correlated with the actual binding data. After the compounds have been
prepared and
CA 02457318 2004-02-20
WO 03/016498 PCT/US02/26418
-29-
screened, the predicted binding energy and the observed Kd values are
correlated for
each compound.
The results are used to develop a predictive scoring scheme, which weighs
various factors (steric, electrostatic) appropriately. The above strategy
allows rapid
evaluation of a number of scaffolds with varying sizes and shapes of different
functional groups for the high ranked compounds. In this manner, a further
data set of
representations of organic compounds comprising compounds which are chemically
related to the organic compounds which rank high in the hierarchy can be
compared to
the numerical representations of the molecular interaction site to determine a
further
hierarchy ranked in accordance with the ability of the organic compounds to
form
physical interactions with the molecular interaction site. In this manner, the
further
data set of representations of the three dimensional structures of compound
which are
related to the compounds ranked high in the hierarchy are produced and have,
in
effect, been optimized by correlating actual binding with virtual binding. The
entire
cycle can be iterated as desired until the desired number of compounds highest
in the
hierarchy are produced.
Compounds which have been determined to have affinity and specificity for a
target biomolecule, especially a target RNase P RNA or which otherwise have
been
shown to be able to bind to the target RNase P RNA to effect modulation
thereof, can,
2o in accordance with some embodiments of this invention, be tagged or labeled
in a
detectable fashion. Such labeling may include all of the labeling forms known
to
persons of skill in the art such as fluorophore, radiolabel, enzymatic label
and many
other forms. Such labeling or tagging facilitates detection of molecular
interaction
sites and permits facile mapping of chromosomes and other useful processes.
In order that the invention disclosed herein may be more efficiently
understood, examples are provided below. It should be understood that these
examples
are for illustrative purposes only and are not to be construed as limiting the
invention
in any manner. Various modifications of the invention, in addition to those
described
herein, will be apparent to those skilled in the art from the foregoing
description. Such
3o modifications are also intended to fall within the scope of the appended
claims. In
addition, the disclosures of each patent, patent application, and publication
cited or
described in this document are incorporated herein by reference in their
entirety.
CA 02457318 2004-02-20
WO 03/016498 PCT/US02/26418
-30-
EXAMPLES
Example 1: Selection of RNase P RNA
To illustrate the strategy for identifying molecular interaction sites for
small
molecules, the RNase P RNA was used. The structures of the RNase P RNA are
disclosed in Massire et al., J. Mol. Biol.,1998, 279, 773-793. The RNase P RNA
is an
RNA of approximately 375 to 400 nucleotides that folds into several domains.
Example 2: Molecular Interaction Sites In RNase P RNA
Numerous molecular interaction sites have been discovered within RNAse P
i0 RNA. Site 1 comprises a region of RNA comprising a first and second
polynucleotide.
The first polynucleotide comprises about twenty four nucleotides to about
sixty nine
nucleotides, wherein portions of the polynucleotide form a double-stranded RNA
having the following features (5' to 3'): a dangling region comprising from
about one
to about three nucleotides, a first side of a first stem comprising from about
three
nucleotides to about eight nucleotides, a first side of a second stem
comprising from
about three nucleotides to about eight nucleotides, a first terminal loop
comprising
from about three nucleotides to about eight nucleotides, a second side of the
second
stem comprising from about three nucleotides to about eight nucleotides, a
first side of
a third stem comprising from about two nucleotides to about six nucleotides, a
second
2o terminal loop comprising from about two nucleotides to about six
nucleotides, a
second side of the third stem comprising from about two nucleotides to about
six
nucleotides wherein a bulge comprising from about one nucleotide to about
three
nucleotides is optionally present in the second side of the third stem, a
first side of a
fourth stem comprising from about two nucleotides to about six nucleotides
wherein a
bulge comprising from about one nucleotide to about five nucleotides is
optionally
present in the first side of the fourth stem, and a dangling region comprising
from
about one nucleotide to about five nucleotides. The second polynucleotide
comprises
from about eight nucleotides to about twenty two nucleotides, wherein portions
of the
polynucleotide form a double-stranded RNA having the following features (5' to
3'): a
dangling region comprising from about three nucleotides to about eight
nucleotides, a
second side of the fourth stem comprising from about two nucleotides to about
six
nucleotides, and a second side of the first stem comprising from about three
nucleotides to about eight nucleotides.
CA 02457318 2004-02-20
WO 03/016498 PCT/US02/26418
-31-
In regard to site 1, the first polynucleotide preferably comprises forty five
nucleotides, wherein portions of the polynucleotide form a double-stranded RNA
having the following features (5' to 3'): a dangling region comprising two
nucleotides,
a first side of a first stem comprising five nucleotides, a first side of a
second stem
comprising five nucleotides, a first terminal loop comprising five
nucleotides, a
second side of the second stem comprising five nucleotides, a first side of a
third stem
comprising four nucleotides, a second terminal loop comprising four
nucleotides, a
second side of the third stem comprising four nucleotides wherein a bulge
comprising
one nucleotide is present between the third and fourth nucleotide of the
second side of
the third stem, a first side of a fourth stem comprising four nucleotides
wherein a
bulge comprising three nucleotides is present between the second and third
nucleotides of the first side of the fourth stem, and a dangling region
comprising three
nucleotides. Preferably, the first polynucleotide comprises the sequence 5'-
cagggugcc
agguaacgccugggggggaaacccacgaccagugca-3' (SEQ ll~ NO:1) (bolded nucleotides
indicate preferred basepairing). The second polynucleotide preferably
comprises
fourteen nucleotides, wherein portions of the polynucleotide form a double-
stranded
RNA having the following features (5' to 3'): a dangling region comprising
five
nucleotides, a second side of the fourth stem comprising four nucleotides, and
a
second side of the first stem comprising five nucleotides. Preferably, the
second
2o polynucleotide comprises the sequence 5'-gguaaacuccaccc-3' (SEQ )D N0:2)
(bolded
nucleotides indicate preferred basepairing). Site 1 is present in E. coli, as
shown in
Figure 1.
Site 2 comprises a region of RNA comprising a first, second and third
polynucleotide. The first polynucleotide comprises from about six nucleotides
to
about sixteen nucleotides, wherein portions of the polynucleotide form a
double-
stranded RNA having the following features (5' to 3'): a dangling region
comprising
from about one nucleotide to about three nucleotides, and a first side of a
first stem
comprising from about four nucleotides to about ten nucleotides wherein a
bulge
comprising from about one nucleotide to about three nucleotides is optionally
present
3o in the first side of the first stem. The second polynucleotide comprises
from about
thirteen nucleotides to about thirty four nucleotides, wherein portions of the
polynucleotide form a double-stranded RNA having the following features (5' to
3'): a
second side of the first stem comprising from about four to about ten
nucleotides
CA 02457318 2004-02-20
WO 03/016498 PCT/US02/26418
-32-
wherein a bulge comprising from about one nucleotide to about three
nucleotides is
optionally present in the second side of the first stem, a bulge comprising
from about
four nucleotides to about ten nucleotides, a first side of a second stem
comprising
from about three nucleotide to about nine nucleotides, and a dangling region
comprising from about one nucleotide to about two nucleotides. The third .
polynucleotide comprises from about five nucleotides to about thirteen
nucleotides,
wherein portions of the polynucleotide form a double-stranded RNA having the
following features (5' to 3'): a dangling region comprising from about one
nucleotide
to about two nucleotides, a second side of the second stem comprising from
about
l0 three nucleotides to about nine nucleotides, and a dangling region
comprising from
about one nucleotide to about two nucleotides.
In regard to site 2, the first polynucleotide preferably comprises eleven
nucleotides, wherein portions of the polynucleotide form a double-stranded RNA
having the following features (5' to 3'): a dangling region comprising two
nucleotides,
and a first side of a first stem comprising seven nucleotides wherein a bulge
comprising two nucleotides is present between the fifth and sixth nucleotide
of the
first side of the first stem. Preferably, the first polynucleotide comprises
the sequence
5'-aaccgccgaug-3' (SEQ m N0:3) (bolded nucleotides indicate preferred
basepairing). The second polynucleotide preferably comprises twenty three
2o nucleotides, wherein portions of the polynucleotide form a double-stranded
RNA
having the following features (5' to 3'): a second side of the first stem
comprising
seven nucleotides wherein a bulge comprising two nucleotides is present
between the
fifth and sixth nucleotide of the second side of the first stem, a bulge
comprising seven
nucleotides, a first side of a second stem comprising six nucleotides, and a
dangling
region comprising one nucleotide. Preferably, the second polynucleotide
comprises
the sequence 5'-cagguaagggugaaagggugcgg-3' (SEQ ID N0:4) (bolded nucleotides
indicate preferred basepairing). The third polynucleotide preferably comprises
eight
nucleotides, wherein portions of the polynucleotide form a double-stranded RNA
having the following features (5' to 3'): a dangling region comprising one
nucleotide,
a second side of the second stem comprising six nucleotides, and a dangling
region
comprising one nucleotide. Preferably, the third polynucleotide comprises the
sequence 5'-gcgcaccg-3' (SEQ ID N0:5) (bolded nucleotides indicate preferred
basepairing). Site 2 is present in E. coli, as shown in Figure 1.
CA 02457318 2004-02-20
WO 03/016498 PCT/US02/26418
-33-
Site 3 comprises a region of RNA comprising a first and second
polynucleotide. The first polynucleotide comprises from about ten nucleotides
to
about twenty six nucleotides, wherein portions of the polynucleotide form a
double-
stranded RNA having the following features (5' to 3'): a dangling region
comprising
from about one nucleotide to about three nucleotides, a first side of a first
stem
comprising from about two nucleotides to about six nucleotides, a first side
of an
internal loop comprising from about three nucleotides to about nine
nucleotides, a first
side of a second stem comprising from about three nucleotides to about six
nucleotides, and a dangling region comprising from about one nucleotide to
about two
to nucleotides. The second polynucleotide comprises from about ten nucleotide
to about
twenty seven nucleotides, wherein portions of the polynucleotide form a double-
stranded RNA having the following features (5' to 3' ): a second side of the
second
stem comprising from about three nucleotides to about nine nucleotides, a
second side
of the internal loop comprising from about three nucleotides to about seven
nucleotides, a second side of the first stem comprising from about two
nucleotides to
about six nucleotides, and a dangling region comprising from about two
nucleotides to
about five nucleotides.
In regard to site 3, the first polynucleotide preferably comprises nineteen
nucleotides, wherein portions of the polynucleotide form a double-stranded RNA
2o having the following features (5' to 3'): a dangling region comprising two
nucleotides,
a first side of a first stem comprising four nucleotides, a first side of an
internal loop
comprising six nucleotides, a first side of a second stem comprising six
nucleotides,
and a dangling region comprising one nucleotide. Preferably, the first
polynucleotide
comprises the sequence 5'-aaggccaaauagggguuca-3' (SEQ ID N0:6) (bolded
nucleotides indicate preferred basepairing). The second polynucleotide
preferably
comprises eighteen nucleotides, wherein portions of the polynucleotide form a
double-
stranded RNA having the following features (5' to 3'): a second side of the
second
stem comprising six nucleotides, a second side of the internal loop comprising
five
nucleotides, a second side of the first stem comprising four nucleotides, and
a
3o dangling region comprising three nucleotides. Preferably, the second
polynucleotide
comprises the sequence 5'-gaacccggguaggcugcu-3' (SEQ >D N0:7) (bolded
nucleotides indicate preferred basepairing). Site 3 is present in E. coli, as
shown in
Figure 1.
CA 02457318 2004-02-20
WO 03/016498 PCT/US02/26418
-34-
Site 4 comprises a region of RNA comprising a polynucleotide comprising
from about twelve nucleotides to about thirty four nucleotides, wherein
portions of the
polynucleotide form a double-stranded RNA having the following features (5' to
3'): a
first side of a stem comprising from about three nucleotides to about nine
nucleotides
wherein a first side of an internal loop comprising from about two nucleotides
to about
five nucleotides is present in the first side of the stem, a terminal loop
comprising
from about two nucleotides to about six nucleotides, a second side of the stem
comprising from about three nucleotides to about nine nucleotides wherein a
second
side of the internal loop comprising from about one nucleotide to about three
nucleotides is present in the second side of the stem, and a dangling region
comprising
from about one nucleotides to about two nucleotides.
In regard to site 4, the region of RNA preferably comprises a polynucleotide
comprising twenty two nucleotides, wherein portions of the polynucleotide form
a
double-stranded RNA having the following features (5' to 3'): a first side of
a stem
comprising six nucleotides wherein a first side of an internal loop comprising
three
nucleotides is present between the third and fourth nucleotides of the first
side of the
stem, a terminal loop comprising four nucleotides, a second side of the stem
comprising six nucleotides wherein a second side of the internal loop
comprising two
nucleotides is present between the third and fourth nucleotides of the second
side of
the stem, and a dangling region comprising one nucleotide. Preferably, the
polynucleotide comprises the sequence 5'-gccuacgucuucggauauggcu-3' (SEQ ll~
N0:8) (bolded nucleotides indicate preferred basepairing). Site 4 is present
in B.
subtilis, as shown in Figure 2.
Site 5 comprises a region of RNA comprising a first, second, third, fourth and
fifth polynucleotide. The first polynucleotide comprises from about three
nucleotides
to about nine nucleotides, wherein portions of the polynucleotide form a
double-
stranded RNA having the following features (5' to 3'): a first side of a first
stem
comprising from about two nucleotides to about six nucleotides and a first
side of a
second stem comprising from about one nucleotide to about three nucleotides.
The
second polynucleotide comprises from about three nucleotides to about eight
nucleotides, wherein portions of the polynucleotide form a double-stranded RNA
having the following features (5' to 3'): a second side of the second stem
comprising
from about one nucleotide to about three nucleotides and a first side of a
third stem
CA 02457318 2004-02-20
WO 03/016498 PCT/US02/26418
-35-
comprising from about two nucleotides to about five nucleotides. The third
polynucleotide comprises from about seven nucleotides to about eighteen
nucleotides,
wherein portions of the polynucleotide form a double-stranded RNA having the
following features (5' to 3' ): a second side of the third stem comprising
from about
two nucleotides to about five nucleotides wherein a bulge comprising from
about one
nucleotide to about two nucleotides is optionally present in the second side
of the third
stem, a first side of a fourth stem comprising from about one nucleotide to
about three
nucleotides, a bulge comprising from about one nucleotide to about three
nucleotides,
and a first side of a fifth stem comprising from about two nucleotides to
about five
1o nucleotides. The fourth polynucleotide comprises from about eight
nucleotides to
about twenty nucleotides, wherein portions of the polynucleotide form a double-
stranded RNA having the following features (5' to 3'): a second side of the
fifth stem
comprising from about two nucleotides to about five nucleotides, a bulge
comprising
from about three nucleotides to about seven nucleotides, a first side of a
sixth stem
comprising from about one nucleotide to about three nucleotides, and a
dangling
region comprising from about two nucleotides to about five nucleotides. The
fifth
polynucleotide comprises from about five nucleotides to about fifteen
nucleotides,
wherein portions of the polynucleotide form a double-stranded RNA having the
following features (5' to 3'): a dangling region comprising from about one
nucleotide
2o to about three nucleotides, a second side of the sixth stem comprising from
about one
nucleotide to about three nucleotides, a second side of the fourth stem
comprising
from about one nucleotide to about three nucleotides, and a second side of the
first
stem comprising from about two nucleotides to about six nucleotides.
In regard to site 5, the first polynucleotide preferably comprises six
nucleotides, wherein portions of the polynucleotide form a double-stranded RNA
having the following features (5' to 3'): a first side of a first stem
comprising four
nucleotides and a first side of a second stem comprising two nucleotides.
Preferably,
the first polynucleotide comprises the sequence 5'-cgugcc-3' (bolded
nucleotides
indicate preferred basepairing). The second polynucleotide preferably
comprises five
nucleotides, wherein portions of the polynucleotide form a double-stranded RNA
having the following features (5' to 3'): a second side of the second stem
comprising
two nucleotides and a first side of a third stem comprising three nucleotides.
Preferably, the second polynucleotide comprises the sequence 5'-gggca-3'
(bolded
CA 02457318 2004-02-20
WO 03/016498 PCT/US02/26418
-36-
nucleotides indicate preferred basepairing). The third polynucleotide
preferably
comprises ten nucleotides, wherein portions of the polynucleotide form a
double-
stranded RNA having the following features (5' to 3'): a second side of the
third stem
comprising three nucleotides wherein a bulge comprising one nucleotide is
present
between the second and third nucleotides of the second side of the third stem,
a first
side of a fourth stem comprising two nucleotides, a bulge comprising one
nucleotide,
and a first side of a fifth stem comprising three nucleotides. Preferably, the
third
polynucleotide comprises the sequence 5'-ugacggcagg-3' (SEQ >D N0:9) (bolded
nucleotides indicate preferred basepairing). The fourth polynucleotide
preferably
comprises thirteen nucleotides, wherein portions of the polynucleotide form a
double-
stranded RNA having the following features (5' to 3' ): a second side of the
fifth stem
comprising three nucleotides, a bulge comprising five nucleotides, a first
side of a
sixth stem comprising two nucleotides, and a dangling region comprising three
nucleotides. Preferably, the fourth polynucleotide comprises the sequence 5'-
ccuugaaagugcc-3' (SEQ JD NO:10) (bolded nucleotides indicate preferred
basepairing). The fifth polynucleotide preferably comprises ten nucleotides,
wherein
portions of the polynucleotide form a double-stranded RNA having the following
features (5' to 3'): a dangling region comprising two nucleotides, a second
side of the
sixth stem comprising two nucleotides, a second side of the fourth stem
comprising
2o two nucleotides, and a second side of the first stem comprising four
nucleotides.
Preferably, the fifth polynucleotide comprises the sequence 5'-aaaccccucg-3'
(SEQ
ID N0:11) (bolded nucleotides indicate preferred basepairing). Site 5 is
present in B.
subtilis, as shown in Figure 2.
Site 6 comprises a region of RNA comprising a polynucleotide comprising
from about thirteen nucleotides to about thirty four nucleotides, wherein
portions of
the polynucleotide form a double-stranded RNA having the following features
(5' to
3'): a dangling region comprising from about two nucleotides to about five
nucleotides, a first side of a stem comprising from about two nucleotides to
about five
nucleotides, a terminal loop comprising from about six nucleotides to about
sixteen
nucleotides, a second side of the stem comprising from about two nucleotides
to about
five nucleotides, and a dangling region comprising from about one nucleotide
to about
three nucleotides.
CA 02457318 2004-02-20
WO 03/016498 PCT/US02/26418
-37-
In regard to site 6, the region of RNA preferably comprises a polynucleotide
comprising twenty two nucleotides, wherein portions of the polynucleotide form
a
double-stranded RNA having the following features (5' to 3'): a dangling
region
comprising three nucleotides, a first side of a stem comprising three
nucleotides, a
terminal loop comprising eleven nucleotides, a second side of the stem
comprising
three nucleotides, and a dangling region comprising two nucleotides.
Preferably, the
polynucleotide comprises the sequence 5'-aaacccaaauuuugguagggga-3' (SEQ
N0:12) (bolded nucleotides indicate preferred basepairing). Site 6 is present
in B.
subtilis, as shown in Figure 2.
to
CA 02457318 2004-02-20
WO 03/016498 PCT/US02/26418
SEQUENCE LISTING
<110> Ecker, David J.
<120> Molecular Interaction Sites Of RNase P RNA And Methods Of
Modulating The Same
<130> IBIS-0425
<150> 60/314,020
<151> 2001-08-21
<160> 12
<170> PatentIn version 3.1
<210> 1
<211> 45
<212> RNA
<213> Artificial Sequence
<220>
<223> Synthetic Construct
<400> 1
cagggugcca gguaacgccu gggggggaaa cccacgacca gugca 45
<210> 2
<211> 14
<212> RNA
<213> Artificial Sequence
<220>
<223> Synthetic Construct
<400> 2
gguaaacucc accc 14
<210> 3
<211> 11
<212> RNA
<213> Artificial Sequence
<220>
<223> Synthetic Construct
<400> 3
aaccgccgau g 11
<210> 4
<211> 23
<212> RNA
<213> Artificial Sequence
<220>
CA 02457318 2004-02-20
WO 03/016498 PCT/US02/26418
<223> Synthetic Construct
<400> 4
cagguaaggg ugaaagggug cgg 23
<210> 5
<211> 8
<212> RNA
<213> Artificial Sequence
<220>
<223> Synthetic Construct
<400> 5
gcgcaccg 8
<210> 6
<211> 18
<212> RNA
<213> Artificial Sequence
<220>
<223> Synthetic Construct
<400> 6 '
aaggccaaau agggguuc 18
<210> 7
<211> 18
<212> RNA
<213> Artificial Sequence
<220>
<223> Synthetic Construct
<400> 7
gaacccgggu aggcugcu 18
<210> 8
<211> 22
<212> RNA
<213> Artificial Sequence
<220>
<223> Synthetic Construct
<400> 8
gccuacgucu ucggauaugg cu 22
<210> 9
<211> 10
<212> RNA
2
CA 02457318 2004-02-20
WO 03/016498 PCT/US02/26418
<213> Artificial Sequence
<220>
<223> Synthetic Construct
<400> 9
ugacggcagg
<210> 10
<211> 13
<212> RNA
<213> Artificial Sequence
<220>
<223> Synthetic Construct
<400> 10
ccuugaaagu gcc 13
<210> 11
<211> 10
<212> RNA
<213> Artificial Sequence
<220>
<223> Synthetic Construct
<400> 11
aaaccccucg 10
<210> 12
<211> 22
<212> RNA
<213> Artificial Sequence
<220>
<223> Synthetic Construct
<400> 12
aaacccaaau uuugguaggg ga 22
3