Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02498194 2005-03-07
WO 2004/035612 PCT/US2003/027489
COMPOSITION, METHOD AND USE OF
BI-FUNCTIONAL BIOMATERIALS
RELATED APPLICATIONS
This patent application claims benefit of provisional application
no.60/408,528 filed
September 4, 2002 to Belcher et al. which is hereby incorporated by reference
in its entirety.
TECHNICAL FIELD OF THE INVENTION
The present invention relates in general to the field of polymer chemistry,
and more
particularly to biologically modified polymers for use as biomaterials.
STATEMENT OF GOVERNMENT SUPPORT
The United States Federal Government may have certain rights in this
invention. The
subject matter of the application was carned out in part under Federal
Government grant
1 S number from the department of
BACKGROUND OF THE INVENTION
Without limiting the scope of the invention, its background is described in
connection
with the development of biological materials (biomaterials, hereafter) by
incorporating
polymers with organic or biologic substituents (bi-functional biomaterials,
hereafter). A
1
CA 02498194 2005-03-07
WO 2004/035612 PCT/US2003/027489
nucleotide and/or amino acid sequence listing is incorporated by reference of
the material on
computer readable form.
Heretofore, in this field, the means of developing biologic materials has
taken a
variety of approaches, wherein traditionally non-biologic (i.e., non-organic)
materials are
S modified in one or more ways to present biologic features that resemble or
are recognized by
natural biologic tissue (i.e., organ tissue). Modifications have included the
use of protein
adsorption and self assembly, synthesis of novel graft-copolymers with the
desired functional
groups, and direct covalent surface modifications. Ultimately, the goal of
manufacturing
such biologic materials is to create a biomaterial that is flexible enough to
adapt to changes in
molecular design, is easy to synthesize, and can be applied to many different
biologic uses
(e.g., claudication, implantation, transplantation, biologic regeneration,
growth, and as
biologic replacements, modifications, or substitutions).
Recent investigations into creating biologic materials include the use of a
microfabrication technique. Here, proteins and other molecular structures
(including cells
and/or tissue) are attached to the surface of a material that exhibits
biologic properties (e.g.,
binds to one ore more biologic or organic compounds); the attachment is
generally through
nonspecific or specific recognition of the protein or other molecular
structures to the
material. For example, microcontact printing with a PDMS stamp is used to
create
micropatterns on the surface of a material. In the second stage, proteins or
other molecular
structures are adsorbed to the solid surface of the material. The unfortunate
consequence of
using such a technique is that the adsorption is nonuniform and creates
irregular surfaces,
much of which does not exhibit the necessary biologic properties that were
initially desired.
This is because the process is largely dependent upon non-specific
interactions between the
molecular structure and the material surface and these non-specific
interactions result in less
than optimal surfaces with randomly oriented molecules.
Others have engineered polymer surfaces to a material, using engineered
polymers
that may even control the adhesion of molecular structures to the polymer
surface and are
thought to be able to be used to attract one or more cells to the surface
while maintaining the
phenotypic expression of the cells. The drawback is that few polymers really
have suitable
2
CA 02498194 2005-03-07
WO 2004/035612 PCT/US2003/027489
functional groups that are able to covalently attach to a biologic structure.
This fundamental
flaw limits the use of a polymer as a biologic surface unless it is also
modified to become
more attractive to one or more biologic structures (e.g., organic compounds,
biologic
compounds, cells, tissue, etc.). Common approaches to functionally modifying a
polymer
S include introducing reactive groups (e.g., poly(L-lysine)) at existing
polymer surfaces by
incorporating monomer units into the polymer backbone. Such approaches,
however, are
cost prohibitive by requiring complicated synthetic pathways and do not create
uniform
biomaterial surfaces (i.e., a surface containing one or more biologic
structure).
An alternative method is a silanization technique that immobilizes peptides on
the
surface of a material. The method was demonstrated by depositing a silane film
with
terminal functional groups on a titanium oxide surface. In addition, the
resulting surface
could be further modified with different bi-functional linkers, eventually
leading to the
covalent attachment of a peptide sequence such as Arg-Gly-Asp (RGD)-a cellular
recognition sequence used by several biologic proteins. The technique was also
altered using
different silane-like compounds such as aminosilane. Accordingly, a number of
reactions
with bi-functional linkers were performed, including: (a) glutaraldehyde to
yield a linkage
between the aldehyde imine and the peptide amine; and (b) aminosilane with a
mixture of
peptides and carbodiimides to yield a linkage between the amide and peptide
carboxyl
groups. These reactions were limited, however, in their ability to create
specific peptide
attachments at one or more defined sites. Consequently, unordered and
nonuniform surfaces
are produced.
Subsequent surface modification techniques have been used to create biologic
materials with specific binding surfaces. For example, one technique was
developed to
create a neural surface (e.g., similar to the extracellular matrix of nervous
tissue) using a
polymer coupled to peptides. Here, poly(tetrafluroethylene-co-
hexafluoropropylene) was
reduced with sodium naphthalide to introduce carbon-carbon double bonds at the
surface
(e.g., a carbon-like film) and the reduced surface was then further modified
to introduce
hydroxyl groups (e.g., with hydroboration/oxidation) or carboxylic acid groups
(e.g., through
oxidation). The polymer, thus, contains either a hydroxyl (-CHXOH) or
carboxylic acid (-
3
CA 02498194 2005-03-07
WO 2004/035612 PCT/US2003/027489
COOH) surface that could be coupled to one or more peptides. In fact, the
attachment of 5-
and 6-mer peptides was found to promote neurite extension (i.e., modified
growth).
Materials with surfaces that resist protein adsorption and fouling have also
been
developed. These materials may be further modified with biologic components to
promote
specific molecular and/or cellular interactions. Polymers such as polyethylene
glycol) or
PEG that resist protein binding are suitable to use for these modifications.
In addition,
peptides such as those containing RGD sequences (e.g., acrylamidoyl peptides)
may be
incorporated into mixtures of PEG diacrylate to create a peptide-modified
polymer.
Unfortunately, this technique is unable to control the spatial orientation of
peptides on the
material (i.e., polymer) surface and only works with biologic structures of
limited type and
size. This type of modification is limited to polymers that have the ability
for Pegylation,
which can be important for immobilization of peptide via covalent reactions.
As evidenced by the above, current techniques are unable to create biologic
materials
with functional surfaces, that is surfaces that displays properties that allow
for and promote
interactions between the surface and another biologic structure (e.g., nucleic
acid, protein,
cell, tissue, organ, chromophore, etc.). There is a need, therefore, to
develop such a
technique that is both cost-effective and adaptable to one or more biologic
structures to
enable its widespread application.
SUMMARY OF THE INVENTION
The invention disclosed herein is a composition, method and use of a modified,
bi-
functionally-linked biopolymer, wherein the functional linkage is between a
biomaterial
surface and cells or biologic molecules.
The present invention takes advantage of molecular screening methods to
prepare
molecular structures with specific binding motifs and/or binding properties.
These molecular
structures are used as the polymer linkers. More importantly, the present
invention allows
for the self selection and screening of molecular structures that display one
or more
specifically required properties. For example, one can select and prepare
linkers that
specifically bind with high affinity to one or more selected materials.
4
CA 02498194 2005-03-07
WO 2004/035612 PCT/US2003/027489
In one embodiment, the present invention uses peptide screening methods to
prepare
peptide binding motifs with specific binding properties, especially those with
high affinity to
one or more materials. More particularly, the present invention includes a
bifunctional
specificity structure, bifunctional peptide linker or peptide having a first
and a second
binding domain, wherein the first binding domain is selective for a first
biomaterial and the
second binding domain is selective for a second biomaterial. The first binding
domain may
binds specifically to a biopolymer and be selected from, e.g., the peptide
amino acid
sequences of SEQ. ID. NOS.: 1-22. The second binding domain may also be part
of the
same (e.g., a chimeric) or a different peptide that is attached to the first
peptide having the
first binding domain.
Examples of materials or biocompatible materials that may be bound by one or
both
of the domains of the bifunctional specificity structure, bifunctional peptide
linker or
peptides of the present invention include plastic, ceramic, metal, composites,
polymers, and
modifications and/or combinations thereof. Another example of a target for the
present
invention may be a biopolymer comprising one or more chloride doped
polypyrrole subunits,
poly-lactic acid based polymers, poly(lactic acid-co-glycolic acid) based
polymers, magnetic
materials, a biocompatible and/or biodegradable matrix, which may even be
formed into a
sheet. Yet another example is one or more growth factors, e.g., those
biocompatible with
nerve tissue.
In another embodiment of the present invention, synthetic polymers that
exhibit
tissue-specific properties are developed. The resulting "natural" polymer may
then be used
for biologic purposes such as in claudication, implantation, transplantation,
biologic
regeneration, growth, and as biologic replacements (valve, limb, etc).
In yet another embodiment, the surface of a synthetic polymer is modified
using one
or more screening method as presented above. For example, a random
bacteriophage library
that displays and expresses a peptide insert on one portion of the protein
coat is used to
prepare linkers that exhibit one or more of the properties required by one or
more materials.
Where synthetic polymers are used as the material, a peptide that specifically
binds to one or
more polymers is selected. Examples of polymers suitable for use with the
present invention
CA 02498194 2005-03-07
WO 2004/035612 PCT/US2003/027489
are oxidized polypyrrole doped with chlorine (PPyCI) and poly lactic acid-co-
glycolic acid
(PLGA).
In still another embodiment of the present invention, one or more bi-
functionally
linked polymers are synthesized such that one end of the linker (i.e.,
biologic molecule or
structure) binds the polymer and the other end binds another biologic
structure (e.g., nucleic
acid, peptide, protein, chromophore, drug, growth factor, cell, chromophore,
or other organic
molecule). For example, the polymer may include one or more peptides with
polymer
binding domains on one end and a domain that binds to cells, drugs, or growth
factors on the
other end. Several biologic applications, as discussed above, are suitable for
the bi-
functionally linked polymers. Importantly, the physical properties of the
polymer (or other
material that may be used) are not altered. The bi-functionally linked polymer
may be
further shaped or modified for its use in various biologic applications,
including claudication,
implantation, transplantation, biologic regeneration, growth, and as a
biologic replacement
(valve, limb, etc). This type of surface modification method can be applied to
a variety of
synthetic material surface functionalization, and in-turn, selective surface
reactivity.
A method of making the bivalent linker of the present invention includes the
steps of
selecting a peptide that includes a first binding domain peptide from a
library of peptides that
binds selectively to a biomaterial; and a second binding domain with the
peptide that binds
selectively to a target material. The peptide may have a length of about 7 to
about 30 amino
acids. The peptide may be bound and/or selected from a peptide phage display
library and
may include peptides selected from SEQ. 117 NOS: 1-22.
The composition, methods and use of the present invention display clear
advantages
over current techniques used to develop biologic materials. Notable, the
present invention
does not require non-specific adsorption or covalent attachment methods.
Furthermore, the
present invention may be tailored to develop and prepare biologic materials
that exhibit one
or more desired properties.
In another embodiment, the present invention is a modified biologic material
with a
unique surface used to direct tissue regeneration. In one embodiment, the
surface
specifically presents biologic structures in a concentration-dependent
fashion. Such a
6
CA 02498194 2005-03-07
WO 2004/035612 PCT/US2003/027489
presentation when presented as a gradient may be used to guide cellular
activity, growth,
and/or regeneration in a time-dependent manner, e.g., akin to a nerve guidance
channel.
Notably, the present invention, when used, does not require multiple surgical
procedures and
reduces both the cost and surgical-related complications associated with
tissue regeneration
procedures.
In yet another embodiment, the present invention is for tissue engineering
such that a
hybrid or bi-functional biologic material is created. The bi-functional
biologic material may
present one or more biologic properties (i.e., through the linkage of biologic
structures) at its
surface in a specific or nonspecific pattern. The hybrid-biomaterial is
designed to behave or
exhibit properties similar to native tissue or a native organ. Applications of
the hybrid or bi-
functional biologic material include its use in tissue regeneration and/or as
a bioreactor or
biosensor, as well as targeted drug delivery.
The present invention provides a bifunctional specificity structure
comprising: a
peptide linker comprising a first binding domain and a second binding domain,
wherein the
first binding domain is selective for specific binding with high affinity to a
first biomaterial
which is an electrically conductive polymer or a biodegradable material, and
the second
binding domain is selective for specific binding with high affinity to a
second biomaterial.
The present invention also provides a bifunctional peptide linker comprising:
a first
binding domain and a second binding domain, wherein the first binding domain
is selective
for a synthetic biocompatible polymer surface and the second binding domain is
selective for
a material.
BRIEF DESCRIPTION OF THE DRAWINGS
For more complete understanding of the features and advantages of the present
invention,
reference is now made to the detailed description of the invention along with
the
accompanying FIGURES.
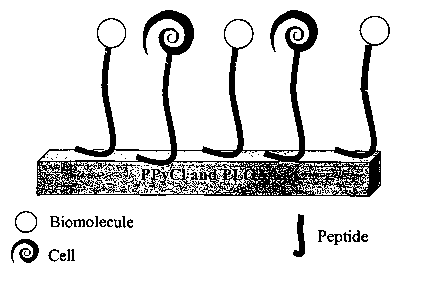
FIGURE 1 is a microscopic illustration of a bi-functional linker connecting
the material and
biomolecules in accordance with the present invention;
7
CA 02498194 2005-03-07
WO 2004/035612 PCT/US2003/027489
FIGURE 2 is a microscopic illustration of a bi-functional system used inside a
host;
FIGURE 3 is a macroscopic illustration of nerve cell axon being guided through
the nerve
guidance channel using a biomaterial with bi-functional linkers that contain
biomolecules in
a concentration gradient on the surface of the material in accordance with the
present
S invention;
FIGURE 4 is an illustration of the screening (biopanning) process used for
selecting one or
more peptides that recognize the surface of a material;
FIGURE 5 includes the peptide sequences from biopanning rounds 3 through 5
obtained from
PPyCI-specific phage;
FIGURE 6 depicts the predominant sequence for PPyCI as THRTSTLDYFVI,
determined by
comparing the percent amino acid occurrence per position for the 12 amino acid
positions, where
the maximum percent in each column corresponds to the highest amino acid
occurrence for that
position within the peptide;
FIGURE 7 shows that the percent amino acid group per position for PPyCI gives
a value that
can be compared to the consensus sequence and the overall group occurrence
(relative to the
combinatorial library of peptides expressed on the pIII);
FIGURE 8 is an example of an amplification study of phage selected for PpyCl,
where the
PPyCI sequence is THRTSTLDYFVI (SEQ ID NO.:1) and the random sequence is
IEHPKTPDSHSR (SEQ ID N0.:4);
FIGURE 9 is an example of a binding affinity study of phage selected for
PPyCI., where
PPyCI sequence.is THRTSTLDYFVI (SEQ m NO.:1) and the random sequence is
IEHPKTPDSHSR (SEQ ID N0.:4);
FIGURE 10 are reflectance image of (A) PPyCI with phage, (B) PPyCI-specific
phage at 1°-
2°, (C) random phage at 1°-2°, (D) WT at 1°-
2°, (E) 1°-2°, (F) 2°, and (G) mounting media,
wherein PPyCI sequence is THRTSTLDYFVI (SEQ ID NO.:1) and the random sequence
is
IEHPKTPDSHSR (SEQ ID N0.:4);
8
CA 02498194 2005-03-07
WO 2004/035612 PCT/US2003/027489
FIGURE 11 includes peptide sequences from biopan rounds 2 through 4 obtained
from PLGA-
specific phage in accordance with the present invention;
FIGURE 12 shows that the predominant sequence for PLGA is SFPDTYVRVKPA (PLGA-
1;
SEQ ID N0.:7), as determined by comparing the percent amino acid occurrence
per position for
the 12 positions, wherein the maximum percent in each column corresponds to
the greatest
occurring amino acid for that position;
FIGURE 13 shows that the percent amino acid group per position for PLGA-1
gives a value that
can be compared to the consensus sequence and the overall amino acid group
occurrence (relative
to the combinatorial library of peptides expressed on the plll);
FIGURE 14 includes the peptide sequences from biopan rounds 3 through 5
obtained from
PLGA-specific phage;
FIGURE 15 shows that the predominant sequence for PLGA is KPLHSNKYYDRY (PLGA-
2;
SEQ ID NO.:15), as determine by comparing the percent amino acid occurrence
per position for
the 12 positions, wherein the maximum percent in each column corresponds to
the greatest
occurnng amino acid for that position;
FIGURE 16 shows that the percent amino acid group per position for PLGA-2
gives a value
that can be compared to the consensus sequence and the overall percentage of
amino acid
group occurrence (relative to the combinatorial library of peptides expressed
on the pIII);
FIGURE 17 is an example of an amplification study of phage selected for PLGA,
where the
PLGA-1 sequence is SFPDTYVRVKPA (SEQ ID N0.:7), the PLGA-2 sequence is
KPLHSNKYYDRY (SEQ ID NO.:15), and the random sequence is IEHPKTPDSHSR (SEQ
ID N0.:4);
FIGURE 18 shows the binding affinity of phage selected for PLGA, where the
PLGA-1
sequence is SFPDTYVRVKPA (SEQ ID N0.:7), the PLGA-2 sequence is
KPLHSNKYYDRY (SEQ ID NO.:15), and the random sequence is IEHPKTPDSHSR (SEQ
117 N0.:4) in accordance with the present invention; and
9
CA 02498194 2005-03-07
WO 2004/035612 PCT/US2003/027489
FIGURE 19 include AFM images of (A) PLGA-1 phage bound to a material after
several washes,
where the scale bar represents 1 pm, and (B) WT on PLGA, where samples are 4
~m x 4 ~m with
a z-scale of 20 run.
FIGURE 20 demonstrates that T59 peptide (SEQ ID NO. l, THRTSTLDYFVI)
(synthesized
from the sequence from the T59 phage) binds to PPyCI and not to PPyPSS or to
polystyrene
(PS). Binding was studied using biotinylated T59 peptide and streptavidin-FITC
labeling. (a)
control substrate in which no T59 peptide was added. (b) 15 pM peptide bound
to 0.5 x 0.5
cm2 PPyCI substrate. (c) 15 pM peptide bound to 0.5 x 0.5 cm2 PPyPSS
substrate. (d) 15 ~M
peptide bound to 0.5 x 0.5 cm2 polystyrene (PS) substrate. All samples were
incubated with
equal concentrations of streptavidin-FITC. Bar, 10 Vim.
FIGURE 21 shows PPyCI-bound T59 peptide (SEQ 117 NO. 1) is stable under in
serum-
containing medium. Binding was studied using biotinylated T59 peptide and
streptavidin-
FITC labeling. (a) control substrate in which no T59 peptide was added, 0.5 x
0.5 cm2 PPyCI
substrates (b) and (c) were incubated with 15 pM peptide, the samples were
then placed in
serum-containing media (pH 7.4 and 15% serum) for 3 hr (b), 7 days (c), and 3
weeks (d).
Bar, 10 pm.
FIGURE 22 depicts that T59 peptide (SEQ m NO. 1) modified with GRGDS promotes
PC12 cell adhesion in serum-free environment on PPyCI. Incubation of 106
cells/sample, (a)
in serum-free media without T59-RGD on PPyCI, and (b) 60 pM of T59-RGD peptide
was
interacted on 0.75 x 0.75 cm2 PPyCI surface, in serum-free media cell adhesion
was
promoted. Bar, 10 Vim.
CA 02498194 2005-03-07
WO 2004/035612 PCT/US2003/027489
DETAILED DESCRIPTION OF THE INVENTION
While the making and using of various embodiments of the present invention are
discussed in detail below, it should be appreciated that the present invention
provides many
S applicable inventive concepts that may be embodied in a wide variety of
specific contexts.
The specific embodiment discussed herein are merely illustrative of specific
ways to make
and use the invention and do not delimit the scope of the invention. Various
modifications
and combinations of the illustrative embodiments, as well as other embodiments
of the
invention, will be apparent to persons skilled in the art upon reference to
the description. It is
therefore intended that the appended claims encompass any such modifications
or
embodiments.
All technical and scientific terms used herein have the same meaning as
commonly
understood by one of ordinary skill in the art to which this invention
belongs, unless defined
otherwise.
1 S To facilitate the understanding of this invention, a number of terms are
defined
below. Terms defined herein have meanings as commonly understood by a person
of
ordinary skill in the areas relevant to the present invention. Terms such as
"a," "an," and
"the" are not intended to refer to only a singular entity, but include the
general class of which
a specific example is used for illustration. The terminology herein is used to
describe
specific embodiments of the invention, but their usage does not limit the
invention, except as
outlined in the claims.
The following are terms as they apply to this application. As used herein
"material"
is a substance with a surface that may come in contact with other materials
and/or molecules.
Materials may be microfabricated and may be made of a single compound, layered
compounds, or a mixture of compounds of one or more molecules or chemicals
such as
polymers or polymer-blends, plastics, glass, metals, semiconductors, organic
or inorganic
compounds, and combinations, thereof. When the material is layered, the
"surface" layer is
one that will come in contact with one or more biologic structures.
11
CA 02498194 2005-03-07
WO 2004/035612 PCT/US2003/027489
As used herein, "biologic materials" also referred to as "biomaterials" are
materials
that exhibit, exert, or mimic biologic properties, such that they are able to
bind, contact,
react, combine, and/or interact in a manner that mimics, minors, or resembles
cellular,
prokaryotic, and eukaryotic biologic activity, process, reaction, interaction,
or encounter.
Biologic materials may include a material to which a biologic structure has
been attached or
to which a biologic structure is in contact with, wherein the contact is
charged, covalent,
polarizable, electrostatic interaction, fluxional, or through a molecular
interaction such as
hydrogen bonding.
"Biologic structures," as used herein, are structures that are of biologic
origin,
generally considered to be organic or carbon-containing compounds with
functional groups
such as amino, carboxyl, thiol or hydroxyl. Examples of biologic structures
include nucleic
acids, peptides, proteins, chromophores, cells, cytokines, cofactors, growth
factors, tissues,
organs, fatty acids, sugars, organic polymers and other simple or complex
carbon-containing
molecules, and combinations thereof. Biologic structures may be structures
with a biologic
backbone that also contain organic or inorganic modifications (e.g.,
modifications including
but not limited to those that incorporate additional charge, structure,
polarizability, hydrogen
bonding, electrostatic interaction, and fluxionality to the biologic
backbone). These biologic
structures, also referred to as biomolecules, are complex molecules with some
biologic
activity and can include all of the examples used for biologic structures as
well as other
complex molecules such as drugs. For the present invention, the terms
biomolecule and
biologic structure are used interchangeably. Biologic structures may be
produced,
synthesized or engineered by biologic or nonbiologic processes. In one
embodiment,
biologic structures such as one or more host cells (e.g., tissue culture cells
or clones, bacterial
cells or bacteriophage) are used to express other biologic structures.
The present invention involves the development of unique and/or improved
interactions between a first biologic structure, a material, and second
biologic structure.
FIGURES 1-3 demonstrate examples of these interactions shown at the
microscopic and
macroscopic level. Specifically, such interactions include a biologic
"linker," as used herein
to refer to a biologic structure that is able to contact a biomaterial and a
second biologic
structure to create a larger complex. The contact may be of any type that
biologic structures
12
CA 02498194 2005-03-07
WO 2004/035612 PCT/US2003/027489
encounter, such as covalent, electrical, electrostatic, hydrogen bonding,
polar, magnetic, etc.
FIGURE 1 is an example of a linker that is a peptide and its interaction with
a biomaterial on
one end and one or more different biologic structures at the other end, where
the biologic
structures in this example are both biomolecules and cell.
The linkers used may be short or long biologic structures. They may be the
native
structure of ones that are synthesized, engineered or expressed in another
biologic structure
such as a cell. In one embodiment, the linkers are engineered and/or expressed
by a different
biologic structure. For example, bacteriophage can express or display biologic
structures
(e.g., or phage display) as a virus that is genetically engineered with one or
more random
biologic structures or molecules, such as a peptide or drug. In one case, the
biologic
molecule is a random peptide of a specified length expressed as a portion of
the virus'
exterior coat.
The advantage of using an expression system to obtain biologic structures is
that large
amounts of the biologic structures (e.g., libraries) are provided (i.e.,
displayed on the phage)
enabling the rapid identification of structures that are specific to one or
more materials and/or
active in one or more particular biologic environments. Thus, specific
biologic structures
(e.g., peptides) that recognize one or more selected materials can be
identified, a method akin
to surface or material engineering. Furthermore, the material has become a
biologic material
(biomaterial) and, when a bi-functional biologic structure is used, one with
the ability to
contact another biologic structure or biologic material, then a bi-functional
biomaterial is
created as recognized by the present invention. Applications for bi-functional
biomaterials
include tissue engineering, tissue replacement, transplantation, biologic
growth,
differentiation or development, as examples. Further examples of compositions
and methods
of making bi-functional biomaterials of the present invention are presented
below.
Using Phage Display to Identifying Material-Specific Biologic Structures.
A filamentous virus (i.e., bacteriophage) may be used to produce large amounts
of
one or more biologic structures. Commercially-available libraries that contain
random
assortments of biologic structures with certain qualities (e.g., length,
innate structure,
species) may be used. For example, bacteriophage libraries (also referred to
herein, as phage
13
CA 02498194 2005-03-07
WO 2004/035612 PCT/US2003/027489
libraries) have been developed that include an assortment of biologic
structures such as
peptides of specific lengths (e.g., 12 amino acid linear, 7 amino acid linear,
or 7 amino acid
constrained where cysteines are at the ls' and 9th position on the peptide to
create a loop by
the disulfide linkage between the two cysteines) on the minor coat protein
(pIII) of the M13
coliphage. Another benefit of using a large library such as this is that after
finding that one
or more specific biologic structures (e.g., peptides) that can contact or bind
a selected
material, the library can be used to find the specific amino acids involved in
contacting or
binding to the material. An example of the process used to find specific
biologic structures
and/or characteristics about them, also referred to as a screening method, is
shown in
FIGURE 4. Phage display libraries and experimental methods for using them in
biopanning
are further described, for example, in the following U.S. patent publications
to Belcher et al.:
(1) "Biological Control of Nanoparticle Nucleation, Shape, and Crystal Phase";
2003/0068900 published April 10, 2003; (2) "Nanoscale Ordering of Hybrid
Materials Using
Genetically Engineered Mesoscale Virus"; 2003/0073104 published April 17,
2003; (3)
"Biological Control of Nanoparticles"; 2003/0113714 published June 19, 2003;
and (4)
"Molecular Recognition of Materials"; 2003/0148380 published August 7, 2003.
The present invention was exemplified by a series of working examples. In one
embodiment of the present invention, a Ph.D.-12TM Phage Display Peptide
Library Kit (New
England Biolabs, Beverly, MA) was used to screen biologic structures (e.g.,
peptides). The
kit contains a library with approximately 109 different 12-amino acid linear
peptide inserts
fused to the pIII coat protein of M13 coliphage. An initial volume of 1pL of
the phage-
display library (in solution and corresponding to a 1x10'2 phage/~L) was used
to begin
screening against one or more materials, a process referred herein as
biopanning.
Biopanning took place in 1mL of Tris-buffered saline (TBS) containing 0.1%
(vol/vol)
Tween-20 (0.1% TBS-T); each material was incubated with the library for at
least about 1
hour at room temperature. Materials were then washed (several times) with 1 mL
of 0.1
TBS-T to discard non-specific phage. To disrupt any phage binding that was not
specific to
the surface of the material, 500 p,L of glycine-HCl (pH 2) was added to the
above mixture for
at least about 9 minutes at room temperature. The solution was then collected
and brought to
a neutral pH with Tris-HCl (pH 9). Half of the volume of the solution was then
introduced
14
CA 02498194 2005-03-07
WO 2004/035612 PCT/US2003/027489
(at a 1:100 dilution with growth media) to Escherichia coli (E.coli) ER2837
bacteria (New
England Biolabs, Beverly, MA) that had been cultured at least about overnight.
The phage-bacterial solution was incubated at least about 5 hours in a shaker
at 37
degrees Centigrade (allows virus to infect bacteria). Bacteria were collected
by
centrifugation (at least about 14,000 rpm for 10 minutes) and phage were
precipitated with
polyethylene glycol) or PEG for at least about 15 minutes at a refrigerated
temperature (4
degrees Centigrade). A second centrifugation (at least about 10,000 rpm for 15
minutes)
followed and the pellet was resuspended in 200 ~,L of TBS. Meanwhile, the
concentration of
phage was also calculated (generally from a sample of phage-bacterial solution
and/or from a
sample of the phage solution when incubating with the material). The
techniques used are
those well known to one of ordinary skill in the art of molecular biology and
includes plating
the phage or allowing a various concentrations of phage solutions to infect a
known amount
of bacteria. When using the infection technique, bacteria with lacZ gene may
be used and
plated in the presence and absence of isopropylthio-~3-D-galactoside (IPTG)
and 5-Bromo-4-
1 S chloro-3-hydroxyindolyl-(3-D-galactose (X-gal) for visual determination of
bacterial growth
on "titer plates." The phage concentration may then be determined by the
following:
Concentration of phage from titer plate (pfu/~L) x (1 ~1/lE-6L) x (S copies of
pIII/1 pfu) x (1
mole/6.023X1023 molecules) (1)
where, pfu = plaque forming unit.
Hi-Throughput Screening of Material-Specific Biologic Structures.
Several biopanning rounds are generally used to determine material-specific
biologic
structures and their material-specific contact or binding regions. For each
biopanning round,
the phage concentration is used to determine the amount (as volume) used in
the next round
of biopanning against the material. A fresh piece of material was used for the
next screening,
where the phage amount was at least about 109 pfu. Multiple rounds of
biopanning follow,
generally at least about five rounds to determine the consensus sequence
involved in binding
the material.
CA 02498194 2005-03-07
WO 2004/035612 PCT/US2003/027489
From the 3rd to the 5th round of biopanning, blue plaques were picked and each
amplified separately, 1:100 in growth media with an overnight culture of E.
coli and allowed
to grow (e.g., amplify) for 5 hours. Bacteria were then separated by
centrifugation for 30
seconds and 500 ~L of the phage solution was precipitated for 10 minutes at
room
temperature with PEG, followed by centrifugation for 10 minutes to pellet the
phage. The
pellet was suspended in a solution of NaI (ruptures the phage protein coat)
and ethanol
(approximately 250 ~L) was used to precipitate DNA from the phage.
Precipitated DNA was
suspended in at least about 60 ~L chemical-free, filtered water and the
nucleotide sequences
obtained and translated into peptide sequences (N-terminus to C-terminus) as
shown in
FIGURE 4. Non-genetically engineered phage (e.g., naturally occurring or wild
type [WT])
lacking a peptide insert on the pIII protein coat will appear as clear plaques
during
biopanning (i.e., when plated on titer plates in the presence of IPTG and X-
gal).
Following the above method and after several rounds of screening or
biopanning, a
consensus region of the biologic structures (e.g., consensus peptide or amino
acid sequences)
will be found and will represent the preferred or common regions involved in
contacting or
binding of the material. For rapid analysis, several steps of the above method
may be
automated and without undue experimentation, as is well-known to one of
ordinary skill in
the art of molecular biology.
Examples of Materials for Developing Bi-Functional Biomaterials
In general, ideal materials for the present invention are those that may
contact a
biologic structure and form an interaction that is more than a non-specific
interaction-a
interaction well-known to one of ordinary skill in the art of physiology.
Examples of
materials for the present invention include plastic, ceramic, metal, other
composites,
polymers, and modifications and/or combinations thereof. The material may be
one that is
shaped, blended, or deposited onto another surface.
A preferred embodiment of the invention comprises use of electrically
conductive
polymers including synthetic electrically conducting polymers in biopanning
experiments.
Electrically conductive polymers are known in the art of nerve regeneration:
(1) U.S. Patent
16
CA 02498194 2005-03-07
WO 2004/035612 PCT/US2003/027489
No. 5,843,741 to Wong et al. (December l, 1998) "Method for Altering the
Differentiation of
Anchorage Dependent Cells on an Electrically Conductive Polymer"; (2) U.S.
Patent No.
6,095,148 to Shastri et al. (August l, 2000) "Neuronal Stimulation using
Electrically
Conductive Polymers". For example, the polymer can comprise a conjugated
polymer
backbone, resulting in electron delocalization and low energy optical
transitions, and these
types of polymers are known in the art as conducting polymers. Conducting
polymers are an
important class of materials because of their potential applications in
electrical, optical, and
sensing devices, as well as biological and biomedical applications.
Prototypical electronic
conducting polymers include polyacetylene, polydiacetylene, poly(phenylene
vinylene)
(PPV), poly-para-phenylene, polypyrrole, polyaniline, polythiophene, and the
like. Doping
can be used for conducting polymers such as polyaniline and polypyrrole to
improve their
conductivities, as well as their solubilities in water. Self doped sulfonated
polyaniline
(SPAN) and doped polypyrrole (PPy), for example, have charged backbones and
have high
solubilities in water. PPV can be made with use of water-soluble precursors as
well which
can be used with doping agents.
Patent literature which describes a variety of conducting and semiconducting
polymers includes: (a) 4,929,389 to Aldissi ("Water-Soluble Conductive
Polymers"); (b)
5,294,372 and 5,401,537 to Kochem et al. ("Aqueous Dispersions of
Intrinsically
Electroconductive Polyalkoxythiophenes, a Process for their Preparation and
their Use"); (c)
5,670,607 to Chen ("Miscible Forms of Electrically Conductive Polyaniline");
(d) 5,569,798
to Wudl et al. ("Self Doped Polymers"); (e) 5,648,453 and 5,688,873 to Saida
et al.
("Electroconductive Polymer and Process for Producing the Polymer"); (fj
5,968,417 to
Viswanathan ("Conducting Compositions of Matter"); and (g) 6,534,329 to Heeger
et al.
17
CA 02498194 2005-03-07
WO 2004/035612 PCT/US2003/027489
("Visible Light Emitting Diodes Fabricated from Soluble Semiconductor
Polymers"), and are
each hereby incorporated by reference for their entire teachings including
synthesis and
characterization. These patents, for example, describe covalently linking
Bronsted acid
groups to polymer backbones, zwitterionic structures, self doping, doping with
acceptors and
S donors which oxidize or reduce the polymer chain, cycling between neutral
and ionic states,
stability, and pi-conjugation of electronic systems which provides
semiconducting or
conducting behavior. In addition, the many applications of conducting polymers
are
described.
Electrically conductive polymers are also described in, for example, Concise
Encyclopedia of Polymer Science, J.I. Kroschwitz, Ex. Ed., John Wiley, 1990,
pages 298-
300, which is hereby incorporated by reference. The polymers are described as
having
conjugated pi-electron backbones which can provide properties such as, for
example, low
energy optical transitions, low ionization potentials, and high electron
affinities. They can be
oxidized or reduced more readily than conventional polymers. Doping of the
following types
of conductive polymers is described: polyacetylene, polyp-phenylene), polyp-
phenylene
sulfide), polypyrrole, and polythiophene.
Additional conducting polymers and their use in patterning on various
substrates is
described in U.S. Patent No. 5,976,284 to Calvert et al. ("Patterning
Conducting Polymer
Surfaces and Process for Preparing the Same and Devices Containing the Same").
This '284
patent teaches that, in principle, any polymer having an electrical
conductivity of at least
sigma >10-3 S/cm, preferably at least sigma > 10-1 S/cm, can be used as the
conducting
polymer. Also, conducting polymers are described in Chapter 11 of Organic
Conductors, J.
P. Farger, Ed. Marcel Dekker, NY, N.Y., 1994, which is incorporated herein by
reference.
Conducting polymers include, e.g;, cis and trans polyacetylenes (PA),
polydiacetylenes
(PDA), polyparaphenylenes (PPP), polypyrroles (PPy), polythiophenes (PT),
polybithiophenes, polyisothianaphthene, polyphenylenevinylenes (PPV),
18
CA 02498194 2005-03-07
WO 2004/035612 PCT/US2003/027489
polythienylvinylenes (PTV), polyphenylenesulfide (PPS), and polyaniline
(PAni), and the
structures of these polymers are shown in the '284 patent. In these
structures, it is to be
understood that H atoms may be replaced by substituents, such as Cl_~8 -alkyl,
or phenyl or
groups containing ionic groups such as carboxylate or sulfonate. These groups
may be
attached directly or through ester, ether, or amide links. In general,
substitution worsens the
electrical conductivity of the conducting polymer, but may enhance features
such as
solubility or orientation at the air/water interface, for example. Other
references which
further describe the synthesis and properties of these conducting polymers
include: M. F.
Combarel et al, C. R. Acad. Sci. Ser. C, vol. 262, p. 459 (1966); L. T. Yu et
al, J. Polym. Sci.
Symp. C, vol. 16, p. 2931 (1967); M. Doriomedoff et al, J. Chim. Phys.
(Paris), vol. 68, p. 39
(1971); T. Ito et al, J. Polym. Sci. Chem. Ed., vol. 12, p. 11 (1974); H.
Shirakawa et al,
Chem. Commun., p. 578 (1977); C. K. Chiang et al, Phys. Rev. Lett., vol. 39,
p. 1098 (1977);
P. J. Nigrey et al, Chem. Commun., p. 594 (1979); A. G. MacDiarmid et al,
Synth. Metals,
vol. 1, p. 101 (1980); D. M. Ivory et al, J. Chem. Phys., vol. 71, p. 1506
(1979); A. F. Diaz et
al, Chem. Commun., p. 635 (1979); K. K. Kanazawa et al, Chem. Commun., p. 854
(1979);
G. Tourillon et al, J. Electroanal. Chem., vol. 135, p. 173 (1982); E. M.
Genies et al, Synth.
Metals, vol. 36, p. 139 (1990); H. W. Gibson et al, J. Am. Chem. Soc., vol.
105, p. 4417
(1983); M. C. Dos Santos et al, Phys. Rev. Lett., vol. 62, p. 2499 (1989);
Synth. Metals, vol.
29, p. E321 (1989); H. Kiess, ed., Conjugated Conducting Polymers, Springer
Series in Solid
State Sciences, Vol. 102, Springer-Verlag, Berlin, 1992.
For example, chlorine doped polypyrrole (PPyCI) is an electrically conductive
material; an oxidized version of polypyrrole has been used as a substrate for
nerve
regeneration. The advantage of this type of material is that it can be
electropolymerized to
form sheets or other shapes of interest.
Materials may also be those that are biodegradable. For example, poly (lactic
acid-
co-glycolic acid) (PLGA) is easy to prepare, its surface area can by
controlled as well as its
degradation rate. See, e.g. Sustained and Controlled Release Drug Delivery
Systems, Ed.
J.R. Robinson, 1978 including discussion at page 328, incorporated herein by
reference in its
entirety.
Electropolymerization of Polypyrrole
19
CA 02498194 2005-03-07
WO 2004/035612 PCT/US2003/027489
Polypyrrole was oxidized enabling current to pass through. The addition of
chloride
as radical anions or "dopants" provide charge neutrality along the highly
conjugated
backbone. In one embodiment of the present invention, a PPyCI film is
electrochemically
deposited on indium tin oxide (ITO)-conductive borosilicate glass (Delta
Technologies, Still
Water, MN). The ITO glass, shaped as slides, may be cleaned before use by
sonication in
hexane, methanol, and dichloromethane, 5 minutes each.
Electrochemical deposition of PPyCL was made with a three-electrode setup
consisting of a saturated calomel reference electrode, platinum gauze counter
electrode, and
an ITO slide as the working electrode. The polymer was deposited at a constant
potential of
720 mV (versus the saturated calomel reference) from an aqueous solution of
0.1 M pyrrole
monomer (Fisher, Scientific, Palatine, IL) containing 0.1 M NaCI (Fisher,
Scientific,
Palatine, IL) as the dopant. A Pine Instruments AFRDES bipotentiostat was used
as the DC
voltage source. Film thickness ranged from 30-40 ~m as determined by
integrating current
over time. The thickness was controlled by the passage of charge based on the
standard
value of 50 mC/cm2. The charge passing through the working electrode was
measured with a
current integrator (IT001, Cypress Systems, Inc.) coupled to a multimeter
(Sperry, DM-8A)
for digital display. Films were rinsed with sterile water and dried in a
desiccator for at least
about two days before use.
Material-Specific Biologic Structures: Selection against a Material
Biologic structures that select for a specific material may be further
analyzed to
determine how or where the contact to the material occurs. In one embodiment,
biologic
structures such as peptides are sequenced to determine consensus binding
regions. The
biologic structures may be obtained after rounds of biopanning or through
other methods of
attachment in which nonspecific interactions are eliminated (e.g., material is
washed to
remove unbound biologic structures such as peptides). The methods are well
known to one
of ordinary skill in the art of molecular biology.
In one embodiment, phage expressed peptides were allowed to bind to PPyCI and
plaques grown after biopan rounds 3 through 5 were sequenced. Results of the
sequencing
(using one-letter amino acid abbreviations) are shown in FIGURE 5 in which a
predominant
CA 02498194 2005-03-07
WO 2004/035612 PCT/US2003/027489
sequence (SEQ ID NO.:1) was found; however additional statistical analysis
should be
performed in order to verify that it is a consensus binding region. In one
embodiment of the
present invention, functional group reactivity is performed, where amino acid
side chains are
grouped together, i.e., basic, acidic, hydrophobic, hydroxyl, aromatic, amide,
methionine, and
proline (one or more amino acids, such as cysteine, may left out when there is
a lack of occurrence
in the sequenced samples). The analysis shows the types of biologic structure
(peptide)--biologic
structure (peptide) interactions as well as biologic structure (peptide}-
material interactions that
occur.
FIGURE 6 shows an example of the statistical analysis in which the percent
amino
acid group per position is displayed. For example, in FIGURE 6, hydroxyl amino
acid
groups appear two time more often as compared to their presence in the parent
combinatorial
peptide library. The consensus region is represented in the last line of the
FIGURE 6 as
THRTSTLDYFV (SEQ ID NO.:1). Additionally, the consensus binding region was
analyzed
for amino acid fimctional group reactivity (FIGURE 7) and helps to illustrate
possible interactions
between fi~nctional groups and the material (i.e., between the biologic
structure and the surface of
the material).
Material-Specific Biologic Structures: Verification of Specificity
Some biologic structures, especially those produced in large amounts through
the help
of another biologic structures (e.g., expressed through host cells such as
bacteria or
bacteriophage), may contain consensus regions that are not only the result of
interactions
between the material but are consensus regions based on the expression system
or method
use to produce the biologic structure. In one embodiment of the present
invention, the
biologic structure consensus region is verified, especially that it is not a
consensus region
resulting from cell growth during biopanning (e.g., amplification). For
example, if a
modified host multiplies better than a naturally occurnng host, then there is
a possibility that
the modified host containing the consensus region was selected because of its
ability to grow,
not because of material-specific interaction.
Verification includes expressing the specific consensus region (or entire
biologic
structure containing the consensus region) in the host, growing the host and
comparing the
21
CA 02498194 2005-03-07
WO 2004/035612 PCT/US2003/027489
number grown to that obtain from a non-modified host. For example, in one
embodiment, a
peptide containing the material consensus region for interacting with PPyCI
was displayed on
phage and amplified as previously described. Titer counts of these phage were
compared to
the amplification of random phage and WT (those not allowed to interact with
or raised
against PPyCI) as shown in FIGURE 8.
FIGURE 8 shows that PPyCI-specific phage (PPyCI bar) amplified to an average
count of 8~2 during a 10-~ dilution, at a concentration of 8~2x10' pfu/pL, or
0.66~0.05 nM,
as obtained from equation (1). The randomly selected engineered phage
amplified to an
average phage count of 7~2 during a 10-7 dilution, at a concentration of
0.58~0.05 nM. WT
phage amplied to an average phage count of 2~2 during a 10-7 dilution or a
concentration of
0.17~0.05 nM. Because the growth pattern was similar for each group analyzed,
PPyCI-
specific phage are found to express a PPyCI-specific consensus region and not
a growth or
expression-related consensus region.
Determining the Biologic Structure-Material Interaction
The consensus region of the biologic structure is presumed to undergo a type
of
specific interaction with the material. The interaction may be any of the
interactions
previously described (e.g., covalent, electrical, electrostatic, hydrogen
bonding, polar,
magnetic, etc., as examples). Several methods are available to determine the
type of
interaction that occurs between the biologic structure and the material and to
determine that
the interaction is specific. Methods are those readily apparent to one of
ordinary skill in the
art of molecular biology and some examples are discussed below.
Titer counts. The use of titer count as a binding study for the peptide on
PPyCI is semi-
quantitative and provides a relative binding comparison of phage counts per
PPyCI-specific
phage, randomly selected engineered phage or WT phage. Initial amounts of
1x10$ pfu
phage interacted with the PPyCI. PPyCI samples were then washed at least about
three times
with 1 mL of 0.1 % TBS-T to remove unbound phage. Elution of bound phage with
S00 ~L
Glycine-HCl (pH 2.2) for 9 minutes was used to disrupt phage bound to the
surface. Titer
counts were obtained from consensus peptide phage experiments and compared to
titer
22
CA 02498194 2005-03-07
WO 2004/035612 PCT/US2003/027489
counts of WT and random peptide phage (and used to compare the binding ability
of PPyCI-
specific phage, randomly selected engineered phage, and WT by comparing the
amount of
phage that could be eluted off the surface of PPyCI). Using 500 pL of glycine-
HCl (pH 2.2),
the titer count method showed that PPyCI-specific phage bind more successfully
than the
other phage (FIGURE 9). Random selected engineered phage had the lowest
recovery with
an average count of 29 phage. WT had an average recovery of 57 counts. PPyCI-
specific
phage had a recovery rate (phage bound to the surface) of 124 counts.
Immunochemistry. With the immunochemistry technique, fluorescently-labeled
phage bound
to the surface of PPyCI are visualized microscopically and enables the number
of -material
bound phage to be quantified. A biotinylated antibody to the M13 bacteriophage
specific to
pVIII (Anti-fd Bacteriophage-Biotin Conjugate from rabbit, Sigma-Aldrich
Corp., St. Louis,
MO) and the biotin-streptavidin interaction were used to attach fluorescein-
labeled-
streptavidin (Exaplha, Boston, MA) to phage. Phage were visualized on the
material using
fluorescence microscopy. Phage were at a concentration of 1x104 pfu/~,L and
allowed to
interact with 1 cm x 0.5 cm sample of PPyCI for at least about 1 hour. The
material (PPyCL)
was then washed at least about three times with 1 mL of 0.1% TBS-T to remove
unbound
phage from the material. A primary anti-body (1°), at a dilution of
1:400 (antibody:4%
Bovine Serum Albumin [BSA] in TBS at pH 7.5) was added to the material for at
least about
1 hr at room temperature. Samples were washed at least about two times with 1
mL of TBS
(pH 7.5). A secondary antibody (2°) of fluorescein-labeled-streptavidin
at a dilution of 1:200
(fluorescein-labeled-streptavidin:4% BSA in TBS at pH 7.5) was added to the
material for
at least about 30 minutes at room temperature in the dark. Material was then
washed at least
about two times with 1 mL of TBS (pH 7.5) and visualized after mounting on
microscope
slides. The images are shown in FIGURE 10 using a Leica TCS 4D confocal
microscope
equipped with differential interference contrast optics and a Kr/Ar mixed gas
laser with a
selected excitation wavelength of 488nm (for fluoroscein) and emission was
collected
through a 40X oil immersion objective (Microscopy Laboratory of the Institute
for Cellular
and Molecular Biology, University of Texas, Austin, TX).
From FIGURE l OB. PPyCI-phage are shown to have specific interaction with the
PPyCI surface. FIGURES 10B-G show the high intensity fluorescence that is the
PPyCI-
23
CA 02498194 2005-03-07
WO 2004/035612 PCT/US2003/027489
specific interaction (with little random binding). Random phage and WT were
used to verify
that the peptide sequence on PPyCI-specific phage was specific to PPyCI. The
peptide
sequence that was expressed on the random phage was different than that of the
peptide
sequence displayed on PPyCI-phage, and also has lower intensity, suggesting
that the PPyCI-
specific phage bind specifically to PpyCl, while the random phage has no
specific
interaction. For example, comparison of FIGURES l OB (PPyCI-specific phage)
and
FIGURE lOD (WT) that the interaction is specific (e.g., higher intensity of
fluorescence with
PPyCI-specific peptides). FIGURES lOEG show that the amount of fluorescence
from the
antibody, fluoroscein-labeled-streptavidin, and mounting media is minimal
compared to the
intensity of labeled phage. All samples were imaged using the same intensity
of laser light
and exposure times, except for the reflectance image (FIGURE l0A) which was
not imaged
with the laser but with a 100 W Hg lamp.
The present invention demonstrates that the surface of a material may be
modified to
encourage an interaction with a biologic structure and can be used to create a
bi-functional
biomaterial. From the methods such as those using immunochemistry, the
interaction
between material and biologic structure is found to be specific. In one
embodiment, a
peptide sequence of THRTSTLDYFVI is the consensus region that specifically
interacts with
the material, PPyCI. Further embodiments include spatially controlling the
concentration of
biologic structure on the material surface and including one or more biologic
structures as
linkers. For example, when used with neural tissue, a biomaterial of the
present invention
can include biologic structures such as neural cells and neural-specific
biomolecules such as
nerve growth factor or other neural-acting agents or drugs. The present
invention will further
expand the possibilities of the tissue engineering industry. In yet another
embodiment of the
present invention, the material is one that can be modified over time (either
synthetically or
naturally) such as those that are biodegradable. Additional working examples
are presented
below.
Example Using a Biodegradable Material
Some materials may be controlled, such as their rate of degradation,
immunogenic
response, etc., and may, thus serve as improved bi-functional biomaterials.
Controlling the
24
CA 02498194 2005-03-07
WO 2004/035612 PCT/US2003/027489
rate of degradation enables one to engineer suitable biomaterials for tissue-
related
applications. In one embodiment of the present invention, a material, such a
biodegradable
PLGA is cast in a form. Casting methods that are used are those readily
apparent to one of
ordinary skill in the art of polymer chemistry. In one example, the material
is solvent cast.
In another embodiment, the material is cast into a film that may be at least
about 80-150 ~m
thick with a smooth.surface.
PLGA is generally used in an 85:15 percent ratio (lactic acid:glycolic acid)
or PLGA
(85:15). The PLGA film is constructed by adding 1 mL dichloromethane (Fisher
Scientific,
Palatine, IL) to 100 mg PLGA (Polysciences Inc., Warrington, PA ) to yield a
100 mg/mL
concentration. The mixture is stirred to homogeneity and dried overnight in a
PyrexTM 100
mL glass beaker for solvent evaporation. Film thicknesses of at least about
150-80 ~m were
determined by the relative concentrations of PLGA and solvent, such as
dichloromethane.
PLGA films may be stored in a UV-protected desiccant for at least about 1
month. Many
forms of PLGA undergo hydrolysis of the ester bonds or oxidize over time.
PLGA is a material that is biologically compatible; however, it does not have
the
ability to interact specifically with one or more select biologic structures.
The present
invention presents methods to identify biologic structures that are specific
to PLGA.
Biologic Structures Specific for Degradable Materials
Biologic structures such as peptides that were specific to PLGA were obtained
from
the phage selected after the 2"a or later biopanning rounds. Results of
peptide sequences
obtained from biopanning rounds 2 through 4 are illustrated in FIGURE 11.
More thorough analysis was performed on the peptide sequences obtained from
the
first screening of PLGA or PLGA-1 (FIGURE 12). The percent of the amino acid
groups per
position was determined and compared with the consensus sequence and the
percent
occurrence of the amino acid group in the combinatorial library of peptides
(FIGURE 13).
The consensus regions as shown in FIGURE 12 for PLGA-1 is SFPDTYVRVKPA (SEQ ID
N0.:7).
CA 02498194 2005-03-07
WO 2004/035612 PCT/US2003/027489
The percent amino acid group per position gives a relatively good confirmation
of the
consensus sequence, but it does not predict the consensus sequence completely.
For
example, the 4th position has relatively high values for amino acids that act
as Lewis-bases
and amino acids that contain a hydrophobic reactive group that seem to compete
for the 4th
position with acidic reactive groups. This is also the case for the 8'h
position with high
values for hydrophobic and hydroxyl reactive groups that do not match the
basic reactive
group in the consensus sequence. We were also interested in the regions of
hydroxyl reactive
groups in the beginning of the sequence and hydrophobic reactive groups at the
end of the
sequence of the peptide. This is consistent with common protein structure with
the
hydrophobic section of the peptide coiled in the interior of the tertiary
peptide structure and
the hydroxyl section on the exterior of the peptide in aqueous solution. A
more thorough
analysis is needed to determine the binding of the peptide to the surface of
the PLGA and
eventually determining what role, if any, the hydroxyl and hydrophobic amino
acids have in
the binding of the peptide to PLGA.
A second peptide screening was performed on PLGA to determine similarities in
peptide sequences. The second screening of PLGA with the 12-mer library of
peptides was
conducted for PLGA-2 (using a similar method as for the screening of PLGA-1
with the 12-
mer library of peptides). The results from the second screening for PLGA are
shown in
FIGURE 14. With this second screening of PLGA, the percent of the amino acid
groups per
position was determined and compared with the consensus sequence and the
percent
occurrence of the amino acid group in the combinatorial library of peptides
(FIGURES 15
and 16). The consensus regions as shown in FIGURE 15 for PLGA-2 is
KPLHSNKYYDRY
(SEQ ID NO.:15).
The sequences obtained for PLGA-1 (SFPDTYVRVKPA) and PLGA-2
(KPLHSNKYYDRY) show similarities such as the hydroxyl reactive groups
localized on
one end and the single aspartic acid in the peptide.
Verification that One ore More Biologic Structures are Specific to a
Biodegradable Material
The material-specific phage were further analyzed to verify that the consensus
regions for PLGA-1 and PLGA-2 were specific to the material and not to host
variables such
26
CA 02498194 2005-03-07
WO 2004/035612 PCT/US2003/027489
as growth or biopanning. PLGA-1 and PLGA-2 were compared to the random and WT
phage.
The amplification study for PLGA shows that the PLGA-1 and PLGA-2-specific
phage were able to amplify to the same level as the random-phage library or WT
(FIGURE
17) and were, thus, specific interactions. FIGURE 17 shows that PLGA-1 phage
amplifies
the most with the average phage count of 8~2 during a 10-7 dilution, denoting
a concentration
of 0.66~0.05 nM. PLGA-1 and PLGA-2 counts are almost identical; 5~2 during a
10-7
dilution for PLGA-2 (a concentration of 0.42~0.05 nM). Random phage amplified
to a count
of 7~2 during a 10-7 dilution (at concentration of 0.58~0.05 nM) and WT
amplified to a count
of 2~1 (same dilution) at a concentration of 0.17~0.05 nM.
Determining the Interaction between Biologic Structure and Biodegradable
Material
The interaction between a specific biologic structure and a biodegradable
material
enables one to recognize and later modify the material and or biologic
structure as needed.
The use of titer counts (FIGURE 18) and immunochemistry are just a few
examples of
methods available to determine the interaction between PLGA and peptide. As an
alternative, fluorescent immunochemistry was not used because PLGA auto
fluoresces at the
same emission wavelength as fluorescein (520 nm). Instead, visualization using
atomic force
microscopy (AFM) was used (FIGURE 19).
Titer counts. At least about 1x10$ pfu of phage were added to the material
(e.g., PLGA).
The material was washed at least about five times with 1 mL of 0.1% TBS-T. Non-
specific
phage was removed by eluting material with 500 pLglycine-HCl (pH 2.2) for at
least about
nine minutes. For comparison titer counts were obtained from PLGA-predominant
phage,
WT, and random phage.
FIGURE 18 shows that random phage have the lowest recovery with an average
count of 1~1 phage, because the random phage is not specific for PLGA. PLGA-1
was found
to bind better than the random phage (average count of 5~2 phage). PLGA-2
affinity was
higher (average count of 39~4 phage) and WT had an average count of 72~10
phage.
27
CA 02498194 2005-03-07
WO 2004/035612 PCT/US2003/027489
AFMImaging. Qualitative analysis of phage interactions with the material were
performed
using AFM. PLGA interacted with 1x10$ pfu of PLGA-1 and WT for at least about
1 bout at
room temperature. The material was then washed at least about five times with
1 mL of 0.1%
TBS-T and then mounted on AFM discs for visualization. The AFM was equipped
with a
Digital Instruments Bioscope mounted on a Zeiss Axiovert 100s-2tv, operating
in tip
scanning mode with a G scanner. Images were taken in air using tapping mode.
Etched
silicon probes with 125-pm cantilevers were used with spring constants of at
least about 20-
100 N/m driven near their resonant frequency (200-400 kHz). Scan rates were of
the order of
1-5 pm/s. Images were leveled using a first-order plane fit to remove sample
tilt.
PLGA-1-specific phage were selected and qualitatively analyzed for binding
specificity.
FIGURES 19A and B show the AFM images. Peptides that are expressed on the
phage
permit binding to the material via a specific molecular recognition event. If
the phage did
not have the ability to bind to the material, they would have been removed
during washing
with 0.1 % TBS-T. FIGURE 19B shows the absence of phage on the WT sample
indicating
that any binding is nonspecific.
The present invention may be used to fmd material-specific biologic structures
subsequently used for tissue engineering applications or as drug delivery
vehicles in
mammals, as examples of their use. The biomaterial with a bi-functional linker
may be used
to dominate or merely represent one or more biologic structures of interest at
the material
surface. The bi-functional linker, such as a peptide or other biologic
structure, is one with
two "sticky" ends, one of which binds to the material and the other to another
biologic
structures or, alternatively, to another biologic structure. Layered
biomaterials may be
subsequently constructed if so needed. Importantly, many of all parts of the
present
invention may be automated with ease.
Independent synthetic peptide or peptide analogs with specific recognition and
binding to a biomaterial can also be made with or without linker groups that
can be
conjugated to RGB or other recognition units based upon the peptide sequences
identified
through, for example, phage display screening. Additionally, the independent
peptides may
be made through chemical or biological synthetic routes and would allow the
same function,
28
CA 02498194 2005-03-07
WO 2004/035612 PCT/US2003/027489
or enhance function, as seen during viral discovery of such peptides.
References in the
literature describe examples where a peptide identified as a positive binding
recognition
sequence when bound to a virus can be then used without the virus. See, e.g.,
(1) Rozinov M.N., Nolan G.P., Evolution of peptides that modulate the spectral
qualities of
bound, small-molecule fluorophores. Chem Biol. 1998 Dec;S(12):713-28.
(2) Venkatesh N, Zaltsman Y, Somjen D, Gayer B, Boopathi E, Kasher R, Kulik T,
Katchalski-Katzir E, Kohen F. A synthetic peptide with estrogen-like activity
derived from a
phage-display peptide library. Peptides. 2002 Mar;23(3):573-80.
(3) Petrenko VA, Vodyanoy VJ. Phage display for detection of biological threat
agents. J
Microbfiol Methods. 2003 May;53(2):253-62.
Additional working example data were obtained for the polypyrrole system. A
peptide was synthesized whose sequence was determined from the T59 phage (ie.,
SEQ IN
NO. 1). Binding assays were performed to determine if the peptide on its own
binds to
PpyCl without the presence of the phage. The results are provided in Figure 20
and
demonstrate that the T59 peptide binds specifically to PPyCI when compared to
PPyPSS
(polystyrene sulfonate)doped polypyrrole, and polystyrene (PS).
In addition, T59 peptide binding stability in serum-containing medium was also
evaluated in working examples using immunofluorescence (note that all previous
experiments were done in saline buffer). The results, presented in Figure 21,
illustrate that a
significant fraction of T59 peptides remained bound to PPyCI even after 3
weeks of
incubation in serum-containing medium, suggesting a strong and stable
interaction between
the T59 peptide and the PPyCI surface.
Finally, PPyCI-specific T59 peptide was modified at the C-terminus with the
cell
adhesion promoting laminin-derived peptide, RGD, to study cell adhesion in
working
examples. The results shown in Figure 22 illustrate that T59-RGD modified
PPyCI promoted
cell adhesion when compared to unmodified PPyCI. These studies demonstrated
the use of
29
CA 02498194 2005-03-07
WO 2004/035612 PCT/US2003/027489
T59 peptide for biomimetic design of PPyCI for tissue engineering applications
where the
control of cell adhesion and migration is important.
The M.A. thesis by Kiley Preston-Halfmann Miller entitled "Fabrication of
Novel
Interactive Biomaterials via Peptide Integration for Tissue Engineering
Applications"
(University of Texas) is hereby incorporated by reference in its entirety.
This thesis includes
sections on: Introduction (Chapter 1); Peptide Selection (Chapter 2); Chlorine
Doped
Polypyrrole (Chapter 3); Poly(lactic acid-co-glycolic acid) (Chapter 4); and
Conclusions
(Chapter 5). Citation to 36 references is provided on pages 55-57 which
provides further
guidance in the practice of the present invention.
While this invention has been described in reference to illustrative
embodiments, the
descriptions are not intended to be construed in a limiting sense. Various
modifications and
combinations of the illustrative embodiments, as well as other embodiments of
the invention,
will be apparent to persons skilled in the art upon reference to the
description. It is therefore
intended that the appended claims encompass any such modifications or
embodiments.
CA 02498194 2005-03-07
WO 2004/035612 PCT/US2003/027489
1/7
SEQUENCE LISTING
<110> Belches; Angela M.
Schmidt, Christine E.
Miller, Kiley
<120> COMPOSITION, METHOD AND USE OF BI-FUNCTIONAL BIOMATERIALS
<130> 119927-1065
<140> N/A
<141> 2002-09-03
<160> 22
<170> PatentIn version 3.1
<210> 1
<211> 12
<212> PRT
<213> artificial sequence
<220>
<223> peptide
<400> 1
Thr His Arg Thr Ser Thr Leu Asp Tyr Phe Val Ile
1 5 10
<210> 2
<211> 12
<212> PRT
<213> artificial sequence
<220>
<223> peptide
<400> 2
Thr Ile Lys Met His Thr Leu Ser Tyr Thr Gly Leu
1 5 10
<210> 3
<211> 12
<212> PRT
<213> artificial sequence
<220>
<223> peptide
<900> 3
Ser His Lys Tyr Pro Lys Pro Pro Tyr Phe His Trp
1 5 10
CA 02498194 2005-03-07
WO 2004/035612 PCT/US2003/027489
2/7
SEQUENCE LISTING
<110> Belches; Angela M.
Schmidt, Christine E.
Miller, Kiley
<120> COMPOSITION, METHOD AND USE OF BI-FUNCTIONAL BIOMATERIALS
<130> 119927-1065
<140> N/A
<141> 2002-09-03
<160> 22
<170> PatentIn version 3.1
<210> 1
<211> 12
<212> PRT
<213> artificial sequence
<220>
<223> peptide
<400> 1
Thr His Arg Thr Ser Thr Leu Asp Tyr Phe Val Ile
1 5 10
<210> 2
<211> 12
<212> PRT
<213> artificial sequence
<220>
<223> peptide
<400> 2
Thr Ile Lys Met His Thr Leu Ser Tyr Thr Gly Leu
1 5 10
<210> 3
<211> 12
<212> PRT
<213> artificial sequence
<220>
<223> peptide
<900> 3
Ser His Lys Tyr Pro Lys Pro Pro Tyr Phe His Trp
1 5 10
CA 02498194 2005-03-07
WO 2004/035612 PCT/US2003/027489
3/7
<210> 4
<211> 12
<212> PRT
<213> artificial sequence
<220>
<223> peptide
<400> 4
Ile Glu His Pro Lys Thr Pro Asp Ser His Ser Arg
1 5 10
<210> 5
<211> 12
<212> PRT
<213> artificial sequence
<220>
<223> peptide
<900> 5
Val Phe Thr Ala Pro Ala Arg Leu Ile Thr Pro Leu
1 5 10
<210> 6
<211> 12
<212> PRT
<213> artificial sequence
<220>
<223> peptide
<900> 6
Ser Gly His Met Gln Pro Val Thr Arg Pro Pro Ala
1 5 10
<210> 7
<211> 12
<212> PRT
<213> artificial sequence
<220>
<223> peptide
<900> 7
Ser Phe Pro Asp Thr Tyr Val Arg Val Lys Pro Ala
1 5 10
CA 02498194 2005-03-07
WO 2004/035612 PCT/US2003/027489
4/7
<210> 8
<211> 12
<212> PRT
<213> artificial sequence
<220>
<223> peptide
<400> 8
Ile Pro His Ser Gln Val Thr Met Arg Gl.y Leu Pro
1 5 10
<210> 9
<211> 12
<212> PRT
<213.> artificial sequence
<220>
<223> peptide
<400> 9
Thr Ser Met Gln.Leu Ser Met Glu His Lys Leu Ser
1 5 10
<210> 10
<211> 12
<212> PRT
<213> artificial sequence
<220>
<223> peptide
<400> 10
His Phe Asn Val Arg His Thr Ile Pro Thr His Leu
1 5 10
<210> 11
<211> 12
<212> PRT
<213> artificial sequence
<220>
<223> peptide
<900> 11
Met Pro Thr Thr Trp.Ser Thr Thr Leu Gln Tyr His
1 5 10
CA 02498194 2005-03-07
WO 2004/035612 PCT/US2003/027489
5/7
<210> 12
<211> 12
<212> PRT
<213> artificial sequence
<220>
<223> peptide
<900> 12
Ser Thr Asn Leu Asp Pro Gly Leu Ala -Pro Leu Pro
1 5 10
<210> 13
<211> 12
<212> PRT.
<213> artificial sequence
<220>
<223> peptide
<400> 13
Gly Gln Ala His Tyr Lys Ile Ala Thr Gly Glu Ala
1 5 10
<210> 14
<211> 12
<212> PRT
<213> artificial sequence
<220>
<223> peptide
<400> 14
Ile Lys Pro His Met Pro Pro Ser Asp Trp Pro Ser
1 5 10
<210> 15
<211> 12
<212> PRT
<213> artificial sequence
<220>
<223> peptide
<400> 15
Lys Pro Leu His Ser Asn Lys Tyr Tyr Asp Arg Tyr
1 5 10
<210> 16
CA 02498194 2005-03-07
WO 2004/035612 PCT/US2003/027489
6/7
<211> 12
<212> PRT
<213> artificial sequence
<220>
<223> peptide
<400> 16
Thr Ser Lys Leu Pro Thr Trp Val Leu Thr Ser Ser
1 5 10
<210> 17
<211> 12
<212> PRT
<213> artificial sequence
<220>
<223> peptide
<400> 17
Phe-Asn Pro His Gln Phe Ile Lys Pro Pro Lys Lys
1 5 10
<210> 18
<211> 12
<212> PRT
<213> artificial sequence
<220>
<223> peptide
<400> 18
Val Ala Ala Pro Ala Lys Ala Thr Met Ser Ser Thr
1 5 10
<210> 19
<211> 12
<212> PRT
<213> artificial sequence
<220>
<223> peptide
<400> 19
Asn His His His Gln Pro Leu Ala Arg Asn Gln Ser
1 5 10
<210>. 20
<211> 12
CA 02498194 2005-03-07
WO 2004/035612 PCT/US2003/027489
7/7
<212> PRT
<213> artificial sequence
<220>
<223> peptide
<400> 20
Lys Pro Ala Ser Phe Glu Lys Val Leu Asp Ser Val
1 5 10
<210> 21
<211> 12
<212> PRT
<213> artificial sequence
<220>
<223> peptide
<400> 21
Met His His His Gln Pro Leu Ala Arg Met Gln Ser
1 5 10
<210> 22
<211> 12
<212> PRT
<213> artificial sequence
<220>
<223> peptide
<900> 22
Lys Ile Ala Leu Met Pro Trp Pro Ser Val Ser Met
10