Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02503468 2005-04-26
1
DETECTION AND IDENTIFICATION OF ENTEROVIRUSES
FIELD OF THE DISCLOSURE
[001] This disclosure relates to methods of amplifying enterovirus (EV)
nucleic
5 acid molecules and to methods of detecting an EV infection in a subject.
BACKGROUND
[002] Enteroviruses (EVs) (genus Enterovirus, family Picornaviridae)
constitute a
broad range of pathogens etiologically responsible for a wide range of
diseases in
10 both humans and in other animals. Enteroviruses are small RNA viruses
that
contain positive, single stranded RNA as the genome. Five groups are found
within
the enteroviruses: coxsackievirus A, coxsackievirus B, echovirus, poliovirus,
and the
numbered enteroviruses. Most EV infections are asymptomatic or result in only
mild symptoms, such as non-specific febrile illness or mild upper respiratory
15 symptoms (for example, the common cold). However, enteroviruses can also
cause
a wide variety of other clinical illnesses, including acute hemorrhagic
conjunctivitis,
aseptic meningitis, undifferentiated rash, acute flaccid paralysis,
myocarditis, and
neonatal sepsis-like disease.
[003] Molecular diagnostic tests to detect EV in clinical specimens usually
target
20 highly conserved sites in the 5' non-translated region (5'-NTR),
allowing detection
of all members of the genus (Romero, J.R., Arch. Path. & Lab. Med. 123:1161-
69,
1999). These tests are genus-specific and provide an EV-positive or EV-
negative
result but cannot be used to identify the serotype.
[004] Molecular diagnostic tests that target the EV VP1 capsid gene, which
25 correlates with serotype determined by antigenic methods (Oberste et
al.," Virol.
73:1941-48, 1999), can provide both virus detection and identification
(Oberste et
al., I Clin. Microbiol. 38:1170-74, 2000 and Oberste etal., Clin. Virol.
26:375-
77, 2003). However, the identification of serotype, particularly from clinical
specimens, is problematic because the virus titer is very low in original
specimens.
30 As a result, non-specific amplification can out-compete virus-specific
amplification.
CA 02503468 2013-06-21
63198-1483
2
Additionally, highly degenerate, inosine-containing primers used in diagnostic
tests
to broaden specificity to include all serotypes (Casas et aLõ I. Med. ViroL
65:138-48,
2001) often result in non-specific amplification of host cell nucleic acids
that
obscure the virus-specific product (Rose et al.,NucL Acids. Res. 26:1628-35,
1998).
To overcome these limitations additional molecular diagnostic methods are
needed.
SUMMARY OF THE DISCLOSURE
[005] Methods that allow the detection and identification of EVs have been
developed and are described herein. The methods include detecting the presence
of
an EV amplicon containing at least a portion of the EV VP1 encoding sequence
and
sequencing the EV amplicon, and permit the diagnosis and identification of the
EV
serotype involved in a enterovirus infection. The provided methods are useful
in
detecting the presence of an EV in a sample and/or diagnosing an EV infection
in a
subject.
[006] This disclosure also provides isolated nucleic acid molecules, which
nucleic
acid molecules have a nucleotide sequence as set forth in SEQ ID NO: 11, SEQ
ID
NO: 12, SEQ ID NO: 13, or SEQ ID NO: 14. In disclosed examples, these nucleic
acid molecules are EV-specific primers for the detection and identification of
EV
infection. Also described herein are kits that include one or more nucleic
acid
cDNA primers that hybridize to an EV VP1 encoding sequence, a first PCR
nucleic
acid primer pair, wherein the first forward PCR primer hybridizes to an EV VP3
encoding sequence and the first reverse PCR primer hybridizes to an EV VP1
sequence, and a second PCR nucleic acid primer pair, wherein both the second
forward and reverse PCR primers hybridize to an EV WI encoding sequence.
CA 02503468 2015-06-03
, .
63198-1483
2a
[006a} This disclosure as claimed relates to:
- a method of detecting an enterovirus (EV) RNA sequence in a sample,
comprising: contacting the sample with one or more nucleic acid cDNA primers
comprising a
sequence as set forth in any one of SEQ ID NOs: 1-4, that hybridize to an EV
VP1 encoding
sequence; reverse transcribing EV cDNA from the EV RNA sequence; amplifying at
least a
portion of the EV cDNA using a first nucleic acid primer pair, wherein the
first forward
primer comprises a sequence as set forth in SEQ ID NO: 5 and hybridizes to an
EV VP3
encoding sequence and the first reverse primer comprises a sequence as set
forth in SEQ ID
NO: 6 and hybridizes to an EV VP1 encoding sequence, thereby generating an EV
amplicon;
amplifying at least a portion of the EV amplicon using a second nucleic acid
primer pair,
wherein the second forward primer comprises a sequence as set forth as SEQ ID
NO: 11 or
SEQ ID NO: 13, and the second reverse primer comprises a sequence set forth as
SEQ ID
NO: 12 or SEQ ID NO: 14, and wherein both the second forward and reverse
primers
hybridize to an EV VP1 encoding sequence; and determining whether an amplified
EV
amplicon is present, thereby detecting an EV nucleic acid sequence in the
sample; and
- a method of detecting an enterovirus (EV) in a biological sample,
comprising:
extracting EV RNA contained in the sample; contacting the EV RNA with one or
more
nucleic acid cDNA primers comprising a sequence as set forth in any one of SEQ
ID
NOs: 1-4, that hybridize to an EV VP1 encoding sequence; reverse transcribing
EV cDNA
from the EV RNA; amplifying at least a portion of the EV cDNA using a first
nucleic acid
primer pair, wherein the first forward primer comprises a sequence as set
forth as SEQ ID
NO: 5 and hybridizes to an EV VP3 encoding sequence and the first reverse
primer comprises
a sequence set forth as SEQ ID NO: 6 and hybridizes to an EV VP1 encoding
sequence,
thereby generating an EV amplicon; amplifying at least a portion of the EV
amplicon using a
second nucleic acid primer pair, wherein the second forward primer comprises a
sequence as
set forth as SEQ ID NO: 11 or SEQ ID NO: 13, and the second reverse primer
comprises a
sequence set forth as SEQ ID NO: 12 or SEQ ID NO: 14, and wherein both the
second
forward and reverse primers hybridize to an EV VP1 encoding sequence; and
determining
CA 02503468 2015-06-03
63198-1483
2b
whether an amplified EV amplicon is present, thereby detecting an EV in the
biological
sample.
[007] The foregoing and other features and advantages will become
more apparent
from the following detailed description of several embodiments, which proceeds
with
reference to the accompanying figures.
CA 02503468 2005-04-26
,
3
BRIEF DESCRIPTION OF THE FIGURES
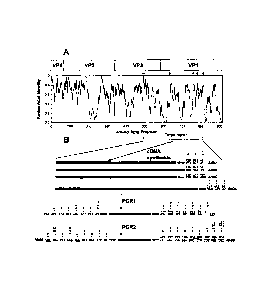
[008] FIGS. IA-1B illustrate the location of the primers used in the COnsensus
DEgenerate Hybrid Oligonucleotide Primer (CODEHOP) VP1 reverse
transcription/semi-nested polymerase chain reaction (RT-snPCR). FIG. lA is a
similarity plot of the aligned capsid amino acid sequences of 64 enterovirus
prototype strains. Sequence identity scores were calculated within each six
residue
window and the window progressively moved across the alignment in one-residue
increments, with the identity score plotted versus position at the center of
the
window. Positions of the four mature EV capsid proteins, VP4, VP2, VP3, and
VP1
are shown at the top. Orientation and approximate position of the cDNA primers
(open arrowheads) and PCR primers (filled arrowheads) are shown directly above
the plot. FIG. 1B illustrates the amino acid motifs used in primer design and
the
steps in the CODEHOP VP1 RT-snPCR assay. Consensus amino acid motifs are
shown. Asterisks indicate that the residue directly above the asterisk is
present at
that position in at least 90% of EV prototype strains. When only a single
residue is
shown, it is present in all prototype strains. Primer sequences are shown
directly
below the amino acid motif sequences. Primers AN32, AN33, AN34, and AN35
(SEQ ID NOs: 1-4, respectively); primer 224 (SEQ ID NO: 5); primer 222 (SEQ ID
NO: 6); primer AN89 (SEQ ID NO: 11); and primer AN88 (SEQ ID NO: 12). IUB
ambiguity codes: R = A or G; Y = C or T; W = A or T; N = A, C, G, or T; M = A
or
C; I = Inosine.
[009] FIG. 2 illustrates the amplification of 101 EV reference strains by VP1
RT-
snPCR. Primers AN32, AN33, AN34, and AN35 (SEQ ID NOs: 1-4, respectively)
were used for cDNA synthesis. Primers 224 (SEQ ID NO: 5) and 222 (SEQ ID NO:
6) were used in the first PCR, and primers AN89 (SEQ ID NO: 11) and AN88 (SEQ
ID NO: 12) were used in the second PCR. The strains tested were CVAl-Tomkins,
CVA2-Fleetwood, CVA3-Olson, CVA4-High Point, CVA5-Swartz, CVA6-Gdula,
CVA7-Parker, CVA8-Donovan, CVA9-Griggs, CVA10-Kowalik, CVA11-Belgium-
1, CVA12-Texas-12, CVA13-Flores, CVA14-G-14, CVA15-G-9, CVA16-G-10,
CVA17-G12, CVA18, G-13, CVA19-NIH-8663, CVA20-IH-35, CVA21-Coe,
CVA22-Chulman, CVA24-Joseph, CVA24-EH24-70 (CVA24v), CVB1-Japan,
CVB2-Ohio-1, CVB3-Nancy, CVB4-NB, CVB5-Faulkner, CVB6-Schmitt, El-
CA 02503468 2005-04-26
4
Farouk, E2-Cornelis, E3-Morrisey, E4-Dutoit (E4D), E4-Shropshire (E4S), E4-
Pesacek (E4P), E5-Noyce, E6-D'Amori (E6D), E6-Cox (E6'), E6-Burgess, (E6"),
E6-Charles (E6C), E7-Wallace, E8-Bryson, E9-Hill, Ell-Gregory, Ell-Silva
(El 1'), E12-Travis, E13-Del Carmen, E14-Tow, E15-CH96-51, E16-Harrington,
E17-CHHE-29, El 8-Metcalf, E19-Burke, E21-Farina, E24-DeCamp,
E25-JV-4, E26-Coronel, E27-Bacon, E29-JV-10, E30-Bastianni (E30B), E30-Frater
(E30F), E30-Giles (E30G), E30-PR-17 (E30P), E31-Caldwell, E32-PR-10, E33-
Toluca-3, E34-DN-19, EV68-Fermon, EV69-Toluca-1, EV70-J670/71, EV71-BrCr,
PV1-Mahoney, PV2-Lansing, PV3-Leon, PV1-Sabin, PV2-Sabin, PV3, Sabin,
EV73-CA55-1988, EV74-10213, EV75-10219, EV76-10226, EV79-10244, EV80-
10246, EV81-10248, EV82-10249, EV83-10251, EV84-10603, EV85-10353, EV86-
10354, EV87-10396, EV88-10398, EV89-10359, EV90-10399, EV91-10406, EV92-
10408, EV96-10358, EV97-10355, EV100-10500, and EV101-10361. Reference
strains for EV77-78 and EV93-95 were not included. Other numbers are missing
due to reclassification (for example, CVA23 is a variant of E9; El0 is
reovirus 1,
genus Orthoreovirus, family Reoviridae; E28 is human rhinovirus 1A, genus
Rhinovirus, family Picornaviridae; EV72 is human hepatitis A virus, genus
Hepatovirus, family Picornaviridae). Also, E8 is a variant of El and E34 is a
variant of CVA24. For each reaction, 10 jil of each semi-nested PCR2 product
was
analyzed by electrophoresis on a 1.5% agarose gel, containing 0.5 micrograms
ethidium bromide per milliliter. Lanes at the ends of each row are DNA size
markers. The negative control reaction, using uninfected cell culture RNA
(CC), is
shown in the bottom row.
[010] FIGS. 3A-3C illustrate the sensitivity of VP! RT-snPCR and a show a
comparison of VP! RT-snPCR with 5'-NTR RT-snPCR. FIG. lA illustrates the
amplification of RNA extracted from 10-fold serial dilutions of an EV68 virus
stock.
FIG. 1B illustrates the amplification of 10-fold serial dilutions of VP3-VP1
sRNA.
FIG. 1C shows a comparison of VP1 RT-snPCR (top) with 5'-NTR RT-snPCR
(bottom) using 10-fold serial dilutions.
[011] FIG. 4 illustrates the amplification of RNA extracted directly from
original
clinical specimens using VP! RT-snPCR. For each reaction, 50 I of each semi-
__ __________________________________
CA 02503468 2005-04-26
nested PCR2 product was analyzed and gel purified by electrophoresis on a 1.5%
agarose gel, containing 0.5 micrograms ethidium bromide per milliliter. The
specimens tested were cerebrospinal fluid (CSF), stool, rectal swab (RS),
nasopharyngeal swab (NPS), eye (conjunctival) swab (ES), serum, and postmortem
5 liver tissue.
SEQUENCE LISTING
[012] The nucleic and amino acid sequences listed in the accompanying sequence
listing are shown using standard letter abbreviations for nucleotide bases,
and three
letter code for amino acids, as defined in 37 C.F.R. 1.822. Only one strand of
each
nucleic acid sequence is shown, but the complementary strand is understood as
included by any reference to the displayed strand. In the accompanying
sequence
listing:
[013] SEQ ID NOs: 1-4 show the nucleic acid sequence of several EV-specific
reverse oligonucleotide primers for cDNA synthesis.
[014] SEQ ID NOs: 5-14 show the nucleic acid sequence of several EV-specific
oligonucleotide primers (forward and reverse) for DNA amplification.
[015] SEQ ID NO: 15 shows the nucleic acid sequence of the non-degenerate
"clamp" portion of primer AN89.
[016] SEQ ID NO: 16 shows the nucleic acid sequence of the non-degenerate
"clamp" portion of primer AN88.
[017] SEQ ID NOs: 17 and 18 show the nucleic acid sequence of a pair of EV-
specific oligonucleotide primers (sense and antisense) for generation of a
synthetic
RNA standard.
[018] SEQ ID NOs: 19-32 show the amino acid sequence of several conserved
amino acid sequences from which EV-specific oligonucleotide primers were
derived
by back-translation.
CA 02503468 2005-04-26
6
DETAILED DESCRIPTION
I. Abbreviations
BAL: bronchoalveolar lavage
degrees Celsius
cDNA: complementary DNA
CSF: cerebrospinal fluid
DTT: dithiothreitol
ES: eye (conjunctival) swab
EV: enterovirus
g: gram
min: minute(s)
ml: milliliter
NPS: nasopharyngeal swab
PCR: polymerase chain reaction
RS: rectal swab
RT-snPCR: reverse transcription semi-nested polymerase chain reaction
microgram(s)
microliter(s)
s: second(s)
IL Terms
[019] Unless otherwise noted, technical terms are used according to
conventional
usage. Definitions of common terms in molecular biology may be found in
Benjamin Lewin, Genes VII, published by Oxford University Press, 2000 (ISBN
019879276X); Kendrew etal. (eds.), The Encyclopedia of Molecular Biology,
published by Blackwell Publishers, 1994 (ISBN 0632021829); and Robert A.
Meyers (ed.), Molecular Biology and Biotechnology: a Comprehensive Desk
Reference, published by Wiley, John & Sons, Inc., 1995 (ISBN 0471186341); and
other similar references.
[020] In order to facilitate review of the various embodiments of this
disclosure,
the following explanations of specific terms are provided:
[021] Amplification of or amplifying a nucleic acid sequence: Increasing the
amount of (number of copies of) a nucleic acid sequence, wherein the increased
sequence is the same as or complementary to the existing nucleic acid
template. The
resulting amplification products are called "amplicons." An example of
CA 02503468 2005-04-26
7
amplification is the polymerase chain reaction (PCR). Other examples of
amplification techniques include reverse-transcription PCR (RT-PCR), semi-
nested
RT-PCR (RT-snPCR), strand displacement amplification (see U.S. Patent No.
5,744,311), transcription-free isothermal amplification (see U.S. Patent No.
6,033,881), repair chain reaction amplification (see WO 90/01069), ligase
chain
reaction amplification (see EP-A-320 308), gap filling ligase chain reaction
amplification (see U.S. Patent No. 5,427,930), coupled ligase detection and
PCR
(see U.S. Patent No. 6,027,889), and NASBATM RNA transcription-free
amplification (see U.S. Patent No. 6,025,134).
[022] The products of amplification may be characterized by, for instance,
electrophoresis, restriction endonuc lease cleavage patterns, hybridization,
ligation,
and/or nucleic acid sequencing, using standard techniques.
[023] Animal: Living multi-cellular vertebrate organisms, a category that
includes, for example, mammals and birds. The term mammal includes both human
and non-human mammals. Similarly, the term "subject" includes both human and
veterinary subjects, for example, humans, non-human primates, dogs, cats,
horses,
and cows.
[024] Antisense and sense: Double-stranded DNA (dsDNA) has two strands, a
5' to 3' strand, referred to as the plus strand, and a 3' to 5' strand,
referred to as the
minus strand. Because RNA polymerase adds nucleic acids in a 5' to 3'
direction,
the minus strand of the DNA serves as the template for the RNA during
transcription. Thus, the RNA formed will have a sequence complementary to the
minus strand, and identical to the plus strand (except that the base uracil is
substituted for thymine).
[025] Antisense molecules are molecules that are specifically hybridizable or
specifically complementary to either RNA or the plus strand of DNA. Sense
molecules are molecules that are specifically hybridizable or specifically
complementary to the minus strand of DNA.
CA 02503468 2005-04-26
8
[026] Detect: To determine the existence or presence of something. For
example,
to determine whether an enterovirus or an EV nucleic acid sequence is present
in a
sample (such as a biological sample), or to determine if an amplicon is
present
following amplification.
[027] Electrophoresis: Electrophoresis refers to the migration of charged
solutes
or particles in a liquid medium under the influence of an electric field.
Electrophoretic separations are widely used for analysis of macromolecules. Of
particular importance is the identification of proteins and nucleic acid
sequences.
Such separations can be based on differences in size and/or charge. Nucleotide
sequences have a uniform charge and are therefore separated based on
differences in
size. Electrophoresis can be performed in an unsupported liquid medium, but
more
commonly the liquid medium travels through a solid supporting medium. The most
widely used supporting media are gels, such as, polyacrylamide and agarose
gels
(used, for example, in capillary gel electrophoresis and slab gel
electrophoresis).
[028] Sieving gels (for example, agarose) impede the flow of molecules. The
pore
size of the gel determines the size of a molecule that can flow freely through
the gel.
The amount of time to travel through the gel increases as the size of the
molecule
increases. As a result, small molecules travel through the gel more quickly
than
large molecules and thus progress further from the sample application area
than
larger molecules, in a given time period. Such gels are used for size-based
separations of nucleotide sequences.
[029] Fragments of linear DNA migrate through agarose gels with a mobility
that
is inversely proportional to the logio of their molecular weight. By using
gels with
different concentrations of agarose, different sizes of DNA fragments can be
resolved. Higher concentrations of agarose facilitate separation of small
DNAs,
while low agarose concentrations allow resolution of larger DNAs.
[030] Hybridization: Oligonucleotides and their analogs hybridize by hydrogen
bonding, which includes Watson-Crick, Hoogsteen or reversed Hoogsteen hydrogen
bonding,. between complementary bases. "Complementary" refers to the base
CA 02503468 2005-04-26
9
pairing that occurs between two distinct nucleic acid sequences or two
distinct
regions of the same nucleic acid sequence.
[031] "Specifically hybridizable," "specifically hybridizes" and "specifically
complementary" are terms which indicate a sufficient degree of complementarity
such that stable and specific binding occurs between an oligonucleotide and
its DNA
or RNA target. An oligonucleotide need not be 100% complementary to its target
DNA or RNA sequence to be specifically hybridizable. An oligonucleotide is
specifically hybridizable when there is a sufficient degree of complementarity
to
avoid non-specific binding of the oligonucleotide to non-target sequences
under
conditions in which specific binding is desired, or under conditions in which
an
assay is performed.
[032] Hybridization conditions resulting in particular degrees of stringency
will
vary depending upon the nature of the hybridization method of choice and the
composition and length of the hybridizing nucleic acid sequences. Generally,
the
temperature of hybridization and the ionic strength (especially the Na +
and/or MC
concentration) of the hybridization buffer will determine the stringency of
hybridization. Calculations regarding hybridization conditions required for
attaining
particular degrees of stringency are discussed by Sambrook et al. (ed.),
Molecular
Cloning: A Laboratory Manual, 2n1 ed., vol. 1-3, Cold Spring Harbor Laboratory
Press, Cold Spring Harbor, NY, 1989, chapters 9 and 11; and Ausubel et al.
Short
Protocols in Molecular Biology, 4th ed., John Wiley & Sons, Inc., 1999.
[033] The following is an exemplary set of hybridization conditions for PCR
and
is not meant to be limiting:
Low stringency annealing conditions
Denaturation: 95 C for 30 seconds
Annealing: 42 C for 30 seconds
Extension: 60 C for 45 seconds
High stringency annealing conditions
Denaturation: 95 C for 30 seconds
Annealing: 60 C for 20 seconds
Extension: 72 C for 15 seconds
_____________________________ _ _
CA 02503468 2005-04-26
[034] Isolated or purified: An "isolated" or "purified" biological component
(such as a nucleic acid, peptide or protein) has been substantially separated,
produced apart from, or purified away from other biological components in the
cell
of the organism in which the component naturally occurs, that is, other
chromosomal
5 and extrachromosomal DNA and RNA, and proteins. Nucleic acids, peptides
and
proteins that have been "isolated" thus include nucleic acids and proteins
purified by
standard purification methods. The term also embraces nucleic acids, peptides
and
proteins prepared by recombinant expression in a host cell as well as
chemically
synthesized nucleic acids or proteins.
10 [035] The term "isolated" or "purified" does not require absolute
purity; rather, it
is intended as a relative term. Thus, for example, an isolated biological
component
is one in which the biological component is more enriched than the biological
component is in its natural environment within a cell. Preferably, a
preparation is
purified such that the biological component represents at least 50%, such as
at least
70%, at least 90%, at least 95%, or greater of the total biological component
content
of the preparation.
[036] Label: A detectable compound or composition that is conjugated or
otherwise attached directly or indirectly to another molecule to facilitate
detection of
that molecule. Specific, non-limiting examples of labels include radioactive
isotopes, enzyme substrates, co-factors, ligands, chemiluminescent or
fluorescent
markers or dyes, haptens, and enzymes. Methods for labeling and guidance in
the
choice of labels appropriate for various purposes are discussed, for example,
in
Sambrook et al. (ed.), Molecular Cloning: A Laboratory Manual, 2nd ed., vol. 1-
3,
Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, 1989 and Ausubel
etal. Short Protocols in Molecular Biology, 4th ed., John Wiley & Sons, Inc.,
1999.
[037] Nucleic acid sequence (or polynucleotide): A deoxyribonucleotide or
ribonucleotide polymer in either single or double stranded form, and unless
otherwise limited, encompasses known analogues of natural nucleotides that
hybridize to nucleic acids in a manner similar to naturally occurring
nucleotides, and
includes polynucleotides encoding full length proteins and/or fragments of
such full
CA 02503468 2005-04-26
=
11
length proteins which can function as a therapeutic agent. A polynucleotide is
generally a linear nucleotide sequence, including sequences of greater than
100
nucleotide bases in length. In one embodiment, a nucleic acid is labeled (for
example, biotinylated, fluorescently labeled or radiolabled nucleotides).
[038] Nucleotide: "Nucleotide" includes, but is not limited to, a monomer that
includes a base linked to a sugar, such as a pyrimidine, purine or synthetic
analogs
thereof, or a base linked to an amino acid, as in a peptide nucleic acid
(PNA). A
nucleotide is one monomer in an oligonucleotide/polynucleotide. A nucleotide
sequence refers to the sequence of bases in an oligonucleotide/polynucleotide.
[039] The major nucleotides of DNA are deoxyadenosine 5'-triphosphate (dATP
or A), deoxyguanosine 5'-triphosphate (dGTP or G), deoxycytidine 5'-
triphosphate
(dCTP or C) and deoxythymidine 5'-triphosphate (dTTP or T). The major
nucleotides of RNA are adenosine 5'-triphosphate (ATP or A), guanosine 5'-
triphosphate (GTP or G), cytidine 5'-triphosphate (CTP or C) and uridine 5'-
triphosphate (UTP or U). Inosine is also a base that can be integrated into
DNA or
RNA in a nucleotide (dITP or ITP, respectively).
[040] Oligonucleotide: A nucleic acid molecule generally comprising a length
of
300 bases or fewer. The term often refers to single-stranded
deoxyribonucleotides,
but it can refer as well to single- or double-stranded ribonucleotides,
RNA:DNA
hybrids and double-stranded DNAs, among others. The term "oligonucleotide"
also
includes oligonucleosides, that is, an oligonucleotide minus the phosphate. In
some
examples, oligonucleotides are about 7 to about 50 bases in length, for
example, 8,
9, 10, 15, 20, 25, 30, or 35 bases in length. Other oligonucleotides are about
40 or
about 45 bases in length.
[041] Oligonucleotides may be single-stranded, for example, for use as probes
or
primers, or may be double-stranded, for example, for use in the construction
of a
mutant gene. Oligonucleotides can be either sense or antisense
oligonucleotides.
An oligonucleotide can be modified as discussed herein in reference to nucleic
acid
molecules. Oligonucleotides can be obtained from existing nucleic acid sources
(for
CA 02503468 2005-04-26
12
example, genomic or cDNA), but can also be synthetic (for example, produced by
laboratory or in vitro oligonucleotide synthesis).
[042] Polypeptide: A polymer in which the monomers are amino acid residues
which are joined together through amide bonds. When the amino acids are alpha-
amino acids, either the L-optical isomer or the D-optical isomer can be used.
The
terms "polypeptide" or "protein" as used herein are intended to encompass any
amino acid sequence and include modified sequences such as glycoproteins. The
term "polypeptide" is specifically intended to cover naturally occurring
proteins, as
well as those which are recombinantly or synthetically produced. The term
"residue" or "amino acid residue" includes reference to an amino acid that is
incorporated into a peptide, polypeptide, or protein.
[043] Primers and probes: Primers are short nucleic acid molecules, for
instance
DNA oligonucleotides 7 nucleotides or more in length, for example that
hybridize to
contiguous complementary nucleotides or a sequence to be amplified. Longer DNA
oligonucleotides may be about 15, 20, 25, 30 or 50 nucleotides or more in
length.
Primers can be annealed to a complementary target DNA strand by nucleic acid
hybridization to form a hybrid between the primer and the target DNA strand,
and
then the primer extended along the target DNA strand by a DNA polymerase
enzyme. Primer pairs (that is, forward/sense and reverse/antisense) can be
used for
amplification of a nucleic acid sequence, for example, by the PCR or other
nucleic-
acid amplification methods known in the art.
[044] A probe includes an isolated nucleic acid sequence attached to a
detectable
label or other reporter molecule. Typical labels include radioactive isotopes,
enzyme substrates, co-factors, ligands, chemiltuninescent or fluorescent
agents,
haptens, and enzymes. Methods for labeling and guidance in the choice of
labels
appropriate for various purposes are discussed, for example, in Sambrook et
al.
(ed.), Molecular Cloning: A Laboratory Manual, 2nd ed., vol. 1-3, Cold Spring
Harbor Laboratory Press, Cold Spring Harbor, NY, 1989 and Ausubel et al. Short
Protocols in Molecular Biology, 4th ed., John Wiley & Sons, Inc., 1999.
CA 02503468 2005-04-26
13
[045] Methods for preparing and using nucleic acid primers and probes are
described, for example, in Sambrook et al. (ed.), Molecular Cloning: A
Laboratory
Manual, 2nd ed., vol. 1-3, Cold Spring Harbor Laboratory Press, Cold Spring
Harbor, NY, 1989; Ausubel et al. Short Protocols in Molecular Biology, 4th
ed.,
John Wiley & Sons, Inc., 1999; and Innis et al. PCR Protocols, A Guide to
Methods
and Applications, Academic Press, Inc., San Diego, CA, 1990. Amplification
primer pairs can be derived from a known sequence, for example, by using
computer
programs intended for that purpose such as Primer (Version 0.5, C 1991,
Whitehead
Institute for Biomedical Research, Cambridge, MA). One of ordinary skill in
the art
will appreciate that the specificity of a particular probe or primer increases
with its
length. Thus, in order to obtain greater specificity, probes and primers can
be
selected that comprise at least 20, 25, 30, 35, 40, 45, 50, 75, 90 or more
consecutive
nucleotides of a target nucleotide sequence.
[046] Recombinant: A recombinant nucleic acid is one that has a sequence that
is not naturally occurring or has a sequence that is made by an artificial
combination
of two otherwise separated segments of sequence. This artificial combination
can be
accomplished by chemical synthesis or, more commonly, by the artificial
manipulation of isolated segments of nucleic acids, for example, by genetic
engineering techniques.
[047] Sample: A portion, piece, or segment that is representative of the whole
from which the sample is obtained. This term encompasses any material,
including
for instance samples obtained from an animal, a plant, or the environment.
[048] An "environmental sample" includes a sample obtained from inanimate
objects or reservoirs within an indoor or outdoor environment. Environmental
samples include, but are not limited to: soil, water, dust, and air samples;
bulk
samples, including building materials, furniture, and landfill contents; and
other
reservoir samples, such as animal refuse, harvested grains, and foodstuffs. It
is to be
understood that environmental samples can and often do contain biological
components.
= CA 02503468 2005-04-26
14
[049] A "biological sample" is a sample obtained from a subject, and may also
be
referred to as a "clinical specimen." As used herein, a biological sample
includes all
samples useful for detection of enterovirus infection in subjects, including,
but not
limited to: cells, tissues, and bodily fluids, such as blood; derivatives and
fractions
of blood (such as serum); extracted galls; biopsied or surgically removed
tissue,
including tissues that are, for example, unfixed, frozen, fixed in formalin
and/or
embedded in paraffin; tears; milk; skin scrapes; surface washings;
oropharyngeal
wash; urine; sputum; cerebrospinal fluid; prostate fluid; semen; pus; bone
marrow
aspirates; bronchoalveolar lavage (BAL); saliva; nasopharyngeal swabs, eye
swabs,
cervical swabs, vaginal swabs, and rectal swabs; and stool and stool
suspensions.
[050] As used herein, the singular terms "a," "an," and "the" include plural
referents unless context clearly indicates otherwise. Similarly, the word "or"
is
intended to include "and" unless the context clearly indicates otherwise.
Also, as
used herein, the term "comprises" means "includes." Hence "comprising A or B"
means including A, B, or A and B. It is further to be understood that all base
sizes
or amino acid sizes, and all molecular weight or molecular mass values, given
for
nucleic acids or polypeptides are approximate, and are provided for
description.
Although methods and materials similar or equivalent to those described herein
can
be used in the practice or testing of the present invention, suitable methods
and
materials are described below. In case of conflict, the present specification,
including explanations of terms, will control. In addition, the materials,
methods,
and examples are illustrative only and not intended to be limiting.
Hi Overview of Several Embodiments
[051] Provided herein are isolated nucleic acid molecules, which nucleic acid
molecules have a nucleotide sequence as set forth in SEQ ID NO: 11, SEQ ID NO:
12, SEQ ID NO: 13, or SEQ ID NO: 14. In certain embodiments, these nucleic
acid
molecules are EV-specific primers for the detection and identification of EV
infection.
[052] Also provided herein in various embodiments is a method of detecting an
EV RNA sequence in a sample. In one embodiment, the method includes (i)
CA 02503468 2005-04-26
contacting the sample with one or more nucleic acid cDNA primers that
hybridize to
an EV VP1 encoding sequence, (ii) reverse transcribing EV cDNA from the EV
RNA sequence, (iii) amplifying at least a portion of the EV cDNA using a first
nucleic acid primer pair, wherein the first forward primer hybridizes to an EV
VP3
5 encoding sequence and the first reverse primer hybridizes to an EV VP1
encoding
sequence, thereby generating an EV amplicon, (iv) amplifying at least a
portion of
the EV amplicon using a second nucleic acid primer pair, wherein both the
second
forward and reverse primers hybridize to an EV VP1 encoding sequence, and (v)
determining whether an amplified EV amplicon is present, thereby detecting an
EV
10 nucleic acid sequence in the sample.
[053] In a specific, non-limiting example of the provided method, the
amplification includes a polymerase chain reaction amplification. In another
specific, non-limiting example, determining whether an amplified EV amplicon
is
present includes gel electrophoresis and visualization of the amplified EV
amplicon,
15 capillary electrophoresis and detection of the amplified EV amplicon,
and/or
hybridization of a labeled probe to the amplified EV amplicon. In yet another
specific, non-limiting example of the provided method, the one or more nucleic
acid
cDNA primers include a sequence as set forth in any one of SEQ ID NOs: 1-4,
the
first forward primer includes a sequence as set forth in SEQ ID NO: 5, the
first
reverse primer includes a sequence as set forth in SEQ ID NO: 6, the second
forward
primer includes a sequence as set forth in any one of SEQ ID NOs: 7-11 and 13,
and
the second reverse primer includes a sequence as set forth in any one of SEQ
ID
NOs: 6, 12 and 14.
[054] In a further specific example of the provided method, the method
includes
extracting EV RNA from the sample. In yet a further specific example of the
provided method, the method includes electrophoresing and sequencing at least
a
portion of the amplified EV amplicon, wherein the method of detecting an EV
RNA
sequence in a sample includes a method of identifying the EV in the sample.
[055] A method of detecting an EV in a biological sample is also described
herein.
This method includes (i) extracting EV RNA contained in the sample, (ii)
contacting
CA 02503468 2005-04-26
16
the EV RNA with one or more nucleic acid cDNA primers that hybridize to an EV
VP1 encoding sequence (iii) reverse transcribing EV cDNA from the EV RNA, (iv)
amplifying at least a portion of the EV cDNA using a first nucleic acid primer
pair,
wherein the first forward primer hybridizes to an EV VP3 encoding sequence and
the first reverse primer hybridizes to an EV VP1 encoding sequence, thereby
generating an EV amplicon, (v) amplifying at least a portion of the EV
amplicon
using a second nucleic acid primer pair, wherein both the second forward and
reverse primers hybridize to an EV VP1 encoding sequence, and (vi) determining
whether an amplified EV amplicon is present, thereby detecting an EV in the
biological sample.
[056] In a specific, non-limiting example of the provided method, the
amplification includes a polymerase chain reaction amplification. In another
specific, non-limiting example, determining whether an amplified EV amplicon
is
present includes gel electrophoresis and visualization of the amplified EV
amplicon,
capillary electrophoresis and detection of the amplified EV amplicon, and/or
hybridization of a labeled probe to the amplified EV amplicon. In yet another
specific, non-limiting example of the provided method, the one or more nucleic
acid
cDNA primers include a sequence as set forth in any one of SEQ ID NOs: 1-4,
the
first forward primer includes a sequence as set forth in SEQ ID NO: 5, the
first
reverse primer includes a sequence as set forth in SEQ ID NO: 6, the second
forward
primer includes a sequence as set forth in any one of SEQ ID NOs: 7-11 and 13,
and
the second reverse primer includes a sequence as set forth in any one of SEQ
ID
NOs: 6, 12 and 14.
[057] In yet a further specific example of the provided method, the method
includes electrophoresing and sequencing at least a portion of the amplified
EV
amplicon, wherein the method of detecting an EV in a biological sample
includes a
method of identifying the EV in the sample. In another specific, non-limiting
example, the biological sample contains free EV particles and/or EV infected
cells.
[058] Kits are also disclosed herein that include one or more nucleic acid
cDNA
primers that hybridize to an EV VP1 encoding sequence, a first PCR nucleic
acid
CA 02503468 2005-04-26
,
17
primer pair, wherein the first forward PCR primer hybridizes to an EV VP3
encoding sequence and the first reverse PCR primer hybridizes to an EV VP1
sequence, and a second PCR nucleic acid primer pair, wherein both the second
forward and reverse PCR primers hybridize to an EV VP1 encoding sequence.
[059] In one embodiment, the one or more nucleic acid cDNA primers include a
sequence as set forth in any one of SEQ ID NOs: 1-4. In another embodiment,
the
first forward PCR primer includes a sequence as set forth in SEQ ID NO: 5 and
the
first reverse PCR primer includes a sequence as set forth in SEQ ID NO: 6. In
still
another embodiment, the second forward PCR primer includes a sequence as set
forth in any one of SEQ ID NOs: 7-11 and 13 and the second reverse PCR primer
includes a sequence as set forth in any one of SEQ ID NOs: 6, 12 and 14. In
yet
another embodiment, the one or more nucleic acid cDNA primers include a
sequence as set forth in any one of SEQ ID NOs: 1-4, the first forward PCR
primer
includes a sequence as set forth in SEQ ID NO: 5, the first reverse PCR primer
includes a sequence as set forth in SEQ ID NO: 6, the second forward PCR
primer
includes a sequence as set forth in SEQ ID NO: 11, and the second reverse PCR
primer includes a sequence as set forth in SEQ ID NO: 12.
IV. Synthesis of Oligonucleotide Primers and Probes
[060] In vitro methods for the synthesis of oligonucleotides are well known to
those of ordinary skill in the art; such methods can be used to produce
primers and
probes for the disclosed methods. The most common method for in vitro
oligonucleotide synthesis is the phosphoramidite method, formulated by
Letsinger
and further developed by Caruthers (Caruthers et al., Chemical synthesis of
deoxyoligonucleotides, in Methods Enzymol. 154:287-313, 1987). This is a non-
aqueous, solid phase reaction carried out in a stepwise manner, wherein a
single
nucleotide (or modified nucleotide) is added to a growing oligonucleotide. The
individual nucleotides are added in the form of reactive 3'-phosphoramidite
derivatives. See also, Gait (Ed.), Oligonucleotide Synthesis. A practical
approach,
IRL Press, 1984.
CA 02503468 2005-04-26
,
,
18
[061] In general, the synthesis reactions proceed as follows: A
dimethoxytrityl or
equivalent protecting group at the 5' end of the growing oligonucleotide chain
is
removed by acid treatment. (The growing chain is anchored by its 3' end to a
solid
support such as a silicon bead.) The newly liberated 5' end of the
oligonucleotide
chain is coupled to the 3'-phosphoramidite derivative of the next
deoxynucleoside to
be added to the chain, using the coupling agent tetrazole. The coupling
reaction
usually proceeds at an efficiency of approximately 99%; any remaining
unreacted 5'
ends are capped by acetylation so as to block extension in subsequent
couplings.
Finally, the phosphite triester group produced by the coupling step is
oxidized to the
phosphotriester, yielding a chain that has been lengthened by one nucleotide
residue.
This process is repeated, adding one residue per cycle. See, for example, U.S.
Patent Nos. 4,415,732, 4,458,066, 4,500,707, 4,973,679, and 5,132,418.
Oligonucleotide synthesizers that employ this or similar methods are available
commercially (for example, the PolyPlex oligonucleotide synthesizer from Gene
Machines, San Carlos, CA). In addition, many companies will perform such
synthesis (for example, Sigma-Genosys, The Woodlands, TX; Qiagen Operon,
Alameda, CA; Integrated DNA Technologies, Coralville, IA; and TriLink
BioTechnologies, San Diego, CA).
V. Detection and Identification of Enteroviruses
[062] A major application of the EV-specific primers presented herein is in
the
area of detection and diagnostic testing for EV infection. Methods for
screening a
subject to determine if the subject is infected with an EV are disclosed
herein.
[063] One such method includes providing a sample, which sample includes an
EV or an EV nucleic acid (such as RNA), and providing an assay for detecting
in the
sample the presence of the EV or EV RNA sequence. Suitable samples include all
biological samples useful for detection of viral infection in subjects,
including, but
not limited to, cells, tissues (for example, lung, liver and kidney), bodily
fluids (for
example, blood, serum, urine, saliva, sputum, and cerebrospinal fluid), bone
marrow
aspirates, BAL, oropharyngeal wash, nasopharyngeal swabs, eye swabs, cervical
swabs, vaginal swabs, rectal swabs, stool, and stool suspensions. Additional
suitable
samples include all environmental samples useful for detection of viral
presence in
CA 02503468 2005-04-26
19
the environment, including, but not limited to, a sample obtained from
inanimate
objects or reservoirs within an indoor or outdoor environment. The detection
in the
sample of the EV or EV RNA sequence may be performed by a number of
methodologies, non-limiting examples of which are outlined below.
[064] In one embodiment, detecting in the sample the presence of an EV or EV
RNA sequence includes the extraction of EV RNA. RNA extraction relates to
releasing RNA from a latent or inaccessible form in a virion, cell or sample
and
allowing the RNA to become freely available. In such a state, it is suitable
for
effective amplification by reverse transcription and the use of, for example,
PCR.
Releasing RNA may include steps that achieve the disruption of virions
containing
viral RNA, as well as disruption of cells that may harbor such virions.
Extraction of
RNA is generally carried out under conditions that effectively exclude or
inhibit any
ribonuclease activity that may be present. Additionally, extraction of RNA may
include steps that achieve at least a partial separation of the RNA dissolved
in an
aqueous medium from other cellular or viral components, wherein such
components
may be either particulate or dissolved.
[065] One of ordinary skill in the art will know suitable methods for
extracting
RNA from a sample; such methods will depend upon, for example, the type of
sample in which the EV RNA is found. For example, the RNA may be extracted
using guanidinium isothiocyanate, such as the single-step isolation by acid
guanidinium isothiocyanate-phenol-chloroform extraction of Chomczynski et al.
(Anal. Biochem. 162:156-59, 1987). Alternatively, an EV virion may be
disrupted
by a suitable detergent in the presence of proteases and/or inhibitors of
ribonuclease
activity. Additional exemplary methods for extracting RNA are found, for
example,
in World Health Organization, Manual for the virological investigation of
polio,
World Health Organization, Geneva, 2001.
[066] Enterovirus RNA is subjected to reverse transcription to prepare a
cognate
cDNA that encompasses the region of the genome chosen for detecting and
identifying the EV serotype (for example, the region encoding VP1). In one
embodiment, a set of random oligonucleotide primers is used, such that certain
of
CA 02503468 2005-04-26
the primers in the set will hybridize to the EV RNA and yield one or more cDNA
molecules from the virus encompassing the required serotype-specific
nucleotide
sequence. In another embodiment, gene-specific primers based on conserved
amino
acid motifs in aligned sequences of known EV serotypes (such as SEQ ID NOs: 1-
4)
5 are used for reverse transcription. Subsequently, the EV cDNA is
amplified using a
suitable amplification protocol to generate an EV amplicon. Any nucleic acid
amplification method can be used. In one specific, non-limiting example, PCR
is
used to amplify the EV cDNA. In another non-limiting example, RT-PCR can be
used to amplify the EV cDNA. In an additional non-limiting example, RT-snPCR
10 can be used to amplify the EV cDNA. RT-snPCR refers to a pair of PCRs
(PCR1
and PCR2) that is initiated with cDNA that has been reverse transcribed from
RNA,
and is run in series, each with a pair of primers flanking the same sequence.
The
first PCR (PCR1) amplifies a sequence, such as an EV cDNA sequence. The second
primer pair (semi-nested primers) for the second PCR (PCR2) bind at one end of
and
15 within the first PCR product and produce a second PCR product that is
shorter than
the first one (see FIG. 1B). Techniques for reverse transcription and nucleic
acid
amplification are well-known to those of skill in the art.
[067] In some embodiments, pairs of EV-specific primers are utilized in the
RT-
snPCR amplification reaction. Specific, non-limiting examples of EV-specific
20 primers include, but are not limited to: 224 (SEQ ID NO: 5), 222 (SEQ ID
NO: 6),
187 (SEQ ID NO: 7), 188 (SEQ ID NO: 8), 189 (SEQ ID NO: 9), 292 (SEQ ID NO:
10), AN89 (SEQ ID NO: 11), AN88 (SEQ ID NO: 12), AN79 (SEQ ID NO: 13),
and AN78 (SEQ ID NO: 14).
[068] Enterovirus amplicons obtained following nucleic acid amplification can
be
sequenced to determine the nucleotide sequence in each. Procedures that can be
used for sequencing include the methods of Maxam and Gilbert (Meth. Enzymol.
65:499-566, 1980) and Sanger et al. (Proc. Natl. Acad. Sci. USA 74:5463-67,
1977).
Sequencing methods are also discussed, for example, in Sambrook et al. (ed.),
Molecular Cloning: A Laboratory Manual, 2"d ed., vol. 1-3, Cold Spring Harbor
Laboratory Press, Cold Spring Harbor, NY, 1989. The method of Sanger etal.
involves the use of different 2',3'dideoxynucleotide chain terminators in each
of
CA 02503468 2005-04-26
21
four template-driven DNA polymerase reactions, and is readily implemented in
automated sequencing instruments, such as those of MJ Research, Inc. (San
Francisco, CA), Stratagene (La Jolla, CA), or Applied Biosystems (Foster City,
CA).
[069] Enterovirus amplicon sequences can be compared with the sequences of EV
reference strains (such as VP1 sequence of EV reference strains), including at
least
one representative of each recognized serotype, in order to identify
(serotype) the
EV. One of ordinary skill in the art will know suitable methods for comparing
sequences. For example, script-driven sequential pairwise comparison using the
program Gap (Wisconsin Sequence Analysis Package, version 10.2, Accelrys,
Inc.,
San Diego, CA) can be used to compare EV amplicon sequences to EV reference
strains.
[070] The subject matter of the present disclosure is further illustrated by
the
following non-limiting Examples.
EXAMPLES
Example 1
Primer Design and Construction
[071] This example describes the design and construction of primers for
amplification and identification of EVs, particularly EVs in clinical
specimens.
[072] Consensus degenerate primers for the cDNA and PCR1 steps were designed
from conserved amino acid motifs in the aligned capsid sequences of the 64 EV
serotype prototype strains (Table 1; FIG. 1). The four cDNA primers (AN32,
AN33,
AN34, and AN35) were designed to anneal to conserved sites downstream of the
reverse PCR primer; these sites encode the motifs, WQT (AN32, SEQ ID NO: 1),
WQS (AN33, SEQ ID NO: 2 and AN35, SEQ ID NO: 4), and YDG (AN34, SEQ ID
NO: 3) (Table 2; FIG. 1). PCR1 forward primer 224 (SEQ ID NO: 5) was designed
to target the site in VP3 encoding the highly conserved motif AMLGTH(I/L/M)
(SEQ ID NO: 19) (Table 2; FIG. 1), while PCR1 reverse primer 222 (SEQ ID NO:
CA 02503468 2005-04-26
22
6) targets a conserved motif M(F/Y)(I/V)PPG(A/G) (SEQ ID NO: 20), near the
middle of VP! (Table 2; FIG. 1; Oberste et al., J. Clin. Microbiol. 38:1170-
74,
2000). These two motifs are conserved among all EV serotypes. The degenerate
primer design approach was used for the PCR1 primers to broaden the
specificity of
amplification, allowing amplification of all EV serotypes and increasing the
absolute
concentration of virus-specific product to be used as template for PCR2. The
presence of inosine residues in positions of four-fold codon degeneracy
reduces the
overall degeneracy of the PCR1 primers but also results in decreased
thermostability
of the primer-template helix. As a result, low stringency annealing conditions
are
required for PCR1.
[073] One pair of internal primers used in PCR2 included forward internal
primer
292 (SEQ ID NO: 10), which targets the conserved motif (Q/T)A(A/V)ETG (SEQ
ID NO: 21), paired with reverse primer 222 (SEQ ID NO: 6) (Table 2; FIG. 1).
Additional internal primers used in PCR2 were designed without the use of
inosine
residues, using the CODEHOP strategy (Rose et al., Nucl. Acids. Res. 26:1628-
35,
1998), and with target sites encoding conserved motifs in VP1. These included
forward PCR2 primer AN89 (SEQ ID NO: 11), which targets the conserved motif
PALTA(A/V)E(I/T)G (SEQ ID NO: 22), paired with reverse PCR2 primer AN88
(SEQ ID NO: 12), which targets the conserved motif M(F/Y)(1N)PPGGPV (SEQ
ID NO: 23) (Table 2; FIG. 1). The consensus clamp and increased length both
contribute to the increased thermostability of primers AN88 (SEQ ID NO: 12)
and
AN89 (SEQ ID NO: 11), compared with, for example, primers 292 (SEQ ID NO:
10) and 222 (SEQ ID NO: 6).
Table 1. Reference strains of 64 recognized EV serotypes, 15 additional
variant
reference strains (italicized) and 22 proposed new serotypes (EV73 Henderson ¨
EV101).
Strain dbGAP % Source and Date Comments
nuc. identity
CA1 Tompkins 100 SM 9+ 4/6/81
CA2 Fleetwood 99 SM 10+ 5/6/81
CA3 Olsen 100 SM 8+ 4/6/81
CA4 High Point 79 SM 1+ 4/6/81
CA5 Swartz 100 SM 9+ 5/6/81
CA6 Gadula 100 SM 4+ 4/6/81
CA7 AB-IV 99 RD2 2/14/84
CA 02503468 2005-04-26
23
Strain dbGAP % Source and Date Comments
nuc. identity
CA8 Donovan 100 SM 10+ 5/6/81
CA9 Griggs 98 MK 2+ 3/16/82
CA10 Kowalik 100 SM 8+ 4/13/81
CAI 1 Belgium-1 100 SM 9+ 5/6/81
CA12 Texas-12 100 SM 7+ 5/13/81
CA13 Flores 99 SM 9+ 4/14/81
CA14 G-14 84 RD 8/28/02
CA15 G-9 100 SM 1 7/6/81
CA16 G-10 95 3/30/81
CA17 G-I2 100 SM 9+ 6/8/81
CA18 G-I3 100 SM 7+ 7/16/74
CA19 8663 100 SM 9+ 4/14/81
CA20 IH-35 99 SM 10+ 8/17/81
CA21 Kuykendall 95 RD3 8/14/02
CA22 Chulman 100 SM 8+ 7/6/81
CA24 Joseph 100 SM 10+ 3/5/90
CA24v EH24 78 RD 9/21/83 Comparison to
CA24 Joseph
CBI Conn-5 78 MK 14+-LLCMK2 1 4/30/99
CB2 Ohio-I 99 MK 12+-BGM 1 7/2/84
CB3 Nancy 98 HELF 2-MK 8/12/80
CB4 JVB 81 MK 4+-LLCMK2 1 6/19/91
CB5 Faulkner 75 LLCMK2 2 1/18/98
CB6 Schmitt 100 MK 10+ 5/2/79
El Farouk 99 MK 2+ 5/6/81
E2 Cornelis 100 MK 14+ 5/2/79
E3 Morrisey 100 MK 14+ 5/6/81
E4 Du Toil 82 MK 6+ 2/16/82 Comparison to
Pesacek
E4 Shropshire 82 MK 6+ 1/9/80 Comparison to
Pesacek
E4 Pesacek 100
E5 Noyce 99 MK 14+ 2/16/82
E6 D'Amori 100 MK 14+ 4/30/85
E6' Cox 77 MK 6+ 2/16/82
E6" Burgess 98 MK 5+ 3/1/82
E6 Charles 100
E7 Wallace 98 MK 19+ 8/19/86
E8 Bryson 76 MK 19+ 3/1/82 Comparison to El
E9 Hill 97 MK 20+-LLCMK2 1 6/14/91
Eli Gregory 100 MK 11+ 10/22/84
Eli' Silva 75 MK 7+ 5/6/81 Comparison to
Gregory
E12 Travis 100 MK 14+ 2/6/84
E13 Del Carmen 99 MK 15+ 3/1/82
E14 Tow 100 MK 15+ 3/1/82
EIS CH96-51 100 MK13+ 5/6/81
El6 Harrington 99 MK19+ 4/25/84
El7 CHHE-29 100 MK 17+ 7/30/81
El 8 Metcalf 99 MK 18+ 3/16/82
El9 Burke 100 MK 20+ 5/12/81
E20 JV-1 100 MK 1+ 5/5/76
CA 02503468 2005-04-26
24
Strain dbGAP % Source and Date Comments
nue. identity
E21 Farina 100 MK 18+ HELF 2 8/18/75
E24 De Comp 100 MK 9-BGM 3/76
E25 JV-4 100 MK 13+ 6/24/81
E26 Coronel 100 MK 13+ 6/24/81
E27 Bacon 100 MK 13+ 7/25/88
E29 JV-I0 100
E30 Bastianni 99 MK 13+ 8/27/84
E30 Frater 88 HELF 1 7/30/81 Comparison to
Gregory
E30 Giles 77 HELF 1 10/14/82 Comparison to
Gregory
E30 PR-17 76 Comparison to
Gregory
E31 Caldwell 100 MK 6+ 3/16/82
E32 PR-10 100 MK 6+ 5/12/81
E33 Toluca-3 100 MK 6+ 5/12/81
E34 DN- 19 80 HELF 1 9/7/84 Comparison to
CA24 Joseph
EV68 Fermon 98 HELF 2 7/7/78
EV69 Toluca-1 100 I-IELF 5 2/22/82
EV70 J670/71 100 HELF 4 9/7/84
EV71 BrCr 100 HELF 2 5/21/99
PV1 Mahoney 100
PVI Sabin 98 Comparison to
Mahoney
PV2 Lansing 84
PV2 Sabin 81
PV3 Leon 100
PV3 Sabin 98 Comparison to
PV3 Leon
EV73 Henderson 100
EV74 100
EV75 100
EV76 99
EV79 100
EV80 100
EV81 100
EV82 100
EV83 100
EV84 100
EV85 100
EV86 100
EV87 100
EV88 100
EV89 100
EV90 100
EV91 99
EV92 100
EV96 89
EV97 99
EV100 100
EV101 99
CA 02503468 2005-04-26
Table 2. Primers
Primer Sequence Amino acid motif Gene
Location'
AN32 GTYTGCCA (SEQ ID NO: 1) WQT VP! 3009-
3002
AN33 GAYTGCCA (SEQ ID NO: 2) WQS VP1 3009-
3002
AN34 CCRTCRTA (SEQ ID NO: 3) YDG VP! 3111-
3104
AN35 RCTYTGCCA (SEQ ID NO: 4) WQS VP! 3009-
3002
224 GCIATGYTIGGIACICAYRT AMLGTH(I/L/M) VP3 1977-
1996
(SEQ ID NO: 5) (SEQ ID NO: 19)
222 CICCIGGIGGIAYRWACAT M(F/Y)(IN)PPG(A/G) VP1 2969-2951
(SEQ ID NO: 6) (SEQ ID NO: 20)
187 ACIGCIGYIGARACIGGNCA TA(A/V)ETGH VP1 2612-
2631
(SEQ ID NO: 7) (SEQ ID NO: 21)
188 ACIGCIGTIGARACIGGNG TAVETG(AN) VP! 2612-
2630
(SEQ ID NO: 8) (SEQ ID NO: 22)
189 CARGCIGCIGARACIGGNGC QAAETGA VP1 2612-
2631
(SEQ ID NO: 9) (SEQ ID NO: 23)
292 MIGCIGYIGARACNGG (Q/T)A(AN)ETG VP! 2612-
2627
(SEQ ID NO: 10) (SEQ ID NO: 24)
AN89 CCAGCACTGACAGCAGYNGARAYNGG PALTA(A/V)E(I/T)G VP1 2602-2627
(SEQ ID NO: 11) (SEQ ID NO: 25)
AN88 TACTGGACCACCIGGNGGNAYRWACATb M(F/Y)(I/V)PPGGPV VP! 2977-2951
(SEQ ID NO: 12) (SEQ ID NO: 26)
AN79 GAAGTACCAGCACTGACAGCAGYI EVPALTA(AN)E(IMG VP! 2596-2627
GARAYNGG (SEQ ID NO: 27)
(SEQ ID NO: 13)
AN78 CTGTITGGTACTGGACCACCTGG 'VWM(F/Y)(IN)PPGGPV VP1 2969-2951
IGGIAYRWACAT (SEQ ID NO: 28)
(SEQ ID NO: 14)
AN232 CCAGCACTGACAGCAb PALTA VP1 2602-
2616
(SEQ ID NO: 15) (SEQ ID NO: 29)
AN233 TACTGGACCACCTGGb PGGPV VP1 2977-
2963
(SEQ ID NO: 16) (SEQ ID NO: 30)
AN230 AATTAACCCTCACTAAAGGGAGAAGATA RYYTHW VP3 1993-
2010
TTATACTCAYTGG` (SEQ ID NO: 17) (SEQ ID NO: 31)
AN231 GTCAGCTGGGTTTATNCCRTA YGINPAD VP1 3069-
3049
(SEQ ID NO: 18) (SEQ ID NO: 32)
a Location relative to the genome of PVI -Mahoney (GenBank accession number
J02281), except for
5 AN230 and AN23 1 , whose locations are relative to the genome of EV68-
Fermon (GenBank
accession number AY426531).
AN232 is the non-degenerate "clamp" portion of AN89 and AN233 is the non-
degenerate clamp
portion of AN88. Within the AN88 and AN89 sequences, these clamp regions are
indicated by italic
type.
10 c The T3 RNA polymerase promoter sequence is underlined.
RIB ambiguity codes: R = A or G; Y = C or T; W = A or T; N = A, C, G, or T; M
= A or C; I =
Inosine.
Example 2
15 Detection and Identification of Enteroviruses
[074] This example describes how enterovirus-specific nucleic acids can be
amplified and detected using specific primers.
[075] Nucleic acid from all 64 EV serotype reference strains, 15 additional
reference strains for some serotypes and 22 proposed new serotypes (Table 1;
101
CA 02503468 2005-04-26
26
strains total) was extracted with the QIAamp Viral RNA Mini Kit (Qiagen, Inc.,
Valencia, CA), which was used according to the manufacturer's instructions.
Eluted
RNAs were dried passively in a bench top desiccator under vacuum. The dried
RNA was resuspended in 16 I of sterile nuclease-free water and stored at ¨20
C
until use.
[076] Synthesis of cDNA was carried out in a 10 I reaction containing 5 1 of
RNA, 100 mM each dNTP (Amersham Biosciences, Piscataway, NJ), 2 I of 5X
reaction buffer (Invitrogen, Carlsbad, CA), 0.01 M dithiothreitol (DTI), 1
pmol
each cDNA primer (AN32 (SEQ ID NO: 1), AN33 (SEQ ID NO: 2), AN34 (SEQ ID
NO: 3), and AN35 (SEQ ID NO: 4); Table 2), 20 U of RNasin (Promega Corp.,
Madison, WI), and 100 U of Superscript II reverse transcriptase (Invitrogen,
Carlsbad, CA). Following incubation at 22 C for 10 min, 45 C for 45 min, and
95
C for 5 min, the entire 10 Al RT reaction was then used in the first PCR
reaction (50
Al final volume) (PCR1), consisting of 5 1 of 10X PCR buffer (Roche Applied
Science, Indianapolis, IN), 200 AM each dNTP, 50 pmol each of primers 224 (SEQ
ID NO: 5) and 222 (SEQ ID NO: 6) (Table 2), and 2.5 U of Taq DNA polymerase
(Roche Applied Science, Indianapolis, IN), with 40 cycles of amplification (95
C
for 30 s, 42 C for 30 s, 60 C for 45 s). One microliter of the first PCR
reaction was
added to a second PCR reaction (PCR2) for semi-nested amplification. PCR2
contained 40 pmol each of primers AN89 (SEQ ID NO: 11) and AN88 (SEQ ID
NO: 12) (Table 2), 200 AM each dNTP, 5 1 of 10X FastStart Taq buffer (Roche
Applied Science, Indianapolis, IN), and 2.5 U of FastStart Taq DNA polymerase
(Roche Applied Science, Indianapolis, IN) in a final volume of 50 1. The
FastStart
Taq polymerase was activated by incubation at 95 C for 6 min prior to 40
amplification cycles of 95 C for 30 s, 60 C for 20 s, and 72 C for 15 s.
Reaction
products were separated and visualized on 1.2% agarose gels, containing 0.5
pug
ethidium bromide per ml, and purified from the gel by using the QIAquick Gel
Extraction Kit (Qiagen, Inc., Valencia, CA). The resulting DNA templates were
sequenced with the Big Dye Terminator v1.1 Ready Reaction Cycle Sequencing Kit
on an ABI Prism 3100 automated sequencer (both from Applied Biosystems, Foster
City, CA), using primers AN89 (SEQ ID NO: 11) and AN88 (SEQ ID NO: 12) or
primers AN232 (SEQ ID NO: 15) and AN233 (SEQ ID NO: 16) (Table 2).
CA 02503468 2005-04-26
27
[077] Amplicon sequences were compared with the VP1 sequences of EV
reference strains, including at least one representative of each recognized
serotype,
by script-driven sequential pairwise comparison using the program Gap
(Wisconsin
Sequence Analysis Package, version 10.2, Accelrys, Inc., San Diego, CA), as
described by Oberste et al. (J. Gen. ViroL 86:445-51, 2005; J. Gen. ViroL
85:3205-
12, 2004; 1 Clin. Virol. 26:375-77, 2003). In cases where the result was not
unequivocal (highest score less than 75% or second-highest score greater than
70%),
deduced amino acid sequences were compared using a similar method.
[078] All 64 EV serotype reference strains, 15 additional reference strains
for
some serotypes and 22 proposed new serotypes (Table 1; 101 strains total) were
successfully amplified and sequenced using the CODEHOP VP1 RT-snPCR
procedure (FIG. 2). Slight variations in the sizes of the PCR products were
observed
due to VP1 gene length differences in the different serotypes, as described by
Oberste et aL (J. ViroL 73:1941-48, 1999; J. Clin. ViroL 26:375-77, 2003). All
87
clinical isolates (Table 3) tested were also successfully amplified,
sequenced, and
identified by comparing the nucleic acid sequence to an EV reference strain
VP1
sequence database (see Example 4 herein). In all cases, the serotype based on
the
VP1 RT-snPCR amplicon was identical to the serotype previously determined by
neutralization or by VP1 sequencing using different primers and conventional
PCR
(Oberste etal., I Clin. Microbiol. 38:1170-74, 2000; Oberste etal., J. Clin.
MicrobioL 37:1288-93, 1999).
Table 3. Clinical Isolates
EV Country Year Serotype VP1 %
Nucleotide % Protein ID
Code RT- ID
snPCR
10052 USA-TX 1992 CA14 CA14 82.7 NA
10053 USA-AZ 1994 CA14 CA14 83.5 NA
10055 TAI 1984 CA16 CA16 77.3 NA
10056 USA-PA 1989 CA16 CA16 78.1 NA
10057 USA-GA 1995 CA16 CA16 78.8 NA
10058 USA-TX 1995 CA16 CA16 77.7 NA
10061 MOR 1983 CA20 CA20 81.5 NA
10062 MOR 1983 CA20 CA20 81.5 NA
10063 USA-MD 1986 CA21 CA21 93.1 NA
10064 GUT 1988 CA21 CA21 76 NA
10066 USA-WA 1989 CA21 CA21 91.5 NA
10067 USA-AZ 1994 CA21 CA21 90.6 NA
10068 USA-GA 1995 CA21 CA21 92.5 NA
10069 USA-TX 1996 CA21 CA21 91.6 NA
CA 02503468 2005-04-26
28
EV Country Year Serotype VP! % Nucleotide %
Protein ID
Code RT- ID
snPCR
10070 USA-GA 1984 CA24 CA24 73.7 NA
10072 USA-GA 1993 CA9 CA9 85.8 NA
10073 USA-GA 1996 CA9 CA9 84.9 NA
10074 USA-MD 1984 CB2 CB2 83 NA
10077 USA-FL 1992 CB2 CB2 81.3 NA
10078 USA-NC 1995 CB2 CB2 83.8 NA
10080 BRA 1988 CB3 CB3 78.6 NA
10082 BRA 1988 CB3 CB3 78.8 NA
10083 BRA 1988 CB3 CB3 78.6 NA
10084 PER 1989 CB3 CB3 76.6 NA
10085 USA-NM 1993 CB3 CB3 75.8 NA
10086 USA-NH 1997 CB3 CB3 75.3 NA
10088 HON 1988 CB4 CB4 81 NA
10090 USA-MD 1986 CB5 C135 97.2 NA
10091 USA-PA 1988 CI35 C135 84 NA
10092 MEX 1988 CBS CBS 83.6 NA
10093 USA-ME 1993 C135 C135 93.4 NA
10096 USA-WA 1992 Ell Ell Ell 76;E19 Ell 89;E19
72.3; E5 70.4 77.6; E5 70.1
10097 USA-GA 1992 Eli Eli Eli 77.6; E19 Eli 89.7; E19
72; CA9 70 78.5; CA9
75.7
10098 USA-FL 1993 Ell Ell Ell 76.9; El9 Ell 89.7; El9
71.3; E7 70.1 78.5; E7 75.7
10099 USA-VA 1995 Ell Ell Ell 77.6; El9 Ell 88.8; El9
71.7; E7 70.1 77.6; E7 75.7
10100 PER 1998 Eli Ell Ell 81.3; El9 Ell 95.3; El9
72.9 80.4
10101 ELS 1988 El2 E12 El2 79.4; E3 E12 99; E3
73.3; E14 71.7 86.4; E14 70.9
10102 USA-VA 1986 E13 El3 E13 73.5; EV69 E3 87.9; EV69
71 83.2
10103 USA-TX 1995 E13 E13 El3 72.6; EV69 El3 87.9;
73.2 EV69 83.2
10105 USA-OR 1985 El8 E18 81.6 NA
10106 USA-SC 1987 El8 E18 80.7 NA
10107 USA-MD 1988 El8 E18 80.4 NA
10108 USA-OK 1989 El8 E18 80.7 NA
10109 USA-CT 1996 El8 E18 81.2 NA
10110 USA-TX 1997 E18 El8 81.6 NA
10111 USA-RI 1994 E21 E21 E21 80.6; E30 E21 97.2; E30
71.9 81.5
10112 USA-NC 1983 E24 E24 78.9 NA
10114 USA-NC 1984 E25 E25 79.7 NA
10115 HON 1986 E25 E25 79.1 NA
10116 USA-MD 1992 E25 E25 E25 80.1; EV73 E25 91.6;
70.8 EV73 73.1
10117 USA-MO 1993 E25 E25 79.9 NA
10118 USA-OR 1993 E25 E25 E25 79.8; EV73 E25 93.6;
70.4 EV73 73.6
10119 USA-MN 1994 E25 E25 E25 81; EV73 E25 94.4;
70.2 EV73 74
10120 PER 1988 E29 E29 78.4 NA
10122 USA-MT 1987 E3 E3 E3 83.8; El2 E3 98.1; El2
73.2 85
CA 02503468 2005-04-26
s
29
EV Country Year Serotype VP! %
Nucleotide % Protein ID
Code RT- ID
snPCR
10123 USA-MD 1988 E3 E3 E3 84.7; El2 E3 99.1; El2
74.5; E14 70.4
85.9; E14 74.8
10124 USA-WA 1994 E3 E3 E3 82.1; E 12 73 E3 94.7;
El2
82.1
10125 USA-OR 1991 E30 E30 E30 81.5; E21 E30 89.8;
E21
71.6
77.8
10126 USA-AR 1994 E30 E30 E30 82.4; E21 E30 90.7;
E21
71
78.7
10127 USA-GA 1993 E30 E30 E30 81.8; E21 E30 90.7;
E21
71.6
78.7
10128 USA-VA 1995 E30 E30 E30 82.4; E21 E30 90.7;
E21
71
78.7
10129 PER 1998 E33 E33 76.9 NA
10131 USA-PA 1988 E4 E4 81.6 NA
10132 USA-WA 1993 E4 E4 82.6 NA
10133 USA-CT 1996 E5 E5 84.9 NA
10134 USA-WA 1991 E6 E6 78.1 NA
10135 USA-NM 1995 E6 E6 77.6 NA
10136 PER 1998 E6 E6 81 NA
10137 USA-GA 1993 E7 E7 E7 78.2; El9 E7 95.3; El9
70.7
75.7
10138 USA-GA 1993 E7 E7 E7 78.2; El9 E7 95.3; El9
70.7
75.7
10139 PER 1998 E7 E7 78.2 NA
10140 USA-NC 1992 E9 E9 79 NA
10141 USA-AR 1995 E9 E9 E9 80.4; E5 E9 94.3; E5
71.4; E14 71.4
71.4; E14 73.3
10142 USA-W1 1995 E9 E9 E9 78; E14 72; E9 96.2;
E14
E571.1
74.5; E5 73.6
10144 USA-TX 1989 EV71 EV71 84.7 NA
10146 USA-MD 1987 EV71 EV71 81
NA
10147 USA-OK 1989 EV71 EV71 84.9 NA
10148 USA-NM 1990 EV71 EV71 83.3 NA
10149 USA-NM 1994 EV71 EV71 84.4
NA
10150 USA-CT 1994 EV71 EV71 85 NA
10151 USA-MD 1995 EV71 EV71 83 NA
10152 USA-CA 1990 HRV2 HRV2 92.2
NA
10153 USA-OK 1985 UNT EV EV75 99.7 NA
10154 USA-VA 1986 UNT EV EV75 EV75 86.6;E33 EV75
100;
70.1
E33 72.9
10155 USA-CT 1987 UNT EV EV75 84 NA
10156 USA-CT 1987 UNT EV EV75 83.2 NA
10157 USA-OK 1988 UNT HRV HRV31 89.2 NA
NA: not applicable; UNT: untypable.
CA 02503468 2005-04-26
Example 3
Assay Sensitivity
[079] This example describes the sensitivity of EV detection/identification
methods using specific primers to amplify enterovirus-specific nucleic acids.
5 [080] Sensitivity was tested by two methods. Sensitivity relative to cell
culture
infectivity was measured using a titered stock of the EV68 prototype strain,
Fermon.
Serial 10-fold dilutions of the EV68-Fermon stock were made in Hank's balanced
salt solution, and RNA from 100 I of each dilution was extracted with the
QIAamp
Viral RNA Mini Kit (Qiagen, Inc., Valencia, CA). RNAs representing from 104
cell
10 culture infectious dose 50% endpoint units (CCID50) to le CCID50 per 5
I were
tested with the VP! RT-snPCR assay. The VP! RT-snPCR assay detected RNA
extracted from as little as 0.01 CCID50 per 5 I of EV68-Fermon (FIG. 3A),
indicating that the assay is at least 100-fold more sensitive than cell
culture (since 1
CCID50 defines the cell culture endpoint).
15 [081] Absolute sensitivity was measured by using an in vitro-transcribed
synthetic
RNA standard derived from EV68-Fermon. To make the synthetic RNA standard,
RT-PCR primers were designed to flank the VP3-VP1 RT-snPCR assay cDNA
product. The sense primer AN230 (SEQ ID NO: 17) contains the 23-base T3 RNA
polymerase promoter at the 5' end, and it was used with the antisense primer
AN231
20 (SEQ ID NO: 18) (Table 2) in a two-step RT-PCR. cDNA was made with
SuperScript II RT (Invitrogen, Carlsbad, CA) according to the kit
instructions, using
10 pmol AN231 (SEQ ID NO: 18) to prime the cDNA. PCR was performed with
FastStart Taq (Roche Applied Science, Indianapolis, IN), using the
manufacturer's
10X buffer with MgC12, 2 I of cDNA, 200 M each dNTP, and 20 pmol each of
25 AN230 (SEQ ID NO: 17) and AN231 (SEQ ID NO: 18) primers, in a final
reaction
volume of 50 I. The thermocycler program consisted of 40 cycles of 95 C for
30
s, 55 C for 40 s, and 72 C for 40 s. The PCR product was purified using the
High
Pure PCR Product Purification Kit (Roche Applied Science, Indianapolis, IN)
according to the manufacturer's instructions. Purified PCR product was
quantitated
30 spectrophotometrically, and 1 g of PCR product was used as template for
in vitro
RNA transcription, using the MEGAscript High Yield Transcription Kit (Ambion,
CA 02503468 2005-04-26
31
Inc., Austin, TX) according to the manufacturer's protocol. The VP3-VP1 single-
stranded, positive-sense standard RNA product (VP3-VP1 sRNA; 1082 nt) was
digested with DNase Ito remove template DNA and then purified with the QIAamp
Viral RNA Mini Kit (Qiagen, Inc., Valencia, CA). The manufacturer's
instructions
were followed, except no carrier tRNA was added to the kit's lysis buffer. The
purified VP3-VP1 sRNA was quantitated spectrophotometrically, and the
concentration was calculated in units of RNA molecules per microliter. Two
separate lots of the VP3-VP1 sRNA were synthesized and diluted to contain from
104 copies to 1 copy per 5 pi and then tested in two separate experiments with
the
VP1 RT-snPCR assay. As few as 10 copies of the in vitro-transcribed VP3-VP1
sRNA produced a detectable gel band in two independent experiments, indicating
a
low limit of absolute sensitivity (FIG. 3B).
[082] The sensitivity of the VP1 RT-snPCR assay was also compared to that of a
5'NTR RT-snPCR assay (Nix etal., Neurol. 62:1372-77, 2004) by serially
diluting
RNA extracted from a recent EV68 clinical isolate and running both the VP1 and
5'NTR RT-snPCR assays in parallel using the same diluted RNA preparations. The
diluted EV68 clinical isolate RNA was amplified from the 10-1 - 10-7 dilutions
with
the 5'NTR RT-snPCR assay, and from the 104 - 104 dilutions with the VP1 RT-
snPCR assay (FIG. 3C).
Example 4
Application to Clinical Specimens
[083] This example demonstrates the application of methods of using specific
primers to amplify and identify enteroviruses from clinical specimens.
[084] To demonstrate the clinical application of the VP1 RT-snPCR method, RNA
was extracted from original clinical specimens obtained from patients with a
number
of different enteroviral illnesses. The specimens and associated illnesses
included
cerebrospinal fluid (aseptic meningitis), stool (aseptic meningitis), rectal
swab
= (febrile rash), nasopharyngeal swab (mild upper respiratory illness),
conjunctival
swab (acute hemorrhagic conjunctivitis), serum (febrile rash), and postmortem
liver
tissue (neonatal sepsis-like illness).
CA 02503468 2005-04-26
32
[085] Stool suspensions were prepared by adding 5 ml of phosphate-buffered
saline, 1 g of glass beads (Corning Inc., Corning, NY), and 0.5 ml of
chloroform to
1 g of stool, shaking the mixture vigorously for 20 min in a mechanical
shaker, and
centrifugation at 1500 g for 20 min at 4 C (World Health Organization, Manual
for
the virological investigation of polio, World Health Organization, Geneva,
2001).
For rectal swab samples, the fluid was centrifuged at 13,000 g for 1 min at
room
temperature to remove solids, and the supernatant was transferred to a fresh
tube.
For fecal specimens (stool suspensions or clarified rectal swab supernatants),
140 I
of the specimen extract was combined with an equal volume of Vertrel XF
(Miller-
Stephenson Chemical Co., Danbury, CT), shaken vigorously, and then centrifuged
at
13,000 g for 1 min at room temperature. The aqueous phase was transferred to a
fresh tube. Other specimen types, including cerebrospinal fluid, virus
isolates, and
supernatants from nasopharyngeal, oropharyngeal, and conjunctival swab
samples,
were processed without pretreatment. Twenty micrograms of proteinase K (Roche
Applied Science, Indianapolis, IN) was added to 140 I of each liquid specimen
or
fecal extract, and then incubated for 30 min at 37 C. Nucleic acid was
extracted
from the digested specimen with the QIAamp Viral RNA Mini Kit (Qiagen, Inc.,
Valencia, CA), which was used according to the manufacturer's instructions.
Eluted
RNAs were dried passively in a bench top desiccator under vacuum. The dried
RNA was resuspended in 16 I of sterile nuclease-free water and stored at ¨20
C
until use. Synthesis of cDNA and RT-PCR amplification were carried out as
described herein in Example 2.
[086] From each of these RNA templates, a specific product was amplified by
VP1 RT-snPCR (FIG. 4). In each assay, the tested RNA represents the equivalent
of
approximately 45 I of original specimen fluid or 10 g of stool. Following
gel
purification, the EV present in each specimen was identified by amplicon
sequencing and comparison to a database of EV VP1 sequences. All of the
amplification products yielded clean, readable sequences, including those with
weak
or multiple bands (for example, rectal swab and liver). The identified EVs
were E30
(cerebrospinal fluid), CVA1 (stool), E9 (rectal swab), CVA9 (nasopharyngeal
swab), CVA24 (conjunctival swab), CVA10 (serum), and Ell (liver).
CA 02503468 2005-04-26
33
[087] While this disclosure has been described with an emphasis upon preferred
embodiments, it will be obvious to those of ordinary skill in the art that
variations of
the preferred embodiments may be used and it is intended that the disclosure
may be
practiced otherwise than as specifically described herein. Accordingly, this
disclosure includes all modifications encompassed within the spirit and scope
of the
disclosure as defined by the claims below.
CA 02503468 2005-05-11
34
SEQUENCE LISTING
(1) GENERAL INFORMATION:
(i) APPLICANT: THE GOVERNMENT OF THE UNITED STATES OF AMERICA AS
REPRESENTED BY THE SECRETARY OF THE DEPARTMENT OF
HEALTH AND HUMAN SERVICES, CENTERS FOR DISEASE
CONTROL AND PREVENTION
(ii) TITLE OF INVENTION: DETECTION AND IDENTIFICATION OF ENTEROVIRUSES
(iii) NUMBER OF SEQUENCES: 32
(iv) CORRESPONDENCE ADDRESS:
(A) ADDRESSEE: SMART & BIGGAR
(B) STREET: P.O. BOX 2999, STATION D
(C) CITY: OTTAWA
(D) STATE: ONT
(E) COUNTRY: CANADA
(F) ZIP: KlP 5Y6
(v) COMPUTER READABLE FORM:
(A) MEDIUM TYPE: Floppy disk
(B) COMPUTER: IBM PC compatible
(C) OPERATING SYSTEM: PC-DOS/MS-DOS
(D) SOFTWARE: ASCII (text)
(vi) CURRENT APPLICATION DATA:
(A) APPLICATION NUMBER: CA
(B) FILING DATE: 26-APR-2005
(C) CLASSIFICATION:
(vii) PRIOR APPLICATION DATA:
(A) APPLICATION NUMBER:
(B) FILING DATE:
(viii) ATTORNEY/AGENT INFORMATION:
(A) NAME: SMART & BIGGAR
(B) REGISTRATION NUMBER:
(C) REFERENCE/DOCKET NUMBER: 63198-1483
(ix) TELECOMMUNICATION INFORMATION:
(A) TELEPHONE: (613)-232-2486
(B) TELEFAX: (613)-232-8440
(2) INFORMATION FOR SEQ ID NO.: 1:
(i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 8
(B) TYPE: nucleic acid
(C) STRANDEDNESS:
(D) TOPOLOGY:
(ii) MOLECULE TYPE: DNA
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Artificial Sequence
(ix) FEATURE
(C) OTHER INFORMATION: Oligonucleotide cDNA primer
(xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 1:
GTYTGCCA 8
(2) INFORMATION FOR SEQ ID NO.: 2:
(i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 8
(B) TYPE: nucleic acid
(C) STRANDEDNESS:
(D) TOPOLOGY:
(ii) MOLECULE TYPE: DNA
CA 02503468 2005-05-11
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Artificial Sequence
(ix) FEATURE
(C) OTHER INFORMATION: Oligonucleotide cDNA primer
(xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 2:
GAYTGCCA 8
(2) INFORMATION FOR SEQ ID NO.: 3:
10 (i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 8
(B) TYPE: nucleic acid
(C) STRANDEDNESS:
(D) TOPOLOGY:
(ii) MOLECULE TYPE: DNA
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Artificial Sequence
(ix) FEATURE
(C) OTHER INFORMATION: Oligonucleotide cDNA primer
20 (xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 3:
CCRTCRTA 8
(2) INFORMATION FOR SEQ ID NO.: 4:
(i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 9
(B) TYPE: nucleic acid
(C) STRANDEDNESS:
(D) TOPOLOGY:
30 (ii) MOLECULE TYPE: DNA
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Artificial Sequence
(ix) FEATURE
(C) OTHER INFORMATION: Oligonucleotide cDNA primer
(xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 4:
RCTYTGCCA 9
(2) INFORMATION FOR SEQ ID NO.: 5:
(i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 20
(B) TYPE: nucleic acid
(C) STRANDEDNESS:
(D) TOPOLOGY:
(ii) MOLECULE TYPE: DNA
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Artificial Sequence
(ix) FEATURE
(C) OTHER INFORMATION: Oligonucleotide amplification primer
(ix) FEATURE
(A) NAME/KEY: misc_feature
(B) LOCATION: (3)..(3)
(C) OTHER INFORMATION: n = inosine
(ix) FEATURE
(A) NAME/KEY: misc_feature
(3) LOCATION: (9)..(9)
(C) OTHER INFORMATION: n = inosine
(ix) FEATURE
(A) NAME/KEY: misc_feature
(B) LOCATION: (12)..(12)
CA 02503468 2005-05-11
36
(C) OTHER INFORMATION: n = inosine
(ix) FEATURE
(A) NAME/KEY: misc_feature
(B) LOCATION: (15)..(15)
(C) OTHER INFORMATION: n = inosine
(xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 5:
GCNATGYTNG GNACNCAYRT 20
(2) INFORMATION FOR SEQ ID NO.: 6:
(i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 19
(B) TYPE: nucleic acid
(C) STRANDEDNESS:
(D) TOPOLOGY:
(ii) MOLECULE TYPE: DNA
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Artificial Sequence
(ix) FEATURE
(C) OTHER INFORMATION: Oligonucleotide amplification primer
(ix) FEATURE
(A) NAME/KEY: misc_feature
(B) LOCATION: (2)..(2)
(C) OTHER INFORMATION: n = inosine
(ix) FEATURE
(A) NAME/KEY: misc_feature
(B) LOCATION: (5)..(5)
(C) OTHER INFORMATION: n = inosine
(ix) FEATURE
(A) NAME/KEY: misc_feature
(B) LOCATION: (8)..(8)
(C) OTHER INFORMATION: n = inosine
(ix) FEATURE
(A) NAME/KEY: misc_feature
(B) LOCATION: (11)..(11)
(C) OTHER INFORMATION: n = inosine
(xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 6:
CNCCNGGNGG NAYRWACAT 19
(2) INFORMATION FOR SEQ ID NO.: 7:
(i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 20
(B) TYPE: nucleic acid
(C) STRANDEDNESS:
(D) TOPOLOGY:
(ii) MOLECULE TYPE: DNA
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Artificial Sequence
(ix) FEATURE
(C) OTHER INFORMATION: Oligonucleotide amplification primer
(ix) FEATURE
(A) NAME/KEY: misc_feature
(B) LOCATION: (3)..(3)
(C) OTHER INFORMATION: n = inosine
(ix) FEATURE
(A) NAME/KEY: misc_feature
(B) LOCATION: (6)..(6)
(C) OTHER INFORMATION: n = inosine
(ix) FEATURE
CA 02503468 2005-05-11
37
(A) NAME/KEY: misc_feature
(B) LOCATION: (9)..(9)
(C) OTHER INFORMATION: n = inosine
(ix) FEATURE
(A) NAME/KEY: misc_feature
(B) LOCATION: (15)¨(15)
(C) OTHER INFORMATION: n = inosine
(ix) FEATURE
(A) NAME/KEY: misc_feature
(B) LOCATION: (18)..(18)
(C) OTHER INFORMATION: n = g, a, t, or c
(xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 7:
ACNGCNGYNG ARACNGGNCA 20
(2) INFORMATION FOR SEQ ID NO.: 8:
(i) SEQUENCE CHARACTERISTICS
00 LENGTH: 19
(E) TYPE: nucleic acid
(C) STRANDEDNESS:
(D) TOPOLOGY:
(ii) MOLECULE TYPE: DNA
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Artificial Sequence
(ix) FEATURE
(C) OTHER INFORMATION: Oligonucleotide amplification primer
(ix) FEATURE
(A) NAME/KEY: misc_feature
(B) LOCATION: (3)..(3)
(C) OTHER INFORMATION: n = inosine
(ix) FEATURE
(A) NAME/KEY: misc_feature
(B) LOCATION: (6)..(6)
(C) OTHER INFORMATION: n = inosine
(ix) FEATURE
(A) NAME/KEY: misc_feature
(B) LOCATION: (9)..(9)
(C) OTHER INFORMATION: n = inosine
(ix) FEATURE
(A) NAME/KEY: misc_feature
(B) LOCATION: (15)..(15)
(C) OTHER INFORMATION: n = inosine
(ix) FEATURE
(A) NAME/KEY: misc_feature
(B) LOCATION: (18)..(18)
(C) OTHER INFORMATION: n = g, a, t, or c
(xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 8:
ACNGCNGTNG ARACNGGNG 19
(2) INFORMATION FOR SEQ ID NO.: 9:
(i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 20
(B) TYPE: nucleic acid
(C) STRANDEDNESS:
(D) TOPOLOGY:
(ii) MOLECULE TYPE: DNA
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Artificial Sequence
(ix) FEATURE
CA 02503468 2005-05-11
38
(C) OTHER INFORMATION: Oligonucleotide amplification primer
(ix) FEATURE
(A) NAME/KEY: misc_feature
(B) LOCATION: (6)..(6)
(C) OTHER INFORMATION: n = inosine
(ix) FEATURE
(A) NAME/KEY: misc_feature
(B) LOCATION: (9)..(9)
(C) OTHER INFORMATION: n = inosine
(ix) FEATURE
(A) NAME/KEY: misc_feature
(B) LOCATION: (15)..(15)
(C) OTHER INFORMATION: n = inosine
(ix) FEATURE
(A) NAME/KEY: misc_feature
(B) LOCATION: (18)..(18)
(C) OTHER INFORMATION: n = g, a, t, or c
(xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 9:
CARGCNGCNG ARACNGGNGC 20
(2) INFORMATION FOR SEQ ID NO.: 10:
(i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 16
(B) TYPE: nucleic acid
(C) STRANDEDNESS:
(D) TOPOLOGY:
(ii) MOLECULE TYPE: DNA
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Artificial Sequence
(ix) FEATURE
(C) OTHER INFORMATION: Oligonucleotide amplification primer
(ix) FEATURE
(A) NAME/KEY: misc_feature
(B) LOCATION: (2)¨(2)
(C) OTHER INFORMATION: n = inosine
(ix) FEATURE
(A) NAME/KEY: misc_feature
(B) LOCATION: (5)..(5)
(C) OTHER INFORMATION: n = inosine
(ix) FEATURE
(A) NAME/KEY: misc_feature
(13) LOCATION: (8)..(8)
(C) OTHER INFORMATION: n = inosine
(ix) FEATURE
(A) NAME/KEY: misc_feature
(B) LOCATION: (14)¨(14)
(C) OTHER INFORMATION: n = g, a, t, or c
(xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 10:
MNGCNGYNGA RACNGG 16
(2) INFORMATION FOR SEQ ID NO.: 11:
(i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 26
(B) TYPE: nucleic acid
(C) STRANDEDNESS:
(D) TOPOLOGY:
(ii) MOLECULE TYPE: DNA
(vi) ORIGINAL SOURCE:
CA 02503468 2005-05-11
39
(A) ORGANISM: Artificial Sequence
(ix) FEATURE
(C) OTHER INFORMATION: Oligonucleotide amplification primer
(ix) FEATURE
(A) NAME/KEY: misc_feature
(B) LOCATION: (18)..(18)
(C) OTHER INFORMATION: n = g, a, t, or c
(ix) FEATURE
(A) NAME/KEY: misc_feature
(B) LOCATION: (24)..(24)
(C) OTHER INFORMATION: n = g, a, t, or c
(xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 11:
CCAGCACTGA CAGCAGYNGA RAYNGG 26
(2) INFORMATION FOR SEQ ID NO.: 12:
(i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 27
(B) TYPE: nucleic acid
(C) STRANDEDNESS:
(D) TOPOLOGY:
(ii) MOLECULE TYPE: DNA
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Artificial Sequence
(ix) FEATURE
(C) OTHER INFORMATION: Oligonucleotide amplification primer
(ix) FEATURE
(A) NAME/KEY: misc_feature
(B) LOCATION: (16)..(16)
(C) OTHER INFORMATION: n = g, a, t, or c
(ix) FEATURE
(A) NAME/KEY: misc_feature
(B) LOCATION: (19)..(19)
(C) OTHER INFORMATION: n = g, a, t, or c
(xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 12:
TACTGGACCA CCTGGNGGNA YRWACAT 27
(2) INFORMATION FOR SEQ ID NO.: 13:
(i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 32
(B) TYPE: nucleic acid
(C) STRANDEDNESS:
(D) TOPOLOGY:
(ii) MOLECULE TYPE: DNA
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Artificial Sequence
(ix) FEATURE
(C) OTHER INFORMATION: Oligonucleotide amplitication primer
(ix) FEATURE
(A) NAME/KEY: misc_feature
(B) LOCATION: (24)..(24)
(C) OTHER INFORMATION: n = inosine
(ix) FEATURE
(A) NAME/KEY: misc_feature
(B) LOCATION: (30)..(30)
(C) OTHER INFORMATION: n = g, a, t, or c
(xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 13:
GAAGTACCAG CACTGACAGC AGYNGARAYN GG 32
CA 02503468 2005-05-11
(2) INFORMATION FOR SEQ ID NO.: 14:
(i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 35
(B) TYPE: nucleic acid
(C) STRANDEDNESS:
(D) TOPOLOGY:
(ii) MOLECULE TYPE: DNA
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Artificial Sequence
10 (ix) FEATURE
(C) OTHER INFORMATION: Oligonucleotide amplification primer
(ix) FEATURE
(A) NAME/KEY: misc_feature
(B) LOCATION: (24)..(24)
(C) OTHER INFORMATION: n = inosine
(ix) FEATURE
(A) NAME/KEY: misc_feature
(B) LOCATION: (27)..(27)
(C) OTHER INFORMATION: n = inosine
20 (xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 14:
CTGTTTGGTA CTGGACCACC TGGNGGNAYR WACAT 35
(2) INFORMATION FOR SEQ ID NO.: 15:
(i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 15
(B) TYPE: nucleic acid
(C) STRANDEDNESS:
(D) TOPOLOGY:
30 (ii) MOLECULE TYPE: DNA
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Artificial Sequence
(ix) FEATURE
(C) OTHER INFORMATION: Oligonucleotide amplification primer
(xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 15:
CCAGCACTGA CAGCA 15
(2) INFORMATION FOR SEQ ID NO.: 16:
40 (i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 15
(B) TYPE: nucleic acid
(C) STRANDEDNESS:
(D) TOPOLOGY:
(ii) MOLECULE TYPE: DNA
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Artificial Sequence
(ix) FEATURE
(C) OTHER INFORMATION: Oligonucleotide amplification primer
(xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 16:
TACTGGACCA CCTGG 15
(2) INFORMATION FOR SEQ ID NO.: 17:
(i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 28
(B) TYPE: nucleic acid
(C) STRANDEDNESS:
(D) TOPOLOGY:
(ii) MOLECULE TYPE: DNA
CA 02503468 2005-05-11
41
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Artificial Sequence
(ix) FEATURE
(C) OTHER INFORMATION: Oligonucleotide amplification primer
(xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 17:
AATTAACCCT CACTAAAGGG AGAAGATA 28
(2) INFORMATION FOR SEQ ID NO.: 18:
(i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 21
(B) TYPE: nucleic acid
(C) STRANDEDNESS:
(D) TOPOLOGY:
(ii) MOLECULE TYPE: DNA
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Artificial Sequence
(ix) FEATURE
(C) OTHER INFORMATION: Oligonucleotide amplification primer
(ix) FEATURE
(A) NAME/KEY: misc_feature
(B) LOCATION: (16)..(16)
(C) OTHER INFORMATION: n = g, a, t, or c
(xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 18:
GTCAGCTGGG TTTATNCCRT A 21
(2) INFORMATION FOR SEQ ID NO.: 19:
(i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 7
(B) TYPE: amino acid
(C) STRANDEDNESS:
(D) TOPOLOGY:
(ii) MOLECULE TYPE: polypeptide
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Artificial Sequence
(ix) FEATURE
(C) OTHER INFORMATION: Conserved enterovirus amino acid motif used in
primer design
(ix) FEATURE
(A) NAME/KEY: MISC_FEATURE
(B) LOCATION: (7)¨(7)
(C) OTHER INFORMATION: Xaa = Ile, Leu or Met
(xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 19:
Ala Met Leu Gly Thr His Xaa
5
(2) INFORMATION FOR SEQ ID NO.: 20:
(i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 7
(B) TYPE: amino acid
(C) STRANDEDNESS:
(D) TOPOLOGY:
(ii) MOLECULE TYPE: polypeptide
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Artificial Sequence
(ix) FEATURE
(C) OTHER INFORMATION: Conserved enterovirus amino acid motif used in
primer design
CA 02503468 2005-05-11
42
(ix) FEATURE
(A) NAME/KEY: misc_feature
(B) LOCATION: (2)..(2)
(C) OTHER INFORMATION: Xaa = Phe or Tyr
(ix) FEATURE
(A) NAME/KEY: misc_feature
(B) LOCATION: (3)..(3)
(C) OTHER INFORMATION: Xaa = Ile or Val
(ix) FEATURE
(A) NAME/KEY: MISC_FEATURE
(B) LOCATION: (7)..(7)
(C) OTHER INFORMATION: Xaa = Ala or Gly
(xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 20:
Met Xaa Xaa Pro Pro Gly Xaa
1 5
(2) INFORMATION FOR SEQ ID NO.: 21:
(i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 7
(B) TYPE: amino acid
(C) STRANDEDNESS:
(D) TOPOLOGY:
(ii) MOLECULE TYPE: polypeptide
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Artificial Sequence
(ix) FEATURE
(C) OTHER INFORMATION: Conserved enterovirus amino acid motif used in
primer design
(ix) FEATURE
(A) NAME/KEY: MISC_FEATURE
(B) LOCATION: (3)..(3)
(C) OTHER INFORMATION: Xaa = Ala or Val
(xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 21:
Thr Ala Xaa Glu Thr Gly His
1 5
(2) INFORMATION FOR SEQ ID NO.: 22:
(i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 7
(B) TYPE: amino acid
(C) STRANDEDNESS:
(D) TOPOLOGY:
(ii) MOLECULE TYPE: polypeptide
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Artificial Sequence
(ix) FEATURE
(C) OTHER INFORMATION: Conserved enterovirus amino acid motif used in
primer design
(ix) FEATURE
(A) NAME/KEY: MISC_FEATURE
(B) LOCATION: (7)..(7)
(C) OTHER INFORMATION: Xaa = Ala or Val
(xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 22:
Thr Ala Val Glu Thr Gly Xaa
1 5
(2) INFORMATION FOR SEQ ID NO.: 23:
CA 02503468 2005-05-11
43
(i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 7
(B) TYPE: amino acid
(C) STRANDEDNESS:
(D) TOPOLOGY:
(ii) MOLECULE TYPE: polypeptide
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Artificial Sequence
(ix) FEATURE
(C) OTHER INFORMATION: Conserved enterovirus amino acid motif used in
primer design
(xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 23:
Gin Ala Ala Glu Thr Gly Ala
1 5
(2) INFORMATION FOR SEQ ID NO.: 24:
(i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 6
(B) TYPE: amino acid
(C) STRANDEDNESS:
(D) TOPOLOGY:
(ii) MOLECULE TYPE: polypeptide
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Artificial Sequence
(ix) FEATURE
(C) OTHER INFORMATION: Conserved enterovirus amino acid motif used in
primer design
(ix) FEATURE
(A) NAME/KEY: misc_feature
(B) LOCATION: (1)..(1)
(C) OTHER INFORMATION: Xaa = Gin or Thr
(ix) FEATURE
(A) NAME/KEY: MISC_FEATURE
(B) LOCATION: (3)¨(3)
(C) OTHER INFORMATION: Xaa = Ala or Val
(xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 24:
Xaa Ala Xaa Glu Thr Gly
1 5
(2) INFORMATION FOR SEQ ID NO.: 25:
(i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 9
(B) TYPE: amino acid
(C) STRANDEDNESS:
(D) TOPOLOGY:
(ii) MOLECULE TYPE: polypeptide
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Artificial Sequence
(ix) FEATURE
(C) OTHER INFORMATION: Conserved enterovirus amino acid motif used in
primer design
(ix) FEATURE
(A) NAME/KEY: misc_feature
(B) LOCATION: (6)..(6)
(C) OTHER INFORMATION: Xaa = Ala or Val
(ix) FEATURE
(A) NAME/KEY: MISC_FEATURE
(B) LOCATION: (8)..(8)
CA 02503468 2005-05-11
44
(C) OTHER INFORMATION: Xaa = Ile or Thr
(xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 25:
Pro Ala Leu Thr Ala Xaa Glu Xaa Gly
1 5
(2) INFORMATION FOR SEQ ID NO.: 26:
(i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 9
(B) TYPE: amino acid
(C) STRANDEDNESS:
(D) TOPOLOGY:
(ii) MOLECULE TYPE: polypeptide
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Artificial Sequence
(ix) FEATURE
(C) OTHER INFORMATION: Conserved enterovirus amino acid motif used in
primer design
(ix) FEATURE
(A) NAME/KEY: misc_feature
(B) LOCATION: (2)..(2)
(C) OTHER INFORMATION: Xaa = Phe or Tyr
(ix) FEATURE
(A) NAME/KEY: MISC_FEATURE
(B) LOCATION: (3)..(3)
(C) OTHER INFORMATION: Xaa = Ile or Val
(xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 26:
Met Xaa Xaa Pro Pro Gly Gly Pro Val
1 5
(2) INFORMATION FOR SEQ ID NO.: 27:
(i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 11
(B) TYPE: amino acid
(C) STRANDEDNESS:
(D) TOPOLOGY:
(ii) MOLECULE TYPE: polypeptide
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Artificial Sequence
(ix) FEATURE
(C) OTHER INFORMATION: Conserved enterovirus amino acid motif used in
primer design
(ix) FEATURE
(A) NAME/KEY: misc_feature
(B) LOCATION: (8)..(8)
(C) OTHER INFORMATION: Xaa = Ala or Val
(ix) FEATURE
(A) NAME/KEY: MISC_FEATURE
(B) LOCATION: (10)..(10)
(C) OTHER INFORMATION: Xaa = Ile or Thr
(xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 27:
Glu Val Pro Ala Leu Thr Ala Xaa Glu Xaa Gly
1 5 10
(2) INFORMATION FOR SEQ ID NO.: 28:
(i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 11
(B) TYPE: amino acid
CA 02503468 2005-05-11
(C) STRANDEDNESS:
(D) TOPOLOGY:
(ii) MOLECULE TYPE: polypeptide
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Artificial Sequence
(ix) FEATURE
(C) OTHER INFORMATION: Conserved enterovirus amino acid motif used in
primer design
(ix) FEATURE
10 (A) NAME/KEY: misc_feature
(B) LOCATION: (4)..(4)
(C) OTHER INFORMATION: Xaa = Phe or Tyr
(ix) FEATURE
(IQ NAME/KEY: MISC_FEATURE
(B) LOCATION: (5)..(5)
(C) OTHER INFORMATION: Xaa = Ile or Val
(xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 28:
Val Trp Met Xaa Xaa Pro Pro Gly Gly Pro Val
1 5 10
(2) INFORMATION FOR SEQ ID NO.: 29:
(i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 5
(B) TYPE: amino acid
(C) STRANDEDNESS:
(D) TOPOLOGY:
(ii) MOLECULE TYPE: polypeptide
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Artificial Sequence
(ix) FEATURE
(C) OTHER INFORMATION: Conserved enterovirus amino acid motif used in
primer design
(xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 29:
Pro Ala Leu Thr Ala
1 5
(2) INFORMATION FOR SEQ ID NO.: 30:
(i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 5
(B) TYPE: amino acid
(C) STRANDEDNESS:
(D) TOPOLOGY:
(ii) MOLECULE TYPE: polypeptide
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Artificial Sequence
(ix) FEATURE
(C) OTHER INFORMATION: Conserved enterovirus amino acid motif used in
primer design
(xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 30:
Pro Gly Gly Pro Val
1 5
(2) INFORMATION FOR SEQ ID NO.: 31:
(i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 6
(B) TYPE: amino acid
(C) STRANDEDNESS:
CA 02503468 2005-05-11
46
(D) TOPOLOGY:
(ii) MOLECULE TYPE: polypeptide
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Artificial Sequence
(ix) FEATURE
(C) OTHER INFORMATION: Conserved enterovirus amino acid motif used in
primer design
(xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 31:
Arg Tyr Tyr Thr His Trp
1 5
(2) INFORMATION FOR SEQ ID NO.: 32:
(i) SEQUENCE CHARACTERISTICS
(A) LENGTH: 7
(B) TYPE: amino acid
(C) STRANDEDNESS:
(D) TOPOLOGY:
(ii) MOLECULE TYPE: polypeptide
(vi) ORIGINAL SOURCE:
(A) ORGANISM: Artificial Sequence
(ix) FEATURE
(C) OTHER INFORMATION: Conserved enterovirus amino acid motif used in
primer design
(xi) SEQUENCE DESCRIPTION: SEQ ID NO.: 32:
Tyr Gly Ile Asn Pro Ala Asp
1 5