Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02705213 2010-05-07
WO 2009/059430
PCT/CA2008/001985
MICROFLUIDIC DEVICE AND METHOD OF USING SAME
[0001] This application claims priority to U.S. Provisional
Application 60/996,236 filed
November 7, 2007, which is herein incorporated by reference in its entirety.
FIELD OF THE INVENTION
[0002] The present invention relates to a microfluidic device.
BACKGROUND OF THE INVENTION
[0003] Current microfluidic devices ("chips") and systems allow for
semi-automated and
automated manipulation of small volumes, generally a few nanolitres. The
miniaturization of
sample processing and assaying provides for both increased speed and
sensitivity, as well as an
economy of scale ¨ less reagent, sample and space is required to run the assay
and obtain the
desired information, and the decreased handling of individual samples and
reagents reduces
opportunity for error. Microfluidic devices and systems have been adapted, or
proposed for use
with a variety of chemical and biochemical analyses including protein
crystallography, cell free
protein synthesis, gas chromatography, cell separation, electrophoresis,
polymerase chain
reaction (PCR) and the like.
[0004] The advent of PCR and other nucleic acid-based methodologies,
and completion
of the sequencing of the human genome has led to various nucleic acid based
diagnostics ant
tests which continue to demand more precise and sensitive analytical tools.
Knowledge of gene
expression, polymorphisms, mutations (heritable or otherwise) and the like
have been translated
into improvements in health care - this in turn has demanded more precise and
sensitive
analytical tools. Earlier detection and diagnosis of genetic disease, cancer
and infection provides
immediate beneficial impact ¨ early stages of disease may be treated more
efficiently, and often
with a greater degree of success, thus greatly improving subject outcome and
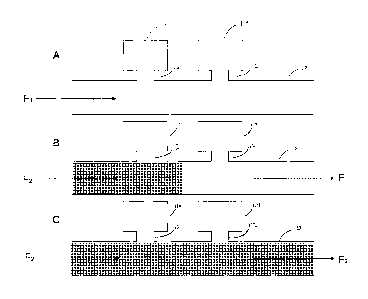
quality of life.
[0005] Quantitative real-time PCR (RT-PCR) is a current 'gold
standard' for detection of
relatively rare polymorphisms, however it requires a difference of ¨20% or
greater to be present
in the sample before the difference is reliably detected. As PCR is an
exponential technique, it is
inherently very sensitive and in principal allows for the detection of single
molecules. However,
any non-specific amplification or contamination may lead to false positives,
thereby making it
very difficult to reliably detect rare sequences. This is particularly true
when target sequences are
very similar to other species that may be present at higher levels, and thus
may limit the
sensitivity of an assay in detecting rare molecular species - the presence of
a single nucleotide
CA 02705213 2010-05-07
WO 2009/059430
PCT/CA2008/001985
polymorphism in rare population of cancer cells may be invisible due to high
background of
normal molecules with nearly identical sequence. Additionally, the real-time
monitoring of an
exponential reaction has high dynamic range but limited discriminatory power -
differences
below approximately 20% in the relative abundance of two sequences may be
difficult to detect.
In many applications a far greater sensitivity may be required. As an example,
the detection of
fetal aneuploidy from circulating blood would require accurate detection of
allelic differences of
approximately 1 ¨ 6 % Lo et al., 1998. Am J Human Genetics).
[0006] Digital PCR was initially described by Vogelstein and Kinzler
in 1999 (Proc. Natl
Acad. Sci USA 96:9236-9241). Digital PCR techniques provide for amplification
of single
molecules, however macroscopic implementations in microlitre volume reactors
using
conventional 96 and 384 well plates may have to overcome non-specific
amplification,
contamination, high reagent costs, and modest numbers of reactions. Performing
quantitative
analysis by digital PCR therefore requires the reliable amplification of
single molecules with low
false positive rates ¨ something that typically requires careful optimization
in microlitre volume
reactors. In addition, the precision of analysis by digital PCR is dependant
on the number of
reactions. Reliable detection of a 1% difference would require hundreds of
thousands to millions
of reactions. This is not practical using existing methods.
[0007] Emerging microfluidic technologies provide increased
sensitivity through small
volume compartmentalization, high scalability, and economy of scale, thereby
allowing for the
full power of digital PCR to be realized.
[0008] Scaling down the size of an assay brings more than just an
economy of scale -
smaller samples may be used and more tests run on one sample is the most
obvious benefit,
however biological assays that were impractical, or not possible using
'conventional' volumes
and samples sizes may also be enabled. In some cases, the conventional assays
may simply be
scaled down ¨ for example use of 1 ul volumes instead of 100 ul; while in
other cases, significant
modification may be needed, in either the way the assay is set up, the data
analysed or in other
respects. Zhang et al., 2006 (Biotechnology Advances 24:243-284) reviews PCR
microfluidic
devices for DNA amplification and various methods, materials and techniques
that may lend
themselves to microfluidics applications.
[0009] The compartmentalization of solutions into a large number of small-
volume
reactions is useful in many fields. Microfluidic devices comprising valves and
fluid channels
- 2 -
CA 02705213 2010-05-07
WO 2009/059430
PCT/CA2008/001985
allow for a planar (2-dimensional) emulsion to be formed with a regular
spatial arrangement.
This regular array has the advantage that it is possible to track or image
each droplet over time.
This is particularly important in assays that require time-monitoring of a
readout (for example
real time polymerase chain reaction). Furthermore, such methods allow for
precisely defined
arrays in which every drop has substantially the same volume.
[0010] Microfluidic digital PCR using compartmentalization by valves
has been
employed in multigene analysis of environmental bacteria (Warren et al., 2006.
PNAS
103:17807-17812), and for transcription factor profiling of hematopoetic cells
(Ottesen et al,
2006. Science 314:1464-1467). The microfluidic devices used in these
experiments provide a
compartmentalized array of approximately 9000 individual 1 OnL reactions.
[0011] The achievable density and minimum volume of PCR reactions in
current
microfluidic devices may be subject to practical limitations. As the density
of the array is
increased, the volume of the individual reaction chambers is decreased, and
valves may occupy
too much space to be feasible. A maximum density at which microfluidic valves
may be reliably
fabricated (typically ¨ 2500cm2, based on a 50x50 chamber array with a pitch
of about 200p,m
provides an upper limit to the number of individual valve-defined reaction
chambers that may be
fabricated (Thorsen et al., 2002. Science 298:580-584 and 2) the minimum
volume of a PCR
reaction that can be implemented in a gas permeable elastomer device without
excessive reagent
evaporation during thermocycling (¨ 1 nL). Density and scale are particularly
important in
digital PCR since accurate measurements require the compartmentalization of a
single sample
into thousands to millions of individual reactions, making it expensive in
terms of both device
area and reagent consumption. New methods for dramatically increasing assay
density and
reducing assay volume are therefore the central issue in realizing the full
potential of this
technique.
[0012] While a variety of materials are known and used in microfluidics
applications,
silicone rubber materials (e.g. polydimethylsiloxane , or PDMS) are preferred
for the ease of
handling and suitability to monolithic construction of microfluidic devices,
PDMS exhibits a
high gas permeability, making it possible to fill dead-end structures. In
addition, PDMS has a
high permeability to water vapour. Thus, some processes that are carried out
in PDMS-
constructed microfluidic devices - particularly where elevated temperatures
are required, such as
- 3 -
CA 02705213 2010-05-07
WO 2009/059430
PCT/CA2008/001985
PCR may suffer from drying out of the small aqueous reaction volume due to
rapid evaporation,
and the reactions may fail.
[0013] Attempts to overcome this limitation have employed the use of
external hydration
methods. While this manages to reduce evaporation rates somewhat, the sample
volume sizes
that are able to be used successfully are still limited by evaporation even
using this hydration.
This limits the ability of such systems to incorporate very small volume
sample sizes (e.g. on the
order of picolitres or smaller) that would enable high density arrays ¨ and
thus large sample
numbers to improve the utility of the devices.
[0014] US 7,118,910 discloses a microfluidic device for performing
PCR assays, and
further discloses use of fluid-filled guard channels in a microfluidic device
to reduce evaporation
of fluid from the device.
[0015] US 6,555,389 discloses a method for compensation for
evaporation in a
microfluidic device by replenishing fluid lost by evaporation from a reservoir
of fluid, via
capillary channels.
SUMMARY OF THE INVENTION
[0016] The present invention relates to a microfluidic device. The
present invention also
relates to a microfluidic device comprising a plurality of reaction chambers
in fluid
communication with a flow channel formed in an elastomeric substrate, a vapor
barrier for
preventing evaporation from the plurality of reaction chambers, and a
continuous phase fluid for
isolation of each of the plurality of reaction chambers. Methods for digital
quantification of a
target nucleic acid, using the microfluidic device are also provided.
[0017] The present invention provides for a microfluidic device that
provides an
improvement in the density of reaction chambers by three orders of magnitude,
achieving
densities of up to about 10,000,000 reaction chambers per square inch (about
1.5 x 106/cm2)
while reducing the volume of each assay by a commensurate amount. The device
provides for
assay compartmentalization in pL-volume emulsion arrays without the need for
valves to
segregate individual reaction chambers.
[0018] In accordance with one aspect of the present invention, there
is provided a
microfluidic device comprising a plurality of reaction chambers in fluid
communication with a
flow channel formed in an elastomeric substrate, a vapor barrier for
preventing evaporation from
- 4 -
CA 02705213 2010-05-07
WO 2009/059430
PCT/CA2008/001985
the plurality of reaction chambers, and a continuous phase fluid for isolation
of each of the
plurality of reaction chambers.
[0019] In accordance with one aspect of the present invention, there
is provided a
microfluidic device comprising a plurality of reaction chambers in fluid
communication with a
flow channel formed in an elastomeric substrate; and a vapor barrier applied
coplanar to the
plurality of reaction chambers and separated from the reaction chambers by a
layer of elastomer.
[0020] In accordance with another aspect of the invention, the
microfluidic device further
comprises a plurality of valves disposed along the flow channel, each of the
plurality of valves
comprising one or more control channels intersecting the flow channel.
[0021] In accordance with another aspect of the invention, the valves are
positioned at
first and second ends of the flow channels.
[0022] In accordance with another aspect of the invention, the
reaction chambers are of
picolitre or femtolitre volume.
[0023] In accordance with another aspect of the invention, the
reaction chamber is a
blind reaction chamber.
[0024] In accordance with another aspect of the invention, the
reaction chamber has one
or more pre-deposited reagents.
[0025] In accordance with another aspect of the invention, the
reaction chambers are
present at a density of 5000 or greater chambers/cm2.
[0026] In accordance with another aspect of the present invention, there is
provided a
method of compartmentalizing a fluid in a microfluidic device, comprising a
plurality of reaction
chambers in fluid communication with a flow channel with a first fluid;
flushing the flow
channel with a second fluid and displacing the first fluid from the flow
channel and not from the
reaction chambers, the second fluid being immiscible with the first fluid;
wherein the first fluid
in each of the plurality of reaction chambers is isolated from the first fluid
in each of the other
reaction chambers by the second fluid in the flow channel.
[0027] In accordance with one aspect of the invention, there is
provided a method for
digital quantification of a target nucleic acid, the method comprising filling
a plurality of reaction
- 5 -
CA 02705213 2010-05-07
WO 2009/059430
PCT/CA2008/001985
chambers in fluid communication with a flow channel of a micro fluidic array,
with a first fluid
comprising a target nucleic acid, ; flushing the flow channel with a second
fluid and displacing
the first fluid from the flow channel and not from the reaction chambers, the
second fluid being
immiscible with the first fluid; wherein the first fluid in each of the
plurality of reaction
chambers is isolated from the first fluid in each of the other reaction
chambers by the second
fluid in the flow channel; and incubating the microfluidic array.
[0028] In accordance with another aspect of the invention, the
reaction chambers are of
picolitre or femtolitre volume.
[0029] In accordance with another aspect of the invention, the first
fluid is a aqueous
solution.
[0030] In accordance with another aspect of the invention, the second
fluid is a non-
aqueous fluid.
[0031] In accordance with another aspect of the invention, the first
fluid comprises a
reaction mix for PCR.
[0032] In accordance with another aspect of the invention, the first fluid
comprises a
sample.
[0033] In accordance with another aspect of the invention, the
reaction chambers are
blind reaction chambers.
[0034] In accordance with another aspect of the invention, the
reaction chambers have
one or more pre-deposited reagents.ln accordance with another aspect of the
invention, the target
nucleic acid is at a suitable concentration in the first fluid to provide less
than about one target
nucleic acids per reaction chamber, averaged over the plurality of reaction
chambers.
[0035] In accordance with another aspect of the invention, the target
nucleic acid is at a
suitable concentration in the first fluid to provide less than about 0.5
target nucleic acids per
reaction chamber, averaged over the plurality of reaction chambers.
[0036] In accordance with another embodiment of the invention, the
step of incubating
comprises thermocycling.
- 6 -
CA 02705213 2010-05-07
WO 2009/059430
PCT/CA2008/001985
[0037] In accordance with another aspect of the invention, there is
provided a
microfluidic device comprising a plurality of reaction chambers in connection
with a flow
channel formed in an elastomeric substrate; and a fluid barrier preventing
fluid communication
between two or more reaction chambers.
[0038] In accordance with another aspect of the invention, the device
further comprises a
vapor barrier applied coplanar to the plurality of reaction chambers and
separated from the
reaction chambers by a layer of elastomer.
[0039] This summary of the invention does not necessarily describe
all features of the
invention. Other aspects, features and advantages of the present invention
will become apparent
to those of ordinary skill in the art upon review of the following description
of specific
embodiments of the invention.
BRIEF DESCRIPTION OF THE DRAWINGS
[0040] These and other features of the invention will become more
apparent from the
following description in which reference is made to the appended drawings
wherein:
[0041] Figure 1 shows a schematic representation of a fabrication process
for a
microfluidic device.
[0042] Figure 2 shows a schematic of a microfluidic array according
to an embodiment
of the invention. (a) A microfluidic array featuring 1,000,000 reaction
chambers in an area of 1"
x 1" (2.54 cm x 2.54 cm). (b) Inset of A showing a sample loading region. (c)
Inset of B showing
microfabricated reaction chambers and feed channels. (d) Three dimensional
rendering of
reaction chamber geometry. In this example, the chamber is 10 microns per side
at the base and
40 microns high for a total volume of 4 picoliters (pL). (e) A microfluidic
array as in (a), lacking
valve channels. (f) Inset of D. (g) A microfluidic array as in A, lacking
valve channels and
comprising a hydration channel.
[0043] Figure 3 shows a schematic representation of isolation of a fluid in
reaction
chambers. In (A), the channel and reaction chambers are filled with a first
fluid (F1- dots). In (B),
a second fluid (F2- checks) is introduced to flow into the channel, the second
fluid displaces the
first fluid from the channel, however the first fluid remains in the reaction
chamber (is "broken
off' by the flow of the second fluid) In (C), the first fluid is displaced
from the channel and
- 7 -
CA 02705213 2010-05-07
WO 2009/059430
PCT/CA2008/001985
remains only in the reaction chambers, and in a portion of the reaction
chamber access channel.
Direction of fluid flow is indicated by the arrows.
[0044] Figure 4 shows a statistical plot of the scaling of precision
and specificity with
volume (A) Numerical calculation of the separation in the measured mean of two
alleles varying
by 1% using digital PCR as a function of the number of chambers. Difference is
normalized by
the expected standard deviation (sigma) as determined by stochastic binomial
noise (sampling
noise). Calculation was performed for template concentration corresponding to
positive
amplification in 50% of reaction chambers. 5 sigma separation is achieved at
approximately
1,000,000 chambers. (B) Numerical calculation of the ratio of the efficiency
of specific and non-
specific amplification as a function of chamber volume for the detection of a
rare species present
at a frequency of one in a million.
[0045] Figure 5 shows a fluorescent image of GAPDH amplification from
genomic
DNA loaded at 10 copies per chamber (positive control) and no template control
(negative
control) samples. The narrow lines outline a set of reaction chambers
connected the same flow
channel. The thick line shows the boundary of the vapour barrier. Edge effect
of failed
amplification due to sample dehydration is visible at the bottom of the
device. PCR reactions in
positive control reaction chambers (regions A, D and E) comprised target DNA
and necessary
reagents for amplification, while the negative control reaction chambers in
regions B and C
lacked target DNA. Bright regions indicate successful PCR amplification of
target DNA.
[0046] Figure 6 shows an amplification plot of a PCR reaction in a
microfluidic device
according to the present invention.
[0047] Figure 7 shows the results of the detection of 3% increase of
chromosome 21
(C21) with respect to chromosome 9 (C9), using a planar emulsion array of the
present invention.
A genomic DNA sample was spiked with 6% trisomy 21 DNA ultimately leading ot
an increase
of 3% of C21 relative to C9 (solid diamond). Pure genomic DNA from a
chromosomally normal
individual has a 1:1 ratio of C21:C9 (solid square).
[0048] Figure 8 shows the results of an assay detecting different
ratios of wild-type to
V617 mutants in JAK2. Differing amounts of mutant plasmid were added to a
constant amount
of wild-type (fill factor of 0.5). (A) shows the obtained ratio (dark bar) and
the theoretical ratio
(light bar). (B) shows the same ratios as in A, but for the 0.1% and control
samples.
- 8 -
CA 02705213 2010-05-07
WO 2009/059430
PCT/CA2008/001985
DETAILED DESCRIPTION
[0049] The present invention relates to a microfluidic device
comprising a plurality of
reaction chambers in fluid communication with a flow channel formed in an
elastomeric
substrate, a vapor barrier overlaying the plurality of reaction chambers, and
a continuous phase
fluid for isolation of each of the plurality of reaction chambers. Methods for
digital
quantification of a target nucleic acid, using the microfluidic device are
also provided.
[0050] A "flow channel" refers generally to a path though which a
fluid may flow.
[0051] A "reaction chamber", a "reaction well" or a "well" is a
bounded region where
a chemical or enzymatic reaction occurs. It may be bounded by a wall and one
or more
valves. Alternately, a reaction chamber may be bounded by walls and a fluid
barrier. A
'wall' of a reaction chamber includes all surfaces ¨ fluid, solid or semi-
solid - bounding the
chamber. A reaction chamber is connected to one or more flow channels; the
connection
comprising an opening that allows for filling and/or emptying of the reaction
chamber via the
one or more flow channels. The plurality of reaction chambers in a
microfluidic device
according to some embodiments of the invention may be arranged in a regular
array ¨ this
arrangement may be referred to as a microfluidic array.
[0052] An "isolated reaction chamber" is a reaction chamber that is
not in fluid
communication with another reaction chamber or flow channel. The fluid
communication
may be prevented by a valve, for example, or by a fluid barrier. The fluid
barrier may be, for
example, formed by a fluid immiscible with the material (usually another
fluid) in the
reaction chamber.
[0053] A "blind reaction chamber" is one with a single access port or
opening that
allows for entrance of a fluid, but lacks a separate exit.
[0054] A "dead-end fill" is a method of filling dead-end or blind reaction
chambers with
a fluid under pressure. When a fluid is initially injected into a channel
structure, it will follow
the path of least resistance, and leave some regions unfilled, or partially
filled. The gas-
permeability of some elastomeric materials used in microfluidic fabrication
may be exploited to
allow for dead-end channels to be filled. By closing all of the exit valves
and injecting the fluid
under pressure (up to about 300 psi or about 200kPa), the pressurized fluid
fills the channels and
- 9 -
CA 02705213 2010-05-07
WO 2009/059430
PCT/CA2008/001985
compressing any gas (air) in the chambers or channels of the device. The
pressurized gas will
diffuse out through the elastomer, leaving behind dead-end or blind reaction
chambers filled with
the fluid.
[0055] A "reagent" refers broadly to any agent used in a reaction,
other than the analyte
(for example, the cell, metabolite, or nucleic acid being analyzed). Examples
of reagents for a
nucleic acid amplification reaction include, but are not limited to, buffers,
surfactants, metal
ions, DNA or RNA polymerase, reverse transcriptase, kinases or phosphatases,
primers,
template or target nucleic acid, nucleotides, labels, dyes, nucleases and the
like. Reagents for
enzyme reactions include, for example, substrates, cofactors, buffer, metal
ions, inhibitors
and activators. Reagents for cell-based reactions include, but are not limited
to, the above
reagents for enzyme reactions or nucleic acid detection, as well as cells,
cell specific dyes, or
ligands (e.g., agonists and antagonists) that bind to cellular receptors. A
surfactant such as
TWEEN-20 may be included in the reaction mixture to reduce adsorption of
reagents to the
elastomer surface.
[0056] A primer is a short (generally 10-30 nucleotides) nucleic acid or
oligonucleotide
that is annealed to a target nucleic acid and extended by a nucleic acid
polymerase. A primer
may be perfectly complementary to the target nucleic acid (100% match between
all nucleotides
of the primer and target nucleic acid), or may be substantially complementary
(less than 100%)
so that the primer hybridizes selectively at a site of the target nucleic
acid.
[0057] A probe is a nucleic acid sequence that specifically hybridizes with
a target
nucleic acid. The hybridization is detectable, for example by a fluorescent
label or dye that has
modified fluorescent properties only with formation of the double stranded
configuration (for
example a DNA staining dye) or by fluorescence (for example a molecular
beacon). A probe
may be of any suitable length to provide sufficient complementarity to the
target nucleic acid
sequence and anneal under the reaction conditions. Probes used in
hybridization may include
double-stranded DNA, single-stranded DNA and RNA oligonucleotides, locked
nucleic acid
(LNA) probes, and peptide nucleic acids. Hybridization methods for the
identification of single
nucleotide polymoiphisms or other mutations involving a few nucleotides are
described in the
U.S. Pat. 6,270,961; 6,025,136; and 6,872,530. Suitable hybridization probes
for use in
accordance with the invention include oligonucleotides, polynucleotides, LNAs
and PNAs in a
-10 -
CA 02705213 2010-05-07
WO 2009/059430
PCT/CA2008/001985
range of lengths, from about 6 to about 400 nucleotides, from about 20 to
about 200
nucleotides, or from about 30 to about 100 nucleotides in length..
[0058] An oligonucleotide is a variable length nucleic acid, which
may be useful as
probes, primers for the detection and/or amplification of specific nucleic
acids. Numerous
methods are known in the art for synthesizing oligonucleotides - see, for
example, Bonora GM.
et al. Nucleic Acid Res.(1990) 18(11):3155-9; Lashkari DA. et al. PNAS (1995)
92(17):7912-5;
McGall G. et al. PNAS (1996) 93(24):13555-60; Albert TJ. et al. Nucleic Acid
Res.(2003)
31(7):e35; Gao X. et al. Biopolymers (2004) 73(5):579-96; and Moorcroft MJ. et
al. Nucleic
Acid Res.(2005) 33(8):e75). Gait, pp. 1-22; Atkinson et al., pp. 35-81; Sproat
et al., pp. 83-115;
and Wu et al., pp. 135-151, in Oligonucleotide Synthesis: A Practical
Approach, M. J. Gait, ed.,
1984, IRL Press, Oxford; or Molecular Cloning: a Laboratory Manual 3' edition.
Sambrook and
Russell. CSHL Press, Cold Spring Harbour, New York.
[0059] Some nucleic acids or olignucleotides may incorporate monomers
that provide for
altered or improved enzymatic stability, or conformational restriction in the
oligonucleotide. For
example, bicyclic nucleosides may provide conformational restriction to the
oligonucleotide, and
may provide varying hybridization or stability profiles compared to unmodified
oligonucleotides.
An LNA nucleoside is an example of a bicyclic nucleoside, having a 2'-4'
cyclic linkage as
described in US 6,268,490, US 6,794,499, US 7,034,133 (each of which are
incorporated herein
by reference). Methods of synthesis and polymerization of nucleic acid
polymers comprising
LNA monomers are described in, for example, WO 99/14226, WO 00/56746, WO
00/56748,
WO 01/25248, WO 0148190, WO 02/28875, WO 03/006475, WO 03/09547, WO
2004/083430,
US 6,268,490, US 6,794,499, U.S. Pat. No. 7,034,133.
[0060] A "one-pot" process or reaction is a process carried out in a
single reaction
chamber without multiple separate steps of addition and reaction of reagents.
In a one-pot
reaction, the reagents necessary for the reaction are admixed together in a
single reaction
medium, substantially at the same time. For example, a one-pot PCR reaction
may combine the
reagents for both the amplification reaction and detection in a single
reaction chamber ¨ both
amplification and detection are carried out in the same reaction chamber. A
reaction mix
comprising all reagents necessary for amplification and detection of a
specific target nucleic acid
(including target nucleic acid, primers, probe, polymerase, nucleotides,
buffer, salts and the like)
may be combined, and this single mixture injected into a microfluidic device
of the present
-11 -
CA 02705213 2010-05-07
WO 2009/059430
PCT/CA2008/001985
invention. All reaction chambers are filled with the reaction mixture, the
chambers isolated and
the thermal cycling performed. Detection of the specific amplified product may
be performed as
thermal cycling is occurring, or afterwards, or both. As a variant on a one-
pot process, a reagent
may be placed in an array or sub-array of reaction chambers of a microfluidic
device of the
present invention during synthesis of the device or afterward. A reaction
mixture comprising all
remaining reagents necessary for amplification is injected into a microfluidic
device of the
present invention, with the final admixing of the primers and probe with the
reaction mix
occurring in the reaction chamber. Again, detection of the specific amplified
product may be
performed as thermal cycling is occurring, or both.
[0061] "Fluid communication" - Two or more elements of a microfluidic
device (for
example a flow channel, injection port, exit port, via, reaction chamber,
inlet or other space
defined by a boundary) are in fluid communication if there exists a continuous
path connecting
the two that does not leave the fluid. Fluid communication may be mediated by
a barrier, valve,
pump or other control device to allow for controlled interruption of the fluid
communication and
isolate one or more elements of the microfluidic device. Fluid communication
between two or
more elements having within them a first fluid of a first phase may also be
mediated by an
immiscible fluid of a second phase. When the immiscible fluid is introduced
into one of the
elements, displacing the first fluid. As an example, in an emulsion, fluid
communication of
droplets of a dispersed phase is interrupted or prevented by the continuous
phase of the emulsion.
[0062] In some embodiments of the present invention, an immiscible
fluid may be the
barrier or valve (a "fluid barrier" or "fluid valve") that mediates or
prevents fluid
communication between droplets in the reaction chambers of the microfluidic
device of the
present invention.
[0063] Without wishing to be bound by theory, the methods provided herein
exploit the
surface tension between a first fluid, one or more channel walls or reaction
chamber walls and a
second fluid that is immiscible with the first fluid, to create a regular
array of micro-scale
droplets whose position and size are precisely defined by the structure of the
microfluidic
channel.
[0064] An emulsion is formed when two immiscible fluids are combined and
one fluid (a
dispersed phase) is suspended in the other (a continuous phase) as droplets or
colloids. The
- 12 -
CA 02705213 2010-05-07
WO 2009/059430
PCT/CA2008/001985
continuous phase forms a barrier between droplets, and if the emulsion is
stabilized (coalescing
of the droplets is prevented) each of the droplets may be an isolated reaction
vessel. Use of a
surfactant is one way to stabilize an emulsion. Another is to exploit the
surface tension between
the dispersed phase, one or more channel or reaction chamber walls, and
continuous phase, in
combination with a chambered microfluidic device that allows for a regular
array of micro-scale
droplets whose position and size are precisely defined by the structure of the
microfluidic device,
and thus are not allowed to come into contact.
[0065] The architecture of a microfluidic device according to some
embodiments of the
invention comprises a linear array of microfluidic channels that are connected
to a series of
nanoliter (nL), picoliter (pL) or femtoliter (fL) volume chambers through
access channels. The
first fluid, for example an assay mixture comprising all required components
for PCR
amplification is first introduced into the flow channels under pressure,
causing it to dead-end fill
all the reaction chambers. Ambient air is forced into the bulk of the gas-
permeable elastomer
device and out of the reaction chamber. Template molecules loaded at limiting
dilution are
randomly distributed through the array of chambers according to Poisson
statistics. Once the
chambers are filled, the second fluid is injected through the flow channels to
displace the first
fluid. Without wishing to be bound by theory, the advance of the phase
interface past each
chamber creates a surface tension-driven instability at the channel
constriction, resulting in the
separation of a droplet of aqueous solution whose position and volume is
precisely defined by the
geometry of the chamber. These aqueous droplets remain confined to the large
cross-section of
the chambers due to surface tension, allowing for precise localization of each
drop during
thermocycling.
[0066] Multilayer soft lithography (MSL) is a well-known fabrication
technique in soft
elastomer processing that allows for facile and robust fabrication of
microfluidic devices having
hundreds to thousands of microscopic reaction chambers, valves, pumps, fluidic
logic elements
and other components. Xia & Whitesides, 1998 (Angewandte Chemie-International
Edition
37:551-575; herein incorporated by reference) describe and review procedures,
material and
techniques for soft lithography, including MSL.
[0067] Briefly, the general idea of multilayer soft lithography (MSL)
is to iteratively
stack layers of elastomers, for example PDMS, of varying thickness on top of
each other. Thin
and thick layers of PDMS with stoichiometric ratios respectively less and
higher than 10:1 are
- 13 -
CA 02705213 2010-05-07
WO 2009/059430
PCT/CA2008/001985
formed on separate wafers. Photoresist patterns previously made on the wafers
will define the
microfluidic channels of the device. The thick layer is then peeled away from
the wafer and
placed on top of the thin wafer. After baking, the excess components in each
layer will bond and
form a PDMS 'chip' composed of two layers of channels. Methods of working with
elastomers
and applying them in microfluidic applications are known in the art; see, for
example, U.S. Pat.
No. 6,929,030; Scherer et al. Science 2000, 290, 1536-1539; Unger et al.
Science 2000, 288, 113-
116; McDonald et al. Acc. Chem. Res. 2002, 35, 491-499; Thorsen, T. et al,.
Science 2002, 298,
580-584; Liu, J. et al. Anal. Chem. 2003, 75, 4718-4723; Rolland et al. 2004
JACS 126:2322-
2323, PCT publications WO 02/43615 and WO 01/01025.
[0068] Various soft polymers, generally referred to as elastomers may be
used in
microfluidic devices and systems. Elastomers may be generally characterized by
a wide range of
thermal stability, high lubricity, water repellence and physiological
inertness. Other desirable
characteristics of elastomers may vary with the application. It is within the
ability of one of skill
in the art to select a suitable elastomer or combination of elastomers for the
desired purpose.
Examples of elastomers include silicone, polydimethylsiloxane (PDMS),
photocurable
perfluoropolyethers (PFPEs), fluorosilicones, polyisoprene, polybutadiene,
polychloroprene,
polyisobutylene, polyurethanes, poly(styrene-butadiene-styrene), vinyl-silane
crosslinked
silicones, and the like. Elastomers may be optically clear, or may be opaque,
or have varying
degrees of transparency. In some embodiments of the invention, it may be
desirable to use a
bio compatible elastomer. PDMS is one of the first developed and more widely
used elastomers
in soft lithography applications. Where PDMS is described as the elastomer
used in various
embodiments of the invention, it is for exemplary purposes only, and the
choice of alternate
elastomers is within the knowledge of one skilled in the art. A variety of
elastomers suitable for
use in microfluidic applications, and their various properties and examples of
applications are
described in U.S.Patent No. 6,929,030.
[0069] Photoresist patterns laid out on a silicon wafer or other
suitable support provide a
mold for casting the layers. Generally, photoresists may be categorized as
positive or negative.
Positive photoresist are capable of very fine resolutions. They are highly
soluble in alkaline
solutions such as KOH; however, photosensitive dissolution inhibitors such as
diazonaphthaquinone (DQ) are typically used to block this effect. A
photoreaction with
ultraviolet (UV) light destroys the DQ and allows the photoresist to be
dissolved by the
developer solution. The idea of processing this type of photoresist is that
all sections exposed to
- 14 -
CA 02705213 2010-05-07
WO 2009/059430
PCT/CA2008/001985
UV light will be removed. An example of a positive photoresist is SPR220-7
(Shipley Company
LLC).
[0070] Negative photoresist generally comprises a non-photosensitive
substrate, a
photosensitive cross-linking agent, and a coating solvent. Upon exposure to UV
light, the cross-
linking agent is activated and causes a hard epoxy to form. The remaining
unexposed sections of
the photoresist are washed away with the developer solution. SU8 (MicroChem)
is an example of
a negative photoresist that may be used in both MSL molds. In addition, SU8 as
an epoxy is very
strong and can resist subsequent photolithography processes. Detailed methods
and techniques
for working with particular photoresists are available from the various
manufacturers, and are not
addressed further herein. Examples of particular photoresists are for
illustrative purposes only,
and are not to be considered as limiting of the present invention.
[0071] Other components may be incorporated into the microfluidic
device during
fabrication - micron-scale valves, pumps, channels, fluidic multiplexers,
perfusion chambers and
the like may be integrated during MSL. Methods of making and integrating such
components
are described in, for example, US Patent Nos. 7,144,616, 7,113,910, 7,040,338,
6,929,030,
6,899,137, 6,408,878, 6,793,753, 6,540,895; US Patent Applications
2004/0224380,
2004/0112442; PCT Applications WO 2006/060748.
[0072] Evaporation is an obstacle to be overcome when performing
assays in picoliter
volume ranges. During the fabrication of a microfluidic device, a vapor
barrier may be
incorporated into the device in proximity to the reaction chambers and
channels where fluid will
be placed or conveyed, and fluid loss is not desired. A vapor barrier may
comprise any suitable
material. A suitable material is, preferably, optically transparent,
impermeable to water vapor,
unreactive with the substrate comprising the microfluidics device and fluids
used in the assay
mixture or immiscible oil or fluid used to displace the assay mixture in the
flow channels, and
able to tolerate the temperature ranges used in fabricating the microfluidic
array and in the assays
where they are used. In addition it is desirable to use materials that may be
bonded to the
elastomer used. Good bonding may be a property of the elastomer or may be
achieved through
the application of one or more adhesive layers or through modification of the
material surface
(for example, by exposure to an oxygen plasma). An example of a suitable
material is an acetate,
polyethylene, or polyethylene-based sheet, polyxylylene polymers (Parylene TM
N, C, D) others
include glass, silicon, quartz, mica. As well, other polymeric material may be
used, these
- 15 -
CA 02705213 2010-05-07
WO 2009/059430
PCT/CA2008/001985
polymeric materials may be deposited as a prefabricated film or by using other
polymer
processing techniques such as depositing a liquid that will set as a film
(examples include resin
formulations, polymeric solutions, precursor mixes, etc), or by vapor
deposition of a polymer
(such as Parylene), or by evaporation where the material condenses on the
surface, or by spraying
of an aerosol, or the like.
[0073] The vapor barrier is applied or positioned during fabrication
of the microfluidic
device coplanar with at least one side of the array. The vapor barrier is
separated from one wall
of the reaction chambers of the array by a layer of elastomer of sufficient
thickness to absorb gas
forced into the elastomer by dead-end filling. This thickness may be, for
example, from about 10
to about 500 gm, or from about 50 to about 250 gm, or from about 75 to about
225 gm, or from
about 100 to about 200 gm, or from about 125 to about 175 gm. Without wishing
to be bound
by theory, this geometry provides for establishment of a substantially-two
dimensional gradient
of water vapor so that transport (evaporation) occurs only through the sides
of the array.
[0074] In a preferred example, the array is flanked on both planar
sides by a vapor
barrier. In some embodiments of the invention, a microfluidic array is
positioned between a
silicon substrate (the bottom of the array) and an acetate or polyethylene
sheet on top of the array,
separated from For a device employing two vapor barriers configured as
described, the barriers
may be separated by any suitable distance, which will vary depending on the
height of the
reaction chambers and the presence of any additional layers of flow channels,
valves, etc
positioned above or below the array of reaction chambers.
[0075] A simple, schematic representation of a fabrication process is
illustrated in Figure
1. (A) Mold features 120 is cast on a wafer 121, using standard soft
lithographic techniques. (B)
Liquid PDMS is spun on top of the wafer and partially hardened, so that its
thickness is slightly
higher than the height of the features. (C) A vapour barrier 123 is applied on
top of the PDMS
layer 122, and covered by a second layer 124 of PDMS. (D) Following de-gassing
and baking,
the device is peeled away from the mold, leaving a negative imprint on the
cured PDMS.
[0076] Referring to Figures 2a-d, a schematic diagram of device
according to one
embodiment of the invention is illustrated generally at 100. Figure 2b shows
inset A of Figure 2a;
Figure 2c shows inset B of Figure 2d; Figure 2d shows a three dimensional
rendering of a
reaction chamber 114, indicated at inset C of Figure 2c. This exemplary device
comprises 13
primary flow channels 103 in fluid communication with a cascading series of
flow channels 105,
- 16-
CA 02705213 2010-05-07
WO 2009/059430
PCT/CA2008/001985
107, 109 for loading reaction chambers 114. Each primary flow channel services
a sub-array and
may be loaded with a fluid comprising a reaction mixture, sample or the like,
and may be
independent of flow channels, thus enabling more than one sample, or more than
one reaction on
the same sample to be loaded and processed on the same device.
[0077] Valve flow channels 102a, 102b intersect the primary flow channel
103, flanking
the array of reaction chambers. Flow of fluid from injection port 104 is
regulated by valve 108;
valve 108 is opened or closed by fluid pressure in valve flow channel 102a, b.
All valves along
valve flow channel 102a may be operated simultaneously by injection port 106a;
all valves along
valve flow channel 102b may be operated simultaneously by injection port 106b.
Reaction
chamber 114 is in fluid communication with reaction chamber flow channel 110
via reaction
chamber access channel 112. In the embodiment illustrated in Figure 2c and 2d,
reaction
chamber 114 is a blind chamber, having a single filling port and no separate
exit port. Reaction
chamber 114 as exemplified in Figure 2d is taller than it is wide, with the
access flow channel at
the bottom of the reaction chamber. Other configurations of reaction chambers
are possible. For
example, reaction chamber such as that shown in Figure 2d, may have a small
'footprint' area
(width x length) and taller than it is wide or long. Such a 'vertical'
configuration may provide
for a larger sample volume and maximize the planar density of the chambers. A
larger reaction
volume may allow for inclusion of a greater quantity of fluorophore (or other
detectable label or
signal producing reagent) in a reaction chamber, thus a stronger signal for
detection).
[0078] Referring to Figure 2e and 2f, a schematic diagram of an alternate
embodiment of
the present invention, lacking valve channels 102a, b, valves 108 and valve
flow channel inlets
106 a, b is shown generally at 130. Figure 2f shows inset D of Figure 2e; the
inset B of Figure 2f
is as illustrated in Figure 2c. Flow channels 103, 105, 107, 109 for loading
reaction chambers
114 are as previously addressed.
[0079] Hydration channels filled and operated separately from the valve
flow channels
and primary fluid channels may be included around the periphery of the array
to hydrate reaction
chambers near the edge of the array. Referring to Figure 2g, a schematic
diagram of an alternate
embodiment of the present invention is shown generally at 140. This embodiment
comprises
injection ports 104 and flow channels as illustrated in Figures 2a-d and e-f,
and further comprises
hydration channel 142 and ports 144. Hydration channel 142 is formed in a
layer above or below
the primary flow channel 103 and separated from flow channels and reaction
chambers,
- 17 -
CA 02705213 2010-05-07
WO 2009/059430
PCT/CA2008/001985
including primary flow channel 103, by a sufficient thickness of elastomer so
that fluid flow and
pressure in the hydration channel does substantially restrict fluid flow in
primary flow channel
103, or flow channels 105, 107 or 109 (not shown). The term "substantially
restrict" (or similar
terms) is to be taken to indicate that the fluid flow is not reduced in, our
or through the flow
channel or reaction chamber by more than 40%, typically less than 30%,
preferably less than 20%
or more preferably less than 10%, as compared to fluid flow in, to or through
the flow channel or
reaction site under the conditions when the hydration channel does not contain
a fluid, or is under
pressure. Hydration channel 142 may have an internal diameter about the same
as that of flow
channel 103, 105, 107 or 109.
[0080] One of skill in the relevant art will appreciate that the various
features disclosed
herein may be combined in some embodiments of the invention. For example, a
microfluidic
device may comprise a valve flow channel as illustrated in Figures 2a and b,
as well as a
hydration channel. Other configurations of the hydration channel are also
contemplated; for
example, a series of separate hydration channels may be positioned along an
edge of the
microfluidic device.
[0081] Devices according to some embodiments of the present invention
may be
fabricated to comprise a plurality of separate injection ports that each
interface with a separate
section or sub-array ¨ the high density of the array allows for multi-sample
analysis using spatial
multiplexing on a single device. For example, an array of approximately
1,000,000 reactions
may be divided into 40 independent sections, each comprised of approximately
25,000 reaction
chambers. A separate injection port for interfacing with each independent sub-
array allows for
implementation of multi-sample analysis.
[0082] A preloading strategy may be employed to spatially multiplex
separate assays
within defined regions of the array. Briefly, one or more valves may be used
to isolate sections of
the array into which distinct reagent sets (for example, distinct primer
and/or probe sets) in
solution may be dead-end loaded. Flow channels may be subsequently flushed
with air to
remove excess reagent, and the device incubated at about 80 C to evaporate
water from the
reaction mixes, leaving dried primers and probes in defined sections of the
array. Using such a
preloaded device, a sample comprising a target nucleic acid in solution may be
introduced to all
reaction chambers of the array and analyzed for a plurality of target nucleic
acids (for example,
- 18 -
CA 02705213 2010-05-07
WO 2009/059430
PCT/CA2008/001985
for an array having 40 independent sections, up to 40 separate target nucleic
acids may be
analyzed).
[0083] In the device exemplified in Figure 2a-d, valve flow channels
provide for valves
positioned at the edges of the device. As illustrated in Figures 2e-g, a
valveless embodiment of a
microfluidic device may also be employed. In such embodiments, fluid flow in,
out and through
flow channels or hydration channels may be controlled by valves external to
the microfluidic
device.
[0084] To load the reaction chambers with a reaction mixture (a first
fluid) ,the reaction
chambers (114) are dead-end filled with a first fluid (F1, Figure 3) injected
through injection port
104. All reaction chambers are thus in fluid communication with the flow
channel via the
reaction chamber access channel. Subsequently, a second fluid (F2) is injected
into the injection
port ¨ F2 displaces F1 as it flows through the flow channels.
[0085] The first fluid may be an aqueous solution, such as a reaction
mixture for use in
an assay. As an example, for a polymerase chain reaction (PCR) assay, the
first fluid may
comprise all of the reagents necessary for PCR ¨ template nucleic acid,
nucleotides, primers a
suitable nucleic acid polymerase, buffer, salts. Depending on the intended use
of the assay, the
first fluid may further comprise a dye, a label, a probe or other reagents for
detecting the product
of the PCR assay. The second fluid is immiscible with the first fluid, and is
also compatible
with the material the microfluidic device is constructed from. The second
fluid may be an oil, for
example a fluorinated oil, that is compatible with the PDMS elastomer from
which the device is
fabricated. By 'compatible' it is meant that the second fluid does not
interact or react with the
device in an undesirable manner ¨ for example, the elastomer does not absorb
the fluid or swell
when exposed to the fluid, and the physical properties of the device are
unaltered (no substantive
change in elasticity or rigidity). Examples of second fluids that may be used
with a microfluidic
device comprising PDMS include perfluorohexane (FluorinertTm), fluorinated
silicone oils,
fluorinated oils (for example, FC40, FC43, FC70, FC-72, FC-77, FC-84, FC-87 or
the like,
available from 3M or other suppliers) and high molecular weight oils such as
paraffin oil.
[0086] The specific dimension and density of the reaction chamber and
other features of a
microfluidics device according to the present invention may be constrained
only by the
limitations of the multilayer soft lithography (MSL) methods themselves.
Current MSL
techniques are routinely capable of fabricating features as small as 1 tm. For
example, a
- 19 -
CA 02705213 2010-05-07
WO 2009/059430
PCT/CA2008/001985
microfluidic device of the present invention may comprise an array of reaction
chambers
numbering from about 50,000 to about 108 or any amount therebetween; or from
about 100,000
to about 5 x 106 or any amount therebetween; or from about 200,000 to about 2
x 106 or any
amount therebetween; or from about 250,000 to about 106 or any amount
therebetween,
depending on the overall size of the device.
[0087] The density of reaction chambers may be expressed in terms of
the quantity per
unit area. For example some devices may have a density of reaction chambers
from about 1000
to about 2x 106 per square centimeter, or any amount therebetween; from about
5000 to about 106
per square centimeter or any amount therebetween; from about 10000 to about
500,000 or any
amount therebetween. For example, the density of reaction chambers may be
about1000, 2000,
3000, 4000, 5000, 6000, 7000, 8000, 9000, 10000, 15000, 20000, 25000, 30000,
35000, 40000,
45000, 50000, 100,000, 150,000, 200,000, 250,000, 300,000, 350000, 400,000,
450,000,
500,000, 550,000, 600,000, 650,000, 700,000, 750,000, 800,000, 850,000,
900,000, 950,000,
1,000,000, 1,050,000, 1,100,000, 1,150,000, 1,200,000, 1,250,000, 1,300,000,
1,350,000,
1,400,000, 1,450,000, 1,500,000, 1,550,000, 1,600,000, 1,650,000, 1,700,000,
1,750,000,
1,800,000, 1,850,000, 1,900,000, 1,950,000, 2,000,000 per square centimeter.
[0088] The density at which reaction chambers may be placed in the
microfluidic array
may vary also. As an example, an array of 5 gm3reaction chambers with a pitch
of about 10 gm
would represent a total density of 106 reaction chambers per square centimetre
(about 125 fL per
chamber). The pitch of the chambers will vary with the intended use of the
device; greater
separation may be necessary for reactions that require a greater volume (for
example, if a whole
cell is to be placed in the reaction chamber, or if the target molecules
cannot be sufficiently
concentrated with convenient methods). For some microfluidic arrays, the pitch
may have to be
determined empirically - determination of the optimal pitch is within the
ability of one skilled in
the art. As an example, the pitch may be from about 1 to about 1000 gm, or any
amount
therebetween, from about 2 to about 80 gm or any amount therebetween, from 5
to about 60 gm
or any amount therebetween, from about 10 to about 40 gm or any amount
therebetween, or from
about 20 to about 50 m or any amount therebetween. For example, the pitch may
be 1, 2, 3, 4,
5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25,
26,
27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45,
46, 47, 48, 49, 50, 55, 60,
65, 70, 75, 80, 85, 90, 95 or 100 gm.
- 20 -
CA 02705213 2010-05-07
WO 2009/059430
PCT/CA2008/001985
[0089] Compartmentalization of a single particle within a droplet of
a small volume
allows for the effective concentration of this particle, cell, or molecule to
be high. One method of
compartmentalization is use of an emulsion where aqueous droplets are
separated on at least one
side by a second liquid phase such as oil . Microfluidic devices comprising
fluid channels with
defined microchambers (reaction chambers) according to some embodiments of the
invention
allow for a planar (2-dimensional emulsion) to be formed with a regular
spatial arrangement ¨
valves are not required to define the reaction chambers, thus the limits they
may impose on pitch
and density of reaction chambers in an array of reaction chambers is removed.
This
concentration enhancement, when combined with amplification methods, allows
for the
enumeration of single molecules ¨ digital quantification. Without wishing to
be bound by theory,
the enhancement of concentration of biological matter through such
compartmentalization
provides a means by which to increase detection and assay sensitivity. The
concentration of the
particle may be expressed relative to the average number per well of an array.
For example, in an
array of reaction chambers where each well has a an average of 1 particle,
some reaction
chambers may contain zero, some may contain one, and some may contain two or
more particles.
As another example, in an array of reaction chambers where each well has an
average of 0.1
particles, most reaction chambers would be expected to have zero particles,
about 10% would be
expected to have one particle, and a very few may have two or more particles.
According to
some embodiments of the invention, the particle may be provided at an average
concentration per
reaction chamber of about 0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.7, 0.8, 0.9, 1.0,
1.1, 1.2, 1.3, 1.4, 1.5, 1.6,
1.7, 1.8, 1.9, 2.0 , 2.1, 2.2, 2.3, 2.4, 2.5, 2.6, 2.7, 2.8, 2.9, 3.0, 3.1,
3.2, 3.3, 3.4, 3.5, 3.6, 3.7, 3.8,
3.9, 4.0 , 4.1, 4.2, 4.3, 4.4, 4.5, 4.6, 4.7, 4.8, 4.9, 5.0, or any amount
therebetween.
[0090] A particle may be any discrete material which can be flowed
through a micro scale
system. Example particles include beads, nucleic acids, target nucleic acids,
proteins, biological
cells, molecules and the like. For example, polymer beads (e.g., polystyrene,
polypropylene,
latex, nylon and many others), silica or silicon beads, clay or clay beads,
ceramic beads, glass
beads, magnetic beads, metallic beads, inorganic compound beads, and organic
compound beads
can be used. An enormous variety of particles are commercially available,
e.g., those typically
used for chromatography (see, e.g., the 1999 Sigma "Biochemicals and Reagents
for Life
Sciences Research" Catalog from Sigma (Saint Louis, Mo.), e.g., pp. 1921-2007;
The 1999
Suppleco "Chromatography Products" Catalogue, and others), as well as those
commonly used
for affinity purification (e.g., Dynabeads.TM. from Dynal, as well as many
derivitized beads,
e.g., various derivitized Dynabeads.TM. (e.g., the various magnetic
Dynabeads.TM., which
- 21 -
CA 02705213 2010-05-07
WO 2009/059430
PCT/CA2008/001985
commonly include coupled reagents) supplied e.g., by Promega, the Baxter
Immunotherapy
Group, and many other sources).
[0091] For example, the compartmentalization of 1 microliter volume
of solution
containing a single DNA molecule has a concentration of 106 L-1. The
compartmentalization of
this solution into 1,000,000 droplets each with a volume of 1 picoliter
results in 999,999 droplets
with a concentration of 0, and 1 droplet with an effective concentration of
1012 L-1 - an
enhancement of 1,000,000 times. In another example, microfluidic devices of
the present
invention may be employed to compartmentalize a cell within a reaction chamber
(for example a
volume of about 100 pL). Isolation of a single cell allows for the cell to
exert a large influence
on its environment, and to secrete molecules to be concentrated sufficiently
for detection.
[0092] Picoliter (pL) volume droplets have large surface to volume
ratios - reliable
amplification of single molecules may be challenged by reagent evaporation
during
thermocycling, a phenomena that is particularly acute in devices made from
PDMS which is
known to have very high vapor permeability. This evaporation may be pronounced
in planar
emulsion arrays where significant gradients occur in the direction normal to
the plane. To
control this evaporation, a vapor barrier may be embedded in the device, and
flanking the array.
Vapor loss in a gradient normal to the plane of the array is thereby
eliminated, allowing for
successful amplification of target nucleic acid in pL volume reactions. A
"picoliter volume"
describes generally a small volume of about 1 to about 1000 pL, or any amount
therebetween;
from about 1 to about 500 pL, or any amount therebetween; from about 1 to
about 200 pL or any
amount therebetween; from about 1 to about 100 pL, or any amount therebetween;
or from about
1 to about 50 pL, or any amount therebetween. For example, the volume of the
reaction mixture
formed by the microfluidic devices of the present invention, or the volume of
reaction mixture
used in methods of the present invention may be 1, 2, 3, 4, 5, 6, 7, 8, 9, 10,
11, 12, 13, 14, 15,
16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34,
35, 36, 37, 38, 39,
40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 55, 60, 65, 70, 75, 80, 85, 90,
95, 100, 110, 120,
130, 140, 150, 160, 170, 180, 190, 200, 210, 220, 230, 240, 250, 260, 270,
280, 290, 300, 310, 320, 330, 340, 350, 400, 450, 500, 550, 600, 650, 700,
750,
800, 850, 900, 950, or 1000 pL.
- 22 -
CA 02705213 2010-05-07
WO 2009/059430
PCT/CA2008/001985
[0093] Sub-picoliter reaction volumes are also practical and useful
according to the
various embodiments of the invention. As discussed, conventional lithographic
fabrication
techniques can achieve features as small as 1 um and are thus capable of
defining chambers with
volumes in the femtoliter to picoliter scale. For example, a 1 m3 chamber in a
microfluidic array
would have a volume of about 1 femtoliter (fL); a chamber of about 5 pm3 would
have a volume
of about 125 fL. A sub-picoliter volume describes generally a small volume of
about 1 to about
1000 fL, or any amount therebetween; from about 10 to about 500 fL, or any
amount
therebetween; or from about 100 to about 500 fL or any amount therebetween.
For example, the
volume of the reaction mixture formed by the microfluidic devices of the
present invention, or
the volume of reaction mixture used in methods of the present invention may be
about 1, 5, 10,
15, 20, 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 105,
110, 115, 120, 125,
130, 135, 140, 145, 150, 155, 160, 165, 170, 175, 180, 185, 190, 195, 200,
250, 300, 350, 400,
450, 500, 550, 600, 650, 700, 750, 800, 850, 900, 950, or 1000 fL.
[0094] The reaction chambers may alternately be described by their
volume in cubic
micrometers, or similar dimensions, rather than by the droplet volume. It will
be within the
ability of one skilled in the art to determine the necessary conversion
between units, for example
from cubic micrometers to picoliters, or the like.
[0095] In addition to limiting or preventing evaporation of the
reaction mix in the
reaction chamber, the reaction conditions are controlled.
[0096] If the device is to be utilized in temperature controlled reactions
(for example,
incubating at one or more temperatures for a period of time, either as a
single incubation or a
series of cyclical temperatures, for example thermocycling as employed in PCR
or other
amplification or extension reactions), the elastomeric device may be fixed to
a support, such as a
glass slide or silicon wafer. The device may be placed in a temperature and/or
environment
controlled incubator, or for a thermocycling reaction, the device may be
placed on any number of
thermocycling plates. Devices for thermal cycling are known and available.
Generally, a
microfluidic device comprising an array of reaction chambers may be placed on
a thermal cycling
plate to effect the thermal cycling of the reaction. A variety of such thermal
cycling plates are
available from commercial sources, for example, BioRad, Thermo, Eppendorf,
Techne, Applied
Bio systems, and others. Alternatively, the substrate of the device may
contain active heating and
-23-
CA 02705213 2010-05-07
WO 2009/059430
PCT/CA2008/001985
temperature sensing elements to provide the necessary thermocycling
conditions. The fabrication
of heaters and temperature sensors in a silicon substrate for instance is well-
known in the art.
[0097] For PCR, thermocycling requires precise control of both
temperature and the time
at which the reaction is incubated at a particular temperature. The
temperature ranges for
denaturation, annealing and extension of the template or target nucleic acid
vary depending on
the particular amplification reaction performed, complementarity of the
primers employed, the
composition of the target nucleic acid and the particular polymerase selected.
Other amplification
methods may not necessitate multiple rounds of thermal cycling, but may still
benefit from
thermal and temporal control of reaction conditions. For example, various
isothermal nucleic
acid amplification strategies such as rolling circle amplification are known
and are envisioned
with the present invention.
[0098] The ability to monitor the thermal profile of the device and
reaction chambers
may also be useful. Sensors may be incorporated into the microfluidic device
(e.g. a
thermocouple, or a thermistor, or a pyroelectric sensor. Temperature may be
monitored by use
of an infrared camera, or by use of thermochromic materials. These and other
methods for
monitoring temperature profiles within a microfluidic device are described in
U.S. Patent No.
7,118,910.
[0099] Applications of the microfluidic devices and methods according
to some
embodiments of the invention are numerous and varied. Uses or applications,
and methods
comprising uses or applications of the microfluidic devices and methods
described herein are not
limited to any particular application or use thereof. In some embodiments of
the invention, the
following uses and applications are contemplated.
[00100] Samples, assays and assay conditions
[00101] A myriad of assays and uses exist to which microfluidic
devices according to
some embodiments of the present invention may be applied. A review of such
uses, and
references and disclosures of methods and protocols is provided in PCT
Publication WO
00/50172. Selected assays are exemplified herein for illustrative purposes,
and are not to be
considered limiting.
[00102] Single-cell assays - Microfluidic-based single cell analyses
may be used for a
variety of purposes, for example cell sorting, nucleic acid purification and
amplification, patch-
- 24 -
CA 02705213 2010-05-07
WO 2009/059430
PCT/CA2008/001985
clamping, calcium flux measurements, and whole cell electrophoresis.
Additionally, the capture
and imaging of one or a few single cells in microfabricated devices may be
used to monitor
cellular response to varying concentrations of one or a few chemical stimuli.
[00103] A microfluidic device, or a system comprising such a device,
may provide cell-
handling capabilities for sorting and capturing a subpopulation of cells in a
format which allows
for subsequent stimulation and analysis. Such a system may allow for selection
of a single cell
from a population, the capture of this cell at any position within an
addressable array of
chambers, the application of one or more reaction conditions, and imaging of
each reaction
chamber. This functionality may provide an instrument for chemical genetics
studies of a
plurality of single cells. Examples of cell-based microfluidic assays are
described in, for
example PCT Publication WO 98/00231 and WO 98/45481. Cell-based microfluidic
assays may
be useful for screening of binding and/or internalization of cell ligands, for
example, receptor
ligands, drugs, cofactors and the like.
[00104] Particles within a microfluidic array according to some
embodiments of the
invention may present a solid or semi-solid surface for any of a variety of
linking chemistries,
allowing the incorporation of biological and chemical components of interest
into the particle
members of the arrays. A wide variety of organic and inorganic polymers, both
natural and
synthetic may be employed as the material for the solid surface. Illustrative
polymers include
polyethylene, polypropylene, poly(4-methylbutene), polystyrene,
polymethacrylate, poly(ethylene
terephthalate), rayon, nylon, poly(vinyl butyrate), polyvinylidene difluoride
(PVDF), silicones,
polyformaldehyde, cellulose, cellulose acetate, nitrocellulose, and the like.
A wide variety of
linking chemistries are available for linking molecules to a wide variety of
solid or semi-solid
particle support elements. One of skill in the relevant art will be aware of
and may easily select
appropriate chemistries, depending on the intended application.
[00105] Protein structural studies - A microfluidic device according to
some embodiments
of the invention may be useful for structural biology applications, for
example protein
crystallography. Methods and techniques useful for such applications are
described in, for
example Hansen & Quake, 2003. Current Opinion in Structural Biology 113:538-
544; Hansen et
al, 2006. J Am Chem Soc. 128:3142-3143; U.S.Patent no. 7,217,321.
[00106] The microfluidic devices according to some embodiments of the
invention may be
adapted for uses comprising screening other biological components as well,
including cells,
-25-
CA 02705213 2010-05-07
WO 2009/059430
PCT/CA2008/001985
antibodies, antibody ligands, proteins, peptides, and the like. The presence
or absence of such
cells, antibodies and antibody ligands are also known to correlate with
desirable or undesirable
features. A limiting dilution of a sample comprising may be applied to the
microfluidic device
and flowed into the reaction chambers, and the chambers isolated by a fluid
barrier as described.
Prior to application, the sample may be combined with a marker such as a
labeled antibody (e.g.
with a fluorescent tag) or similar marker. Following isolation, the individual
chambers may be
interrogated for the presence or absence of the biological component, by
screening for the
presence of the marker. Examples of screening assays, immunoassays, protein
identification
assays and the like are described in, for example U.S. Patent No. 6,632,655.
[00107] Sequencing, identification of polymorphisms - A microfluidic device
according to
some embodiments of the invention may be used for nucleic acid sequencing,
particularly
generation of sequencing libraries. The DNA to be sequenced (target nucleic
acid) is provided
with a polymerase and a primer and other necessary reagents, and is then
exposed to one type of
DNA base (A, C, T, or G) at a time in order to rapidly assay for base
incorporation. A variety of
techniques for determining the sequence of a nucleotide or nucleotides are
described infra. In
some embodiments of the invention, the microfluidic device of the invention
may be used for
sequencing nucleic acids. The devices of the invention optionally include
reagents (which may be
part of the array or flowed into contact with the array, e.g. in a reagent
train) for performing a
biological or chemical assay. The reaction mix for sequencing may comprise
some or all of
nucleotides, polymerases, dNTP, ddNTP, dNTP analogues, fluorescent dNTPõ or a
fluorescent
dNTP, inorganic phosphate, ATP, a thermostable polymerase, an endonuclease, an
exonuclease,
a phosphatase, an intercalating dye, a labeled probe, a reducing agent, Mg, a
molecular
crowding agent, e.g., PEG, a buffer, a salt, DTT, BSA, a detergent or
surfactant, chemicals to
inhibit or enhance electroosmotic flow (e.g., polyacrylamide) or the like.
[00108] Other methods of analysis of target nucleic acids are described
infra.
[00109] A target nucleic acid is a nucleic acid comprising one or more
sequences of
interest. The presence of a target nucleic acid in a sample or reaction
mixture may be detected,
and depending on the assay design, may also be quantified. A target nucleic
acid may be
obtained from a biological sample (a "sample").
[00110] A "polymorphism" is the occurrence of two or more forms of a target
nucleic acid
in a population. Polymorphic sites may be at known positions within a nucleic
acid sequence or
- 26 -
CA 02705213 2010-05-07
WO 2009/059430
PCT/CA2008/001985
may be determined to exist in a target nucleic acid using the methods
described herein. A
polymorphism may be alternately described as a sequence variation or sequence
variant; if
occurring in DNA, a 'DNA sequence variation'; if occurring in RNA, an 'RNA
sequence
variation'. A single nucleotide polymorphism, or SNP, is a polymorphism
consisting of a single
base change.
[00111] Tissue samples may be obtained by, for example, curettage,
needle aspiration
biopsy or needle (core) biopsy, incisional biopsy for sampling tissue, or
excisional biopsy, which
may entail total removal of the tissue of interest. Alternatively, other
bodily samples that contain
genetic material, may be used, for example hair, blood, plasma, serum, sputum,
urine, stool,
semen, amniotic fluid, chorionic villi or other fetal or embryonic tissue
using methods known in
the art.
[00112] DNA and RNA may be isolated from a biological sample, either
separately or
together, by any of a variety of methods known in the art. Choice of the
method may depend on
the nucleic acid to be assayed (DNA or RNA), the method used to assay, and the
like Methods
for the isolation of DNA and RNA from biological samples are known in the art,
for example
Sambrook J. et al. "Molecular Cloning", Cold Spring Harbor Laboratory Press
(1989) and
Ausubel, FM. et al., "Current Protocols in Molecular Biology", John Wiley &
Sons, Inc. (1994),
Botwell, DDL. Anal. Biochem. (1987) 162:463-465); U.S. Pat. # 5,130,423; U.S.
Pat. #
5,945,515; U.S. Pat. # 5,989,431; U.S. Pat. # 5,128,247. In some cases, such
as analysis of single
cells, it may not be necessary to purify DNA or RNA prior to analysis
[00113] Specific sequences of a target nucleic acid in a sample may be
amplified by any of
several methods, known in the art, for example Touchdown PCR, Reverse
Transcription
Polymerase Chain Reaction (RT-PCR), Polymerase Chain Reaction (PCR), Inverse
PCR,
Transcription Mediated Amplification (TMA), Nested PCR, Ligase chain reaction
(LCR),
Nucleic Acid Sequence Based Amplification (NASBA) and the like. See, for
example, Compton
J. 1991. Nature. 350:91-92; Malek et al., 1994. Methods Mol Biol 28:253-60;
Innis et al. (eds.)
PCR Protocols: A Guide to Methods and Applications, pp. 60-66, San Diego, CA,
Academic
Press, Inc., 1990; Roux K., 1994. Biotechniques 16:812-4; HeckerK and Roux,
L., 1996;
Biotecluliques 20:478-85; Ochman et al., 1988. Genetics 120:621-3; U.S. Patent
No. 5,299,491.
Selection of these parameters, methodologies and reagents, and methods of
optimizing them for
particular reaction conditions is within the ability of one skilled in the
art.
-27 -
CA 02705213 2010-05-07
WO 2009/059430
PCT/CA2008/001985
[00114] The high density reaction chamber arrays of the microfluidic
devices of the
present invention also allows analysis of multiple target nucleic acids, or
multiple samples, or
both, on a single device. For example, an array of 1,000,000 reactions can be
divided into 40
independent sections each comprised of 25,000 digital PCR chambers. Inclusion
of separate
injection ports that interface with independent sub-arrays allow for
implementation of multi-
sample analysis. In another example, a preloading strategy may be employed to
spatially
multiplex separate assays within defined regions of the array. Microvalves may
be used to
isolate sections of the array into which distinct primer and probe sets may be
dead-end loaded as
described. Flow channels may be subsequently be flushed with air to remove
excess assay mix
and the preloaded devices incubated (e.g. at about 70-80 C) to dehydrate the
reagents in the
preloaded assay mixes, leaving dried primers and probes in defined section.
Following this
dehydration step, a reaction mix comprising one or more target nucleic acids
and other reagents
necessary for PCR (but not primers and probes, which have been preloaded) is
injected into the
one or more sub-arrays of the microfluidic device as described above, and the
dehydrated
reagents are rehydrated. The flow channels are subsequently flushed to isolate
the reaction
chambers and the reactions thermally cycled. Amplification products may be
monitored during,
or following the amplification reaction.
[00115] In another example, two or more amplification products may be
detected and
quantified in a single reaction. This may be achieved by incorporating
multiple sequence-specific
fluorescent probes labeled with spectrally distinct fluorophores in a reaction
mix. Multiplexing
in this manner increases the number of target nucleic acids that may be
assayed in a single
sample and has an important role in quantitative analysis of one or more
target nucleic acids by
enabling the normalization of results by a reference nucleic acid.
[00116] Detection of the target nucleic acid, or the product of its
amplification is also
useful. A variety of strategies may be used with the microfluidics devices and
methods described
herein; selection of an appropriate system is also dependent on the
particulars of the reaction, the
compatibility of the detection system and reagents with the product of the
reaction conditions, the
scale of the number of reactions (for example, number, density of reaction
chambers), and
whether the target nucleic acid, or product of its amplification is monitored
during the assay, or
as an endpoint. Signal types to be detected may include those from
fluorophores, chromophores,
chemiluminescence, colorimetric reactions, radioactivity, substrates from
enzymatic reactions
and the like.
-28-
CA 02705213 2010-05-07
WO 2009/059430
PCT/CA2008/001985
[00117] Exemplary methodologies for detecting signals include confocal
laser scanning
microscopy, resonance energy transfer (RET), fluorescent resonance energy
transfer (FRET),
bioluminescent resonance energy transfer (BRET), scintillation detection,
fluorescence
correlation spectroscopy, light scattering, light absorption (UV or visible),
reflectivity and the
like.
[00118] The presence of a target nucleic acid or the product of its
amplification may be
determined by, for example, uptake of a dye specific for double-stranded
nucleic acids, or by the
hybridization of a portion of the amplified nucleic acid to a probe.
[00119] DNA-staining dyes that fluoresce only when bound to double-
stranded nucleic
acids may be useful for detection of amplification products. Examples of such
dyes include
SYBR Green I, II, SYBR Gold, YO (Oxazole Yellow), TO (Thiazole Orange),
PicoGreen (PG)
(available from Molecular Probes, Invitrogen), ethidium bromide, propidium
iodide, Hoechst
33258, Hoechst 33342, DAPI and the like. Further discussion relating to the
uses of DNA-
staining dyes may be found in, for example, Glazer et al., 1997. Curr Opin
Biotechnol 8:94-102;
and Glazer, AN and Rye, HS. 1992. Nature 359:859-61, Higuchi et al.,
BioTechnology 10:413-
417.
[00120] A "molecular beacon" is a single stranded oligonucleotide
that, unless hybridized
to a target nucleic acid, will exist in a hairpin configuration. A first end
of the oligonucleotide
has attached a fluorescent dye, and a second end has attached a quencher
molecule. When in the
hairpin configuration, the fluorescence is quenched by the proximity of the
quencher molecule
and no fluorescence is observed. Once the oligonucleotide 'beacon' hybridizes
to the target
nucleic acid, the fluorescent dye is sufficiently separated from the quencher
molecule, and
fluorescence is detectable. By monitoring emission changes in the dye,
indirect monitoring of
the amplification product is possible. Further details sufficient to guide one
of skill in the art
may be found in, for example, PCT Publication WO 95/13399, Tyagi S et al.
1998. Nat
Biotechnol 1:49-53; Tyagi S and Kramer FR. 1996 Nat Biotechnol 3:303-8; and
Marras SA et
al., 1999. Genet Anal 5-6:151-6.
[00121] Fluorescence energy resonance transfer (FRET) is a distance-
dependent
interaction between a donor fluorophore and acceptor fluorophore pair,
selected so that the
emission spectrum of the donor overlaps with the excitation spectrum of the
acceptor. When the
fluorophores are brought within sufficient proximity, excitation of the donor
fluorophore by a
- 29 -
CA 02705213 2010-05-07
WO 2009/059430
PCT/CA2008/001985
first defined wavelength is transferred from the donor to the acceptor, and
fluorescence at a
second defined wavelength may be detected. For detection of nucleic acid
hybridization, a
specific probe is labeled with a one member of the donor/acceptor pair, and a
nucleotide is
labeled with the other member of the donor/acceptor pair. When free in
solution (for example,
before the nucleic acid polymerization, or hybridization occurs), the
donor/acceptor pair are
sufficient distant to prevent the energy transfer. As the polymerization
reaction proceeds, labeled
nucleotides will be incorporated into the molecule sufficiently close to the
labeled primer so that
the energy transfer may take place, and the fluorescence detected. Examples of
methods that may
be used to detect polymorphisms in a target nucleic acid are described in, for
example, U.S.
Patent No. 6,500,650, U.S. Patent No. 5,945,283 U.S. Publication 2004/0005613
and WO
97/22719.
[00122] Real-time quantitative PCR may be used to determine the
quantity of a target
nucleic acid sequence in a sample by measuring the amount of amplification
product formed
during and/or after the amplification. The commercially available TaqManTm
assay (Applied
Biosystems) is based on the 5' nuclease activity of Taq polymerase that
displaces and cleaves the
oligonucleotide probes hybridized to the target DNA generating a fluorescent
signal. It is
necessary to have two probes that differ at the polymorphic site wherein one
probe is
complementary to the 'normal' sequence and the other to the mutation of
interest. These probes
have different fluorescent dyes attached to the 5' end and a quencher attached
to the 3' end when
the probes are intact the quencher interacts with the fluorophor by
fluorescence resonance energy
transfer (FRET) to quench the fluorescence of the probe. During the PCR
annealing step the
hybridization probes hybridize to target DNA. In the extension step the 5'
fluorescent dye is
cleaved by the 5' nuclease activity of Taq polymerase, leading to an increase
in fluorescence of
the reporter dye. Mismatched probes are displaced without fragmentation. The
presence of a
mutation in a sample is determined by measuring the signal intensity of the
two different dyes.
See also U.S. Paten t No. 5,210,015, 5,487,972.
[00123] Invasive cleavage methods employ an oligonucleotide called an
InvaderTM probe
and sequence-specific probes to anneal to the target DNA with an overlap of
one nucleotide.
When the sequence-specific probe is complementary to the polymorphic base,
overlaps of the 3'
end of the invader oligonucleotide form a structure that is recognized and
cleaved by a Flap
endonuclease releasing the 5' arm of the allele specific probe. Further
details sufficient to guide
- 30 -
CA 02705213 2010-05-07
WO 2009/059430
PCT/CA2008/001985
one of ordinary skill in the art is provided by, for example, Neri B. et al.
2000. Advances in
Nucleic Acid and Protein Analysis 3826:117-125, U.S. Patent No. 6,964,848.
[00124] Assays comprising the use of a Scorpion probe system may also
be useful. Such
probes and systems are described in, for example Nucleic Acids Research 2000
28:3752-3761
and PCT Publication WO 99/66071.
[00125] Primer extension reactions (i.e. mini sequencing, nucleotide-
specific extensions,
or simple PCR amplification) may also be useful in sequence discrimination
reactions to identify
a polymorphism in a target nucleic acid. For example, in a mini sequencing
reaction, a primer
anneals to its target nucleic acid (usually DNA) immediately upstream of the
SNP and is
extended with a single nucleotide complementary to the polymorphic site. Where
the nucleotide
is not complementary, no extension occurs.
[00126] Oligonucleotide ligation assays (OLA) require two sequence-
specific probes and
one common ligation probe per SNP. The common ligation probe hybridizes
adjacent to a
sequence-specific probe and when there is a perfect match of the appropriate
sequence-specific
probe, the ligase joins both the sequence-specific and the common probes.
Where there is not a
perfect match the ligase is unable to join the sequence-specific and common
probes. The
annealing of the probes may be detected by, for example, an enzyme immunoassay
(Villahermosa
ML. J Hum Virol (2001) 4(5):238-48; Romppanen EL. Scand J Clin Lab Invest
(2001)
61(2):123-9; Iannone MA. et al. Cytometry (2000) 39(2):131-40).
[00127] Ligation-Rolling Circle Amplification (L-RCA) has also been
successfully used
for genotyping single nucleotide polymorphisms as described in Qi X. et al.
Nucleic Acids Res
(2001) 29(22):E116.
[00128] Sanger sequencing (Sanger et al., 1977 PNAS 74:5463-5467)
employs a DNA
polymerase to synthesize sequence-dependent fragments of various lengths. The
lengths of the
fragments are determined by the random incorporation of dideoxynucleotide base-
specific
terminators. These fragments can then be separated in a gel, visualized, and
the sequence
determined. Numerous improvements have been made to refine the above methods
and to
automate the sequencing procedures. Similarly, RNA sequencing methods are also
known, see,
for example Zimmern D. and Kaesberg P. Proc. Natl. Acad. Sci. USA (1978)
75(9):4257-4261)
and Mills DR. and Kramer FR. (Proc. Natl. Acad. Sci. USA (1979) 76(5):2232-
2235) Direct
-31-
CA 02705213 2010-05-07
WO 2009/059430
PCT/CA2008/001985
chemical methods for sequencing RNA are also known (Peattie DA. Proc. Natl.
Acad. Sci. USA
(1979) 76(4):1760-1764). Other methods include those of Donis-Keller et al.
(1977, Nucl. Acids
Res. 4:2527-2538), Simoncsits A. et al. (Nature (1977) 269(5631):833-836),
Axelrod VD. et al.
(Nucl. Acids Res.(1978) 5(10):3549-3563), and Kramer FR. and Mills DR. (Proc.
Natl. Acad.
Sci. USA (1978) 75(11):5334-5338).
[00129] Nucleic acid sequences can also be read by stimulating the
natural fluoresce of a
cleaved nucleotide with a laser while the single nucleotide is contained in a
fluorescence
enhancing matrix (U.S. Pat. # 5,674,743); In a mini sequencing reaction, a
primer that anneals to
target DNA adjacent to a SNP is extended by DNA polymerase with a single
nucleotide that is
complementary to the polymorphic site. This method is based on the high
accuracy of nucleotide
incorporation by DNA polyrnerases. There are different technologies for
analyzing the primer
extension products. For example, the use of labeled or unlabeled nucleotides,
ddNTP combined
with dNTP or only ddNTP in the mini sequencing reaction depends on the method
chosen for
detecting the products.
[00130] Template-directed methods ¨ In one example, a template-directed dye-
terminator
incorporation with fluorescent polarization-detection (TDI-FP) method is
described by Freeman
BD. et al. (J Mol Diagnostics (2002) 4(4):209-215) for large scale screening;
5' nuclease assay
may also be used for genotyping single nucleotide polymorphisms (Aydin A. et
al. Biotechniques
(2001) (4):920-2, 924, 926-8.); Polymerase proofreading methods may be used to
determine
SNPs identities as described in WO 0181631; Detection of single base pair DNA
mutations by
enzyme-amplified electronic transduction is described in Patolsky F et al. Nat
Biotech. (2001)
19(3):253-257.
[00131] Sequence-specific PCR methods have also been successfully used
for genotyping
single nucleotide polymorphisms (Hawkins JR. et al. Hum Mutat (2002) 19(5):543-
553).
Alternatively, a Single-Stranded Conformational Polymorphism (SSCP) assay or a
Cleavase
Fragment Length Polymorphism (CFLP) assay may be used to detect polymorphisms
as
described herein.
[00132] Applications of microfluidic devices
[00133] Microfluidic devices comprising arrays of picoliter or
femtoliter reaction
chambers as described are useful for biological assays and diagnostics. For
example, digital
- 32 -
CA 02705213 2010-05-07
WO 2009/059430
PCT/CA2008/001985
quantification of nucleic acids using hundreds of thousands, to millions, of
reactions provides
unique capabilities in 1) the discrimination of very small allelic imbalances
with high statistical
power, 2) the co-localization of target sequences on a single molecule over a
long genomic
distance and 3) the detection of rare events in a highly homologous genetic
background.
[00134] The dynamic range of a digital PCR assay may be described as the
range of
concentrations over which the number of expected positive reactions increases
linearly with
template concentration. Without wishing to be bound by theory, the
distribution of molecules
throughout the array is random, so that a linear response is observed until
the mean occupation of
each chamber approaches one, resulting in saturation. The dynamic range of a
digital array
therefore scales with the total number of chambers. Picolitre volume
compartmentalization
provides an effective enhancement in concentration that may be used to detect
rare sequences
within a large background of high sequence homology. To illustrate this point
consider the task
of detecting 10 copies of a single nucleotide polymorphism (SNP) in a
background of wild-type
at a relative concentration of 1 copy per million (10 copies of the SNP in a
background of 10
million wild type). In such a situation even a minute amount of non-specific
amplification of the
wild type sequence may result a false positive, making detection exceedingly
difficult or
impossible by any PCR method. However, the partitioning of this sample into
1,000,000
chambers results in an average of 10 copies of the wild-type sequence per
reaction chamber. The
majority of these reaction chambers will have none of the target sequence but
exactly 10 reaction
chambers (with high probability) will contain a single copy of the SNP. In
such a situation the
background may be reduced by a factor of one million and detection of the SNP
may be achieved
with an assay capable of discriminating only 1 in ten molecules. Figure 4
shows the volume
dependence of the required relative efficiencies of the specific and non-
specific reactions
required to detect a single molecule in a background of 1 million molecules
having high
sequence homology. For example, with a 30 cycle PCR reaction and a required
signal to noise
ratio of 10, as defined by the ratio of specific to non-specific amplicons,
the required relative
reaction efficiency in a 1 L reactor is approximately 6x. By comparison, if
this same sample is
divided into 100,000 reactors having volumes of 10 pL the requisite ratio of
reaction efficiencies
would be 1.4.
[00135] By reducing the reaction volume to a few picolitres and dispersing
the nucleic
acid in the sample to be present at an average of less than one per reaction,
a high effective
- 33 -
CA 02705213 2010-05-07
WO 2009/059430
PCT/CA2008/001985
concentration is provided. This also has the advantage of reducing non-
specific amplification and
competitive reactions, which would otherwise increase the background.
[00136] Digital PCR provides for the ability to use optically
multiplexed PCR to measure
the frequency of genetic rearrangements that result in two target sequences
being present on the
same DNA molecule. As molecules are loaded into multiple partitions they
distribute themselves
randomly and independently. If the sample contains a rare subset of molecules
that harbour two
target sequences, the co-localization of amplification within the reaction
chambers will be higher
than expected by chance, indicating the presence of this species. The
statistical power of this
analysis improves with the total number of chambers so that in the case of
very large arrays it is
possible to detect minority species that posses both target sequences
independent of the exact
fusion site. This capability should allow for the detection of a variety of
rare genetic
rearrangements including gene fusions, transpositions, alternative splicing,
and inversions. For
example, such an approach may be used for the detection of variants of the
well-characterized
BCR/ABL fusion oncogene using a single two-colour multiplexed assay. The
fusion of BCR on
22q11 with ABL on 9q34 is a well-known chromosomal aberration that is present
in 95% of cases
of chronic myeloid leukemia (CML) and in approximately 25% of cases of acute
lymphoblastic
leukemia (ALL) in adults and approximately 2-5% of ALL cases in children
(Burmeister et al.,
2000. Leukemia 14:1850). The detection of common fusion transcripts by RT-qPCR
is routinely
used as the gold standard for diagnosis and prognosis. However, the large
number of possible
breakpoints creates difficulties in designing primers that detect all
transcript variants due to the
prohibitively long amplicons required to bridge internal exons, rule out the
use of a single set of
primers. In the vast majority of fusions, exons El (BCR) and All (ABL) flank
the fusion site.
A multiplexed assay in which two primer sets were used to independently detect
exons El and
All using FRET probes reporting in two separate colours (FAM and Cal Orange)
may be used to
test for the frequency of co-localization in samples of genomic DNA spiked
with serial dilutions
of cloned cDNA of cells comprising a BCR/ABL fusion (e.g. chronic mylogenous
leukemia
primary cells, or a cell line having a similar fusion), to determine the
minimum fraction of
fusions that give rise to statistically significant enrichment of co-
localization.
[00137] It will be appreciated by a person of skill in the art that
the numerical designations
of the positions of a polymorphism within a sequence are relative to the
specific sequence. Also
the same positions may be assigned different numerical designations depending
on the way in
which the sequence is numbered and the sequence chosen. Furthermore, sequence
variations
- 34 -
CA 02705213 2010-05-07
WO 2009/059430 PCT/CA2008/001985
such as insertions or deletions, may change the relative position and
subsequently the numerical
designations of particular nucleotides at and around a mutational site.
[001381 Table 1: Sequences
SEQ Sequence Description
ID
NO:
1 CCGGTGCCACCTGTGGTCCACCTGACCCTCCGCCAGGCC Response curve
GGCGACGACTTCTCCCGCCGCTACCGCCGCGACTTCGCC template
GAGATGTCCAGCCAGCTGC (BCL2)
2 GCCACCTGTGGTCCACCT Response curve
primer
3 TGGACATCTCGGCGAAGTCG response curve
primer
4 FAM-CGACGACTTCTCCCGCCGCT-BHQ FAM-BHQ
labeled probe
CACATTTGGAGGGCACAAAAGTGAGAAGCCGGCTCTGC human ABL11
CTCGGAAGAGGGCAGGGGAGAACAGGTCTGACCAGGTG template
ACCCGAGGCACAGTAACGCCTCCCCCCAGGCTGGTGAA
AAAGAATGAGGAAGCTGCTGATGAGGTCTTCAAAGACA
TCATGGAGTCCAGCCCGGGCTCCAGCCCGCCCAACCTGA
CTCCAAAACCCCTCCGGC
6 ATTTGGAGGGCACAAAAGTG ABL11 primer
7 GGGGGAGGCGTTACTGTG ABL11 primer
8 ACAGGTCTGACCAGGTGACC CalOrange
labeled probe
9 ATCGAGCAGGAGCTGGAG AIRE primer
TGCCGGTCATAGCTCTTCTT AIRE primer
11 GCTCCAGG AIRE LNA
probe 1
12 ATTTGGAGGGCACAAAAGTG ABL primer
13 AGGGGTTTTGGAGTCAGGTT ABL primer
14 ACAGGTCTGACCAGGTGACC CalOrange
labelled probe
for ABL
<none>
16 <none>
17 AAGCTTTCTCACAAGCATTTGGTTT forward JAK
primer
- 35 -
CA 02705213 2010-05-07
WO 2009/059430
PCT/CA2008/001985
18 AGAAAGGCATTAGAAAGCCTGTAGTT reverse JAK
primer
19 TCTCCACAGACACATAC wild-type
JAK
probe (VIC
label)
20 TCCACAGAAACATAC JAK probe
for
polymorphism
(FAM label)
21 TCCTCAGAACGTTGATGGCAG Jak2fwd
22 ATTGCTTTCCTTTTTCACAAGAT Jalc2rev
Experimental methods
[00139] Chip Fabrication: The microfluidic polydimethylsiloxane (PDMS)
devices were
fabricated using standard multilayer soft lithography. Standard lithography is
used to pattern the
photo resist by utilizing 20,000 dpi transparency masks (CAD/Art Services).
Chroma masks were
generated for the production of the MegaPixel mold (University of Alberta
Nanofab). Negative
molds were made using soft lithography SU8-2025 was used for the control layer
yielding a
channel thickness of 25 micrometers, while SU8-100 was used to create the
cubical reaction
chambers. Channels 10 urn high connect the reaction chambers, fabricated with
SU8-5. SPR-220
was spun to achieve a thickness of 10 urn was used to create the section of
the flow lines that was
intersected by valves (Unger M.A. et al., 2000. Science 288:113).
[00140] Briefly, a layer of elastomer (5:1 GE RTV) was spin-coated
onto a mold
(250rpm x lmin) and partially hardened. A vapor barrier (IPA cleaned
transparency) was
positioned over the area of the reaction chambers and covered with a ¨7 mm
layer of elastomer
(5:1 GE RTV) and degassed, and baked for 90 mm at 80 C to harden the
elastomer, and allowed
to cool for 1 hr. The hardened layer of reaction chambers was peeled of the
mold and aligned to
a control layer of elastomer control layer (20:1 GE RTV previously spin-coated
onto a support
matrix - 500 RPM x 30 sec + 1875 RPM x 90sec, followed by baking for 45min at
80 C). The
two layers were baked for 1 hour at 80 C to bind, cooled and peeled from the
support matrix.
Ports were punched in the device, and the device mounted to a base layer (20:1
GE RTV, spin-
coated as described). The final assembly was baked overnight at 80 C to harden
the elastomer.
[00141] The devices are composed of two layers of PDMS bonded to a
silicon wafer, with
a push up geometry. The flow layer was obtained by first spinning a layer of
PDMS (5:1 RTV
A:13) at 820 RPM. Polyethylene transparency slides for water vapor control is
then positioned on
-36-
CA 02705213 2010-05-07
WO 2009/059430
PCT/CA2008/001985
top of the relevant features and about 40 g of PDMS are poured on top of the
entire silicon wafer,
which is later baked for one hour. For the control layer the wafer is coated
with PDMS (10:1
RTV A:B) and spun at 1800 RPM and successively baked for 45 minutes. Control
and flow
layers are then aligned and baked together.
[00142] Chip Operation: The sample to be analyzed is first mixed with all
reagents
required for the reaction (primers, probes, dNTPs, polymerase, MgC12) and is
then injected into a
microfluidic channel structure featuring the large array of lithographically
defined chambers. The
PCR reaction chambers are firstly pressure filled with PCR reaction mix. The
main flow channel
is then pressurized with oil.
[00143] The geometry of the chambers ensures high surface tension and
requires high
pressure applied (about 10 PSI) for the fluid to enter. The geometry helps
later
compartmentalization as after the chambers are filled with aqueous fluid, the
pressurization of
the flow line results in displacement of the solution by oil only in the
connecting channels but not
in the chambers themselves.
[00144] PCR Assay and Analysis: Unless otherwise noted, all PCR reactions
performed
using the microfluidic devices of the present invention were run with ABI TAQ
FAST master
mix, following manufacturer's instructions, with a concentration of probe at
500 nM for FAM
probes and 250 nM for Cal Orange probes. A surfactant (0.1% TWEEN-20) was
added to the
reaction mix. Each primer was at a concentration of 750 nM. The thermocycling
protocol
included a 20 second hot start and 40 cycles of 95 degrees for 1 second and 60
degrees for 30
seconds. Thermal cycling and imaging were performed using a BioMark TM System
(Fluidigm).
End point measurements were obtained and the images were analyzed using
Matlab. The
template (target DNA) comprised a portion of the human BCL2 gene according to
SEQ ID NO:
1, synthesized and PAGE purified by IDT (Integrated DNA Technologies). This
template had
previously demonstrated 100% PCR efficiency by Real Time PCR. The primers used
are
GCCACCTGTGGTCCACCT (SEQ ID NO: 2) and TGGACATCTCGGCGAAGTCG (SEQ ID
NO: 3) and the probe FAM-CGACGACTTCTCCCGCCGCT-BHQ (SEQ ID NO: 4), all
synthesized from IDT.
[00145] To monitor device-to-device variation a control was used in a
different color assay
as follows. A template from the human ABL11 gene (SEQ ID NO: 5) was
synthesized and
PAGE purified by IDT. The following primers were synthesized from IDT:
-37-
CA 02705213 2010-05-07
WO 2009/059430
PCT/CA2008/001985
ATTTGGAGGGCACAAAAGTG (SEQ ID NO: 6) and GGGGGAGGCGTTACTGTG (SEQ ID
NO: 7). The following probe in CalOrange was synthesized by Biosearch
Technologies:
ACAGGTCTGACCAGGTGACC (SEQ ID NO: 8).
[00146] Trisomy 21 differentiation: A region on the AIRE gene on
chromosome 21 was
targeted with the following primers and probe: ATCGAGCAGGAGCTGGAG (SEQ ID NO:
9),
TGCCGGTCATAGCTCTTCTT (SEQ ID NO: 10) and LNA probe 1 (GCTCCAGG) (SEQ ID
NO: 11) from Roche Diagnostics.
[00147] The ABL gene on chromosome 9 was targeted with the following
primers and
probe: ATTTGGAGGGCACAAAAGTG (SEQ ID NO: 12), AGGGGTTTTGGAGTCAGGTT
(SEQ ID NO: 13)(synthesized by IDT) and CalOrange-ACAGGTCTGACCAGGTGACC-BHQ1
(SEQ ID NO: 14) from Biosearch Technologies. Genomic DNA was spiked with 6% of
pure
trisomy 21 DNA, obtained from the Child and Family Research Institute,
Vancouver, BC. The
final DNA sample used in the digital PCR reaction was set to obtain about 20%
fill factor. A
device with 90,000 chambers was used for this application.
[00148] JAK2 Assay:
[00149] Jak2 wildtype and mutant plasmids were created from amplicons
derived from a
heterozygous patient. Briefly, a 453bp target was amplified using primers,
Jak2fwd
TCCTCAGAACGTTGATGGCAG (SEQ ID NO: 21), and Jak2rev
ATTGCTTTCCTTTTTCACAAGAT (SEQ ID NO: 22). The conditions for amplification were
50 C annealing temperature at 3.5mM MgC12 concentration starting with 10Ong of
genomic
DNA. PCR products were run on a 7% polyacrylamide get to verify correct size
fragments.
Subcloning was achieved using a TOPO TA subcloning kit (Invitrogen), following
manufacturer's instructions. Briefly, one ul of PCR product was combined with
lul of buffer
solution and lul of linearized, activated vector and 3u1 of dH20 incubated at
room temperature
for 5 minutes and then placed on ice. Four p,1 of the cloning reaction was
mixed with one aliquot
of comptent cells provided with the kit and incubated on ice for 5 minutes.
Fifty IA of the
transfection reaction was plated on a LB ampicillin plate and grown overnight
at 37 C. Ten
single colonies were picked into LB amp media for overnight growth and
subsequent DNA
isolation.
- 38 -
CA 02705213 2010-05-07
WO 2009/059430
PCT/CA2008/001985
[00150] Plasmid DNA was isolated using Qiagen Midi plasmid
purification kit. DNA
from the ten single colonies were individually test in a Taqman based allelic
discrimination assay
(Applied Biosystems). One each of a Jak2 wildtype (plasmid A) and Jak2 mutant
(plasmidB)
were further isolated for on going studies.
[00151] The plasmids were used as template with a constant background of
wildtype (wt
or WT) and increasing amounts of mutant (mt or MT) (from 1:1 wt:mt to 1:1000).
Primers and
probes at a final concentration of 750 nM were used in the reaction, with the
following
sequences:
Forward primer AAGCTTTCTCACAAGCATTTGGTTT (SEQ ID NO: 17)
Reverse primer: AGAAAGGCATTAGAAAGCCTGTAGTT (SEQ ID NO: 18)
Probes (MGB)
WT TCTCCACAGACACATAC (VIC) (SEQ ID NO: 19)
MT TCCACAGAAACATAC (FAM) (SEQ ID NO: 20)
[00152] The concentration of wildtype was about 0.5 plastnids per
chamber. A device with
90,000 chambers and five inlet lines was used for this application. 40 cycles
of PCR were applied
and the number of positive chambers was counted by using a Matlab program.
Example 1:
Amplification of target nucleic acid in a large scale microfluidic emulsion
array
[00153] A device incorporating selected coverage by a vapour barrier was
used to
demonstrate the effects of the evaporation during a PCR experiment. A
microfluidic device
having the chamber geometry illustrated in Figure 2d with a chamber density of
1chamber/1600um2 was designed with 5 separate chamber arrays in order to test
different
samples on the same chip. The PCR solution was introduced into the device and
compartmentalized as outlined in Figure 3. A standard TaqMan assay for GAPDH
along with
TaqMan mastermix (Applied Biosystems) was used to monitor the progress of the
reaction with a
concentration of template DNA of about 10 copies per reaction chamber. The
reaction was
allowed to progress over 40 cycles and the end point picture is shown in
Figure 5.
- 39 -
CA 02705213 2010-05-07
WO 2009/059430
PCT/CA2008/001985
[00154] The photomicrograph of Figure 5 shows the results of one
experiment. Reaction
chambers with positive controls covered by the vapor barrier (positive control
regions A and D)
were successful in amplification of the target nucleic acid, while reaction
chambers with positive
controls outside the region covered by the vapor barrier (a portion of
Positive control region E)
were not successful in amplification.
[00155] By incorporating a vapor barrier into the microfluidic device,
we have
demonstrated reliable single molecule amplification in arrays of up to 90,000
reaction chambers,
having volumes of about 30 picolitres.
Example 2
Single-copy amplification of target DNA
[00156] By diluting the initial DNA (target nucleic acid) such that
each reaction chamber
contains only one molecule of DNA or none, it was possible to quantitatively
identify the initial
amount of DNA present in the sample. Only a reaction chamber with a copy of
the target nucleic
acid will provide a fluorescent signal for detection, as each chamber that has
an optical signal
indicates the presence of a target nucleic acid prior to PCR amplification.
[00157] As described, the microfluidic chip was loaded by connecting
the fluid lines to a
pressurized reservoir of PCR solution containing diluted DNA. After all the
chambers were
filled, FluorinertTM oil was pressurized in the main flow lines thereby
displacing the fluid
everywhere except in the chambers, and the chip ready to be thermocycled. As
the PCR reaction
proceeds, images of the chip were taken at each cycle. It is therefore
possible to see the DNA
product (by means of the fluorescent signal) increasing through each cycle.
This is demonstrated
in Figure 6 .The reaction chamber was 30 micrometers per side. As the cycle
number advances,
signal from the negative control reaction chambers remains at background,
while the signal from
positive controls increases exponentially.
[00158] We have demonstrated that, by running digital PCR on a chip with a
million
reaction chambers it is possible to count DNA molecules very accurately due to
the highly
increased dynamic range. In particular this type of analysis is essential for
counting rare gene
expression products against a known control. Due to the possibility of using
several different
probes it is possible to quantitatively identify the expression profile over
several genes.
- 40 -
CA 02705213 2010-05-07
WO 2009/059430
PCT/CA2008/001985
Example 3:
Discrimination of small allelic imbalances: Non-Invasive Detection of Fetal
Trisomy 21
[00159] Minor imbalances in chromosome number were detected using
digital PCR
techniques employing the microfluidic arrays described herein. In order to
determine a 1.5%
increase in C21 in a sample, approximately 900,000 PCR reactions were required
for 5 sigma
certainty:
12c21 nC15 = = k-o- = k\ln,21 e = 1.5%
nc2i = NchambP(1+ 6) nc21 =m(2 + 6)
p(1 + s)
Chamb
k = ___________________ =5 => N = 895 570chambers
2+e
[00160] M is the number of genomic equivalents (1 genome set = two of
each
chromosome). The sampling noise sigma is the square root of the number of
chromosome 21. As
discussed, if conventional PCR techniques are used, this number of reactions
is prohibitively
large and effectively impossible. Use of a microfluidics array comprising 106
reaction chambers
condenses this number of reactions into a scale that is possible and
effective.
[00161] Copy number of chromosomes 21 (C21) and 9 (C9) were determined
using digital
detection of the AIRE gene and ABL genes known to be present at single copy on
chromosomes
21 and 9 respectively. Measurements over 90,000 digital chambers of samples
spiked with 6%
trisomy 21 DNA, displayed the expected 3% enrichment of the AIRE gene when
normalized by
counts obtained in normal genomic samples (Figure 7). These measurements
represent the
highest discriminatory power of any PCR technology reported to date and may be
extended in a
straight forward manner to the detection of differences below 1%.
Interestingly, measurements
of AIRE and ABL in normal genomic DNA displayed consistent differences in the
absolute
number of positive reaction chambers.
- 41 -
CA 02705213 2010-05-07
WO 2009/059430
PCT/CA2008/001985
Example 4
Detection of rare sequences in large homologous background: JAK2 assay
[00162] As a proof of concept plasmid samples containing SNP mutations
in the Jak2 gene
(V617F) were analyzed. The V617F mutation is specific for several
myeloproliferative
disorders, such as polycythemia vera, essential thrombocythemia, idiopathic
myelofibrosis and
several leukemias. Plasmids containing the wild-type Jak2 gene or the V617F
variant were tested
with probes specific for the SNP region while using common primers. A probe in
FAM was used
to test the SNP while a probe in VIC was used to the V617F mutant. Samples
containing
different amounts of mutant over a constant wild type background were tested,
and the assays
were able to detect ratios as small as 1:1000 mutant to wildtype, compared to
a minimum of
5:100 as reported by literature (Bosdquet et al., 2006. Hum Pathol 37:1458).
These results are
shown in figure 8A,B.
[00163] Picolitre volume compartmentalization provided an effective
enhancement in
concentration that may be used to detect rare sequences within a large
background of high
sequence homology. The detection of a few copies of a single nucleotide
polymorphism (SNP) in
a background of wild-type at a relative concentration of 1 copy per thousand
(1 copy of the SNP
in a background of 1000 wild type) is not easily detected with non-digital
techniques. In such a
situation, even a minute amount of non-specific amplification of the wild type
sequence will
result a false positive, making detection exceedingly difficult or impossible
by any PCR method.
However, the partitioning of this sample into 100,000 chambers resulted in an
average of 100
copies of the wild-type sequence per reaction chamber. The majority of these
reaction chambers
had none of the target nucleic acid, but 100 reaction chambers (with high
probability) contained
a single copy of the target nucleic acid comprising the SNP. In such a
situation the background is
thus reduced by 4 orders of magnitude, and detection of the SNP may be
achieved with an assay
capable of discriminating only 1 in ten molecules.
Example 5
Application of microfluidic arrays to generation of sequencing libraries
[00164] Sequencing libraries may be generated for use with automated
sequencing
machines or systems that rely on sequencing by extension or sequencing by
ligation. A key step
- 42 -
CA 02705213 2015-08-21
in these sequencing technologies is the localized amplification of single
molecules prior to
sequencing. In this step single molecules of the DNA template to be sequenced
may be isolated
from their neighbours and subject to amplification using polymerase chain
reaction or rolling
cycle amplification. As an example, a microfluidic device according to the
present invention
may be loaded with micron scale beads at a concentration chosen to give
several beads per
chamber. A device comprising about 10,000,000 reaction chambers may be loaded
with a
suspension of beads in a PCR reaction mix comprising target nucleic acid at a
concentration of
about 0.1 to about 0.5. A sufficient quantity of beads is provided to average
about 5 beads per
chamber. Based on Poisson statistics this loading would result in
approximately 1,000,000
chambers each having sample in them with approximately 95% of these containing
single
templates, and nearly all of them having at least one bead (average of 5).
After amplification the
beads may be removed from the chambers. Examples of methods for removing the
beads from
the chambers include a wash step or peeling the microfluidic device off the
support (e.g glass).
The beads may be recovered for sequencing. This would in a library of
approximately
5,000,000 beads, of which about 95% are attached to a unique sequence,
representing a total
sample size of approximately 1,000,000 molecules. The size of these libraries
may further be
increased by the fabrication of devices having more chambers or by the use of
multiple devices.
[00165] One or more currently preferred embodiments have been described by
way of
example. It will be apparent to persons skilled in the art that a number of
variations and
modifications can be made without departing from the scope of the invention as
defined in the
claims.
- 43 -