Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02740747 2011-05-20
4459 1012-001 CA - Application.docx PATENT APPLICATION
AOCRLS/1b(jt) Docket No. 4459.1012-001
May 19, 2011
-1-
CHARGED CONJUGATED POLYELECTROLYTES WITH
APTAMER-FUNCTIONALIZED SILICA NANOPARTICLES
BACKGROUND OF THE INVENTION
Protein detection and quantification are of vital importance in both basic
discovery research and clinical diagnosis. Enzyme immunosorbent assay (ELISA)
is a
widely used immunoassay and requires antibodies to be immobilized on a
substrate to
capture antigens and the secondary antibodies. Despite its high sensitivity,
ELISA
requires tedious protein modification and is limited by the availability of
commercial
antibodies. Although alternative assays have been developed for protein
detection using
aptamers as the recognition elements, most of these assays require the
modification of
aptamers with fluorescent dyes or other reporter groups, which are expensive
and can
impair the original affinity and specificity of the aptamer toward target
proteins.
Furthermore, the fluorescence signal of these dyes can be greatly affected by
proteins in
biological media.
Therefore, there remains a need for CPE-based assays that can be used for real-
sample detection (i.e., target detection in mixed protein samples and/or in
biological
media).
SUMMARY OF THE INVENTION
One embodiment of the invention is a compound of Structural Formula (I),
wherein the values and alternative values for the variables are as defined in
the Detailed
Description of the Invention.
Another embodiment of the invention is a method of detecting a target in a
sample, comprising functionalizing a solid support with a ligand; incubating
the ligand-
functionalized solid support with a sample; incubating the sample with a
charged
1092269.1
CA 02740747 2011-05-20
4459.1012-000
-2-
conjugated polyelectrolyte (CPE) or charged conjugated oligoelectrolyte (COE);
and
detecting the fluorescence of the solid support, thereby detecting the target.
Yet another embodiment of the invention is a method of detecting a target in a
sample, comprising functionalizing a surface of a solid support with a charged
ligand,
thereby creating a charge on the surface of the solid support; incubating the
ligand-
functionalized solid support with a sample, whereupon binding of the target,
the charge
on the surface of the solid support switches; incubating the sample with a
conjugated
polyelectrolyte (CPE) or a conjugated oligoelectrolyte (COE) that has a
complementary
charge to the charge of the target-bound surface; and detecting the
fluorescence of the
solid support, thereby detecting the target.
The compounds of the invention possess high photoluminescence quantum
yields in biological media, low cytotoxicity, and excellent environmental
stability and
photostability, and can be used in biosensor and bioimaging applications.
BRIEF DESCRIPTION OF THE DRAWINGS
The foregoing will be apparent from the following more particular description
of
example embodiments of the invention, as illustrated in the accompanying
drawings.
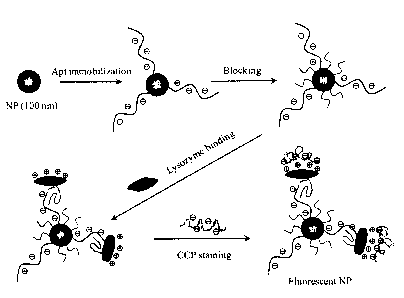
FIG. 1 is a schematic illustration of CPE-based, label-free protein detection.
FIG. 2 is an absorbance spectrum of PFVSO3 in water at [RU] = 4 M
(excitation at 428 nm).
FIG. 3 is a graph depicting the photoluminescence intensity (triangle) and
percentage of unbound lysozyme (square) as a function of surface density of
aptamers
on silica nanoparticle (NP) surface.
FIG. 4 is a photoluminescence (PL) spectrum of polymer-stained NPs incubated
with (a) 20 g/mL lysozyme; (b) a mixture of 20 gg/mL each for BSA, thrombin,
and
trypsin; or (c) a mixture of (a) and (b) followed by subsequent staining with
1 M
PFVSO3Na in 15 mM PBS at pH = 7.4 (excitation at 428 nm).
FIG. 5 is a PL spectra of polymer-stained NPs incubated with increasing
concentrations of lysozyme in 15 mM PBS at pH = 7.4 (excitation at 428 nm).
1092269.1
CA 02740747 2011-05-20
4459.1012-000
-3-
FIG. 6 is the calibration curves for lysozyme detection plotted as PL
intensity as
a function of lysozyme concentration (each data point represents the average
value of
six independent experiments with error bars indicated).
FIG. 7 depicts the synthetic route to P2.
FIG. 8 depicts the synthetic route to P4.1.
DETAILED DESCRIPTION OF THE INVENTION
A description of example embodiments of the invention follows.
The invention generally relates to a heterogeneous assay that uses charged
CPEs
or charged COEs with biofunctionalized nanoparticles (NPs) for label-free and,
optionally, naked-eye detection of proteins.
As used herein, the singular forms "a," "an" and "the" include plural
referents
unless the context clearly dictates otherwise. Thus, for example, reference to
"a
biomolecule" can include a plurality of biomolecules. Further, the plurality
can
comprise more than one of the same biomolecule or a plurality of different
biomolecules.
As used herein, "conjugated polyelectrolyte," "conjugated oligoelectrolyte,"
"CPE" and "COE" refer to fluorescent macromolecules with electron-delocalized
backbones and water-soluble side chains. CPEs and COEs combine the light-
harvesting
properties of conjugated polymers with the electrostatic behavior of
electrolytes,
providing unique opportunities for construction of sensory and imaging
materials.
As used herein, "oligo" refers to a monomer unit repeating ten or less times
in
the chain. For example, "oligo(ethylene oxide)" refers to an ethylene oxide
repeat unit
[e.g., -(CH2CH2O)õ ], wherein n is 1-10; 2-10; 2-5; 5-10; 2-8; 2-6; or 3-6.
As used herein, "poly" refers to a monomer unit repeating ten or more times in
the chain. For example, "poly(ethylene oxide)" refers to an ethylene oxide
repeat unit
[e.g., -(CH2CH2O)õ], wherein n is greater than 10. Specifically, n is 10-100,
10-200;
10-50; or 50-100.
1092269.1
CA 02740747 2011-05-20
4459.1012-000
-4-
In some embodiments of the invention, the CPEs and COEs are functionalized
with polyhedral oligomeric silsesquioxanes (POSS). As used herein, "polyhedral
oligomeric silsesquioxanes" or "POSS" are a category of polycyclic compounds,
which
consist of a silicon/oxygen cage surrounded by tunable organic substitution
groups.
Due to the nano-scaled dimension and facile modification of substitution
groups, POSS
serve as organic-inorganic nanobuilding blocks for the construction of
fluorescent
nanomaterials. Functionalization with POSS can minimize self-quenching of CPEs
and
COEs, which can be desirable for optical applications.
A first embodiment of the invention a CPE or COE represented by Structural
Formula (I):
R' R3
R R2
M
(I), or a salt thereof,
wherein:
R and R2 are each independently -(OCH2CH2)pOCH3 or -(CH2CH2O)pCH3,
wherein p is an integer between 1 and 100, inclusive;
R' and R3 are each independently hydrogen or a charged side group;
in is an integer between 2 and 50, inclusive; and
T and T' are each independently a terminating group.
In a first aspect of the first embodiment, the CPE or COE is represented by
Structural Formula (I), or a salt thereof, with the proviso that the CPE or
COE is not
represented by the following structural formula:
1092269.1
CA 02740747 2011-05-20
4459.1012-000
-5-
NaO3S SO3Na
T- T'
O 10-20
OO O
O
wherein the values and
alternative values for the remaining variables are as described in the first
embodiment.
In a second aspect of the first embodiment, R and R2 are each
-(OCH2CH2)pOCH3 or -(CH2CH2O)pCH3. Specifically, R and R2 are each
-(CH2CH2O)pCH3. More specifically, p is an integer between 1 and 50,
inclusive,
between, 1 and 25, inclusive, between 1 and 10, inclusive, or between 1 and 5,
inclusive. The values and alternative values for the remaining variables are
as described
in the first embodiment, or first aspect thereof.
In a third aspect of the first embodiment, R' and R3 are each independently a
charged side group, wherein the values and alternative values for the
remaining
variables are as described in the first embodiment, or first or second aspects
thereof.
In a fourth aspect of the first embodiment, R' and R3 are each a charged side
group. Specifically, R' and R3 are each an anionic side group. Alternatively,
R' and R3
are each a cationic side group. The values and alternative values for the
remaining
variables are as described in the first embodiment, or first through third
aspects thereof.
In a fifth aspect of the first embodiment, in is an integer between 2 and 10,
inclusive, or 20 and 30, inclusive. Specifically, in is an integer between 2
and 10,
inclusive. Alternatively, in is an integer between 20 and 30, inclusive. The
values and
alternative values for the remaining variables are as described in the first
embodiment,
or first through fourth aspects thereof.
1092269.1
CA 02740747 2011-05-20
4459.1012-000
-6-
In some embodiments of the invention, the charged side group can be a cationic
alkyl side group, a cationic oligo(ethylene oxide) side group or a cationic
poly(ethylene
oxide) side group. As used herein, "a cationic alkyl side group" is a (C 1-C
15)alkyl that
includes a moiety, such as an amine, that confers a positive charge. As used
herein,
"cationic oligo(ethylene oxide) side group" and "cationic poly(ethylene oxide)
side
group" refer to a polymer of ethylene oxide that includes a moiety, such as an
amine,
that confers a positive charge. The amine can be a primary, a secondary, a
tertiary or a
quaternary amine. Specifically, the amine is a quaternary amine.
Alternatively, the
amine is a protonated amine.
In some embodiments of the invention, the charged side group can be an anionic
alkyl side group, an anionic oligo(ethylene oxide) side group or an anionic
poly(ethylene oxide) side group. As used herein, "anionic alkyl side group"
refers to a
(C 1-C 15)alkyl that includes a moiety, such as a phosphonate, a sulfonate or
a
carboxylate, that confers a negative charge. As used herein, "anionic
oligo(ethylene
oxide) side group" and "anionic poly(ethylene oxide) side group" refer to a
polymer of
ethylene oxide that includes a moiety, such as a phosphonate, a sulfonate or a
carboxylate, that confers a negative charge.
In some embodiments of the invention, the charged side groups are selected
from the group consisting of -(CH2)õN(R2)3X, -(OCH2CH2)õ N(R2)3X,
-(CH2CH2O)gCH2CH2N(R2)3X, -(CH2)õ X', -(OCH2CH2)õ X', -(OCH2CH2)nOX',
-(CH2CH2O)õX' and -(CH2CH2O)gCH2CH2X', wherein R2 is (C 1-C6)alkyl, n is an
integer between 2 and 13, inclusive, q is an integer between 1 and 12,
inclusive, X is an
anionic counterion and X' is -CO2Y, -SO3Y or -P03Y2, wherein Y is hydrogen or
a
cationic counterion.
In some embodiments of the invention, the charged side groups are selected
from the group consisting of -(CH2),,N(R2)3X, -(OCH2CH2)õ N(R2)3X and
-(CH2CH2O)gCH2CH2N(R2)3X, wherein R2 is (Cl-C6)alkyl, n is an integer between
2
and 13, inclusive, q is an integer between 1 and 12, inclusive, and X is an
anionic
counterion. Specifically, R2 is methyl or ethyl.
1092269.1
CA 02740747 2011-05-20
4459.1012-000
-7-
In some embodiments of the invention, the charged side groups are selected
from the group consisting of -(CH2)õX', -(OCH2CH2)õX', -(OCH2CH2)õ OX',
-(CH2CH2O)õX' and -(CH2CH2O)gCH2CH2X', wherein n is an integer between 2 and
13,
inclusive, q is an integer between 1 and 12, inclusive, and Xis -CO2Y, -SO3Y
or
-PO3Y2, wherein Y is hydrogen or a cationic counterion. Specifically, X is -
SO3Y or
-P03Y2. More specifically, Xis -SO3Y. Alternatively, Y is a cationic
counterion.
In a second embodiment of the invention, the CPE or COE is represented by
Structural Formula (II):
R' R3
H3C~0 O)CH3
P P M
(II), or a salt thereof,
wherein the values and alternative values for the variables are as described
in the first
embodiment, or aspects thereof.
In a first aspect of the second embodiment, p is 3, wherein the values and
alternative values for the variables are as described in the first embodiment,
or aspects
thereof or the second embodiment.
In a second aspect of the second embodiment, R' and R3 are each an anionic
alkyl side group, wherein the values and alternative values for the variables
are as
described in the first embodiment, or aspects thereof, or the second
embodiment, or first
aspect thereof.
In a third aspect of the second embodiment, p is 3 and R' and R3 are each
-(CH2)nSO3Y, wherein n is 4 and Y is sodium, wherein the values and
alternative values
for the variables are as described in the first embodiment, or aspects thereof
or the
second embodiment, or first or second aspects thereof.
1092269.1
CA 02740747 2011-05-20
4459.1012-000
-8-
In a fourth aspect of the second embodiment, the CPE or COE is represented by
Structural Formula (II), or a salt thereof, with the proviso that the CPE or
COE is not
represented by the following structural formula:
Na03S S3Na
T- T'
0 10-20
0 0
% O
wherein the values
and alternative values for the remaining variables are as described in the
first
embodiment, or aspects thereof, or the second embodiment, or first through
third
aspects thereof.
In a third embodiment of the invention, the CPE or COE is functionalized with
POSS and is represented by the following structural formula:
R R
Linker Ar
A A
0-Si-0-S1
A. ' 0. 0 O J R R
Si-. SI~A
O(A~~ l A= Linker ArO-Si10/ OSi\ CPO
3i\O.-Si A A R R
A: Conjugated Oligelectrolyte Linker .. A r =
R: Cationic Side Group or a salt
thereof, wherein:
Ar is an optionally substituted aromatic group;
1092269.1
CA 02740747 2011-05-20
4459.1012-000
-9-
Linker is a single bond, double bond, triple bond or -CR'2-; wherein each R'
is
independently hydrogen, halogen, hydroxy, amino, (C 1 -C6)alkyl, (CI-
C6)alkenyl, (C1-C6)alkynyl, or (C1-C6)alkoxy; wherein the alkyl, alkenyl,
alkynyl or alkoxy may be optionally substituted with halogen, hydroxy,
(C1-C4)alkoxy or amino;
each R is independently hydrogen, a cationic alkyl side group or a cationic
oligo
or poly(ethylene oxide) group.
In a first aspect of the third embodiment, Linker is a single bond, double
bond,
triple bond, -CH2- or -CH2CH2-, wherein the values and alternative values for
the
variables are as described in the third embodiment or in the fourth
embodiment, or
aspects thereof.
In a second aspect of the third embodiment, the values and alternative values
for
the variables are as described in the fourth embodiment or aspects thereof.
In a fourth embodiment, the CPE or COE is functionalized with POSS and is
represented by the following structural formula:
A
A~,Si ' R'- Ar' - Linker ~' Ar
0, -0-SI
O-S1
A- '
O-SI-OrSi
ASi o
.~~Si A A= R'-` Ar' Linker Ar
A: Conjugated Oligelectrolyte
R-Ar - Linker_ Ar ora
salt thereof, wherein:
Ar
each is independently selected from:
R R N,R OR R
\ / or RO R
1092269.1
CA 02740747 2011-05-20
4459.1012-000
-10-
each Ar is independently an optionally substituted aromatic group;
each R is independently a cationic, anionic, or neutral alkyl group or a
cationic,
anionic, or neutral oligo or poly(ethylene oxide) group;
each Linker is a single bond, double bond, triple bond, -CH2- or -CH2CH2-; and
each R' is independently a terminating group.
In a first aspect of the fourth embodiment, Ar is fluorene, benzene, biphenyl,
pyridine, bipyridinium, triphenylamine, anthracene, thiophene, carbazole, or
benzothiadiazole. Optional substituents include those defined by R. The values
and
alternative values for the remaining variables are as described in the third
embodiment,
or aspects thereof, or in the fourth embodiment.
In a second aspect of the fourth embodiment, each R is independently selected
from the group consisting of hydrogen, -(CH2)nNMe3X; -(CH2)õNEt3X;
-(CH2CH2O)gCH2CH2NMe3X and -(CH2CH2O)gCH2CH2NEt3X, wherein X is an
anionic counterion, n is an integer between 2 and 13, inclusive, and q is an
integer
between 1 and 12, inclusive. Specifically, each R is independently selected
from the
group consisting of hydrogen, -(CH2)nNMe3X and -(CH2CH2O)gCH2CH2NMe3X,
wherein X is an anionic counterion, n is an integer between 2 and 13,
inclusive, and q is
an integer between 1 and 12, inclusive. The values and alternative values for
the
remaining variables are as described in the third embodiment, or aspects
thereof, or in
the fourth embodiment, or first aspect thereof.
In a third aspect of the fourth embodiment, the POSS-functionalized CPE or
COE is represented by the following structural formula:
1092269.1
CA 02740747 2011-05-20
4459.1012-000
-11-
R
R
R R
\ I ~ /
R \ I / R
R R R
\ ~ R I i
R
si-10-' R R
R R ip!p g.0
Sip Si-0/ OSi R
_-Si
R
R R
R R
R
R R
i
R
R
\ R Q
R = {CFh)s (CH3)3Br
In a fourth aspect of the fourth embodiment, the POSS-functionalized CPE or
COE is represented by the following structural formula:
/I
R RR \ /
N
R S
R - N-S ,N \ N R R
Ni /S
I \ i
S. ~ Oi-O_Si\
N\ N Sid O-S\
O \ 1 N
-Si- Si N-S
Si\O-Si'O
R R S-N N~
N~ \ N. S-N
R
N
R R
R R
R = -(CH2)6N(CH3)3Br
O O
1092269.1
CA 02740747 2011-05-20
4459.1012-000
- 12-
In a fifth aspect of the fourth embodiment, R is an anionic group selected
from
-(CH2)õX', -(OCH2CH2)õX', -(OCH2CH2)nOX', -(CH2CH2O)õX' and
-(CH2CH2O)gCH2CH2X', wherein Xis selected from -SO3Y, -P03Y2, and -CO2Y, n is
an integer between 2 and 13, inclusive, q is an integer between 1 and 12,
inclusive, and
Y is a cationic counterion. The values and alternative values for the
remaining
variables is as described in the fourth embodiment, or aspects thereof, or the
fifth
embodiment, or the first through fourth aspects thereof.
In a fifth embodiment of the invention, the CPE or COE is a hyperbranched CPE
(HCPE). Specifically, the HCPE is represented by structural formula (III):
T'
Ar T
M
Q-~
T,. R' R3
(III),
or a salt thereof, wherein:
R' and R3 are each independently hydrogen or a charged side group;
in is an integer between 2 and 50, inclusive;
Ar is an optionally substituted monocyclic or polycyclic aromatic ring system
or
an optionally substituted monocyclic or polycyclic heteroaromatic ring
system; and
T, T' and T" are each independently a terminating group.
As used herein, "hyperbranched conjugated polyelectrolyte" or "HCPE" refers
to a CPE which has a densely branched structure and a large number of end
groups.
In a first aspect of the fifth embodiment, Ar is fluorene, benzene, biphenyl,
thiophene, benzothiadiazole, 4,7-di(thien-5'-yl)-2,1,3-benzothiadiazole,
pyridine,
bipyridinium, triphenylamine, anthracene or carbazole. Specifically, Ar is
benzothiadiazole. The values and alternative values for the remaining
variables are as
described in the first or second embodiments, or aspects thereof, or the fifth
embodiment.
1092269.1
CA 02740747 2011-05-20
4459.1012-000
-13-
In a second aspect of the fifth embodiment, T, T' and T" are each -CCH,
wherein
the values and alternative values for the remaining variables are as described
in the first
or second embodiments, or aspects thereof, or the fifth embodiment, or the
first aspect
thereof.
In a third aspect of the fifth embodiment, Wand R3 are each a charged side
group. Specifically, the charged side groups are selected from the group
consisting of
-(CH2)õN(R2)3X, -(OCH2CH2)õN(R2)3X and -(CH2CH2O)gCH2CH2N(R2)3X, wherein R2
is (C 1-C6)alkyl, n is an integer between 2 and 13, inclusive, q is an integer
between 1
and 12, inclusive, and X is an anionic counterion. The values and alternative
values for
the remaining variables are as described in the first or second embodiments,
or aspects
thereof, or the fifth embodiment t, or the first or second aspects thereof.
In a fourth aspect of the fifth embodiment, the HCPE is represented by the
following structural formula:
T' N\ S, N
T R'= R3 = (CH2)6N(CH3)3Br
M
T"
R' R3
wherein
the values and alternative values for the remaining variables are as described
in the first
or second embodiments, or aspects thereof, or the fifth embodiment, or the
first through
third aspects thereof.
In a fifth aspect of the fifth embodiment, in is an integer between 2 and 30,
inclusive, wherein the values and alternative values for the remaining
variables are as
described in the first or second embodiments, or aspects thereof, or the fifth
embodiment, or the first through fourth aspects thereof.
In a sixth aspect of the fifth embodiment, the values and alternative values
for
the variables are as described in the first, second or sixth embodiments, or
aspects
thereof.
1092269.1
CA 02740747 2011-05-20
4459.1012-000
-14-
A sixth embodiment of the invention is a molecular brush represented by
structural formula (IV):
T - \ / \Ar T
R' R3
M
(IV), or a salt thereof; wherein:
R' and R3 are each independently hydrogen or a charged side group;
m is an integer between 2 and 50, inclusive;
Ar is an optionally substituted monocyclic or polycyclic aromatic ring system
or
an optionally substituted monocyclic or polycyclic heteroaromatic ring
system; and
T and T' are each independently a terminating group.
As used herein, "molecular brush" refers to a CPE or COE with densely grafted
side chains on a linear polymeric backbone.
In a first aspect of the sixth embodiment of the invention, the values and
alternative values for the variables are as defined in the first, second or
fifth
embodiments, or aspects thereof.
In a second aspect of the sixth embodiment, T and T are each independently
hydrogen, halo, -CH=CH2 or -CH2CH3, wherein the values and alternative values
for
the variables are as defined in the first, second or fifth embodiments, or
aspects thereof,
or the sixth embodiment, or the first aspect thereof.
In a third aspect of the sixth embodiment, the CPE or COE is represented by
the
following structural formula:
1092269.1
CA 02740747 2011-05-20
4459.1012-000
-15-
N\S, N
T T' R'= R3 = (CH2)6N(CH3)3Br
R' R3
wherein
the values and alternative values for the variables are as defined in the
first, second or
fifth embodiments, or aspects thereof, or the sixth embodiment, or the first
or second
aspects thereof.
In a fourth aspect of the sixth embodiment, Ar is an optionally substituted
monocyclic or polycyclic (C6-C12)aromatic ring system or an optionally
substituted
monocyclic or polycyclic (C6-C 12)heteroaromatic ring system, wherein the
values and
alternative values for the remaining variables are as described in the first,
second, or
fifth embodiments, or aspects thereof, of the sixth embodiment, or the first
through third
aspects thereof.
In a fifth aspect of the sixth embodiment, the CPE or COE is not represented
by
the following structural formula:
Na03S S3Na
0 10-20
Of 0 01O
wherein the values and
alternative values for the remaining variables are as described in the first,
second, or
fifth embodiments, or aspects thereof, of the sixth embodiment, or the first
through
fourth aspects thereof.
1092269.1
CA 02740747 2011-05-20
4459.1012-000
-16-
In a sixth aspect of the sixth embodiment, in is an integer between 2 and 10,
inclusive, or 20 and 30, inclusive, wherein the values and alternative values
for the
remaining variables are as described in the first, second, or fifth
embodiments, or
aspects thereof, or the sixth embodiment, or the first through fifth aspects
thereof.
As used herein, "terminating group" refers to the functional group left at
each
end of a polymer upon termination of the polymerization reaction. Non-limiting
examples of terminating groups include hydrogen, halo, -CH=CH2, -CCH and
-CH2CH3.
"Alkyl" means an optionally substituted saturated aliphatic branched or
straight-chain monovalent hydrocarbon radical having the specified number of
carbon
atoms. Thus, "(C1-C6) alkyl" means a radical having from 1-6 carbon atoms in a
linear
or branched arrangement. "(C1-C6)alkyl" includes, for example, methyl, ethyl,
propyl,
iso-propyl, n-butyl, tent-butyl, pentyl and hexyl.
"Alkenyl" refers to a straight or branched aliphatic group with at least one
double bond. Typically, alkenyl groups have from 2 to 12 carbon atoms, from 2
to 8,
from 2 to 6, or from 2 to 4 carbon atoms. Examples of alkenyl groups include
ethenyl
(-CH=CH2), n-2-propenyl (allyl, -CH2CH=CH2), pentenyl, hexenyl, and the like.
"Alkynyl" refers to a straight or branched aliphatic group having at least 1
site
of alkynyl unsaturation. Typically, alkynyl groups contain 2 to 12, 2 to 8, 2
to 6 or 2 to
4 carbon atoms. Examples of alkynyl groups include ethynyl (-C=CH), propargyl
(-CH2C=CH), pentynyl, hexynyl, and the like.
As used herein, "halogen" refers to fluorine, chlorine, bromine or iodine.
"Halogen" and "halo" are used interchangeably herein.
"Alkoxy" means an alkyl radical attached through an oxygen linking atom.
"(C1-C3)alkoxy" includes methoxy, ethoxy and propoxy.
"Aryl" or "aromatic" means an aromatic monocyclic or polycyclic (e.g.,
bicyclic
or tricyclic) carbocyclic ring system. Thus, "(C5-C14)aryl" is a (5-14)-
membered
monocylic or bicyclic system. Aryl systems include, but are not limited to,
phenyl,
naphthalenyl, fluorenyl, indenyl, azulenyl, and anthracenyl.
1092269.1
CA 02740747 2011-05-20
4459.1012-000
-17-
"Hetero" refers to the replacement of at least one carbon atom in a ring
system
with at least one heteroatom selected from N, S and 0. "Hetero" also refers to
the
replacement of at least one carbon atom in an acyclic system. A hetero ring
system or a
hetero acyclic system may have, for example, 1, 2 or 3 carbon atoms replaced
by a
heteroatom.
"Heteroaryl" means a monovalent heteroaromatic monocyclic or polycyclic
(e.g., bicylic or tricyclic) ring radical. A heteroaryl contains 1, 2, 3 or 4
heteroatoms
independently selected from N, 0 and S. Thus, "(C5-C14)heteroaryl" refers to a
(5-14)-
membered ring system, wherein at least one carbon atom has been replaced with
at least
one heteroatom selected from N, S and 0. Heteroaryls include, but are not
limited to
furan, oxazole, thiophene, 1,2,3-triazole, 1,2,4-triazine, 1,2,4-triazole,
1,2,5-thiadiazole
1,1-dioxide, 1,2,5-thiadiazole 1-oxide, 1,2,5-thiadiazole, 1,3,4-oxadiazole,
1,3,4-
thiadiazole, 1,3,5-triazine, imidazole, isothiazole, isoxazole, pyrazole,
pyridazine,
pyridine, pyridine-N-oxide, pyrazine, pyrimidine, pyrrole, tetrazole, and
thiazole.
"Bicycloheteroaryl," as used herein, refers to bicyclic heteroaryl rings, such
ase
bicyclo[4.4.0] and bicyclo[4.3.0] fused ring systems containing at least one
aromatic
ring and 1 to 4 heteroatoms independently selected from N, 0 and S. In some
embodiments of the invention, the first ring is a monocyclic heterocyclyl
(such as
dioxolane) and the second ring is a monocyclic aryl (such as phenyl) or a
monocyclic
heteroaryl (such as pyridine). Examples of bicyclic heteroaryl rings include,
but are not
limited to, indole, quinoline, quinazoline, benzothiophene, benzofuran, 2,3-
dihydrobenzofuran, benzodioxole, benzimidazole, indazole, benzisoxazole,
benzoxazole
and benzothiazole.
Each aryl and heteroaryl is optionally and independently substituted.
Exemplary substituents include halogen, (C1-C3)alkoxy, (C1-C3)alkylthio,
hydroxy, (C5-
C14)aryl, (C5-C14)heteroaryl, (C3-C1s)cycloalkyl, (C3-C15)heterocyclyl, amino,
(C1-
Cs)alkylamino, (C1-C5)dialkylamino, thio, oxo, (C1-C5)alkyl, (C5-C14)aryl(C1-
C5)alkyl,
(C5-C14)heteroaryl(C1-Cs)alkyl, nitro, cyano, sulfonato, phosphonato,
carboxylate,
hydroxyl(C1-C5)alkyl and halo(C1-C5)alkyl.
1092269.1
CA 02740747 2011-05-20
4459.1012-000
- 18-
"Anionic counterion," as used herein, refers to a negatively charged ion.
Examples of anionic counterions include, but are not limited to, halide,
trifluoroacetate,
acetate, benzenesulfonate, benzoate, perchlorate, sulfonate, bicarbonate,
carbonate,
citrate, mesylate, methylsulfate, nitrate, phosphate/diphosphate, sulfate,
trifluoromethanesulfonate, tetrafluoroborate, ammonium hexafluorophosphate and
tetrakis[3,5,-bis(trifluoromethyl)phenyl]borate. Specifically, the anionic
counterion is
halide, tetrafluoroborate, trifluoromethanesulfonate, ammonium
hexafluorophosphate or
tetrakis[3,5,-bis(trifluoromethyl)phenyl]borate. More specifically, the halide
is bromide
or iodide. Yet more specifically, the halide is bromide.
"Cationic counterion," as used herein, refers to a positively charged ion.
Specifically, the cationic counterion is sodium, lithium or potassium. More
specifically,
the cationic counterion is sodium or potassium.
One embodiment of the invention is illustrated in FIG. 1. FIG. 1 depicts the
functionalization of NPs [e.g., silica NPs, polystyrene NPs,
poly(methylmethacrylate)
NPs], with a ligand, such as an aptamer, to yield ligand-functionalized NPs.
These
ligand-functionalized NPs can be further treated with a blocking agent, such
as
ethanolamine, to generate blocked NPs. Upon incubation with a sample
containing a
target, such as a protein (e.g., lysozyme), the blocked NPs specifically bind
the target.
Binding of the target switches the charge of the NPs. For example, if the NPs
were
initially negatively-charged, upon binding of the target, the NPs will be
positively-
charged. A fluorescent CPE that has a complementary charge to the target can
be added
to the NP-treated sample to yield CPE/target/ligand complexes on the surface
of the NP,
giving rise to fluorescent NPs after removal of excess CPE, which can be
accomplished,
for example, by a wash-centrifugation-redispersion process. Since no binding
takes
place between the ligand and non-specific proteins, the surface charge on the
ligand-
functionalized NPs that are not bound to the target remains the same as that
of the CPE.
The CPE is thus electrostatically repelled from NPs not bound to the target
and, as a
result, NPs not bound to the target remain non-fluorescent. By taking
advantage of the
1092269.1
CA 02740747 2011-05-20
4459.1012-000
-19-
recognition-induced switching of surface charge, label-free, naked-eye protein
detection
can be realized.
"Biomolecule," as used herein, refers to a natural or synthetic molecule for
use
in biological systems. Examples of biomolecules include, but are not limited
to,
proteins, peptides, enzyme substrates, pharmaceuticals, ligands, hormones,
antibodies,
antigens, haptens, carbohydrates, oligosaccharides, polysaccharides, nucleic
acids,
aptamer, fragments of DNA, fragments of RNA and mixtures thereof.
"Ligand," as used herein, refers to a molecule that specifically binds to a
biomolecule, such as a target. Examples of ligands include, but are not
limited to,
aptamers [e.g., anti-lysozyme aptamer (5'-NH2-ATC TAC GAA TTC ATC AGG GCT
AAA GAG TGC AGA GTT ACT TAG; SEQ. ID. NO. 1), anti-thrombin aptamer (5'-
NH2-GGT TGG TGT GGT TGG; SEQ. ID. NO. 2)] and antibodies (e.g., anti-
thrombin).
Aptamers are oligonucleic acid or peptide molecules that bind to a specific
target molecule. More specifically, aptamers can be classified as: DNA or RNA
aptamers, consisting of (usually short) strands of oligonucleotides or peptide
aptamers,
consisting of a short variable peptide domain, attached at both ends to a
protein
scaffold. An aptamer to be immobilized on the solid support is selected based
upon its
ability to bind the biological molecule of interest.
"Target," as used herein, refers to a biomolecule that specifically binds to
another biomolecule. Examples of targets include, but are not limited to, a
protein, a
peptide, an enzyme, an oligosaccharide, a polysaccharide, a fragment of DNA
and a
fragment of RNA. In some embodiments of the invention, target proteins (e.g.,
lysozyme, thrombin) bind ligands (e.g., anti-lysozyme aptamer, anti-thrombin
aptamer).
As used herein, "functionalized" refers both to (1) the covalent attachment of
a
ligand to a nanoparticle, as might be achieved, for example, by chemical
reaction, and
to (2) the noncovalent attachment of a ligand to a nanoparticle, as might be
achieved,
for example, by surface adsorption. In some embodiments, a surface of a solid
support
(e.g., NP) is functionalized with a ligand.
1092269. I
CA 02740747 2011-05-20
4459.1012-000
-20-
The compounds according to the present invention may be in free form or in the
form of salts. These salts may be obtained by reacting the respective
compounds with
acids and bases. Examples of such salts include but are not limited to
hydrochloride,
hydrobromide, hydroiodide, hydrofluoride, nitrate, sulfate, bisulfate,
pyrosulfate,
sulfite, bisulfite, phosphate, acid phosphate, monohydrogenphosphate,
dihydrogenphosphate, metaphosphate, pyrophosphate, isonicotinate, acetate,
trifluoroacetate, propionate, caprylate, isobutyrate, lactate, salicylate,
citrate, tartrate,
oxalate, malonate, suberate, sebacate, mandelate, chlorobenzoate,
methylbenzoate,
dinitrobenzoate, phthalate, phenylacetate, malate, pantothenate, bitartrate,
ascorbate,
succinate, maleate, gentisinate, fumarate, gluconate, glucuronate, saccharate,
formate,
benzoate, glutamate, methanesulfonate, ethanesulfonate, benzenesulfonate, p-
toluenesulfonate and pamoate [i.e., l,l'-methylene-bis-(2-hydroxy-3-
naphthoate)] salts.
Certain compounds of the invention can form salts with various amino acids.
Suitable
base salts include, but are not limited to, aluminium, calcium, lithium,
magnesium,
potassium, sodium, zinc, and diethanolamine, N,N'-dibenzylethylenediamine,
chloroprocaine, choline, dicyclohexylamine, ethylenediamine, N-
methylglucamine, and
procaine salts.
CPEs undergo a photophysical property change upon interaction with proteins.
For example, the emission intensity, emission maximum, and/or the absorption
maximum, as well as the associated fluorescence and absorbance profiles, can
change
upon interaction with proteins. (See (a) Ambade, A. V., et al., S. Polym. Int.
2007, 56,
474-481. (b) Ho, H. A., et al., Ace. Chem. Res. 2008, 41, 168-178. (c) Li, K.;
Liu, B.
Polym. Chem. 2010, 1, 252-259.)
Water solubility of CPEs is achieved through introduction of charged
hydrophilic functionalities to the macromolecular backbone. Good water
solubility
minimizes polymer interchain aggregation, which leads to less fluorescence
quenching
and greater fluorescence intensity in aqueous solution. (See (a) Khan, A., et
al., Chem.
Commun. 2005, 584-586. (b) Lee, K. W., et al.,Chem. Commun. 2006, 1983-1985;
the
entire teachings of which are incorporated herein by reference). In addition,
good
1092269.1
CA 02740747 2011-05-20
4459.1012-000
-21 -
polymer water solubility can minimize nonspecific interactions between CPEs
and the
nanoparticles, thereby decreasing any background signal.
One embodiment of the present invention is a method of detecting a target in a
sample, comprising: functionalizing a solid support with a ligand; incubating
the ligand-
functionalized solid support with a sample; incubating the sample with a CPE
or COE;
and detecting the fluorescence of the solid support, thereby detecting the
target.
Specifically, the CPE or COE is a charged CPE or COE.
A sample can be, for example, a cellular lysate, a biomolecule, a cell, a
mixture
of biomolecules, or a mixture thereof. A sample can be in the form of a
solution in
buffer, for example, and can include biological media.
As used herein, "incubating the sample with a CPE or COE" means the sample
and the CPE or COE are present in the same container or in the same solution
and may
come into contact. Incubating the sample with the CPE or COE includes adding
the
CPE or COE, either in suspension or as a solid, to the sample.
In some embodiments, the method further includes isolating the solid support
from the sample. In other embodiments, the method further includes isolating
the solid
support from the sample and washing the solid support. Isolating the solid
support from
the sample and/or washing the solid support can occur before detecting the
fluorescence
of the solid support.
Suitable solid supports include nanoparticles (NPs) or solid-state substrates
(e.g.,
paper, glass, quartz). Silica NPs, in particular, can be easily
functionalized, are
chemically inert, and are easily separable from biological media. The chemical
modification of silica NPs can be accomplished chemically using reactive
functional
groups (e.g., cyanuric chloride, aldehyde, and NHS ester) (see, for example,
Steinberg,
G., et al., Biopolymers 2004, 73, 597-605; Kato, N.; Caruso, F. J. Phys. Chem.
B 2005,
109, 19604-19612; and Liang, Y, et al., Talanta 2007, 72, 443-449, the entire
teachings of each are incorporated herein by reference). Meanwhile, the high
density of
silica (1.96 g/cm3) facilitates easy separation of NPs from biological media
via
centrifugation-washing-redispersing circles. Such a method can help to
eliminate
1092269.1
CA 02740747 2011-05-20
4459.1012-000
-22-
nonspecific proteins, while retaining the bound target, and can promote the
trace
detection of a target in biological samples. In addition, silica NPs of 100 nm
in diameter
are transparent in dilute solutions, and their optical properties do not
interfere with those
of fluorescent dyes or CPEs.
Aptamer-functionalized silica NPs can be an effective platform for selectively
capturing a target, such as lysozyme or thrombin, and effectively isolating
the target via
centrifugation-washing-redispersing circles. Lysozyme binding to aptamer-
functionalized silica NPs switches the surface charges of Apt-NP from negative
to
partially positive, which subsequently allows for CPE binding, which can be
detected as
blue-green fluorescence by, for example, the naked eye or a fluorescence
spectrometer.
Moreover, the linear intensity increase of polymer emission as a function of
lysozyme
concentration allows the accurate quantification of lysozyme in the
concentration range
of 0 to approximately 22.5 M with a limit of detection of approximately 0.36
g/mL.
The high quantum yield and good water solubility of CPEs also enables naked-
eye
lysozyme detection with picomole sensitivity.
In a specific embodiment, aptamer-functionalized silica nanoparticles (NPs)
have been synthesized to capture lysozyme, resulting in a switching of the
surface
charge from negative to partially positive. The aptamer/protein binding event
can be
monitored by fluorescence spectroscopy. Upon its addition, PFVSO3 binds to and
"stains" the protein/aptamer/NP complexes via an electrostatic interaction.
The blue-
green fluorescence of PFVSO3 can be observed in the presence of lysozyme by
the
naked eye, while no fluorescence is obtained for NPs treated with a non-
specific
mixture of proteins.
One embodiment of the invention is a method of detecting a target in a sample,
comprising functionalizing a surface of a solid support with a charged ligand,
thereby
creating a charge (e.g., a positive or negative charge) on the surface of the
solid support;
incubating the ligand-functionalized solid support with a sample, whereupon
binding of
the target, the charge on the surface of the solid support switches (e.g.,
from positive to
negative or from negative to positive); incubating the sample with a
conjugated
1092269.1
CA 02740747 2011-05-20
4459.1012-000
- 23 -
polyelectrolyte (CPE) or a conjugated oligoelectrolyte (COE) that has a
complementary
charge to the charge of the target-bound surface (i.e., if the target-bound
surface is
negatively charged, the CPE or COE is positively charged and visa versa); and
detecting
the fluorescence of the sample, thereby detecting the target.
In some embodiments, the ligand is a charged ligand. As used herein, "charged
ligand" refers to a ligand having a net positive or net negative charge under
the
conditions of the assay. Typically, the conditions are neutral conditions or
neutral pH.
Proteins, CPEs and COEs can also be described as "charged" if they have a net
positive
or net negative charge under the conditions of the assay.
In a specific embodiment, the biological molecule to be detected is lysozyme,
which has an isoelectric point (pI) of 11.0, and is, therefore, positively
charged at neutral
pH. Lysozyme is a ubiquitous protein serving as the "body's own antibiotic" by
cleaving acetyl groups in the polysaccharide walls of many bacteria.
Therefore, the
lysozyme level in blood is regarded as the clinical index for many diseases
such as HIV,
myeloid leukemia, etc. (see (a) Vocadlo, D. J., et al., Nature 2001, 412, 835-
838. (b)
Lee-Huang, S. et al., Proc. Natl. Acad. Sci. U &A. 1999, 96, 2678-268 1, the
teachings
of each are herein incorporated by reference).
One embodiment of the invention is a label-free, naked-eye lysozyme detection
method using aptamer-functionalized silica NPs as the recognition element to
capture a
target and an anionic conjugated polymer as "a polymeric stain" to transduce a
signal.
EXEMPLIFICATION
Example 1. Label-free, Naked-eye detection of Lysozyme Using CPEs
Antilysozyme aptamer (5'-NH2-ATC TAC GAA TTC ATC AGG GCT AAA
GAG TGC AGA GTT ACT TAG SEQ ID NO.: 1) was ordered from Sigma-Genosys.
Hen egg white lysozyme, BSA, and human trypsin were ordered from Sigma-
Aldrich.
Human R-thrombin was ordered from HTI.
Instrumentation. The NMR spectra were collected on a Bruker ACF400 (400
MHz). The absorption spectra of aptamer and lysozyme were measured using a UV-
vis
1092269.1
CA 02740747 2011-05-20
4459.1012-000
-24-
spectrometer (Shimadzu, UV- 1700, Japan). The photoluminescence spectra were
recorded on a fluorometer (Perkin-Elmer, LS-55) equipped with a xenon lamp
excitation
source and a Hamamatsu (Japan) 928 PMT, using 90 angle detection for solution
samples. The size of silica NPs was calculated using a field emission scanning
electron
microscope (FE-SEM JEOLJSM-6700 F) after coating a thin Pt layer via a
platinum
coater. The zeta-potential of the NPs was measured using a zeta-potential
analyzer
(ZetaPlus, Brookhaven Instruments Corp.) at room temperature.
Synthesis and Characterization of PFVSO3. 2,7-Dibromo-9,9-bis(2-(2-(2-
methoxyethoxy)-ethoxy)ethyl)fluorene was synthesized according to our previous
report. (See, for example, (a) Pu, K. Y., et al., Adv. Funct. Mater. 2008, 18,
1321-1328;
(b) Wang, F. K.; Bazan, G. C., J. Am. Chem. Soc. 2006,128,15786-15792; (c) Pu,
K.
Y., et al., Chem. Mater. 2009, 21, 3816-3822, the entire teachings of which
are
incorporated herein by reference.)
9,9-Bis(2-(2-(2-methoxyethoxy)ethoxy)ethyl)-2,7-divinylfluorene (1). 2,7-
dibromo-9,9-bis(2-(2-(2-methoxyethoxy)-,ethoxy)ethyl)fluorene (1.23 g, 2.0
mmol),
tributylvinyltin (1.33 g, 4.2 mmol), PdC12(PPh3)2 (56 mg, 0.09 mmol), 2,6-di-
tert-
butylphenol (8 mg, 38 mmol), and toluene (20 mL) were mixed in a 50-mL flask.
The
reaction mixture was stirred and heated at 100 C for 24 hours under nitrogen.
After
cooling to room temperature, the mixture was diluted with ether, treated with
an aqueous
solution of HF (approximately 10%), and stirred for 12 hours. The mixed
solution was
then filtered to remove the solids, and the filtrate was dried over anhydrous
MgSO4. The
solvent was removed under reduced pressure, and the residue was
chromatographed on
silica gel using hexanes/ethyl acetate (1:1) as eluent to give 1 (0.70 g, 68%)
as a blue
liquid. 1HNMR (500 MHz, CDC13, 6 ppm):7.60(d, 2H, J=7.8 Hz), 7.44 (s, 2 H),
7.39 (d,
2 H, J=7.7 Hz), 6.78 (dd, 2 H, J=10.9 Hz, J=17.6 Hz), 5.80 (d, 2 H,
J=17.5Hz),5.27(d,
1092269.1
CA 02740747 2011-05-20
4459.1012-000
-25-
2H, J=10.9 Hz),3.51 (dd,4H, J = 3.4 Hz, J = 5.9 Hz), 3.46 (dd, 4 H, J = 3.3
Hz, J =6.0
Hz), 3.39 (t, 4 H, J = 3.2 Hz), 3.33 (s, 6 H), 3.21 (t, 4 H, J= =3.3 Hz), 2.76
(t, 4 H, J =
Hz), 2.40 (t, 4 H, J =5.17 Hz). 13CNMR (125 MHz, CDC13, S ppm): 149.50,
139.96,
137.00, 136.83, 125.82, 120.69, 119.85, 113.54, 71.83, 70.43, 70.39, 69.96,
66.98,
58.96, 50.96, 39.75.
2,7-Dibromo-9,9-bis(4-sulfonatobutyl)fluorene disodium (2). 2,7-
Dibromofluorene (4 g, 12 mmol) and tetrabutylammoium bromide (80 mg) were
dissolved in a mixture of a 50 wt % aqueous solution of sodium hydroxide (8
mL) and
dimethyl sulfoxide (DMSO) (60 mL). A solution of 1,4-butane sultone (4 g, 29
mmol) in
DMSO (20 mL) was added dropwise into the mixture under nitrogen. After
stirring at
room temperature for 4 hours, the reaction mixture was precipitated into
acetone to
afford the crude product. The product was collected by filtration, washed with
ethanol,
recrystallized twice from acetone/water, and dried under vacuum at 60 C for
24 hours
to yield 2 as white needle crystals (4.3 g, 58.6%). 'H NMR (500 MHz, CD3OD, 6
ppm):
7.68 (d, J=8.11 Hz, 2 H), 7.63 (d, 2 H, J = 1.45 Hz), 7.52 (dd, 2 H, J = 1.42,
8.08 Hz),
2.68-2.47 (m, 4 H), 2.22-2.00 (m, 4 H), 1.62 (td, 4 H, J =7.83, J=7.83,
J=15.65 Hz,),
0.67 (td, 4 H, J= 7.83, J=7.83, J=15.65 Hz). 13C NMR (125 MHz, CD3OD, 6 ppm):
153.39, 140.68, 131.61, 127.38, 122.74, 122.52, 52.37, 40.76, 26.19, 24.25. MS
(MALDI-TOF): m/z 619.89 [M-Na]. (See, for example, Huang, F., et al., Polymer
2005, 46, 12010-12015, the entire teachings of which are incorporated herein
by
reference.)
Poly[9,9-bis(2-(2-(2-methoxyethoxy)ethoxy)ethyl)fluorenevinylene-alt-9,9-
bis(4-sulfonatobutyl)fluorenevinylene Sodium Salt] (PFVSO3). 1 (216 mg, 0.423
mmol), 2 (271 mg, 0.423 mmol), Pd(OAc)2 (4.0 mg, 0.018 mmol), and P(o-tolyl)3
(30
mg, 0.098 mmol) were placed in a round-bottomed flask. A mixture of DMF (3.0
mL),
H2O (1.0 mL), and triethylamine (1.5 mL) was added to the flask, and the
reaction
vessel was degassed. The mixture was vigorously stirred at 110 C for 12
hours. The
mixture was filtered through a 0.22 m syringe driven filter unit, and the
filtrate was
poured into acetone. The precipitate was collected and washed with acetone and
then
1092269.1
CA 02740747 2011-05-20
4459.1012-000
-26-
dried under vacuum for 24 hours to afford PFVSO3 (328 mg, 78%, Mn=15000) as
yellow fibers. 'H NMR (500 MHz, CD3OD, 6 ppm): 7.87-7.51(m, 12 H), 7.38 (br, 4
H),
3.54-3.39 (m, 12 H), 3.36 (br, 4 H), 3.27-3.13 (m, 6 H), 2.90 (br, 4 H), 2.57
(br, 8 H),
2.20 (br, 4 H), 1.63 (br, 4 H), 0.76 (br, 4H). 13C NMR (125 MHz, CD3OD, 6
ppm):
150.90, 149.97, 140.69, 140.00, 137.01, 128.63, 128.25, 126.13, 125.81,
120.86, 120.45,
119.71. 119.58, 71.45, 69.95, 69.91, 69.85, 69.82, 69.50, 57.74, 54.67, 51.18,
42.01,
39.20, 25.00.
Comparison of the integrated areas between the peak at 5.95 ppm and the peak
at
0.76 ppm revealed that the number-average degree of polymerization (DP) of
PFVSO3 is
approximately 15. Thus, the number-average molecular weight is approximately
15,000.
The water solubility of PFVSO3 is approximately 20 mg/mL at 24 C.
The absorbance and photoluminescence (PL) spectra of PFVSO3 in water are
depicted in FIG. 2. The polymer concentration based on repeat unit (RU) is 4
M.
PFVSO3 has an absorption maximum at 428 nm and a shoulder peak at 455 nm,
while
its emission maximum is at 475 nm. While not wishing to be bound by any
particular
theory, the blue-green emission of PFVSO3 is attributed to the introduction of
CdC bond
to the polymer backbone, which elongates the effective conjugated length
relative to that
of polyfluorene. The PL quantum yield of PFVSO3 in water is 0.56 and was
measured
using quinine sulfate in O.1M H2SO4 (quantum yield = 0.55) as the reference.
The high
water solubility provided by the terminal sulfonate groups and the ethylene
oxide side
chains is thought to be responsible for the high quantum yield of PFVSO3 in
aqueous
solution. (See Mikroyannidis, J. A.; Barberis, V. P. J. Polym. Sci., Part A:
Polym. Chem.
2007, 45, 1481-1491.)
Preparation of anti-Lysozyme Aptamer-Functionalized Silica NPs. The bare
silica NPs were synthesized according to a modified Stober method, which
yielded
uniform NPs with a diameter of approximately 100 nm. (See Stober, W., et al.,
J.
Colloid Interface Sci. 1968, 26,62-69, the entire teachings of which are
incorporated
herein by reference.) On the basis of the NP size and the density of silica
(1.96 g cm 3),
it can be estimated that 1.0 mg of the synthesized NPs contained approximately
1 x 1012
1092269.1
CA 02740747 2011-05-20
4459.1012-000
-27-
NPs. Modification of the silica NP surface involved two steps. (See Wang, Y.
S.; Liu, B.
Anal. Chem. 2007, 79, 7214-7220, the entire teachings of which are
incorporated herein
by reference). First, the silica NP was reacted with 3-
aminopropyltriethoxysilane
(APTES) to generate amino groups on the NP surface. Then, the amino-
functionalized
NPs were treated with 2,4,6-trichloro-1,3,5-triazine to produce a triazine-
covered surface
for subsequent aptamer immobilization. After chemical modification, the
triazine-
functionalized silica NPs (1 mg) were dispersed in immobilization buffer (20.1
mM
boric acid, 1.4 mM sodium tetraborate decahydrate, 1.2 M NaCl pH 8.5, 25 L).
In heterogeneous assays, the kinetic and thermodynamic binding process of the
analyte can be significantly influenced by the density of the recognition
element on the
solid support. (See, for example, (a) Peterson, A. W., et al., Nucleic Acids
Res. 2001, 29,
5163-5168; (b) Gong, P.; Levicky, R., Proc. Natl. Acad. Sci. U.S.A. 2008, 105,
5301-
5306; (c) Herne., T. M.; Tarlov., M. J., J. Am. Chem. Soc. 1997, 119, 8916-
8920, the
entire teachings of which are incorporated herein by reference.) Previous
studies have
shown that aptamer-target binding can be inhibited by densely-packed aptamers
on gold
rod electrodes due to cross-hybridization of individual aptamer sequences
(See, for
example, White, R. J., et al., Langmuir 2008, 24, 10513-10518, the entire
teachings of
which are incorporated herein by reference.)
To study the effect of aptamer density on lysozyme detection, different
concentrations of aptamers, ranging from 2 to 36 gM were incubated with silica
NPs (1
mg) to prepare Apt-NPs with different aptamer densities on the NP surface. The
surface
density, expressed as "number of aptamers per NP", was determined by the ratio
of the
total number of immobilized aptamers to the total number of silica NPs in
solution.
Various aliquots of NH2-aptamer solution (100 M) from 0.5 to 9 L were
subsequently
added into the NP suspension and incubated at room temperature for 14 hours.
The NP
suspension was centrifuged, and the supernatant was collected for absorbance
measurements. The aptamer-immobilized NPs were washed with immobilization
buffer.
The number of immobilized aptamer molecules on the silica NPs was calculated
from
the absorbance difference at 260 nm between the aptamer solution before
1092269.1
CA 02740747 2011-05-20
4459.1012-000
-28-
immobilization and the supernatant after immobilization and NP removal. The
surface
density was calculated to be in a range of 30 Apt/NP to 510 Apt/NP.
To minimize nonspecific absorption of proteins on NPs, ethanolamine was used
to block the free triazine sites on the NP surface after aptamer
immobilization. (See, for
example, Wang, Y. S.; Liu, B. Chem. Commun. 2007, 34, 3553-3555; Frederix, F.,
et
al., Biochem. Biophys. Methods 2004, 58, 67-74; the teachings of which are
incorporated
herein by reference.) Blocking was carried out by redispersing the Apt-NPs (1
mg) in
blocking buffer (4 M ethanolamine, 20 mM Tris-HCI, 100 mM NaC1, 5 mM MgC12, pH
= 8.5, 200 L) and incubating the resulting mixture for 1 hour at room
temperature. The
NP suspension was then centrifuged and washed with washing buffer (20 mM Tris-
HCI,
100 mM NaCl, 5 mM MgCl2, pH = 8.5).
Optimization of Assay. Aptamer-functionalized NPs (2 mg) with different probe
densities were incubated with the same concentration of lysozyme (20 g/mL),
then
washed. The lysozyme bound aptamer-NPs (lysozyme/Apt-NPs) were subsequently
treated with 10 M PFVSO3 based on repeat unit (RU) for 5 minutes, which was
followed by washing to remove excess polymer. The PL intensity of the final NP
suspension was plotted as a function of aptamer surface density, and the
results are
shown in FIG. 3. The PL intensity significantly decreases with increased
surface
aptamer density, which could be ascribed to insufficient binding of lysozyme
to aptamer
at elevated surface density. (See Cheng, A. K. H., et al., Anal. Chem. 2007,
79, 5158-
5164, the entire teachings of which are incorporated herein by reference). At
low
surface density, aptamers have more space which favors their G-quartet folding
structure
for lysozyme binding. However, in the case of high surface density,
steric/conformational effects could hamper the specific binding between
lysozyme and
the aptamer. To further confirm this hypothesis, the adsorbed lysozyme was
monitored
according to the UV difference at 280 nm between the same lysozyme solution
before
incubation and the supernatant solution after incubation with different Apt-
NPs and NP
removal. As shown in FIG. 3, the percentage of unbound lysozyme increases with
increased aptamer density on NPs, which verifies that more lysozyme molecules
are
1092269.1
CA 02740747 2011-05-20
4459.1012-000
-29-
captured by Apt-NPs at a low surface density. The optimum surface density was
approximately 60 aptamers per NP (60 Apt-NP), where the polymer stained Apt-NP
PL
intensity reached the maximum, which is beneficial for effective lysozyme
quantification.
To understand the surface charge change upon aptamer/lysozyme/PFVSO3
interaction, the zeta-potentials of 60 Apt-NP, lysozyme/Apt-NPs (2 mg of 60
Apt-NP
upon incubation with 20 gg/mL of lysozyme, followed by washing with washing
buffer
and redispersion), and PFVSO3/lysozyme/Apt-NP (the obtained lysozyme/Apt-NPs
upon further treatment with 1 gM PFVSO3 followed by washing with water and
redispersion) were measured. 60 Apt-NP possess a negative zeta-potential value
of -
39.34 1.55 mV, due to the large amount of negatively-charged aptamers on NP
surface. The capture of lysozyme shifts the zeta potential from -39.35 to -
14.96 f 0.88
mV, due to the presence of positively charged lysozyme molecules on NP
surface.
Staining with PFVSO3 results in an increase in zeta-potential from -14.96 to -
35.75 +
1.44 mV due to self-assembly between PFVSO3 and lysozyme on NPs. This data
confirms that the NP surface charge changes in the recognition event, which
plays a vital
role in lysozyme detection.
Lysozyme Detection Using Blocked Apt-NPs. Various volumes of lysozyme
(1.5 mg/mL) were added to the Apt-NPs (0.2 mg) in lysozyme reaction buffer (20
mM
Tris-HCI, 100 mM NaCl, 5 mM MgC12, pH = 8.5, 100 L) to yield final lysozyme
concentrations from 0 to 37.5 g/mL. The resulting mixtures were incubated for
30
minutes at room temperature. Free lysozyme was removed and the NPs were washed
with washing buffer three times. The lysozyme-associated NPs were redispersed
in
Milli-Q TM (Millipore Corp.) water (100 .tL), and PFVSO3 (100 M, 1 L) was
added.
The mixture was incubated for 5 minutes. Excess PFVSO3 was washed away by a
centrifugation-washing-redispersion process with washing buffer (100 mL, 3
times).
The collected NPs were redispersed in 15 mM PBS buffer (pH = 7.4) for
fluorescence
measurements.
1092269.1
CA 02740747 2011-05-20
4459.1012-000
-30-
Parallel experiments were conducted using a mixture of BSA (20 g/mL),
thrombin (20 g/mL), and trypsin (20 gg/mL) to examine the assay specificity.
BSA,
human thrombin, and trypsin have pI values of 4.7, 7.0-7.6, and 10.5,
respectively,
with net negative, neutral, and positive charges on the protein surface under
the
experimental conditions. The 60 Apt-NP (0.2 mg) was incubated with lysozyme
(20
g/mL) as well as a mixture of interference proteins (20 gg/mL BSA, 20 gg/mL
thrombin, and 20 gg/mL trypsin) in binding buffer (20 mM Tris-HCI, 100 mM
NaCl, 5
mM MgC12, pH = 8.5), followed by polymer staining ([RU] =1 M) for 5 minutes
and
NP washing with washing buffer (20 mM Tris-HCI, 100 mM NaCl, 5 mM MgC12, pH
= 8.5). The PL spectra of the redispersed NPs are shown in FIG. 4. Intense
polymer
emission at 475 nm is only witnessed in the presence of lysozyme due to the
recognition-induced switching of lysozyme/Apt-NP charge, followed by PFVSO3
self-
assembly due to electrostatic interaction. No polymer fluorescence was
observed in the
presence of interference proteins. The nonspecific absorption of foreign
proteins (e.g.,
positively charged trypsin) was largely avoided by blocking the NPs with
ethaholamine
and washing the NPs.
As such, PFVSO3 hardly stains negatively charged Apt-NPs due to electrostatic
repulsion in our experimental conditions and NPs remain nonfluorescent. In
addition,
the fluorescent signal from 60 Apt-NPs upon incubation with the mixture of
lysozyme
and interference proteins (20 pg/mL each) after washing is shown in curve c of
FIG. 4.
The polymer signal obtained from lysozyme in protein mixtures is almost the
same as
that from the pure lysozyme. The specific recognition of lysozyme in protein
mixtures
not only indicates the effectiveness of aptamer-protein binding but also
highlights the
intelligent target capture and interference isolation of the silica NP sensing
platform.
To demonstrate lysozyme quantification, different concentrations of lysozyme
(ranging from 0 to 37.5 g/mL) were incubated with 60 Apt-NP suspension for 30
minutes. The lysozyme/Apt-NPs were then stained with 1 M PFVSO3 for 5
minutes,
then washed. The PL spectra of polymer-stained NPs are shown in FIG. 5. The PL
intensities of the NPs progressively increase with increased lysozyme
concentrations.
1092269.1
CA 02740747 2011-05-20
4459.1012-000
-31 -
This is due to increased positive charge on the Apt-NP surface in the presence
of higher
lysozyme concentrations, which enables increased numbers of negatively charged
PFVSO3 to self-assemble on the NPs. In addition, the fluorescence of the NP
suspension upon treatment with lysozyme and PFVSO3 can be monitored by the
naked
eye. The intensity of the blue-green fluorescence of PFVSO3 gradually
increases in the
presence of increased concentrations of lysozyme, which allows clear naked-eye
discrimination of lysozyme with a limit of detection (LOD) as low as 1.5 gg/mL
(10
pmol).
The calibration curve for lysozyme detection is shown in FIG. 6. The PL
intensity of the NP suspension increases linearly with lysozyme concentration
and
finally saturates at a lysozyme concentration of approximately 22.5 gg/mL. The
LOD is
estimated to be 0.36 gg/mL (2.4 pmol, based on R y from six independent
measurements)
using a standard fluorometer, which is more sensitive to aptamer-based
electrochemical
and fluorescent arrays and is similar to that obtained from a standard of
ELISA. (See
Vidal, M. L., et al., Agric. Food Chem. 2005, 53, 2379- 2385, the entire
teachings of
which are incorporated herein by reference). However, the strategy of using
Apt-NP as a
platform for lysozyme detection reduces the bonding affinity (Kd) of the
aptamer to its
target. The apparent Kd in our assay is approximately 9 g/ mL (approximately
625 nM),
which is estimated from the lysozyme concentration that induces half-maximum
signal
in FIG. 6. Similar to that of aptamer-immobilized gold assays, this Kd value
is 20-fold
larger compared to that measured in solution (31 nM). (See, for example, Cox,
J. C.;
Ellington, A. D., Bioorg. Med. Chem. 2001, 9, 2525-2531, the entire teachings
are
incorporated herein by reference). The large Kd on the NP surface is
detrimental to
assay sensitivity, could be the result of. (1) the steric hindrance induced by
the folded
aptamer upon binding to lysozyme which prevents the adjacent aptamers from
folding
into G-quartet structure; (2) the binding of lysozyme on the Apt-NP surface
hampers
subsequent aptamer/lysozyme binding due to electrostatic repulsion.
Although quite a few strategies have been reported for lysozyme detection,
very
few allow label-free and visible detection and quantification of lysozyme in
real time.
1092269.1
CA 02740747 2011-05-20
4459.1012-000
-32-
Example 2. Synthesis of POSSFF and POSSFBT
Synthesis of 2-(9, 9-bis(6-bromohexyl)fluoren-2 yl)-4, 4, 5, 5-tetramethyl-1,
3,2-
dioxaborolane (1). 2-Bromo-9,9-bis- (6-bromohexyl)fluorene (4.54 g, 7.95
mmol),bis(pi
nacolatodiboron) (3.02 g, 11.93 mmol), and potassium acetate (2.94 g,29.82
mmol) were
placed in a 100-mL round bottom flask. Anhydrous dioxane (80 mL) and
[PdC12(dppf)]
(0.20 g,0.24 mmol) were added to the flask and the reaction vessel was
degassed. The
mixture was stirred at 80 C for 12 h under nitrogen. After the mixture had
been cooled
to room temperature, dioxane was removed by rotary evaporation. The residue
was
extracted with dichloromethane, and the organic phase was washed with water
and
brine, and dried over magnesium sulfate. The solvent was removed and the
residue was
purified by silica gel column chromatography (dichloromethane/hexane=1:2) to
afford 2.
Synthesis of 2, 7-dibromo-9, 9-bis(6-bromohexyl)fluorene (2). 2,7-
Dibromofluorene (1.23 g, 5 mmol) was added to a mixture of aqueous potassium
hydroxide (100 mL, 50 w%), tetrabutylammonium bromide (0.330 g, 1 mmol), and
1,2-
bis(2-bromoethoxy)ethane (13.9 g, 50 mmol) at 75 C. After 15 min, the mixture
was
cooled to room temperature. After extraction with CH2C12, the combined organic
layers
were washed successively with water, aqueous HCl (1 M), water, and brine and
then
dried over Na2SO4. After removal of the solvent and the excess 1,2-bis(2-
bromoethoxy)ethane, the residue was purified by silica gel column
chromatography
using hexane and dichloromethane (1:2) as the eluent, and recrystallized from
ethanol
and CH2C12 (5:1) to afford M2 as white needle crystals (1.50 g, 48.0%).
Synthesis of 2-(7-bromo-9, 9-bis(6-bromohexyl)fluorenyl)-9,9-bis(6-
bromohexyl)fluorene (3). 1 (2.84 g, 4.60 mmol), 2 (4.5 g, 6.9 mmol), Pd(PPh3)4
(53 mg,
0.046 mmol), potassium carbonate (4.43, 32.0 mmol) were placed in a 100 mL
round
bottom flask. A mixture of water (12 mL) and toluene (30 mL) was added to the
flask
and the reaction vessel was degassed. The mixture was vigorously stirred at 90
C for 2
days. After it was cooled to room temperature, dichloromethane was added to
the
reaction mixture. The organic portion was separated and washed with brine
before
drying over anhydrous MgSO4. The solvent was evaporated off, and the solid
residues
1092269.1
CA 02740747 2011-05-20
4459.1012-000
-33-
were purified by column chromatography on silica gel using
dichloromethane/hexane
(1:5) as eluent to afford 3.
Synthesis of 2-(7-bromo-bis(6-N,N,N-trimethylammonium)hexyl)fluorenyl)-bis(6-
N, N, N-trimethylammonium) hexyl)fluorene (4). Condensed trimethylamine (-5
mL) was
added dropwise to a solution of 3 (1 g, 0.94 mmol) in THE (10 mL) at -78 C.
The
mixture was allowed to warm to room temperature. The precipitate was
redissolved by
the addition of water (10 mL). After the mixture was cooled to -78 C,
additional
trimethylamine (-3 mL) was added. The mixture was stirred at room temperature
for 24
h. After removal of the solvent, acetone was added to precipitate 4 (1.2 mg,
98%) as
white powders.
Synthesis of 4-(9,9-bis(6-bromohexyl)-9H fluoren-2 yl)-7-
bromobenzothiadiazole (7). 2-(9,9-bis(6-bromohexyl)- fluoren-2-yl)-4,4,5,5-
tetramethyl-
1,3,2-dioxaborolane (6) (2.84 g, 4.60 mmol), 4,7-dibromobenzothiadiazole (2.16
g, 7.36
mmol), Pd(PPh3)4 (53 mg, 0.046 mmol), potassium carbonate (4.43, 32.0 mmol)
were
placed in a 100 mL round bottom flask. A mixture of water (12 mL) and toluene
(30
mL) added to the flask and the reaction vessel was degassed. The mixture was
vigorously stirred at 90 C for 2 days. After it was cooled to room
temperature,
dichloromethane was added to the reaction mixture. The organic portion was
separated
and washed with brine before drying over anhydrous MgSO4. The solvent was
evaporated off, and the solid residues were purified by column chromatography
on silica
gel using dichloromethane/hexane (1:5) as eluent to afford as grassy liquid.
1H NMR
(500 MHz, CD3OD, 6 ppm): 8.0-7.87 (m, 3 H), 7.85 (d, 1 H, J = 7.84), 7.77 (d,
1 H, J =
7.26), 7.66 (d, 1 H, J = 7.57), 7.45-7.30 (m, 3 H), 3.27 (t, 4 H, J = 6.84,
6.84), 2.14-1.97
(m, 4 H), 1.74-1.62 (m, 4 H), 1.32-1.18 (m, 4 H), 1.17-1.04 (m, 4 H), 0.83-
0.66 (m, 4
H). 13C NMR (125 MHz, CD3OD, 6 ppm): 154.00, 153.35, 152.83, 150.90, 141.76,
140.50, 135.37, 134.49, 132.31, 128.24, 128.05, 127.58, 127.08, 123.79,
122.91, 120.13,
119.89, 112.81, 55.16, 40.12, 33.92, 32.60, 29.04, 27.73, 23.61. MS (MALDI-
TOF):
m/z 707.37 [M]+.
1092269.1
CA 02740747 2011-05-20
4459.1012-000
-34-
Synthesis of 4-(9, 9-bis(6-N, N,N-trimethylammonium)hexyl)fluorenyl)-7-
bromobenzothiadiazole (8). Synthesis of Condensed trimethylamine ('5 mL) was
added
dropwise to a solution of 2 (1 g, 0.94 mmol) in THE (10 mL) at -78 C. The
mixture was
allowed to warm to room temperature. The precipitate was redissolved by the
addition of
water (10 mL). After the mixture was cooled to -78 C, additional
trimethylamine (-3
mL) was added. The mixture was stirred at room temperature for 24 h. After
removal of
the solvent, acetone was added to precipitate 3 (1.4 mg, 99%) as yellow
powders. 1H
NMR (500 MHz, CD3OD, 6 ppm): 8.38-8.26 (m, 2 H), 8.26-8.19 (m, 1 H), 8.19-8.12
(m, 1), 8.12-8.00 (m, 2 H), 7.79-7.56 (m, 3 H), 3.53-3.42 (m, 4 H), 3.09 (3,
18 H), 2.55-
2.42 (m, 4 H), 1.95-1.72 (m, 4 H), 1.53-1.31 (m, 8 H), 1.12-0.78 (m, 4H). (13C
NMR
(125 MHz, CD3OD, 6 ppm): 155.28, 154.50, 152.26, 152.055, 143.31, 142.18,
136.97,
135.38, 134.03, 129.73, 128.93, 128.46, 125.18, 124.33, 121.35, 121.05,
113.78, 67.81,
55.58, 53.68, 41.19, 30.35, 26.98, 24.92, 23.75.
Synthesis ofPOSSFF. Octavinyl POSS (5) (11.4 mg, 0.018 mmol), 4 (187 mg,
0.144 mmol), Pd(OAc)2 (3.2 mg, 14.4 mol), and P(o-tolyl)3 (24 mg, 78.4 mol)
were
placed in a 25 mL round bottom flask. A mixture of DMF (1 mL), and
triethylamine (0.5
mL) was added to the flask and the reaction vessel was degassed. The mixture
was
vigorously stirred at 100 C for 36 h. It was then filtered and the filtrate
was poured into
acetone. The precipitate was collected and washed with acetone, and was
redissolved in
water. The solution was filtered through a 0.22 m syringe driven filter to
give limpid
solution. Finally, the product was purified by dialysis against Milli-Q water
using a 3.5
kDa molecular weight cutoff dialysis membrane for 5 days. After freeze-drying,
POSSFF (74 mg, 45%) was obtained as light yellow powders.
Synthesis of POSSFBT. Octavinyl POSS (5) (11.4 mg, 0.018 mmol), 8 (119 mg,
0.144 mmol), Pd(OAc)2 (3.2 mg, 14.4 gmol), and P(o-tolyl)3 (24 mg, 78.4 mol)
were
placed in a 25 mL round bottom flask. A mixture of DMF (1 mL), and
triethylamine (0.5
mL) was added to the flask and the reaction vessel was degassed. The mixture
was
vigorously stirred at 110 C for 36 h. It was then filtered and the filtrate
was poured into
acetone. The precipitate was collected and washed with acetone, and was
redissolved in
1092269.1
CA 02740747 2011-05-20
4459.1012-000
-35-
water. The solution was filtered through a 0.22 m syringe driven filter to
give limpid
solution. Finally, the product was purified by dialysis against Milli-Q water
using a 3.5
kDa molecular weight cutoff dialysis membrane for 5 days. After freeze-drying,
POSSBT (96 mg, 73%) was obtained as yellow fibers. 'H NMR (500 MHz, CD3OD, 6
ppm): 8.47 (s, 1 H), 8.43 (d, 2 H), 8.31 (d, 1 H), 8.25 (d, 2 H), 7.74-7.76
(m, 2 H), 7.83-
7.74 (m, 1 H), 7.74-7.63 (m, 2 H), 3.54-3.38 (m, 4 H), 3.09 (s, 18 H), 2.57-
2.39 (m, 4
H), 1.95-1.80 (m, 4 H), 1.54-1.40 (m, 8 H), 1.13-0.95 (m, 4 H).
This unimolecular nanoparticle has a good water-solubility (-23 mg/mL at 24
C), as a result of its high charge density on its nanoglobular surface. The
morphology
and size of POSSFBT were studied by high-resolution transmission electron
microscopy
(HR- TEM). Spherical nanoparticles with an average diameter of 3.3 + 0.5 nm
were
observed, which coincides well with the single-molecular size of POSSFBT.
POSS compounds containing catonic, anionic or neutral R groups on either Ar or
Ar' can be synthesized by the similar method as that used to synthesize POSSFF
and
POSSFBT.
Example 3. Synthesis of P2.
The synthesis of P2 is depicted in FIG. 7.
Synthesis of 4-(9, 9'-Bis(6-bromohexyl)fluorenyl)-7-bromobenzothiadiazole (2).
2-(9,9-Bis(6-bromohexyl)fluorenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (1)
(2.84 g,
4.60 mmol), 4,7-dibromobenzothiadiazole (2.16 g, 7.36 mmol), Pd(PPh3)4 (53 mg,
0.046
mmol), potassium carbonate (4.43 g, 32.0 mmol) were placed in a 100 mL round
bottom
flask. A mixture of water (12 mL) and toluene (30 mL) were added to the flask
and the
reaction vessel was degassed. The mixture was vigorously stirred at 90 C for
2 days.
After it was cooled to room temperature, dichloromethane was added to the
reaction
mixture. The organic portion was separated and washed with brine before drying
over
anhydrous MgSO4. The solvent was evaporated off, and the solid residues were
purified
by column chromatography on silica gel using dichloromethane/hexane (1 : 5) as
eluent
to afford 2 as grassy yellow liquid (2 g, 62%). 'H NMR (500 MHz, CD3C1, 6
ppm): 8.0-
7.87 (m, 3 H), 7.85 (d, 1 H, J= 7.84 Hz), 7.77 (d, 1 H, J= 7.26 Hz), 7.66 (d,
1 H, J=
1092269.1
CA 02740747 2011-05-20
4459.1012-000
-36-
7.57 Hz), 7.45-7.30 (m, 3 H), 3.27 (t, 4 H, J= 6.84 Hz), 2.14-1.97 (m, 4 H),
1.74-1.62
(m, 4 H), 1.32-1.18 (m, 4 H), 1.17-1.04 (m, 4 H), 0.83-0.66 (m, 4 H). 13C NMR
(125
MHz, CD3Cl, 6 ppm): 154.00, 153.35, 152.83, 150.90, 141.76, 140.50, 135.37,
134.49,
132.31, 128.24, 128.05, 127.58, 127.08, 123.79, 122.91, 120.13, 119.89,
112.81, 55.16,
40.12, 33.92, 32.60, 29.04, 27.73, 23.61. MS (MALDI-TOF): m/z 707.37 [M]+.
Synthesis of 4-Bromo- 7-(7-bromo-9,9 '-bis(6-bromohexyl)fluorenyl)
benzothiadiazole (3). 2 (0.80 g, 1.14 mmol) was dissolved in dichloromethane
(20 mL)
and cooled in an ice bath. Bromine liquid (0.45 g, 2.72 mmol) was then added
slowly.
After stirring at 45 C for 12 h, the reaction was quenched with sodium
sulfite solution.
Dichloromethane was added, and the organic portion was separated and washed
with
brine before drying over anhydrous MgSO4. The solvent was evaporated off, and
the
solid residues were purified by column chromatography on silica gel using
dichloromethane/hexane (1 : 5) as eluent to afford 3 as yellow crystals (0.81
g, 90%). 'H
NMR (500 MHz, CD3C1, 6 ppm): 7.95 (d, 1H, J= 7.75 Hz), 7.91 (dd, 1 H, J= 1.33,
7.89
Hz), 7.88 (s, 1 H), 7.81 (d, 1 H, J= 7.88 Hz), 7.64 (dd, 2 H, J= 8.12, 13.86
Hz), 7.50
(m, 2 H), 3.28 (t, 4 H, J= 6.70 Hz), 2.0 (m, 4 H), 1.67 (m, 4 H), 1.23 (m, 4
H), 1.11 (m,
4 H), 0.73 (td, 4 H, J= 7.74, 15.61 Hz). 13C NMR (125 MHz, CD3C1, 6 ppm):
153.98,
153.14, 150.46, 140.60, 139.54, 135.86, 134.20, 132.29, 130.30, 128.46,
128.13, 126.23,
123.83, 121.50, 120.04, 113.04, 55.51, 40.05, 33.96, 32.61, 29.00, 27.74,
23.60. MS
(MALDI-TOF): m/z 785.44 [M]+.
Synthesis of 4-(9,9 '-Bis(6-bromohexyl)- 7-((trimethylsilyl)ethynyl)fluorenyl)-
7-
((trimethylsilyl)ethynyl)benzothiadiazole (4). A solution of trimethylsilyl
acetylene
(1.08 g, 1.55 mL, 11.0 mmol, d = 0.695 g/mL) in diisopropylamine ((iPr)2NH)
(20.0
mL) was slowly added to a solution of 3 (3.9 g, 5.0 mmol), (Ph3P)2PdC12 (0.175
g, 0.25
mmol), and Cul (0.047 g, 0.25 mmol) in (iPr)2NH (50.0 mL) under nitrogen at
room
temperature. The reaction mixture was then stirred at 70 C for 8 h. The
solvent was
removed under reduced pressure, and the residue was chromatographed on silica
gel
using hexane as eluent to give 4 (2.8 g, 65%) as yellow crystals. 1H NMR (500
MHz,
CD3C1, 6 ppm): 7.94 (m, 2 H), 7.87 (d, 1 H, J= 7.39 Hz), 7.81 (d, 1 H, J= 7.85
Hz),
1092269.1
CA 02740747 2011-05-20
4459.1012-000
-37-
7.73 (d, I H, J= 7.28 Hz), 7.69 (d, 1 H, J= 7.82 Hz), 7.50 (d, 1 H, J= 7.86
Hz), 7.47
(s, 1 H), 3.26 (t, 4 H, J = 6.79 Hz), 2.00 (m, 4 H), 1.66 (m, 4 H), 1.21 (m, 4
H), 1.09 (m,
4 H), 0.70 (td, 4 H, J= 7.70, 15.16 Hz), 0.36 (s, 9 H), 0.30 (s, 9 H). 13C NMR
(125
MHz, CD3C1, d ppm): 155.41, 153.20, 151.10, 150.87, 141.01, 140.91, 136.19,
135.16,
133.82, 131.43, 128.51, 127.27, 126.27, 123.86, 123.85, 121.85, 120.23,
119.85, 115.58,
106.05, 101.84, 100.52, 94.46, 55.27, 40.09, 33.90, 32.64, 29.00, 27.76,
23.57, 0.10,
0.04. MS (MALDI-TOF): m/z 819.70 [M]+.
Synthesis of 4-(9, 9'-Bis(6-bromohexyl)-7-ethynyljluorenyl)-7-
ethynylbenzothiadiazole (5). A KOH aqueous solution (3.0 mL, 20.0%) was
diluted
with methanol (15.0 mL) and added to a stirred solution of 4 (2.1 g, 2.5 mmol)
in THE
(20.0 mL). The mixture was stirred at room temperature for 6 h and extracted
with
hexane. The organic fraction was washed with water and dried over sodium
sulfate. The
crude product was chromatographed on silica gel using hexane as the eluent.
Recrystallization of the product from methanol gave 5 (1.6 g, 92%) as yellow
crystals.
1H NMR (500 MHz, CD3C1, 6 ppm): 7.98 (dd, 1 H, J= 1.47, 7.87 Hz), 7.94 (s, 1
H),
7.91 (d, 1 H, J = 7.34 Hz), 7.84 (d, 1 H, J = 7.90 Hz,), 7.76 (d, 1 H, J =
7.47 Hz,), 7.72
(d, 1 H, J= 7.80 Hz), 7.53 (dd, 1 H, J= 1.10, 7.63 Hz,), 7.50 (s, 1 H), 3.64
(s, 1 H),
3.27 (t, 1 H, J= 6.74, 6.74 Hz), 3.17 (s, 1 H), 2.03 (m, 4 H), 1.66 (m, 4 H),
1.22 (m, 4
H), 1.10 (m, 4 H), 0.71 (td, 4 H, J= 7.72, 15.20 Hz). 13C NMR (125 MHz, CD3C1,
6
ppm): 155.61, 153.16, 151.15, 150.97, 141.24, 140.97, 136.15, 135.69, 133.98,
131.46,
128.55, 127.25, 126.55, 123.91, 120.84, 120.31, 120.07, 114.48, 84.52, 83.70,
79.55,
77.47, 55.27, 40.06, 33.88, 32.61, 29.02, 27.75, 23.60. MS (MALDI-TOF): m/z
673.01
[M]+.
Synthesis of Neutral Hyperbranched Conjugated Polymer (P0). A Schlenk tube
charged with 5 (100 mg, 0.15 mmol) was degassed with three vacuum-nitrogen
cycles.
A solution of cyclopentadienylcobaltdicarbonyl (CpCo(CO)2) in anhydrous
toluene (1.5
mL, 0.01 M) was then added to the tube, and the system was further frozen,
evacuated,
and thawed three times to remove oxygen. The mixture was vigorously stirred at
65 C
under irradiation with a 200 W Hg lamp (operating at 100 V) placed close to
the tube for
1092269.1
CA 02740747 2011-05-20
4459.1012-000
-38-
8 h. After the mixture was cooled to room temperature, it was dropped into
methanol
(100 mL) through a cotton filter. The precipitate was collected and
redissolved in
tetrahydrofuran. The resultant solution was filtered through 0.22 m filter,
and poured
into hexane to further precipitate the product. After dried in vacuum at 40
C, PO was
obtained as brown powders (65 mg, 65%). 'H NMR (500 MHz, CDC13, 6 ppm): 8.50-
7.30 (m, 8 H), 7.20 (br, 1 H), 3.67 (s, 0.20 H), 3.30 (br, 4 H), 3.20 (s, 0.20
H), 2.0 (br, 4
H), 1.70 (br, 4 H), 1.42-1.06 (m, 8 H), 0.77 (br, 4 H). 13C NMR (125 MHz,
CDC13, 6
ppm): 155.41, 154.34, 153.73, 153.06, 151.10, 150.97, 150.91, 150.08, 141.43,
140.50,
137.87, 134.02, 131.45, 129.04, 128.53, 128.23, 126.54, 125.30, 123.97,
120.68, 120.30,
119.98, 84.60, 83.30, 80.88, 77.92, 55.27, 40.10, 33.91, 32.64, 29.06, 27.77,
23.65. Mn =
6700, MH,IMõ = 1.8.
Synthesis of Cationic HCPE (PI). Trimethylamine (2 mL) was added dropwise
to a solution of PO (50 mg) in THE (10 mL) at -78 C. The mixture was stirred
for 12 h,
and then allowed to warm to room temperature. The precipitate was redissolved
by the
addition of methanol (8 mL). After the mixture was cooled to -78 C,
additional
trimethylamine (2 mL) was added, and the mixture was stirred at room
temperature for
24 h. After removal of the solvent, acetone was added to precipitate P1 as
brown
powders (55 mg, 95%). 'H NMR (500 MHz, CD3OD, 6 ppm): 8.77-7.35 (m, 9 H), 3.63
(s, 0.20 H), 3.28 (br, 4 H), 3.05 (s, 18 H), 2.05 (br, 4 H), 1.58 (br, 4 H),
1.20 (br, 8 H),
0.77 (br, 4 H). 13C NMR (125 MHz, CD3OD, 6 ppm): 155.49, 154.10, 150.97,
141.91,
141.37, 140.70, 138.15, 134.00, 133.43, 131.07, 130.22, 128.54, 128.32,
126.23, 125.97,
123.89, 121.27, 121.13, 119.92, 87.08, 80.08, 66.31, 55.21, 52.20, 39.52,
28.73, 25.38,
23.29, 22.17.
Synthesis of Core-Shell HCPE (P2). P1 (30 mg, 0.05 mmoL alkyne) and N3-
PEG-NH2 (140 mg, 0.25 mmoL) were dissolved in DMF (5 mL). The mixture was
degassed, and then N,N,N',N",N"'-pentametyldiethylenetriamine (PMDETA) (12 mg,
0.0825 mmoL) and CuBr (11.8 mg, 0.0825 mmoL) were added. After reaction at 65
C
under nitrogen for 24 h, the reaction mixture was cooled to room temperature
and
filtered through 0.22 pm syringe driven filter. The filtrate was precipitated
into diethyl
1092269.1
CA 02740747 2011-05-20
4459.1012-000
-39-
ether to give red powders. The crude product was redissolved in water and
further
purified by dialysis against Mill-Q water using a 3.5 kDa molecular weight
cutoff
dialysis membrane for 3 days. After freeze-drying, P2 (45 mg, 78%) was
obtained as
brown fibers. 'H NMR (500 MHz, d(-DMS ), 6 ppm): 8.60-7.05 (m, 10.8 H), 4.56-
3.40
(m, 145 H), 3.00-2.65 (m, 8 H), 2.47-1.70 (m, 22 H), 1.66-0.78 (m, 12 H), 0.56
(br, 4
H).
Example 4. Synthesis of Molecular Brush (P4.1).
Molecular brushes are unique macromolecules with densely grafted side chains
on a linear polymeric backbone. Although several "grafting from" methods
including
nitroxyl radical mediated polymerization (NRMP) and atom transfer radical
polymerization (ATRP) have been utilized to synthesize neutral conjugated
polymer
based molecular brushes, the resultant polymers share the drawbacks of
incapability of
further biofunctionalization. In comparison, "grafting onto" strategy is more
versatile as
it offers a facile way to modify the brush prior to attachment onto the
backbone, while
the brush density is strongly limited by the grafting chemical reaction used.
Fortunately,
the Huisgen 1,3-dipolar cycloaddition reaction between organic azides and
alkynes,
known as click chemistry, recently emerged as an advanced chemistry
technology,
allowing post-polymerization with nearly quantitative yield, mild reaction
condition, and
broad tolerance towards various functional groups. In light of these
considerations, the
"grafting onto" strategy based on click chemistry is adopted to synthesize the
surface-
amenable CPE-g-PEG molecular brush.
The synthetic route toward the CPE-g-PEG molecular brush and its folic acid
(FA)-functionalized derivative is shown in FIG. 8. 9,9-Bis(6'-bromohexyl)-2,7-
diviny1fluorene (2.1), was synthesized in 78% yield by heating the mixture of
2,7-
dibromo-9,9-bis(6'-bromohexyl)-fluorene (1.1) and tributylvinyltin in toluene
at 100 C
for 24 h using PdC12(PPh3)2/2,6-di-tert-butylphenol as catalyst. Treatment of
2.1 with
dimethylamine in THE afforded the divinyl monomer, 9,9-bis(6'-(N,N-
dimethylamino)hexyl)-2,7-divinylfluorene (3.1). After successful determination
of the
chemical structure of 3.1 by NMR and mass spectrum, it was polymerized with
4,7-
1092269.1
CA 02740747 2011-05-20
4459.1012-000
-40-
dibromobenzothiadiazole (4.1) via a Pd(OAc)2/P(o-tolyl)3 catalyzed Heck
coupling
reaction in the mixture of DMF/TEA (2 : 1) at 100 C to afford the neutral
polymer,
poly[9,9-bis(6'-(N,N-dimethylamino)hexyl))fluorenyldivinylene-alt-4,7-
(2',1',3',-
benzothiadiazole) dibromide] (P1.1). Quaternization of P1.1 with 4-bromobut-1-
yne in
the mixture of THF/DMF/DMSO at 55 C gave the clickable cationic polymer,
poly[9,9-
bis(N-(but-3'-ynyl)-N,N-dimethylamino)hexyl))fluorenyldivinylene-alt-4,7-
(2',1',3',-
benzothiadiazole) dibromide] (P2.1). This polymer precursor has alkyne groups
at the
end of the side chains, which allows for subsequent click reaction with azide
compounds. The click reaction was carried out in DMF between P2.1 and azide-
functionalized monodispersed PEG-NH2 (N3-PEG-NH2) at 65 C using N,N,N',N",N"'-
pentametyldiethylenetriamine (PMDETA) and CuBr as the catalyst, leading to the
CPE-
g-PEG (P3.1). Finally, coupling reaction between the amine groups of P3.1 and
y-
carboxylic acid of FA using dicyclohexylcarbodiimide (DCC) and N-
hydroxysuccinimide (NHS) as the catalyst in DMSO gave the FA-functionalized
CPE-g-
PEG (P4.1). The cationic polymers P2.1, P3.1 and P4.1 were purified by micro-
filtration, precipitation, and finally dialysis against Mill-Q water using a
3.5 kDa
molecular weight cutoff dialysis membrane for 3 days.
The chemical structures of these polymers were determined by 1H NMR spectra.
As compared to P1.1, a new resonance peak at 3.08 ppm appears in the 'H NMR
spectrum of P2.1, which is assigned to the alkyne protons. The integral ratio
of the peak
at 3.08 ppm to that at 2.64 ppm (corresponding to the methylene protons near
the 9-
position of fluorene) is close to 0.48, indicating that the degree of
quaternization is
--96%. The successful click reaction is verified by the presence of a single
resonance
peak at 8.00 ppm in the 'H NMR spectrum of P3.1, which corresponds to the
proton
next to the nitrogen atom of the triazole group. Comparison of the integrated
areas
between the multiple peaks ranging from 4.56 to 3.40 ppm (assigned to the
methylene
protons of PEG) and the peak at 0.56 ppm (assigned to the methylene protons
secondly
close to the 9-position of fluorene) reveals a high PEG graft efficiency of -
90%, which
is attributed to the high activity of the click reaction using PMDETA/CuBr as
the
1092269.1
CA 02740747 2011-05-20
4459.1012-000
-41-
catalyst. After FA functionalization of P3.1, the 1H NMR spectrum becomes more
complicated for P4.1. Nevertheless, the characteristic proton resonance peak
of FA
located at 8.66 ppm is separated from those of the conjugated backbone.
Thereby, the
molar percentage of FA in P4.1 is calculated to be -60%.
Synthesis of 9,9-Bis(6'-(N,N-dimethylamino)hexyl)-2, 7-divinylfluorene (3.1):
Dimethylamine solution (5 mL, 5.6 M in absolute ethanol) was added dropwise to
a
solution of 2.1 (500 mg, 0.92 mmol) in THE (8 mL) at room temperature. After
stirring
for 12 h, additional dimethylamine solution (3 mL) was added, and the mixture
was
stirred at room temperature for 12 h. The solvent was then removed under
reduced
pressure, and the residual was washed by hexane and methanol to afford 3.1
(370 mg,
85%) as white powders. 'H NMR (500 MHz, CDC13, S ppm): 7.61 (d, 2 H, J= 7.78
Hz),
7.39 (d, 2 H, J= 7.12 Hz), 7.35 (s, 2 H), 6.79 (dd, 2 H, J= 10.85, 17.54 Hz),
5.82 (d, 2
H, J= 17.54 Hz), 5.27 (d, 2 H, J= 10.85 Hz), 2.14 (s, 12 H), 2.10 (m, 4 H),
1.96 (m, 4
H), 1.27 (m, 4 H), 1.08 (m, 8 H), 0.65 (m, 4 H). 13C NMR (125 MHz, CDC13, 6
ppm):
151.25, 140.72, 137.40, 136.51, 125.30, 120.48, 119.72, 113.04, 59.79, 54.88,
45.46,
40.33, 29.90, 27.59, 27.07, 23.68. EIMS (m/z): 472.30 (M).
Synthesis ofPoly[9,9-bis(6'-(~A;N-climethylamino)hexyl))fluorenyldivinylene-
alt-
4, 7-(2 ,1, 3 , -benzothiadiazole) dibromide] (P1.1): A Schlenk tube was
charged with 3.1
(100 mg, 0.212 mmol), 4.1 (62 mg, 0.212 mmol), Pd(OAc)2 (2 mg, 9 mmol), and
P(o-
tolyl)3 (15 mg, 49 mol) before it was sealed with a rubber septum. The
Schlenk tube
was degassed with three vacuum-argon cycles to remove air. Then, DMF (1.6 mL)
and
triethylamine (0.8 mL) was added to the Schlenk tube and the mixture was
frozen,
evacuated, and thawed three times to further remove air. The polymerization
was carried
out at 100 C under vigorous stir for 12 h. It was then filtered through 0.22
gm syringe
driven filter and the filtrate was poured into diethyl ether. The precipitate
was collected
and washed with methanol and acetone, and then dried under vacuum for 24 h to
afford
P1.1 (108 mg, 81%) as red fibers. 'H NMR (500 MHz, CDC13, 6 ppm): 8.14 (br 4
H),
7.93-7.36 (m, 8 H), 2.30 (br, 4 H), 2.13 (s, 12 H), 2.00 (br, 4 H), 1.30 (br,
4 H), 1.12 (br,
8 H), 0.73 (br, 4 H). 13C NMR (125 MHz, CDC13, d ppm): 154.05, 151.68, 141.21,
1092269.1
CA 02740747 2011-05-20
4459.1012-000
-42-
136.71, 133.89, 129.43, 127.04, 126.43, 123.92, 121.25, 120.18, 59.77, 55.17,
45.41,
40.51, 29.98, 27.59, 27.17, 23.82. Mõ = 9500, MWIMõ = 2.1.
Synthesis ofPoly[9,9-bis(N-(but-3' ynyl)-N,N-
dimethylainilio)hexyl))fluorenyldivinylene-alt-4, 7-(2 ;1, 3, -
benzothiadiazole) dibromide]
(P2.1): 4-Bromobut-l-yne (2 mL) was added to P1.1 (50 mg) in THE (5 mL) and
DMF
(5 mL), and the mixture was stirred at 55 C for 2 h. Then, DMSO (5 mL) was
added to
dissolve the precipitate. After reaction for 48 h, THE and methanol was
removed under
reduced pressure. The residual solution was then poured into acetone to give
the crude
product as dark red powders. The product was further purified by dialysis
against Mill-Q
water using a 3.5 kDa molecular weight cutoff dialysis membrane for 3 days.
After
freeze-drying, P2.1 (56 mg, 78%) was obtained as red fibers. 'H NMR (500 MHz,
d7-
DMF, 6 ppm): 8.53 (br, 4 H), 8.36-8.18 (m, 6 H), 7.99 (br, 2 H), 3.65 (br,
3.84 H), 3.42
(br, 4 H), 3.20 (br, 3.84 H), 3.08 (t, 1.92 H), 2.95 (br, 12 H), 2.44 (br, 4
H), 1.79 (br, 4
H), 1.32 (br, 8 H), 0.88 (br, 4 H).
Synthesis of PFVBT-g-PFG (P3.1): P2.1 (30 mg, 0.05 mmoL alkyne) and N3-
PEG-NH2 (140 mg, 0.25 mmoL) were dissolved in DMF (5 mL). The mixture was
degassed, and then N,N,N',N",N"'-pentametyldiethylenetriamine (PMDETA) (12 mg,
0.0825 mmoL) and CuBr (11.8 mg, 0.0825 mmoL) were added. After reaction at 65
C
under nitrogen for 24 h, the reaction mixture was cooled to room temperature
and
filtered through 0.22 m syringe driven filter. The filtrate was precipitated
into diethyl
ether to give red powders. The crude product was redissolved in water and
further
purified by dialysis against Mill-Q water using a 3.5 kDa molecular weight
cutoff
dialysis membrane for 3 days. After freeze-drying, P3.1 (45 mg, 78%) was
obtained as
red powders. 1H NMR (500 MHz, d6-DMSO, 6 ppm): 8.60-7.05 (m, 10.8 H), 4.56-
3.40
(m, -145 H), 3.00-2.65 (m, 8 H), 2.47-1.70 (m, -22 H), 1.66-0.78 (m, 12 H),
0.56 (br, 4
H).
Synthesis of PFVBT-g-PEG-FA (P4.1): The carboxylic acid group of FA (16.5
mg, 0.0335 mmol) dissolved in DMSO (0.8 mL) was pre-activated with DCC (8.25
mg,
1092269.1
CA 02740747 2011-05-20
4459.1012-000
- 43 -
0.04 mmol) and NHS (7.5 mg, 0.065 mmol) at room temperature. In the reaction,
dicyclohexylurea was formed and removed by filtration. Although FA has a- and
y-
carboxylic acid groups, y-carboxylic acid was primarily activated in the
DCC/NHS
reaction due to its higher reactivity. P3.1 (12 mg, 0.02 mmoL -NH2) was added
to the
NHS-activated FA solution. The reaction was kept at room temperature for 48 h.
The
product was further purified by dialysis against Mill-Q water using a 3.5 kDa
molecular
weight cutoff dialysis membrane for 3 days. After freeze-drying, P4.1 (22 mg,
72%) was
obtained as red powders. 'H NMR (500 MHz, d6-DMSO, 6 ppm): 8.66 (s, 1.2), 8.13-
6.56 (m, 13 H), 5.57 (br, 2.4), 4.50-2.60 (m, 157 H), 2.3-1.44 (m, 27 H), 1.36-
0.93 (m,
12 H), 0.76 (br, 4 H).
The relevant teachings of all patents, published applications and publications
cited herein are incorporated by reference in their entirety.
While this invention has been particularly shown and described with references
to example embodiments thereof, it will be understood by those skilled in the
art that
various changes in form and details may be made therein without departing from
the
scope of the invention encompassed by the appended claims.
1092269.1