Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02766627 2012-01-23
DEMANDES OU BREVETS VOLUNIINEUX
. LA PRESENTE PARTIE DE CETTE DEVIANDE OU CE BREVETS
COMPREND PLUS D'UN TOME.
CECI EST LE TOME _____________________ DE ____
NOTE: Pour les tomes additionels, veillez contacter le Bureau Canadien des
Brevets.
JUMBO APPLICATIONS / PATENTS
THIS SECTION OF THE APPLICATION / PATENT CONTAINS MORE
THAN ONE VOLUME.
THIS IS VOLUME I OF 2)
NOTE: For additional volumes please contact the Canadian Patent Office.
CA 02766627 2012-01-23
52620-2D
OPTIMIZED Fc VARIANTS AND METHODS FOR THEIR GENERATION
[001] This application is a division of application 2,524,399 filed March 26,
2004.
FIELD OF THE INVENTION
[002] The present invention relates to novel optimized Fc variants,
engineering methods for their
generation, and their application, particularly for therapeutic purposes.
BACKGROUND OF THE INVENTION
[003] Antibodies are immunological proteins that bind a specific antigen. In
most mammals,
including humans and mice, antibodies are constructed from paired heavy and
light polypeptide
chains. Each chain is made up of individual immunoglobulin (Ig) domains, and
thus the generic term
immunoglobulin is used for such proteins. Each chain is made up of two
distinct regions, referred to
as the variable and constant regions. The light and heavy chain variable
regions show significant
sequence diversity between antibodies, and are responsible for binding the
target antigen. The
constant regions show less sequence diversity, and are responsible for binding
a number of natural
proteins to elicit important biochemical events. In humans there are five
different classes of
antibodies including IgA (which includes subclasses IgA1 and IgA2), IgD, IgE,
IgG (which includes
subclasses IgG1, IgG2, lgG3, and IgG4), and IgM. The distinguishing features
between these
antibody classes are their constant regions, although subtler differences may
exist in the V region.
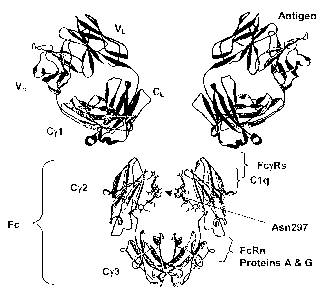
Figure 1 shows an IgG1 antibody, used here as an example to describe the
general structural
features of immunoglobulins. IgG antibodies are tetrameric proteins composed
of two heavy chains
and two light chains. The IgG heavy chain is composed of four immunoglobulin
domains linked from
N- to C-terminus in the order VH-Cy1-Cy2-Cy3, referring to the heavy chain
variable domain, constant
gamma 1 domain, constant gamma 2 domain, and constant gamma 3 domain
respectively. The IgG
light chain is composed of two immunoglobulin domains linked from N- to C-
terminus in the order VL
CL, referring to the light chain variable domain and the light chain constant
domain respectively.
[004] The variable region of an antibody contains the antigen binding
determinants of the molecule,
and thus determines the specificity of an antibody for its target antigen. The
variable region is so
named because it is the most distinct in sequence from other antibodies within
the same class. The
majority of sequence variability occurs in the complementarity determining
regions (CDRs). There are
6 CDRs total, three each per heavy and light chain, designated VH CDR1, VH
CDR2, VH CDR3, VL
CDR1, VL CDR2, and VL CDR3. The variable region outside of the CDRs is
referred to as the
framework (FR) region. Although not as diverse as the CDRs, sequence
variability does occur in the
FR region between different antibodies. Overall, this characteristic
architecture of antibodies provides
1
CA 02766627 2012-01-23
WO 2004/099249
PCT/US2004/009298
a stable scaffold (the FR region) upon which substantial antigen binding
diversity (the CDRs) can be
explored by the immune system to obtain specificity for a broad array of
antigens. A number of high-
resolution structures are available for a variety of variable region fragments
from different organisms,
some unbound and some in complex with antigen. The sequence and structural
features of antibody
variable regions are well characterized (Morea et aL, 1997, Biophys Chem 68:9-
16; Morea of at,
2000, Methods 20:267-279), and the conserved features of antibodies have
enabled the development
of a wealth of antibody engineering techniques (Maynard etal., 2000, Annu Rev
Biomed Eng 2:339-
376). For example, it is possible to graft the CDRs from one antibody, for
example a murine antibody,
onto the framework region of another antibody, for example a human antibody.
This process, referred
to in the art as "humanization", enables generation of less immunogenic
antibody therapeutics from
nonhuman antibodies. Fragments comprising the variable region can exist in the
absence of other
regions of the antibody, including for example the antigen binding fragment
(Fab) comprising VH-C71
and VH-CL, the variable fragment (Fv) comprising VH and VL, the single chain
variable fragment (scFv)
comprising Vry and VL linked together in the same chain, as well as a variety
of other variable region
fragments (Little et at, 2000, Immunol Today 21:364-370).
[005] The Fc region of an antibody interacts with a number of Fc receptors and
ligands, imparting
an array of important functional capabilities referred to as effector
functions. For IgG the Fc region, as
shown in Figure 1, comprises Ig domains Cy2 and Cy3 and the N-terminal hinge
leading into Cy2. An
important family of Fc receptors for the IgG class are the Fc gamma receptors
(FcyRs). These
receptors mediate communication between antibodies and the cellular arm of the
immune system
(Raghavan et al., 1996, Annu Rev Cell Dev Biol 12:181-220; Ravetch of aL,
2001, Annu Rev Immunol
19:275-290). In humans this protein family includes FcyRI (CD64), including
isoforms FcyRla, FcyR1b,
and FcyRIc; FcyRII (CD32), including isoforms FcyRIla (including allotypes
H131 and R131), FcyRIlb
(including FcyRIlb-1 and FcyRIlb-2), and FcyRlIc; and FcyRIII (CD16),
including isoforms FcyRIlla
(including allotypes V158 and F158) and FcyRIllb (including allotypes FcyR111b-
NA1 and FcyR111b-
NA2) (Jefferis et at., 2002, Immunol Lett 82:57-65). These receptors typically
have an extracellular
domain that mediates binding to Fc, a membrane spanning region, and an
intracellular domain that
may mediate some signaling event within the cell. These receptors are
expressed in a variety of
immune cells including monocytes, macrophages, neutrophils, dendritic cells,
eosinophils, mast cells,
platelets, B cells, large granular lymphocytes, Langerhans' cells, natural
killer (NK) cells, and yy T
cells. Formation of the Fc/FcyR complex recruits these effector cells to sites
of bound antigen,
- typically resulting in signaling events within the cells and important
subsequent immune responses
such as release of inflammation mediators, B cell activation, endocytosis,
phagocytes's, and cytotoxic
attack. The ability to mediate cytotoxic and phagocytic effector functions is
a potential mechanism by
which antibodies destroy targeted cells. The cell-mediated reaction wherein
nonspecific cytotoxic
cells that express FcyRs recognize bound antibody on a target cell and
subsequently cause lysis of
the target cell is referred to as antibody dependent cell-mediated
cytotoxicity (ADCC) (Raghavan et
2
CA 02766627 2012-01-23
WO 2004/099249
PCT/US2004/009298
al., 1996, Annu Rev Cell Dev Blot 12:181-220; Ghetie et aL, 2000, Annu Rev
Immunol 18:739-766;
Ravetch of at., 2001, Annu Rev Immunol 19:275-290). The cell-mediated reaction
wherein
nonspecific cytotoxic cells that express FcyRs recognize bound antibody on a
target cell and
subsequently cause phagocytosis of the target cell is referred to as antibody
dependent cell-mediated
phagocytosis (ADCP). A number of structures have been solved of the
extracellular domains of
human FcyRs, including FcyRIla (pdb accession code 1H9V)(Sondermann et aL,
2001, J Mol Bidl
309:737-749) (pdb accession code 1FCG)(Maxwell et aL, 1999, Nat Struct Biol
6:437-442), FcyR111)
(pdb accession code 2FCB)(Sondermann et aL, 1999, Embo J 18:1095-1103); and
FcyRIllb (pdb
accession code 1E4J)(Sondermann et al., 2000, Nature 406:267-273.). All FcyRs
bind the same
region on Fc, at the N-terminal end of the Cy2 domain and the preceding hinge,
shown in Figure 2.
This interaction is well characterized structurally (Sondermann et al., 2001,
J Mol Blot 309:737-749),
and several structures of the human Fc bound to the extracellular domain of
human FcyRIllb have
been solved (pdb accession code 1E4K)(Sondermann of aL, 2000, Nature 406:267-
273.) (pdb
accession codes 111S and 11IX)(Radaev etal., 2001, J Blot Chem 276:16469-
16477), as well as has
the structure of the human IgE Fc/FccRla complex (pdb accession code
1F6A)(Garman of aL, 2000,
Nature 406:259-266).
[006] The different IgG subclasses have different affinities for the FcyRs,
with 1gG1 and IgG3
typically binding substantially better to the receptors than IgG2 and IgG4
(Jefferis et al., 2002,
Immune! Lett 8257-65). All FcyRs bind the same region on 1gG Fc, yet with
different affinities: the
high affinity binder FcyRI has a Kd for IgG1 of 10 M-1, whereas the low
affinity receptors FcyR11 and
FcyRIII generally bind at 10-6 and i05 respectively. The extracellular domains
of FcyRIlla and
FcyRIllb are 96% identical, however FcyRIllb does not have a intracellular
signaling domain.
Furthermore, whereas FcyRI, FcyRIla/c, and FcyRIlla are positive regulators of
immune complex-
triggered activation, characterized by having an intracellular domain that has
an immunoreceptor
tyrosine-based activation motif (ITAM), FcyRilb has an immunoreceptor tyrosine-
based inhibition motif
(ITIM) and is therefore inhibitory. Thus the former are referred to as
activation receptors, and FcyRIlb
is referred to as an inhibitory receptor. The receptors also differ in
expression pattern and levels on
different immune cells. Yet another level of complexity is the existence of a
number of FcyR
polymorphisms in the human proteome. A particularly relevant polymorphism with
clinical significance
is V158/F158 FcyRIlla. Human IgG1 binds with greater affinity to the V158
allotype than to the F158
allotype. This difference in affinity, and presumably its effect on ADCC
and/or ADCP, has been
shown to be a significant determinant of the efficacy of the anti-CD20
antibody rituximab (Rituxan , a
registered trademark of IDEC Pharmaceuticals Corporation). Patients with the
V158 allotype respond
favorably to rituximab treatment; however, patients with the lower affinity
F158 allotype respond poorly
(Cartron of al., 2002, Blood 99:754-758). Approximately 10-20% of humans are
V158N158
homozygous, 45% are V158/F158 heterozygous, and 35-45% of humans are F158/F158
homozygous
3
CA 02766627 2012-01-23
WO 2004/099249
PCT/1_182004/009298
(Lehrnbecher et aL, 1999, Blood 94:4220-4232; Cartron at al., 2002, Blood
99:754-758). Thus 80-
90% of humans are poor responders, that is they have at least one allele of
the F158 FcyRIlla.
[007] An overlapping but separate site on Fc, shown in Figure 1, serves as the
interface for the
complement protein C1q. In the same way that Fc/FcyR binding mediates ADCC,
Fc/C1q binding
mediates complement dependent cytotoxicity (CDC). C1q forms a complex with the
serine proteases
C1r and Cis to form the Cl complex. C1q is capable of binding six antibodies,
although binding to
two IgGs is sufficient to activate the complement cascade_ Similar to Fc
interaction with FcyRs,
different IgG subclasses have different affinity for C1q, with IgG1 and IgG3
typically binding
substantially better to the FcyRs than IgG2 and IgG4 (Jefferis at al., 2002,
Immunol Lett 82:57-65).
There is currently no structure available for the Fc/C1q complex; however,
mutagenesis studies have
mapped the binding site on human IgG for C1q to a region involving residues
D270, K322, K326,
P329, and P331, and E333 (ldusogie etal., 2000, J Immunol 164:4178-4184;
Idusogie etal., 2001, J
Immunol 166:2571-2575).
[008] A site on Fc between the Cy2 and C73 domains, shown in Figure 1,
mediates interaction with
the neonatal receptor FcRn, the binding of which recycles endocytosed antibody
from the endosome
back to the bloodstream (Raghavan et aL, 1996, Annu Rev Cell Day 810/ 12:181-
220; Ghetie et al,
2000, Ahnu Rev Immune! 18:739-766). This process, coupled with preclusion of
kidney filtration due
to the large size of the full length molecule, results in favorable antibody
serum half-lives ranging from
one to three weeks. Binding of Fc to FcRn also plays a key role in antibody
transport. The binding
site for FcRn on Fc is also the site at which the bacterial proteins A and G
bind. The tight binding by
these proteins is typically exploited as a means to purify antibodies by
employing protein A or protein
G affinity chromatography during protein purification. Thus the fidelity of
this region on Fc is important
for both the clinical properties of antibodies and their purification.
Available structures of the rat
Fc/FcRn complex (Martin et al., 2001, Mo/ Cell 7:867-877), and of the
complexes of Fc with proteins A
and G (Deisenhofer, 1981, Biochemistry 20:2361-2370; Sauer-Eriksson et al.,
1995, Structure 3:265-
278; Tashiro at al., 1995, Curr Opin Struct Biol 5:471-481) provide insight
into the interaction of Fc
with these proteins.
[0091 A key feature of the Fc region is the conserved N-linked glycosylation
that occurs at N297,
shown in Figure 1. This carbohydrate, or oligosaccharide as it is sometimes
referred, plays a critical
structural and functional role for the antibody, and is one of the principle
reasons that antibodies must
be produced using mammalian expression systems. While not wanting to be
limited to one theory, it
is believed that the structural purpose of this carbohydrate may be to
stabilize or solubilize Fc,
determine a specific angle or level of flexibility between the Cy3 and Cy2
domains, keep the two Cy2
domains from aggregating with one another across the central axis, or a
combination of these.
Efficient Fc binding to FcyR and C1q requires this modification, and
alterations in the composition of
the N297 carbohydrate or its elimination affect binding to these proteins
(Umalia at al., 1999, Nat
iotechnol 17:176-180; Davies at al., 2001, Biotechnol Bioeng 74:288-294;
Mimura etal., 2001, J Biol
4
CA 02766627 2012-01-23
WO 2004/999249
PCPUS2004/009298
Chem 276:45539-45547.; Radaev et al., 2001, J Biol Chem 276:16478-16483;
Shields of aL, 2001, J
Biol Chem 276:6591-6604; Shields et aL, 2002, J Blot Chem 277:26733-26740;
Simmons of al., 2002,
J Immunol Methods 263:133-147). Yet the carbohydrate makes little if any
specific contact with
FcyRs (Radaev et al., 2001, J Biol Chem 276:16469-16477), indicating that the
functional role of the
N297 carbohydrate in mediating Fc/FcyR binding may be via the structural role
it plays in determining
the Fc conformation. This is supported by a collection of crystal structures
of four different Fc
glycoforms, which show that the composition of the oligosaccharide impacts the
conformation of Cy2
and as a result the Fe/RIR interface (Krapp et at., 2003, J Mol Biol 325:979-
989).
[010] The features of antibodies discussed above - specificity for target,
ability to mediate immune
effector mechanisms, and long half-life in serum - make antibodies powerful
therapeutics. Monoclonal
antibodies are used therapeutically for the treatment of a variety of
conditions including cancer,
inflammation, and cardiovascular disease. There are currently over ten
antibody products on the
market and hundreds in development. In addition to antibodies, an antibody-
like protein that is finding
an expanding role in research and therapy is the Fc fusion (Chamow et aL,
1996, Trends Biotechnol
14:52-60; Ashkenazi et al., 1997, Curr Opin Immunol 9:195-200). An Fc fusion
is a protein wherein
one or more polypeptides is operably linked to Fc. An Fc fusion combines the
Fc region of an
antibody, and thus its favorable effector functions and pharmacokinetics, with
the target-binding
region of a receptor, ligand, or some other protein or protein domain. The
role of the latter is to
mediate target recognition, and thus ills functionally analogous to the
antibody variable region.
Because of the structural and functional overlap of Fc fusions with
antibodies, the discussion on
antibodies in the present invention extends directly to Fc fusions.
[011] Despite such widespread use, antibodies are not optimized for clinical
use. Two significant
deficiencies of antibodies are their suboptimal anticancer potency and their
demanding production
requirements. These deficiencies are addressed by the present invention
[012] There are a number of possible mechanisms by which antibodies destroy
tumor cells,
including anti-proliferation via blockage of needed growth pathways,
intracellular signaling leading to
apoptosis, enhanced down regulation and/or turnover of receptors, CDC, ADCC,
ADCP, and
promotion of an adaptive immune response (Gregg of al., 1999, Curr Opin
Immunol 11:541-547;
Glennie of al., 2000, Immunol Today 21:403-410). Anti-tumor efficacy may be
due to a combination of
these mechanisms, and their relative importance in clinical therapy appears to
be cancer dependent.
Despite this arsenal of anti-tumor weapons, the potency of antibodies as anti-
cancer agents is
unsatisfactory, particularly given their high cost. Patient tumor response
data show that monoclonal
antibodies provide only a small improvement in therapeutic success over normal
single-agent
cytotoxic chemotherapeutics. For example, just half of all relapsed low-grade
non-Hodgkin's
lymphoma patients respond to the anti-CD20 antibody rituximab (McLaughlin et
al., 1998, J Clin Oncol
16:2825-2833). Of 166 clinical patients, 6% showed a complete response and 42%
showed a partial
response, with median response duration of approximately 12 months.
Trastuzumab (HerceptinO, a
CA 02766627 2012-01-23
WO 2004/099249
PCT/US2004/009298
registered trademark of Genentech), an anti-HER2/neu antibody for treatment of
metastatic breast
cancer, has less efficacy. The overall response rate using trastuzumab for the
222 patients tested
was only 15%, with 8 complete and 26 partial responses and a median response
duration and survival
of 9 to 13 months (Cobleigh etal., 1999, J Clin Onco/ 17:2639-2648). Currently
for anticancer
therapy, any small improvement in mortality rate defines success. Thus there
is a significant need to
enhance the capacity of antibodies to destroy targeted cancer cells.
[013] A promising means for enhancing the anti-tumor potency of antibodies is
via enhancement of
their ability to mediate cytotoxic effector functions such as ADCC, ADCP, and
CDC. The importance
of FcyR-mediated effector functions for the anti-cancer activity of antibodies
has been demonstrated
in mice (Clynes et at, 1998, Proc Nat! Aced Sci U S A 95:652-656; Clynes
etal., 2000, Nat Med
6:443-446), and the affinity of interaction between Fc and certain FcyRs
correlates with targeted
cytotoxicity in cell-based assays (Shields et at, 2001, J Bin! Chem 276:6591-
6604; Presta et at,
2002, Biochem Soc Trans 30:487-490; Shields et al., 2002, J Biol Chem
277:26733-26740).
Additionally, a correlation has been observed between clinical efficacy in
humans and their allotype of
high (V158) or low (F158) affinity polymorphic forms of FcyRIlla (Cartron et
a/., 2002, Blood 99:754-
758). Together these data suggest that an antibody with an Fc region optimized
for binding to certain
FcyRs may better mediate effector functions and thereby destroy cancer cells
more effectively in
patients. The balance between activating and inhibiting receptors is an
important consideration, and
optimal effector function may result from an Fe with enhanced affinity for
activation receptors, for
example FcyRI, FcyRIla/c, and FcyRIlla, yet reduced affinity for the
inhibitory receptor FcyRIlb.
Furthermore, because FcyRs can mediate antigen uptake and processing by
antigen presenting cells,
enhanced Fc/FcyR affinity may also improve the capacity of antibody
therapeutics to elicit an adaptive
immune response.
[014] Mutagenesis studies have been carried out on Fc towards various goals,
with substitutions
typically made to alanine (referred to as alanine scanning) or guided by
sequence homology
substitutions (Duncan etal., 1988, Nature 332:563-564; Lund et at, 1991, J
/mmuno/147:2657-2662;
Lund et at, 1992, Mo/ /mmuno/ 29:53-59; Jefferis etal., 1995, Immunol Lett
44:111-117; Lund etal.,
1995, Faseb J 9:115-119; Jefferis et at, 1996, Immunol Lett 54:101-104; Lund
et at, 1996, J Immunol
157:4963-4969; Armour et at, 1999, Eur J Immunol 29:2613-2624; Shields et aL,
2001, J Biol Chem
276:6591-6604; Jefferis etal., 2002, Immunol Lett 82:57-65) (US 5,624,821; US
5,885,573; PCT WO
00/42072; PCT WO 99/58572). The majority of substitutions reduce or ablate
binding with FcyRs.
\ However some success has been achieved at obtaining Fc variants with
higher FcyR affinity. (See for
example US 5,624,821, and PCT WO 00/42072). For example, Winter and colleagues
substituted the
human amino acid at position 235 of mouse IgG2b antibody (a glutamic acid to
leucine mutation) that
increased binding of the mouse antibody to human FcyRI byl 00-fold (Duncan et
a/., 1988, Nature
332:563-564) (US 5,624,821). Shields eta!, used alanine scanning mutagenesis
to map Fc residues
important to FcyR binding, followed by substitution of select residues with
non-alanine mutations
6
CA 02766627 2012-01-23
WO 2004/099249
PCT/US2004/009298
(Shields et a/., 2001, J Biol Chem 276:6591-6604; Presta etal., 2002, Biochem
Soc Trans 30:487-
490) (PCT WO 00/42072). Several mutations disclosed in this study, including
S298A, E333A, and
K334A, show enhanced binding to the activating receptor FcyRIlla and reduced
binding to the
inhibitory receptor FcyRIlb. These mutations were combined to obtain double
and triple mutation
variants that show additive improvements in binding. The best variant
disclosed in this study is a
8298A/E333A/K334A triple mutant with approximately a 1.7-fold increase in
binding to F158 FcyRIlla,
a 5-fold decrease in binding to FcyRIlb, and a 2.1-fold enhancement in ADCC.
[015] Enhanced affinity of Fc for FcyR has also been achieved using engineered
glyeoforrns
generated by expression of antibodies in engineered or variant cell lines
(Umaria of al., 1999, Nat
Blotechnol 17:176-180; Davies et aL, 2001, Biotechnol Bioeng 74:288-294;
Shields of a/., 2002, J Biol
Chem 277:26733-26740; Shinkawa of aL, 2003, J Biol Chem 278:3466-3473). This
approach has
generated substantial enhancements of the capacity of antibodies to bind
FcyRIlla and to mediate
ADCC. Although there are practical limitations such as the growth efficiency
of the expression strains
under large scale production conditions, this approach for enhancing Fc/FcyR
affinity and effector
function is promising. Indeed, coupling of these alternate glycoform
technologies with the Fc variants
of the present invention may provide additive or synergistic effects for
optimal effector function.
[016] Although there is a need for greater effector function, for some
antibody therapeutics reduced
or eliminated effector function may be desired. This is often the case for
therapeutic antibodies
whose mechanism of action involves blocking or antagonism but not killing of
the cells bearing target
antigen. In these cases depletion of target cells is undesirable and can be
considered a side effect.
For example, the ability of anti-CD4 antibodies to block CD4 receptors on T
cells makes them
effective anti-inflammatories, yet their ability to recruit FcyR receptors
also directs immune attack
against the target cells, resulting in T cell depletion (Reddy of al, 2000, J
Immunol 164:1925-1933).
Effector function can also be a problem for radiolabeled antibodies, referred
to as radioconjugates,
and antibodies conjugated to toxins, referred to as immunotoxins. These drugs
can be used to
destroy cancer cells, but the recruitment of immune cells via Fc interaction
with FcyRs brings healthy
immune cells in proximity to the deadly payload (radiation or toxin),
resulting in depletion of normal
lymphoid tissue along with targeted cancer cells (Hutchins of a/., 1995, Proc
Nat! Aced Sci U S A
92:11980-11984; White etal., 2001, Annu Rev Med 52:125-145). This problem can
potentially be
circumvented by using IgG isotypes that poorly recruit complement or effector
cells, for example IgG2
and IgG4. An alternate solution is to develop Fc variants that reduce or
ablate binding (Alegre etal.,
1994, Transplantation 57:1537-1543; Hutchins et al., 1995, Proc Nat! Aced Sc!
U S A 92:11980-
11984; Armour etal., 1999, Eur J Immunol 29:2613-2624; Reddy etal., 2000, J
Immune! 164:1925-
1933; Xu at al., 2000, Cell Immune! 200:16-26; Shields et aL, 2001, J Biol
Chem 276:6591-6604) (US
6,194,551; US 5,885,573; PCT WO 99/58572). A critical consideration for the
reduction or elimination
of effector function is that other important antibody properties not be
perturbed. Fc variants should be
engineered that not only ablate binding to FcyRs and/or Clq, but also maintain
antibody stability,
7
CA 02766627 2012-01-23
WO 2004/0992-19
PCT/US2004/009298
solubility, and structural integrity, as well as ability to interact with
other important Fc ligands such as
FcRn and proteins A and G.
[017] The present invention addresses another major shortcoming of antibodies,
namely their
demanding production requirements (Garber, 2001, Nat Biotechnol 19:184-185;
Dove, 2002, Nat
Biotechnol 20:777-779). Antibodies must be expressed in mammalian cells, and
the currently
marketed antibodies together with other high-demand biotherapeutics consume
essentially all of the
available manufacturing capacity. With hundreds of biologics in development,
the majority of which
are antibodies, there is an urgent need for more efficient and cheaper methods
of production. The
downstream effects of insufficient antibody manufacturing capacity are three-
fold. First, it dramatically
raises the cost of goods to the producer, a cost that is passed on to the
patient. Second, it hinders
industrial production of approved antibody products, limiting availability of
high demand therapeutics
to patients. Finally, because clinical trials require large amounts of a
protein that is not yet profitable,
the insufficient supply impedes progress of the growing antibody pipeline to
market.
[018] Alternative production methods have been explored in attempts at
alleviating this problem.
Transgenic plants and animals are being pursued as potentially cheaper and
higher capacity
production systems (Chadd etal., 2001, Curr Opin Biotechno/ 12:188-194). Such
expression
systems, however, can generate glycosylation patterns significantly different
from human
glycoproteins. This may result in reduced or even lack of effector function
because, as discussed
above, the carbohydrate structure can significantly impact FcyR and complement
binding. A
potentially greater problem with nonhuman glycoforms may be immunogenicity;
carbohydrates are a
key source of antigenicity for the immune system, and the presence of nonhuman
glycoforms has a
significant chance of eliciting antibodies that neutralize the therapeutic, or
worse cause adverse
immune reactions. Thus the efficacy and safety of antibodies produced by
transgenic plants and
animals remains uncertain. Bacterial expression is another attractive solution
to the antibody
production problem. Expression in bacteria, for example E. coil, provides a
cost-effective and high
capacity method for producing proteins. For complex proteins such as
antibodies there are a number
of obstacles to bacterial expression, including folding and assembly of these
complex molecules,
proper disulfide formation, and solubility, stability, and functionality in
the absence of glycosylation
because proteins expressed in bacteria are not glycosylated. Full length
unglycosylated antibodies
that bind antigen have been successfully expressed in E. coil (Simmons et al.,
2002, J lmmunol
Methods 263:133-147), and thus, folding, assembly, and proper disulfide
formation of bacterially
expressed antibodies are possible in the absence of the eukaryotic chaperone
machinery. However
the ultimate utility of bacterially expressed antibodies as therapeutics
remains hindered by the lack of
glycosylation, which results in lack effector function and may result in poor
stability and solubility. This
will likely be more problematic for formulation at the high concentrations for
the prolonged periods
demanded by clinical use.
8
CA 02766627 2012-01-23
WO 2004/099249 PCT/1.152004/009298
[019] An aglycosylated Fc with favorable solution properties arid the capacity
to mediate effector
functions would be significantly enabling for the alternate production methods
described above. By
overcoming the structural and functional shortcomings of aglycosylated Fc,
antibodies can be
produced in bacteria and transgenic plants and animals with reduced risk of
immunogenicity, and with
effector function for clinical applications in which cytotoxicity is desired
such as cancer. The present
invention describes the utilization of protein engineering methods to develop
stable, soluble Fc
variants with effector function. Currently, such Fc variants do not exist in
the art.
[020] In summary, there is a need for antibodies with enhanced therapeutic
properties Engineering
of optimized or enhanced Fc variants is a promising approach to meeting this
need. Yet a substantial
obstacle to engineering Fc variants with the desired properties is the
difficulty in predicting what amino
acid modifications, out of the enormous number of possibilities, will achieve
the desired goals,
coupled with the inefficient production and screening methods for antibodies.
Indeed one of the
principle reasons for the incomplete success of the prior art is that
approaches to Fc engineering have
thus far involved hit-or-miss methods such as alanine scans or production of
glycoforms using
different expression strains. In these studies, the Fc modifications that were
made were fully or partly
random in hopes of obtaining variants with favorable properties. The present
invention provides a
variety of engineering methods, many of which are based on more sophisticated
and efficient
techniques, which may be used to overcome these obstacles in order to develop
Fc variants that are
optimized for the desired 'properties. The described engineering methods
provide design strategies to
guide Fc modification, computational screening methods to design favorable Fc
variants, library
generation approaches for determining promising variants for experimental
investigation, and an array
of experimental production and screening methods for determining the Fc
variants with favorable
properties.
SUMMARY OF THE INVENTION
[021] The present invention provides Fc variants that are optimized for a
number of therapeutically
relevant properties. These Fc variants are generally contained within a
variant protein, that preferably
comprises an antibody or a Fc fusion protein.
[022] It is an object of the present invention to provide novel Fc positions
at which amino acid
modifications may be made to generate optimized Fc variants. Said Fc positions
include 230, 240,
244, 245, 247, 262, 263, 266, 273, 275, 299, 302, 313, 323, 325, 328, and 332,
wherein the
numbering of the residues in the Fc region is that of the EU index as in
Kabat. The present invention
describes any amino acid modification at any of said novel Fc positions in
order to generate an
optimized Fc variant.
[023] It is a further object of the present invention to provide Fc variants
that have been screened
computationally. A computationally screened Fc variant is one that is
predicted by the computational
screening calculations described herein as having a significantly greater
potential than random for
9
CA 02766627 2012-01-23
WO 2004/099249
PCT/US2004/009298
being optimized for a desired property. In this way, computational screening
serves as a prelude to or
surrogate for experimental screening, and thus said computationally screened
Fc variants are
considered novel.
[024] It is a further object of the present invention to provide Fc variants
that have been
characterized using one or more of the experimental methods described herein.
In one embodiment,
said Fc variants comprise at least one amino acid substitution at a position
selected from the group
consisting of: 230, 233, 234, 235, 239, 240, 241, 243, 244, 245, 247, 262,
263, 264, 265, 266, 267,
269, 270, 272, 273, 274, 275, 276, 278, 283, 296, 297, 298, 299, 302, 313,
318, 320, 323, 324, 325,
326, 327, 328, 329, 330, 331, 332, 333, 334, and 335, wherein the numbering of
the residues in the
Fc region is that of the EU index as in Kabat. In one embodiment, said Fc
variants comprise at least
one amino acid substitution at a position selected from the group consisting
of: 221, 222, 224, 227,
228, 230, 231, 223, 233, 234, 235, 236, 237, 238, 239, 240, 241, 243, 244,
245, 246, 247, 249, 250,
258, 262, 263, 264, 265, 266, 267, 268, 269, 270, 271, 272, 273, 274, 275,
276, 278, 280, 281, 283,
285, 286, 288, 290, 291, 293, 294, 295, 296, 297, 298, 299, 300, 302, 313,
317, 318, 320, 322, 323,
324, 325, 326, 327, 328, 329, 330, 331, 332, 333, 334, 335 336 and 428õ
wherein the numbering of
the residues in the Fc region is that of the EU index as in Kabat. In a
preferred embodiment, said Fc
variants comprise at least one substitution selected from the group consisting
of P230A, E233D,
1234D, 1234E, L234N, L234Q, L2341, L234H, 1234Y, L234I, L234V, 1234F, L23D,
L235S, L235N,
L235Q, L235T, L235H,L235Y, L235I, L235V, L235F, S239D, S239E, S239N, S239Q,
S239F, S239T,
5239H, S239Y, V240I, V240A, V240T, V240M, F241W, F241L, F241Y, F241E, F241R,
F243W,
F243L F243Y, F243R, F2430, P244H, P245A, P247V, P247G, V262I, V262A, V262T,
V262E, V263I,
V263A, V263T, V263M, V264L, V264I, V264W, V2641, V264R, V264F, V264M, V264Y,
V264E,
D265G, D265N, D265Q, D265Y, D265F, D265V, D265I, 0265L, D265H, D265T, V266I,
V266A,
V266T, V266M, S2670, S267L, S267T, S267H, S267D, 8267N, E269H, E269Y, E269F,
E269R,
E269T, E269L, E269N, 0270Q, D270T, D270H, E272S, E272K, E2721, E272Y, V273I,
K274T,
K274E, K274R, K274L, K274Y, F275W, N276S, N276E, N276R, N2761, N276Y, Y278T,
Y278E,
Y278K, Y278W, E283R, Y296E, Y296Q, Y296D, Y296N, Y296S, Y296T, Y296L, Y296I,
Y296H,
N2975, N297D, N297E, A298H, T299I, 1299L, T299A, T299S, T299V, T299H, T299F,
1299E, V3021,
W313F, E318R, K320T, K320D, K320I, K322T, K322H, V323I, S324T, S324D, S324R,
S3241, 5324V,
S324L, S324Y, N325Q, N325L, N325I, N325D, N325E, N325A, N325T, N325V, N325H,
K326L,
K326I, K326T, A327N, A327L, A327D, A327T, L328M, L328D, L328E, L328N, L328Q,
L328F, L328I,
L328V, L3281, L328H, L328A, P329F, A330L, A330Y, A330V, A330I, A330F, A330R,
A330H, A3303,
A330W, A330M, P331V, P331H, I332D, 1332E, I332N, 1332Q, I332T, I332H, 1332Y,
1332A, E3331,
E333H, E3331, E333Y, K334I, K334T, K334F, T335D, T335R, and T335Y, wherein the
numbering of
the residues in the Fc region is that of the EU index as in Kabat. In a mostly
preferred embodiment,
said Fc variants are selected from the group consisting of V264L, V264I,
F241W, F241L, F243W,
F243L, F241UF243L/V2621N2641, F241W/F243W, F241W/F243WN262AN264A, F241LN262I,
F243LN2641, F243LN2621N264W, F241Y/F243YIV262TN264T, F241E/F243RN262EN264R,
CA 02766627 2012-01-23
WO 2004/099249
PCT/1152004/009298
F241E/F243QN262TN264E, F241R/F243QN262TN264R, F241E/F243YN262TN264R, L328M,
L328E, L328F, 1332E, L328M/I332E, P244H, P245A, P247V, W313F,
P244H/P245A/P247V, P247G,
V2641/1332E, F241E/F243RN262EN264R/1332E, F241E/F243QN262TN264E/1332E,
F241R/F243QN262TN264R/I332E, F241E/F243YN262TN264R/1332E, S298A/I332E,
S239E/1332E,
S2390/I332E, S239E, D265G, D265N, S239E/D265G, S239E/D265N, S239E/D265Q,
Y296E,
Y296Q, 12991, A327N, S267Q/A327S, S267L/A327S , A327L, P329F, A330L, A330Y,
I332D, N297S,
N297D, N297S/I332E, N2970/I332E, N297E/I332E, D265Y/N2970/I332E,
D265Y/N297D/T299L/1332E, D265F/N297E/1332E, L3281/1332E, L328Q/1332E, 1332N,
13320, V2641,
V264F, V240I, V263I, V266I, T299A, T299S, T299V, N325Q, N325L, N325I, S239D,
S239N, S239F,
S239D/1332D, S239D/1332E, S239D/1332N, S239D/13320, 8239E/1332D, S239E/I332N,
S239E/I3320, S239N/I332D, S239N/1332E, S239N/I332N, S239N/I332Q, S239Q/13320,
S2390/I332N, S2390/I332Q, Y296D, Y296N, F241Y/F243YN262TN264T/N297D/I332E,
A330Y/1332E, V2641/A330Y/I332E, A330U1332E, V2641/A330L/1332E, L234D, L234E,
L234N, L234Q,
L234T, L234H, L234Y, L234I, L234V, 1234F, L235D, L235S, L235N, 12350,
1235T,1235H, L235Y,
12351, 1235V, L235F, S239T, S239H, 3239Y, V240A, V240T, V240M, V263A, V263T,
V263M,
V264M, V264Y, V266A, V266T, V266M, E269H, E269Y, E269F, E269R, Y296S, Y296T,
Y296L,
Y296I, A298H, T299H, A330V, A330I, A330F, A330R, A330H, N325D, N325E, N325A,
N3251,
N325V, N325H, L328D/I332E, L328E/I332E, L328N/I332E, L3280/1332E, L328V/I332E,
L328T/I332E, L328H11332E, L3281/I332E, L328A, 1332T, 1332H, I332Y, 1332A,
S239EN2641/1332E,
S2390N2641/1332E, S239EN2641/A330Y/I332E, S239EN2641/S298A/A330Y/1332E,
S2390/N297D/I332E, S239E/N297D/1332E, S239D/D265V/N297D/1332E,
S239D/D2651/N2970/1332E, S239D/D265L/N297D/1332E, S239D/D265F/N297D/1332E,
S239D/D265Y/N297D/1332E, S2390/D265H/N297D/1332E, S239D/D2651/N297D/1332E,
V264E/N2970/1332E, Y2960/N297D/1332E, Y296E/N297D/I332E, Y296N/N297D/1332E,
Y2960/N297D/1332E, Y296H/N297D/I332E, Y296T/N297D/I332E, N297D/T299V/I332E,
N2970/T2991/I332E, N297D/T299UI332E, N2970/1299F/1332E, N297D/1299H/1332E,
N297D/1299E/I332E, N297D/A330Y/1332E, N297D/S298A/A330Y/I332E,
S239D/A330Y/I332E,
S239N/A330Y/1332E, S239D/A330U1332E, S239N/A330L/1332E, V2641/S298A/1332E,
S239D/S298A/1332E, S239N/S298N1332E, S239D1V2641/1332E, S239DN2641/S298N1332E,
S239DN2641/A3301/1332E, L328N, 1328H, 8239D/1332E/A3301,
N297D/1332E/S239D/A330L,
P230A, E233D, P230A/E2330, P230A/E23313/1332E, S267T, S267H, S267D, S267N,
E2691, E2691,
E269N, D2700, D270T, D270H, E272S, E272K, E2721, E272Y, V273I, K274T, K274E,
K274R,
K274L, K274Y, F275W, N276S, N276E, N276R, N276L, N276Y, Y278T, Y278E, Y278K,
Y278W,
E283R, V3021, E318R, K320T, K320D, K320I, K3221, K322H, V323I, S3241, 8324D,
S324R, S324I,
S324V, S324L, S324Y, K3261, K326I, K326T, A327D, A3271, A3306, A330W, A330M,
P331V,
P331H, E333T, E333H, E3331, E333Y, K334I, K334T, K334F, 1335D, T335R,
T335Y,12341/235D,
V2401N2661, S239D/A330Y/I332E/L2341, 8239D/A330Y/1332E/1235D,
S239D/A330Y/1332EN2401,
S239D/A330Y/1332EN264T, S239D/A330Y/1332EN2661, S239D/A330Y/I332EJK326E,
11
CA 02766627 2012-01-23
WO 2004/099249 PCT/IJS2004/009298
S2390/A330Y/1332E/K326T, S2390/N297D/1332E/A330Y,
S239D/N297D/1332EJA330Y/F241S/F243HN262TN2641, S239D/N297D/1332E/L235D, and
S239D/N2970/1332E/K326E, wherein the numbering of the residues in the Fc
region is that of the EU
index as in Kabat.
[025] It is a further object of the present invention to provide Fc variants
that are selected from the
group consisting of D221K, D221Y, K222E, K222Y, T223E, T223K, H224E, H224Y,
T225E, 1225,
T225K, T225W, P227E, P227K, P227Y, P227G, P228E, P228K, P228Y, P228G, P230E,
P230Y,
P230G, A231E, A231K, A231Y, A231P, A231G, P232E, P232K, P232Y, P232G, E233N,
E233Q,
E233K, E233R, E233S, E2331,.E233H, E233A, E233V, E233L, E2331, E233F, E233M,
E233Y,
E233W, E233G, L234K, L234R, L234S, L234A, L234M, L234W, L234P, L234G, L235E,
L235K,
L235R, L235A, L235M, L235W, L235P, L235G, G236D, G236E, G236N, G2360, G236K,
G236R,
G236S, G236T, G236H, G236A, G236V, G236L, G236I, G236F, G236M, G236Y, G236W,
G236P,
G237D, G237E, G237N, G2370, G237K, G237R, G237S, G237T, G237H, G237V, G237L,
G237I,
G237F, G237M, G237Y, G237W, G237P, P238D, P238E, P238N, P2380, P238K, P238R,
P238S,
P2381, P238H, P238V, P238L, P238I, P238F, P238M, P238Y, P238W, P238G, S2390,
8239K,
S239R, 8239V, S239L, S239I, S239M, S239W, S239P, S239G, F241D, F241E, F241Y,
F243E,
K246D, K246E, K246H, K246Y, D2490, D249H, D249Y, R255E, R255Y, E258S, E258H,
E258Y,
T260D, T260E, T260H, T260Y, V262E, V262F, V264D, V264E, V264N, V264Q, V264K,
V264R,
V264S, V264H, V264W, V264P, V264G, 02650, 0265K, D265R, D265S, D265T, 0265H,
D265V,
D265L, 0265I, D265F, D265M, D265Y, D265W, 0265P, S267E, 82670, S267K, 8267R,
S267V,
8267L, 8267I, 8267F, S267M, S267Y, 8267W, S267P, H268D, H268E, H2680, H268K,
H268R,
H268T, H268V, H268L, H268I, H268F, H268M, H268W, H268P, H268G, E269K, E269S,
E269V,
E2691, E269M, E269W, E269P, E269G, 0270R, D270S, 0270L, D270I, 0270F, 0270M,
0270Y, .
0270W, D270P, 0270G, P271D, P271E, P271N, P2710, P271K, P271R, P271S, P271T,
P271H,
P271A, P271V, P271L, P271I, P271F, P271M, P271Y, P271W, P271G, E272D, E272R,
E272T,
E272H, E272V, E272L, E272F, E272M, E272W, E272P, E272G, K2740, K274N, K274S,
K274H,
K274V, K274I, K274F, K274M, K274W, K274P, K274G, F275L, N276D, N276T, N276H,
N276V,
N2761, N276F, N276M, N276W, N276P, N276G, Y278D, Y278N, Y278Q, Y278R, Y278S,
Y278H,
Y278V, Y278L, Y278I, Y278M, Y278P, Y278G, 0280K, 0280L, 0280W, 0280P, 0280G,
G281D,
G281K, G281Y, G281P, V282E, V282K, V282Y, V282P, V282G, E283K, E283H, E283L,
E283Y,
E283P, E283G, V284E, V284N, V284T, V284L, V284Y, H285D, H285E, H2850, H285K,
H285Y,
- H285W, N286E, N286Y, N286P, N286G, K288D, K288E, K288Y, K290D, K290N, K290H,
K290L,
K290W, P291D, P291E, P2910, P291T, P291H, P291 I, P291G, R2920, R292E, R292T,
R292Y,
E293N, E293R, E293S, E293T, E293H, E293V, E293L, E2931, E293F, E293M, E293Y,
E293W,
E293P, E293G, E294K, E294R, E294S, E294T, E294H, E294V, E294L, E2941, E294F,
E294M,
E294Y, E294W, E294P, E294G, 02950, 0295E, 0295N, 0295R, 0295S, 0295T, 0295H,
Q295V,
\0295I, 0295F, 0295M, 0295Y, Q295W, 0295P, 0295G, Y296K, Y296R, Y296A, Y296V,
Y296M,
Y296G, N2970, N297K, N297R, N297T, N297H, N297V, N297L, N297I, N297F, N297M,
N297Y,
12
\
CA 02766627 2017-01-04
, -
52620-2D
N297W, N2970, N2970, 62980, 6298E, 82980,8298K, S298R, S2981, 8298F, S298M,
S298Y,
S298W, 1299D, T299E,1299N, T2990, 1299K, 1299R, 1299L, 1299F, 1299M, 1299Y,
1299W,
12991', T2990, Y3000, Y300E, Y300N, Y3000, Y300K, Y300R, Y3008, Y300T, Y300H,
Y300A,
Y300V, Y300M, Y300W, Y300P, Y3000, R301D, R301E, R301H, R301Y, V303D, V303E,
V303Y,
S304D, 8304N, S3041, 83041-1, 53041, V305E, V3051, V305Y, K317E, 1(3170,
E316Q, E318H,
*E318L, E318Y,K320N, 1<3208, K320H, K320V, 1(3201, K320F, K320Y, 1<320W,
K320P, 1<320G,
K322D, K322S, K322V, K3221, K322F, K322Y, 1<322W, K32213, 1(3220, 8324H,
8324F, 5324M,
8324W, S324P, S3240, N325K, N325R, N325S, N325F, N325M, N325Y, N326W, N325P,
N3250,
K326P, A327E; A327K, A327R, A327H, A327V, A327I, A327F, A327M, A327Y, A327W,
A327P,
L3280, 13280, 1328K, 1328R, 13288, 13281, L328V, 1328I, 1328Y, 1328W, L328P,
L3280, P329D,
P329E, P329N, P3290, P329K, P329R, P3295, P3291, P329H, P329V, P329L, P3291,
P329M,
P329Y, P329W, P3290, A330E, A330N, A3301, A330P, A3300, P331D, P3310, P331R,
P3311,
P331L, P3311, P331F, P331M, P331Y, P331W, 1332K, 1332R,13326,
1332V,13321,1332F,1332M,
= 1332W,1332P,=13320, E333L, E333F, E333M, E33312; 1(33412,1335N, 1335S,
13351-1, 1335V, 1335L,
13351, 1335F, 1335M, 1335W, T335P,13350,1336E,1336K,1336Y, B337E, 5337N, and
8337H, =
wherein the numbering of the residues in the Fc reglarls that of the EU Index
as In Kabat.
[026] It is a further object of some embodiments of the present
invention to provide an Fc variant that binds ,
vWth greater affinity to one or MOle FcyRs. In one embodiment, said Fc
variants have affinity for an FcyR that is
. more than 1-fold greater than that of the parent Fc polypeptide. In an
alternate embodiment, said Fc
variants have affinity for an FcyR that Is more than 5-fold greater than that
of the parent Fc
polypeptIde. In a preferred embodiment, said Fe variants have affinity for an
FcyR that is between 5-
fold and 300-fold greater than that of the parent Fc poIapUda. In one
embodiment, said Fc variants =
= comprise at least one amino acid substitutional a position selected from
the group consisting Of: 230,
233, 234, 235, 239, 240, 243, 264, 266, 272, 274,=275, 276, 278, 302, 318,
324, 325, 326, 328, 330, '
332, and 335, wherein the numbering of the residues in the Fc region Is that
of the ELI index as in
Kabat. In a preferred embodiment, said Fe variants comprise at least one amino
acid substitution
selected from the group consisting of: P230A, E2330, L234E, 1234Y, L234I,
L2350, L235S, L235Y,
= L235I, S239D, 8239E, 5239N, 82390, S2391, V240I, V240M, F2431, V2641,
V2641, V264Y, V266I,
E272Y, K274T, K274E, K274R, K274L, K274Y, F275W, N276L, Y2781, V302I, E318R,
33240, ,
= S3241, 8324V, N3251, K326I, K3261, 1328M, 13281; L328Q, L3280, 1.328V,
13281, A330Y, A330L,
A330I, I332D, 1332E, 1332N, 13320, T3350, 1335R; and 1335Y, wherein the
numbering of the
=
residues In the Fo region is that of the EU Index as in Kabat. In a mostly
preferred embodiment, said =
Fc variants are selected from the group consisting of V264I, F24311V2641,
1.328M, 1332E,
= L328M/1332E,.V2641/1332E, 8298A/1332E, 8239E/1332E, 8239011332E, S239E,
A330Y,1332D,
L3281/1332E, 13280/1332E, V264T, V240I, V266I, 82390,82390/13320,
S23913/1332E,
= 3239D/I332N, S2390/1332Q, $239E/13320, 5239E/1332N, 8239E/13320,
8239N/1332D,
5239N/1332E, 8239Q/1332D, A330Y/1332E, V2641/A330Y/1332E, A3301./1332E,
V2641/A330U1332E,
= 1234E, 1234Y, 1234I, 1235D, L235S, L235Y, L235I, 82391, V240M, V264Y,
A3301, N325T,
= 13
=
=
CA 02766627 2017-01-04
52620-2D
=
L328D/1332E, 1328V/1332E, L328T/I332E, L3281/1332E, S239E1V2641/1332E,
52390/V2641/1332E,
S239EN2641/A330Y/1332E, S239D/A330Y/I332E, S239N/A330Y/1332E,
S239D/A330U1332E,
3239N/A330L/1332E, V2641/S298A/1332E, S239p/S298A/1332E, S239N/S298A/1332E,
8239DN2641/1332E, S2390N2641/S298A/1332E, S239D/V2641/A330L/1332E,
S2390/1332E/A3301,
P230A, P230A/E2330/1332E, E272Y, K274T, K274E, K274R, K274L, K274Y, F275W,
N276L, Y278T,
V302I, E318R, S324D, S324I, 8324V, K326I, K326T, 7335D, T335R, T335Y,
V240IN2661,
S239D/A330Y/1332E/L2341, S2390/A330Y/1332E/L235D, 8239D/A330Y/1332E/V2401,
S2390/A330Y/1332EN2641, S239D/A330Y/I332E/K326E, and 8239D/A330Y/1332E/K3261,
wherein
the numbering of the residues In the Fc region Is that of the EU Index as in
Kabat.
[027] __________________ It isa further objectd some embodt nenis &he
rxEsent invention to povtle Fc variant that have a FcyRIII a-
fold:FcyRilb-fold ratio greater than 1:1. In one embodiment, said Fc variants
have a FcyRIlla-
fold:FcyRilb-fold ratio greater than 11:1. In a preferred embodiment, said Fc
variants have a FcyRIlia-
fold:Fcylillb-fold ratio between 11:1 and 86:1. In one embodiment, said Fc
variants comprise at least
one amino acid substitution at a position selected from the group consisting
of: 234, 235, 239, 240,
264, 296, 330, and 1332, wherein the numbering of the residues in the Fc
region is that of the EU
index as in Kabat. In a preferred embodiment, said Fc variants comprise at
least one amino acid
substitution selected from the group consisting of: L234Y, L234I, L2351,
S239D, S239E, S239N,
6239Q, V240A, V240M, V264I, V264Y, Y296Q, A330L, A330Y, A330I, I332D, and
1332E, wherein the
numbering of the residues in the Fc region is that of the EU index as in
Kabat. In a mostly preferred
embodiment, said Fc variants are selected from the group consisting of: 1332E,
V2641/I332E,
S239E/I332E, S2390/1332E, Y296Q, A330L, A330Y, 1332D, S239D, 8239D/1332E,
A330Y/I332E,
V2641/A330Y/1332E, A330U1332E, V2641/A330U1332E, L234Y, 12341, 1.2351, V240A,
V240M, V264Y,
A3301, 82390/A330L/1332E, S239D/8298A/1332E, S239N/S298A/1332E,
S239D/V2641/1332E,
8239DN2641/8296A/I332E, and 3239DN2641/A330U1332E, wherein the numbering of
the residues
in the Fc region is that of the EU index as in Kabat. -
[028] It is a further object of some embodiments of the present invention
to provide Fc variants that mediate effector
function more effectively in the presence of effector cells. In one
embodiment, said Fc vaiants
mediate ADCC that is greater than that mediated by the parent Fc polypeptide.
In a preferred
embodiment, said Fc variants mediate ADCC that Is more than 6-fold greater
than that mediated by
the parent Fc polypeptide. In a mostly preferred embodiment, said Fc variants
mediate ADCC that is
between 5-fold and 1000-fold greater than that mediated by the parent Fc
polypeptide. In one
embodiment, said Fc variants comprise at least one amino acid substitution at
a position selected
from the group consisting of: 230, 233, 234, 235, 239,.240, 243, 264, 266,
272, 274, 275, 276, 278,
302, 318, 324, 325, 326, 328, 330, 332, and 335; wherein the numbering of the
residues in the Fc
region is that of the EU index as In Kabat.. In a preferred embodiment, said
Fc variants comprise at
least one amino acid substitutions selected from the group consisting of:
P230A, E233D, L234E,
L234Y, L234I, L235D, L2358, L235Y, L235I, 8239D, S239E, S239N, 8239Q, 8239T,
V240I, V240M,
14
=
=
CA 02766627 2017-01-04
52620-2D
F2431, V264I, V264T, V264Y, V266I, E272Y, K274T, K274E, K274R, K274L, K274Y,
F275W, N276L,
Y2781, V3021, E318R, S3240, S3241, S324V, N3251, 1<3261, K326T, L328M, L3281,
L328Q, L3280,
L328V, L328T, A330Y, A330L, A330I, 1332D, 1332E, I332N, I332Q, 7335D, T335R,
and T335Y,
wherein the numbering of the residues in the Fc region is that of the EU index
as In Kabat. In a
mostly preferred embodiment, said Fc variants are selected from the group
consisting of: V2641,
F243UV2641, 1328M, 1332E, 1328M/1332E, V2641/1332E, 5298A/1332E, 8239E/1332E,
S239Q/1332E,
S239E, A330Y, 13320, L3281/1332E, L328Q/1332E, V264T, V2401, V2661, S2390,
S2390/13320,
82390/1332E, S239D/1332N, S239D/1332Q, S239E/I332D, S239E/1332N, 6239E/1332Q,
S239N/I332D, S239N/1332E, 8239Q/13320, A330Y/1332E, V2641/A330Y/I332E,
A330U1332E,
V2641/A330111332E, L234E, L234Y, L234I, L235D, 1235S, L235Y, L235I, S2391,
V240M, V264Y,
A3301, N325T, L328D/1332E, L328V/1332E, L328T/I332E, L3281/I332E,
S239E1V2641/1332E,
S239QA/2641/1332E, S239EN2641/A330Y/I332E, S239D/A330Y/1332E,
S239N/A330Y/I332E,
8239D/A330L/1332E, S239N/A330U1332E, V2641/S298A/1332E, S239D/S298A/1332E,
8239N/S298A/1332E, S239D/V2641/1332E, S2390N2641/3298A/1332E,
S2390N2641/A330U1332E,
S239D/I332E/A3301, P230A, P230A/E2330/1332E, E272Y, K2741, K274E, 1(274R,
K274L, K274Y,
F275W, N276L, Y278T, V302I, E318R, S3240, S324I, S324V, K326I, K326T, T3350,
1335R, 1335Y,
V240IN2661, S2390/A330Y/1332E/L2341, 8239121/A330Y/1332E/1235D,
5239D/A330Y/1332E1V2401,
82390/A330Y/1332EN2641, 8239D/A330Y/1332E/K326E, and S239D/A330Y/1332E1K3261,
wherein
the numbering of the residues In the Fc region is that of the EU index as in
Kabat.
[029 It is a
further oiled of some embozli-nents of the present invention to provide Fc
vaiants that bind with
weaker affinity to one or more FcyRs. In one embodiment said Fc variants
comprise at least one ant* acid
substitution at a position selected from the group consisting of: 230, 233,
234, 235, 239, 240, 241,
243, 244, 245, 247, 262, 263, 264, 265, 266, 267, 269, 270, 273, 276, 278,
283, 296, 297, 298, 299,
313, 323, 324, 325, 327, 328, 329, 330, 332, and 333, wherein the numbering of
the residues In the
Fc region is that of the EU index as In Kabat. In a preferred embodiment, said
Fc variants comprise
an amino acid substitution at a position selected from the group consisting
of: P230A, E233D, L234D,
L234N, L234Q, L234T, L234H, L234V, L234F, L234I, 1235N, L235Q, L235T, L235H,
1235V, L235F,
L235D, S239E, 5239N, 82390, S239F, 8239H, 8239Y, V240A, V2401, F241W, F241L,
F241Y,
F241E, F241R, F243W, F243L F243Y, F243R, F243Q, P244H, P245A, P247V, P247G,
V262I,
V262A, V262T, V262E, V263I, V263A, V263T, V263M, V264L, V264I, V264W, V2641,
V264R,
V264F, V264M, V264E, D265G, 0265N, 02650, 0265Y, 0265F, 0265V, D265I, 0265L,
0265H,
0265T, V266A, V266T, V266M, S2670, S267L, E269H, E269Y, E269F, E269R, E2691,
E269L,
E269N, 0270Q, D2701, D270H, V2731, N276S, N276E, N276R, N276Y, Y278E, Y278W,
E283R,
Y296E, Y296Q, Y296D, Y296N, Y2968, Y2967, Y296L, Y296I, Y296H, N297S, N2970,
N297E,
A298H, T299I, T299L, 1299A, T299S, T299V, 12991-I, T299F, T299E, W313F, V3231,
S324R, S324L,
S324Y, N3250, N3251, N325I, N325D, N325E, N325A, N325V, N325H, A327N, A327L,
L328Iv1,
328E, L328N, L3280, A327D, A327T, 1328F; L328H, 1328A, 1328N, 1328H, P329F,
A330L, A330V,
A330F, A330R, A330H, 1332N, I3320, I332T, 1332H, I332Y, I332A, E3331, and
E333H, wherein the
CA 02766627 2017-01-04
52620-2D
numbering of the residues in the Fc region is that of the EU index as in
Kabat. In a mostly preferred
embodiment, said Fc variants are selected from the group consisting of: V264L,
F241W, F241L,
F243W, F243L, F241UF243LN262IN2641, F241W/F243W, F241W/F243WN262AN264A,
F241UV262I, F243UV26211V264W, F241Y/F243YN262TN264T, F241E/F243RN262EN264R,
F241E/F243QN262TN264E, F241R/F243Q/V262TN264R, F241E/F243YN262TN264R, L328M,
L328E, L328F, P244H, P245A, P247V, W313F, P244H/P245A/P247V, P247G,
F241E/F243RN262EN264R/I332E, F241E/F243YN262TN264R/1332E, D265G, D265N,
S239E/D265G, S239E/D265N, S239E/D2650, Y296E, Y296Q, T299I, A327N,
S267Q/A327S,
S2671JA3278 , A327L, P329F, A330L, N297S, N297D, N2978/1332E,1332N, I332Q,
V264F, V263I,
1299A, T2998, T299V, N325Q, N325L, N325I, S239N, S239F, 5239N/1332N,
5239N/1332Q,
8239Q/1332N, S239Q/I332Q, Y296D, Y296N, L234D, 1.234N, L234Q, L234T, L234H,
L234V, L234F,
L235N, L235Q, L2351, L235H, L235V, L235F, S239H, 5239Y, V240A, V263T, V263M,
V264M,
V266A, V2661, V266M, E269H, E269Y, E269F, E269R, Y296S, Y296T, Y296L, Y296I,
A298H,
T299H, A330V, A330F, A330R, A330H, N325D, N325E, N325A, N325V, N325H,
L328E/I332E,
L328N/1332E, L328Q/1332E, L328H/1332E, L328A, 13327,1332H, I332Y, 1332A,
L328N, L328H,
.E233D, P230A/E233D, E2691, E269L, E269N, D270Q, 0270T, D270H, V273I, N2765,
N276E,
N276R, N276Y, Y278E, Y278W, E283R, V323I, S324R, S324L, 8324Y, A327D, A3271,
E3331,
E333H, and L234I/L235D, wherein the numbering of the residues in the Fc region
is that of the EU
index as in Kabat.
[030] It is a kirther object of some embodiments dire present invention to
provide Fc vaiants that mediate ADCC in
the presence of effector cells iESS effectively. In one embodiment, sad Fc
variants comprise at least one
amino acid substitution at a position selected from the group consisting of:
230, 233, 234, 235, 239,
240, 241, 243, 244, 245, 247, 262, 263, 264, 265, 266, 267, 269, 270, 273,
276, 278, 283, 296, 297,
298, 299, 313, 323, 324, 325, 327, 328, 329, 330, 332, and 333, wherein the
numbering of the
residues in the Fc region is that of the EU Index as in Kabat. In a preferred
embodiment, said Fc
variants comprise at least one amino acid substitution at a position selected
from the group consisting
. of: P230A, E233D, L234D, L234N, L234Q, L234T, L234H, L234V, L234F, L234I,
L235N, L235Q,
L2351, L235H, L235V, 1235F, L235D, S239E, 5239N, 5239Q, 8239F, 8239H, 8239Y,
V240A,
V240T, F241W, F241L, F241Y, F241E, F241R, F243W, F243L F243Y, F243R, F243Q,
P244H,
P245A, P247V, P247G, V262I, V262A, V2621, V262E, V263I, V263A, V263T, V263M,
V264L, V264I,
V264W, V264T, V264R, V264F, V264M, V264E, D265G, D265N, D265Q, D265Y,
D265F,D265V,
D265I, D265L, D265H, D2651, V266A, V2661, V266M, S267Q, S267L, E269H, E269Y,
E269F,
E269R., E2691, E269L, E269N, D270Q, D2701, D270H, V273I, N276S, N276E, N276R,
N276Y,
Y278E, Y278W, E283R, Y296E, Y296Q, Y296D, Y296N, Y2965, Y296T, Y296L, Y296I,
Y296H,
N297S, N297D, N297E, A298H, T2991, 1299L, T299A, T299S, 1299V, T299H, T299F,
T299E,
W313F, V323I, S324R, S324L, 8324Y, N325Q, N325L, N325I, N325D, N325E, N325A,
N325V,
N325H, A327N, A327L, L328M, 328E, L328N, L328Q, A327D, A3271, L328F, L328H,
L328A, L328N,
L328H, P329F, A330L, A330V, A330F, A330R, A330H, 1332N, I332Q, I332T, I332H,
I332Y, I332A,
16
CA 02766627 2017-01-04
52620-2D
E333T, and E333H, wherein the numbering of the residues in the Fc region is
that of the EU index as
In Kabat. In a mostly preferred embodiment, said Fc variants are selected from
the group consisting
of: V264L, F241W, F241L, F243W, F243L, F241UF243UV262IN2641, F241W/F243W,
F241W/F243WN262A/V264A, F241UV2621, F243UV2621N264W, F241Y/F243YN262TN264T,
F241E/F243RN262E/V264R, F241E/F243QN262TN264E, F241R/F243QN262TN264R,
F241E/F243YN262TN264R, L328M, L328E, L328F, P244H, P245A, P247V, W313F,
P244H/P245A/P247V, P247G, F241E/F243R/V262E/V264R/I332E,
F241E/F243YN262TN264R/1332E, D265G, D265N, 5239E/D265G, 5239E/0265N,
S239E/0265Q,
Y296E, Y296Q, T2991, A327N, S267Q/A3275, S267UA327S A327L, P329F, A330L,
N297S,
N2970, N2978/I332E, I332N, I332Q, V264F, V263I, T299A, T299S, T299V, N325Q,
N325L, N325I,
S239N, S239F, 5239N/1332N, S239N/13320, 5239Q/1332N, S239Q/13320, Y296D,
Y296N, L2340,
L234N, L234Q, L2341, L234H, L234V, L234F, 1235N; L2350, L2357, L235H, L235V,
L235F, S239H,
S239Y, V240A, V263T, V263M, V264M, V266A, V266T, V266M, E269H, E269Y, E269F,
E269R,
Y296S, Y296T, Y296L, Y296I, A298H, 1299H, A330V, A330F, A330R, A330H, N325D,
N325E,
N325A, N325V, N325H, 1328E/I332E, L328N/I332E, 1_328Q/1332E, L328H/I332E,
L328A, I332T,
1332H,1332Y,1332A, L328N, L328H, E233D, P230A/E233D, E269T, E269L, E269N,
0270Q, D270T,
0270H, V273I, N2768, N276E, N276R, N276Y, Y278E, Y278W, E283R, V323I, S324R,
S324L,
S324Y, A327D, A327T, E333T, E333H, and L2341/L235D, wherein the numbering of
the residues in
the Fc region is that of the EU index as in Kabat.
[031] It is a
further object of some embodiments of the present invention to provide Fc
variants that have
improved function and/or solution properties as compared to the aglycosylated
form of the parent Fc
polypeptide. Improved functionality herein includes but is not limited to
binding affinity to an Fc ligand.
Improved solution properties herein includes but is not limited to stability
and solubility. In one
embodiment, said aglycosylated Fc variants bind to an FcyR with an affinity
that is comparable to or
better than the glycosylated parent Fc polypeptide. In an alternate
embodiment, said Fc variants bind
to an FcyR with an affinity that Is within 0.4-fold of the glycosylated form
of the parent Fc polypeptide.
In one embodiment, said Fc variants comprise at least one amino acid
substitution at a position
selected from the group consisting of: 239, 241, 243, 262, 264, 265, 296, 297,
330, and 332, wherein
the numbering of the residues in the Fc region is that of the EU index as in
Kabat. In a preferred
embodiment, said Fc variants comprise an amino acid substitution selected from
the group consisting
of: S2390, S239E, F241Y, F243Y, V2621, V264T, V264E, 0265Y, 0265H, 0265V,
D265I, Y296N,
N2970, A330Y, and 1332E, wherein the numbering of the residues in the Fc
region is that of the EU
index as in Kabat. In a mostly preferred embodiment, said Fc variants are
selected from the group
consisting of: N297D/I332E, F241Y/F243YN262TN264T/N297D/1332E,
S2390/N297D/1332E,
S239E/N297D/1332E, S239D/D265Y/N297D/1332E, S2390/D265H/N297D/I332E,
V264E/N297D/1332E, Y296N/N297D/1332E, N297D/A330Y/I332E,
S239D/0265V/N2970/1332E,
8239D/D265I/N297D/1332E, and N297DIS298A/A330Y/1332E, wherein the numbering of
the residues
in the Fc region is that of the EU index as In Kabat.
17
81624197
[032] The present invention also provides methods for engineering optimized
Fc variants. It is an object of the present invention to provide design
strategies that
may be used to guide Fc optimization. It is a further object of the present
invention to
provide computational screening methods that may be used to design Fc
variants. It
is a further object of the present invention to provide methods for generating
libraries
for experimental testing. It is a further object of some embodiments of the
present
invention to provide experimental production and screening methods for
obtaining
optimized Fc variants.
[033] The present invention provides isolated nucleic acids encoding the Fc
variants described herein. The present invention provides vectors comprising
said
nucleic acids, optionally, operably linked to control sequences. The present
invention
provides host cells containing the vectors, and methods for producing and
optionally
recovering the Fc variants.
[034] The present invention provides novel antibodies and Fc fusions that
comprise the Fc variants disclosed herein. Said novel antibodies and Fc
fusions may
find use in a therapeutic product.
[035] The present invention provides compositions comprising antibodies and
Fc fusions that comprise the Fc variants described herein, and a
physiologically or
pharmaceutically acceptable carrier or diluent.
[036] The present invention contemplates therapeutic and diagnostic uses
for
antibodies and Fc fusions that comprise the Fc variants disclosed herein.
[036a] In one aspect, the invention provides a polypeptide comprising an
Fc variant of a parent Fc polypeptide, said parent Fc polypeptide comprising
an
Fc region, wherein said Fc variant comprises an amino acid modification in the
Fc region of said parent Fc polypeptide at position 328, wherein said Fc
variant
exhibits a decrease in affinity for FcrRIlla as compared to the parent Fc
polypeptide, and wherein numbering is according to the EU index.
18
CA 2766627 2018-10-01
CA 02766627 2014-02-27
52620-2D
[036b] In another aspect, the invention provides a pharmaceutical
composition comprising a polypeptide as described above and a pharmaceutically
acceptable carrier.
[036c] In another aspect, the invention provides use of the
polypeptide as
described above to produce a medicament.
BRIEF DESCRIPTION OF THE DRAWINGS
[037] Figure 1. Antibody structure and function. Shown is a model of a full
length human IgG1 antibody, modeled using a humanized Fab structure from pdb
accession code 10E1 (James eta!, 1999, J Mc/ Bic)/ 289:293-301) and a human
IgG1
Fc structure from pdb accession code 1DN2 (DeLano et al., 2000, Science
287:1279-
1283). The flexible hinge that links the Fab and Fc regions is not shown. IgG1
is a
homodimer of heterodimers, made up of two light chains and two heavy chains.
The Ig
domains that comprise the antibody are labeled, and include VL and CL for the
light
chain, and VH, Cgarnmal (Cy1), Cgamma2 (C72), and Cgamma3 (Cy3) for the heavy
chain. The Fc region is labeled. Binding sites for relevant proteins are
labeled,
including the antigen binding site in the variable region, and the binding
sites for Fc7Rs,
FcRn, C1q, and proteins A and G in the Fc region.
[038] Figure 2. The Fc/FcyRIllb complex structure 11IS. Fc is shown as a
gray
ribbon diagram, and FcyRIllb is shown as a black ribbon. The N297 carbohydrate
is
shown as black sticks.
[039] Figure 3. The amino acid sequence of the heavy chain of the antibody
alemtuzumab (Campath , a registered trademark of Ilex Pharmaceuticals LP),
illustrating positions numbered sequentially (2 lines above the amino acid
sequence)
and positions numbered according to the EU index as in Kabat (2 lines below
the
amino acid sequence). The approximate beginning of Ig domains VH1, Cyl , the
hinge,
Cy2 and C73 are also labeled above the sequential numbering. Polymorphisms
have
been observed at a number of Fc positions, including but not limited to Kabat
18a
CA 02766627 2012-01-23
WO 2004/099249 PCT/US2004/009298
270, 272, 312, 315, 356, and 358, and thus slight differences between the
presented sequence and
sequences in the prior art may exist.
[040] Figure 4. Experimental library residues mapped onto the Fc/FcyRIllb
complex structure 11IS.
Fc is shown as a gray ribbon diagram, and FcyRIllb is shown as a black ribbon.
Experimental library
residues are shown in black, the N297 carbohydrate is shown in grey.
[041] Figure 5. The human IgG1 Fc sequence showing positions relevant to the
design of the Fc
variant experimental library. The sequence includes the hinge region, domain
Cy2, and domain C73.
Residue numbers are according to the EU index as in Kabat. Positions relevant
to the experimental
library are underlined. Because of observed polymorphic mutations at a number
of Fc positions, slight
differences between the presented sequence and sequences in the literature may
exist.
[042] Figure 6. Expression of Fc variant and wild type (WT) proteins of
alemtuzumab in 293T cells.
Plasmids containing alemtuzumab heavy chain genes (WT or variants) were co-
transfected with
plasmid containing the alemtuzumab fight chain gene. Media were harvested 5
days after
transfection. For each transfected sample, 10u1 medium was loaded on a SOS-
PAGE gel for Western
analysis. The probe for Western was peroxidase-conjugated goat-anti human 1gG
(Jackson lmmuno-
Research, catalog # 109-035-088). WT: wild type alemtuzumab; 1-10: alemtuzumab
variants. H and
=
L indicate antibody heavy chain and light chain, respectively.
[043) Figure 7. Purification of alemtuzumab using protein A chromatography. WT
alemtuzumab
proteins was expressed in 293T cells and the media was harvested 5 days after
transfection. The
media were diluted 1:1 with PBS and purified with protein A (Pierce, Catalog
#20334). 0: original
sample before purification; FT: flow through; E: elution; C: concentrated
final sample. The left picture
shows a Simple Blue-stained SDS-PAGE gel, and the right shows a western blot
labeled using
peroxidase-conjugated goat-anti human IgG.
[044] Figure 8. Production of deglycosylated antibodies. Wild type and
variants of alemtuzumab
were expressed in 293T cells and purified with protein A chromatography.
Antibodies were incubated
with peptide-N-glycosidase (PNGase F) at 37 C for 24h. For each antibody, a
mock treated sample (-
PNGase F) was done in parallel. WT: wild-type alemtuzumab; #15, #16, #17, #18,
#22: alemtuzumab
variants F241E/F243RN262EN264R, F241E/F243Q/V262TN264E, F241R/F2430N262TN264R,
F241E/F243YN262TN264R, and 1332E respectively. The faster migration of the
PNGase F treated
versus the mock treated samples represents the deglycosylated heavy chains.
[045] Figure 9. Alemtuzumab expressed from 293T cells binds its antigen. The
antigenic C052
peptide, fused to GST, was expressed in E. coil BL21 (DE3) under IPTG
induction. Both uninduced
and induced samples were run on a SDS-PAGE gel, and transferred to PVDF
membrane. For
western analysis, either alemtuzumab from Sotec (a-CD52, Sotec) (final
concentration 2.5ng/u1) or
media of transfected 293T cells (Campath, Xencor) (final alemtuzumab
concentration approximately
0.1-0.2ng/u1) were used as primary antibody, and peroxidase-conjugated goat-
anti human IgG was
19
CA 02766627 2012-01-23
WO 2004/099249 PCT/US2004/009298
used as secondary antibody. M: pre-stained marker; U: un-induced sample for
GST-CD52; induced
sample for GST-CD52.
[046] Figure 10. Expression and purification of extracellular region of human
V158 FcyRIlla.
Tagged FcyRIlla was transfected in 293T cells, and media containing secreted
FcyRIlla were
harvested 3 days later and purified using affinity chromatography. 1: media;
2: flow through; 3: wash;
4-8: serial elutions. Both simple blue-stained SDS-PAGE gel and western result
are shown. For the
western blot, membrane was probed with anti-GST antibody.
[047] Figure 11. Binding to human V158 FcyRIlla by select alemtuzumab Fc
variants from the
experimental library as determined by the AlphaScreen."'" assay, described in
Example 2. In the
presence of competitor antibody (Fc variant or WT alemtuzumab) a
characteristic inhibition curve is
observed as a decrease in luminescence signal. Phosphate buffer saline (PBS)
alone was used as
the negative control. The binding data were normalized to the maximum and
minimum luminescence
signal for each particular curve, provided by the baselines at low and high
antibody concentrations
respectively. The curves represent the fits of the data to a one site
competition model using nonlinear
regression. These fits provide 1050s for each antibody, illustrated for WT and
S239D by the dotted
lines.
[048] Figures 12. AlphaScreen TM assay showing binding of select alemtuzumab
Fc variants to =
human FcyR1lb. The binding data were normalized to the upper and lower
baselines for each
particular antibody, and the curves represent the fits of the data to a one
site competition model. PBS
was used as a negative control.
[049] Figures 13a and 13b. AlphaScreen TM assay showing binding of select
alemtuzumab (Figure
13a) and trastuzumab (Figure 13b) Fc variants to human Va1158 FcyRIlla. The
binding data were
normalized to the upper and lower baselines for each particular antibody, and
the curves represent
the fits of the data to a one site competition model.. PBS was used as a
negative control.
[050] Figures 14a and 14b. AlphaScreen TM assay measuring binding to human
V158 FcyRIlla by
select Fc variants in the context of trastuzumab. The binding data were
normalized to the upper and
lower baselines for each particular antibody, and the curves represent the
fits of the data to a one site
competition model. PBS was used as a negative control.
[051] Figures 15a and 15b. AlphaScreenTM assay measuring binding to human V158
FcyRIlla by
select Fc variants in the context of rituximab (Figure 15a) and cetuximab
(Figure 15b). The binding
data were normalized to the upper and lower baselines for each particular
antibody, and the curves
represent the fits of the data to a one site competition model. PBS was used
as a negative control.
[052] Figures 16a ¨ 16b. AlphaScreenTm assay comparing binding of select
alemtuzumab Fc
variants to human V158 FcyRIlla (Figure 16a) and human FcyRIlb (Figure 16b).
The binding data
were normalized to the upper and lower baselines for each particular antibody,
and the curves
CA 02766627 2012-01-23
WO 2004/099249
PCT/11S2004/009298
represent the fits of the data to a one site competition model. PBS was used
as a negative control.
[053] Figure 17. AlphaScreen TM assay measuring binding to human V158 FcyRIlla
by select Fc
variants in the context of trastuzumab. The binding data were normalized to
the upper and lower
baselines for each particular antibody, and the curves represent the fits of
the data to a one site
competition model. PBS was used as a negative controL
[054] Figures 18. AlphaScreen Tm assay showing binding of select alemtuzumab
Fc variants to
human R131 FcyRIla. The binding data were normalized to the upper and lower
baselines for each
particular antibody, and the curves represent the fits of the data to a one
site competition model.
[055] Figures 19a and 19b. AlphaScreen TM assay showing binding of select
alemtuzumab Fc
variants to human V158 FcyRIlla. The binding data were normalized to the upper
and lower baselines
for each particular antibody, and the curves represent the fits of the data to
a one site competition
model. PBS was used as a negative control. -
[056] Figure 20. AlphaScreen T" assay showing binding of aglycosylated
alemtuzumab Fc variants
to human V158 FcyRIlla. The binding data were normalized to the upper and
lower baselines for
each particular antibody, and the curves represent the fits of the data to a
one site competition model.
PBS was used as a negative control.
[057] Figure 21. AlphaScreen Tm assay comparing human V158 FcyRIlla binding by
select
alemtuzumab Fc variants in glycosylated (solid symbols, solid lines) and
deglycosylated (open
symbols, dotted lines). The binding data were normalized to the upper and
lower baselines for each
particular antibody, and the curves represent the fits of the data to a one
site competition model.
[058] Figures 22a - 22b. AlphaScreen TM assay showing binding of select
alemtuzumab Fc variants
to the V158 (Figure 22a) and F158 (Figure 22b) allotypes of human FcyRIlla.
The binding data were
normalized to the upper and lower baselines for each particular antibody, and
the curves represent
the fits of the data to a one site competition model. PBS was used as a
negative control.
[059] Figures 23a - 23d. Figures 23a and 23b show the correlation between SPR
Kd's and
AlphaScreen TM IC50's from binding of select alemtuzumab Fc variants to V158
FcyRIlla (Figure 23a)
and F158 FcyRIlla (Figure 23b). Figures 23c and 23d show the correlation
between SPR and
AlphaScreen 1M fold-improvements over WT for binding of select alemtuzumab Fc
variants to V158
FcyRIlla (Figure 23c) and F158 FcyRIlla (Figure 23d). Binding data are
presented in Table 63. The
lines through the data represent the linear fits of the data, and the r2
values indicate the significance of
these fits.
[060] Figures 24a - 24b. Cell-based ADCC assays of select Fc variants in the
context of
alemtuzumab. ADCC was measured using the DELFIAO EuTDA-based cytotoxicity
assay (Perkin
Elmer, MA), as described in Example 7, using DoHH-2 lymphoma target cells and
50-fold excess
human PBMCs. Figure 24a is a bar graph showing the raw fluorescence data for
the indicated
=
21
CA 02766627 2012-01-23
WO 2004/099249 PCT/US2004/009298
alemtuzumab antibodies at 10 ng/ml. The PBMC bar indicates basal levels of
cytotoxicity in the
absence of antibody. Figure 24b shows the dose-dependence of ADCC on antibody
concentration for
the indicated alemtuzumab antibodies, normalized to the minimum and maximum
fluorescence signal
for each particular curve, provided by the baselines at low and high antibody
concentrations
respectively. The curves represent the fits of the data to a sigmoidal dose-
response model using
nonlinear regression.
[061] Figures 25a - 25c. Cell-based ADCC assays of select Fc variants in the
context of
trastuzumab. ADCC was measured using the DELFIA EuTDA-based cytotoxicity
assay, as
described in Example 7, using BT474 and Sk-Br-3 breast carcinoma target cells
and 50-fold excess
human PBMCs. Figure 25a is a bar graph showing the raw fluorescence data for
the indicated
trastuzumab antibodies at 1 ng/ml. The PBMC bar indicates basal levels of
cytotoxicity in the
absence of antibody. Figures 25b and 250 show the dose-dependence of ADCC on
antibody
concentration for the indicated trastuzumab antibodies, normalized to the
minimum and maximum
fluorescence signal for each particular curve, provided by the baselines at
low and high antibody
concentrations respectively. The curves represent the fits of the data to a
sigmoidal dose-response
model using nonlinear regression.
[062] Figures 26a - 26c. Cell-based ADCC assays of select Fc variants in the
context of rituximab.
ADCC was measured using the DELFIA0 EuTDA-based cytotoxicity assay, as
described in Example
7, using WIL2-S lymphoma target cells and 50-fold excess human PBMCs. Figure
26a is a bar graph
showing the raw fluorescence data for the indicated rituximab antibodies at 1
ng/ml. The PBMC bar
indicates basal levels of cytotoxicity in the absence of antibody. Figures 26b
and 26c show the dose-
dependence of ADCC on antibody concentration for the indicated rituximab
antibodies, normalized to
the minimum and maximum fluorescence signal for each particular curve,
provided by the baselines at
low and high antibody concentrations respectively. The curves represent the
fits of the data to a
sigmoidal dose-response model using nonlinear regression.
[063] Figures 27a - 27b. Cell-based ADCC assay of select trastuzumab (Figure
27a) and rituximab
(Figure 27b) Fc variants showing enhancements in potency and efficacy. Both
assays used
homozygous F158/F158 FcyRIlla PBMCs as effector cells at a 25-fold excess to
target cells, which
were Sk-Br-3 for the trastuzumab assay and WIL2-S for the rituximab assay.
Data were normalized
according to the absolute minimal lysis for the assay, provided by the
fluorescence signal of target
cells in the presence of PBMCs alone (no antibody), and the absolute maximal
lysis for the assay,
provided by the fluorescence signal of target cells in the presence of Triton
X1000, as described in
Example 7.
[064] Figure 28. Cell-based ADCC assay of select trastuzumab Fc variants
against different cell
lines expressing varying levels of the Her2/neu target antigen. ADCC assays
were run as described
in Example 7, with various cell lines expressing amplified to low levels of
Her2/neu receptor, including
Sk-Br-3 (1x106 copies), Sk0V3 (-1x105), OVCAR3(-1x104), and MCF-7 (-3x103
copies). Human
22
CA 02766627 2012-01-23
WO 2004/099249 PCT/US2004/009298
PBMCs allotyped as homozygous F158/F158 FcyRIlla were used at 25-fold excess
to target cells.
The bar graph provides ADCC data for WT and Fc variant against the indicated
cell lines, normalized
to the minimum and maximum fluorescence signal provided by minimal lysis
(PBMCs alone) and
maximal lysis (Triton X1000).
[065] Figure 29. Cell-based ADCC assays of select Fc variants in the context
of trastuzumab using
natural killer (NK) cells as effector cells and measuring LDH release to
monitor cell lysis. NK cells,
allotyped as heterozygous V158/F158 FcyRIlla, were at an 8-fold excess to Sk-
Br-3 breast carcinoma
target cells, and the level of cytotoxicity was measured using the LDH
Cytotoxicity Detection Kit,
according to the manufacturer's protocol (Roche Diagnostics GmbH, Penzberg,
Germany). The
graph shows the dose dependence of ADCC on antibody concentration for the
indicated trastuzumab
antibodies, normalized to the minimum and maximum fluorescence signal for each
particular curve,
provided by the baselines at low and high antibody concentrations
respectively. The curves represent
the fits of the data to a sigmoidal dose-response model using nonlinear
regression.
[066] Figure 30. Cell-based ADCP assay of select variants. The ADCP assay was
carried out as
described in Example 8, using a co-labeling strategy coupled with flow
cytometry. Differentiated
macrophages were used as effector cells, and Sk-Br-3 cells were used as target
cells. Percent
phagocytosis represents the number of co-labeled cells (macrophage + Sk-Br-3)
over the total
= number of Sk-Br-3 in the population (phagocytosed + non-phagocytosed).
[067] Figures 31a ¨ 31c. Capacity of select Fc variants to mediate binding and
activation of
complement. Figure 31a shows an AlphaScreenTm assay measuring binding of
select alemtuzumab
=
Fc variants to C1g. The binding data were normalized to the upper and lower
baselines for each
particular antibody, and the curves represent the fits of the data to a one
site competition model.
Figures 31b and 31c show a cell-based assay measuring capacity of select
rituximab Fc variants to
mediate CDC. CDC assays were performed using Alamar Blue to monitor lysis of
Fc variant and WT
rituximab -opsonized WIL2-S lymphoma cells by human serum complement (Quidel,
San Diego, CA).
The dose-dependence on antibody concentration of complement-mediated lysis is
shown for the
indicated rituximab antibodies, normalized to the minimum and maximum
fluorescence signal for each
particular curve, provided by the baselines at low and high antibody
concentrations respectively. The
curves represent the fits of the data to a sigmoidal dose-response model using
nonlinear regression.
[068] Figure 32. AlphaScreenim assay measuring binding of select alemtuzumab
Fc variants to
bacterial protein A, as described in Example 10. The binding data were
normalized to the upper and
lower baselines for each particular antibody, and the curves represent the
fits of the data to a one site
competition model. PBS was used as a negative control.
[069] Figure 33. AlphaScreenThl assay measuring binding of select alemtuzumab
Fc variants to
human FcRn, as described in Example 10. The binding data were normalized to
the upper and lower
23
CA 02766627 2012-01-23
WO 2004/099249 PCT/US2004/009298
baselines for each particular antibody, and the curves represent the fits of
the data to a one site
competition model. PBS was used as a negative control.
[070] Figures 34a and 34b. AlphaScreenTM assay measuring binding of select
alemtuzumab
(Figure 34a) and trastuzumab (Figure 34b) Fc variants to mouse FcyRIII, as
described in Example 11.
The binding data were normalized to the upper and lower baselines for each
particular antibody, and
the curves represent the fits of the data to a one site competition model. PBS
was used as a negative
control.
[071] Figure 35. Cell-based ADCC assays of select Fc variants in the context
of trastuzumab using
mouse PBMCs as effector cells. ADCC was measured using the DELFIA EuTDA-based
cytotoxicity
assay using Sk-Br-3 breast carcinoma target cells and 8-fold excess mouse
PBMCs. The bar graph
shows the raw fluorescence data for the indicated trastuzumab antibodies at 10
ng/ml. The PBMC
bar indicates basal levels of cytotoxicity in the absence of antibody, and TX
indicates complete cell
lysis in the presence of Triton X1000.
[072] Figure 36. AlphaScreenTu assay measuring binding to human V158 FcyRIlla
by select
trastuzumab Fc variants expressed in 293T and CHO cells, as described in
Example 12. The binding
data were normalized to the upper and lower baselines for each particular
antibody, and the curves
represent the fits of the data to a ohe site competition model. PBS was used
as a negative control. =
[073] Figures 37a ¨ 37b. Synergy of Fc variants and engineered glycoforms.
Figure 37a presents
an AlphaScreen TM assay showing V158 FcyRIlla binding by WT and Fc variant
(V209,
S239/1332E/A330L) trastuzumab expressed in 2931, CHO, and Lec-13 CHO cells.
The data were
normalized to the upper and lower baselines for each antibody, and the curves
represent the fits of
the data to a one site competition model. PBS was used as a negative control.
Figure 37b presents a
cell-based ADCC assay showing the ability of 239T, CHO, and Lec-13 CHO
expressed WT and V209
trastuzumab to mediate ADCC. ADCC was measured using the DELFIAO EuTDA-based
cytotoxicity
assay as described previously, with Sk-Br-3 breast carcinoma target cells. The
data show the dose-
dependence of ADCC on antibody concentration for the indicated trastuzumab
antibodies, normalized
to the minimum and maximum fluorescence signal for each particular curve,
provided by the baselines
at low and high antibody concentrations respectively. The curves represent the
fits of the data to a
sigmoidal dose-response model using nonlinear regression.
[074] Figures 38a ¨ 38c. Sequences showing improved anti-CD20 antibodies. The
light and heavy
chain sequences of rituximab are presented in Figure 38a and Figure 38b
respectively, and are taken
from translated Sequence 3 of US 5,736,137. Relevant positions in Figure 38b
are bolded, including
S239, V240, V2641, E272, K274, N297, S298, K326, A330, and 1332. Figure 38c
shows the improved
anti-CD20 antibody heavy chain sequences, with variable positions designated
in bold as Xi, X21 X37
X4, X5, X6, X7, X5, Zl, and Z2. The table below the sequence provides possible
substitutions for these
positions. The improved anti-CD20 antibody sequences comprise at least one non-
WT amino acid
24
CA 02766627 2012-01-23
WO 2004/09249
PCT/U52004/009298
selected from the group of possible substitutions for X1, X2, X3, X4, X5, X6,
X7, and X6. These
improved anti-CD20 antibody sequences may also comprise a substitution Z1
and/or Z2. These
positions are numbered according to the EU index as in Kabat, and thus do not
correspond to the
sequential order in the sequence.
DETAILED DESCRIPTION OF THE INVENTION
[075] In order that the invention may be more completely understood, several
definitions are set
forth below. Such definitions are meant to encompass grammatical equivalents.
[076] By "ADCC" or "antibody dependent cell-mediated cvtotoxicity" as used
herein is meant the
cell-mediated reaction wherein nonspecific cytotoxic cells that express FeyRs
recognize bound
antibody on a target cell and subsequently cause lysis of the target cell.
[077] By "ADCP" or antibody dependent cell-mediated phagocytosis as used
herein is meant the
cell-mediated reaction wherein nonspecific cytotoxic cells that express FcyRs
recognize bound
antibody on a target cell and subsequently cause phagocytosis of the target
cell.
[078] By "amino acid modification" herein is meant an amino acid substitution,
insertion, and/or
deletion in a polypeptide sequence. The preferred amino acid modification
herein is a substitution.
By "amino acid substitution" or "substitution" herein is meant the replacement
of an amino acid at a
particular position in a parent polypeptide sequence with another amino acid,.
For example, the
substitution I332E refers to a variant polypeptide, in this case an Fe
variant, in which the isoleucine at
position 332 is replaced with a glutamic acid.
[079] By "antibody" herein is meant a protein consisting of one or more
polypeptides substantially
encoded by all or part of the recognized immunoglobulin genes. The recognized
immunoglobulin
genes, for example in humans, include the kappa (K), lambda (X.), and heavy
chain genetic loci, which
together comprise the myriad variable region genes, and the constant region
genes mu (u), delta (5),
gamma (y), sigma (6), and alpha (a) which encode the IgM, IgD, IgG, IgE, and
IgA isotypes
respectively. Antibody herein is meant to include full length antibodies and
antibody fragments, and
may refer to a natural antibody from any organism, an engineered antibody, or
an antibody generated
recombinantly for experimental, therapeutic, or other purposes as further
defined below. The term
"antibody" includes antibody fragments, as are known in the art, such as Fab,
Fab', F(ab)2, Fv, scFv,
or other antigen-binding subsequences of antibodies, either produced by the
modification of whole
antibodies or those synthesized de novo using recombinant DNA technologies.
Particularly preferred
are full length antibodies that comprise Fc variants as described herein. The
term "antibody"
comprises monoclonal and polyclonal antibodies. Antibodies can be antagonists,
agonists,
neutralizing, inhibitory, or stimulatory.
[080] The antibodies of the present invention may be nonhuman, chimeric,
humanized, or fully
human. For a description of the concepts of chimeric and humanized antibodies
see Clark at al.,
2000 and references cited therein (Clark, 2000, Immune! Today 21:397-402).
Chimeric antibodies
CA 02766627 2012-01-23
WO 2004/099249
PCT/US2004/009298
comprise the variable region of a nonhuman antibody, for example VH and VL
domains of mouse or
rat origin, operably linked to the constant region of a human antibody (see
for example U.S. Patent
No. 4,816,567). In a preferred embodiment, the antibodies of the present
invention are humanized.
By "humanized' antibody as used herein is meant an antibody comprising a human
framework region
(FR) and one or more complementarity determining regions (CDR's) from a non-
human (usually
mouse or rat) antibody. The non-human antibody providing the CDR's is called
the "donor" and the
human immunoglobulin providing the framework is called the "acceptor".
Humanization relies
principally on the grafting of donor CDRs onto acceptor (human) VL and VH
frameworks (Winter US
5225539). This strategy is referred to as "CDR grafting". "Backmutation" of
selected acceptor
framework residues to the corresponding donor residues is often required to
regain affinity that is lost
in the initial grafted construct (US 5530101; US 5585089; OS 5693761; US
5693762; US 6180370;
US 5859205; US 5821337; US 6054297; US 6407213). The humanized antibody
optimally also will
comprise at least a portion of an immunoglobulin constant region, typically
that of a human
immunoglobulin, and thus will typically comprise a human Fc region. Methods
for humanizing non-
human antibodies are well known in the art, and can be essentially performed
following the method of
Winter and co-workers (Jones et al., 1986, Nature 321:522-525; Riechmann et
at.,1988, Nature
332:323-329; Verhoeyen et at, 1988, Science, 239:1534-1536). Additional
examples of humanized
murine monoclonal antibodies are also known in the art, for example antibodies
binding human
protein C (O'Connor etal., 1998, Protein Eng 11:321-8), interleukin 2 receptor
(Queen etal., 1989,
Roc Nail Aced Sci, USA 86:10029-33), and human epidermal growth factor
receptor 2 (Carter etal.,
1992, Proc Nat! Aced Sci USA 89:4285-9). In an alternate embodiment, the
antibodies of the present
invention may be fully human, that is the sequences of the antibodies are
completely or substantially
human. A number of methods are known in the art for generating fully human
antibodies, including
the use of transgenic mice (Bruggennann of at, 1997, Curt Opin Blotechnol
8:455-458) or human
antibody libraries coupled with selection methods (Griffiths et at., 1998,
Curt Opin Biotechnol 9:102-
108).
10811 Specifically included within the definition of "antibody" are
aglycosylated antibodies. By
"aglycosylated antibody" as used herein is meant an antibody that lacks
carbohydrate attached at
position 297 of the Fc region, wherein numbering is according to the EU system
as in Kabat. The
aglycosylated antibody may be a deglycosylated antibody, that is an antibody
for which the Fc
carbohydrate has been removed, for example chemically or enzymatically.
Alternatively, the
aglycosylated antibody may be a nonglycosylated or unglycosylated antibody,
that is an antibody that
was expressed without Fc carbohydrate, for example by mutation of one or
residues that encode the
glycosylation pattern or by expression in an organism that does not attach
carbohydrates to proteins,
for example bacteria.
[082] Specifically included within the definition of "antibody" are full-
length antibodies that contain
an Fc variant portion. .By "full length antibody" herein is meant the
structure that constitutes the
natural biological form of an antibody, including variable and constant
regions. For example, in most
26
CA 02766627 2012-01-23
WO 20041099249 PCT/US2004/009298
mammals, including humans and mice, the full length antibody of the IgG class
is a tetramer and
consists of two identical pairs of two immunoglobulin chains, each pair having
one light and one heavy
chain, each light chain comprising immunoglobulin domains VL and CL, and each
heavy chain
comprising immunoglobulin domains VH, Cyl, Cy2, and Cy3. In some mammals, for
example in
camels and llamas, IgG antibodies may consist of only two heavy chains, each
heavy chain
comprising a variable domain attached to the Fc region. By "IgG" as used
herein is meant a
polypeptide belonging to the class of antibodies that are substantially
encoded by a recognized
immunoglobulin gamma gene. In humans this class comprises IgG1, IgG2, IgG3,
and IgG4. In mice
this class comprises IgG1, IgG2a, IgG2b, IgG3.
[083] By "amino acid" and "amino acid identity" as used herein is meant one of
the 20 naturally
occurring amino acids or any non-natural analogues that may be present at a
specific, defined
position. By "protein" herein is meant at least two covalently attached amino
acids, which includes
proteins, polypeptides, oligopeptides and peptides. The protein may be made up
of naturally occurring
amino acids and peptide bonds, or synthetic peptidomimetic structures, i.e.
"analogs", such as
peptoids (see Simon et al., 1992, Proc Nat! Acad Sci USA 89(20):9367)
particularly when LC
peptides are to be administered to a patient. Thus "amino acid", or "peptide
residue", as used herein
means both naturally occurring and synthetic amino acids. For example,
homophenylalanine, citrulline
and noreleucine are considered amino acids for the purposes of the invention.
"Amino acid" also
includes imino acid residues such as proline and hydroxyproline. The side
chain may be in either the
= (R) or the (S) configuration. In the preferred embodiment, the amino
acids are in the (S) or L-
configuration. If non-naturally occurring side chains are used, non-amino acid
substituents may be
used, for example to prevent or retard in vivo degradation.
[084] By "computational screening method" herein Is meant any method for
designing one or more
mutations in a protein, wherein said method utilizes a computer to evaluate
the energies of the
interactions of potential amino acid side chain substitutions with each other
and/or with the rest of the
protein. As will be appreciated by those skilled in the art, evaluation of
energies, referred to as energy
calculation, refers to some method of scoring one or more amino acid
modifications. Said method
may involve a physical or chemical energy term, or may involve knowledge-,
statistical-, sequence-
based energy terms, and the like. The calculations that compose a
computational screening method
are herein referred to as "computational screening calculations".
[085] By "effector function" as used herein is meant a biochemical event that
results from the
interaction of an antibody Fc region with an Fc receptor or ligand. Effector
functions include but are
not limited to ADCC, ADCP, and CDC. By "effector cell" as used herein is meant
a cell of the immune
system that expresses one or more Fc receptors and mediates one or more
effector functions.
Effector cells include but are not limited to monocytes, macrophages,
neutrophils, dendritic cells,
eosinophils, mast cells, platelets, B cells, large granular lymphocytes,
Langerhans' cells, natural killer
(NK) cells, and yy T cells, and may be from any organism including but not
limited to humans, mice,
27
CA 02766627 2012-01-23
WO 2004/099249
PCT/US2004/009298
rats, rabbits, and monkeys. By "library" herein is meant a set of Fc variants
in any form, including but
not limited to a list of nucleic acid or amino acid sequences, a list of
nucleic acid or amino acid
substitutions at variable positions, a physical library comprising nucleic
acids that encode the library
sequences, or a physical library comprising the Fc variant proteins, either in
purified or unpurified
form.
[086] By "Fc", 'Fe region", FC polypeptide", etc. as used herein is meant an
antibody as defined
herein that includes the polypeptides comprising the constant region of an
antibody excluding the first
constant region immunoglobulin domain. Thus Fc refers to the last two constant
region
immunoglobulin domains of IgA, IgD, and IgG, and the last three constant
region immunogtobulin
domains of IgE and IgM, and the flexible hinge N-terminal to these domains.
For IgA and IgM Fc may
include the J chain. For IgG, as illustrated in Figure 1, Fc comprises
immunoglobulin domains
Cgamma2 and Cgamma3 (C12 and Cy3) and the hinge between Cgammal (Cy1) and
Cgamma2
(Cy2). Although the boundaries of the Fc region may vary, the human IgG heavy
chain Fc region is
usually defined to comprise residues C226 or P230 to its carboxyl-terminus,
wherein the numbering is
according to the EU index as in Kabat. Fc may refer to this region in
isolation, or this region in the
context of an antibody, antibody fragment, or Fc fusion. An Fc may be an
antibody, Fc fusion, or an
protein or protein domain that comprises Fc. Particularly preferred are Fc
variants, which are non-
naturally occurring variants of an Fc.
[087] By "Fc fusion" as used herein is meant a protein wherein one or more
polypeptides is
operably linked to an Fc region or a derivative thereof. Fc fusion is herein
meant to be synonymous
with the terms "immunoadhesin", "Ig fusion", "Ig chimera", and "receptor
globulin" (sometimes with
dashes) as used in the prior art (Charnow et aL, 1996, Trends Blotechnot 14:52-
60; Ashkenazi et aL,
1997, Curr Opin Immune' 9:195-200). An Fc fusion combines the Fc region of an
immunoglobulin
with a fusion partner, which in general can be any protein or small molecule.
The role of the non-Fc
part of an Fc fusion, i.e. the fusion partner, is to mediate target binding,
and thus it is functionally
analogous to the variable regions of an antibody. Virtually any protein or
small molecule may be
linked to Fc to generate an Fc fusion. Protein fusion partners may include,
but are not limited to, the
target-binding region of a receptor, an adhesion molecule, a ligand, an
enzyme, a cytokine, a
chemokine, or some other protein or protein domain. Small molecule fusion
partners may include any
therapeutic agent that directs the Fc fusion to a therapeutic target. Such
targets may be any
molecule, preferrably an extracellular receptor, that is implicated in
disease. Two families of surface
receptors that are targets of a number of approved small molecule drugs are G-
Protein Coupled
Receptors (GPCRs), and ion channels, including K+, Na+, Ca+ channels. Nearly
70% of all drugs
currently marketed worldwide target GPCRs. Thus the Fc variants of the present
invention may be
fused to a small molecule that targets, for example, one or more GABA
receptors, purinergic
receptors, adrenergic receptors, histaminergic receptors, opiod receptors,
chemokine receptors,
glutamate receptors, nicotinic receptors, the 5HT (serotonin) receptor, and
estrogen receptors. A
28
CA 02766627 2012-01-23
WO 2004/099249
PCTAIS2004/009298
fusion partner may be a small-molecule mimetic of a protein that targets a
therapeutically useful
target. Specific examples of particular drugs that may serve as Fc fusion
partners can be found in L.
S. Goodman et al., Eds., Goodman and Gilman's The Pharmacological Basis of
Therapeutics
(McGraw-Hill, New York, ed. 9, 1996). Fusion partners include not only small
molecules and proteins
that bind known targets for existing drugs, but orphan receptors that do not
yet exist as drug targets.
The completion of the genome and proteome projects are proving to be a driving
force in drug
discovery, and these projects have yielded a trove of orphan receptors. There
is enormous potential
to validate these new molecules as drug targets, and develop protein and small
molecule therapeutics
that target them. Such protein and small molecule therapeutics are
contemplated as Fc fusion
partners that employ the Fc variants of the present invention. A variety of
linkers, defined and
described below, may be used to covalently link Fc to a fusion partner to
generate an Fc fusion.
[088] By 'Fe gamma receptor" or "FcyR" as used herein is meant any member of
the family of
proteins that bind the IgG antibody Fc region and are substantially encoded by
the FcyR genes. In
humans this family includes but is not limited to FcyRI (C064), including
isoforms FeyMa, FcyRib, and
FcyRIc; FcyRII (CD32), including isoforms FcyRIla (including allotypes H131
and R131), FcyRIlb
(including FcyRIlb-1 and Fc7RIlb-2), and Fc7RI1c; and PcyRIII (CD16),
including isoforms FcyRIlla
(including allotypes V158 and F158) and FcyRIllb (including allotypes FcyRillb-
NA1 and FcyR111b-
NA2) (Jefferis etal., 2002, Immunol Lett 82:57-65), as well as any
undiscovered human FcyRs or
FcyR isoforms or allotypes. An FcyR may be from any organism, including but
not limited to humans,
mice, rats, rabbits, and monkeys. Mouse FcyRs include but are not limited to
FcyRI (CD64), FcyRII
(CD32), FcyRIII (CD16), and FcyRIII-2 (CD16-2), as well as any undiscovered
mouse FcyRs or FcyR
isoforms or allotypes.
[089] By "Fc ligand" as used herein is meant a molecule, preferably a
polypeptide, from any
organism that binds to the Fc region of an antibody to form an Fc-ligand
complex. Fc ligands include
but are not limited to FcyRs, FcyRs, FcyRs, FcRn, Cl q, C3, mannan binding
lectin, mannose receptor,
staphylococcal protein A, streptococcal protein G, and viral FcyR. Fc ligands
also include Fc receptor
homologs (FcRH), which are a family of Fc receptors that are homologous to the
FcyRs (Davis et al.,
2002, Immunological Reviews 190:123-136). Fc ligands may include undiscovered
molecules that
bind Fe.
[090] By "IgG" as used herein is meant a polypeptide belonging to the class of
antibodies that are
substantially encoded by a recognized immunoglobulin gamma gene. In humans
this class comprises
IgG1, IgG2, IgG3, and IgG4. In mice this class comprises IgG1, IgG2a, IgG2b,
IgG3. By
"immunoglobulin (le)" herein is meant a protein consisting of one or more
polypeptides substantially
encoded by immunoglobulin genes. lmmunoglobulins include but are not limited
to antibodies.
lmmunoglobulins may have a number of structural forms, including but not
limited to full length
antibodies, antibody fragments, and individual immunoglobulin domains. By
"immuno_gtobulin (Ig)
29
CA 02766627 2012-01-23
WO 2004/099249
PCT/US2004/009298
domain" herein is meant a region of an immunoglobulin that exists as a
distinct structural entity as
ascertained by one skilled in the art of protein structure. Ig domains
typically have a characteristic 0-
sandwich folding topology. The known 1g domains in the IgG class of antibodies
are VH, Cyl , Cy2,
Cy3, VI., and CL.
[091] By "parent polypeptide" or "precursor polypeptide" (including Fc parent
or precursors) as
used herein is meant a polypeptide that is subsequently modified to generate a
variant. Said parent
polypeptide may be a naturally occurring polypeptide, or a variant or
engineered version of a naturally
occurring polypeptide. Parent polypeptide may refer to the polypeptide itself,
compositions that
comprise the parent polypeptide, or the amino acid sequence that encodes it
Accordingly, by "parent
Fc polypeptide" as used herein is meant an unmodified Fc polypeptide that is
modified to generate a
variant, and by "parent antibody" as used herein is meant an unmodified
antibody that is modified to
generate a variant antibody.
[092] As outlined above, certain positions of the Fc molecule can be altered.
By "position" as used
herein is meant a location in the sequence of a protein. Positions may be
numbered sequentially, or
according to an established format, for example the EU index as in Kabat. For
example, position 297
is a position in the human antibody IgGl. Corresponding positions are
determined as outlined above,
generally through alignment with other parent sequences.
[093] By "residue" as used herein is meant a position in a protein and its
associated amino acid
identity. For example, Asparagine 297 (also referred to as Asn297, also
referred to as N297) is a
residue in the human antibody IgG1 _
[094] By "target antigen" as used herein is meant the molecule that is bound
specifically by the
variable region of a given antibody. A target antigen may be a protein,
carbohydrate, lipid, or other
chemical compound.
[095] By "target cell" as used herein is meant a cell that expresses a target
antigen.
[096] By "variable region" as used herein is meant the region of an
immunoglobulin that comprises
one or more Ig domains substantially encoded by any of the VK, VA., and/or VH
genes that make up
the kappa, lambda, and heavy chain immunoglobulin genetic loci respectively.
[097] By "variant polypeptide" as used herein is meant a polypeptide sequence
that differs from that
of a parent polypeptide sequence by virtue of at least one amino acid
modification. Variant
polypeptide may refer to the polypeptide itself, a composition comprising the
polypeptide, or the amino
sequence that encodes it. Preferably, the variant polypeptide has at least one
amino acid
modification compared to the parent polypeptide, e.g. from about one to about
ten amino acid
modifications, and preferably from about one to about five amino acid
modifications compared to the
parent. The variant polypeptide sequence herein will preferably possess at
least about 80%
homology with a parent polypeptide sequence, and most preferably at least
about 90% homology,
more preferably at least about 95% homology. Accordingly, by "Fc variant" as
used herein is meant
CA 02766627 2012-01-23
WO 2004/099249
PCT/US2004/009298
an Fc sequence that differs from that of a parent Fc sequence by virtue of at
least one amino acid
modification. An Fc variant may only encompass an Fc region, or may exist in
the context of an
antibody, Fc fusion, or other polypeptide that is substantially encoded by Fc.
Fc variant may refer to
the Fc polypeptide itself, compositions comprising the Fc variant polypeptide,
or the amino acid
sequence that encodes it. In a preferred embodiment, the variant proteins of
the invention comprise
an Fc variant, as described herein, and as such, may comprise an antibody (and
the corresponding
derivatives) with the Fc variant, or an Fc fusion protein that comprises the
Fc variant. In addition, in
some cases, the Fc is a variant as compared to a wild-type Fc, or to a
"parent' variant.
[098] For all positions discussed in the present invention, numbering of an
immunoglobulin heavy
chain is according to the EU index (Kabat et al., 1991, Sequences of Proteins
of Immunological
Interest, 5th Ed., United States Public Health Svice, National Institutes of
Health, Bethesda). The "EU
index as in Kabat" refers to the residue numbering of the human IgG1 EU
antibody.
[099] The Fc variants of the present invention may be optimized for a variety
of properties.
Properties that may be optimized include but are not limited to enhanced or
reduced affinity for an
FcyR. In a preferred embodiment, the Fc variants of the present invention are
optimized to possess
enhanced affinity for a human activating FcyR, preferably FcyRI, FcyRIla,
FcyRIlc, FcyRIlla, and
FcyRIllb, most preferably FcyMita. In an alternately preferred embodiment, the
Fc variants are
optimized to possess reduced affinity for the human inhibitory receptor
FcyRIlb. These preferred
embodiments are anticipated to provide antibodies and Fc fusions with enhanced
therapeutic
properties in humans, for example enhanced effector function and greater anti-
cancer potency. In an
alternate embodiment, the Fc variants of the present invention are optimized
to have reduced or
ablated affinity for a human FcyR, including but not limited to FcyRI,
FcyRIla, FcyRllb, FcyRIlc,
FcyRIlla, and FcyRIllb. These embodiments are anticipated to provide
antibodies and Fc fusions with
enhanced therapeutic properties in humans, for example reduced effector
function and reduced
toxicity. Preferred embodiments comprise optimization of Fc binding to a human
FcyR, however in
alternate embodiments the Fc variants of the present invention possess
enhanced or reduced affinity
for FeyRs from nonhuman organisms, including but not limited to mice, rats,
rabbits, and monkeys. Fc
variants that are optimized for binding to a nonhuman FcyR may find use in
experimentation. For
example, mouse models are available for a variety of diseases that enable
testing of properties such
as efficacy, toxicity, and pharmacokinetics for a given drug candidate. As is
known in the art, cancer
cells can be grafted or injected into mice to mimic a human cancer, a process
referred to as
xenografting. Testing of antibodies or Fc fusions that comprise Fc variants
that are optimized for one
or more mouse FcyRs, may provide valuable information with regard to the
efficacy of the antibody or
Fc fusion, its mechanism of action, and the like. The Fc variants of the
present invention may also be
optimized for enhanced functionality and/or solution properties in
aglycosylated form. In a preferred
embodiment, the aglycosylated Fc variants of the present invention bind an Fc
ligand with greater
affinity than the aglycosylated form of the parent Fc polypeptide. Said Fc
ligands include but are not
31
CA 02766627 2012-01-23
WO 2004/099249 PCT/1152004/009298
limited to FoyRs, Cl q, FcRn, and proteins A and G, and may be from any source
including but not
limited to human, mouse, rat, rabbit, or monkey, preferably human. In an
alternately preferred
embodiment, the Fc variants are optimized to be more stable and/or more
soluble than the
aglycosylated form of the parent Fc polypeptide. An Fc variant that is
engineered or predicted to
display any of the aforementioned optimized properties is herein referred to
as an "optimized Fc
variant".
[100] The Fc variants of the present invention may be derived from parent Fc
polypeptides that are
themselves from a wide range of sources. The parent Fc polypeptide may be
substantially encoded
by one or more Fc genes from any organism, including but not limited to
humans, mice, rats, rabbits,
camels, llamas, dromedaries, monkeys, preferably mammals and most preferably
humans and mice.
In a preferred embodiment, the parent Fc polypeptide composes an antibody,
referred to as the
parent antibody. The parent antibody may be fully human, obtained for example
using transgenic
mice (Bruggemann et al., 1997, Carr Opin Biotechnol 8:455-458) or human
antibody libraries coupled
with selection methods (Griffiths et al., 1998, Curr Opin Biotechnol 9:102-
108). The parent antibody
need not be naturally occurring. For example, the parent antibody may be an
engineered antibody,
including but not limited to chimeric antibodies and humanized antibodies
(Clark, 2000, Immunol
Today 21:397-402). The parent antibody may be an engineered variant of an
antibody that is
substantially encoded by one or more natural antibody genes. In one
embodiment, the parent
antibody has been affinity matured, as is known in the art. Alternatively, the
antibody has been
modified in some other way, for example as described in USSN 10/339788, filed
on March 3, 2003.
['101] The Fc variants of the present invention may be substantially encoded
by immunoglobulin
genes belonging to any of the antibody classes. In a preferred embodiment, the
Fc variants of the
present invention find use in antibodies or Fc fusions that comprise sequences
belonging to the IgG
class of antibodies, including IgG1, IgG2, IgG3, or IgG4. In an alternate
embodiment the Fc variants
of the present invention find use in antibodies_ or Fc fusions that comprise
sequences belonging to the
IgA (including subclasses IgAl and IgA2), IgD, IgE, IgG, or IgM classes of
antibodies. The Fc
variants of the present invention may comprise more than one protein chain.
That is, the present
invention may find use in an antibody or Fc fusion that is a monomer or an
oligomer, including a
homo- or hetero-oligomer.
[102] In a preferred embodiment, the antibodies of the invention are based on
human sequences,
and thus human sequences are used as the "base" sequences, against which other
sequences, such
as rat, mouse, and monkey sequences are compared. In order to establish
homology to primary
sequence or structure, the amino acid sequence of a precursor or parent Fc
polypeptide is directly
compared to the human Fc sequence outlined herein. After aligning the
sequences, using one or
more of the homology alignment programs known in the art (for example using
conserved residues as
between species), allowing for necessary insertions and deletions in order to
maintain alignment (i.e.,
avoiding the elimination of conserved residues through arbitrary deletion and
insertion), the residues
32
CA 02766627 2012-01-23
WO 2004/099249 PCT/11S2004/009298
equivalent to particular amino acids in the primary sequence of human Fc are
defined. Alignment of
conserved residues preferably should conserve 100% of such residues. However,
alignment of
greater than 75% or as little as 50% of conserved residues is also adequate to
define equivalent
residues (sometimes referred to as "corresponding residues"). Equivalent
residues may also be
defined by determining homology at the level of tertiary structure for an Fc
polypeptide whose tertiary
structure has been determined. Equivalent residues are defined as those for
which the atomic
coordinates of two or more of the main chain atoms of a particular amino acid
residue of the parent or
precursor (Non N, CA on CA, C on C and 0 on 0) are within 0.13 rim and
preferably 0.1 rim after
alignment. Alignment is achieved after the best model has been oriented and
positioned to give the
maximum overlap of atomic coordinates of non-hydrogen protein atoms of the Fc
polypeptide.
[103] The Fc variants of the present invention may be combined with other Fc
modifications,
including but not limited to modifications that alter effector function or
interaction with one or more Fc
ligands. Such combination may provide additive, synergistic, or novel
properties in antibodies or Fc
fusions. In one embodiment, the Fc variants of the present invention may be
combined with other
known Fc variants (Duncan et aL, 1988, Nature 332:563-564; Lund et aL, 1991, J
Immunol 147:2657-
2662; Lund et al., 1992, Mol lmmunol 29:53-59; Alegre etal., 1994,
Transplantation 57:1537-1543;
Hutchins of aL, 1995, Proc Nat! Aced Sci U S A 92:11980-11984; Jefferis et
a/., 1995, Immunol Lett
44:111-117; Lund etal., 1995, Faseb J 9:115-119; Jefferis etal., 1996, Immunol
Lett 54:101-104;
Lund of aL, 1996, J Immune! 157:4963-4969; Armour of aL, 1999, Eur J Immunol
29:2613-2624;
Idusogie etal., 2000, J Immunol 164:4178-4184; Reddy etal., 2000, J Immunol
164:1925-1933; Xu et
aL, 2000; Cell Immunol 200:16-26; Idusogie et al., 2001, J Immunol 166:2571-
2575; Shields et al.,
2001, J Biol Chem 276:6591-6604; Jefferis etal., 2002, Immune! Lett 82:57-65;
Presta etal., 2002,
Biochem Soc Trans 30:487-490; Hinton et al., 2004, J Biol Chem 279:6213-6216)
(US 5,624,821; US
5,885,573; US 6,194,551; PCT WO 00/42072; PCT WO 99/58572; US 2004/0002587
Al). In an
alternate embodiment, the Fc variants of the present invention are
incorporated into an antibody or Fc
fusion that comprises one or more engineered glycoforms. By "engineered
glvcoforrn" as used herein
is meant a carbohydrate composition that is covalently attached to an Fc
polypeptide, wherein said
carbohydrate composition differs chemically from that of a parent Fc
polypeptide. Engineered
glycoforms may be useful for a variety of purposes, including but not limited
to enhancing or reducing
effector function. Engineered glycoforms may be generated by a variety of
methods known in the art
(Umafia of at., 1999, Nat Biotechnol 17:176-180; Davies etal., 2001,
Biotechnol Bioeng 74:288-294;
Shields et al., 2002, J Biol Chem 277:26733-26740; Shinkawa et aL, 2003, J
Blot Chem 278:3466-
3473); (US 6,602,684; USSN 10/277,370; USSN 10/113,929; PCT WO 00/61739A1; PCT
WO
01/29246A1; PCT WO 02/31140A1; PCT WO 02/30954A1); (Potelligentim technology
[Biowa, Inc.,
Princeton, NJ]; GlycoMAb7m glycosylation engineering technology [GLYCART
biotechnology AG,
Zurich, Switzerland]). Many of these techniques are based on controlling the
level of fucosylated
and/or bisecting oligosaccharides that are covalently attached to the Fc
region, for example by
expressing an Fc polypeptide in various organisms or cell lines, engineered or
otherwise (for example
33
CA 02766627 2012-01-23
WO 20114/099249
PCT/US2004/009298
Lec-13 CHO cells or rat hybridoma YB2/0 cells), by regulating enzymes involved
in the glycosylation
pathway (for example FUT8 [0,6-fucosyltranserase] and/or 61-4- N-
acetylglucosaminyltransferase III
[GnT111]), or by modifying carbohydrate(s) after the Fc polypeptide has been
expressed. Engineered
glycoform typically refers to the different carbohydrate or oligosaccharide;
thus an Fc polypeptide, for
example an antibody or Fc fusion, may comprise an engineered glycoform.
Alternatively, engineered
glycoform may refer to the Fc polypeptide that comprises the different
carbohydrate or
oligosaccharide. Thus combinations of the Fc variants of the present invention
with other Fc
modifications, as well as undiscovered Fc modifications, are contemplated with
the goal of generating
novel antibodies or Fc fusions with optimized properties.
[104] The Fc variants of the present invention may find use in an antibody. By
"antibody of the
present invention" as used herein is meant an antibody that comprises an Fc
variant of the present
invention. The present invention may, in fact, find use in any protein that
comprises Fc, and thus
application of the Fc variants of the present invention is not limited to
antibodies. The Fc variants of
the present invention may find use in an Fc fusion. By "Fc fusion of the
present invention" as used
herein refers to an Fc fusion that comprises an Fc variant of the present
invention. Fc fusions may
comprise an Fc variant of the present invention operably linked to a cytokine,
soluble receptor
domain, adhesion molecule, ligand, enzyme, peptide, or other protein or
protein domain, and include
but are not limited to Fc fusions described in US 5,843,725; US 6,018,026; US
6,291,212; US
6,291,646; US 6,300,099; US 6,323,323; PCT WO 00/24782; and in (Chamow etal.,
1996, Trends
Biotechno114:52-60; Ashkenazi et at., 1997, Curr Opin Immune! 9:195-200.
[105] Virtually any antigen may be targeted by the antibodies and fusions of
the present invention,
including but not limited to the following list of proteins, subunits,
domains, motifs, and epitopes
belonging to the following list of proteins: CD2; CD3, CD3E, CD4, CD11, CD11a,
CD14, CD16,
CD18, CD19, CD20, CO22, CD23, CD25, CD28, CD29, CD30, CD32, CD33 (p67
protein), CD38,
CD40, CD4OL, CD52, CD54, CD56, CD80, CD147, GD3, IL-1, IL-1R, IL-2, 1L-2R, IL-
4, IL-5, IL-6, IL-
6R, IL-8, IL-12, IL-15, IL-18, IL-23, interferon alpha, interferon beta,
interferon gamma; TNF-alpha,
TNFbeta2, TNFc, TNFalphabeta, TNF-RI, INF-R11, FasL, CD27L, CD3OL, 4-1BBL,
TRAIL, RANKL,
TWEAK, APRIL, BAFF, LIGHT, VEGI, OX4OL, TRAIL Receptor-1, Al Adenosine
Receptor,
Lymphotoxin Beta Receptor, TACI, BAFF-R, EPO; LFA-3, ICAM-1, ICAM-3, EpCAM,
integrin betal ,
integrin beta2, integrin alpha4/beta7, integrin a1pha2, integrin alpha3,
integrin alpha4, integrin a1pha5,
integrin alpha6, integrin alphav, alphaVbeta3 integrin, FGFR-3, Keratinocyte
Growth Factor, VLA-1,
VLA-4, L-selectin, anti-Id, E-selectin, HLA, HLA-DR, CTLA-4, T cell receptor,
B7-1, B7-2, VNRintegrin,
TGFbetal , TGFbeta2, eotaxinl , BLyS (B-lymphocyte Stimulator), complement C5,
IgE, factor VII,
CD64, CBL, NCA 90, EGFR (ErbB-1), Her2/neu (ErbB-2), Her3 (ErbB-3), Her4 (ErbB-
4), Tissue
Factor, VEGF, VEGFR, endothelin receptor, VLA-4, Hapten NP-cap or NIP-cap, T
cell receptor
alpha/beta, E-selectin, digoxin, placental alkaline phosphatase (PLAP) and
testicular PLAP-like
alkaline phosphatase, transferrin receptor, Carcinoembryonie antigen (CEA),
CEACAM5, HMFG
34
CA 02766627 2012-01-23
WO 2004/099249
PCT/US2004/009298
PEM, mucin MUC1, MUC18, Heparanase 1, human cardiac myosin, tumor-associated
glycoprotein-72
(TAG-72), tumor-associated antigen CA 125, Prostate specific membrane antigen
(PSMA), High
molecular weight melanoma-associated antigen (HMW-MAA), carcinoma-associated
antigen,
Gcoprotein Ilb/Illa (GPIlb/111a), tumor-associated antigen expressing Lewis Y
related carbohydrate,
human cytomegalovirus (HCMV) gH envelope glycoprotein, HIV gp120, HCMV,
respiratory syncital
virus RSV F, RSVF Fgp, VNRintegrin, IL-8, cytokeratin tumor-associated
antigen, Hee B gp120, CMV,
gplIbIlla, HIV IIIB gp120 V3 loop, respiratory syncytial virus (RSV) Fgp,
Herpes simplex virus (HSV)
gD glycoprotein, HSV gB glycoprotein, HCMV gB envelope glycoprotein, and
Clostridium perfringens
toxin.
[106] One skilled in the art will appreciate that the aforementioned list of
targets refers not only to
specific proteins and biomolecules, but the biochemical pathway or pathways
that comprise them.
For example, reference to CTLA-4 as a target antigen implies that the ligands
and receptors that
make up the T cell co-stimulatory pathway, including CTLA-4, B7-1, B7-2, CD28,
and any other
undiscovered ligands or receptors that bind these proteins, are also targets.
Thus target as used
herein refers not only to a specific biomolecule, but the set of proteins that
interact with said target
and the members of the biochemical pathway to which said target belongs. One
skilled in the art will
further appreciate that any of the aforementioned target antigens, the ligands
or receptors that bind
them, or other members of their corresponding biochemical pathway, may be
operably linked to the
Fc variants of the present invention in order to generate an Fc fusion. Thus
for example, an Fc fusion
that targets EGFR could be constructed by operably linking an Fc variant to
EGF, TGFa, or any other
ligand, discovered or undiscovered, that binds EGFR. Accordingly, an Fc
variant of the present
invention could be operably linked to EGFR in order to generate an Fc fusion
that binds EGF, TGFa,
or any other ligand, discovered or undiscovered, that binds EGFR. Thus
virtually any polypeptide,
whether a ligand, receptor, or some other protein or protein domain, including
but not limited to the
aforementioned targets and the proteins that compose their corresponding
biochemical pathways,
may be operably linked to the Fc variants of the present invention to develop
an Fc fusion.
[107] A number of antibodies and Fc fusions that are approved for use, in
clinical trials, or in
development may benefit from the Fc variants of the present invention. Said
antibodies and Fc
fusions are herein referred to as "clinical products and candidates". Thus in
a preferred embodiment,
the Fc variants of the present invention may find use in a range of clinical
products and candidates.
For example, a number of antibodies that target CD20 may benefit from the Fc
variants of the present
invention. For example the Fc variants of the present invention may find use
in an antibody that is
substantially similar to rituximab (Rituxan , 1DEC/Genentech/Roche) (see for
example US
5,736,137), a chimeric anti-CD20 antibody approved to treat Non-Hodgkin's
lymphoma; HuMax-
CD20, an anti-CD20 currently being developed by Genmab, an anti-CD20 antibody
described in US
5,500,362, AME-133 (Applied Molecular Evolution), hA20 (1mmunomedics, Inc.),
and HumaLYM
(Intracel). A number of antibodies that target members of the family of
epidermal growth factor
CA 02766627 2012-01-23
WO 2004/099249
PCT/US2004/009298
receptors, including EGFR (ErbB-1), Her2/neu (ErbB-2), Her3 (ErbB-3), Her4
(ErbB-4), may benefit
from the Fc variants of the present invention. For example the Fc variants of
the present invention
may find use in an antibody that is substantially similar to trastuzumab
(HerceptinO, Genentech) (see
for example US 5,677,171), a humanized anti-Her2/neu antibody approved to
treat breast cancer;
pertuzumab (rhuMab-2C4, Omnitarglm), currently being developed by Genentech;
an anti-Her2
antibody described in US 4,753,894; cetuximab (Erbitux0, lmclone) (US
4,943,533; PCT WO
96/40210), a chimeric anti-EGFR antibody in clinical trials for a variety of
cancers; ABX-EGF (US
6,235,883), currently being developed by Abgenix/Immunex/Amgen; HuMax-EGFr
(USSN
10/172,317), currently being developed by Genmab; 425, EMD55900, EMD62000, and
EMD72000
(Merck KGaA) (US 5558864; Murthy et al. 1987, Arch Biochem Biophys. 252(2):549-
60; Rodeck et
al., 1987, J Ce11 Biochem. 35(4):315-20; Kettleborough et al., 1991, Protein
Eng. 4(7):773-83); ICR62
(Institute of Cancer Research) (PCT WO 95/20045; Modjtahedi et at., 1993, J.
Cell Biophys. 1993,
22(1-3):129-46; Modjtahedi et al., 1993, Br J Cancer. 1993, 67(2):247-53;
Modjtahedi et al, 1996, Br J
Cancer, 73(2):228-35; Modjtahedi et al, 2003, Int J Cancer, 105(2):273-80);
TheraCIM hR3 (YM
Biosciences, Canada and Centro de Immunologia Molecular, Cuba (US 5,891,996;
US 6, 506,883;
Mateo eta!, 1997, Immunotechnology, 3(1):71-81); mAb-806 (Ludwig Institue for
Cancer Research,
Memorial Sloan-Kettering) (Jungbluth et al. 2003, Proc Nati Acad Sci U S A.
100(2):639-44); KSB-102
(KS Biomedix); MR1-1 (IVAX, National Cancer Institute) (PCT WO 0162931A2); and
SC100
(Scancell) (PCTWO 01/88138). In another preferred embodiment, the Fc variants
of the present
invention may find use in alemtuzumab (Campath , Millenium), a humanized
monoclonal antibody
currently approved for treatment of B-cell chronic lymphocytic leukemia. The
Fc variants of the
present invention may find use in a variety of antibodies or Fc fusions that
are substantially similar to
other clinical products and candidates, including but not limited to muromonab-
CD3 (Orthoclone
0KT30), an anti-CD3 antibody developed by Ortho Biotech/Johnson & Johnson,
ibritumomab
tiuxetan (Zevaline), an anti-0O20 antibody developed by IDEC/Schering AG,
gemtuzumab
ozogamicin (Mylotarg0), an anti-CD33 (p67 protein) antibody developed by
Celltech/Wyeth, alefacept
(Arnevive0), an anti-LFA-3 Fc fusion developed by Biogen), abciximab
(ReoPro0), developed by
Centocor/Lilly, basiliximab (Simulect0), developed by Novartis, palivizumab
(Synagis0), developed by
Medlmmune, infliximab (Remicade0), an anti-TNFalpha antibody developed by
Centocor,
adalimumab (Humira0), an anti-TNFalpha antibody developed by Abbott,
Humicadelm, an anti-
TNFalpha antibody developed by Celltech, etanercept (Enbrel0), an anti-
TNFalpha Fc fusion
developed by lmmunex/Amgen, ABX-CBL, an anti-CD147 antibody being developed by
Abgenix,
ABX-18, an anti-IL8 antibody being developed by Abgenix, ABX-MA1, an anti-
MUC18 antibody being
developed by Abgenix, Pemtumomab (R1549,90Y-muHMFG1), an anti-MUC1 In
development by
Antisoma, Therex (R1550), an anti-MUC1 antibody being developed by Antisoma,
AngioMab
(AS1405), being developed by Antisoma, HuBC-1, being developed by Antisoma,
Thioplatin (AS1407)
being developed by Antisoma, Antegren0 (natalizumab), an anti-alpha-4-beta-1
(VLA-4) and alpha-4-
beta-7 antibody being developed by Biogen, VLA-1 mAb, an anti-VLA-1 integrin
antibody being
36
CA 02766627 2012-01-23
WO 20041099249
PCT11JS2004/009298
developed by Biogen, LTBR mAb, an anti-lymphotoxin beta receptor (LTBR)
antibody being
developed by Biogen, CAT-152, an anti-TGF02 antibody being developed by
Cambridge Antibody
Technology, J695, an anti-IL-12 antibody being developed by Cambridge Antibody
Technology and
Abbott, CAT-192, an anti-TGF61 antibody being developed by Cambridge Antibody
Technology and
Genzyme, CAT-213, an anti-Eotaxin1 antibody being developed by Cambridge
Antibody Technology,
LymphoStat-BT" an anti-Blys antibody being developed by Cambridge Antibody
Technology and
Human Genome Sciences Inc., TRAIL-RImAb, an anti-TRAIL-R1 antibody being
developed by
Cambridge Antibody Technology and Human Genome Sciences, Inc., Avastinni
(bevacizumab,
rhuMAb-VEGF), an anti-VEGF antibody being developed by Genentech, an anti-HER
receptor family
antibody being developed by Genentech, Anti-Tissue Factor (ATF), an anti-
Tissue Factor antibody
being developed by Genentech, Xolairn1(0malizumab), an anti-IgE antibody being
developed by
Genentech, RaptivaTM (Efalizumab), an anti-CD1la antibody being developed by
Genentech and
Xoma, MLN-02 Antibody (formerly LDP-02), being developed by Genentech and
Millenium
Pharmaceuticals, HuMax CD4, an anti-CD4 antibody being developed by Genmab,
HuMax-IL15, an
anti-IL15 antibody being developed by Genmab and Amgen, HuMax-Inflam, being
developed by
Genmab and Medarex, HuMax-Cancer, an anti-Heparanase I antibody being
developed by Genmab
and Medarex and Oxford GcoSciences, HuMax-Lymphoma, being developed by Genmab
and
Amgen, HuMax-TAC, being developed by Genmab, IDEC-131, and anti-CD4OL antibody
being
developed by IDEC Pharmaceuticals, IDEC-151 (Clenoliximab), an anti-CD4
antibody being
developed by IDEC Pharmaceuticals, IDEC-114, an anti-CD80 antibody being
developed by IDEC
Pharmaceuticals, IDEC-152, an anti-CD23 being developed by IDEC
Pharmaceuticals, anti-
macrophage migration factor (MIF) antibodies being developed by IDEC
Pharmaceuticals, BEC2, an
anti-idiotypic antibody being developed by lmclone, IMC-1C11, an anti-KDR
antibody being developed
by ImoIone, DC101, an anti-flk-1 antibody being developed by Imolone, anti-VE
cadherin antibodies
being developed by !melone, CEACideTM (labetuzumab), an anti-carcinoembryonic
antigen (CEA)
antibody being developed by lmmunomedics, LymphoCidel" (Epratuzumab), an anti-
CD22 antibody
being developed by Immunomedics, AFP-Cide, being developed by Immunomedics,
MyelomaCide,
being developed by lmmunomedics, LkoCide, being developed by lmmunomedics,
ProstaCide, being
developed by Immuhomedics, MDX-010, an antl-CTLA4 antibody being developed by
Medarex, MDX-
060, an anti-CD30 antibody being developed by Medarex, MDX-070 being developed
by Medarex,
MDX-018 being developed by Medarex, Osidemn' (IDM-1), and anti-Her2 antibody
being developed
by Medarex and lmmuno-Designed Molecules, HuMaxT"-CD4, an anti-CD4 antibody
being developed
by Medarex and Genmab, HuMax-IL15, an anti-IL15 antibody being developed by
Medarex and
Genmab, CNTO 148, an anti-TNFa antibody being developed by Medarex and
Centocor/J&J, CNTO
1275, an anti-cytokine antibody being developed by Centocor/J&J, MOR101 and
MOR102, anti-
intercellular adhesion molecule-1 (ICAM-1) (CD54) antibodies being developed
by MorphoSys,
M0R201, an anti-fibroblast growth factor receptor 3 (FGFR-3) antibody being
developed by
MorphoSys, Nuvion (visilizumab), an anti-CD3 antibody being developed by
Protein Design Labs,
37
CA 02766627 2012-01-23
WO 2004/099249 PCT/US2004/009298
HuZAFTm, an anti-gamma interferon antibody being developed by Protein Design
Labs, Anti-0501
Integrin, being developed by Protein Design Labs, anti-IL-12, being developed
by Protein Design
Labs, ING-1, an anti-Ep-CAM antibody being developed by Xoma, and MLN01, an
anti-Beta2 integrin
antibody being developed by Xoma.
11081 Application of the Fc variants to the aforementioned antibody and Fc
fusion clinical products
and candidates is not meant to be constrained to their precise composition.
The Fc variants of the
present invention may be incorporated into the aforementioned clinical
candidates and products, or
into antibodies and Fc fusions that are substantially similar to them. The Fc
variants of the present
invention may be incorporated into versions of the aforementioned clinical
candidates and products
that are humanized, affinity matured, engineered, or modified in some other
way. Furthermore, the
entire polypeptide of the aforementioned clinical products and candidates need
not be used to
construct a new antibody or Fc fusion that incorporates the Fc variants of the
present invention; for
example only the variable region of a clinical product or candidate antibody,
a substantially similar
variable region, or a humanized, affinity matured, engineered, or modified
version of the variable
region may be used. .In another embodiment, the Fc variants of the present
invention may find use in
an antibody or Fc fusion that binds to the same epitope, antigen, ligand, or
receptor as one of the
aforementioned clinical products and candidates.
[109] The Fc variants of the present invention may find use in a wide range of
antibody and Fc =
fusion products. In one embodiment the antibody or Fc fusion of the present
invention is a
therapeutic, a diagnostic, or a research reagent, preferably a therapeutic.
Alternatively, the antibodies
and Fc fusions of the present invention may be used for agricultural or
industrial uses. In an alternate
embodiment, the Fc variants of the present invention compose a library that
may be screened
experimentally. This library may be a list of nucleic acid or amino acid
sequences, or may be a
physical composition of nucleic acids or polypeptides that encode the library
sequences. The Fc
variant may find use in an antibody composition that is monoclonal or
polyclonal. The antibodies and
Fc fusions of the present invention may be agonists, antagonists,
neutralizing, inhibitory, or
stimulatory. In a preferred embodiment, the antibodies and Fc fusions of the
present invention are
used to kill target cells that bear the target antigen, for example cancer
cells. In an alternate
embodiment, the antibodies and Fe fusions of the present invention are used to
block, antagonize, or
agonize the target antigen, for example for antagonizing a cytokine or
cytokine receptor. In an
alternately preferred embodiment, the antibodies and Fc fusions of the present
invention are used to
block, antagonize, or agonize the target antigen and kill the target cells
that bear the target antigen.
[110] The Fc variants of the present invention may be used for various
therapeutic purposes. In a
preferred embodiment, the Fc variant proteins are administered to a patient to
treat an antibody-
related disorder. A "patient" for the purposes of the present invention
includes both humans and other
animals, preferably mammals and most preferably humans. Thus the antibodies
and Fc fusions of the
present invention have both human therapy and veterinary applications. In the
preferred embodiment
38
CA 02766627 2012-01-23
WO 2004/099249 PCTMS2004/009298
the patient is a mammal, and in the most preferred embodiment the patient is
human. The term
"treatment" in the present invention is meant to include therapeutic
treatment, as well as prophylactic,
or suppressive measures for a disease or disorder. Thus, for example,
successful administration of
an antibody or Fc fusion prior to onset of the disease results in treatment of
the disease. As another
example, successful administration of an optimized antibody or Fc fusion after
clinical manifestation of
the disease to combat the symptoms of the disease comprises treatment of the
disease. "Treatment"
also encompasses administration of an optimized antibody or Fc fusion protein
after the appearance
of the disease in order to eradicate the disease. Successful administration of
an agent after onset
and after clinical symptoms have developed, with possible abatement of
clinical symptoms and
perhaps amelioration of the disease, comprises treatment of the disease. Those
"in need of
treatment" include mammals already having the disease or disorder, as well as
those prone to having
the disease or disorder, including those in which the disease or disorder is
to be prevented. By
"antibody related disorder" or "antibody responsive disorder" or "condition"
or "disease" herein are
meant a disorder that may be ameliorated by the administration of a
pharmaceutical composition
comprising an antibody or Fc fusion of the present invention. Antibody related
disorders include but
are not limited to autoimmune diseases, immunological diseases, infectious
diseases, inflammatory
diseases, neurological diseases, and oncological and neoplastic diseases
including cancer. By
"cancer" and "cancerous" herein refer to or describe the physiological
condition in mammals that is
typically characterized by unregulated cell growth. Examples of cancer include
but are not limited to
carcinoma, lymphoma, blastoma, sarcoma (including liposarcoma), neuroendocrine
tumors,
mesothelioma, schwanoma, meningioma, adenocarcinoma; melanoma, and leukemia or
lymphoid
malignancies. More particular examples of such cancers include squamous cell
cancer (e.g epithelial
squamous cell cancer), lung cancer including small-cell lung cancer, non-small
cell lung cancer,
adenocarcinoma of the lung and squamous carcinoma of the lung, cancer of the
peritoneum,
hepatocellular cancer, gastric or stomach cancer including gastrointestinal
cancer, pancreatic cancer,
glioblastoma, cervical cancer, ovarian cancer, liver cancer, bladder cancer,
hepatoma, breast cancer,
colon cancer, rectal cancer, colorectal cancer, endometrial or uterine
carcinoma, salivary gland
carcinoma, kidney or renal cancer, prostate cancer, vulva) cancer, thyroid
cancer, hepatic carcinoma,
anal carcinoma, penile carcinoma, testicular cancer, esophagael cancer, tumors
of the biliary tract, as
well as head and neck cancer. Furthermore, the Fc variants of the present
invention may be used to
treat conditions including but not limited to congestive heart failure (CHF),
vasculitis, rosecea, acne,
eczema, myocarditis and other conditions of the myocardium, systemic lupus
erythematosus,
diabetes, spondylopathies, synovial fibroblasts, and bone marrow stroma; bone
loss; Paget's disease,
osteoclastoma; multiple myeloma; breast cancer; disuse osteopenia;
malnutrition, periodontal
disease, Gaucher's disease, Langerhans' cell histiocytosis, spinal cord
injury, acute septic arthritis,
osteomalacia, Cushing's syndrome, monoostotic fibrous dysplasia, polyostotic
fibrous dysplasia,
periodontal reconstruction, and bone fractures; sarcoidosis; multiple myeloma;
osteolytic bone
cancers, breast cancer, lung cancer, kidney cancer and rectal cancer; bone
metastasis, bone pain'
.39
CA 02766627 2012-01-23
=
WO 2()04/099249
PCT/US2004/009298
management, and humoral malignant hypercalcemia, ankylosing spondylitisa and
other
spondyloarthropathies; transplantation rejection, viral infections,
hematologic neoplasisas and
neoplastic-like conditions for example, Hodgkin's lymphoma; non-Hodgkin's
lymphomas (Burkitt's
lymphoma, small lymphocytic lymphoma/chronic lymphocytic leukemia, mycosis
fungoides, mantle
cell lymphoma, follicular lymphoma, diffuse large B-cell lymphoma, marginal
zone lymphoma, hairy
cell leukemia and lymphoplasmacytic leukemia), tumors of lymphocyte precursor
cells, including B-
=
cell acute lyrnphoblastic leukemia/lymphoma, and T-cell acute lymphoblastic
leukemia/lymphoma,
thymoma, tumors of the mature T and NK cells, including peripheral T-cell
leukemias, adult T-cell
leukemiaff-cell lymphomas and large granular lymphocytic leukemia, Langerhans
cell histocytosis,
myeloid neoplasias such as acute myelogenous leukemias, including AML with
maturation, AML
without differentiation, acute promyelocytic leukemia, acute myelomonocytic
leukemia, and acute
monocytic leukemias, myelodysplastic syndromes, and chronic myeloproliferative
disorders, including
chronic myelogenous leukemia, tumors of the central nervous system, e.g.,
brain tumors (glioma,
neuroblastoma, astrocytoma, medulloblastoma, ependyrnoma, and retinoblastoma),
solid tumors
(nasopharyngeal cancer, basal cell carcinoma, pancreatic cancer, cancer of the
bile duct, Kaposi's
sarcoma, testicular cancer, uterine, vaginal or cervical cancers, ovarian
cancer, primary liver cancer
or endometrial cancer, and tumors of the vascular system (angiosarcoma and
hemagiopericytoma),
osteoporosis, hepatitis, HIV, AIDS, spondyloarthritis, rheumatoid arthritis,
inflammatory bowel
diseases (IBD), sepsis and septic shock, Crohn's Disease, psoriasis,
schleraderma, graft versus host
=
disease (GVHD), allogenic islet graft rejection, hematologic malignancies,
such as multiple myeloma
= (MM), myelodysplastic syndrome (MDS) and acute myelogenous leukemia
(AML), inflammation
associated with tumors, peripheral nerve injury or demyelinating diseases.
[111] In one embodiment, an antibody or Fe fusion of the present invention is
administered to a
patient having a disease involving inappropriate expression of a protein.
Within the scope of the
present invention this is meant to include diseases and disorders
characterized by aberrant proteins,
due for example to alterations in the amount of a protein present, the
presence of a mutant protein, or
both. An overabundance may be due to any cause, including but not limited to
overexpression at the
molecular level, prolonged or accumulated appearance at the site of action, or
increased activity of a
protein relative to normal. Included within this definition are diseases and
disorders characterized by
a reduction of a protein. This reduction may be due to any cause, including
but not limited to reduced
expression at the molecular level, shortened or reduced appearance at the site
of action, mutant
forms of a protein, or decreased activity of a protein relative to normal.
Such an overabundance or
reduction of a protein can be measured relative to normal expression,
appearance, or activity of a
protein, and said measurement may play an important role in the development
and/or clinical testing
of the antibodies and Fe fusions of the present invention.
[112] In one embodiment, an antibody or Fe fusion of the present invention is
the only
therapeutically active agent administered to a patient. Alternatively, the
antibody or Fe fusion of the
present invention is administered in combination with one or more other
therapeutic agents, including
CA 02766627 2012-01-23
WO 2004/099249 PCT/US2004/009298
but not limited to oytotoxic agents, chemotherapeutic agents, cytokines,
growth inhibitory agents, anti-
hormonal agents, kinase inhibitors, anti-angiogenic agents, cardioprotectants,
or other therapeutic
agents. Such molecules are suitably present in combination in amounts that are
effective for the
purpose intended. The skilled medical practitioner can determine empirically
the appropriate dose or
doses of other therapeutic agents useful herein. The antibodies and Fc fusions
of the present
invention may be administered concomitantly with one or more other therapeutic
regimens. For
example, an antibody or Fc fusion of the present invention may be administered
to the patient along
with chemotherapy, radiation therapy, or both chemotherapy and radiation
therapy. In one
embodiment, the antibody or Fc fusion of the present invention may be
administered in conjunction
with one or more antibodies or Fc fusions, which may or may not comprise an Fc
variant of the
present invention.
[113] In one embodiment, the antibodies and Fc fusions of the present
invention are administered
with a chemotherapeutic agent. By "chemotherapeutic agent" as used herein is
meant a chemical
compound useful in the treatment of cancer. Examples of chemotherapeutic
agents include but are
not limited to alkylating agents such as thiotepa and cyclosphosphamide
(CYTOXANTm); alkyl
sulfonates such as busulfan, improsulfan and piposulfan; aziridines such as
benzodopa, carboquone,
meturedopa, and uredopa; ethylenimines and methylamelamines including
altretamine,
triethylenemeramine, trietylenephosphoramide, triethylenethiophosphaoramide
and
trimethylolomelamine; nitrogen mustards such as chlorambucil, chlornaphazine,
cholOphosphamide,
estramustine, ifosfamide, mechlorethamine, mechlorethamine oxide
hydrochloride, melphalan,
novembichin, phenesterine, prednimustine, trofosfamide, uracil mustard;
nitrosureas such as
carmustine, chlorozotocin, fotemustine, lomustine, nimustine, ranimustine;
antibiotics such as
aclacinomysins, actinomycin, authramycin, azaserine, bleomycins, cactinomycin,
calicheamicin,
carabicin, caminomycin, carzinophilin, chromomycins, dactinomycin,
daunorubicin, detorubicin, 6-
diazo-5-oxo-L-norleucine, doxorubicin, epirubicin, esorubicin, idarubicin,
marcellomycin, mitomycins,
mycophenolic acid, nogalamycin, olivomycins, peplomycin, pothromycin,
puromycin, quelamycin,
rodorubicin, streptonigrin, streptozocin, tubercidin, ubenimex, zinostatin,
zorubicin; anti-metabolites
such as methotrexate and 5-fluorouracil (5-FU); folic acid analogues such as
denopterin,
methotrexate, pteropterin, trimetrexate; purine analogs such as fiudarabine, 6-
mercaptopurine,
thiamiprine, thioguanine; pyrimidine analogs such as ancitabine, azacitidine,
6-azauridine, carmofur,
cytarabine, dideoxyuridine, doxifluridine, enocitabine, floxuridine, 5-FU;
androgens such as
calusterone, dromostanolone propionate, epitiostanol, mepitiostane,
testolactone; anti-adrenals such
as aminoglutethimide, mitotane, trilostane; folic acid replenisher such as
frolinic acid; aceglatone;
aldophosphamide glycoside; aminolevulinic acid; amsacrine; bestrabucil;
bisantrene; edatraxate;
defofamine; demecolcine; diaziquone; elformithine; elliptinium acetate;
etoglucid; gallium nitrate;
hydroxyurea; lentinan; lonidamine; mitoguazone; mitoxantrone; mopidamol;
nitracrine; pentostatin;
phenamet; pirarubicin; podophyllinic acid; 2-ethylhydrazide; procarbazine;
PSK.D; razoxane;
sizofuran; spirogermanium; tenuazonic acid; triaziquone; 2, 2',2"-
trichlorotriethylamine; urethan;
41
CA 02766627 2012-01-23
= WO
2004/099249 PCT/US2004/009298
vindesine; dacarbazine; mannomustine; mitobronitol; mitolactol; pipobroman;
gacytosine; arabinoside
("Ara-C"); cyclophosphamide; thiotepa; taxanes, e.g. paclitaxel (TAXOL ,
Bristol-Myers Squibb
Oncology, Princeton, N.J.) and docetaxel (TAXOTERE , Rhne-Poulenc Rorer,
Antony, France);
chlorambucil; gemcitabine; 6-thioguanine; mercaptopurine; methotrexate;
platinum analogs such as
cisplatin and carboplatin; vinblastine; platinum; etoposide (VP-16);
ifosfamide; mitomycin C;
mitoxantrone; vincristine; vinorelbine; navelbine; novantrone; teniposide;
daunomycin; aminopterin;
xeloda; ibandronate; CPT-11; topoisomerase inhibitor RFS 2000;
difluoromethylornithine (DMF0);
retinoic acid; esperamicins; capecitabine; thymidylate synthase inhibitor
(such as Tomudex); cox-2
inhibitors, such as celicoxib (CELEBREX ) or MK-0966 (VIOXX0); and
pharmaceutically acceptable
salts, acids or derivatives of any of the above. Also included in this
definition are anti-hormonal
agents that act to regulate or inhibit hormone action on tumors such as anti
estrogens including for
example tamoxifen, raloxifene, aromatase inhibiting 4(5)-imidazoles, 4-
hydroxytamoxifen, trioxifene,
keoxifene, LY 117018, onapristone, and toremifene (Fareston); and anti-
androgens such as flutamide,
nilutamide, bicalutamide, leuprolide, and goserelin; and pharmaceutically
acceptable salts, acids or
derivatives of any of the above.
[114] A chemotherapeutic or other cytotoxic agent may be administered as a
prodrug. By "prodruq"
as used herein is meant a precursor or derivative form of a pharmaceutically
active substance that is
less cytotoxic to tumor cells compared to the parent drug and is.capable.of
being enzymatically
activated or converted into the more active parent form. See, for example
Wilman, 1986, Biochemical
Society Transactions, 615th Meeting Belfast, 14:375-382; and Stella etal.,
"Prodrugs: A Chemical
Approach to Targeted Drug Delivery," Directed Drug Delivery, Borchardt et al.,
(ed.): 247-267,
Humana Press, 1985. The prodrugs that may find use with the present invention
include but are not
limited to phosphate-containing prodrugs, thiophosphate-containing prodrugs,
sulfate-containing
prodrugs, peptide-containing prodrugs, D-amino acid-modified prodrugs,
glycosylated prodrugs, beta-
lectern-containing prodrugs, optionally substituted phenoxyacetamide-
containing prodrugs or
optionally substituted phenylacetamide-containing prodrugs, 5-fluorocytosine
and other 5-fluorouridine
prodrugs which can be converted into the more active cytotoxic free drug.
Examples of cytotoxic
drugs that can be derivatized into a prodrug form for use with the antibodies
and Fc fusions of the
present invention include but are not limited to any of the aforementioned
chemotherapeutic agents.
[115] The antibodies and Fc fusions of the present invention may be combined
with other
therapeutic regimens. For example, in one embodiment, the patient to be
treated with the antibody or
Fc fusion may also receive radiation therapy. Radiation therapy can be
administered according to
protocols commonly employed in the art and known to the skilled artisan. Such
therapy includes but
is not limited to cesium, iridium, iodine, or cobalt radiation. The radiation
therapy may be whole body
irradiation, or may be directed locally to a specific site or tissue in or on
the body, such as the lung,
bladder, or prostate. Typically, radiation therapy is administered in pulses
over a period of time from
about 1 to 2 weeks. The radiation therapy may, however, be administered over
longer periods of
time. For instance, radiation therapy may be administered to patients having
head and neck cancer
42
CA 02766627 2012-01-23
WO 2004/099249 PCT/US2004/009298
for about 6 to about 7 weeks. Optionally, the radiation therapy may be
administered as a single dose
or as multiple, sequential doses. The skilled medical practitioner can
determine empirically the
appropriate dose or doses of radiation therapy useful herein. In accordance
with another embodiment
of the invention, the antibody or Fc fusion of the present invention and one
or more other anti-cancer
therapies are employed to treat cancer cells ex vivo. It is contemplated that
such ex vivo treatment
may be useful in bone marrow transplantation and particularly, autologous bone
marrow
transplantation. For instance, treatment of cells or tissue(s) containing
cancer cells with antibody or
Fc fusion and one or more other anti-cancer therapies, such as described
above, can be employed to
deplete or substantially deplete the cancer cells prior to transplantation in
a recipient patient. It is of
course contemplated that the antibodies and Fc fusions of the invention can be
employed in
combination with still other therapeutic techniques such as surgery.
[116] In an alternate embodiment, the antibodies and Fc fusions of the present
invention are
administered with a cytokine. By "cytokine" as used herein is meant a generic
term for proteins
released by one cell population that act on another cell as intercellular
mediators. Examples of such
cytokines are lymphokines, monokines, and traditional polypeptide hormones_
Included among the
cytokines are growth hormone such as human growth hormone, N-methionyl human
growth hormone,
and bovine growth hormone; parathyroid hormone; thyroxine; insulin;
proinsulin; relaxin; prorelaxin;
glycoprotein hormones such as follicle stimulating hormone (FSH), thyroid
stimulating hormone
(TSH),. and luteinizing hormone (LH); hepatic growth factor; fibroblast growth
factor; prolactin;
placental lactogen; tumor necrosis factor-alpha and -beta; mullerian-
inhibiting substance; mouse
gonadotropin-associated peptide; inhibin; activin; vascular endothelial growth
factor; integrin;
thrombopoletin (TP0); nerve growth factors such as NGF-beta; platelet-growth
factor, transforming
growth factors (TGFs) such as TGF-alpha and TGF-beta; insulin-like growth
factor-I and -II;
erythropoietin (EPO); osteoinductive factors; interferons such as interferon-
alpha, beta, and -gamma;
colony stimulating factors (CSFs) such as macrophage-CSF (M-CSF); granulocyte-
macrophage-CSF
(GM-CSF); and granulocyte-CSF (G-CSF); interleukins (Ls) such as IL-1, IL-
1alpha, IL-2, IL-3, IL-4,
IL-5,11-6, I1-7, IL-8, I1-9, IL-10, IL-11, IL-12; IL-15, a tumor necrosis
factor such as TNF-alpha or
TNF-beta; and other polypeptide factors including LIF and kit ligand (KL). As
used herein, the term
cytokine includes proteins from natural sources or from recombinant cell
culture, and biologically
active equivalents of the native sequence cytokines.
[117] A variety of other therapeutic agents may find use for administration
with the antibodies and
Fc fusions of the present invention. In one embodiment, the antibody or Fa
fusion is administered
with an anti-angiogenic agent. By "anti-anoioaenic agent" as used herein is
meant a compound that
blocks, or interferes to some degree, the development of blood vessels. The
anti-angiogenic factor
may, for instance, be a small molecule or a protein, for example an antibody,
Fc fusion, or cytokine,
that binds to a growth factor or growth factor receptor involved in promoting
angiogenesis. The
preferred anti-angiogenic factor herein is an antibody that binds to Vascular
Endothelial Growth Factor
(VEGF). In an alternate embodiment, the antibody or Fc fusion is administered
With a therapeutic
43
CA 02766627 2012-01-23
WO 2004/099249 PCT/US2004/009298
=
agent that induces or enhances adaptive immune response, for example an
antibody that targets
CTLA-4. In an alternate embodiment, the antibody or Fc fusion is administered
with a tyrosine kinase
inhibitor. By "tyrosine kinase inhibitor" as used herein is meant a molecule
that inhibits to some extent
tyrosine kinase activity of a tyrosine kinase. Examples of such inhibitors
include but are not limited to
quinazolines, such as PD 153035, 11-(3-chforoanilino) quinazoline;
pyridopyrimidines;
pyrimidopyrimidines; pyrrolopyrimidines, such as CGP 59326, CGP 60261 and CGP
62706;
pyrazolopyrimidines, 4-(phenylamino)-7H-pyrrolo(2,3-d) pyrimidines; curcumin
(diferuloyl methane,
4,5-bis (4-fluoroanilino)phthalimide); tyrphostines containing nitrothiophene
moieties; PD-0183805
(Wamer-Lambert); antisense molecules (e.g. those that bind to ErbB-encoding
nucleic acid);
quinoxalines (US 5,804,396); tryphostins (US 5,804,396); ZD6474 (Astra
Zeneca); PTK-787
(Novartis/Schering A G); pan-ErbB inhibitors such as C1-1033 (Pfizer);
Affinitac (ISIS 3521; Isis/Lilly);
imatinib mesylate (ST1571,Gleevec0; Novartis); PKI 166 (Novartis); GW2016
(Glaxo SmithKline); C1-
1033 (Pfizer); EKB-569 (Wyeth); Semaxinib (Sugen); ZD6474 (AstraZeneca); PTK-
787
(Novartis/Schering AG); INC-1C11 (Imclone); or as described in any of the
following patent
publications: US 5,804,396; PCT WO 99/09016 (American Cyanimid); PCT WO
98/43960 (American
Cyanamid); PCT WO 97/38983 (Warner-Lambert); PCT WO 99/06378 (Warner-Lambert);
PCT WO
99/06396 (Wamer-Lambert); PCT WO 96/30347 (Pfizer, Inc); PCT WO 96/33978
(AstraZeneca); PCT
W096/3397 (AstraZeneca); PCT WO 96/33980 (AstraZeneca), gefitinib (IRESSATM,
Z01839,
AstraZeneca), and OSI-774 (TarcevaTm, OSI Pharmaceuticals/Genentech).
[118] A variety of linkers may find use in the present invention to generate
Fc fusions (see definition
above) or antibody- or Fc fusion- conjugates (see definition below). By
"linker", linker sequence",
"spacer", "tethering sequence" or grammatical equivalents thereof, herein is
meant a molecule or
group of molecules (such as a monomer or polymer) that connects two molecules
and often serves to
place the two molecules in a Preferred configuration. A number of strategies
may be used to
covalently link molecules together. These include, but are not limited to
polypeptide linkages between
N- and C-termini of proteins or protein domains, linkage via disulfide bonds,
and linkage via chemical
cross-linking reagents. In one aspect of this embodiment, the linker is a
peptide bond, generated by
recombinant techniques or peptide synthesis. Choosing a suitable linker for a
specific case where
two polypeptide chains are to be connected depends on various parameters,
including but not limited
to the nature of the two polypeptide chains (e.g., whether they naturally
oligomerize), the distance
between the N- and the C-termini to be connected if known, and/or the
stability of the linker towards
proteolysis and oxidation. Furthermore, the linker may contain amino acid
residues that provide
flexibility. Thus, the linker peptide may predominantly include the following
amino acid residues: Gly,
Ser, Ala, or Thr. The linker peptide should have a length that is adequate to
link two molecules in
such a way that they assume the correct conformation relative to one another
so that they retain the
desired activity. Suitable lengths for this purpose include at least one and
not more than 30 amino
acid residues. Preferably, the linker is from about 1 to 30 amino acids in
length, with linkers of 1, 2, 3,
4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18 19 and 20 amino acids in
length being preferred. In
44
CA 02766627 2012-01-23
WO 2004/099249
PCT/US2004/009298
addition, the amino acid residues selected for inclusion in the linker peptide
should exhibit properties
that do not interfere significantly with the activity of the polypeptide.
Thus, the linker peptide on the
whole should not exhibit a charge that would be inconsistent with the activity
of the polypeptide, or
interfere with internal folding, or form bonds or other interactions with
amino acid residues in one or
more of the monomers that would seriously impede the binding of receptor
monomer domains. Useful
linkers include glycine-serine polymers (including, for example, (GS)n,
(GSGGS)n (GGGGS)n and
(GGGS)n, where n is an integer of at least one), glycine-alanine polymers,
alanine-serine polymers,
and other flexible linkers such as the tether for the shaker potassium
channel, and a large variety of
other flexible linkers, as will be appreciated by those in the art. Glycine-
serine polymers are preferred
since both of these amino acids are relatively unstructured, and therefore may
be able to serve as a
neutral tether between components. Secondly, serine is hydrophilic and
therefore able to solubilize
what could be a glebular glycine chain. Third, similar chains have been shown
to be effective in
joining subunits of recombinant proteins such-as single chain antibodies.
Suitable linkers may also be
identified by screening databases of known three-dimensional structures for
naturally occurring motifs
that can bridge the gap between two polypeptide chains. In a preferred
embodiment, the linker is not
immunogenic when administered in a human patient. Thus linkers may be chosen
such that they
have low immunogenicity or are thought to have low immunogenicity. For
example, a linker may be
chosen that exists naturally in a human. In a preferred embodiment the linker
has the sequence of
= the hinge region of an antibody, that is the sequence that links the
antibody Fab and Fc regions;
alternatively the linker has a sequence that comprises part of the hinge
region, or a sequence that is
substantially similar to the hinge region of an antibody. Another way of
obtaining a suitable linker is
by optimizing a simple linker, e.g., (Gly4Ser)n, through random mutagenesis.
Alternatively, once a
suitable polypeptide linker is defined, additional linker polypeptides can be
created to select amino
acids that more optimally interact with the domains being linked. Other types
of linkers that may be
used in the present invention include artificial polypeptide linkers and
inteins. In another embodiment,
disulfide bonds are designed to link the two molecules. In another embodiment,
linkers are chemical
cross-linking agents. For example, a variety of bifunctional protein coupling
agents may be used,
including but not limited to N-succinimidy1-3-(2-pyridyldithiol) propionate
(SPDP), succinimidy1-4-(N-
maleimidomethyl) cyclohexane-1-carboxylate, iminothiolane (IT), bifunctional
derivatives of
imidoesters (such as dimethyl adipimidate HCL), active esters (such as
disuccinimidyl suberate),
aldehydes (such as glutareldehyde), bis-azido compounds (such as bis (p-
azidobenzoyl)
hexanediamine), bis-diazonium derivatives (such as bis-(p-diazoniumbenzoyI)-
ethylenediamine),
diisocyanates (such as tolyene 2,6-diisocyanate), and bis-active fluorine
compounds (such as 1,5-
difluoro-2,4-dinitrobenzene). For example, a ricin immunotoxin can be prepared
as described in
Vitetta et al., 1971, Science 238:1098. Chemical linkers may enable chelation
of an isotope. For
example, Carbon-14-labeled 1-isothiocyanatobenzy1-3-methyldiethylene
triaminepentaacetic acid
(MX-DTPA) is an exemplary chelating agent for conjugation of radionucleotide
to the antibody (see
PCT WO 94/11026). The linker may be cleavable, facilitating release of the
cytotoxic drug in the cell.
CA 02766627 2012-01-23
WO 2004/099249 PCT/US2084/809298
For example, an acid-labile linker, peptidase-sensitive linker, dimethyl
linker or disulfide-containing
linker (Cheri et al., 1992, Cancer Research 52:127-131) may be used.
Alternatively, a variety of
nonproteinaceous polymers, including but not limited to polyethylene glycol
(PEG), polypropylene
glycol, polyoxyalkylenes, or copolymers of polyethylene glycol and
polypropylene glycol, may find use
as linkers, that is may find use to link the Fc variants of the present
invention to a fusion partner to
generate an Fc fusion, or to link the antibodies and Fc fusions of the present
invention to a conjugate.
[119] In one embodiment, the antibody or Fc fusion of the present invention is
conjugated or
operably linked to another therapeutic compound, referred to herein as a
conjugate. The conjugate
may be a cytotoxic agent, a chemotherapeutic agent, a cytokine, an anti-
angiogenic agent, a tyrosine
kinase inhibitor, a toxin, a radioisotope, or other therapeutically active
agent. Chemotherapeutic
agents, cytokines, anti-angiogenic agents, tyrosine kinase inhibitors, and
other therapeutic agents
have been described above, and all of these aforemention therapeutic agents
may find use as
antibody or Fc fusion conjugates. In an alternate embodiment, the antibody or
Fc fusion is conjugated
or operably linked to a toxin, including but not limited to small molecule
toxins and enzymatically
active toxins of bacterial, fungal, plant or animal origin, including
fragments and/or variants thereof.
Small molecule toxins include but are not limited to calicheamicin, maytansine
(US 5,208,020),
trichothene, and CC1065. In one embodiment of the invention, the antibody or
Fc fusion is
conjugated to one or more maytansine molecules (e.g. about 1 to about 10
maytansine molecules per
antibody molecule). Maytansine may, for example, be converted to May-SS-Me
which may be
reduced to May-SH3 and reacted with modified antibody or Fc fusion (Chari
etal., 1992, Cancer
Research 52: 127-131) to generate a maytansinoid-antibody or maytansinoid-Fc
fusion conjugate.
Another conjugate of interest comprises an antibody or Fc fusion conjugated to
one or more
calicheamicin molecules. The calicheamicin family of antibiotics are capable
of producing double-
stranded DNA breaks at sub-picomolar concentrations. Structural analogues of
calicheamicin that
may be used include but are not limited to yll, 0(.21, ot3, N-acetyl-y11,
PSAG, and 011, (Hinman et aL,
1993, Cancer Research 53:3336-3342; Lode et aL, 1998, Cancer Research 58:2925-
2928) (US
5,714,586; US 5,712,374; US 5,264,586; US 5,773,001). Dolastatin 10 analogs
such as auristatin E
(AE) and monomethylauristatin E (MMAE) may find use as conjugates for the Fc
variants of the
present invention (Doronina et al., 2003, Nat Biotechnol21(7):778-84;
Francisco etal., 2003 Blood
102(4):1458-65). Useful enyzmatically active toxins include but are not
limited to diphtheria A chain,
nonbinding active fragments of diphtheria toxin, exotoxin A chain (from
Pseudomonas aeruginosa),
ricin A chain, abrin A chain, modeccin A chain, alpha-sarcin, Aleurites fordii
proteins, dianthin
proteins, Phytolaca americana proteins (PAPI, PAPII, and PAP-S), momordica
charantia inhibitor,
curcin, crotin, sapaonaria officinalis inhibitor, gelonin, mitogellin,
restrictocin, phenomycin, enomycin
and the tricothecenes. See, for example, PCT WO 93/21232. The present
invention further
contemplates a conjugate or fusion formed between an antibody or Fc fusion of
the present invention
and a compound with nucleolytic activity, for example a ribonuclease or DNA
endonuclease such as a
deoxyribonudease (DNase).
46
CA 02766627 2012-01-23
WO 2004/099249
PCT/US2004/009298
[120] In an alternate embodiment, an antibody or Fc fusion of the present
invention may be
conjugated or operably linked to a radioisotope to form a radioconjugate. A
variety of radioactive
isotopes are available for the production of radioconjugate antibodies and Fc
fusions. Examples
include, but are not limited to, At211, 1131, 1125, y90, Re186, Re188, sm153,
B1212, P32, and radioactive
isotopes of Lu.
[121] In yet another embodiment, an antibody or Fc fusion of the present
invention may be
conjugated to a "receptor' (such streptavidin) for utilization in tumor
pretargeting wherein the
antibody-receptor or Fc fusion-receptor conjugate is administered to the
patient, followed by removal
of unbound conjugate from the circulation using a clearing agent and then
administration of a "ligand"
(e.g. avidin) which is conjugated to a cytotoxic agent (e.g. a
radionucleotide). In an alternate
embodiment, the antibody or Fc fusion is conjugated or operably linked to an
enzyme in order to
employ Antibody Dependent Enzyme Mediated Prodrug Therapy (ADEPT). ADEPT may
be used by
conjugating or operably linking the antibody or Fc fusion to a prodrug-
activating enzyme that converts
a prodrug (e.g. a peptidyl chemotherapeutic agent, see PCT WO 81/01145) to an
active anti-cancer
drug. See, for example, PCT WO 88/07378 and US 4,975,278. The enzyme component
of the
immunoconjugate useful for ADEPT includes any enzyme capable of acting on a
prodrug in such a
way so as to covert it into its more active, cytotoxic form. Enzymes that are
useful in the method of
this invention include but are not limited to alkaline=phosphatase useful for
converting phosphate-
containing prodrugs into free drugs; arylsulfatase useful for converting
sulfate-containing prodrugs into
free drugs; cytosine deaminase useful for converting non-toxic 5-
fluorocytosine into the anti-cancer
drug, 5-fluorouracil; proteases, such as serratia protease, therrnolysin,
subtilisin, carboxypeptidases
and cathepsins (such as cathepsins B and L), that are useful for converting
peptide-containing
prodrugs into free drugs; D-alanylcarboxypeptidases, useful for converting
prodrugs that contain D-
amino acid substituents; carbohydrate-cleaving enzymes such as .beta.-
galactosidase and
neurarnimidase useful for converting glycosylated prodrugs into free drugs;
beta-lactamase useful for
converting drugs derivatized with .alpha.-lactams into free drugs; and
penicillin amidases, such as
penicillin V amidase or penicillin G amidase, useful for converting drugs
derivatized at their amine
nitrogens with phenoxyacetyl or phenylacetyl groups, respectively, into free
drugs. Alternatively,
antibodies with enzymatic activity, also known in the art as "abzymes", can be
used to convert the
prodrugs of the invention into free active drugs (see, for example, Massey,
1987, Nature 328: 457-
458). Antibody-abzyme and Fc fusion-abzyme conjugates can be prepared for
delivery of the abzyme
to a tumor cell population.
[122] Other modifications of the antibodies and Fc fusions of the present
invention are
contemplated herein. For example, the antibody or Fc fusion may be linked to
one of a variety of
nonproteinaceous.polymers, e.g., polyethylene glycol (PEG), polypropylene
glycol, polyoxyalkylenes,
or copolymers of polyethylene glycol and polypropylene glycol.
47
CA 027 66627 2012-01-23
WO 2004/099249 PCT/US2004/009298
[123] Pharmaceutical compositions are contemplated wherein an antibody or Fc
fusion of the
present invention and and one or more therapeutically active agents are
formulated. Formulations of
the antibodies and Fc fusions of the present invention are prepared for
storage by mixing said
antibody or Fc fusion having the desired degree of purity with optional
pharmaceutically acceptable
carriers, excipients or stabilizers.(Remington's Pharmaceutical Sciences 16th
edition, Osol, A.
Ed.,1980), in the form of lyophilized formulations or aqueous solutions.
Acceptable carriers,
excipients, or stabilizers are nontoxic to recipients at the dosages and
concentrations employed, and
include buffers such as phosphate, citrate, acetate, and other organic acids;
antioxidants including
ascorbic acid and methionine; preservatives (such as octadecyldimethylbenzyl
ammonium chloride;
hexamethonium chloride; benzalkonium chloride, benzethonium chloride; phenol,
butyl orbenzyl
alcohol; alkyl parabens such as methyl or propyl paraben; catechol;
resorcinol; cyclohexanol; 3-
pentanol; and m-cresol); low molecular weight (less than about 10 residues)
polypeptides; proteins,
such as serum albumin, gelatin, or immunoglobulins; hydrophilic polymers such
as
polyvinylpyrrolidone; amino acids such as glycine, glutamine, asparagine,
histidine, arginine, or lysine;
monosaccharides, disaccharides, and other carbohydrates including glucose,
mannose, or dextrins;
chelating agents such as EDTA; sugars such as sucrose, mannitol, trehalose or
sorbitol; sweeteners
and other flavoring agents; fillers such as microcrystalline cellulose,
lactose, corn and other starches;
binding agents; additives; coloring agents; salt-forming counter-ions such as
sodium; metal
complexes (e.g. Zn-protein complexes); and/or lion-ionic surfactants such as
TWEENTm,
PLURONICSTm or polyethylene glycol (PEG). In a preferred embodiment, the
pharmaceutical
composition that comprises the antibody or Fc fusion of the present invention
is in a water-soluble
form, such as being present as pharmaceutically acceptable salts, which is
meant to include both acid
and base addition salts. "Pharmaceutically acceptable acid addition salt"
refers to those salts that
retain the biological effectiveness of the free bases and that are not
biologically or otherwise
undesirable, formed with inorganic acids such as hydrochloric acid,
hydrobromic acid, sulfuric acid,
nitric acid, phosphoric acid and the like, and organic acids such as acetic
acid, propionic acid, glycolic
acid, pyruvic acid, oxalic acid, maleic acid, malonic acid, succinic acid,
fumaric acid, tartaric acid, citric
acid, benzoic acid, cinnamic acid, mandelic acid, methanesulfonic acid,
ethanesulfonic acid, p-
toluenesulfonic acid, salicylic acid and the like. "Pharmaceutically
acceptable base addition salts"
include those derived from inorganic bases such as sodium, potassium, lithium,
ammonium, calcium,
magnesium, iron, zinc, copper, manganese, aluminum salts and the like.
Particularly preferred are
the ammonium, potassium, sodium, calcium, and magnesium salts. Salts derived
from
pharmaceutically acceptable organic non-toxic bases include salts of primary,
secondary, and tertiary
amines, substituted amines including naturally occurring substituted amines,
cyclic amines and basic
ion exchange resins, such as isopropylamine, trimethylamine, diethylamine,
triethylamine,
tripropylamine, and ethanolamine. The formulations to be used for in vivo
administration are
preferably sterile. This is readily accomplished by filtration through sterile
filtration membranes or
other methods.
48
CA 02766627 2012-01-23
WO 2004/099249 PCT/US2004/009298
[124] The antibodies and Fc fusions disclosed herein may also be formulated as
immunoliposomes.
A liposome is a small vesicle comprising various types of lipids,
phospholipids and/or surfactant that is
useful for delivery of a therapeutic agent to a mammal. Liposomes containing
the antibody or Fc
fusion are prepared by methods known in the art, such as described in Epstein
at al., 1985, Proc Nat!
Aced Sc! USA, 82:3688; Hwang et al., 1980, Proc Nat/Aced Sci USA, 77:4030; US
4,485,045; US
4,544,545; and PCT WO 97/38731. Liposomes with enhanced circulation time are
disclosed in US
5,013,556. The components of the liposome are commonly arranged in a bilayer
formation, similar to
the lipid arrangement of biological membranes. Particularly useful liposomes
can be generated by the
reverse phase evaporation method with a lipid composition comprising
phosphatidylcholine,
cholesterol and PEG-derivatized phosphatidylethanotamine (PEG-PE). Liposomes
are extruded
through filters of defined pore size to yield liposomes with the desired
diameter. A chemotherapeutic
agent or other therapeutically active agent is optionally contained within the
liposome (Gabizon et al.,
1989, J National Cancer lnst 81:1484).
[1251 The antibodies, Fc fusions, and other therapeutically active agents may
also be entrapped in
microcapsules prepared by methods including but not limited to coacervation
techniques, interfacial
polymerization (for example using hydroxymethylcellulose or gelatin-
microcapsules, or poly-
(methylmethacylate) microcapsules), colloidal drug delivery systems (for
example, liposomes, albumin
microspheres, microemulsions, nano-particles and nanocapsules), and
macroemulsions. Such
techniques are disclosed in Remington's Pharmaceutical Sciences 16th edition,
Osol, A. Ed., 1980.
Sustained-release preparations may be prepared. Suitable examples of sustained-
release
preparations include semipermeable matrices of solid hydrophobic polymer,
which matrices are in the
form of shaped articles, e.g. films, or microcapsules. Examples of sustained-
release matrices include
polyesters, hydrogels (for example poly(2-hydroxyethyl-methacrylate), or
poly(vinylalcohol)),
polylactides (US 3,773,919), copolymers of L-glutamic acid and gamma ethyl-L-
glutamate, non-
degradable ethylene-vinyl acetate, degradable lactic acid-glycolic acid
copolymers such as the
LUPRON DEPOTTy (which are injectable rnicrospheres composed of lactic acid-
glycolic acid
copolymer and leuprolide acetate), poly-D-(-)-3-hydroxybutyric acid, and
ProLease0 (commercially
available from Alkermes), which is a microsphere-based delivery system
composed of the desired
bioactive molecule incorporated into a matrix of poly-DL-lactide'-co-glycolide
(PLG).
[126] The concentration of the therapeutically active antibody or Fc fusion of
the present invention
in the formulation may vary from about 0.1 to 100 weight %. In a preferred
embodiment, the
concentration of the antibody or Fc fusion is in the range of 0.003 to 1.0
molar. In order to treat a
patient, a therapeutically effective dose of the antibody or Fc fusion of the
present invention may be
administered. By "therapeutically effective dose" herein is meant a dose that
produces the effects for
which it is administered. The exact dose will depend on the purpose of the
treatment, and will be
ascertainable by one skilled in the art using known techniques. Dosages may
range-from 0.01 to 100
mg/kg of body weight or greater, for example 0.1, 1, 10, or 50 mg/kg of body
weight, with 1 to
10mg/kg being preferred. As is known in the art, adjustments for antibody or
Fc fusion degradation,
49
CA 02766627 2012-01-23
WO 2004/099249
PCT/US2004/009298
systemic versus localized delivery, and rate of new protease synthesis, as
well as the age, body
weight, general health, sex, diet, time of administration, drug interaction
and the severity of the
condition may be necessary, and will be ascertainable with routine
experimentation by those skilled in
the art.
[127] Administration of the pharmaceutical composition comprising an antibody
or Fc fusion of the
present invention, preferably in the form of a sterile aqueous solution, may
be done in a variety of
ways, including, but not limited to orally, subcutaneously, intravenously,
intranasally, intraotically,
transdermally, topically (e.g., gels, salves, lotions, creams, etc.),
intraperitoneally, intramuscularly,
intrapulmonary (e.g., AERxiD inhalable technology commercially available from
Aradigm, or Inhanceim
pulmonary delivery system commercially available from Inhale Therapeutics),
vaginally, parenterally,
rectally, or intraocularly. In some instances, for example for the treatment
of wounds, inflammation,
etc., the antibody or Fc fusion may be directly applied as a solution or
spray. As is known in the art,
the pharmaceutical composition may be formulated accordingly depending upon
the manner of
introduction.
Engineering Methods
[128] The present invention provides engineering methods that may be used to
generate Fc
variants. A principal obstacle that has hindered previous attempts at Fc
engineering is that only
random attempts at modification have been possible, due in part to the
inefficiency of engineering
strategies and methods, and to the low-throughput nature of antibody
production and screening. The
present invention describes engineering methods that overcome these
shortcomings. A variety of
design strategies, computational screening methods, library generation
methods, and experimental
production and screening methods are contemplated. These strategies,
approaches, techniques, and
methods may be applied individually or in various combinations to engineer
optimized Fc variants.
Design Strategies
[129] The most efficient approach to generating Fc variants that are optimized
for a desired
property is to direct the engineering efforts toward that goal. Accordingly,
the present invention
teaches design strategies that may be used to engineer optimized Fc variants.
The use of a design
strategy is meant to guide Fc engineering, but is not meant to constrain an Fc
variant to a particular
optimized property based on the design strategy that was used to engineer it.
At first thought this may
seem counterintuitive; however its validity is derived from the enormous
complexity of subtle
interactions that determine the structure, stability, solubility, and function
of proteins and protein-
protein complexes. Although efforts can be made to predict which protein
positions, residues,
interactions, etc. are important for a design goal, often times critical ones
are not predictable. Effects
on protein structure, stability, solubility, and function, whether favorable
or unfavorable, are often
unforeseen. Yet there are innumerable amino acid modifications that are
detrimental or deleterious to
proteins. Thus often times the best approach to engineering comes from
generation of protein
CA 02766627 2012-01-23
WO 2004/099249 PCT/US2004/009298
variants that are focused generally towards a design goal but do not cause
detrimental effects. In this
way, a principal objective of a design strategy may be the generation of
quality diversity. At a
simplistic level this can be thought of as stacking the odds in one's favor.
As an example,
perturbation of the Fc carbohydrate or a particular domain-domain angle, as
described below, are
valid design strategies for generating optimized Fc variants, despite the fact
that how carbohydrate
and domain-domain angles determine the properties of Fc is not well
understood. By reducing the
number of detrimental amino acid modifications that are screened, i.e. by
screening quality diversity,
these design strategies become practical. Thus the true value of the design
strategies taught in the
present invention is their ability to direct engineering efforts towards the
generation of valuable Fc
variants. The specific value of any one resulting variant is determined after
experimentation.
[130] One design strategy for engineering Fc variants is provided in which
interaction of Fc with
some Fc ligand is altered by engineering amino acid modifications at the
interface between Fc and
said Fc ligand. Fc ligands herein may include but are not limited to FeyRs,
Gig, FcRn, protein A or G,
and the like. By exploring energetically favorable substitutions at Fc
positions that impact the binding
interface, variants can be engineered that sample new interface conformations,
some of which may
improve binding to the Fc ligand, some of which may reduce Fc ligand binding,
and some of which
may have other favorable properties. Such new interface conformations could be
the result of, for
example, direct interaction with Fc ligand residues that form the interface,
or indirect effects caused by
the amino acid modifications such as perturbation of side chain or backbone
conformations. Variable
positions may be chosen as any positions that are believed to play an
important role in determining
the conformation of the interface. For example, variable positions may be
chosen as the set of
residues that are within a certain distance, for example 5 Angstroms (A),
preferrably between 1 and
A, of any residue that makes direct contact with the Fc ligand.
[131] An additional design strategy for generating Fc variants is provided in
which the conformation
of the Fc carbohydrate at N297 is optimized. Optimization as used in this
context is meant to includes
conformational and compositional changes in the N297 carbohydrate that result
in a desired property,
for example increased or reduced affinity for an FcyR. Such a strategy is
supported by the
observation that the carbohydrate structure and conformation dramatically
affect Fc/FcyR and Fc/C1q
binding (Umatia at al., 1999, Nat Biotechnol 17:176-180; Davies at aL, 2001,
Biotechnol Bioeng
74:288-294; Mimura etal., 2001, J Bib, Chem 276:45539-45547.; Radaev at aL,
2001, J Slol Chem
276:16478-16483; Shields etal.. 2002, J Biol Chem 277:26733-26740; Shinkawa at
al., 2003, J Bidl
Chem 278:3466-3473). However the carbohydrate makes no specific contacts with
FcyRs. By
exploring energetically favorable substitutions at positions that interact
with carbohydrate, a quality
diversity of variants can be engineered that sample new carbohydrate
conformations, some of which
may improve and some of which may reduce binding to one or more Fc ligands.
While the majority of
mutations near the Fe/carbohydrate interface appear to alter carbohydrate
conformation, some
51
CA 02766627 2012-01-23
WO 2004/099249 PCT/US2004/009298
mutations have been shown to alter the glycosylation composition (Lund et aL,
1996, J Immune!
157:4963-4969; Jefferis et aL, 2002, Immund Lett 82:57-65).
[132] Another design strategy for generating Fc variants is provided in which
the angle between the
0y2 and C73 domains is optimized Optimization as used in this context is meant
to describe
conformational changes in the Cy2-Cy3 domain angle that result in a desired
property, for example
increased or reduced affinity for an FcyR. This angle is an important
determinant of Fc/FcyR affinity
(Radaev et at., 2001, J Blot Chem 276:16478-16483), and a number of mutations
distal to the
Fc/FcyR interface affect binding potentially by modulating it (Shields et al.,
2001, J Biol Chem
276:6591-6604). By exploring energetically favorable substitutions positions
that appear to play a key
role in determining the Cy2-Cy3 angle and the flexibility of the domains
relative to one another, a
quality diversity of variants can be designed that sample new angles and
levels of flexibility, some of
which may be optimized for a desired Fc property.
[133] Another design strategy for generating Fc variants is provided in which
Fc is reengineered to
eliminate the structural and functional dependence on glycosylation. This
design strategy involves the
optimization of Fc structure, stability, solubility, and/or Fc function (for
example affinity of Fc for one or
more Fc ligands) in the absence of the N297 carbohydrate. In one approach,
positions that are
exposed to solvent in the absence of glycosylation are engineered such that
they are stable,
structurally consistent with Fc structure, and have no tendency to aggregate.
The C72 is the only
unpaired Ig domain in the antibody (see Figure 1): Thus the N297 carbohydrate
covers up the
exposed hydrophobic patch that would normally be the interface for a protein-
protein interaction with
another Ig domain, maintaining the stability and structural integrity of Fc
and keeping the 0y2 domains
from aggregating across the central axis. Approaches for optimizing
aglycosylated Fc may involve but
are not limited to designing amino acid modifications that enhance
aglycoslated Fc stability and/or
solubility by incorporating polar and/or charged residues that face inward
towards the C12-Cy2 dimer
axis, and by designing amino acid modifications that directly enhance the
aglycosylated Fc/FcyR
interface or the interface of aglycosylated Fc with some other Fc ligand.
[134] An additional design strategy for engineering Fc variants is provided in
which the
conformation of the Cy2 domain is optimized Optimization as used in this
context is meant to
describe conformational changes in the Cy2 domain angle that result in a
desired property, for
example increased or reduced affinity for an FcyR. By exploring energetically
favorable substitutions
at Cy2 positions that impact the C12 conformation, a quality diversity of
variants can be engineered
that sample new Cy2 conformations, some of which may achieve the design goal.
Such new Cy2
conformations could be the result of, for example, alternate backbone
conformations that are sampled
by the variant. Variable positions may be chosen as any positions that are
believed to play an
important role in determining C72 structure, stability, solubility,
flexibility, function, and the like. For
example, Cy2 hydrophobic core residues, that is Cy2 residues that are
partially or fully sequestered
52
CA 02766627 2012-01-23
WO 2004/099249
PCT/US2004/009298
from solvent, may be reengineered. Alternatively, noncore residues may be
considered, or residues
that are deemed important for determining backbone structure, stability, or
flexibility.
[135] An additional design strategy for Fc optimization is provided in which
binding to an FcyR,
complement, or some other Fc ligand is altered by modifications that modulate
the electrostatic
interaction between Fc and said Fc ligand. Such modifications may be thought
of as optimization of
the global electrostatic character of Fc, and include replacement of neutral
amino acids with a
charged amino acid, replacement of a charged amino acid with a neutral amino
acid, or replacement
of a charged amino acid with an amino acid of opposite charge (i.e. charge
reversal). Such
modifications may be used to effect changes in binding affinity between an Fc
and one or more Fc
ligands, for example FcyRs. In a preferred embodiment, positions at which
electrostatic substitutions
might affect binding are selected using one of a variety of well known methods
for calculation of
electrostatic potentials. In the simplest embodiment, Coulomb's law is used to
generate electrostatic
potentials as a function of the position in the protein. Additional
embodiments include the use of
Debye-Huckel scaling to account for ionic strength effects, and more
sophisticated embodiments such
as Poisson-Boltzmann calculations. Such electrostatic calculations may
highlight positions and
suggest specific amino acid modifications to achieve the design goal. In some
cases, these
substitutions may be anticipated to variably affect binding to different Fc
ligands, for example to
enhance binding to activating FcyRs while decreasing binding affinity to
inhibitory FeyRs.
Computational Screening
[136] A principal obstacle to obtaining valuable Fc variants is the difficulty
in predicting what amino
acid modifications, out of the enormous number of possibilities, will achieve
the desired goals. Indeed
one of the principle reasons that previous attempts at Fc engineering have
failed to produce Fc
variants of significant clinical value is that approaches to Fc engineering
have thus far involved hit-or-
miss approaches. The present invention provides computational screening
methods that enable
quantitative and systematic engineering of Fc variants. These methods
typically use atomic level
scoring functions, side chain rotamer sampling, and advanced optimization
methods to accurately
capture the relationships between protein sequence, structure, and function.
Computational
screening enables exploration of the entire sequence space of possibilities at
target positions by
filtering the enormous diversity which results. Variant libraries that are
screened computationally are
effectively enriched for stable, properly folded, and functional sequences,
allowing active optimization
of Fc for a desired goal. Because of the overlapping sequence constraints on
protein structure,
stability, solubility, and function, a large number of the candidates in a
library occupy "wasted"
sequence space. For example, a large fraction of sequence space encodes
unfolded, misfolded,
incompletely folded, partially folded, or aggregated proteins. This is
particularly relevant for Fc
engineering because Ig domains are small beta sheet structures, the
engineering of which has proven
extremely demanding (Quinn etal., 1994, Proc Natl Aced Sc/USA 91:8747-8751;
Richardson et al.,
2002, Proc Nat! Acad Sci U S A 99:2754-2759). Even seemingly harmless
substitutions on the
53
CA 02766627 2012-01-23
WO 2004/099249
PCT/US2004/009298
surface of a beta sheet can cause severe packing conflicts, dramatically
disrupting folding equilibrium
(Smith et aL, 1995, Science 270:980-982); incidentally, alanine is one of the
worst beta sheet formers
(Minor et al., 1994, Nature 371:264-267). The determinants of beta sheet
stability and specificity are
a delicate balance between an extremely large number of subtle interactions.
Computational
screening enables the generation of libraries that are composed primarily of
productive sequence
space, and as a result increases the chances of identifying proteins that are
optimized for the design
goal. In effect, computational screening yields an increased hit-rate, thereby
decreasing the number
of variants that must be screened experimentally. An additional obstacle to Fc
engineering is the
need for active design of correlated or coupled mutations. For example, the
greatest Fc/FcyR affinity
enhancement observed thus far is S298A/E333A/K334A, obtained by combining
three better binders
obtained separately in an alanine scan (Shields at aL, 2001, J Blot Chem
276:6591-6604).
Computational screening is capable of generating such a three-fold variant in
one experiment instead
of three separate ones, and furthermore is able to test the functionality of
all 20 amino acids at those
positions instead of just alanine. Computational screening deals with such
complexity by reducing the
combinatorial problem to an experimentally tractable size.
[137] Computational screening, viewed broadly, has four steps: 1) selection
and preparation of the
protein template structure or structures, 2) selection of variable positions,
amino acids to be
considered at those positions, and/or selection of rotamers to model
considered amino acids, 3)
energy calculation, and 4) combinatorial optimization. In more detail, the
process of computational*
screening can be described as follows. A three-dimensional structure of a
protein is used as the
starting point. The positions to be optimized are identified, which may be the
entire protein sequence
or subset(s) thereof. Amino acids that will be considered at each position are
selected. In a preferred
embodiment, each considered amino acid may be represented by a discrete set of
allowed
conformations, called rotamers. interaction energies are calculated between
each considered amino
acid and each other considered amino acid, and the rest of the protein,
including the protein backbone
and invariable residues. In a preferred embodiment, interaction energies are
calculated between
each considered amino acid side chain rotamer and each other considered amino
acid side chain
rotamer and the rest of the protein, including the protein backbone and
invariable residues. One or
more combinatorial search algorithms are then used to identify the lowest
energy sequence and/or
low energy sequences.
[138] In a preferred embodiment, the computational screening method used is
substantially similar
to Protein Design Automation@ (PDAO) technology, as is described in US
6,188,965; US 6,269,312;
US 6,403,312; USSN 09/782,004; USSN 09/927,790; USSN 10/218,102; PCT WO
98/07254; PCT
WO 01/40091; and PCT WO 02/25588. In another preferred embodiment, a
computational screening
method substantially similar to Sequence Prediction Algorithm TM (SPA)
technology is used, as is
described in (Raha et aL, 2000, Protein Sci 9:1106-1119), USSN 09/877,695, and
USSN 10/071,859.
In another preferred embodiment, the computational screening methods described
in USSN
54
CA 02766627 2012-01-23
WO 2004099249
PCT/US2004/009298
10/339788, filed on March 3, 2003, entitled "ANTIBODY OPTIMIZATION", are used.
In some
embodiments, combinations of different computational screening methods are
used, including
combinations of PDAO technology and SPAT" technology, as well as combinations
of these
computational methods in combination with other design tools. Similarly, these
computational
methods can be used simultaneously or sequentially, in any order.
[139] A template structure is used as input into the computational screening
calculations. By
"template structure" herein is meant the structural coordinates of part or all
of a protein to be
Optimized. The template structure may be any protein for which a three
dimensional structure (that is,
three dimensional coordinates for a set of the protein's atoms) is known or
may be calculated,
estimated, modeled, generated, or determined. The three dimensional structures
of proteins may be
determined using methods including but not limited to X-ray crystallographic
techniques, nuclear
magnetic resonance (NMR) techniques, de novo modeling, and homology modeling.
If optimization is
desired for a protein for which the structure has not been solved
experimentally, a suitable structural
model may be generated that may serve as the template for computational
screening calculations.
Methods for generating homology models of proteins are known in the art, and
these methods find
use in the present invention. See for example, Luo, et al. 2002, Protein Sci
11: 1218-1226, Lehmann
& Wyss, 2001, Curr Opin Biotechnol 12(4):371-5.; Lehmann et al., 2000, Biochim
Biophys Acta
1543(2):408-415; Rath & Davidson, 2000, Protein Sci, 9(12):2457-69; Lehmann
etal., 2000, Protein
Eng 13(1):49-57; Desjarlais & Berg, 1993, Proc Nail Acad Sci USA 90(6):2256-
60; Desjarlais & Berg,
1992, Proteins 12(2):101-4; Henikoff & Henikoff, 2000, Adv Protein Chem 54:73-
97; Henikoff &
Henikoff, 1994, J Mol Biol 243(4):574-8; Morea et al., 2000, Methods 20:267-
269. Protein/protein
complexes may also be obtained using docking methods. Suitable protein
structures that may serve
as template structures include, but are not limited to, all of those found in
the Protein Data Base
compiled and serviced by the Research Collaboratory for Structural
Bioinformatics (RCSB, formerly
the Brookhaven National Lab).
[140] The template structure may be of a protein that occurs naturally or is
engineered. The
template structure may be of a protein that is substantially encoded by a
protein from any organism,
with human, mouse, rat, rabbit, and monkey preferred. The template structure
may comprise any of a
number of protein structural forms. In a preferred embodiment the template
structure comprises an
Fc region or a domain or fragment of Fc. In an alternately preferred
embodiment the template
structure comprises Fc or a domain or fragment of Fc bound to one or more Fc
ligands, with an
FdFcyR complex being preferred. The Fc in the template structure may be
glycosylated or
unglycosylated. The template structure may comprise more than one protein
chain. The template
structure may additionally contain nonprotein components, including but not
limited to small
molecules, substrates, cofactors, metals, water molecules, prosthetic groups,
polymers and
carbohydrates. In a preferred embodiment, the template structure is a
plurality or set of template
proteins, for example an ensemble of structures such as those obtained from
NMR. Alternatively, the
CA 02766627 2012-01-23
WO 20041099249
PCT/US2004/0092911
set of template structures is generated from a set of related proteins or
structures, or artificially
created ensembles_ The composition and source of the template structure
depends on the
engineering goal. For example, for enhancement of human Fc/FcyR affinity, a
human Fc/FcyR
complex structure or derivative thereof may be used as the template structure.
Alternatively, the
uncomplexed Fc structure may be used as the template structure. If the goal is
to enhance affinity of
a human Fc for a mouse FcyR, the template structure may be a structure or
model of a human Fc
bound to a mouse FcyR.
[141] The template structure may be modified or altered prior to design
calculations. A variety of
methods for template structure preparation are described in US 6,188,965; US
6,269,312; US
6,403,312; USSN 09/782,004; USSN 09/927,790; USSN 09/877,695; USSN 10/071,859,
USSR
10/218,102; PCT WO 98/07254; PCT WO 01/40091; and PCT WO 02/25588. For
example, in a
preferred embodiment, explicit hydrogens may be added if not included within
the structure. In an
alternate embodiment, energy minimization of the structure is run to relax
strain, including strain due
to van der Weals clashes, unfavorable bond angles, and unfavorable bond
lengths. Alternatively, the
template structure is altered using other methods, such as manually, including
directed or random
perturbations. It is also possible to modify the template structure during
later steps of computational
screening, including during the energy calculation and combinatorial
optimization steps. In an
alternate embodiment, the template structure is not modified before or during
computational screening
calculations.
[142] Once a template structure has been obtained, variable positions are
chosen. By "variable
position" herein is meant a position at which the amino acid identity is
allowed to be altered in a
computational screening calculation. As is known in the art, allowing amino
acid modifications to be
considered only at certain variable positions reduces the complexity of a
calculation and enables
computational screening to be more directly tailored for the design goal. One
or more residues may
be variable positions in computational screening calculations. Positions that
are chosen as variable
positions may be those that contribute to or are hypothesized to contribute to
the protein property to
be optimized, for example Fc affinity for an FcyR, Fc stability, Fc
solubility, and so forth. Residues at
variable positions may contribute favorably or unfavorably to a specific
protein property. For example,
a residue at an Fc/FcyR interface may be involved in mediating binding, and
thus this position may be
varied in design calculations aimed at improving Fc/FcyR affinity. As another
example, a residue that
has an exposed hydrophobic side chain may be responsible for causing
unfavorable aggregation, and
thus this position may be varied in design calculations aimed at improving
solubility. Variable
positions may be those positions that are directly involved in interactions
that are determinants of a
, particular protein property. For example, the FcyR binding site of Fc may
be defined to include all
residues that contact that particular FycR. By "contacr herein is meant some
chemical interaction
between at least one atom of an Fc residue with at least one atom of the bound
FcyR, with chemical
interaction including, but not limited to van der Weals interactions, hydrogen
bond interactions,
56
CA 02766627 2012-01-23
WO 2004/099249 PCT/US2004/909298
electrostatic interactions, and hydrophobic interactions. In an alternative
embodiment, variable
positions may include those positions that are indirectly involved in a
protein property, i.e. such
positions may be proximal to residues that are known to or hypothesized to
contribute to an Fc
property. For example, the FcyR binding site of an Fc may be defined to
include all Fc residues within
a certain distance, for example 4 - 10 A, of any Fc residue that is in van der
Waals contact with the
FcyR. Thus variable positions in this case may be chosen not only as residues
that directly contact
the FcyR, but also those that contact residues that contact the FcyR and thus
influence binding
indirectly. The specific positions chosen are dependent on the design strategy
being employed.
[143] One or more positions in the template structure that are not variable
may be floated. By
"floated position" herein is meant a position at which the amino acid
conformation but not the amino
acid identity is allowed to vary in a computational screening calculation. In
one embodiment, the
floated position may have the parent amino acid identity. For example, floated
positions may be
positions that are within a small distance, for example 5 A, of a variable
position residue. In an
alternate embodiment, a floated position may have a non-parent amino acid
identity. Such an
embodiment may find use in the present invention, for example, when the goal
is to evaluate the
energetic or structural outcome of a specific mutation.
[144] Positions that are not variable or floated are fixed. By "fixed
position" herein is meant a
position at which the amino acid identity and the conformation are held
constant in a computational
screening calculation. Positions that may be fixed include residues that are
not known to be or
hypothesized to be involved in the property to be optimized. In this case the
assumption is that there
is little or nothing to be gained by varying these positions. Positions that
are fixed may also include
positions whose residues are known or hypothesized to be important for
maintaining proper folding,
structure, stability, solubility, and/or biological function. For example,
positions may be fixed for
residues that interact with a particular Fc ligand or residues that encode a
glycosylation site in order to
ensure that binding to the Fc ligand and proper glycosylation respectively are
not perturbed.
Likewise, if stability is being optimized, it may be beneficial to fix
positions that directly or indirectly
interact with an Fc ligand, for example an FcyR, so that binding is not
perturbed. Fixed positions may
also include structurally important residues such as cysteines participating
in disulfide bridges,
residues critical for determining backbone conformation such as proline or
glycine, critical hydrogen
bonding residues, and residues that form favorable packing interactions.
[145] The next step in computational screening is to select a set of possible
amino acid identities
that will be considered at each particular variable position. This set of
possible amino acids is herein
referred to as "considered amino acids" at a variable position. "Amino acids"
as used herein refers to
the set of natural 20 amino acids and any nonnatural or synthetic analogues.
In one embodiment, all
20 natural amino acids are considered. Alternatively, a subset of amino acids,
or even only one
=
amino acid is considered at a given variable position. As will be appreciated
by those skilled in the
art, there is a computational benefit to considering only certain amino acid
identities at variable
57
CA 02766627 2012-01-23
WO 2004/099249 PCT/US2004/009298
positions, as it decreases the combinatorial complexity of the search.
Furthermore, considering only
certain amino acids at variable positions may be used to tailor calculations
toward specific design
strategies. For example, for solubility optimization of aglycosylated Fc, it
may be beneficial to allow
only polar amino acids to be considered at nonpolar Fc residues that are
exposed to solvent in the
absence of carbohydrate. Nonnatural amino acids, including synthetic amino
acids and analogues of
natural amino acids, may also be considered amino acids. For example see Chin
et aL, 2003,
Science, 301(5635):964-7; and Chin et aL, 2003, Chem Bio/.10(6):511-9.
[146] A wide variety of methods may be used, alone or in combination, to
select which amino acids
will be considered at each position. For example, the set of considered amino
acids at a given
variable position may be chosen based on the degree of exposure to solvent.
Hydrophobic or
nonpolar amino acids typically reside in the interior or core of a protein,
which are inaccessible or
nearly inaccessible to solvent. Thus at variable core positions it may be
beneficial to consider only or
mostly nonpolar amino acids such as alanine, valine, isoleucine, leucine,
phenylalanine, tyrosine,
tryptophan, and methionine. Hydrophilic or polar amino acids typically reside
on the exterior or
surface of proteins, which have a significant degree of solvent accessibility.
Thus at variable surface
positions it may be beneficial to consider only or mostly polar amino acids
such as alanine, serine,
threonine, aspartic acid, asparagine, glutamine, glutamic acid, arginine,
lysine and histidine. Some
positions are partly exposed and partly buried, and are not clearly protein
core or surface positions, in
a sense serving as boundary residues between core and surface residues. Thus
at such variable
boundary positions it may be beneficial to consider both nonpolar and polar
amino acids such as
alanine, serine, threonine, aspartic acid, asparagine, glutamine, glutamic
acid, arginine, lysine
histidine, valine, isoleucine, leucine, phenylalanine, tyrosine, tryptophan,
and methionine.
Determination of the degree of solvent exposure at variable positions may be
by subjective evaluation
or visual inspection of the template structure by one skilled in the art of
protein structural biology, or by
using a variety of algorithms that are known in the art. Selection of amino
acid types to be considered
at variable positions may be aided or determined wholly by computational
methods, such as
calculation of solvent accessible surface area, or using algorithms that
assess the orientation of the
Ca-Cp vectors relative to a solvent accessible surface, as outlined in US
6,188,965; 6,269,312; US
6,403,312; USSN 09/782,004; USSN 09/927,790; USSN 10/218,102; PCT WO 98/07254;
PCT WO
01/40091; and PCT WO 02/25588. In one embodiment, each variable position may
be classified
explicitly as a core, surface, or boundary position or a classification
substantially similar to core,
surface, or boundary.
[147] In an alternate embodiment, selection of the set of amino acids allowed
at variable positions
may be hypothesis-driven. Hypotheses for which amino acid types should be
considered at variable
positions may be derived by a subjective evaluation or visual inspection of
the template structure by
one skilled in the art of protein structural biology. For example, if it is
suspected that a hydrogen
bonding interaction may be favorable at a variable position, polar residues
that have the capacity to
58
CA 02766627 2012-01-23
WO 2004/099249 PCT/US2004/009298
form hydrogen bonds may be considered, even if the position is in the core.
Likewise, if it is
suspected that a hydrophobic packing interaction may be favorable at a
variable position, nonpolar
residues that have the capacity to form favorable packing interactions may be
considered, even if the
position is on the surface. Other examples of hypothesis-driven approaches may
involve issues of
backbone flexibility or protein fold. As is known in the art, certain
residues, for example proline,
glycine, and cysteine, play important roles in protein structure and
stability. Glycine enables greater
backbone flexibility than all other amino acids, proline constrains the
backbone more than all other
amino acids, and cysteines may form disulfide bonds. It may therefore be
beneficial to include one or
more of these amino acid types to achieve a desired design goal.
Alternatively, it may be beneficial to
exclude one or more of these amino acid types from the list of considered
amino acids.
[148] In an alternate embodiment, subsets of amino acids may be chosen to
maximize coverage.
In this case, additional amino acids with properties similar to that in the
template structure may be
considered at variable positions. For example, if the residue at a variable
position in the template
structure is a large hydrophobic residue, additional large hydrophobic amino
acids may be considered
at that position. Alternatively, subsets of amino acids may be chosen to
maximize diversity. In this
case, amino acids with properties dissimilar to those in the template
structure may be considered at
variable positions. For example, if the residue at a variable position in the
template is a large
hydrophobic residue, amino acids that are small, polar, etc. may be
considered.
59
CA 02766627 2012-01-23
WO 2004/099249 PCT/US2004/009298
(1491 As is known in the art, some computational screening methods require
only the identity of
considered amino acids to be determined during design calculations. That is,
no information is
required concerning the conformations or possible conformations of the amino
acid side chains.
Other preferred methods utilize a set of discrete side chain conformations,
called rotamers, which are
considered for each amino acid. Thus, a set of rotamers may be considered at
each variable and
floated position. Rotamers may be obtained from published rotamer libraries
(see for example, Lovel
et al., 2000, Proteins: Structure Function and Genetics 40:389-408; Dunbrack &
Cohen, 1997, Protein
Science 6:1661-1681; DeMaeyer eta)., 1997, Folding and Design 2:53-66; Tuffery
et aL, 1991, J
Biomol Struct Dyn 8:1267-1289, Ponder & Richards, 1987, J Mel 8/0/ 193:775-
791). As is known in
the art, rotamer libraries may be backbone-independent or backbone-dependent.
Rotamers may also
be obtained from molecular mechanics or ab in/ti calculations, and using other
methods. In a
preferred embodiment, a flexible rotamer model is used (see Mendes et al.,
1999, Proteins: Structure,
Function, and Genetics 37:530-543). Similarly, artificially generated rotamers
may be used, or
augment the set chosen for each amino acid and/or variable position. In one
embodiment, at least
one conformation that is not low in energy is included in the list of
rotamers. In an alternate
embodiment, the rotamer of the variable position residue in the template
structure is included in the
list of rotamers allowed for that variable position. In an alternate
embodiment, only the identity of
each amino acid considered at variable positions is provided, and no specific
conformational states of
each amino acid are used during design calculations. That is, use of rotamers
is not essential for
computational screening.
[150] Experimental information may be used to guide the choice of variable
positions and/or the
choice of considered amino acids at variable positions. As is known in the
art, mutagenesis
experiments are often carried out to determine the role of certain residues in
protein structure and
function, for example, which protein residues play a role in determining
stability, or which residues
make up the interface of a protein-protein interaction. Data obtained from
such experiments are
useful in the present invention. For example, variable positions for Fc/FcyR
affinity enhancement
could involve varying all positions at which mutation has been shown to affect
binding. Similarly, the
results from such an experiment may be used to guide the choice of allowed
amino acid types at
variable positions. For example, if certain types of amino acid substitutions
are found to be favorable,
similar types of those amino acids may be considered. In one embodiment,
additional amino acids
with properties similar to those that were found to be favorable
experimentally may be considered at
variable positions. For example, if experimental mutation of a variable
position at an Fc/FcyR
interface to a large hydrophobic residue was found to be favorable, the user
may choose to include
additional large hydrophobic amino acids at that position in the computational
screen. As is known in
the art, display and other selection technologies may be coupled with random
mutagenesis to
generate a list or lists of amino acid substitutions that are favorable for
the selected property. Such a
list or lists obtained from such experimental work find use in the present
invention. For example,
positions that are found to be invariable in such an experiment may be
excluded as variable positions
CA 02766627 2012-01-23
WO 2004/099249
PCT/US2004/009298
in computational screening calculations, whereas positions that are found to
be more acceptable to
mutation or respond favorably to mutation may be chosen as variable positions.
Similarly, the results
from such experiments may be used to guide the choice of allowed amino acid
types at variable
positions. For example, if certain types of amino acids arise more frequently
in an experimental
selection, similar types of those amino acids may be considered. In one
embodiment, additional
amino acids with properties similar to those that were found to be favorable
experimentally may be
considered at variable positions. For example, if selected mutations at a
variable position that resides
at an Fc,/FcyR interface are found to be uncharged polar amino acids, the user
may choose to include
additional uncharged polar amino acids, or perhaps charged polar amino acids,
at that position.
[151] Sequence information may also be used to guide choice of variable
positions and/or the
choice of amino acids considered at variable positions. As is known in the
art, some proteins share a
common structural scaffold and are homologous in sequence. This information
may be used to gain
insight into particular positions in the protein family. As is known in the
art, sequence alignments are
often carried out to determine which protein residues are conserved and which
are not conserved.
That is to say, by comparing and contrasting alignments of protein sequences,
the degree of
variability at a position may be observed, and the types of amino acids. that
occur naturally at positions
may be observed. Data obtained from such analyses are useful in the present
invention. The benefit
of using sequence information to choose variable positions and considered
amino acids at variable
positions are several fold. For choice of variable positions, the primary
advantage of using sequence
information is that insight may be gained into which positions are more
tolerant and which are less
tolerant to mutation. Thus sequence information may aid in ensuring that
quality diversity, i.e.
mutations that are not deleterious to protein structure, stability, etc., is
sampled computationally. The
same advantage applies to use of sequence information to select amino acid
types considered at
variable positions. That is, the set of amino acids that occur in a protein
sequence alignment may be
thought of as being pre-screened by evolution to have a higher chance than
random for being
compatible with a protein's structure, stability, solubility, function, etc.
Thus higher quality diversity is
sampled computationally. A second benefit of using sequence information to
select amino acid types
considered at variable positions is that certain alignments may represent
sequences that may be less
immunogenic than random sequences. For example, if the amino acids considered
at a given
variable position are the set of amino acids which occur at that position in
an alignment of human
protein sequences, those amino acids may be thought of as being pre-screened
by nature for
generating no or low immune response if the optimized protein is used as a
human therapeutic.
[152] The source of the sequences may vary widely, and include One or more of
the known
databases, including but not limited to the Kabat database (Johnson & Wu,
2001, Nucleic Acids Res
29:205-206; Johnson & Wu, 2000, Nucleic Acids Res 28:214-218), the 1MGT
database (IMGT, the
international ImMunoGeneTics information system(); Lefranc et al., 1999,
Nucleic Acids Res 27:209-
212; Ruiz et aL, 2000 Nucleic Acids Re. 28:219-221; Lefranc et aL, 2001,
Nucleic Acids Res 29:207-
61
CA 02766627 2012-01-23
WO 2004/099249 PCT/US2004/009298
209; Lefranc et aL, 2003, Nucleic Acids Res 31:307-310), and VBASE, SwissProt,
GenBank and
Entrez, and EMBL Nucleotide Sequence Database. Protein sequence information
can be obtained,
compiled, and/or generated from sequence alignments of naturally occurring
proteins from any
organism, including but not limited to mammals. Protein sequence information
can be obtained from
a database that is compiled privately. There are numerous sequence-based
alignment programs and
methods known in the art, and all of these find use in the present invention
for generation of sequence
alignments of proteins that comprise Fc and Fc ligands.
[153] Once alignments are made, sequence information can be used to guide
choice of variable
positions. Such sequence information can relate the variability, natural or
otherwise, of a given
position. Variability herein should be distinguished from variable position.
Variability refers to the
degree to which a given position in a sequence alignment shows variation in
the types of amino acids
that occur there. Variable position, to reiterate, is a position chosen by the
user to vary in amino acid
identity during a computational screening calculation. Variability may be
determined qualitatively by
one skilled in the art of bioinformatics. There are also methods known in the
art to quantitatively
determine variability that may find use in the present invention. The most
preferred embodiment
measures Information Entropy or Shannon Entropy. Variable positions can be
chosen based on
sequence information obtained from closely related protein sequences, or
sequences that are less
closely related.
[154] The use of sequence information to choose variable positions finds broad
use in the present
invention_ For example, if an Fc/FcyR interface position in the template
structure is tryptophan, and
tryptophan is observed at that position in greater than 90% of the sequences
in an alignment, it may
be beneficial to leave that position fixed. In contrast, if another interface
position is found to have a
greater level of variability, for example if five different amino acids are
observed at that position with
frequencies of approximately 20% each, that position may be chosen as a
variable position. In
another embodiment, visual inspection of aligned protein sequences may
substitute for or aid visual
inspection of a protein structure. Sequence information can also be used to
guide the choice of amino
acids considered at variable positions. Such sequence information can relate
to how frequently an
amino acid, amino acids, or amino acid types (for example polar or nonpolar,
charged or uncharged)
occur, naturally or otherwise, at a given position. In one embodiment, the set
of amino acids
considered at a variable position may comprise the set of amino acids that is
observed at that position
in the alignment. Thus, the position-specific alignment information is used
directly to generate the list
of considered amino acids at a variable position in a computational screening
calculation. Such a
strategy is well known in the art; see for example Lehmann & Wyss, 2001, Curr
Opin Biotechnol
12(4):371-5; Lehmann etal., 2000, Biochim Biophys Acta 1543(2):408-415; Rath &
Davidson, 2000,
Protein Sci, 9(12):2457-69; Lehmann et al., 2000, Protein Eng 13(449-57;
Desjarlais & Berg, 1993,
Proc Nati Aced Sci USA 90(6):2256-60; Desjarlais & Berg, 1992, Proteins
12(2):101-4; Henikoff &
Henikoff, 2000, Adv Protein Chem 54:73-97; Henikoff & Henikoff, 1994, J Mol
Bio1243(4):574-8. In
62
CA 02766627 2012-01-23
WO 2004/099249
PCT/US2004/009298
an alternate embodiment, the set of amino acids considered at a variable
position or positions may
comprise a set of amino acids that is observed most frequently in the
alignment. Thus, a certain
criteria is applied to determine whether the frequency of an amino acid or
amino acid type warrants its
inclusion in the set of amino acids that are considered at a variable
position. As is known in the art,
sequence alignments may be analyzed using statistical methods to calculate the
sequence diversity at
any position in the alignment and the occurrence frequency or probability of
each amino acid at a
position. Such data may then be used to determine which amino acids types to
consider. In the
simplest embodiment, these occurrence frequencies are calculated by counting
the number of times
an amino acid is observed at an alignment position, then dividing by the total
number of sequences in
the alignment. In other embodiments, the contribution of each sequence,
position or amino acid to the
counting procedure is weighted by a variety of possible mechanisms. In a
preferred embodiment, the
contribution of each aligned sequence to the frequency statistics is weighted
according to its diversity
weighting relative to other sequences in the alignment. A common strategy for
accomplishing this is
the sequence weighting system recommended by Henikoff and Henikoff (Henikoff &
Henikoff, 2000,
Adv Protein Chem 54:73-97; Henikoff & Henikoff, 1994, J Mot Blot 243:574-8. In
a preferred
embodiment, the contribution of each sequence to the statistics is dependent
on its extent of similarity
to the target sequence, i.e. the template structure used, such that sequences
with higher similarity to
the target sequence are weighted more highly. Examples of similarity measures
include, but are not
limited to, sequence identity, BLOSUM similarity score, PAM matrix similarity
score, and BLAST
score. In an alternate embodiment, the contribution of each sequence to the
statistics is dependent
on its known physical or functional properties. These properties include, but
are not limited to,
thermal and chemical stability; contribution to activity, and solubility. For
example, when optimizing
aglycosylated Fc for solubility, those sequences in an alignment that are
known to be most soluble
(for example see Ewert et al., 2003, J Mot Bib! 325:531-553), will contribute
more heavily to the
calculated frequencies.
[155] Regardless of what criteria are applied for choosing the set of amino
acids in a sequence
alignment to be considered at variable positions, use of sequence information
to choose considered
amino acids finds broad use in the present invention. For example, to optimize
Fc solubility by
replacing exposed nonpolar surface residues, considered amino acids may be
chosen as the set of
amino acids, or a subset of those amino acids which meet some criteria, that
are observed at that
position in an alignment of protein sequences. As another example, one or more
amino acids may be
added or subtracted subjectively from a list of amino acids derived from a
sequence alignment in
order to maximize coverage. For example, additional amino acids with
properties similar to those that
are found in a sequence alignment may be considered at variable positions. For
example, if an Fc
position that is known to or hypothesized to bind an FcyR is observed to have
uncharged polar amino
acids in a sequence alignment, the user may choose to include additional
uncharged polar amino
acids in a computational screening calculation, or perhaps charged polar amino
acids, at that position.
63
CA 02766627 2012-01-23
WO 2004/099239 PCT/U52004/009298
[156] In one embodiment, sequence alignment information is combined with
energy calculation, as
discussed below. For example, pseudo energies can be derived from sequence
information to
generate a scoring function. The use of a sequence-based scoring function may
assist in significantly
reducing the complexity of a calculation. However, as is appreciated by those
skilled in the art, the
use of a sequence-based scoring function alone may be inadequate because
sequence information
can often indicate misleading correlations between mutations that may in
reality be structurally
conflicting. Thus, in a preferred embodiment, a structure-based method of
energy calculation is used,
either alone or in combination with a sequence-based scoring function. That
is, preferred
embodiments do not rely on sequence alignment information alone as the
analysis step.
[157] Energy calculation refers to the process by which amino acid
modifications are scored. The
energies of interaction are measured by one or more scoring functions. A
variety of scoring functions
find use in the present invention for calculating energies. Scoring functions
may include any number
of potentials, herein referred to as the energy terms of a scoring function,
including but not limited to a
van der Weals potential, a hydrogen bond potential, an atomic solvation
potential or other salvation
models, a secondary structure propensity potential, an electrostatic
potential, a torsional potential, and =
an entropy potential. At least one energy term is used to score each variable
or floated position,
although the energy terms may differ depending on the position, considered
amino acids, and other
considerations. In one embodiment, a scoring function using one energy term is
used. In the most
preferred embodiment, energies are calculated using a scoring function that
contains more than one
energy term, for example describing van der Weals, salvation, electrostatic,
and hydrogen bond
interactions, and combinations thereof. In additional embodiments, additional
energy terms include
but are not limited to entropic terms, torsional energies, and knowledge-based
energies.
[158] A variety of scoring functions are described in US 6,188,965; US
6,269,312; US 6,403,312;
USSN 09/782,004; USSN 09/927,790; USSN 09/877,695; USSN 10/071,859, USSN
10/218,102; PCT
WO 98/07254; PCT WO 01/40091; and PCT WO 02/25588. As will be appreciated by
those skilled in
the art, scoring functions need not be limited to physico-chemical energy
terms. For example,
knowledge-based potentials may find use in the computational screening
methodology of the present
invention. Such knowledge-based potentials may be derived from protein
sequence and/or structure
statistics including but not limited to threading potentials, reference
energies, pseudo energies,
homology-based energies, and sequence biases derived from sequence alignments.
In a preferred
embodiment, a scoring function is modified to include models for
immunogenicity, such as functions
derived from data on binding of peptides to MHC (Major Htocompatability
Complex), that may be used
to identify potentially immunogenic sequences (see for example USSN
09/903,378; USSN
10/039,170; USSN 60/222,697; USSN 10/339788; PCT WO 01/21823; and PCT WO
02/00165). In
one embodiment, sequence alignment information can be used to score amino acid
substitutions. For
example, comparison of protein sequences, regardless of whether the source of
said proteins is
human, monkey, mouse, or otherwise, may be used to suggest or score amino acid
mutations in the
computational screening methodology of the present invention. In one
embodiment, as is known in
64
CA 02766627 2012-01-23
= WO
2004/099249 PCPUS2004/009298
=
the art, one or more scoring functions may be optimized or "trained" during
the computational
analysis, and then the analysis re-run using the optimized system. Such
altered scoring functions
may be obtained for example, by training a scoring function using experimental
data. As will be
appreciated by those skilled in the art, a number of force fields, which are
comprised of one or more
energy terms, may serve as scoring functions. Force fields include but are not
limited to ab initio or
quantum mechanical force fields, semi-empirical force fields, and molecular
mechanics force fields.
Scoring functions that are knowledge-based or that use statistical methods may
find use in the
present invention. These methods may be used to assess the match between a
sequence and a
three-dimensional protein structure, and hence may be used to score amino acid
substitutions for
fidelity to the protein structure. In one embodiment, molecular dynamics
calculations may be used to
computationally screen sequences by individually calculating mutant sequence
scores.
[159] There are a variety of ways to represent amino acids in order to enable
efficient energy
calculation. In a preferred embodiment, considered amino acids are represented
as rotamers, as
described previously, and the energy (or score) of interaction of each
possible rotamer at each
variable and floated position with the other variable and floated rotamers,
with fixed position residues,
and with the backbone structure and any non-protein atoms, is calculated. In a
preferred
embodiment, two sets of interaction energies are calculated for each side
chain rotamer at every
variable and floated position: the interaction energy between the rotamer and
the fixed atoms (the
"singles" energy), and the interaction energy between the variable and floated
positions rotamer and
all other possible rotamers at every other variable and floated position (the
"doubles" energy). In an
alternate embodiment, singles and doubles energies are calculated for fixed
positions as well as for
variable and floated positions. In an alternate embodiment, considered amino
acids are not
represented as rotamers.
[160] An important component of computational screening is the identification
of one or more
sequences that have a favorable score, i.e. are low in energy. Determining a
set of low energy
sequences from an extremely large number of possibilities is nontrivial, and
to solve this problem a
combinatorial optimization algorithm is employed. The need for a combinatorial
optimization algorithm
is illustrated by examining the number of possibilities that are considered in
a typical computational
screening calculation. The discrete nature of rotamer sets allows a simple
calculation of the number
of possible rotameric sequences for a given design problem. A backbone of
length n with m possible
rotamers per position will have mn possible rotamer sequences, a number that
grows exponentially
with sequence length. For very simple calculations, it is possible to examine
each possible sequence
in order to identify the optimal sequence and/or one or more favorable
sequences. However, for a
typical design problem, the number of possible sequences (up to 1080 or more)
is sufficiently large that
examination of each possible sequence is intractable. A variety of
combinatorial optimization
algorithms may then be used to identify the optimum sequence and/or one or
more favorable
sequences. Combinatorial optimization algorithms may be divided into two
classes: (1) those that are
guaranteed to return the global minimum energy configuration if they converge,
and (2) those that are
CA 02766627 2012-01-23
WO 2004/099249
PCT/US2004/009298
not guaranteed to return the global minimum energy configuration, but which
will always return a
solution. Examples of the first class of algorithms include but are not
limited to Dead-End Elimination
(DEE) and Branch & Bound (B&B) (including Branch and Terminate) (Gordon &
Mayo, 1999,
Structure Fold Des 7:1089-98). Examples of the second class of algorithms
include, but are not
limited to, Monte Carlo (MC), self-consistent mean field (SCMF), Boltzmann
sampling (Metropolis et
al., 1953, J Chem Phys 21:1087), simulated annealing (Kirkpatrick at aL, 1983,
Science, 220:671-
680), genetic algorithm (GA), and Fast and Accurate Side-Chain Topology and
Energy Refinement
(FASTER) (Desmet, et at., 2002, Proteins, 48:31-43). A combinatorial
optimization algorithm may be
used alone or in conjunction with another combinatorial optimization
algorithm.
[161] In one embodiment of the present invention, the strategy for applying a
combinatorial
optimization algorithm is to find the global minimum energy configuration. In
an alternate
embodiment, the strategy is to find one or more low energy or favorable
sequences. In an alternate
embodiment, the strategy is to find the global minimum energy configuration
and then find one or
more low energy or favorable sequences. For example, as outlined in USSN
6,269,312, preferred
embodiments utilize a Dead End Elimination(DEE) step and a Monte Carlo step.
In other
embodiments, tabu search algorithms are used or combined with DEE and/or Monte
Carlo, among
other search methods (see Modern Heuristic Search Methods, edited by V.J.
Rayward-Smith et aL,
1996, John Wiley & Sons Ltd.; USSN 10/218,102; and PCT WO 02/25588). In
another preferred
embodiment, a genetic algorithm may be used; see for example USSN 09/877,695
and USSN
10/071,859. As another example, as is more fully described in US 6,188,965; US
6,269,312; US
6,403,312; USSN 09/782,004; USSN 09/927,790; USSN 10/218,102; PCT WO 98/07254;
PCT WO
01/40091; and PCT WO 02/25588, the global optimum may be reached, and then
further
computational processing may occur, which generates additional optimized
sequences. In the
simplest embodiment, design calculations are not combinatorial. That is,
energy calculations are
used to evaluate amino acid substitutions individually at single variable
positions. For other
calculations it is preferred to evaluate amino acid substitutions at more than
one variable position. In
a preferred embodiment, all possible interaction energies are calculated prior
to combinatorial
optimization. In an alternatively preferred embodiment, energies may be
calculated as needed during
combinatorial optimization.
Library generation
[162] The present invention provides methods for generating libraries that may
subsequently be
screened experimentally to single out optimized Fc variants. By "library" as
used herein is meant a
set of one or more Fc variants. Library may refer to the set of variants in
any form. In one
embodiment, the library is a list of nucleic acid or amino acid sequences, or
a list of nucleic acid or
amino acid substitutions at variable positions. For example, the examples used
to illustrate the
present invention below provide libraries as amino acid substitutions at
variable positions. In one
embodiment, a library is a list of at least one sequence that are Fc variants
optimized for a desired
66
CA 02766627 2012-01-23
WO 2904/099249 PCT/US2004/009298
property. For example see, Filikov et aL, 2002, Protein Sci 11:1452-1461 and
Luo etal., 2002,
Protein Sci 11:1218-1226. In an alternate embodiment, a library may be defined
as a combinatorial
list, meaning that a list of amino acid substitutions is generated for each
variable position, with the
implication that each substitution is to be combined with all other designed
substitutions at all other
variable positions. In this case, expansion of the combination of all
possibilities at all variable
positions results in a large explicitly defined library. A library may refer
to a physical composition of
polypeptides that comprise the Fc region or some domain or fragment of the Fc
region. Thus a library
may refer to a physical composition of antibodies or Fc fusions, either in
purified or unpurified form. A
library may refer to a physical composition of nucleic acids that encode the
library sequences. Said
nucleic acids may be the genes encoding the library members, the genes
encoding the library
members with any operably linked nucleic acids, or expression vectors encoding
the library members
together with any other operably linked regulatory sequences, selectable
markers, fusion constructs,
and/or other elements. For example, the library may be a set of mammalian
expression vectors that
encode Fc library members, the protein products of which may be subsequently
expressed, purified,
and screened experimentally. As another example, the library may be a display
library. Such a
library could, for example, comprise a set of expression vectors that encode
library members operably
linked to some fusion partner that enables phage display, ribosome display,
yeast display, bacterial
surface display, and the like.
[163] The library may be generated using the output sequence or sequences from
computational
screening. As discussed above, computationally generated libraries are
significantly enriched in
stable, properly folded, and functional sequences relative to randomly
generated libraries. As a result,
computational screening increases the chances of identifying proteins that are
optimized for the
design goal. The set of sequences in a library is generally, but not always,
significantly different from
the parent sequence, although in some cases the library preferably contains
the parent sequence. As
is known in the art, there are a variety of ways that a library may be derived
from the output of
computational screening calculations. For example, methods of library
generation described in US
6,403,312; USSN 09/782,004; USSN 09/927,790; USSN 10/218,102; PCT WO 01/40091;
and PCT
WO 02/25588 find use in the present invention. In one embodiment, sequences
scoring within a
certain range of the global optimum sequence may be included in the library.
For example, all
sequences within 10 kcal/mol of the lowest energy sequence could be used as
the library. In an
alternate embodiment, sequences scoring within a certain range of one or more
local minima
sequences may be used. In a preferred embodiment, the library sequences are
obtained from a
filtered set. Such a list or set may be generated by a variety of methods, as
is known in the art, for
example using an algorithm such as Monte Carlo, B&B, or SCMF. For example, the
top 103 or the top
105 sequences in the filtered set may comprise the library. Alternatively, the
total number of
sequences defined by the combination of all mutations may be used as a cutoff
criterion for the
library. Preferred values for the total number of recombined sequences range
from 10 to 1029,
particularly preferred values range from 100 to 109. Alternatively, a cutoff
may be enforced when a
67
CA 02766627 2012-01-23
= WO 2004/099249
PCT/US2004/009298
predetermined number of mutations per position is reached. In some
embodiments, sequences that
do not make the cutoff are included in the library. This may be desirable in
some situations, for
instance to evaluate the approach to library generation, to provide controls
or comparisons, or to
sample additional sequence space. For example, the parent sequence may be
included in the library,
even if it does not make the cutoff.
[164] Clustering algorithms may be useful for classifying sequences derived by
computational
screening methods into representative groups. For example, the methods of
clustering and their
application described in USSN 10/218,102 and PCT WO 02/25588, find use in the
present invention.
Representative groups may be defined, for example, by similarity. Measures of
similarity include, but
are not limited to sequence similarity and energetic similarity. Thus the
output sequences from
computational screening may be clustered around local minima, referred to
herein as clustered sets of
sequences. For example, sets of sequences that are close in sequence space may
be distinguished
from other sets. In one embodiment, coverage within one or a subset of
clustered sets may be
maximized by including in the library some, most, or all of the sequences that
make up one or more
clustered sets of sequences. For example, it may be advantageous to maximize
coverage within the
one, two, or three lowest energy clustered sets by including the majority of
sequences within these
sets in the library. In an alternate embodiment, diversity across clustered
sets of sequences may be
sampled by including within a library only a subset of sequences within each
clustered set. For
example, all or most of the clustered sets could be broadly sampled by
including the lowest energy
sequence from each clustered set in the library.
[165] Sequence information may be used to guide or filter computationally
screening results for
generation of a library. As discussed, by comparing and contrasting alignments
of protein sequences,
the degree of variability at a position and the types of amino acids which
occur naturally at that
position may be observed. Data obtained from such analyses are useful in the
present invention.
The benefits of using sequence information have been discussed, and those
benefits apply equally to
use of sequence information to guide library generation. The set of amino
acids that occur in a
sequence alignment may be thought of as being pre-screened by evolution to
have a higher chance
than random at being compatible with a protein's structure, stability,
solubility, function, and
immunogenicity. The variety of sequence sources, as well as the methods for
generating sequence
alignments that have been discussed, find use in the application of sequence
information to guiding
library generation. Likewise, as discussed above, various criteria may be
applied to determine the
importance or weight of certain residues in an alignment. These methods also
find use in the
application of sequence information to guide library generation. Using
sequence information to guide
library generation from the results of computational screening finds broad use
in the present invention.
In one embodiment, sequence information is used to filter sequences from
computational screening
output. That is to say, some substitutions are subtracted from the
computational output to generate
the library. For example the resulting output of a computational screening
calculation or calculations
may be filtered so that the library includes only those amino acids, or a
subset of those amino acids
68
CA 02766627 2012-01-23
WO 2004/099249 PCT/U52004/009298
that meet some criteria, for example that are observed at that position in an
alignment of sequences.
In an alternate embodiment, sequence information is used to add sequences to
the computational
screening output. That is to say, sequence information is used to guide the
choice of additional amino
acids that are added to the computational output to generate the library. For
example, the output set
of amino acids for a given position from a computational screening calculation
may be augmented to
include one or more amino acids that are observed at that position in an
alignment of protein
sequences. In an alternate embodiment, based on sequence alignment
information, one or more
amino acids may be added to or subtracted from the computational screening
sequence output in
order to maximize coverage or diversity. For example, additional amino acids
with properties similar
to those that are found in a sequence alignment may be added to the library.
For example, if a
position is observed to have uncharged polar amino acids in a sequence
alignment, additional
uncharged polar amino acids may be included in the library at that position.
[166] Libraries may be processed further to generate subsequent libraries. In
this way, the output
from a computational screening calculation or calculations may be thought of
as a primary library.
This primary library may be combined with other primary libraries from other
calculations or other
libraries, processed using subsequent calculations, sequence information, or
other analyses, or
processed experimentally to generate a subsequent library, herein referred to
as a secondary library.
As will be appreciated from this description, the use of sequence information
to guide or filter libraries,
discussed above, is itself one method of generating secondary libraries from
primary libraries.
Generation of secondary libraries gives the user greater control of the
parameters within a library.
This enables more efficient experimental screening, and may allow feedback
from experimental
results to be interpreted more easily, providing a more efficient
design/experimentation cycle.
[167] There are a wide variety of methods to generate secondary libraries from
primary libraries.
For example, USSN 10/218,102 and PCT WO 02/25588, describes methods for
secondary library
generation that find use in the present invention. Typically some selection
step occurs in which a
primary library is processed in some way. For example, in one embodiment a
selection step occurs
wherein some set of primary sequences are chosen to form the secondary
library. In an alternate
embodiment, a selection step is a computational step, again generally
including a selection step,
wherein some subset of the primary library is chosen and then subjected to
further computational
analysis, including both further computational screening as well as techniques
such as "in silico"
shuffling or recombination (see, for example US 5,830,721; US 5,811,238; US
5,605,793; and US
5,837,458, error-prone PCR, for example using modified nucleotides; known
mutagenesis techniques
including the use of multi-cassettes; and DNA shuffling (Cramer' etal., 1998,
Nature 391:288-291;
Coco et aL, 2001, Nat Biotechnol 19:354-9; Coco etal., 2002, Nat Biotechnol,
20:1246-50),
heterogeneous DNA samples (US 5,939,250); ITCHY (Ostermeier of aL, 1999, Nat
Biotechnol
17:1205-1209); StEP (Zhao of al., 1998, Nat Biotechnol 16:258-261), GSSM (US
6,171,820 and US
5,965,408); in vivo homologous recombination, ligase assisted gene assembly,
end-complementary
PGR, profusion (Roberts & Szostak, 1997, Proc Nati Aced Sc! USA 94:12297-
12302); yeast/bacteria
69
CA 02766627 2012-01-23
= WO
200.1/099249 PCT/US2004/009298
surface display (Lu of al., 1995, Biotechnology 13:366-372); Seed & Aruffo,
1987, Proc Natl Acad Sc!
USA 84(10):3365-3369; Boder & Wittrup, 1997, Nat Biotechno/ 15:553-557). In an
alternate
embodiment, a selection step occurs that is an experimental step, for example
any of the library
screening steps below, wherein some subset of the primary library is chosen
and then recombined
experimentally, for example using one of the directed evolution methods
discussed below, to form a
secondary library. In a preferred embodiment, the primary library is generated
and processed as
outlined in US 6,403,312.
[168] Generation of secondary and subsequent libraries finds broad use in the
present invention. In
one embodiment, different primary libraries may be combined to generate a
secondary or subsequent
library. In another embodiment, secondary libraries may he generated by
sampling sequence
diversity at highly mutatable or highly conserved positions. The primary
library may be analyzed to
determine which amino acid positions in the template protein have high
mutational frequency, and
which positions have low mutational frequency. For example, positions in a
protein that show a great
deal of mutational diversity in computational screening may be fixed in a
subsequent round of design
calculations. A filtered set of the same size as the first would now show
diversity at positions that
were largely conserved in the first library. Alternatively, the secondary
library may be generated by
varying the amino acids at the positions that have high numbers of mutations,
while keeping constant
the positions that do not have mutations above a certain frequency.
[169] This discussion is not meant to constrain generation of libraries
subsequent to primary
libraries to secondary libraries. As will be appreciated, primary and
secondary libraries may be
processed further to generate tertiary libraries, quaternary libraries, and so
on. In this way, library
generation is an iterative process. For example, tertiary libraries may be
constructed using a variety
of additional steps applied to one or more secondary libraries; for example,
further computational
processing may occur, secondary libraries may be recombined, or subsets of
different secondary
libraries may be combined. In a preferred embodiment, a tertiary library may
be generated by
combining secondary libraries. For example, primary and/or secondary libraries
that analyzed
different parts of a protein may be combined to generate a tertiary library
that treats the combined
parts of the protein. In an alternate embodiment, the variants from a primary
library may be combined
with the variants from another primary library to provide a combined tertiary
library at lower
computational cost than creating a very long filtered set. These combinations
may be used, for
example, to analyze large proteins, especially large multi-domain proteins, of
which Fc is an example.
Thus the above description of secondary library generation applies to
generating any library
subsequent to a primary library, the end result being a final library that may
screened experimentally
to obtain protein variants optimized for a design goal. These examples are not
meant to constrain
generation of secondary libraries to any particular application or theory of
operation for the present
invention. Rather, these examples are meant to illustrate that generation of
secondary libraries, and
subsequent libraries such as tertiary libraries and so on, is broadly useful
in computational screening
methodology for library generation.
CA 02766627 2012-01-23
= WO
2004/099249 PCT/US2004/009298
Experimental Production and Screening
[170] The present invention provides methods for producing and screening
libraries of Fc variants.
The described methods are not meant to constrain the present invention to any
particular application
or theory of operation. Rather, the provided methods are meant to illustrate
generally that one or
more Fc variants or one or more libraries of Fc variants may be produced and
screened
experimentally to obtain optimized Fc variants. Fc variants may be produced
and screened in any
context, whether as an Fc region as precisely defined herein, a domain or
fragment thereof, or a
larger polypeptide that comprises Fc such as an antibody or Fc fusion. General
methods for antibody
molecular biology, expression, purification, and screening are described in
Antibody Engineering,
edited by Duebel & Kontermann, Springer-Verlag, Heidelberg, 2001; and Hayhurst
& Georgiou, 2001,
Cuff Opin Chem Biol 5:683-689; Maynard & Georgiou, 2000, Annu Rev Biomed Eng
2:339-76;
Antibodies: A Laboratory Manual by Harlow & Lane, New York: Cold Spring Harbor
Laboratory Press,
1988.
[171] In one embodiment of the present invention, the library sequences are
used to create nucleic
acids that encode the member sequences, and that may then be cloned into host
cells, expressed
and assayed, if desired. Thus, nucleic acids, and particularly DNA, may be
made that encode each
member protein sequence. These practices are carried out using well-known
procedures. For
example, a variety of methods that may find use in the present invention are
described in Molecular
Cloning - A Laboratory Manual, 3rd Ed. (Maniatis, Cold Spring Harbor
Laboratory Press, New York,
2001), and Current Protocols in Molecular Biology (John Wiley & Sons). As will
be appreciated by
those skilled in the art, the generation of exact sequences for a library
comprising a large number of
sequences is potentially expensive and time consuming. Accordingly, there are
a variety of
techniques that may be used to efficiently generate libraries of the present
invention. Such methods
that may find use in the present invention are described or referenced in US
6,403,312; USSN
09/782,004; USSN 09/927,790; USSN 10/218,102; PCT WO 01/40091; and PCT WO
02/25588.
Such methods include but are not limited to gene assembly methods, PCR-based
method and
methods which use variations of PCR, ligase chain reaction-based methods,
pooled oligo methods
such as those used in synthetic shuffling, error-prone amplification methods
and methods which use
oligos with random mutations, classical site-directed mutagenesis methods,
cassette mutagenesis,
and other amplification and gene synthesis methods. As is known in the art,
there are a variety of
commercially available kits and methods for gene assembly, mutagenesis, vector
subcloning, and the
like, and such commercial products find use in the present invention for
generating nucleic acids that
encode Fc variant members of a library.
[172] The Fc variants of the present invention may be produced by culturing a
host cell transformed
with nucleic acid, preferably an expression vector, containing nucleic acid
encoding the Fc variants,
under the appropriate conditions to induce or cause expression of the protein.
The conditions
appropriate for expression will vary with the choice of the expression vector
and the host cell, and will
71
CA 02766627 2012-01-23
= WO
2004/099249 PCT/US2004/009298
be easily ascertained by one skilled in the art through routine
experimentation. A wide variety of
appropriate host cells may be used, including but not limited to mammalian
cells, bacteria, insect
cells, and yeast For example, a variety of cell lines that may find use in the
present invention are
described in the ATCC cell line catalog, available from the American Type
Culture Collection.
[173] In a preferred embodiment, the Fc variants are expressed in mammalian
expression systems,
including systems in which the expression constructs are introduced into the
mammalian cells using
virus such as retrovirus or adenovirus. Any mammalian cells may be used, with
human, mouse, rat,
hamster, and primate cells being particularly preferred. Suitable cells also
include known research
cells, including but not limited to Jurkat T cells, NIH3T3, CHO, COS, and 293
cells. In an alternately
preferred embodiment, library proteins are expressed in bacterial cells.
Bacterial expression systems
are well known in the art, and include Escherichia colt (E. coil), Bacillus
subtilis, Streptococcus
cremoris, and Streptococcus lividans. In alternate embodiments, Fc variants
are produced in insect
cells or yeast cells. In an alternate embodiment, Fc variants are expressed in
vitro using cell free
translation systems. In vitro translation systems derived from both
prokaryotic (e.g. E. co/i) and
eukaryotic (e.g. wheat germ, rabbit reticulocytes) cells are available and may
be chosen based on the
expression levels and functional properties of the protein of interest. For
example, as appreciated by
those skilled in the art, in vitro translation is required for some display
technologies, for example
ribosome display. In addition, the Fc variants may be produced by chemical
synthesis methods.
074] The nucleic acids that encode the Fc variants of the present invention
may be incorporated
into an expression vector in order to express the protein. A variety of
expression vectors may be
utilized for protein expression. Expression vectors may comprise self-
replicating extra-chromosomal
vectors or vectors which integrate into a host genome. Expression vectors are
constructed to be
compatible with the host cell type. Thus expression vectors which find use in
the present invention
include but are not limited to those which enable protein expression in
mammalian cells, bacteria,
insect cells, yeast, and in in vitro systems. As is known in the art, a
variety of expression vectors are
available, commercially or otherwise, that may find use in the present
invention for expressing Fc
variant proteins.
[175] Expression vectors typically comprise a protein operably linked with
control or regulatory
sequences, selectable markers, any fusion partners, and/or additional
elements. By "operably linked"
herein is meant that the nucleic acid is placed into a functional relationship
with another nucleic acid
sequence. Generally, these expression vectors include transcriptional and
translational regulatory
nucleic acid operably linked to the nucleic acid encoding the Fc variant, and
are typically appropriate
to the host cell used to express the protein. In general, the transcriptional
and translational regulatory
sequences may include promoter sequences, ribosomal binding sites,
transcriptional start and stop
sequences, translational start and stop sequences, and enhancer or activator
sequences. As is also
known in the art, expression vectors typically contain a selection gene or
marker to allow the selection
72
CA 02766627 2014-02-27
52620-2D
of transformed host cells containing the expression vector. Selection genes
are well known in the art
and will vary with the host cell used.
[1761 Fc variants may be operably linked to a fusion partner to enable
targeting of the expressed
protein, purification, screening, display, and the like. Fusion partners may
be linked to the Fc variant
sequence via a linker sequences. The linker sequence will generally comprise a
small number of
amino acids, typically less than ten, although longer linkers may also be
used. Typically, linker
sequences are selected to be flexible and resistant to degradation. As will be
appreciated by those
skilled in the art, any of a wide variety of sequences may be used as linkers.
For example, a common
linker sequence comprises the amino acid sequence GGGGS. A fusion partner may
be a targeting or
signal sequence that directs Fc variant protein and any associated fusion
partners to a desired cellular
location or to the extracellular media. As is known in the art, certain
signaling sequences may target a
protein to be either secreted into the growth media, or into the periplasmic
space, located between the
inner and outer membrane of the cell. A fusion partner may also be a sequence
that encodes a
peptide or protein that enables purification and/or screening. Such fusion
partners include but are not
limited to polyhistidine tags (His-tags) (for example H6 and H10 or other tags
for use with Immobilized .
Metal Affinity Chromatography (IMAC) systems (e.g. Ni+2affinity columns)), GST
fusions, MBP
fusions, Strep-tag, the BSP biotinylation target sequence of the bacterial
enzyme BirA, and epitope
tags which are targeted by antibodies (for example c-myc tags, flag-tags, and
the like). As will be
appreciated by those skilled in the art, such tags may be useful for
purification, for screening, or both.
' For example, an Fc variant may be purified using a His-tag by
immobilizing it to a Ni+2 affinity column,
and then after purification the same His-tag may be used to immobilize the
antibody to a N1+2 coated
plate to perform an ELISA or other binding assay (as described below). A
fusion partner may enable
the use of a selection method to screen Fc variants (see below). Fusion
partners that enable a variety
of selection methods are well-known in the art, and all of these find use in
the present invention. For ,
example, by fusing the members of an Fc variant library to the gene III
protein, phage display can be
employed (Kay et al., Phage display of peptides and proteins: a laboratory
manual, Academic Press,
San Diego, CA, 1996; Lowman et al., 1991, Biochemistry 30:10832-10838; Smith,
1985, Science
228:1315-1317). Fusion partners may enable Fc variants to be labeled.
Alternatively, a fusion
partner may bind to a specific sequence on the expression vector, enabling the
fusion partner and
associated Fc variant to be linked covalently or noncovalently with the
nucleic acid that encodes them.
For example, USSN 09/642,574; USSN 10/080,376; USSN 09/792,630; USSN
10/023,208; USSN
09/792,626; USSN 10/082,671; USSN 09/953,351; USSN 10/097,100; PCT WO
00/22906; PCT WO 01/49058; PCT WO 02/04852; PCT WO 02/04853; PCT WO 02/08023;
PCT WO
01/28702; and PCT WO 02/07466 describe such a fusion partner and technique
that may find use in
the present invention.
[177) The methods of introducing exogenous nucleic acid into host cells are
well known in the art,
and will vary with the host cell used. Techniques include but are not limited
to dextran-mediated
transfection, calcium phosphate precipitation, calcium chloride treatment,
polybrene mediated
73
CA 02766627 2012-01-23
WO 2004/099249
PCT/US2004/009298
transfection, protoplast fusion, electroporation, viral or phage infection,
encapsulation of the
polynucleotide(s) in liposomes, and direct microinjection of the DNA into
nuclei. In the case of
mammalian cells, transfection may be either transient or stable.
[178] In a preferred embodiment, Fc variant proteins are purified or isolated
after expression.
Proteins may be isolated or purified in a variety of ways known to those
skilled in the art. Standard
purification methods include chromatographic techniques, including ion
exchange, hydrophobic
interaction, affinity, sizing or gel filtration, and reversed-phase, carried
out at atmospheric pressure or
at high pressure using systems such as FPLC and HPLC. Purification methods
also include
electrophoretic, immunological, precipitation, dialysis, and chromatofocusing
techniques.
Ultrafiltration and diafiltration techniques, in conjunction with protein
concentration, are also useful.
As is well known in the art, a variety of natural proteins bind Fc and
antibodies, and these proteins can
find use in the present invention for purification of Fc variants. For
example, the bacterial proteins A
and G bind to the Fc region. Likewise, the bacterial protein L binds to the
Fab region of some
antibodies, as of course does the antibody's target antigen. Purification can
often be enabled by a
particular fusion partner. For example, Fc variant proteins may be purified
using glutathlone resin if a
GST fusion is employed, Ni+2affinity chromatography if a His-tag is employed,
or immobilized anti-flag
antibody if a flag-tag is used. For general guidance in suitable purification
techniques, see Protein
= Purification: Principles and Practice, 3rd Ed., Scopes, Springer-Verlag,
NY, 1994. The degree of
purification necessary will vary depending on the screen or use of the Fc
variants. In some instances
no purification is necessary. For example in one embodiment, if the Fc
variants are secreted,
screening may take place directly from the media. As is well known in the art,
some methods of
selection do not involve purification of proteins. Thus, for example, if a
library of Fc variants is made
into a phage display library, protein purification may not be performed.
[179] Fc variants may be screened using a variety of methods, including but
not limited to those
that use in vitro assays, in vivo and cell-based assays, and selection
technologies. Automation and
high-throughput screening technologies may be utilized in the screening
procedures. Screening may
employ the use of a fusion partner or label. The use of fusion partners has
been discussed above.
By "labeled" herein is meant that the Fc variants of the invention have one or
more elements,
isotopes, or chemical compounds attached to enable the detection in a screen.
In general, labels fall
Into three classes: a) immune labels, which may be an epitope incorporated as
a fusion partner that is
recognized by an antibody, b) isotopic labels, which may be radioactive or
heavy isotopes, and c)
small molecule labels, which may include fluorescent and colorimetric dyes, or
molecules such as
biotin that enable other labeling methods. Labels may be incorporated into the
compound at any
position and may be incorporated in vitro or in vivo during protein
expression.
[180] In a preferred embodiment, the functional and/or biophysical properties
of Fc variants are
screened in an in vitro assay. In vitro assays may allow a broad dynamic range
for screening
properties of interest. Properties of Fc variants that may be screened include
but are not limited to
74
CA 02766627 2012-01-23
WO 2004/099249
PCT/US2004/009298
stability, solubility, and affinity for Fc ligands, for example FcyRs.
Multiple properties may be
screened simultaneously or individually. Proteins may be purified or
unpurified, depending on the
requirements of the assay. In one embodiment, the screen is a qualitative or
quantitative binding
assay for binding of Fc variants to a protein or nonprotein molecule that is
known or thought to bind
the Fc variant. In a preferred embodiment, the screen is a binding assay for
measuring binding to the
antibody's or Fc fusions' target antigen. In an alternately preferred
embodiment, the screen is an
assay for binding of Fc variants to an Fc ligand, including but are not
limited to the family of FcyRs,
the neonatal receptor FcRn, the complement protein Cl q, and the bacterial
proteins A and a Said
Fc ligands may be from any organism, with humans, mice, rats, rabbits, and
monkeys preferred.
Binding assays can be carried out using a variety of methods known in the art,
including but not
limited to FRET (Fluorescence Resonance Energy Transfer) and BRET
(Bioluminescence Resonance
Energy Transfer) -based assays, AlphaScreen"' (Amplified Luminescent Proximity
Homogeneous
Assay), Scintillation Proximity Assay, EUSA (Enzyme-Linked Immunosorbent
Assay), SPR (Surface
Plasmon Resonance, also known as BIACORE ), isothermal titration calorimetry,
differential
scanning calorimetry, gel electrophoresis, and chromatography including gel
filtration. These and
other methods may take advantage of some fusion partner or label of the Fc
variant. Assays may
employ a variety of detection methods including but not limited to
chromogenic, fluorescent,
luminescent, or isotopic labels.
[1811 The biophysical properties of Fc variant proteins, for example stability
and solubility, may be
screened using a variety of methods known in the art. Protein stability may be
determined by
measuring the thermodynamic equilibrium between folded and unfolded states.
For example, Fc
variant proteins of the present invention may be unfolded using chemical
denaturant, heat, or pH, and
this transition may be monitored using methods including but not limited to
circular dichroism
spectroscopy, fluorescence spectroscopy, absorbance spectroscopy. NMR
spectroscopy, calorimetry,
and proteolysis. As will be appreciated by those skilled in the art, the
kinetic parameters of the folding
and unfolding transitions may also be monitored using these and other
techniques. The solubility and
overall structural integrity of an Fc variant protein may be quantitatively or
qualitatively determined
using a wide range of methods that are known in the art. Methods which may
find use in the present
invention for characterizing the biophysical properties of Fc variant proteins
include gel
electrophoresis, chromatography such as size exclusion chromatography and
reversed-phase high
performance liquid chromatography, mass spectrometry, ultraviolet absorbance
spectroscopy,
fluorescence spectroscopy, circular dichroism spectroscopy, isothermal
titration calorimetry,
differential scanning calorimetry, analytical ultra-centrifugation, dynamic
light scattering, proteolysis,
and cross-linking, turbidity measurement, filter retardation assays,
immunological assays, fluorescent
dye binding assays, protein-staining assays, microscopy, and detection of
aggregates via ELISA or
other binding assay. Structural analysis employing X-ray crystallographic
techniques and NMR
spectroscopy may also find use. In one embodiment, stability and/or solubinty
may be measured by
determining the amount of protein solution after some defined period of time.
In this assay, the
CA 02766627 2012-01-23
WO 2004/099249 PCT/1JS2004/009298
protein may or may not be exposed to some extreme condition, for example
elevated temperature,
low pH, or the presence of denaturant. Because function typically requires a
stable, soluble, and/or
well-folded/structured protein, the aforementioned functional and binding
assays also provide ways to
perform such a measurement. For example, a solution comprising an Fc variant
could be assayed for
its ability to bind target antigen, then exposed to elevated temperature for
one or more defined periods
of time, then assayed for antigen binding again. Because unfolded and
aggregated protein is not
expected to be capable of binding antigen, the amount of activity remaining
provides a measure of the
Fc variant's stability and solubility.
[182] In a preferred embodiment, the library is screened using one or more
cell-based or in vivo
assays. For such assays, Fc variant proteins, purified or unpurified, are
typically added exogenously
such that cells are exposed to individual variants or pools of variants
belonging to a library. These
assays are typically, but not always, based on the function of an antibody or
Fc fusion that comprises
the Fc variant; that is, the ability of the antibody or Fc fusion to bind a
target antigen and mediate
some biochemical event, for example effector function, ligand/receptor binding
inhibition, apoptosis,
and the like. Such assays often involve monitoring the response of cells to
antibody or Fc fusion, for
example cell survival, cell death, change in cellular morphology, or
transcriptional activation such as
cellular expression of a natural gene or reporter gene. For example, such
assays may measure the
ability of Fc variants to elicit ADCC, ADCP, or CDC: For some assays
additional cells or components,
that is in addition to the target cells, may need to be added, for example
example serum complement,
or effector cells such as peripheral blood monocytes (PBMCs), NK cells,
macrophages, and the like.
Such additional cells may be from any organism, preferably humans, mice, rat,
rabbit, and monkey.
Antibodies and Fc fusions may cause apoptosis of certain cell lines expressing
the antibody's target
antigen, or they may mediate attack on target cells by immune cells which have
been added to the
assay. Methods for monitoring cell death or viability are known in the art,
and include the use of dyes,
immunochemical, cytochemical, and radioactive reagents. For example, caspase
staining assays
may enable apoptosis to be measured, and uptake or release of radioactive
substrates or fluorescent
dyes such as alamar blue may enable cell growth or activation to be monitored.
In a preferred
embodiment, the DELFIA EuTDA-based cytotoxicity assay (Perkin Elmer, MA) is
used.
Alternatively, dead or damaged target cells may be monitoried by measuring the
release of one or
more natural intracellular proteins, for example lactate dehydrogenase.
Transcriptional activation may
also serve as a method for assaying function in cell-based assays. In this
case, response may be
monitored by assaying for natural genes or proteins which may be upreguiated,
for example the
release of certain interleukins may be measured, or alternatively readout may
be via a reporter
construct. Cell-based assays may also involve the measure of morphological
changes of cells as a
response to the presence of an Fc variant. Cell types for such assays may be
prokaryotic or
eukaryotic, and a variety of cell lines that are known in the art may be
employed.
[183] Alternatively, cell-based screens are performed using cells that have
been transformed or
76
CA 02766627 2012-01-23
WO 21)04/099249 PCT/US2004/009298
transfected with nucleic acids encoding the Fc variants. That is, Fc variant
proteins are not added
exogenously to the cells. For example, in one embodiment, the cell-based
screen utilizes cell surface
display. A fusion partner can be employed that enables display of Fe variants
on the surface of cells
(Witrrup, 2001, Curr Opin Biotechnol, 12:395-399). Cell surface display
methods that may find use in
the present invention include but are not limited to display on bacteria
(Georgiou et aL, 1997, Nat
Biotechnol 15:29-34; Georgiou et al., 1993, Trends Biotechnol 11:6-10; Lee
etal., 2000, Nat
Biotechnol 18:645-648; Jun etal., 1998, Nat Biotechnol 16:576-80), yeast
(Boder & Wittrup, 2000,
Methods Enzymol 328:430-44; Boder & Wittrup, 1997, Nat Biotechnol 15:553-557),
and mammalian
cells (Whitehorn ef aL, 1995, 810/technology 13:1215-1219). In an alternate
embodiment, Fc variant
proteins are not displayed on the surface of cells, but rather are screened
intracellularly or in some
other cellular compartment. For example, periplasmic expression and cytometric
screening (Chen et
al., 2001, Nat Biotechnol 19: 537-542), the protein fragment complementation
assay (Johnsson &
Varshaysky, 1994, Proc Nall Acad Sc! USA 91:10340-10344.; Pelletier et aL,
1998, Proc Nat! Aced
Sci USA 95:12141-12146), and the yeast two hybrid screen (Fields & Song, 1989,
Nature 340:245-
246) may find use in the present invention. Alternatively, if a polypeptide
that comprises the Fc
variants, for example an antibody or Fc fusion, imparts some selectable growth
advantage to a cell,
this property may be used to screen or select for Fc variants.
[184] As is known in the art, a subset of screening methods are those that
select for favorable
members of a library. Said methods are herein referred to as "selection
methods", and these methods
find use in the present invention for screening Fc variant libraries. When
libraries are screened using
a selection method, only those members of a library that are favorable, that
is which meet some
selection criteria, are propagated, isolated, and/or observed. As will be
appreciated, because only the
most fit variants are observed, such methods enable the screening of libraries
that are larger than
those screenable by methods that assay the fitness of library members
individually. Selection is
enabled by any method, technique, or fusion partner that links, covalently or
noncovalently, the
phenotype of an Fc variant with its genotype, that is the function of an Fc
variant with the nucleic acid
that encodes it. For example the use of phage display as a selection method is
enabled by the fusion
of library members to the gene III protein. In this way, selection or
isolation of variant proteins that
meet some criteria, for example binding affinity for an FcyR, also selects for
or isolates the nucleic
acid that encodes it. Once isolated, the gene or genes encoding Fc variants
may then be amplified.
This process of isolation and amplification, referred to as panning, may be
repeated, allowing
favorable Fc variants in the library to be enriched. Nucleic acid sequencing
of the attached nucleic
acid ultimately allows for gene identification.
[185] A variety of selection methods are known in the art that may find use in
the present invention
for screening Fc variant libraries. These include but are not limited to phage
display (Phage display of
peptides and proteins: a laboratory manual, Kay et a/., 1996, Academic Press,
San Diego, CA, 1996;
Lowman etal., 1991, Biochemistry 30:10832-10838; Smith, 1985, Science 228:1315-
1317) and its
77
CA 02766627 2014-02-27
52620-2D
derivatives such as selective phage infection (Malmborg etal., 1997, J Mol
Biol 273:544-551),
selectively infective phage (Krebber at al., 1997, J Mol 8/0/268:619-630), and
delayed infectivity
panning (Benhar of al., 2000, J Mot Biol 301:893-904), cell surface display
(Witrrup, 2001, Curr Opin
Biotechnol, 12:395-399) such as display on bacteria (Georgiou etal., 1997, Nat
Biotechnol 15:29-34;
Georgiou et al., 1993, Trends Biotechnol 11:6-10; Lee etal., 2000, Nat
Biotechnol 18:645-648; Jun of
al., 1998, Nat Biotechnol 16:576-80), yeast (Bader & Wittrup, 2000, Methods
Enzymol 328:430-44;
Bader & Wittrup, 1997, Nat Biotechnol 15:553-557), and mammalian cells
(Whitehom etal., 1995,
B(0/technology 13:1215-1219), as well as in vitro display technologies
(Amstutz etal., 2001, Curt Opin
Biotechnol 12:400-405) such as polysome display (Mattheakis of a/., 1994, Proc
Nat! Aced Sc! USA
91:9022-9026), ribosome display (Hanes etal., 1997, Proc Nat! Aced Sc! USA
94:4937-4942), mRNA
display (Roberts & Szostak, 1997, Proc Nat! Acad Sc! USA 94:12297-12302;
Nemoto et al., 1997,
FEBS Lett 414:405-408), and ribosome-inactivation display system (Zhou at al.,
2002, J Am Chem
Soc 124, 538-543)
[186] Other selection methods that may find use in the present invention
include methods that do
not rely on display, such as in vivo methods including but not limited to
periplasnnic expression and
cytometric screening (Chen etal., 2001, Nat Biotechnol 19:537-542), the
protein fragment
complementation assay (Johnsson & Varshaysky, 1994, Proc Nat! Aced Sc! USA
91:10340-10344;
Pelletier etal., 1998, Proc Nat! Aced Sc! USA 95:12141-12146), and the yeast
two hybrid screen
(Fields & Song, 1989, Nature 340:245-246) used in selection mode (Visintin et
aL, 1999, Proc Nat!
Acad Sci USA 96:11723-11728). In an alternate embodiment, selection is enabled
by a fusion partner
that binds to a specific sequence on the expression vector, thus linking
covalently or noncovalently
the fusion partner and associated Fc variant library member with the nucleic
acid that encodes them.
For example, USSN 09/642,574; USSN 10/080,376; USSN 09/792,630; USSN
10/023,208; USSN
09/792,626; USSN 10/082,671; USSN 09/953,351; USSN 10/097,100; PCT WO
00/22906; PCT WO 01/49058; PCT WO 02/04852; PCT WO 02/04853; PCT WO 02/08023;
PCT WO
01/28702; and PCT WO 02/07466 describe such a fusion partner and technique
that may find use in
the present invention. In an alternative embodiment, in vivo selection can
occur if expression of a
polypeptide that comprises the Fc variant, such as an antibody or Fc fusion,
imparts some growth,
reproduction, or survival advantage to the cell.
[187] A subset of selection methods referred to as "directed evolution"
methods are those that
Include the mating or breading of favorable sequences during selection,
sometimes with the
incorporation of new mutations. As will be appreciated by those skilled in the
art, directed evolution
methods can facilitate identification of the most favorable sequences in a
library, and can increase the
diversity of sequences that are screened. A variety of directed evolution
methods are known in the art
that may find use in the present invention for screening Fc variant libraries,
including but not limited to
DNA shuffling (PCT WO 00/42561 A3; PCT WO 01/70947 A3), exon shuffling (US
6,365,377;
Kolkman & Stemmer, 2001, Nat Biotechnol 19:423-428), family shuffling (Crameri
etal., 1998, Nature
78
CA 02766627 2012-01-23
WO 2004/099249
PCT/US2004/009298
391:288-201; us 6,376,246), RACHITTTm (Coco etal., 2001, Nat Bietechnol 19:354-
359; PCT WO
02/06469), STEP and random priming of in vitro recombination (Zhao et aL,
1998, Nat Biotechnol
16:258-261; Shao etal., 1998, Nucleic Acids Res 26:681-683), exonuclease
mediated gene assembly
(US 6,352,842; US 6,361,974), Gene Site Saturation MutagenesisTm (US
6,358,709), Gene
ReassemblyTM (US 6,358,709), SCRATCHY (Lutz et al., 2001, Proc Nat! Acad Sci
USA 98:11248-
11253), DNA fragmentation methods (Kikuchi of al., Gene 236:159-167), single-
stranded DNA
shuffling (Kikuchi etal., 2000, Gene 243:133-137), and AMEsystem II directed
evolution protein
engineering technology (Applied Molecular Evolution) (US US 5,824,514; US
5,817,483; US
5,814,476; US 5,763,192; US 5,723,323).
[188] The biological properties of the antibodies and Fc fusions that comprise
the Fc variants of the
present invention may be characterized in cell, tissue, and whole organism
experiments. As is know
in the art, drugs are often tested in animals, including but not limited to
mice, rats, rabbits, dogs, cats,
pigs, and monkeys, in order to measure a drug's efficacy for treatment against
a disease or disease
model, or to measure a drug's pharmacokinetics, toxicity, and other
properties. Said animals may be
referred to as disease models. Therapeutics are often tested in mice,
including but not limited to nude
mice, SCID mice, xenograft mice, and transgenic mice (including knockins and
knockouts). For
example, an antibody or Fc fusion of the present invention that is intended as
an anti-cancer
therapeutic may be tested in a mouse cancer model, for example a xenograft
mouse. In this method,
a tumor or tumor cell line is grafted onto or injected into a mouse, and
subsequently the mouse is
treated with the therapeutic to determine the ability of the antibody or Fc
fusion to reduce or inhibit
cancer growth. Such experimentation may provide meaningful data for
determination of the potential
of said antibody or Fc fusion to be used as a therapeutic. Any organism,
preferably mammals, may
be used for testing. For example because of their genetic similarity to
humans, monkeys can be
suitable therapeutic models, and thus may be used to test the efficacy,
toxicity, pharmacokinetics, or
other property of the antibodies and Fc fusions of the present invention.
Tests of the antibodies and
Fc fusions of the present invention in humans are ultimately required for
approval as drugs, and thus
of course these experiments are contemplated. Thus the antibodies and Fc
fusions of the present
invention may be tested in humans to determine their therapeutic efficacy,
toxicity, pharmacokinetics,
and/or other clinical properties.
= EXAMPLES
[189] Examples are provided below to illustrate the present invention. These
examples are not
meant to constrain the present invention to any particular application or
theory of operation.
[190] For all positions discussed in the present invention, numbering is
according to the EU index
as in Kabat (Kabat et al., 1991, Sequences of Proteins of Immunological
Interest, 5th Ed., United
States Public Health Service, National Institutes of Health, Bethesda). Those
skilled in the art of
antibodies will appreciate that this convention consists of nonsequential
numbering in specific regions
of an immunoglobulin sequence, enabling a normalized reference to conserved
positions in
79
CA 02766627 2012-01-23
WO 2004/099249 PCT/US2004/009298
immunoglobulin families. Accordingly, the positions of any given
immunoglobulin as defined by the
EU index will not necessarily correspond to its sequential sequence. Figure 3
shows the sequential
and EU index numbering schemes for the antibody alemtuzumab in order to
illustrate this principal
more clearly. It should also be noted that polyrnorphisms have been-observed
at a number of Fc
positions, including but not limited to Kabat 270, 272, 312, 315, 356, and
358, and thus slight
differences between the presented sequence and sequences in the scientific
literature may exist.
[191] Example 1. Computational Screening and Design of Fc Libraries
Computational screening calculations were carried out to design optimized Fc
variants. Fc variants
were computationally screened, constructed, and experimentally investigated
over several
computation/experimention cycles. For each successive cycle, experimental data
provided feedback
into the next set of computational screening calculations and library design.
All computational
screening calculations and library design are presented in Example 1. For each
set of calculations, a
table is provided that presents the results and provides relevant information
and parameters.
[192] Several different structures of Fc bound bound to the extracellular
domain of FcyRs served as
template structures for the computational screening calculations. Publicly
available Fc/FcyR complex
structures included pdb accession code 1E4K (Sondermann etal., 2000, Nature
406:267-273.), and
pdb accession codes 11IS and 11IX (Radaev etal., 2001, J Biol Chem 276:16469-
16477). The
extracellular regions of FcyRIllb and FcyRIlla are 96% identical, and
therefore the use of the
Fc/Fe-yRIllb structure is essentially equivalent to use of FcyRilla.
Nonetheless, for some calculations,
a more precise Fc/FeyRilla template structure was constructed by modeling a
D129G mutation in the
111S and 1E4K structures (referred to as D129G 11IS and D129G 1E4K template
structures). In
addition, the structures for human Fc bound to the extracellular domains of
human FcyRIlb, human
F158 Fel/Rifle, and mouse FcyRIII were modeled using standard methods, the
available FcyR
sequence information, the aforementioned Fc./FcyR structures, as well as
structural information for
unbound complexes (pdb accession code 1H9V)(Sondermann etal., 2001, J Mol
Bic:4309:737-749)
(pdb accession code 1FCG)(Maxwell et at., 1999, Nat Struct Bid l 6:437-442),
FcyRIlb (pdb accession
code 2FCB)(Sondermann etal., 1999, Embo J 18:1095-1103), and FcyRIllb (pdb
accession code
1E4J)(Sondermann et al., 2000, Nature 406:267-273.).
[193] Variable positions and amino acids to be considered at those positions
were chosen by visual
inspection of the aforementioned Fc/FcyR and FcyR structures, and using
solvent accessibility
information and sequence information. Sequence information of Fcs and FcyRs
was particularly
useful for determining variable positions at which substitutions may provide
distinguishing affinities
between activating and inhibitory receptors. Virtually all Cy2 positions were
screened
computationally. The Fc structure is a homodimer of two heavy chains (labeled
chains A and B in the
11IS, 111X, and 1E4K structures) that each include the hinge and Cy2-Cy3
domains (shown in Figure
2). Because the FcyR (labeled chain C in the 111S, 1 I IX, and 1E4K
structures) binds asymmetrically to
CA 02766627 2012-01-23
WO 2004/099249
PCT/US2004/009298
the Fc homodimer, each chain was often considered separately in design
calculations. For some
calculations, Fc and/or FOR residues proximal to variable position residues
were floated, that is the
amino acid conformation but not the amino acid identity was allowed to vary in
a protein design
calculation to allow for conformational adjustments. These are indicated below
the table for each set
of calculations when relevant. Considered amino acids typically belonged to
either the Core, Core
XM, Surface, Boundary, Boundary XM, or All 20 classifications, unless noted
otherwise. These
classifications are defined as follows: Core = alanine, valine, isoleucine,
leucine, phenylalanine,
tyrosine, tryptophan, and methionine; Core XM = alanine, valine, isoleucine,
leucine, phenylalanine,
tyrosine, and tryptophan; Surface = alanine, serine, threonine, aspartic acid,
asparagine, glutamine,
glutamic acid, arginine, lysine and histidine; Boundary = alanine, serine,
threonine, aspartic acid,
asparagine, glutamine, glutamic acid, arginine, lysine, histidine, valine,
isoleucine, leucine,
phenylalanine, tyrosine, tryptophan, and methionine; Boundary XM = Boundary =
alanine, serine,
threonine, aspartic acid, asparagine, glutamine, glutamic acid, arginine,
lysine, histidine, valine,
isoleucine, leucine, phenylalanine, tyrosine, and tryptophan; All 20 = all 20
naturally occurring amino
acids.
[194] The majority of calculations followed one of two general types of
computational screening
methods. In one method, the conformations of amino acids at variable positions
were represented as
a set of backbone-independent side chain rotamers derived from the rotamer
library of Dunbrack &
Cohen (Dunbrack et al., 1997, Protein Sci 6:1661-1681). The energies of all
possible combinations of
the considered amino acids at the chosen variable positions were calculated
using a force field
containing terms describing van der Weals, solvation, electrostatic, and
hydrogen bond interactions,
and the optimal (ground state) sequence was determined using a Dead End
Elimination (DEE)
algorithm. As will be appreciated by those in the art, the predicted lowest
energy sequence is not
necessarily the true lowest energy sequence because of errors primarily in the
scoring function,
coupled with the fact that subtle conformational differences in proteins can
result in dramatic
differences in stability. However, the predicted ground state sequence is
likely to be close to the true
ground state, and thus additional favorable diversity can be explored by
evaluating the energy of
sequences that are close in sequence space and energy around the predicted
ground state. To
accomplish this, as well as to generate a diversity of sequences for a
library, a Monte Carlo (MC)
algorithm was used to evaluate the energies of 1000 similar sequences around
the predicted ground
state. The number of sequences out of the 1000 sequence set that contain that
amino acid at that
variable position is referred to as the occupancy for that substitution, and
this value may reflect how
favorable that substitution is. This computational screening method is
substantially similar to Protein
Design Automation (PO/VD) technology, as described in US 6,188,965; US
6,269,312; US
6,403,312; USSN 09/782,004; USSN 09/927,790; USSN 10/218,102; PCT WO 98/07254;
PCT WO
01/40091; and PCT WO 02/25588, and for ease of description, is referred to as
PDAO technology
throughout the examples. Tables that present the results of these calculations
provide for each
variable position on the designated chain (column 1) the amino acids
considered at each variable
81
CA 02766627 2012-01-23
WO 2004/099249
PCT/1JS2004/009298
=
position (column 2), the WT Fc amino acid identity at each variable position
(column 3), the amino
acid identity at each variable position in the DEE ground state sequence
(column 4), and the set of
amino acids and corresponding occupancy that are observed in the Monte Carlo
output (column 5).
For example in the first row of Table 1 below, when position 328 was varied
using boundary amino
acids as the set of variable residues for that position, L occurred 330 times
in the top 1000 sequence,
M occurred 302 times, etc.
[195] Other calculations utilized a genetic algorithm (GA) to screen for low
energy sequences, with
energies being calculated during each round of "evolution" for those sequences
being sampled. The
conformations of amino acids at variable and floated positions were
represented as a set of side chain
rotamers derived from a backbone-independent rotamer library using a flexible
rotamer model
(Mendes et al., 1999, Proteins 37:530-543). Energies were calculated using a
force field containing
terms describing van der Waals, solvation, electrostatic, and hydrogen bond
interactions. This
calculation generated a list of 300 sequences which are predicted to be low in
energy. To facilitate
analysis of the results and library generation, the 300 output sequences were
clustered
computationally into 10 groups of similar sequences using a nearest neighbor
single linkage
hierarchical clustering algorithm to assign sequences to related groups based
on similarity scores
(Diamond, 1995, Acta Clyst D51:127-135). That is, all sequences within a group
are most similar to
all other sequences within the same group and less similar to sequences in
other groups. The lowest
energy sequence from each of these ten clusters are used as a representative
of each group, and are
presented as results. This computational screening method is substantially
similar to Sequence
Prediction Algorithmim (SPA) technology, as described in (Raha et al., 2000,
Protein Sci 9:1106-
1119); USSN 09/877,695; and USSN 10/071,859, and for ease of description, is
referred to as SPATM
technology throughout the examples. Tables that present the results of these
calculations provide for
each variable position on the designated chain (column 1) the amino acids
considered at each
variable position (column 2), the WT Fc amino acid identity at each variable
position (column 3), and
the amino acid identity at the variable positions for the lowest energy
sequence from each cluster
group (columns 4-13).
[196] Computational screening was applied to design energetically favorable
interactions at the
Fc/FcyR interface at groups of variable positions that mediate or potentially
mediate binding with
FcyR. Because the binding interface involves a large number of Fc residues on
the two different
chains, and because FcyRs bind asymmetrically to Fc, residues Were grouped in
different sets of
interacting variable positions, and designed in separate sets of calculations.
In many cases these
sets were chosen as groups of residues that were deemed to be coupled, that is
the energy of one or
more residues is dependent on the identity of one or more other residues.
Various template
structures were used, and in many cases calculations explored substitutions on
both chains. For
many of the variable position sets, calculations were carried out using both
the FDA and SPATIA
technology computational screening methods described. The results of these
calculations and
82
CA 02766627 2012-01-23
WO 2004/099249 PCT/US2004/009298
relevant are presented in Tables 1 ¨ 30 below. Relevant parameters and
information are presented
below each table, including the computational screening method used, the
template structure used,
whether or not that structure had carbohydrate atoms, and any residues that
may have been floated.
For example, Table 2 presents results from a PDAC) calculation in which
residues 120, 132, and 134
on chain C (the FcyRIllb receptor) were floated.
[197] Included within the compositions of the invention are antibodies that
have any of the listed
amino acid residues in the listed positions, either alone or in any
combination (note preferred
combinations are listed in the claims, the summary and the figures). One
preferred combination is the
listed amino acid residues in the listed positions in a ground state
(sometimes referred to herein as
the "global solution", as distinguished from the wild-type). In addition,
combinations between SPATM
proteins, both within tables and between tables, are also included. It should
be noted that residues
not listed in a given table are implied to have not been varied, and thus
remain wild-type. For
example, in the SPA TI" calculation results presented in Table 4, column 4
(representing cluster 1)
indicates a protein with the six listed amino acids at the six listed
positions (e.g. column 4 is a single
protein with a wild-type sequence except for 239E, 265G, 267S, 269Y, 270T and
299S). Thus, each
of these individual proteins are included within the invention. Alternatively,
residue positions and
particular amino acids at those residue positions may be combined between
columns within a table, or
between tables. Furthermore, it should be noted that although each table
indicates the presence or
absence of carbohydrate, the presence or absence of said atoms in the
computational screening
calculation is not meant to imply that Fc variants designed by such
calculations should be applicable
to only aglycosylated or glycosylated Fc. Thus although the calculations in
Table 1 were run without
carbohydrate atoms present in the template structure, the resulting predicted
substitutions may be
favorable in a glycosylated or aglycosylated antibody or Fc fusion.
83
CA 02766627 2012-01-23
WO 2004/099249
PCT/US2004/009298
Table 1
Considered Ground Sequences Around
Position VVT
Amino Acids State Ground State
L:330 M:302 E:111 K:62 A:45 0:39 0:36 S:30
328 A Boundary L
T:28 N:10 R:7
R:247 K:209 0:130 H:95 E:92 T:59 D:51 N:51
332A Surface 1
S:42 A:24
L:321 M:237 T:166 K:73 R:72 S:55 0:20 D:17
328 B Boundary L
E:13 A:12 V:10 N:4
E:269 0:180 R:145 K:111 0:97 T:78 N:65 S:28
332B Surface 1
A:14 H:13
PDA technology, 111S template structure; - carbohydrate
Table 2
Considered Ground Sequences Around
Position WT
Amino Acids State Ground State
239 A Surface S K E:349 D:203 k:196 k.95 Q:63 S:63 N:10 R:1
265 A Boundary XM D D D:616 N:113 1:110 E:104 S:25 A:23
0:9
299 A Boundary XM T I 1:669 H:196 V:135
327 A Boundary XM A S A:518 S:389 N:67 D:26
Q:314 R:247 N:118 1:115 A:63 E:55 D:34 S:22 -
265 B Boundary XM D Q
K:21 V:11
PDA technology; 111S template structure; 4- carbohydrate; floated 120 C, 132
C, 134 C
=
Table 3
Considered ' Ground Sequences Around
Position WT
Amino Acids State Ground State
239 A Surface - S E E:872 Q:69 0:39 K:16 A:4
265A Boundary XM D Y Y:693 H:111 E:69 D:62.F:29 K:19
R:14 W:2 Q:1
267 A BoUndary XM S 8 S:991 A:9
269 A Core XM E F F:938 E:59 Y:3
E:267 T:218 K:186 D:89 0:88 R:46 6:34 N:29
270A Surface
H:23 A:20
299 A Boundary XM T H H:486 T:245 K:130 E:40 S:39 D:27
Q:27 A:4 N:2
PDA technology; 11IS template structure; - carbohydrate; floated 120 C, 122
C. 132 C, 133 C,
134C
Table 4
Considered
Position WT 1 2 3 4 5 6 7 8 9 10
Amino Acids
239A Surface SEQQQEE EQE E
265A All 20 DGGGGGGGGGG
, .
267A All 20 S.SSSSSS S SS S
269A Core EYYAAVY A A A A
270A Surface D TSA.ST T
I AA A
299A All 20 I SS S S S S_S S S S
84
CA 02766627 2012-01-23
WO 2004/09249
PCT/US2004/009298
=
SPA technology; ills template structure; + carbohydrate; floated
120 C, 122 C, 132 C, 133 C,
134 C
CA 02766627 2012-01-23
= WO
2004/099249 PCT/US2004/009298
Table 5
Considered Ground Sequences Around
Position VVT
Amino Acids State Ground State
T:195 V:131 1:112 W:107 K:85 F:66 Y:56 E:52
235 A Boundary XM L
6:38 S:37 1:34 R:29 H:26 N:23 D:9
N:322 D:181 R:172 K:76 Y:70 6:59 E:48 S:40
296 A Surface Y N
11-20 T=11 _ . . .
298A Surface S _ T T:3.70 R:343 K:193 A:55 S:39
235 B Boundary XM L L L:922 1:78
PDACItechnology; 1IIS template structure; - carbohydrate; floated 119 C, 128
C, 157 C
Table 6
Considered
Position WT 1 2 3 4 5 6 7 8 9 10
Amino Acids
235A All 20 L SSPSSS S SS S
296A Surface Y 000E E 0 E
298A Surface SSKKKKS SS K S
235 B All 20 L ,K K,K L ,L L L L _I K ,
SPA TM technology; 111S template structure; + carbohydrate; floated 119 C, 128
C, 157 C
Table 7
Considered Ground Sequences Around
Position WT
________ Amino Acids State Ground State
K:402 E:282 H:116 T:67 R:47 6:39 D:26 A:11
239 B Surface
S 7 N:3
Y 341 W:283 1:236 V:77 F:36 H:9 T:7 E:4 K:4
265 B Boundary XM D W
-A 2 D:1
=
3276 Boundary XM A R R f338 K: 86 H:35 E:12 T:10 6:7 A:6 D:3
N:3
323 B Core XM L L L1000
329 6 Core XIVI P P P:801 A:199
330 B Core X1Y1 = A Y Y:918 F:42 L:22 A:18
332 B Surface I 1 1:792 E:202 0:5 K:1
PDA0 technology; 11IS template structure; - carbohydrate; floated 88 C, 90 C,
113 C, 114 C, 116 C,
160 C, 161 C
Table 8
Considered I
Position WT 1 2 3 4 5 6 7 8 9 10
Amino Acids
239 B Surface =SDTEEEE EEE E
- 265 B All 20 DiG G K C K G" GK K
G
327 B A1120 ,A,KMLINL KILL
328 B Core LMMMLAMIML L
329B Core P P PP P'P,P P P P P
3306 Core A L A A _A A A A A A_ A_
332B Surface I I Q 1 _ 1 Q Q _ E b I
SPAN technology; 111S template structure; + carbohydrate; floated 88 C, 90 C,
113 C, 114 C,
116 C, 160 C, 161 C
CA 02766627 2012-01-23
,52620-2D
Table 9
Position Considered WT 1 2 3 4 5 6 7 8 9 10
Amino Acids
_ ¨
239 A Surface SQQQEQEQ EQQ
265A All 20 DGGGGGGG GG
299A All 20 TSSASSSS SS S
327A All 20 A ASSSSSS S A S
265B All 20 ONGGOGGG GGG
SPA"' technology; lliS template structure; -carbohydrate; floated 120 C, 132
C. 134 C
Table 10
Considered Ground Sequences Around
Position VVT
Amino Acids State Ground State
K Y:401 1:260
F:151 1:82 K:63 H:17 Q:11 W:7 R:3
234 A Boundary XM I
T:2 E:2 V:1
235 A Boundary XM L L W:777 L:200 K:12 Y:5 1:3 F:2
V:1
W:427 Y:203 L:143 F:74 1:59 E:32 K:23 V:14
234 B Boundary XM L
D:10 T:7 H:4 R:4
W:386 Y:380 F:135 K:38 L:26 E:15 0:12 H:8 R:4
235 6 Boundary XM L
T:2
MAID technology; D1290 1E4K template structure; - carbohydrate; floated 113 C,
116 C. 132 C.
155 C, 157 C
Table 11
Position Considered 1121111.11111111111 6 Effs 9 10
Amino Acids
234 A All 20 1lalla112111811111111101= G G
= 235 A m
20 Mill[1110.1011M1111.11111MEIMERM L
234 B All 20 L
11:61=118111011C1111611 G G 1111111 G
235 B All 20 inimai A Ei A
Aims S A A
SPAT?" technology; D129G 1E4K template structure; + carbohydrate; floated
113G. 116 C, 132 C,
155 C, 157 C
Table 12
Considered Ground Sequences Around
Position WT
Amino Acids State Ground State
E:235 S:122 D:94 0:93 A:74 K:70 L:67 1:63
239A Boundary XM S
N:57 R:51 1:29 V:18 W:15 H:12
1688 E:121 K:43 0:41 A:33 D:26 8:14 1:14
328 A = Boundary XM L
N:12 R:8
=
1:155 W:95 1182 K:79 E:74 0:69 H:67 V:63 R:57
332 A Boundary XM I
T:57 D:45 S:43 N:42 A:35 F:19 Y:18
PDA technology; 01290 if IS template structure; - carbohydrate; floated 120 C
87
CA 02766627 2012-01-23
52620-2D
Table 13
Considered
Position Amino Acids WT 1 2 3 4 5 6 7 8 9 10
239A All 20 S 'LEEQ.EE K K K K
328A A1120 L L Q L 0 K L L 0 K L
332A A1120 I KKLQAK L Q A Q
SPA Th" technology; 0129G 11IS template structure; + carbohydrate; floated 120
C
Table 14
Considered Ground Sequences Around
Position VVT
Amino Acids State Ground State
R:1951:169 L:126 V:91 K:89 E:61 H:521:50
239 B Boundary XM S
0:42 N:35 S:34 0:30 A:26
L:671 T:165 K:40 S:38 E:28 R:17 Q:17 V:11 A:8
328 B Boundary XM L
D:5
1:387 E:157 L:151 V:78 0:63 K:50 R:33 1:29
332 B Boundary XM 1 1
0:25 A:12 N:8 S:6 W:1
PDAO technology; D129G 11IS template structure; - carbohydrate; floated 90 C,
160 C, 161 C
Table 15
Considered
Position WT 1 2 3 4 5 6 7 8 9 10
Amino Acids
239B AII20 STLILLLILL L
328B All 20 LMRMDTML QD L
33213 All 20 1 IDQQQL L T Q L
SPATu technology, D129G 11IS template structure; + carbohydrate; floated 90 C,
160 C. 161 C
Table 16
Considered Ground Sequences Around
Position VVT
Amino Acids State Ground State
1:164 S:159 L:156 E:86 W:76 K:71 0:65 A:52
239B Boundary XM S T
R:43 H:38 Q:38 N:31 1:14 V:7
= 1:556 E:114 T.:84 k:80 S:69 0:36 A:31 D:15
3288 .= Boundary XM L L= R:11 N:4
1:188 W:177 E:97 1:94 1:59 Q:57 V:54 K:52 F:51
332 B Boundary XM 1
0:34 H:33 S:27 R:26 N:18 A:17 Y:16
PDAO technology; 0129G 1E4K template structure; - carbohydrate; floated 117 C
Table 17
Considered
Position
Amino Acids WT 1 2 3 4 5 6 7 8 9 10
239 B All 20 S PSPEL L L L L L
3288 All 20. LKKKKKIK K K L
33213 All 20 1 S S E L E L L L
SPA Tu technology; 0129G 1E4K template structure; + carbohydrate; floated 117
C
=
88
CA 02766627 2012-01-23
,52620-2D
Table 18
= Considered Ground
Sequences Around
Position WT
Amino Acids State Ground State
K:196 L:171 1:146 E:88 V:76 R:75 T:50 H:45
239 A Boundary XM S
13:43 Q:39 8:30 N:22 A:19
328 A Boundary XM L W L:517 1:230 W:164 H:40 K:29
E:11 R:5 T:4
1:283 L:217 E:178 Q:81 V:64 D:47 T:35 K:27
332 A Boundary XM I
W:18 R:12 A:10 Y:7 N:7 F:6 S:5 H:3
PDA technology; 01290 1E4K template structure; - carbohydrate; floated 87 C,
157 C, 158 C
Table 19
Considered
Position WT 1 2 3 4 5 6 7 8 9 10
Amino Acids
239A A1120 SFQETPP T PPP
328A All 20 L KR_RKK_MfR K MR
332 A All 20 _ILLIIEIEE I 1
SPA Tm technology; D129G 1E4K template structure; + carbohydrate atoms;
floated 87 C, 157 C,
158 C
Table 20
Considered Ground Sequences Around
Position WT
Amino Acids State Ground State
240 A Core + Thr _ V V V:698 M:162 T:140
263 A Core + 'Thr V V V:966 T:34
266 A Core + Thr V V V:983 T:17
325A _ Boundary N N N:9431:40 A:17
328 A _ Boundary I L L:610 M:363 K:27
332A Glu I E E:1000
PDA technology; 0129G 11IS template structure; - carbohydrate; floated 273 A,
275 A. 302 A,
323 A, 134C
Table 21
Considered
Position VVT 1 2 3 4 5 6 7 8 9 10
________________ Amino Acids
240A All 20 V VAVVVV V V V V
263A All 20 V VVVVVV V V V V
266A A1120 V IVIITV V V V I
325A Ali 20 NANNNQT TQN T
328A . All 20 L K K ,L., K L K L L I I.
332A Glu 1 DIDDDD_D
ID 0 0 D
SPAT"' technology; D129G 111S template structure; + carbohydrate; floated 273
A, 275 A, 302 A,
323 A, 134 C
89
CA 02766627 2012-01-23
52620-2D
Table 22
Position
Considered Ground Sequences Around
WT
Amino Acids State Ground State
240 B Core + Thr V V V:7131:287
263 B Core + Thr V V V:992 1:8
266 B Core + Thr V V V:976 1:24
325B Boundary N N N:453 .r296 A:116 D:96 5:30 V:9
328 B Boundary L L L:623 M:194 1:100 R:72 K:11
332B Glu E E:1000
PDAO technology; D129G 11IS template structure; - carbohydrate; floated 273 B,
275 B, 302 B,
323 [3, 161 C
Table 23
Considered
Position WT 1 2 3 4 5 6 7 8 9 10
Amino Acids
240 B All 20 V ATATTA ATIT
26313 AII20 V VAATTV V T A T
266 8 All 20 _V _ V. V _V, V V V V V I V
325 B All 20 NNKKNKK NNNN
328 B All 20 L R L L L L L L L L'L
_
332A _ Glu I DODDIDDDDDD
SPA T14 technology; D129G 11IS template structure; + carbohydrate; floated 273
B, 275 B, 302 B,
323 B. 161 C
Table 24
Considered Ground Sequences Around
Position WT
Amino Acids State Ground State
240 B Core + Thr V kt V:715 M:271 1:12 1:2
263 13 Core + Thr V V V:992 1:8
266 B Core + Thr V V V:996 T:4
325 B Boundary - N N N:651 T:232 0:64 A:53
328 B ¨ Boundary L M M:556 L:407 K:37
-
332 B Glu 1 E E:1000
PDACI technology; 0129G 1E4K template structure; - carbohydrate; floated 273
B, 275 13, 302 B,
323 B, 131 C
Table 25
Considered
Position WT 1 2 3 4 5 6 7 8 9 10
Amino Acids_
240 B All 20 V TATAAA ATAA
_
263 B All 20 VTWTTAT T IL L
266 8 All 20 VLATT _ VL _ T
T L V
_
325 B All 20 ___________________________________ N ANA
AN A A A A A
328 B A1120 LIKLILLL L LL
_
332A Glu I DDDODDDDDD
SPATM technology; D129G 1E4K template structure; + carbohydrate; floated 273
B, 275 B. 302 B,
323 E3, 131 C
= 90
CA 02766627 2012-01-23
52620-2D
Table 26
. _________________________________________________________________
Considered Ground Sequences Around
Position WT
Amino Acids State Ground State
240 A Core + Thr V V V:876 T:109 M:15
263 A Core + Thr V V V:913 T:87
266 A Core + Thr -7 V V V:969 T:31
325 A Boundary _ N V V:491 N:236 1:187 A:35 D:32
S:19
328 A _ Boundary _1 L 1:321 W:290 M:271 F:49 K:46
R:23
-332 A Gin 1 E E:1000
PDA technology; 0129d 1E4K template structure; - carbohydrate; floated 273 A.
275 A, 302 A,
323 A, 158 C
Table 27
¨ =
Considered
Position WT 1 2 3 4 5 6 7 8 9 10
Amino Acids
240 A All 20 = V ATAATT A A A T
. _
263A A1120 VT TVVT V L L=V T
_ _ _ _
266A AII20 VVVVVVV V V V V
325A A1120 NCZNQQ0OQ 0 N N
328 A All 20 LKMKKKK K K K K
332A dlu 1 .D 0 0-0 D D 0 D 0-D
SPAIN technology; 0129G 1E4K template structure; + carbohydrate; floated 273
A, 275 A, 302 A,
323 A, 158 C
[198] Computational screening calculations were aimed at designing Fc variants
to optimize the
conformation of the N297 carbohydrate and the 0y2 domain. By exploring
energetically favorable
substitutions at positions that interact with carbohydrate, variants can be
engineered that sample new,
potentially favorable carbohydrate conformations. Fc residues F241, F243,
V262, and V264 mediate
the Fe/carbohydrate interaction and thus are target positions. The results of
these design calculations
are presented in Table 28.
Table 28
Considered Ground Sequences Around
Position WT
Amino Acids State Ground State
241 A Core F Y Y:172 M:162 1:144 F:140 W:110 1:97 A:91
V:84
243A Core F Y _ Y:211 L:204 W199 F:160 M:141 A:85
262 A Core V _ M M:302 1:253 V:243 A:202 . -
264A Core V
1:159 M:152 V:142 L:140 W:136 F:120 Y:104
A:47
PDA technology, 1US template structure; - carbohydrate
[1991 Computational screening calculations were aimed at designing Fc variants
to optimize the
angle between the C73 and C12 domains. Residues P244, P245. P247, and W313,
which reside at
91
CA 02766627 2012-01-23
52620-2D
=
the Cy2/Cy3 interface, appear to play a key role in determining the C72-Cy3
angle and the flexibility of
the domains relative to one another. By exploring energetically favorable
substitutions at these
positions, variants can be designed that sample new, potentially favorable
angles and levels of
flexibility. The results of these design calculations are presented in Table
29.
Table 29
Considered Ground Sequences Around
Position WT
Amino Acids State Ground State
K164 H:152 R:110 M:100 S:92 N:57 A:54 D:50
244 A Boundary
Q:49 1:46 E:37 V:30 L:27 W:23 F:9
245 A Boundary P A k491 S:378 N:131
V:156 T:125 K:101 E:87 Q:79 R:78 S:76 A:72
247 A Boundary
0:72 H:60 M:47 N:47
W:359 F:255 Y:128 M:114 H:48 K:291:24 A:11
313 A Boundary W
E:10 V:10 S:9 Q:3
PDA@ technology; 11IS template structure; - carbohydrate
[200] In addition to the above calculations using PDAO and SPA"' computational
screening
methods, additional calculations using solely an electrostatic potential were
used to computationally
screen Fc variants. Calculations with Coulomb's law and Debye-Huckel scaling
highlighted a number
of positions in the Fc for which amino acid substitutions would favorably
affect binding to one or more
FcyRs, including positions for which replacement of a neutral amino acid with
a negatively charged
amino acid may enhance binding to FcyRIlla, and for which replacement of a
positively charged
=
amino acid with a neutral or negatively charged amino acid may enhance binding
to FcyRIlla. These
results are presented in Table 30.
Table 30
Replacement of a + residue Replacement of a neutral residue
with a - residue with a - residue
H268 S239
K326 Y29e
= K334 A327 .
1332
Coulomb's law and Debye-Huckel scaling; 11I3 template structure; +
carbohydrate
[201] Computational screening calculations were carried out to optimize
aglycosylated Fc, that is to
optimize Fc structure, stability, solubility, and Fc/FcyR affinity in the
absence of the N297
carbohydrate. Design calculations were aimed at designing favorable
substitutions in the context of
92
CA 02766627 2012-01-23
52620-2D
the aglycosylated Fc template structure at residue 297, residues proximal to
it, residues at the
Fc./FcyR interface, and residues at the Fc/carbohydrate interface. Variable
positions were grouped in
different sets of interacting variable positions and designed in separate sets
of calculations, and
various template structures were used. For many of the variable position sets,
calculations were
carried out using both the PDA and SPA Tu computational screening methods.
The results of these
calculations and relevant information are presented in Tables 31 - 53 below.
Table 31
Considered Ground Sequences Around
Position WT
Amino Acids State Ground State
265 A Boundary XIV' D V Y:531 F:226 W:105 H:92 K:21
0:16 E:6 T:3
A:235 S:229 0:166 E:114 N:92 Y:57 F:55 Q:25
297 A Boundary XM N
H:10 T:7 K:6 L:3 R:1
L:482 le:186 F:131 T;55 S:51 K:31 H:22 A:18
299A Boundary Xfv1 T
E:14 Q:10
1:299 K147 V:85 R:82 W:71 N:65 0:35 E:35
297 B Boundary XM N
Q:34 S:32 L:31 H:30 T:28 A:26
PDA technology; 11IS template structure; - carbohydrate; floated 122 C,
129.C, 132G. 155 C -
Table 32
Considered
Position WT 1 2 3 4 5 6 7 8 9 10
Amino Acids
265A All 20 DGGGGGG G GC G
297A M20 N_A_T A EK K A A N N
299 A All 20 T _ S K S K F F F F _ F S
2978 A1120 N K K K K K K K K K K
SPA PA technology, 11IS template structure; - carbohydrate; floated 122C, 129
C, 132 C, 155 C
Table 33
Considered Ground Sequences Around
Position WT
-Amino Acids State Ground State
239 A Surface S E E:928 Q:65 0:7
265 A Boundary XM 0 W W:709 Y:248 F:43
H:449 Y:146 E:17 0:89 K:64 N:32 1:30 R:25
296 A Surface
Q:23 S:5
=
297A Surface N E E:471 H:189 D:102 T:97 K:96 R:22 Q:15
S:8
298 A Boundary XM 8 R R:353 T275 K:269 A:56 S:-38
E5 0:2 H:2
299 A Boundary XM- T F Y:398 F:366 L:217 H.:15 K4
PDA technology; 0129G 11IS. template structure; - carbohydrate; floated 120
C, 122 C. 128 C,
132 C, 155 C
93
CA 02766627 2012-01-23
,5220-2D
Table 34
Considered
Position
Amino Acids WT 1 2 3 4 5 6 7 8 9 10
239A All 20 S
EQQEQQ0000
265A All 20 DGGGGGGG
G GG
296A All 20
YDONNQNNNQN
297A M20 N
AANADD E NNE
298A A1120
SKKKSKK K K SK
299A AII20
_TSYFSYF K F SK
SPAT" technology; D1290 11IS template structure; - carbohydrate; floated 120
C, 122 C, 128 C,
132 C, 155 C
Table 35
Considered Ground Sequences Around
Position WT
Amino Acids State Ground State
E:417 T:122 D:117 0:94 R:84 S:63 K:47 H:29
2398 Surface
N:19 A:8
265 B Boundary XM D W W865 Y:79 F:55 K:1
Y:549 H:97 0:80 S:75 N:48 E:45 K:32 R:30 0:28
2968 Surface
A:16
Ft:265 H:224 E:157 K:154 Q:75 D:47 T:34 N:24
297 B Surface
S:13 A:7
298 B Boundary XM S V V:966 D:101:8 A:8 N:4 3:4
299 B Boundary XM T Y Y:667 F:330 H:3
PDA technology; D129G 1E4K template structure; - carbohydrate; floated 117 C,
119 C, 125 C,
129 C, 152 C
Table 36
=
Considered
Position WT 1 2 3 4 5 6 7 8 1 9 10
Amino Acids
2398. All 20
SSREKSSEEEK
265B All 20 DADKYAAF F K Y
296B AII20 _Y
AA_AAA A A A A A
2978 AII20 N
TSITEE,E S E E
2988 All 20 S
GGGGGGGGGO
299B All 20 T LFE,EYF V F Y
Y
SPAT"' technology; D129G 1E4K template structure; - carbohydrate; floated 117
C, 119 C, 125 C,
129 C, 152C
94
CA 02766627 2012-01-23
52620-2D
Table 37
Considered Ground Sequences Around
Position WT
Amino Acids State Ground State
239 A Surface S E E:868 0:92 D:38 K:1 N:1
265 A Boundary XM D W W:575 Y:343 F:66 H:15 K:1
296 A Surface Y H H:489 Y:103 R:98 K:97 Q:64 0:63 T:41
N:38 E:7
297A Asp N D 0:1000
298 A Boundary XM _ S R R:340 K:262 T:255 A:59
S:57 E:11 Q:10 H:6
299 A Boundary XM T F Y:375 F:323 L:260 H:24 K:18
PDA technology; 0129G 11IS template structure; - carbohydrate; floated 120 C,
122 C, 128 C,
132 C, 155 C
Table 38
Considered
Position WT 1 2 3 4 5 6 7 8 9 10
Amino Acids _
239A. AII20 S,EQEE_E E E E Q E
265A Al120 DGGGGGGOGGG
296A A1120 Y ENQENQ 0 00 N
297A Asp NDDDDDDDDDD
298A A1120 S= K_S K 5K K K S K K
299A A]120 T SK-YSFF F F F K
SPATM technology; D129G 11IS template structure; - carbohydrate; floated 120
C, 122 C, 128 C,
132 C, 155 C
Table 39
Considered Ground Sequences Around
Position VVT
Amino Acids State Ground State
E E:318 0:123 T:109 b:108 R:93 S:89 K:69
N:40
2398 Surface
H:38 A:13
265 B Bounaary XM D W W:745 Y:158 F:85 K:9 E:1 R:1 H:1
Y:390 11:127 S:83 R:81 K:78 N:65 0:55 E:49
296 B Surface
0:44 A:26 T:2
297 B Asp N D 0:1000
298 B Boundary XM S V V:890 T:35 A:29 0:19 8:16 N:10 E:1
299 B Boundary XM T Y Y:627 F:363 H:10
FDA technology; Di 29G 1E4K template structure; - carbohydrate; floated 117
C. 119 C, 125 C,
129C, 152C
Table 40
Considered
Position WT 1 2 3 4 5 6 7 8 9 10
Amino Acids
239B All 20 SKEEQEK QEK Q
265B All 20 OFKKAKYWK L F
296 B All 20 Y A A A AA A A,A A A
2978 Asp WOODED() DE) D
298 6 All 20 SGGGGGGGGGG
2998 All 20 T YYYYYYF F Y Y
CA 02766627 2012-01-23
.52620-2D
SPAT" technology; 0129G 1E4K template structure; - carbohydrate; floated 117
C. 119 C, 125 C,
129 C. 152 C
=
=
= 96
CA 02766627 2012-01-23
52620-2D
=
Table 41
Considered Ground Sequences Around
Position WT
Amino Acids State Ground State
E:312 L:148 D:102 0:98 K:641:61 8:57 A:44
239 A Boundary XM S
T:39 N:29 R:23 V:18 W:5
W:363 Y:352 F:139 H:77 K:39 R:14 0:11 E:4
265 A Boundary XM D
0:1
297A Asp N D D:1000
Y:309 F:224 1:212 H:96 K:92 E:28 Q:20 R:16
299 A Boundary XM T
T:2 al
PDA technology; 0129G 11IS template structure; - carbohydrate; floated 120 C,
122 C, 132 C.
155C
Table 42
- Considered
Position
Amino Acids WT 1 2 3 4 5 6 7 8 9 10
2'39A All 20 SELLLEE E Q I E
265A All 20 DGGGGGG G GO
97B AsP NDDDDDDDDDD
299 A All 20 _ T S K _ l< F F F K F K F
SPAT" technology; 0129G 11IS template structure; - carbohydrate; floated 120
C, 122 C, 132 C,
155 C
Table 43
Considered Ground Sequences Around
Position WT
Amino Acids State Ground State
L:194 T:122 8:120 E:111 0:79 K:71 A:62 0:57
239 B Boundary XM S
R:43 11:43 N:37 1:24 W:24 V:13
Y:248 W:233 F:198 K:84 D:57 E:55 H:42 R:28
265B Boundary XM D
0:20 A:10 T:10 N:8 S:7
297B Asp N D 0:1006
299 B Boundary-XM T Y Y:493 F:380 11:76 T:31 E:10
D:4 A:3 S:3
PDA technology; D129G 1E4K template structure; - carbohydrate; floated 117 C,
119 C, 129 C,
152 C
Table 44
Considered Position WT 1 2 3
4 5 6 7 8 9 10
Amino Acids
= 239 B All 20 - S R E P_L 1 F P
P 1 L
265B All 20 DDKSFSY A M A D
297B. Asp NIDDIDIDDDDIDDD
2998 A1120 T "(V Y YE V V YY
SPA"' technology; 0129G 1E4K template structure; - carbohydrate; floated 117
C, 119 C, 129 C,
152C
97
CA 02766627 2012-01-23
52620-2D
Table 45
Considered Ground = Sequences Around
Position WT
Amino Acids State Ground State
E
E:251 L:125 0:120 0:112 8:73 K:651:61 A:58
239 A Boundary XM S
T:45 N:35 R:28 V:23 W:4
Y:216 H:153 K:135 0:109 W:104 F:86 R:54 T:38
265 A Boundary XM
E:29 0:22 A:21 N:17 8:13 L:3
297A Asp N D D:1000
PDA technology; D129G 11IS template structure; - carbohydrate; floated 299 A,
120 C, 122 C,
132 C, 155 C
Table 46
Considered
Position WT 1 2 3 4 5 6 7 8 9 10
Amino Acids
239 A All 20 S SLELQQ E QQ E
265A All 20 DGGGGGGGGG G
297A Asp NDDDDDDDDD D
SPA' technology; 0129G I IIS template structure; - carbohydrate; floated 299
A, 120 C, 122 C,
132 C, 155 C
Table 47
Considered Ground Sequences Around
Position VVT
Amino Acids State Ground State
L:158 S:137 T:125 E:115 0:86 K:75 A:62 0:56
239 B Boundary XM S
1-1:43 R:39 N:35 W:30 1:24 V:15
Y:188 W:159 F:156 0:122 K:71 E:71 H:61 0:44
265B Boundary XM
R:39 fic24 S:22 N:19 T:18
2978 Asp N D 0:1000
PDA technology; 0129G 1E4K template structure; - carbohydrate; floated 299 B,
117 C, 119 C,
129 C. 152 C
Table 48
Considered
Position WT 1 2 3 4 5 6 7 8 9 10
Amino Acids
2398 All 20 S S E,P,P E,S P L F L
, 2658 All 20 DAkAMKF VD F F
297B Asp 1\1 ODD!) D D'b 0 0 D
SPAT"' technology; 0129G 1E4K template structure; - carbohydrate; floated 299
8, 117 C, 119 C,
129 C, 152 C
98
CA 02766627 2012-01-23
52620-2D
Table 49
Considered Ground Sequences Around
Position WT
Amino Acids State Ground State
297A , Asp ¨ N D D:1000
T:123 Y:64 H:64 K:64 0:64 F:64 R:63 0:63 E:63
299 A Boundary XM T
5:63 1:63 N:62 1:57 A:54 V:52 W:17
MAID technology; 0129G 11IS template structure; - carbohydrate; floated 239 A,
265 A, 120 C,
122 C, 132 C, 155 C
Table 50
Considered
Position WT 1 2 3 4 5 6 7 8 9 10
Amino Acids
297A Asp NDDIDDCID 0 0 00
299A M20 TKKKKFF K K K K
SPAT" technology; D129G 11IS template structure; - carbohydrate; floated 239
A, 265 A, 120 C,
1220. 132C, 155 C
Table 51
Position Considered WT Ground Sequences Around
Amino Acids State Ground State
2976 Asp N D D:1000
1-1.23 F:64 Y:64 H:64 S:63 N:61 0:61 0:61 E:60
299 B Boundary XM T
K:58 V:57 A:57 R:54 1:52 L:51 W:50
PDA technology; 0129G 1E4K template structure; - carbohydrate; floated 239 B,
2656, 117 C,
119 C, 129 C, 152 C
Table 52
Considered
Position WT 1 2 3 4 5 6 7 8 9 10
Amino Acids
297B As N_DDDD DO D
299 B All 20 T Y V µ11-V Y V'1( Y
SPAT" technology; 0129G 1E4K template structure; - carbohydrate; floated 239
B, 265 B, 117 C,
119C, 129C, 152C
[202] Computational screening calculations were carried out to optimize
aglycosylated Fc by
designing favorable substitutions at residues that are exposed to solvent in
the absence of
glycosytation such that they are stable, maintain Fc structure, and have no
tendency to aggregate.
The N297 carbohydrate covers up the exposed hydrophobic patch that would
normally be the
interface for a protein-protein interaction with another Ig domain,
maintaining the stability and
structural integrity of Fc and keeping the C72 domains from aggregating across
the central axis. Key
residues for design are F241, F243, V262, and V264, which reside behind the
carbohydrate on Cy2,
99
CA 02766627 2012-01-23
-52620-2D
in addition to residues such as L328,1332, and 1336, which are exposed
nonpolar residues facing
inward towards the opposed Cy2 domain, that were considered in previously
presented calculations.
The importance of these Cy2 residues is supported by noting that the
corresponding residues in the
Cy3 domain by sequence alignment either mediate the nonpolar interaction
between the two Cy3
domains or are buried in the Cy3 core. The results of these design
calculations are presented in
Table 53.
Table 53
Considered wT Ground Sequences Around
Position
Amino Acids State Ground State
E:190 R:172 K:138 H:1171:93 0:91 D:85 S:49
241 A Surface
N:49 A:16
R:190 H:164 0:152 E:149 K:92 T:71 0:64 N:58
243A Surface
S:42 A:18
D:416 E:164 N:138 0:87 T:83 R:44 S:32 K:24
262A Surface V
All H:1
264 A S urface R:368 H:196 K:147 E:108 0:68 T:34 N:33 D:25
V
S:15 A:6
POND technology-, 111S template structure: - carbohydrate
[203] In a final set of calculations, a SPA Tm computational screening method
was applied to
evaluate the replacement of all chosen variable positions with all 20 amino
acids. The lowest energy
rotamer conformation for all 20 amino acids was determined, and this energy
was defined as the
energy of substitution for that amino acid at that variable position. These
calculations thus provided
an energy of substitution for each of the 20 amino acids at each variable
position. The calculations
used various template structures including different Fc/FcyRIllb complexes
(Ills, 1IIX, 1E4K), a
modeled Fc/FcyRIlb complex, and uncomplexed Fc (10N2), and thus were useful
for a variety of
design goals aimed at both glycosylated and aglycosylated Fc, including
optimization of Fc/FcyR
affinity, C1q affinity, Fc stability, Fc solubility, carbohydrate
conformation, and hinge conformation.
Furthermore, because these calculations provide energies for both favorable
and unfavorable
substitutions, they guide substitutions that may enable differential binding
to activating versus
inhibitory FcyRs. Various template structures were used, and calculations
explored substitutions on
both chains. The results of these calculations and relevant parameters and
information are presented'
in Tables 54 - 60 below. Column 1 lists the variable positions on chain A and
B of the template
structure. Column 2 lists the wild-type amino acid identity at each variable
position. The remaining 20
columns provide the energy for each of the natural 20 amino acids (shown in
the top row). All
substitutions were normalized with respect to the lowest energy substitution,
which was set to 0
energy. For example in Table 54, for L235 on chain A, serine is the lowest
energy substitution, and
=
=
100
CA 02766627 2012-01-23
.52620-2D
1235A is 0.9 kcal/mol less stable than 1235S. Extremely high energies were set
to 20 kcal/mol for
energies between 20 - 50 kc,alknol, and 50 kcal/mold for energies greater than
50 kcal/mot
Favorable substitutions may be considered to be the lowest energy substitution
for each position, and
substitutions that have small energy differences from the lowest energy
substitution, for example
substitutions within 1-2, 1-3, 1-5, or 1-10 kcal/mol.
Table 54
PosWTACD E F_GH 1 KLMNP.QRS.-T V W Y
235A L 0.92.8 2.8 1.5 3.2 3.2 3.4 4.9 1.62.1 3.2 0.9. 0.3 1.3 0.7 0 1.7 4.3
6.5 3.2
236A G 0 1.9 5.1 6:7 10 2.34.3 17.2 5.7 20 4.6 3.212.65.6 6.1 0.6 6.2 12.0 6.7
20
237 A- G 20 20 20 50 50 0 50 50 20 50 20 20 50 50 50 20 20 50 50 50
239A S 0.24.32.6 0 12.84.5 6.9 11.3 t70.1 2.1 1.7 7.9 1.2 2.6 0.3 5.7 11.0 20
20
265A D 9.08.1 -6:3- 7.8 5.1 0 7.3 50 8.29.9 7.7 6.0 50 9.0 8.5 7.8 20 50 20
5.8
267A S 2.13.3 7.3 1.4 50 7.3 20 20 0.92.2 5.0 4.8 0 2.2 3.1 2.9. 20 20 50 50 ,
269 A' E 0.52.1 1.3 0.6 1.6 3.9 2.0 1.2 1.11.3 2.7 0 50 0.6 1.1 0.3 0.8
1.0,5.6 1.2_
= 270A D 0.32.8 2.3 2.0 4.0 4.0 3.4 2.4 -1.2 0 _2.3 2.1 20 2.0 2.3 1.4 1.8
4.2 5.4 6.0
296A Y 2.72.0 1.4 0, 50 0 50 4.6 2.12.4 3.3 1.2 50 0.2 1.5 1.3 4.6 4.4 ,
16.318.2
298A S 0.72.4 6.7 3.4 20 3.9 20 6.7 0 4.1 1.4 4.1 50 1.8 1.1 0.2 2.2 6.3 17.8
20
299 A. T 0.62.811.510.1 20 6.1_20 10.7 7.1 20 4.3 6.8_ 50 _6.312.0 0 3.0 7.1
14.820
23413 L 2.13.2 4.1 .4.2 1.6 5.3 0.1 _0.7 0.6 1.0 2.0 1.7 50 2.8 0.3 2.3 1.7
2.6 13.0 0
235 6: L. 0.62.3 2.5 0.7 5.4 4.8 1.4 3.6 0.1 0 2.0 1.716.90.5 1.2 bi 0.7 5.3
6.8 5.5
236 B G 3.11.3 4.4 8.2 5.2 0 1.9 20 3.120 4.1 2.7 50 3.716.01.2 20 20 20 11.3
237 B G 26 50. 50 50 50 0 50 50 50 50 50 50 50 _ 50 50 50' 50 50 50 60
2398 s 0.92.4 3.4 1.8 5.4 5.6 2.7 3.0 0.9 0 2.0 1.6 50 1.8 1.8 1.4 1.4 5.1 20
5.3
2658 0 4.55.1 4.6 4.64.9 0 3.8 .9.0 2.02.5 4.1 2.1 50 4.5 5.1 4.4 5.9 _ 9.2
11.4 5.8
327B A 1.83.4 4.7 3.9 20 7.0 20 20 0.8 0 1.9 1.5 20 3.0 2.6 3.2 20 20 20 20
328B L 3.73.6 4 3.7 50 8.4
6.8 50 3.8 0.2.1 4.1 _ 50 _ 3.6 8.1 4.9 3.0 12.5 50 50
3298 P '3.48.6 20 20 50 8.0 16.8 50 20 20 16.920 0 20 20 1.317.1 16.5 50 50
330 H A 0.52.0 2.6 0.5 2.4 3.8 1_4 4.2 0 2.0 2.2 0.6.- 200.1 0.6 0_9_0.3 5.1
8.0 2_7
332 8- I 1.52.7 1.2 fa 11.96.812..9 1.2 2.9 0 1.4 1.7 50 1..3 4.9 1.8 1i 3.0
20 201
SPATu technology; 11IS template structure; + carbohydrate atoms, no floated
positions
=
101
CA 02766627 2012-01-23
,
-52620-2D
,
=
Table 55
Pos IWT-A CDEF GH I KLM N P Q R'S T V ' W Y
235A L 0.92.8 2.6 1.7 3.3_3.3 3.4 5.0 1.6 2.1 , 3.3 1.00.3 1.4 1.8 0 1.93.6
6.6 , 3.3
236A G '0 1.7 5.2 6.0 11.3 2.3 4.4_17.2 5.8 19.0 4.9 .38.2,,5.6 6.0 0.8 5.6
11.8 6.6 20
237A G 20 20 20 sb 50_ 0 50_ 50 20 50 20 20_50 50 50 20 20 50 50 , 50
.
238A 0 8.68.010.513.4 6.44 0 5.0 50 12.411.3-9.7 4.3 3212.4 2C8.6 50 50 20 8.4
239A S 0.14.2 2.5 0 20 4.59.0 10.8 1.8 0.2 2.1 1.8 9.1 1.3 2.5 0.3 5.7 10.7 20
19.7
240A V 1.32.4. 2.3_ 6.3 20 7.2 2051 , _ 10_8 6.25.7 .01.1k9.5 13.12.5,0.5 0 20
: 20
241 A F_J0.11.6 1.2_ 0.3 0.2 4.1 1.2 10 1.3 0.12.1 1.414.7 0.5 1.1 0.1 0
8.33.6 0_4
242A L 3.03.4 5.5 8.3 14.4 8.5 11.1- 3.3 13.9 2.2 2.7 .5 0.9 7.9 17.13.8 2.3 0
20 17.5
l
243A
F 1.62.2 2.7 0.2 1.4 5.6'2.5 0 2.2 2.0 l 3.0 2.310.2 0.5 1.6 1.3 0.9 1.2 5.3
1.6
244A P 1.21.8 3.8 0.8 10.2 3.8 -4.6 20 0.2 2.942.0 .8 2.0 0.9 1.7 0 19.320
7.6412.2
245A P 3.9 20'420 120 20 19.1 20 20 20:20 20 20 0 20 20 8.0 20 50 , 20 20
246A K t32.72.0 2.0 2.9 5.72.92 1.4 ' 1A 1.543.1 1.2 0 11.2 1.5 1.741:4 1.2
5.43.0
247 A P 1.22.1 0.3 0.7 4.0 3,913,7 1.8 1.6 ' 1.7 3.3 00.5 0.9 1.5 0.7 1.1 1.3
6.9 ' 3.7
248A K 0.92.7 1.5 0.8 3,1 4.7 3.4 3.3 2.0 1_9 2.6 1.20.6 1.5 2.3 0.7 0 2.5'5$
2.7
249 A D 1.23.7 1.e 0 2047.3 ,19.7 50 , 1.7 20 2.2 1.4 20 1.5 3.4 2.518.3 50 ,
20 20
250A T 0 1.8 3.8 ' 5.8 5046.0 20 14.5 , 6.3 6.3 0.3 3.2, 50 8.7 9.3 1.8, 1.3
1.9 20 ' 50
251 A L 1.11.91.20.5 5.8 , 5.1 1.9 5.6 0.9 0.7 .2.4 1.4,50 0 1.4 0.5 0.8 6.9 ;
8.9 5.8
252A M 0.31.2 0.6 0 3.0_ 3.8 3.43.9 1.0 0.3 2.2 1.317.4 0.1 1.1 0.1 0.2 4.6
,4.2 3.3
253A I 0.71.7,1.1 0.2 1.8 3.5 .2.2 2.0 0.3 1.2 0.8 0.8 0.3 0 1.1 0.3 0.5 2.8
2.4 1.9
254A S j0.71.7 0.4 0.7-'2.2 3.62.0 0.341.241i 2.4 0 20 0.3 1.2 0,3T0=8 0.7 3.8
1.9
255A R1.4.8 2.4 2.50.2 5.4 1.1 `17.0 1.0 2.2 1.5 1.7 50 2.1 0 2.3 50 17.2 4.0
0.5,
256A T 0.61.8 1.2 1.1 2.7 3.4 2.1 1.4 0.7 1_5 2.4 0 _0.4 0.1 0.2 0.4 0.9 1.2
5.6 2.7
257A P , 0 7.8 20 12950 6.2 50 20 12.312.814.4 20 0.113,1 20 2.916.0 20 50 50
258A E 0 ,1.6 4.8 2.6 , 1.0-44.372.2714.8-4.4 6.2 , 3.2 2.9110.4 7.4 6.0 1_0
6_2 17.6 20 1.0
259A V 3.94.3 5.1 8.7 20 10.3,6.8 2.3 ,9.6 2.8 6.2 '.1 50 9.2 205.2 2.1 0 20
20
260 A T 1.72.33.3 1.1 20 j6.648.6 _ 0 0.2 1.8-2.8 1.8 1.1 0.8 0.9 1.7 0.4 1.9
7.1 20
261A C 0 20 20 20 20 3.9 ' 20 20 4_20 20 ,20 20 50 20 20 3.620 20 20 ,20
262A V 1.93.2 0 3.3 20 7.2 20 8.3 2.9 2.9 2.2 1.6 50 3.8 5.2 3.4 3.0 1.7 20 20
263A V 2.7 6.0 17.4 20 8.8 20 10 7.1 7.616.95.2 50 1.9.817.72.8 1.4 0 20 20
264A V 1.93.3 2.8 2.2 0 6.4 2.1 0.7 2.6 0.9 2.7 2.1 2.32.6 2.7 .2 1.1 0.6
3.9 , 0.1
265A D *.08.1 5.9 8.6 5.3 0 7.3 50 7_9 9.7 7.5 5.5 50 10.2 8.6 7.9 20 , 50 20
5.7
266A V '.95.3 7.1 12.1 20 11.2 20 0.4 12.2 20 8.8 7.1 50 12_2 20 6.1 3.8 0 20
20
267A S 2.33.5 7.2 1.3 50 7.4 20 20 _0.7 1.4 3.9 ,.7 0 2.3 3.i,3.020 20 50 50
68A H 1.21.9 2.2 1.5 3.7 5.0 4.9 0.4 0.5 3.7 2.7 1.7 0 -1.4 1.7 1.1 0.2 0.9
6.1 3.7
_. .
269 A E ,0.31.941.3 0.5 1.343.7-1.941.1 0.8 1.2 2.5 0 50 0.6 0.8 0.2 0.6. 0.7
4.01.0
270 A, D 0.22.6 2.1 ' 1.95.2 3.93.1 2.1 '1.2 0 2.2 1.9 20 11.9 1.8 1.2 1.7 4.1
5.1 7.0
271A P 0 5.38.1 9.3 20 3.1 9.1 20 6.0 9.5_ 5.3 7.3 5.9 _5.9 5.9 1.6 4.1 15.2
20 20 "
272A CI 0.81.9 0.9 1.2 3.0 3.23.7 3.7 1.6 1.8 3.2 0.3 50 1.1 1.6 0 _1.0
3.5,4.0 3.4_
273A V1.22.91.8, 20 20 7.1 20 6.8 20 20 , 20 0 2.8 20 20 2.11.4 1.7 20 20
274A K 0.41.8 1.4 0.8 ,1.9 3.9 2.4_1.4 0.7 1.1 2.9 1.9 20 0 0.1 0 0,40.7 3.3
2.3 .
275A F 8.09.510.3 9.5 0 _13.5 5.1 10.1 6.2 6.3 6.0 *.1 6.1 9.1 15.19.67.2 6.1
13.5 4.3-
276 A N 1.32.4 2.4 2.2 0.85.1 0.8_ 1.20.6 2.3 _2.5 1.8 50 1.6 2.5 1.2 0 '0.3
4.24_3.6'
277A W_5.57.4,8.4 6.4 15A 11.2 3.28.2 1.9_3.9 3.6 ..6 3.5 5.5 15.46.9_6.1 14.1
0 20
278-A Y 1_62.7 3.-9 1.6 _1.0_ 7.33.417.71.4 7.5 2.1 0 50 1.9 2.22.6 9.9 20
15.8-1.4
279A V 3.14.1 4.0 2.2 20 8.1 9.7 8.5 0 1.4 3.1 .320 1.9 4.6-4.3 3.4 4.2 20 20
280A 0_1.82.6 2.7 0.2 11.5 2.9 8.8 20 3.4 3.2_28 .8 50 0 3.7 0.6 618
12.711.911.4
281 A G 50 50_ 56 50 50 0 50 56 50 50 50 50 50 50 50 -50 50 50 50 50
282 Al V 0.92.1 1.6_ 1.1 2.9- 4.2 3.5_1.4 1.5 1.8 3.6 1.418.9 0.5 1.0,0 0.6
0.9 ' 4.7 3.1
283 Al E 0.71.6 0.70.5 1.0 4.4 1.4 0.4 1.2,1.8 1.9 0 0.4 , 0.6 , 1.5 .4,0.3
1.2 4.1 0.9
.
.
102
CA 02766627 2012-01-23
t.2620-20
(284 Al V 012.2 3.1 t2_ 20 15.0 20 1_4.010.7 2.6, 0.8 2.6 50 _0.8 0.7 -0.810.1
-1.5 20 20
103
CA 02766627 2012-01-23
- 52620-2D
Table 55 (continued)
PoswItJACDE=F GH 1 K LMNPQRST VWY
+65 IRENE 20131 3.61112.6 3.0 0.7 ii 0.2 0.8 mai 4.9 4.0
r 86 , 4 4.7 2.1 0.7 1.8 0.6 1.2 0.7 I=
-- 52 2.7
OE A 32 2.6 5.8 m10.4110 9.1 EKE 0 Ego 3.6 50 2.6 gem 1.9 031 9.1 10.4
88 = IN 2.6 2.011 3.0 3.4 3.8 El 1.4 gil 2.5 0.3 50 Bei 1.3 0E12.0 4.5 3.6
89 ' I 1.9 4.7 3.1 3.6 2.9 0.4 1.6 2.1
8.2 2.0 0 = 12.0 3.9 3.2
mmHg 0.5 0.6 3.0 OD 3.0 muzugupg 0 50 0.7 2.0 mom 5.6 I03
91 = P = 3. 1.8 0.5 1.9 1 8 0.1 1 0.7 0 2.9#
0.9 .3 2.6 0.9
+92 = R 2.2 3.1 0.8 5.9 8.0 5.0 = 8.4 0.2 0.4 1.3 4.
8.3 5:
93 ' E + 6.5 9.0 17.9 6.3 I 13. 50 : 5.5 15.114.5 20 50
14.517.1
94 ' E 1. 2 2.1 0.7 8.1 2.8 3.3 2.0 2.6 1.8 2.8 1.0 50 1.3 1.3 I 0 3.4
11.210.2
r 96 , Q 50 50 50 50 50 0 50 50 50 50 50 50 50 50 50 50 50 50
50
+96 = Y '.8 2.3 1.1 0.4 50 0 50 4.6 2.2 2.3 3.1 0.9 50 0.2 1.8 4.7 4.8
18.2 20
+97 ,N 0 6.5 8.4 5.3 20 3.4 20 13.82.7 20 4.8 9.3 50 4.4 4.4 L6 15 20
20
+98 ' S I. 2.4 5,7 2.2 20 3.7 20 6.2 0.9 9.2 1.8 3.3 50 1.7 2.1 2.2 7.3
15.6 20
+99 = T 1.9 3.4 6.0 3.1 1.0 7.1 2.9 3.1 0 2.7 2.6 3.6 50 2.2 2.5 2.2 5.4
3.6 1.4
4 00 ' Y .: 2.9 2.7 4.5 20 4.0 7.5 13.1 1.2 0 2.2 2.3 50 3.3 4.0 . 1.1 1.1
11.0 2.4
4,01 , R 14.0 3 5 3.8 2.8 0.8 3.4 1.8 0 1.3 0.72.6 2.5 50 2.6 2.3s * i .a 0.9
9.8 1.8
02 , V +.7 4.6 6.7 3 9 2.8 8.9 1.2 6.9 2.7 2.0 2.2 4 8 50 4.7 3.2 , 7.73.8 0
8.4
SE V 0 NI 3.3 1.0 6.7 4.5. 5.3 14 ill 3.1 Mi2 3.1 1.0 2 2.9$R 2.9 iii 6.2
, S 0 10.820 20 6.2 20 20 20 *16.. 50 20 16.6
17_9 20
EMEIMOIMEINE 0-3 EOM 0 0-9 lal 2 8 El 3 9 NOME 0 9 0 0.8 0.8
Il 406 = 6.2 7.1 5.9 2.8 10.4 3.4 13.7 3.0 0
6.0 50 5.9 . 5.3 11.4 9.6 10.3
07 = 3 2 3.8 2.2 6.5 5.5 4.2 0.5 0.3 4_2
12.2 0 1.9 0.9 1 2 6.2 6.5
408 5.5 6.5 8.0 50 7.9 20 4.5 20
5.5 == 7.6 50 7.7 I 0.7 5.9 50 50
09 L 1. 2 7
0.7 0.7 1.3 4.6 2.7 0.7 1.7 1.0 2.8 0 1.6 0.7 1.3 I 0.6 0.5 5.0 2.1
H +.0 2 6 0.9 4.1 50 5.6 0.2 6 8 4.0 7.1 4.0 0 0.2 4.9 10 0 2.5 6.4 50 50
1 . Q I .
2.5 1.6 .6 2.5 4.3 1.6 1.4 0.6 0.9 2 9 0.9 .7 0.8 0.9 I 0.3 2.2 4.6 2.0
12 N .4 5.1 5.9 1.3 20 0 20 10 3.4 4.8 3.3 7.1 50 2.7 3.9 , 3.2 11.9 20 20
13 W = 6 6.4 5.5 5.6 1.1 10.8 5.0 11.1 5.8 5.2 7.6 5.4 50 4.8 12.9. I 3.8 6.6
0 2.6
14 L + 1 2 9
4 3 2 2 5 7 6 7 9 5 4 0 7 0 1.7.2.3 50 1 6 1 6 4 4 7 6 3 8 0 6 0
315 D 4.3 1 4
1.5 0 1 3 3 4.2 1 9 .1 8 0 8 0.5 1.8 0.6 50 0 0.7' 0.9 2.4 6 2 3.7
316 G 50 50 50 50 50 0 50 50 50 50 50 50 50 50 50 I 50 50 50 50
4 17 K 0 14.1 8.417.9 50 5.0 50 20 8.5 12.512. 20 15.917.213.5+ 9.2 20 50 50
18. E +.0 3.0 2.7 1.7 2.7 6.7 2.6 0 1.1 1.6 1.3 1.7 20 1.4 2.6' 1.3 0 6.1
9.5
319 Y .9 4.4 3.9 3.4 0 8.8
1.8 20 0.5 5.2 0.7 3.2 50 3.1 5.6 3.6 20 20 0.2
Emma 3.0 su B 7.8 20 9.4 0 0.6 Eism 50 2.4 1.9 maim 20 20
00LiEl 0 m 20 18.8 6.9 20 20
20 20 10.4 20 50 19.6 20 EE 8.7 18.3 20 20
i 4 22 , K + 0 2.5 3.5 2.8 2.7 6.4 2.1 0.2 0.1 1.2 2.7 2.7 50 2.1 0 1.6 0.9
14.' 2,8
323 , V 1. 2.8 7.3 11.9 20 8.1 20 6.0 9.6 20 4.9 8.5 50 13.6 20 1.6 0 20
20
24 , S 9.0 2 1 0.6 0 1.9 4 9 3.9 1.5 2.8 0.7 1.9 0.9 50 0.8 2 9 + 1 9 2 1 3 8
2 5
425 ''N +.8 3 9 8.4 3.0 20 6.3 20 0 7.7 20 6.2 1.6 13.4 0.5 20 0 .3 20
20 .
26 ' K 1.0 2.7 3.0 1.6 3.7 4. 3.1 3.2 .7 2.43.7 1.2 0 0.61.4 0 1.9 2.6 5.6 3.6
27 , A I.' 2.8 5.8 3.1 20 6.3 16.714.7 2.8 20 2.5 5.3 20 1.3 4.1 0 5.2 13.7 20
20
328 , L ,..4 6.3 7.0 4.1 50 8:6 20 50 5.7 0 7.1 6.0 50 3.7 8.2 ... 50 50 20 50
329 = P 1.4 2..5 0.9 0.6 4.0 3.4 3.3 1.7 1.9 2.5 3.6 0 0.3 0,7 1.5 4.1 1.1 1.1
6.2 3.6
330 = A I.' 2.0 1,3 0.7 3.4 3.8 3.0 2.0 1.4 2.0 3.4 0 20 0.6 1.2 1.2 0.6 1.9
7.03.4
31 , P 50 50 50 50 50 0 50 50 50 50 50 50 50 50 50 50 50 50 50 50
332 = 1 1.9 3.7 4.6 1.7 5.0 7.0 1.9 3.8 1.8 0 2.5 3.9 20 0.8 2.4 .3 2.6 4.4 20
5.9
333A E 0 3.1 3.2 0.8 4.1 4.4 4.2 16.9 3.6 2.8 .2.8 2.5 1.6 1.3 3.2 1.3 1.4
7_7 4.0 4.8
104
CA 02766627 2012-01-23
.52620-2D
334tJ K 1.712.912.51 0 1.0161 3.3_1.011.5 0.5[3.5 1.5 4.4 0.1 2.72.2 0.9 1.3
4.9 1.13,
105
CA 02766627 2012-01-23
' 52620-2D
Table 55 (continued)
PosVVTACDEF GH I K LMNPQRSTVWY
335 A T 1.5 3.2 ,. 2.7 42 4.9 4.1 20 2.1 3.1 3.0 1.2 0 2.3 2.8 1.4 1.4 7.3 5.1
4.5
336A =i 1.5 20
IN 6.8 0.7 3.4 7.8 2.5 32.20 2.8 1.4 0.7 0.6 0 20 20
337 BBE 11.5 10.1 5.5 50
9.9 7.0 7.9 5.0 50 11.4 12.7 4 5 2.3 50 19.3 10.6
338A K .0 2.7 2.3 2.2 4.6 5.9 2.4 50 0 2.1 1.9 1.0 50 1.5 0.9 0.7 10.3 50 5.4
4.9
339A A 1 0 2 50.8 1.1 4.4 3.7 3.7 2.1 1.8 2.6 3.6 0 0.8 0.6 1.6 '.6 0.9 2.4
6.8 3.8
340A K 1.3 2 42 3 2.0 1.7 4.1 2.3 1.9 0 2.3 1.8 1.0 1.9 0.9 1.3 0.5 0.8 1.7
4.9 2.4
2328 P 1.3111 2.2 4.1 2.9 3.6 1.8 2.110 3 . 9 1 1151g1 1.6 0.7 1.4 3.0 6.2 4.1
2338 E 0.5 -0.5 2.6 3.7 2.9 4.4 1.4 3.2 0.6
1.6 0 1.2 6.9 5.5 2.6
1 2348 L 2.9 = 4.8 4.9 2.0 6.1 0.8 1.5 0 1.9 2.7 2.6 20 3.6 1.2 3.1 2.5 3.4
13.4 0.5
2358 L 0.6 2.4 0 9 5.7 4.91.4 3.7 0 0 9 1 9
17.3 0.6 1 4 # 8 0 7 5.2 7.8 5.3
236B G 3.6 5.1 11.8
6.8 0 2.8 20 5.0 20 4.5 3.5 50 5.5 19.9 .6 20 20 20 4.1
237 B G 20 50 50 50 0 50 50 50 50 50 50 50 50 50 50 50 50 50 50
2388 P 3.5 ,.7 8.5 4.2 20 9.8 20 0 5.6 9.6 4.6 8.1 1.3 5.8 20 ,.= 4.4 1.3 20
20
2398 S 1.0 . 3.4 2.0 7.2 5.7 3.1 3.1 0.6 0 2.0 1.9 50 1.7 1.1 1.5 1.5 5.2 20
5.2
240B V 1.1 2.3 7.0 11.9 20 6.5 20 8.1 12.7 20 12.07.6 0 11.6 20 1.2 1.9 0.8 20
20
241 B F 0 2.0 1.4 0.8 1.0 4.0 2.0 6.5 1.1 0.6 2.3 0.2 50 0.3 .1.5 0.1 0.9 5.7
4.1 1.1
2428 L 2 23 36 5 6.6 6.9 7.9 4.3 0 8.7 3.9 4.8 5.3 0 9.1 6.8 2.9 1.1 0.5 20
8.7
2438 F 0.8 2 6 1 9 1.7 0.8 4.9 2.0 3.6 1.2 0.8 2.5 0 50 1.6 2.7 0.1 1.8 3.9
4.3 1.0
244B P 1.12.1 , 0 1.1 11.9 3.5 5.4 20 1.4 3.2 3.0 2.0 1.8 1.2 1.3 0 19.6 20
9.1 11.0
245 B P 3.2 20 20 20 20 8.6 20 20 20 20 20 20 0 20 20 . 0 20 50 20 20
246 B K 0 52.6 1.4 1.2 2.1 4.41.6 1.6 0.6 0.9 1.4 2.5Ø2 0.3 0.2 0.3 '.1 0
2.0 4.9 2.4
2478 P 0.8 2.5 4 .7 1.0 3.6 3.9 2.6 6.2 1.8 2.1 2.9 6.3 0 0.8 1.5 6,3 0.7 9.
6.6 3.4
2488 K 0.2 2.2 4 2 0.6 2.2 4.1 2.5 2.4 1.7 1.0 2.2 0 1.3 0.8 1.7 6.5 O. "
.84.7 2.3
249B D 2.8 3.3 0 4.6 10.1 8.2 6.5 50 4.6 6.2 4.4 I. 50 4.7 6.3 3.5 6.1 50 20
7.2
250 B T 0 2.24.9 2.8 20 6.3 20 2.2 4.3 3.2 3.0 9.2 50 3.4 4.9 1.3 2.3 3.1 20
20
2518 L 0 2.41.6 1.2 5.6 3.6 2.2 7.4 1.2 0.6 2.3 0.5 50 0.6 1.8 0.4 2.5 8.7 8.2
5.9
252B M 1.32.40.8 0 1.8 5.7 2.3 0.6 1.6 0.6 2.5 1.0 50 1.0 1.6 1.5 1.3 0.8 5.1
1.6
253B I 1.63.02.0 1.2 3.7 4.5 3.5 2.9 0.8 2.4 2.4 1.1 1.0 0 1.5 1.2 1.4 3.4 4.4
3.6
2548 S 1.0 1.50.8 0.6 3.8 3.8 3.2 0.5 1.9 2.5 3.1 0.3 6.2 0.5 1.7 0 0.1 1.1
5.5 3.7
2558 Ft 0.9 2 02.0 1.7 0 5.4 1.4 20 1.0 1.6 1.3 0.8 50 1.4 1.1 1.5 20 20 3.7
0.8
256B T 0.6 2.0 1.8 1 2.5 3.7
1.9,1.6 1.0 1.4 2.2 0 1.2 0.5 0.1 1.8 1.2 1.2 5.5 2.4
25 'TB P 2.5 20 20 20 50 9.0 50 20 20 20 20 20 0 20 204.820 20 50 50
2588 E 1.52.4 . 1.4 2.7 6.4 4:2 0 0.2 5.4 2.4 1.1 50 1.3 2.52.2 1.1 1.019.1
3.0
259 BElliog6.3ng 20 9.3 20 0 8 8.9 m5.6 50 6.2 20 , 5BEIM, 20 20
260 BEI am 1.9 20 4.9 20
0.6 not 2.8 1.4 3.9 0.2 ER 2.6 0.4 0.1 20 20
261 B 20 20 2.6
20 20 20 20 20 20 20 20 20 2.0 16.6 20 20 20
262 BEIM i 2.4 8.1 7.2 3.8 1.8 3.5 8.6 3.4 2.7 50 3.2 4.8 2.9 1.9 0 14.7 9.1
.
263 8 V 4.7 11.2
20 9.1 20 15.0 13.7 2.8 20 5.4 50 13.0 20 3.6 2.1 0 20 20
2648 V 4.6 2.7
8.6 6.8 6.6 0 1.8 1.8 3.7 3.6 10.1 3.0 2.2 2.6 2.2 1.0 12 7 20
265B 6 4.8 4.7
5.0 0 3.8 8.5 .8 2.6 4.1 1.8 50 4.5 5.3 4.5 6.0 9,212.2 5.6
266 B V 7.2 12.7
20 12.0 20 2.1 20 20 20 5.7 50 18.3 20 5.9 4.7 0 50 50
2678 S 6.2 3.8 0
7.4 1.0 50 1.0 0.3 3.2 3.2 0.5 1.5 0.8 3.3 11.6 50 6.3 50
268 B C12.6 go 4.1 4.9 6.0 1.8 2.6 0 2.5 3.8 2.6 3.4 HD 1.8 3.8 7.8 mg
2698 0.42.4 1.7 0.8 2.8 2.6 1.0
1.0 1.6 3.0 0 12.8 0.5 0.7 I 0.7 I . 5.1 2.7
270B = 0 1.6 1.1 7.3 4.8 2.6 20 3.8 14.5 3.8 1.2 5.9 6.3 2.1 0 1.9
16.3 5.6
271 B 1.13.3 5.6 3.4 4.1 4.2 20 1.9 3.6 3.9
3.3 7.4 2.7 0 2.2 4.8 4.4
272 B = 0.9 1.9 1.0 0.6 3.0 3.9 ISIMIHREEM 4.9 0 1.4 ES 0.6 1112 3.9 mg
273B V 1E4.8 6.2 8.3 20 9.2 4.6 E87.4 50
10.6 20 2.0 0 MC 20 20
= 106
CA 02766627 2012-01-23
. 52620-2D _
Table 55 (continued)
Pos ACDEF GH 1 K LMNPQRST VWY
2748 K 011.60.4 0.9 1.7 3.8 1.8 1.9 0.4 0.5 .40.3 15.60.1 o 0 0.2 1.6 2.2 1.9
2758
F 5.7 .0 8.4 9.2 0 11.2 3.5 9.2 7.9 5.7 7.0 4.1 9.7 12 3 . 9 4.5 3.3 10.3
5.0
276 B N 0 ..2 6.9 6.4 20 4.7 2.1 20 9.3 10 3.8 50
6.4 9.2 2.8 20 20 20 20
2778 W8. '10 10.6 9 2 2.6 14.2 7.4 12.7 6.7 7.4. 10.8 6.8 9.3 11.9 9.7 8.0
14.4 0 15.9
278 8 Y o . 17.4 4.0 50 5.1 50 20 2.8 20 12.6
11.04.4 2.0 1.8 2.5 19.8 20 4.2
279 8 V 3.13.5 4.2 2.9 20 8.5 13.9 0.4 0 2.9 3.4 20
1.4 4.0 , .2 2.4 1.2 20 20
280 8 0 0.53.0 2.1 1.5 6.7 3.1 4.7 12.6 2.9 1.6 1.6 20
1.4 3.1 0 2.7 5.5 8.1 7.3
281 B G 5 6 5.8 5.5 4.8 7.9 0 7.2 6.5 5.3 5.7 3.4 50
4.1 5.3 3.6 3.2 6.4 10.37.6
282 8 V 1 = 1.1
0.6 2.9 4.1 2.1 1.3 1.0 1.4 .9 0.2 50 0 0.7 0 0.4 0.7 6 2.8
283 13 E 1 = 4.3
1.7 6.7 4.2 5.2 2.9 0.5 4.4 '.3 2.5 0 1 1.6 1.0 1.5 3.9 7.9 6.7
284 B V f 2.5
1.1 20 5.9 20 1.1 1.2 6 2 1.8 2.4 50 1.5 3 3 0 1.5 1.8 20 20
2858 H 2.1
1.7 2.4 3.4 1.2 1.8 0.7 2.3 .7 0 1.6 0.9 0.8 0.5 Ø4 20 5.8 2.4
286 8 N 1.0 1 3.0
3.1 2.6 0.8 2.0 1.9 2.9 0 50 0.4 1.6 1.1 0.5 2.9 4.9 3.0
287 8 A EgE 6.1 EEll 0 82 3.0 10 2E1116.54 5 0.3 ngii8 1 9.1 mum 3.4 0.8
288 8E10.41 9 1.9 0 2.9 3.5 2.9 2.5 1.8 ME 0.9 15.4 0 611m 0.9 3.8 5.9
289 8 0.11.5 3.7 1.4 2.7 3.9 2.6 1.8 0 ii
1 5 1.8 1.1 0.4 2.3 3.4
29013 1.9 0.8
0.5 2.4 0.8 2.7 3.0 1.3 1.3 = 0.2 50 0.7 1.5 0 0.6 2.9 5.0 2.7
291 13 1.2 2.5 0.5 3.9 4.6 3.4 0.7 0 3.4 1.1
0.6 0.1 1.1 1. 0.9 1.5 3 2 2 6
292 8 1 1.8 = 3.3 1.2 4.9 3.6 6.8 3.1 2.0 2.4 2.2
16.61.5 1.8 0.1 0 3.2 7.6 5.2
293 8 0 I 4.1 2.8 7.3 3.6 5.8 5.8 2.6 4.5
2.2 1.3 2,2 2.5 0 1.2 7.8 7.0 6.9
294 Bum" 3.9 811 8.3 6.8 4.4 5.6 3.6 Egag 4.1 0 ai 5.0 2.1 2.9 5.0 6.7 11.9
295 8 Q 1.9 0.6 2.8 3.1 8.0 1.4 2.2 3 4 1.0 0.4 I 1.1 0 3.9
6.6 6 3.5
296 8 Y 1.2 1.2 4.1
3.5 1.1 1.8 2.7 3.5 0 20 I= 1.9 1.2 1.4 1.3 6.4 4.0
297B = 1 10.1 6.0 7.3
16.7 6.6 0 5.1 4.6 7.3 20 4,2 3.6 4.1 7.9 18 0 15.0
988 S 3.5 2.5 3.7 2.4 3.0 0 1.8 2.3 0.4 50 1.0
0.9 2.2 3.3 5.5 2.0
2998 EIE/1 11.1 20 4.8 7.5
6.9 20 1 4.8 50 9.8 17 9 0.3 1.3 5.8 EI 20
300 B 4.3 20 8.6 12.2 0 4.3 6.5 50 4.0 3.8
4.3 3.6 9.1 6.4
3018 R De1.8 1.1 20 5.8 mg 0.3
5.0 2.0 1.6 14.10.6 0.4 1 8E1417.9 20
302 1311114.81DEEll 0.2 9.6 pn 0.5 2.6 memo 9.6 SE 0.6 DB 20 0.2
303 BE= 0 0.1 1.0 20 5.0 IBEE11116110 43: 2.0 8.6 EC 4.7 10.5 ign 20 20
3048EN 8.2 20 20 7.6 20 7.6 20 20 20 6.3 50 Ijii 20 0 27 3.8 20 20
3058 1 3.3 .1
20 4.6 20 3.2 1.1 11 0 1 5 1.8 50 1 = 2.0 0.6 0 0.7 20 20
306 8111116.8 6.3 11110.4 11.1 7.8 4.2 3.0 0 3 81113.41114.111 4.3 6.0 20 12
3078 3.0 2.7 4.1 5.2 3.0 1.6 1.9 3.1 3.4 0
1.9 1.4 2.0 4.4 4.3
308 BEI 0 0.6 7.6 20 20 6.6 20 EI16.1 15.1 20 12.4E1 20 20 1.2 3.6 4.3 20 20
309B 1.43.0 2-.2 1.1 3.0 6.0 3.5 2.4
1.7 3.6 0.2 = 1.6 2.3 1,814.3 20 5.1 3.3
310 BIE16 2 9ffl 4.9 20 6.8 4.4 4.8 3.1 15.01 0 gam 7.0 1.8 1.6 3.8 20 20
311 8 Q=
pg 0.7
MEE 2.4 12.6 0.6 0.9 E 0.6 ME 0.8 0.21E18.8 4.6 2.0
el
312 B 0 1.0 0.2 0.3 6.0 5.4 eg12.0EMI 2.9 6 0.9 EDE=
3.8 8.0 7.8
3 3 B W 6.6 7.3 5.4 0 6.2 20
4,0 5.2 4.3 8Ø 50 .. 8.9 6 6 17 2 20 2.1 0.9
3 4 B L 2.2 3.1 0 6.4 = 1.5
2.1 0.6 0.2 1 1 9 50 0.8 1.0 1.7 0.9 .3.1 3.7 11.3
..
3158 D'2.3 2.4 0.7 6.0 2.3
4.8 2.2 1.0 2.9 1.8 50 1.02.2 4.2 0 4.5 8.5 6.8
31.6 B G I 50 50 50 50 50 50
50 50 50 50 50 50 50 50 50 50 50 50
3178 EINIII 2.8 1.2 111 0.6 13.9 0 4.8 1.6161511 0.9 113.8 10.1 20 Fli
3188 1 1.0 7.0 8.2 0.4 0 5.7 1.7 1 1.6 1 0 1.0 3.8
319 Bum 8.5 8.8 0 gm 3.9 meg 5.4 Egg 50 9.01ME 5.8 3.9 20 re
320 8111804.383 4.3 20 8.6 15.0 1.4 0 11.63.6 6.6 50 2.9 2.4 4.0m2.0 20 20
321B C 0 6.5 20 20 20 6.6 20 20 20 20 20 20 19.7 20 20 3.1 11.2 20 20 20.
107
CA 02766627 2012-01-23
-2620-2D
Table 55 (continued)
Pos 'WI' A C D F G Fi 1K L MN PQRST_V W Y
3228 K 2.3,3.23.5 1.8, 20 7.9 20 1.1 0.6 4.9 3.7 2.2 50-0.9 0.3-3.3 1.6 0 20
20
3238 V 4.04.66.9 8.1 20 10.620 9.0 17.17.98.1 10.550 8.7 20 5.6 4.6 0 20 20_i
325B N 3.45.19.0 4.7 20 8.2 20 16.6 16.6 20 20 _ 0 50-'6.3 20 4.6 8.8 17.8 20
20
3268 k 0.32.12.0 0.9 ,1.0 3.5 2.0- 2.9 0.9 2.9 2.8 0.1 4.4 0_41.1-0.1 3.2 2.1_
5.2 0.7 =
327 8, A 1.93.34.7 3.5 20 7.0 20 20_ 0.3 0. 1.9_ 1.9 203.0'243.3 20 20 20 20_
328 8, L 3.73.63.8 4.4 50 8.4- 7.0 50 3_8 0 2.6 4.0 50 4.28.7_,-4.8 2_9 12.3
50 50
3298 P 3.38.5 20 20 50 8.0 16.550 18.5 20 14.7 20 0 20201.417.116.450 50
33013 A 0.52.02.8 0.5 2.4 3.9 1.2 -4.0 0 2.0 2.1 0.8 20 0 0.50.8,0.2 4.6 8.2
2.6_
3316 P 1.73.86.4 10.-i 20 4.7 11.010.1 7.5 20 5.5 5.0 0 7.67.4,2.6 20 10.117.6
20_4
332B 1 1.72,91.3-1.7 14.87.0 1391.7 3.1 0 1.7 1.7 -501.8,5.32.0,1.9 3.4. 20 20
333B E ,1.92.51.9r 0 -8.9_5.98.2-1.2 3.0 6.4 3.4 2.0 l-3.1_1.1,2.3,1.6 1.6
_1.6 8:0 9.3
334 fi K 20 8.3 12.1 1.5 2.6 5.3 3.714.3 , 50 1,9 0 3.4 1.8 1.4-,
9.9 20
335B T 02.17.2 7.04.2 0.4 3.3 17.3 6.5_7.7 5.2 5.5 3.5 7.05.7 0.2 5.5 11.5 5.2
3.1_
336B I 0.51.62.1 0.7 20 :5.0 6.1 0 -1.3 5.3 2.1 1.8 200.63.11.1 0.8 0.7 19.4
20
337B S -1.12.14.0 2.0 3.1 _3.2 2.0 50 0 1.60.9 1.9 15.8-1.12.21.4, 50 50 5.5
3.9
3388 K0.62.33.0 3.0 94 5.3 10.6 2.2 _1.1 0 3.2 _1.5 16_22.72_71.1 2.813.5 8.1
11.61
3398 A 1.12.41.2 0.8 , 4.3 3.6 3.7 2.6 1.8 2.6 3.5 0 2.3 0.51.i0.2" 0.8 2.0
6.7 3.8
340 8 K O.92.01.40.8 3.0 3.4 2.9 2.1 0.8 2.5 2.3 _0.2 1.00.11.2 0 0.5 2.0 5.5
3.2
SPAT"' technology; 11IS=template structure; - carbohydrate, no floated
positions
=
=
108
CA 02766627 2012-01-23
b2620-2D
Table 56
Pos1NTACDE F GH I KIM N P QRSTV W Y
239 A S 0.24.621 0 20 4.6 14.5 11.0 1.9 0.3 2.0 1.9 8.1 1.4 2.6 0.4 5.7 11.6
20 20
240 A V 1.52.42.4 6.9 20 7.4 20 5_1 9.9 59 5.5 2.4 1.1 12.3 13.1 2.60.5 0 20
20
263A V 2.3 2.8 6.3 16.5 20 8.8 20 9.6 7.3 7.3 15.3 4.8 50 16.4 17.4 2.8 1.4 0
20 20
264A V 1.83.1 .6 1.8 0 6.3 1.9 0.6 2.4 0.8 2.7 2.1 1.6 2.3 2.7 2.3 1.1 0.5 3.5
0
266A V 4.9 5.2 6.9 12.3 20 11.1 20 0.8 11.920 8.5 6.6 50 12.5 20 6.1 3.7 0 20
20
296A Y 3.42.7 1.1 0 50 0.7 50 5.0 3.6 3.5 4.2 0.9 50 0.9 2.9 2.2 5.3 5.5 16.1
18.4
299A T p.73.29.9 10.4 20 6.2 20 10.7 6.7 20 4.1 12.9 50 5.9 11.8 0 2_5 8.2
13.3 20
325A N 2.5 3.5 7.7 .2.5 20 8.0 20 0 6.1 20 7.8 1.2 12.8 0.8 20 2.7 0 1.0 20 20
328 A L 6.1 6.3 7.1 4.2 50 8.8 20 50 4.6 0 7.2 6.1 50 4.0 8.3 6.7 50 50 20 50
330A A 0.9 1.81.2 0 2.5 4.0 2.9 1.7 1.2 1.6 2.8 0 20 0.4 1.0 0.20.5 1.7 6.2
2.9
332A I 1.93.84.6 1.3 5.1 7.1 1.8 3.4 0.2 0 2.6 3.8 20 0.6 2.4 2.3 2.5 4.2 20
5.6
2398 S 1.02.43.5 2.0 61 5.6 2.9 3.1 0.3 0 1.9 2.1 50 1.5 1.8 1.41_4 5.2 20 4_2
2406 V 0.3 2.46.9 11.7 20 6.6 20 8.3 12.3 20 14.2 7.4 0 13.4 20 1.3 1.9 0.9 20
20
2638 V 2.4 3.94.5 12.5 20 9.3 20 15.8 17.1 .1 20 5.3 50 13.8 20 3.9 2.2 0 20
20
2646 V 2.23.24.8 2.7 7.4 6.9 6.0 0 1.9 1.9 3.8 3.7 9.9 3.1 2.2 2.72.4 0.9
14718.2
266 B V 5.4 5.5 7. 13.2 20 12.1 20 2.6 20 20 20 5.4 50 16.1 20 6.0 4.7 0 50 50
2968 Y 1.5 2.7 1.3 1.2 4.0 4.1. 3.6 1.1 1.9 2.6 3.5 0 20 0.7 1.8 1.11.4 1.3
6.5 4.2
2996 T 0 2.2 7.510.2 20 4.8 26 7.75.8 2010,3 5.1 50 10.2 18.4 0.3 1.1 5.4 20
20
3256 N 3.4 5.18.6.5.0 20 8.2 20 16.7 20 20 20 0 191 6.3 20 4.68.6 18.2 20 20
3288 L 3.6 3.5 3.8 3.9 50 8.3 7.0 50 2.9 0 1.9 3.8 50 3.4 8.4 4.7 219 12.5 50
50
330B A 0.7 2.1 2.9 0.7 2.7 4.0 1.4 4.8 b 2.2 2.3 0.8 20 0.2 0.8 1.10.2 4.7.
7.8 3.2
3326 1 1.8 2.9 1.2 1.8 13.5 7.0 9.9 1.7 3.2 0 1.7 1.9 50 1.2 5.4 2.02.0 3.3 20
20
SPA"' technology; 0129G 11IS template structure; + carbohydrate
= =
=
=
109
CA 02766627 2012-01-23
52620-2D
Table 57
PasVVTAC D E.F G H I K L.M_N P,Q R S T V-W Y
= 239A S 1.23.5 1.7 0 20 5.8 11.0 6.6 2.9 3.9 3.9 2.7 8.5 1.3 2.7 0.63.5
5.4 20 20
240A V 1.22.4 6.0 14.0 20 7.1 20 6.7 9.4 10.1 7.5 4.4 1.8 14.8 20 2.00.4 0 20
20
263A V 0 0.4 1.0 8.7 20 6.9 4.4 11.7 4.9 16.019.20.8 50 11.7 20 1.40.1 1.0 20
20
264A v 2.93.7 6.3 2.8 11.6 7.6 13.2 0 3.2 3.4 4.1 4.2 7.1 2.9 3.4 3.1 1.9 0.8
12.8 16.3
266 A, V 4.85.9 6.8 9.5 50 10.3 20 3.5 12.7 12.2412.7,4.1 50 )1.9 11.9 5.2 2.9
0 50 50
296A Y 0.82.0 1.5 0.1 0.2 3.4 1.5 6.6 1.7 0.6 1.8 1.2 2.6 0 _1.6 0.22.5 5.6
3.8 0
299A T 1.93.7 7.5 0 20 7.9 14.2 2.9 0.8 3.4 4.4 2.3 50 1.9 3.0 3.54.1 3.3 20
20
325A N 1.01.4 3.1 2.8 20 7.4 20 8.5 7.7 10.4 6.1_2.8 15.4 5.4 20 0 0.1 3.8 20
20
328A 1 2.55.3 4.0 1.9 50 7.5 20 20 1.6 0.2 0 2.9 50 0.4 4.83.22.9 7.0 50 50
330A A 0.92.1 1.8 1.2 2.4 2.7 3.1 3.1 1.4 2.1 3.5 0.5 20 0.8 1.0_ 0 0.5 2.9
5.2 2.9
332A I 2.93.7 3.9 0.9 6.1 7.8 2.5 0 2.7 0.8 2.8 3.5 50 0.7 3.7 2.92.5 1.0 8.1
6.9
2398 S 1.93.1 3.0 1.9 1.5 672 2.3 14.1 1.8 1.4 2.9 1.8 0 1.9 3.2 1.92.3 7.7
6.6 15.8
240B V 0.5 1.7 5.0 13.3 20 6.6 20 1.2 12.4 12.1 8.8 4.6 6.3 2.0 20 1.00.2 0 20
20
263B V ,2.9 3.2, 6.4 18.2 10.1 9.2 6.9 12.8 6:0 20 10.35.7 50 17.5 20 3.242:2
0 20 20
2648 V 2.93.6 4.4 3.0 8.8 7.1 6.2 0 2.3 1.9 4.5 3.4 1.7 3.2 3.5 '3.5 2.0 0.9
12.0 16.4
266B V 4.44.62.6 6.6 20 10/ 20 0 _ 4.9 1.7 8,515.6 50 6.0 12.4 5.3 4.6 1.5 20
50
296 B Y 0 7.1 6.7 7.2 20 0.1. 18.6 50 7.0 2.7 6.6 6.8 50 7.2 9.3 2.3 50 50 20
14.1
299.8 T 0 3.2 10.4 6_0 20 5.5 20_15.9 3.2 5.9 4.4 6.4 50 5.7 9.4 1.21.4 13.7
20 20
325B N 1.42.5 5.0 0 20 7.0 , 20 20 1.02.2 1.0 0.3.1-.9 1.1 20 2.65.1 20 20 20
3288 L 0.4 1.3.5.6 0 50 4.5 50 50 1.9 .2.4_ 2.4 8.3 50 0.8 16.4 1.0 1.i 50 50
50
330B A 0.6 1.4 2.5 0.9 3.1 2.5 1.2 20 0 2.4 2.1 0.3 20 0.4 0.6 0 4.0 20 13.5
3.4
332B I 4.3 5.3 5.7 0 11.4 9.3 4.3 2.5 5.8 2.0 4.0 6.517.9 3.7 5.9 4.64.2 3.7
20 11.6
SPA"' technology; D129G 11IX template structure; + carbohydrate
=
=
110
CA 02766627 2012-01-23
52620-2D
Table 58
PosVYT'A.CDEFGH 1K I MNPQRSTVWY
239A S 122.3 2.2 1.8, 7.9 5.5 7.6 0.5 0.2 1.8 2.6 1.4_ 0.9 1.3_1.9 1.50.80 8.6
9.6_
240 A V 0.72.9 6.8 4.3 206.5 20 0 10.7 20 3.1_ 9.1 2.1 7.7 20 1.4 1.1 2.4 20
20
263 A V 1.12.9 4.6 18.8 20 8.4 5.8 15.1 2.3 14.5 2.1 3.2 50 20 15.0 3.6 1.2 0
20 20
_
264A V 2.73.3 3.6 1.5 13.96.7 5.9 0 2.3 4.9- 3.7 3.2 1_9 2.5 3.0 3.0 25 0.7
'19.9 19.0
266A V 3.53.5 5.7 12.4 20 10 20 5.7_ 6.3 7.8 7.4 5.2_ 5016.6 20 4.2 1.7 0 20
50
296 A Y 2.6-50 50 50 50 0 50 50 18.5 18.0 50 50_ 50 50 50 50. 50 -50 50 13.6
299 A4. T 0.20.7 6.6 1.2 20 5.6 9.6 16 0.8 1.5 1.8 4.8_ 50 1.0 -9.2 0 0 1.6 20
_20
325A N p13.6 7.3 2.4 20 7_7 20 20 20 10 13i3.6 50_ 0 . 20 4.69.7 20 20 20
328 A 1 0.6 0 1.5 5.4 50 1.6,50 50 3.1 4.2 9.6 1.4 50 6.9 9.50.6 0.1 50 50 50
330A A 1.92.5 4.1 2.6 4.5 ,4.1 3.0 3.2 1.0 2.7 3.5 2.1 20 2.4- 2.6 1.3 0 3.9
7.6 5.3
332A 1 42.3,3.512.2, 0.8 , 20 6.8 9.6 0 3.4 0.2 2.6 2.8 14.5 3.3 4.6 2.6
1.30.9 10.5 20
239 B'S 1.43.6 2.5 1.4 16.85.8 6.2 5.0 2.5 1.4 2.0 3.8 0.3 0.5 2.4 0 1.65.3 20
19.5
2408 V 0 2.6 12.8 18.6 20 5.7 20 12.710.4 20 8.5 15.1 3.1 20 , 20 1.00.22.4
20 20
2638 V 1.1 2.4 3.6 20 20/.8 17.7 11..8 4.5 20 6.3 3.3 50 20 _20 3.21.2 0 20 20
264B V 3.34.0 5.0 2.9 14,27.5 4.8 0 2.6 3.6 4.6 3.5 1.7 3.1 4.1 3.9 2.9 1.3
6.9 20
266 B V 2.9 3.3 4.9 11.3 50 9.5'20 20 20 7.9 15.0 4.5 50 4.9 "--20 1.9 0 3.6
50 50
296 Y .8502 50 50 50 0
50 , 50 17.7_18.7 50 50 50 50 50 50 50 50 50 11.3
2998 T 0 3,812.6 9.2 20 5.9 20 _7.3 4.8 3.2 4.3 8.0 50 1.2.3 8.8 0.22.14.4 20
20
325B N 0.32.0 5.5 2.2 , 50 6.1 26 0 -10.5 15.5 14.6 1.3 10 2.4 _ 20 2.32.0 1.0
20 50
3288 L 5.45.7 7.3 4.4 50 9.8 20 50 2.5 0 5.1 5.9 50 2.8 7-4 6.1 6.4 50 50 50
3308 A 0.6 1.4 3.2 1.3 3.9 3.2 2.7 4.0 1.3 3.7 3.1 0.7 _ 20 0.6 1-3 0 0.44.2
8.2 3.6
3328 1 1.93.1 2.7 1.7 5.2 6.9 3.1 0.4 ,1.3 0 1.9 2.67.7 1.3 2.2 2.3 1.6 2.0
10.4 5.6
SPAT"' technology; 0129G 1E4K template structure: + carbohydrate
=
=
111
= =
CA 02766627 2012-01-23
52620-2D
,
Table 59
1 Pos ACDEFGH I K LMNPQRS T VWY
+39 = 1.4 2.6
3.1 1.0 20 5.7 4.8 3.4 2.0 1.2 2.6 1.. 4.8 0 2.1 1.3 2.1 3:3 13.819.6
P'46 ' 2.9 3.5 3.7 4.6 20 8.2 10.8 0 9.1 3.2 5.4 4 .1 4.8 5.5 17. 4.0 1.8
1.2 20 20
063 ' 3.6 4.9
6.2 8.7 20 9.9 20 3.7 4.2 0.5 6.7 ..1 50 9.5 20 5.1 3.6 b 20 20
064 ' 1.8 2.8
3.3 2.0 2.9 6.2 3.1 0 2.4 0.8 3.0 +.4 6.1 1.4 2.8 2.4 1.9 0.8 10.4 2.2
ME 4.4 m 4.9 El 20 10.6 20 1.0 gm 4.8 9.1 1 50 7.9 12.6 5.8 pig 0 20 20
+96. ' Y 1.2 2.9 0.7 1.4 3.1 = 2.7 2.4
2.3 1.9 2.2 = 1.6 1.4 3.0 0.9 1.0 3.5 6.0 2.6
'99 = T 0 2.6 6.0 11. 20 20 20 6.0
20 4.4 II 50 14 113. 0.9 3.8 15.1 15.0 20
4 25 ' N 5.2 7.0 6.6 6.9 50 20 1.3 14
13.513.9 I 5.0 6.0 20 6.0 4.6 3.2 20 50
4 28 = L 4.8 5.5 7.6 3.2 20 S 20 50 5.1 0 8.5 50 3.5
8.2 5.5 13 = 50 20 50
30 * A 0.9 1.8 1.1 0.9 3.5 4.0 3.0 2.3 1.2 1.6 2.8 0 14. 0.9 1.1 0.1 0.4 2.0
6.4 3.2
32 = I 5.3 6.4 6.7 4.8 8.2 9.9 5.2 3.1 0 3.6 5.2 ..8 20 3.5 4.6 5.5 4.8 4.0
11.2 7.1
+ 39 B S 0.7 2.3 2.6 2.0 5.3 5.1 3.3 1.7 0 0 2.0 1.:
15. 0.9 0.8 0.7 0.7 3.3 8.2 6.0
94013 V 2.3 3.0 4.1 7.3. 20 8.1 20 5.1 20 11 810.* 3.8 2.0 17.1 20 3.6 1.3 0
20 20
+63 : V 3.2. 4.3 7.3 8.3 20 9.6 20 13. 8.5 0.6 20 ..1 50. 8.5 .20 4.6 4.0 0 20
20
064 :, V 2.1 3.2 3.7 2.7 17. 6.6 11." 0 2.0 0.8 3.5 0 7.8 2.0 1.5 2.5 1.3 1.0
13.9 20
066 B V 5.0 5.0 5.2 16.3 20 11.4 20 2.3 20 14 317.3 .1 50 11.6 20 5.4 3.9 0 20
20
+96 : Y 0.9 2.3 1.0 0.5 2.7 3.7 2.5 1.2 1.3 2.1 3.0 0 7.0 0.4 1.1 0.3 0.8 1.8
6.0 2.4
+99 : T 1.1 2.2 7.6 5.4 20 6.4 12.: 1.8 3.9 17.5 6.9 4.= 20 4.6 10. 0.8 0 1.9
20 20
4 25 : N 10.111. 13.111.2 20 15.7 20 8.6 14. 17.1 20 0 16.110.6 20 11.110.10.5
20 20
28 : L 2.9 4.1 4.8 3.5 50 8.5 1.7 9.6 1.5 0 1.5 . 50 3.3 2.0 3.3 1.9 5.2 50 50
30B A 0.1 2.0 1.4 1.8 1.6 4.0 3.0 2.0 0.5 0.5 2.6 a 20 Ø7 2.0 0.3 0.6 2.1
4.4 2.4
_. . .
32.6 I 3.4 4.4 3.5 3.1 6.1 8.2 4.1 0 3.3 1.3 3.3 ,.I15. Ø8 2.1 3.9 2.7 1.1
20 6.1
SPAT '4 technology; Fc/FcyRIlb model template structure; - carbohydrate
= . 112
CA 02766627 2012-01-23
,52620-2D
Table 60
Pos ACDE.F GH I K.LM,NPQRS T VWY
037 G 1.0 3.0
5.9 1.8_ 5.0 4.7 3.1 1.4 1.4 4.2 3.7 2.5 0.5 1.3_ 1.5 0 1.2 1.9_ 7.5 5.6
+38 P 3.3 5.29.7 4.6 20 9.2 20 0.4 19.. 5.0 8.2 7.3 0 6.1 20 1.3 3.7 2.1 20 20
+39 S 0.6 1.8 3.3_ 1.3 1.8 4.7 0.5 2.1 0 0.8 2.6 0.2 8.4 0.9 0.5 0.3 1.32.5
4.7 1.8
+40 V 1:3 2.3
2.7 8.2 20, 7.0 20 6.1 8.7. 0 9.22.2 0.3 7.4 20 2.0 0 0.4 20 26
+41 = F 0 2.1 2.4 0.5 1.0 4.0 2.6 7.5 1.7 0.9 2.8-0.7 10 0.7 1.4 0 0.9 6.9
3.90.9
042 L 317 4.7
6.6 6.7 20 9.2 20 13.013.8 0 7.0_5.7 3.6 9.2 20 4.8, 4.7 3.4 20 20
+43 F 0.8 2.0 1.0 0.2 0.3 4.9 1.5 4.0 1.5 0.8 2.20.350 0 0.5 0.5 0.5 4.0 4.3_
0_6
+44 P + .8
3.7_5.5 3.2 6.4 2.93.6 0 0.1 4.0 3.7 3.8-0.3 1.1 0.9 1.4- 1.50.9 7.7 6.9
945A P +.5 20 20 20 20 7_7 20 50 20 20 20 20 0 20 20 3.4 19.2-50 20 _20
'46A 'K +.1 3.5 2.8' 2.5 4.0 6.5,,4.5 3.4 2.7 1.9 4.1 0 1.2 2.7 2.3 2.2- 2.5
2.9 6.8 3.7
'47A P +.4 5.0 6.6 7.8 50 7.5 20 10 9.6 _50 6.59.5 0 7.0 8.4 0.1 0.64.6 20 _50
#48 K + .0 4.0
4.8 4.2 20 _6.9 9.6 3.3 0 2.7 3.0- 4.1 2.4 3:1 3.0 2.1 0.8 2.0 15.2 20
'49 AD 4.3 4.2-.3.3 0.9 4.8 9.34.0 50 3.1 0 3.2,3.1 50 1.94.1 4.4 8.1 50 6.3
5.3
+50 A T 1.11.5 3.0 8.0 20 6.7,20 2.55.4 9.5 5.722 50 7,310.7 1.6 0.8 0 20 .20
051 A L 4.14.6 6.3 6.1 2.3 9.5 1.5 7.2,3.1 1.3 5.4 4.7 50 5.2 4.4 4.8 2.1 -2.4
50 0
+ 52 A M 4_0 3.55.3 2.8 20 9.4 20 4.4 0.6 5.0- 0 4.7 50 2.9114.4 0.9 6.7 6.0
20 20
r 53 A I = .9 1.8-3.9 3.3 4.0 5.5 6.2 1.3_ 0 0.9 1.8 2.3 50 2.6 2.7 1.2 1.22.0
5.8 4.7
+ 54 A S 0 2.4 5.5- 3.4 8.1 3.9 8.5 4.2 3.3 1.75.0 2.1 50 4.4 4.6 0.2 5.1 610
9.08.9
r 55 A R +.2 3.8 , 5.4 .3.9 7.1 7.3 1.6 15.8 0 0.5 1.5 3.2 50 2.0,0.6 2.0 1.4
1.4.5 5.4-- 20
956 T 1.1 2.3
.1.7 1.5 3.2 3.9 2.5 3.1-71.1 2.0 2.7 0 12.3 , 0.4 0.3 0.741.612.4 6.0 3.0
r 57 A P +.1 9.1 20. 16.1!50 8.5 50 20 20 20 20 20 0 18.620 4.1 2() 20 50 50
= r 58 A E 0 1.62.3 0.6 0.9 4.2 2.1 9.4 2.4 3.5 1.9 1.9 14.7 2.2 2.7 0.9
4.1 10.4 20 1.0
959J \V 4.3 7.0
11.4 20 10.4-20 3.2 14.014.910.6 7.7 50 12.2 20 5.1 .1.8 0 20 _20
960A T +.6 2.8 -3.2 0 20 7.3 5.6 1.7 3.1 5.53.5 2.7 4.5- 1.1 3.0 2_5 2.0 2_0
14.0 20
961 A6 0 18.6 20 20 20_ 3.8 20 20_ 20 20 20 20 50 20, 20 4.2 20 20 20 20
962A V 1.8 1.5_1.3 11.1_20 ,j.1_20 5.8 14.64 20 20 3.6 50. 20 16.- 2.1 1.9 0
20_ 20
#63A V = .5 5.1 6.5. 14.1 20 10.620 8.0 6.113:2 10.9 5.7 50 14.418. 6.0 3.6 0
20 20
964A V +.6 3.2,3.7 2.0 14.0 7.0 9.0 0 2.6 2.5- 3.8 2.3 10_4 1.9)2.4 2.6 1.6
0_5 15.918.-2-
65 µ' D 1.4 2.8 2.7 1.6 1.8 2.5 2.2 18.5, 0 0.1 2.5 1..1 50 0.5 0.8 1.6
11.8)9.0 4.9 2.1
r 66 A V = .8 5_1 5.0 14.2 2011.5 20 0.2 20 9.8 20 5.4 50 19.9'20 6.34.2 0 50
20
+67 A S '.64.9 '5_7 3.3 h-1.6 7_2 2.9 0.5 , 1.0 2.0 3.7 3.7 0 2_2 1.5 1.9 1.3
2.6 5.6 2.3
968A H 1.5 1.9 2.62.6 414 4.82.7 1.7, 0 2.8 1.9 0.6 0.5: 0.9 1.0 0.7 1.8
1.67.3 4.6 .
969A E 1_3 2.0 0.8 0.6 2.8 3.62.7 1.51.6.1.42.9 0 6.8 0.5 -1.5 0.1 ,_0.3 -1.3
5.3 2.7
'70A N 0 1.4 1.9 3.8 0.7 3.9 2.2 6.8 -2.4 3.5 2.7 1.6 13_3 3.2 3.0 0.9 1.2 5.1
2.2 1.0
971 A P 1.9 2.2. 5.7 6.5 3.0 5.5 2.9 12.85.4 15.15.6 4.2 0 8.014.2 1.3 2.97.5
4.3 2.6
972A E 1.7 1.8 _0.3 0.4 3.0 3.6 ,2.6 1./ 1.4 2.0 2.9 ,0.3 11.8 0.2_ 1.6 0 _0.9
_1.2 4.4_ 2.8
973A V 4.1 4.0 2.1 15.5 20 8.9 20 0.5 10.6 7.1 12.4 1.2 50 20 20 1.4 0 0.7 20
20
r 74 A K 0.62.1 1.5 0.9 2.24.3 2.6 1.6 1.3 ,1.2 2.9, 0.9 50 0 0.6 0.6 1.1 1.4
2.8 2.2
975A F _1 8.6 -10.1 8.0 _ 0 -12.14.2 7.7 4.7 5.576_8_ 8.3_ 7.0 7_1 11.= 8.1
7.3 7.4 10.3 1.8
976A N 1.5 1.8 1.3 0 20 5.4 20 12.6 2.6 3.2 3.0 0.2 18,7 1_31-1_8 1.1 6.3 10.8
20 20
+77 A W =.311.112.310.7 4.1 15.0 8.6 10.27.3 7.8-7.9.-11.4 7.6 9.5:14.610.2
9.6 1.1.3 0 20 =
978A Y 0.1 1.9. 6.1. 0 2.7 5.5 4.5 16.4 1.316.3j1.5 3.3 50 0.3 _2.2 0:5 P
10,8.6 1.0
'79A V 4.2 4.4 5.1 3.1 20 8.2-19.7 0.3 0 1.9 3.1_13.8, 20 0.5 3.6 3.8 -2.3 1_6
20 20
980A 03.63.5. 0 2.7 12.5 4.0 9.9 17.6 3.8_2.8,3.9 0.5,50 3.1,-3.5 3.1 8.9
13_113.713.0
981 AG 50 5050 50 50 0 150 50_ 50 50 50 50_ 50 50,50 50 19.2,50 50 50
#82A V 0.511.8 1.8 0.8,2.2 4.0 2.2 0.70.9 1.3,3.0 0.4 50 0.4 0.5 0.1 0.5 0
5.42.5
983A E0.0 112 4.9 0 7.9 4.44.2 2.2 1.3 4.7 1.33.1 0.5 0.5,1.3 0.6 1.6 3.08.3
7.2
_
984=A V 2.1,2.6 4.4 2.6 15.0 6.7- 4.4 2.0 1.8 6.3 2.6 2.7 15.1 2.4_ 1.6 2.6
1.3 0 , 1.116.1
985A H 1.0 2.3 1.9_ 1.3 2.8 2.8 2.6 2.0 1.6 2.2 2.8 0 1.8 0.90.9 0.3 0.5 2.3
5_1 2.3_
=
113
CA 02766627 2012-01-23
,52620-2D
Table 60 (continued)
PosiVTACDEF GH I KLMNPQRST VWY
286 A N 0.9 1.7 0.7 0= .1 3.3 3.4 3.2 0 0.9 2.1 2.3 0.2 6.6 0.4 0.9 1.7 0.7
0.8 5.4 3.4
287.A A 2:4 4.3 5:1 8.4 0 _8.4 3.1 11.26.6 17.4 4.4 _1.3 12_9 8.3 8.6 .1 4.5
9.9 1.4 2.0
288A K 0.7 1.9 2_0 0.9 2.7 3.9 2.6 1.01.3 1.9 2.2 0.4 5_0 0.5 0.6 0 0.5 1.8
6.0 2.6
289 i-r-"T 1.0 1.7 2.2_0.5, 6.6, 4.7 2.3 0.5 0.30.5 1.6 _1.2 _0_8 0.60.7 0 0:2
019 6.2 7.0
290A K 1.1 2.6 2.6 1.6 3.8, 5.6 , 5.4 3_1 0.80.4 2.2 1.5 , 50 1.01.3 0.6 0 2.9
6:3 4.5
291A P 1.0 2_4 1_8 1.7 4.5 3.6 3.4 2.6 1.62.6 3.5 0 ,0.90.9_1.3 0 1.02.4
7.64.1
292A R ,115 3.2 3.2 1.9 1.7_ 5.2 3.0 1.8 1.0 0 1.6 1.9 0.9 1.3 1.6 1.5 1.6 2.5
4.3 1.9
293=A E 0.6 2.1 1.5 0.4 8.0 4.3 -5.5 ,3.2 14 Ø8 2.2 12163 0 2.2 1.1,0.8 r3.3
10.6,8.3
1294 A E 2.3 2.5 0.5 0.5 5.0 5.8 3.5 4.1 1.5 2.02.7 0 1.6 0.9 -1.3 1.9 1.6 2.4
9.05.4
1295 A,:,6 3.1 3.2 3.8 2.8 17.6 7.8 12.8 9.7 1.3 0 2.3 3.3, 50 1.6 3.6 .8 7.9
10.1 20 _20
296 A, '(3.33.5 3.6 -2.3 4.7 0 .3.5 50 2.6 3.6 4.7 1.7 50 2.1 2.5 1.814.0 50
7.7_4.7
97A N 1.4 2.3t 3.2 1.4 4.44.33.0 0.60.7 2.2 2.4 1.1 50 0.9 0 0.8 1.1 1.5 7.3
4.3
298A S6.5 7.1 6.9_6.9 11.6 0 8.6 L 50 5.6 7.2 -8.5 5.2-50 ,6.2 6.3 = .412.0 50
12.510.6
299 T 0.3
2.2,3.0 20 , 0.5 4.4 0.5 5.1 1,516.2 3.30.1 50,20 , 3.4 s.7 0 0.4 50 1.7
300 Ay 4.1 4_8 5_1 5:2 0 1,9_8 2.2 11.9 3.3- 2.6 3.7 4.4 50 5.4 7.9 .7
7.212.T 5.8 0.4
301A R '1.6 2.4 1.1 0.6 r, 20 6.5 20 Jim 0 3.5 1.7 '-0.5 , 50 0.3 0.9 .53.1
9.820 20
302 Ai V 3.2 4.0 5.0 -,4.0 26 9.6 5.8 0.3 1.6 2.3'2.7 3.6 10.8 2.9- 3.9 .62.1
0 0 20 20
303 A V 0.9 to 2_0 0 20 5.0 20 30441244627 1648 4.9 1.3 0.7 1 .0, 20 20
04A S 1.1 4.1 9.2 20
7.8 20 7.6. 6.9 20-19.3 ,2.8 2011.2 9.4 0 2.54.1 20 20
305A V 1.6 2.1 1.8 4.1 20 6.3 , 20 1.0 3:9 2.9 3.0 0.6 12.6 4.5 3.8 1.8 1.0 0
20 _20
306A L 5.1 p.8 6.7 ,7.0 1.7 11.5 4.9 3.8 3.9 0 6.3 6.0 15.0 7:3 15.4..9 4.2
5.6 20 _2.5
307A T 1_5 3.0 2.6 1.6 1.1 5.5 3.0 6.2 1.7 1.0 3.0 1.8 0 1.6 2.4 1.6 1.0 0.8
9.51.4
308 A V 4.0 4.512.8 7.2 20 10.2 20 0 171,5.0 20 10.2' 50 13.4 20 .4 4.4 1.7
2020
3O9 AL 1.3 2.8 1.8 1= .7 3.2 5.5 3.3 0.8 1.5 1Ø 3.2-r 0 0.3- 1.3 -0.4 1.6
1_3 oil. 5_8 3.4
310&H 1.6 2.2 2.1_ 4.0 18.6 6.3 4.2 3.5 3.2 ,6.7 3.4 1.1 0 3.7- 7.5 1:7 0.5
3,314:8,18.9
311 A 0 0.2 1.61.4 0.7 1.9 3.9 1.6 -0.4 0.7 0.7 1.9 0 -0.7_ 0.9 1.2 1.5 0 1.2
2.6 1.8
312&D 0 1.6 0.6 0.9 20 :5_3 11.3 8.9 2.4 2.3,2.5, 1.1 50,3.0 3.0 1.6 5.5_9.6
16.020
313 A W 4.0-5.5 7.0 .5.1 0 10 5.1-11.0 2.7 4.2 2.7 6.6 50 ,4.8, 6.3 17 4.59.9
1.30.9
314 A L 2.8 4.3 5.8 3.7 20 7.6 7.3 4.2 1:8 0 2.9 4.0 50 3.2 2.5 .9 3.6 5.7
17.5, 20
315A N 0 5,13.1 3.4 11.1 2_6 11.31.64 3.4 3.4_ 4.811.9 50 3,34.6 1.78.9 50
13.012.0
316 A G 11.4 10 111 8.9-16.6- 0 1275085 _8.8 ,_9A 9.5 50 8.8 9.2 4.2 50 , 50 1-
1.916.6
317 A-K 3.1 -it.a 8.1 5.6 6= .5 µ8.4 2.5 7.3_ 0 5.5. 3.5- 5.0 50 3.6 1:5 3.1
5.2 6.8, 20-7.4
18A E 15228 11 20 5.8 9620 19_59 _3.4- 2.2 17.5' 1.6 Z7 1.1 0 34 20 20
319A Y 7.0 7.9 9.4 9:9 12.84.6 3.2
6.06.6 _7.1 8.2 20'1-8.9 12. .7 5.8 _-3.8_ 8.7 0.9
320A K = 1.8 2.9 6.7 -1.8 20_ 7.1 20_ 1.1 0 _8.1 2.2_ 4.2 8.4 1.7 1.4 1.5 0.6
1.0 _20 20
321A c 0 3.920 20 20 6.1 20 20 _20 20 20 20_20 20 20 9.2 8.618.8 20 20
322A K 2.43.36.i ,3.0 20 8Ø 201:1 0.4 16.9 2.5 4.1 , 50 2.8 3.0 4.1 1.6 0 20
_20
323 A V 3.5 (4.3 8.0 9= .0 20 9.9 20 -4.7 10.2 20 .5.7-8.7_50_9.3 20 = :9 2.5
_ 0 20 20
[324 A S 0.4,2.00.9 0 0.2 5.0 _2.5 1.3 1.3- 0 2.40.4 50 1.0 õ2.6 1.7 1.3 1.3
8.7 -0.3
,325A N 4.9 5.9 6.3 6.0 20 10.4t20- 1.714.Q11.3 20 0 13.3 6.3 20 3.7 3.4 20
20
26A K 1.6 3.6 2.4 2.4 3= .8 4.9 3.6 10.6 1.4 2,9 4.2 1.1 0 1.7 -2.1 1.5 4.5
9.4 5.1 3.3
327A A 2.7 3.85.5 2.8 11.6 7.1 9.53.2 1.23.74.3 3.820 3.2 2.8 1.8 0 3.3
18.312:6
328A L 2.53.7t4.6 1.7' 5= 07.420 5.7 1.8_ 0 10.474.3 501.7 6.09.98.35.9 50 50.
329A P , 0.8 2.2 1.2 1.24.0 3.3 3,3 2.9 1..6 2.5 3.50.1 0.30.6 1.4 0_1.1 2.2
6.63.7
330A A 0.3,1.6 2.0 1.33.4 3:2 2.3 2.4 1.3 2.5 3.0_ 0.3 20_ 0.51.1 00.1 2.2_6.0
3.4
331 A P 1.53.5 8.0 10.4 6.6 6.3_54-_9.3 8.1 12.6i6.4. 6.1 , 0 _8.0_7.9 1.4 6.4
4.61,6.5 _6.8
332A I , 2.473.5 2.6_ 2.2 7.9 7.2 _4.0 1.3 2:3 0.72.9 2.8 50, 0 3.7 1.8 1.3
12.69.4
333A E 2.0 2.7 2.6 0 8.0 6.3 7.63.1 3.5 5.9 3.0_2.9 3..6_1.1-2.9 1.7 1.21_4.8
8.0 , 8.4
114
CA 02766627 2012-01-23
-52620-2D
,
,
Table 60 (continued)
_
Pos ACDEFGH 1 K LAA_NPQ[RST VWY
334A K 0:1 3.4 3.5 2.0 7.1 7.1 3.52.0 2.2 3.0 3.0 2.5 2.8- 1.5 0 0.4 1.6
1.54.1 7.9
335P T 1.5 1.0 2.1 0.3 4_6 4.6 4.1, 6.6 0.2, 4.0 2.1 1.22.1 1.0 0.2 1.8 0 0.9
5.1 4.8
336A I 1.4 1.4 2.2 0.9 13.9 4.8 4.7 0 1.3 4.3 1.6 1.3 20 0.1 2.6 1.1 0.4
0.312.313.'
37A S 1.30.8 4.0113.520 3.1 20 50 9.9 9.35.6 5.0 12.7 6.7 7.3 0 10.550 20 20
338 A K , .7 8.3 7.1 7.6 20 9.8 20 5.5 0 2.3 5.9 5.5 5.86.4 4.6 ..4, 6.8 6.9
20 20
= I ,
339P A 1.4 2.7 3.1 2.2 2.3 5.5 3.3 0 0.5 0.2 2.7 1.5 1.0 0.8 0.4 1. 1.2 0.4
6.6 2.4
_
340P K 1.2 3.0 2.0_1.5 1.9 3.9 2.5 2.6 0.2 2.2 2.1 1.0 1.0 0 , 1.1 1. a 1.2
3.1 ,7.8 2.2
237 E G 1.62.2 1.7 0:5 5.0 4.22.5 0.3 0.3 4.1 1.7-0.5 0_3 0 0.3 6.4 0.3 1.07.3
5.4
238 B 13- .0 5.3 8.9 5.4 20 9.1 20 1.114,4 3.6 11.6 7.3 0 7.8 20 1.= 4.7 2.7
20 20
239 e S 1.7 2.6 2.9, 1.1 2.2 4.8 0.5 2.9 0 0.8 2.5 0.2 9.3 -6.8 0.3 0.4 1.7
2.7 L4.8 2.1
pa E V 0_0 3.0 3.4 5.3 20 7.7 20 ,6.4 11.3 0 10.22.7 1.39.7 20 0.4 0.6 0.7 20
20
241 e F 1.32.0 2.7 0.2 1.5 4.3 3.2 5.7 1.3-1.2 2.4 ,1.5 8.5 1.2 1.9 0 1.3 5.3
4.3 1.3
242 e 't. , .0 5.1 '7,0 6.5 20 9.5 20 12.913.6, 0 8.7 5.9 3.9 10.619.* ..1 5.6
4.2 20 20
243B F 1.71.8 1.2 0 0.2 4.9 1.54.8 1.4 0.8, 2.3, 0.4 50 0 0.5'. 0.6 4.5 4.3
0.7
244 e P 0:1 3.1 5.4 2.8 7.0 3.5 3.7_ 0.7 0 _3.6 3.3_3.5 0.3 1.0 1.2 1.4 1.4-
0.5 7.3 7.2
245 E P 0.1 20 _20 20 20 7.320 50 20 20_20 20,0 20 -20 '.119.420 20 ' 20
246 E K 1.4 2.8 _1.8.2.0 3.4 ,5.8 3.9_2.3 1.8 1.23.2 0 0.3 2.1_ 1.5 1." 1.7
1.96.0 3.1
247 E4 0 0.4 5.0 8.4 7.4 20 7.5 1-8.5 8.7 9.4 , 20 7.9 12.4 0 6.3 9.1 I.= 0.8
3.0 20 20
;248 E K '.0'4.1 4.6'3.7 20 6.8 6.1 3.7 , 0 2.2 2.9 3.8 2.1 ,2.7 2.5'. 1.2
2.1 20 20
1
24913 b 4.2 4.2 3.1 1.2 3.2,9.1 4.0 50 2.9 0 3.3 3.8 50 1.64.1 ,. i_' 8.4
50 6.6 4.6
250 B T 1.11.4 3.1- 7.4 20 6.720 2.9A5.9 8.9,6.1 2.2, 50 , 6.7- 6.1 1., 0.6 0
20 120
. 251 B L , .045.6 6.9 7.9-3.5 i10.4 1.9 4.9 5.0 2.1 3.35.3 50 49.3 ' 5.4 ..*
3.3,4.5' 50 0 . ,.
252 e' M c _0 3.6 '5.6 29 20 , 9_2 20 6.0 1.0 4.9_ 0 ,,4.2 50 2.4 14.11. 6.9
7.7 20 20
25313_1 1.51213 416 3.5 4.6 6.1 6.8 1.7 0 1.6 2.2 3.6 50 3.3 24 1_6 1.5 2.3
6_5 5.3
254B S 0-2.8 6..1 3_4 8.54.1 -9.9 4.54.2 1.5 3.93.1 -50 3.4- 5.1 0.3 5.6 6.6
10 8.7
255 6 R 1.8 3.7 5.0 3.7 3.5 6.8 0.8 14.--i 0.1 0 _1.5 2.6 so 1.6 0 1.7 1:0
13.6 4.8 ' 20
256 6 T 1.02.3 1.6 1_4 311 3.7 2.33.5, 1.2 1.9 2.6 0 2.4 0.5 0.2 1.7 1.5 ,2.8
5.9 2.9
257 B P 0.610.4 20420 50 8.4 50 20 20 17.3 20 20 0 16.5 20 . 20 20 50 50
R588 E 6 Ito , i.7 0.8 0.8 3.9 2.01-12.0 2.4 4.01.8 2.1 16.2 29 3.0 1. 4.5
11.7 20 1.6
591V , .1 4.5 7,09.1 20 101 20 ,1.5 12.214.415.5 7.5 50 8.820 . 2.0 0 20 20
26013 T 0.4 2.7 3.0 _ 0 20,7.2 7.2 1.63.0 5.3 3.5 21 4.1 0.3 1.3 e . -12.0 1.6
15.9 20
p61 E C 0 18.020 20 20 4.0,20 20- 20"-20 20 20 _50 20 20 . 20 20 20 20
262 B V 1.6 1.5 0.8 20 20 7.0 20 8.4 16.8_20 203.5 5014.919.'1.9 1.6 0 20 20
263 B V .5 5.2 5.5 18.4 20 10.920 10.9 4.6 3.5 8.9- 4.7 50 -15.319. . 3.1 0 20
20
26413 V '.63.0 3.7 1612.8 7.1 12.2 0 2.3-3.1_ 4.0 2.55.0 1.53.1 0.4 1.6 0.420
20
265 B D 1.4 2.7. 2.4 1.3-2.0 2.32.3 50- 1.0 0 2.40.9 500.3 0.7 1.. 11.418.0
5.0 2.2
266 Li V , .9 5.4 7.0 15.9 20 11.6 20 2.7 20 20 19.7 6.2 50 17.2 20 Xi 4.1 0
'50 50
26713 S 0.4 4.6 L4_3- 4.3 1.3 7.1_2.8 1.3 0.91.7 3.62.7 0 1.4 1.9'.3 1.8 3.2 -
5.2 2.5
2688 H 1.2 3.3 4.0 2.9
0.1.,2.9 3.81 2.4_6.9 5.5 2.0 1:7 0 4.92.7 1_8 3.2 0.9 1.1 1.4 1.2 2.8 8.46.4
2698 t 1.8 2.5 ,0.9 - 1.2 2.0 2.2- 3.2 0 5.3 0.8 1.6 4.8 1.0 1.8
5.6 3.1
70B N 6 1.3 0.9 3.9 2.9- 3.874.8 5.7 3.5 1.2- 3.8 2.9 -15.5 4.0 '3.5 1.11.4
7.0 6.6 3.1
271 B P 1.6 2.5 6.0 7.8 3.1 5.6 4.2_16.4-5.7 -16.9 6.0 4.0 0 7.6 5.3 1.3
2.9.6.2 5.0 3.6
272 B E 1.7 1.7, 0.1 0 3.0 3.7 -2.8 1.0 1.6 1.8 3.1 0.6 17.4 0.1 1.01. 1.00.6
3.7 3.0
2738 v '.85.5 .5.1.16.6 20 10.8 20 3.1 9.6 7.2_ 9.8 4.2 56 20 , 20 1. 0 2.5
20 20
274 BL K 1.8_2.4 1.6 1.02.3 4.52.7 1.5 1.5 1.4 3.1 0.6 50_ 0 0.5 1. 1.3 1.1
2.8 2.4
2758 F ..98.5 9.98.0 0 12.1-3.7 8.2 4.8 5.5 6.8_8.0 7.1,7.2 13.114. 7.1 7.4
10.7 2.0
27613 N 1.4-1.6 1.2 0 20 5.3 20 9.1 2.'7, 3.1_ 2.7 0.1 19.61.1 1.71. 5.9- 8.8
20 20
277 B W 4.910.911.610.6 4.8 14.6 7.8 10.1 6.3 7.6 7.5 10.9 6.89.314.010 9.5
11.0 0 20
2788 i' 1.8 2.1 5.6 0 1.4 6.3 3.908.3,1.3- 2.7 1.7 3.0 18.1 0.3 2.41. 0.3 10.3
7.5 0.3
115
CA 02766627 2012-01-23
52620-2D
2798 V 3.8 4.9 5.9 3.8 20 8.8 16.0 1.0 0 2.8 3.6 4.5 20 1.0 41 4.5 2.9 2.0 20
20
280803.53.4 0 1.8 - 12.4 3.9 -9.8 17.03.6 2_3 4.00.2 503i 3.6 3.0 8.8
12.113.912.7
. =
=
=
116
CA 02766627 2012-01-23
52620-2D
Table 60 (continued)
PosWTAC----DEFGH I K LMNPQRSTVWY
_ õ
281 B G 50 50 50 50 50 0 50 50 50 50 50 50 50 50 50_50 50 50 50 50
82B V 0.41.8 1.7 0.8 2.2 3.9 2.1 0.8 0.9 1.32.9 0.2 500.3 0.3 00.3 0 5.62.4
283 B E 0_91.2 4.9 0 7.8 4.7 4.3 1.7 1.5 4.720 3.2 0.4 0.7 0.8 0.6 1.5 2.7
8.3 7.5
DIM .7 5.0 2.6 16.4 7.0 5.5 3.1 2.0 7.0 2.9 3.2 50 2.6 2.2 2.71.4 0 13.3 20
85 B H 0.69.0 1.8 1.0 2.4 2.5 2.3 2.0 1.4 1.9 2.9_ 0 1.6 0.5 0.7 0 0.2 2.2,
4.8 1.9
286B N 1.01.8 0.9 0.2 3.4 3.5 3.1 0 1.412.2 2.4 0.2 8.3 0.4 1.0 0.7 0.8 1.0
5.4 34
287 8, A 2.5 .3 5.5 8.5 0 8.4 3.2 13.3 6.7.17.7 4.6 1.1 14.1 8.2 9.1_3.5 3.2
10.7 1.1 _1.4
28813, K 0.61.7 1.6 0.8 2.5 3.7 2.5 1.0 1.4 1.7 2.0,0.3 5.7 0.6 0.4 0 0.5 1.7
5.8 2.3
2898 T 0.91.5 2.1 0.5 5.4 , 4.6 2.3 0 0.2 0.2 1.3_1.1 0.7 0.5 0.2 0.2 6.2 0.4
6.7 5.4
260 B, K 0.72.4_2.5 1.2 3.2 5.2 5.2 2.4 0.2 0.1 1.8 1.3_ 50 0.6 1.2 0.7 0 2.4
5.7 4.3
291 BP 1.09.5 2.0 1:7 ,4.5 3.6 3.4 2.5 1.6 2.6 3.4 0.2 0.8 0.7 1.2 0 0.8 2.6
7.6 3.9
92 B R 1.83.5 3.5 2.2 1.6 5.5 2:8, 2.2 1.1 0 1.8 2.4 1.3 1.6 1.5 1.8 2.0 2.6
.4.5 2.0
2938 E 0.72.2 1.6 0.6 8.6 4.5 4.7 2.8 1.5 0.9 2.4 1.1 15.9 0 2.3 0.2 0.9 2.8
9.3 8.6
294B E .19.2 0.5 0.4 5.2 5.7 3.3 2.8 1.3 3.0 2.4 0 1.3 0:9 1.31.5 1.3 2.2 8.4
6.2
295 B Q .33.1 4.0 3.1 18.8 7.6 13.1 8.3 1.2 0 2.4 3.4 50 1.8 376-3.6 7.2 8.7
20 18.4
296 B Y 4.2 ..4 4.5 3.6 5.6 0 4.4 50 3.4 4.4 5.5 2.5 50 2.9 3.3 2.611.3 50 8.5
5.4
B N 1.3 .1 3.1 1.4 4.0 4.2_ 2.7 0.3 1.3 2.1 2.3 0.9 50 1.0 0 0.8 0.8 1.3 7.0
4.1
98 S 5.5 ..0 6.3 5.7 9.7 0 7.5 50 5.7 6.3 7.8 4.5 50 5.5 5.4 3.3 9.6 50
11.9 9.6
299 B T 1.1 .1 3.5 15.2 0.9 5.9 0 6.3 1.4 10.9 2.3_ 0.9 50 15.0 1.8 1.8 1.1
2.3 20 1.6
300 B Y .7 4.0 3.8 2.9 8.8 2.3 10.5 2.0 1.5 2.8 3.2 50 -4.3 5.9 4.6 3.1
10.9 5.7 0
301 B R 1.59:3 1.4 0.3 20 6.4 20 6.8 0.1 3.8 1.7 0.7 50 0 0.6 2.4 2.7 7.1 20
20
5.8 3.9 20 9.3 7.0 0.2 1.8 3.4 219 4.8 20 2.8 5,0 3.7 2.1 0 20 20
3038 V 0.211.2 1.2 1.2 20 5.3 20 5.8 3.9 11.4 4.9 2.3 0.9 5.36.40.4 0 0.2 20
20
3048 S 1.01.9 4.1 10.8 20 7.8 20 8.3 7.4 20 12.12.6 16:414_112.0 0 1.7 3:6 20
õ 20
3058 V 1.51.8 1.7 3.9 20, 6.2 20 0.7 õ3.2 4.2 2.9 0.714,7 4.2 3_3 1_7 0.9 0 20
20
3068 L 5.27.1 6.7 7.3 1.5 11:64.9 3.7 5.1 0 6.0 ,6.0 12.26.9 14.66.2 4.5 5.5
20 1.9
3079 T hl.63.02.5 1.9 1.1 5.5 3.0 0.21.6 1.2 3.0 2.1 o 1.7 '2.3 1.7 1.1 0.8
9.7 1.5
308B V 5.15.812.7 7:4 20 11.3 20 0 19.1 6.1 '20,11.7 50 10.8 20 5.6 4.5 2.5 20
20
36913 L 1.32.8 1.9 1.7 3.2 5.4 3.3,0.9 1.5 1.0 3.2 0 0.2,1.2 0.4 1.6 1.3 0.8
5.8 3.3
3108 H 1.72.4 2.5 3.8 13.1 6.4 5.5 3.6 3.6 7.5 3.7 1.2 0 4.1 10.51.2 0.6 4.6
11.613.6
Mall11.6 1.1 0.5 0.9 3.7 1.6 0.4 0.6 0.6 1.8 0 1.7 0.8 1.0 0.4 0 1.4 2.3 1.0
312 B D 0 1.7 0.8 4.6 20 5.3 20 11.0 3.1_ 2.9, 2.7 1.5 50 6.8 4.0 0.5 7.1 9.7
20 20
313 B W 4.45.6 7.2 5.7 0 10.35.3 9.3 3:0 4.3 2.9 7.7 50 5.1 7.1 6_0 4_8 7.2
1.6 1.4
314B L 2.8.4 5.73.7 20 7.6 8.3 4.5 1.6 0 ,3.0 4.0 50 3.4 4.0 3,9 3.6 5.9 17.5
20
3158 0 0 7.4 3.9 5.2 12.9 2.1 12.014.7 4.5 3.3 5.6 2.5 50 5.8 5.9
2.011.418.013.711.8
316 B G8.97.7 9.2 6.9 13.3 0 10.8 56 6.7 6.8 7.0 7.5 50 6.8 7.7 6.9 50 50 10
13.6
3178 K 2.6.3 7.6 5.96.7 7.42.3 7.5 0 ,4.8 3.4 4.4 50 3.2, 1.3 2.2 6.6 6.4 20
7.6
318 B E 1.79.6 2.9 1.6 20 6.0 9.6 1.8 2.2 6.1 3.7 2.4 13.4 1.7 3.0 1.1 0 3.4
16.3 20
3198 Y `6.9 .8 9.3-10.1 0 12.7 4.7 3.1 6.2 7.1 6.9 8.0 50 9.5 13.17.4 5.5 3.6
10.4 0.8
3208 K 1.72.9 6.7 1.9 , 20 7.0 20 0.6 0 _ 8.9 2.0 3.9 12.. 1.8 1.2 1.6 0.7 1.1
20 20
321 B d 0 .5 20 20 20 _6.2 20 20 20, 20 _ 20 .20 20 20 20 2.3 9.1 19.2 20 20
=
322 B K 2.8 .8 6.2 3.3 20 8.4, 20 1.8 0.8,16.3 3.0 4_6 50 2.7 2.7 3_5 2.0 0 20
20
323 B V 3.54.4 8.8 7.6- 20 9,8 20 5.4 9.2 20 6.0 9.2 50 9.8 19.44.8 2_7 0 20
20
324 B S 0.52.5 1.4 0.6 0.4 5.2 2.7 .3.5 0 0.2 2.4 1.3 11.1 1.0 2.7 0.4 1.2 3.4
2.5 1.2
325 B N 4.15.5 6.9, 5.7 20 9.6 20 1.0 10.611:217.0 0.4.11.5 5.5 16.53.1 0 2.1,
20 50
326 B K p.92.8 1.8 1.8 2:6 4.4 2.5 4.2 .1.3 2.9 3.5 0.1 0 1.2 -1.3 0.7 2.4 3.4
6.4 2.4
3278 A 3.1 .4 6.1 3.2 10.3 7.4 4.0, 7.0 2.8 2.54.5 3.9_20 3.5 3.71.4 0 2.8
10.312.2
328 B L 4.7 .4 6.6 3.3 20 9.8 20 50 4.1 b 15.1 6.0 50 3.7, 6.2 5.618.4 .50 20
50
3298 P 0.62.1 0.9 tp 3.8 3.2 2.8 2.2 1.3 2.4 3.2 0_0.4 0.3 0.9 0 0.6 1.6 6.4
3.7
= 117
CA 02766627 2012-01-23
52620-2D
33013 A 10.41.811.511.2[3.4133 2.712.11_1.311.9_3.0 0 200.6 t3 0.1 0.3 1.8 6.0
3.7
=
=
=
118
CA 02766627 2012-01-23
52620-2D
Table 60 (continued)
PosWTACD E_F.G 1-1.IK 1._õM_N,P_ORST V W Y
331 B P 1.6 3.6 7.8 10.6 7.3 .6.5- 4.68.9 7.7 13.6.6.3, 5.7 0 8.3 7.77 1.75.3
5.2 6.5 8.0
33213- I 2.0 3.0 2.6 0.9 5.8 , 6.92.6 0 2.1 0.1 , 2.5 2.6 50 0 2.4 2.3 -1.3
0.9 15.3 6.6
33313 E 2:2 2.8 2.7 0 8.1 .6.4- 7.8 3.4 3.6 6.1. 3.3, 2.6 3.6 1.2 3.0 2:4 1.4
4.9 _8.4 8.8
-3346 K 2.2 3.4 412 2.1 10.4 7.2 4.1 1.7 1.8: 3.1 2.9 2.6 2.8 1.5 0 2.7 1.6
1.4 5.5 10.8
3356 T 0.51.1 ti 0.8 4.8 4.6 4.4 0.5 0.3 3.7_2.20.8 1.7 _1. -i 0.6 0.6 0 0.8
5.2 5.1
336 B, I 9.7 1.52.5 1.0 18.4 5.0 5.4 0.17,1 .9- 4.6 1.9 1.5 20 0 3.0 1.510.5
0.4 14.2 19.6
33713 S 0.4 1.1 4.9 10.6, 20 3.4 20 50 7.9 11.05.1 3.6 12.8 6.2 7.0 0 4.6 50
20 20
338 B K 4.5 8.2 7.4 8.0 20 9.6 26 5.3 0 2.0 5.7 6.0 5.8 6.8 4.9 5.4 6.4 6.7 20
20
339 B A 1.5 2.8 3.0 2.1 2.4 5.5 33 0 0.4 0.22.81.4 1.5 0.9 0.7 1.5 1.1 0.5 6.6
2.6
34613 K 1.02.71.7 1.3 1.7 3.7 21 2.4 0 7_1 1.90.6 1.0 0 1.00.2 1.02.3 7.0 1.8
SPA TN technology; 1DN2 template structure; + carbohydrate
[204] The results of the design calculations presented above in Tables 1 -60
were used to
construct a series of Fc variant libraries for experimental production and
screening. Experimental
libraries were designed in successive rounds of computational and experimental
screening. Design of
subsequent Fc libraries benefitted from feedback from prior libraries, and
thus typically comprised
combinations of Fc variants that showed favorable properties in the previous
screen. The entire set of
Fc variants that were constructed and experimentally tested is shown in Table
61. In this table, row 1
lists the variable positions, and the rows that follow indicate the
amino.acids at those variable
positions for WT and the Fc variants. For example, variant 18 has the
following four mutations:
F241E, F243Y, V2621, and V264R. The variable position residues that compose
this set of Fc
variants are illustrated structurally in Figure 4, and are presented in the
context of the human IgG1 Fc
sequence in Figure 5.
=
=
=
119
CA 02766627 2012-01-23
52620-2D
Table 61
:Variant Substitution(s) Variant, Substitution(s)
1 V264A 50 _ Y296Q
2 V2641 51 S298T
3 V264I 52 S298N
4 F241W 53 T2991
F241L 54 -"'A327S
6 F243W 55 -A327N
7 F243L 56 S2670/A327S
8 F24111F243LN2621/V2641 57 S267UA327S
9 F241 W/F243W 58 _A3271
F241W/F243WN262AN264A 59 I-3329F
11 F2411V2621 60 A3301
12 F243UV2641 61 A330Y
13 F2431V2621N264W 62 1332D
14 F241Y/F243YN262TN264T 63 N2978
F241 E/F243R/V262EN264R 64 N297D
16 F241E/F243Q/V262T/V264E 65 N2978/1332E
17 F241R/F243a/V262TN264R 66 N2971Dii332E
la F241 E/F243YN262TN264R 67 N297E/1332E
19 L328M 68 0265Y/N 297D/1332 E
1328E __________________________ 69 0265Y/N297D/T299U1332E
21 L328F 70 0265F/N297E/1332E
22 1332E ' 71 13281/1332E
23 ,1328M/1332E. 72 13280/1332E
24 P244H 73 I332N
P245A 74 1332Q __
26 P247V 75 V264T
27 W313F 76 V264F __________________
28 P244H/P245A/P247V 77 V2401
29 P247G 78 V2631
V2641/1332E 79 V2661
31 F241E/F243RN262EN264R/1332E 80 T299A
32 F241E/F243Q/V262TN264EJI332E 81 72993
33 F241R/F243Q/V262TN264R/1332E 82 T299V
34 F241E/F243YN262TN264R/1332E 83 N325Q
S298A 84 N3251
36 S298A11332E 85 , N3251
37 S298A/E333A/K334A 86 S2390
41 S239E/1332E 87 S239N
42 S2390/1332E 88 S239F
43 S239E 89 1 S2390/1332D
44 0265G 90 8239D/1332E
D265N 91 3239D/1332N
46 S239 EID265G 92 3239D/13320
47 S239E/D265N 93 5239E/13320
48 S239E/0265Q 94 -S239E/1332N
49 Y296E 95 S239EJ13320
=
120
CA 02766627 2012-01-23
52620-2D
Table 61 (continued)
Variant Substitution(s) Variant Substitution(s)
96 S239N/13320 141 V264Y
97 S239N/1332E , 142 V266A
98 S239N/1332N 143 _V2661
99 S239N/1332Q 144 V266M
100 S2396/13320 145 E269H
101 S2390/1332N 146 E269Y __
102 5239-Q/1332Q 147 E269F
1.03 K326E 148 E269R
104 Y2960 149 Y296S
105 NI/296N 150 Y296T
106 N2970/1332E/F241Y/F243YN262TN2641 151 Y296L
. -
107 1332E1A330Y 152 Y2961 .
108 .13320/2641/A330Y 153 ,-S298H
= 1.09 1332E/A330L 154 , 1299H
1.10 1332EN2641/A330L 155 A330V
111 , L2340 156 A3301
112 L.234E 157 A330F __
- 113 L234N 158 A330R
114 L2340 159 A330H
115 L2341 160 N325D
116 L234H 161 N325E
117 L234Y 162 N325A _________________
118 12341 163 N325T
116. L234V 164 N325V
7 120 1234F 165 N325H
121 L2350 166 ,L3280/1332E __________
122 L2355 167 L328E/1332E
123 L235N 168 , L328N/I332E
124 L-235Q 169 L328Q/1332E
125 L235T 170 L328V/1332E
126 .1.235H , 171 13281/1332E
127 L235Y __________________________ 172 L328H/1332E
128 L235I , 173 L3281/1332E
L
129 1235V 174 L328A
130 L235F 175 13321
131 3239T, 176 133211
132 S239H 177 I332Y
133 S239Y 178 I332A
134 V240A 179 V2641/1332E/S239E
=
135 V2401 180 V2641/1332E/82390
136 V240M 181 V2641/1332EJS239E/A330Y
= 137 V263A 182
V2641/1332E/S239E/A330Y/S298A
138 V2631 183 N2970/1332E/82390
139 V263M 184 N297D/1332EJS239E
140 V264M 185 N297D/1332E/S2390/0265V
=
121
CA 02766627 2012-01-23
52620-2D
Table 61 (continued)
Variant Substitution(s) Variant Substitution(s)
186 N297D/1332E/S2390/02651 231 E269N
187 N2970/1332E/S239D/0265L 232 02700
188 N297D/1332E/S23913/0265E 233 02701
189 N297131/1332E/S2390/0265Y 234 D270H
190 N2970/1332E/S239D/0265H 235 E272S
191 N2970/1332E/S2390/0265T 236 E272K
192 N2970/1332EN264E 237 E2721
193 N2970/1332E/Y2960 238 E272Y
194 N2970/1332E/Y296E 239 V2731
195 N2970/1332E/Y296N 240 K274T
196 N297011332E/Y2960 241 K274E
197 N2970/1332E/Y296H 242 K274R
198 N2970/1332E/Y296T 243 K274L
199 N2970/1332EfT299V 244 K274Y
200 N2970/1332E/T2991 245 F275W
201 N2970/1332E1T299L 246 N276S
202 N2970/1332E/T299F 247 N276E
203 N2970/1332EfT299H 248 N276R
204 N297D/1332E/T299E 249 N276L
205 N2970/1332E/A330Y 250 N276Y
206 N2970/1332E/S298A/A330Y 251 Y2781
207 S239D/1332E/A330Y 252 Y278E
208 S239N/1332E/A330Y 253 Y278K
209 S2390/1332E/A330L 254 Y278W
210 S239N/1332E/A330L 255 E283R
211 1332EN2641/S298A 256 V3021
212 1332E1S239D/S298A 257 E318R
213 1332E1S239N/S298A 258 K320T
214 S2390/1332EJV2641 259 K3200
215 S2390/1332EJV2641/S298A 260 K320I
216 S239D/1332EN2641/A330L 261 K322T
217 L328N 262 K322H
218 L328H 263 V323I
219 S2390/1332E/A3301 264 S3241
220 N2970/1332E/S2390/A330L 265 S324D
221 P230A 266 S324R
222 E2330 267 S3241
223 P230NE2330 268 S324V
224 P230A/E2330/1332E 269 S324L
225 S2671 270 S324Y
226 S267H 271 K326L
227 S2670 272 K326I
228 S267N 273 K3261
229 E2691 274 A3270
230 E269L 275 A327T
122 =
CA 02766627 2012-01-23
52620-2D
Table 61 (continued)
Variant Substitution(s) Variant Substitution(s)
276 A330S 290 T335Y
277 A330W 291 L2341/L235D
278 A330M 292 V2401N2661
279 P331V 293 _ S239D/A330Y/1332E/1_2341
280 P331H 294 S239D/A330Y/1332E1L2350
281 E333T 295 S239D/A330Y/1332EN2401
282 E333H 296 S2390/A330Y/1332EN2641
283 E3331 297 S2390/A330Y/1332EN2661
284 E333Y 298 S239b/A330Y/1332E/K326E
285 K3341 299 S239D/A330Y/1332E/k3261
286 K334T 300 S2390/N2970/1332E/A330Y
S239D/N29713/1332E/A330Y
287 K334F 301
/F241S/F243HN262TN2641
288 T3350 302 S239D/N2970/1332E/L235D
289 T335R 303 S239D/N2970/1332E/K326E
[205] Example 2: Experimental production and screening of Fc libraries
The majority of experimentation on the Fc variants was carried out in the
context of the anti-cancer
antibody alemtuzumab (Campath0, a registered trademark of Ilex Pharmaceuticals
LP).
Alemtuzumab binds a short linear epitope within its target antigen CD52 (Hale
et a/., 1990, Tissue
Antigens 35:118-127; Hale, 1995, lmmunotechnology 1:175-187). Alemtuzumab has
been chosen as
the primary engineering template because its efficacy is due in part to its
ability to recruit effector cells
(Dyer et at., 1989, Blood 73:1431-1439; Friend et al., 1991, Transplant Proc
23:2253-2254; Hale et
aL, 1998. Blood 92:4581-4590; Glennie et aL, 2000, Immunol Today 21:403-410),
and because
production and use of its antigen in binding assays are relatively
straightforward. In order to evaluate
the optimized Fc variants of the present invention in the context of other
antibodies, select Fc variants
were evaluated in the anti-CD20 antibody rituximab (Rituxan , a registered
trademark of (DEC
Pharmaceuticals Corporation), the anti-Her2 antibody trastuzumab (HerceptinO,
a registered
trademark of Genentech), and the anti-EGER antibody cetuximab (Erbitux0, a
registered trademark of
!melone). The use of alemtuzumab, rituximab, and trastuzumab for screening
purposes is not meant
to constrain the present invention to any particular antibody.
[206] The IgG1 full length light (VL-CI) and heavy (VH-Cyl -Cy2-Cy3) chain
antibody genes for
alemktzumab, rituximab, and trastuzumab were constructed with convenient end
restriction sites to
facilitate subcloning. The genes were ligated into the mammalian expression
vector pcDNA3.1Zeo
(lnvitrogen). The VH-Cy1-Cy2-Cy3 clone in pcDNA3.1zeo was used as a template
for mutagenesis of
the Fc region. Mutations were introduced into this clone using PCR-based
mutagenesis techniques.
Fc variants were sequenced to confirm the fidelity of the sequence. Plasmids
containing heavy chain
gene (Vui-Cy1-Cy2-Cy3) (wild-type or variants) were co-transfected with
plasmid containing light chain
gene (V1-C1) into 293T cells. Media were harvested 5 days after transfection.
Expression of
123
CA 02766627 2012-01-23
52620-2D
immunoglobulin was monitored by screening the culture supernatant of
transfectomas by western
using peroxidase-conjugated goat-anti human IgG (Jackson ImmunoResearch,
catalog It 109-035-
088). Figure 6 shows expression of wild-type alemtuzumab and variants 1
through 10 in 2931 cells.
Antibodies were purified from the supernatant using protein A affinity
chromatography (Pierce,
Catalog #20334. Figure 7 shows results of the protein purification for WT
alemtuzumab. Antibody Fc
variants showed similar expression and purification results to WT. Some Fc
variants were
deglyc,osylated in order to determine their solution and functional properties
in the absence of
carbohydrate. To obtain deglycosylated antibodies, purified alemtuzumab
antibodies were incubated
with peptide-N-glycosidase (PNGase F) at 37 C for 24h. Figure 8 presents an
SOS PAGE gel
confirming deglycosylation for several Fc variants and WT alemtuzumab.
[207] In order to confirm the functional fidelity of alemtuzumab produced
under these conditions,
the antigenic C052 peptide, fused to GST, was expressed in E.c,oli BL21 (0E3)
under IPTG induction.
Both un-induced and induced samples were run on a SOS PAGE gel, and
transferred to PVDF
membrane. For western analysis, either alemtuzumab from Sotec (final
concentration 2.5ng/u1) or
media of transfected 2931 cells (final alemtuzumab concentration about 0.1-
0.2ng/u1) were used as
primary antibody, and peroxidase-conjugated goat-anti human IgG was used as
secondary antibody.
Figure 9 presents these results. The ability to bind target antigen confirms
the structural and
functional fidelity of the expressed alemtuzumab. Fc variants that have the
same variable region as
WT alemtuzumab are anticipated to maintain a comparable binding affinity for
antigen.
[208] In order to screen for Fc/FcyR binding, the extracellular regions of
human V158 FcyRIlla,
human F158 FcyRIlla, human FcyRIlb, human FcyRIla, and mouse FcyRIII, were
expressed and
purified. Figure 10 presents an SOS PAGE gel that shows the results of
expression and purification
of human V158 FcyRIlla. The extracellular region of this receptor was obtained
by PCR from a clone
obtained from the Mammalian Gene Collection (MGC:22630). The receptor was
fused with
glutathione S-Transferase (GST) to enable screening. Tagged FcyRIlla was
transfected in 293T cells,
and media containing secreted FcyRIlla were harvested 3 days later and
purified. For western
analysis, membrane was probed with anti-GST antibody.
[209] Binding affinity to Fc-yRIlla and FcyRIlb was measured for all designed
Fc variants using an
AlphaScreen Mt assay (Amplified Luminescent Proximity Homogeneous Assay
(ALPHA), PerkinElmer,
Wellesley, MA), a bead-based non-radioactive luminescent proximity assay.
Laser excitation of a =
= donor bead excites oxygen, which if sufficiently close to the acceptor
bead generates a cascade of -
chemiluminescent events, ultimately leading to fluorescence emission at 520-
620 nm. The
AlphaScreenIN assay was applied as a competition assay for screening Fc
variants. WT
alemtuzumab antibody was biotinylated by standard methods for attachment to
streptavidin donor
beads, and GST-tagged FcyR was bound to glutathione chelate acceptor beads. In
the absence of
competing Fc variants, WT antibody and FcyR interact and produce a signal at
520-620 nm. Addition
of untagged Fc variant competes with the WT Fc/FcyR interaction. reducina
fluorescence
=
124
CA 02766627 2012-01-23
52620-2D
quantitatively to enable determination of relative binding affinities. All Fc
variants were screened for
V158 FcyRIlla binding using the AlphaScreen."' assay. Fc variants were
screened in the context of
either alemtuzumab or trastuzumab, and select Fc variants were also screened
in the context of
rituximab and cetuximab. Select Fc variants were subsequently screened for
binding to FcyRIlb, as
well as other FcyRs and Fc ligands.
[210] Figure 11 shows AlphaScreenTM data for binding to human V158 FcyRIlla by
select Fc
variants. The binding data were normalized to the maximum and minimum
luminescence signal for
each particular curve, provided by the baselines at low and high antibody
concentrations respectively.
The data were fit to a one site competition model using nonlinear regression,
and these fits are
represented by the curves in the figure. These fits provide the inhibitory
concentration 50% (IC50)
(i.e. the concentration required for 50% inhibition) for each antibody,
illustrated by the dotted lines in
Figure 11, thus enabling the relative binding affinities of Fc variants to be
quantitatively determined.
Here, WT alemtuzumab has an IC50 of (4.63x10-9)x(2) = 9.2 nM, whereas S2390
has an IC50 of
(3.98x10-10)x(2) = 0.8.nM. Thus S239D alemtuzumab binds 9.2 nM 10.8 nM = 11.64-
fold more tightly
than WT alemtuzumab to human V158 FcyRIlla. Similar calculations were
performed for the binding
of all Fc variants to human V158 FcyRIlla. Select Fc variants were also
screened for binding to
human FcyRIlb, and examples of these AlphaScreen Tm binding data are shown in
Figure 12. Table
62 presents the fold-enhancement or fold-reduction relative to the parent
antibody for binding of Fc
variants to human V158 FcyRIlla (column 3) and human FcyRIlb (column 4), as
determined by the
AlphaScreen TM assay. For these data, a fold above 1 indicates an enhancement
in binding affinity,
and a fold below 1 indicates a reduction in binding affinity relative to WT
Fc. Data for 1-206 and 217-
218 were obtained in the context of alemtuzumab, except for those indicated
with an asterix (*), which
were tested in the context of trastuzumab. All data for 207-216 and 219-303
were obtained in the
context of trastuzumab.
125
CA 02766627 2012-01-23
52620-2D
Table 62
Variant Substitution(s) FcyRIlla
FcyRIlb Fcyllla-fold
Fold Fold Fcyllb-fold
1 V264A 0.53
2 V264L 0.56
3 V2641 1.43
4 F241W 0.29
F241L 0.26
6 F243W 0.51
7 F243L 0.51
8 F241UF243UV2621N2641 0.09
9 F241W/F243W 0.07
F241W/F243WN262A/V264A 0.04
. 11 F2411V2621 0.06
12 F2431/V2641 1.23
13 F243UV2621N264W 0.02
14 F241Y/F243YN262TN264T 0.05
F241E/F243R/V262EN264R 0.05
16 F241E/F2430N262TN264E 0.07
17 F241R/F243QN262TN264R 0.02
18 F241E/F243YN262TN264R 0.05
19 L328M 0.21
L328 E 0.12
21 L328F 0.24 .
22 1332E 6.72 3.93 1.71
23 L328M/1352E 2.60
24 P244H 0.83
P245A 0.25
26 P247V 0.53
27 W313F 0.88
28 P244 H/P245A/P247V 0.93
29 P247G 0.54
V2641/1332E 12.49 1.57* 7.96
31 F241E/F243R/V262EN264R/1332E 0.19
32 F241E/F2430/V262TN264 E/1332E
33 F241R/F243QN262T/V264R/1332E
34 F241E/F243YN2621N264R/1332E 0.10
S298A 2.21
36 S298A/I332E 21.73
37 8298A/E333A/K334A 2.56
41 S239E/1332E 5.80 3.49 1.66
42 S2390/1332E 6.60 4.68 1.41
43 S239E 10.16
44 . D265G <0.02
D265N = <0.02
46 S239E/D265G <0.02
47 S239E/0265N 0_02
48 S239E/D265Q 0.05
49 Y296E 0.73 1.11 0.66
Y2960 0.52 _ 0.43 1.21
126
CA 02766627 2012-01-23
52620-2D
Table 62 (continued)
FcyRIlla FtyRIlb :
Variant Substitution(s)
Fold Fold Fcyllb-fo Id
51 S29131 0.94 <0.02
52 S298N 0.41 <0.02
53 T299I <0.02
54 A327S 0.23 0.39 0.59
55 A327N 0.19 1.15 0.17
56 S2670/A327S 0.03
57 3267UA327S <0.02
58 A327L 0.05
59 P329F <0.02
60 A330L 0.73 0.38 1.92
61 A330Y 1.64 0.75 2.19
62 13320 17.80 3.34 5.33
63 N297S <0.02
64 N2970 <0.02
65 N2978/1332E <0.02
66 N2970/1332E 0.08 <0.02
67 N297E/I332E <0.02
68 0265Y/N29713/1332E <0.02
69 0265Y/N297D/1299LJ1332E <0.02
70 D265F/N297E/1332E <0.02
71 L3281/I332E 7.03 .
= 72 L3280/1332E 1.54
73 I332N 0.39
74 I332Q 0.37
75 V2641 2.73
76 V264F 0.16
77 V240I 3.25
78 V263I 0.10
79 V266I 1.86
80 T299A 0.03
81 T299S 0.15
82 T299V <0.02
83 N3250 <0.02
84 N325L <0.02
85 N325I <0.02
86 S239D 11.64 4.47* 2.60
87 S239N <0.02
88 S239F 0.22 <0.02
89 S2390/13320 14.10
90 S2390/133.2E 56.10 19.71* 2.85
_91 S2390/1332N 7:19
92 S2390/1332Q = 9.28
93 S239E/13320 9.33
94 S239E/1332N 11.93
95 S239 E/I332Q 3.80
96 S239N/I332D 3.08
97 3239N/1332E 14.21
98 S239N/1332N 0.43
127
CA 02766627 2012-01-23
52620-2D
Table 62 (continued)
Variant Substitution(s) FcyRIlla FcyRIIb Fcyllla-fold :
Fold Fold Fcyllb-fold
99 3239N/13320 0.56
100 S239Q/I332D 5.05
101 S239Q/I332N 0.39
102 S239Q/13320 0.59
103 K326E 3.85
104 Y296D 0.62
105 Y296N 0.29
F241Y/F243YN262TN264T/
106
N297D/I332E 015 =
107 A330Y/1332E 1202 4.40 2.73
108 V2641/A330Y/1332E 12.00 3.54 3.39
109 A3301J1332E 10.34 2.03 5.09
110 V2641/A3301J1332E 11.15 1.79 6.23
111 L234D 0.21
112 L234E 1.34 2.21 0.61
113 L234N 0.56 1.39 0.40
114 L2340 0.37
115 L234T 0.35
116 L234H 0.33
117 L234Y 1_42 1.08 1.31
118 L2341 1.55 1.14 1.36
119 L234V 0.38 =
120 L234F 0.30
121 L2350 106 3.63 . 0.46
122 L235S 1.25
123 L235N 0.40
124 L2350 0.51
125 L235T 0.52
126 L235H 0.41
127 L235Y 1.19 10.15 0.12
128 L2351 1.10 0.94 1.17
129 1_235V 0.48
130 1235F 0.73 3.53 0.21
131 S239T 1.34
132 S239H 0.20
133 S239Y 0.21
134 V240A 0.70 0.14 5.00
135 V240T
136 V240M 2.06 1.38 1_49
137 V263A
138 V263T 0.43 =
139 V263M 0.05
140 V264M 0.26
141 V264Y 102 0.27 3.78
142 V266A <0.02
143 V266T 0.45
144 V266M 0.62
145 E269H <0.02
146 E269Y 0.12
128
CA 02766627 2012-01-23
52620-2D
Table 62 (continued)
Variant Substitution(s)
FcyRIlla FcyRIlb Fcillla-fold
:
Fold Fold Fcyllb-fold
147 E269F 0.16
148 E269R 0.05
149 Y296S 0.12
150 Y296T <0.02
151 Y2961 0.22
152 Y296I 0.09
153 A298H = 0.27
154 T299H <0.02
155 A330V 0.43
156 A3301 1.71 0_02 85,5
157 A330F 0.60
158 A330R <0.02
159 A330H 0.52
160 N325D 0.41
161 N325E <0_02
162 N325A 0.11
163 N325T 1.10
164 N325V 0.48
165 N325H 0.73
166 1328D/1332E 1.34
167 L328E/1332E 0.20
168 L328N/1332E <0.02
169 13280/1332E 0.70
170 1328V/1332E 2.06
171 13281/1332E 1.10
172 1328H/1332E <0.02
173 13281/1332E 3.49
174 1328A 0.20
175 I332T 0.72
176 I332H 0.46
177 1332Y 0.76
178 I332A 0.89
179 8239E/V2641/1332E 15.46
= 180 S239QN2641/1332E 2.14
181 S239EN2641/A330Y/1332E 8.53
182 S239EN2641/5298A/A330Y/1332E
183 S2390/N297D/1332E 0.28
184 S239E/N2970/1332E 0.06
185 S2390/0265V/N297D/I332E 0.03
186 S239D/02651/N2970/1332E 0.01
187' S239D/0265UN297D/1332E. <0.02
= 188 3239D/0265F/N2970/1332E
<0.02
189 S239D/D265Y/N297D/1332E 0.02
190 S2390/026511/N2970/1332E 0.04
191 S239D/0265T/N297D/1332E <0.02
192 V264E/N297D/1332E 0.05
193 Y2960/N297D/I332E
194 Y296E/N2970/1332E <0.02
129
CA 02766627 2012-01-23
52620-2D
Table 62 (continued)
Fc-712111a FcyRI1b :
Variant Substitution(s)
Fold Fold Fcyllb-fold
195 Y296N/N297D/1332E 0.04
196 Y296Q/N29713/1332E <0.02
197 Y296H/N297D/1332E <0.02
198 Y2961/N297D/1332E <0.02
199 N297D/1299V/1332E <0.02
200 N297D/12991/1332E <0.02
201 N2970fT299U1332E <0.02
202 N297D/T299F/I332E <0.02
203 N2970/T299H/1332E <0.02
204 N297D/1299E/1332E <0.02
205 N297D/A330Y/I332E 0.43
206 N297D/S298A/A330Y/1332E 0.16
207 S2390/A330Y/1332E 129.58
208 8239N/A330Y/1332E 14.22
209 S239D/A330L/1332E 138.63 7.50 18.48
210 S239N/A330U1332E 12.95
211 V2641/S298A/1332E 16.50 =
212 S239D/S298A/1332E 295.16 6.16 47.92
213 S239N/S298A/1332E 32.14 5.15 6.24
214 S239D/V2641/1332E 36.58 14.39 2.54
215 8239DN2641/S298A/1332E
216 62390N2641/A3301J1332E
217 L328N 0.59
218 L328H <0.02 .
219 S2390/1332E/A3301 59.1
220 N2970/1332E/S2390/A330L
221 P230A 1.09
222 E2330 0.85
223 P230A/E2330 0.92
224 P230A/E2330/1332E 1.87
225 S267T
226 S267H
227 S267D
228 S267N
229 E269T <0.02
230 E269L <0.02
231 E269N <0.02
232 0270Q <0.02
233 0270T <0.02
234 0270H <0.02
235 . E272S
236 E272K =
237 E2721
238 E272Y 8.70
239 V2731 0.79
240 K2741 1.41
241 K274E = 6.11
242 K274R 1.41
243 K274L 1.09
244 K274Y 1.06
130
CA 02766627 2012-01-23
= 52620-2D
245 I F275W I 1.11
131
CA 02766627 2012-01-23
52620-2D
Table 62 (continued)
FcyRIlla FcyRIlb Fcyllla-fold :
= Variant Substitution(s)
Fold Fold Fcyllb-fold
246 N2768 0.41
247 N276E 0.87
248 N276R 0.66
249 N276L 1.07
250 N276Y 0.56
251 Y278T 1.87
252 Y278E 0.90
253 Y278K
254 Y278W 0.41
255 E283R 0.67
256 V3021 1.01
257 E318R 1.06
258 K320T
259 K3200
260 K3201
261 K322T
262 K322H
263 V3231 0.83
264 S324T
265 S3240 1.07
266 8324R = 0.71
267 8324I 1.15
268 8324V 1.17
269 S324L <0.02
270 S324Y 0.98
271 K326L
272 K3261 1.43
273 K326T 1.88
274 A3270 <0.02
275 A3277 <0.02
276 A330S
277 A330W
278 A330M
279 P331V
280 P331H
281 E3337 0.78
282 E333H 0.75
283 E3331
284 E333Y
285 K3341
286 K334T
287 K334F =
288 T3350 2.79
289 1335R 2.58
290 T335Y 1.56
291 L2341/1_2350 0.07
292 V2401N2661 1.72
293 S2390/A330Y/1332E/L2341 22.39
294 S2390/A330Y/1332E/L235D 7.04
295 82390/A330Y/1332E/V2401 27_97
132
CA 02766627 2012-01-23
,52620-2D
Table 62 (continued)
Variant Substitution(s) FcyRIlla FcyR1lb
Fcyllla-fold :
Fold Fold Fcyllb-
fold
296 S2390/A330Y/1332EN2641 17.72
297 S2390/A330Y/1332EN2661
298 S2390/A330Y/1332E/K326E 64.14
=
299 S2390/A330Y/1332E/K326T 59.03
300 S239D/N2970/1332E/A330Y <0.02
3239D/N29713/1332E/A330Y/
301 <0.02
F241S/F243HN2621N264T
302 5239D/N2970/1332E/L2350
303 8239D/N297D/1332E/K326E
[211] Example 3: Selectively enhanced binding to FcyRs
A number of promising Fc variants with optimized properties were obtained from
the FcyRIlla and
FcyRIlb screen. Table 62 provides Fc variants that bind more tightly to
FcyRIlla, and thus are
candidates for improving the effector function of antibodies and Fc fusions.
These include a number
of variants that comprise substitutions at 239, 264, 272, 274, 330, and 332.
Figtires 13a and 13b
show AlphaScreenlm binding data for some of these Fc variants. The majority of
these Fc variants
provide substantially greater FcyRIlla binding enhancements over
6298A/E333A/K334A.
.(2121 Select Fc variants were screened in the context of multiple antibodies
in order to investigate
the breadth of their applicability. AlphaScreenlm data for binding of select
Fc variants to human V158
FcyRIlla in the context of trastuzumab, rituximab, and cetuximab are shown in
Figures 14a, 14b, 15a,
and 15b. Together with the data for alemtuzumab in Figure 13, the results
indicate consistent binding
enhancements regardless of the antibody context, and thus that the Fc variants
of the present
invention are broadly applicable to antibodies and Fc fusions.
[2131 Fc variants have been obtained that show differentially enhanced binding
to FcyRIlla over
FcyRIlb. As discussed, optimal effector function may result from Fc variants
wherein affinity for
activating FcyRs is greater than affinity for the inhibitory FcyRIlb.
AlphaScreen TM data directly
comparing binding to FcyRIlla and FcyRIlb for two Fc variants with this
specificity profile are shown in
Figures 16a and 16b. This concept can be defined quantitatively as the fold-
enhancement or -
= reduction of the activating FyR (Table 62, column 3) divided by the fold-
enhancement Or -reduction of
the inhibitory FcyR (Table 62, column 4), herein referred to as the FcyRIlla-
fold:FcyRIlb-fold ratio.
This value is provided in Column 5 in Table 62. Table 62 shows that Fc
variants provide this
specificity profile, with a FcyRIlla-fold:FcyRlib-fold ratio as high as 86:1_
[214] Some of the most promising Fc variants of the present invention for
enhancing effector
function have both substantial increases in affinity for FcyRIlla and
favorable FcyRIlla-fold:FcyRIlb-fold
133
CA 02766627 2012-01-23
- 52620-2D
ratios. These include, for example, S239D11332E (FcyRIlla-fold = 56, FcyRIlla-
fold:FcyRIlb-fold = 3),
S239D/A330Y/I332E (FcyRilla-fold = 130), S239D/A330L/1332E (FcyRIlla-fold =
139, FcyRIlla-
fold:Fc)'RIlb-fold = 18), and S239D/S298A/I332E (FcyRIlla-fold = 295, FcyRIlla-
fold:FeyRIlb-fold = 48).
Figure 17 shows AlphaScreen TM binding data for these and other Fc variants to
human V158
FcyRIlla.
[215] Because there are a number of FcyRs that contribute to effector
function, it may be worthwhile ,
to additionally screen Fc variants against other receptors. Figure 18 shows
AlphaScreen TM data for
binding of select Fc variants to human R131 FcyRIla. As can be seen, those
aforementioned variants
with favorable binding enhancements and specificity profiles also show
enhanced binding to this
activating receptor. The use of FcyRIlla, FcyRI lb, and FcyRlIc for screening
is not meant to constrain
experimental testing to these particular FcyRs; other FcyRs are contemplated
for screening, including
but not limited to the myriad isoforms and allotypes of FcyRI, FcyRII, and
FcyRIII from humans, mice,
rats, monkeys, and the like, as previously described.
[216] Taken together, the FcyR binding data provided in Figures 11 ¨ 18 and
Table 62 indicate that
a number of substitions at positions 234, 235, 239, 240, 243, 264, 266, 272,
274, 278, 325, 328, 330,
and 332 are promising candidates for improving the effector function of
antibodies and Fc fusions.
Because combinations of some of these substitutions have typically resulted in
additive or synergistic
binding improvements, it is anticipated that as yet unexplored combinations of
the Fc variants
provided in Table 62 will also provide favorable results. Thus
all*combinations of the Fc variants in
Table 62 are contemplated. Likewise, combinations of any of the Fc variants in
Table 62 with other
discovered or undiscovered Fc variants may also provide favorable properties,
and these
combinations are also contemplated. Furthermore, it is anticipated from these
results that other
substitutions at positions 234, 235, 239, 240, 243, 264, 266, 325, 328, 330,
and 332 may also provide
favorable binding enhancements and specificities, and thus substitutions at
these positions other than
those presented in Table 62 are contemplated.
[217] Example 4: Reduced binding to FcyRs
As discussed, although there is a need for greater effector function, for some
antibody therapeutics,
reduced or eliminated effector function may be desired. Several Fc variants in
Table 62 substantially
reduce or ablate FcyR binding, and thus may find use in antibodies and Fc
fusions wherein effector
function is undesirable. AlphaScreen TM binding data for some examples of such
variants are shown
in Figures 19a and 19b. These Fc variants, as well as their use in
combination, may find use for
eliminating effector function when desired, for example in antibodies and Fc
fusions whose
mechanism of action involves blocking or antagonism but not killing of the
cells bearing target antigen.
[218] Example 5: Aglycosylated Fc variants
134
CA 02766627 2012-01-23
52620-2D
As discussed, one goal of the current experiments was to obtain optimized
aglycosylated Fc variants.
Several Fc variants provide significant progress towards this goal. Because it
is the site of
glycosylafion, substitution at N297 results in an aglycosylated Fc_ Whereas
all other Fc variants that
comprise a substitution at N297 completely ablate FcyR binding, N297D/I332E
has significant binding
affinity for FcyRIlla, shown in Table 62 and illustrated in Figure 20. The
exact reason for this result is
uncertain in the absence of a high-resolution structure for this variant,
although the computational
screening predictions suggest that it is potentially due to a combination of
new favorable FeircyR
interactions and favorable electrostatic properties. Indeed other
electrostatic substitutions are
envisioned for further optimization of aglycosylated Fc. Table 62 shows that
other aglycosylated Fc
variants such as S2390/N297D/I332E and N2970/A330Y/I332E provide binding
enhancements that
bring affinity for FcyRIlla within 0.28- and 0.43-fold respectively of
glycosylated WT alemtuzumab.
Combinations of these variants with other Fc variants that enhance FcyR
binding are contemplated,
with the goal of obtaining aglycosylated Fc variants that bind one or more
FcyRs with affinity that is
approximately the same as or even better than glycosylated parent Fc. An
additional set of promising
Fc variants provide stability and solubility enhancements in the absence of
carbohydrate. Fc variants
that comprise substitutions at positions 241. 243. 262, and 264. positions
that do not Mediate FyR
binding but do determine the interface between the carbohydrate and Fe. ablate
FyR binding,
presumably because they perturb the conformation of the carbohydrate. In
deglycosylated form,
however, Fc variants F241E/F243R/V262E/V264R, F241E/F243QN262TN264E,
F241R/F24301V262TN264R. and F241E/F243Y/V262TN264R show stronger binding to
FcyRIlla
than in glycosylated form, as shown by the AlphaScreenTh data in Figure 21.
This result indicates
that these are key positions for optimization of the structure, stability,
solubility, and function of
aglycosylated Fc. Together these results suggests that protein engineering can
be used to restore
the favorable functional and solution properties of antibodies and Fc fusions
in the absence of
carbohydrate, and pave the way for aglycosylated antibodies and Fc fusions
with favorable solution
properties and full functionality that comprise substitutions at these and
other Fc positions.
[2191 Example 6. Affinity of Fc variants for polymorphic forms of FcyRIlla
As discussed above, an important parameter of Fc-mediated effector function is
the affinity of Fc for
both V158 and F158 polymorphic forms of FcyRIlla. AlphaScreen TM data
comparing binding of select
variants to the two receptor allotypes are shown in Figure 22a (V158 FcyRIlla)
and Figure 22b (F158
= FcyRIlla)., As can be seen, all variants improve binding to both FcyRIlla
allotypes. These data
indicate that those Fc variants of the present invention with enhanced
effector function will be broadly
applicable to the entire patient population, and that enhancement to clinical
efficacy will potentially be
greatest for the low responsive patient population who need it most.
[2201 The FcyR binding affinities of these Fc variants were further
investigated using Surface
Plasmon Resonance (SPR) (Biacore, Uppsala, Sweden). SPR is a sensitive and
extremely
quantitative method that allows for the measurement of binding affinities of
protein-protein
135
CA 02766627 2012-01-23
52620-2D
interactions, and has been used to effectively measure Fc/FcyR binding (Radaev
et at, 2001, J Blot
Chem 276:16478-16483). SPR thus provides an excellent complementary binding
assay to the
Alpha&menu" assay. His-tagged V158 FcyRIlla was immobilized to an SPR chip,
and WT and Fe
variant alemtuzumab antibodies were flowed over the chip at a range of
concentrations. Binding
constants were obtained from fitting the data using standard curve-fitting
methods. Table 63 presents
dissociation constants (Kd) for binding of select Fc variants to V158 FcyRIlla
and F158 FcyRIlla
obtained using SPR, and compares these with 1050s obtained from the
AlphaScreenTM assay. By
dividing the Kd and IC50 for each variant by that of WT alemtuzumab, the fold-
improvements over WT
(Fold) are obtained.
Table 63
SPR SPR
V158 F158 AlphaScreenlm AlphaScreenl'"
Fcy_RIlla FcyRIlla V158 FcyRIlla F158 FcyRIlla
Kd Kd IC50 1050
(nM)
Fold (nM) Fold (nM) Fold (nM) Fold
WT 68 730 6.4 17.2
V26.41 64 1.1 550 1.3 4.5 1.4 11.5 1_5
I332E 31 2.2 72 10.1 1.0 6.4 2.5 6.9
V264111332E 17 4.0 52 14.0 0.5 12.8 1.1 15.6
S298A ___________ 52 1.3 285 2.6 2.9 2.2 12.0 1.4
S298A/E333A/
39 1.7 156 4.7 2.5 2.6 7.5 2.3
K334A
[221] The SPR data corroborate the improvements to FcyRIlla affinity observed
by AlphaScreen 114
assay. Table 63 further indicates the superiority of V2641/1332E and 1332E
over S298A and
S298A/E333A/K334A; whereas S298A/E333A/K334A improves Fc binding to V158 and
F158 FcyRIlla
by 1.7-fold and 4.7-fold respectively, 1332E shows binding enhancements of 2.2-
fold and 10.1-fold
respectively, and V264I/1332E shows binding .enhancements of 4.0-fold and 14-
fold respectively. Also
worth noting is that the affinity of V2641/I332E for F158 FcyRIlla (52 nM) is
better than that of WT for
the V158 allotype (68 nM), suggesting that this Fc variant, as well as those
with even greater
Improvements in binding, may enable the clinical efficacy of antibodies for
the low responsive patient
population to achieve that currently possible for high responders. The
correlation between the SPR
= and AlphaScreenlm binding measurements are shown in Figures 23a - 23d.
Figures 23a and 23b =
show the Kd - IC50 correlations for binding to V158 FcyRIlla and F158 FcyRIlla
respectively, and
Figures 23c and 23d show the fold-improvement correlations for binding to V158
FcyRIlla and F158
RIRIlla respectively. The good fits of these data to straight lines (r2 =
0.9,12 :=-=-= 0.84, r2 = 0.98, and 12
= 0.90) support the accuracy the AlphaScreenn" measurements, and validate its
use for determining
the relative FcyR binding affinities of Fc variants.
136
CA 02766627 2012-01-23
52620-2D
[222] SPR data were also acquired for binding of select trastuzumab Fc
variants to human V158
FcyRIlla, F158 FcyRilla, and FcyRIlb. These data are shown in Table 64. The Fc
variants tested
show substantial binding enhancements to the activating receptor FcyRIlla,
with over 100-fold tighter
binding observed for interaction of S2390/1332E/S298A with F158 FcyRIlla.
Furthermore, for the best
FcyR(lla binders, F158 FcyRIlla/FcyRIlb ratios of 3 ¨ 4 are observed.
Table 64
SPR SPR SPR
V158 FcyRIlla F158 FcyRIlla FcyRIlb
Kd Kd IC50
Fold Fold Fold
OM) (nM) (nM)
WT 363.5 503 769
V2641/I332E 76.9 4.7 252 2.0 756 1.0
V2641/1332E/
113.0 3.2 88 5.7 - 353 2.2
A3301..
S230D/1332EI
8.2 44.3 8.9 56.5 46 16.7
A330t.
S239D/1332E/
8.7 41.8 4.9 102.7 32 24.0
S298A
S239D/1332E/
12.7 28.6 6.3 79.8 35 22.0
V2641/A330L
[223] Example 7. ADCC of Fc variants
In order to determine the effect on effector function, cell-based ADCC assays
were performed on
select Fc variants. ADCC was measured using the DELFIA EuTDA-based
cytotoxicity assay (Perkin
Elmer, MA) with purified human peripheral blood monocytes (PBMCs) as effector
cells. Target cells
were loaded with BATDA at 1x106 cells/nil, washed 4 times and seeded into 96-
well plate at 10,000
cells/well. The target cells were then opsonized using Fc variant or WT
antibodies at the indicated
final concentration. Human PBMCs, isolated from buffy-coat were added at the
indicated fold-excess
of target cells and the plate was incubated at 37 C for 4 firs. The co-
cultured cells were centrifuged at
500xg, supernatants were transferred to a separate plate and incubated with Eu
solution, and relative
fluorescence units were measured using a Packard Fusion TM o'-FP HT reader
(Packard Biosciences,
IL)_ Samples were run in triplicate to provide error estimates (n=3, +/-
S.D.). PBMCs were allotyped
for the V158 or F158 FcyRIlla allotype using PCR.
[22/11 ADCC assays were run on Fc variant and WT alemtuzumab using DoHH-2
lymphoma target
cells. Figure 24a is a bar graph showing the ADCC of these proteins at 10
ng/ml antibody. Results
show that alemtuzumab Fc variants 1332E, V264I, and 1332EA/264I have
substantially enhanced
ADCC compared to WT alemtuzumab, with the relative ADCC enhancements
proportional to their
137
CA 02766627 2012-01-23
52620-2D
binding improvements to FcyRIlla as indicated by AlphaScreen."' assay and SPR.
The dose
=
dependence of ADCC on antibody concentration is shown in Figure 24b. The
binding data were
normalized to the minimum and maximum fluorescence signal for each particular
curve, provided by
the baselines at low and high antibody concentrations respectively. The data
were fit to a sigmoidal =
dose-response model using nonlinear regression, represented by the curve in
the figure. The fits
enable determination of the effective concentration 50% (EC50) (i.e. the
concentration required for
50% effectiveness), which provides the relative enhancements to ADCC for each
Fc variant. The
EC50s for these binding data are analogous to the 1050s obtained from the
AlphaScreen,'"
competition data, and derivation of these values is thus analogous to that
described in Example 2 and
Figure 11. In Figure 24b, the log(EC50)s. obtained from the fits to the data,
for WT, V2641/I332E, and
52390/1332E alemtuzumab are 0.99, 0.60, and 0.49 respectively, and therefore
their respective
EC50s are 9.9, 4.0, and 3Ø Thus V2641/I332E and S239E/1332E provide a 2.5-
fold and 3.3-fold
enhancement respectively in ADCC over WT alemtuzumab using PBMCs expressing
heterozygous
V158/F158.FcyRIlla. These data are summarized in Table 65 below.
Table 65
log(EC50) EC50 (ng/ml) Fold Improvement Over WT
WT 0.99 9_9
V2641/1332E 0.60 - 4.0 2.5
S23913/1332E 0.49 3.0 3.3
{2251 In order to determine whether these ADCC enhancements are broadly
applicable to
antibodies, select Fc variants were evaluated in the context of trastuzumab
and rituximab. ADCC
assays were run on Fc Variant and WT trastuzumab using two breast carcinoma
target cell lines
8T474 and Sk-Br-3. Figure 25a shows a bar graph illustrating ADCC at 1 ng/m1
antibody. Results
indicate that V264I and V2641/I332E trastuzumab provide substantially enhanced
ADCC compared to
WT trastuzumab, with the relative ADCC enhancements proportional to their
binding improvements to
FcyRIlla as indicated by AlphaScreenTh assay and SPR. Figures 25b and 25c show
the dose
dependence of ADCC on antibody concentration for select Fc variants. The EC50s
obtained from the
fits of these data and the relative fold-improvements in ADCC are provided in
Table 66 below.
Significant ADCC improvements are observed for I332E trastuzumab when combined
with A330L and
A330Y. Furthermore, S239D/A330U1332E provides a substantial ADCC enhancement,
greater than
300-fold for PBMCs expressing homozygous F158/F158 FcyRIlla, relative to WT
trastuzumab and
S298A/E333A/K334A, consistent with the FcyR binding data observed by the
AlphaScreen."4 assay
and SPR.
138
CA 02766627 2012-01-23
52620-2D
Table 66
log(EC50) EC50 (ng/ml) Fold Improvement Over WT
Figure 25b
WT 1.1 11.5
1332E - 0_34 2_2 5.2
A330Y/1332E -0.04 0.9 12.8
A330U1332E 0.04 1.1 10.5
Figure 25d
WT -0.15 0.71
hS298A/E333A/K334A -0.72 0.20 3.6
S23913/A330111332E -2.65 0.0022 323
[2261 ADCC assays were run on V2641/I332E, WT, and 8298A/0333A/K334A rituximab
using
WIL2-S lymphoma target cells. Figure 26a presents a bar graph showing the ADCC
of these proteins
at 1 ng/mt antibody. Results indicate that V2641/1332E rituximab provides
substantially enhanced
ADCC relative to WI rituximab, as well as superior ADCC to S298A/0333A/K334A.
consistent with
the FcyRIlla binding improvements observed by AlphaScreenTU assay and SPR.
Figures 26b and
26c show the dose dependence of ADCC on antibody concentration for select Fc
variants. The
EC5Os obtained from the fits of these data and the relative fold-improvements
in ADCC are provided
in Table 67 below. As can be seen S239D/1332F1A330L rituximab provides greater
than 900-fold
enhancement in EC50 over WT for PB/VICs expressing homozygous F158/F158
FcyRIlla. The
differences in ADCC enhancements observed for alemtuzumab, trastuzumab, and
rituximab are likely
due to the use of different PBMCs, different antibodies, and different target
cell lines.
Table 67
log(EC50) EC50 (ng/ml) Fold Improvement Over WT
_
Figure 26b -
WT 0.23 1.7
=
S298A/E333A/K334A -0.44 0.37 4_6
V2641/1332E -0.83 0.15 11.3
= Figure 26c
, =
WT 0.77 5.9
S239D/1332E/A3301 -2.20 0.0063 937
[2271 Thus far, ADCC data has been normalized such that the lower and upper
baselines of each
Fc polypeptide are set to the minimal and maximal fluorescence signal for that
specific Fc
139
CA 02766627 2012-01-23
, 52620-2D
polypeptide, typically being the fluorescence signal at the lowest and highest
antibody concentrations
respectively. Although presenting the data in this matter enables a
straightforward visual comparison
of the relative EC50s of different antibodies (hence the reason for presenting
them in this way),
important information regarding the absolute level of effector function
achieved by each Fc
polypeptide is lost. Figures 27a and 27b present cell-based ADCC data for
trastuzumab and
rituximab respectively that have been normalized according to the absolute
minimal lysis for the
assay, provided by the fluorescence signal of target cells in the presence of
PBMCs alone (no
antibody), and the absolute maximal lysis for the assay, provided by the
fluorescence signal of target
cells in the presence of Triton X1000. The graphs show that the antibodies
differ not only in their
EC50, reflecting their relative potency, but also in the maximal level of ADCC
attainable by the
antibodies at saturating concentrations, reflecting their relative efficacy.
Thus far these two terms,
potency and efficacy, have been used loosely to refer to desired clinical
properties. In the current
experimental context, however, they are denoted as specific quantities, and
therefore are here
explicitly defined. By "potencV. as used in the current experimental context
is meant the EC50 of an
antibody or Fc fusion. By "efficacy" as used in the current experimental
context is meant the maximal
possible effector function of an antibody or Fc fusion at saturating levels.
In addition to the substantial
enhancements to potency described thus far, Figures 27a and 27b show that the
Fc variants of the
present invention provide greater than.100% enhancements in efficacy over WT
trastuzumab and
rituximab_
[2281 A critical parameter governing the clinical efficacy of anti-cancer
antibodies is the expression
level of target antigen on the surface of tumor cells. Thus a major clinical
advantage of Fc variants
that enhance ADCC may be that it enables the targeting of tumors that express
lower levels of
antigen. In To test this hypothesis, WT and Fe variant trastuzumab antibodies
were tested for their
ability to mediate ADCC against different cell lines expressing varying levels
of the Her2/neu target
antigen. ADCC assays were run with various cell lines expressing amplified to
low levels of Her2/neu
receptor, including Sk-Br-3 (1x106 copies), Sk0V3 (-1x106), OVCAR3(-1x104),
and MCF-7 (-3x103
copies), using the DELFIA EuTDA Cytotoxicity kit (PerkinElmer, Boston, MA).
Target cells were
loaded with BATDA in batch for 25 minutes, washed multiple times with medium
and seeded at
10,000 cells per well in 96-well plates. Target cells were opsonized for 15
minutes with various
antibodies and concentrations (final conc. ranging from 100 ng/ml to .0316
ng/ml in 1/2 log steps,
including no treatment control). Human PBMCs, isolated from buffy-coat and
allotyped as
= homozygous F158/F158 FcyRIlla were then added to opsonized cells at 25-
fold excess and co-
cultured at 37 C for 4 hrs. Thereafter, plates were centrifuged, supernatants
were removed and
treated with Eu3+ solution, and relative fluorescence units (correlating to
the level of cell lysis) were
measured using a Packard FusionTM a-FP HT reader (PerkinElmer, Boston, MA).
The experiment
was carried out in triplicates. Figure 28 shows the ADCC data comparing WT and
Fc variant
trastuzumab against the four different Her2/neu` cell lines_ The S239D/1332E
and
S2390/1332E/A330L variants provide substantial ADCC enhancements over WT
trastuzumab at high,
140
CA 02766627 2012-01-23
52620-2D
moderate, and low expression levels of target antigen. This result suggests
that the Fc variants of the
present invention may broaden the therapeutic window of anti-cancer
antibodies.
[2291 Natural killer (NK) cells are a subpopulation of cells present in PBMCs
that are thought to play
a significant role in ADCC. Select Fc variants were tested in a cell-based
ADCC assay in which
natural killer (NK) cells rather than PBMCs were used as effector cells. In
this assay the release of
endogenous lactose dehydrogenase (LDH), rather than EuTDA, was used to monitor
cell lysis. Figure
29 shows that the Fc variants show substantial ADCC enhancement when NK cells
are used as
effector cells. Furthermore, together with previous assays, the results
indicate that the Fc variants of
the present invention show substantial ADCC enhancements regardless of the
type of effector cell or
the detection method used.
[2301 Example 8. ADCP of Fc Variants
Another important FcyR-mediated effector function is ADCP. Phagocytosis of
target cancer cells may
not only lead to the immediate destruction of target cells, but because
phagocytosis is a potential
mechanism for antigen uptake and processing by antigen presenting cells,
enhanced ADCP may also
improve the capacity of the antibody or Fc fusion to elicit an adaptive immune
response. The ability of
the Fc variants of the present invention to mediate ADCP was therefore
investigated. Monocytes
were isolated from heterozygous V158/F158 FcyRIlla PBMCs using a Percoll
gradient_ After one
week in culture in the presence of 0.1 ng/ml, differentiated macrophages were
detached with
EDTA/PBS- and labeled with the lipophilic fluorophore, PKH26, according to the
manufacturers
protocol (Sigma, St Louis, Mo). Sk-Br-3 target cells were labeled with P1(I-
167 (Sigma, St Louis, Mo),
seeded in a 96-well plate at 20,000 cells per well, and treated with
designated final concentrations of
WT or Fc variant trastuzumab. PKH26-labeled macrophages were then added to the
opsonized,
labeled Sk-Br-3 cells at 20,000 cells per well and the cells were co-cultured
for 18 hrs before
processing cells for analysis of dual label flow cytometry. Percent
phagocytosis was determined as
the number of cells co-labeled with PKH76 and PKH26 (macrophage + Sk-Br-3)
over the total number
of Sk-Br-3 in the population (phagocytosed + non-phagocytosed) after 10,000
counts. Figure 30
shows data comparing WT and Fc variant trastuzumab at various antibody
concentrations. The
results indicate that the S2390/1332E/A330L variant provides a significant
enhancement in ADCP
over WT trastuzumab.
[2311 Example 9. Complement binding and activation by Fc variants
=
Complement protein C1g binds to a site on Fc that is proximal to the FcyR
binding site, and therefore
it was prudent to determine whether the Fc variants have maintained their
capacity to recruit and
activate complement. The AlphaScreen assay was used to measure binding of
select Fc variants
to the complement protein C1g. The assay was carried out with biotinylated WT
alemtuzumab
antibody attached to streptavidin donor beads as described in Example 2, and
using C1g coupled
directly to acceptor beads. Binding data of V2641, 1332E, S239E, and
V264I/1332E rituximab shown in
-
141
CA 02766627 2012-01-23
52620-2D
Figure 31a indicate that C1q binding is uncompromised. Cell-based CDC assays
were also
performed on select Fe variants to investigate whether Fc variants maintain
the capacity to activate
complement. Alamar Blue was used to monitor lysis of Fc variant and WT
rituximab-opsonized WIL2-
S lymphoma cells by human serum complement (Quidel, San Diego, CA). The data
in Figure 31b
show that CDC is uncompromised for the Fc variants S239E, V264I, and
V2641/I332E rituximab. In
contrast, Figure 31c shows that CDC of the Fe variant S2390/1332E/A3301_ is
completely ablated,
whereas the S23913/1332E variant mediates CDC that is comparable to WT
rituximab. These results
indicate that protein engineering can be used to distinguish between different
effector functions. Such
control will not only enable the generation of antibodies and Fc fusions with
properties tailored for a
desired clinical outcome, but also provide a unique set of reagents with which
to experimentally
investigate effector function biology.
[232] Example 10. Protein A and FcRn binding by Fc variants
As discussed, bacterial proteins A and G and the neonatal Fc receptor FcRn
bind to the Fc region
between the Cy2 and Cy3 domains. Protein A is frequently employed for antibody
purification, and
FcRn plays a key role in antibody pharmacokinetics and transport. It was
therefore important to
investigate the ability of the Fc variants of the present invention to bind
protein A and FcRn. The
AlphaScreen Tm assay was used to measure binding of select Fc variants to
protein A and human
FcRn using biotinylated WT alemtuzumab antibody attached to streptavidin donor
beads as described
in Example 2, and using protein A and FcRn coupled directly to acceptor beads.
The binding data are
shown in Figure 32 for protein A and Figure 33 for FcRn. The results indicate
that the Cy2-Cy3 hinge
region is unaffected by the Fc substitutions, and importantly that the
capacity of the Fc variants to bind
protein A and FcRn is uncompromised.
[233] Example 11. Capacity of Fc variants to bind mouse FcyRs
Optimization of Fc to nonhuman FcyRs may be useful for experimentally testing
Fc variants in animal
models_ For example, when tested in mice (for example nude mice, SCID mice,
xenograft mice,
and/or transgenic mice), antibodies and Fc fusions that comprise Fc variants
that are optimized for
one or more mouse FcyRs may provide valuable information with regard to
clinical efficacy,
mechanism of action, and the like. In order to evaluate whether the Fc
variants of the present
invention may be useful in such experiments, affinity of select Fc variants
for mouse FcyRIII was
measured using the AlphaScreenrm assay. The AlphaScreenTM assay was carried
out using
biotinylated WT alemtuzumab attached to streptavidin donor beads as described
in Example 2, and
GST-tagged mouse FcyRIII bound to glutathione chelate acceptor beads,
expressed and purified as
described in Example 2. These binding data are shown in Figures 34a and 34b in
the context of
= alemtuzumab and trastuzumab respectively. Results show that some Fc
variants that enhance
binding to human FcyRIlla also enhance binding to mouse FcyR111. The
enhancement of mouse
effector function by the Fc variants was investigated by performing the
aforementioned cell-based
ADCC assays using mouse rather than human PBMC's. Figure 35 shows that the
142
CA 02766627 2012-01-23
- 52620-2D
S239D/1332E1A330L trastuzumab variant provides substantial ADCC enhancement
over WT in the
presence of mouse immune cells. This result indicates that the Fc variants of
the present invention,
or other Fc variants that are optimized for nonhuman FeyRs, may find use in
experiments that use
animal models.
12341 Example 12. Validation of Fc variants expressed in CHO cells
Whereas the Fc variants of the present invention were expressed in 2931 cells
for screening
purposes, large scale production of antibodies is typically carried out by
expression in Chinese
Hamster Ovary (CHO) cell lines. In order to evaluate the properties of CHO-
expressed Fc variants,
select Fc variants and WT alemtuzumab were expressed in CHO cells and purified
as described in
Example 2. Figure 36 shows AlphaScreen a' data comparing binding of CHO- and
2931- expressed
Fc variant and WT alemtuzumab to human V158 FcyRIlla. The results indicate
that the Fc variants of
the present invention show comparable FcyR binding enhancements whether
expressed in 293T or
CHO.
[235] Example 13. Enhancement of Fc variants in Fucose Minus Strain.
Combinations of the Fc variants of the present invention with other Fc
modifications are contemplated
with the goal of generating novel antibodies or Fc fusions with optimized
properties. It may be
beneficial to combine the Fc Variants of the present invention with other Fc
modifications, including
modifications that alter effector function or interaction with one or more Fc
ligands. Such combination
may provide additive, synergistic, or novel properties in antibodies or Fc
fusions. For example, a
number of methods exist for engineering different glycoforms of Fc that alter
effector function.
Engineered glycoforms may be generated by a variety of methods known in the
art, many of these
techniques are based on controlling the level of fucosylated and/or bisecting
oligosaccharides that are
covalently attached to the Fc region_ One method for engineering Fc glycoforms
is to express the Fc
polypeptide in a cell line that generates altered glycoforms, for example Lec-
13 CHO cells. In order to
investigate the properties of Fc variants combined with engineered glycoforms,
WT and V209
(82390/1332E/A330L) trastuzumab were expressed in Lec-13 CHO cells and
purified as described
above. Figure 37a shows AlphaScreenT" binding data comparing the binding to
human V158
FcyRIlla by WT and V209 trastuzumab expressed in 293T. CHO, and Lec-13 cells.
The results show
that there is substantial synergy between the engineered glycoforms produced
by this cell line and the
Fc variants of the present invention. The cell-based ADCC assay, shown in
Figure 37b, supports this
result. Together these data indicate that other Fc modifications, particularly
engineered glycoforms,
may be combined with the Fc variants of the present invention to generate
antibodies and Fc fusions
with optimized effector functions.
[236] Example 14. Therapeutic application of Fc variants
A number of Fc variants described in the present invention have significant
potential for improving the
therapeutic efficacy of anticancer antibodies. For illustration purposes, a
number of Fc variants of the
. -
=
143
CA 02766627 2012-01-23
- 52620-2D
k
present invention have been incorporated into (lie sequence of the antibody
rituximab. The WT
rituximab light chain and heavy chain, described in US 5,736,137, are provided
in Figures 38a and
38b. The improved anti-0O20 antibody sequences are provided in Figure 38c. The
improved anti-
0O20 antibody sequences comprise at least non-WT amino acid selected from the
group consisting of
X1, X2, X3, X4, Xs. X6, X7, and Xg. These improved anti-CD20 antibody
sequences may also comprise
a substitution Z1 and/or Z2. The use of rituximab here is solely an example,
and is not meant to
constrain application of the Fc variants to this antibody or any other
particular antibody or Fc fusion.
[237] Example 15. A complete structure/function analysis Fc 1 Fc ligand
specificity
It is clear from the results of these experiments that protein engineering is
a powerful tool for mining
Fc substitutions that significantly alter its biological function and
specificity. Given the profound
clinical value of antibodies and Fc fusions, the implication is that the
protein engineering methods of
the present invention can be used to tune the clinical properties of these
important biotherapeutics.
Such capability, however, demands a more complete understanding of the
relationship between the
structure and function of Fc and Fc ligands. In addition, the lack of
available information on the
determinants of Fc I Fc ligand specificity means that it is not possible to
actively design Fc variants
with all desired properties as target goals. Thus it is likely that, despite
the aggressive experimental
effort described in the present invention, there are therapeutically useful Fc
variants that have not
been mined, and biochemical properties of Fc variants that remain
undiscovered. Equally important
to obtaining new Fc variants for biotherapeu tic application is the ability to
improve the predictiveness
of the design method, thereby permitting variants to be identified even more
efficiently. Towards
these goals, a more thorough characterization of Fc / Fc ligand biology was
carried out. This
included: 1) an expansion of the primary screen to include all relevant Fc
ligands, and 2) an increase
in the number of Fc variants to explore a greater set of substitutions at all
relevant Fc positions.
Together this broadened approach will enable a more thorough mining of useful
Fc variants, provide a
greater understanding_of Fc / Fc ligand specificity and biology, and provide a
greater data set to
enable a rigorous quantitative assessment of the predictiveness of the design
methods.
[238] Expansion of the primary screen
In order to better characterize the structural and functional determinants of
Fc specificity, the primary
screen was expanded to include all relevant Fc ligands. Thus all Fc variants
are tested in parrallel for
binding to FcyR1, FcyRIla, FcyRIlb, FeyRIlc, FcyRIlla (Va1158 isoform), FcRn,
and C1q. The =
AlphaScreenTm assay was used as described above. All Fc variants were screened
in the context of
either alemtuzumab or trastuzumab according to Table 62. Table 68 shows an
example of the
parrallel screen for a set of substitutions at Fc positions 234 and 235. In
this table, light grey indicates
that Fc variant! Fc ligand affinity is 0.5-fold or less than WT, medium grey
indicates that Fc variant /
Fc ligand affinity is within 0.5 ¨ 2.0 of WT, dark grey indicates that Fe
variant! Fc ligand affinity is
increased by 2-fold Or greater, and white indicates that the Fc variant / Fc
ligand interaction was not
measured or that the data did not allow an accurate determination of affinity.
Thus Fc variants are
144
CA 02766627 2012-01-23
52620-2D
_
grouped as those that significantly decrease, those that do not substantially
alter, and those that
significantly increase binding to a given Fc ligand. Visualization of the data
in this way provides a
structure/function map of Fc, enabling a straightforward interpretation of the
results for each position
such that useful and interesting variants can be efficiently identified, and
such that predictiveness of
the design method can be assessed in a practical manner.
Table 68
Variant Substitution(s) FcyRI FcyRlla Fcy1211b FcyRlIc FcyFUlla FcRn C1q
111 L2340 ftlig;rricIfti
) 441
112 L234E h:.r.W;tiE0-71,::Api;i16.5Z:-;,ulr' ''.t-orgt=
=
113 1234N
ir,p.::1:1;Y7....7.0F40...ZY' '1141-S11rtoO$j'-
114 L2340 tP,
115 L234T r 41 .99; '
116 L234H 41j'
41# 11'4) = =
117 L234Y ;,..01'3,4
1 441:VAA,Ogzele TON'
18 ' L234I
119 L234V Eb441.14.4.;51237* - .1t4'
120 L234 F [111.4.-:`29m1-4,1004.AT 49, 35 02
121 12350 F*0-41?.10,PROPL.-VAMIlle.'.7Vtli
122 L235S NOZ00.14:1*Agria*k4A,,01
123 I-2351\1L003 l56 07l 1034 rl 10 t6'
124 12350 t4438'I 08 89I4
125 1235T k0:10,
416.3.0;e1093k4:72!;;;,:::,Q:16:,K;0 9:\49,130-.:
126 1235H ofb-6:ViiTIA. .
127 L235Y P"Ø0104;%i 7-'0Y 1
j"..444.,.)454rAilteA
- .
128 12351 0 :9 .24,4 ow. it:,
129 L235V 0 22.ti4f1,41.-M41ktge-W:71(V,y-e. .1%/.
130 1235F C.4,43'f
UMNT'...7.414E,PAigg.,0710t..iõ,V
[239] A number of substitutions at positions 234 and 235 show differenct
specificities for binding to
the various Fc ligands. Although the differences in some cases are subtle, the
results indicate that it
is indeed possible to engineer Fc specificity for different Fc ligands, even
at at the FcyR interface
where a number of highly homologous receptors bind to the same site. Other Fc
variants that provide
more distinct affinity differences are presented in Table 69.
Table 69
Variant Substitution(s) FcyRI FcyRIla FcyRIlb FcyR(Ic FcyRfIla FcRn C1q
107 A330Y/I 332E st 33# k izzzoottle.wt
,F1"4õ?
109 A3301/1332E ..õ7*.VICV:91
167 L320E/I332E 09tj7.7.:!t 0.31
171 L3281/I332E 142rirn.:"Vii0X.!:g:41rA/V.:A40a01111-70,1,
174 L328A 11;Sieg4:3;.4:õ
[240] These data show even more convincingly that it is possible to tune Fc
for Fc ligand specificity,
often by using very subtle mutational differences. For example, the
A330Y/I332E variant enhances
145
CA 02766627 2012-01-23
- 52620-2D
binding to all FcyRs, particularly FcyRIlla, as well as FcRn, while
maintaining binding to Cl q.
However the A3001/1332E variant shows enhanced binding to FcyR1 and FcyRIlla,
but has WT affinity
for the FcyRII's. In contrast, mutations at L328 provide preferential
enhancement of the FcyR11's over
FcyRI and FcyRilla. In the case of the L328E/I332E variant, affinity for all
FcyRIrs is increased,
whereas L328T/I332E provides a clear enhancement specificity profile of
FcyRfic > FcyRIlb > FcyRIla.
In contrast, L328A significantly enhances binding to FcyRIla, but provides WT
affinity for all other
FcyR's including FcyRilb and FcyRlic. It is clear from these results that very
subtle mutational
differences can provide substantial differences in specificity. Accordingly,
collections of Fc variants
such as these will not only enable the generation of antibodies and Fc fusions
that have effector
function tailored for the desired outcome, but they also provide a unique set
of reagents with which to
experimentally investigate and characterize effector function biology.
[2411 Expansion of the Fc variant set
Because of the incomplete information concerning the structural and functional
determinants of Fc
Fc ligand interaction, it has not been possible to actively engineer Fc for
all desired optimization goals.
The distinct specificity differences observed in Tables 68 and 69 to the
various FcyRs were due more
to the aggressive screening approach of the present invention; these Fc
variants were not actively
designed with their particular properties as the target goals due to the lack
of structural information for
binding of Fc to the different FcyRs, as well as the lack of understanding of
how the structure and
flexibility of the hinge impacts FcyR binding. Indeed the decision to explore
a large number and
variety of substitutions at these positions 234 and 235 was based on the
knowledge that they are near
the Fc/FcyR binding site, that mutations at these positions affect FcyR
binding, and that according to
computational screening calculations a large number and variety of
substitutions are permissible at
these positions. Overall, the lack of structural information on the
determinants of Fc/FcyR specificity,
the lack of high-resolution structural information for the Fc/Clq complex, and
the inability to account
for indirect affects of substitutions on Fc / Fc ligand binding, together make
it a certainty that all of the
interesting and potentially useful Fc variants will not be explored using the
current engineering
methods. In order to fully mine useful Fc variants, as well as to obtain a
more complete picture of the
structural and function determinants of Fc / Fc ligand interaction, the set of
Fc variants was expanded
to explore a broader set of mutations. All Fc positions at or near the binding
sites for FcyR's and Cl q,
chosen by visual inspection of the available structures and using the
information provided by the
= -results of previous Fc variant screening, were saturated such that all
substitutions were constructed .
that have not been tested previously. At Fc positions significantly distal to
the FcyR and Clq binding
sites, a subset of select substitutions were designed based on predicted
energies in previously
described computational screening calculations, and based on available data
from existing Fc
variants. This new set of Fc variants, 576 total, is presented in Table 70.
=
146
CA 02766627 2012-01-23
,52620-2D
Table 70
Position WT Substitution(s) Variant
221 D KY 801-802
222 K EY 513-514
223 T EK 803-804
224 H EY 805-806
225 T EKW 807-809
227 P EKYG 705-708
228 P EKYG 709-712
230 P EYG 609-611
231 A EKYPG 612-616
232 P EKYG 321-324
233 E NQKRSTHAVLIFMYWG 617-632
234 L KRSAMWPG 417-424
235 L EKRAMWPG 425-432
236 G DENQKRSTHAVLIFMYWP 713-730
237 G DENQKRSTHVLIFMYWP 731-747
238 P DENQKRSTHVLIFMYWG 748-764
239 S QKRVLIMWPG 325-334
241 F DEY 335-337
243 F E 515
246 K - DEHY 810-813
249 D QHY 814-816
255 R EY 817-818
258 E SHY 819-821
260 T DEHY 822-825
262 V EF 826-827
264 V DENQKRSHWPG 433-443
265 D QKRSTHVLIFMYWP 444-457
267 S EQKRVLIFMYWP 338-349
268 H DEQKRTVLIFMWPG 350-363
269 E KSVIMWPG 765-772
270 D RSLIFMYWPG 516-525
271 P DENQKRSTHAVL1FMYWG 526-543
272 E DRTHVLFMWPG 633-643
274 K DNSHVIFMWPG 644-654
275 F L 828
276 N DTHVIFMWPG 655-664
278 Y DNQRSHVLIMPG 665-676
=
280 D KLWPG 544-548
= 281 G DKYP 829-
832
282 V EKYPG 833-837
283 E KHLYPG 838-843
284 V ENTLY 844-848
285 H DEQKYW 773-778
286 N EYPG 779-782
288 K DEY 783-785
290 K DNHLW 549-553
=
147
CA 02766627 2012-01-23
,52620-2D
Table 70 (continued)
Position WT Su bstitution(s) Variant
291 p D.EGTHIG 849-855
292 R DETY 786-789
293 E NRSTHVLIFMYWPG 554-567
294 E KRSTHVLIFMYWPG 568-581
295 Q DENRSTHVIFMYWPG 582-596
296 Y KRAVMG 597-602
297* N QKRTHVLIFMYWPG 856-869
298 S DEQKRIFMYW 364-373
299 T DENQKRLFMYWPG 374-386
300 Y DENQKRSTHAVMWPG 387-401
301 R DEHY 870-873
303 V D E Y 874-876
304 S DNTHL 877-881
305 V E T Y 882-884
317 K E Q 885-886
318 E QHLY 887-890
320 K NSHVLFYWPG 677-686
322 K DSVIFYWPG 687-695
324 S HFMW PG 603-608
325 N KRSFMYWPG 696-704
326 K P 458
327 A EKRHVIFMYWP 459-469
328 L DOKRSTVIYWPG 470-481
329 P DENQKRSTHVLIMYWG 482-497
330 A EN TPG 402-406
331 P DQRTLIFMYW 498-507
332 I KRSVLFMWPG 407-416
333 E LFMP 508-511
334 K P 512
335 T NSHVL1FMWPG 790-800
336 I E K Y 891-893
337 S E N H 894-896
* Substitutions at 297 were made in the context of S2390/I332E
[241] Whereas particular embodiments of the invention have been described
above for purposes of
= illustration, itwill be appreciated by those skilled in the art that
numerous variations of the details may
be made without departing from the invention as described in the appended
claims.
148
CA 02766627 2012-01-23
=
DEMANDES OU BREVETS VOLUMINEUx
. LA PR SENTE PARTIE DE CETTE DENLA.NDE OU CE BREVETS
COMPREND PLUS D'UN TOME.
CECI EST LE TOME _____________________ DE a
NOTE: Pour les tomes additionels, veillez contacter te Bureau Canadien des
Brevets.
=
JUMBO APPLICATIONS PATENTS
THIS SECTION OF THE APPLICATION / PATENT CONTAINS MORE
THAN ONE VOLUME.
THIS IS VOLUME I OF _______________________________ .
NOTE: For additional volumes please contact the Canadian Patent Office.