Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02773064 2012-03-02
WO 2011/033194
PCT/FR2010/000625
-1-
NOUVEAU PROCEDE DE SYNTHESE DE L'IVABRADINE
ET DE SES SELS D'ADDITION
A UN ACIDE PHARMACEUTIQUEMENT ACCEPTABLE
La présente invention concerne un procédé de synthèse de l'ivabradine de
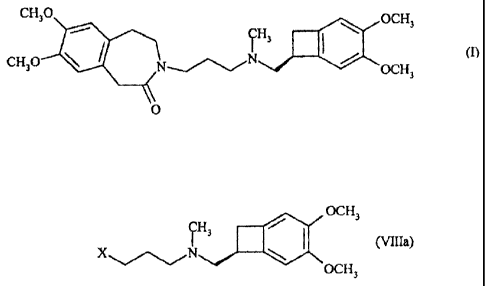
formule (I) :
CH30
0013
ILI3 = CH30 OCH3 (I)
0
ou 3- 3 -[ { [(75)-3,4-diméthoxybicyclo [4.2.0] octa-1,3 ,5-trién-7-yl]
méthyl } (méthypamino]
propyl } -7,8-diméthoxy-1,3,4,5-tétrahydro-2H-3-benzazépin-2-one,
de ses sels d'addition à un acide pharmaceutiquement acceptable et de leurs
hydrates.
L'ivabradine, ainsi que ses sels d'addition à un acide pharmaceutiquement
acceptable, et plus
particulièrement son chlorhydrate, possèdent des propriétés pharmacologiques
et
thérapeutiques très intéressantes, notamment des propriétés bradycardisantes,
qui rendent ces
composés utiles dans le traitement ou la prévention des différentes situations
cliniques
d'ischémie myocardique telles que l'angine de poitrine, l'infarctus du
myocarde et les troubles
du rythme associés, ainsi que dans les différentes pathologies comportant des
troubles du
rythme, notamment supra-ventriculaires, et dans l'insuffisance cardiaque.
La préparation et l'utilisation en thérapeutique de l'ivabradine et de ses
sels d'addition à un
acide pharmaceutiquement acceptable, et plus particulièrement de son
chlorhydrate, ont été
décrits dans le brevet européen EP 0 534 859.
Ce brevet décrit la synthèse du chlorhydrate de l'ivabradine à partir du
composé de formule
(II) :
CA 02773064 2012-03-02
WO 2011/033194 PCT/FR2010/000625
-2-
MISOCH3
(0)
CH3HN OCH3
qui est mis en réaction avec le composé de formule (III) :
CH 0
3 =(III)
CH30
0
pour conduire au composé de formule (IV) :
CH30
CH30
OCH3
CH3
(IV)
OCH3
0
dont l'hydrogénation catalytique conduit à l'ivabradine, qui est alors
transformée en son
chlorhydrate.
L'inconvénient de cette voie de synthèse est de conduire à l'ivabradine avec
un rendement
inférieur à 17 % sur l'ensemble des trois étapes.
Compte-tenu de l'intérêt pharmaceutique de ce composé, il était important de
pouvoir y
accéder avec un procédé de synthèse performant, conduisant à l'ivabradine avec
un bon
rendement.
La demande internationale WO 2008/065681 divulgue un mode de préparation du
chlorhydrate de l'ivabradine dans lequel le composé de formule (II) :
CA 02773064 2012-03-02
WO 2011/033194 PCT/FR2010/000625
-3-
OCH3
(ID
CH3HN OCH3
est mis en réaction avec le 1-bromo-3-chloropropane en présence de carbonate
de potassium
pour conduire au composé de formule (V) :
3
I OCH3
CH
(V)
Cl
OCH3
lequel, sans avoir été purifié au préalable, est mis en réaction avec le
composé de formule
(VI) :
CH30
NH (VI)
CH30
O
en présence de tert-butylate de potassium dans le diméthylsulfoxyde,
pour conduire à l'ivabradine de formule (I) qui est ensuite convertie en son
chlorhydrate, sans
avoir été purifiée au préalable.
Le rendement global de cette voie de synthèse n'est pas mentionné dans la
demande WO
2008/065681.
Or, la Demanderesse a trouvé qu'il n'était pas possible de préparer le
chlorhydrate de
l'ivabradine en reproduisant le mode opératoire décrit dans la demande
W02008/065681.
La présente invention concerne un procédé de synthèse de l'ivabradine de
formule (I), de ses
sels d'addition à un acide pharmaceutiquement acceptable et de leurs hydrates
:
CA 02773064 2012-03-02
WO 2011/033194 PCT/FR2010/000625
-4-
CH30
OCH3
CH3
(i)
CH30 41b Mie OCH3
0
caractérisé en ce que l'on soumet le composé de formule (VIII) :
OCH3
CH
3
OCH3
dans laquelle X représente un atome d'halogène, un groupement mésylate ou un
groupement
tosylate,
à une réaction d'alkylation par le composé de formule (IX) :
CH30si A
NH
(IX)
CH30 0
dans laquelle A représente H2C-CH2 ou HC=CH,
en présence d'une base,
dans un solvant organique,
pour conduire au composé de formule (VII) :
CH30
CH30 11, OCH3
A CH3 le
(VII)
NN
OCH3
0
dans laquelle A est tel que défini précédemment,
puis,
CA 02773064 2012-03-02
WO 2011/033194
PCT/FR2010/000625
-5-
dans le cas où A représente H2C-CH2, l'ivabradine de formule (I), cas
particulier
des composés de formule (VII) et produit de la réaction d'alkylation du
composé
de formule (VIII) par le composé de formule (IX), est isolée et purifiée puis
peut
être transformée en ses sels d'addition à un acide pharmaceutiquement
acceptable,
choisi parmi les acides chlorhydrique, bromhydrique, sulfurique, phosphorique,
acétique, trifluoroacétique, lactique, pyruvique, malonique, succinique,
glutarique, fiunarique, tartrique, maléïque, citrique, ascorbique, oxalique,
méthanesulfonique, benzènesulfonique et camphorique, et en leurs hydrates,
= dans le cas où A représente CH=CH, le composé de formule (IV), produit de
la
réaction d'alkylation du composé de formule (VIII) par le composé de formule
(IX), est soumis à une réaction d'hydrogénation catalytique pour conduire à
l'ivabradine de formule (I), qui est isolée et purifiée puis peut être
transformée en
ses sels d'addition à un acide pharmaceutiquement acceptable, choisi parmi les
acides chlorhydrique, bromhydrique, sulfurique, phosphorique, acétique,
trifluoroacétique, lactique, pyruvique, malonique, succinique, glutarique,
fumarique, tartrique, maléïque, citrique,
ascorbique, oxalique,
méthanesulfonique, benzènesulfonique et camphorique, et en leurs hydrates.
Parmi les bases pouvant être utilisées pour effectuer la réaction d'alkylation
du composé de
formule (VIII) par le composé de formule (IX), on peut citer à titre non
limitatif l'hydrure de
sodium, le tert-butylate de potassium, le méthanolate de sodium, l'hydroxyde
de potassium,
l'hydroxyde de sodium, le carbonate de potassium ou le carbonate de césium.
La base préférentiellement utilisée pour effectuer la réaction d'alkylation du
composé de
formule (VIII) par le composé de formule (IX) est le tert-butylate de
potassium.
Parmi les solvants pouvant être utilisés pour effectuer la réaction
d'alkylation du composé de
formule (VIII) par le composé de formule (IX), on peut citer à titre non
limitatif le
tétrahydrofurane, le 1,4-dioxane, le diméthylsulfoxyde, le tert-butanol, le
N,N-
diméthylformamide, le N,N-diméthylacétamide ou la N-méthylpyrrolidone.
Le solvant préférentiellement utilisé pour effectuer la réaction d'alkylation
du composé de
formule (VIII) par le composé de formule (IX) est le diméthylsulfoxyde.
CA 02773064 2012-03-02
WO 2011/033194 PCT/FR2010/000625
-6-
Les composés de formule (Villa), cas particuliers des composés de formule
(VIII) pour
lesquels X représente un atome de brome ou d'iode, un groupement mésylate ou
un
groupement tosylate, sont des produits nouveaux, utiles comme intermédiaires
de synthèse
dans l'industrie chimique ou pharmaceutique, notamment dans la synthèse de
l'ivabradine, de
ses sels d'addition à un acide pharmaceutiquement acceptable et de leurs
hydrates, et font à ce
titre partie intégrante de la présente invention.
Les exemples ci-dessous illustrent l'invention.
Liste des abréviations utilisées :
DMF : N,N-diméthylformamide
DMSO : diméthylsulfoxyde
IR : infrarouge
Les points de fusion (PF) ont été mesurés au banc Kiffler.
EXEMPLE 1: Chlorhydrate de la 3-13-[{1(7S)-3,4-diméthoxybieyelo[4.2.0Jocta-
1,3,5-
trién-7-yll m éthyl} (méthyl)amino] p ropy1}-7,8-dim éthoxy-1,3,4,5-
tétrahydro-2H-3-benzazépin-2-one
Stade 1: 3-chloro-N-W7S)-3,4-diméthoxybicyclo[4.2.0]oeta-1,3,5-trién-7-
yllméthy1}-N-
méthylpropan-1-amine
A une solution de (1S)-4,5-diméthoxy-1-(méthyl aminométhyp-benzocyclobutane
(10 g; 48
mmol) dans du 1-bromo-3-chloropropane (30 mL) est ajouté du carbonate de
potassium (9,9
g; 72 mmol). Après une nuit de contact à température ambiante, le milieu
réactionnel est
versé sur un mélange d'eau distillée (30 mL) et de dichlorométhane (30 mL).
Après
décantation, la phase organique est extraite par HC1 2N puis la phase aqueuse
est amenée à
pH 9-10 après traitement par une solution aqueuse d'ammoniaque à 28%. La phase
organique,
obtenue après extraction de la phase aqueuse basique par du dichlorométhane,
est lavée par de
l'eau distillée puis séchée sur MgSO4. Le résidu obtenu après concentration
sous pression
CA 02773064 2012-03-02
WO 2011/033194 PCT/FR2010/000625
-7-
réduite est purifié par chromatographie sur silice (dichlorométhane/acétate
d'éthyle : 80/20) et
7,7 g de produit du titre sont obtenus sous la forme de cristaux.
Rendement = 56%
PF = 42-45 C
Stade 2 : 3-{3-1{R7S)-3,4-diméthoxybicyclo[4.2.01octa-1,3,5-trién-7-yl]
méthyl}(méthyl)amino]propy1}-7,8-diméthoxy-1,3,4,5-tétrahydro-2H-3-benzazépin-
2-
one
A une solution de 7,8-diméthoxy-1,3,4,5-tetrahydro-2H-3-benzazépin-2-one (2,6
g; 11,75
mmol) dans du DMSO est ajouté à température ambiante du tert-butylate de
potassium (1,49
g; 13,3 mmol). Après 1 heure de contact à température ambiante, est ajoutée
une solution de
3,5 g (12,3 mmol) du produit obtenu au stade précédent dans du DMSO (4,7 mL).
Après une
nuit de contact à température ambiante, le milieu réactionnel est versé sur de
l'eau distillée
(100 mL) puis la phase aqueuse est extraite par de l'acétate d'éthyle. Les
phases organiques
jointes sont lavées par de l'eau distillée puis séchées sur MgSO4. Après
concentration sous
pression réduite, le résidu obtenu est purifié par chromatographie sur silice
(dichlorométane/éthanol/NH4OH 28% : 95/5/0.5) et 3,65 g du produit du titre
sont obtenus
sous la forme d'une huile (pureté HPLC : 98%) et engagés à l'étape suivante.
Rendement = 66%
IR (pur) : y = 2787, 1645, 1246-1206, 832 cm-1.
Stade 3: Chlorhydrate de la 3-{31{1(7S)-3,4-diméthoxybieyelo[4.2.01octa-1,3,5-
trién-7-
yllméthyl}(méthyl)amino]propy11-7,8-diméthoxy-1,3,4,5-tétrahydro-2H-3-
benzazépin-2-
one
A une solution du produit obtenu au stade précédent (3,6 g; 7,6 mmol) dans
l'acétonitrile (40
mL) est ajouté à température ambiante de l'éther chlorhydrique 1N (12 mL).
Après une nuit de
contact, la suspension est refroidie à 0 C puis filtrée. 3 g de produit du
titre sont obtenus sous
la forme de cristaux blancs.
Rendement = 78%
CA 02773064 2012-03-02
WO 2011/033194 PCT/FR2010/000625
-8-
PF = 125-128 C
EXEMPLE 2: Chlorhydrate de la 3-{3-1{1(7S)-3,4-diméthoxybicyclo[4.2.01octa-
1,3,5-
trién-7-yliméthyl}(méthypamino]propy1}-7,8-diméthoxy-1,3,4,5-
tétrahydro-2H-3-benzazépin-2-one
Stade 1: 3-chloro-N-{1(7S)-3,4-diméthoxybicyclo[4.2.0]octa-1,3,5-trién-7-
yl]méthy1}-N-
méthylpropan-1-amine
Le produit du titre est préparé en suivant le mode opératoire décrit au stade
1 de l'exemple 1.
Stade 2 : 3-{3-1{1(7S)-3,4-diméthoxybicyclo[4.2.01octa-1,3,5-trién-7-yl]
méthyl}(méthypaminolpropy1}-7,8-diméthoxy-1,3-dihydro-2H-3-benzazépin-2-one
A une solution de 7,8-diméthoxy-1,3-dihydro-2H-3-benzazépin-2-one (2,94 g;
13,4 mmol)
dans du DMSO (12 mL) est ajouté à température ambiante du tert-butylate de
potassium (1,7
g; 15,15 mmol). Après 30 minutes de contact à température ambiante, est
ajoutée une
solution de 4 g (14,1 mmol) du produit obtenu au stade précédent dans du DMSO
(10 mL).
Après une nuit de contact à température ambiante, le milieu réactionnel est
versé sur de l'eau
distillée (100 mL) puis la phase aqueuse est extraite par de l'acétate
d'éthyle. Les phases
organiques jointes sont lavées par de l'eau distillée puis séchées sur MgSO4.
Après
concentration sous pression réduite, 6,2 g de produit du titre sont obtenus
sous la forme d'une
huile (pureté HPLC : 88%) et engagés à l'étape suivante.
Rendement = 87%
IR (pur) : v = 2788, 1656, 1510-1401, 836-760 cm'I.
Stade 3 : 3-13-[{[(7S)-3,4-diméthoxybicyclo 14.2.01 octa-1,3,5-trién-7-yl]
méthyl} (méthyl)
aminolpropy11-7,8-diméthoxy-1,3,4,5-tétrahydro-2H-3-benzazépin-2-one
Dans un autoclave de 250 mL 4 g du produit obtenu au stade précédent et 2 g de
Pd(OH)2
20% humide à 50% sont ajoutés à une solution d'éthanol (90 mL) et d'acide
acétique (10 mL).
Après 5 h de contact à température ambiante sous une pression d'hydrogène de 5
bars, le
milieu réactionnel est filtré sur célite. Le résidu obtenu après concentration
sous pression
CA 02773064 2012-03-02
WO 2011/033194 PCT/FR2010/000625
-9-
réduite est repris dans du dichlorométhane (100 mL) puis lavé par une solution
aqueuse
saturée en bicarbonate de sodium. L'huile obtenue après séchage de la phase
organique sur
MgSO4 puis concentration sous pression est purifiée par chromatographie sur
silice
(dichlorométhane/éthanol/NH4OH 28%: 95/5/0,5) et 2,6 g de produit du titre
sont obtenus
sous la forme d'une huile.
Rendement = 74%
IR (pur) : y = 2788, 1646, 1519-1461, 1245-1105 cm-1.
Stade 4: chlorhydrate de la 3-(34{[(7S)-3,4-diméthoxybicyclo[4.2.01octa-1,3,5-
trién-7-
yl] m éthyl} (méthypamino] propy1}-7,8-dim éthoxy-1,3,4,5-tétrahydro-2H-3-b
enzazépin-2-
one
A une solution du produit obtenu au stade précédent (2.6 g; 5,5 mmol) dans
l'acétonitrile (25
mL) est ajouté à température ambiante de l'éthanol chlorhydrique 2,9N (3 mL).
Après une nuit
de contact, la suspension est filtrée et 2,2 g de produit du titre sont
obtenus sous la forme de
cristaux blancs.
Rendement = 79%
PF = 123-125 C
EXEMPLE COMPARATIF : Reproduction du mode opératoire décrit dans la demande
WO 2008/065681
Stade 1: 3-chloro-N-W7S)-3,4-diméthoxybicyclo[4.2.01octa-1,3,5-trién-7-
yllméthyl)-N-
méthylpropan-l-amine
A une solution de (1S)-4,5-diméthoxy-1-(méthyl aminométhyl)-benzocyclobutane
(10 g; 48
mmol) dans du 1-bromo-3-chloropropane (30 mL) est ajouté du carbonate de
potassium (9,9
g; 72 mmol). Après une nuit de contact à température ambiante, le milieu
réactionnel est
versé sur un mélange d'eau distillée (30 mL) et de dichlorométhane (30 mL).
Après
décantation, la phase organique est extraite par HC1 2N puis la phase aqueuse
est amenée à
pH 9-10 après traitement par une solution aqueuse d'ammoniaque à 28%. La phase
organique,
CA 02773064 2012-03-02
WO 2011/033194 PCT/FR2010/000625
-10-
obtenue après extraction de la phase aqueuse basique par du dichlorométhane,
est lavée par de
l'eau distillée puis séchée sur MgSO4. Après concentration sous pression
réduite, le produit du
titre est obtenu avec un rendement massique brut de 82% et une pureté de 56%.
Le brut
réactionnel contient encore 40% de (1S)-4,5-diméthoxy-1-(méthyl aminométhyl)-
benzocyclobutane.
Stade 2 : 3-{3-11[(7S)-3,4-diméthoxybicyclo [4.2.0] octa-1,3,5-trién-7-yl]
méthyl}(méthyl)amino]propy1}-7,8-diméthoxy-1,3,4,5-dihydro-2H-3-benzazépin-2-
one
A une solution de 7,8-diméthoxy-1,3,4,5-tetrahydro-2H-3-benzazépin-2-one (2,6
g; 11,75
mmol) dans du DMSO est ajouté à température ambiante du tert-butylate de
potassium (1,49
g; 13,3 mmol). Après 1 heure de contact à température ambiante, est ajoutée
une solution de
3,5 g (12,3 mmol) du produit obtenu au stade précédent dans du DMSO (4,7 mL).
Après une
nuit de contact à température ambiante, le milieu réactionnel est versé sur de
l'eau distillée
(100 mL) puis la phase aqueuse est extraite par de l'acétate d'éthyle. Les
phases organiques
jointes sont lavées par de l'eau distillée puis séch'ées sur MgSO4. Après
concentration sous
pression réduite le composé du titre est obtenu avec un rendement brut de
96,8% et une pureté
de 55%.
Stade 3: Chlorhydrate de la 3-{3-[{[(75)-3,4-diméthoxybieyelo[4.2.0]octa-1,3,5-
trién-7-
yl] méthyl} (méthyDamino] p ropy1}-7,8-dim éthoxy-1,3,4,5-tétrahyd ro-2H-3-b
enzazépin-2-
one
A une solution de 5 g du produit brut obtenu au stade précédent dans
l'acétonitrile (15 mL),
est ajouté une solution d'acide chlorhydrique 6N dans l'isopropanol. Après une
nuit de contact
à température ambiante, le chlorhydrate du composé du titre n'a pas précipité
et n'est donc pas
isolable. A partir du composé brut obtenu au stade précédent, il a été
impossible d'obtenir le
produit du titre en suivant le mode opératoire décrit dans la demande WO
2008/065681.