Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02779198 2012-04-26
WO 2011/053281 PCT/US2009/062250
RECOMBINANT PRODUCTION OF AUTHENTIC HUMAN PROTEINS
USING HUMAN CELL EXPRESSION SYSTEMS
FIELD OF THE INVENTION
The present invention relates to human expression systems for
recombinantly producing authentic human proteins.
SUMMARY OF THE INVENTION
Aspects of the present invention include novel expression cassettes
and vectors for efficiently producing authentic recombinant human proteins
from stable cultures of novel human cell lines. Also contemplated by the
present invention are authentic recombinant proteins produced by these novel
methods and from these novel components, as well as antibodies raised
against those authentic recombinant proteins. The recombinantly-produced,
authentic human proteins of the present invention may also be used in gene
therapy, and the vectors disclosed herein may also be employed in gene
therapy to express human proteins in vivo or in vitro/ex vivo in order to
treat a
particular disease.
It is to be understood that the vectors and cell lines of the present
invention can be used together and also independently from one another.
That is, a novel vector of the present invention may be introduced into a
novel
cell line of the present invention, such as HZ-293TS (described below), and
within that cell express a particular encoded human protein, such as a
cytokine. It is equally true, however, that any vector may be expressed in a
cell line of the present invention. Similarly, any of the vectors disclosed
herein
may be expressed in different human cell lines, not only in those described
herein.
CHIC_4513901 1 1
CA 02779198 2012-04-26
WO 2011/053281 PCT/US2009/062250
An aspect of the present invention is an expression cassette that
facilitates the expression of an authentic human protein in human cells, which
comprises in a 5'- to 3'-orientation the following sequences:
(1) a cytomegalovirus (CMV) enhancer element;
(2) a human promoter sequence, wherein the promoter is selected
from the group consisting of (i) a human 13-actin promoter, (ii) a human serum
albumin promoter, and (iii) a human fibrinogen promoter;
(3) a human globin gene intron; and
(4) a signal peptide, such as immunoglobulin superfamily 8 signal
peptide or alpha-fibrinogen signal peptide.
A desired polynucleotide, which encodes a human protein or fragment
thereof of interest, is operably linked to and inserted downstream of the
signal
peptide sequence. The expression cassette may also further comprise a
polyadenylation signal sequence and/or a termination site sequence, such
that the desired polynucleotide is operably linked to and inserted between the
signal peptide and the polyadenylation/termination signal sequence.
Alternatively, the desired polynucleotide itself may comprise a termination
signal sequence or a polyadenylation signal sequence to aid appropriate
termination of transcription.
In whatever arrangement and permutation of these regulatory
elements, it is clear to the skilled person that expression of this cassette
will
produce a fusion protein comprising the protein or polypeptide encoded by the
desired polynucleotide linked to the signal peptide. Thus, a signal
peptide/human protein may be produced according to the present invention
and then the signal peptide cleaved away from the fusion protein to leave the
authentic human protein intact. In this respect, the human cell machinery
recognizes the cleavage site during the protein synthesis. For instance, the
cell's Golgi apparatus and signal peptides interact during the protein
synthesis
CHIC_4513901 1 2
CA 02779198 2012-04-26
WO 2011/053281 PCT/US2009/062250
at which point the signal peptide is separated from the fusion protein.
Accordingly, when the protein is secreted to the extracellular space the
signal
peptide is already separated.
An expression cassette of the present invention therefore may
comprise the following operably linked sequences: (1) a CMV immediate early
enhancer sequence; (2) a human 13-actin promoter; (3) a globin gene intron;
(4) an alpha-fibrinogen signal peptide; (5) a desired polynucleotide; and (6)
a
polyadenylation/termination sequence. The skilled artisan is aware that other
permutations of these elements can differ and exchanged with other
sequences. For instance, as indicated above, an expression cassette may be
constructed that employs a human serum albumin promoter or a human
fibrinogen promoter in the cassette in place of the human 13-actin promoter.
Thus, an expression cassette of the present invention may comprise the
following operably linked sequences: (1) a CMV immediate early enhancer
sequence; (2) a human 13-actin promoter; (3) a globin gene intron;
(4) immunoglobulin superfamily 8 signal peptide; (5) a desired polynucleotide;
and (6) a polyadenylation/termination sequence.
Accordingly, in one aspect of the present invention is an expression
vector comprising a desired polynucleotide operably linked to a CMV
immediate early enhancer, a beta-actin promoter sequence, a human globin
gene intron sequence, and an immunoglobulin superfamily 8 signal peptide,
wherein the desired polynucleotide is operably linked to, and positioned
downstream of, the signal peptide.
Another aspect of the present invention is an expression vector
comprising a desired polynucleotide operably linked to a CMV immediate
early enhancer, a beta-actin promoter sequence, a human globin gene intron
sequence, and an alpha-fibrinogen signal peptide, wherein the desired
polynucleotide is operably linked to, and positioned downstream of, the signal
peptide.
CHIC_4513901 1 3
CA 02779198 2012-04-26
WO 2011/053281 PCT/US2009/062250
In this regard, the present invention is not limited to the use of only
human genetic elements in the expression cassette. For instance, the
promoter and gene intron sequence, as well as the signal peptide can be from
the genome of other species. For instance, the sequences can be from any
mammal, reptile, bird, fish, insect, bacterial, fungus, yeast, virus, or
amphibian. Thus, in one embodiment of the present invention, the expression
cassette comprises nucleotide sequences for a promoter, intron, and signal
peptide from a species other than human, such as from mouse, monkey, ape,
rat, cat, dog, rabbit, gerbil, hamster, guinea pig, pig, cattle, or sheep.
In another embodiment, the signal sequence encodes a sequence that
helps transport the transcribed RNA molecule product to the cell's Golgi body
or to the extracellular space.
Another aspect of the present invention is a method for producing a
protein in a cell, comprising introducing an expression vector of the present
invention into a cell, wherein the cell may be from the same species as the
sequences of the vector cassette, wherein the gene sequence in the cassette
is transcribed and translated in the cell to produce the protein that it
encodes.
In one embodiment, the expression vector is introduced into a cell that is
from
the same species as the species from which the promoter, intron, and signal
peptide sequences are obtained from. In one embodiment, both the cell and
the sequences of the cassette are from any mammal, reptile, bird, fish,
insect,
bacterial, fungus, yeast, virus, or amphibian. Thus, in one embodiment of the
present invention, the cell and sequences of the expression cassette are both
from a species selected from the group consisting of human, mouse, monkey,
ape, rat, cat, dog, rabbit, gerbil, hamster, guinea pig, pig, cattle, and
sheep.
In one embodiment, the cell is a human cell. In another embodiment, the
human cell is a human kidney cell. In one embodiment, the human kidney cell
is an HEK 293 cell. A sequence of the expression cassette of the present
invention includes a nucleic acid sequence of interest, or a desired
polynucleotide, operably linked downstream of the signal peptide and
CHIC_4513901 1 4
CA 02779198 2012-04-26
WO 2011/053281 PCT/US2009/062250
operably linked thereby to the enhancer, promoter, and intron so that the
nucleic sequence of interest or the desired polynucleotide is expressed.
Examples of desired polynucleotides include, but are not limited to, those
disclosed in (1)-(13) of the Sequences listing below, as well as those listed
in
Table 5. Thus, examples of human cytokines encoded by the desired
polynucleotide of the present invention include but are not limited to Activin
A,
Activin B, Activin A/2xINHbA, Activin B/2xINHbB, AMH/MIS, Artemin, BDNF,
BMP2, BMP15/GDF9B, BMP2/BMP2A, BMP3/0steogenin, BMP4,
BMP4/BMP2B, BMP5, BMP7/0P-1, BMP1, BMP10, BMP15/GDF9B, B-NGF,
Cystatin C, Delta 1, EGF, Erythropoietin (EPO), FGF acidic, FGF basic,
FGF10, FGF5, FGF7, FGF8b, FLT3 ligand, G-CSF, GDF15, GDF2/BMP9,
GDF3, GDF5/BMP14, GDF8/myostatin, GDF9, GDNF, GM-CSF, HGF, HGH,
IFN-a2A, IFN-a2B, IFN-y, IFN-131, IGF I, IGF II, IGF 11v1 , IGF IIv2, IL1 0,
IL1 1,
IL12, IL15, IL1 7/IL1 7A, IL1 7F, IL1 1 IL2, IL23, IL27, IL28A/IFN-lambda-2,
IL28B/IFN-lambda-3, IL29/IFN-lambda-1, IL113, IL2 IL3, IL32, IL35, IL4, IL5,
IL6, IL7, IL8, IL9, Inhibin A/INHa&INHbA, Inhibin B/INHa&INHbB, Inhibin
C/INHa&INHbC, Inhibin E/INHa&INHbE, LEFTYB, LEFTY1/LeftyB, M-CSF,
mouse CSF, mouse SCF, NODAL, Noggin, NT3 (neurotrophin3), Oncostatin
M, PDGFa, PDGF13, Persephin, SCF, SDF1 a, SHH, Somatotropin, TGF 131,
TGF 132, TGF 133, TGF134/LEFTY2/LeftyA, TNF a, TP0a, VEGF121aa,
VEGF165aa, WIF1, WNT1, Wnt10A, Wnt106/12, Wnt11, Wnt16, Wnt2,
Wnt2B/13, Wnt3, Wnt3A, Wnt4, Wnt5A, Wnt5B, Wnt6, Wnt7A, Wnt7B,
Wnt8B, and Wnt9A/14. A desired polynucleotide of the present invention may
encode EPO, G-CSF, GM-CSF, IL-2, IL-4, IL-6, M-CSF, Noggin, SCF,
Somatotropin, TGFB1, TNFa, or VEGF-165.
One aspect of the present inventive method comprises isolating the
produced protein from the cell. In one embodiment, the method further
comprises raising an antibody against the isolated protein. Thus, another
aspect of the present invention is an antibody raised against an epitope of
the
protein produced by any one of the inventive methods disclosed herein. In
one embodiment, the antibody is a polyclonal or monoclonal antibody.
CHIC_4513901 1 5
CA 02779198 2012-04-26
WO 2011/053281 PCT/US2009/062250
Another aspect of the present invention is a cell, which expresses the
cassette of any of the expression vectors disclosed herein. In one
embodiment, the cell is from the same species as sequences in the cassette.
In one embodiment, the cell is a human cell.
Another aspect of the present invention is a method for inducing an
immune response in an individual, comprising expressing the cassette of any
of the expression vectors disclosed herein in cells of the individual, wherein
expression of the gene sequence produces a protein or RNA product that
induces an immune response in the individual, and wherein the species of the
individual is the same as the species of the sequences in the cassette.
Another aspect of the present invention is an expression vector,
comprising a cassette which comprises (A) a human promoter sequence, (B)
a human signal sequence, (C) a human gene sequence, and (D) a human
polyadenylation signal sequence, wherein all of the sequences are operably
linked.
In one embodiment, the sequence of an exemplary CMV enhancer
element is shown in SEQ ID NO: 27, which is the human CMV immediate
early (1E) enhancer. The sequence of an exemplary human promoter
sequence is shown in SEQ ID NO: 28 and is the human beta-actin promoter
sequence. The sequence of an exemplary human globin gene intron is
shown in SEQ ID NO: 29. Examples of signal peptides includes the human
fibrinogen alpha chain signal peptide shown in SEQ ID NO: 30, and the
human immunoglobulin superfamily member 8 precursor signal shown in SEQ
ID NO: 31.
Examples of expression cassettes that include these elements
operably linked to one another are shown in SEQ ID NOs: 32 and 33.
Any desired polynucleotide or nucleic acid of interest can be inserted
downstream of the signal peptide so that it is operably linked to the signal
CHIC_4513901 1 6
CA 02779198 2012-04-26
WO 2011/053281 PCT/US2009/062250
peptide and the regulatory elements, such as the CMV IE enhancer, human
promoter, and human globin intron sequences, of the expression cassette. A
cassette of the present invention may further comprise a cleavage site
sequence between the globin gene intron and the signal peptide so that post-
transcriptional processing generates a protein that comprises the desired
protein linked at its 5'-end to the signal peptide. The signal peptide may be
a
signal peptide that transports the protein to the extracellular space. A
desired
polynucleotide of the present invention includes polynucleotides that encode
cytokines. Thus, one aspect of the present invention is an isolated,
recombinant human cytokine that is authentically glycosylated and
comparable in structure to the same cytokine that is native, endogenously-
expressed in vivo from a human cell.
In one embodiment of the present invention, the recombinant human
cytokine is comparable to the native cytokine because the recombinant
human cytokine comprises sugar chains that are terminated with human-
specific N-acetylneuraminic acids. In one embodiment, the sugar chains are
covalently attached to the surface of the cytokine. In another embodiment of
the present invention a sugar chain comprises at least one of N-
acetylglucosamine, fucose, mannose, and galactose moities. In another
embodiment, the authentically glycosylated cytokine only comprises sugar
chains from a human cell and no sugar chain moieties derived from any non-
human cell.
In one embodiment, the recombinant human cytokine that is encoded
by a desired polynucleotide of the present invention is selected from the
group
consisting of Activin A, Activin B, Activin A/2xINHbA, Activin B/2xINHbB,
AMH/MIS, Artemin, BDNF, BMP2, BMP15/GDF9B, BMP2/BMP2A,
BMP3/0steogenin, BMP4, BMP4/BMP2B, BMP5, BMP7/0P-1, BMP1,
BMP10, BMP15/GDF9B, 13-NGF, Cystatin C, Delta 1, EGF, Erythropoietin
(EPO), FGF acidic, FGF basic, FGF10, FGF5, FGF7, FGF8b, FLT3 ligand, G-
CSF, GDF15, GDF2/BMP9, GDF3, GDF5/BMP14, GDF8/myostatin, GDF9,
CHIC_4513901 1 7
CA 02779198 2012-04-26
WO 2011/053281 PCT/US2009/062250
GDNF, GM-CSF, HGF, HGH, IFN-a2A, IFN-a2B, IFN-y, IFN-131, IGF I, IGF II,
IGF 11v1, IGF 11v2, IL10, IL11, IL12, IL15, IL17/1L17A, IL17F, IL1 131L2,
IL23,
IL27, IL28A/IFN-lambda-2, IL28B/IFN-lambda-3, IL29/IFN-lambda-1, IL1B, IL2
IL3, IL32, IL35, IL4, IL5, IL6, IL7, IL8, IL9, Inhibin A/INHa&INHbA, Inhibin
B/INHa&INHbB, Inhibin C/INHa&INHbC, Inhibin E/INHa&INHbE, LEFTYB,
LEFTY1/LeftyB, M-CSF, mouse CSF, mouse SCF, NODAL, Noggin, NT3
(neurotrophin3), Oncostatin M, PDGFa, PDGFI3, Persephin, SCF, SDF1a,
SHH, Somatotropin, TGF 01, TGF 02, TGF 03, TGF134/LEFTY2/LeftyA, TNF a,
TP0a, VEGF121aa, VEGF165aa, WIF1, WNT1, Wnt10A, Wnt106/12, Wnt11,
Wnt16, Wnt2, Wnt2B/13, Wnt3, Wnt3A, Wnt4, Wnt5A, Wnt5B, Wnt6, Wnt7A,
Wnt7B, Wnt8B, and Wnt9A/14. See also the "List of Cloned Cytokine Genes"
at Table 5. The recombinant human cytokine that is encoded by a desired
polynucleotide of the present invention may also be selected from the group
of cytokines listed in Table 5. In another embodiment, the human
recombinant cytokine is selected from the group consisting of EPO, G-CSF,
GM-CSF, IL-2, IL-4, IL-6, M-CSF, Noggin, SCF, Somatotropin, TGFB1, TNFa,
and VEGF-165.
Another aspect of the present invention is a recombinant method for
producing an authentic human cytokine, comprising (1) transfecting a human
cell that is able to survive on serum-free medium with any one of the
expression vectors disclosed herein that comprises (i) a desired
polynucleotide that encodes a human cytokine sequence and (ii) an antibiotic
resistance gene; (2) (i) selecting those transfected cells that survive
exposure
to medium containing the antibiotic to which the antibiotic gene is resistant
to,
(ii) transferring those cells that survive antibiotic exposure to a liquid
culture
that has low-serum concentration, (3) reducing the serum concentration to 0%
over a period of time; and (4) isolating the human cytokine expressed from
cells that grow in the serum-free medium liquid culture, wherein the isolated
human cytokine is biologically active. In another aspect of the present
invention, the human cells can be co-transfected with one vector that
comprises the desired polynucleotide that comprises the cytokine sequence,
CHIC_4513901 1 8
CA 02779198 2012-04-26
WO 2011/053281 PCT/US2009/062250
and another distinct vector that comprises the antibiotic resistance gene
expression cassette. Thus, the present invention contemplates the use of one
vector (that contains both the cytokine sequence and the antibiotic resistance
gene sequence) or the use of two vectors (one containing the cytokine gene
sequence, the other containing the antibiotic resistance gene sequence), for
transfecting the human cells of the present invention.
In one embodiment, the desired polynucleotide comprises a sequence
that encodes a signal peptide secretory sequence in frame with the sequence
that encodes the human cytokine. In another embodiment, the desired
polynucleotide is operably linked to a promoter and a terminator.
In one embodiment, the serum-free medium comprises one or more
antibiotics. In one embodiment, an antibiotic is neomycine (G418),
hygromycine, zeocin, or blasticidine. In another embodiment, the step of
reducing the serum concentration to 0% occurs during a period of time that is
one day to several weeks. In one embodiment, the volume of liquid culture is
1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22,
23,
24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42,
43,
44, 45, 46, 47, 48, 49, 50 liters, or more than 50 liters of culture, or any
integer
in between.
In another embodiment, the step of isolating the human cytokine
comprises centrifuging an aliquot of the serum-free liquid cell culture and
capturing the human cytokine from the supernatant into which it is secreted.
In one embodiment, the step of isolating the human cytokine further
comprises returning the human cells that have been pelleted from the
centrifugation step to the same or different serum-free liquid cell culture
vessel.
In one embodiment, the supernatant from which the secreted human
cytokines are isolated contains no more than about 1%, about 2%, about 3%,
about 4%, about 5%, about 6%, about 7%, about 8%, about 9%, about 10%,
CHIC_4513901 1 9
CA 02779198 2012-04-26
WO 2011/053281
PCT/US2009/062250
about 11%, about 12%, about 13%, about 14%, about 15%, about 16%, about
17%, about 18%, about 19%, or about 20% of contaminating material or
protein that is not the secreted human cytokine.
In one embodiment, the purity of a sample of supernatant comprising
the secreted human cytokines is at least about 99% pure, at least about 98%
pure, at least about 97% pure, at least about 96% pure, at least about 95%
pure, at least about 94% pure, at least about 93% pure, at least about 92%
pure, at least about 91% pure, at least about 90% pure, at least about 89%
pure, at least about 88% pure, at least about 87% pure, at least about 86%
pure, at least about 85% pure, at least about 84% pure, at least about 83%
pure, at least about 82% pure, at least about 81% pure, or at least about 80%
pure.
In one embodiment, the supernatant of the cell culture consists
essentially of the expressed and secreted human cytokine protein.
In another embodiment, the supernatant of the cell culture consists of
no other biologically active protein other than the expressed and secreted
human cytokine protein.
Another aspect is an isolated, recombinant human cytokine produced
by the recombinant method. In one embodiment, that isolated, recombinant
human cytokine is selected from the group consisting of Activin A, Activin B,
Activin A/2xINHbA, Activin B/2xINHbB, AMH/MIS, Artemin, BDNF, BMP2,
BMP15/GDF9B, BMP2/BMP2A, BMP3/0steogenin, BMP4, BMP4/BMP2B,
BMP5, BMP7/0P-1, BMP1, BMP10, BMP15/GDF9B, B-NGF, Cystatin C,
Delta 1, EGF, Erythropoietin (EPO), FGF acidic, FGF basic, FGF10, FGF5,
FGF7, FGF8b, FLT3 ligand, G-CSF, GDF15, GDF2/BMP9, GDF3,
GDF5/BMP14, GDF8/myostatin, GDF9, GDNF, GM-CSF, HGF, HGH, IFN-
a2A, IFN-a2B, IFN-y, IFN-131, IGF I, IGF II, IGF 11v1 , IGF IIv2, IL1 0, IL1
1, IL12,
IL15, IL1 7/IL1 7A, IL1 7F, IL1 1 IL2, IL23, IL27, IL28A/IFN-lambda-2,
IL28B/IFN-lambda-3, IL29/IFN-lambda-1, IL113, IL2 IL3, IL32, IL35, IL4, IL5,
CHIC_4513901 1 10
CA 02779198 2012-04-26
WO 2011/053281
PCT/US2009/062250
IL6, IL7, IL8, IL9, Inhibin A/INHa&INHbA, Inhibin B/INHa&INHbB, Inhibin
C/INHa&INHbC, Inhibin E/INHa&INHbE, LEFTYB, LEFTY1/LeftyB, M-CSF,
mouse CSF, mouse SCF, NODAL, Noggin, NT3 (neurotrophin3), Oncostatin
M, PDGFa, PDGF13, Persephin, SCF, SDF1a, SHH, Somatotropin, TGF 131,
TGF 132, TGF 133, TGF134/LEFTY2/LeftyA, TNF a, TP0a, VEGF121aa,
VEGF165aa, WIF1, WNT1, Wnt10A, Wnt106/12, Wnt11, Wnt16, Wnt2,
Wnt2B/13, Wnt3, Wnt3A, Wnt4, Wnt5A, Wnt5B, Wnt6, Wnt7A, Wnt7B,
Wnt8B, and Wnt9A/14. See also the "List of Cloned Cytokine Genes" at
Table 5.
In one embodiment, none of the human cytokine proteins produced by
the methods disclosed herein comprises a non-human sugar chain. That is,
the human cytokine proteins of the present invention only comprises
glycosylated sugar chains that have been covalently bound to their protein
surface by enzymes and substrates available only in human cells.
Another aspect of the present invention is an antibody that is raised
against any one of the isolated recombinant human cytokines disclosed
herein. In one embodiment, the antibody is a monoclonal antibody. In
another embodiment, it is a polyclonal antibody.
Another aspect of the present invention is a kit, comprising at least one
of any isolated recombinant human cytokine or antibody disclosed herein.
Accordingly, to elaborate on the above embodiments, an aspect of the
present invention is an expression vector, comprising a cassette which
comprises the following operably linked expression elements:
(A) a cytomegalovirus enhancer element sequence;
(B) a human promoter sequence selected from the group consisting
of (i) a human actin promoter sequence, (ii) a human serum albumin promoter
sequence, and (iii) a human fibrinogen promoter sequence;
CHIC_4513901 1 11
CA 02779198 2012-04-26
WO 2011/053281
PCT/US2009/062250
(C) a human globin gene intron sequence; and
(D) a human signal peptide sequence.
In one embodiment, the expression vector further comprises a desired
polynucleotide positioned downstream of the signal peptide sequence and
operably linked to elements (A), (B), (C), and (D) of claim 1. In another
embodiment, either (i) the desired polynucleotide is operably linked to a
termination signal sequence that is located in the expression vector
downstream of the human signal peptide sequence, or (ii) the desired
polynucleotide itself comprises a termination signal sequence.
In one embodiment, the human promoter sequence is a functional
human beta-actin promoter sequence. In another embodiment, the CMV
enhancer element is the CMV immediate early enhancer sequence. In
another embodiment, the signal peptide sequence is (A) a sequence that
comprises intron 1 of the human immunoglobulin A gene and a cleavage
recognition sequence positioned toward the 3'-end of the intron sequence,
(B) alpha-fibrinogen signal peptide, or (C) an immunoglobulin superfamily 8
signal peptide.
In one embodiment, the desired polynucleotide encodes a cytokine. In
another embodiment the encoded cytokine is selected from the group
consisting of Activin A, Activin B, Activin A/2xINHbA, Activin B/2xINHbB,
AMH/MIS, Artemin, BDNF, BMP2, BMP15/GDF9B, BMP2/BMP2A,
BMP3/0steogenin, BMP4, BMP4/BMP2B, BMP5, BMP7/0P-1, BMP1,
BMP10, BMP15/GDF9B,13-NGF, Cystatin C, Delta 1, EGF, Erythropoietin
(EPO), FGF acidic, FGF basic, FGF10, FGF5, FGF7, FGF8b, FLT3 ligand, G-
CSF, GDF15, GDF2/BMP9, GDF3, GDF5/BMP14, GDF8/myostatin, GDF9,
GDNF, GM-CSF, HGF, HGH, IFN-a2A, IFN-a2B, IFN-y, IFN-131, IGF I, IGF II,
IGF 11v1, IGFIlv2, IL10, IL11, IL12, IL15, IL17/1L17A, IL17F, IL1 131L2, IL23,
IL27, IL28A/IFN-lambda-2, IL28B/IFN-lambda-3, IL29/IFN-lambda-1, IL1B, IL2
IL3, IL32, IL35, IL4, IL5, IL6, IL7, IL8, IL9, Inhibin A/INHa&INHbA, Inhibin
CHIC_4513901 1 12
CA 02779198 2012-04-26
WO 2011/053281 PCT/US2009/062250
B/INHa&INHbB, Inhibin C/INHa&INHbC, Inhibin E/INHa&INHbE, LEFTYB,
LEFTY1/LeftyB, M-CSF, mouse CSF, mouse SCF, NODAL, Noggin, NT3
(neurotrophin3), Oncostatin M, PDGFa, PDGFI3, Persephin, SCF, SDF1a,
SHH, Somatotropin, TGF 01, TGF 02, TGF 03, TGF134/LEFTY2/LeftyA, TNF a,
TP0a, VEGF121aa, VEGF165aa, WIF1, WNT1, Wnt10A, Wnt106/12, Wnt11,
Wnt16, Wnt2, Wnt2B/13, Wnt3, Wnt3A, Wnt4, Wnt5A, Wnt5B, Wnt6, Wnt7A,
Wnt7B, Wnt8B, and Wnt9A/14. See also the "List of Cloned Cytokine Genes"
at Table 5. In another embodiment, the encoded cytokine is selected from the
group consisting of EPO, G-CSF, GM-CSF, IL-2, IL-4, IL-6, M-CSF, Noggin,
SCF, Somatotropin, TGFB1, TNF a, and VEGF-165.
In one embodiment, an expression vector of the present invention
("pHZhag") comprises operably linked nucleotide sequences for (i) an hCMV
1E, (ii) a human beta-actin promoter, and (iii) a human beta-globin intron.
In one embodiment, an expression vector of the present invention
("pHZA") comprises operably linked nucleotide sequences for (i) an hCMV
promoter, and (ii) a human fibrinogen subunit A signal peptide.
In one embodiment, an expression vector of the present invention
("pHZI") comprises operably linked nucleotide sequences for (i) an hCMV
promoter, and (ii) a human Ig superfamily 8 signal peptide.
In one embodiment, an expression vector of the present invention
("pHZhagA") comprises operably linked nucleotide sequences for (i) an hCMV
1E, (ii) a human beta-actin promoter, (iii) a human beta-globin intron, and
(iv) a
human fibrinogen subunit A signal peptide;
In one embodiment, an expression vector of the present invention
("pHZhagl") comprises operably linked nucleotide sequences for (i) an hCMV
1E, (ii) a human beta-actin promoter, (iii) a human beta-globin intron, and
(iv) a
human Ig superfamily 8 signal peptide.
CHIC_4513901 1 13
CA 02779198 2012-04-26
WO 2011/053281 PCT/US2009/062250
In one embodiment, an expression vector of the present invention
("pHZhag-TGFB1") comprises operably linked nucleotide sequences for (i) an
hCMV 1E, (ii) a human beta-actin promoter, (iii) a human beta-globin intron,
and (iv) TGFB1.
In one embodiment, an expression vector of the present invention
("phZhagl-TGFB1") comprises operably linked nucleotide sequences for (i) an
hCMV 1E, (ii) a human beta-actin promoter, (iii) a human beta-globin intron,
(iv) a human Ig superfamily 8 signal peptide, and (v) TGFB1.
In any of these expression vectors a desired polynucleotide may be
incorporated downstream, and operably linked to, the signal peptide of the
particular expression vector, where the desired polynucleotide encodes a
protein, such as a human protein.
Thus, another aspect of the present invention is a recombinant method
for producing an authentic human protein, comprising introducing any one of
the expression vectors of the present invention into a human cell, wherein the
desired polynucleotide encodes a human protein and wherein expression of
the desired polynucleotide in the human cell produces an authentic human
protein.
In one embodiment, the authentic human protein has a similar size,
structure, molecular weight, glycosylation pattern, and post-transcriptional
modifications to that of a native version of the same human protein. The
skilled person is aware of various assays and tests, such as chromatographic,
gel, genetic, protein analyses, crystallography, and compositional analyses
useful for characterizing proteins; a readily available one being protein gel
electrophoresis of the recombinantly-produced protein of the present invention
against a native, endogenous version of the same protein run alongside each
other and a protein marker lane.
CHIC_4513901 1 14
CA 02779198 2012-04-26
WO 2011/053281 PCT/US2009/062250
In one embodiment, the desired polynucleotide encodes a cytokine. In
another embodiment the encoded cytokine is selected from the group
consisting of Activin A, Activin B, Activin A/2xINHbA, Activin B/2xINHbB,
AMH/MIS, Artemin, BDNF, BMP2, BMP15/GDF9B, BMP2/BMP2A,
BMP3/0steogenin, BMP4, BMP4/BMP2B, BMP5, BMP7/0P-1, BMP1,
BMP10, BMP15/GDF9B, B-NGF, Cystatin C, Delta 1, EGF, Erythropoietin
(EPO), FGF acidic, FGF basic, FGF10, FGF5, FGF7, FGF8b, FLT3 ligand, G-
CSF, GDF15, GDF2/BMP9, GDF3, GDF5/BMP14, GDF8/myostatin, GDF9,
GDNF, GM-CSF, HGF, HGH, IFN-a2A, IFN-a2B, IFN-y, IFN-131, IGF I, IGF II,
IGF 110 , IGFIlv2, IL10, IL11, IL12, IL15, IL17/1L17A, IL17F, IL1 131L2, IL23,
IL27, IL28A/IFN-lambda-2, IL28B/IFN-lambda-3, IL29/IFN-lambda-1, IL1B, IL2
IL3, IL32, IL35, IL4, IL5, IL6, IL7, IL8, IL9, Inhibin A/INHa&INHbA, Inhibin
B/INHa&INHbB, Inhibin C/INHa&INHbC, Inhibin E/INHa&INHbE, LEFTYB,
LEFTY1/LeftyB, M-CSF, mouse CSF, mouse SCF, NODAL, Noggin, NT3
(neurotrophin3), Oncostatin M, PDGFa, PDGFI3, Persephin, SCF, SDF1a,
SHH, Somatotropin, TGF 01, TGF 02, TGF 03, TGF134/LEFTY2/LeftyA, TNF a,
TP0a, VEGF121aa, VEGF165aa, WIF1, WNT1, Wnt10A, Wnt106/12, Wnt11,
Wnt16, Wnt2, Wnt2B/13, Wnt3, Wnt3A, Wnt4, Wnt5A, Wnt5B, Wnt6, Wnt7A,
Wnt7B, Wnt8B, and Wnt9A/14. See also the "List of Cloned Cytokine Genes"
at Table 5. In another embodiment, the encoded cytokine is selected from the
group consisting of EPO, G-CSF, GM-CSF, IL-2, IL-4, IL-6, M-CSF, Noggin,
SCF, Somatotropin, TGFB1, TNF a, and VEGF-165.
In one embodiment, the human cell is HZ-293T5 which is a human
kidney embryonic cell line derived from HEK293T, and adapted according to
the present invention, and was deposited in accordance with the Budapest
Treaty with the American Type Culture Collection (ATCC ) at the ATCC IP,
Licensing and Services 10801 University Boulevard, Manassas, Virginia
20110-2209, USA, bearing the ATCC biological deposit accession number of
PTA-10165, on July 1, 2009 (date of receipt by the ATCC ), by HumanZyme,
Inc.. 2201 W. Campbell Park Dr., Chicago, Illinois 60612. References
elsewhere in this specification to this deposited cell line are cited in
CHIC_4513901 1 15
CA 02779198 2012-04-26
WO 2011/053281 PCT/US2009/062250
correspondence to the "ATCC PTA-10165" patent deposit designation
number.
In respect of all designated States to which action is possible and to
the extent that it is legally permissible under the law of the designated
State, it
is requested that a sample of the deposited biological material be made
available only be the issue thereof to an independent expert, in accordance
with the relevant patent legislation, e.g., EPC Rule 32(1); U.K. Patent Rules
2007, Schedule 1, Paragraph 6; Australian Regulation 3.25(3) and generally
similar provisions mutatis mutandis for any other designated State.
In one embodiment, the recombinant method further comprises
isolating the authentic human protein from the human cell.
Another aspect of the present invention is a recombinantly-produced,
authentic human protein produced by a human cell that expresses the desired
polynucleotide in any one of the expression vectors of the present invention,
wherein the desired polynucleotide encodes a human protein, and wherein
expression of the desired polynucleotide produces an authentic human
protein. In one embodiment, the desired polynucleotide encodes a cytokine.
In another embodiment the encoded cytokine is selected from the group
consisting of Activin A, Activin B, Activin A/2xINHbA, Activin B/2xINHbB,
AMH/MIS, Artemin, BDNF, BMP2, BMP15/GDF9B, BMP2/BMP2A,
BMP3/0steogenin, BMP4, BMP4/BMP2B, BMP5, BMP7/0P-1, BMP1,
BMP10, BMP15/GDF9B,13-NGF, Cystatin C, Delta 1, EGF, Erythropoietin
(EPO), FGF acidic, FGF basic, FGF10, FGF5, FGF7, FGF8b, FLT3 ligand, G-
CSF, GDF15, GDF2/BMP9, GDF3, GDF5/BMP14, GDF8/myostatin, GDF9,
GDNF, GM-CSF, HGF, HGH, IFN-a2A, IFN-a2B, IFN-y, IFN-131, IGF I, IGF II,
IGF 110, IGFIlv2, IL10, IL11, IL12, IL15, IL17/1L17A, IL17F, IL1 131L2, IL23,
IL27, IL28A/IFN-lambda-2, IL28B/IFN-lambda-3, IL29/IFN-lambda-1, IL1B, IL2
IL3, IL32, IL35, IL4, IL5, IL6, IL7, IL8, IL9, Inhibin A/INHa&INHbA, Inhibin
B/INHa&INHbB, Inhibin C/INHa&INHbC, Inhibin E/INHa&INHbE, LEFTYB,
CHIC_4513901 1 16
CA 02779198 2012-04-26
WO 2011/053281 PCT/US2009/062250
LEFTY1/LeftyB, M-CSF, mouse CSF, mouse SCF, NODAL, Noggin, NT3
(neurotrophin3), Oncostatin M, PDGFa, PDGFI3, Persephin, SCF, SDF1a,
SHH, Somatotropin, TGF 01, TGF 02, TGF 03, TGF134/LEFTY2/LeftyA, TNF a,
TP0a, VEGF121aa, VEGF165aa, WIF1, WNT1, Wnt10A, Wnt106/12, Wnt11,
Wnt16, Wnt2, Wnt2B/13, Wnt3, Wnt3A, Wnt4, Wnt5A, Wnt5B, Wnt6, Wnt7A,
Wnt7B, Wnt8B, and Wnt9A/14. See also the "List of Cloned Cytokine Genes"
at Table 5. In another embodiment, the encoded cytokine is selected from the
group consisting of EPO, G-CSF, GM-CSF, IL-2, IL-4, IL-6, M-CSF, Noggin,
SCF, Somatotropin, TGFB1, TNF a, and VEGF-165.
In one embodiment, the recombinantly-produced, authentic human
protein is produced from the HZ-293T5 human cell line deposited as ATCC
PTA-10165.
In one embodiment, the glycosylation pattern of the authentic human
protein is similar to the native, endogenous version of that human protein. In
a further embodiment, the size, structure, and molecular weight of the
authentic human protein is similar to the size and molecular weight of the
native, endogenous version of that human protein. In one embodiment, the
authentic human protein is TGFB. In another embodiment, the desired
polynucleotide encodes the noggin protein and wherein expression of the
desired polynucleotide produces in the cell a disulfide-bonded noggin dimer.
Another aspect of the present invention is an antibody raised against
an epitope of any one of the authentic human proteins produced by the
recombinant methods described herein. In one embodiment, the antibody is a
polyclonal or monoclonal antibody. In another embodiment, the antibody is a
monoclonal antibody raised against G-CSF.
Another aspect of the present invention is a stable human cell line
called HZ-293T5 (a human kidney embryonic cell line adapted to proliferate
on serum-free medium), deposited under and bearing the ATCC biological
deposit accession number of PTA-10165. In one embodiment of the present
CHIC_4513901 1 17
CA 02779198 2012-04-26
WO 2011/053281 PCT/US2009/062250
invention, the viability of cells of the HZ-293TS cell line is longer than
that of
other HEK293 cell types. That is, the HZ-293TS cells remain viable for 3, 4,
5, 6, 7, 8, 9, 10, 11, 12, 13, or 14, or more than 14 days viable compared to
other HEK293 cell types.
Another aspect of the present invention is any human cell that
expresses any of the expression vectors disclosed herein. In one
embodiment, the human cell is HZ-293TS deposited under and bears the
ATCC biological deposit accession number of PTA-10165.
Another aspect of the present invention is a method for treating a
condition associated with cell growth, cell proliferation, cell
differentiation, or
inflammation, comprising administering (A) any one of the recombinantly-
produced authentic human proteins described herein or obtainable by the
methods described herein, or (B) any one of the antibodies disclosed herein
or obtainable by the methods described herein, to an individual who has a
condition associated with cell growth, proliferation, differentiation, or
inflammation. In one embodiment, the method comprises administering an
inventive antibody raised against TNFa to an individual with a condition
associated with inflammation or arthritis.
In one embodiment, the method comprises administering an inventive
antibody raised against VEGF to an individual with a condition associated with
cancer or cell proliferation.
One aspect of the present invention is the use of an antibody raised
against an epitope of any one of the authentic human proteins of the present
inventino as a diagnostic agent for detecting that human protein from a
sample. In one embodiment, the antibody is used as a diagnostic agent in an
ELISA assay to detect the human protein.
CHIC_4513901 1 18
CA 02779198 2012-04-26
WO 2011/053281
PCT/US2009/062250
In another embodiment, the sample is a human tissue sample, human
cell sample, human blood sample, or a human bodily fluid sample. In a
further embodiment, the human protein is a cytokine.
Another aspect of the present invention is a method for treating a
condition associated with cell growth, cell proliferation, cell
differentiation, or
inflammation, comprising administering a vector that expresses a desired
polynucleotide which encodes a protein to an individual who has a condition
associated with cell growth, proliferation, differentiation, or inflammation,
wherein the expressed protein in a cell of the individual helps to treat the
condition.
BRIEF DESCRIPTION OF THE DRAWINGS
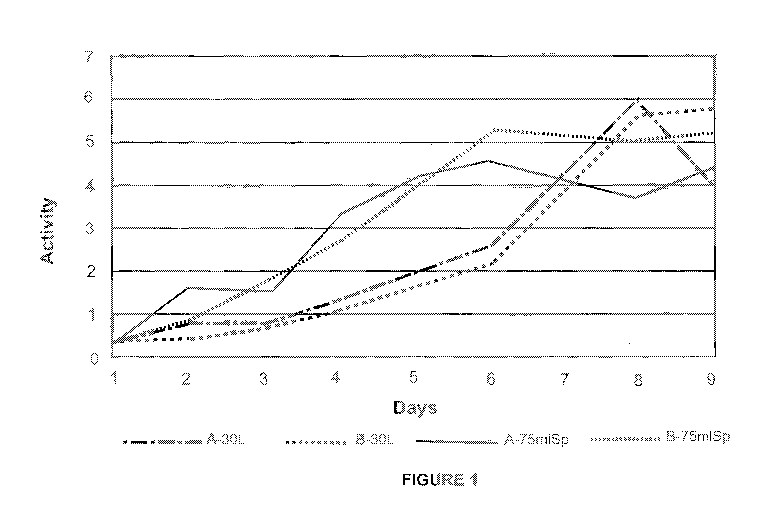
Figure 1: Protein activity for alkaline phosphastase over the course of 9
days between two different volumes of human cell culture (75 ml vs 30 liters).
Figure 2: SDS-PAGE gel showing high expression of cytokines in the
inventive stable human cell lines for expression of TGFB1 (lane 1), G-CSF
(lane 2), IL113 (lane 3), and VEGF165 (lane 4).
Figure 3: SDS-PAGE gel showing efficient tag-free purification for
somatotropin and GM-CSF.
Figure 4: Graph depicting a GM-CSF stability assay after expression in
the inventive human (cell) cells and non-human cells (square).
Figure 5: SDS-PAGE gel evidencing the expression of active and
mature TGFB1, which is notoriously difficult to express.
Figure 6: Graphs and corresponding gels depict the activity and purity
(as indicated on SDS gel staining) of cytokines produced according to the
present inventive method. These examples convey impressive activity and
purity data for GM-CSF (Figure 6A), IL-4 (Figure 6B), somatotrophin (Figure
CHIC_4513901 1 19
CA 02779198 2012-04-26
WO 2011/053281
PCT/US2009/062250
6C), TGF-131 (Figure 6D), VEGF-165 (Figure 6E), TNF-a (Figure 6F), M-CSF
(Figure 6G), IL-6 (Figure 6H, EPO (Figure 61), IL-2 (Figure 6J), SCF (Figure
6K), Noggin (Figure 6L), and G-CSF (Figure 6M).
Figure 7: SDS-PAGE gel analyses of purified cytokines from the
inventive human cell system compared with cytokines from non-human cell
system: (A) EPO, Noggin; (B) G-CSF, SCF, GM-SCF, Somatotropin; (C) IL-2,
TGF-131, IL-4, and TNFa; and (D) IL6, VEGF165, and M-CSF.
Figure 8: Western blot on purified cytokines from the inventive human
cell system compared with cytokines from non-human cell system: (A) EPO,
Noggin; (B) GM-CSF, SCF, IL-2, Somatotropin; (C) IL4, TNFa, IL6, and
VEGF165.
Figure 9: Comparison of VEGF165 activity in human and non-human
cells.
Figure 10: Comparison of IL-4 activity in human and non-human cells.
Figure 11: Example of TGF131 expression from HEK293T using (a)
pHZsec vector (pHZ-TGFb1) or (b) pHZhag vector (pHZhag-TGFb1). TGF131
latency-associated peptide (LAP) and mature TGF131 are indicated by red
arrows. Both cell lines transfected with pHZhag-TGF131 and pHZ-TGF131 was
treated with extra 20mM glucose at day 3 (A), day 4 (B), or day 5 (C) and no
added (D) in the culture medium. Gels were run the samples of day 3 (d3)
and day 10 (d10) for each treatment. For both LAP and mature TGF131,
pHZhag-TGF131 in (b) shows 2-3 fold higher expression than pHZ-TGF131 in
(a).
Figure 12: Cell growth curves of HEK293T cells transfected with pHZ-
TGF131 (a) and pHZhag-TGF131 (c). Viability (3/0) over the culture period is
presented for pHZ-TGF131 (b) and pHZhag-TGF131 (d). HEK293T cells
transfected with pHZhag-TGF131 were more than 60% viable even at day 10
(d) where as cells with pHZ-TGF131 were below 60% viable at day 8 (b).
CHIC_4513901 1 20
CA 02779198 2012-04-26
WO 2011/053281 PCT/US2009/062250
Higher viability of pHZhag-TGF131 transfected cells was also reflected in much
less background proteins on the gel in Figure 1(b).
Figure 13: TGF131 expression from HEK293T cells. (a) Expression test
with another promoter or signal peptides: Lane 1: mock vector (negative
control); Lane 2: pHZsec vector (CMV promoter and mlg kappa signal
peptide); Lane 3: hag promoter; Lane 4: hFbgA signal peptide; Lane 5: hFbgB
signal peptide; Lane 6: hFbgG signal peptide; Lane 7: hIg8 signal peptide.
There was no expression with hFbgB and hFbgG signal peptides. (b)
Comparison of TGF-131 expression using mock vector (negative control), pHZ-
TGF-131 (Lanes 2 and 3; murine Ig kappa signal peptide) or pHZA-TGF-131
(Lanes 4 and 5; human FbgA signal peptide).
Figure 14: Vector schematics.
Figure 15: Human FGF8b expression from HEK293T (a) and HZ-
293TS (b). Two to three fold higher expression of FGF8b from HZ-293TS
than that from HEK293T was unexpected observation. As the cells in
suspension their viable cell density (VCD) reached up to 5 ¨ 6 million cells
per
milliliter as in the tables below the gels.
Figure 16: Comparative gene expression results for different vectors
expressing TGF-131. Comparison of TGF-b1 expression level at day 5 from
suspension 293T culture using different vectors. 1, negative control; 2, hCMV
promoter with murine IgK signal peptide; 3, hag promoter with murine IgK
signal peptide; 4, hCMV promoter with human fibrinogen alpha chain signal
peptide; 5, hCMV promoter with human Ig superfamily 8 signal peptide.
Latency-associated peptide (LAP) and mature TGF-b1 are indicated by
arrows. All the cells are about three million cells/ml and 90% viability. Lane
1
= control; Lane 2- pHZ-TGF-131; Lane 3 = pHZhag-TGF-131 (hCMV IE + 13-
actin promoter + 13-globin intron + TGF-131); Lane 4 = pHZA-TGF-131 (hCMV
promoter + fibrinogen subunit A signal peptide + TGF-131); Lane 5 = pHZI-
CHIC_4513901 1 21
CA 02779198 2012-04-26
WO 2011/053281
PCT/US2009/062250
TGF-131 (hCMV IE + 13-actin promoter + 13-globin intron + human Ig
superfamily 8 signal peptide + TGF-131).
Figure 17: Efficient tag-free purification. Purified recombinant human
(A) GM-CSF, (B) IL-4, (C) IL-6, and (D) Noggin from engineered human cells.
All cytokines are expressed as native proteins without tag. Efficient
purification protocols were developed to yield authentic cytokines with native
heterogeneous glycosylation (A ¨ D) and a disulfide bond (D).
Figure 18: Production of difficult to express cytokines. Purified
recombinant human IL-23 (A) and mature TGF-131, -132, and -133 (B) from the
human cell expression system. Human cell expression system efficiently
expressed cytokines that are very difficult to express or not properly
expressed in non-human cells due to its complexity of glycosylated and
disulfide linked subunits (A: IL-23) or due to a delicate conversion process
from latency-associated peptide (LAP) complex to mature form (B: TGF-131, -
132, and -133).
Figure 19: Recombinantly-produced, authentic human VEGF165 a
homodimer. VEGF165 expressed from E. co/land engineered human 293
cells. The E. coli expressed protein is lack of glycosylation and a mixture of
monomer and dimer (19kDa and 38kDa, respectively), whereas human cell
expressed protein is fully glycosylated homodimer of 45kDa.
Figure 20: More authentic: human EPO vs CHO EPO. Recombinant
EPO expressed from human cells exhibits apparent molecular mass of 34kDa
on SDS-PAGE as native human serum EPO (Skibeli et al. 2001 Blood
98:3626) whereas CHO EPO exhibits apparent molecular mass of 40kDa (A).
Human EPO contains substantially high content of neutral glycans compared
to CHO EPO (B). (C) Acidic glycan structures of human EPO and CHO EPO.
The most abundant glycans in human EPO are tetra-antennary complex
types whereas those in CHO EPO are enlongated bi-antennary complex
CHIC_4513901 1 22
CA 02779198 2012-04-26
WO 2011/053281
PCT/US2009/062250
types. Symbols are G1cNAc (=), HexNAc (0), Mannose (.), Hexose ( ),
Fucose (A), NeuAc (*).
Figure 21: IL23 expressed in human cells of the present invention is
500-fold more potent than the cytokine produced from insect cells. Authentic
IL-23 expressed in human cells 100x more potent for induction of human
Th17 cells. (A) IL-23 activities determined by the dose-dependent secretion of
IL-17 from mouse splenocytes activated with 10 ng/ml PMA. (B) IL-23
activities determined by the dose-dependent secretion of IL-17 from human
CD4+ T cells stimulated with 2 mg/ml plate bound anti-CD3 and 1 mg/ml
soluble anti-CD28 in the presence of Th17 polarizing cytokines.
Figure 22: Efficient differentiation of Th17 cells with authentic cytokines
expressed in human cells. Whole CD4+ T cells isolated from a healthy donor
were stimulated with 2 gg/mL plate bound anti-CD3 and 1 iug/mL soluble anti-
CD28 in the presence of Th17 polarizing cytokines, including a titration of IL-
23. After 5 days, supernatants were harvested for measurement of IL-17 by
ELISA. See (A)-(G). Figure 22 (H) depicts flow cytometry analysis on Human
Th17 Differentiation by Authentic human TGF-131. See Example 17 below.
Figure 23: Cost and time-saving G4 protocol for dendritic cell
differentiation. (A) The profile of DC generation of selected cytokines and
chemokines demonstrated that DCs generated in HZ G4 DC medium (5 ng/ml
GM-CSF (authentic human, expressed in human cells) and IL-4 (authentic
human, expressed in human cells) without medium replacement) showed a
similar profile to DCs in EC G4 DC medium (50 ng/ml E. coli GM-CSF and IL-
4 with twice medium replacement) before and after maturation by
lipopolysaccharide (LPS). (B) Allogenic MLR of DCs generated and matured
in HZ G4 DC medium appeared to be even better than DCs in EC G4 DC
medium.
Figure 24: : Noggin Expressed in Human Cells of the Present Invention
Treated HES Cells Are OCT3/4 Positive. Treated with recombinantly-
CHIC_4513901 1 23
CA 02779198 2012-04-26
WO 2011/053281 PCT/US2009/062250
produced, authentic human Noggin at 10-20pg/ml, human embryonic stem
(hES) cells consistently expressed 0ct3/4, which is a marker for
undifferentiated ES cells. Western blot analysis of 0ct3/4. 1, Negative
control;
2, Positive control; 3, authentic Noggin 10pg/m1 treatment; 4, authentic
Noggin
20pg/m1treatment.
Figure 25: G-CSF antibodies will more accurately monitor human
biology. Recombinant human G-CSF expressed from human cells according
to the present invention is authentically glycosylated and presents unique
epitopes that may not exist in the protein expressed from non-human cells
(A). In Western blot a monoclonal antibody (HZ mAb1) raised against a
unique epitope recognize only human cell G-CSF (B) while another antibody
(HZ mAb2) against a common epitope recognize both (C).
Figure 26: New Cell Line (HZ-293T5). (A) New cell line HZ-293T5, a
293T cell line adapted to suspension culture in a serum-free chemically
defined medium. (B) Comparison of FGF8b expression level from 1, 293T and
2, HZ-293T5. (C) Comparison of TGF-131 expression level from 1, 293T and 2,
HZ-293T5. In both cases protein expression level was 2-fold higher from HZ-
293T5 cell line compare to that from 293T cell line.
Figure 27: Signal Cleavage Schematic.
DETAILED DESCRIPTION
A. GENERAL CONSIDERATIONS
CYTOKINES AND THE PRESENT INVENTION
Cytokines are a group of proteins and polypeptides that organisms use
as signaling molecules. Most cytokines are glycoproteins less than 30 kDa in
size and bind to specific, high-affinity cell surface receptors. Due to their
central role in the immune system, cytokines are involved in a variety of
immunological, inflammatory and infectious diseases and widely used in
research, diagnostics and therapeutics. Currently, these proteins are
CHIC_4513901 1 24
CA 02779198 2012-04-26
WO 2011/053281 PCT/US2009/062250
predominantly produced in non-human cells, such as E coli, and therefore
lack authenticity, as described below, due to the absence of relevant
glycosylation patterns. Furthermore, a number of important cytokines are not
commercially available due to inadequate proteolytic processing or other post-
translational modification that occur in such non-human cell expression
systems. See Table 1. Authentic in this regard intimates that the
recombinant human cytokine that is expressed according to the present
inventive stable human cell expression method conforms to its native
endogenous counterpart that is normally expressed in the human cell in vivo
and reproduces certain features that are associated with that native
counterpart. That is, for example, a recombinant human IFNa that is
expressed using the present inventive stable human cell method, looks like,
and has essentially the same structure, biological activity, size, molecular
weight, folding patterns, and glycosylation patterns as, the native human
IFNa. Thus, a human protein that is expressed by the present inventive
method may be regarded as an "authentic protein," or "authentic cytokine" or
a "recombinant, authentic" protein and so on. Its in vivo counterpart may be
regarded as the native or endogenous cytokine or protein. Such "authentic"
features of the proteins and cytokines expressed by the inventive stable
human cell expression system are disclosed in more detail below. By
contrast, a human IFNa protein that is expressed from an E. co//cell, or from
a
yeast or fungal cell, or from an insect cell, or from a non-human mammalian
cell, such as in a Chinese Hamster Ovary cell, is not considered to be an
"authentic" human IFNa protein in this context. Accordingly, an "authentic"
recombinantly-produced protein of the present invention is one that is highly
similar to one or more features and properties of the native version of the
protein, such as, but not limited to the same structure, biological activity,
size,
molecular weight, protein folding patterns, dimerization properties, disulfide-
bonding properties, and surface-bound glycosylation patterns.
(a) Cytokine Families
CHIC_4513901 1 25
CA 02779198 2015-12-22
It is possible to group many cytokines together based on structural
similarities that each constituent member shares other members, as well as
other similarities based on their respective primary amino acid sequences.
See Chapter 1 of The Cytokine Handbook, Volumes 1 and 2, Fourth Edition,
Eds. Thomson & Lotze.
According to shared structural features, therefore, the
following "families" of individual cytokines can be grouped as follows:
(1)12/IL-4: representative members include IL-2, IL-4, IL-5 GM-CSF.
(2) IL-6/1L12: representative members include IL-6 and IL-12.
(3) Interferons ¨ a/{3: representative members include IFN-a (many
subtypes), IFN-13, IFN-co, and IFN-r.
(4) Tumor necrosis factors: representative members include TNF-a,
LT-a (TNF-13), LT-0, Fas ligand, CD40 ligand, TRAIL, BAFF, APRIL, RANK,
and LIGHT.
(5) IL-10: representative members include IL-10, IL-19, IL-20, IL-22,
and 1L24.
(6) IL-17: representative members include IL-17 and IL-25.
(7) Interleukin-1: representative members include IL-la, IL-113, IL-1
receptor antagonist, and IL-18.
(8) TGF-f3: representative members include TGF-13, bone
morphogenetic proteins, lnhibins, and Activins.
(9) Chemokines: representative members include CXC subfamily
(CXCLI-16), CC subfamily (CCLI-28), C subfamily (CLI/Lymphotactin), and
CX3C subfamily (CX3CLI/Fractalkin).
26
CA 02779198 2015-12-22
Most cytokines are simple polypeptides or glycoproteins. Some of
them can form dimmers and some are produced transiently and induce
biological responses and cellular cascades by binding to specific high
affinity
cell surface receptors. Phenotypically, such responses include increases or
decreases in the rate of cell proliferation, changes in cell differentiation
state,
and changes in the expression of some differentiated functions. Thus, for
example, interleukin-1 (IL-1) activates T cells; IL-2 stimulates proliferation
of
antigen-activated T and B cells; IL-4, IL-5, and IL-6, stimulate proliferation
and
differentiation of B cells; Interferon gamma (IFN7) activates macrophages; and
IL-3, IL-7 and Granulocyte Monocyte Colony-Stimulating Factor (GM-CSF)
stimulate hematopoiesis. Table 2
highlights some additional information about
the source, target, and function of certain cytokines.
Cytokines can exist as monomers and dimers. With respect to the
latter, cytokines can exist as homodimers, where two monomers of the same
cytokine are joined together via a disulfide bond, or as heterodimers, such as
in the case for IL12, 11_23, IL27, and IL35. In the native human cell in vivo,
two
different cytokine genes are expressed from the genome and joined together
via disulfide bond formation between appropriate residues on each cytokine.
In artificial recombinant systems, these genes have to be expressed
separately in different cultures, the resultant cytokine proteins extracted
and
isolated and then linked together in a further bonding step. According to the
present invention, however, a stable human cell can be transfected with both
desired cytokine genes and, because the resultant protein product in each
case is authentic with respect to folding, epitope-presentation, and
glycosylation, the two cytokines will naturally come together and form the
heterodimer via disulfide bond linkage. Hence, the supernatant in that case
will contain already-formed heterodimers.
27
CA 02779198 2012-04-26
WO 2011/053281 PCT/US2009/062250
The present invention is not limited to these particular cytokines or to
polynucleotides that encode these particular proteins. Rather, an aspect of
the invention is the recombinant expression of cytokine mutants, homologs,
splice variants, or isomers that either are known to exist or are created to
determine its effect on a certain cytokine parameter. Thus, the present
invention also encompasses the recombinant production of authentic cytokine
variants that comprise one or more amino acid substitutions, deletions,
insertions, or splice junctions that differ from a native cytokine sequence,
or
which represents a mutated cytokine sequence that might be associated with
some disease or disorder. A cytokine DNA sequence may be engineered to
comprise such mutations and used for research purposes to ascertain the
effect of that, or those, mutations on cytokine function or downstream in the
relevant cytokine pathway. All of these proteins and mutants and variants
therefore can be designed into a polynucleotide that is cloned into an
expression cassette of the present invention so that it can be expressed in
the
human expression system of the present invention and subsequently used as
described herein.
(b) Functions and associated Diseases, such as Asthma and
Allergies
Each cytokine can have multiple functions depending upon the cell that
produces it and the target cell(s) upon which it acts, which may be on distant
target cells (endocrine), on target cells adjacent to those that produce them
(paracrine) or on the same cell that produces the cytokine (autocrine).
Of the families identified above, there are four major categories of
cytokines:
(1) Interferons: interferon alpha (IFNa) is produced by the buffy coat
layer from white blood cells and is used in treatment of a variety of
malignant
and immune disorders. Interferon beta (IFNB) is produced by fibroblasts and
is currently being evaluated in the treatment of multiple sclerosis.
Interferon
CHIC_4513901 1 28
CA 02779198 2012-04-26
WO 2011/053281
PCT/US2009/062250
gamma (IFNy) is produced by activated T cells and is an important
immunoregulatory molecule, particularly in allergic diseases.
(2) Colony stimulating factors: includes granulocyte colony stimulating
factor (G-CSF), macrophage colony stimulating factor (M-CSF), and
granulocyte-macrophage colony stimulating factor (GM-CSF), as well as
Interleukin (IL) -3, which can stimulate a variety of hematopoietic precursors
and is being evaluated as a therapy in aplastic anemia and bone marrow
transplantation; and c-Kit ligand (stem cell factor), which has recently been
demonstrated as a cytokine necessary to cause the differentiation of bone
marrow stem cells into their various precursor elements for eventual
differentiation into RBC, WBC and megakaryocytes (platelets).
(3) Tumor necrosis factors (TNFs): TNFa is produced by activated
macrophages and TNF13 is produced by activated T cells (both TH and CTL).
These molecules seem to be involved in the pathogenesis of septic shock.
TNFs can be useful clinically for treating human tumors.
(4) Interleukins: produced by a variety of cell types such as monocytes
and macrophages, T cells, B cells and even non-leucocytes. Major
interleukins that are involved in allergies are IL-4, IL-5, IL-10 and IFNy. IL-
4
causes a switch to IgE production by differentiating B cells. IFNy can inhibit
that switch and prevent the production of specific IgE. IL-10 can actually
inhibit the activity of IFNy, allowing the original IL-4 to proceed in the IgE
cascade. Thus, an allergic response can be viewed as an allergen-specific
production of excess IL-4 and/or IL-l0, lack of adequate IFNy production or
both. Eosinophilic inflammation, a major component of allergic reactions, is
under control of IL-5 and TNFa.
Accordingly, recombinantly-produce authentic human cytokines, or
antibodies raised against them, can be administered to individuals with such
disorders or diseases in which the function of the particular cytokine is
implicated, for instance, IFNB for treatment of multiple sclerosis.
CHIC_4513901 1 29
CA 02779198 2012-04-26
WO 2011/053281 PCT/US2009/062250
3. GLYCOSYLATION AND THE PRESENT INVENTION
Most proteins undergo post-translational modification, which can alter
their physical and chemical properties, e.g., MW, pl, folding, stability, and
biological activity. Glycosylation is the most prevalent type of post-
translational modification, with estimations that 80% of all plasma proteins
are
glycosylated and the major part of the most important known human natural
interferon alpha species are glycoproteins.
Glycoproteins are oligosaccharides or sugar chains that are covalently
linked to proteins. The attachment of such sugar chains is performed in vivo
by specific glycotransferases, which are highly sensitive to stimuli within
the
cell. The carbohydrate components of the glycoproteins affect the
functionality of the molecule because they affect protein folding, oligomer
assembly, and secretion, as well as solubility and aggregation of the
expressed protein. These polysaccharide sugar chains therefore have
various functions which culminate in conferring appropriate bioactivity and
stability of the protein. Furthermore, certain protein-bound glycans are
abundant in the nucleus and cytoplasm, where they appear to serve as
regulatory switches.
The two most common classes of such sugar chains that are covalently
attached to expressed proteins in vivo, are (1) N-glycans sugar chains which
are covalently linked to a protein's asparagine residue within the consensus
"Asn-X-Ser/Thr"; and which share a common pentasaccharide core region
and fall into three classes, (a) high-mannose-type, (b) complex-type, and (c)
hybrid-type; and (2) 0-glycans which are linked to the protein via an N-
acetylgalactosamine (GaINAc) to a serine or threonine residue in the protein.
The corresponding glycosylation pathways that attach the sugar chains to the
expressed protein in these ways typically occur in the cytosol, endoplasmic
reticulum and the Golgi complex and involve glycosidases and
glycosyltransferases to facilitate those attachments.
CHIC_4513901 1 30
CA 02779198 2012-04-26
WO 2011/053281
PCT/US2009/062250
While not being bound to any particular theory of mechanistic action, a
reason why a human protein that is expressed in the present inventive human
cell system is superior to a counterpart that is expressed in a non-human cell
lies in its authentically-glycosylated surface and appropriately-folded
tertiary
structure, for the reasons described above. That is, in an in vivo human cell
environment, the post-translation modification machinery of the human cell
attaches oligosaccharide or glycan sugar chains to the outer surface of the
endogenously expressed protein so that the natively-produced protein is
covered in an array of covalently attached sugar chains, which also helps to
delay clearance of the protein from the bloodstream via the kidney renal
system. Hence, the half-life of circulatory "authentic" human proteins is
increased compared to the same protein expressed from a non-human cell.
Accordingly, while a non-human cell might be able express a human
polynucleotide sequence from an introduced expression vector, the non-
human cell environment does not lend itself to such post-translational
modification. Consequently, the resultant and expressed protein necessarily
lacks authenticity with respect its in vivo human counterpart that is properly
folded and glycosylated. For this reason, the use of human proteins that have
been expressed and purified from non-human cells in research, diagnostics,
and therapeutic ends is inferior and undesirable.
(a) Human Vs. Non-Human Patterns
The non-human cell expression systems, e.g., bacterial, yeast, fungi,
insect, and non-human mammalian systems, do not produce authentic human
proteins. Common bacterial expression systems, such as E. coli cells, for
instance, do not glycosylate recombinant mammalian proteins. Yeast and
fungal expression systems can express human DNA sequences but the
resultant glycosylation patterns from yeast and fungal cells are significantly
different from the glycosylation processing of human cells. For instance,
yeast and fungal cells attach non-human high-mannose sugar chains to the
recombinantly-expressed human protein. The mannose chains may be
CHIC_4513901 1 31
CA 02779198 2012-04-26
WO 2011/053281 PCT/US2009/062250
immunogenic and the protein cleared much quicker from the system via the
renal pathways. Insect cell expression systems are like yeast and fungi,
although the length of the mannose chains that become attached to an insect-
expressed protein are typically shorter than those attached in the yeast
system. See Zopf and Vergis, Pharmaceutical Visions, Neose Technologies,
Inc (www.neose.com), for more details.
As for non-human mammalian cell expression systems, the Chinese
Hamster Ovary cells are the most commonly used by those practicing in this
field of art. The CHO cells glycosylate differently to human cells. The CHO
glycan structure is not human-like. In CHO cells, for instance, an expressed
IFN-y includes substituted fucose residues and high mannose oligosaccharide
chains; in transgenic mice cells, the IFN-y has variant N-glycan structures
and
in insect cells, the IFN-y has tri-mannosyl core structures.
Transgenic animals also are used to produce human proteins, such as
in goat milk, but similar problems exist, such as underglycosylation and the
addition of non-human sialic acid (N-glycolylneuraminic acid).
By contrast, the N-glycans on human proteins have a specific order
that terminate in N-acetylneuraminic acid. Accordingly, an aspect of the
present invention is the recombinant expression of a human protein that
comprises only human sugar chains. That is, an "authentic" protein of the
present invention comprises one or more combinations of (1) N-
acetylglucosamine, (2) fucose, (3) mannose, and (4) galactose chains which
are terminated with N-acetylneuraminic acid. See Table 1 and Figure 4.
Accordingly, the human cell environment provides the appropriate and correct
building blocks and mechanisms for processing a recombinantly-expressed,
authentic human protein. Thus, in the context of human proteins and human
cytokines, a recombinantly-produced, authentic human cytokine/protein that
has an authentic glycosylation pattern includes, but is not limited to, a
recombinant protein that comprises one or more combinations of (1) N-
CHIC_4513901 1 32
CA 02779198 2012-04-26
WO 2011/053281
PCT/US2009/062250
acetylglucosamine, (2) fucose, (3) mannose, and (4) galactose, sugar chains
which are terminated with N-acetylneuraminic acid.
In some cases, not every individual cytokine protein that is expressed
according to the present invention will comprise the same glycosylation
pattern or extent of glycosylation as other cytokines isolated from an
expression run. Thus, there may be subpopulations of an expressed cytokine
that contain more or fewer attached human sugar chains, or which are
processed to contain longer or shorter attached sugar chains, than others
expressed from a particular human cell culture. This is not detrimental but
rather approximates the state of glycosylation in vivo. Thus, when visualized
on a protein gel, a smear around a predominantly-stained band may appear.
In the present expression system, the smear does not represent protein
contamination or degraded proteins or debris, but rather the distribution in
protein size molecular weight is attributable to the extent of glycosylation
and
the number or respective sizes of the sugar chains attached to the
recombinantly expressed protein. Thus, the more extensive is glycosylation,
or the attachment of longer sugar chains, will increase the total apparent
molecular weight of the glycosylated protein or cytokine.
B. OVERVIEW
The present invention therefore relates to a rapid and scalable method
for producing authentic human proteins from stable cultures of human cells in
vitro. The human cell expression system of the present invention is made to
be more receptive to the introduction of a novel expression vector that
encodes a human protein, as well as to suspension media that facilitates
subsequent expression of that protein-encoding polynucleotide.
1. ADVANTAGES
The human proteins produced using the inventive human cells and
methods, possess and exhibit more authentic "human-like" properties and
CHIC_4513901 1 33
CA 02779198 2012-04-26
WO 2011/053281
PCT/US2009/062250
structures than those that are expressed from non-human cells, particularly
with respect to protein folding and post-translational modifications, such as
proteolytic cleavage processing and glycosylation. For these reasons, a
human protein that is expressed using the present methods and reagents
exhibits a biological activity and circulatory half-life that more closely
approximate its endogenous, naturally-expressed native form. Furthermore,
the recombinantly-expressed human protein is more structurally comparable
to the native protein in terms of folding and normally-available disulfide
bond-
forming residues and is consequently less immunogenic than its non-human
cell-expressed counterpart. Likewise, the human cell, because it contains the
appropriate human enzymes, can more readily glycosylate the expressed
recombinant protein since its cellular environment and cytoplasmic organelles
are ideally suited, unlike non-human cell systems, to recognize and
manipulate human proteins. Accordingly, the glycosylation pattern and state
of recombinant cytokines expressed in the stable human cell cultures of the
present invention are glycosylated as though they had been produced in vivo
endogenously.
Furthermore, because the surfaces of the recombinantly-expressed
protein are more similar to the native surfaces, it possesses more authentic
surface epitopes that are ideal for producing high-affinity antibodies. Such
high-affinity antibodies therefore are very useful in therapy, diagnostics,
and
Research & Development studies. Since they have been raised against
authentic recombinantly-expressed proteins and are therefore can recognize
the endogenous, naturally-expressed counterpart proteins more specifically, a
smaller quantity of antibody may be administered to an individual or used in
an in vitro method. For that reason, a fraction of the dosage of a particular
antibody raised against an authentic protein of the present invention may be
used to accomplish a treatment that otherwise requires a much more
concentrated dose, as explained in detail elsewhere herein. Not only does
the use of lower concentrations of such antibodies mean that the frequency of
dosage may be reduced, but also that the incidence of side effects can also
CHIC_4513901 1 34
CA 02779198 2012-04-26
WO 2011/053281 PCT/US2009/062250
be reduced or eliminated entirely. Furthermore, the present human cell line,
methods, and compositions make it possible to express human proteins,
especially certain cytokines such as TGFB1, that have hitherto been
notoriously difficult, if not impossible, to express in such other non-human
cells. See the Examples and Figure 5, which evidence production of TGFB1,
for instance, using the present inventive method.
As explained herein, the human cell cultures of the present invention
are stable. See "Methodology" below and the Examples. That is, the same
culture of cells can be used repeatedly to express and subsequently secrete
into the supernatant any desired human protein, such as a human cytokine.
The human cells are stable because they have been selected for their ability
to grow in serum-free media using antibiotic selection techniques. The
significance of this is many-fold: the same volume of human cells can be
sustained for long periods of time unlike in existing systems which are unable
to be replenished. This means that the human cells of a spun-down aliquot of
the culture can be returned to the stock culture and allowed to re-
proliferate.
By contrast, because the human cell systems of the prior art are not
stable, a volume of culture represents only a one-time opportunity to harvest
the expressed proteins. That is, once the cells have been lysed or collected
by centrifugation to isolate the protein-containing supernatant, the human
cells of the prior art are for all intents and purposes dead and unusable.
Accordingly, the re-usability of the present human cells is highly appealing.
Not only does this mean that the same volume of cells can be reused
time and again, but that the inventive method is adaptable to large-scale
processing. Since the stability of the human cells is established early on,
then
the human cells are able to survive in any volume of culture medium. See
Figure 1. For instance, it is possible to maintain a viable culture of human
cells in 1,2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20,
21,
22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40,
41,
42, 43, 44, 45, 46, 47, 48, 49, 50 liters, or more than 50 liters of culture,
or any
CHIC_4513901 1 35
CA 02779198 2012-04-26
WO 2011/053281
PCT/US2009/062250
integer in between. Accordingly, the present inventive method allows for
commercial scale production of recombinant proteins, such as recombinantly-
expressed cytokines, at yields that are commercially desirable.
Thus, the present inventive human cell expression system lends itself
to scalability, and thereby to producing increased yields of an authentically-
produced protein per unit time. See Figure 2. Because the cell culture uses
serum-free media, the resultant supernatant contains essentially only the
secreted expressed recombinant protein. Consequently, the purity of the end
product, i.e., of the recombinantly-expressed protein, is very high.
Consequently, the concentration and bioactivity of the isolated expressed
protein are high and the possibility of contamination with cellular debris and
other non-desirable proteins is low.
The purity of the expressed human cytokine and the supernatant into
which it is secreted also are important factors in establishing accurate and
authentic biological activity and for ensuring that antibodies that are raised
against it are also highly specific with high binding affinities in and of
themselves. The use of serum-free media therefore for growing a stable
human cell culture is very helpful in this regard because there is little, if
any,
contaminating protein or cell debris in the expressed cytokine eluate or
supernatant.
Along the same lines, because the recombinant protein is secreted
from the human cells and into serum-free media, it is relatively
straightforward
to therefore isolate the expressed and secreted protein. That is, it is not
necessary to employ a "His-Tag" isolation scheme to isolate the expressed
protein from the supernatant. See Figure 3. A His-tag is typically a stretch
of
six histidine amino acid residues which are engineered into the expression
cassette so that the expressed protein is a fusion including the six residues,
which can subsequently become bound to partnering residues that are coated
in the inside of a column. Accordingly, because it is unnecessary to use such
CHIC_4513901 1 36
CA 02779198 2012-04-26
WO 2011/053281 PCT/US2009/062250
a tag to isolate the expressed proteins of the present invention, an
additional
step toward producing the authentic human protein is eliminated.
Thus, the inventive method is scalable to commercial levels, employs a
re-usable, self-perpetuating human cell culture suspended in serum-free
media, which eliminates contamination and increases purity yields of the
expressed recombinant protein. That resultant recombinant protein is highly
authentic with respect to its folding and glycosylation state such that the
recombinantly-expressed authentic protein is highly similar to its endogenous,
native counterpart.
2. METHODOLOGY
(a) Preparation of human cells
Human cells are plated onto a Petri dish as a monolayer on serum-free
medium. The cells that survive on the serum-free medium are then placed
into serum-containing medium to produce a working cell bank. Serum-free
media includes, but is not limited to 293 SFM II, CD 293, FreeStyle 293,
Hybridoma-SFM (Invitrogen), and Ex-Cell 293 (JRH Biosciences).
(b) Transfection
An appropriate concentration of plasmid DNA and transfectant is added
to a Petri dish containing a confluent layer of cells. Useful transfectants
include, but are not limited to FuGene 6, FuGene HD (from Roche);
Lipofectamin, Lipofectamin 2000, 293fectin (Invitrogen), and
Polyethyleneimine. The plasmid DNA is engineered to comprise an antibiotic
resistance gene to aid in the selection of appropriately transfected human
cells. See the sections which follow below.
(c) Selection of transfected cells after exposure to antibiotics
The transfected cells of step (b) are harvested by centrifugation after
trypsin treatment (which detaches cells from the Petri dish plate), and then
CHIC_4513901 1 37
CA 02779198 2012-04-26
WO 2011/053281 PCT/US2009/062250
resuspended in serum medium that contains a certain concentration of
antibiotics, e.g., 400 ug/ml or 800 ug/ml, of an antibiotic such as neomycine
(G418), hygromycine, zeocin, or blasticidine, for a period of time. Surviving
cell colonies can then be harvested and exposed to another round of
antibiotics for another period of time.
(d) Cell adaptation
The cells that survive antibiotic treatment are then resuspended in
serum-low medium that includes, for instance, 1% serum, and antibiotics, to
determine which cells are stable enough to maintain viability and growth in
liquid culture. This "adaptation" period can take a few weeks, after which
time
the cells are transferred to serum-free medium that contains only antibiotics,
and again left to "adapt" for a period of time.
Such a suspension of adapted cells therefore represents a stable
culture of human cells that can withstand the rigors of transfection and
antibiotic selection. Furthermore, those cells can be continuously grown and
used to inoculate larger volumes of culture or also can be cryo-banked in
fresh medium for future inoculations of culture.
(e) Producing cytokines from suspended, antibiotic-resistant human
cells
The plasmid DNA expresses a cytokine gene or encoding
polynucleotide that is secreted from the antibiotic-resistant human cells that
are continuously growing in the large volume culture suspension of (d).
Aliquots of that large suspension can therefore be centrifuged gently to
pellet
the human cells and separate the supernatant which will contain the desired
cytokine. Since the suspension medium in which the human cells are growing
is serum-free, or protein-free, there is little, if any, contaminating
cellular
material, such as other proteins or cellular debris. Accordingly, the
supernatant that contains the secreted cytokine is relatively pure.
CHIC_4513901 1 38
CA 02779198 2012-04-26
WO 2011/053281 PCT/US2009/062250
The human cell pellet can then be resuspended and reintroduced into
the same or different suspension or media to restart the expression process.
Thus, the whole system is repeatable and not, unlike prior art expression
systems, a one-time usable suspension of cells.
(f) Yield
The yield of protein that can be produced by the present inventive
method can depend on the volume of human cell culture used to express the
desired polynucleotide construct, the cytokine and its size, and the
constituent
base of the culture medium itself. Yield can be affected by changing certain
components of the culture medium, temperature, pH, or by including glycan
precursors in the mix to facilitate glycosylation mechanisms. The yield of
expressed human cytokine therefore from the human cell expression system
of the present invention can be from 1-500 mg/liter, or more than 500
mg/liter.
Accordingly, the present invention contemplates yields of cytokines from the
human cell expression system of the present invention of about 1 mg/liter,
about 2 mg/liter, about 3 mg/liter, about 4 mg/liter, about 5 mg/liter, about
6
mg/liter, about 7 mg/liter, about 8 mg/liter, about 9 mg/liter, about 10
mg/liter,
about 20 mg/liter, about 30 mg/liter, about 40 mg/liter, about 50 mg/liter,
about
60 mg/liter, about 70 mg/liter, about 80 mg/liter, about 90 mg/liter, about
100
mg/liter, about 120 mg/liter, about 140 mg/liter, about 160 mg/liter, about
180
mg/liter, about 200 mg/liter, about 220 mg/liter, about 240 mg/liter, about
260
mg/liter, about 280 mg/liter, about 300 mg/liter, about 320 mg/liter, about
340
mg/liter, about 360 mg/liter, about 380 mg/liter, about 400 mg/liter, about
420
mg/liter, about 440 mg/liter, about 460 mg/liter, about 480 mg/liter, or about
500 mg/liter or more than about 500 mg/liter. The invention also
contemplates yields between any of these concentrations.
Thus, a yield of the present invention may be between about 10-50
mg/liter, or between about 50-100 mg/liter, or between about 100-150 mg/liter,
or between about 150-200 mg/liter, or between about 200-250 mg/liter, or
between about 250-300 mg/liter, or between about 300-350 mg/liter, or
CHIC_4513901 1 39
CA 02779198 2012-04-26
WO 2011/053281 PCT/US2009/062250
between about 350-400 mg/liter, or between about 400-450 mg/liter, or
between about 450-500 mg/liter, or between about 500-550 mg/liter, or
between about 550-600 mg/liter, or between about 600-650 mg/liter, or
between about 650-700 mg/liter, or between about 700-750 mg/liter, or
between about 750-800 mg/liter, or between about 800-850 mg/liter, or
between about 850-900 mg/liter, or between about 900-950 mg/liter, or
between about 950-1,000 mg/liter.
In other words, a yield of recombinant-produced, authentic proteins of
the present invention made according to the methods disclosed herein may
be 2-fold, 3-fold, 4-fold, 5-fold, 6-fold, 7-fold, 8-fold, 9-fold, or 10-fold
greater
than the yield obtained from other cell expression systems.
3. VECTOR DESIGN
An aspect of the present invention is a novel expression cassette that
comprises
(1) a cytomegalovirus enhancer element, such as the CMV
immediate early enhancer element (CMV I E);
(2) a human promoter sequence selected from the group consisting
of (i) a human actin promoter, (ii) a human serum albumin promoter, and (iii)
a
human fibrinogen promoter.
(3) a human globin gene intron; and
(4) a signal peptide, such as immunoglobulin superfamily 8 signal
peptide or an alpha-fibrinogen signal peptide.
The expression vector may also comprise a nucleic acid sequence of
interest or a desired polynucleotide operably linked to the above-described
elements (1)-(4), positioned downstream of the signal peptide, so that it can
be subsequently expressed in a cell.
CHIC_4513901 1 40
CA 02779198 2012-04-26
WO 2011/053281 PCT/US2009/062250
A desired polynucleotide sequence may be one that encodes a desired
cytokine or cytokine fragment. The expression cassette also may comprise a
sequence for an antibiotic resistance gene, which may be cloned between the
regulatory elements used to express the cytokine polynucleotide, or which
may be operably linked to its own promoter and terminator independently from
the expression cassette that is used to express the cytokine-signal peptide
fusion protein.
Thus, expression vectors of the present invention include, for instance,
the following:
1. pHZhag comprising operably linked nucleotide sequences for (i)
an hCMV 1E, (ii) a human beta-actin promoter, and (iii) a human beta-globin
intron;
2. pHZA comprising operably linked nucleotide sequences for (i)
an hCMV promoter, and (ii) a human fibrinogen subunit A signal peptide;
3. pHZI comprising operably linked nucleotide sequences for (i) an
hCMV promoter, and (ii) a human Ig superfamily 8 signal peptide;
4. pHZhagA comprising operably linked nucleotide sequences for
(i) an hCMV 1E, (ii) a human beta-actin promoter, (iii) a human beta-globin
intron, and (iv) a human fibrinogen subunit A signal peptide;
5. pHZhagl comprising operably linked nucleotide sequences for
(i) an hCMV 1E, (ii) a human beta-actin promoter, (iii) a human beta-globin
intron, and (iv) a human Ig superfamily 8 signal peptide;
6. pHZhag-TGFB1 comprising operably linked nucleotide
sequences for (i) an hCMV 1E, (ii) a human beta-actin promoter, (iii) a human
beta-globin intron, and (iv) TGFB1; and
CHIC_4513901 1 41
CA 02779198 2012-04-26
WO 2011/053281 PCT/US2009/062250
7. phZhagl-TGFB1 comprising operably linked nucleotide
sequences for (i) an hCMV 1E, (ii) a human beta-actin promoter, (iii) a human
beta-globin intron, (iv) a human Ig superfamily 8 signal peptide, and (v)
TGFB1.
Control expression cassettes include:
1. pCAG comprising operably linked nucleotide sequences for (i)
hCMV 1E, (ii) chicken beta-actin promoter, and (iii) rabbit beta-globin
intron;
and
2. pHZsec comprising operably linked nucleotide sequences for (i)
hCMV promoter, and (ii) mlg-kappa leader.
(a) Cloning
Cloning of cytokines can be accomplished by various methods
available to one skilled in the art of genetic engineering. For example, total
RNA or poly-A RNA can be purified from human tissues samples abundant in
particular cytokine expression (for example lymphocytes) and used as a
template for gene specific RT-PCR. Additionally, pre-made cDNA libraries
can be purchased from commercial sources and PCR can be employed to
amplify the cytokine cDNA directly. Still further, synthetic oligonucleotides
can
be constructed to create a synthetic gene for the cytokine based on sequence
information available in National Center for Biotechnology Information with
their gene accession numbers. Additionally, full length cDNA clones can be
obtained from, for example, the IMAGE clone consortium (image.IInl.gov/) or
Openbiosystems (Huntsville, AL). The full length cytokine cDNA clones were
obtained from Openbiosystems (Huntsville, AL). Gene accession numbers of
cloned cytokines are presented in Table 3.
A cytokine typically has a signal peptide at its N-terminal which can be
identified by numerous tools available to one skilled in the art; for example
using Swiss-Prot protein knowledgebase. Some cytokines have variants due
CHIC_4513901 1 42
CA 02779198 2012-04-26
WO 2011/053281 PCT/US2009/062250
to different transcription that can be also identified by the available tools.
Sequences of the secreted cytokines are listed in Subsection 10 above. To
facilitate rapid cloning of cytokines with different signal peptides, it is
possible
to engineer an expression cassette so that the expressed cytokine contains a
signal peptide. For instance, pSecTag2c (Invitrogen, Carlsbad CA), which is
suitable for the production of secreted recombinant protein in mammalian
cells (for example, CHO and HEK293), can be engineered into a vector of the
present invention by site directed mutagenesis using appropriate cloning and
restriction digest strategies to integrate the DNA encoding the signal peptide
in the correct reading frame orientation so that it is properly transcribed
and
translated along with the co-joined cytokine sequence. For instance, it is
possible to introduce a Srf I restriction site in frame with the IgK leader
sequence (see Figure 27) using mutagenesis primers SecTag2c-srflf
(TCCACTGGTGACGCGCCCGGGCCGGCCAGGCGCGCC) (SEQ. ID. NO
34) and SecTag2c-srflr
(GGGGCGCCTGGCCGGCCCGGGCGCGTCACCAGTGGA) (SEQ. ID. NO:
35).
4. HUMAN CELLS
The types of human cells that can be used to express a protein of the
present invention includes human stem cells, human precursor cells, human
kidney cells, human retina-derived PER-C6 cell, a human embryonal kidney
cell line. Particularly useful human cell lines include but are not limited to
HEK 293 cells and derivatives thereforeof, such as HEK 293T, HEK 293S,
and HEK 293 EBNA.
In particular the present invention provides a novel human cell line
denoted as HZ-293T5, which was adapted herein to serum-free and
chemically-defined media. See Example 2. The cell line named HZ-293T5 is
deposited under and bears the ATCC biological deposit accession number of
PTA-10165.
CHIC_4513901 1 43
CA 02779198 2012-04-26
WO 2011/053281 PCT/US2009/062250
5. PRODUCTS
(a) Cytokines
According to the present inventive methods, it is possible to conduct
medium and large scale production of human cytokines from human cells.
The inventive method has been successful in producing more than 60 tag-free
cytokines, including difficult-to-express proteins of the TGF-B superfamily.
Using GM-CSF, IL4 and VEGF165 as examples, for instance, it is
demonstrated herein that highly-authentic glycosylated cytokines can be
expressed and isolated from human cells and used as highly preferred
reagents for subsequent development of treatments of inflammation, cancer,
stem cell research, and raising of antibodies. Furthermore, not only are the
human cytokines that are produced by the present inventive technology more
natively glycosylated but the appropriate disulfide bonds that are sometimes
necessary to create a fully functional complex are also intact. This is unlike
the situation in certain non-human cells, where disulphide bonding between
expressed monomers is not possible.
Being able to produce highly authentic human proteins without the use
fo purification- or isolation-required tags, such as histamine-tags is a
highly
desirable feature of the present invention. The present inventive method and
vector system permit the expression of human proteins that are highly
authentic in terms of their structural, biochemical, and functional identities
to
their native, endogenous human versions; meaning that the polynucleotides
that encode the authentic protein sequences of the present invention do not
necessarily require the incorporation of histidine-encoding residues to aid in
purification and isolation of the subsequently expressed protein. Thus, an
aspect of the present invention is a method for recombinantly producing
authentic human proteins without the use of peptidic tags, and the authentic
proteins themselves which are "tag-free." Hence, one aspect of an "authentic"
protein of the present invention is that it is tag-free. However, this is not
meant to exclude the possible use of tags, such as his-tags in conjunction
CHIC_4513901 1 44
CA 02779198 2012-04-26
WO 2011/053281 PCT/US2009/062250
with the present inventive methods and vectors. That is, the present invention
also encompasses the recombinant production of an authentic protein in
which a tag, such as a his-tag, is incorporated as a fusion protein to the N-
or
C-terminus of the authentic protein to aid in its identification,
purification, and
isolation.
It is highly desirable to express the following cytokines in the stabilized
human cell expression system of the present invention: Activin A, Activin B,
Activin A/2xINHbA, Activin B/2xINHbB, AMH/MIS, Artemin, BDNF, BMP2,
BMP15/GDF9B, BMP2/BMP2A, BMP3/0steogenin, BMP4, BMP4/BMP2B,
BMP5, BMP7/0P-1, BMP1, BMP10, BMP15/GDF9B, B-NGF, Cystatin C,
Delta 1, EGF, Erythropoietin (EPO), FGF acidic, FGF basic, FGF10, FGF5,
FGF7, FGF8b, FLT3 ligand, G-CSF, GDF15, GDF2/BMP9, GDF3,
GDF5/BMP14, GDF8/myostatin, GDF9, GDNF, GM-CSF, HGF, HGH, IFN-
a2A, IFN-a2B, IFN-y, IFN-131, IGF I, IGF II, IGF 11v1 , IGF IIv2, IL1 0, IL11,
IL12,
IL15, IL17/1L17A, IL17F, IL1 1 IL2, IL23, IL27, IL28A/IFN-lambda-2,
IL28B/IFN-lambda-3, IL29/IFN-lambda-1, IL113, IL2 IL3, IL32, IL35, IL4, IL5,
IL6, IL7, IL8, IL9, Inhibin A/INHa&INHbA, Inhibin B/INHa&INHbB, Inhibin
C/INHa&INHbC, Inhibin E/INHa&INHbE, LEFTYB, LEFTY1/LeftyB, M-CSF,
mouse CSF, mouse SCF, NODAL, Noggin, NT3 (neurotrophin3), Oncostatin
M, PDGFa, PDGF13, Persephin, SCF, SDF1 a, SHH, Somatotropin, TGF 131,
TGF 132, TGF 133, TGF134/LEFTY2/LeftyA, TNF a, TP0a, VEGF121aa,
VEGF165aa, WIF1, WNT1, Wnt10A, Wnt106/12, Wnt11, Wnt16, Wnt2,
Wnt2B/13, Wnt3, Wnt3A, Wnt4, Wnt5A, Wnt5B, Wnt6, Wnt7A, Wnt7B,
Wnt8B, and Wnt9A/14. See also the "List of Cloned Cytokine Genes" at
Table 5. See also the list of cytokines listed in Table 5. Any of those
cytokines may be expressed in the human cell expression system of the
present invention too.
The Examples below and the corresponding figures demonstrate
successful expression of highly pure, authentically-glycosylated bioactive
cytokines, namely EPO, G-CSF, GM-CSF, IL-2, IL-4, IL-6, M-CSF, Noggin,
CHIC_4513901 1 45
CA 02779198 2012-04-26
WO 2011/053281 PCT/US2009/062250
SCF, Somatotropin, TGF81, TNFa, and VEGF-165. See Figures 6A-M, 7A-D,
and 8A-C for graphical and gel data evidencing recombinant expression of
these cytokines and their respective bioactivities and comparisons against
cytokines expressed from other cell systems.
(b) Antibodies
Another benefit attributable to authentically glycosylated, folded, and
phosphorylated proteins of the present invention is that they are excellent
reagents for raising highly specific antibodies.
Monoclonal antibodies are useful for assaying for the presence of a
particular cytokine or for isolation and purification of the proteins to which
they
specifically bind. Accordingly, monoclonal antibodies are useful for
diagnostic
assays, detection assays, and purification protocols. Because the
recombinant cytokines produced by the present inventive human expression
system are more authentically glycosylated and folded than had they been
expressed in non-human cells, any antibodies that are raised against them
will have a higher affinity toward endogenous cytokines in the human body or
in a human biological sample.
Monoclonal antibodies can be made according to known methods
using recombinant human interferon(s) used as specific antigen(s). One or
more antigens can be injected at one time. For instance by using interferons
made from E. coli, and from interferons made from transfected according to
previous methods described in this invention using transfected human cell
producing the identical interferon(s). Cross board analytic testing of several
of
the clones producing antibodies selected from recombinant interferons made
from E. co//when compared to antibodies selected from clones immunized
with authentic recombinant human interferon, made from transfected human
cells. It also is possible to obtain a cell line which is able to produce
monoclonal antibodies by fusing mouse myeloma cells to spleen cells
followed by subsequent selection of clones capable of producing the desired
CHIC_4513901 1 46
CA 02779198 2012-04-26
WO 2011/053281 PCT/US2009/062250
antibody. See KOhler & Milstein, Nature (1975), 256 (5517):495-497; and
KOhler et al., Eur J Immunol. (1976), 6:292-295. Panels of monoclonal
antibodies produced against epitopes can be screened for various properties;
i.e., for isotype and epitope affinity. An alternative technique involves
screening phage display libraries where, for example the phage express scFv
fragments on the surface of their coat with a large variety of complementarity
determining regions (CDRs). This technique is well known in the art.
Polyclonal antibodies using antigens made from the secretion of
glycosylated recombinant human cytokines can produce antibodies that
besides recognizing the protein part of the molecule, also recognizing the
glycosylated part of the particular recombinant human interferon. Polyclonal
antibodies may be raised by immunizing various species of animals, such as
rabbits, goats, sheep, or other animals, by immunizing the animals with the
authentic recombinant human interferon, made using the method
encompassed in this invention by repeated injections of microgram to mg
amounts of one of these interferons as antigen, often together with Freunds
Incomplete adjuvant. Examples of polyclonal antibodies ("pAbs")
encompassed by the present invention include, but are not limited to, pABs for
EPO, FLt3, G-CSF, GM-CSF, M-CSF, IFN-a2A, IL-4, and IL-6.
As used herein, an "antibody" refers to a protein consisting of one or
more polypeptides substantially encoded by immunoglobulin genes or
fragments of immunoglobulin genes. Antibodies may exist as intact
immunoglobulins or as a number of fragments, including those well-
characterised fragments produced by digestion with various peptidases. While
various antibody fragments are defined in terms of the digestion of an intact
antibody, one of skill will appreciate that antibody fragments may be
synthesised de novo either chemically or by utilising recombinant DNA
methodology. Thus, the term antibody, as used herein also includes antibody
fragments either produced by the modification of whole antibodies or
synthesised de novo using recombinant DNA methodologies. Antibody
CHIC_4513901 1 47
CA 02779198 2015-12-22
fragments encompassed by the use of the term "antibodies" include, but are
not limited to, Fab, Fab', F(ab')2, scFv, Fv, dsFy diabody, and Fd fragments.
Furthermore, the antibodies and fragments thereof may be humanised
antibodies, for example as described in EP-A-239400.
Examples of antibodies prepared according to the present invention
include polyclonal antibodies raised against recombinantly-produced,
authentic human IL-2, IL-4, IL-6, EPO, G-CSF, GM-CSF, M-CSF, FLT3L, and
IFN-a2A. Monoclonal antibodies that are being made include, but are not
limited to, monoclonal antibodies against recombinantly-produced, authentic
human IL-2, IL-4, IL-6, EPO, G-CSF, TGF-131, IFN-a, and IL-17.
(c) Kinases
It is also highly desirable to express the following human kinase genes
in the stabilized human cell expression system of the present invention so as
to obtain recombinantly-produced, authentic human kinases, including but not
limited to: AKT1, AKT2, AMPK1, ATM, Aurora A, BTK3, CDK6, ERK5, Fyn-1,
GRK5, JNK1, LYN, MAPKAPK2, MAPKAPK3, MEKK3, MKK3, MKK4, mTOR,
P38-a, P70S6K2, PDK1, PKC-B, PKC-y, PTEN, SYK, and Zap70.
(d) Other proteins
The present invention is not limited to producing only cytokines and
antibodies raised against those cytokines. Other human proteins can be
expressed according to the present invention using the inventive stable
human cell line, including, but not limited to kinases and other enzymes.
Thus, the present invention also encompasses the recombinant production of
authentic human plasma proteins selected from the group consisting of
albumin, IgG, IgA, IgM, IgD, IgE, alpha-1-proteinase inhibitors, blood pro-
coagulation proteins, blood anti-coagulation proteins, thrombolytic agents,
anti-angiogenic proteins, alpha.-2-antiplasmins, C-1 esterase inhibitors,
48
CA 02779198 2012-04-26
WO 2011/053281 PCT/US2009/062250
apolipoproteins, HDL, LDL, Fibronectin, beta-2-glycoprotein I, fibrinogens,
plasminogens, plasmin, plasminogen activators, plasminogen inhibitors,
plasma protease inhibitors, anti-thrombin III, streptokinases, inter-alpha-
trypsin inhibitors, alpha.-2-macroglobulin, amyloid protein, ferritins, pre-
albumin, GC-globulin, haemopexin, C3-complement, transferrin, urokinase,
alpha.-1-acid-glycoprotein, coagulation, anti-coagulation factor(s) (such as
Factor II, Factor V, Factor VII, Factor VIII, von Willebrand factor, Factor
VIII--
von Willebrand factor complex, Factor IX, Factor X, Factor XI, Cl inhibitor,
protein C and Protein S), extracellular membrane proteins, or extracellular
domains of receptors.
7. PURIFICATION
Purification of the authentic-like recombinant is tailor made to the
expression of cells, and human cells such as HEK cells, and other related
human cells after having expanded the necessary amount of the targeted
interferon genes in coli bacteria culture. The cells used for the particular
interferons are chosen and is tailor made to the particular vector, where one
has chosen the correct human-component originated promoter. The particular
human cell selected for individual interferons necessitates thorough selection
of the optimal human cell line(s). Due to the fact that the cells are adapted
to
cell suspension culture serum-free medium, purification methods applied to
the harvested medium are easier to design when compared to serum
containing cell culture medium.
For instance, the supernatant of the serum-free medium can be
collected by centrifugation and the cell pellet resuspended in fresh serum-
free
medium for further production of the cytokine. Expression of cytokines can be
readily identified on SDS-PAGE gel analysis using Coomassie stain by known
molecular weight or via traditional Western blot analysis. To capture the
cytokine, the supernatant was at first loaded on an immobilized metal affinity
chromatography (IMAC) column. Based on their properties some cytokines
were bound on IMAC column while some were found in flow through. As next
CHIC_4513901 1 49
CA 02779198 2012-04-26
WO 2011/053281 PCT/US2009/062250
purification step cytokine fraction pool was loaded on an ion exchange
chromatography (IEX) column after buffer exchange to a proper buffer
condition. Finally as polishing step cytokine fraction pool was loaded on
another !EX or different affinity chromatography (for example Heparin resin)
column.
(b) Purity
(i) Cytokine purity levels
After up to three chromatography steps cytokines were more than 95%
pure judged by Coomassie stain on SDS-PAGE gel (Figure 7) and by
Western blot with available antibodies (Figure 8). Pure cytokines then were
quantified using known methods available to one skilled in the art (for
example Bradford assay, Coomassie stain on the gel, and OD280nm). After
the quantification cytokines were analyzed endotoxin level by endotoxin
detection kit from Lonza (Allendale, NJ) according to manufacturer's manual,
aliquoted based on required amounts and lyophilized for commercialization.
(ii) Low contaminants
Endotoxins are frequent contaminants in the cytokines prepared from
bacterial and other expression systems. Even low levels of endotoxins can be
toxic to cells or organisms and must be removed. The industry standard
reported value for commercially supplied cytokines is <1.0 EU/pg. The
present inventive methods are able to produce proteins, such as various
different human cytokines, with ultra-low levels of endotoxin contamination.
Cytokines, as disclosed below, have been prepared according to the present
invention which yield ten times to one thousand times less endotoxin than the
levels reported in standard commercial preparations.
8. ASSAYS
CHIC_4513901 1 50
CA 02779198 2015-12-22
Many different assay systems are available for ascertaining cytokine
"purity," "activity" and for quantifying concentrations of cytokines or for
detecting cells that express them. Cytokine bioassays measure, for instance:
(i) cell proliferation induced by cytokines, (ii) chemotaxis, (iii)
cytotoxicity, (iv)
capacity to induce colony formation, (v) cellular degranulation, or (vi) the
induction of secretion of further cytokines or other compounds.
(a) Activity
As mentioned above, standard assays for determining cytokine activity
include (a) cytokine-induced proliferation of indicator cell lines; (b)
cytokine-
induced apoptosis; (c) cytokine-induced protection from viral infection; and
(4)
cytokine-induced cytokine production. Details for each particular method are
well known to the skilled artisan and can be found at "Cytokine Bioassays".
Biological responses induced by cytokines show saturation kinetics,
which can be used to quantitate their amounts from dose-response curves.
These assays involve the use of primary cell cultures and, more frequently,
established cell lines that depend upon the presence of (a) particular
cytokine(s) for their growth or survival or that respond to a given cytokine
in a
particular way. (See www.copewithcytokines.de/cope.cgi?key=bioassays).
One method is the MTT assay, a quantitative colorimetric method that
determines the effect of a cytokine on cell proliferation. The MTT assay
utilizes the yellow tetrazolium salt [3-(4,5-Dimethylthiazol-2-y1)-2,5-
diphenyltetrazolium-bromide] which is metabolized by mitochondrial succinic
dehydrogenase activity of proliferating cells to yield a purple formazan
reaction product, which can be detected using a colorimeter.
Other cytokine assays include (1) immunoassays which generally
measure immunoreactivities and are useful indicators, therefore, of the
presence of cytokines and include (a) radioimmunoassays,
51
CA 02779198 2012-04-26
WO 2011/053281 PCT/US2009/062250
(b) immunoradiometric assays, and (c) enzyme-linked immunosorbent
assays. These particular assays require cytokine-specific antibodies or
labeled cytokines, receptors, or antibodies. Accordingly, the high-affinity
antibodies created by the present invention are ideally useful for
implementing
such immunoassays, especially since the synthesis of some cytokines in vivo
often occurs at such low levels that it is difficult their presence by
standard
immunoassay techniques; (2) the Cytometric Bead Array combines sandwich
immunoassays with flow cytometry for simultaneous measurement of the
characteristics of multiple particles and has been adapted to the simultaneous
determination of a variety of cytokines in small volumes; (3) immunological
assays are commercially available for many cytokines; (4) the in vivo cytokine
capture assay is a method allowing the determination of in vivo concentrations
of cytokines; (5) radioreceptor assays measure concentrations of cytokines by
displacing ligands from cell-bound receptors; (6) the reverse hemolytic plaque
assay is an adaptation of a immunoglobulin-secreting cell plaque assay that
can be used to detect individual cells that secrete cytokines and to determine
the amounts of a particular cytokine secreted by this cell; (7) the cell blot
assay also allows visualization of release of cytokines by producer cells; (8)
the kinase receptor activation assay which exploits the fact that ligand
binding
to receptors can cause tyrosine phosphorylation of the receptor; hence,
activity can be inferred by measuring the amount of receptor phosphotyrosine
rather than cell proliferation or cell survival; (9) Factor-dependent cell
line
assays are assays in which cells respond in a particular way to individual
cytokines or freshly isolated cells; (10) cytokine immunotrapping is an assay
for studying the kinetics of production and consumption or degradation of
cytokines; (11) RT-PCR quantification of cytokine mRNA using probes and
PCR primers can be designed to anneal and amplify cytokine genes or their
alternatively spliced variants, e.g., the splice variants of HGF, designated
as
HGF/NK1, HGF/NK2, HGF/NK4, for example, can be amplified or detected
using variant-specific primers or probes.
CHIC_4513901 1 52
CA 02779198 2012-04-26
WO 2011/053281 PCT/US2009/062250
In particular, biological activity of an expressed cytokine can be
measured by ED50 on the dose-dependant cytotoxicity (for example, TNFa),
stimulation of the proliferation (for example, IL-2), or inhibition of other
cytokine induced proliferation on effective cells (for example, TGF-(31) based
on the nature of the cytokine.
(b) Comparative Assays
Also provided herein are side-by-side gel comparisons of cytokines that
have been expressed by the inventive system alongside the same cytokines
expressed in non-human cells to demonstrate the differences in purity and
glycosylation state between the two protein products. See Figures 7 and 8.
9. EXEMPLARY THERAPEUTIC USES OF RECOMBINANTLY-
PRODUCED AUTHENTIC CYTOKINES
As detailed in the preceding passages and sections, the cytokines that
are expressed from the present inventive stable human cell expression
system have a number of advantages and benefits over cytokines that are
produced in other cell systems, such as more "human-like" or more
"authentic" folding, glycosylation, phosphorylation state, epitope-
presentation,
and native dimerization characteristics. Accordingly, the actual protein
structure produced according to the present inventive method is more similar
to its native, endogenous counterpart. This translates into significant
advantages in the context of therapeutic uses for these authentic recombinant
cytokines. See also Table 2 below for correlations between various cytokines
and function. The skilled person is able to correlate particular disorders and
diseases with abnormal or malfunctioning cytokine activity and thereby devise
a therapy to compensate or rectify that abnormality as is discussed in more
detail below.
Cytokines that are expressed in non-human cell systems are not
authentic, e.g., they lack human-specific glycosylation patterns, are
covalently
CHIC_4513901 1 53
CA 02779198 2015-12-22
linked to non-human sugar chains, or are incorrectly folded. Furthermore,
those cell cultures cannot be replenished, i.e., the culture of cells cannot
be
re-used after the recombinant protein has been expressed and isolated from
it. For these reasons, such non-authentic cytokines have low potency, short
half-life and can induce allergies and undesirable immunogenic responses
from the body.
By contrast, the cytokines of the present invention are authentic and
human-like. Therefore, the cytokines produced by the present invention have
a higher potency and long half-life which means, in realistic and practical
terms, that a lesser dosage of the cytokine needs to be administered to an
individual in need of it. The lower dose and longer half-life also means that
the frequency of dosing can be reduced. Furthermore, because the present
cytokines are more human-like, they are less likely to induce allergies or be
immunogenic. Overall, therefore, the individual who needs to receive
treatment can be entered into a course of treatment that requires him or her
to
take fewer pills or tablet formulations of the cytokine, or injections of a
cytokine preparation, and not so frequently, without fear of side-effects
attributable to immunogenic responses triggered by present non-authentic
cytokines. See, for instance, the potential side-effects and problems
associated with currently available drugs such as Roferon which is
formulated to contain recombinant IFN-02a.
In this regard, because the present invention provides for the
recombinant production of highly authentic human cytokine proteins, dosage
amounts of the cytokine that are required for a particular use, are
dramatically
lower than the dosage amounts required for the same cytokine produced from
different constructs in different cell types. For instance, the recombinant
expression of highly authentic human leukocyte interferon requires only 3 to 5
million IU, as compared to 40 million units per dose required to elicit the
same
54
CA 02779198 2015-12-22
effect from non-authentic human leukocyte interferon (based on yield and
authenticity analyses of the presently produced cytokines). This represents a
log-10 higher efficacy than the currently produced recombinant human
interferons alpha. Accordingly, the side effects produced by interferon alpha
are minimal, if any, at 3-5 million units; whereas the 40 million units used
to
produce the same effect as 3 -5 million units however, produces very serious
side effects, including serious neurological side effects.
The skilled person is aware of the various in vivo and ex vivo
therapeutic uses for the present inventive authentic human cytokines. For
instance, the present invention encompasses, and is not limited to, the use
of:
(1) recombinantly-produced, authentic interferons for treating multiple
sclerosis, immune mediated disease, cancer, autoimmune diseases (such as
lupus, asthma, and Crohn's Disease), and viral hepatitis;
(2) recombinantly-produced, authentic interleukins for treating
autoimmunity, cancer; IL-6 (Tocilizumab) for treating Castleman's Disease;
IL10, IL-11, IL-12, and IL-13 for treating immune cell modulation; IL-21 and
Its
Receptor for preparing cancer models in mice; IL-1 for treating autoimmune
disease (such as rheumatoid arthiritis and psoriasis); IL -4 Receptor and IL-5
for treating asthma; IL-8 for treating psoriasis; and IL-12, IL-13, IL-17, and
IL-
18 for treating TH1-mediated autoimmunity;
(3) recombinantly-produced, authentic TNF-cc and inhibitors thereof in
TM
FDA-approved and follow-on drugs such as but not limited to Enbrel
TM
(Etanercept), Remicade (lnfliximab), Golimumab (ONTO 148), Humira
TM
(Adalimumab), Cimzia (Certolizumab Pegol), Additional TNF-a Antibody
Inhibitors in Autoimmunity and Inflammation; and Tumor Necrosis Factor-a for
treating cancer and infection.
(4) recombinantly-produced, authentic chemokines for treating
autoimmune diseases; cancer; CCR2 for use as an anti-inflammatory drug.
CA 02779198 2015-12-22
(5) recombinantly-produced, authentic proteins for development of
therapeutics for disorders concerning growth and colony stimulating factors,
such as, but not limited to, VEGF for treating angiogenesis, angiogenesis
TM
antibodies (Avastin and Lucentis), VEGF Antagonists Macugen, Multi-kinase
TM
Inhibitor Sutent, and Nexavar, VEGF Monoclonals and Inhibitors, VEGF Gene
TM
Therapy; Hepatocyte Growth Factor; Platelet-derived Growth Factor; Gleevec
and other Protein Tyrosine Kinase Inhibitors; Fibroblast Growth Factor; TGF
Binding Proteins, and Inhibitors of TGF Beta Signaling; Insulin-like Growth
Factor; Keratinocyte Growth Factor; rhKGF for treating oral mucositis;
connective tissue growth factor (CTGF); Colony Stimulating Factors (CSF);
Erythropoietin; Thrombopoietin; Angiopoietin; Bone Morphogenetic Proteins
and Growth Differentiation Factors; rhBMP-2, rhBMP-7, and OPG; Growth
and Differentiation Factors GDF; rhGDF-5, rhBMP, Inhibition of Myostatin;
and neurotrophic factors for treating development disorders.
The recombinantly-produced, authentic human proteins of the present
invention may be formulated into drugs, vaccines, liposomes, or delivered as
proteins directly into an individual in vivo or administered to cells or cell
cultures in vitro or ex vivo. For instance, the skilled person is aware of a
number of approaches have been used to modify peptides for therapeutic
application. One approach is to link the peptides or proteins to a variety of
polymers, such as polyethylene glycol (PEG) and polypropylene glycol (PPG).
See for example U.S. Pat. Nos. 5,091,176, 5,214,131 and 5,264,209.
A recombinantly-produced, authentic human protein of the present
invention may therefore be suitably formulated in pharmaceutical preparations
for the oral or parenteral administration. Formulations for parenteral
administration include but are not limited to injectable solutions or
suspensions and liquids for infusions. For the preparation of the parenteral
forms, an effective amount of the active ingredient is dissolved or suspended
in a sterile carrier, optionally adding excipients such as solubilizers,
isotonicity
56
CA 02779198 2015-12-22
agents, preservatives, stabilizers, emulsifiers or dispersing agents, and
subsequently distributed in sealed vials or ampoules.
The present invention also contemplates conjugates of the authentic
human proteins produced by the methods disclosed herein. For instance, an
authentic human protein can be combined with a pharmaceutically acceptable
carrier, diluent or excipient to produce a pharmaceutical composition (which
may be for human or animal use). Suitable carriers and diluents include
isotonic saline solutions, for example phosphate-buffered saline. Details of
excipients may be found in The Handbook of Pharmaceutical Excipients, 2nd
Edn, Eds Wade & Weller, American Pharmaceutical Association.
A composition of the invention may also be
administered by direct injection. Thus, a therapeutic composition of the
present invention may be formulated for parenteral, intramuscular,
intravenous, subcutaneous, intraocular, oral or transdermal administration.
The preparation of human proteins, such as cytokines, in liposomal form also
can improve the biological activity thereof.
A therapeutic authentic protein of the present invention may be
formulated such that administration daily, weekly or monthly will provide the
desired daily dosage. The skilled person will appreciate that such a
, 20 therapeutic composition may be conveniently formulated for
administrated
less frequently, such as every 2,4, 6, 8, 10 or 12 hours.
The authentic proteins of the present invention also can be employed
in gene therapy of these and other diseases by administering the authentic
human protein directly to a patient or individual, or to the patient's or
individual's cells. The treated cells may then be reintroduced into the
patient
or individual. Thus, a desired polynucleotide encoding an authentic human
protein of the present invention may be administered directly as a naked
nucleic acid construct, and may also be linked to flanking sequences
homologous to the host cell genome that facilitate incorporation of the
internal
expression cassette and, hence desired polynucleotide, into the host cell
57
CA 02779198 2012-04-26
WO 2011/053281
PCT/US2009/062250
genome. The individual can be any mammal, reptile, bird, fish, or amphibian.
In one embodiment, the individual recipient of gene therapy is a human. Any
vector of the present invention can be used to express a desired protein, such
as a desired therapeutic cytokine, directly in a cell in vivo and thereby
provide
a gene therapy approach to treating a particular disease, e.g., a cytokine-
related disease or disorder.
The present invention contemplates the use of any of the vectors
disclosed herein in the gene therapy treatment of a variety of diseases and
disorders. For instance, the present invention contemplates the use of any of
the vectors disclosed herein to express in vivo the CFTR gene (cystic fibrosis
transmembrane conductance regulator) for the treatment of cystic fibrosis;
genes for factors VIII and IX, deficiency of which is responsible for
hemophilia
A and B), respectively; genes called E1A and P53 that cause cancer cells to
undergo cell death or revert to normal; AC6 gene which increases the ability
of the heart to contract and may help in heart failure; and VEGF, a gene that
induces the growth of new blood vessels (angiogenesis) of use in blood
vessel disease. Thus, in one embodiment of the present invention, a vector
contains an expression cassette with a desired polynucleotide that encodes a
therapeutic protein gene selected by the group consisting of the CFTR gene,
the factor VIII gene, the factor IX gene, the E1A gene, the P53 gene, the AC6
gene, and the VEGF gene.
Gene diseases that can be treated with gene therapy using any vector
of the present invention includes but is not limited to the expression of one
or
more genes for the treatment of Spinocerebella ataxia type 1, Huntington s
disease, familial hypercholesterolemia (FH), AIDS, cancers such as
melanoma, or skin cancer, involves introducing a gene with an anticancer
protein called tumor necrosis factor (TNF) into test tube samples of the
patient's own cancer cells, which are then reintroduced into the patient;
brain
cancer, the approach is to insert a specific gene that increases the cancer
cells' susceptibility to a common drug used in fighting the disease; and
CHIC_4513901 1 58
CA 02779198 2012-04-26
WO 2011/053281 PCT/US2009/062250
prostate cancer and cervical cancer cells; and Gaucher disease is an
inherited disease caused by a mutant gene that inhibits the production of an
enzyme called glucocerebrosidase.
A vector of the present invention also can be used in gene therapy to
solve problems associated with surgical procedures, such as balloon
angioplasty, a procedure which induces the body's immune system to
cascade and cause restenosis. Gene therapy using any of the vectors
described herein can express gene reduce this overactive healing response.
Thus examples of gene therapies that can be treated by introducing a
vector of the present invention into a patient, wherein the vector expresses
an
appropriate gene to counter or treat the genetic disease include but are not
limited to (A) gene transfer therapy for Treating Children and Adults With
Limb
Girdle Muscular Dystrophy Type 2D (LGMD2D), wherein the vector encodes
and expresses in vivo rAAV1.tMCK.human-alpha-sarcoglycan in patients with
LGMD2D; (B) gene therapy for treating HIV-1 Infected Patients with any of the
vectors disclosed herein that expresses GX-12; (C) gene therapy treatment of
prostate cancer using Ad5-yCD/mutTKSR39rep-ADP, RTVP-1, Ad.hIL-12,
FP253/Fludarabine, or Ad5-CMV-NIS Gene; (D) gene therapy treatment of
Leber Congenital Amaurosis using tgAAG76 (rAAV 2/2.hRPE65p.hRPE65);
(E) gene therapy treatment of Sickle Cell Anemia or Thalaassemia; (F) gene
therapy treatment of pleural malignancies using BG00001 (adenoviral-
mediated interferon-beta); (G) gene therapy treatment of chronic
granulomatous disease using phagocyte oxidase subunit transduced CD34
hematopoietic stem cells; (H) gene therapy treatment of in transit melanoma
using INGN 241; (I) gene therapy treatment of malignant gliomas using AdV-
tk; (J) gene therapy treatment of Bilateral Idiopathic Parkinson's Disease
using ProSavin; (K) gene therapy treatment of metastatic breast cancer using
adenovirus-mediated human interleukin-12; (L) gene therapy treatment of
patients who have received a left ventricular assist device using
CHIC_4513901 1 59
CA 02779198 2012-04-26
WO 2011/053281 PCT/US2009/062250
AAV6.SERCA2a; and (M) gene therapy treatment of Leber congenital
amaurosis caused by RPE65 mutations by using rAAV2-hRPE65
Accordingly, any of these genes can be engineered into any one of the
vectors disclosed herein and then that vector introduced into cells of an
individual wherein the vector expresses the encoded protein the presence of
which in the cell acts to counteract, correct, or otherwise treat the genetic
basis or bases of the underlying disease. The vector that encodes the
therapeutic protein may be introduced into cells that have been cultured
outside of the individual or may be administered directly into the individual
and
the encoded protein expressed in vivo. Thus, the present invention
contemplates both in vivo and ex vivo/in vitro gene therapy methodologies for
treating a particular disease.
Thus, one aspect of the present invention is the use of a vector
selected from the group consisting of pHZhag, pHZA, pHZA, pHZI, pHZhagA,
and pHZhagl in gene therapy wherein the vector expresses a therapeutic
protein from a particular expression cassette inserted into the vector. In one
embodiment, the pHZhag vector comprises operably linked nucleotide
sequences for (i) an hCMV 1E, (ii) a human beta-actin promoter, and (iii) a
human beta-globin intron. In another embodiment, the pHZA vector
comprises operably linked nucleotide sequences for (i) an hCMV promoter,
and (ii) a human fibrinogen subunit A signal peptide. In another embodiment,
the pHZI vector comprises operably linked nucleotide sequences for (i) an
hCMV promoter, and (ii) a human Ig superfamily 8 signal peptide. In another
embodiment, the pHZhagA vector comprises operably linked nucleotide
sequences for (i) an hCMV 1E, (ii) a human beta-actin promoter, (iii) a human
beta-globin intron, and (iv) a human fibrinogen subunit A signal peptide. In
another embodiment, the pHZhagl vector comprises operably linked
nucleotide sequences for (i) an hCMV 1E, (ii) a human beta-actin promoter,
(iii) a human beta-globin intron, and (iv) a human Ig superfamily 8 signal
peptide.
CHIC_4513901 1 60
CA 02779198 2012-04-26
WO 2011/053281
PCT/US2009/062250
Uptake of naked nucleic acid vectors, such as these, which contain the
inventive expression cassettes by mammalian cells can be enhanced by
several known transfection techniques for example those including the use of
transfection agents. Examples of these agents include cationic agents (for
example calcium phosphate and DEAE-dextran) and lipofectants (for example
lipofectam.TM. and transfectam.TM.). If desired, nucleic acid constructs may
also mixed with the transfection agent to produce a composition. An
expression vector of the present invention may also be a pharmaceutically
acceptable carrier or diluent to produce a pharmaceutical composition.
Suitable carriers and diluents include isotonic saline solutions, for example
phosphate-buffered saline. The composition may be formulated for
parenteral, intramuscular, intravenous, subcutaneous, intraocular or
transdermal administration.
The routes of administration and dosage regimens described are
intended only as a guide since the skilled artisan will be able to readily
determine the optimum route of administration and dosage regimens for any
particular individual and condition.
Accordingly, the present invention also therefore encompasses the use
of any of the recombinantly-produced, authentic proteins of the present
invention in the preparation of a medicament that is useful for treating a
particular disease or disorder, such as any of those disclosed herein. Hence,
the present invention encompasses the use of a recombinantly-produced,
authentic human cytokine for preparation of a medicament for treating cancer
and disease abnormalities concerning cell growth, cell proliferation, cell
differentiation, and inflammation. An aspect of the present invention
therefore
is the use of a recombinantly-produced, authentic human cytokine for
preparation of a medicament for treating cancer. An aspect of the present
invention therefore is the use of a recombinantly-produced, authentic human
cytokine for preparation of a medicament for treating a cell proliferation-
associated disorder or disease. An aspect of the present invention is also the
CHIC_4513901 1 61
CA 02779198 2012-04-26
WO 2011/053281 PCT/US2009/062250
use of a recombinantly-produced, authentic human cytokine for preparation of
a medicament for treating a cell growth-associated disorder or disease.
Another aspect of the present invention is the use of a recombinantly-
produced, authentic human cytokine for preparation of a medicament for
treating a cell differentiation-associated disorder or disease. An aspect of
the
present invention is the use of a recombinantly-produced, authentic human
cytokine for preparation of a medicament for treating an inflammation-
associated disorder or disease.
10. EXEMPLARY THERAPEUTIC USES OF ANTIBODIES
RAISED AGAINST RECOMBINANTLY-PRODUCED AUTHENTIC
CYTOKINES
Overexpression of certain cytokines are known to be associated with
certain diseases and abnormal pathological states. For instance, too much
TNFa is associated with inflammation and arthritis. Accordingly, antibodies
that block TNFa activity and binding to ligands and receptors helps to
alleviate
problems associated with inflammation and arthritis. Likewise, blocking VEGF
cytokine activity via specific antibody binding is an effective mechanism for
treating cancer and undesirable cell proliferation by reducing angiogenesis.
Since the cytokines of the present invention are more human-like and
therefore present more authentic epitopes at their surface than cytokines
expressed in other cell systems, then the antibodies that are raised against
them also will be more authentic with respect to their ability to recognize
and
target them as antigens in vivo. Thus, antibodies raised against
recombinantly-produced, authentic human proteins of the present invention
have a higher sensitivity and higher specificity than antibodies that have not
been raised against the recombinantly-produced, authentic human proteins of
the present invention.
Thus, the antibodies of the present invention, which are raised against
the cytokines produced by the inventive method, have higher specificity and
CHIC_4513901 1 62
CA 02779198 2012-04-26
WO 2011/053281
PCT/US2009/062250
higher binding affinity than antibodies raised against cytokines produced
elsewhere.
As in the case for the cytokines, since the antibodies have a higher
affinity and specificity, it is possible to use a lower concentration or dose
in
any therapeutic regime that requires administering an antibody to combat a
particular cytokine-pertinent disease or disorder.
The skilled person is aware that the present recombiantly-produced,
authentic human proteins can be formulated into therapeutic anticancer
antibodies, such as those currently available. That is, an antibody raised
against the relevant recombiantly-produced, authentic human protein of the
present invention can be formulated into antibody-based drugs such as, but
not limited to, Panorex0 (edrecolomab), Rituxane (rituximab), Herceptine
(traztuzumab), Mylotarge (gentuzumab), Campathe (alemtuzumab),
ZevalinTM (ibritumomab), ErbituxTM (cetuximab), and AvastinTM (bevicizumab).
Antibodies raised against the recombinantly-produced, authentic
human proteins of the present invention, or fragments of such antibodies, can
be useful as immunoconjugated anticancer antibodies.
Antibodies of the present invention also are useful for treating
cardiovascular disorders, infectious diseases, and inflammatory Diseases;
and thus formulated into drugs such as those currently available for treating
such disorders and diseases, such as RaptivaTM (efalizumab), Remicadee
(infliximab), HumiraTM (adalimumab), and XolairTM (omalizumab).
Antibodies of the present invention also can be useful in the context of
transplantation, such as in drugs like Orthoclone OKT30 (muromomab-CD3),
SimulectO (basiliximab), and Zenapaxe (daclizumab).
Accordingly, the present invention also therefore encompasses the use
of any of antibodies (monoclonal or polyclonal), raised against any one of the
recombinantly-produced, authentic proteins of the present invention, in the
CHIC_4513901 1 63
CA 02779198 2012-04-26
WO 2011/053281 PCT/US2009/062250
preparation of a medicament that is useful for treating a particular disease
or
disorder, such as any of those disclosed herein. Hence, the present invention
encompasses the use of an antibody raised against one or more epitopes of a
recombinantly-produced, authentic human cytokine for preparation of a
medicament for treating cancer and disease abnormalities concerning cell
growth, cell proliferation, cell differentiation, and inflammation. An aspect
of
the present invention therefore is the use of an antibody raised against an
recombinantly-produced, authentic human cytokine for preparation of a
medicament for treating cancer. An aspect of the present invention is the use
of an antibody raised against a recombinantly-produced, authentic human
cytokine for preparation of a medicament for treating a cell proliferation-
associated disorder or disease. An aspect of the present invention is also the
use of an antibody raised against a recombinantly-produced, authentic human
cytokine for preparation of a medicament for treating a cell growth-associated
disorder or disease. Another aspect of the present invention is the use of an
antibody raised against a recombinantly-produced, authentic human cytokine
for preparation of a medicament for treating a cell differentiation-associated
disorder or disease. An aspect of the present invention is the use of an
antibody raised against a recombinantly-produced, authentic human cytokine
for preparation of a medicament for treating an inflammation-associated
disorder or disease.
11. EXEMPLARY EX VIVO AND DIAGNOSTIC USES OF
ANTIBODIES AND RECOMBINANTLY-PRODUCED AUTHENTIC
CYTOKINES
The skilled person also is aware that the authentic proteins of the
present invention can be useful in a diagnostic environment too and in
screening assays for testing candidate drugs and substances with and without
exposure to a particular authentic human protein. Antibodies raised against
recombinantly-produced, authentic human proteins of the present invention
have a higher sensitivity and higher specificity than antibodies that have not
CHIC_4513901 1 64
CA 02779198 2015-12-22
been raised against the recombinantly-produced, authentic human proteins of
the present invention. One such diagnostic tool is the cytokine bead array.
See Lambeck etal., Clinical Cancer Research 13, 2385, (April 15, 2007).
That method permits the
simultaneous measurement of multiple cytokines in a small volume of serum,
such as by using a LINCOplex kit and related protocol (Linco research, St.
Charles, MO).
Basically, a diagnostic or detection method of the present invention can
entail comparing the expression levels of one or more cytokines from a
sample taken from an individual against known amounts of control cytokines,
such as in titration comparative studies like ELISA assays and the cytokine
bioassays and activity assays described in subsection 8 above. According to
the present invention, the authentic human proteins, such as recombinantly-
produced, authentic human cytokines, can be used as highly sensitive
controls since they are highly similar in size, structure, and molecular
weight
to native cytokines. Thus, a comparison of an unknown sample against a
known concentration of an authentic human cytokine of the present invention
will be highly sensitive and accurate. Depending on the results of that
comparison of cytokine levels, a conclusion can be made on whether or not
the invidual's expression level for one or more cytokines is abnormal and, if
so, whether that abnormal expression level is indicative of, or diagnostic of,
a
particular disease or disorder.
Accordingly, the recombinant authentic proteins and antibodies raised
against them according to the present invention can be used in a number of
different arrays and multiplexed arrays in diagnostic assays. For instance,
the
present invention includes the use of authentic proteins and antibodies of the
present invention in Multiplex Immunoassay Designs, such as Sandwich
assays, Antigen-down assays, Competitive assays, and Reverse-phase
assays; in Array Substrates, such as in 96-well plates, on membranes, coated
on to slides, deposited onto flow-through chips, and adhered to porous
filters;
CA 02779198 2015-12-22
in Array Fabrication, where the protein or antibody is contact printed onto a
surface, or used in non-contact dispensing; in Detection Methodologies, such
as colorimetric, fluorescent, chemiluminescent, surface plasmon resonance
imaging techniques; in Array Processing, such as in microfluidic manipulation
and surface acoustic wave processing; and in Image Analysis and Data
Acquisition methods.
The antibodies raised against the authentic proteins of the present
invention also are useful for producing diagnostic kits for detecting
cytokines
and related antigens in vitro tests, such as in ELISA assays and histological
analyses on human tissue sections. Accordingly, the present invention
encompasses kits and reagents that include one or more antibodies that have
been raised against one or more of the cytokines produced by the inventive
human cell system.
Antibodies that block binding of cytokines to their specific receptors
and neutralize their effects ("neutralizing antibodies") can be very useful in
studies of cytokine function in particular disease states. In vitro bioassays
using neutralizing anti-mouse and anti-human cytokine antibodies therefore
are useful for determining the effectiveness of a particular antibody in
neutralizing cytokine-Induced cell proliferation, apoptosis, viral protection,
and
inappropriate cytokine production.
For example, using a reference standard calibrated against the World
Health Organization natural interferon beta standard (Second International
Standard for Interferon, Human Fibroblast GB 23 902 531), authentic
recombinant human interferon beta can be tested and can be compared to a
recombinant interferon-beta1a product called Rebif0 has a specific activity of
approximately 270 million international units (MIU) of antiviral activity per
mg
of interferon beta-la determined specifically by an in vitro cytopathic effect
bioassay using WISH cells and Vesicular Stomatitis virus. Rebif 8.8 mcg, 22
66
CA 02779198 2012-04-26
WO 2011/053281 PCT/US2009/062250
mcg and 44 mcg contains approximately 2.4 MIU, 6 MIU or 12 MIU,
respectively, of antiviral activity using this method.
As mentioned in a preceding subsection, both monoclonal and
polyclonal antibodies can be raised against the authentic cytokines produced
by the present inventive method. This means, that it is possible to raise
antibodies that specifically recognize and target one cytokine in a highly
effective manner, as well as antibodies that recognize and target closely
related cytokine homologs and variants that might have been produced in vivo
by various splicing mechanisms.
With respect to diagnostic kits, the inventive authentic cytokines
against which high affinity and specific antibodies were raised, can
themselves be used for the creation of titration standards in such kits like
those used for conducting ELISA assays or other immunological kits. Thus,
the standard curve, when using an authentic cytokine and the antibody that
was raised against it, is a much more accurate measurement of antibody-to-
antigen binding, than if the antibody was titrated against a cytokine prepared
from non-human cells. Thus, the present invention contemplates kits that not
only include authentic antibodies, and fragments thereof, but also aliquots of
the authentic cytokines produced by the present inventive method as
standards against which the antibody binding standard curve can be created
in such assays. The sensitivity of antibody-antigen binding will be high.
Thus,
detection and quantification of the presence and amount of a particular
cytokine in a biological sample or tissue section can be readily and more
accurately determined using those authentic cytokine/antibody partners to
create a standard curve against which unknown samples can be accurately
compared and quantified.
The antibodies of the present invention also can be used to prepare
and construct cytokine antibody arrays, which can simultaneously detect, in
one assay, multiple human cytokines from a variety of sources, including cell
lysates, conditioned media, patient sera, plasma, and urine. Such an array,
CHIC_4513901 1 67
CA 02779198 2012-04-26
WO 2011/053281 PCT/US2009/062250
especially when using the antibodies of the present invention, has a high
sensitivity making it possible to detect cytokine proteins at very low
concentrations, such as in the pg/ml range.
The authentic human proteins and antibodies of the present invention
also can be used in screening assays to identify candidate substances,
chemicals, compounds, and other proteins that interact with them in some
way. For instance, a recombinantly-produced, authentic human cytokine of
the present invention can be isolated and purified and then exposed directly
to
a particular candidate substance, and subsequently monitored under any one
of a variety of assays known to the skilled person, and as identified in the
Assay subsections above, to determine whether any interaction has occurred
between the protein and the candidate substance. See also for example
Table 4 which relates the results of one such screening assay utilizing a
recombinantly-produced kinase, p38a to identity and record inhibitor 1050
values when exposed to various compounds. That experiment is described
elsewhere herein in more detail but is mentioned here as an example in which
a protein or antibody of the present invention can be used in an ex vivo-style
screening assay. This current study demonstrates that the properties of the
human protein kinase p38 a produced in human cells are distinct from the
non-human cell version. Using human kinases with high authenticity for drug
screening will allow researchers to avoid pursuing false negative leads and
missing promising targets.
The proteins and antibodies produced by the present inventive method
and vectors also can be used as reagents in and of themselves. That is, a
recombinantly-produced, authentic cytokine of the present invention can be
used, for instance, in cell cultures by adding the cytokine to help promote
cell
growth and viability during culturing; and, as described above, a vector of
the
present invention can be used in gene therapy regimes to express a particular
protein with the cells of an individual.
CHIC_4513901 1 68
CA 02779198 2015-12-22
The following examples are merely exemplary and in
no way limit the scope of the present invention.
EXAMPLES
EXAMPLE 1: CLONING OF CYTOKINES
Cloning of cytokines can be accomplished by various methods
available to one skilled in the art of genetic engineering. For example, total
RNA or poly-A RNA can be purified from human tissues samples abundant in
particular cytokine expression (for example lymphocytes) and used as a
template for gene specific RT-PCR. Additionally, pre-made cDNA libraries
can be purchased from commercial sources and PCR can be employed to
amplify the cytokine cDNA directly. Still further, synthetic oligonucleotides
can
be constructed to create a synthetic gene for the cytokine based on sequence
information available in National Center for Biotechnology Information with
their gene accession numbers. Additionally, full length cDNA clones can be
obtained from, for example, the IMAGE clone consortium (image.IInl.gov/) or
Openbiosystems (Huntsville, AL). The full length cytokine cDNA clones were
obtained from Openbiosystems (Huntsville, AL). Gene accession numbers
are presented in Table 3.
Cytokines contain signal peptides at their N-terminal that are typically
lost in the secreted forms and that can be identified by numerous tools
available to one skilled in the art (for example Swiss-Prot protein
knowledgebase). Some cytokines have a number of variants, by different
transcription that can be also identified by the available tools. Sequences of
the secreted cytokines are listed in Subsection 10 above.
To facilitate rapid cloning of cytokines with different signal peptides,
pSecTag2c (Invitrogen, Carlsbad CA), which is suitable for the production of
secreted recombinant protein in mammalian cells (for example, CHO and
69
CA 02779198 2015-12-22
HEK293), was modified to pHZsec by site directed mutagenesis (Quick
Change, Stratagene, Carlsbad, CA) to introduce a Srf I restriction site in
frame
with the IgK leader sequence (see schematic below) using mutagenesis
primers SecTag2c-srflf
(TCCACTGGTGACGCGCCCGGGCCGGCCAGGCGCGCC) (SEQ. ID. NO
27) and SecTag2c-srflr
(GGGGCGCCTGGCCGGCCCGGGCGCGTCACCAGTGGA) (SEQ. ID. NO:
33). See Figure 27 for a schematic of the cleavage site.
The secreted forms of the cytokine genes were translationally fused to
the Srfl site in plasmid pHZsec using In-FusionTm PCR Cloning Kits from
Clontech (Mountain View, CA) with designed primers.
VECTORS
(A) pHZsec: conventional CMV expression vector control
Human cytomegalovirus (CMV) promoter-based vectors are typically
used in the design of vectors employed in mammalian cell expression
systems. See, for instance, the Product Notes for Invitrogen's "Mammalian
Expression Systems".
(B) pHZhag: a vector with a human promoter and a human
intron
The pHZhag vector, designed herein, consists of human beta-actin
promoter and human beta-globin intron. The pHZhag vector expresses a
desired polynucleotide or gene sequence at much higher levels than
conventional CMV-based vectors; and, unexpectedly, cells expressing the
pHZhag vector have higher cell viability and yield fewer contaminating
background proteins during purification.
CA 02779198 2012-04-26
WO 2011/053281 PCT/US2009/062250
The expression profile of the cytokine TGF-131 was compared after
expression in human cells using the newly constructed pHZhag vector and the
pHZsec vector. See the expression results for pHZ-TGF-131 and pHZhag-
TGF-131 in Figures 1 1a and 1 lb, respectively. When the cell lines
transfected
with each vector reached a comparable cell density and viability TGF-131
expression level was then compared. The results showed that cells
transfected with the pHZhag vector expressed TGF-131 two to three times fold
higher than the expression level for pHZsec.
In addition to the higher expression there was an unexpected
observation that pHZhag-transfected cells had a higher cell viability during
day 7 to day 9 in contrast to the pHZsec-transfected cells, the viability of
which sharply declined during that same time period.
To elaborate, culture cells are typically harvested at day 7 or day 8
when the cell's viable cell density ("VCD" - million cells per milliliter)
reaches
over 3 million cells/mL (Figure 12a) and viability (3/0) reaches around 60%
(Figure 12b). However, pHZhag-transfected cells kept their viability above
60% even at day 10 (Figure 12d) and had a VCD of over 3 million cells/mL
(Figure 12c). This is a new and unexpected finding with pHZhag vector.
(C) pHZA and pHZI: vectors with human signal peptide
sequences
The next step in the design of high-expression vectors that produce
highly authentic human proteins was the replacement of the murine Ig kappa
chain leader signal peptide sequence, which is widely used in CHO cells, with
a human protein signal peptide. The results shown herein indicate that the
use of a human signal peptide sequence improves expression levels by at
least two fold.
Candidate signal peptide sequences were selected from genes that
encode fibrinogen Aa chain (hFbgA), fibrinogen Bb chain (hFbgB), fibrinogen
CHIC_4513901 1 71
CA 02779198 2012-04-26
WO 2011/053281 PCT/US2009/062250
g chain (hFbgG), and human immunoglobulin superfamily 8 precursor signal
peptide (hIg8). Protein informatics analysis had showed that a good signal
peptide has a high content of Leucine (L). Particularly hIg8 precursor signal
peptide was chosen after comparing the signal peptide to the signal peptides
of human immunoglobulin superfamilies 1, 2, 3, and 6 because it has high
content of Leucine residue (37%) and five consecutive leucines (LLLLL).
Each signal peptide encoding sequence was subcloned into a pHZhag
vector and the expression of TGFb1 monitored. The vector, pHZA, which
contained the fibrinogen Aa chain signal peptide expressed TGF131 well. See
Figure 13a, Lanes 4, 5, and 6. The pHZI vector, expressing the human Ig 8
precursor signal peptide, also expressed TGF131 well. See Figure 13a, Lane
7.
pHZA-TGF131 was cultured to the appropriate cell density and its cell
viability was recorded to be comparable to that of pHZ-TGF131 and pHZhag-
TGF131. The cells expressing pHZI and pHZA-TGF131 had two-fold higher
levels of expression of TGF131 than that for pHZ-TGF131. See Figure 13b.
(D) pHZhagA and pHZhagl: vectors with human promoter,
human intron, and human signal peptide sequences
Vectors were designed herein that comprise a human beta-actin
promoter, a human intron sequence, and either one of the human signal
peptides described above (hFbgA signal peptide or hIg8 signal peptide).
Data comparing pHZhag-TGF-131 and pHZhagl-TGF131 shows about
two fold higher expression of pHZhagl-TGFb1 than that of pHZhag-TGF131
and 4-fold higher than pHZsec vector. The following comparative expression
studies can be performed using the denoted constructs:
Promoter comparison: pHZ-TGF131 vs pHZhag-TGF131;
CHIC_4513901 1 72
CA 02779198 2012-04-26
WO 2011/053281 PCT/US2009/062250
Signal peptide comparison: pHZ-TGF131 vs pHZI-TGF131 and pHZhag-
TGF131 vs pHZhagl-TGF131 or pHZhagA-TGF131; and
Promoter and signal peptide combination comparison: pHZ-TGF131 vs
pHZhagl-TGF131 or pHZhagA-TGF131.
See Figure 14 for schematic representations of these vectors.
EXAMPLE 2: PREPARING HEK CELLS
Human embryonic kidney cells were placed on a 100mm Petri dish with
the recommended medium (typically DMEM medium with 10% bovine calf
serum/2mM L-Glutamine/10mM HEPES/1x MEM non-essential amino acid).
Cells were kept in the manner for 3 to 4 passages to select the population
that
attached the plate. The cells attached on the plate were then exposed to
various serum-free media (293 SFM II, CD 293, FreeStyle 293, Hybridoma-
SFM (from Invitrogen); Ex-Cell 293 (JRH Biosciences)), for 4 days in the plate
to select the cells that grow or survive in the serum-free medium. Those cells
were then placed back in the 10% serum medium to produce working cell
bank.
(A) Cell line adapted to serum-free and chemically defined
media
HZ-293T5: HEK293T (or 293T) is a HEK293 cell genetic variant
(ATCC CRL-11268). This cell line was adapted to serum-free and chemically
defined media using the manufacturer's reagents (Invitrogen). Adapting
HEK293T cells to suspension and completely serum-free and chemically
defined media is novel. HZ-293T5 reaches at least about 5 to 6 million cells
per milliliter (VCD) in suspension culture (Figure 15b, table). The human cell
line which is denoted as HZ-293T5 is deposited under, and bears the ATCC
biological deposit accession number of PTA-10165, deposited on July 1,
2009, by HumanZyme, Inc., as described in detail elsewhere in this
specification.
CHIC_4513901 1 73
CA 02779198 2015-12-22
HZ-293TS cell lines can be cultured in serum-media as adherent on
the plates, as well as in serum-free and chemically defined media, as
suspension in the shaking flasks, spinners, and bioreactors. This feature
allows HZ-293TS cells to be plated as monolayer in serum media and
subsequently transfected. Transfected cells then can go to suspension
directly or can be selected on the plate prior to the suspension culture like
a
shuttle system.
There have been three 293 cell lines reported available for suspension
TM
culture: HEK293S, FreeStyle293, 293EBNA. Among those three HEK293S
lo and FreeStyle293 are of HEK293 cell line that has been known to have
much
less recombinant protein expression than 293T and 293EBNA that are
capable for episomal amplification of the plasmid of interest. Further
advantage of HZ-293TS over and FreeStlye293 and 293EBNA is that the
selected cells stably expressing the recombinant of interest have versatile
scalability and continuous culture (more than 20 passages without losing
expression level) while transfected in suspension 293EBNA and FreeStyle293
are mostly for single harvest within a short period of time. To evaluate what
is
the characteristic of HZ-293TS in terms of recombinant protein expression
level human FGF8b cytokine was transfected to HZ-293TS using the shuttle
way and its expression level was compared with that from current HEK293T
system in following section 2.
HZ-293S: HEK293 cell line (ATCC CRL-1573) adapted to the serum-
free and chemically defined media. This cell line has not yet been tested for
recombinant protein production.
HZ-293E6NA: 293EBNA cell line (Invitrogen R6200&7) adapted to the
serum-free and chemically defined media. Although 293EBNA has episomal
amplification ability, like HEK293T, it requires OriP (EBV origin of
replication)
in the plasmids that limited further use of this cell line. This cell line has
not
yet been tested for recombinant protein expression.
74
CA 02779198 2015-12-22
(B) Application of HZ-293TS to recombinant cytokine
production
Because of its easiness of maintaining in suspension culture and
adaptation to monolayer culture, the HZ-293TS cell line was tested for its
recombinant protein production capability compared to that of HEK293T for
expressingh human FGF8b cytokine-encoding plasmid. When both cell lines
reach a comparable cell density and viability, the cytokine expression in the
HZ-293TS cells was unexpectedly 2 to 3 fold higher than that of 293T. Thus,
HZ-293TS has become a very different cell line from 293T and that can be the
reason for higher recombinant cytokine expression.
Other proteins have been expressed at much higher levels in the
inventive HZ-293TS cells than in HEK293T cells, including but not limited to
expression of Activin A, FGFbasic, IL-23,
VEGF165, and TGFB1. See
for example the comparative data presented in the Figures for FGFbasic and
TGFB1.
EXAMPLE 3: TRANSFECTION
One day before transfection a confluent 100mm dish of the cells was
passed to 5 dishes (70 - 80% confluency). A 100mm dish transfection
requires 500u1DMEM medium without supplements, bug plasmid DNA, and
zo a certain amount of transfectant (FuGene 6, FuGene HD (from Roche);
TM
Lipofectamin, Lipofectamin 2000, 293fectin (lnvitrogen); Polyetheleneimine),
according to the manufacturer's manual. Mixture of transfection materials
was added to the culture dish in drop wise.
EXAMPLE 4: ANTIBIOTICS SELECTION
Forty-eight hours later the transfected cells were harvested by
centrifugation after trypsin treatment. The cell pellet was resuspended in 5m1
fresh 10% serum medium. One hundred micro liters of resuspended cells
was added into a six well plate with 2m1 of the serum medium with certain
CA 02779198 2015-12-22
concentration of antibiotics (for example, 400 ug/ml or 800 ug/ml) (neomycine
(G418), hygromycine, zeocin, blasticidine).
Media in the six-well plate was changed with fresh including antibiotics
every 3 to 4 days later for two weeks by then transfected cells grow as cell
colonies while untransfected ones died out. Cell colonies were harvested with
trypsin treatment and tranfered into new plate with fresh serum medium
including a quarter concentration of antibiotics and grown for two weeks.
EXAMPLE 5: SUSPENSION ADAPTATION
Once the selected cells were confluently grown in the plate the cells
were trypsin treated to detach from the plate. Two plate amounts of cells
were harvested after trypsin treatment and resuspended in 10m1 of serum-free
medium (for example CD 293) including 1% serum and antibiotics.
Adaptation may take up to 8 weeks and the medium was changed every 3 to
4 days depending on the cell condition. Later the cells were transferred to
serum-free medium and antibiotics only. This adaptation takes up to 4 weeks
and the medium was changed every 3 to 4 days depending on the cell
condition. Once suspension adapted cells were continuously grown to larger
scales for production and cryo-banked in fresh medium plus 10% DMSO for
future production,
EXAMPLE 6: TRANSFECTION AND ESTABLISHMENT OF
STABLE HEK 293 CELLS EXPRESSING CYTOKINES
Cytokine expression plasmids were transfected into 293 cell lines, or
derivatives thereof (e.g., HEK 293T, HEK 293S, and HEK 293 EBNA) that are
serum-free adapted in house, in 100mm Petri dishes using transfectant
TM
FuGene 6 (Roche) according to manufacturer's recommendations.
Transfected cells were grown in DMEM medium, supplemented with 10%
BCS, 1% MEM non-essential amino acids, 1% penicillin-streptomycin and
2mM L-glutamine. Forty-eight hours later the transfected cells were
76
CA 02779198 2012-04-26
WO 2011/053281 PCT/US2009/062250
harvested by centrifugation after trypsin treatment. The cell pellet was
resuspended in 5m1 fresh 10% serum medium. One hundred micro liters of
resuspended cells was added into a six well plate with 2m1 of the serum
medium with certain concentration of antibiotics (for example, 400 pg/ml or
800 pg/ml zeocin).
Media in the six-well plate was changed with fresh including antibiotics
every 3 to 4 days later for two weeks by then transfected cells grow as cell
colonies while untransfected ones died out. Cell colonies were harvested with
trypsin treatment and tranfered into new plate with fresh serum medium
including a quarter concentration of antibiotics and grown for two weeks.
Once the selected cells were confluently grown in the plate the cells
were trypsin treated to detach from the plate. Two plate amounts of cells
were harvested after trypsin treatment and resuspended in 10m1 of serum-free
medium (for example CD 293) including 1% serum and antibiotics.
Adaptation takes up to 8 weeks and the medium was changed every 3 to 4
days depending on the cell condition. Later the cells were transferred to
serum-free medium and antibiotics only. This adaptation takes up to 4 weeks
and the medium was changed every 3 to 4 days depending on the cell
condition. Once suspension adapted cells were continuously grown to larger
scales for production and cryo-banked in fresh medium plus 10% DMSO for
future production.
EXAMPLE 7: PURIFICATION OF CYTOKINES
Human cytokines are modified by complicated post-translational
molecular mechanisms such as glycosylation, phosphorylation, and
multimerization, and are subjected to a complicated cleavage and conversion
process that modifies their intrinsic physicochemical properties based on bare
amino acid sequences and that make their purification difficult without any
tag
in particular. The present invention encompasses the development of efficient
purification scheme utilizing up to three steps of conventional chromatography
CHIC_4513901 1 77
CA 02779198 2012-04-26
WO 2011/053281
PCT/US2009/062250
and yielding >95% purity on SDS-PAGE. The purification scheme is
consisted of capture with immobilized metal ion affinity chromatography
followed by purification and polishing with ion exchange chromatography.
After 6 days growth, the supernatant of the serum-free medium was
collected by centrifugation and the cell pellet was resuspended in fresh
serum-free medium for further production of the cytokine. Expression of
cytokines was identified on SDS-PAGE gel with Coomassie stain by known
molecular weight or on PVDF membrane with Western blot. To capture the
cytokine, the supernatant was at first loaded on an immobilized metal affinity
chromatography (IMAC) column. Based on their properties some cytokines
were bound on IMAC column while some were found in flow through. As next
purification step cytokine fraction pool was loaded on an ion exchange
chromatography (IEX) column after buffer exchange to a proper buffer
condition. Finally as polishing step cytokine fraction pool was loaded on
another !EX or different affinity chromatography (for example Heparin resin)
column.
After up to three chromatography steps cytokines were more than 95%
pure judged by Coomassie stain on SDS-PAGE gel (Figure 7) and by
Western blot with available antibodies (Figure 8). Pure cytokines then were
quantified using known methods available to one skilled in the art (for
example Bradford assay, Coomassie stain on the gel, and OD280nm). After
the quantification cytokines were analyzed endotoxin level by endotoxin
detection kit from Lonza (Allendale, NJ) according to manufacturer's manual,
aliquoted based on required amounts and lyophilized for commercialization.
GM-CSF, IL-4, and IL-6 recombinantly-produced using the inventive
human cell expression system are glycoproteins with different degrees of
glycosylation. The disclosed and inventive, efficient purification scheme
yielded GM-CSF with a range of heterogeneous glycosylation (Fig. 17A), and
IL-4 and IL-6 with distinct glycosylation (Fig. 17B&C) in high purity. The
CHIC_4513901 1 78
CA 02779198 2012-04-26
WO 2011/053281 PCT/US2009/062250
purification scheme was also applied to purify authentic homodimer Noggin
purification (Fig. 17D).
EXAMPLE 8: ACTIVITY ASSAY
Biological activity of the cytokine was measured by ED50 on the dose-
dependant cytotoxicity (for example, TNFa), stimulation of the proliferation
(for
example, IL-2), or inhibition of other cytokine induced proliferation on
effective
cells (for example, TGF-B1) based on the nature of the cytokine. See Figures
6A-M.
EXAMPLE 9: COMPARATIVE ANALYSIS OF CYTOKINES
EXPRESSED FROM THE INVENTIVE HUMAN CELL SYSTEM AND NON-
HUMAN CELLS
Experiments were conducted to compare the purity and extent of
glycosylation between cytokines expressed according to the present inventive
method and those that were expressed in non-human cells. Figures 7 and 8
depict side-by-side comparisons of SDS-PAGE gel analyses of different
cytokines in non-reduced and reduced human cells as compared against non-
reduced and reduced non-human cells. The cytokines that were compared in
such fashion include EPO, Noggin, G-CSF, SCF, GM-CSF, Somatotropin, IL-
2, TGFB1, IL-4, TNFa, IL-6, VEGF-165, and M-CSF. Figure 8 shows the
comparative results of Western Blot analyses of those same cytokines
between the inventive human expression system and non-human expression
system. This data show that the stable human cell culture of the present
invention expressed cytokines that have molecular weights and authentic
tertiary structures and activities that are comparable to native human
cytokines and distinct from the sizes and structures of cytokines expressed
from non-human cells.. Furthermore, the antibodies raised against the
authentic cytokines, due to the presentation of authentic epitope surfaces,
have high affinity binding properties.
CHIC_4513901 1 79
CA 02779198 2012-04-26
WO 2011/053281 PCT/US2009/062250
EXAMPLE 10: VEGF165 AND IL-4 COMPARATIVE DATA
VEGF165 plays a prominent role in normal and pathological
angiogenesis. It has been demonstrated that inhibition of VEGF activity by
treatment with a monoclonal antibody specific for VEGF can suppress tumor
growth in vivo. Currently, commercially available VEGF165 protein reagents
are produced from non-human cells including E coli and insect cells. The
inventive method disclosed herein has been use to produce VEGF165 from
engineered human 293 cells. Figure 9 shows a comparison of activities of
VEGF165 expressed between E. co/land human cells the molecular mass of
the E. coli expressed protein in monomer is 18 kD. This compares with the
inventive VEGF165 which migrates as a band of 28kD due to glycosylation.
See Figure 9 for comparative activities of VEGF in human and non-human cell
systems.
The bioactivity of the VEGF165 produced by the inventive stable
human cells was determined by its ability to induce proliferation of human
umbilical vein endothelial cells, indicating that that VEGF165 is 6-fold more
active than the E coli expressed protein.
IL-4 plays a critical role in the development of allergic inflammation and
asthma. Currently, commercially available IL4 protein reagents are produced
from E. coli with a molecular mass of 14 kD (Fig. 7C). This compares with the
IL4 from human cells which migrates as a major band of 19 kD due to
glycosylation. The biological activity of IL-4 was determined by the dose-
dependent stimulation of the proliferation of human TF-1 cells. As shown in
Figure 10, IL4 has 4-fold higher potency than the E. coli expressed cytokine.
See Figure 10 for comparative activities of IL4 in human and non-human cell
systems.
Cytokines produced in E. coli are not glycosylated and may expose
cryptic or normally hidden epitopes. Hence, antibodies may have different
affinities for native human proteins compared to the E. coli produced
proteins.
CHIC_4513901 1 80
CA 02779198 2012-04-26
WO 2011/053281 PCT/US2009/062250
Indeed, Western blot analysis shows the monoclonal antibodies raised
against a full length protein from insect cells recognize the VEGF165 protein
from E. co/las well as other highly reactive species that may correspond to
micro-aggregates (Figure 8C). In contrast, only one band is seen with the
human cell version. The monoclonal antibodies raised against a full length
protein from E. coli recognize the protein from E. coli under both reducing
and
non-reducing conditions. In contrast, only the protein under non-reducing
conditions is detected with the human cell version.
EXAMPLE 11: AUTHENTIC TGF-B1
Transforming growth factors beta (TGF-13) are highly pleiotropic
cytokines that act as cellular switches and regulate immune function,
proliferation and epithelial-mesenchymal transition. These proteins are
produced as precursors. A furin-like convertase processes the proprotein to
generate an N-terminal latency-associated peptide (LAP) and a C-terminal
mature TGF-B. Disulfide-linked homodimers of LAP and TGF-13 remain non-
covalently associated after secretion, forming the small latent TGF-13
complex.
Covalent linkage of LAP to latent TGF-13 binding proteins create large latent
complex that may interact with the extracellular matrix. Commercially
available TGF-13 proteins are produced as a recombinant protein expressed in
CHO cells or as purified native protein from human platelets. Due to complex
post-proteolytic modifications, TGF-13 yield is low and the products are not
available in economic bulk quantity. The efficient and inventive human-cell
based expression system has been herein developed for scalable production
of various human cytokines and produces highly authentic human TGF-131, 132
and 133 proteins from engineered human 293 cells. The proteins are highly
purified disulfide-linked dimers of 25 kD that can be cost-effectively
produced
in large scale (Fig. 18B).
IL-17-producing CD4+ T cells (Th-17 cells) have been identified as a
unique subset of Th cells that develop along a pathway that is distinct from
the Th1, and Th2-cell differentiation pathways. This finding has provided
CHIC_4513901 1 81
CA 02779198 2012-04-26
WO 2011/053281 PCT/US2009/062250
exciting new insights into immunoregulation, host defense and the
pathogenesis of autoimmune diseases. Recently it has been shown that IL1 (3,
IL6 and IL23 are important in driving human Th17 differentiation. However,
TGF-I31, which is important for the differentiation of murine Th17 cells, is
reported to be not required, and even inhibits human Th17 differentiation
(McGeachy & Cua (2008) Immunity 28:445, Chen & O'Shea (2008) Cytokines
41:71). In this study, whole CDE4+ cells isolated from a healthy donor were
stimulated with 10 pg/mL plate bound anti-CD3 and 10 pg/ml soluble anti-
CD28 in the presence of Th17 polarizing cytokines from the inventive human
cell expression system and from an insect cell or bacterial expression system.
After 5 days supernatants were harvested for measurement of IL-17 by
ELISA.
The results show that recombinantly-produced, authentic human IL1 13,
IL6 and IL23 are significantly more effective in inducing IL-17 secretion.
More
importantly, it demonstrates the recombinantly-produced, authentic human
TGF-I31 is also effective in enhancing the effect. In contrast, this cytokine
from
insect cells only showed marginal effect. The results indicate that by using
more authentic cytokines, it is possible to more effectively induce Th17 cell
differentiation and lead to more accurate scientific understanding of human
biological process. A separated study performed under the same condition
with TGF-I31, TGF-I32, and TGF-33 (Fig 18B) from the inventive human cell
expression system demonstrated that all three biologically relevant cytokines
can effectively induce Th17 cell polarization (Fig 22E-G). Furthermore, in
another study, among commercially available TGF-31 cytokines only authentic
TGF-31 Fig. 22H) matched human platelet derived native TGF-31 ('positive
control' in Fig. 22H) in differentiation of a naïve T cell (Th0) to Th17 cells
presented by flow cytometry analysis on the population of IL-17 and INF-y
producing cells (Fig. 22H).
EXAMPLE 12: AUTHENTIC VEGF
CHIC_4513901 1 82
CA 02779198 2012-04-26
WO 2011/053281 PCT/US2009/062250
VEGF165 is a member of the cysteine-knot growth factor superfamily.
This cytokine stimulates proliferation and survival of endothelial cells, and
promote angiogenesis and vascular permeability. Expressed in vascularized
tissues, VEGF165 plays a prominent role in normal and pathological
angiogenesis. It has been demonstrated that inhibition of VEGF165 activity by
treatment with a monoclonal antibody specific for VEGF165 can suppress
tumor growth in vivo.
Currently, commercially available VEGF165 proteins are produced
from non-human cells including E. co/land insect cells. Authentic VEGF165
has herein been produced from engineered human 293 cells. The E. coli
expressed protein is a mixture of monomer and dimer and has a molecular
mass of 18 and 38 kD in SDS-PAGE. This compares with the recombinantly-
produced, authentic human VEGF165 which migrates as a glycosylated band
of 45kD due to glycosylation and dimerization (Fig. 19). The bioactivity of
the
recombinantly-produced, authentic human VEGF165 was determined by its
ability to induce proliferation of human umbilical vein endothelial cells.
These
results indicate that the recombinantly-produced, authentic human VEGF165
is 10-fold more active than the E. coli expressed protein under the same
bioassay condition: ED50 of lng/mL for the recombinantly-produced, authentic
human VEGF protein vs 10 ng/mL for E. coli expression (Fig. 9).
EXAMPLE 13: AUTHENTIC EPO
Erythropoietin (EPO) is a 34 kD glycoprotein hormone which is related
to thrombopeietin. This protein promotes erythrocyte formation by preventing
the apoptosis of early erythroid precursors. It has been shown glycosylation
of
EPO is required for biological activities in vivo. Currently, commercially
available recombinant human EPO proteins are produced from CHO cells.
These recombinant proteins differ from the native human EPO by having
higher apparent molecular mass of 40 kD on SDS-PAGE gel (Fig. 20A) and
lower content of neutral glycans (Fig. 20B). Recombinantly-produced,
authentic human EPO has herein been produced from engineered human 293
CHIC_4513901 1 83
CA 02779198 2012-04-26
WO 2011/053281 PCT/US2009/062250
cells. Similar to the native human protein in the literature (Skibeli et al.
(2001)
Blood 98:3626), recombinantly-produced, authentic human EPO exhibits a
lower apparent molecular mass and substantially higher content of neutral
glycans. Furthermore, recombinantly-produced, authentic human EPO has
more abundant and diverse glycan profiles than the CHO cell produced
version. The most abundant glycans in the recombinantly-produced,
authentic human EPO are tetra-anten nary complex types whereas those in
CHO EPO are elongated bi-antennary complex types (Fig. 20C).
EXAMPLE 14: AUTHENTIC IL-23
Currently, commercially available recombinant IL-23 cytokine is
produced as a heterodimeric or fusion protein from an insect cell expresion
system. Recombinantly-produced, authentic human IL-23 has been herein
produced in a stable cell culture of engineered human HEK293 cells. The
protein is expressed as a disulfide-linked heterodimer of 55 kD and, due to
the
scalability of the stable culture, can be cost-effectively produced and
efficiently purified (Fig. 18A).
IL-17-producing CD4+ T cells (Th17 cells) have been identified as a
unique subset of T helper cells that develop along a pathway that is distinct
from the Th1 and Th2-cell differentiation pathways. This finding has provided
exciting new insights into immunoregulation, host defense and the
pathogenesis of autoimmune diseases. Recently it has been shown that TGF-
131, IL-1I3, IL-6 and IL-23 are important in driving human Th17
differentiation
(Chen & O'Shea (2008) Cytokines 41:71). The bioactivities of IL-23 from
human and insect cells were first determined by the dose-dependent
secretion of IL-17 from mouse splenocytes activated with 10 ng/ml PMA,
which shows that recombinantly-produced, authentic human IL-23 is ten fold
more active (Fig. 21A). The activities were further assayed with human CD4+
cells which were isolated from a healthy donor and stimulated with 10 pg/ml
plate bound anti-CD3 and 10 pg/ ml soluble anti-CD28 in the presence of
CHIC_4513901 1 84
CA 02779198 2012-04-26
WO 2011/053281 PCT/US2009/062250
Th17 polarizing cytokines. After 5 days supernatants were harvested for
measurement of IL-17 by ELISA. The results show that recombinantly-
produced, authentic human IL-23 is 100-fold more potent for inducing IL-17
secretion in two independent studies, maximum induction was achieved with
0.1ng/m1 of the recombinantly-produced, authentic human IL-23 vs long/m1
with insect cell-produced IL-23 (Fig. 21B). These results demonstrate that
authentic human cell expressed cytokines can induce Th17 cell differentiation
at physiologically relevant concentrations.
IL-23 is a glycosylated hetero dimer protein of IL12p40 and IL23p19.
Currently, commercially available recombinant IL-23 cytokine is produced as a
heterodimeric or fusion protein from an insect cell expression system.
Authentic IL-23 has been herein produced in a stable cell culture of
engineered human HEK293 cells. The protein is expressed as authentic
disulfide-linked dimer of 55 kD and, due to the scalability of the stable
culture,
can be cost-effectively produced. (Fig. 18A).
EXAMPLE 15: AUTHENTIC GM-CSF AND IL-4 ENABLE MEDIUM-
CHANGE-FREE DIFFERENTIATION OF DENDRITIC CELLS
Purified human peripheral blood monocytes were cultured in either G4
DC medium (as specified below) at 5x105cells/m1 in humidified air containing
5% CO2 at 37 C for a total of 7 days. HZ G4 DC was used at 5 ng/ml of
recombinantly-produced, authentic human GM-CSF and IL-4 without medium
replacement whereas EC G4 DC was used routinely (50 ng/ml of E. co//GM-
CSF and IL-4 with 50% medium replacement on day 3 and day 5). On day 6,
Lipopolysaccharide (LPS) was added to half of the wells to induce DC
maturation while the other half of the wells were used as sham treatment. At
the end of the culture (7 days), the supernatants were harvested for cytokine
measurement, while the resulting DCs were analyzed for the surface markers
by flow cytometry, the antigen uptake by phagocytosis of FITC-dextran, and
the antigenpresenting capacity by allogeneic MLR.
CHIC_4513901 1 85
CA 02779198 2012-04-26
WO 2011/053281 PCT/US2009/062250
The profile of DC generation of selected cytokines and chemokines
was measured by Pierce Cytokine Array. The data indicate that DCs
generated in the presence of HZ G4 DC (5 ng/ml without medium
replacement) showed a similar profile of cytokines and chemokines as DCs
generated in the presence of EC G4 DC (50 ng/ml with medium replacement)
before and after maturation (Fig. 23A). DCs differentiated in the presence of
HZ G4 DC or EC G4 DC before or after LPS maturation were cultured in
triplicate with allogeneic human peripheral blood T cells at various ratios
for 5
days. The cultures were pulsed with 3H-TdR (0.5 uCi/well) for the last 18 h
before cell harvest. The proliferation of T lymphocytes was measured by beta
scintillation counting. As shown by figure 23B, DCs differentiated in the
presence of either HZ G4 DC or EC G4 DC showed similar low capacities to
stimulated the proliferation of allogeneic T cells in particular when DC:T
ratio
was low. After LPS induced maturation, DCs differentiated under both
conditions increased their capacity to stimulate the proliferation of
allogeneic
T cells. DCs generated in the presence of HZ G4 DC seemed to be even
better than DCs generated in the presence of EC G4 DC in this regard (Fig.
23B). Therefore, DCs differentiated in the presence of HZ G4 DC had similar
or better antigen-presenting capacity than DCs differentiated in the presence
of EC G4 DC.
EXAMPLE 16: AUTHENTIC NOGGIN
Noggin is a secreted homodimeric glycoprotein that is an antagonist of
bone morphogenetic proteins (BMPs). During culture of human embryonic
stem (hES) cells without feeder layer or conditioned medium (but with addition
of FGF basic), the addition of Noggin allows the stem cells to maintain their
undifferentiated, pluripotent state. Commercially available Noggin products
are produced in a variety of forms none of which are authentic: non-
glycosylated protein expressed in E coli; glycosylated Fc-fusion protein
expresed in NSO, for example. Recombinantly-produced, authentic human
Noggin has herein been produced in a stable, engineered human 293 cell
CHIC_4513901 1 86
CA 02779198 2015-12-22
expression system. The protein is expressed as an authentic glycosylated,
disulfide-linked dimer. The recombinantly-produced, authentic human
homodimer Noggin expresses so effectively and consistently 0ct3/4, which is
a marker for undifferentiated hES cells, as at the concentration of 10pg/m1
treatment (Fig. 24, lane 3) and 20pg/m1treatment (Fig. 24, lane 4) compared
to negative and positive controls (Figs. 24, lanes 1 and 2). See Wang et al.,
Biochem. Biophys. Res. Comm., 330:934-942 (2005), and ltsykson etal., Mol.
Cell. Neurosci. 30:24-36 (2005).
EXAMPLE 17: MONOCLONAL ANTIBODIES TO AUTHENTIC G-
CSF
Cytokines produced in E. colt are not glycosylated and may expose
cryptic or normally hidden epitopes. Similarly, cytokines produced in SF9 or
CHO cells have post-translational modifications which are not human-like.
Because of these factors, antibodies may have different affinities depending
on whether they were created from human cell expressed protein antigens or
non-human cell expressed protein antigens.
Several monoclonal antibodies against G-CSF from the inventive
human cell expression system have been raised which have a higher
apparent molecular mass of 22-25kD due to its glycosylation compare to that
of 18kD from E. colt on SDS-PAGE gel (Fig 25A). Among the monoclonal
antibodies HZmAb G-CSF-1 recognizes only G-CSF from the human cells
and not G-CSF from E. co/i(Fig. 25B) whereas HZmAb G-CSF-2 recognizes
both G-CSFs (Fig. 25C). These results indicate that recombinant cytokines
from the human cell expression system are highly preferred antigens to raise
antibodies that can selectively detect unique epitope sites of human serum
cytokines as well as to use as standards in ELISA assays.
EXAMPLE 18: OTHER PROTEINS THAT CAN BE EXPRESSED IN
THE HUMAN CELL SYSTEM OF THE PRESENT INVENTION
87
CA 02779198 2012-04-26
WO 2011/053281 PCT/US2009/062250
The present inventive human expression system is not limited to the
expression of human cytokines. Human proteins other than human cytokines
can be expressed in the stable human cell expression system of the present
invention. For instance, human kinases, and human phosphatases, and other
human proteins and enzymes can also be authentically expressed in the
present human cell expression system to produce recombinant, authentic
human-like kinases, phosphatases, proteins, and enzymes.
Due to their critical role in intracellular communication, dysregulation of
protein kinases has been implicated in as many as 400 human diseases
including cancer, diabetes, heart disease, neurological disorders and
rheumatoid arthritis. Hence, protein kinases are important for drug design and
screening. Currently, kinases are predominantly produced in non-human cells
(e.g. E coli or insect cells) many of which require protein truncation and/or
in
vitro activation steps, due to the limitations of the expression system. It is
possible, according to the present invention to express human protein kinases
which are full length and in vivo activated. Using p38a as an example, it has
been demonstrated herein that the properties and inhibition profiles of the
human protein kinases produced in the human cell system of the present
invention are differentiated from versions of the same kinase that were
produced in non-human cell systems.
Recombinantly-produced, authentic human p38a was produced and
activated in human cells in the presence of arsenite according to the present
invention. Sample kinases from Vendor A and B were expressed and purified
from E. coli, in vitro activated by MKK6, and repurified. SDS PAGE analysis
shows that p38a produced in the human cell expreseeion system is pure with
a dominant band of 60 kD and minor band of endogenous human GST of 23
kD. This was confirmed by MS analysis and no other contaminant proteins
were found. The Km.ATP for the authentic p38a is 109 12 i.IM while the Km
was 212 26 i.IM for the Vendor preparation. The Km of 120 i.IM was found
with Vendor B enzyme. The 1050 values were determined for 14 known
CHIC_4513901 1 88
CA 02779198 2012-04-26
WO 2011/053281 PCT/US2009/062250
kinase inhibitors. See Table 4. While the 1050 values for SB-202190 (the
known p38a selective inhibitor) for both p38a preparations were similar (0.02
i.tM and 0.03 i.tM respectively), there is clearly a difference in the
sensitivity to
the inhibitors between the two preparations. See Table 4. The Vendor A
preparation was only sensitive to AMP-PNP (a non-hydrolysable ATP analog).
Yet, the protein was 7-fold less sensitive than p38a, which is consistent with
its higher Km. p38a on the other hand, had measurable 1050 values against
staurosporine, K252a, Ro 31-8220, KT5720, and SB-202190. The inhibition
profile of Vendor A kinase is comparable to that of Vendor B. See Table 4.
CHIC_4513901 1 89
CA 02779198 2012-04-26
WO 2011/053281
PCT/US2009/062250
TABLES
TABLE 1
Expression E. con Insect cell CHO cell Human cell
system
Protein folding + ++ +++ ++++
Phosphorylation ++ +++ ++++
Proteolytic + +++ ++++
processing
Glycosylation - Poor Not human-like Authentic
CHIC 4513901.1 90
CA 02779198 2012-04-26
WO 2011/053281 PCT/US2009/062250
TABLE 2 CYTOKINES AND THEIR ACTIVITIES
,
Cytokine Producing Target Cell Function
............. Cell
GM-CSF Th cells progenitor cells growth and differentiation
of
monocytes and DC
i
IL-la monocytes Th cells co-stimulation
IL-18 macrophages B cells maturation and
proliferation
B cells, DC NK cells, various Activation, inflammation,
acute
phase response, fever :
IL-2 Th1 cells activated T and B cells, NK cells growth,
proliferation,
.................................................. activation
1
IL-3 Th cells stern cells growth and differentiation
,
NK cells mast cells growth and histamine
release
,
'IL-4 Th2 cells activated B cells proliferation and
differentiation
IgGi and IgE synthesis
macrophages MHC Class II _______________________________________________ .
T cells proliferation
IL-5 Th2 cells activated B cells proliferation and
differentiation
IgA synthesis
. ....................................................................... ;
IL-6 monocytes activated B cells differentiation into
plasma cells
macrophages ,plasma cells antibody secretion
Th2 cells stem cells differentiation i
stromal cells various acute phase response
IL-7 marrow stroma stem cells differentiation into
progenitor B
thymus stroma and T cells :
IL-8 macrophages neutrophils chemotaxis ,
endothelial
cells
IL-10 Th2 cells macrophages cytokine production
......................................................................... i
B cells activation
IL-12 macrophages activated Tc cells differentiation into CTL
,
,
B cells (with IL-2)
......................... NK cells activation
IFN-a leukocytes various viral replication
MHC I expression
IFIA-B fibroblasts various viral replication ,
,
MHC I expression
IFN-y Th1 cells, various Viral replication
Tc cells, NK macrophages MHC expression
cells activated B cells ____ faclass switch to IgG2a
Th2 cells proliferation
......................... macrophages pathogen
eliminati7,,,,,,,,,,,,,,,,,,,,,,,,
MIP-1 a macrophages monocytes, T cells __ chemotaxis
MIP-18 Imphocytes monocytes, T cells chemotaxis
,
TGF-8 T cells, monocytes, macropha9es __ chemotaxis
_________________________________________________________________________ ,
, monocytes activated macrophages IL-1 synthesis
,
,
,
activated B cells I9A synthesis
various proliferation
INFa macrophages, macrophages _______ CAM and cytokine
expression
mast cells, NK tumor cells cell death
INF-8 Th1 and Tc phagocytes phagocytosis, NO
production
.. cells tumor cells , cell death
CHIC 4513901.1 91
CA 02779198 2012-04-26
WO 2011/053281
PCT/US2009/062250
TABLE 3. GENE ACCESSION NUMBERS FOR EXEMPLARY CYTOKINES
Gene Accession Gene Accession
Cytokine Cytokine
Number Number
Erythropoietin BC093628 Noggin BC034027
G-CSF NM 000759 SCF BC074725
GM-CSF BC113999 Somatotropin BC075012
IL-2 BC066255 TGF- 1 BC001180
IL-4 BC067514 TNF BCO28148
IL-6 BC015511 VEGF165 NM 003376
M-CSF BCO21117
CHIC_4513901 1 92
CA 02779198 2012-04-26
WO 2011/053281
PCT/US2009/062250
TABLE 4 INHIBITOR IC50 VALUES
Human Cell E coli-I E coli-II
IC50 pM IC50 pM IC50 pM
Staurosporine 0.14 >10 >10
H-9 dihydrochloride >10 >10 >10
AMP-PNP 257 1806 >2000
HA-1077 dihydrochlorie >10 >10 >10
Rottlerin >200 >200 >200
H89 Dihydrochloride >10 >10 >10
5-iodotubercidin >10 >10 >10
K252a 0.005 >10 >10
Ro 32-0432 >10 >10 >10
Ro 31-8220 9.9 >10 >10
GF 109203X >10 >10 >10
KT5720 5.0 >10 >10
Imatinib mesylate >10 >10 >10
SB 0.02 0.03 0.01
CHIC_4513901 1 93
CA 02779198 2012-04-26
WO 2011/053281 PCT/US2009/062250
TABLE 5 LIST OF CLONED CYTOKINE GENES
Activin A/2xINHbA GDF5 / BMP14 IL3 TGF 02
Activin B/2xINHbB GDF8/myostatin IL32 TGF f33
AMH/MIS GDF9 IL35 TGF04 / LEFTY2 /
LeftyA
Artemin GDNF IL4 TNF a
BDNF GM-CSF IL5 TP0a
BMP15 / GDF9B HGF IL6 VEGF121aa
BMP2 / BMP2A IFN a2A IL7 VEGF165aa
BMP3 / Osteogenin IFN ct2B IL8 WIF1
BMP4 / BMP2B IFN y IL9 WNT1
BMP5 IFN 01 Inhibin A/INHa&INHbA WntlOA
BMP7 / OP-1 IGF I Inhibin B/INHa&INHbB Wntl0B/12
0-NGF IGF II Inhibin C/INHa&INHbC Wntll
Cystatin C IGF IIvl Inhibin E/INHa&INHbE Wnt16
Delta 1 IGF IIv2 LEFTY1 / LeftyB Wnt2
EGF IL10 M-CSF Wnt2B/13
Erythropoietin (Epo) IL11 NODAL Wnt3
FGF acidic IL12 Noggin Wnt3A
FGF basic IL15 NT3 (neurotrophin3) Wnt4
FGF10 IL17 Oncostatin M Wnt5A
FGF5 IL17F PDGF a Wnt5B
FGF7 IL10 PDGF f3 Wnt6
FGF8b IL2 Persephin Wnt7A
FLT3 ligand IL23 SCF Wnt7B
G-CSF IL27 SDF la Wnt8B
GDF15 IL28A/IFNk2 SHH Wnt9A/14
GDF2 / BMP9 IL28B/IFN22 Somatotropin
GDF3 IL29/IFNk1 TGF 01
CHIC 4513901.1 94
CA 02779198 2012-04-26
WO 2011/053281
PCT/US2009/062250
SEQUENCES
(1) VEGF165
DNA sequence is SEQ ID NO: 1.
Amino acid sequence is SEQ ID NO: 2.
gcacccatggcagaaggaggagggcagaatcatcacgaagtggtgaagttcatggatgtc
APMAEGGGQNHHEVVKFMDV
tatcagcgcagctactgccatccaatcgagaccctggtggacatcttccaggagtaccct
YQRSYCHPIETLVDIFQEYP
gatgagatcgagtacatcttcaagccatcctgtgtgcccctgatgcgatgcgggggctgc
DEIEYIFKPSCVPLMRCGGC
tgcaatgacgagggcctggagtgtgtgcccactgaggagtccaacatcaccatgcagatt
CNDEGLECVPTEESNITMQI
atgcggatcaaacctcaccaaggccagcacataggagagatgagcttcctacagcacaac
MRIKPHQGQHIGEMSFLQHN
aaatgtgaatgcagaccaaagaaagatagagcaagacaagaaaatccctgtgggccttgc
KCECRPKKDRARQENPCGPC
tcagagcggagaaagcatttgtttgtacaagatccgcagacgtgtaaatgttcctgcaaa
SERRKHLFVQDPQTCKCSCK
aacacagactcgcgttgcaaggcgaggcagcttgagttaaacgaacgtacttgcagatgt
NTDSRCKARQLELNERTCRC
gacaagccgaggcggtgataa
DKPRR- -
(2) G-CSF
DNA sequence is SEQ ID NO: 3.
Amino acid sequence is SEQ ID NO: 4.
acccccctgggccctgccagctccctgccccagagcttcctgctcaagtgcttagagcaa
TPLGPASSLPQSFLLKCLEQ
gtgaggaagatccagggcgatggcgcagcgctccaggagaagctgtgtgccacctacaag
VRKIQGDGAALQEKLCATYK
ctgtgccaccccgaggagctggtgctgctcggacactctctgggcatcccctgggctccc
LCHPEELVLLGHSLGIPWAP
ctgagcagctgccccagccaggccctgcagctggcaggctgcttgagccaactccatagc
LSSCPSQALQLAGCLSQLHS
ggccttttcctctaccaggggctcctgcaggccctggaagggatctcccccgagttgggt
GLFLYQGLLQALEGISPELG
cccaccttggacacactgcagctggacgtcgccgactttgccaccaccatctggcagcag
PTLDTLQLDVADFATTIWQQ
atggaagaactgggaatggcccctgccctgcagcccacccagggtgccatgccggccttc
MEELGMAPALQPTQGAMPAF
gcctctgctttccagcgccgggcaggaggggtcctggttgcctcccatctgcagagcttc
ASAFQRRAGGVLVASHLQSF
ctggaggtgtcgtaccgcgttctacgccaccttgcccagccctgataa
LEVSYRVLRHLAQP- -
(3) M-CSF
DNA sequence is SEQ ID NO: 5.
Amino acid sequence is SEQ ID NO: 6.
gaggaggtgtcggagtactgtagccacatgattgggagtggacacctgcagtctctgcag
EEVSEYCSHMIGSGHLQSLQ
cggctgattgacagtcagatggagacctcgtgccaaattacatttgagtttgtagaccag
RLIDSQMETSCQITFEFVDQ
gaacagttgaaagatccagtgtgctaccttaagaaggcatttctcctggtacaagacata
EQLKDPVCYLKKAFLLVQDI
atggaggacaccatgcgcttcagagataacacccccaatgccatcgccattgtgcagctg
MEDTMRFRDNTPNAIAIVQL
CHIC_4513901 1 95
CA 02779198 2012-04-26
WO 2011/053281
PCT/US2009/062250
caggaactctctttgaggctgaagagctgcttcaccaaggattatgaagagcatgacaag
QELSLRLKSCFTKDYEEHDK
gcctgcgtccgaactttctatgagacacctctccagttgctggagaaggtcaagaatgtc
ACVRTFYETPLQLLEKVKNV
tttaatgaaacaaagaatctccttgacaaggactggaatattttcagcaagaactgcaac
FNETKNLLDKDWNIFSKNCN
aacagctttgctgaatgctccagccaaggccatgagaggcagtccgagggatcctgataa
NSFAECSSQGHERQSEGS- -
(4) IL-2
DNA sequence is SEQ ID NO: 7.
Amino acid sequence is SEQ ID NO: 8.
gcacctacttcaagttctacaaagaaaacacagctacaactggagcatttactgctggat
APTSSSTKKTQLQLEHLLLD
ttacagatgattttgaatggaattaataattacaagaatcccaaactcaccaggatgctc
LQMILNGINNYKNPKLTRML
acatttaagttttacatgcccaagaaggccacagaactgaaacatcttcagtgtctagaa
TFKFYMPKKATELKHLQCLE
gaagaactcaaacctctggaggaagtgctaaatttagctcaaagcaaaaactttcactta
EELKPLEEVLNLAQSKNFHL
agacccagggacttaatcagcaatatcaacgtaatagttctggaactaaagggatctgaa
RPRDLISNINVIVLELKGSE
acaacattcatgtgtgaatatgctgatgagacagcaaccattgtagaatttctgaacaga
TTFMCEYADETATIVEFLNR
tggattaccttttgtcaaagcatcatctcaacactgacttgataa
WITFCQSIISTLT- -
(5) Somatotropin
DNA sequence is SEQ ID NO: 9.
Amino acid sequence is SEQ ID NO: 10.
ttcccaaccattcccttatccaggctttttgacaacgctatgctccgcgcccatcgtctg
FPTIPLSRLFDNAMLRAHRL
caccagctggcctttgacacctaccaggagtttgaagaagcctatatcccaaaggaacag
HQLAFDTYQEFEEAYIPKEQ
aagtattcattcctgcagaacccccagacctccctctgtttctcagagtctattccgaca
KYSFLQNPQTSLCFSESIPT
ccctccaacagggaggaaacacaacagaaatccaacctagagctgctccgcatctccctg
PSNREETQQKSNLELLRISL
ctgctcatccagtcgtggctggagcccgtgcagttcctcaggagtgtcttcgccaacagc
LLIQSWLEPVQFLRSVFANS
ctggtgtacggcgcctctgacagcaacgtctatgacctcctaaaggacctagaggaaggc
LVYGASDSNVYDLLKDLEEG
atccaaacgctgatggggaggctggaagatggcagcccccggactgggcagatcttcaag
IQTLMGRLEDGSPRTGQIFK
cagacctacagcaagttcgacacaaactcacacaacgatgacgcactactcaagaactac
QTYSKFDTNSHNDDALLKNY
gggctgctctactgcttcaggaaggacatggacaaggtcgagacattcctgcgcatcgtg
GLLYCFRKDMDKVETFLRIV
cagtgccgctctgtggagggcagctgtggcttctagtaa
QCRSVEGSCGF- -
(6) TGFb1
DNA sequence is SEQ ID NO: 11.
Amino acid sequence is SEQ ID NO: 12.
ctatccacctgcaagactatcgacatggagctggtgaagcggaagcgcatcgaggccatc
LSTCKTIDMELVKRKRIEAI
cgcggccagatcctgtccaagctgcggctcgccagccccccgagccagggggaggtgccg
CHIC_4513901 1 96
CA 02779198 2012-04-26
WO 2011/053281 PCT/US2009/062250
RGQILSKLRLASPPSQGEVP
cccggcccgctgcccgaggccgtgctcgccctgtacaacagcacccgcgaccgggtggcc
PGPLPEAVLALYNSTRDRVA
ggggagagtgcagaaccggagcccgagcctgaggccgactactacgccaaggaggtcacc
GESAEPEPEPEADYYAKEVT
cgcgtgctaatggtggaaacccacaacgaaatctatgacaagttcaagcagagtacacac
RVLMVETHNEIYDKFKQSTH
agcatatatatgttcttcaacacatcagagctccgagaagcggtacctgaacccgtgttg
SIYMFFNTSELREAVPEPVL
ctctcccgggcagagctgcgtctgctgaggctcaagttaaaagtggagcagcacgtggag
LSRAELRLLRLKLKVEQHVE
ctgtaccagaaatacagcaacaattcctggcgatacctcagcaaccggctgctggcaccc
LYQKYSNNSWRYLSNRLLAP
agcgactcgccagagtggttatcttttgatgtcaccggagttgtgcggcagtggttgagc
SDSPEWLSFDVTGVVRQWLS
cgtggaggggaaattgagggctttcgccttagcgcccactgctcctgtgacagcagggat
RGGEIEGFRLSAHCSCDSRD
aacacactgcaagtggacatcaacgggttcactaccggccgccgaggtgacctggccacc
NTLQVDINGFTTGRRGDLAT
attcatggcatgaaccggcctttcctgcttctcatggccaccccgctggagagggcccag
IHGMNRPFLLLMATPLERAQ
catctgcaaagctcccggcaccgccgagccctggacaccaactattgcttcagctccacg
HLQSSRHRRALDTNYCFSST
gagaagaactgctgcgtgcggcagctgtacattgacttccgcaaggacctcggctggaag
EKNCCVRQLYIDFRKDLGWK
tggatccacgagcccaagggctaccatgccaacttctgcctcgggccctgcccctacatt
WIHEPKGYHANFCLGPCPYI
tggagcctggacacgcagtacagcaaggtcctggccctgtacaaccagcataacccgggc
WSLDTQYSKVLALYNQHNPG
gcctcggcggcgccgtgctgcgtgccgcaggcgctggagccgctgcccatcgtgtactac
ASAAPCCVPQALEPLPIVYY
gtgggccgcaagcccaaggtggagcagctgtccaacatgatcgtgcgctcctgcaagtgc
/GRKPKVEQLSNMIVRSCKC
agctgataa
S - -
(7) TNFa
DNA sequence is SEQ ID NO: 13.
Amino acid sequence is SEQ ID NO: 14.
gtcagatcatcttctcgaaccccgagtgacaagcctgtagcccatgttgtagcaaaccct
/RSSSRTPSDKPVAHVVANP
caagctgaggggcagctccagtggctgaaccgccgggccaatgccctcctggccaatggc
QAEGQLQWLNRRANALLANG
gtggagctgagagataaccagctggtggtgccatcagagggcctgtacctcatctactcc
VELRDNQLVVPSEGLYLIYS
caggtcctcttcaagggccaaggctgcccctccacccatgtgctcctcacccacaccatc
QVLFKGQGCPSTHVLLTHTI
agccgcatcgccgtctcctaccagaccaaggtcaacctcctctctgccatcaagagcccc
SRIAVSYQTKVNLLSAIKSP
tgccagagggagaccccagagggggctgaggccaagccctggtatgagcccatctatctg
CQRETPEGAEAKPWYEPIYL
ggaggggtcttccagctggagaagggtgaccgactcagcgctgagatcaatcggcccgac
GGVFQLEKGDRLSAEINRPD
tatctcgactttgccgagtctgggcaggtctactttgggatcattgccctgtgataa
YLDFAESGQVYFGIIAL- -
(8) IL6
CHIC 4513901.1 97
CA 02779198 2012-04-26
WO 2011/053281
PCT/US2009/062250
DNA sequence is SEQ ID NO: 15.
Amino acid sequence is SEQ ID NO: 16.
gccccagtacccccaggagaagattccaaagatgtagccgccccacacagacagccactc
APVPPGEDSKDVAAPHRQPL
acctcttcagaacgaattgacaaacaaattcggtacatcctcgacggcatctcagccctg
TSSERIDKQIRYILDGISAL
agaaaggagacatgtaacaagagtaacatgtgtgaaagcagcaaagaggcactggcagaa
RKETCNKSNMCESSKEALAE
aacaacctgaaccttccaaagatggctgaaaaagatggatgcttccaatctggattcaat
NNLNLPKMAEKDGCFQSGFN
gaggagacttgcctggtgaaaatcatcactggtcttttggagtttgaggtatacctagag
EETCLVKIITGLLEFEVYLE
tacctccagaacagatttgagagtagtgaggaacaagccagagctgtgcagatgagtaca
YLQNRFESSEEQARAVQMST
aaagtcctgatccagttcctgcagaaaaaggcaaagaatctagatgcaataaccacccct
KVLIQFLQKKAKNLDAITTP
gacccaaccacaaatgccagcctgctgacgaagctgcaggcacagaaccagtggctgcag
DPTTNASLLTKLQAQNQWLQ
gacatgacaactcatctcattctgcgcagctttaaggagttcctgcagtccagcctgagg
DMTTHLILRSFKEFLQSSLR
gctcttcggcaaatgtagtaa
ALRQM- -
(9) Erythropoietin (Epo)
DNA sequence is SEQ ID NO: 17.
Amino acid sequence is SEQ ID NO: 18.
gccccaccacgcctcatctgtgacagccgagtcctggagaggtacctcttggaggccaag
APPRLICDSRVLERYLLEAK
gaggccgagaatatcacgacgggctgtgctgaacactgcagcttgaatgagaatatcact
EAENITTGCAEHCSLNENIT
gtcccagacaccaaagttaatttctatgcctggaagaggatggaggtcgggcagcaggcc
/PDTKVNFYAWKRMEVGQQA
gtagaagtctggcagggcctggccctgctgtcggaagctgtcctgcggggccaggccctg
/EVWQGLALLSEAVLRGQAL
ttggtcaactcttcccagccgtgggagcccctgcagctgcatgtggataaagccgtcagt
LVNSSQPWEPLQLHVDKAVS
ggccttcgcagcctcaccactctgcttcgggctctgggagcccagaaggaagccatctcc
GLRSLTTLLRALGAQKEAIS
cctccagatgcggcctcagctgctccactccgaacaatcactgctgacactttccgcaaa
PPDAASAAPLRTITADTFRK
ctcttccgagtctactccaatttcctccggggaaagctgaagctgtacacaggggaggcc
LFRVYSNFLRGKLKLYTGEA
tgcaggacaggggacagatgataa
CRTGDR- -
(10) GM-CSF
DNA sequence is SEQ ID NO: 19.
Amino acid sequence is SEQ ID NO: 20.
gcacccgcccgctcgcccagccccagcacgcagccctgggagcatgtgaatgccatccag
APARSPSPSTQPWEHVNAIQ
gaggcccggcgtctcctgaacctgagtagagacactgctgctgagatgaatgaaacagta
EARRLLNLSRDTAAEMNETV
gaagtcatctcagaaatgtttgacctccaggagccgacctgcctacagacccgcctggag
EVISEMFDLQEPTCLQTRLE
ctgtacaagcagggcctgcggggcagcctcaccaagctcaagggccccttgaccatgatg
LYKQGLRGSLTKLKGPLTMM
gccagccactacaagcagcactgccctccaaccccggaaacttcctgtgcaacccagatt
CHIC_4513901 1 98
CA 02779198 2012-04-26
WO 2011/053281
PCT/US2009/062250
ASHYKQHCPPTPETSCATQI
atcacctttgaaagtttcaaagagaacctgaaggactttctgcttgtcatcccctttgac
ITFESFKENLKDFLLVIPFD
tgctgggagccagtccaggagtgataa
CWEPVQE- -
(11) IL4
DNA sequence is SEQ ID NO: 21.
Amino acid sequence is SEQ ID NO: 22.
cacaagtgcgatatcaccttacaggagatcatcaaaactttgaacagcctcacagagcag
HKCDITLQEIIKTLNSLTEQ
aagactctgtgcaccgagttgaccgtaacagacatctttgctgcctccaagaacacaact
KTLCTELTVTDIFAASKNTT
gagaaggaaaccttctgcagggctgcgactgtgctccggcagttctacagccaccatgag
EKETFCRAATVLRQFYSHHE
aaggacactcgctgcctgggtgcgactgcacagcagttccacaggcacaagcagctgatc
KDTRCLGATAQQFHRHKQLI
cgattcctgaaacggctcgacaggaacctctggggcctggcgggcttgaattcctgtcct
RFLKRLDRNLWGLAGLNSCP
gtgaaggaagccaaccagagtacgttggaaaacttcttggaaaggctaaagacgatcatg
/KEANQSTLENFLERLKTIM
agagagaaatattcaaagtgttcgagctgataa
REKYSKCSS- -
(12) Noggin
DNA sequence is SEQ ID NO: 23.
Amino acid sequence is SEQ ID NO: 24.
cagcactatctccacatccgcccggcacccagcgacaacctgcccctggtggacctcatc
QHYLHIRPAPSDNLPLVDLI
gaacacccagaccctatctttgaccccaaggaaaaggatctgaacgagacgctgctgcgc
EHPDPIFDPKEKDLNETLLR
tcgctgctcgggggccactacgacccaggcttcatggccacctcgccccccgaggaccgg
SLLGGHYDPGFMATSPPEDR
cccggcgggggcgggggtgcagctgggggcgcggaggacctggcggagctggaccagctg
PGGGGGAAGGAEDLAELDQL
ctgcggcagcggccgtcgggggccatgccgagcgagatcaaagggctagagttctccgag
LRQRPSGAMPSEIKGLEFSE
ggcttggcccagggcaagaagcagcgcctaagcaagaagctgcggaggaagttacagatg
GLAQGKKQRLSKKLRRKLQM
tggctgtggtcgcagacattctgccccgtgctgtacgcgtggaacgacctgggcagccgc
WLWSQTFCPVLYAWNDLGSR
ttttggccgcgctacgtgaaggtgggcagctgcttcagtaagcgctcgtgctccgtgccc
FWPRYVKVGSCFSKRSCSVP
gagggcatggtgtgcaagccgtccaagtccgtgcacctcacggtgctgcggtggcgctgt
EGMVCKPSKSVHLTVLRWRC
cagcggcgcgggggccagcgctgcggctggattcccatccagtaccccatcatttccgag
QRRGGQRCGWIPIQYPIISE
tgcaagtgctcgtgctagtaa
CKCSC- -
(13) SCF
DNA sequence is SEQ ID NO: 25.
Amino acid sequence is SEQ ID NO: 26.
gaagggatctgcaggaatcgtgtgactaataatgtaaaagacgtcactaaattggtggca
CHIC_4513901 1 99
CA 02779198 2012-04-26
WO 2011/053281 PCT/US2009/062250
EGICRNRVTNNVKDVTKLVA
aatcttccaaaagactacatgataaccctcaaatatgtccccgggatggatgttttgcca
NLPKDYMITLKYVPGMDVLP
agtcattgttggataagcgagatggtagtacaattgtcagacagcttgactgatcttctg
SHCWISEMVVQLSDSLTDLL
gacaagttttcaaatatttctgaaggcttgagtaattattccatcatagacaaacttgtg
DKFSNISEGLSNYSIIDKLV
aatatagtggatgaccttgtggagtgcgtgaaagaaaactcatctaaggatctaaaaaaa
NIVDDLVECVKENSSKDLKK
tcattcaagagcccagaacccaggctctttactcctgaagaattctttagaatttttaat
SFKSPEPRLFTPEEFFRIFN
agatccattgatgccttcaaggactttgtagtggcatctgaaactagtgattgtgtggtt
RSIDAFKDFVVASETSDCVV
tcttcaacattaagtcctgagaaagattccagagtcagtgtcacaaaaccatttatgtta
SSTLSPEKDSRVSVTKPFML
ccccctgttgcagccagctcccttaggaatgacagcagtagcagtaataggaaggccaaa
PPVAASSLRNDSSSSNRKAK
aatccccctggagactccagcctacactgataa
NPPGDSSLH- -
SEQ ID NO: 27
Human CMV immediate early (IE) enhancer from a CMV vector
GTTGACATTGATTATTGACTAGTTATTAATAGTAATCAATTACGGGGTCATTAGTTCATAGCCCATATA
TGGAGTTCCGCGTTACATAACTTACGGTAAATGGCCCGCCTGGCTGACCGCCCAACGACCCCCGCCCAT
TGACGTCAATAATGACGTATGTTCCCATAGTAACGCCAATAGGGACTTTCCATTGACGTCAATGGGTGG
ACTATTTACGGTAACGCCAATAGGGACTTTCCATTGACGTCAATGGGTGGACTATTTACGGTAAACTGC
CCACTTGGCAGTACATCAAGTGTATCATATGCCAAGTACGCCCCCTATTGACGTCAATGACGGTAAATG
GCCCGCCTGGCATTATGCCCAGTACATGACCTTATGGGACTTTCCTACTTGGCAGTACATCTACGTATT
AGTCATCGCTATTACCATGG
SEQ ID NO: 28
Human beta-actin promoter
CCGGGCCCAGCACCCCAAGGCGGCCAACGCCAAAACTCTCCCTCCTCCTCTTCCTCAATCTCGCTCTCG
CTCTTTTTTTTTTTCGCAAAAGGAGGGGAGAGGGGGTAAAAAAATGCTGCACTGTGCGGCGAAGCCGGT
GAGTGAGCGGCGCGGGGCCAATCAGCGTGCGCCGTTCCGAAAGTTGCCTTTTATGGCTCGAGCGGCCGC
GGCGGCGCCCTATAAAACCCAGCGGCGCGACGCGCCACCACCGCCGAGACCGCGTCCGCCCCGCGAGCA
CAGAGCCTCGCCTTTGCCGATCCGCCGCCCGTCCACACCCGCCGCCAGGTAAGCCCGGCCAGCCGACCG
GGGCAGGCGGCTCACGGCCCGGCCGCAGGCGGCCGCGGCCCCTTCGCCCGTGCAGAGCCGCCGTCTGGG
CCGCAGCGGGGGGCGCATGGGGGGGGAACCGGACCGCCGTGGGGGGCGCGGGAGAAGCCCCTGGGCCTC
CGGAGATGGGGGACACCCCACGCCAGTTCGGAGGCGCGAGGCCGCGCTCGGGAGGCGCGCTCCGGGGGT
GCCGCTCTCGGGGCGGGGGCAACCGGCGGGGTCTTTGTCTGAGCCGGGCTCTTGCCAATGGGGATCGCA
GGGTGGGCGCGGCGGAGCCCCCGCCAGGCCCGGTGGGGGCTGGGGCGCCATTGCGCGTGCGCGCTGGTC
CTTTGGGCGCTAACTGCGTGCGCGCTGGGAATTGGCGCTAATTGCGCGTGCGCGCTGGGACTCAAGGCG
CTAACTGCGCGTGCGTTCTGGGGCCCGGGGTGCCGCGGCCTGGGCTGGGGCGAAGGCGGGCTCGGCCGG
AAGGGGTGGGGTCGCCGCGGCTCCCGGGCGCTTGCGCGCACTTCCTGCCCGAGCCGCTGGCCGCCCGAG
GGTGTGGCCGCTGCGTGCGCGCGCGCCGACCCGGCGCTGTTTGAACCGGGCGGAGGCGGGGCTGGCGCC
CGGTTGGGAGGGGGTTGGGGCCTGGCTTCCTGCCGCGCGCCGCGGGGACGCCTCCGACCAGTGTTTGCC
TTTTATGGTAATAACGCGGCCGGCCCGGCTTCCTTTGTCCCCAATCTGGGCGCGCGCCGGCGCCCCCTG
GCGGCCTAAGGACTCGGCGCGCCGGAAGTGGCCAGGGCGGGGGCGACCTCGGCTCACAGCGCGCCCGGC
TATTCTCGCAG
SEQ ID NO: 29
CHIC 4513901.1 100
CA 02779198 2012-04-26
WO 2011/053281 PCT/US2009/062250
Human beta-globin intron
CTTTTGCTAATCATGTTCATACCTCTTATCTTCCTCCCACAGCTCCTGGGCAACGTGCTGGTCTGTGTG
CTGGCCCATCACTTTGGCAAAGAATTC
SEQ ID NO: 30
Human fibrinogen alpha chain signal peptide (cleavage site is between
nucleotides t and g of tgac at the 3'-end)
atgttttccatgaggatcgtctgcctggtcctaagtgtggtgggcacagcatggactgac
MFSMRIVCLVLSVVGTAWTD
SEQ ID NO: 31
Human immunoglobulin superfamily member 8 precursor signal peptide
(cleavage site is between nucleotides c and g of cgac at the 3'-end)
atgggcgccctcaggcccacgctgctgccgccttcgctgccgctgctgctgctgctaatg
MGALRPTLLPPSLPLLLLLM
ctaggaatgggatgctgggccgac
LGMGCWAD
SEQ ID NO: 32
HumanZyme vector-1
GTTGACATTGATTATTGACTAGTTATTAATAGTAATCAATTACGGGGTCATTAGTTCATAGCCCATATA
TGGAGTTCCGCGTTACATAACTTACGGTAAATGGCCCGCCTGGCTGACCGCCCAACGACCCCCGCCCAT
TGACGTCAATAATGACGTATGTTCCCATAGTAACGCCAATAGGGACTTTCCATTGACGTCAATGGGTGG
ACTATTTACGGTAACGCCAATAGGGACTTTCCATTGACGTCAATGGGTGGACTATTTACGGTAAACTGC
CCACTTGGCAGTACATCAAGTGTATCATATGCCAAGTACGCCCCCTATTGACGTCAATGACGGTAAATG
GCCCGCCTGGCATTATGCCCAGTACATGACCTTATGGGACTTTCCTACTTGGCAGTACATCTACGTATT
AGTCATCGCTATTACCATGGCCGGGCCCAGCACCCCAAGGCGGCCAACGCCAAAACTCTCCCTCCTCCT
CTTCCTCAATCTCGCTCTCGCTCTTTTTTTTTTTCGCAAAAGGAGGGGAGAGGGGGTAAAAAAATGCTG
CACTGTGCGGCGAAGCCGGTGAGTGAGCGGCGCGGGGCCAATCAGCGTGCGCCGTTCCGAAAGTTGCCT
TTTATGGCTCGAGCGGCCGCGGCGGCGCCCTATAAAACCCAGCGGCGCGACGCGCCACCACCGCCGAGA
CCGCGTCCGCCCCGCGAGCACAGAGCCTCGCCTTTGCCGATCCGCCGCCCGTCCACACCCGCCGCCAGG
TAAGCCCGGCCAGCCGACCGGGGCAGGCGGCTCACGGCCCGGCCGCAGGCGGCCGCGGCCCCTTCGCCC
GTGCAGAGCCGCCGTCTGGGCCGCAGCGGGGGGCGCATGGGGGGGGAACCGGACCGCCGTGGGGGGCGC
GGGAGAAGCCCCTGGGCCTCCGGAGATGGGGGACACCCCACGCCAGTTCGGAGGCGCGAGGCCGCGCTC
GGGAGGCGCGCTCCGGGGGTGCCGCTCTCGGGGCGGGGGCAACCGGCGGGGTCTTTGTCTGAGCCGGGC
TCTTGCCAATGGGGATCGCAGGGTGGGCGCGGCGGAGCCCCCGCCAGGCCCGGTGGGGGCTGGGGCGCC
ATTGCGCGTGCGCGCTGGTCCTTTGGGCGCTAACTGCGTGCGCGCTGGGAATTGGCGCTAATTGCGCGT
GCGCGCTGGGACTCAAGGCGCTAACTGCGCGTGCGTTCTGGGGCCCGGGGTGCCGCGGCCTGGGCTGGG
GCGAAGGCGGGCTCGGCCGGAAGGGGTGGGGTCGCCGCGGCTCCCGGGCGCTTGCGCGCACTTCCTGCC
CGAGCCGCTGGCCGCCCGAGGGTGTGGCCGCTGCGTGCGCGCGCGCCGACCCGGCGCTGTTTGAACCGG
GCGGAGGCGGGGCTGGCGCCCGGTTGGGAGGGGGTTGGGGCCTGGCTTCCTGCCGCGCGCCGCGGGGAC
GCCTCCGACCAGTGTTTGCCTTTTATGGTAATAACGCGGCCGGCCCGGCTTCCTTTGTCCCCAATCTGG
GCGCGCGCCGGCGCCCCCTGGCGGCCTAAGGACTCGGCGCGCCGGAAGTGGCCAGGGCGGGGGCGACCT
CGGCTCACAGCGCGCCCGGCTATTCTCGCAGCTTTTGCTAATCATGTTCATACCTCTTATCTTCCTCCC
ACAGCTCCTGGGCAACGTGCTGGTCTGTGTGCTGGCCCATCACTTTGGCAAAGAATTCatgttttccat
gaggatcgtctgcctggtcctaagtgtggtgggcacagcatggactgacGCGCCCGGGCCGGCCAGGCG
CGCGCGCCGTACGTACGAAGCTTGGTACCGAGCTCGGATCCACTCCAGTGTGGTGGAATTCTGCAGATA
TCCAGCACAGTGGCGGCCGCTCGAGGAGGGCCCGAACAAAAACTCATCTCAGAAGAGGATCTGA
SEQ ID NO: 33
CHIC 4513901.1 101
CA 02779198 2012-04-26
VIM) 20111053281 PCT/US2009/062250
HumanZyme vector-2 (underline = hCMV IE Enhancer; bold = human beta-
actin promoter; italics = human immunoglobulin superfamily member 8
precursor signal peptide)
GTTGACATTGATTATTGACTAGTTATTAATAGTAATCAATTACGGGGTCATTAGTTCATAGCCCATATA
TGGAGTTCCGCGTTACATAACTTACGGTAAATGGCCCGCCTGGCTGACCGCCCAACGACCCCCGCCCAT
TGACGTCAATAATGACGTATGTTCCCATAGTAACGCCAATAGGGACTTTCCATTGACGTCAATGGGTGG
ACTATTTACGGTAACGCCAATAGGGACTTTCCATTGACGTCAATGGGTGGACTATTTACGGTAAACTGC
CCACTTGGCAGTACATCAAGTGTATCATATGCCAAGTACGCCCCCTATTGACGTCAATGACGGTAAATG
GCCCGCCTGGCATTATGCCCAGTACATGACCTTATGGGACTTTCCTACTTGGCAGTACATCTACGTATT
AGTCATCGCTATTACCATGGCCGGGCCCAGCACCCCAAGGCGGCCAACGCCAAAACTCTCCCTCCTCCT
CTTCCTCAATCTCGCTCTCGCTCTTTTTTTTTTTCGCAAAAGGAGGGGAGAGGGGGTAAAAAAATGCTG
CACTGTGCGGCGAAGCCGGTGAGTGAGCGGCGCGGGGCCAATCAGCGTGCGCCGTTCCGAAAGTTGCCT
TTTATGGCTCGAGCGGCCGCGGCGGCGCCCTATAAAACCCAGCGGCGCGACGCGCCACCACCGCCGAGA
CCGCGTCCGCCCCGCGAGCACAGAGCCTCGCCTTTGCCGATCCGCCGCCCGTCCACACCCGCCGCCAGG
TAAGCCCGGCCAGCCGACCGGGGCAGGCGGCTCACGGCCCGGCCGCAGGCGGCCGCGGCCCCTTCGCCC
GTGCAGAGCCGCCGTCTGGGCCGCAGCGGGGGGCGCATGGGGGGGGAACCGGACCGCCGTGGGGGGCGC
GGGAGAAGCCCCTGGGCCTCCGGAGATGGGGGACACCCCACGCCAGTTCGGAGGCGCGAGGCCGCGCTC
GGGAGGCGCGCTCCGGGGGTGCCGCTCTCGGGGCGGGGGCAACCGGCGGGGTCTTTGTCTGAGCCGGGC
TCTTGCCAATGGGGATCGCAGGGTGGGCGCGGCGGAGCCCCCGCCAGGCCCGGTGGGGGCTGGGGCGCC
ATTGCGCGTGCGCGCTGGTCCTTTGGGCGCTAACTGCGTGCGCGCTGGGAATTGGCGCTAATTGCGCGT
GCGCGCTGGGACTCAAGGCGCTAACTGCGCGTGCGTTCTGGGGCCCGGGGTGCCGCGGCCTGGGCTGGG
GCGAAGGCGGGCTCGGCCGGAAGGGGTGGGGTCGCCGCGGCTCCCGGGCGCTTGCGCGCACTTCCTGCC
CGAGCCGCTGGCCGCCCGAGGGTGTGGCCGCTGCGTGCGCGCGCGCCGACCCGGCGCTGTTTGAACCGG
GCGGAGGCGGGGCTGGCGCCCGGTTGGGAGGGGGTTGGGGCCTGGCTTCCTGCCGCGCGCCGCGGGGAC
GCCTCCGACCAGTGTTTGCCTTTTATGGTAATAACGCGGCCGGCCCGGCTTCCTTTGTCCCCAATCTGG
GCGCGCGCCGGCGCCCCCTGGCGGCCTAAGGACTCGGCGCGCCGGAAGTGGCCAGGGCGGGGGCGACCT
CGGCTCACAGCGCGCCCGGCTATTCTCGCAGCTTTTGCTAATCATGTTCATACCTCTTATCTTCCTCCC
ACAGCTCCTGGGCAACGTGCTGGTCTGTGTGCTGGCCCATCACTTTGGCAAAGAATTCatgggcgccct
caggcccacgctgctgccgccttcgctgccgctgctgctgctgctaatg
ctaggaatgggatgctgggccgacGCGCCCGGGCCGGCCAGGCGCGCGCGCCGTACGTACGAAGCTTGG
TACCGAGCTCGGATCCACTCCAGTGTGGTGGAATTCTGCAGATATCCAGCACAGTGGCGGCCGCTCGAG
GAGGGCCCGAACAAAAACTCATCTCAGAAGAGGATCTGA
CHIC 4513901.1 102