Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
81662840
- 1 -
SMALL MOLECULE INIIIBITORS OF USP1 DEUBIQUITINATING ENZYME
ACTIVITY
10
BACKGROUND OF THE INVENTION
Ubiquitin is a small protein consisting of 76 amino acids that is important in
the
regulation of protein function in the cell. Ubiquitination and
cleubiquitination are
enzymatically mediated processes by which ubiquitin is covalently bound to or
unbound
from a target protein. These processes have been implicated in the regulation
of the cell
cycle, apoptosis, the marking of transmembrane proteins such as receptors for
removal,
regulation of DNA transcription and repair, and other important functions.
Proteins are
targeted for degradation by the proteasome in the cell by being "tagged" with
three Or
more ubiquitin molecules (polyubiquitination). The binding of a single
ubiquitin
molecule (monoubiquitination) does not generally target the monoubiquitinated
protein
for degradation. Rather, it may trigger activities such as DNA repair and gene
silencing,
among other functions. Huang and D'Andrea (2006) Mol Cell Biol, 7:323-34.
Deubiquitination allows ubiquitin to be recycled and restores the function of
the
deubiquitinated proteins. Ubiquitin molecules are cleaved from a protein by
deubiquitinating enzymes, which are cysteine proteases that operate through an
active
site thiol. There are approximately 95 different deubiquitinating enzymes in
human
cells. Huang et al. (2006) Nature Cell Riot. 8(4):339-47. Among them,
Ubiquitin
Specific Protease 1 (USP1 ), also known as ubiquitin specific peptidase 1 and
as ubiquitin
CA 2797719 2017-10-10
CA 02797719 2012-10-26
WO 2011/137320
PCT/US2011/034514
_ 2 _
carboxyl terminal hydrolase 1, has been found to regulate the repair of DNA
damage
induced by DNA cross-linking agents, which include agents such as cisplatin,
mitomycin
C (MMC), diepoxybutane (DEB), ionizing radiation (IR), and ultraviolet (UV)
radiation.
USP1 has been shown to deubiquitinate monoubiquitinated Fanconi anemia
group complementation group D2 (FANCD2-Ub), a protein that in its
monoubiquitinated
form mediates DNA repair from the damage induced by the aforementioned agents.
Nijman et al. (2005) Mol Cell 17:331-39. USP1 also has been shown to
deubiquitinate
monoubiquitinated proliferating cell nuclear antigen (PCNA-Ub), a protein that
in its
monoubiquitinated form activates DNA translesion synthesis, a polymerase-
mediated
bypass of DNA lesions. Huang et al. (2006) Nature Cell Biol, 8(4):339-47.
Recently it was reported that USP1 forms a complex with and is activated by
USP1 associated factor 1 (UAF1), also known as WD repeat domain 48. Cohn et
al.
(2007) Mol Cell 28:786-97: US 2008/0167229 Al. The active USP1/IJAF1 complex
controls cellular levels of monubiquitinated FANCD2. Even more recently, it
was
reported that UAF1 also separately forms complexes with and activates two
additional
deubiquitinating enzymes, ubiquitin specific protease 12 (USP12) and ubiquitin
specific
protease 46 (USP46). Cohn et al. (2009) J Biol Chem. 284(8):5343-51.
Co-owned US 2008/0167229 discloses three compounds, f3-lapachone, Biomol
AP401 (propidium iodide), and RK-682, as potential small molecule inhibitors
of
USP1/UAF1 complex-mediated deubiquitinase activity.
SUMMARY OF THE INVENTION
The invention provides certain small molecule inhibitors of USP1 activity as
well
as compositions and methods related to the inhibitors useful for the
inhibition of USP1
activity in vitro and in vivo. These small molecule inhibitors of USP1
activity are
structurally distinct from13-lapachone, Biomol AP401 (propidium iodide), and
RK-682.
The invention further provides, in part, compositions and methods useful for
the
treatment, prevention, and characterization of cancer based on the ability to
inhibit USP1
activity in cancer cells.
It has surprisingly been discovered according to the present invention that
inhibition of USP1 activity, which inhibits deubiquitination of
monoubiquitinated
FANCD2 (FANCD2-Ub), results in reduced DNA repair activity in cells, including
CA 02797719 2012-10-26
WO 2011/137320
PCT/US2011/034514
- 3 -
cancer cells. This was unexpected because FANCD2-Ub is the active form of
FANCD2.
Inhibition of USP1 activity thus can render cancer cells, including cancer
cells otherwise
resistant to treatment with DNA cross-linking agents, susceptible to treatment
with DNA
cross-linking agents.
The deubiquitinating enzyme USP1 and its associated factor UA141 regulate the
Fanconi Anemia (FA)-BRCA DNA repair pathway by catalyzing the deubiquitination
of
mono-ubiquitinated FANCD2. Inactivation of USP1/UAF1 results in elevated level
of
mono-ubiquitinated FANCD2, disruption of the FA-BRCA pathway, and cellular
hypersensitivity to DNA crosslinking agents, such as mitomycin C and
cisplatin.
Targeting USPI/UAFI with a small-molecule inhibitor therefore enhances the
efficacy
of DNA crosslinking agents in cancer therapy.
An aspect of the invention is a pharmaceutical composition including a small
molecule inhibitor of ubiquitin specific protease 1 (IJSP1) according to
Formula I:
_____________________________________________ R2
x>
0
Formula I
wherein
Xis 0, S, or NR3;
n is 0, 1, 2, 3, or 4;
each occurrence of 121 is independently hydrogen; halogen; cyclic or acyclic,
substituted or unsubstituted, branched or unbranched aliphatic; cyclic or
acyclic,
substituted or unsubstituted, branched or unbranched heteroaliphatic;
substituted or
unsubstituted, branched or unbranched acyl; substituted or unsubstituted aryl;
substituted
or unsubstituted heteroaryl; -OR"; -C(=0)RA; -C(=0)N(RA)2; -CO2RA; -CN; -SCN; -
SRA; -SORA; -SO2RA; -NO2; -N3; -N(RA)2; -NHC(=0)RA; -NRAC(=0)N(RA)2; -
.. OC(=0)0RA; -0C(=0)RA; -0C(=0)N(RA)2; -NRAC(=0)0RA; or -C(RA)3; wherein each
occurrence of RA is independently a hydrogen, a protecting group, an aliphatic
moiety, a
heteroaliphatic moiety, an acyl moiety; an aryl moiety; a heteroaryl moiety;
alkoxy;
CA 02797719 2012-10-26
WO 2011/137320
PCT/US2011/034514
- 4 -
aryloxy; alkylthio; arylthio; amino, alkylamino, dialkylamino, heteroaryloxy;
or
heteroarylthio moiety;
R2 is hydrogen; halogen; cyclic or acyclic, substituted or unsubstituted,
branched
or unbranched aliphatic; cyclic or acyclic, substituted or unsubstituted,
branched or
unbranched heteroaliphatic; substituted or unsubstituted, branched or
unbranched acyl;
substituted or unsubstituted aryl; substituted or unsubstituted heteroaryl; -
ORB; -
C(=0)RB; -C(=0)N(RB)2; -CO2RB; -CN; -SCN; -SRB; -SORB; -SO2RB; -NO2; -N3; -
N(RB)1; -NHC(=0)RB; -NRBC(=0)N(RB)1; -0C(=0)ORB; -0C(=0)RB; -0C(=0)N(RB)2;
-NRBC(=0)ORB; or -C(RB)3; wherein each occurrence of RB is independently a
hydrogen, a protecting group, an aliphatic moiety, a heteroaliphatic moiety,
an acyl
moiety; an aryl moiety; a heteroaryl moiety; alkoxy; aryloxy; alkylthio;
arylthio; amino,
alkylamino, dialkylamino, heteroaryloxy; or heteroarylthio moiety;
R3 is independently a hydrogen, a protecting group, an aliphatic moiety, a
heteroaliphatic moiety, an acyl moiety; an aryl moiety; a heteroaryl moiety;
alkoxy;
aryloxy; alkylthio; arylthio; amino, alkylamino, dialkylamino, heteroaryloxy;
or
heteroarylthio moiety;
or a pharmaceutically acceptable salt thereof,
and a pharmaceutically acceptable carrier.
In one embodiment according to this and other aspects of the invention, the
small
molecule inhibitor of USP1 according to Formula I is the compound of Formula
IV
(527):
0
N\
0
0
Formula IV (527),
or a pharmaceutically acceptable salt thereof.
In one embodiment the pharmaceutical composition further includes a DNA
cross-linking agent, for example chemotherapeutic agents including alkylating
agents,
cisplatin, and mitomycin C.
In one embodiment the pharmaceutical composition further includes a poly
(adenosine diphosphate (ADP)-ribose) polymerase (PARP) inhibitor.
CA 02797719 2012-10-26
WO 2011/137320
PCT/US2011/034514
- 5 -
An aspect of the invention is a pharmaceutical composition including a small
molecule inhibitor of USP1 according to Formula II:
0
R3 R3
Formula II
wherein
n is 0, 1, 2, 3, or 4;
each occurrence of 121 is independently hydrogen; halogen; cyclic or acyclic,
substituted or unsubstituted, branched or unbranched aliphatic; cyclic or
acyclic,
substituted or unsubstituted, branched or unbranched heteroaliphatic;
substituted or
unsubstituted, branched or unbranched acyl; substituted or unsubstituted aryl;
substituted
or unsubstituted heteroaryl; -OR'; -C(=0)RA: -CO?RA; -C(=0)N(RA)2; -CN; -SCN; -
SRA; -SORA; -SO2RA; -NO2; -N3; -N(RA)2; -NHC(=0)RA; -NRAC(=0)N(RA)2; -
0C(=0)0RA; -0C(=0)RA; -0C(=0)N(RA)2; -NRAC(=0)0RA; or -C(RA)3; wherein each
occurrence of RA is independently a hydrogen, a protecting group, an aliphatic
moiety, a
heteroaliphatic moiety, an acyl moiety; an aryl moiety; a heteroaryl moiety;
alkoxy;
aryloxy; alkylthio; arylthio; amino, alkylamino, dialkylamino, heteroaryloxy;
or
heteroarylthio moiety;
R2 is hydrogen; a nitrogen-protecting group; cyclic or acyclic, substituted or
unsubstituted, branched or unbranched aliphatic; cyclic or acyclic,
substituted or
unsubstituted, branched or unbranched heteroaliphatic; substituted or
unsubstituted,
branched or unbranched acyl; substituted or unsubstituted aryl; substituted or
unsubstituted heteroaryl; -C(=0)RB; -C(=0)N(RB)2; -0O2RB; or -C(RB)3; wherein
each
occurrence of RB is independently a hydrogen, an aliphatic moiety, a
heteroaliphatic
moiety, an acyl moiety; an aryl moiety; a heteroaryl moiety; alkoxy; aryloxy;
alkylthio;
arylthio; amino, alkylamino, dialkylamino, heteroaryloxy; or heteroarylthio
moiety;
each occurrence of R3 is independently hydrogen; halogen; cyclic or acyclic,
substituted or unsubstituted, branched or unbranched aliphatic; cyclic or
acyclic,
substituted or unsubstituted, branched or unbranched heteroaliphatic;
substituted or
unsubstituted, branched or unbranched acyl; substituted or unsubstituted aryl:
substituted
CA 02797719 2012-10-26
WO 2011/137320
PCT/US2011/034514
- 6 -
or unsubstituted heteroaryl; -ORc; -C(=0)Rc; -C(=0)N(Rc)2; -0O2Rc; -CN; -SCN; -
SRC; -SORc; -SO2Rc; -NO2; -N3; -N(Rc)2; -NHC(=0)Rc; -NRcC(=0)N(Rc)2; -
0C(=0)0Rc; -0C(=0)Rc; -0C(=0)N(Rc)2; -NRcC(=0)0Rc; or -C(Rc)3; wherein each
occurrence of RC is independently a hydrogen, a protecting group, an aliphatic
moiety, a
heteroaliphatic moiety, an acyl moiety; an aryl moiety; a heteroaryl moiety;
alkoxy;
aryloxy; alkylthio; arylthio; amino, alkylamino, dialkylamino, heteroaryloxy;
or
heteroarylthio moiety; wherein both occurrences of R3 may optionally be taken
together
with the intervening carbon atoms to form an optionally substituted cyclic
moiety or may
be =0, =S, or =NRc;
or a pharmaceutically acceptable salt thereof,
and a pharmaceutically acceptable carrier.
In one embodiment according to this and other aspects of the invention, the
small
molecule inhibitor of I JSP1 according to Formula II is the compound of
Formula V
(947):
0
1 )
N"--0
Formula V (947),
or a pharmaceutically acceptable salt thereof.
In one embodiment the pharmaceutical composition further includes a DNA
cross-linking agent, for example chemotherapeutic agents including alkylating
agents,
cisplatin, and mitomycin C.
In one embodiment the pharmaceutical composition further includes a PARP
inhibitor.
An aspect of the invention is a pharmaceutical composition including (a) a
small
molecule inhibitor of USP1 according to Formula III (933):
0
NH2
Ci
0
Formula III (933),
CA 02797719 2012-10-26
WO 2011/137320
PCT/US2011/034514
- 7 -
or a pharmaceutically acceptable salt thereof, (b) a DNA cross-linking agent,
and (c) a
pharmaceutically acceptable carrier.
An aspect of the invention is a pharmaceutical composition including (a) a
small
molecule inhibitor of USP1 according to Formula III (933):
0
NH2
CI
0
Formula III (933),
or a pharmaceutically acceptable salt thereof, (b) a PARP inhibitor, and (c) a
pharmaceutically acceptable carrier.
In one embodiment, any of foregoing pharmaceutical compositions is formulated
for targeted delivery to a cancer cell.
In another aspect the invention is a method for inhibiting USP1-mediated
deubiqitination of a ubiquitinated substrate. The method includes the step of
contacting
the ubiquitinated substrate with a small molecule inhibitor of USP1 according
to
Formula I:
_____________________________________________ R2
x>
Formula I
as set forth above, or a pharmaceutically acceptable salt thereof, in an
amount effective
to inhibit USP1-mediated deubiqitination of the ubiquitinated substrate.
An aspect of the invention is a method for inhibiting USP1-mediated
deubiqitination of a ubiquitinated substrate. The method includes the step of
contacting
the ubiquitinated substrate with a small molecule inhibitor of USP1 according
to
Formula II:
CA 02797719 2012-10-26
WO 2011/137320
PCT/US2011/034514
-8-
0
(R1)õ,-,
0
R3 R3
Formula IT
as set forth above, or a pharmaceutically acceptable salt thereof, in an
amount effective
to inhibit USP1-mediated deubiqitination of the ubiquitinated substrate.
An aspect of the invention is a method for inhibiting USP1-mediated
deubiqitination of a ubiquitinated substrate. The method includes the step of
contacting
the ubiquitinated substrate with a small molecule inhibitor of USP1 according
to
Formula III (933):
0
NH2
Ci
0
Formula III (933),
or a pharmaceutically acceptable salt thereof, in an amount effective to
inhibit USP1-
mediated deubiqitination of the ubiquitinated substrate.
An aspect of the invention is a method for treating a subject having a cancer.
The method includes the step of administering to a subject having cancer in
need of such
treatment a small molecule inhibitor of USP1 according to Formula 1:
(R1 fI111IIIIIIIIT11II x> __ R2
Formula I
as set forth above, or a pharmaceutically acceptable salt thereof, in an
amount effective
to treat the cancer.
In one embodiment the method further includes the step of administering to the
subject a DNA cross-linking agent.
CA 02797719 2012-10-26
WO 2011/137320
PCT/US2011/034514
- 9 -
In one embodiment the method further includes the step of administering to the
subject a PARP inhibitor.
In one embodiment the method further includes the step or steps of
administering
to the subject a DNA cross-linking agent and a PARP inhibitor.
An aspect of the invention is a method for treating a subject having a cancer.
The
method includes the step of administering to a subject having cancer in need
of such
treatment a small molecule inhibitor of USP1 according to Formula II:
(R1> jNR2
R3 R3
Formula II
as set forth above, or a pharmaceutically acceptable salt thereof, in an
amount effective
to treat the cancer.
In one embodiment the method further includes the step of administering to the
subject a DNA cross-linking agent.
In one embodiment the method further includes the step of administering to the
subject a PARP inhibitor.
In one embodiment the method further includes the step or steps of
administering
to the subject a DNA cross-linking agent and a PARP inhibitor.
An aspect of the invention is a method for treating a subject having a cancer.
The
method includes the step of administering to a subject having cancer in need
of such
treatment a small molecule inhibitor of ubiquitin specific protease 1 (USP1)
according to
Formula III:
0
NH2
CI
Formula III (933),
or a pharmaceutically acceptable salt thereof, in an amount effective to treat
the cancer.
CA 02797719 2012-10-26
WO 2011/137320
PCT/US2011/034514
- 10 -
In one embodiment the method further includes the step of administering to the
subject a DNA cross-linking agent.
In one embodiment the method further includes the step of administering to the
subject a PARP inhibitor.
In one embodiment the method further includes the step or steps of
administering
to the subject a DNA cross-linking agent and a PARP inhibitor.
An aspect of the invention is a method for sensitizing a cancer to a DNA cross-
linking agent. The method includes the step of contacting a cancer with a
small
molecule inhibitor of USP1 according to Formula I:
>
(R1), ____________________ iIIIIiiiIIIIIIiiiiiI: R2
/0
Formula I
as set forth above, or a pharmaceutically acceptable salt thereof, in an
amount effective
to sensitize the cancer to a DNA cross-linking agent.
An aspect of the invention is a method for sensitizing a cancer to a DNA cross-
linking agent. The method includes the step of contacting a cancer with a
small
molecule inhibitor of IJSP1 according to Formula II:
(R1) -
n I
0
R3 R3
Formula IT
as set forth above, or a pharmaceutically acceptable salt thereof, in an
amount effective
to sensitize the cancer to a DNA cross-linking agent.
An aspect of the invention is a method for sensitizing a cancer to a DNA cross-
linking agent. The method includes the step of contacting a cancer with a
small
molecule inhibitor of USP1 according to Formula III (933):
CA 02797719 2012-10-26
WO 2011/137320
PCT/US2011/034514
- 11 -
0
NH2
CI
0
Formula III (933),
or a pharmaceutically acceptable salt thereof, in an amount effective to
sensitize the
cancer to a DNA cross-linking agent.
An aspect of the invention is a method to identify a cancer that is responsive
to
USP1 inhibition. The method includes the steps of contacting cancer cells from
a cancer
with a small molecule inhibitor of USP1 according to Formula I:
(R1)n-! R2
>
x
Formula I
as set forth above, or a pharmaceutically acceptable salt thereof; and
measuring USP1
activity in the cancer cells contacted with the small molecule inhibitor of
USP1, wherein
reduced USP1 activity in the cancer cells contacted with the small molecule
inhibitor of
USP1 relative to control USP1 activity in the cancer cells not contacted with
the small
molecule inhibitor of USP1 identifies the cancer as a cancer that that is
responsive to
ITSP1 inhibition.
An aspect of the invention is a method to identify a cancer that is responsive
to
USP1 inhibition. The method includes the steps of contacting cancer cells from
a cancer
with a small molecule inhibitor of USP1 according to Formula II:
0
R3 R3
Formula II
CA 02797719 2012-10-26
WO 2011/137320
PCT/US2011/034514
- 12 -
as set forth above, or a pharmaceutically acceptable salt thereof; and
measuring USP1
activity in the cancer cells contacted with the small molecule inhibitor of
USP1, wherein
reduced USP1 activity in the cancer cells contacted with the small molecule
inhibitor of
USP1 relative to control USP1 activity in the cancer cells not contacted with
the small
.. molecule inhibitor of USP1 identifies the cancer as a cancer that that is
responsive to
USP1 inhibition.
An aspect of the invention is a method to identify a cancer that is responsive
to
USP1 inhibition. The method includes the steps of contacting cancer cells from
a cancer
with a small molecule inhibitor of USP1 according to Formula III:
0
NH2
CI
0
Formula III (933),
or a pharmaceutically acceptable salt thereof; and measuring USP1 activity in
the cancer
cells contacted with the small molecule inhibitor of USP1, wherein reduced
USP1
activity in the cancer cells contacted with the small molecule inhibitor of
USP1 relative
to control USP1 activity in the cancer cells not contacted with the small
molecule
inhibitor of USP1 identifies the cancer as a cancer that that is responsive to
USP1
inhibition.
An aspect of the invention is a method to identify a cancer that is responsive
to
DNA cross-linking therapy together with USP1 inhibition. The method includes
the
steps of contacting cancer cells from a cancer with a DNA cross-linking agent
and a
small molecule inhibitor of USP1 according to Formula I:
>(R1), x __ R2
0
Formula I
as set forth above, or a pharmaceutically acceptable salt thereof; and
measuring
proliferation of the cancer cells contacted with the DNA cross-linking agent
and the
CA 02797719 2012-10-26
WO 2011/137320
PCT/US2011/034514
- 13 -
small molecule inhibitor of USP1, wherein reduced proliferation of the cancer
cells
contacted with the DNA cross-linking agent and the small molecule inhibitor of
USP1
relative to control proliferation of the cancer cells contacted with the DNA
cross-linking
agent but not the small molecule USP1 inhibitor identifies the cancer as a
cancer that is
responsive to DNA cross-linking therapy together with USP1 inhibition.
An aspect of the invention is a method to identify a cancer that is responsive
to
DNA cross-linking therapy together with USP1 inhibition. The method includes
the
steps of contacting cancer cells from a cancer with a DNA cross-linking agent
and a
small molecule inhibitor of USP1 according to Formula II:
(R1>n-
0
R3 R3
Formula II
as set forth above, or a pharmaceutically acceptable salt thereof; and
measuring
proliferation of the cancer cells contacted with the DNA cross-linking agent
and the
small molecule inhibitor of USP1, wherein reduced proliferation of the cancer
cells
contacted with the DNA cross-linking agent and the small molecule inhibitor of
USP1
relative to control proliferation of the cancer cells contacted with the DNA
cross-linking
agent but not the small molecule USP1 inhibitor identifies the cancer as a
cancer that is
responsive to DNA cross-linking therapy together with USP1 inhibition.
An aspect of the invention is a method to identify a cancer that is responsive
to
DNA cross-linking therapy together with USP1 inhibition. The method includes
the
steps of contacting cancer cells from a cancer with a DNA cross-linking agent
and a
small molecule inhibitor of USP1 according to Formula III:
0
NH2
CI
0
Formula III (933),
81662840
- 14 -
or a pharmaceutically acceptable salt thereof; and measuring proliferation of
the cancer cells
contacted with the DNA cross-linking agent and the small molecule inhibitor of
USP1,
wherein reduced proliferation of the cancer cells contacted with the DNA cross-
linking agent
and the small molecule inhibitor of USP1 relative to control proliferation of
the cancer cells
contacted with the DNA cross-linking agent but not the small molecule USP1
inhibitor
identifies the cancer as a cancer that is responsive to DNA cross-linking
therapy together with
USP1 inhibition.
According to one aspect of the present invention, there is provided a
pharmaceutical
composition for use in sensitizing a cancer to a DNA cross-linking agent
comprising a small
molecule inhibitor of ubiquitin specific protease 1 (USP1) according to
Formula I:
0
) _______________________________________________ R2
X
Formula I
wherein X is 0, S. or NR3; R2 is aryl or heteroaryl; R3 is independently a
hydrogen, a
protecting group, an aliphatic moiety, a heteroaliphatic moiety, an acyl
moiety; an aryl
moiety; a heteroaryl moiety; alkoxy; aryloxy; alkylthio; arylthio; amino,
alkylamino,
dialkylamino, heteroaryloxy; or heteroarylthio moiety; or a pharmaceutically
acceptable salt
thereof, and a pharmaceutically acceptable carrier.
According to another aspect of the present invention, there is provided an ex
vivo or
in vitro method to identify a cancer that is responsive to ubiquitin specific
protease I (USP1)
inhibition, comprising contacting cancer cells from a cancer with a small
molecule inhibitor of
USP1 according to Formula I:
CA 2797719 2019-03-25
81662840
- 14a -
o
) _______________________________________________ R2
X
Formula I
wherein X is 0, S, or NR3; R2 is aryl or heteroaryl; R3 is independently a
hydrogen, a
protecting group, an aliphatic moiety, a heteroaliphatic moiety, an acyl
moiety; an aryl
moiety; a heteroaryl moiety; alkoxy; aryloxy; alkylthio; arylthio; amino,
alkylamino,
dialkylamino, heteroaryloxy; or heteroarylthio moiety; or a pharmaceutically
acceptable salt
thereof; and measuring USP1 activity in the cancer cells contacted with the
small molecule
inhibitor of USP1, wherein reduced USP1 activity in the cancer cells contacted
with the small
molecule inhibitor of USP1 relative to control USP1 activity in the cancer
cells not contacted
with the small molecule inhibitor of USP1 identifies the cancer as a cancer
that that is
responsive to USP1 inhibition.
According to still another aspect of the present invention, there is provided
an ex vivo
or in vitro method to identify a cancer that is responsive to DNA cross-
linking therapy
together with ubiquitin specific protease 1 (USP1) inhibition, comprising
contacting cancer
cells from a cancer with a DNA cross-linking agent and a small molecule
inhibitor of USP1
according to Formula I:
) _______________________________________________ R2
X
Formula I
CA 2797719 2019-03-25
81662840
- 14b -
wherein X is 0, S. or NR3; R2 is aryl or heteroaryl; R3 is independently a
hydrogen, a
protecting group, an aliphatic moiety, a heteroaliphatic moiety, an acyl
moiety; an aryl
moiety; a heteroaryl moiety; alkoxy; aryloxy; alkylthio; arylthio; amino,
alkylamino,
dialkylamino, heteroaryloxy; or heteroarylthio moiety; or a pharmaceutically
acceptable salt
thereof; and measuring proliferation of the cancer cells contacted with the
DNA cross-linking
agent and the small molecule inhibitor of USP1, wherein reduced proliferation
of the cancer
cells contacted with the DNA cross-linking agent and the small molecule
inhibitor of USP1
relative to control proliferation of the cancer cells contacted with the DNA
cross-linking agent
but not the small molecule USP1 inhibitor identifies the cancer as a cancer
that is responsive
to DNA cross-linking therapy together with USP1 inhibition.
BRIEF DESCRIPTION OF THE DRAWINGS
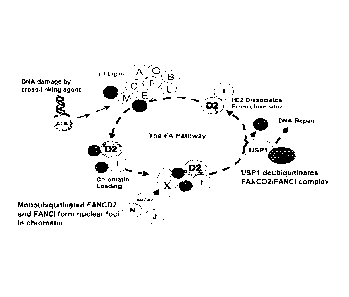
FIG. 1 is a schematic drawing depicting the Fanconi anemia (FA) DNA repair
pathway. Ub, ubiquitin: D2, FANCD2; 1, FANCI.
FIG. 2 is a composite figure depicting small molecule inhibition of USP1
activity
in vitro. Ub-Vs, ubiquitin vinyl sulfone; Ub-Ald, ubiquitin aldehyde:
LDN_0082527 and
C527, the compound of Formula IV (527); C933, the compound of Formula III
(933); C009,
the compound of Formula VI (009); C947, the compound of Formula V (947).
FIG. 3 is a composite figure depicting dose-dependent blockade of USP1
activity
(lanes 1-5) and dose-dependent accumulation of ubiquitinated FANCD2 and FANCI
in
response to the compound of Formula IV (527). C527, the compound of Formula IV
(527);
Ub-Vs, ubiquitin vinyl sulfone; Ub-Ald, ubiquitin aldehyde.
FIG. 4 is a composite figure depicting sensitization of HeLa cells to the DNA
cross-
linking agent mitomycin C by the compound of Formula IV (527). C527, the
compound of
Formula IV (527); MMC, mitomycin C.
Fig. 5 is a composite figure depicting time-course and dose-dependent C527
inhibition of USP1/UAF1 activity. Fig. 5A demonstrates that C527 inhibits USP1
activity in a time-dependent manner. Purified USP1/UAF1 complex was incubated
with
CA 2797719 2019-03-25
81662840
- 14c -
1 M C527 or DMSO for indicated time, followed by the addition of Ub-AMC at
0.5 M final concentration. The fluorescence at 535 nM was measured to
indicate the
enzymatic activity of USP I. Fig. 5B shows that C527 inhibits USP1 activity in
a dose-
dependent manner C527 at indicated concentrations was incubated with USP1/UAF1
for
3 h and the reaction was as described in Fig. 5A. Fig. 5C shows the IC50 of
C527 against
USP/UAF1 complex.
CA 2797719 2019-03-25
CA 02797719 2012-10-26
WO 2011/137320
PCT/US2011/034514
- 15 -
Fig. 6 is a composite figure showing that C527 is a pan-deubiqutinating enzyme
inhibitor in vivo. Ubiquitin-AMC assay was performed using purified (Fig. 6A)
USP12/UAF1/WDR20 (Fig. 6B) USP5 and (Fig. 6C) UCH-Li. Enzymes were
incubated with DMSO or C527 at indicated concentrations for 3 h, followed by
the
addition of Ub-AMC at 0.5 juM final concentration. The fluorescence at 535 nM
was
measured to indicate the enzymatic activity. Fig. 6D is a summary showing the
IC50 of
C527 against the indicated enzymes.
Fig. 7 is a composite figure depicting preferential inhibition of the activity
of
native USP1/UAF1 by C527. IIeLa cell extracts were incubated with ubiquitin-
aldehyde
(Ub-Ald, 5 M) or C527 at indicated concentrations for 4 h, followed by the
addition of
0.5 uM Ub-Vs for 40 mm. The reaction was split into two aliquots and subjected
to
immunoblotting for HA or USP1, respectively.
Fig. 8 is a composite figure showing that C527 inhibits homologous
recombination repair. Fig. 8A shows that C527 inhibits DR-GFP reporter for
homologous recombination repair activity. U20S-DRGFP cells were transfected
with I-
SCE-1 and then exposed to C527 at the indicated concentration for 24 h. Cells
were then
subjected to flow cytometry analysis. The percentage of GFP positive cells was
normalized by solvent vehicle treated group. Fig. 8B shows that C527 has
minimal
inhibition of NHEJ activity. HeLa cells, integrated with the NHEJ reporter,
were treated
with C527 and analyzed as described in (Fig. 8A). Fig. 8C shows that C527
inhibits
camptothecin-induced RAD51 foci formation. HeLa cells were pre-treated with
DMSO
or C527 at the indicated concentration and then exposed to camptothecin (CPT)
for 1 hr.
RAD51 foci were detected using immunofluorescence.
Fig. 9 is a composite figure showing that C527 causes DNA damage and inhibits
the proliferation of tumor cells. Fig. 9A shows that C527 causes DNA damage in
tumor
cells. HeLa cells were treated with C527 at the indicated concentrations for
24 hrs, and
cells were subjected to immunoblotting. Mitomycin C (MMC, 1 uM) was used as a
positive control. Fig. 9B shows that C527 inhibits the proliferation of tumor
cells. HeLa
cells were seeded in a 6-well plate and treated with C527 at the indicated
concentrations.
Cell proliferation was evaluated using a clonogenic assay.
Figure 10 is a composite figure showing that C527 sensitizes tumor cells to
DNA
damaging agents. HeLa cells were plated in 96-well plates and treated with 1
uM C527
CA 02797719 2012-10-26
WO 2011/137320
PCT/US2011/034514
- 16 -
for 24 h, followed by treatment with mitomycin C (MMC, 0.25 iM), camptothcin
(CPT,
0.1 uM), or etoposide for 4 more days. Cell viability was assessed using the
MTT assay.
DETAILED DESCRIPTION OF THE INVENTION
The invention is based, at least in part, on the discovery by the instant
inventors
of certain small molecule inhibitors of I JSP1 activity. These small molecule
inhibitors of
USP1 activity inhibit the deubiquitinating activity of USP1, both alone and in
combination with UAE1, both in vitro and in vivo. Furthermore, it has now
unexpectedly
been discovered by the inventors that inhibition of USPI activity can render
cancer cells,
including cancer cells otherwise resistant to treatment with DNA cross-linking
agents,
susceptible to treatment with DNA cross-linking agents.
Ubiquitin is a 76 amino acid protein that can be covalently attached to other
proteins, targeting them for degradation or regulating their function. The
amino acid
sequence of the human form of ubiquitin is given by SEQ ID NO:1:
1 MQIFVKILTG KTITLEVEPS CTIENVKAKT QDKEGIPPDQ QRLIFAGKQL ECGRTLSDYN
61 IQKESTLHLV IRLRGG
Ubiquitin is covalently attached to other proteins by a ubiquitin transferase
enzyme, and
it is released from a ubiquitinated protein by a deubiquitinating enzyme.
Ubiquitination and deubiquitination regulate a number of essential biological
processes such as gene transcription, DNA replication, and DNA repair. Pickart
et al.
(2004) Biochim Biophys Acta 1695:55-72. Ubiquitin modifications can be divided
into
three principal types. First, monoubiquitination may alter the activity of the
substrate, as
has been described for the FANCD2 protein of the Fanconi anemia pathway and
the
PCNA protein involved in translesion synthesis. Kennedy et al. (2005) Genes
Dev
19:2925-40; Hoegh et al. (2002) Nature 419:135-41. Second, polyubiquitination
through
K48 linkage typically targets the protein substrate for degradation by the
proteasome.
Third, polyubiquitination through K63 linkage can alter the activity of the
protein
substrate by modifying its protein-protein interaction properties.
There are at least 95 putative deubiquitinating enzymes in humans. Nijman et
al.
(2005) Cell 123:773-86. This family of deubiquitinating enzymes is divided
into five
subfamilies, including the ubiquitin specific protease (USP) subfamily (58
members), the
CA 02797719 2012-10-26
WO 2011/137320
PCT/US2011/034514
- 17 -
otubain protease (OTU) subfamily and the JABl/MPN/Mov34 metalloprotease (JAMM)
subfamily (14 members each), the Josephine domain protease (MID) subfamily (5
members), and the ubiquitin C-terminal hydrolase (UCH) subfamily (4 members).
The
exact biological function for many of these enzymes is currently unknown.
However, for
those enzymes whose function has been uncovered, it has become apparent that
regulation of their activities is essential for integrity of the pathways they
regulate.
Ubiquitin-specific protease 1 (USP1) is a cysteine protease with
deubiquitinase
activity. USP1 cleaves ubiquitin from monoubiquitinated and polyubiquitinated
protein
substrates, including FANCD2 and PCNA. Huang et al. (2006) Nature Cell Biol.
8(4):339-47. The amino acid sequence for the human form of USP1 is given by
SEQ ID
NO:2:
1 MPGVIPSESN GLSRGSPSKIK NRLSLKFFQK KETKRALDFT DSQENEEKAS EYRASEIDQV
61 VPAAQSSPIN CEKRENLLPF VGLNNLGNTC YLNSILQVLY FCPGFKSGVK HLFNIISRKK
121 EALKDEANQK DKGNCKEDSL ASYELICSLQ SLIISVEQLQ ASYLLNPEKY TDELATQPRR
181 LLNTLRELNP MYEGYLQHDA QEVLQCILGN IQETCQLLKK EEVKNVAELP TKVEEIPHPK
241 EEMNGINSIE MDSMRHSEDF KEKLPKGNGK RKSDTEFGNM KKKVKLSKEH QSLEENQRQT
301 RSKRKATSDT LESPPKIIPK YISENESPRP SQKKSRVKIN WLKSATKQPS ILSKFCSLGK
361 ITINQGVKGQ SKENECDPEE DLGKCESDNT TNGCGLESPG NTVTPVNVNE VKPINKGEEQ
421 IGFELVEKLF QGQLVLRTRC LECESLTERR EDFUISVPV QEDELSKVEE SSEISPEPKT
481 EMKTLRWAIS QFASVERIVG EDKYFCENCH HYTEAERSLL FDKMPEVITI HLKCFAASGL
541 EFDCYGGGLS KINTPLLTPL KLSLEEWSTK PTNDSYGLFA VVMHSGITIS SGHYTA3VKV
601 TDLNSLELDK GNFVVDQMCE IGKPEPLNEE EARGVVENYN DEEVSIRVGG NTQPSKVLNK
661 KNVEAIGLLG GQKSKADYEL YNKASNPDKV ASTAFAENRN SETSDTTGTH ESDRNKESSD
721 QTGINISGFE NKISYVVQSL KEYEGKWLLF DDSEVKVTEE KDFLNSLSPS TSPTSTPYLL
781 FYKKL
Fanconi anemia (FA) is a rare chromosome instability syndrome characterized by
aplastic anemia in childhood, susceptibility to leukemia and cancer, and
hypersensitivity
of FA cells to interstrand DNA cross-linking agents such as cisplatin and
melphalan.
There are thirteen Fanconi anemia genes, and their corresponding proteins fall
into
several classes of enzymes and structural proteins, including a ubiquitin
ligase,
monoubiquitinated proteins, a helicase, and one with both helicase and
nuclease motifs.
Since FA patients share a characteristic clinical and cellular phenotype, it
has been
assumed that the thirteen FA proteins cooperate in a common DNA repair
pathway.
CA 02797719 2012-10-26
WO 2011/137320
PCT/US2011/034514
- 18 -
Indeed, the FA proteins work in concert to control the monoubiquitinated state
of the
FANCD2 and FANCI proteins (see FIG. 1) and the downstream functions of the
pathway.
As shown in FIG. 1, after DNA damage, the Ataxia Telangiectasia and Rad3-
.. related kinase (AIR) activates FA core complex (FANCA/B/C/E/14/Ci/L/M).
r[he FA
core complex then functions as an E3 ubiquitin ligase and monoubiquitinates
FANCD2
and FANCI. The monoubiquitinated FANCD2/FANCI complex is then targeted to
chromatin where it forms a complex with additional FA proteins and other DNA
repair
proteins. A protein complex of USP1 and UAF1 then deubiquitinates FANCD2/FANCI
.. complex, allowing their release from chromatin. USP1 is also required for
localization
of FANCD2/FANCI into DNA repair foci.
Although mutations in the thirteen FA genes account for most, if not all,
cases of
FA, additional genes related to the FA-BRCA pathway may also be important. For
example, disruption of USP1 in mice yields the FA phenotype, and knockdown of
either
USP1 or UAF1 also disrupts the FA-BRCA pathway and causes hypersensitivity to
DNA
cross-linking agents.
USP1 associated factor 1 (UAF1) is a WD repeat endosomal protein that has been
described to form a heterodimeric complex with USP1 and thereby to enhance the
deubiquitinase activity of USP1. This protein has been previously shown to
play a role
in the downregulation of the T lymphocyte receptor. Park et al. (2002)
Immunity 17:221-
33. The amino acid sequence for human UAF1 is given by SEQ ID NO:3:
1 MAAHHRQNTA GRRKVQVSYV IRDEVEKYNR NGVNALQLDP ALNRLFTAGR DSIIRIWSVN
61 QHKQDPYIAS MEHHTDWVND IVLCCNGKTL ISASSDTTVK VWNAHKGFCM STLRTHKDYV
121 KADAYAKDKE LVASAGLDRO IFLWDVNTLT ALTASNNTVT TSSLSGNKDS IYSLAMNOLG
181 TIIVSGSTEK VLRVWDPRTC AKLMKLKGHT DNVKALLLNR DGTQCLSGSS DGTIRLWSLG
241 QQRCIATYRV HDEGVWALQV NDAFTHVYSG GRDRKIYCTD LRNPDIRVLI CEEKAPVLKM
301 ELDRSADPPP AIWVATTKST VNKWTLKGIH NFRASGDYDN DCTNPITPLC TQPDQVIKGG
361 ASIIQCHILN DKRHILTNDT NNNVAYWDVL KACEVEDLGK VDPEDEIKHR FKMVYVPNWF
421 SVDLKTGMLT ITLDESDCFA AWVSAKDAGF SSPDGSDPKL NIGGLLLQAL LEYWPRTHVN
481 PMDEEENEVN HVNGEQENRV QKGNGYFQVP PHTPVIFGEA GGRILFALLC RDSGGETESM
541 LLNETVPQWV IDITVDKNMP KFNKIPFYLQ PHASSGAKTL KKDRLSASDM LQVRKVMEHV
601 YEKIINLDNE SQTTSSSNNE KPGEQEKEED IAVLAEEKIE LLCQDQVLDP NMDLRTVKHF
661 IWKSGGDLTL HYRQKST
CA 02797719 2012-10-26
WO 2011/137320
PCT/US2011/034514
- 19 -
Proliferating cell nuclear antigen (PCNA) is a DNA replication sliding clamp
protein that can form part of a DNA polymerase complex. Upon
monoubiquitination,
PCNA can interact with any of several different DNA polymerases to form a
complex
which carries out either DNA replication or DNA repair, particularly
translesion DNA
synthesis. Huang & D'Andrea (2006) Mol Cell Biol. 7:323-34. As used herein,
"translesion DNA synthesis" is a form of DNA repair in which specialized,
damage-
tolerant DNA polymerases (such as Pol eta) bypass DNA lesions that would
normally
stall replication of a DNA strand, allowing for later repair of the bypassed
lesion. These
DNA lesions can occur upon cellular exposure to ionizing radiation,
ultraviolet light, or
DNA-disrupting chemical agents. The amino acid sequence of the human form of
PCNA is given by SEQ ID NO:4:
1 MFEARLVQGS ILKKVLEALK DLINEACWDI SSSGVNLQSM DSSHVALVQL TLRSEGFDTY
El RCDRNLAMGV NLTSMSKILK CAGNEDIITL RAEDNADTLA LVFEAPNQEK VSDYEMKLMD
121 LDVEQIGIPE QEYSCVVKMP SGEFARICRD LSHIGDAVVI SCAKDGVKFS ASGELGNGNI
181 KLSQTSNVDK EEEAVTIEMN EPVQLTFALR YLNFFTKATP LSSTVTLSMS ADVPLVVEYK
241 IADMGHLKYY LAPKIEDEEG S
Using an initial in vitro screening assay described in the Examples, certain
small
molecule inhibitors of USP1 were identified from a library of approximately
150,000
compounds. Small molecule inhibitors of USPI so identified include the
compounds of
Formula III (933), Formula IV (527), Formula V (947), and Formula VI (009).
0
NH2
CI
0
Formula III (933)
0
N\
0
0
QF
CA 02797719 2012-10-26
WO 2011/137320
PCT/US2011/034514
- 20 -
Formula IV (527)
0
0
Formula V (947)
0
401 OMe
0 OMe
Formula VI (009)
The compounds of Formula IV and V are disclosed herein as representative
examples of individual classes of compounds according to Formula I (and
related
Formula IA) and Formula II, respectively (see below).
As used herein, an "inhibitor of USP1" refers to an agent that decreases
deubiquitinase activity of USP1. As used herein, the term "deubiquitinase
activity of
USP1" and, equivalently, "USP1 activity", refers to the action of USP1 to
remove
ubiquitin from a ubiquitinated substrate. Deubiquitinase activity can be
measured in
vitro or in vivo, and it can be measured directly or indirectly.
Deubiquitinase activity can
be measured by a number of assays including assays that measure the
deubiquitination of
FANCD2-Ub or PCNA-Ub, natural targets of USP1 deubiquitination. Alternatively,
deubiquitinase activity of USP I can be determined by measuring the
deubiquitination of
a test molecule such as ubiquitin-7-amido-4-methylcoumarin (AMC-Ub), which
produces a fluorescently detectable signal upon the cleavage of ubiquitin. A
USP1
polypeptide is deemed to have deubiquitinase activity where the level of
deubiquitinated
FANCD2, PCNA, or AMC in the presence of USP1 is at least 10% greater than the
level
of deubiquitinated FANCD2, PCNA, or AMC from a control sample (e.g., sample
from
the same tissue, or a separate aliquot of a cellular sample) in the absence of
USP1.
Alternatively a USP1 polypeptide is deemed to have deubiquitinase activity
where the
level of ubiquitinated FANCD2-Ub, PCNA-Ub, or AMC-Ub is decreased by at least
10% in the presence of USP1 relative to a control sample in the absence of
USP1. In one
CA 02797719 2012-10-26
WO 2011/137320
PCT/US2011/034514
- 21 -
embodiment an increase in USP1 activity in response to an agent as used herein
refers to
any detectable increase in the production of a deubiquitinated substrate
(e.g.,
deubiquitinated AMC) in the presence of the agent relative to in the absence
of the agent,
such as a 0.5% increase, a 1% increase or decrease, 2%, 3-5%, 5-10%, 10-20%,
20-40%,
40-80%, 90%, or 100% or more increase in the production of a deubiquitinated
substrate.
In one embodiment a decrease in USP1 activity in response to an agent as used
herein
refers to any detectable decrease in the production of a deubiquitinated
substrate (e.g.,
deubiquitinated AMC) in the presence of the agent relative to in the absence
of the agent,
such as a 0.5% decrease, a 1% decrease, 2%, 3-5%, 5-10%, 10-20%, 20-40%, 40-
80%,
90%, or 100% decrease in the production of a deubiquitinated substrate.
As used herein, a "small molecule" refers to an organic molecule of molecular
weight of less than 1500 Dalions. Small molecules of the invention exclude
short
interfering RNA or short interfering hairpin RNA.
Short interfering RNA ("siRNA") is a double-stranded RNA molecule that is
generally 17 to 25 base pairs in length, one of whose strands contains a
sequence
(antisense sequence) that is complementary to a segment of a target messenger
RNA.
siRNA associates with an RNA-induced silencing complex in the cell, which then
binds
to a complementary region of a target messenger RNA and inactivates it.
Short interfering hairpin RNA ("shRNA") is a ribonucleic acid containing sense
and antisense sequences from a target gene connected by a loop; it can be
expressed in
mammalian cells from a vector. Transcribed shRNA is transported from the
nucleus into
the cytoplasm, where it is processed, where it can decrease the expression of
a gene with
complementary sequences by RNA interference (RNAi).
As used herein, RNA interference ("RNAi") refers to a selective intracellular
degradation of RNA by means of an RNA-induced silencing complex (RISC). RNAi
occurs in cells naturally to remove foreign RNAs (e.g.. viral RNAs). RNAi
proceeds via
fragments cleaved from free double-stranded RNA molecules (such as viral RNA)
which
direct the degradative mechanism to other similar RNA sequences. Introduction
of the
double-stranded RNA into a cell triggers the degradation of the double-
stranded RNA
into shorter siRNA strands. These siRNAs then associate with RNA-induced
silencing
complexes, leading to the unwinding of the siRNAs into single strands, which
then
associate with complementary regions of messenger RNA and prevent the
expression of
CA 02797719 2012-10-26
WO 2011/137320
PCT/US2011/034514
_ 77 _
the corresponding proteins. The use of synthetic siRNA to "direct RNA
interference
(RNAi) against expression" of a target gene refers to the reduction of the
expression of
the target gene by entry of the synthetic siRNA into the natural RNAi
mechanism at the
same point as natural siRNA created from double-stranded RNA, e.g., viral RNA,
would
enter that mechanism. That is, synthetic siRNA associate with the RISC,
unwind, and
then associate with complementary mRNA regions of target transcripts, which
are then
degraded.
The compounds of Formula III (933) and Formula V (947) are known. For
example, a method for synthesizing the compound of Formula V (947) is
disclosed in
Sondhi et al. (2008) Letts Org Chem 5:51-4, but this reference does not
disclose a use for
this compound.
In contrast, the compounds of Formula IV (527) and Formula VI (009) appear to
be novel compounds.
The invention in one aspect provides the compound of Formula IV (527).
The invention in one aspect provides the compound of Formula VI (009).
The invention provides a pharmaceutical composition that includes (a) a small
molecule inhibitor of USP1 selected from the group consisting of a compound of
Formula I, a compound of Formula IA, a compound of Formula II, the compound of
Formula IV (527), the compound of Formula V (947), and a combination thereof,
and (b)
a pharmaceutically acceptable carrier. In one embodiment the pharmaceutical
composition includes the compound of Formula IV (527) and a pharmaceutically
acceptable carrier. In one embodiment the pharmaceutical composition includes
the
compound of Formula V (947) and a pharmaceutically acceptable carrier.
As used herein, a "pharmaceutically acceptable carrier" refers to one or more
compatible solid or liquid filler, diluents or encapsulating substances which
are suitable
for administration to a human or other vertebrate animal. The term "carrier"
denotes an
organic or inorganic ingredient, natural or synthetic, with which the active
ingredient is
combined to facilitate the application. The components of the pharmaceutical
compositions also are capable of being commingled with the compounds of the
present
invention, and with each other, in a manner such that there is no interaction
which would
substantially impair the desired pharmaceutical efficacy.
CA 02797719 2012-10-26
WO 2011/137320
PCT/US2011/034514
- 23 -
A pharmaceutical composition of the invention can be prepared by placing an
effective amount of at least one active agent of the invention, such as a
compound of any
one of Formulas I-TV, in a pharmaceutically acceptable carrier.
The invention also provides a pharmaceutical composition that includes (a) a
small molecule inhibitor of USP1 selected from the group consisting of a
compound of
Formula I, a compound of Formula IA, a compound of Formula II, the compound of
Formula III (933), the compound of Formula IV (527), the compound of Formula V
(947), and any combination thereof; (b) a DNA cross-linking agent; and (c) a
pharmaceutically acceptable carrier. In one embodiment the small molecule
inhibitor of
USP1 is the compound of Formula III (933). In one embodiment the small
molecule
inhibitor of USP1 is the compound of Formula IV (527). In one embodiment the
small
molecule inhibitor of USP1 is the compound of Formula V (947).
As used herein, a "DNA cross-linking agent" refers to a chemical compound or
radiation that induces DNA cross-linking in a cell when applied to the cell.
In one
embodiment a DNA cross-linking agent is a chemical compound. Such compounds
include certain chemotherapeutic agents, such as cisplatin, carboplatin,
oxaliplatin,
alkylating agents, mitomycin C, as well as certain carcinogenic chemicals,
such as
diepoxybutane. Cisplatin (cis-diamminedichloroplatinum(II)) and related
compounds
carboplatin and oxaliplatin forms DNA cross-links as monoadduct, interstrand
cross-link,
intrastrand cross-link or DNA protein cross-link. Alkylating agents include
cyclophosphamide, chlorambucil, melphalan, ifosfainide, uramustine, and
bendamus tine.
DNA cross-linking radiation includes ionizing radiation and ultraviolet (UV)
radiation. Ionizing radiation consists of subatomic particles or
electromagnetic waves
that are energetic enough to detach electrons from atoms or molecules,
ionizing them.
The occurrence of ionization depends on the energy of the impinging individual
particles
or waves. Generally, particles or photons with energies above a few electron
volts (eV)
are ionizing. Examples of ionizing particles are energetic alpha particles,
beta particles,
and neutrons. The ability of an electromagnetic wave (photons) to ionize an
atom or
molecule depends on its frequency. Radiation on the short-wavelength end of
the
electromagnetic spectrum, such as high frequency ultraviolet, x-rays, and
gamma rays, is
ionizing. Ionizing radiation comes from radioactive materials, x-ray tubes,
and particle
accelerators.
CA 02797719 2012-10-26
WO 2011/137320
PCT/US2011/034514
- 94 -
The manner of exposure to DNA cross-linking agents can be deliberate or
accidental. Examples of deliberate exposure include therapeutic chemotherapy
and
radiotherapy. Examples of accidental exposure include occupational exposure to
chemicals or ionizing radiation, frequent air travel, and nuclear accidents
and nuclear
warfare.
In one embodiment the pharmaceutical composition includes a DNA cross-
linking agent selected from the group consisting of cisplatin, carboplatin,
oxaliplatin, an
alkylating agent, and mitomycin C. Any combination of such DNA cross-linking
agents
is also embraced by the invention.
The invention provides a pharmaceutical composition including (a) a small
molecule inhibitor of USP1 selected from the group consisting of a compound of
Formula I, a compound of Formula IA, a compound of Formula II, the compound of
Formula III (933), the compound of Formula IV (527), the compound of Formula V
(947), and any combination thereof; (b) a poly (adenosine diphosphate (ADP)-
ribose)
polymerase (PARP) inhibitor; and (c) a pharmaceutically acceptable carrier. In
one
embodiment the small molecule inhibitor of USP1 is the compound of Formula III
(933).
In one embodiment the small molecule inhibitor of USP1 is the compound of
Formula
IV (527). In one embodiment the small molecule inhibitor of USP1 is the
compound of
Formula V (947).
Poly(ADP ribose) polymerase (PARP) is a key signaling enzyme involved in
triggering repair of single-strand DNA damage. PARP actually includes a family
of at
least 17 members, including PARP-1 (also known as PARP1 and PARP I) and PARP-2
(also known as PARP2 and PARP II). One important function of PARP-1 is
assisting in
the repair of single-strand DNA nicks. It binds sites with single strand
breaks through its
N-terminal zinc fingers and recruits XRCC1, DNA ligase III, DNA polymerase
beta and
a kinase to the nick, a process called base excision repair (BER). PARP-2 has
been
shown to oligomerize with PARP1 and therefore is also implicated in BER. The
oligomerization has also been shown to stimulate PARP catalytic activity. PARP-
1 is
also known for its role in transcription through remodeling of chromatin by
PARylating
histones and relaxing chromatin structure, thus allowing transcription complex
to access
genes. Several forms of cancer are more dependent on PARP than regular cells.
making
PARP an attractive target for chemotherapeutic cancer therapy.
CA 02797719 2012-10-26
WO 2011/137320
PCT/US2011/034514
- 25 -
Since PARP acts to repair DNA nicks by BER, whereas the FA-BRCA pathway
acts to repair DNA defects by a different mechanism, homologous recombination
(HR),
it is believed that the combined inhibition of the FA-BRCA pathway and of PARP
may
be significantly more potent than inhibition of either one alone.
As used herein, a "PARP inhibitor" refers to a pharmacological agent that
reduces DNA repair by PARP. PARP inhibition has been demonstrated to
selectively
kill tumor cells lacking components of the homologous recombination (HR) DNA
repair
pathway while sparing normal cells. Known defects in HR repair include the
well-
characterized hereditary BRCA1 and BRCA2 mutations in breast and ovarian
cancer, as
well as nonhereditary BRCA mutations.
Several PARP inhibitors are known in the art and are currently under
development, including BSI 201 (BiPar Sciences; 4-iodo-3-nitrobenzamide),
olaparib
(AZD-2281; Astra Zeneca) (4-[(3-[(4-cyclopropylcarbonyl)piperazin-4-
ylicarbonyl) -4-
fluorophenyllmethyl(2H)phthalazin-1-one), AB T-888 (veliparib) (2-[(R)-2-
/5 methylpyrrolidin-2-y1]-1H-benzimidazole-4-carboxamide), CEP-9722
(Cephalon), MK-
4827, KU-0059436 (AZD-2281), LT-673, and 3-aminobenzamide.
In one embodiment a PARP inhibitor is a PARP-1 inhibitor.
In one embodiment the pharmaceutical composition including (a) a small
molecule inhibitor of USP1, (b) a PARP inhibitor, and (c) a pharmaceutically
acceptable
carrier, further includes a DNA cross-linking agent.
It should also be understood that, in addition to compounds of Formula III,
Formula IV, and Formula V, and pharmaceutical compositions including said
compounds, the invention also embraces structurally related active compounds
and
pharmaceutical compositions including said structurally related active
compounds. Such
structurally related active compounds are believed to include compounds
according to
Formula I, Formula IA, and Formula II, below.
Definitions of specific functional groups and chemical terms are described in
more detail below. For purposes of this invention, the chemical elements are
identified
in accordance with the Periodic Table of the Elements, CAS version, Handbook
of
Chemistry and Physics, Tith Ed., inside cover, and specific functional groups
are
generally defined as described therein. Additionally, general principles of
organic
chemistry, as well as specific functional moieties and reactivity, are
described in Organic
81662840
- 96 -
Chemisiry, Thomas Sorrell, University Science Books, Sausalito: 1999.
Certain compounds of the present invention may exist in particular geometric
or
stereoisomeric forms. The present invention contemplates all such compounds,
including cis- and trans-isomers, 1?- and S-enantiomers, diastereomers, (D)-
isomers, (L)-
isomers, the racemic mixtures thereof, and other mixtures thereof, as falling
within the
scope of the invention. Additional asymmetric carbon atoms may be present in a
substituent such as an alkyl group. All such isomers, as well as mixtures
thereof, are
intended to be included in this invention.
Isomeric mixtures containing any of a variety of isomer ratios may be utilized
in
accordance with the present invention. For example, where only two isomers are
combined, mixtures containing 50:50, 60:40, 70:30, 80:20, 90:10, 95:5, 96:4,
97:3, 98:2,
99:1, or 100:0 isomer ratios are all contemplated by the present invention.
Those of
ordinary skill in the art will readily appreciate that analogous ratios are
contemplated for
more complex isomer mixtures.
If, for instance, a particular enantiomer of a compound of the present
invention is
desired, it may be prepared by asymmetric synthesis, or by derivation with a
chiral
auxiliary, where the resulting diastereomeric mixture is separated and the
auxiliary group
cleaved to provide the pure desired enantiomers. Alternatively, where the
molecule
contains a basic functional group, such as amino, or an acidic functional
group, such as
carboxyl, diastereomeric salts are formed with an appropriate optically-active
acid or
base, followed by resolution of the diastereomers thus formed by fractional
crystallization or chromatographic means well known in the art, and subsequent
recovery
of the pure enantiomers.
One of ordinary skill in the art will appreciate that the synthetic methods,
as
described herein, utilize a variety of protecting groups. By the term
"protecting group,"
as used herein, it is meant that a particular functional moiety, e.g., 0, 5,
or N, is
temporarily blocked so that a reaction can be carried out selectively at
another reactive
site in a multifunctional compound. In certain embodiments, a protecting group
reacts
selectively in good yield to give a protected substrate that is stable to the
projected
reactions; the protecting group should be selectively removable in good yield
by readily
available, preferably non-toxic reagents that do not attack the other
functional groups;
CA 2797719 2017-10-10
CA 02797719 2012-10-26
WO 2011/137320
PCT/US2011/034514
_ 97 _
the protecting group forms an easily separable derivative (more preferably
without the
generation of new stereogenic centers); and the protecting group has a minimum
of
additional functionality to avoid further sites of reaction. As detailed
herein, oxygen,
sulfur, nitrogen, and carbon protecting groups may be utilized. Hydroxyl
protecting
groups include methyl, methoxylmethyl (MOM), methylthiomethyl (M'I'M), t-
butylthiomethyl, (phenyldimethylsilyl)methoxymethyl (SMOM), benzyloxymethyl
(BOM), p-methoxybenzyloxymethyl (PMBM), (4-methoxyphenoxy)methyl (p-AOM),
guaiacolmethyl (GUM), t-butoxymethyl, 4-pentenyloxymethyl (POM), siloxymethyl,
2-
methoxyethoxymethyl (MEM), 2,2,2-trichloroethoxymethyl, bis(2-
chloroethoxy)methyl,
2-(trimethylsilyl)ethoxymethyl (SEMOR), tetrahydropyranyl (THP), 3-
bromotetrahydropyranyl, tetrahydrothiopyranyl, 1-methoxycyclohexyl, 4-
methoxytetrahydropyranyl (MTHP), 4-methoxytetrahydrothiopyranyl, 4-
methoxytetrahydrothiopyranyl S,S-dioxide, 1-R2-chloro-4-methyl)pheny11-4-
methoxypiperidin-4-y1 (CTMP), 1,4-dioxan-2-yl, tetrahydrofuranyl,
tetrahydrothiofuranyl, 2,3,3a,4,5,6,7,7a-octahydro-7,8,8-trimethy1-4,7-
methanobenzofuran-2-yl, 1-ethoxyethyl, 1-(2-chloroethoxy)ethyl, 1-methyl-l-
methoxyethyl, 1-methyl-l-benzyloxyethyl, 1-methy1-1-benzyloxy-2-fluoroethyl,
2,2,2-
trichloroethyl, 2-trimethylsilylethyl, 2-(phenylselenyeethyl, t-butyl, allyl,
p-
chlorophenyl, p-methoxyphenyl, 2,4-dinitrophenyl, benzyl, p-methoxybenzyl, 3,4-
dimethoxybenzyl, o-nitrobenzyl, p-nitrobenzyl, p-halobenzyl, 2,6-
dichlorobenzyl, p-
cyanobenzyl, p-phenylbenzyl, 2-picolyl, 4-picolyl, 3-methyl-2-picoly1N-oxido,
diphenylmethyl,p,p'-dinitrobenzhydryl, 5-dibenzosuberyl, triphenylmethyl, a-
naphthyldiphenylmethyl, p-methoxyphenyldiphenylmethyl, di(p-
methoxyphenyl)phenylmethyl, tri(p-methoxyphenyl)methyl, 4-(4'-
bromophenacyloxyphenyl)diphenylmethyl, 4,4',4"-tris(4,5-
dichlorophthalimidophenyl)methyl, 4,4',4"-tris(levulinoyloxyphenyl)methyl,
4.4',4"-
tris(benzoyloxyphenyl)methyl, 3-(imidazol-1-yl)bis(4',4"-
dimethoxyphenyl)methyl, 1,1-
bis(4-methoxypheny1)-1'-pyrenylmethyl, 9-anthryl, 9-(9-phenyl)xanthenyl, 9-(9-
phenyl-
10-oxo)anthryl, 1,3-benzodithiolan-2-yl, benzisothiazolyl S,S-dioxido,
trimethylsilyl
(TMS), triethylsilyl (TES), triisopropylsilyl (TIPS), dimethylisopropylsilyl
(IPDMS),
diethylisopropylsilyl (DEIPS), dimethylthexylsilyl, t-butyldimethylsilyl
(TBDMS), t-
butyldiphenylsily1 (TBDPS), tribenzylsilyl, tri-p-xylylsilyl, triphenylsilyl,
CA 02797719 2012-10-26
WO 2011/137320
PCT/US2011/034514
- 28 -
diphenylmethylsily1 (DPMS), t-butylmethoxyphenylsilyl (TBMPS), formate,
benzoylformate, acetate, chloroacetate, dichloroacetate, trichloroacetate,
trifluoroacetate,
methoxyacetate, triphenylmethoxyacetate, phenoxyacetate, p-
chlorophenoxyacetate, 3-
phenylpropi on ate, 4-oxopentanoate (levulinate), 4,4-
(ethylenedithio)pentanoate
(levulinoyldithioacetal), pivaloate, adamantoate, crotonate, 4-
methoxycrotonate,
benzoate, p-phenylbenzoate, 2,4,6-trimethylbenzoate (mesitoate), alkyl methyl
carbonate, 9-fluorenylmethyl carbonate (Fmoc), alkyl ethyl carbonate, alkyl
2,2,2-
trichloroethyl carbonate (Troc), 2-(trimethylsilyl)ethyl carbonate (TMSEC), 2-
(phenylsulfonyl) ethyl carbonate (Psec), 2-(triphenylphosphonio) ethyl
carbonate (Peoc),
alkyl isobutyl carbonate, alkyl vinyl carbonate alkyl allyl carbonate, alkyl p-
nitrophenyl
carbonate, alkyl benzyl carbonate, alkyl p-methoxybenzyl carbonate, alkyl 3,4-
dimethoxybenzyl carbonate, alkyl o-nitrobenzyl carbonate, alkyl p-nitrobenzyl
carbonate, alkyl S-benzyl thiocarbonate, 4-ethoxy-1-napththyl carbonate,
methyl
dithiocarbonate, 2-iodobenzoate, 4-azidobutyrate, 4-nitro-4-methylpentanoate,
0-
(dibromomethyl)benzoate, 2-formylbenzenesulfonate, 2-(methylthiomethoxy)ethyl,
4-
(methylthiomethoxy)butyrate, 2-(methylthiomethoxymethyl)benzoate, 2,6-dichloro-
4-
methylphenoxyacetate, 2,6-dichloro-4-(1,1,3,3-tetramethylbutyl)phenoxyacetate,
2,4-
bis(1,1-dimethylpropyl)phenoxyacetate, chlorodiphenylacetate, isobutyrate,
monosuccinoate, (E)-2-methyl-2-butenoate, o-(methoxycarbonyl)benzoate, a-
naphthoate,
nitrate, alkyl N,N,N',N'-tetramethylphosphorodiamidate, alkyl N-
phenylcarbamate,
borate, dimethylphosphinothioyl, alkyl 2,4-dinitrophenylsulfenate, sulfate,
methanesulfonate (mesylate), benzylsulfonate, and tosylate (Ts). For
protecting 1,2- or
1,3-diols, the protecting groups include methylene acetal, ethylidene acetal,
1-t-
butylethylidene ketal, 1-phenylethylidene ketal, (4-methoxyphenyl)ethylidene
acetal,
2,2,2-trichloroethylidene acetal, acetonide, cyclopentylidene ketal,
cyclohexylidene
ketal, cycloheptylidene ketal, benzylidene acetal, p-methoxybenzylidene
acetal, 2,4-
dimethoxybenzylidene ketal, 3,4-dimethoxybenzylidene acetal, 2-
nitrobenzylidene
acetal, methoxymethylene acetal, ethoxymethylene acetal, dimethoxymethylene
ortho
ester, 1-methoxyethylidene ortho ester, 1-ethoxyethylidine ortho ester, 1,2-
dimethoxyethylidene ortho ester, a-methoxybenzylidene ortho ester, 1-(N,N-
dimethylamino)ethylidene derivative, ct-(N,N'-dimethylamino)benzylidene
derivative, 2-
oxacyclopentylidene ortho ester, di-t-butylsilylene group (DTBS), 1,3-(1,1,3,3-
CA 02797719 2012-10-26
WO 2011/137320
PCT/US2011/034514
- 29 -
tetraisopropyldisiloxanylidene) derivative (TIPDS), tetra-t-butoxydisiloxane-
1,3-
diylidene derivative (TBDS), cyclic carbonates, cyclic boronates, ethyl
boronate, and
phenyl boronate. Amino-protecting groups include methyl carbamate, ethyl
carbamante,
9-fluorenylmethyl carbamate (Fmoc), 9-(2-sulfo)fluorenylmethyl carbamate, 9-
(2,7-
dibromo)fluoroenylmethyl carbamate, 2,7-di-t-buty1-19-(10,10-dioxo-10,10,10,10-
tetrahydrothioxanthyl)]methyl carbamate (DBD-Tmoc), 4-methoxyphenacyl
carbamate
(Phenoc), 2,2,2-trichloroethyl carbamate (Troc), 2-trimethylsilylethyl
carbamate (Teoc),
2-phenylethyl carbamate (hZ), 1-(1-adamanty1)-1-methylethyl carbamate (Adpoc),
1,1-
dimethy1-2-haloethyl carbamate, 1,1-dimethy1-2,2-dibromoethyl carbamate (DB-t-
B OC),
/0 1,1-dimethy1-2,2,2-trichloroethyl carbamate (TCBOC), 1-methyl-1-(4-
biphenylyl)ethyl
carbamate (Bpoc), 1-(3,5-di-t-butylpheny1)-1-methylethyl carbamate (t-Bumeoc),
2-(2'-
and 4'-pyridyBethyl carbamate (Pyoc), 2-(/V,N-dicyclohexylcarboxamido)ethyl
carbamate, t-butyl carbamate (BOC), 1-adamantyl carbamate (Adoc), vinyl
carbamate
(Voc), allyl carbamate (Alloc), 1-isopropylally1 carbamate (Ipaoc), cinnamyl
carbamate
(Coc), 4-nitrocinnamyl carbamate (Noc), 8-quinoly1 carbamate, N-
hydroxypiperidinyl
carbamate, alkyldithio carbamate, benzyl carbamate (Cbz), p-methoxybenzyl
carbamate
(Moz), p-nitobenzyl carbamate, p-bromobenzyl carbamate, p-chlorobenzyl
carbamate,
2,4-dichlorobenzyl carbamate, 4-methylsulfinylbenzyl carbamate (Msz), 9-
anthrylmethyl
carbamate, diphenylmethyl carbamate, 2-methylthioethyl carbamate, 2-
.. methylsulfonylethyl carbamate, 2-(p-toluenesulfonyl)ethyl carbamate, 1241,3-
dithianylbnethyl carbamate (Dmoc), 4-methylthiophenyl carbamate (Mtpc), 2,4-
dimethylthiophenyl carbamate (Bmpc), 2-phosphonioethyl carbamate (Peoc), 2-
triphenylphosphonioisopropyl carbamate (Ppoc), 1,1-dimethy1-2-cyanoethyl
carbamate,
m-chloro-p-acyloxybenzyl carbamate, p-(dihydroxyboryl)benzyl carbamate, 5-
.. benzisoxazolylinethyl carbamate, 2-(trifluoromethyl)-6-chromonylmethyl
carbamate
(Tcroc), m-nitrophenyl carbamate, 3,5-dimethoxybenzyl carbamate, o-nitrobenzyl
carbamate, 3,4-dimethoxy-6-nitrobenzyl carbamate, phenyl(o-nitrophenyl)methyl
carbamate, phenothiazinyl-(10)-carbonyl derivative, N'-p-
toluenesulfonylaminocarbonyl
derivative. N'-phenylaminothiocarbonyl derivative, t-amyl carbamate, S-benzyl
thiocarbamate, p-cyanobenzyl carbamate, cyclobutyl carbamate, cyclohexyl
carbamate,
cyclopentyl carbamate, cyclopropylmethyl carbamate, p-decyloxybenzyl
carbamate, 2,2-
dimethoxycarbonylvinyl carbamate, o-(N,N-dimethylcarboxamido)benzyl carbamate,
CA 02797719 2012-10-26
WO 2011/137320
PCT/US2011/034514
- 30 -
1,1-dimethy1-3-(N,N-dimethylcarboxamido)propyl carbamate, 1,1-dimethylpropynyl
carbamate, di(2-pyridyl)methyl carbamate, 2-furanylmethyl carbamate, 2-
iodoethyl
carbamate, isoborynl carbamate, isobutyl carbamate, isonicotinyl carbamate, p-
(pr-
methoxyphenylazo)benzyl carbamate, 1 -methylcyclobutyl carbamate, 1-
methylcyclohexyl carbamate, 1-methyl-1-cyclopropylmethyl carbamate, 1-methy1-1-
(3 ,5-dimethoxyphenyl)ethyl carbamate, 1-methyl- 1-(p-phenylazophenyl)ethyl
carbamate,
1-methyl-l-phenylethyl carbamate, 1-methyl-1-(4-pyridyl)ethyl carbamate,
phenyl
carbamate, p-(phenylazo)benzyl carbamate, 2,4,6-tri-t-butylphenyl carbamate, 4-
(trimethylammonium)benzyl carbamate. 2,4,6-trimethylbenzyl carbamate,
formamide,
acetamide, chloroacetamide, trichloroacetamide, trifluoroacetamide,
phenylacetamide, 3-
phenylpropanamide, picolinamide, 3-pyridylcarboxamide, N-benzoylphenylalanyl
derivative, benzamide, p-phenylbenzamide, o-nitophenylacetamide, o-
nitrophenoxyacetamide, acetoacetamide, (N'-
dithiobenzyloxycarbonylamino)acetamide,
3-(p-hydroxyphenyl)propanamide, 3-(o-nitrophenyl)propanamide, 2-methyl-2-(o-
nitrophenoxy)propanamide, 2-methyl-2-(o-phenylazophenoxy)propanamide, 4-
chlorobutanamide, 3-methy1-3-nitrobutanamide, o-nitrocinnamide, N-
acetylmethionine
derivative, o-nitrobenzamide, o-(benzoyloxymethyl)benzamide, 4,5-dipheny1-3-
oxazolin-2-one, N-phthalimide, N-dithiasuccinimide (Dts), N-2,3-
diphenylmaleimide, N-
2,5-dimethylpyrrole, N-1,1,4,4-tetramethyldisilylazacyclopentane adduct
(STABASE),
5-substituted 1,3-dimethy1-1,3,5-triazacyclohexan-2-one, 5-substituted 1,3-
dibenzyl-
1,3,5-triazacyclohexan-2-one, 1-substituted 3,5-dinitro-4-pyridone, N-
inethylamine, N-
allylamine, N42-(trimethylsilyl)ethoxy]methylamine (SEM), N-3-
acetoxypropylamine,
N-(1-isopropy1-4-nitro-2-oxo-3-pyroolin-3-yl)amine, quaternary ammonium salts,
N-
benzylamine, N-di(4-methoxyphenyl)methylamine, N-5 -dibenzosuberylamine, N-
triphenylmethylamine (Tr), N-1(4-methoxyphenyl)diphenylinethyljamine (MMTr), N-
9-
phenylfluorenylamine (PhF), N-2,7-dichloro-9-fluorenylmethyleneamine, N-
ferrocenylmethylamino (Fcm), N-2-picolylamino N'-oxide, N- 1,1-
dimethylthiomethyleneamine, N-benzylideneamine, N-p-methoxybenzylideneamine, N-
diphenylmethyleneamine, N-[(2-pyridyl)mesityllmethyleneamine, N-(N ',N
dimethylaminomethylene)amine, N,N' -isopropylidenediamine, N-p-
nitrobenzylideneamine, N-salicylideneamine, N-5-chlorosalicylideneamine, N-(5-
chloro-
2-hydroxyphenyl)phenylmethyleneamine, N-cyclohexylideneamine, N-(5,5-dimethy1-
3-
81662840
- 31 -
oxo-l-cyclohexenyliamine, N-borane derivative, N-diphenylborinic acid
derivative, N-
[phenyl(pentacarbonylchrornium- or tungsten)carbonyl]amine, N-copper chelate,
N-zinc
chelati,µ, N-nitroamine, N-nitrosoamine, amine N-oxide, diphenylphosphinamide
(I )pp),
dimethylthiophosphinamide (Mpt), diphenylthiophosphinamide (Ppt), chalky]
phosphoramidates, dibenzyl phosphorarnidate, diphenyl phosphoramidate,
benzenesulfenamidc, o-nitrobenzencsulfenamide (Nps), 2,4-
dinitrobenzenesulfenamidc,
pentachlorobenzenesulfenamide, 2-nitro-4-inethoxybenzenesulfenamide,
triphenylmethylsulfenamide, 3-nitropyridinesulfenamide (Npys), p-
toluenesulfonamide
(Ts), benzenesulfonamide, 2,3,6,-trimethy1-4-methoxybenzenesulfonamide (Mtr),
2,4,6-
/0 trimethoxybenzenesulfonamide (Mtb), 2,6-dimethy1-4-
methoxybenzenesulfonamide
(Pme), 2,3,5,6-tetramethy1-4-methoxybenzenesulfonamide (Mte), 4-
methoxybenzenesulfonamide (Mbs), 2,4,6-trimethylbenzenesulfonamide (Mts), 2,6-
dimethoxy-4-methylbenzenesulfonamicie (iMds), 2,2,5,7,8-pentamethylchroman-6-
sulfonamide (Pinc), methanesulfonamicle (Ms), (3-
trimethylsilylethanesulfonamide
(SES), 9-anthracenesulfonamide, 4-(4',8'-
diniethoxynaphthylmethyl)benzenesulfonamide
(DNMBS), benzylsulfonamide, trifluoromethylsulfonamide, and
phenacylsulfonamide.
Exemplary protecting groups are detailed herein. However, it will be
appreciated that
the present invention is not intended to be limited to these protecting
groups; rather; a
variety of additional equivalent protecting groups can be readily identified
using the
above criteria and utilized in the method of the present invention,
Additionally, a variety
of protecting groups are described in Protective Groups in Organic Synthesis,
Third Ed.
Greene, T.W. and Wuts, PG., Eds., John Wiley & Sons, New York: 1999.
It will be appreciated that the compounds, as described herein, may be
substituted
with any number of substituents or functional moieties. In general, the term
"substittted"
whether preceded by the term "optionally" or not, and substituents contained
in formulas
of this invention, refer to the replacement of hydrogen radicals in a given
structure with
the radical of a specified substituent. When more than one position in any
given
structure may be substituted with more than one substituent selected from a
specified
group, the substituent may be either the same or different at every position.
As used
herein, the term "substituted" is contemplated to include all permissible
substituents of
organic compounds. In a broad aspect, the permissible substituents include
acyclic and
CA 2797719 2017-10-10
CA 02797719 2012-10-26
WO 2011/137320
PCT/US2011/034514
- 32 -
cyclic, branched and unbranched, carbocyclic and heterocyclic, aromatic and
nonaromatic substituents of organic compounds. Heteroatoms such as nitrogen
may
have hydrogen substituents and/or any permissible substituents of organic
compounds
described herein which satisfy the valencies of the heteroatoms. Furthermore,
this
invention is not intended to be limited in any manner by the permissible
substituents of
organic compounds. Combinations of substituents and variables envisioned by
this
invention are preferably those that result in the formation of stable
compounds useful in
the treatment, for example, of infectious diseases or proliferative disorders.
The term
"stable", as used herein, preferably refers to compounds which possess
stability
sufficient to allow manufacture and which maintain the integrity of the
compound for a
sufficient period of time to be detected and preferably for a sufficient
period of time to be
useful for the purposes detailed herein.
The term "aliphatic," as used herein, includes both saturated and unsaturated,
straight chain (i.e., unbranched), branched, acyclic, cyclic, or polycyclic
aliphatic
hydrocarbons, which are optionally substituted with one or more functional
groups. As
will be appreciated by one of ordinary skill in the art, "aliphatic" is
intended herein to
include, but is not limited to, alkyl, alkenyl, alkynyl, cycloalkyl,
cycloalkenyl, and
cycloalkynyl moieties. Thus, as used herein, the term "alkyl" includes
straight, branched
and cyclic alkyl groups. An analogous convention applies to other generic
terms such as
"alkenyl," "alkynyl," and the like. Furthermore, as used herein, the terms
"alkyl,"
"alkenyl," "alkynyl," and the like encompass both substituted and
unsubstituted groups.
In certain embodiments, the alkyl, alkenyl, and alkynyl groups employed in the
invention contain 1-20 aliphatic carbon atoms. In certain other embodiments,
the alkyl,
alkenyl, and alkynyl groups employed in the invention contain 1-10 aliphatic
carbon
atoms. In yet other embodiments, the alkyl, alkenyl, and alkynyl groups
employed in the
invention contain 1-8 aliphatic carbon atoms. In still other embodiments, the
alkyl,
alkenyl, and alkynyl groups employed in the invention contain 1-6 aliphatic
carbon
atoms. In yet other embodiments, the alkyl, alkenyl, and alkynyl groups
employed in the
invention contain 1-4 carbon atoms. Illustrative aliphatic groups thus
include, but are not
limited to, for example, methyl, ethyl, n-propyl, isopropyl, cyclopropyl, -CH2-
cyclopropyl, vinyl, allyl, n-butyl, sec-butyl, isobutyl, tert-butyl,
cyclobutyl, -CH2-
cyclobutyl, n-pentyl, sec-pentyl, isopentyl, tert-pentyl, cyclopentyl, -CH2-
cyclopentyl, n-
CA 02797719 2012-10-26
WO 2011/137320
PCT/US2011/034514
- 33 -
hexyl, sec-hexyl, cyclohexyl, -CH2-cyclohexyl moieties and the like, which
again, may
bear one or more substituents. Alkenyl groups include, but are not limited to,
for
example, ethenyl, propenyl, butenyl, 1-methyl-2-buten-1-yl, and the like.
Representative
alkynyl groups include, but are not limited to, ethynyl, 2-propynyl
(propargyl), 1 -
propynyl, and the like.
Some examples of substituents of the above-described aliphatic (and other)
moieties of compounds of the invention include, but are not limited to
aliphatic;
heteroaliphatic; aryl; heteroaryl; arylalkyl; heteroarylalkyl; alkoxy;
aryloxy;
heteroalkoxy; heteroaryloxy; alkylthio; arylthio; heteroalkylthio;
heteroarylthio; -F; -Cl;
-Br; -I; -OH; -NO2; -CN; -CF3; -CH2CF3; -CHC12; -CH,OH; -CH2CH2OH; -CH2NH2: -
CH2S02CH3: -C(0)R; -0O2(Rx); -CON(R)2; -0C(0)R; -0CO2Rx; -000N(R)2; -
N(R)2; -S(0)2R; -NRx(CO)Rx wherein each occurrence of Rx independently
includes,
but is not limited to, aliphatic, heteroaliphatic, aryl. heteroaryl,
arylalkyl, or
heteroarylalkyl, wherein any of the aliphatic, heteroaliphatic, arylalkyl, or
heteroarylalkyl substituents described above and herein may be substituted or
unsubstituted, branched or unbranched, cyclic or acyclic, and wherein any of
the aryl or
heteroaryl substituents described above and herein may be substituted or
unsubstituted.
Additional examples of generally applicable substituents are illustrated by
the specific
embodiments described herein.
The term "amino," as used herein, refers to a primary (-NH2), secondary (-
NHR),
tertiary (-NRxRy), or quaternary (-N+RxRyRz) amine, where Rx, Ry and R, are
independently an aliphatic, alicyclic, heteroaliphatic, heterocyclic, aryl, or
heteroaryl
moiety, as defined herein. Examples of amino groups include, but are not
limited to,
methylamino, dimethylamino, ethylamino, diethylamino, diethylaminocarbonyl,
methylethylamino, iso-propylamino, piperidino, trimethylamino, and
propylamino.
The term "alkylamino" refers to a group having the structure -NHR', wherein R'
is aliphatic, as defined herein. In certain embodiments, the aliphatic group
contains 1-20
aliphatic carbon atoms. In certain other embodiments, the aliphatic group
contains 1-10
aliphatic carbon atoms. In yet other embodiments, the aliphatic group employed
in the
invention contain 1-8 aliphatic carbon atoms. In still other embodiments, the
aliphatic
group contains 1-6 aliphatic carbon atoms. In yet other embodiments, the
aliphatic group
contains 1-4 aliphatic carbon atoms. Examples of alkylamino groups include,
but are not
CA 02797719 2012-10-26
WO 2011/137320
PCT/US2011/034514
- 34 -
limited to, methylamino, ethylamino, n-propylamino, iso-propylamino,
cyclopropylamino, n-butylamino, tert-butylamino, neopentylamino, n-
pentylamino,
hexylamino, cyclohexylamino, and the like.
The term "dialkylamino" refers to a group having the structure -NRR ', wherein
R
and R' are each an aliphatic group, as defined herein. R and R' may be the
same or
different in an dialkyamino moiety. In certain embodiments, the aliphatic
groups
contains 1-20 aliphatic carbon atoms. In certain other embodiments, the
aliphatic groups
contains 1-10 aliphatic carbon atoms. In yet other embodiments, the aliphatic
groups
employed in the invention contain 1-8 aliphatic carbon atoms. In still other
.. embodiments, the aliphatic groups contains 1-6 aliphatic carbon atoms. In
yet other
embodiments, the aliphatic groups contains 1-4 aliphatic carbon atoms.
Examples of
dialkylamino groups include, but are not limited to, dimethylainino, methyl
ethylamino,
diethylamino, methylpropylamino, di(n-propyl)amino, di(iso-propyl)amino,
di(cyclopropyl)amino, di(n-butyl)amino, di(tert-butyl)amino,
di(neopentyl)amino, di(n-
.. pentyl)amino, di(hexyl)amino, di(cyclohexyl)amino, and the like. In certain
embodiments, R and R' are linked to form a cyclic structure. The resulting
cyclic
structure may be aromatic or non-aromatic. Examples of cyclic diaminoalkyl
groups
include, but are not limted to, aziridinyl, pyrrolidinyl, piperidinyl,
morpholinyl, pyrrolyl,
imidazolyl, 1,3,4-trianolyl, and tetrazolyl.
In general, the terms "aryl" and "heteroaryl," as used herein, refer to stable
mono-
or polycyclic, heterocyclic, polycyclic, and polyheterocyclic unsaturated
moieties having
preferably 3-14 carbon atoms, each of which may be substituted or
unsubstituted.
Substituents include, but are not limited to, any of the previously mentioned
substituents,
i.e., the substituents recited for aliphatic moieties, or for other moieties
as disclosed
.. herein, resulting in the formation of a stable compound. In certain
embodiments of the
present invention, "aryl" refers to a mono- or bicyclic carbocyclic ring
system having
one or two aromatic rings including, but not limited to, phenyl, naphthyl,
tetrahydronaphthyl, indanyl, indenyl, and the like. In certain embodiments of
the present
invention, the term "heteroary1,- as used herein, refers to a cyclic aromatic
radical having
from five to ten ring atoms of which one ring atom is selected from S, 0, and
N; zero,
one, or two ring atoms are additional heteroatoms independently selected from
S, 0, and
N; and the remaining ring atoms are carbon, the radical being joined to the
rest of the
CA 02797719 2012-10-26
WO 2011/137320
PCT/US2011/034514
- 35 -
molecule via any of the ring atoms, such as, for example, pyridyl, pyrazinyl,
pyrimidinyl,
pyrrolyl, pyrazolyl, imidazolyl, thiazolyl, oxazolyl, isooxazolyl,
thiadiazolyl,oxadiazolyl,
thiophenyl, furanyl, quinolinyl, isoquinolinyl, and the like. In certain
embodiments, the
term "aryl" or "heteroaryl" refers to a planar ring having p-orbitals
perpendicular to the
plane of the ring at each ring atom, and satisfying the Huckel rule where the
number of
pi electrons in the ring is (4n+2) wherein n is an integer.
It will also be appreciated that aryl and heteroaryl moieties, as defined
herein may
be attached via an alkyl or heteroalkyl moiety and thus also include -
(alkyl)aryl, -
(heteroalkyl)aryl, -(heteroalkyl)heteroaryl, and -(heteroalkyl)heteroaryl
moieties. Thus,
as used herein, the phrases "aryl or heteroaryl moieties" and "aryl,
heteroaryl, ¨
(alkyl)aryl, -(heteroalkyl)aryl, -(heteroalkyl)heteroaryl, and
¨(heteroalkyl)heteroaryl" are
interchangeable. It will be appreciated that aryl and heteroaryl groups can be
unsubstituted or substituted, wherein substitution includes replacement of
one, two,
three, or more of the hydrogen atoms thereon independently with any one or
more of the
following moieties including, but not limited to: aliphatic; heteroaliphatic;
aryl;
heteroaryl; arylalkyl; heteroarylalkyl; alkoxy; aryloxy; heteroalkoxy;
heteroaryloxy;
alkylthio; arylthio; heteroalkylthio; heteroarylthio; -F; -Cl; -Br; -I; -OH; -
NO2; -CN; -
CF3; -CH2CF3; -C11C12; -CH2OH; -CH2CH2OH; -CH2NH2; -CI-19S02CH3; -C(0)R; -
CO2(Rx); -CON(R)2; -0C(0)R; -000/Rx; -000N(R.)2; -N(R)2; -S(0)9R.: -
NR,(CO)Rx, wherein each occurrence of R), independently includes, but is not
limited to,
aliphatic, heteroaliphatic, aryl, heteroaryl, arylalkyl, or heteroarylalkyl,
wherein any of
the aliphatic, heteroaliphatic, arylalkyl, or heteroarylalkyl substituents
described above
and herein may be substituted or unsubstituted, branched or unbranched, cyclic
or
acyclic, and wherein any of the aryl or heteroaryl substituents described
above and herein
may be substituted or unsubstituted. Additional examples of generally
applicable
substitutents are illustrated by the specific embodiments shown in the
Examples that are
described herein.
The term "heteroaliphatic," as used herein, refers to aliphatic moieties that
contain one or more oxygen, sulfur, nitrogen, phosphorus, or silicon atoms,
e.g., in place
of carbon atoms. Heteroaliphatic moieties may be branched, unbranched, cyclic
or
acyclic and include saturated and unsaturated heterocycles such as morpholino,
pyrrolidinyl, etc. In certain embodiments, heteroaliphatic moieties are
substituted by
CA 02797719 2012-10-26
WO 2011/137320
PCT/US2011/034514
- 36 -
independent replacement of one or more of the hydrogen atoms thereon with one
or more
moieties including, but not limited to aliphatic; heteroaliphatic; aryl;
heteroaryl;
arylalkyl; heteroarylalkyl; alkoxy; myloxy; heteroalkoxy; heteroaryloxy;
alkylthio;
arylthio; heteroalkylthio; heteroarylthio; -F; -Cl; -Br; -1; -OH; -NO2; -CN: -
CF3; -
CH2CF3; -CHC12; -CH,OH; -CH2CH2OH; -CH2NH2; -CH2S02CH3; -C(0)R; -0O2(R.);
-CON(R)2; -0C(0)R; -0CO2Rx; -000N(Rx)2; -N(R)2; -S(0)2R; -NR(CO)R,
wherein each occurrence of R, independently includes, but is not limited to,
aliphatic,
heteroaliphatic, aryl, heteroaryl, arylalkyl, or heteroarylalkyl, wherein any
of the
aliphatic, heteroaliphatic, arylalkyl, or heteroarylalkyl substituents
described above and
herein may be substituted or unsubstituted, branched or unbranched, cyclic or
acyclic,
and wherein any of the aryl or heteroaryl substituents described above and
herein may be
substituted or unsubstituted. Additional examples of generally applicable
substitu tents
are illustrated by the specific embodiments described herein.
The terms "halo" and "halogen" as used herein refer to an atom selected from
fluorine, chlorine, bromine, and iodine.
In certain embodiments, the compound of the invention or for use in the
invention is of the formula:
I ) (R
X ___________________________________________ R2
Formula I
wherein
Xis 0, S, or NR3;
n is 0, 1, 2, 3, or 4;
each occurrence of RI is independently hydrogen; halogen; cyclic or acyclic,
substituted or unsubstituted, branched or unbranched aliphatic; cyclic or
acyclic,
substituted or unsubstituted, branched or unbranched heteroaliphatic;
substituted or
unsubstituted, branched or unbranched acyl; substituted or unsubstituted aryl;
substituted
or unsubstituted heteroaryl; -OR'; -C(=0)RA; -C(=0)N(RA)2; -CO2RA; -CN; -SCN; -
SRA; -SORA; -SO2RA; -NO2; -N3; -N(RA)2; -NHC(=0)RA; -NRAC(=0)N(RA)2; -
81662840
- 37 -
0C(=0)0RA; -0C(=0)RA; -0C(=0)N(RA)2; -NRAC(=0)0RA; or -C(RA)3; wherein each
occurrence of RA is independently a hydrogen, a protecting group, an aliphatic
moiety, a
heteroaliphatic moiety, an acyl moiety; an aryl moiety; a heteroaryl moiety;
alkoxy;
aryloxy; alkylthio; arylthio; amino, alkylamino, dialkylamino, heteroaryloxy;
or
heteroarylthio moiety;
R2 is hydrogen; halogen; cyclic or acyclic, substituted or unsubstituted,
branched
or unbranched aliphatic; cyclic or acyclic, substituted or unsubstituted,
branched or
unbranched heteroaliphatic; substituted or unsubstituted, branched or
unbranched acyl;
substituted or unsubstituted aryl; substituted or unsubstituted heteroaryl; -
ORB; -
C(=-0)RB ; -C(=0)N(RB)); -0O2RB; -CN; -SCN; -SRB; -SORB; -SORB; -NO-); -N3; -
N(RB)1; -NHC(=0)RB; -NRBC(=0)N(RB)2; -0C(=0)ORB; -0C(=0)RB; -0C(=0)N(RB)2;
-NRBC(=0)ORB; or -C(RB)3; wherein each occurrence of RB is independently a
hydrogen, a protecting group, an aliphatic moiety, a heteroaliphatic moiety,
an acyl
moiety; an aryl moiety; a heteroaryl moiety; alkoxy; aryloxy; alkylthio;
arylthio; amino,
alkylamino, dialkylamino, heteroaryloxy; or heteroarylthio moiety;
R3 is independently a hydrogen, a protecting group, an aliphatic moiety, a
heteroaliphatic moiety, an acyl moiety; an aryl moiety; a heteroaryl moiety;
alkoxy;
aryloxy; alkylthio; arylthio; amino, alkylamino, dialkylamino, heteroaryloxy;
or
heteroarylthio moiety; and pharmaceutically acceptable salts thereof.
In one embodiment, a compound of Formula I is the compound of Formula IV
(527):
0
N\
0
0
Certain embodiments of Formula I are disclosed in WO 97/21684 and WO
97/21710.
WO 97/21684 and WO 97/21710 disclose compounds in accordance with Formula I
are
useful for treating diseases related to venous insufficiency and inflammatory
edema,
In certain embodiments, a compound of Formula I is any one of the following
compounds: 1,2-dimethy1-111-naphthr2,3-dlimidazole-4,9-dione (Registry No.
4572-59-
2, Ryan Scientific, Inc., Mt, Pleasant, SC); 1,2,3,4-tetrahydro-
CA 2797719 2017-10-10
CA 02797719 2012-10-26
WO 2011/137320
PCT/US2011/034514
- 38 -
naphthI2',3':4,5limidazo[1,2-1]pyrazine-6,11-dione (Registry No. 132545-27-8,
Ryan
Scientific, Inc.); 2-(pentafluoropheny1)-1H-naphtho[2,3-d]imidazole-4,9-dione
(Registry
No. 418805-36-4, ChemBridge Corp., San Diego, CA); 2-(4-fluoropheny1)-1-(4-
methylpheny1)-1H-naphth[2,3-dlimidazole-4,9-dione (Registry No. 325731-50-8,
Ryan
Scientific, Inc.); and 1-pheny1-1H-naphth[2,3-d_limidazole-4,9-dione (Registry
No.
15030-17-8, TimTec, Inc., Newark, DE).
In certain embodiments, the compound is in a reduced form of the formula:
oR4
I
(R _________________________________________ IR2
x)
OR4
Formula IA
/0 wherein
n, R1, R2, and X are defined as above; and
each occurrence of R4 is independently hydrogen, an oxygen protecting group,
cyclic or acyclic, substituted or unsubstituted, branched or unbranched
aliphatic; cyclic
or acyclic, substituted or unsubstituted, branched or unbranched
heteroaliphatic;
substituted or unsubstituted, branched or unbranched acyl; substituted or
unsubstituted
aryl; substituted or unsubstituted heteroaryl; -C(=O)RD; -CO,RD; -C(=0)N(RD)2;
or -
C(R1)3; wherein each occurrence of RD is independently a hydrogen, an
aliphatic moiety,
a heteroaliphatic moiety, an acyl moiety; an aryl moiety; a heteroaryl moiety;
alkoxy;
aryloxy; alkylthio; arylthio; amino, alkylamino, dialkylamino, heteroaryloxy;
or
heteroarylthio moiety; and pharmaceutically acceptable salts thereof.
In certain embodiments, X is 0. In certain embodiments, X is S. In certain
embodiments, X is NH. In certain embodiments, X is NR3.
In certain embodiments, n is 0. In certain embodiments, n is 1. In certain
embodiments, n is 2. In certain embodiments, n is 3. In certain embodiments, n
is 4.
In certain embodiments, at least one R' is halogen. In certain embodiments, at
least one R3 is C1_6 alkyl. In certain embodiments, at least one 123 is ¨ORA.
In certain
embodiments, at least one Rl is _N(RA)2. In certain embodiments, at least one
121 is ¨
CA 02797719 2012-10-26
WO 2011/137320
PCT/US2011/034514
- 39 -
NO2. In certain embodiments, at least one Rl is ¨CN. In certain embodiments,
at least
one RI is ¨SCN.
In certain embodiments, R2 is substituted or unsubstituted aryl. In certain
embodiments, R2 is unsubstituted aryl. In certain embodiments, R2 is
substituted aryl. In
certain embodiments, R2 is substituted or unsubstituted phenyl. In certain
embodiments,
R2 is unsubstituted phenyl. In certain embodiments, R2 is substituted phenyl.
In certain
embodiments, R2 is ortho-substituted phenyl. In certain embodiments, R2 is
meta-
substituted phenyl. In certain embodiments, R2 is para-substituted phenyl. In
certain
embodiments, R2 is phenyl substituted with a halogen. In certain embodiments,
R2 is
/0 substituted or unsubstituted heteroaryl. In certain embodiments, R2 is
unsubstituted
heteroaryl. In certain embodiments, R2 is substituted heteroaryl. In certain
embodiments, R2 is 5-membered, substituted or unsubstituted heteroaryl. In
certain
embodiments, R2 is 6-membered, substituted or unsubstituted heteroaryl.
In certain embodiments, the compound of the invention or for use in the
invention is of the formula:
(R1), I
0
R3 R3
Formula II
wherein
n is 0, 1, 2, 3, or 4;
each occurrence of 121 is independently hydrogen; halogen; cyclic or acyclic,
substituted or unsubstituted, branched or unbranched aliphatic; cyclic or
acyclic,
substituted or unsubstituted, branched or unbranched heteroaliphatic;
substituted or
unsubstituted, branched or unbranched acyl; substituted or unsubstituted aryl;
substituted
or unsubstituted heteroaryl; -ORA; -C(=0)RA; -CO,RA; -C(=0)N(RA)2; -CN; -SCN; -
SRA; -SORA; -SO2RA; -NO2; -N3; -N(RA)2; -NHC(=0)RA; -NRAC(=0)N(RA)2; -
0C(=0)0RA; -0C(=0)RA; -0C(=0)N(RA)2; -NRAC(=0)0RA; or -C(RA)3; wherein each
occurrence of RA is independently a hydrogen, a protecting group, an aliphatic
moiety, a
heteroaliphatic moiety, an acyl moiety; an aryl moiety; a heteroaryl moiety;
alkoxy;
CA 02797719 2012-10-26
WO 2011/137320
PCT/US2011/034514
- 40 -
aryloxy; alkylthio; arylthio; amino, alkylamino, dialkylamino, heteroaryloxy;
or
heteroarylthio moiety;
R2 is hydrogen; a nitrogen-protecting group; cyclic or acyclic, substituted or
unsubstituted, branched or unbranched aliphatic; cyclic or acyclic,
substituted or
unsubstituted, branched or unbranched heteroaliphatic; substituted or
unsubstituted,
branched or unbranched acyl; substituted or unsubstituted aryl; substituted or
unsubstituted heteroaryl; -C(=0)RB; -C(=0)N(RB)2; -CO,RB; or -C(RB)3; wherein
each
occurrence of RB is independently a hydrogen, an aliphatic moiety, a
heteroaliphatic
moiety, an acyl moiety; an aryl moiety; a heteroaryl moiety; alkoxy; aryloxy;
alkylthio;
arylthio; amino, alkylamino, dialkylamino, heteroaryloxy; or heteroarylthio
moiety;
each occurrence of R3 is independently hydrogen; halogen; cyclic or acyclic,
substituted or unsubstituted, branched or unbranched aliphatic; cyclic or
acyclic,
substituted or unsubstituted, branched or unbranched heteroaliphatic;
substituted or
unsubstituted, branched or unbranched acyl; substituted or unsubstituted aryl;
substituted
or unsubstituted heteroaryl; -ORc; -C(=0)Rc; -C(=0)N(Rc)2; -0O2Rc; -CN; -SCN; -
SRC; -SORc; -S02Rc; -NO2; -N3; -N(Rc)2; -NHC(=0)1(c; -NRcC(=0)N(Itc)2; -
0C(=0)0Rc; -0C(=0)Rc; -0C(=0)N(Rc)2; -NRcC(=0)0Rc; or -C(Rc)3; wherein each
occurrence of Rc is independently a hydrogen, a protecting group, an aliphatic
moiety, a
heteroaliphatic moiety, an acyl moiety; an aryl moiety; a heteroaryl moiety;
alkoxy;
aryloxy; alkylthio; arylthio; amino, alkylamino, dialkylamino, heteroaryloxy;
or
heteroarylthio moiety; wherein both occurrences of R3 may optionally be taken
together
with the intervening carbon atoms to form an optionally substituted cyclic
moiety or may
be =0, =S, or =NRc; and pharmaceutically acceptable salts thereof.
In certain embodiments, n is 0. In certain embodiments, n is 1. In certain
embodiments, n is 2. In certain embodiments, n is 3. In certain embodiments, n
is 4.
In certain embodiments, at least one le is halogen. In certain embodiments, at
least one le is Ci_6 alkyl. In certain embodiments, at least one le is ¨OR''.
In certain
embodiments, at least one Rl is ¨N(RA)2. In certain embodiments, at least one
Rl is ¨
NO2. In certain embodiments, at least one RI is ¨CN. In certain embodiments,
at least
one le is ¨SCN.
In certain embodiments, R2 is hydrogen. In certain embodiments, R2 is
substituted or unsubstituted, branched or unbranched acyl. In certain
embodiments. R2 is
CA 02797719 2012-10-26
WO 2011/137320
PCT/US2011/034514
- 41 -
cyclic or acyclic, substituted or unsubstituted, branched or unbranched
aliphatic. In
certain embodiments, R2 is cyclic or acyclic, substituted or unsubstituted,
branched or
unbranched heteroaliphatic. In certain embodiments, R2 is substituted or
unsubstituted
aryl. In certain embodiments, R2 is substituted or unsubstituted heteroaryl.
In certain
.. embodiments, R2 is optionally substituted heteroarylalkyl. In certain
embodiments, R2 is
optionally substituted arylalkyl. In certain embodiments, R2 is
¨C1-12-heteroaryl. In certain embodiments, R2 is of the formula: . In
certain embodiments, R2 is of the formula: . In
certain embodiments,
R2 is of the formula: . In certain embodiments, R2 is ¨CH2-aryl.
In certain embodiments, both R3 are hydrogen. In certain embodiments, at least
one R3 is halogen. In certain embodiments, at least one R3 is fluorine. In
certain
embodiments, at least one R3 is C1_6 alkyl. In certain embodiments, at least
one R3 is ¨
ORc. In certain embodiments, at least one R3 is ¨N(Rc)1.
In one embodiment, a compound of Formula II is the compound of Formula V
/5 (947):
0
0
As noted above, a method for synthesizing the compound of Formula V (947) is
disclosed in Sondhi et al. (2008) Letts Org Chem 5:51-4, but this reference
does not
.. disclose a use for this compound.
In certain embodiments, a compound of Formula II is any one of the following
compounds: 2-(2-furanylmethyl)-7-methoxy-1,3(2H,4H)-isoquinolinedione
(Registry
No. 328039-10-7, Ryan Scientific, Inc.); 2-benzy1-1,3(21-1,411)-
isoquinolinedione
(Registry No. 21640-31-3, Key Organics Ltd., Camelford, United Kingdom); 2-(4-
.. methoxybenzy1)-1,3(2H,4H)-isoquinolinedione (Registry No. 217493-71-5, Key
Organics, Ltd.); 2-(2-phenylethyl)-1,3(2H,4H)-isoquinolinedione (Registry No.
53558-
CA 02797719 2012-10-26
WO 2011/137320
PCT/US2011/034514
- 42 -
67-1, ChemBridge Corp.); 3-(2-furanylmethyl)-2,4,(1H,3H)-quinazolinedione
(Registry
No. 436855-78-6, Ryan Scientific, Inc.); 3-(2-furanylmethyl)-1-methy1-
2,4,(1H,3H)-
quinazolinedione (Registry No. 531504-02-6, Enamine, Kiev, Ukraine); 2-(2-
furylmethyl)-1H-isoindole-1,3(2H)-dione (Registry No. 4667-83-8, ChemBridge
Corp.);
2-(2-(3,4-dimethoxyphenyl)ethyl)isoquinoline-1,3(2H,4H)-dione (Registry No.
198139-
89-8, Sigma-Aldrich, St. Louis, MO); and 2-benzo[1,31dioxo1-5-ylmethy1-4H-
isoquinoline-1,3-dione (Registry No. 565193-20-6, Ryan Scientific, Inc.).
It should be understood that, in addition to compounds of Formula III (933),
Formula IV (527), and Formula V (947), and likewise related compounds as
disclosed
above, and pharmaceutical compositions including said compounds, the invention
also
embraces pharmaceutically acceptable salts of said compounds and
pharmaceutical
compositions including said pharmaceutically acceptable salts of said
compounds. As
used herein, a "pharmaceutically acceptable salt" refers to an acid or base
form of a
compound, usually in combination with a counter ion, that is suitable for use
in
pharmacy. When used in medicine the salts should be pharmaceutically
acceptable, but
non-pharmaceutically acceptable salts may conveniently be used to prepare
pharmaceutically acceptable salts thereof. Such salts include, but are not
limited to,
those prepared from the following acids: hydrochloric, hydrobromic, sulphuric,
nitric,
phosphoric, maleic, acetic, salicylic, p-toluene sulphonic, tartaric, citric,
methane
sulphonic, formic, malonic, succinic, naphthalene-2-sulphonic, and benzene
sulphonic.
Also, such salts can be prepared as alkaline metal or alkaline earth salts,
such as sodium,
potassium or calcium salts of the carboxylic acid group. Pharmaceutically
acceptable
salts are well known in the art and are the subject of numerous reviews and
monographs
such as P. H. Stahl and C. G. Weimuth, editors, Handbook of Pharmaceutical
Salts:
Properties, Selection and Use, Weinheim/Zilrich:Wiley-VCH/VHCA, 2002.
The invention further provides pharmaceutical compositions of the invention
formulated for targeted delivery to a cancer cell. As used herein, a "cancer
cell" refers to
a living cell in or isolated from a cancer. "Cancer" as used herein refers to
an
uncontrolled growth of cells. Cancers which migrate from their original
location and
seed vital organs can eventually lead to the death of the subject through the
functional
deterioration of the affected organs. Cancers include, but are not limited to,
basal cell
carcinoma, biliary tract cancer; bladder cancer; bone cancer; brain and CNS
cancer;
CA 02797719 2012-10-26
WO 2011/137320
PCT/US2011/034514
- 43 -
breast cancer; cervical cancer; choriocarcinoma; colon and rectum cancer;
connective
tissue cancer; cancer of the digestive system; endometrial cancer; esophageal
cancer; eye
cancer; cancer of the head and neck; gastric cancer; intra-epithelial
neoplasm; kidney
cancer; larynx cancer; leukemia; liver cancer; lung cancer (e.g. small cell
and non-small
cell); lymphoma including Hodgkin's and non-Hodgkin's lymphoma; melanoma;
myeloma; neuroblastoma; oral cavity cancer (e.g., lip, tongue, mouth, and
pharynx);
ovarian cancer; pancreatic cancer; prostate cancer; retinoblastoma;
rhabdomyosarcoma;
rectal cancer; renal cancer; cancer of the respiratory system; sarcoma; skin
cancer;
stomach cancer; testicular cancer; thyroid cancer; uterine cancer; cancer of
the urinary
system, as well as other carcinomas and sarcomas.
Certain cancer cells express cancer antigens on their cell surface, thereby
making
it possible to target delivery of an agent to the cancer cells. A "cancer
antigen" as used
herein is a compound, such as a peptide or protein, associated with a tumor or
cancer cell
surface and which is capable of provoking an immune response when expressed on
the
surface of an antigen presenting cell in the context of an MHC molecule. Some
of these
antigens are encoded, although not necessarily expressed, by normal cells.
These
antigens can be characterized as those which are normally silent (i.e., not
expressed) in
normal cells, those that are expressed only at certain stages of
differentiation and those
that are temporally expressed such as embryonic and fetal antigens. Other
cancer
antigens are encoded by mutant cellular genes, such as oncogenes (e.g.,
activated ras
oncogene), suppressor genes (e.g., mutant p53), fusion proteins resulting from
internal
deletions or chromosomal translocations. Still other cancer antigens can be
encoded by
viral genes such as those carried on RNA and DNA tumor viruses. Cancer
antigens can
be prepared from cancer cells either by preparing crude extracts of cancer
cells, for
example, as described in Cohen, et al., 1994, Cancer Research, 54:1055, by
partially
purifying the antigens, by recombinant technology, or by de novo synthesis of
known
antigens. Cancer antigens include but are not limited to antigens that are
recombinantly
expressed, an immunogenic portion of, or a whole tumor or cancer. Such
antigens can
be isolated or prepared recombinantly or by any other means known in the art.
A pharmaceutical composition formulated for targeted delivery to a cancer cell
includes a delivery vehicle that contains or is coated with an active agent,
such as a small
molecule USP1 inhibitor of the invention, linked to a targeting agent that is
specific for a
CA 02797719 2012-10-26
WO 2011/137320
PCT/US2011/034514
- 44 -
cancer antigen or other target molecule of interest that is present on the
cancer cell. A
delivery vehicle can be a nanosphere, microsphere, emulsome, liposome, or
virosome,
any of which can be prepared using methods well known in the art. A targeting
agent
can be an antigen-specific antibody or antigen-binding fragment thereof, a
receptor, or
counter-receptor. As an example, a receptor-counter-receptor pair can be a
growth factor
receptor and the corresponding growth factor that binds to the growth factor
receptor. In
one embodiment, the targeting agent can include biotin or a biotinylated
molecule that
binds to avidin or streptavidin that is present on the surface of a cancer
cell. In one
embodiment, the targeting agent can include avidin or streptavidin that binds
to biotin or
a biotinylated molecule that is present on the surface of a cancer cell.
The invention provides a method for inhibiting USP1-mediated deubiqitination
of
a ubiquitinated substrate, whereby the ubiquitinated substrate is contacted
with a small
molecule inhibitor of IJSP1 selected from the group consisting of a compound
of
Formula I, a compound of Formula IA, a compound of Formula II, the compound of
Formula III (933), the compound of Formula IV (527), the compound of Formula V
(947), and any combination thereof, in an amount effective to inhibit USP1-
mediated
deubiqitination of the ubiquitinated substrate. In one embodiment the small
molecule
inhibitor of USP1 is the compound of Formula III (933). In one embodiment the
small
molecule inhibitor of USP1 is the compound of Formula IV (527). In one
embodiment
the small molecule inhibitor of USP1 is the compound of Formula V (947). The
method
can be performed in vitro or in vivo. In one embodiment the ubiquitinated
substrate is an
isolated ubiquitinated substrate. A substrate or other compound is "isolated"
when it is
substantially separated from other cellular components with which it is found
in nature.
In one embodiment an isolated substrate is a substrate that has been purified
from a
natural source. In one embodiment the isolated substrate is expressed
artificially by a
host cell and then separated from the host cell.
In one embodiment the substrate is a natural substrate of USP1. For example,
in
one embodiment the substrate is ubiquitinated FANCD2 (FANCD2-Ub). In one
embodiment the substrate is ubiquitinated PCNA (PCNA-Ub).
In one embodiment the substrate is an artificial substrate of USP1. For
example,
in one embodiment the substrate is ubiquitinated AMC (AMC-Ub).
CA 02797719 2012-10-26
WO 2011/137320
PCT/US2011/034514
- 45 -
The invention provides a method for treating a subject having cancer, whereby
a
small molecule inhibitor of USP1 selected from the group selected from the
group
consisting of a compound of Formula I, a compound of Formula IA, a compound of
Formula II, the compound of Formula III (933), the compound of Formula IV
(527), the
compound of Formula V (947), and any combination thereof, is administered to a
subject
having cancer in need of such treatment in an amount effective to treat the
cancer. In one
embodiment the small molecule inhibitor of USP1 is the compound of Formula III
(933).
In one embodiment the small molecule inhibitor of USP1 is the compound of
Formula
IV (527). In one embodiment the small molecule inhibitor of USP1 is the
compound of
Formula V (947).
As used herein, a "subject having a cancer" refers to a living mammal with a
detectable cancer. In one embodiment the subject is a human. Methods for
detecting
cancer are well known in the art. Such methods include biopsy, blood smear,
bone
marrow biopsy, X-ray, CAT scan, magnetic resonance imaging (MRI), radionuclide
scanning, polymerase chain reaction (PCR), and the like.
The compound can be administered to the subject using any suitable route of
administration. Such routes of administration can include oral, intravenous,
intramuscular, intraperitoneal, intravesical, intracisternal, other direct
injection (e.g., into
a tumor), topical, aerosol to lung, rectal, vaginal, and other mucosal.
As used herein, an "effective amount" is an amount that is sufficient to
realize a
desired biological effect. Combined with the teachings provided herein, by
choosing
among the various active compounds and weighing factors such as potency,
relative
bioavailability, patient body weight, severity of adverse side-effects and
preferred mode
of administration, an effective prophylactic or therapeutic treatment regimen
can be
planned which does not cause substantial toxicity and yet is effective to
treat the
particular subject. The effective amount for any particular application can
vary
depending on such factors as the disease or condition being treated, the
particular active
agent being administered, the size of the subject, or the severity of the
disease or
condition. One of ordinary skill in the art can empirically determine the
effective
amount of a particular active agent and/or other therapeutic agent without
necessitating
undue experimentation. It is preferred generally that a maximum dose be used,
that is,
the highest safe dose according to sound medical judgment. Multiple doses per
day may
CA 02797719 2012-10-26
WO 2011/137320
PCT/US2011/034514
- 46 -
be contemplated to achieve appropriate systemic levels of compounds.
Appropriate
systemic levels can be determined by, for example, measurement of the
patient's peak or
sustained plasma level of the drug. "Dose" and "dosage" are used
interchangeably
herein.
For any compound described herein the therapeutically effective amount can be
initially determined from animal models. A therapeutically effective dose can
also be
determined from human data for active agents which have been tested in humans
and for
compounds which are known to exhibit similar pharmacological activities, such
as other
related active agents. Higher doses may be required for enteral administration
as
compared to parenteral administration. The applied dose can be adjusted based
on the
relative bioavailability and potency of the administered compound. Adjusting
the dose
to achieve maximal efficacy based on the methods described above and other
methods as
are well-known in the art is well within the capabilities of the ordinarily
skilled artisan.
Generally, daily oral doses of active compounds will be from about 0.01
milligrams/kg per day to 1000 milligrams/kg per day. It is expected that oral
doses in the
range of 0.5 to 50 milligrams/kg, in one or several administrations per day,
will yield the
desired results. Dosage may be adjusted appropriately to achieve desired drug
levels,
local or systemic, depending upon the mode of administration. For example, it
is
expected that intravenous administration would be from an order to several
orders of
.. magnitude lower dose per day. In the event that the response in a subject
is insufficient
at such doses, even higher doses (or effective higher doses by a different,
more localized
delivery route) may be employed to the extent that patient tolerance permits.
Multiple
doses per day are contemplated to achieve appropriate systemic levels of
compounds.
As used herein, the term "treat" means to reduce or ameliorate a disease or
condition by a detectable amount or degree. The term "treat" as used herein
refers to
both complete and partial treatment. In the context of cancer, the term
"treat" as used
herein thus can refer to complete or partial remission of a cancer. For
example, treating a
tumor may be manifest as a halted or slowed progression in the size or volume
of a
tumor, a decrease in the size or volume of a tumor, or complete resolution of
a tumor.
The method can be combined with at least one additional cancer treatment
agent.
For example, in one embodiment the method further includes administering to
the
subject a DNA cross-linking agent. In one embodiment the method further
includes
CA 02797719 2012-10-26
WO 2011/137320
PCT/US2011/034514
- 47 -
administering to the subject a PARP inhibitor. In one embodiment the method
further
includes administering to the subject a DNA cross-linking agent and a PARP
inhibitor.
The additional cancer treatment agent can be administered either essentially
simultaneously or sequentially with respect to the administering of the small
molecule
USP1 inhibitor. When the administering is sequential, either agent can be
administered
before the other. Alternatively or in addition, the administering of the small
molecule
USP1 inhibitor and the administering of the at least one additional cancer
treatment agent
can be at least partially overlapping in time or non-overlapping in time.
Furthermore, the
administering of the small molecule USP1 inhibitor and the administering of
the at least
one additional cancer treatment agent can be accomplished using the same or
different
routes of administration, as may be appropriate.
It is believed that cancers that do not express USP1 are particularly
sensitive to
DNA cross-linking agents. It is also believed that cancers that do not express
IJAF1 are
particularly sensitive to DNA cross-linking agents. Conversely, it is believed
that
cancers that do express USP1 are resistant to DNA cross-linking agents. It is
also
believed that cancers that do express USP 1 and UA141 are particularly
resistant to DNA
cross-linking agents. A cancer that does express USP1 can therefore be
sensitized to
DNA cross-linking agents by contacting the cancer with a USP1 inhibitor.
The invention provides a method for sensitizing a cancer to a DNA cross-
linking
agent, whereby a cancer is contacted with a small molecule inhibitor of USP1
selected
from the group selected from the group consisting of a compound of Formula I,
a
compound of Formula IA, a compound of Formula II, the compound of Formula III
(933), the compound of Formula IV (527), the compound of Formula V (947), and
any
combination thereof, in an amount effective to sensitize the cancer to a DNA
cross-
linking agent. In one embodiment the small molecule inhibitor of USP1 is the
compound
of Formula III (933). In one embodiment the small molecule inhibitor of IJSP1
is the
compound of Formula IV (527). In one embodiment the small molecule inhibitor
of
USP1 is the compound of Formula V (947). The method can be performed in vitro
or in
vivo. In one embodiment the cancer expresses USP1. In one embodiment the
cancer
expresses both USP1 and UAF1.
As used herein, to "sensitize" means to make more susceptible. A cancer or
cancer cell is sensitized to a DNA cross-linking agent according to the method
when it is
CA 02797719 2012-10-26
WO 2011/137320
PCT/US2011/034514
- 48 -
made more susceptible to the DNA cross-linking agent than it was, or otherwise
would
have been, without the contacting with the small molecule USP1 inhibitor.
In one embodiment the DNA cross-linking agent is a chemotherapy agent
selected from the group consisting of cisplatin, carboplatin, oxaliplatin,
alkylating
agents, mitomycin C, and any combination thereof. In one embodiment the DNA
cross-
linking agent is cisplatin.
In one embodiment the DNA cross-linking agent is ionizing radiation, for
example, X-ray therapy, cobalt-60 gamma ray irradiation, cesium-137 gamma ray
irradiation, iridium-192 gamma ray irradiation, other external beam
radiotherapy, and
/0 brachytherapy.
The invention provides a method to identify a cancer that is responsive to
USP1
inhibition, whereby cancer cells from a cancer are contacted with a small
molecule
inhibitor of IJSPI selected from the group consisting of a compound of Formula
I, a
compound of Formula IA, a compound of Formula II, the compound of Formula III
.. (933), the compound of Formula IV (527), the compound of Formula V (947),
and any
combination thereof; and measuring USP1 activity in the cancer cells contacted
with the
small molecule inhibitor of USP1, wherein reduced USP1 activity in the cancer
cells
contacted with the small molecule inhibitor of USP1 relative to control USP1
activity in
the cancer cells not contacted with the small molecule inhibitor of USP1
identifies the
cancer as a cancer that that is responsive to USP1 inhibition. In one
embodiment the
small molecule inhibitor of USP1 is the compound of Formula III (933). In one
embodiment the small molecule inhibitor of USP1 is the compound of Formula IV
(527).
In one embodiment the small molecule inhibitor of USP1 is the compound of
Formula V
(947). The method can be performed in vitro or in vivo. Alternatively or in
addition,
individual steps of the method can be performed, independent of each other, in
vitro or in
vivo. In vitro methods and, likewise, in vitro method steps, can be performed
in a cell-
free assay or in a cell-based assay.
The term "cancer cells from a cancer" as used herein can refer to cancer cells
isolated from a cancer or to cancer cells in situ in a cancer.
In one embodiment the method is a method to identify a subject having a cancer
that is responsive to USP1 inhibition, whereby cancer cells from a subject
having a
cancer are contacted with a small molecule inhibitor of USP1 selected from the
group
CA 02797719 2012-10-26
WO 2011/137320
PCT/US2011/034514
- 49 -
consisting of a compound of Formula I, a compound of Formula IA, a compound of
Formula II, the compound of Formula III (933), the compound of Formula IV
(527), the
compound of Formula V (947), and any combination thereof; and measuring USP1
activity in the cancer cells contacted with the small molecule inhibitor of
USP1, wherein
reduced USP1 activity in the cancer cells contacted with the small molecule
inhibitor of
USP1 relative to control USP1 activity in the cancer cells not contacted with
the small
molecule inhibitor of USP1 identifies the subject as a subject having a cancer
that that is
responsive to USP1 inhibition.
The term "cancer cells from a subject having a cancer" as used herein can
refer to
.. cancer cells isolated from a subject having a cancer or to cancer cells in
situ in subject
having a cancer.
USP1 activity can be measured by any of the assays described herein. For
example, activity can be measured by measuring the level of deubiquitinated
target
protein in response to USP1/UAF1 (that is, for example, the conversion of AMC-
Ub to
.. AMC). In one embodiment, an assay to measure USP1 deubiquitination activity
includes the steps of contacting a preparation comprising USP1 (and
optionallyUAF1)
with a ubiquitinated target, e.g., AMC-Ub, and measuring fluorescence
emission.
Cleavage of ubiquitin from AMC-Ub releases the fluorogenic AMC moiety, which
can
be detected as an increase in fluorescence emission at 460 nm (X,=380 nm).
Dang et al.
(1998) Biochemistry 37:1868-79.
For measurement of USP1 activity in a cell-based assay, levels of
ubiquitinated
FANCD2-Ub and unubiquitinated FANCD2, or levels of ubiquitinated PCNA-Ub and
unubiquitinated PCNA can be measured, for example by immunoblotting whole cell
lysates with anti-FANCD2 antibody (e.g., sc-20022; Santa Cruz Biotechnology,
Inc.) or
anti-PCNA antibody (e.g., sc-56; Santa Cruz Biotechnology, Inc.),
respectively.
Assays to determine the deubiquitinase activity of USP1 can be performed in a
cell-based assay, for example, in which USP1 (and optionally UAF1) are
recombinantly
expressed in a cell (such as E. coli or SF9 insect cells) along with the
target, or in a cell-
free assay in which, for example, USP1 (or a USP1/UAF1 complex) is contacted
in vitro
with a candidate inhibitor in the presence of a ubiquitinated target.
The invention provides a method to identify a cancer that is responsive to
combined DNA cross-linking therapy and I JSP1 inhibition, whereby cancer cells
from a
CA 02797719 2012-10-26
WO 2011/137320
PCT/US2011/034514
- 50 -
cancer are contacted with a DNA cross-linking agent and a small molecule
inhibitor of
USP1 selected from the group consisting of a compound of Formula I, a compound
of
Formula IA, a compound of Formula II, the compound of Formula III (933), the
compound of Formula IV (527), the compound of Formula V (947), and any
combination thereof; and proliferation of the cancer cells contacted with the
DNA cross-
linking agent and the small molecule inhibitor of USP1 is measured, wherein
reduced
proliferation of the cancer cells contacted with the DNA cross-linking agent
and the
small molecule inhibitor of USP1 relative to control proliferation of the
cancer cells
contacted with the DNA cross-linking agent but not the small molecule USP1
inhibitor
identifies the cancer as a cancer that is responsive to DNA cross-linking
therapy together
with USP1 inhibition. In one embodiment the small molecule inhibitor of USP1
is the
compound of Formula III (933). In one embodiment the small molecule inhibitor
of
I TSP1 is the compound of Formula IV (527). In one embodiment the small
molecule
inhibitor of USP1 is the compound of Formula V (947). The method can be
performed
in vitro or in vivo. Alternatively or in addition, individual steps of the
method can be
performed, independent of each other, in vitro or in vivo. In vitro methods
and, likewise,
in vitro method steps, can be performed in a cell-free assay or in a cell-
based assay.
In one embodiment the method is a method to identify a subject having a cancer
that is responsive to combined DNA cross-linking therapy and USP1 inhibition,
whereby
cancer cells from a subject having a cancer are contacted with a DNA cross-
linking agent
and a small molecule inhibitor of USP1 selected from group consisting of a
compound of
Formula I, a compound of Formula IA, a compound of Formula II, the compound of
Formula III (933), the compound of Formula IV (527), the compound of Formula V
(947), and any combination thereof; and proliferation of the cancer cells
contacted with
the DNA cross-linking agent and the small molecule inhibitor of USP1 is
measured,
wherein reduced proliferation of the cancer cells contacted with the DNA cross-
linking
agent and the small molecule inhibitor of USP1 relative to control
proliferation of the
cancer cells contacted with the DNA cross-linking agent but not the small
molecule
USP1 inhibitor identifies the subject as a subject having a cancer that is
responsive to
DNA cross-linking therapy together with USP1 inhibition.
The term "combined DNA cross-linking therapy and USP1 inhibition" as used
herein refers to any combination of DNA cross-linking therapy and USP1
inhibition.
CA 02797719 2012-10-26
WO 2011/137320
PCT/US2011/034514
- 51 -
"DNA cross-linking therapy" refers to the administration to a subject of at
least one
DNA cross-linking agent, as defined herein. "USP1 inhibition" refers to the
administration to a subject of at least one small molecule USP1 inhibitor. In
one
embodiment a small molecule USP1 inhibitor is a small molecule USP1 inhibitor
of the
invention. The administration of the DNA cross-linking agent and the USP1
inhibitor
can be accomplished either essentially simultaneously or sequentially with
respect to the
administering of the USP1 inhibitor. When the administering is sequential,
either agent
can be administered before the other. Alternatively or in addition, the
administering of
the small molecule USP1 inhibitor and the administering of the at least one
DNA cross-
linking agent can be at least partially overlapping in time or non-overlapping
in time.
Furthermore, the administering of the small molecule USP1 inhibitor and the
administering of the at least one DNA cross-linking agent can be accomplished
using the
same or different routes of administration, as may be appropriate.
The formulations of the invention are administered in pharmaceutically
acceptable solutions, which may routinely contain pharmaceutically acceptable
concentrations of salt, buffering agents, preservatives, compatible carriers,
adjuvants, and
optionally other therapeutic ingredients.
For use in therapy, an effective amount of the active agent can be
administered to
a subject by any mode that delivers the active agent to the desired surface.
Administering the pharmaceutical composition of the present invention may be
accomplished by any means known to the skilled artisan. Preferred routes of
administration include but are not limited to oral, parenteral, intramuscular,
intranasal,
sublingual, intratracheal, inhalation, ocular, vaginal, and rectal.
For oral administration, the compounds (e.g., compounds of Formula III-V, and
other therapeutic agents) can be formulated readily by combining the active
compound(s)
with pharmaceutically acceptable carriers well known in the art. Such carriers
enable the
compounds of the invention to be formulated as tablets, pills, dragees,
capsules, liquids,
gels, syrups, slurries, suspensions and the like, for oral ingestion by a
subject to be
treated. Pharmaceutical preparations for oral use can be obtained as solid
excipient,
optionally grinding a resulting mixture, and processing the mixture of
granules, after
adding suitable auxiliaries, if desired, to obtain tablets or dragee cores.
Suitable
excipients are, in particular, fillers such as sugars, including lactose,
sucrose, mannitol,
CA 02797719 2012-10-26
WO 2011/137320
PCT/US2011/034514
- 52 -
or sorbitol; cellulose preparations such as, for example, maize starch, wheat
starch, rice
starch, potato starch, gelatin, gum tragacanth, methyl cellulose,
hydroxypropylmethyl-cellulose, sodium carboxymethylcellulose, and/or
polyvinylpyrrolidone (PVP). If desired, disintegrating agents may be added,
such as the
cross-linked polyvinyl pyrrolidone, agar, or alginic acid or a salt thereof
such as sodium
alginate. Optionally the oral formulations may also be formulated in saline or
buffers,
i.e. EDTA for neutralizing internal acid conditions or may be administered
without any
carriers.
Also specifically contemplated are oral dosage forms of the above component or
components. The component or components may be chemically modified so that
oral
delivery of the derivative is efficacious. Generally, the chemical
modification contemplated
is the attachment of at least one moiety to the component molecule itself,
where said moiety
permits (a) inhibition of proteolysis; and (b) uptake into the blood stream
from the stomach
or intestine. Also desired is the increase in overall stability of the
component or
components and increase in circulation time in the body. Examples of such
moieties
include: polyethylene glycol, copolymers of ethylene glycol and propylene
glycol,
carboxymethyl cellulose, dextran, polyvinyl alcohol, polyvinyl pyrrolidone and
polyproline.
Abuchowski and Davis, 1981, "Soluble Polymer-Enzyme Adducts" In: Enzymes as
Drugs,
Hocenberg and Roberts, eds., Wiley-Interscience, New York, NY, pp. 367-383;
Newmark,
et al., 1982, J. Appl. Biochem. 4:185-189. Other polymers that could be used
are poly-1,3-
dioxolane and poly-1,3,6-tioxocane. Preferred for pharmaceutical usage, as
indicated
above, are polyethylene glycol moieties.
For the component (or derivative) the location of release may be the stomach,
the
small intestine (the duodenum, the jejunum, or the ileum), or the large
intestine. One
skilled in the art has available formulations which will not dissolve in the
stomach, yet will
release the material in the duodenum or elsewhere in the intestine.
Preferably, the release
will avoid the deleterious effects of the stomach environment, either by
protection of the
active agent (or derivative) or by release of the biologically active material
beyond the
stomach environment, such as in the intestine.
To ensure full gastric resistance a coating impermeable to at least pH 5.0 is
essential. Examples of the more common inert ingredients that are used as
enteric coatings
are cellulose acetate trimellitate (CAT), hydroxypropylmethylcellulose
phthalate (HPMCP),
CA 02797719 2012-10-26
WO 2011/137320
PCT/US2011/034514
- 53 -
HPMCP 50, HPMCP 55, polyvinyl acetate phthalate (PVAP), Eudragit L30D,
Aquateric,
cellulose acetate phthalate (CAP), Eudragit L, Eudragit S, and Shellac. These
coatings
may be used as mixed films.
A coating or mixture of coatings can also be used on tablets, which are not
intended
for protection against the stomach. This can include sugar coatings, or
coatings which
make the tablet easier to swallow. Capsules may consist of a hard shell (such
as gelatin) for
delivery of dry therapeutic i.e. powder; for liquid forms, a soft gelatin
shell may be used.
The shell material of cachets could be thick starch or other edible paper. For
pills, lozenges,
molded tablets or tablet triturates, moist massing techniques can be used.
The therapeutic can be included in the formulation as fine multi-particulates
in the
form of granules or pellets of particle size about 1 mm. The formulation of
the material for
capsule administration could also be as a powder, lightly compressed plugs or
even as
tablets. The therapeutic could be prepared by compression.
Colorants and flavoring agents may all be included. For example, the active
agent
(or derivative) may be formulated (such as by liposome or microsphere
encapsulation) and
then further contained within an edible product, such as a refrigerated
beverage containing
colorants and flavoring agents.
One may dilute or increase the volume of the therapeutic with an inert
material.
These diluents could include carbohydrates, especially mannitol, a-lactose,
anhydrous
lactose, cellulose, sucrose, modified dextrans and starch. Certain inorganic
salts may be
also be used as fillers including calcium triphosphate, magnesium carbonate
and sodium
chloride. Some commercially available diluents are Fast-Flo, Emdex, STA-Rx
1500,
Emcompress and Avicell.
Disintegrants may be included in the formulation of the therapeutic into a
solid
dosage form. Materials used as disintegrates include but are not limited to
starch, including
the commercial disintegrant based on starch, Explotab. Sodium starch
glycolate. Amberlite,
sodium carboxymethylcellulose, ultramylopectin, sodium alginate, gelatin,
orange peel,
acid carboxymethyl cellulose, natural sponge and bentonite may all be used.
Another form
of the disintegrants are the insoluble cationic exchange resins. Powdered gums
may be
used as disintegrants and as binders and these can include powdered gums such
as agar,
Karaya or tragacanth. Alginic acid and its sodium salt are also useful as
disintegrants.
CA 02797719 2012-10-26
WO 2011/137320
PCT/US2011/034514
- 54 -
Binders may be used to hold the therapeutic agent together to form a hard
tablet and
include materials from natural products such as acacia, tragacanth, starch and
gelatin.
Others include methyl cellulose (MC), ethyl cellulose (EC) and carboxymethyl
cellulose
(CMC). Polyvinyl pyrrolidone (PVP) and hydroxypropylmethyl cellulose (HPMC)
could
both be used in alcoholic solutions to granulate the therapeutic.
An anti-frictional agent may be included in the formulation of the therapeutic
to
prevent sticking during the formulation process. Lubricants may be used as a
layer
between the therapeutic and the die wall, and these can include but are not
limited to; stearic
acid including its magnesium and calcium salts, polytetrafluoroethylene
(PTFE), liquid
paraffin, vegetable oils and waxes. Soluble lubricants may also be used such
as sodium
lauryl sulfate, magnesium lauryl sulfate, polyethylene glycol of various
molecular weights,
Carbowax 4000 and 6000.
Glidants that might improve the flow properties of the drug during formulation
and
to aid rearrangement during compression might be added. The glidants may
include starch,
talc, pyrogenic silica and hydrated silicoaluminate.
To aid dissolution of the therapeutic into the aqueous environment a
surfactant
might be added as a wetting agent. Surfactants may include anionic detergents
such as
sodium lauryl sulfate, dioctyl sodium sulfosuccinate and dioctyl sodium
sulfonate. Cationic
detergents might be used and could include benzalkonium chloride or
benzethomium
.. chloride. The list of potential non-ionic detergents that could be included
in the formulation
as surfactants are lauromacrogol 400, polyoxyl 40 stearate, polyoxyethylene
hydrogenated
castor oil 10, 50 and 60, glycerol monostearate, polysorbate 40, 60, 65 and
80, sucrose fatty
acid ester, methyl cellulose and carboxymethyl cellulose. These surfactants
could be
present in the formulation of the active agent or derivative either alone or
as a mixture in
different ratios.
Pharmaceutical preparations which can be used orally include push-fit capsules
made of gelatin, as well as soft, sealed capsules made of gelatin and a
plasticizer, such as
glycerol or sorbitol. The push-fit capsules can contain the active ingredients
in
admixture with filler such as lactose, binders such as starches, and/or
lubricants such as
talc or magnesium stearate and, optionally, stabilizers. In soft capsules, the
active
compounds may be dissolved or suspended in suitable liquids, such as fatty
oils, liquid
paraffin, or liquid polyethylene glycols. In addition, stabilizers may be
added.
CA 02797719 2012-10-26
WO 2011/137320
PCT/US2011/034514
- 55 -
Microspheres formulated for oral administration may also be used. Such
microspheres
have been well defined in the art. All formulations for oral administration
should be in
dosages suitable for such administration.
For buccal administration, the compositions may take the form of tablets or
lozenges formulated in conventional manner.
For administration by inhalation, the compounds for use according to the
present
invention may be conveniently delivered in the form of an aerosol spray
presentation
from pressurized packs or a nebulizer, with the use of a suitable propellant,
e.g.,
dichlorodifluoromethane, trichlorofluoromethane, dichlorotetrafluoroethane,
carbon
dioxide or other suitable gas. In the case of a pressurized aerosol the dosage
unit may be
determined by providing a valve to deliver a metered amount. Capsules and
cartridges of
e.g. gelatin for use in an inhaler or insufflator may be formulated containing
a powder
mix of the compound and a suitable powder base such as lactose or starch.
Also contemplated herein is pulmonary delivery of the active agents (or
derivatives
.. thereof). The active agent (or derivative) is delivered to the lungs of a
mammal while
inhaling and traverses across the lung epithelial lining to the blood stream.
Other reports of
inhaled molecules include Adjei et al., 1990, Pharmaceutical Research, 7:565-
569; Adjei
et al., 1990, International Journal of Pharmaceutics, 63:135-144 (leuprolide
acetate);
Braquet et al., 1989, Journal of Cardiovascular Pharmacology, 13(suppl. 5):143-
146
(endothelin-1); Hubbard et al., 1989, Annals of Internal Medicine, Vol. III,
pp. 206-212 (al-
antitrypsin); Smith et al., 1989, J. Clin. Invest. 84:1145-1146 (a- 1-
proteinase); Oswein et
al., 1990, "Aerosolization of Proteins", Proceedings of Symposium on
Respiratory Drug
Delivery II, Keystone, Colorado, March, (recombinant human growth hormone);
Debs
et al., 1988, J. Immunol. 140:3482-3488 (interferon-g and tumor necrosis
factor alpha) and
Platz et al., U.S. Patent No. 5,284,656 (granulocyte colony stimulating
factor). A method
and composition for pulmonary delivery of drugs for systemic effect is
described in U.S.
Patent No. 5,451,569, issued September 19, 1995 to Wong et al.
Contemplated for use in the practice of this invention are a wide range of
mechanical devices designed for pulmonary delivery of therapeutic products,
including but
not limited to nebulizers, metered dose inhalers, and powder inhalers, all of
which are
familiar to those skilled in the art.
CA 02797719 2012-10-26
WO 2011/137320
PCT/US2011/034514
- 56 -
Some specific examples of commercially available devices suitable for the
practice
of this invention are the Ultravent nebulizer, manufactured by Mallinckrodt,
Inc.,
St. Louis, Missouri; the Acorn II nebulizer, manufactured by Marquest Medical
Products,
Englewood, Colorado; the Vent lin metered dose inhaler, manufactured by Glaxo
Inc.,
Research Triangle Park, North Carolina; and the Spinhaler powder inhaler,
manufactured
by Fisons Corp., Bedford, Massachusetts.
All such devices require the use of formulations suitable for the dispensing
of active
agent (or derivative). Typically, each formulation is specific to the type of
device employed
and may involve the use of an appropriate propellant material, in addition to
the usual
diluents, adjuvants and/or carriers useful in therapy. Also, the use of
liposomes,
microcapsules or microspheres, inclusion complexes, or other types of carriers
is
contemplated. Chemically modified active agent may also be prepared in
different
formulations depending on the type of chemical modification or the type of
device
employed.
Formulations suitable for use with a nebulizer, either jet or ultrasonic, will
typically
comprise active agent (or derivative) dissolved in water at a concentration of
about 0.1 to 25
mg of biologically active active agent per mL of solution. The formulation may
also include
a buffer and a simple sugar (e.g., for active agent stabilization and
regulation of osmotic
pressure). The nebulizer formulation may also contain a surfactant, to reduce
or prevent
surface induced aggregation of the active agent caused by atomization of the
solution in
fonning the aerosol.
Formulations for use with a metered-dose inhaler device will generally
comprise a
finely divided powder containing the active agent (or derivative) suspended in
a propellant
with the aid of a surfactant. The propellant may be any conventional material
employed for
.. this purpose, such as a chlorofluorocarbon, a hydrochlorofluorocarbon, a
hydrofluorocarbon, or a hydrocarbon, including trichlorofluoromethane,
dichlorodifluoromethane, dichlorotetrafluoroethanol, and 1,1,1,2-
tetrafluoroethane, or
combinations thereof. Suitable surfactants include sorbitan trioleate and soya
lecithin.
Oleic acid may also be useful as a surfactant.
Formulations for dispensing from a powder inhaler device will comprise a
finely
divided dry powder containing active agent (or derivative) and may also
include a bulking
agent, such as lactose, sorbitol, sucrose, or mannitol in amounts which
facilitate dispersal of
CA 02797719 2012-10-26
WO 2011/137320
PCT/US2011/034514
- 57 -
the powder from the device, e.g., 50 to 90% by weight of the formulation. The
active agent
(or derivative) should most advantageously be prepared in particulate form
with an average
particle size of less than 10 micrometers ( m), most preferably 0.5 to 5 Lm,
for most
effective delivery to the distal lung.
Nasal delivery of a pharmaceutical composition of the present invention is
also
contemplated. Nasal delivery allows the passage of a pharmaceutical
composition of the
present invention to the blood stream directly after administering the
therapeutic product
to the nose, without the necessity for deposition of the product in the lung.
Formulations
for nasal delivery include those with dextran or cyclodextran.
/0 For nasal administration, a useful device is a small, hard bottle to
which a
metered dose sprayer is attached. In one embodiment, the metered dose is
delivered by
drawing the pharmaceutical composition of the present invention solution into
a chamber
of defined volume, which chamber has an aperture dimensioned to aerosolize and
aerosol
formulation by forming a spray when a liquid in the chamber is compressed. The
chamber is compressed to administer the pharmaceutical composition of the
present
invention. In a specific embodiment, the chamber is a piston arrangement. Such
devices
are commercially available.
Alternatively, a plastic squeeze bottle with an aperture or opening
dimensioned to
aerosolize an aerosol formulation by forming a spray when squeezed is used.
The
opening is usually found in the top of the bottle, and the top is generally
tapered to
partially fit in the nasal passages for efficient administration of the
aerosol formulation.
Preferably, the nasal inhaler will provide a metered amount of the aerosol
formulation,
for administration of a measured dose of the drug.
The compounds, when it is desirable to deliver them systemically, may be
formulated for parenteral administration by injection, e.g., by bolus
injection or
continuous infusion. Formulations for injection may be presented in unit
dosage form,
e.g., in ampoules or in multi-dose containers, with an added preservative. The
compositions may take such forms as suspensions, solutions or emulsions in
oily or
aqueous vehicles, and may contain formulatory agents such as suspending,
stabilizing
and/or dispersing agents.
Pharmaceutical formulations for parenteral administration include aqueous
solutions of the active compounds in water-soluble form. Additionally,
suspensions of
CA 02797719 2012-10-26
WO 2011/137320
PCT/US2011/034514
- 58 -
the active compounds may be prepared as appropriate oily injection
suspensions.
Suitable lipophilic solvents or vehicles include fatty oils such as sesame
oil, or synthetic
fatty acid esters, such as ethyl oleate or triglycerides, or liposomes.
Aqueous injection
suspensions may contain substances which increase the viscosity of the
suspension, such
.. as sodium carboxymethyl cellulose, sorbitol, or dextran. Optionally, the
suspension may
also contain suitable stabilizers or agents which increase the solubility of
the compounds
to allow for the preparation of highly concentrated solutions.
Alternatively, the active compounds may be in powder form for constitution
with
a suitable vehicle, e.g., sterile pyrogen-free water, before use.
The compounds may also be formulated in rectal or vaginal compositions such as
suppositories or retention enemas, e.g., containing conventional suppository
bases such
as cocoa butter or other glycerides.
In addition to the formulations described previously, the compounds may also
be
formulated as a depot preparation. Such long acting formulations may be
formulated
.. with suitable polymeric or hydrophobic materials (for example as an
emulsion in an
acceptable oil) or ion exchange resins, or as sparingly soluble derivatives,
for example,
as a sparingly soluble salt.
The pharmaceutical compositions also may comprise suitable solid or gel phase
carriers or excipients. Examples of such carriers or excipients include but
are not limited
.. to calcium carbonate, calcium phosphate, various sugars, starches,
cellulose derivatives,
gelatin, and polymers such as polyethylene glycols.
Suitable liquid or solid pharmaceutical preparation forms are, for example,
aqueous or saline solutions for inhalation, microencapsulated, encochleated,
coated onto
microscopic gold particles, contained in liposomes, nebulized, aerosols,
pellets for
implantation into the skin, or dried onto a sharp object to be scratched into
the skin. The
pharmaceutical compositions also include granules, powders, tablets, coated
tablets,
(micro)capsules, suppositories, syrups, emulsions, suspensions, creams, drops
or
preparations with protracted release of active compounds, in whose preparation
excipients and additives and/or auxiliaries such as disintegrants, binders,
coating agents,
swelling agents, lubricants, flavorings, sweeteners or solubilizers are
customarily used as
described above. The pharmaceutical compositions are suitable for use in a
variety of
=
81662840
- 59 -
=
drug delivery systems. For a brief review of methods for drug delivery, see
Langer,
Science 249:1527-1533, 1990.
The active agents and optionally other therapeutics may be administered per se
(neat) or in the form of a pharmaceutically acceptable salt. When used in
medicine the
salts should be pharmaceutically acceptable, but non-pharmaceutically
acceptable salts
may conveniently be used to prepare pharmaceutically acceptable salts thereof.
Such
salts include, but are not limited to, those prepared from the following
acids:
hydrochloric, hydrobromic, sulphuric, nitric, phosphoric, maleic, acetic,
salicylic, p-
toluene sulphonic, tartaric, citric, methane sulphonic, formic, malonic,
succinic,
naphthalene-2-sulphonic, and benzene sulphonic. Also, such salts can be
prepared as
alkaline metal or alkaline earth salts, such as sodium, potassium or calcium
salts of the
carboxylic acid group.
Suitable buffering agents include: acetic acid and a salt (1-2% w/v); citric
acid
and a salt (1-3% w/v); boric acid and a salt (0.5-2.5% w/v); and phosphoric
acid and a
salt (0.8-2% w/v). Suitable preservatives include benzalkonium chloride (0.003-
0.03%
w/v); chlorobutanol (0.3-0.9% w/v); parabens (0.01-0.25% w/v) and thimerosal
(0.004-
0.02% w/v).
The therapeutic agent(s), including specifically but not limited to the active
agent,
may be provided in particles. Particles as used herein means nano or
microparticles (or
in some instances larger) which can consist in whole or in part of the active
agent or the
other therapeutic agent(s) as described herein. The particles may contain the
therapeutic
agent(s) in a core surrounded by a coating, including, but not limited to, an
enteric
coating. The therapeutic agent(s) also may be dispersed throughout the
particles. The
therapeutic agent(s) also may be adsorbed into the particles. The particles
may be of any
order release kinetics, including zero order release, first order release,
second order
release, delayed release, sustained release, immediate release, and any
combination
thereof, etc. The particle may include, in addition to the therapeutic
agent(s), any of those
materials routinely used in the art of pharmacy and medicine, including, but
not limited
to, erodible, noneroclible, biodegradable, or nonbiodegradable material or
combinations
thereof. The particles may be microcapsules which contain the active agent in
a solution
or in a semi-solid state. The particles may be of virtually any shape.
CA 2797719 2017-10-10
=
81662840
- 60 -
Both non-biodegradable and biodegradable polymeric materials can be used in
the manufacture of particles for delivering the therapeutic agent(s). Such
polymers may
be natural or synthetic polymers. The polymer is selected based on the period
of time
over which release is desired. Bioadhesive polymers of particular interest
include
bioerodible hydrogels described by H.S. Sawhney, C.P. Pathak and J.A. Hubei]
in
Macromolecules, (1993) 26:581-587.
These include polyhyaluronic acids, casein, gelatin, glutin, polyanhydrides,
polyacrylic
acid, alginate, chitosan, poly(inethyl inethacrylates), poly(ethyl
inethacrylates),
poly(butylmethacrylate), poly(isobutyl methacrylate), poly(hexylmethacrylate),
/0 poly(isodecyl methacrylate), poly(lauryl methacrylate), poly(phenyl
methacrylate),
poly(methyl acrylate), poly(isopropyl acrylate), poly(isobutyl acrylate), and
poly(octaclecyl acrylate).
The therapeutic agent(s) may be contained in controlled release systems. The
term "controlled release" is intended to refer to any drug-containing
formulation in
which the manner and profile of drug release from the formulation are
controlled. This
refers to immediate as well as non-immediate release formulations, with non-
immediate
release formulations including but not limited to sustained release and
delayed release
formulations. The term "sustained release" (also referred to as "extended
release") is
used in its conventional sense to refer to a drug formulation that provides
for gradual
release of a drug over an extended period of time, and that preferably,
although not
necessarily, results in substantially constant blood levels of a drug over an
extended time
period. The term "delayed release" is used in its conventional sense to refer
to a drug
formulation in which there is a time delay between administration of the
formulation and
the release of the drug there from. "Delayed release" may or may not involve
gradual
release of drug over an extended period of time, and thus may or may not be
"sustained
release."
Use of a long-term sustained release implant may be particularly suitable for
treatment of chronic conditions. "Long-term" release, as used herein, means
that the
implant is constructed and arranged to deliver therapeutic levels of the
active ingredient
for at least 7 days, and preferably 30-60 days. Long-tenn sustained release
implants are
well-known to those of ordinary skill in the art and include some of the
release systems
described above.
CA 2797719 2017-10-10
=
81662840
- 61 -
The present invention is further illustrated by the following Examples, which
in
no way should be construed as further limiting.
EXAMPLES
Materials and Methods
Cell Lines, enzymes and compounds
FleLa and U20S-DRGFP cells were grown in Dulbecco's modified Eagle's
medium (Invitrogen) supplemented with 15% fetal bovine serum (Invitrogen) and
penicillin/streptomycin glutamine (Invitrogen). Purified USP5 enzyme was
purchased
from Boston Biochem. UCH-L1 and UCL-113 were as reported previously (Mermerian
et
al, Bioorganic & Medicinal Chemistry Letters, 17:3729). C526 was synthesized
and the
purity validated by high-performance liquid chromatography.
Purification of USP1 and UAF1 protein complex
The co-purification of USP1/UAF'l complex was performed as described
previously (Cohn et al, Mol Cell, 2007). N-tenninal His-tagged LTSP1
GG670/671AA
was expressed using pFASTBac-IlTa vector (Invitrogen), and UAF1 were expressed
using pFASTBac-1 vector (Invitrogen). Each recombinant virus was produced by
transfecting the corresponding bacmid to Sf9 cells. For the purification, cell
pellets were
re-suspended in lysis buffer (50 mM Tris-HC1 [pH 8.01, 150 mM NaCl, 10 in.M
BME, 10
mM imidazolc, 10% glycerol, and 0.2% Triton X-100) and sonicated to lyse.
Lysates
were centrifuged, and the supernatants were incubated with Ni-NTA agarose
resin
(QIAGEN) for 1 hr. The resin was washed extensively, and the proteins were
eluted in
elution buffer (50 rnM Tris-HCl [pH 8.01, 100 mM NaCl, 10 mM BME, 10%
glycerol,
and 250 mM imidazole). Eluted protein was bound to a 5 ml IIiTrap Q-FF
cartridge (GE
Biosciences), washed with washing buffer (50 mM Tris-FIC1 [pH 8.01, 100 mM
KC1, 5
mM DTT, 0.1 mM EDTA, and 10% glycerol), eluted in the same buffer containing
500
mM KCI and stocked at -80 C. After final purification, the protein
concentration was
CA 2797719 2017-10-10
CA 02797719 2012-10-26
WO 2011/137320
PCT/US2011/034514
- 62 -
measured using Bradford assay and the quality of the protein was determined by
SDS-
PAGE and coomassie blue staining.
In Vitro Deubiquitination Assay
The in vitro enzymatic assays were performed using purified proteins from Sf9
cells as described previously (Cohn et al.). The assays using ubiquitin-AMC
(Ub-7-
amido-4-methylcoumarin; Boston Biochem) as substrate was carried out in a
reaction
buffer containing 20 mM HEPES-KOH (pH 7.8), 20 mM NaCl, 0.1 mg/ml ovalbumin,
0.5 mM EDTA and 10 mM dithiothreitol. The fluorescence was measured by
FluoStar
Galaxy Fluorometer (BMG Labtech). For the Ub-vinylsulfone (VS) assay, the
proteins
were incubated with Ub-VS at 0.5 M final concentration for 1 h at 30 C,
followed by
the immunoblotting analysis.
High Throughput Screening
The ubiquitin-Rho based enzyme assay for high throughput screening was
established in a 384-well format. The reaction buffer containing and
USP1/UA141
complex were added in 384 well plates using automated liquid handling robot-
Bio-Tek
Microfill (Bio-Tek Instrments Inc., VT), followed by addition of the compounds
(in
DMSO) from the compound library plates to wells using a pin transfer robotic
system at
a final concentration of 10 tiM. The reaction was composed of 0.1 nM purified
USP1/UAF complex, 75 nM ubiquitin Rhodamine (Ub-Rho, Boston Biochem, U-600)
and 2 ttM ubiquitin. Fluorescence was monitored in a FluoStar Galaxy
Fluorometer
(BMG Labtech). Fluorescence emission at 535 nm (Xex = 485 nm) was measured in
an
automated plate reader Envision 2 (Perkin Elmer, MA).
Clonogenic Assay
Cells were seeded in 6-well plate at 500 cells per well followed by treatment
with
C527. Colonies were allowed to grow for 7-10 days, fixed with a solution
containing
10% Methanol and 10% acetic acid at room temperature for 15 min and then
stained with
1% crystal violet in methanol. Colonies of >50 cells were counted, and the
surviving
fraction was calculated and normalized to untreated control.
CA 02797719 2012-10-26
WO 2011/137320
PCT/US2011/034514
- 63 -
HR Analysis
HR repair activity was analyzed using DR-GFP reporter as previously described
(Pierce AJ et al. Genes Dev, 13: 2633). U20S-DRGFP cells carrying a
chromosomally
integrated single copy of HR repair substrate were used to test the effect of
C527 on HR.
DSB-induced HR results in restoration and expression of GFP and was quantified
by
FACS. Briefly, 24 h after induction of chromosomal double-strand breaks
through the
expression of I-SceI, cells were treated with C527 for 24 h. Cells were
subjected to
FACS analysis and the percentage of GFP-positive cells was quantitated
relative to the
total viable cell number. For each analysis, 100,000 cells were processed.
Immunofluorescence
Cells were pre-extracted with extraction buffer (0.25% Triton-X100 in 200 mM
HEPES at pH 7.4, 50 mM NaC1, 3 mM MgCl2 and 300 mM sucrose) on ice for 2 min
prior to fixation with 4% paraformaldehyde. RAD51 foci were detected using
anti-
RAD51 antibody (Santa Cruz Biotechnology) and visualized using Alexa Fluor 488-
conjugated secondary antibody. The quantification of cells with RAD51 foci was
performed by counting the number of cells with RAD51 foci. At least 200 cells
were
counted from each sample. Data are represented as mean - SD from three
independent
experiments.
MTT assay
HeLa cells were seeded in 96-well plate and treated with C527 for 4 more days.
20 uL MTT (Sigma) at 5 mg/mL was added to each well and cells were left at 37
C for
incubation. 4 h later, the metabolic product was dissolved in a solution
containing 10%
SDS. 5% isopropanol and 0.01mol/L HC1. Optical density at 565 nm was measured
in an
automated plate reader. The percentage of cell survival was determined by
normalizing
to solvent vehicle treated cells.
.. Example 1. Recombinant USP1
Recombinant USP1 protein purified from Sf9 cells was expressed using
pFastflac-HTa vector (Invitrogen) containing an N-terminal His tag. For USP1
and
CA 02797719 2012-10-26
WO 2011/137320
PCT/US2011/034514
- 64 -
USP1/UAF1 complex purification, cell pellets were resuspended in lysis buffer
(50 mM
Tris-HC1, pH 8.0, 150 mM NaCl, 10 mMf3-mercaptoethanol, 10 mM imidazole, 10%
glycerol and 0.2% Triton X-100) and sonicated to lyse. Lysates were
centrifuged and the
supernatants were incubated with Ni-NTA agarose resin (Qiagen) for 1 hour. The
resin
was washed extensively and the proteins eluted in elution buffer (50 mM Tris-
HC1, pH
8.0, 100 mM NaC1, 10 mM f3-mercaptoethanol, 10% glycerol and 250 mM
imidazole).
Eluted protein was bound to a 5 mL HiTrap Q-FF cartridge (GE Biosciences),
washed
with washing buffer (50 mM Tris-HC1, pH 8.0, 100 mM KC1, 5 mM DTT, 0.1 mM
EDTA and 10% glycerol) and eluted in the same buffer containing 500 mM KC1.
to
Example 2. Recombinant UAF1
Recombinant UAF1 protein purified from SD cells was expressed using
pFastBac-1 vector (Invitrogen) with an engineered C-terminal Strep II tag.
Cell pellets
were resuspended in lysis buffer (50 mM Tris-HC1, pH 8.0, 150 mM NaCl, 2 mM
DTT,
10% glycerol), centrifuged, and the clarified lysate was incubated for 1 hour
with the
Strep-Tactin resin (Novagen). Following incubation, the resin was washed
extensively
and the protein eluted in the same buffer containing 2.5 mM desthiobiotin.
Example 3. Purification of Native USP1/UAF1 Complex
USP1 enzyme was purified with associated proteins as a native protein complex
from HeLa cells. A HeLa cell line stably expressing a Flag- and HA-epitope
tagged
fusion protein of USP1 (e-USP1) was generated by retroviral transduction. The
exogenous e-USP1 protein was expressed at levels comparable to the endogenous
protein
and also underwent autocleavage, a feature previously reported for the USP1
protein.
Huang et al. (2006) Nat Cell Biol 8:339-47; Nijman et al. (2005) Mol Cell
17:331-9.
Nuclear extract was prepared from HeLa cells, and the native USP1 complex was
purified by a two-step immunoaffinity purification scheme. Nakatani and
Ogryzko
(2003) Methods Enzymol 370:430-44. SDS-PAGE analysis of the purified complex
demonstrated the presence of multiple polypeptides. No polypeptides were
observed in a
mock purification from untransduced HeLa cells, indicating that all
polypeptides
copurifying with e-USP1 were bona fide subunits of the USP1 complex. Mass
spectrometric analysis of the polypeptides identified full length USP1, the N-
terminal
CA 02797719 2012-10-26
WO 2011/137320
PCT/US2011/034514
- 65 -
cleavage product of USP1, and the C-terminal cleavage product of USP1. A
fourth
major polypeptide, with a molecular weight of 80 kDa, was identified as the
previously
studied p80 protein. Park et al. (2002) Immunity 17:221-33. This protein is
now referred
to as UAF1 (USP1 Associated Factor 1). UAF1 contains 677 amino acids and
harbors 7
or 8 potential WD40-repeats in the N-terminal half and a predicted coiled coil
structure
in the C-terminal half. Tertiary structure prediction using the Phyre
software, available
on the world wide web at sbg.biolc.ac.ukLabout.phyre, suggests the presence of
a
complete propeller structure comprised by the WD40 repeats. The intensities of
Coomassie blue stained USP1 and UAF1 proteins in the SDS-PAGE were nearly
identical, suggesting stoichiometric amounts of the two proteins in the
complex and a
possible functional relationship.
The protein subunits in the USP1 complex were analyzed by immunoblotting,
using antibodies to the USP1 protein (Nijman et al. (2005) Mol Cell 17:331-9)
and newly
generated antibodies against the UAF1 protein. The results confirmed the
presence of
full length USP1, N-terminal USP1, and UAF1 in the USP1 complex, in good
correlation
with the Coomassie blue stain of the complex.
Flag-HA-tagged UAF1 was expressed in HeLa cells and the protein was
immunoprecipitated with associated proteins. Immunoblotting of the
immunoprecipitate
revealed the presence of endogenous USP1 as well as its cleavage product,
confirming
the presence of a native USP1/UAF1 complex. The majority of USP1 protein in
the
HeLa cells was observed to exist as a protein complex with UAF1.
Example 4. Production of a Polyclonal Antibody to UAF1
A polyclonal rabbit antibody was raised against a fragment of the UAF1 protein
consisting of amino acids 400-677. A 6xHis-UAF1 (400-677) fusion protein was
expressed in E. coli and purified over an NTA column. The purified protein was
injected
subcutaneously into a rabbit with Freund's complete adjuvant in the first
injection and
Freund's incomplete adjuvant for the following boost injections.
Example 5. Initial In Vitro Screening to Identify Small Molecule Inhibitors of
USP1
Deubiquitinating Activity
CA 02797719 2012-10-26
WO 2011/137320
PCT/US2011/034514
- 66 -
USP1/UAF1 protein complex serves as an excellent target for high throughput
screening to identify small molecule inhibitors as radioprotective agents. A
baculovirus
mediated SF9 insect cell expression method has been developed for both USP1
and
UAF1. The expressed proteins are purified by affinity, ion exchange, and gel
filtration
chromatography. Briefly, a baculovirus for USP1 and UAF1 was generated using
the
Bac-to-Bac baculovirus expression system (Invitrogen, CA). Full length USP1
contains
an autocleavage site at residue 670-671, where cleavage occurs at GG after a
Ubiquitin
like motif. Huang et al. (2006) Nat Cell Biol. 8:1043-45. In order to purify
the intact
full length protein, two Glycine residues were mutated to Alanines using site
directed
mutagenesis (Stratagene). This modified full length USP1 clone was PCR
amplified and
subcloned into pFastBac-HT vectors (Invitrogen, CA) with N-terminal His tag.
USP1
Associated Factor 1 (UAF1) was also cloned into pFastBac vector, but without
the N-
terminal His tag. The clones were PCR verified and transformed into DH10Bac
cells for
blue-white colony selection. Bacmid DNA from the re-confirmed colony was
extracted
and presence of correct USP1 or UAF1 clones were confirmed by PCR
amplification.
'the bacmid DNA were then transfected into S149 cells using the Cellfectin
reagent
(Invitrogen, CA). P1 virus sets were collected following manufacturer's
protocol and
used to amplify P2 and P3 virus respectively. SF9 cells were co-infected with
USP1 and
UAF1 viruses to co-express these two proteins. A series of virus titer was
used to
optimize the expression level of USP1 and UAF1. Best expression of USP1 and
UAF1
was observed with the virus titers of 1 1 in 2000 1 and 4000 ill of SF9
culture
respectively. Expression level of these proteins at various time points after
infection was
also tested to determine optimum time of expression. Best expression was
achieved at
60 hours after infection. Finally, SF9 suspension culture was infected at cell
density 1.8
million/ml and grown at 28 C on orbital shaker for 60 hours. After
expression, cells
were harvested at 500 g for 7 minutes, washed with 1xPBS, centrifuged again
and stored
at -80 C for purification.
USP1 and UAF1 complex was initially purified by Ni-NTA affinity purification.
SF9 cells were resuspended in a pre-chilled lysis buffer containing 50 mM Tris
pH 8.0,
.. 200 mM NaCl. 10 mM imidazole, 10 mM P-mercaptoethanol, 10% glycerol and
0.2%
Triton X. After sonication, the cell lysate was centrifuged at 20,000 r.p.m.
for 45
minutes at 4 C Ni-NTA resin equilibrated in the lysis buffer and added to the
soluble
CA 02797719 2012-10-26
WO 2011/137320
PCT/US2011/034514
- 67 -
supernatant from the whole cell lysate. 2-3 ml of resin was added to per 2
liter of SF9
culture. The resin was incubated with mixing for 1 hour at 4 C and spun down
resin at
500xg for 5 min. After carefully removing the supernatant, the Ni-NTA resin
was re-
suspended in lysis buffer to wash out any unbound proteins. After spinning
down the
resin again, it was resuspended in wash buffer containing 50 mM Tris pH 8.0,
150 mM
NaCl, 20 mM imidazole, 10 mMI3-mercaptoethanol, 10% glycerol, and 1% Triton X.
After spinning down the resin again, it was washed twice with wash buffer
containing 50
mM Tris pH 8.0, 1000 mM NaC1, 20 mM imidazole, 10 mM P-mercaptoethanol, and
10% glycerol, followed by a final wash with buffer containing 50 mM Tris pH
8.0, 100
mM NaCl, 20 mM imidazole, 10 mM P-mercaptoethanol, and 10% glycerol. The bound
USP1/UAF1 complex was eluted slowly from the column by elution buffer
containing
50 mM Tris pH 8.0, 100 mM NaCl, 250 mM imidazole, 10 mM P-mercaptoethanol, and
10% glycerol. At this stage, eluted USP1/UAF1 complex was further purified by
ion
exchange chromatography using Q-sepharose column. The purified protein complex
was
further purified by gel filtration using S-200 column. Final buffer to be used
in gel
filtration column contained 50 mM Tris pH 8.0, 100 mM KC1, 5 mM DTT, and 0.1
mM
EDTA. After final purification, quality of the protein was checked by SDS-PAGE
and
Coomassie blue staining. Finally, USP1/UAF1 concentration was measured by
Bradford
assay (BioRad, Calif.). 11 mg of protein complex were purified from 2 liter
SF9 culture
at a final concentration of 0.77 mg/ml.
In order to develop a highly sensitive assay with easy readout for high
throughput
detection, the fluorogenic compound Ubiquitin-7-amido-4-inethylcouniarin (AMC-
Ub,
Boston Biochem) was used as a substrate for USP1/UAF1 enzyme complex. USP1
catalyzes the cleavage of AMC-Ub, releasing free AMC moiety, which leads to
increase
in fluorescence emission at 460 nm (X,,õ=380 nm). Dang et al. (1998)
Biochemistry
37:1868-79. Catalytic activity towards AMC-Ub increases 35 fold compared to
USP1
enzyme alone. Initial enzyme assay development was performed in 96-well plates
with
100 Rlreaction volume containing 20 mM HEPES-KOH, 0.1 mg/ml ovalbumin (Sigma),
0.5 mM Ell'I'A, and 10 mM DTT. 2.5 nM USP1/UAF1 enzyme complex was mixed
with the reaction buffer, incubated at 37" C for 10 minutes and then 0.3
AMC-Ub
was added. Fluorescence was measured using a Fluostar Galaxy Fluorometer (BMG
Labtech Inc.).
CA 02797719 2012-10-26
WO 2011/137320
PCT/US2011/034514
- 68 -
Ubiquitin aldehyde (Al-Ub) is a potent covalent inhibitor of deubiquitinating
enzymes, and it inhibits the enzyme by covalently attaching to the active
site. There is a
sharp decrease in enzyme activity upon incubation of USP1/UAF1 complex with 25
nM
of Al-Ub. The significant decrease in signal upon Al-Ub can easily be detected
in high
throughput data collection and data interpretation. Hence, Al-Ub can be used
as positive
control for inhibition in screening assays, including high throughput
screening, and any
potential inhibition signal can be compared to this control.
Once the preliminary enzyme assay was established in 96-well format, it was
optimized for 384 wells for high throughput screening (I ITS) of inhibitors.
The major
points to be considered were the volume of the reaction and amount of AMC-Ub
substrate to be used in the assay. Since the price of AMC-Ub is very high, in
order to
make it a feasible assay for HTS, lowest workable amount of the AMC conjugate
was
used. Reaction volume was minimized to 30 p.1 in order to meet the
recommendation of
ICCB-L screening facility (Harvard). In the preliminary optimization
experiment, a
series of pilot experiments were performed with varying AMC-Ub concentrations
(0.025
1.1M to 0.2 ,M), along with the varying concentration of USP1/UAF1 complex
(0.2 nM
to 3.2 nM). This concentration grid was performed in 384-well plates (Corning,
3711)
using the automated liquid handler present at ICCB-L screening facility. After
incubating the enzyme and buffer at 37D C for 10 minutes, AMC-Ub was added and
fluorescence emission at 460 lint (k,380 tun) was measured in an automated
plate
reader Envision 2 (Perkin Elmer). Fluorescence reading for all time points up
to one
hour was noted and the data for different AMC-Ub concentration range was
plotted with
each USP1/UAF1 enzyme complex concentration. Comparison of fluorescence
emission
in each set showed the minimum optimal AMC-Ub concentration required with
optimum
USP1/UAF1 concentration to obtain significant increase in signal compared to
the
baseline. Based on the results, 0.1 p.M AMC-Ub concentration along with 1.6 nM
USP1/UAF1 concentration produced significantly increased signal from the
baseline and
these optimum concentrations were used for HTS. So by this optimization AMC-Ub
concentration was reduced 3 times (0.3 1,1M to 0.11.tM) and volume of the
reaction was
reduced 3.33 times (100 .1 to 30 p.1). Taken together, the overall use of the
substrate was
reduced 10 times by doing this optimization (3x3.33). Unlike single enzyme
assay, only
one time point can be collected in HTS with thousands of compound screen. This
CA 02797719 2012-10-26
WO 2011/137320
PCT/US2011/034514
- 69 -
optimization step also shows the desirable time point to read fluorescence
signal is 15
minutes after addition of AMC-Ub, as this is before the reaction reaches the
saturation.
After large scale expression-purification of USP1/UAF1 protein complex, high
throughput screening was performed. Buffers and USP1/ITAF1 complex were added
into 384-well plates (Corning, black 3711) using automated liquid handling
robot Bio-
Tek Microfill (Bio-Tek Instruments Inc., VT). This was followed by addition of
compounds to the plate wells using pin transfer robotic system. This includes
a custom
designed Seiko Cartesian robot with pin arrays (V & P Scientific Inc.) and
Zymark
Twister II robotic arm to stack and transfer compound plates and assay plates.
In this
step, 100 !IL of the compounds (in DMS0) from compound library plates was
added to
the 384-well assay plates. After incubating the assay plates at 37 C for 10
minutes,
desired volume of AMC-Ub was added by the automated liquid handler. Finally,
fluorescence emission at 460 nm (Xõ=380 nm) was measured in an automated plate
reader Envision 2 (Perkin Elmer, Mass.). The Z' factor for the assay was
calculated
(Zhang et al. (1999) J Biornol Screen 4:67-73) and, if required, the assay was
further
optimized until Z'>0.5.
The HTS assay was run using the ICCB-L Biomol bioactive library (Harvard
Medical School, Boston, Mass.). ICCB-L has a collection of over 150,000
compounds
in their compound library, which consists of known bioactive libraries,
natural product
libraries, and commercial libraries such as ChemDiv, ChemBridge, etc. Known
bioactive libraries containing Biomol 1CCB known bioactives, N1NDS, and
Prestwick
collections were tested first for potential inhibitors. This set of the
library contains many
FDA-approved drugs and the compounds are known to exhibit low toxicity and
high
cellular retention, making them excellent targets for further cell-based
assay. The
reactions were performed using automated instruments as described above. Among
two
bioactive libraries tested, three compounds, 13-lapachone, Biomol AP401
(propidium
iodide), and RK-682, were previously shown to have substantial low
fluorescence signal
in the range of Al-Ub-treated positive control sets. US 2008/0167229 Al.
Identification
of these compounds by high throughput screening established the validity of
the
experimental setup.
CA 02797719 2012-10-26
WO 2011/137320
PCT/US2011/034514
- 70 -
Results. Four additional small molecule inhibitors of USP1 deubiquitinating
activity were identified from a screen of approximately 150,000 compounds. The
compounds so identified included the following:
0
NHLT)2
Ci
o Formula III (933),
0
o
N\ =0
Formula IV (527),
0
NIC))
0 Formula V (947), and
0
OMe
N
0 OMe
Formula VI (009).
Example 6. Additional In Vitro Screening to Characterize Small Molecule
Inhibitors of
USPI Identified in Initial Screening
Hits from the initial screening were analyzed further in secondary screening.
In
this step, the compounds were re-tested for inhibition using the AMC-Ub in
vitro
enzyme assay. After reconfirming inhibition, different concentration range of
the
inhibitors were tested to show dose dependency of inhibition and IC50
calculation.
In one set of experiments, HeLa cells were incubated in the presence of 50 ,M
of
selected inhibitors including the compound of Formula III (933) and the
compound of
Formula IV (527) for 2, 4, or 6 hours, followed by cell lysis. Ub-aldehyde (Ub-
Ald, a
known USP1 inhibitor) was used as a positive control, and dimethysulfoxide
(DMSO)
was used as a negative control. Lysates were incubated with the LTb-vinyl
sulfone (Ub-
CA 02797719 2012-10-26
WO 2011/137320
PCT/US2011/034514
- 71 -
Vs) reagent for 40 minutes. Proteins were electrophoresed on SDS-PAGE,
transferred to
nitrocellulose, and immunoblotted with the anti-USP1 antibody. Results are
shown in
Fig. 2.
Results. Ub-aldehyde blocked Ub-Vs conjugation of USP1 (lane 4), whereas
DMSO alone did not block conjugation of USP1 (lane 3). Of five putative USP1
inhibitors evaluated (lanes 5-9), the C527 compound was a potent inhibitor of
deubiquitinase activity.
IC50 values for compounds of Formulas determined following 4 hour
preincubation, are shown in Table 1.
Table 1.
Compound IC50 (nM)
Formula III (933) --a
Formula IV (527) 134
Formula V (947) 7,500
Folinula VI (009) 2,400
a No data
Example 7. Cell-Based Screening to Further Characterize Small Molecule
Inhibitors of
USPI Identified in In Vitro Screening
Candidate USP1 inhibitors from initial and secondary screens were then used in
cell-based assays to look for USP1 inhibition in vivo using levels of FANCD2-
Ub and
PCNA-Ub as biomarkers for inhibition. For the cell-based assay, cells were
treated with
the candidate compounds and levels of monoubiquitinated FANCD2 and PCNA were
tested both with and without DNA damage. USP1 inhibition was detected as high
level
of endogenous monoubiquitinated FANCD2 and PCNA. For this assay HeLa and
HEK293T cells were treated with selected concentrations of these compounds and
grown
in Dulbecco's modified Eagle's medium supplemented with 15% heat-inactivated
fetal
calf serum in a humidified 5% CO2 incubator at 37 C. Damage was induced by
both
UV irradiation using Stratalinker (Stratagene) and treatment with mitomycin C
(MMC,
Sigma). After cell lysis, levels of monoubiquitinated FANCD2 and PCNA in whole
cell
lysate was tested by immunoblotting with anti-FANCD2 antibody (sc-20022; Santa
Cruz
CA 02797719 2012-10-26
WO 2011/137320
PCT/US2011/034514
- 72 -
Biotechnology) and anti-PCNA antibody (sc-56: Santa Cruz Biotechnology).
Protection
from chromosome aberrations were also tested in UV/MMC-damaged cells treated
with
candidate USP1 inhibitors using standard chromosome breakage assay. Yang et
al.
(2001) Blood 98:3435-40. Representative results are shown in Fig. 3.
Results. HeLa cells were incubated with the compound of Formula IV (527) for
eight hours, followed by cell lysis and incubation of protein extract with Ub-
vinyl
sulfone. Increasing concentrations of the compound of Formula IV (527)
resulted in a
dose-dependent blockade of USP1 deubiquitinase activity (lanes 3, 4, 5).
Increasing
concentrations of the compound of Formula IV (527) also resulted in a dose-
dependent
increase in FANCD2-Ub levels and FANCI-Ub levels (lanes 7, 8, 9).
Example 8. USP1/UAF1 Inhibitor Sensitizes Hall cells to the DNA Crosslinking
Agent
Mitotnycin C
HeLa cells were pretreated for six hours with selected concentrations (0, 200,
or
500 nM) of mitomycin C (MMC), washed, then exposed to selected concentrations
(0,
10, or 20 M) of the compound of Formula IV (527) for eight hours. Cell
survival was
measured by redox dye. Results are shown in Fig. 4.
Results. Pretreatment with the compound of Formula IV (20 micromolar)
resulted in sensitization of the HeLa cells to the cytotoxic effects of MMC.
Example 9. C527 inhibits USPI/UAF1 in a time-course and dose-dependent manner
The ability of C527, the compound of Formula IV (527) to inhibit USP1/UAF1 in
a time-course and dose-dependent manner (Fig. 5) was confirmed using ubiquitin-
AMC
(Ub-7-amido-4-methylcoumarin), another ubiquitin derivative similar to Ub-
Rhodamine
(Ub-Rho) but having a different emission wavelength, as substrate.
Pretreatment of
USP1/UAF1 with a single dose of C527 (1 uM) for variable time periods resulted
in a
serial increase in inhibition of DUB activity (i.e., fluorescence) (Fig. 5A).
Treatment for
2 h with an increase in dose of C527, ranging from 0.5 to 2 pM, resulted in a
dose-
dependent decrease in DUB activity (Fig. 5B). Based on these results, the IC50
of C527
for the USP1/UAF1 complex was 0.88 0.03 pM (Fig. 5C).
Example 10. C527 is a pan-deubiquitinating enzyme inhibitor in vitro
CA 02797719 2012-10-26
WO 2011/137320
PCT/US2011/034514
- 73 -
To examine the specificity of C527 in USP1/UAF1 inhibition, its ability to
inhibit
other DUB enzymes in vitro was examined. It has been recently showed that UAF1
stimulates not only USP1, but also two other DUB enzymes, USP12 and USP46
(Cohn
MA, Kee Y, Haas W, Gygi SP, D'Andrea AD. UAF1 is a subunit of multiple
deubiquitinating enzyme complexes. J Biol Chem, 2009;284:5343-5351). Moreover,
the
active complex of USP12 contains an additional WD40 subunit (WDR20). C527 also
inhibited the DUB activity of the USP12/UAF1/WDR20 complex, as well as the
USP5
enzyme in vitro (Fig. 6A and 6B). However, the IC50 of C527 for these DUB
enzymes
was slightly higher (Fig. 6D). UCH-L1 and UCII-L3 are members of a different
subclass
of deubiquitinating enzymes, referred to as the ubiquitin-carboxy terminal
hydrolases
(Mermerian AH, Case A, Stein RL, Cuny GD. Structure-activity relationship,
kinetic
mechanism, and selectivity for a new class of obiquitin C-terminal hydrolase-
Li (UCH-
L1) inhibitors. Bioorg Med Chem Lett. 2007;17:3729-3732). Interestingly, C527
had
considerably less inhibitory effect on these deubiquitinating enzymes, even
though they
are also cysteine proteases. Taken together, C527 is a pan-deubiquitinating
enzyme
inhibitor in vitro, though it has some specificity for the USP subfamily of
DUBs,
compared to the UCH subfamily.
The ability of C527 to inhibit the deubiquitinating activity of native USP1
complexes derived from human cells was determined next (Fig. 7). For this
purpose, the
.. HA-tagged ubiquitin-VS, an affinity reagent known to covalently modify and
trap active
DUB enzymes in cell extracts was used. Cell extracts prepared from HeLa cells
were
pretreated with increasing concentrations of C527 for 4 h, followed by the
incubation
with Ub-VS for 40 mm at 30 C. The enzymatic activity of endogenous DUBs was
indicated by the conjugation of Ub-VS, detected as HA-labeling (see Fig. 7,
high
molecular weight bands). Solvent vehicle treated cell extracts showed
increased
enzymatic activity of endogenous DI Ms, as indicated by a whole panel of HA-
labeling
at different molecular weight. Ub-VS conjugation was inhibited by ubiquitin-
aldehyde
(Ub-Ald), a non-specific inhibitor of DUBs (lane 3). In contrast, C527 failed
to inhibit
the Ub-VS labeling of other endogenous DUB enzyme complexes but did inhibit
USP1
binding to Ub-VS in the same reaction in a dose-dependent manner. These data
indicate
that C527 has a preferential inhibition of the native USP1, compared to other
native
DUB s in the extract.
CA 02797719 2012-10-26
WO 2011/137320
PCT/US2011/034514
- 74 -
Example 11. C527 impairs homologous recombination repair
Recent studies have indicated that the Fanconi Anemia pathway is required for
optimal homologous recombination (HR) repair (Nakanishi K, Yang YG, Pierce AJ,
et
al. Human Fanconi anemia monoubiquitination pathway promotes homologous DNA
repair. Proc Natl Acad Sci US A. 2005;102:1110-1115). Disruption of FANCD2
monoubiquitination or of USP1 activity (Oestergaard VH, Langevin F, Kuiken HJ,
et al.
Deubiquitination of FANCD2 is required for DNA crosslink repair. Mol Cell.
2007;28:798-809; Kim JM, Parmar K, Ihiang M, et al. Inactivation of murine
Uspl
results in genomic instability and a Fanconi anemia phenotype. Dev Cell.
2009;16:314-
320) results in decreased HR. A gene conversion assay was used to examine the
effect
of C527 on cellular HR activity (Fig. 8A). Interestingly, C527 caused a dose-
dependent
decreased in gene conversion, based on the measurement of cellular GFP in this
assay,
and inhibition occurred in the 0 to 5 uM range (Fig. 8A). In contrast, C527
did not
inhibit non-homologous end joining (NHEJ) activity under the same condition
(Fig. 8B).
To confirm that the C527 treatment caused HR defect, RAD51 foci formation,
another surrogate marker of cellular HR activity, was tested (Fig. 8C).
Camptothecin
activated the assembly of RAD51 and H2AX foci. C527 treatment inhibited RAD51
foci formation but had a non-detectable effect on H2AX foci formation. Taken
together,
these results further indicate that C527 has HR-inhibitory activity.
Example 12. C527 causes DNA damage and inhibits the proliferation of tumor
cells
Impaired HR integrity may lead to genotoxic effect. Genotoxic agents are known
to directly activate DNA damage response pathways, resulting in FANCD2
monoubiquitination and CHK1 phosphorylation (Fig. 9A). The results indicate
that
C527, like the genotoxic crosslinking agent MMC, can activate markers of the
DNA
damage response. Accordingly, C527 treatment inhibited cell proliferation as
assessed
using clonogenic assay (Fig. 9B).
Example 13. C527 sensitizes tumor cells to DNA damaging agents
Inhibitors of the FA-BRCA pathway are known to sensitize tumor cells to the
cytotoxic effects of ICL-indicating agents. Specifically, it is known that
proteasome
CA 02797719 2012-10-26
WO 2011/137320
PCT/US2011/034514
- 75 -
inhibitors block FANCD2 monoubiquitination, perhaps through depletion of
intracellular
pools of free ubiquitin, resulting in inhibition of HR repair (Chirnomas D,
Taniguchi T,
de la Vega M, et al. Chemosensitization to cisplatin by inhibitors of the
Fanconi
anemia/BRCA pathway. Mol Cancer Ther. 2006;5:952-961). The ability of C527 to
sensitize pretreated human cells to MMC or to other agents was tested. C527
sensitized
cells to MMC and to CPT, consistent with its ability to specifically inhibit
HR in cells
(Fig.10).
DNA Repair inhibitors have recently emerged as a novel class of anti-cancer
agents, Conventional anticancer agents, such as the alkylating agents busulfan
and
cyclophosphamide, kill cancer cells by causing DNA damage. Tumors can become
resistant to conventional therapy by amplifying DNA repair pathways. DNA
repair
inhibitors, such as the newly developed PARP inhibitors, block DNA repair and
sensitize
tumor cells to conventional agnets.
There are several DNA repair pathways in human cells, and each pathway deals
with a specific set of DNA lesions. The Fanconi Anemia (FA-BRCA) pathway,
regulates homologous recombination repair and is require for interstrand DNA
crosslink
repair. A DNA repair inhibitor for this pathway is expected to sensitize tumor
cells to
crosslinking agents, such as cisplatin and mitomycin C.
Several enzymatic steps comprise the FA-BRCA pathway, and each step is a
potential target for inhibitor development. Critical catalytic events in the
FA-BRCA
pathway include 1) the E3 ligase dependent monoubiquitination of FANCD2 and 2)
the
USP1-dependent deubiquitination of FANCD2-Ub. Disruption of either enzymatic
step
results in cisplatin sensitization.
Recent studies have identified small molecule inhibitors of FANCD2
monoubiquitination, and these compounds are currently in preclinical
development
(curcumin and proteasome inhibitor). Small molecule inhibitors of the FA-BRCA
pathway can block HR repair and sensitize tumors to DNA crosslinking agents
such as
cisplatin. The results described herein demonstrate that C527 can inhibit
USP1/UAF1
in vitro in a dose-dependent manner. Cells exposed to C527 exhibited reduced
USP1
activity, increased monoubiquitinated FANCD2 levels, and a defect in
homologous
recombination repair. C527 also enhanced the cellular sensitivity of cancer
cells to
mitomycin C and camptothecin but not to etoposide treatment. The
identification of this
CA 02797719 2012-11-14
76
lead inhibitor against the USP1/UAF1 complex provides a structural basis for
further
development of new anticancer drugs.
Example 14. In vivo activtily of C527
Bortezomib (Velcade) has been shown to inhibit the FA-BRCA pathway.
Preliminary breast cancer xenograft studies with one HR pathway-proficient
breast
tumor line have been conducted. Bortezomib inhibited the FA/BRCA pathway and
the
combination of Vekade plus PARP inhibitor (ABT-888) resulted in enhanced
killing of
this cell line in vivo, compared to either agent alone (data not shown)
Nude mice bearing MDA-MB-231 tumor xenografts are treated with either
vehicle control, ABT-888 (25mg/kg B.W), cisplatin, C527 or combination of ABT-
888
or cysplatin and C527. It is anticipated that C527 will have homologous
recombination
(HR)-inhibiting activity in vivo, and will synergize with PARP inhibitor
and/or Cisplatin
in vivo.
The foregoing written specification is considered to be sufficient to enable
one
skilled in the art to practice the invention. The present invention is not to
be limited in
scope by examples provided, since the examples are intended as a single
illustration of
one aspect of the invention and other functionally equivalent embodiments are
within the
scope of the invention. Various modifications of the invention in addition to
those
shown and described herein will become apparent to those skilled in the art
from the
foregoing description and fall within the scope of the appended claims. The
advantages
and objects of the invention are not necessarily encompassed by each
embodiment of the
invention.
SEQUENCE LISTING IN ELECTRONIC FORM
In accordance with Section 111(1) of the Patent Rules, this
description contains a sequence listing in electronic form in ASCII
text format (file: 64371-1186 Seq 01-NOV-12 vl.txt).
A copy of the sequence listing in electronic form is available from
the Canadian Intellectual Property Office.
The sequences in the sequence listing in electronic form are
reproduced in the following table.
CA 02797719 2012-11-14
76a
SEQUENCE TABLE
<110> Dana-Farber Cancer Institute, Inc.
The Brigham and Women's Hospital, Inc.
<120> SMALL MOLECULE INHIBITORS OF USP1 DEUBIQUITINATING ENZYME
ACTIVITY
<130> 64371-1186
<140> CA national phase of PCT/U52011/034514
<141> 2011-04-29
<150> US 61/329984
<151> 2010-04-30
<160> 4
<170> PatentIn version 3.5
<210> 1
<211> 76
<212> PRT
<213> Homo sapiens
<400> 1
Met Gin Ile Phe Val Lys Thr Leu Thr Gly Lys Thr Ile Thr Leu Glu
1 5 10 15
Val Glu Pro Ser Asp Thr Ile Glu Asn Val Lys Ala Lys Ile Gin Asp
20 25 30
Lys Glu Gly Ile Pro Pro Asp Gin Gin Arg Leu Ile Phe Ala Gly Lys
35 40 45
Gin Leu Glu Asp Gly Arg Thr Leu Ser Asp Tyr Asn Ile Gin Lys Glu
50 55 60
Ser Thr Leu His Leu Val Leu Arg Leu Arg Gly Sly
65 70 75
<210> 2
= <211> 785
<212> PRT
<213> Homo sapiens
<400> 2
Met Pro Gly Val Ile Pro Ser Glu Ser Asn Gly Leu Ser Arg Gly Ser
1 5 10 15
Pro Ser Lys Lys Asn Arg Leu Ser Leu Lys Phe Phe Gin Lys Lys Glu
20 25 30
Thr Lys Arg Ala Leu Asp Phe Thr Asp Ser Gin Glu Asn Glu Glu Lys
35 40 45
Ala Ser Glu Tyr Arg Ala Ser Glu Ile Asp Gin Val Val Pro Ala Ala
50 55 60
Gin Ser Ser Pro Ile Asn Cys Glu Lys Arg Glu Asn Leu Leu Pro Phe
65 70 75 80
CA 02797719 2012-11-14
6b
Val Gly Leu Asn Asn Leu Gly Asn Thr Cys Tyr Leu Asn Ser Ile Leu
85 90 95
Gin Val Leu Tyr Phe Cys Pro Gly Phe Lys Ser Gly Val Lys His Leu
100 105 110
Phe Asn Ile Ile Ser Arg Lys Lys Glu Ala Leu Lys Asp Glu Ala Asn
115 120 125
Gin Lys Asp Lys Gly Asn Cys Lys Glu Asp Ser Leu Ala Ser Tyr Glu
130 135 140
Leu Ile Cys Ser Leu Gin Ser Leu Ile Ile Ser Val Glu Gin Leu Gin
145 150 155 160
Ala Ser Phe Leu Leu Asn Pro Glu Lys Tyr Thr Asp Glu Leu Ala Thr
165 170 175
Gin Pro Arg Arg Leu Leu Asn Thr Leu Arg Glu Leu Asn Pro Met Tyr
180 185 190
Glu Gly Tyr Leu Gin His Asp Ala Gin Glu Val Leu Gin Cys Ile Leu
195 200 205
Gly Asn Ile Gin Glu Thr Cys Gin Leu Leu Lys Lys Glu Glu Val Lys
210 215 220
Asn Val Ala Glu Leu Pro Thr Lys Val Glu Glu Ile Pro His Pro Lys
225 230 235 240
Glu Glu Met Asn Gly Ile Asn Ser Ile Glu Met Asp Ser Met Arg His
245 250 255
Ser Glu Asp Phe Lys Glu Lys Leu Pro Lys Gly Asn Gly Lys Arg Lys
260 265 270
Ser Asp Thr Glu Phe Gly Asn Met Lys Lys Lys Val Lys Leu Ser Lys
275 280 285
Glu His Gin Ser Leu Glu Glu Asn Gin Arg Gin Thr Arg Ser Lys Arg
290 295 300
Lys Ala Thr Ser Asp Thr Leu Glu Ser Pro Pro Lys Ile Ile Pro Lys
305 310 315 320
Tyr Ile Ser Glu Asn Glu Ser Pro Arg Pro Ser Gin Lys Lys Ser Arg
325 330 335
Val Lys Ile Asn Trp Leu Lys Ser Ala Thr Lys Gin Pro Ser Ile Leu
340 345 350
Ser Lys Phe Cys Ser Leu Gly Lys Ile Thr Thr Asn Gin Gly Val Lys
355 360 365
Gly Gin Ser Lys Glu Asn Glu Cys Asp Pro Glu Glu Asp Leu Gly Lys
370 375 380
Cys Glu Ser Asp Asn Thr Thr Asn Gly Cys Gly Leu Glu Ser Pro Gly
385 390 395 400
Asn Thr Val Thr Pro Val Asn Val Asn Glu Val Lys Pro Ile Asn Lys
405 410 415
Gly Glu Glu Gin Ile Gly Phe Glu Leu Val Glu Lys Leu Phe Gin Gly
420 425 430
Gin Leu Val Leu Arg Thr Arg Cys Leu Glu Cys Glu Ser Leu Thr Glu
435 440 445
Arg Arg Glu Asp Phe Gin Asp Ile Ser Val Pro Val Gin Glu Asp Glu
450 455 460
Leu Ser Lys Val Glu Glu Ser Ser Glu Ile Ser Pro Glu Pro Lys Thr
465 470 475 480
Glu Met Lys Thr Leu Arg Trp Ala Ile Ser Gin Phe Ala Ser Val Glu
485 490 495
Arg Ile Val Gly Glu Asp Lys Tyr Phe Cys Glu Asn Cys His His Tyr
500 505 510
Thr Glu Ala Glu Arg Ser Leu Leu Phe Asp Lys Met Pro Glu Val Ile
515 520 525
= . CA 02797719 2012-11-14
76c
Thr Ile His Leu Lys Cys Phe Ala Ala Ser Gly Leu Glu Phe Asp Cys
530 535 540
Tyr Gly Gly Gly Leu Ser Lys Ile Asn Thr Pro Leu Leu Thr Pro Leu
545 550 555 560
Lys Leu Ser Lou Glu Glu Trp Ser Thr Lys Pro Thr Asn Asp Ser Tyr
565 570 575
Gly Leu Phe Ala Val Val Met His Ser Gly Ile Thr Ile Ser Ser Gly
580 585 590
His Tyr Thr Ala Ser Val Lys Val Thr Asp Leu Asn Ser Leu Glu Leu
595 600 605
Asp Lys Gly Asn Phe Val Val Asp Gin Met Cys Glu Ile Gly Lys Pro
610 615 620
Glu Pro Leu Asn Glu Glu Glu Ala Arg Gly Val Val Glu Asn Tyr Asn
625 630 635 640
Asp Glu Glu Val Ser Ile Arg Val Gly Gly Asn Thr Gin Pro Ser Lys
645 650 655
Val Leu Asn Lys Lys Asn Val Glu Ala Ile Gly Leu Leu Gly Gly Gin
660 665 670
Lys Ser Lys Ala Asp Tyr Glu Leu Tyr Asn Lys Ala Ser Asn Pro Asp
675 680 685
Lys Val Ala Ser Thr Ala Phe Ala Glu Asn Arg Asn Ser Glu Thr Ser
690 695 700
Asp Thr Thr Gly Thr His Glu Ser Asp Arg Asn Lys Glu Ser Ser Asp
705 710 715 720
Gin Thr Gly Ile Asn Ile Ser Gly Phe Glu Asn Lys Ile Ser Tyr Val
725 730 735
Val Gin Ser Len Lys Gin Tyr Glu Gly Lys Trp Leu Leu Phe Asp Asp
740 745 750
Ser Glu Val Lys Val Thr Glu Glu Lys Asp Phe Leu Asn Ser Leu Ser
755 760 765
Pro Ser Thr Ser Pro Thr Ser Thr Pro Tyr Leu Leu Phe Tyr Lys Lys
770 775 780
Leu
785
<210> 3
<211> 677
<212> PRT
<213> Homo sapiens
<400> 3
Met Ala Ala His His Arg Gin Asn Thr Ala Gly Arg Arg Lys Val Gin
1 5 10 15
Val Ser Tyr Val Ile Arg Asp Glu Val Glu Lys Tyr Asn Arg Asn Gly
20 25 30
Val Asn Ala Leu Gin Leu Asp Pro Ala Leu Asn Arg Leu Phe Thr Ala
35 40 45
Gly Arg Asp Ser Ile Ile Arg Ile Trp Ser Val Asn Gin His Lys Gin
50 55 60
Asp Pro Tyr Ile Ala Ser Met Glu His His Thr Asp Trp Val Asn Asp
65 70 75 80
Ile Vol Leu Cys Cys Asn Gly Lys Thr Leu Ile Ser Ala Ser Ser Asp
85 90 95
Thr Thr Val Lys Vol Trp Asn Ala His Lys Gly Phe Cys Met Ser Thr
100 105 110
= . CA 02797719 2012-11-14
76d
Leu Arg Thr His Lys Asp Tyr Val Lys Ala Leu Ala Tyr Ala Lys Asp
115 120 125
Lys Glu Leu Val Ala Ser Ala Gly Leu Asp Arg Gin Ile Phe Leu Trp
130 135 140
Asp Val Asn Thr Leu Thr Ala Leu Thr Ala Ser Asn Asn Thr Val Thr
145 150 155 160
Thr Ser Ser Leu Ser Gly Asn Lys Asp Ser Ile Tyr Ser Leu Ala Met
165 170 175
Asn Gin Leu Gly Thr Ile Ile Val Ser Gly Ser Thr Giu Lys Val Leu
180 185 190
Arg Val Trp Asp Pro Arg Thr Cys Ala Lys Leu Met Lys Leu Lys Gly
195 200 205
His Thr Asp Asn Val Lys Ala Leu Leu Leu Asn Arg Asp Gly Thr Gin
210 215 220
Cys Leu Ser Gly Ser Ser Asp Gly Thr Ile Arg Leu Trp Ser Leu Gly
225 230 235 240
Gin Gin Arg Cys lie Ala Thr Tyr Arg Val His Asp Glu Gly Val Trp
245 250 255
Ala Leu Gin Val Asn Asp Ala Phe Thr His Val Tyr Ser Gly Gly Arg
260 265 270
Asp Arg Lys Ile Tyr Cys Thr Asp Leu Arg Asn Pro Asp Ile Arg Val
275 280 285
Leu Ile Cys Glu Glu Lys Ala Pro Val Leu Lys Met Glu Leu Asp Arg
290 295 300
Ser Ala Asp Pro Pro Pro Ala Ile Trp Val Ala Thr Thr Lys Ser Thr
305 310 315 320
Val Asn Lys Trp Thr Leu Lys Gly Ile His Asn Phe Arg Ala Ser Gly
325 330 335
Asp Tyr Asp Asn Asp Cys Thr Asn Pro Ile Thr Pro Leu Cys Thr Gin
340 345 350
Pro Asp Gin Val Ile Lys Gly Gly Ala Ser Ile Ile Gin Cys His Ile
355 360 365
Leu Asn Asp Lys Arg His Ile Leu Thr Lys Asp Thr Asn Asn Asn Val
370 375 380
Ala Tyr Trp Asp Val Leu Lys Ala Cys Lys Val Glu Asp Leu Gly Lys
385 390 395 400
Val Asp Phe Glu Asp Glu Ile Lys Lys Arg Phe Lys Met Val Tyr Val
405 410 415
Pro Asn Trp Phe Ser Val Asp Leu Lys Thr Gly Met Leu Thr Ile Thr
420 425 430
Leu Asp Glu Ser Asp Cys Phe Ala Ala Trp Val Ser Ala Lys Asp Ala
435 440 445
Gly Phe Ser Ser Pro Asp Gly Ser Asp Pro Lys Leu Asn Ile Gly Gly
450 455 460
Leu Leu Leu Gin Ala Leu Leu Glu Tyr Trp Pro Arg Thr His Val Asn
465 470 475 480
Pro Met Asp Glu Glu Glu Asn Glu Val Asn His Val Asn Gly Glu Gin
485 490 495
Glu Asn Arg Val Gin Lys Gly Asn Gly Tyr Phe Gin Val Pro Pro His
500 505 510
Thr Pro Val Ile Phe Gly Glu Ala Gly Gly Arg Thr Leu Phe Arg Leu
515 520 525
Leu Cys Arg Asp Ser Gly Gly Gin Thr Glu Ser Met Leu Leu Asn Glu
530 535 540
Thr Val Pro Gin Trp Val Ile Asp Ile Thr Val Asp Lys Asn Met Pro
545 550 555 560
CA 02797719 2012-11-14
76e
Lys Phe Asn Lys Ile Pro Phe Tyr Leu Gin Pro His Ala Ser Ser Gly
565 570 575
Ala Lys Thr Leu Lys Lys Asp Arg Leu Ser Ala Ser Asp Met Leu Gin
580 585 590
Val Arg Lys Val Met Glu His Val Tyr Glu Lys Ile Ile Asn Leu Asp
595 600 605
Asn Glu Ser Gin Thr Thr Ser Ser Ser Asn Asn Glu Lys Pro Gly Glu
610 615 620
Gin Glu Lys Glu Glu Asp Ile Ala Val Leu Ala Glu Glu Lys Ile Glu
625 630 635 640
Leu Leu Cys Gin Asp Gin Val Leu Asp Pro Asn Met Asp Leu Arg Thr
645 650 655
Val Lys His Phe Ile Trp Lys Ser Gly Gly Asp Leu Thr Leu His Tyr
660 665 670
Arg Gin Lys Ser Thr
675
<210> 4
<211> 261
<212> PRT
<213> Homo sapiens
<400> 4
Met She Glu Ala Arg Leu Val Gin Gly Ser Ile Leu Lys Lys Val Leu
1 5 10 15
Glu Ala Leu Lys Asp Leu Ile Asn Glu Ala Cys Trp Asp Ile Ser Ser
20 25 30
Ser Gly Val Asn Leu Gin Ser Met Asp Ser Ser His Val Ala Leu Val
35 40 45
Gin Leu Thr Leu Arg Ser Glu Gly Phe Asp Thr Tyr Arg Cys Asp Arg
50 55 60
Asn Leu Ala Met Gly Val Asn Leu Thr Ser Met Ser Lys Ile Leu Lys
65 70 75 80
Cys Ala Gly Asn Glu Asp Ile Ile Thr Leu Arg Ala Glu Asp Asn Ala
85 90 95
Asp Thr Leu Ala Leu Val She Glu Ala Pro Asn Gin Glu Lys Val Ser
100 105 110
Asp Tyr Glu Met Lys Leu Met Asp Leu Asp Val Glu Gin Ile Gly Ile
115 120 125
Pro Glu Gin Glu Tyr Ser Cys Val Val Lys Met Pro Ser Gly Glu Phe
130 135 140
Ala Arg Ile Cys Arg Asp Leu Ser His Ile Gly Asp Ala Val Val Ile
145 150 155 160
Ser Cys Ala Lys Asp Gly Val Lys Phe Ser Ala Ser Gly Glu Leu Gly
165 170 175
Asn Gly Asn Ile Lys Leu Ser Gin Thr Ser Asn Val Asp Lys Glu Glu
180 185 190
Glu Ala Val Thr Ile Glu Met Asn Glu Pro Val Gin Leu Thr Phe Ala
195 200 205
Leu Arg Tyr Leu Asn Phe Phe Thr Lys Ala Thr Pro Leu Ser Ser Thr
210 215 220
Val Thr Lou Ser Met Ser Ala Asp Val Pro Leu Val Val Glu Tyr Lys
225 230 235 240
= . CA 02797719 2012-11-14
76f
Ile Ala Asp Met Gly His Leu Lys Tyr Tyr Leu Ala Pro Lys Ile Glu
245 250 255
Asp Glu Glu Gly Ser
260