Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
METHODS AND COMPOSITIONS FOR EVALUATING GENETIC MARKERS
RELATED APPLICATIONS
The present application claims the benefit of and priority to U.S. non-
provisional
application serial number 13/934,093, filed July 2, 2013, which claims the
benefit of and priority
to U.S. provisional application serial number 61/789,164, filed March 15,
2013, the content of
each of which is incorporated by reference herein in its entirety.
SEQUENCE LISTING
The instant application contains a Sequence Listing which has been submitted
in ASCII
format via EFS-Web and is hereby incorporated by reference in its entirety.
Said ASCII copy,
created on March 13, 2014, is named GSGE_002_03W0_Sequence_Listing.txt and is
1,921,826
bytes in size.
FIELD OF INVENTION
The invention relates to methods and compositions for determining genotypes in
patient
samples.
BACKGROUND OF THE INVENTION
Information about the genotype of a subject is becoming more important and
relevant for
a range of healthcare decisions as the genetic basis for many diseases,
disorders, and
physiological characteristics is further elucidated. Medical advice is
increasingly personalized,
with individual decisions and recommendations being based on specific genetic
information.
Information about the type and number of alleles at one or more genetic loci
impacts disease
risk, prognosis, therapeutic options, and genetic counseling amongst other
healthcare
considerations.
For cost-effective and reliable medical and reproductive counseling on a large
scale, it is
important to be able correctly and unambiguously identify the allelic status
for many different
genetic loci in many subjects.
Numerous technologies have been developed for detecting and analyzing nucleic
acid
sequences from biological samples. These technologies can be used to genotype
subjects and
1
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
determine the allelic status of any locus of interest. However, they are not
sufficiently robust and
cost-effective to be scaled up for reliable high throughput analysis of many
genetic loci in large
numbers of patients. The frequency of incorrect or ambiguous calls is too high
for current
technology to manage large numbers of patient samples without involving
expensive and time-
consuming steps to resolve uncertainties and provide confidence in the
information output.
SUMMARY OF THE INVENTION
Aspects of the invention relate to preparative and analytical methods and
compositions
for evaluating genotypes, and in particular, for determining the allelic
identity (or identities in a
diploid organism) of one or more genetic loci in a subject.
Aspects of the invention are based, in part, on the identification of
different sources of
ambiguity and error in genetic analyses, and, in part, on the identification
of one or more
approaches to avoid, reduce, recognize, and/or resolve these errors and
ambiguities at different
stages in a genetic analysis.
According to aspects of the invention, certain types of genetic information
can be under-
represented or over-represented in a genetic analysis due to a combination of
stochastic variation
and systematic bias in any of the preparative stages (e.g., capture,
amplification, etc.),
determining stages (e.g., allele-specific detection, sequencing, etc.), data
interpretation stages
(e.g., determining whether the assay information is sufficient to identify a
subject as homozygous
or heterozygous), and/or other stages.
According to aspects of the invention, error or ambiguity may be apparent in a
genetic
analysis, but not readily resolved without running additional samples or more
expensive assays
(e.g., array-based assays may report no-calls due to noisy/low signal).
According to further
aspects of the invention, error or ambiguity may not be accounted for in a
genetic analysis and
incorrect base calls may be made even when the evidence for them is limited
and/or not
statistically significant (e.g., next-generation sequencing technologies may
report base calls even
if the evidence for them is not statistically significant). According to
further aspects of the
invention error or ambiguity may be problematic for a multi-step genetic
analysis because it is
apparent but not readily resolved in one or more steps of the analysis and not
apparent or
accounted for in other steps of the analysis.
2
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
In some embodiments, sources of error and ambiguity in one or more steps can
be
addressed by capturing and/or interrogating each target locus of interest with
one or more sets of
overlapping probes that are designed to overcome any systematic bias or
stochastic effects that
may impact the complexity and/or fidelity of the genetic information that is
generated.
In some embodiments, sources of error and ambiguity in one or more steps can
be addressed by
capturing and/or interrogating each target locus of interest with at least one
set of probes,
wherein different probes are labeled with different identifiers that can be
used to track the assay
reactions and determine whether certain types of genetic information are under-
represented or
over-represented in the information that is generated.
In some embodiments, errors and ambiguities associated with the analysis of
regions
containing large numbers of sequence repeats are addressed by systematically
analyzing
frequencies of certain nucleic acids at particular stages in an assay (e.g.,
at a to capture,
sequencing, or detection stage). It should be appreciated that such techniques
may be particularly
useful in the context of a standardized protocol that is designed to allow
many different loci to be
evaluated in parallel without requiring different assay procedures for each
locus. In some
embodiments, the use of a single detection modality (e.g., sequencing) to
assay multiple types of
genetic lesions (e.g., point mutations, insertions/deletions, length
polymorphisms) is
advantageous in the clinical setting. In some embodiments of the invention,
methods are
provided that facilitate the use of multiple sample preparation steps in
parallel, coupled with
multiple analytical processes following sequence detection. Thus, in some
embodiments of the
invention, an improved workflow is provided that reduces error and uncertainty
when
simultaneously assaying different types of genetic lesions across multiple
loci in multiple
patients.
In some embodiments, aspects of the invention provide methods for overcoming
preparative and/or analytical bias by combining two or more techniques, each
having a different
bias (e.g., a known bias towards under-representation or over-representation
of one or more types
of sequences), and using the resulting data to determine a genetic call for a
subject with greater
confidence.
It should be appreciated that in some embodiments, aspects of the invention
relate to
multiplex diagnostic methods. In some embodiments, multiplex diagnostic
methods comprise
capturing a plurality of genetic loci in parallel (e.g., one or more genetic
loci from Table 1). In
3
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
some embodiments, the genetic loci possess one or more polymorphisms (e.g.,
one or more
polymorphisms from Table 2) the genotypes of which correspond to disease
causing alleles.
Accordingly, in some embodiments, the disclosure provides methods for
assessing multiple
heritable disorders in parallel. In some embodiments, methods are provided for
diagnosing
multiple heritable disorders in parallel at a pre-implantation, prenatal,
perinatal, or postnatal
stage. In some embodiments, the disclosure provides methods for analyzing
multiple genetic loci
(e.g., a plurality of target nucleic acids selected from Table 1) from a
patient sample, such as a
blood, pre-implantation embryo, chorionic villus or amniotic fluid sample, or
other sample (e.g.,
other biological fluid or tissue sample such as a biopsy sample) as aspects of
the invention are
not limited in this respect.
Other samples may include tumor tissue or circulating tumor cells. In some
embodiments,
a patient sample (e.g., a tumor tissue or cell sample) is mosaic for one or
more mutations of
interest, and thus, may require higher sensitivity than is needed for a
germline mutation analysis.
In some embodiments, a sample comprises cells from a non-host organism (e.g.,
bacterial or viral
infections in a human subject) or a sample for environmental monitoring (e.g.,
bacterial, viral,
fungal composition of a soil, water, or air sample).
Accordingly, in some embodiments, aspects of the methods disclosed herein
relate to
genotyping a polymorphism of a target nucleic acid. In some embodiments, the
genotyping may
comprise determining that one or more alleles of the target nucleic acid are
heterozygous or
homozygous. In further embodiments, the genotyping may comprise determining
the sequence of
a polymorphism and comparing that sequence to a control sequence that is
indicative of a disease
risk. In some embodiments, the polymorphism is selected from a locus in Table
1 or Table 2.
However, it should be appreciated that any locus associated with a disease or
condition of
interest may be used.
In some embodiments, a diagnosis, prognosis, or disease risk assessment is
provided to a
subject based on a genotype determined for that subject at one or more genetic
loci (e.g., based
on the analysis of a biological sample obtained from that subject). In some
embodiments, an
assessment is provided to a couple, based on their respective genotypes at one
or more genetic
loci, of the risk of their having one or more children having a genotype
associated with a disease
or condition (e.g., a homozygous or heterozygous genotype associated with a
disease or
condition). In some embodiments, a subject or a couple may seek genetic or
reproductive
4
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
counseling in connection with a genotype determined according to embodiments
of the
invention. In some embodiments, genetic information from a tumor or
circulating tumor cells is
used to determine prognosis and guide selection of appropriate
drugs/treatments.
It should be appreciated that any of the methods or compositions described
herein may be
used in combination with any of the medical evaluations associated with one or
more genetic loci
as described herein.
In some embodiments, aspects of the invention provide effective methods for
overcoming
challenges associated with systematic errors (bias) and/or stochastic effects
in multiplex genomic
capture and/or analysis (including sequencing analysis). In some embodiments,
aspects of the
invention are useful to avoid, reduce and/or account for variability in one or
more sampling
and/or analytical steps. For example, in some embodiments, variability in
target nucleic acid
representation and unequal sampling of heterozygous alleles in pools of
captured target nucleic
acids can be overcome.
Accordingly, in some embodiments, the disclosure provides methods that reduce
variability in the detection of target nucleic acids in multiplex capture
methods. In other
embodiments, methods improve allelic representation in a capture pool and,
thus, improve
variant detection outcomes. In certain embodiments, the disclosure provides
preparative methods
for capturing target nucleic acids (e.g., genetic loci) that involve the use
of different sets of
multiple probes (e.g., molecular inversion probes MIPs) that capture
overlapping regions of a
target nucleic acid to achieve a more uniform representation of the target
nucleic acids in a
capture pool compared with methods of the prior art. In other embodiments,
methods reduce
bias, or the risk of bias, associated with large scale parallel capture of
genetic loci, e.g., for
diagnostic purposes. In other embodiments, methods are provided for increasing
reproducibility
(e.g., by reducing the effect of polymorphisms on target nucleic acid capture)
in the detection of
a plurality of genetic loci in parallel. In further embodiments, methods are
provided for reducing
the effect of probe synthesis and/or probe amplification variability on the
analysis of a plurality
of genetic loci in parallel.
According to some aspects, methods of analyzing a plurality of genetic loci
are provided.
In some embodiments, the methods comprise contacting each of a plurality of
target nucleic
acids with a probe set, wherein each probe set comprises a plurality of
different probes, each
probe having a central region flanked by a 5' region and a 3' region that are
complementary to
5
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
nucleic acids flanking the same strand of one of a plurality of subregions of
the target nucleic
acid, wherein the subregions of the target nucleic acid are different, and
wherein each subregion
overlaps with at least one other subregion, isolating a plurality of nucleic
acids each having a
nucleic acid sequence of a different subregion for each of the plurality of
target nucleic acids,
and analyzing the isolated nucleic acids.
In other embodiments, methods comprise contacting each of a plurality of
target nucleic
acids with a probe set, wherein each probe set comprises a plurality of
different probes, each
probe having a central region flanked by a 5' region and a 3' region that are
complementary to
nucleic acids flanking the same strand of one of a plurality of subregions of
the target nucleic
acid, wherein the subregions of the target nucleic acid are different, and
wherein a portion of the
5' region and a portion of the 3' region of a probe have, respectively, the
sequence of the 5'
region and the sequence of the 3' region of a different probe, isolating a
plurality of nucleic acids
each having a nucleic acid sequence of a different subregion for each of the
plurality of target
nucleic acids, and analyzing the isolated nucleic acids.
In certain aspects, methods of the invention involve analyzing one or more
genes with
one or more molecular inversion probes provided in Appendix A. Particularly,
those molecular
inversion probes are used to capture various targets or subregions thereof on
a gene selected
from the group consisting of ABCC8, ASPA, BCKDHA, BCKDHB, BLM, CFTR, CLRN1,
DLD, FANCC, G6PC, HEXA, IKBKAP, MCOLN1, PCDH15, and SMPD1. In certain
applications, a set of two or more molecular inversion probes provided in
Appendix A may be
used to tile across different, but overlapping sub-regions of one or more
genes so that one or
more targets on the one or more genes are captured by at least two molecular
inversion probes of
the set. The number of molecular inversion probes used in a set for tile
capture depends on the
amount of overlapping coverage one desires for a certain target. In certain
embodiments, a
portion of one or more genes is captured using one or more molecular inversion
probes in
Appendix A. One or more molecular inversion probes of Appendix A may also be
chosen to
capture particular regions of interest, such as coding or noncoding regions,
of a gene. In
addition, one or more molecular inversion probes may be chosen to capture
regions specific to
certain diseases. The diseases may include, for example, Familial
hyperinsulinism, Canavan
disease, Maple syrup urine disease type la/lb, Bloom syndrome, Cystic
fibrosis, Usher
syndrome type IIIA, Dihydrolipoamide dehydrogenase deficiency, Fanconi anemia
group C,
6
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
Glycogen storage disease type la, Tay-Sachs disease, Familial dysautonomia,
Mucolipidosis
type IV, Usher syndrome type IF, Niemann-Pick disease type A/B.
Aspects of the disclosure are based, in part, on the discovery of methods for
overcoming
problems associated with systematic and random errors (bias) in genome
capture, amplification
and sequencing methods, namely high variability in the capture and
amplification of nucleic
acids and disproportionate representation of heterozygous alleles in
sequencing libraries.
Accordingly, in some embodiments, the disclosure provides methods that reduce
errors
associated with the variability in the capture and amplification of nucleic
acids. In other
embodiments, the methods improve allelic representation in sequencing
libraries and, thus,
improve variant detection outcomes. In certain embodiments, the disclosure
provides preparative
methods for capturing target nucleic acids (e.g., genetic loci) that involve
the use of differentiator
tag sequences to uniquely tag individual nucleic acid molecules. In some
embodiments, the
differentiator tag sequence permit the detection of bias based on the
occurrence of combinations
of differentiator tag and target sequences observed in a sequencing reaction.
In other
embodiments, the methods reduce errors caused by bias, or the risk of bias,
associated with the
capture, amplification and sequencing of genetic loci, e.g., for diagnostic
purposes.
Aspects of the invention relate to providing sequence tags (referred to as
differentiator
tags) that are useful to determine whether target nucleic acid sequences
identified in an assay are
from independently isolated target nucleic acids or from multiple copies of
the same target
nucleic acid molecule (e.g., due to bias in a preparative step, for example,
amplification). This
information can be used to help analyze a threshold number of independently
isolated target
nucleic acids from a biological sample in order to obtain sequence information
that is reliable
and can be used to make a genotype conclusion (e.g., call) with a desired
degree of confidence.
This information also can be used to detect bias in one or more nucleic acid
preparative steps.
In some embodiments, the methods disclosed herein are useful for any
application where
reduction of bias, e.g., associated with genomic isolation, amplification,
sequencing, is
important. For example, detection of cancer mutations in a heterogeneous
tissue sample,
detection of mutations in maternally-circulating fetal DNA, and detection of
mutations in cells
isolated during a preimplantation genetic diagnostic procedure.
Accordingly, in some aspects, methods of genotyping a subject are provided. In
some
embodiments, the methods comprise determining the sequence of at least a
threshold number of
7
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
independently isolated nucleic acids, wherein the sequence of each isolated
nucleic acid
comprises a target nucleic acid sequence and a differentiator tag sequence,
wherein the threshold
number is a number of unique combinations of target nucleic acid and
differentiator tag
sequences, wherein the isolated nucleic acids are identified as independently
isolated if they
comprise unique combinations of target nucleic acid and differentiator tag
sequences, and
wherein the target nucleic acid sequence is the sequence of a genomic locus of
a subject.
In some embodiments, the isolated nucleic acids are products of a
circularization
selection-based preparative method, e.g., molecular inversion probe capture
products. In other
embodiments, the isolated nucleic acids are products of an amplification-based
preparative
methods. In other embodiments, the isolated nucleic acids are products of
hybridization-based
preparative methods.
Circularization selection-based preparative methods selectively convert
regions of
interest (target nucleic acids) into a covalently-closed circular molecule
which is then isolated
typically by removal (usually enzymatic, e.g. with exonuclease) of any non-
circularized linear
nucleic acid. Oligonucleotide probes (e.g., molecular inversion probes) are
designed which have
ends that flank the region of interest (target nucleic acid) and, optionally,
primer sites, e.g.,
sequencing primer sites. The probes are allowed to hybridize to the genomic
target, and enzymes
are used to first (optionally) fill in any gap between probe ends and second
ligate the probe
closed. Following circularization, any remaining (non-target) linear nucleic
acid is typically
removed, resulting in isolation (capture) of target nucleic acid.
Circularization selection-based
preparative methods include molecular inversion probe capture reactions and
'selector' capture
reactions. In some embodiments, molecular inversion probe capture of a target
nucleic acid is
indicative of the presence of a polymorphism in the target nucleic acid.
In amplification-based (e.g., PCR-based or LCR-based, etc.) preparative
methods,
genomic loci (target nucleic acids) are isolated directly by means of a
polymerase chain reaction
or ligase chain reaction (or other amplification method) that selectively
amplifies each locus
using one or more oligonucleotide primers. It is to be understood that primers
will be sufficiently
complementary to the target sequence to hybridize with and prime amplification
of the target
nucleic acid. Any one of a variety of art known methods may be utilized for
primer design and
synthesis. One or more of the primers may be perfectly complementary to the
target sequence.
Degenerate primers may also be used. Primers may also include additional
nucleic acids that are
8
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
not complementary to target sequences but that facilitate downstream
applications, including for
example restriction sites and differentiator tag sequences. Amplification-
based methods include
amplification of a single target nucleic acid and multiplex amplification
(amplification of
multiple target nucleic acids in parallel).
Hybridization-based preparative methods involve selectively immobilizing
target nucleic
acids for further manipulation. It is to be understood that one or more
oligonucleotides
(immobilization oligonucleotides), which comprise differentiator tag
sequences, and which may
be from 15 to 170 nucleotides in length, are used which hybridize along the
length of a target
region of a genetic locus to immobilize it. In some embodiments,
immobilization
oligonucleotides, are either immobilized before hybridization is performed
(e.g.,
Roche/Nimblegen 'sequence capture'), or are prepared such that they include a
moiety (e.g.
biotin) which can be used to selectively immobilize the target nucleic acid
after hybridization by
binding to e.g., streptavidin-coated microbeads (e.g. Agilent `SureSelece).
It should be appreciated that any of the circularization, amplification,
and/or
hybridization based methods described herein may be used in connection with
one or more of the
tiling/staggering, tagging, size-detection, and/or sensitivity enhancing
algorithms described
herein.
In some embodiments, the methods disclosed herein comprise determining the
sequence
of molecular inversion probe capture products, each comprising a molecular
inversion probe and
a target nucleic acid, wherein the sequence of the molecular inversion probe
comprises a
differentiator tag sequence and, optionally, a primer sequence, and wherein
the target nucleic
acid is a captured genomic locus of a subject, and genotyping the subject at
the captured genomic
locus based on the sequence of at least a threshold number of unique
combinations of target
nucleic acid and differentiator tag sequences of molecular inversion probe
capture products.
In some embodiments, the methods disclosed herein comprise obtaining molecular
inversion probe capture products, each comprising a molecular inversion probe
and a target
nucleic acid, wherein the sequence of the molecular inversion probe comprises
a differentiator
tag sequence and, optionally, a primer sequence, wherein the target nucleic
acid is a captured
genomic locus of the subject, amplifying the molecular inversion probe capture
products, and
genotyping the subject by determining, for each target nucleic acid, the
sequence of at least a
threshold number of unique combinations of target nucleic acid and
differentiator tag sequence
9
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
of molecular inversion probe capture products. In certain embodiments,
obtaining comprises
capturing target nucleic acids from a genomic sample of the subject with
molecular inversion
probes, each comprising a unique differentiator tag sequence. In specific
embodiments, capturing
is performed under conditions wherein the likelihood of obtaining two or more
molecular
inversion probe capture products with identical combinations of target and
differentiator tag
sequences is equal to or less than a predetermined value, optionally wherein
the predetermined
value is about 0.05.
In one embodiment, the threshold number for a specific target nucleic acid
sequence is
selected based on a desired statistical confidence for the genotype. In some
embodiments, the
methods further comprising determining a statistical confidence for the
genotype based on the
number of unique combinations of target nucleic acid and differentiator tag
sequences.
According to some aspects, methods of analyzing a plurality of genetic loci
are provided.
In some embodiments, the methods comprise obtaining a plurality of molecular
inversion probe
capture products each comprising a molecular inversion probe and a target
nucleic acid, wherein
the sequence of the molecular inversion probe comprises a differentiator tag
sequence and,
optionally, a primer sequence (e.g., a sequence that is complementary to the
sequence of a
nucleic acid that is used as a primer for sequencing or other extension
reaction), amplifying the
plurality of molecular inversion probe capture products, determining numbers
of occurrence of
combinations of target nucleic acid and differentiator tag sequence of
molecular inversion probe
capture products in the amplified plurality, and if the number of occurrence
of a specific
combination of target nucleic acid sequence and differentiator tag sequence
exceeds a
predetermined value, detecting bias in the amplification of the molecular
inversion probe
comprising the specific combination. In some embodiments, the methods further
comprise
genotyping target sequences in the plurality, wherein the genotyping comprises
correcting for
bias, if detected.
In some embodiments, the target nucleic acid is a gene (or portion thereof)
selected from
Table 1. In some embodiments, the genotyping comprises determining the
sequence of a target
nucleic acid (e.g., a polymorphic sequence) at one or more (both) alleles of a
genome (a diploid
genome) of a subject. In certain embodiments, the genotyping comprises
determining the
sequence of a target nucleic acid at both alleles of a diploid genome of a
subject, wherein in the
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
target nucleic acid comprises, or consists of, a sequence of Table 1, Table 2,
or other locus of
interest.
In some embodiments, aspects of the invention provide methods and compositions
for
identifying nucleic acid insertions or deletions in genomic regions of
interest without
determining the nucleotide sequences of these regions. Aspects of the
invention are particularly
useful for detecting nucleic acid insertions or deletions in genomic regions
containing nucleic
acid sequence repeats (e.g., di- or tri-nucleotide repeats). However, the
invention is not limited to
analyzing nucleic acid repeats and may be used to detect insertions or
deletions in any target
nucleic acid of interest. Aspects of the invention are particularly useful for
analyzing multiple
loci in a multiplex assay.
In some embodiments, aspects of the invention relate to determining whether an
amount
of target nucleic acid that is captured in a genomic capture assay is higher
or lower than
expected. In some embodiments, a statistically significant deviation from an
expected amount
(e.g., higher or lower) is indicative of the presence of a nucleic acid
insertion or deletion in the
genomic region of interest. In some embodiments, the amount is a number of
nucleic acid
molecules that are captured. In some embodiments, the amount is a number of
independently
captured nucleic acid molecules in a sample. It should be appreciated that the
captured nucleic
acids may be literally captured from a sample, or their sequences may be
captured without
actually capturing the original nucleic acids in the sample. For example,
nucleic acid sequences
may be captured in an assay that involves a template-based extension of
nucleic acids having the
region of interest, in the sample.
Aspects of the invention are based on the recognition that the efficiency of
certain capture
techniques is affected by the length of the nucleic acid being captured.
Accordingly, an increase
or decrease in the length of a target nucleic acid (e.g., due to an insertion
or deletion of a
repeated sequence) can alter the capture efficiency of that nucleic acid. In
some embodiments, a
difference in the capture efficiency (e.g., a statistically significant
difference in the capture
efficiency) of a target nucleic acid is indicative of an insertion or deletion
in the target nucleic
acid. It should be appreciated that the capture efficiency for a target
nucleic acid may be
evaluated based on an amount of captured nucleic acid (e.g., number of
captured nucleic acid
molecules) relative to a control amount (e.g., based on an amount of control
nucleic acid that is
11
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
captured). However, the invention is not limited in this respect and other
techniques for
evaluating capture efficiency also may be used.
According to aspects of the invention, evaluating the capture efficiency as
opposed to
determining the sequence of the entire repeat region reduces errors associated
with sequencing
through repeat regions. Repeat sequences often give rise to stutters or skips
in sequencing
reactions that make it very difficult to accurately determine the number of
repeats in a target
region without running multiple sequencing reactions under different
conditions and carefully
analyzing the results. Such procedures are cumbersome and not readily scalable
in a manner that
is consistent with high throughput analyses of target nucleic acids. In some
embodiments, repeat
regions may be longer than the length of the individual sequence read, making
length
determination on the basis of a single read impossible. For example, when
using next-generation
sequencing the repeat regions may be longer than the length of the individual
sequence read,
making length determination on the basis of a single read impossible.
Accordingly, aspects of the
invention are useful to increase the sensitivity of detecting insertions or
deletions in target
regions, particularly target regions containing repeated sequences.
In some embodiments, aspects of the invention relate to capturing genomic
nucleic acid
sequences using a molecular inversion probe (e.g., MIP or Padlock probe)
technique, and
determining whether the amount (e.g., number) of captured sequences is higher
or lower than
expected. In some embodiments, the amount (e.g., number) of captured sequences
is compared to
an amount (e.g., number) of sequences captured in a control assay. The control
assay may
involve analyzing a control sample that contains a nucleic acid from the same
genetic locus
having a known sequence length (e.g., a known number of nucleic acid repeats).
However, a
control may involve analyzing a second (e.g., different) genetic locus that is
not expected to
contain any insertions or deletions. The second genetic locus may be analyzed
in the same
sample as the locus being interrogated or in a different sample where its
length has been
previously determined. The second genetic locus may be a locus that is not
characterized by the
presence of nucleic acid repeats (and thus not expected to contain insertions
or deletions of the
repeat sequence).
In some embodiments, a target nucleic acid region that is being evaluated may
be
determined by the identity of the targeting arms of a probe that is designed
to capture the target
region (or sequence thereof). For example, the targeting arms of a MIP probe
may be designed to
12
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
be complementary (e.g., sufficiently complementary for selective hybridization
and/or
polymerase extension and/or ligation) to genomic regions flanking a target
region suspected of
containing an insertion or deletion. It should be appreciated that two
targeting arms may be
designed to be complementary (e.g., sufficiently complementary for selective
hybridization
and/or polymerase extension and/or ligation) to the two flanking regions that
are immediately
adjacent (e.g., immediately 5' and 3', respectively) to a region of a sequence
repeat on one strand
of a genomic nucleic acid. However, one or both targeting arms may be designed
to hybridize
several bases (e.g., 1-5, 5-10, 10-25, 25-50, or more) upstream or downstream
from the repeat
region in such a way that the captured sequence includes a region of unique
genomic sequence
that on one or both sides of the repeat region. This unique region can then be
used to identify the
captured target (e.g., based on sequence or hybridization information).
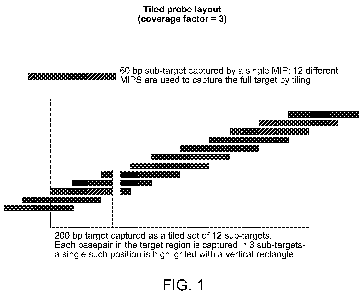
In some embodiments, two or more (e.g., 2, 3, 4, 5, 6, 7, 8, 9, 10 or more)
different loci
may be interrogated in parallel in a single assay (e.g., in a multiplex
assay). In some
embodiments, the ratio of captured nucleic acids for each locus may be used to
determine
whether a nucleic acid insertion or deletion is present in one locus relative
to the other. For
example, the ratio may be compared to a control ratio that is representative
of the two loci when
neither one has an insertion or deletion relative to control sequences (e.g.,
sequences that are
normal or known to be associated with healthy phenotypes for those loci).
However, the amount
of captured nucleic acids may be compared to any suitable control as discussed
herein.
The locus of a captured sequence may be identified by determining a portion of
unique
sequence 5' and/or 3' to the repeat region in the target nucleic acid
suspected of containing a
deletion or insertion. This does not require sequencing the captured repeat
region itself.
However, some or all of the repeat region also could be sequenced as aspects
of the invention are
not limited in this respect.
Aspects of the invention may be combined with one or more sequence-based
assays (e.g.,
SNP detection assays), for example in a multiplex format, to determine the
genotype of one or
more regions of a subject.
In some embodiments, methods of detecting a polymorphism in a nucleic acid in
a
biological sample are provided. In some embodiments, the methods comprise
evaluating the
efficiency of capture at one or more loci and determining whether one or both
alleles at that
13
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
locus contain an insertion or deletion relative to a control locus (e.g., a
locus indicative of a
length of repeat sequence that is associated with a healthy phenotype).
Accordingly, aspects of the invention relate to methods for determining
whether a target
nucleic acid has an abnormal length by evaluating the capture efficiency of a
target nucleic acid
in a biological sample from a subject, wherein a capture efficiency that is
different from a
reference capture efficiency is indicative of the presence, in the biological
sample, of a target
nucleic acid having an abnormal length. It should be appreciated that the term
"abnormal" is a
relative term based on a comparison to a "normal" length. In some embodiments,
a normal
length is a length that is associated with a normal (e.g., healthy or non-
carrier phenotype).
Accordingly, an abnormal length is a length that is either shorter or longer
than the
normal length. In some embodiments, the presence of an abnormal length is
indicative of an
increased risk that the locus is associated with a disease or a disease
carrier phenotype. In some
embodiments, the abnormal length is indicative that the subject is either has
a disease or
condition or is a carrier of a disease or condition (e.g., associated with the
locus). However, it
should be appreciated that the description of embodiments relating to
detecting the presence of
an abnormal length also support detecting the presence of a length that is
different from an
expected or control length.
In some embodiments, aspects of the invention relate to estimating the length
of a target
nucleic acid (e.g., of a sub-target region within a target nucleic acid). In
some embodiments,
aspects of the invention relate to methods for estimating the length of a
target nucleic acid by
contacting the target nucleic acid with a plurality of detection probes under
conditions that
permit hybridization of the detection probes to the target nucleic acid,
wherein each detection
probe is a polynucleotide that comprises a first arm that hybridizes to a
first region of the target
nucleic acid and a second arm that hybridizes to a second region of the target
nucleic acid,
wherein the first and second regions are on a common strand of the target
nucleic acid, and
wherein the nucleotide sequence of the target between the 5' end of the first
region and the 3' end
of the second region is the nucleotide sequence of a sub-target nucleic acid;
and capturing a
plurality of sub-target nucleic acids that are hybridized with the plurality
of detection probes; and
measuring the frequency of occurrence of a sub-target nucleic acid in the
plurality of sub-target
nucleic acids, wherein the frequency of occurrence of the sub-target nucleic
acid in the plurality
of sub-target nucleic acids is indicative of the length of the sub-target
nucleic acid. It should be
14
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
appreciated that methods for estimating a nucleic acid length may involve
comparing a capture
efficiency for a target nucleic acid region to two or more reference
efficiencies for known
nucleic acid lengths in order to determine whether the target nucleic acid
region is smaller,
intermediate, or larger in size than the known control lengths. In some
embodiments, a series of
nucleic acids of known different lengths may be used to provide a calibration
curve for
evaluating the length of a target nucleic acid region of interest.
In some embodiments, the capture efficiency of a target region suspected of
having a
deletion or insertion is determined by comparing the capture efficiency to a
reference indicative
of a normal capture efficiency. In some embodiments, the capture efficiency is
lower than the
reference capture efficiency. In some embodiments, the subject is identified
as having an
insertion in the target region. In some embodiments, the capture efficiency is
higher than the
reference capture efficiency. In some embodiments, the subject is identified
as having a deletion
in the target region. In some embodiments, the subject is identified as being
heterozygous for the
insertion. In some embodiments, the subject is identified as being
heterozygous for the deletion.
In some embodiments of any of the methods described herein (e.g.,
tiling/staggering,
tagging, size-detection, and/or sensitivity enhancement) aspects of the
invention relate to
capturing a sub-target nucleic acid (or a sequence of a sub-target nucleic
acid). In some
embodiments, a molecular inversion probe technique is used. In some
embodiments, a molecular
inversion probe is a single linear strand of nucleic acid that comprises a
first targeting arm at its
5' end and a second targeting arm at its 3' end, wherein the first targeting
arm is capable of
specifically hybridizing to a first region flanking one end of the sub-target
nucleic acid, and
wherein the second targeting arm is capable of specifically hybridizing to a
second region
flanking the other end of the sub-target nucleic acid on the same strand of
the target nucleic acid.
In some embodiments, the first and second targeting arms are between about 10
and about 100
nucleotides long. In some embodiments, the first and second targeting arms are
about 10-20, 20-
30, 30-40, or 40-50 nucleotides long. In some embodiments, the first and
second targeting arms
are about 20 nucleotides long. In some embodiments, the first and second
targeting arms have
the same length. In some embodiments, the first and second targeting arms have
different
lengths. In some embodiments, each pair of first and second targeting arms in
a set of probes has
the same length. Accordingly, if one of the targeting arms is longer, the
other one is
correspondingly shorter. This allows for a quality control step in some
embodiments to confirm
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
that all captured probe/target sequence products have the same length after a
multiplexed
plurality of capture reactions. In some embodiments, a set of probes may be
designed to have the
same length if the intervening region is varied to accommodate any differences
in the length of
either one or both of the first and second targeting arms.
In some embodiments, the hybridization Tms of the first and second targeting
arms are
similar. In some embodiments, the hybridization Tms of the first and second
targeting arms are
within 2-5 C. of each other. In some embodiments, the hybridization Tms of
the first and second
targeting arms are identical. In some embodiments, the hybridization Tms of
the first and second
targeting arms are close to empirically-determined optima but not necessarily
identical.
In some embodiments, the first and second targeting arms of a molecular
inversion probe
have different Tms. For example, the Tm of the first targeting arm (at the 5'
end of the molecular
inversion probe) may be higher than the Tm of the second targeting arm (at the
3' end of the
molecular inversion probe). According to aspects of the invention, and without
wishing to be
bound by theory, a relatively high Tm for the first targeting arm may help
avoid or prevent the
first targeting arm from being displaced after hybridization by the extension
product of the 3' end
of the second targeting arm. It should be appreciated that a reference to the
Tm of a targeting arm
as used herein relates to the Tm of hybridization of the targeting arm to a
nucleic acid having the
complementary sequence (e.g., the region of the target nucleic acid that has a
sequence that is
complementary to the sequence of the targeting arm). It also should be
appreciated that the Tms
of the targeting arms described herein may be calculated using any appropriate
method. For
example, in some embodiments an experimental method (e.g., a gel shift assay,
a hybridization
assay, a melting curve analysis, for example in a PCR machine with a SYBR dye
by stepping
through a temperature ramp while monitoring signal level from an intercalating
dye, for
example, bound to a double-stranded DNA, etc.) may be used to determine one or
more Tms
empirically. In some embodiments, an optimal Tm may be determined by
evaluating the number
of products formed (e.g., for each of a plurality of MIP probes), and
determining the optimal Tm
as the center point in a histogram of Tm for all targeting arms. In some
embodiments, a
predictive algorithm may be used to determine a Tm theoretically. In some
embodiments, a
relatively simple predictive algorithm may be used based on the number of G/C
and A/T base
pairs when the sequence is hybridized to its target and/or the length of the
hybridized product
(e.g., for example, 64.9+41*([G+C]-16.4)/(A+T+G+C), see for example, Wallace,
R. B.,
16
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
Shaffer, J., Murphy, R. F., Bonner, J., Hirose, T., and Itakura, K. (1979)
Nucleic Acids Res
6:3543-3557). In some embodiments, a more complex algorithm may be used to
account for the
effects of base stacking entropy and enthalpy, ion concentration, and primer
concentration (see,
for example, SantaLucia J (1998), Proc Natl Acad Sci USA, 95:1460-5). In some
embodiments
an algorithm may use modified parameters (e.g., nearest-neighbor parameters
for basepair
entropy/enthalpy values). It should be appreciated that any suitable algorithm
may be used as
aspects of the invention are not limited in this respect. However, it also
should be appreciated
that different methodologies may results in different calculated or predicted
Tms for the same
sequences. Accordingly, in some embodiments, the same empirical and/or
theoretical method is
used to determine the Tms of different sequences for a set of probes to avoid
a negative impact
of any systematic difference in the Tm determination or prediction when
designing a set of
probes with predetermined similarities or differences for different Tms.
In some embodiments, the Tm of the first targeting arm may be about 1 C.,
about 2 C.,
about 3 C., about 4 C., about 5 C., or more than about 5 C. higher than
the Tm of the second
targeting arm. In some embodiments, each probe in a plurality of probes (e.g.,
each probe in a set
of 5-10, each probe in a set of at least 10, each probe in a set of 10-50,
each probe in a set of 50-
100, each probe in a set of 100-500, each probe in a set of 500-1,000, each
probe in a set of
1,000-1,500, each probe in a set of 1,500-2,000, each probe in a set of 2,000-
3,000, 3,000-5,000,
5,000-10,000 or each probe in a set of at least 5,000 different probes) has a
unique first targeting
arm (e.g., they all have different sequences) and a unique second targeting
arm (e.g., they all
have different sequences). In some embodiments, for at least 10% of the probes
(e.g., at least
25%, 25%-50%, 50%-75%, 75%-90%, 90%-95% or over 95%, or all of the probes) the
first
targeting arm has a Tm for its complementary sequence that is higher (e.g.,
about 1 C., about 2
C., about 3 C., about 4 C., about 5 C., or more than about 5 C. higher)
than the Tm of the
second targeting arm for its complementary sequence. In some embodiments, each
of the first
targeting arms have similar or identical Tms for their respective
complementary sequences and
each of the second targeting arms have similar or identical Tms for their
respective
complementary sequences (and the first targeting arms have higher Tms than the
second
targeting arms). For example, in some embodiments, the Tm of the first arm(s)
may be about 58
C. and the Tm of the second arm(s) may be about 56 C. In some embodiments,
the Tm of the
first arm(s) may be about 68 C., and the Tm of the second arm(s) may be about
65 C. It should
17
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
be appreciated that in some embodiments the similarity (e.g., within a range
of 1 C., 2 C., 3
C., 4 C., 5 C.) or identity of the Tms for the different targeting arms
should be based either on
empirical data for each arm or based on the same predictive algorithm for each
arm (e.g.,
Wallace, R. B., Shaffer, J., Murphy, R. F., Bonner, J., Hirose, T., and
Itakura, K. (1979) Nucleic
Acids Res 6:3543-3557, SantaLucia J (1998), Proc Natl Acad Sci USA, 95:1460-5,
or other
algorithm).
In some embodiments, the Tm of the first targeting arm of a molecular
inversion probe
(at the 5' end of the molecular inversion probe) is selected to be
sufficiently stable to prevent
displacement of the first targeting arm from its complementary sequence on a
target nucleic acid.
In some embodiments, the Tm of the first targeting arm is 50-55 C., at least
55 C., 55-60 C.,
at least 60 C., 60-65 C., at least 65 C., at least 70 C., at least 75 C.,
or at least 80 C. As
discussed above, it should be appreciated that the for a particular targeting
arm may be
determined empirically or theoretically. Different theoretical models may be
used to determine a
Tm and it should be appreciated that the predicted Tm for a particular
sequence may be different
depending on the algorithm used for the prediction. In some embodiments, each
probe in a
plurality of probes (e.g., each probe in a set of 5-10, each probe in a set of
at least 10, each probe
in a set of 10-50, each probe in a set of 50-100, each probe in a set of 100-
500, or each probe in a
set of at least 500 different probes) has a different first targeting arm
(e.g., different sequences)
but each different first targeting arm has a similar or identical Tm for its
complementary
sequence on a target nucleic acid. It should be appreciated that in some
embodiments the
similarity (e.g., within a range of 1 C, 2 C, 3 C, 4 C, 5 C) or identity of
the Tms for the different
targeting arms should be based either on empirical data for each arm or based
on the same
predictive algorithm for each arm (e.g., Wallace, R. B., Shaffer, J., Murphy,
R. F., Bonner, J.,
Hirose, T., and Itakura, K. (1979) Nucleic Acids Res 6:3543-3557, SantaLucia J
(1998), Proc
Natl Acad Sci USA, 95:1460-5, or other algorithm).
In some embodiments, the sub-target nucleic acid contains a nucleic acid
repeat. In some
embodiments, the nucleic acid repeat is a dinucleotide or trinucleotide
repeat. In some
embodiments, the sub-target nucleic acid contains 10-100 copies of the nucleic
acid repeat in the
absence of an abnormal increase or decrease in nucleic acid repeats. In some
embodiments, the
sub-target nucleic acid is a region of the Fragile-X locus that contains a
nucleic acid repeat. In
some embodiments, one or both targeting arms hybridize to a region on the
target nucleic acid
18
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
that is immediately adjacent to a region of nucleic acid repeats. In some
embodiments, one or
both targeting arms hybridize to a region on the target nucleic acid that is
separated from a
region of nucleic acid repeats by a region that does not contain any nucleic
acid repeats. In some
embodiments, the molecular inversion probe further comprises a primer-binding
region that can
be used to sequence the captured sub-target nucleic acid and optionally the
first and/or second
targeting arm.
In some embodiments, aspects of the invention relate to evaluating the length
of a
plurality of different target nucleic acids in a biological sample. In some
embodiments, the
plurality of target nucleic acids are analyzed using a plurality of different
molecular inversion
probes. In some embodiments, each different molecular inversion probe
comprises a different
pair of first and second targeting arms at each of the 3' and 5' ends. In some
embodiments, each
different molecular inversion probe comprises the same primer-binding
sequence.
In some embodiments, aspects of the invention relate to analyzing nucleic acid
from a biological
sample obtained from a subject. In some embodiments, the biological sample is
a blood sample.
In some embodiments, the biological sample is a tissue sample, specific cell
population, tumor
sample, circulating tumor cells, or environmental sample. In some embodiments,
the biological
sample is a single cell. In some embodiments, nucleic acids are analyzed in
biological samples
obtained from a plurality of different subjects. In some embodiments, nucleic
acids from a
biological sample are analyzed in multiplex reactions. It should be
appreciated that a biological
sample contains a plurality of copies of a genome derived from a plurality of
cells in the sample.
Accordingly, a sample may contain a plurality of independent copies of a
target nucleic acid
region of interest, the capture efficiency of which can be used to evaluate
its size as described
herein.
In some embodiments, aspects of the invention relate to evaluating a nucleic
acid capture
efficiency by determining an amount of target nucleic acid that is captured
(e.g., an amount of
sub-target nucleic acid sequences that are captured). In some embodiments, the
amount of target
nucleic acid that is captured is determined by determining a number of
independently captured
target nucleic acid molecules (e.g., the amount of independently captured
molecules that have the
sequence of the sub-target region). In some embodiments, the amount of target
nucleic acid that
is captured is compared to a reference amount of captured nucleic acid. In
some embodiments,
the reference amount is determined by determining a number of independently
captured
19
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
molecules of a reference nucleic acid. In some embodiments, the reference
nucleic acid is a
nucleic acid of a different locus in the biological sample that is not
suspected of containing a
deletion or insertion. In some embodiments, the reference nucleic acid is a
nucleic acid of known
size and amount that is added to the capture reaction. As described herein, a
number of
independently captured nucleic acid sequences can be determined by contacting
a nucleic acid
sample with a preparation of a probe (e.g., a MIP probe as described herein).
It should be
appreciated that the preparation may comprise a plurality of copies of the
same probe and
accordingly a plurality of independent copies of the target region may be
captured by different
probe molecules. The number of probe molecules that actually capture a
sequence can be
evaluated by determining an amount or number of captured molecules using any
suitable
technique. This number is a reflection of both the number of target molecules
in the sample and
the efficiency of capture of those target molecules, which in turn is related
to the size of the
target molecules as described herein. Accordingly, the capture efficiency can
be evaluated by
controlling for the abundance of the target nucleic acid, for example by
comparing the number or
amount of captured target molecules to an appropriate control (e.g., a known
size and amount of
control nucleic acid, or a different locus that should be present in the same
amount in the
biological sample and is not expected to contain any insertions or deletions).
It should be
appreciated that other factors may affect the capture efficiency of a
particular target nucleic acid
region (e.g., the sequence of the region, the GC content, the presence of
secondary structures,
etc.). However, these factors also can be accounted for by using appropriate
controls (e.g.,
known sequences having similar properties, the same sequences, other genomic
sequences
expected to be present in the biological sample at the same frequency, etc.,
or any combination
thereof).
In some embodiments, aspects of the invention relate to identifying a subject
as having an
insertion or deletion in one or more alleles of a genetic locus if the capture
efficiency for that
genetic locus is statistically significantly different than a reference
capture efficiency.
It should be appreciated that hybridization conditions used for any of the
capture
techniques described herein (e.g., MIP capture techniques) can be based on
known hybridization
buffers and conditions.
In some embodiments, the methods disclosed herein are useful for any
application where
the detection of deletions or insertions is important.
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
In some embodiments, aspects of the invention relate to basing a nucleic acid
sequence
analysis on results from two or more different nucleic acid preparatory
techniques that have
different systematic biases in the types of nucleic acids that they sample.
According to the
invention, different techniques have different sequence biases that are
systematic and not simply
due to stochastic effects during nucleic acid capture or amplification.
Accordingly, the degree of
oversampling required to overcome variations in nucleic acid preparation needs
to be sufficient
to overcome the biases (e.g., an oversampling of 2-5 fold, 5-10 fold, 5-15
fold, 15-20 fold, 20-30
fold, 30-50 fold, or intermediate to higher fold).
According to some embodiments, different techniques have different
characteristic or
systematic biases. For example, one technique may bias a sample analysis
towards one particular
allele at a genetic locus of interest, whereas a different technique would
bias the sample analysis
towards a different allele at the same locus. Accordingly, the same sample may
be identified as
being different depending on the type of technique that is used to prepare
nucleic acid for
sequence analysis. This effectively represents a sensitivity limitation,
because each technique has
different relative sensitivities for polymorphic sequences of interest.
According to aspects of the invention, the sensitivity of a nucleic acid
analysis can be
increased by combining the sequences from different nucleic acid preparative
steps and using the
combined sequence information for a diagnostic assay (e.g., for a making a
call as to whether a
subject is homozygous or heterozygous at a genetic locus of interest).
In some embodiments, the invention provides a method of increasing the
sensitivity of a
nucleic acid detection assay by obtaining a first preparation of a target to
nucleic acid using a
first preparative method on a biological sample, obtaining a second
preparation of a target
nucleic acid using a second preparative method on the biological sample,
assaying the sequences
obtained in both first and second nucleic acid preparations, and using the
sequence information
from both first and second nucleic acid preparations to determine the genotype
of the target
nucleic acid in the biological sample, wherein the first and second
preparative methods have
different systematic sequence biases. In some embodiments, the first and
second nucleic acid
preparations are combined prior to performing a sequence assay. In some
embodiments, separate
sequence assays are performed on the first and second nucleic acid
preparations and the sequence
information from both assays are combined to determine the genotype of the
target nucleic acid
in the biological sample. In some embodiments, the first preparative method is
an amplification-
21
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
based, a hybridization-based, or a circular probe-based preparative method. In
some
embodiments, the second method is an amplification-based, a hybridization-
based, or a circular
probe-based preparative method. In some embodiments, the first and second
methods are of
different types (e.g., only one of them is an amplification-based, a
hybridization-based, or a
circular probe-based preparative method, and the other one is one or the other
two types of
method). Accordingly, in some embodiments the second preparative method is an
amplification-
based, a hybridization-based, or a circular probe-based preparative method,
provided that the
second method is different from the first method. However, in some
embodiments, both methods
may be of the same type, provided they are different methods (e.g., both are
amplification based
or hybridization-based, but are different types of amplification or
hybridization methods, e.g.,
with different relative biases).
In amplification-based (e.g., PCR-based or LCR-based, etc.) preparative
methods,
genomic loci (target nucleic acids) are isolated directly by means of a
polymerase chain reaction
or ligase chain reaction (or other amplification method) that selectively
amplifies each locus
using a pair of oligonucleotide primers. It is to be understood that primers
will be sufficiently
complementary to the target sequence to hybridize with and prime amplification
of the target
nucleic acid. Any one of a variety of art known methods may be utilized for
primer design and
synthesis. One or both of the primers may be perfectly complementary to the
target sequence.
Degenerate primers may also be used. Primers may also include additional
nucleic acids that are
not complementary to target sequences but that facilitate downstream
applications, including for
example restriction sites and identifier sequences (e.g., source sequences).
PCR based methods
may include amplification of a single target nucleic acid and multiplex
amplification
(amplification of multiple target nucleic acids in parallel).
Hybridization-based preparative may methods involve selectively immobilizing
target
nucleic acids for further manipulation. It is to be understood that one or
more oligonucleotides
(immobilization oligonucleotides), which in some embodiments may be from 10 to
200
nucleotides in length, are used which hybridize along the length of a target
region of a genetic
locus to immobilize it. In some embodiments, immobilization oligonucleotides
are either
immobilized before hybridization is performed (e.g., Roche/Nimblegen 'sequence
capture'), or
are prepared such that they include a moiety (e.g., biotin) which can be used
to selectively
22
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
immobilize the target nucleic acid after hybridization by binding to e.g.,
streptavidin-coated
microbeads (e.g., Agilent `SureSelece).
Circularization selection-based preparative methods selectively convert each
region of
interest into a covalently-closed circular molecule which is then isolated by
removal (usually
enzymatic, e.g., with exonuclease) of any non-circularized linear nucleic
acid. Oligonucleotide
probes are designed which have ends that flank the region of interest. The
probes are allowed to
hybridize to the genomic target, and enzymes are used to first (optionally)
fill in any gap
between probe ends and second ligate the probe closed. In some embodiments,
following
circularization, any remaining (non-target) linear nucleic acid can be
removed, resulting in
isolation (capture) of target nucleic acid. Circularization selection-based
preparative methods
include molecular inversion probe capture reactions and 'selector' capture
reactions. However,
other techniques may be used as aspects of the invention are not limited in
this respect. In some
embodiments, molecular inversion probe capture of a target nucleic acid is
indicative of the
presence of a polymorphism in the target nucleic acid.
A variety of methods may be used to evaluate and compare bias profiles of each
preparative technique. Next-generation sequencing may be used to
quantitatively measure the
abundance of each isolated target nucleic acid obtained from a certain
preparative method. This
abundance may be compared to a control abundance value (e.g., a known starting
abundance of
the target nucleic acid) and/or with an abundance determined through the use
of an alternative
preparative method. For example, a set of target nucleic acids may be isolated
by one or more of
the three preparative methods; the target nucleic acid may be observed x times
using the
amplification technique, y times using the hybridization enrichment technique,
and z times using
the circularization selection technique. A pairwise correlation coefficient
may be computed
between each abundance value (e.g., x and y, x and z, and y and z) to assess
bias in nucleic acid
isolation between pairs of preparative methods. Since the mechanisms of
isolation are different
in each approach, the abundances will usually be different and largely
uncorrelated with each
other.
In some embodiments, the invention provides a method of obtaining a nucleic
acid
preparation that is representative of a target nucleic acid in a biological
sample by obtaining a
first preparation of a target nucleic acid using a first preparative method on
a biological sample,
obtaining a second preparation of a target nucleic acid using a second
preparative method on the
23
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
biological sample, and combining the first and second nucleic acid
preparations to obtain a
combined preparation that is representative of the target nucleic acid in the
biological sample.
In some embodiments of any of the methods described herein, a third
preparation of the target
nucleic acid is obtained using a third preparative method that is different
from the first and
second preparative methods, wherein the first, second, and third preparative
methods all have
different systematic sequence biases. In some embodiments of any of the
methods described
herein, the different preparative methods are used for a plurality of
different loci in the biological
sample to increase the sensitivity of a multiplex nucleic acid analysis. In
some embodiments, the
target nucleic acid has a sequence of a gene selected from Table 1.
However, it should be appreciated that a genotyping method of the invention
may include
several steps, each of which independently may involve one or more different
preparative
techniques described herein. In some embodiments, a nucleic acid preparation
may be obtained
using one or more (e.g., 2, 3, 4, 5, or more) different techniques described
herein (e.g.,
amplification, hybridization capture, circular probe capture, etc., or any
combination thereof) and
the nucleic acid preparation may be analyzed using one or more different
techniques (e.g.,
amplification, hybridization capture, circular probe capture, etc., or any
combination thereof) that
are selected independently of the techniques used for the initial preparation.
In some embodiments, aspects of the invention also provide compositions, kits,
devices,
and analytical methods for increasing the sensitivity of nucleic acid assays.
Aspects of the
invention are particularly useful for increasing the confidence level of
genotyping analyses.
However, aspects of the invention may be used in the context of any suitable
nucleic acid
analysis, for example, but not limited to, a nucleic acid analysis that is
designed to determine
whether more than one sequence variant is present in a sample.
In some embodiments, aspects of the invention relate to a plurality of nucleic
acid probes
(e.g., 10-50, 50-100, 100-250, 250-500, 500-1,000, 1,000-2,000, 2,000-5,000,
5,000-7,500,
7,500-10,000, or lower, higher, or intermediate number of different probes).
In some
embodiments, each probe or each of a subset of probes (e.g., 10-25%, 25-50%,
50-75%, 75-90%,
or 90-99%) has a different first targeting arm. In some embodiments, each
probe or each probe of
a subset of probes (e.g., 10-25%, 25-50%, 50-75%, 75-90%, or 90-99%) has a
different second
targeting arm. In some embodiments, the first and second targeting arms are
separated by the
same intervening sequence. In some embodiments, the first and second targeting
arms are
24
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
complementary to target nucleic acid sequences that are separated by the same
or a similar length
(e.g., number of nucleic acids, for example, 0-25, 25-50, 50-100, 100-250, 250-
500, 500-1,000,
1,000-2,500 or longer or intermediate number of nucleotides) on their
respective target nucleic
acids (e.g., genomic loci). In some embodiments, each probe or a subset of
probes (e.g., 10-25%,
25-50%, 50-75%, 75-90%, or 90-99%) includes a first primer binding sequence.
In some
embodiments, the primer binding sequence is the same (e.g., it can be used to
prime sequencing
or other extension reaction). In some embodiments, each probe or a subset of
probes (e.g., 10-
25%, 25-50%, 50-75%, 75-90%, or 90-99%) includes a unique identifier sequence
tag (e.g., that
is predetermined and can be used to distinguish each probe).
In some embodiments, the methods disclosed herein are useful for any
application where
sensitivity is important. For example, detection of cancer mutations in a
heterogenous tissue
sample, detection of mutations in maternally-circulating fetal DNA, and
detection of mutations
in cells isolated during a preimplantation genetic diagnostic procedure.
According to some aspects of the invention, methods of detecting a
polymorphism in a nucleic
acid in a biological sample are provided. In some embodiments, the methods
comprise obtaining
a nucleic acid preparation using a preparative method (e.g., any of the
preparative methods
disclosed herein) on a biological sample, and performing a molecular inversion
probe capture
reaction on the nucleic acid preparation, wherein a molecular inversion probe
capture (e.g., using
a mutation-detection MIP) of a target nucleic acid of the nucleic acid
preparation is indicative of
the presence of a mutation (polymorphism) in the target nucleic acid,
optionally wherein the
polymorphism is selected from Table 2.
According to some aspects of the invention, methods of genotyping a nucleic
acid in a
biological sample are provided. In some embodiments, the methods comprise
obtaining a nucleic
acid preparation using a preparative method on a biological sample, sequencing
a target nucleic
acid of the nucleic acid preparation, and performing a molecular inversion
probe capture reaction
on the biological sample, wherein a molecular inversion probe capture of the
target nucleic acid
in the biological sample is indicative of the presence of a polymorphism in
the target nucleic
acid, genotyping the target nucleic acid based on the results of the
sequencing and the capture
reaction.
In some embodiments of the methods disclosed herein, the target nucleic acid
has a
sequence of a gene selected from Table 1.
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
It should be appreciated that any one or more embodiments described herein may
be used
for evaluating multiple genetic markers in parallel. Accordingly, in some
embodiments, aspects
of the invention relate to determining the presence of one or more markers
(e.g., one or more
alleles) at multiple different genetic loci in parallel. Accordingly, the risk
or presence of multiple
heritable disorders may be evaluated in parallel. In some embodiments, the
risk of having
offspring with one or more heritable disorders may be evaluated. In some
embodiments, an
evaluation may be performed on a biological sample of a parent or a child
(e.g., at a pre-
implantation, prenatal, perinatal, or postnatal stage). In some embodiments,
the disclosure
provides methods for analyzing multiple genetic loci (e.g., a plurality of
target nucleic acids
selected from Table 1 or 2) from a patient sample, such as a blood, pre-
implantation embryo,
chorionic villus or amniotic fluid sample. A patient or subject may be a
human. However,
aspects of the invention are not limited to humans and may be applied to other
species (e.g.,
mammals, birds, reptiles, other vertebrates or invertebrates) as aspects of
the invention are not
limited in this respect. A subject or patient may be male or female. In some
embodiments, in
connection with reproductive genetic counseling, samples from a male and
female member of a
couple may be analyzed. In some embodiments, for example, in connection with
an animal
breeding program, samples from a plurality of male and female subjects may be
analyzed to
determine compatible or optimal breeding partners or strategies for particular
traits or to avoid
one or more diseases or conditions. Accordingly, reproductive risks may be
determined and/or
reproductive recommendations may be provided based on information derived from
one or more
embodiments of the invention.
However, it should be appreciated that aspects of the invention may be used in
connection with any medical evaluation where the presence of one or more
alleles at a genetic
locus of interest is relevant to a medical determination (e.g., risk or
detection of disease, disease
prognosis, therapy selection, therapy monitoring, etc.). Further aspects of
the invention may be
used in connection with detection, in tumor tissue or circulating tumor cells,
of mutations in
cellular pathways that cause cancer or predict efficacy of treatment regimens,
or with detection
and identification of pathogenic organisms in the environment or a sample
obtained from a
subject, e.g., a human subject.
These and other aspects of the invention are described in more detail in the
following
description and non-limiting examples and drawings.
26
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
BRIEF DESCRIPTION OF DRAWINGS
FIG. 1 illustrates a non-limiting embodiment of a tiled probe layout;
FIG. 2 illustrates a non-limiting embodiment of a staggered probe layout;
FIG. 3 illustrates a non-limiting embodiment of an alternating staggered probe
layout;
FIG. 4, panels a), b), and c) depict various non-limiting methods for
combining
differentiator tag sequence and target sequences (NNNN depicts a
differentiator tag
sequence);
FIG. 5 depicts a non-limiting method for genotyping based on target and
differentiator
tag sequences;
FIG. 6 depicts non-limiting results of a simulation of a MIP capture reaction;
FIG. 7 depicts a non-limiting graph of sequencing coverage;
FIG. 8 illustrates that shorter sequences are captured with higher efficiency
that longer
sequences using MIPs;
FIG. 9 illustrates a non-limiting scheme of padlock (MIP) capture of a region
that
includes both repetitive regions (thick wavy line) and the adjacent unique
sequence (thick
strait line);
FIG. 10 illustrates a non-limiting hypothetical relationship between target
gap size and
the relative number of reads of the repetitive region;
FIG. 11A depicts MIP capture of FMR1 repeat regions from a diploid genome;
FIG. 11B depicts preparative methods for biallelic resolution of FMR1 repeat
region
lengths in a diploid genome using MIP capture probes and unique differentiator
tags;
FIG. 11C depicts an analysis of FMR1 repeat region lengths in a diploid
genome;
FIG. 12 is a schematic of an embodiment of an algorithm of the invention;
FIG. 13 illustrates a non-limiting example of a graph of per-target abundance
with MIP
capture; and,
FIG. 14 shows a non-limiting a graph of correlation between two MIP capture
reactions.
FIGS. 15A-B show a SNaPshot validation of a putative Sanger variant call. FIG.
15A
discloses "GM17080" sequences as SEQ ID NO: 6328, 6329, and 6328 and FIG. 15B
discloses the "GM17074" sequences as SEQ ID NO: 6328, 6328, and 6328, all
respectively, in order of appearance.
27
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
FIGS. 16A-16D depict skewed allelic fractions in aneuploid cell line GM18540.
FIG.
16A depicts an IGV view of NGS data from GM18540 for the genotype call of
interest (shown
between vertical lines) (figure 16A discloses SEQ ID NO: 6330-6331). FIG. 16B
depicts bi-
directional Sanger data for the variant-containing region. FIG. 16C depicts a
histogram of allele
ratios for all non-reference genotype calls in chromosome 11 derived from
wholegenome
shotgun sequencing (WGSS) of GM18540 and control sample GM18537. FIG. 16D
depicts
genome-wide relative coverage for GM18540. WGSS coverage data for each of the
25 autosomes was binned into 50 Kb intervals and the log-ratio of the per-
sample mean
normalized values was plotted versus chromosome position. Dashed vertical
lines denote
chromosome boundaries; within a chromosome the ratios are arranged according
to
genomic position.
FIGS. 17A-D depict detection of previously-uncharacterized mutations in
samples from
individuals affected with cystic fibrosis. FIG. 17A depicts IGV of
heterozygous splice site
mutation c.3368-2A>T in sample GM12960 (figure 17A discloses SEQ ID NO: 6332-
6333).
FIG. 17B depicts IGV of heterozygous premature stop codon mutation R1158X in
sample
GM18802 (figure 17B discloses SEQ ID NO: 6334-6335). FIG. 17C depicts Sanger
data
confirming existence of mutation c.3368-2A>T in sample GM12960 (figure 17C
discloses SEQ
ID NO: 6336 and 6336). FIG. 17D depicts Sanger data confirming existence of
mutation
R1158X in sample GM18802 (figure 17D discloses SEQ ID NO: 6337 and 6337).
FIGS. 18A-E depict next-generation DNA sequencing workflow according to
certain
embodiments. FIG. 18B discloses (top panel) SEQ ID NO: 6338-6349, (left panel)
SEQ ID NO:
6338-6343, and (right panel) SEQ ID NO: 6344-6349, all respectively, in order
of appearance.
FIG. 18C discloses SEQ ID NO: 6350-6356, 6353, 6352, 6357, and 6357,
respectively, in order
of appearance. FIG. 18D discloses (left panel) SEQ ID NO: 6352, 6358, 6350,
6352, 6358,
6350, 6359, and 6359, and (right panel) SEQ ID NO: 6360, 6361, 6355, 6360,
6361, 6355,
6362, and 6363, all respectively, in order of appearance. FIG. 18E discloses
(left panel) SEQ ID
NO: 6358, 6352, and 6350, (right panel) SEQ ID NO: 6360, 6361, and 6355, and
(bottom
panel) SEQ ID NO: 6364 and 6364, all respectively, in order of appearance.
FIGS. 19A-D depict data from genotyping by assembly template alignment (GATA).
GATA correctly genotypes insertions and deletions that are undetectable by the
Alignment Only
method. Read from top to bottom, each panel provides tracks for cumulative
depth of coverage
28
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
(vertical grey bars); representative MIP alignments (horizontal grey bars)
with mismatches
(letters), insertions (black bars), and gaps (dashed lines); chromatogram;
reference DNA
andamino acid sequence for FIG. 19A heterozygous BLM c.2207_2212de1insTAGATTC
in
sample GM04408 as well as several alleles in the first exon of SMPD1 (FIG. 19A
discloses SEQ
ID NO: 6365 and 6366) including FIG. 19B a heterozygous 18bp deletion in
sample GM20342
(minus strand) (FIG.19B discloses SEQ ID NO: 6367 and 6368), FIG. 19C a
heterozygous 12bp
insertion and homozygous substitution in sample GM17282 (plus strand) (FIG.
19C discloses
SEQ ID NO: 6369 and 6370), and FIG. 19D compound heterozygous 6 and 12 bp
deletions in
sample GM00502 (minus strand) (FIG. 19D discloses SEQ ID NO: 6369 and 6370).
Chromatogram trace offsets corresponding to specific heterozygous insertion
and deletion
patterns are indicated with slanted lines color coded by reference base. For
clarity offsets are shown for FIGS. 19C-D only.
FIGS. 20A-1, 20A-2, 20A-3, 20B-1, 20B-2 and 20B-3 show NGS detection of allele
dropout in Sanger reactions. FIG. 20A-1 discloses SEQ ID NO: 6371, 6372, and
6372, FIG.
20A-B depicts SEQ ID NO: 6371, 6371, 6372, and FIG. 20A-3 disclosesSEQ ID NO:
6373 and
6374, all respectively, in order of appearance. FIG. 20B-1 discloses SEQ ID
NO: 6371, 6372,
and 6372, FIG. 20B-2 discloses SEQ ID NO: 6371, 6371, 6372, and FIG. 20B-3
discloses SEQ
ID NO: 6373 and 6374, all respectively, in order of appearance.
FIG. 21 diagrams use of methods of the invention to validate a genotyping by
assembly-
templated alignment (GATA) technique.
FIG. 22 illustrates obtaining sequence reads and inserting a simulated
mutation.
FIG. 23 shows standard analysis of sequence reads for comparison to GATA.
FIG. 24 shows analysis by GATA.
DETAILED DESCRIPTION
Aspects of the invention relate to preparative and analytical methods and
compositions
for evaluating genotypes, and in particular, for determining the allelic
identity (or identities in a
diploid organism) of one or more genetic loci in a subject. Aspects of the
invention are based, in
part, on the identification of different sources of ambiguity and error in
genetic analyses, and, in
part, on the identification of one or more approaches to avoid, reduce,
recognize, and/or resolve
these errors and ambiguities at different stages in a genetic analysis.
Aspects of the invention
relate to methods and compositions for addressing bias and/or stochastic
variation associated
29
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
with one or more preparative and/or analytical steps of a nucleic acid
evaluation technology. In
some embodiments, preparative methods can be adapted to avoid or reduce the
risk of bias
skewing the results of a genetic analysis. In some embodiments, analytical
methods can be
adapted to recognize and correct for data variations that may give rise to
misinterpretation (e.g.,
incorrect calls such as homozygous when the subject is actually heterozygous
or heterozygous
when the subject is actually homozygous). Methods of the invention may be used
for any type of
mutation, for example a single base change (e.g., insertion, deletion,
transversion or transition,
etc.), a multiple base insertion, deletion, duplication, inversion, and/or any
other change or
combination thereof.
In some embodiments, additional or alternative techniques may be used to
address loci
characterized by multiple repeats of a core sequence where the length of the
repeat is longer than
a typical sequencing read thereby making it difficult to determine whether a
deletion or
duplication of one or more core sequence units has occurred based solely on a
sequence read.
In some embodiments, increased confidence in an assay result may be obtained
by i) selecting
two or more different preparative and/or analytical techniques that have
different biases (e.g.,
known to have different biases), ii) evaluating a patient sample using the two
or more different
techniques, iii) comparing the results from the two or more different
techniques, and/or iv)
determining whether the results are consistent for the two or more different
techniques. In some
embodiments, if determining in step (iv) indicates that the results are
consistent (e.g., the same)
then increased confidence in the assay result is obtained. In other
embodiments, if determining in
step (iv) indicates that the results are inconsistent (e.g., that the results
are ambiguous) then one
or more additional preparative and/or analytical techniques, which have a
different bias (e.g.,
known to have a different bias) compared with the two or more different
preparative and/or
analytical techniques selected in step (i), are used to evaluate the patient
sample, and the results
of the one or more additional preparative and/or analytical techniques are
compared with the
results from step (ii) to resolve the inconsistency.
In some embodiments, two or more independent samples may be obtained from a
subject
and independently analyzed. In some embodiments, two or more independent
samples are
obtained at approximately the same time point. In some embodiments, two or
more independent
samples are obtained at multiple different time points. In some embodiments,
the use of two or
more independent sample facilitates the elimination, normalization, and/or
quantification of
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
stochastic measurement noise. It is to be appreciated that two or more
independent samples may
be obtained in connection with any of the methods disclosed herein, including,
for example,
methods for pathogen profiling in a human or other animal subjects, monitoring
tumor
progression/regression, analyzing circulating tumor cells, analyzing fetal
cells in maternal
circulation, and analyzing/monitoring/profiling of environmental pathogens.
In some embodiments, one or more of the techniques described herein may be
combined
in a single assay protocol for evaluating multiple patient samples in
parallel.
It should be appreciated that aspects of the invention may be useful for high
throughput, cost-
effective, yet reliable, genotyping of multiple patient samples (e.g., in
parallel, for example in
multiplex reactions). In some embodiments, aspects of the invention are useful
to reduce the
error frequency in a multiplex analysis. Certain embodiments may be
particularly useful where
multiple reactions (e.g., multiple loci and/or multiple patient samples) are
being processed. For
example, 10-25, 25-50, 50-75, 75-100 or more loci may be evaluated for each
subject out of any
number of subject samples that may be processed in parallel (e.g., 1-25, 25-
50, 50-100, 100-500,
500-1,000, 1,000-2,500, 2,500-5,000 or more or intermediate numbers of patient
samples). It
should be appreciated that different embodiments of the invention may involve
conducting two
or more target capture reactions and/or two or more patient sample analyses in
parallel in a single
multiplex reaction. For example, in some embodiments a plurality of capture
reactions (e.g.,
using different capture probes for different target loci) may be performed in
a single multiplex
reaction on a single patient sample. In some embodiments, a plurality of
captured nucleic acids
from each one of a plurality of patient samples may be combined in a single
multiplex analysis
reaction. In some embodiments, samples from different subjects are tagged with
subject-specific
(e.g., patient-specific) tags (e.g., unique sequence tags) so that the
information from each product
can be assigned to an identified subject. In some embodiments, each of the
different capture
probes used for each patient sample have a common patient-specific tag. In
some embodiments,
the capture probes do not have patient-specific tags, but the captured
products from each subject
may be amplified using one or a pair of amplification primers that are labeled
with a patient-
specific tag. Other techniques for associating a patient-specific tag with the
captured product
from a single patient sample may be used as aspects of the invention are not
limited in this
respect. It should be appreciated that patient-specific tags as used herein
may refer to unique tags
that are assigned to identify patients in a particular assay. The same tags
may be used in a
31
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
separate multiplex analysis with a different set of patient samples (e.g.,
from different patients)
each of which is assigned one of the tags. In some embodiments, different sets
of unique tags
may be used in sequential (e.g., alternating) multiplex reactions in order to
reduce the risk of
contamination from one assay to the next and allow contamination to be
detected on the basis of
the presence of tags that are not expected to be present in a particular
assay.
Embodiments of the invention may be used for any of a number of different
settings:
reproductive settings, disease screening, identifying subjects having cancer,
identifying subjects
having increased risk for a disease, stratifying a population of subjects
according to one or more
of a number of factors, for example responsiveness to a particular drug, lack
or not of an adverse
reaction (or risk therefore) to a particular drug, and/or providing
information for medical records
(e.g., homozygosity, heterozygosity at one or more loci). It should be
appreciated that the
invention is not limited to genomic analysis of patient samples. For example,
aspects of the
invention may be useful for high throughput genetic analysis of environment
samples to detect
pathogens.
In some embodiments, the methods disclosed herein are useful for diagnosis of
one or
more heritable disorders. In some embodiments, a heritable disorder that may
be diagnosed with
the methods disclosed herein is a genetic disorder that is prevalent in the
Ashkenazi Jewish
population. In some embodiments, the heritable disorders are selected from: 21-
Hydroxylase-
Defiocient Congenital Adrenal Hyperplasia; ABCC8-Related Hyperinsulinism;
Alpha-
Thalassemia, includes Constant Spring, & MR associated; Arylsulfatase A
Deficiency-
Metyachromatic Leukodystrophy; Biotimidase Deficiency-Holocarboxylase
Synthetase
Deficiency; Bloom's Syndrome; Canavan Disease; CFTR-Related Disorders-cystic
fibrosis;
Citrullinemia Type I; Combined MMA & Homocystinuria-db1C; Dystrophinopathies
(DMD &
BMD); Familial Dysautonomia; Fanconi Anemia-FANCC; Galactosemia-Ci_assical:
Galactokinase Defiency & Galactose Epimerase Deficiency; Gaucher Disease; GJB2-
Related
DFNB 1 Nonsyndromic Hearing Loss and Deafness; Glutaric acidemia Type 1;
Hemoglobinopathies beta-chain disorders; Glycogen Storage Disease Type 1A;
Maple Syrup
Urine Disease; Types 1A, 1B, 2, 3; Medium Chain Acyl-Coenzyme A; Dehydrogenase
Deficiency-MCADD; Methylmalonic Acidemia; Mucolipidosis IV; Nemaline Myopathy;
Nieman-Pick Type A-Acid Sphingomyelinase Deficiency; Non-Ketotic
Hyperglycinemia-
Glycine Encephalopathy; Ornithine Transcarbamylase Deficiency; PKU
Phenylalanine
32
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
Hydroxylase Deficiency; Propionic Acidemia; Short Chain Acyl-CoA Dehydrogenase
Deficiency-SCADD; Smith-Lemli-Opitz Syndrome; Spinal Muscular Atrophy (SMN1)-
SMA;
Tay Sachs-HexA Deficiency; Usher Synbdrome-Type I (Type IB, Type IC, Type ID,
Type IF,
Type IG); X-Linked Mental Retardation ARX-Related Disorders; X-Linked Mental
Retardation
with Cerebellar Cypoplasia and sistinctive Facial Appearance; X-Linked Mental
Retardation;
includes 9, 21, 30, 46, 58, 63, 88, 89; X-linked mental retardation: FM1-
Related Disorders-
FRXA, Fragile X MR; X-linked SMR: Renpenning Syndrome 1; Zellweger Spectrum
disorders¨Peroxisomal Bifunctional Enzyme Deficiencies including Zellweger,
NALD, and/or
infantile Refsums. However, all of these, subsets of these, other genes, or
combinations thereof
may be used.
According to some aspects, the disclosure relates to multiplex diagnostic
methods. In
some embodiments, multiplex diagnostic methods comprise capturing a plurality
of genetic loci
in parallel (e.g., a genetic locus of Table 1). In some embodiments, genetic
loci possess one or
more polymorphisms (e.g., a polymorphism of Table 2) the genotypes of which
correspond to
disease causing alleles. Accordingly, in some embodiments, the disclosure
provides methods for
assessing multiple heritable disorders in parallel.
In some embodiments, methods are provided for diagnosing multiple heritable
disorders
in parallel at a pre-implantation, prenatal, perinatal, or postnatal stage. In
some embodiments, the
disclosure provides methods for analyzing multiple genetic loci (e.g., a
plurality of target nucleic
acids selected from Table 1) from a patient sample, such as a blood, pre-
implantation embryo,
chorionic villus or amniotic fluid sample. A patient or subject may be a
human. However,
aspects of the invention are not limited to humans and may be applied to other
species (e.g.,
mammals, birds, reptiles, other vertebrates or invertebrates) as aspects of
the invention are not
limited in this respect. A subject or patient may be male or female. In some
embodiments, in
connection with reproductive genetic counseling, samples from a male and
female member of a
couple may be analyzed. In some embodiments, for example, in connection with
an animal
breeding program, samples from a plurality of male and female subjects may be
analyzed to
determine compatible or optimal breeding partners or strategies for particular
traits or to avoid
one or more diseases or conditions.
However, it should be appreciated that any other diseases may be studied
and/or risk
factors for diseases or disorders including, but not limited to allergies,
responsiveness to
33
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
treatment, cancer tumor profiling for treatment and prognosis, monitoring and
identification of
patient infections, and monitoring of environmental pathogens.
1. Reducing Representational Bias in Multiplex Amplification Reactions:
In some embodiments, aspects of the invention relate to methods that reduce
bias and
increase reproducibility in multiplex detection of genetic loci, e.g., for
diagnostic purposes.
Molecular inversion probe technology is used to detect or amplify particular
nucleic acid
sequences in potentially complex mixtures. Use of molecular inversion probes
has been
demonstrated for detection of single nucleotide polymorphisms (Hardenbol et
al. 2005 Genome
Res 15:269-75) and for preparative amplification of large sets of exons
(Porreca et al. 2007 Nat
Methods 4:931-6, Krishnakumar et al. 2008 Proc Natl Acad Sci USA 105:9296-
301). One of the
main benefits of the method is in its capacity for a high degree of
multiplexing, because
generally thousands of targets may be captured in a single reaction containing
thousands of
probes. However, challenges associated with, for example, amplification
efficiency (See, e.g.,
Turner E H, et al., Nat. Methods. 2009 Apr. 6:1-2.) have limited the practical
utility of the
method in research and diagnostic settings.
Aspects of the disclosure are based, in part, on the discovery of effective
methods for
overcoming challenges associated with systematic errors (bias) in multiplex
genomic capture and
sequencing methods, namely high variability in target nucleic acid
representation and unequal
sampling of heterozygous alleles in pools of captured target nucleic acids
(e.g., isolated from a
biological sample). Accordingly, in some embodiments, the disclosure provides
methods that
reduce variability in the detection of target nucleic acids in multiplex
capture methods. In other
embodiments, methods improve allelic representation in a capture pool and,
thus, improve
variant detection outcomes. In certain embodiments, the disclosure provides
preparative methods
for capturing target nucleic acids (e.g., genetic loci) that involve the use
of different sets of
multiple probes (e.g., molecular inversion probes MIPs) that capture
overlapping regions of a
target nucleic acid to achieve a more uniform representation of the target
nucleic acids in a
capture pool compared with methods of the prior art. In other embodiments,
methods reduce
bias, or the risk of bias, associated with large scale parallel capture of
genetic loci, e.g., for
diagnostic purposes. In other embodiments, methods are provided for increasing
reproducibility
(e.g., by reducing the effect of polymorphisms on target nucleic acid capture)
in the detection of
a plurality of genetic loci in parallel. In further embodiments, methods are
provided for reducing
34
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
the effect of probe synthesis and/or probe amplification variability on the
analysis of a plurality
of genetic loci in parallel.
In some aspects, the disclosure provides probe sets that comprise a plurality
of different
probes. As used herein, a 'probe' is a nucleic acid having a central region
flanked by a 5' region
and a 3' region that are complementary to nucleic acids flanking the same
strand of a target
nucleic acid or subregion thereof. An exemplary probe is a molecular inversion
probe (MIP). A
'target nucleic acid' may be a genetic locus. Exemplary genetic loci are
disclosed herein in Table
1 (RefSeqGene Column).
While probes have been typically designed to meet certain constraints (e.g.
melting
temperature, G/C content, etc.) known to partially affect
capture/amplification efficiency (Ball et
al (2009) Nat Biotech 27:361-8 AND Deng et al (2009) Nat Biotech 27:353-60), a
set of
constraints which is sufficient to ensure either largely uniform or highly
reproducible
capture/amplification efficiency has not previously been achieved. As
disclosed herein,
uniformity and reproducibility can be increased by designing multiple probes
per target, such
that each base in the target is captured by more than one probe. In some
embodiments, the
disclosure provides multiple MIPs per target to be captured, where each MIP in
a set designed
for a given target nucleic acid has a central region and a 5' region and 3'
region ('targeting arms')
which hybridize to (at least partially) different nucleic acids in the target
nucleic acid
(immediately flanking a subregion of the target nucleic acid). Thus,
differences in efficiency
between different targeting arms and fill-in sequences may be averaged across
multiple MIPs for
a single target, which results in more uniform and reproducible capture
efficiency.
In some embodiments, the methods involve designing a single probe for each
target (a
target can be as small as a single base or as large as a kilobase or more of
contiguous sequence).
It may be preferable, in some cases, to design probes to capture molecules
(e.g., target nucleic
acids or subregions thereof) having lengths in the range of 1-200 bp (as used
herein, a by refers
to a base pair on a double-stranded nucleic acid¨however, where lengths are
indicated in bps, it
should be appreciated that single-stranded nucleic acids having the same
number of bases, as
opposed to base pairs, in length also are contemplated by the invention).
However, probe design
is not so limited. For example, probes can be designed to capture targets
having lengths in the
range of up to 10, 20, 30, 40, 50, 60, 70, 80, 90, 100, 200, 300, 400, 500,
1000, or more bps, in
some cases.
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
It is to be appreciated that the length of a capture molecule (e.g., a target
nucleic acid or
subregion thereof) is selected based upon multiple considerations. For
example, where analysis
of a target involves sequencing, e.g., with a next-generation sequencer, the
target length should
typically match the sequencing read-length so that shotgun library
construction is not necessary.
However, it should be appreciated that captured nucleic acids may be sequenced
using any
suitable sequencing technique as aspects of the invention are not limited in
this respect.
It is also to be appreciated that some target nucleic acids are too large to
be captured with
one probe. Consequently, it may be necessary to capture multiple subregions of
a target nucleic
acid in order to analyze the full target.
In some embodiments, a subregion of a target nucleic acid is at least 1 bp. In
other
embodiments, a subregion of a target nucleic acid is at least 10, 20, 30, 40,
50, 60, 70, 80, 90,
100, 200, 300, 400, 500, 600, 700, 800, 900, 1000 bp or more. In other
embodiments, a
subregion of a target nucleic acid has a length that is up to 10%, 20%, 30%,
40%, 50%, 60%,
70%, 80%, 90%, 95%, or more percent of a target nucleic acid length.
The skilled artisan will also appreciate that consideration is made, in the
design of MIPs,
for the relationship between probe length and target length. In some
embodiments, MIPs are
designed such that they are several hundred basepairs (e.g., up to 100, 200,
300, 400, 500, 600,
700, 800, 900, 1000 bp or more) longer than corresponding target (e.g.,
subregion of a target
nucleic acid, target nucleic acid).
In some embodiments, lengths of subregions of a target nucleic acid may
differ. For
example, if a target nucleic acid contains regions for which probe
hybridization is not possible or
inefficient, it may be necessary to use probes that capture subregions of one
or more different
lengths in order to avoid hybridization with problematic nucleic acids and
capture nucleic acids
that encompass a complete target nucleic acid.
Aspects of the invention involve using multiple probes, e.g., MIPs, to amplify
each target
nucleic acid. In some embodiments, the set of probes for a given target can be
designed to 'tile'
across the target, capturing the target as a series of shorter sub-targets. In
some embodiments,
where a set of probes for a given target is designed to 'tile' across the
target, some probes in the
set capture flanking non-target sequence). Alternately, the set can be
designed to 'stagger' the
exact positions of the hybridization regions flanking the target, capturing
the full target (and in
some cases capturing flanking non-target sequence) with multiple probes having
different
36
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
targeting arms, obviating the need for tiling. The particular approach chosen
will depend on the
nature of the target set. For example, if small regions are to be captured, a
staggered-end
approach might be appropriate, whereas if longer regions are desired, tiling
might be chosen. In
all cases, the amount of bias-tolerance for probes targeting pathological loci
can be adjusted
('dialed in') by changing the number of different MIPs used to capture a given
molecule.
In some embodiments, the 'coverage factor', or number of probes used to
capture a basepair in a
molecule, is an important parameter to specify. Different numbers of probes
per target are
indicated depending on whether one is using the tiling approach (see, e.g.,
FIG. 1) or one of the
staggered approaches (see, e.g., FIG. 2 or 3).
FIG. 1 illustrates a non-limiting embodiment of a tiled probe layout showing
ten captured
sub-targets tiled across a single target. Each position in the target is
covered by three sub-targets
such that MIP performance per base pair is averaged across three probes.
FIG. 2 illustrates a non-limiting embodiment of a staggered probe layout
showing the
targets captured by a set of three MIPs. Each MIP captures the full target,
shown in black, plus
(in some cases) additional extra-target sequence, shown in gray, such that the
targeting arms of
each MIP fall on different sequence. Each position in the target is covered by
three sub-targets
such that MIP performance per basepair is averaged across three probes.
Targeting arms land
immediately adjacent to the black or gray regions shown. It should be
appreciated that in some
embodiments, the targeting arms (not shown) can be designed so that they do
not overlap with
each other.
FIG. 3 illustrates a non-limiting embodiment of an alternating staggered probe
layout
showing the targets captured by a set of three MIPs. Each MIP captures the
full target, shown in
black, plus (in some cases) additional extra-target sequence, shown in gray,
such that the
targeting arms of each MIP fall on different sequence. Each position in the
target is covered by
three sub-targets such that MIP performance per basepair is averaged across
three probes.
Targeting arms land immediately adjacent to the black or gray regions shown.
It should be appreciated that for any of the layouts, the targeting arms on
adjacent tiled or
staggered probes may be designed to either overlap, not overlap, or overlap
for only a subset of
the probes.
In certain embodiments for any of the layouts, a coverage factor of about 3 to
to about 10
is used. However, the methods are not so limited and coverage factors of up to
2, 3, 4, 5, 6, 7, 8,
37
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
9, 10, 20 or more may be used. It is to be appreciated that the coverage
factor selected may
depend the probe layout being employed. For example, in the tiling approach,
for a desired
coverage factor, the number of probes per target is typically a function of
target length, sub-
target length, and spacing between adjacent sub-target start locations (step
size). For example,
for a desired coverage factor of 3, a 200 bp target with a start-site
separation of 20 bp and sub-
target length of 60 bp may be encompassed with 12 MIPs (FIG. 1). Thus, a
specific coverage
factor may be achieved by varying the number of probes per target nucleic acid
and the length of
the molecules captured. In the staggered approach, a fixed-length target
nucleic acid is captured
as several subregions or as `super-targets', which are molecules comprising
the target nucleic
acid and additional flanking nucleic acids, which may be of varying lengths.
For example, a
target of 50 bp can be captured at a coverage factor of 3 with 3 probes in
either a 'staggered'
(FIG. 2) or 'alternating staggered' configuration (FIG. 3).
The coverage factor will be driven by the extent to which detection bias is
tolerable. In
some cases, where the bias tolerance is small, it may be desirable to target
more subregions of
target nucleic acid with, perhaps, higher coverage factors. In some
embodiments, the coverage
factor is up to 2, 3, 4, 5, 6, 7, 8, 9, 10 or more.
In some embodiments, when a tiled probe layout is used, when the target length
is greater
than 1 bp and when a step size (distance between the 5'-end of a target and
the 5' end of its
adjacent target) is less than the length of a target or subregion thereof, it
is possible to compute
probe number for a particular target based on target length (T), sub-target
length (S), and
coverage factor (C), such that probe number=T/(S/C)+(C-1).
In some aspects, the disclosure provides methods to increase the uniformity of
amplification efficiency when multiple molecules are amplified in parallel;
methods to increase
the reproducibility of amplification efficiency; methods to reduce the
contribution of targeting
probe variability to amplification efficiency; methods to reduce the effect on
a given target
nucleic acid of polymorphisms in probe hybridization regions; and/or methods
to simplify
downstream workflows when multiplex amplification by MIPs is used as a
preparative step for
analysis by nucleic acid sequencing.
Polymorphisms in the target nucleic acid under the regions flanking a target
can interfere
with hybridization, polymerase fill-in, and/or ligation. Furthermore, this may
occur for only one
allele, resulting in allelic drop-out, which ultimately decreases downstream
sequencing accuracy.
38
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
In some embodiments, using a set of MIPs having multiple hybridization sites
for the capture of
any given target, the probability of loss from polymorphism is substantially
decreased because
not all targeting arms in the set of MIPs will cover the location of the
mutation.
Probes for MIP capture reactions may be synthesized on programmable
microarrays
because of the large number of sequences required. Because of the low
synthesis yields of these
methods, a subsequent amplification step is required to produce sufficient
probe for the MIP
amplification reaction. The combination of multiplex oligonucleotide synthesis
and pooled
amplification results in uneven synthesis error rates and representational
biases. By synthesizing
multiple probes for each target, variation from these sources may be averaged
out because not all
probes for a given target will have the same error rates and biases.
Multiplex amplification strategies disclosed herein may be used analytically,
as in
detection of SNPs, or preparatively, often for next-generation sequencing or
other sequencing
techniques. In the preparative setting, the output of an amplification
reaction is generally the
input to a shotgun library protocol, which then becomes the input to the
sequencing platform.
The shotgun library is necessary in part because next-generation sequencing
yields reads
significantly shorter than amplicons such as exons. In addition to the bias-
reduction afforded by
the multi-tiled approach described here, tiling also obviates the need for
shotgun library
preparation. Since the length of the capture molecule can be specified when
the probes, e.g.,
MIPs, are designed, it can be chosen to match the readlength of the sequencer.
In this way, reads
can 'walk' across an exon by virtue of the start position of each capture
molecule in the probe set
for that exon.
Exemplary molecular inversion probes are provided in Appendix A. These
molecular
inversion probes are designed to capture targets or sub-regions thereof on one
or more genes
listed in Table 5 (provided in Example 8). In certain applications, the
molecular inversion
probes provided in Appendix A may be used to tile-capture targets or sub-
regions thereof on the
one or more genes provided in Table 5. In particular applications, two or more
of the molecular
inversion probes of Appendix A tile across different, but overlapping sub-
regions of one or more
genes listed in Table 5 so that a target on the gene is capture by both of the
two or more
molecular inversion probes, as exemplified in Figure 1.
In certain embodiments, the molecular inversion probes of Appendix A that are
chosen
for tile-capture a target depends on the desired amount of overlapping
coverage for the target. In
39
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
one example, two or more molecular inversion probes of Appendix A, being in
directly
ascending SEQ ID NO: order and corresponding to a target nucleic acid, will
tile across the
target nucleic acid with a period of 25 base pairs such that every genomic
position of the target
nucleic acid is capture by multiple probes with orthogonal targeting arm
sequences. If less
coverage is desired for a target nucleic acid, one may select, for example,
every other molecular
inversion probes of Appendix A in ascending order that correspond to that
target.
The first and second targeting arms of the molecular inversion probes are
designed to
hybridize to nucleotides upstream and downstream of a capture region of a gene
(i.e. the
targeting arms flank the region to be captured). The capture region may be a
target nucleic acid
or a sub-region thereof. Appendix B lists the capture regions of the genes
that correspond to the
molecular inversion probes listed in Appendix A. Appendix A also specifies the
upstream and
downstream regions of the capture regions corresponding to each targeting arm
of the molecular
inversion probes. The upstream and downstream regions of the capture region
are between the
start position and the end position coordinates, which are relative to the
Human Genome 18 (HG
18).
The molecular inversion probes of Appendix A include a central region flanked
by a 5'
first targeting arm (i.e. ligation arm or left arm) and a 3' second targeting
arm (i.e. extension arm
or right arm). The targeting arm sequences are shown in lowercase letters and
the central region
sequence is shown in uppercase letters. The 5' first targeting arm and the 3'
second targeting
arm of the molecular inversion probes provided in Appendix A include a total
of 40 nucleotides,
and are designed to flank 130 bp capture regions. Some of the molecular
inversion probes listed
in Appendix A are designed to capture the coding regions of the genes, whereas
others are
designed to capture non-coding regions of the genes. The genes listed in Table
5 corresponded
to diseases, and as such, the molecular inversion probes listed in Appendix A
can be utilized to
analyze one or more of the diseases provided in table 5. The molecular
inversion probes
provided in Appendix A are described in more detail in Example 8.
While all of the molecular inversion probes provided in Appendix A may be used
in a
single assay to comprehensively examine several or all of the genomic regions
of the genes
provided in Table 5, one may also select one or more molecular inversion
probes provided in
Appendex A to evaluate one or more targets present in one gene or a
combination of the genes
provided in Table 5. For example, one may choose to only examine the coding
regions of one or
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
more of the genes listed in Table 5, and therefore use the one or more of the
molecular inversion
probes designed to capture those regions. In another example, one may choose
to only examine
the non-coding regions of one or more gene listed in Table 5, and therefore
use the one or more
molecular inversion probes designed to capture those regions. In another
example, one may
choose to only examine a portion of or the entirety of a gene listed in Table
5, and therefore use
the one or more molecular inversion probes design to capture the portion of or
the entirety of that
gene. In another example, one may choose to examine nucleic acid regions
specific to one or
more diseases listed in Table 5, and therefore use the one or more nucleic
acids corresponding to
those diseases. In yet another example, one may chose to examine a portion or
entirety of two or
more of the genes listed in Table 5, and there uses the moleculre inversion
probes specific to
those genes. In yet another example, one may chose to only examine certain
chromosomes with
the molecular inversion probes provided in Appendix A. In all of these
examples, the number of
molecular inversion probes that correspond to the target chosen depends on the
amount of
coverage one desires.
It is understood that one can modify the molecular inversion probes listed in
Appendix A,
while achieving a similar coverage and tile-capture layout as the probes
listed in Appendix A.
For example, the sequence of the central region of the molecular inversion
probes may be
different from the sequence of the central region provided in Appendix A
without changing
capture region of the probe. For molecular inversion probe sets, the sequence
chosen for the
central region is preferably the same across each molecular inversion probe in
a set of probes.
This allows the capture targets to be amplified with a single set of primers.
It is also preferable
that the central region is designed so that it is not complementary to the
target sequences or any
other sequence in the sample.
In addition, it should be appreciated that other molecular inversion probes
than those
listed in Appendix A may be used to tile-capture different regions of the
genes listed in Table 5.
Those molecular inversion probes may include a different first targeting arm,
second targeting
arm, and/or central region from the molecular inversion probes listed in
Appendix A. In a non-
limiting example, a modified molecular inversion probe may include the first
targeting arm
sequence of SEQ ID NO: 300, but have a different sequence for the central
region and the second
targeting arm. The specific sequences and length of the sequences chosen for
the first targeting
41
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
arm, second targeting arm, and/or central region depend on the desired capture
region and
coverage.
In certain embodiments, the molecular inversion probes for tile or staggered
capture are
selected to maximize performance with respect to both capture efficiency and
robustness to
common polymorphisms. In order to determine which probes maximize performance
for a
genomic target, methods of the invention, according to certain aspects,
involve designing all
possible probes capable of targeting a genomic interval and ranking the probes
based on a
number of score tuples or ranking factors. In certain embodiments, the
possible probes are
assigned score tuples including, but not limited to: 1) presence of guanine or
cystosine as the 5'-
most base of the ligation arm, 2) the number of dbSNP (version 130) entries
intersecting
targeting arm sites, 3) the root mean squared deviation of the targeting arms'
predicted melting
temperatures from optimal values derived from empirical studies of
efficiencies. Using any
combination of these score tuples, the possible probes for a certain genomic
interval may be
ranked, and the highest ranking probe for the genomic interval is preferably
chosen for capture.
In certain aspects, method of the invention provide for shearing or
fragmenting genomic
nucleic acid prior to performing capture with a molecular inversion probe (
e.g. capture with one
or more of the molecular inversion probes provided in Appendix A). Fragmenting
the genomic
nucleic acid prior to performing a capture reaction allows for greater
exposure of a target site to a
molecular inversion probe, which reduces failed capture and increases the
percentage of
molecular inversion probes that hybridize to targets within the genome. This
advantageously
yields a target abundance distribution that is significantly more uniform than
if a native high
molecular weight genomic nucleic acid is used. Molecular inversion techniques
involving a
fragmenting step are described in co-owned and co-assigned U.S. Serial Number
13/448,961,
having U.S. Publication No. 2012/0252020, entitled "Capture Reactions."
Fragmenting the nucleic acid can be accomplished by any technique known in the
art.
Exemplary techniques include mechanically fragmenting, chemically fragmenting,
and/or
enzymatically fragmenting. Mechanical nucleic acid fragmentation can be, for
example,
sonication, nebulization, and hydro-shearing (e.g., point-sink shearing).
Enzymatic nucleic acid
fragmenting includes, for example, use of nicking endonucleases or restriction
endonucleases.
The nucleic acid can also be chemically fragmented by performing acid
hydrolysis on the nucleic
acid or treating of the nucleic acid with alkali or other reagents.
42
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
The fragment length can be adjusted based on the sizes of the nucleic acid
targets to be
captured. The nucleic acid fragments can be of uniform length or of a
distribution of lengths. In
certain embodiments, the nucleic acid is fragmented into nucleic acid
fragments having a length
of about 10 kb or 20 kb. In addition, the nucleic acid fragments can range
from between 1 kb to
20 kb, with various distributions.
In certain embodiments, the nucleic acid is also denatured, which may occur
prior to,
during, or after the fragmenting step. The nucleic acid can be denatured using
any means known
in the art, such as pH-based denaturing, heat-based denaturing, formamide or
urea, exonuclease
degradation, or endonuclease nicking. In certain embodiments, the use of pH,
such as in acid
hydrolysis, alone or in combination with heat fragments and either partially
or fully denatures the
nucleic acid. This combined fragmenting and denaturing method can be used to
fragment the
nucleic acid for MIP capture or to fragment captured target nucleic acids or
whole genomic DNA
for shotgun library preparation.
In one aspect, a nucleic acid is fragmented by heating a nucleic acid immersed
in a buffer
system at a certain temperature for a certain period to time to initiate
hydrolysis and thus
fragment the nucleic acid. The pH of the buffer system, duration of heating,
and temperature can
be varied to achieve a desired fragmentation of the nucleic acid. In one
embodiment, after a
genomic nucleic acid is purified, it is resuspended in a Tris-based buffer at
a pH between 7.5 and
8.0, such as Qiagen's DNA hydrating solution. The resuspended genomic nucleic
acid is then
heated to 65 C and incubated overnight (about 16-24 hours) at 65 C. Heating
shifts the pH of
the buffer into the low- to mid- 6 range, which leads to acid hydrolysis. Over
time, the acid
hydrolysis causes the genomic nucleic acid to fragment into single-stranded
and/or double-
stranded products. The above method of fragmenting can be modified by
increasing the
temperature and reducing the heating time. For example, a nucleic acid is
fragmented by
incubating the nucleic acid in the Tris-based buffer at a pH between 7.5 and
8.0 for 15 minutes at
92 C. In addition to adjusting the temperature and the duration of heating,
the pH of the Tris-
based buffer can be adjusted to achieve a desired nucleic acid fragmentation.
Once molecular inversion probes of the invention are hybridized to genomic or
fragmented nucleic acid, the captured target may further be subjected to an
enzymatic gap-filling
and ligation step, such that a copy of the target sequence is incorporated
into a circle. Capture
efficiency of the MIP to the target sequence on the nucleic acid fragment can
be improved by
43
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
lengthening the hybridization and gap-filing incubation periods. (See, e.g.,
Turner EH, et al., Nat
Methods. 2009 Apr 6:1-2.).
The result of molecular inversion probe capture as described above is a
library of circular
target probes, which then can be processed in a variety of ways. In one
aspect, adaptors for
sequencing can be attached during common linker-mediated PCR, resulting in a
library with non-
random, fixed starting points for sequencing. In another aspect, for
preparation of a shotgun
library, a common linker-mediated PCR is performed on the circle target
probes, and the post-
capture amplicons are linearly concatenated, sheared, and attached to adaptors
for sequencing.
Methods for shearing the linear concatenated captured targets can include any
of the methods
disclosed for fragmenting nucleic acids discussed above. In certain aspects,
performing a
hydrolysis reaction on the captured amplicons in the presence of heat is the
desired method of
shearing for library production.
Sequencing may be by any method known in the art. DNA sequencing techniques
include
classic dideoxy sequencing reactions (Sanger method) using labeled terminators
or primers and
gel separation in slab or capillary, sequencing by synthesis using reversibly
terminated labeled
nucleotides, pyrosequencing, 454 sequencing, Illumina/Solexa sequencing,
allele specific
hybridization to a library of labeled oligonucleotide probes, sequencing by
synthesis using allele
specific hybridization to a library of labeled clones that is followed by
ligation, real time
monitoring of the incorporation of labeled nucleotides during a polymerization
step, polony
sequencing, and SOLiD sequencing. Separated molecules may be sequenced by
sequential or
single extension reactions using polymerases or ligases as well as by single
or sequential
differential hybridizations with libraries of probes.
An example of a sequencing technology that can be used is Illumina sequencing.
Illumina
sequencing is based on the amplification of DNA on a solid surface using fold-
back PCR and
anchored primers. Genomic DNA is fragmented, and adapters are added to the 5'
and 3' ends of
the fragments. DNA fragments that are attached to the surface of flow cell
channels are extended
and bridge amplified. The fragments become double stranded, and the double
stranded molecules
are denatured. Multiple cycles of the solid-phase amplification followed by
denaturation can
create several million clusters of approximately 1,000 copies of single-
stranded DNA molecules
of the same template in each channel of the flow cell. Primers, DNA polymerase
and four
fluorophore-labeled, reversibly terminating nucleotides are used to perform
sequential
44
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
sequencing. After nucleotide incorporation, a laser is used to excite the
fluorophores, and an
image is captured and the identity of the first base is recorded. The 3'
terminators and
fluorophores from each incorporated base are removed and the incorporation,
detection and
identification steps are repeated. Sequencing according to this technology is
described in U.S.
Pat. 7,960,120; U.S. Pat. 7,835,871; U.S. Pat. 7,232,656; U.S. Pat. 7,598,035;
U.S. Pat.
6,911,345; U.S. Pat. 6,833,246; U.S. Pat. 6,828,100; U.S. Pat. 6,306,597; U.S.
Pat. 6,210,891;
U.S. Pub. 2011/0009278; U.S. Pub. 2007/0114362; U.S. Pub. 2006/0292611; and
U.S. Pub.
2006/0024681, each of which are incorporated by reference in their entirety.
Sequencing generates a plurality of reads. Reads generally include sequences
of
nucleotide data less than about 150 bases in length, or less than about 90
bases in length. In
certain embodiments, reads are between about 80 and about 90 bases, e.g.,
about 85 bases in
length. In some embodiments, these are very short reads, i.e., less than about
50 or about 30
bases in length. A set of sequence reads can be analyzed by any suitable
method known in the
art. For example, in some embodiments, sequence reads are analyzed by hardware
or software
provided as part of a sequence instrument. In some embodiments, individual
sequence reads are
reviewed by sight (e.g., on a computer monitor). A computer program may be
written that pulls
an observed genotype from individual reads. In certain embodiments, analyzing
the reads
includes assembling the sequence reads and then genotyping the assembled
reads.
In certain embodiments, the sequences obtained using the molecular inversion
probe
techniques of the invention are analyzed using the methods for evaluating of
genetic test, which
are described in co-pending and co-owned U.S. Provisional Serial Number
61/723,508, entitled
"Validation of Genetic Test." The method involves obtaining a plurality of
sequence reads,
introducing a simulated mutation into at least one of the plurality of
sequence reads, and
analyzing the sequence reads to determine if the test identifies the simulated
mutation. To mimic
the expected genotype of a heterozygous carrier, the simulated mutation can be
introduced into
each of those sequence reads that span a location of the mutation with a
probability of 0.5 (e.g.,
into about half of those sequence reads that should contain the location of
the simulated
mutation). The simulated mutation can be introduced by manipulating a data
field in the
sequence read such as, for example, a base sequence field or quality data
field. The sequences
can be manipulated by a computer program. For example, a program can be
written using Java,
Groovy, Python, Perl, or other languages, or a combination thereof, that can
automatically insert
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
simulated mutations into sequence reads. Computer-based methods can be used to
automatically
introduce a number of different simulated mutations into different ones of the
plurality of
sequence reads.
The sequence reads including the manipulated reads are analyzed to detect a
genotype.
Analysis can include any method known in the art, such as de novo assembly,
alignment to a
reference, or a combination thereof. In some embodiments, the sequence reads
are assembled
into a contig. The contig can be aligned to a reference genome. In certain
embodiments,
individual reads are then aligned back to the contig.
Sequence assembly can be done by methods known in the art including reference-
based
assemblies, de novo assemblies, assembly by alignment, or combination methods.
Assembly can
include methods described in U.S. Pat. 8,209,130 titled Sequence Assembly by
Porecca and
Kennedy, the contents of each of which are hereby incorporated by reference in
their entirety for
all purposes. In some embodiments, sequence assembly uses the low coverage
sequence
assembly software (LOCAS) tool described by Klein, et al., in LOCAS-A low
coverage
sequence assembly tool for re-sequencing projects, PLoS One 6(8) article 23455
(2011), the
contents of which are hereby incorporated by reference in their entirety.
Sequence assembly is
described in U.S. Pat. 8,165,821; U.S. Pat. 7,809,509; U.S. Pat. 6,223,128;
U.S. Pub.
2011/0257889; and U.S. Pub. 2009/0318310, the contents of each of which are
hereby
incorporated by reference in their entirety.
In certain embodiments, genetic test of the invention are validated using a
genotyping by
assembly-template alignment (GATA) technique, which is also described in co-
pending and co-
owned U.S. Provisional Serial Number 61/723,508, entitled "Validation of
Genetic Test." FIG.
21 diagrams the validation of a genotyping by assembly-templated alignment
(GATA) technique.
Genetic analysis by GATA-based methods includes obtaining 401 sequence reads
and
assembling 405 the reads into a contig, which is then aligned 409 to a
reference. Differences are
identified by comparison 413. The raw reads are aligned 417 to the contigs and
positional and
variant information is mapped to the reads from the reference via the contig,
allowing
genotyping 421 to produce an observed genotyping. The GATA-based method is
evaluated by
introducing 403 at least one simulated mutation into the reads.
FIG. 22 illustrates obtaining sequence reads and inserting a simulated
mutation. As
shown in FIG. 22, if only wild type sample is sequenced, the raw sequence
reads may only
46
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
include wild type sequence. However, a mutation of interest may be known, for
example, from
the literature or it may be desirable to simply invent a difficult-to-detect
mutation to use in
methods of validating a genetic analysis. Here, a hypothetical 8 base pair
deletion proximal to a
C>A substitution is depicted. As shown in FIG. 22, the raw sequence reads are
edited so that
they include base sequence data, quality data, or both that would arise from
sequencing the
simulated mutation.
FIG. 23 shows an example in which a standard analytical method is performed
for
comparison to a GATA-based method. The standard analysis is demonstrated to
not be able to
detect a mutation. FIG. 23 depicts a workflow in which edited sequence reads
(e.g., as depicted
in FIG. 22) are aligned to a reference genome (here, using BWA and GATK). The
alignment
software properly aligns the wild type sequence reads to the reference genome,
finding a perfect
match and giving a result indicating that the sample is the wild type.
However, the alignment
software finds no valid alignment for the edited sequence reads and is unable
to produce a result.
Due to the fact that the expected genotype of the edited sequence reads is
known a priori (and, in
fact intentionally supplied by editing), an operator is able to identify that
this analysis method¨
alignment of sequence reads to a reference genome¨is incapable of detecting
the mutation. For
comparison, the sequence reads are also analyzed by a GATA-based method.
FIG. 24 shows analysis of sequence reads that include simulated mutations by
GATA. In
step 1, reads are assembled into contigs. Assembly can include any method
including those
discussed below. In step 2, each contig is aligned to a reference genome.
Alignment can be by
any method such as those discussed below, including, e.g., the bwa-sw
algorithm implemented
by BWA. As shown in FIG. 24, both align to the same reference position.
Differences between
the contig and the reference genome are identified and, as shown in FIG. 24,
described by a
CIGAR string.
In step 3, raw reads are aligned to contigs (using any method such as, for
example, BWA
with bwa-short and writing, for example, a CIGAR string). At step 4, raw read
alignments are
mapped from contig space to original reference space (e.g., via position and
CIGAR
information). In step 5, genotyping is performed using the translated, aligned
reads from step 4
(e.g., including raw quality scores for substitutions).
For step 1, reads may be assembled into contigs by any method known in the
art.
Algorithms for the de novo assembly of a plurality of sequence reads are known
in the art. One
47
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
algorithm for assembling sequence reads is known as overlap consensus
assembly. Assembly
with overlap graphs is described, for example, in U.S. Pat. 6,714,874. In some
embodiments, de
novo assembly proceeds according to so-called greedy algorithms, as described
in U.S. Pub.
2011/0257889, incorporated by reference in its entirety. In other embodiments,
assembly
proceeds by either exhaustive or heuristic pairwise alignment. Exhaustive
pairwise alignment,
sometimes called a "brute force" approach, calculates an alignment score for
every possible
alignment between every possible pair of sequences among a set. Assembly by
heuristic multiple
sequence alignment ignores certain mathematically unlikely combinations and
can be
computationally faster. One heuristic method of assembly by multiple sequence
alignment is the
so-called "divide-and-conquer" heuristic, which is described, for example, in
U.S. Pub.
2003/0224384. Another heuristic method of assembly by multiple sequence
alignment is
progressive alignment, as implemented by the program ClustalW (see, e.g.,
Thompson, et al.,
Nucl. Acids. Res., 22:4673-80 (1994)).
With continuing reference to step 1 of FIG. 24, in some embodiments assembly
into
contigs involves making a de Bruijn graph. De Bruijn graphs reduce the
computation effort by
breaking reads into smaller sequences of DNA, called k-mers, where the
parameter k denotes the
length in bases of these sequences. In a de Bruijn graph, all reads are broken
into k-mers (all
subsequences of length k within the reads) and a path between the k-mers is
calculated. In
assembly according to this method, the reads are represented as a path through
the k-mers. The
de Bruijn graph captures overlaps of length k-1 between these k-mers and not
between the actual
reads. By reducing the entire data set down to k-mer overlaps, the de Bruijn
graph reduces the
high redundancy in short-read data sets. Assembly of reads using de Bruijn
graphs is described in
U.S. Pub. 2011/0004413, U.S. Pub. 2011/0015863, and U.S. Pub. 2010/0063742,
incorporated
by reference in their entirety. Assembly of reads into contigs is further
discussed in U.S. Pat.
6,223,128, U.S. Pub. 2009/0298064, U.S. Pub. 2010/0069263, and U.S. Pub.
2011/0257889,
each of which is incorporated by reference herein in its entirety.
2. Reducing Analytical Errors Associated with Bias in Nucleic Acid
Preparations:
In some embodiments, aspects of the invention relate to preparative steps in
DNA
sequencing-related technologies that reduce bias and increase the reliability
and accuracy of
downstream quantitative applications.
48
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
There are currently many genomics assays that utilize next-generation (polony-
based)
sequencing to generate data, including genome resequencing, RNA-seq for gene
expression,
bisulphite sequencing for methylation, and Immune-seq, among others. In order
to make
quantitative measurements (including genotype calling), these methods utilize
the counts of
sequencing reads of a given genomic locus as a proxy for the representation of
that sequence in
the original sample of nucleic acids. The majority of these techniques require
a preparative step
to construct a high-complexity library of DNA molecules that is representative
of a sample of
interest. This may include chemical or biochemical treatment of the DNA (e.g.,
bisulphite
treatment), capture of a specific subset of the genome (e.g., padlock probe
capture, solution
hybridization), and a variety of amplification techniques (e.g., polymerase
chain reaction, whole
genome amplification, rolling circle amplification).
Systematic and random errors are common problems associated with genome
amplification and sequencing library construction techniques. For example,
genomic sequencing
library may contain an over- or under-representation of particular sequences
from a source
genome as a result of errors (bias) in the library construction process. Such
bias can be
particularly problematic when it results in target sequences from a genome
being absent or
undetectable in the sequencing libraries. For example, an under-representation
of particular
allelic sequences (e.g., heterozygotic alleles) from a genome in a sequencing
library can result in
an apparent homozygous representation in a sequencing library. As most
downstream sequencing
library quantification techniques depend on stochastic counting processes,
these problems have
typically been addressed by sampling enough (over-sampling) to obtain a
minimum number of
observations necessary to make statistically significant decisions. However,
the strategy of
oversampling is generally limited to elimination of low-count Poisson noise,
and the approach
wastes resources and increases the expense required to perform such
experiments. Moreover,
oversampling can result in a reduced statistical confidence in certain
conclusions (e.g., diagnostic
calls) based on the data. Accordingly, new approaches are needed for
overcoming bias in
sequencing library preparatory methods.
Aspects of the disclosure are based, in part, on the discovery of methods for
overcoming
problems associated with systematic and random errors (bias) in genome
capture, amplification
and sequencing methods, namely high variability in the capture and
amplification of nucleic
acids and disproportionate representation of heterozygous alleles in
sequencing libraries.
49
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
Accordingly, in some embodiments, the disclosure provides methods that reduce
variability in
the capture and amplification of nucleic acids. In other embodiments, the
methods improve
allelic representation in sequencing libraries and, thus, improve variant
detection outcomes. In
certain embodiments, the disclosure provides preparative methods for capturing
target nucleic
acids (e.g., genetic loci) that involve the use of differentiator tag
sequences to uniquely tag
individual nucleic acid molecules. In some embodiments, the differentiator tag
sequence permits
the detection of bias based on the frequency with which pairs of
differentiator tag and target
sequences are observed in a sequencing reaction. In other embodiments, the
methods reduce
errors caused by bias, or the risk of bias, associated with the capture,
amplification and
sequencing of genetic loci, e.g., for diagnostic purposes.
Aspects of the invention relate to associating unique sequence tags (referred
to as
differentiator tag sequences) with individual target molecules that are
independently captured
and/or analyzed (e.g., prior to amplification or other process that may
introduce bias). These tags
are useful to distinguish independent target molecules from each other thereby
allowing an
analysis to be based on a known number of individual target molecules. For
example, if each of a
plurality of target molecule sequences obtained in an assay is associated with
a different
differentiator tag, then the target sequences can be considered to be
independent of each other
and a genotype likelihood can be determined based on this information. In
contrast, if each of the
plurality of target molecule sequences obtained in the assay is associated
with the same
differentiator tag, then they probably all originated from the same target
molecule due to over-
representation (e.g., due to biased amplification) of this target molecule in
the assay. This
provides less information than the situation where each nucleic acid was
associated with a
different differentiator tag. In some embodiments, a threshold number of
independently isolated
molecules (e.g., unique combinations of differentiator tag and target
sequences) is analyzed to
determine the genotype of a subject.
In some embodiments, the invention relates to compositions comprising pools
(libraries)
of preparative nucleic acids that each comprise "differentiator tag sequences"
for detecting and
reducing the effects of bias, and for genotyping target nucleic acid
sequences. As used herein, a
"differentiator tag sequence" is a sequence of a nucleic acid (a preparative
nucleic acid), which
in the context of a plurality of different isolated nucleic acids, identifies
a unique, independently
isolated nucleic acid. Typically, differentiator tag sequences are used to
identify the origin of a
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
target nucleic acid at one or more stages of a nucleic acid preparative
method. For example, in
the context of a multiplex nucleic acid capture reaction, differentiator tag
sequences provide a
basis for differentiating between multiple independent, target nucleic acid
capture events. Also,
in the context of a multiplex nucleic acid amplification reaction,
differentiator tag sequences
provide a basis for differentiating between multiple independent, primary
amplicons of a target
nucleic acid, for example. Thus, combinations of target nucleic acid and
differentiator tag
sequence (target:differentiator tag sequences) of an isolated nucleic acid of
a preparative method
provide a basis for identifying unique, independently isolated target nucleic
acids. FIG. 4A-C
depict various non-limiting examples of methods for combining differentiator
tag sequence and
target sequences.
It will be apparent to the skilled artisan that differentiator tags may be
synthesized using
any one of a number of different methods known in the art. For example,
differentiator tags may
be synthesized by random nucleotide addition. Differentiator tag sequences are
typically of a
predefined length, which is selected to control the likelihood of producing
unique
target:differentiator tag sequences in a preparative reaction (e.g.,
amplification-based reaction, a
circularization selection-based reaction, e.g., a MIP reaction).
Differentiator tag sequences may
be, up to 5, up to 6, up to 7 up to 8, up to 9, up to 10, up to 11, up to 12,
up to 13, up to 14, up to
15, up to 16, up to 17, up to 18, up to 19, up to 20, up to 21, up to 22, up
to 23, up to 24, up to
25, or more nucleotides in length. For purposes of genotyping, isolated
nucleic acids are
identified as independently isolated if they comprise unique combinations of
target nucleic acid
and differentiator tag sequences, and observance of threshold numbers of
unique combinations of
target nucleic acid and differentiator tag sequences provide a certain
statistical confidence in the
genotype.
During a library preparation process, each nucleic acid molecule may be tagged
with a
unique differentiator tag sequence in a configuration that permits the
differentiator tag sequence
to be sequenced along with the target nucleic acid sequence of interest (the
nucleic acid sequence
for which the library is being prepared, e.g., a polymorphic sequence). The
incorporation of the
nucleic acid comprising a differentiator tag sequence at a particular step
allows the detection and
correction of biases in subsequent steps of the protocol.
A large library of unique differentiator tag sequences may be created by using
degenerate, random-sequence polynucleotides of defined length. The
differentiator tag sequences
51
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
of the polynucleotides may be read at the final stage of the sequencing. The
observations of the
differentiator tag sequences may be used to detect and correct biases in the
final sequencing
read-out of the library. For example, the total possible number of
differentiator tag sequences,
which may be produced, e.g., randomly, is 4N, where N is the length of the
differentiator tag
sequence. Thus, it is to be understood that the length of the differentiator
tag sequence may be
adjusted such that the size of the population of MIPs having unique
differentiator tag sequences
is sufficient to produce a library of MIP capture products in which identical
independent
combinations of target nucleic acid and differentiator tag sequence are rare.
As used herein
combinations of target nucleic acid and differentiator tag sequences, may also
be referred to as
"target:differentiator tag sequences".
In the final readout of a sequencing process, each read may have an additional
unique
differentiator tag sequence. In some embodiments, when differentiator tag
sequences are
distributed randomly in a library, all the unique differentiator tag sequences
will be observed
about an equal number of times. Accordingly, the number of occurrences of a
differentiator tag
sequence may follow a Poisson distribution.
In some embodiments, overrepresentation of target:differentiator tag sequences
in a pool
of preparative nucleic acids (e.g., amplified MIP capture products) is
indicative of bias in the
preparative process (e.g., bias in the amplification process). For example,
target:differentiator tag
sequence combinations that are statistically overrepresented are indicative of
bias in the protocol
at one or more steps between the incorporation of the differentiator tag
sequences into MIPs and
the actual sequencing of the MIP capture products.
The number of reads of a given target:differentiator tag sequence may be
indicative (may
serve as a proxy) of the amount of that target sequence present in the
originating sample. In some
embodiments, the numbers of occurrence of sequences in the originating sample
is the quantity
of interest. For example, using the methods disclosed herein, the occurrence
of differentiator tag
sequences in a pool of MIPs may be predetermined (e.g., may be the same for
all differentiator
tag sequences). Accordingly, changes in the occurrence of differentiator tag
sequences after
amplification and sequencing may be indicative of bias in the protocol. Bias
may be corrected to
provide an accurate representation of the composition of the original MIP
pool, e.g., for
diagnostic purposes.
52
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
According to some aspects, a library of preparative nucleic acid molecules
(e.g., MIPs,
each nucleic acid in the library having a unique differentiator tag sequence,
may be constructed
such that the number of nucleic acid molecules in the library is significantly
larger than the
number prospective target nucleic acid molecules to be captured using the
library. This ensures
that products of the preparative methods include only unique
target:differentiator tag sequence;
e.g., in a MIP reaction the capture step would undersample the total
population of unique
differentiator tag sequences in the MIP library. For example, an experiment
utilizing li_tg of
genomic DNA will contain about -150,000 copies of a diploid genome. For a MIP
library, each
MIP in the library comprising a randomly produced 12-mer differentiator tag
sequence C1.6
million possible unique differentiator tag sequences), there would be more
than 100 unique
differentiator tag sequences per genomic copy. For a MIP library, each MIP in
the library
comprising a randomly produced 15-mer differentiator tag sequence (-1 billion
possible unique
differentiator tag sequences), there would be more than 7000 unique
differentiator tag sequences
per genomic copy. Therefore, the probability of the same differentiator tag
sequence being
incorporated multiple times is incredibly small. Thus, it is to be appreciated
that the length of the
differentiator tag sequence is to be selected based on the amount of target
sequence in a MIP
capture reaction and the desired probability for having multiple, independent
occurrences of
target:differentiator tag sequence combinations.
FIG. 5 depicts a non-limiting method for genotyping based on target and
differentiator
tag sequences. Sequencing reads of target and differentiator tags sequences
are collapsed to make
diploid genotype calls. FIG. 6 depicts non-limiting results of a simulation of
a MIP capture
reaction in which MIP probes, each having a differentiator tag sequence of 15
nucleotides, are
combined with 10000 target sequence copies (e.g., genome equivalents). In this
simulated
reaction, the probability of capturing one or more copies of a target sequence
having the same
differentiator tag sequence is 0.05. The Y axis reflects the number of
observations. The X axis
reflects the number of independent occurrences of target:differentiator tag
combinations. FIG. 7
depicts a non-limiting graph of sequencing coverage, which can help ensure
that alleles are
sampled to sufficient depth (e.g., either 10x or 20x minimum sampling per
allele, assuming 1000
targets). In this non-limiting example, the X axis is total per-target
coverage required, and the Y
axis is the probability that a given total coverage will result in at least
10x or 20x coverage for
each allele.
53
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
The skilled artisan will appreciate that as part of a MIP library preparation
process,
adapters may be ligated onto the ends of the molecules of interest. Adapters
often contain PCR
primer sites (for amplification or emulsion PCR) and/or sequencing primer
sites. In addition,
barcodes may be included, for example, to uniquely identify individual samples
(e.g., patient
samples) that may be mixed together. (See, e.g., USPTO Publication Number US
2007/0020640
Al (McCloskey et al.)
The actual incorporation of the random differentiator tag sequences can be
performed
through various methods known in the art. For example, nucleic acids
comprising differentiator
tag sequences may be incorporated by ligation. This is a flexible method,
because molecules
having differentiator tag sequence can be ligated to any blunt-ended nucleic
acids. The
sequencing primers must be incorporated subsequently such that they sequence
both the
differentiator tag sequence and the target sequence. Alternatively, the
sequencing adaptors can be
synthesized with the random differentiator tag sequences at their 3' end (as
degenerate bases), so
that only one ligation must be performed. Another method is to incorporate the
differentiator tag
sequence into a PCR primer, such that the primer structure is arranged with
the common adaptor
sequence followed by the random differentiator tag sequence followed by the
PCR priming
sequence (in 5' to 3' order). A differentiator tag sequence and adaptor
sequence (which may
contain the sequencing primer site) are incorporated as tags. Another method
to incorporate the
differentiator tag sequences is to synthesize them into a padlock probe prior
to performing a gene
capture reaction. The differentiator tag sequence is incorporated 3' to the
targeting arm but 5' to
the amplification primer that will be used downstream in the protocol. Another
method to
incorporate the differentiator tag sequences is as a tag on a gene-specific or
poly-dT reverse-
transcription primer. This allows the differentiator tag sequence to be
incorporated directly at the
cDNA level.
In some embodiments, at the incorporation step, the distribution of
differentiator tag
sequences can be assumed to be uniform. In this case, bias in any part of the
protocol would
change the uniformity of this distribution, which can be observed after
sequencing. This allows
the differentiator tag sequence to be used in any preparative process where
the ultimate output is
sequencing of many molecules in parallel.
Differentiator tag sequences may be incorporated into probes (e.g., MIPs) of a
plurality
when they are synthesized on-chip in parallel, such that degeneracy of the
incorporated
54
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
nucleotides is sufficient to ensure near-uniform distribution in the plurality
of probes. It is to be
appreciated that amplification of a pool of unique differentiator tag
sequences may itself
introduce bias in the initial pool. However, in most practical cases, the
scale of synthesis (e.g., by
column synthesis, chip based synthesis, etc.) is large enough that
amplification of an initial pool
of differentiator tag sequences is not necessary. By avoiding amplification or
selection steps on
the pool of unique differentiator tag sequences, potential bias may be
minimized.
One example of the use of the differentiator tag sequences is in genome re-
sequencing.
Considering that the raw accuracy of most next-generation sequencing
instruments is relatively
low, it is crucial to oversample the genomic loci of interest. Furthermore,
since there are two
alleles at every locus, it is important to sample enough to ensure that both
alleles have been
observed a sufficient number of times to determine with a sufficient degree of
statistical
confidence whether the sample is homozygous or heterozygous. Indeed, the
sequencing is
performed to sample the composition of molecules in the originating sample.
However, after
multiple reads have been collected for a given locus, it is possible that due
to bias (e.g., caused
by PCR amplification steps), a large fraction of the reads are derived from a
single originating
molecule. This would skew the population of target sequences observed, and
would affect the
outcome of the genotype call. For example, it is possible that a locus that is
heterozygous is
called as homozygous, because there are only a few observations of the second
allele out of
many observations of that locus. However, if information is available on
differentiator tag
sequences, this situation could be averted, because the over-represented
allele would be seen to
also have an over-represented differentiator tag sequence (i.e., the sequences
with the
overrepresented differentiator tag sequence all originated from the same
single molecule).
Therefore, the sequences and corresponding distribution of differentiator tag
sequences can be
used as an additional input to the genotype-calling algorithm to significantly
improve the
accuracy and confidence of the genotype calls.
In some aspects, the disclosure provides methods for analyzing a plurality of
to target
sequences which are genetic loci or portions of genetic loci (e.g., a genetic
locus of Table 1). The
genetic loci may be analyzed by sequencing to obtain a genotype at one or more
polymorphisms
(e.g., SNPs). Exemplary polymorphisms are disclosed in Table 2. The skilled
artisan will
appreciate that other polymorphisms are known in the art and may be
identified, for example, by
CA 02907177 2015-09-15
WO 2014/143994 PCT/US2014/028212
querying the Entrez Single Nucleotide Polymorphism database, for example, by
searching with a
GeneID from Table 1.
TABLE 1
Target Nucleic Acids
Gene Gene Chromosome
name ID Description Gene aliases OMIIVI RefSeqGene map
position
CYP21 CAHl; CPS1; 20191 NG_008337,
1589 cytochrome P450, 6p21.3
A2 CA21H; 0 1
CYP21;
family 21, subfamily A,
CYP21B;
P450c21B;
polypeptide 2
MGC150536;
MGC150537;
CYP21 A2
HI; SUR; HHF1; 60050 NG_008867.
ABCC8 6833 ATP-binding cassette, 11p15.1
MRP8; 9 1
PHHI; SUR1;
sub-family C
ABC36;
HRINS; TNDM2;
(CFTR/MRP),
ABCC8
member 8
SHS; XH2; XNP; 30003 NG_008838.
ATRX 546 alpha Xq13.1-
ATR2; 2 1
thalassemia/mental SFM1; RAD54; q21.1
MRXHF1;
retardation syndrome
RAD54L;
ZNF-HX;
X-linked (RAD54
MGC2094;
homolog, S. cerevisiae) ATRX
56
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
60757 NG_009260.
ARSA 410 arylsulfatase A MLD; ARSA 22q13.31-
4 1
qter;
22q13.33
GLBA; SAP1; 17680 NG_008835.
PSAP 5660 Prosaposin 10q21-
F1100245; 1 1
MGC110993;
q22
PSAP
60901 NG_008019.
BTD 686 Biotinidase BTD 3p25
9 1
60901 NC_000021.
HLCS 3141 holocarboxylase HCS; HLCS 8 21q22.1;
7
synthetase (biotin- 21q22.13
(proprionyl-Coenzyme
A-carboxylase (ATP-
hydrolysing)) ligase)
BS; RECQ2; 60461 NG_007272.
BLM 641 Bloom syndrome, 15q26.1
RECQL2; 0 1
RECQL3;
RecQ helicase-like
MGC126616;
MGC131618;
MGC131620;
BLM
ASP; ACY2; 60803 NG_008399.
ASPA 443 aspartoacylase 17pter-P13
ASPA 4 1
(Canavan disease)
CF; MRP7; 60242 NC 000007.
CFTR 1080 cystic fibrosis 7q31.2
ABC35; 1 12
transmembrane ABCC7;
57
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
CFTR/MRP;
TNR-CFTR;
conductance regulator
dJ76005.1;
(ATP-binding cassette CFTR
sub-family C,
member 7)
ASS; CTLN1; 60347 NG_011542.
ASS1 445 argininosuccinate 9q34.1
ASS1 0 1
synthetase 1
MMACH 60983 NC_000001.
25974 methylmalonic aciduria cb1C; FLJ25671; 1p34.1
C 1 9
DKFZp564I122;
(cobalamin deficiency)
RP11-
291L19.3;
cb1C type, with
MMACWC
homocystinuria
FD; DYS; ELP1; 60372 NG_008788.
IKBKAP 8518 inhibitor of kappa light 9q31
IKAP; 2 1
IK13; TOT1;
polypeptide gene
FLJ12497;
DKFZp781H142
enhancer in B-cclls,
5;
kinase complex- IKBKAP
associated protein
FA3; FAC; 22764 NG_011707.
FANCC 2176 Fanconi anemia, 9q22.3
FACC; 5 1
FLJ14675;
complementation
FANCC
group C
58
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
GKl; GALK; 60431 NG_008079.
GALK1 2584 galactokinase 1 17q24
GALK1 3 1
60699 NC_000009.
GALT 2592 galactose-1-phosphate GALT 9p13
9 10
uridylyltransferase
SDR1E1; 60695 NG_007068.
GALE 2582 UDP-galactose-4- 1p36-p35
FLJ95174; 3 1
epimcrase FLJ97302; GALE
GCB; GBAl; 60646 NG_009783.
GBA 2629 glucosidase, beta; acid 1q21
GLUC; 3 1
(includes GBA
glucosylceramidase)
HID; KID; PPK; 12101 NG_008358.
GJB2 2706 gap junction protein, 13q11-q12
CX26; 1 1
DFNA3; DFNB1;
beta 2, 26 kDa
NSRD1;
DFNA3A;
DFNB1A;
GJB2
GCD; ACAD5; 60880 NG_009292.
GCDH 2639 glutaryl-Cocnzyme A 19p13.2
GCDH 1 1
dehydrogenase
G6PT; GSD1; 23220 NG_011808.
G6PC 2538 glucose-6-phosphatase, 17q21
GSD1a; 0 1
MGCI63350;
catalytic subunit
G6PC
CD113t-C; beta- 14190 NG_000007.
HBB 3043 hemoglobin, beta 11p15.5
globin; 0 3
HBB
59
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
MSU; MSUD1; 60834 NC_000019.
BCKDHA 593 branched chain keto 19q13.1-
OVD1A; 8 8
BCKDE1A;
acid dehydrogenase El, q13.2
FLJ45695;
alpha polypeptide BCKDHA
24861 NG_009775.
BCKDHB 594 branched chain keto ElB; FLJ17880; 6q13-q15
1 1
acid dehydrogenase El dJ279A18.1;
BCKDHB
beta polypeptide
E2; E2B; 24861 NG_011852.
DBT 1629 dihydrolipoamide 1p31
BCATE2; 0 1
branched chain MGC9061; DBT
transacylase E2
E3; LAD; DLDH; 23833 NG_008045.
DLD 1738 dihydrolipoamide 7q31-q32
GCSL; 1 1
dehydrogenase PHE3; DLD
60700 NG_007045.
ACADM 34 acyl-Coenzyme A MCAD; ACAD1; 8 1p31
1
MCADH;
dehydrogenase, C-4 to
FLJ18227;
FLJ93013;
C-12 straight chain
FLJ99884;
ACADM
16678 cblA; 60748 NG_007536.
MMAA methylmalonic aciduria 4q31.22
MGC120010; 1 1
(cobalamin deficiency) MGC120011;
cblA type MGC120012;
MGC120013;
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
MMAA
32662 ATR; cb1B; 60756 NG_007096.
MMAB methylmalonic aciduria 12q24
MGC20496; 8 1
(cobalamin deficiency) MMAB
cb1B type
60905 NG_007100.
MUT 4594 methylmalonyl MCM; MUT 8 6p12.3
1
Coenzyme A mutase
ML4; MLIV; 60524 NC_000019.
MCOLN 57192 mucolipin 1 19p13.3-
MST080; 8 8
TRPML1;
1 p13.2
MSTP080;
TRP-ML1;
TRPM-L1;
MCOLN1
ACTA; ASMA; 10261 NG_006672.
ACTA1 58 actin, alpha 1, skeletal 1q42.13
CFTD; 0 1
MPFD; NEM1;
muscle
NEM2;
NEM3; CFTD1;
CFTDM;
ACTA1
TM3; TRK; 19103 NG_008621.
TPM3 7170 tropomyosin 3 1q21.2
NEM1; TM- 0 1
5; TM30; TM30
nm;
TPMsk3; hscp30;
MGC3261;
FLJ41118;
61
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
MGC14582;
MGC72094;
OK/SW-c1.5;
TPM3
ANM; TNT; 19104 NG_011829.
TNNT1 7138 troponin T type 1 19q13.4
STNT; 1 1
TNTS;
(skeletal, slow)
FLJ98147;
MGC104241;
TNNT1
NEM2; 16165 NG_009382.
NEB 4703 nebulin 2q22
NEB177D; 0 1
FLJ11505;
FLJ36536;
FLJ39568;
FLJ39584;
DKFZp686C1456
;NEB
ASM; NPD; 60760 NG_011780.
SMPD1 6609 sphingomyelin 11p15.4-
SMPD1 8 1
phosphodiesterase 1, p15.1
acid lysosomal
GCE; NKH; 23830 NC_000009.
GLDC 2731 glycine dehydrogenase 9p22
GCSP; 0 10
HYGN1;
(decarboxylating)
MGC138198;
MGC138200;
GLDC
GCE; NKH; 23833 NC_000016.
GCSH 2653 glycine cleavage 16q23.2
GCSH 0 8
62
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
system protein H
(aminomethyl carrier)
GCE; NKH; 23831 NC_000003.
AMT 275 aminomethyltransferase 3p21.2-
GCST; AMT 0 10
p21.1
OCTD; 30046 NG_008471.
OTC 5009 ornithine Xp21.1
MGC129967; 1 1
carbamoyltransferase MGC129968;
MGC138856;
OTC
PH; PKU; PKUl; 61234 NG_008690.
PAH 5053 phenylalanine 12g22-
PAH 9 1
hydroxylase q24.2
DHPR; PKU2; 61267 NG_008763.
DHPR 5860 quinoid 4p15.31
SDR33C1; 6 1
dihydropteridine F1142391; QDPR
reductase
PTPS; F1197081; 26164 NG_008743.
PTS 5805 6- 11q22.3-
PTS 0 1
pyruvoyltetrahydropteri
q23.3
n
synthase
23200 NG_008768.
PCCA 5095 propionyl Coenzyme A PCCA 13q32
0 1
carboxylase, alpha
polypeptide
DKFZp451E113; 23205 NG_008939.
PCCB 5096 propionyl Coenzyme A 3q21-q22
PCCB 0 1
carboxylase, beta
63
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
polypeptide
SCAD; ACAD3; 60688 NG_007991.
ACADS 35 acyl-Coenzyme A 12g22-
ACADS 5 1
dehydrogenase, C-2 to qter
C-3 short chain
60285 NC_000011.
DHCR7 1717 7-dehydrocholesterol SLOS; DHCR7 8 8 11q13.2-
reductase q13.5
SMA; SMN; 60035 NG_008691.
SMNT 6606 survival of motor 5q13
SMAl; 4 1
SMA2; SMA3;
neuron 1, telomeric
SMA4;
SMA@; SMNT;
BCD541;
T-BCD541;
SMN1
TSD;
60686 NG_009017.
HEXA 3073 hexosaminidase A MGC99608; 15g23-
9 1
HEXA
(alpha polypeptide) q24
DFNB2; 27690 NG_009086.
MY07A 4647 myosin VIIA 11q13.5
MYU7A; 3 1
NSRD2; USH1B;
DFNA11;
MYOVIIA;
MY07A
60524 NC_000011.
USH1C 10083 Usher syndrome 1 C PDZ73; AIE-75; 2 8 11p15.1-
(autosomal recessive, DFNB18; PDZ- p14
64
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
45; PDZ-
73; NY-CO-37;
severe)
NY-00-
38; ushlcpst;
PDZ-73/NY-
CO-38; USH1C
USH1D; 60551 NG_008835.
CDH23 64072 cadherin-like 23 10g21-
DFNB12; 6 1
F1100233;
q22
F1136499;
KIAA1774;
KIAA1812;
MGC102761;
DKFZp434P2350
; CDH23
USH1F; 60551 NG_009191.
PCDH15 65217 protocadherin 15 10q21.1
DFNB23; 4 1
DKFZp667A171
1;
PCDH15
12459 SANS; 60769 NG_007882.
SANS Usher syndrome 1G 17q25.1
0 ANKS4A; 6 1
F1133924;
(autosomal recessive)
USH1G
17030 ISSX; PRTS; 30038 NG_008281.
ARX aristaless related Xp21
2 MRX29; 2 1
MRX32;
homeobox
MRX33;
MRX36;
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
MRX38;
MRX43;
MRX54;
MRX76;
MRX87;
MRXS1; ARX
OPN1; MRX60; 30012 NG_008960.
OPHN1 4983 oligophrenin 1 Xq12
OPHN1 7 1
MRXJ; SMCX; 31469 NG_008085.
JAR1DIC 8242 lysine (K)-specific Xp11.22-
MRXSJ; 0 1
XE169;
demethylase 5C p11.21
JARID1C;
DXS1272E;
KDM5C
JM23; MRX9; 30049 NG_008879.
FTSJ1 24140 FtsJ homolog 1 Xp11.23
SPB1; 9 1
TRM7; CDLIV;
(E. coli)
MRX44;
FTSJ1
CRT; CT1; 30003 NC 000023.
SLC6A8 6535 solute carrier family 6 Xq28
CRTR; 6 9
MGC87396;
(neurotransmitter
SLC6A8
transporter, creatine),
member 8
MRX; MRX90; 30018 NC_000023.
DLG3 1741 discs, large homolog 3 Xq13.1
NEDLG; 9 9
NE-D1g;
(Drosophila)
SAP102; SAP-
66
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
102; KIAA1232;
DLG3
A15; MXS1; 30009 NG_009160.
TM4SF2 7102 letraspanin 7 Xp11.4
CD231; 6 1
MRX58; CCG-
B7;
TM4SF2;
TALLA-1;
TM4SF2b;
DXS1692E;
TSPAN7
MRX89; 31499 NG_008238.
ZNF41 7592 zinc finger protein 41 Xp11.23
MGC8941; 5 1
ZNF41
ACS4; FACL4; 30015 NG_008053.
FACL4 2182 acyl-CoA synthetase Xq22.3-
LACS4; 7 1
MRX63;
long-chain family q23
MRX68; ACSL4
member 4
SHS; MRX55; 30046 NC_000023.
PQBP1 10084 polyglutamine binding Xp11.23
MRXS3; 3 9
MRXS8;
protein 1
NPW38;
RENS1; PQBP1
60213 NG_008341.
PEX1 5189 peroxisomal biogenesis ZWS1; PEX1 6 1 7q21.2
factor 1
PAF1; PEX2; 17099 NG_008371.
PXMP3 5828 peroxisomal membrane 8q21.1
PMP3; 3 1
67
CA 02907177 2015-09-15
WO 2014/143994 PCT/US2014/028212
PAF-1; PMP35;
protein 3, 35 kDa
RNF72;
PXMP3
PAF2; PAF-2; 60149 NG_008370.
PEX6 5190 peroxisomal biogenesis 6p21.1
PXAAA1; 8 1
factor 6 PEX6
60285 NG_008342.
PEX10 5192 peroxisomal biogenesis NALD; RNF69; 1p36.32
9 1
MGC1998;
factor 10
PEX10
60175 NG_008447.
PEX12 5193 peroxisomal biogenesis PAF-3; PEX12 8 1 17q12
factor 12
PXR1; PTS1R; 60041 NG_008448.
PEX5 5830 peroxisomal biogenesis 12p13.31
PTS1-BP; 4 1
FLJ50634;
factor 5
FLJ50721;
FLJ51948; PEX5
FLJ20695; 60866 NG_008339.
PEX26 55670 peroxisomal biogenesis 22q11.21
PEX26M1T; 6 1
Pex26pM1T;
factor 26
PEX26
The mutations listed in Table 2 are documented polymorphisms in several
disease-associated
genes (CFTR is mutated in cystic fibrosis, GBA is mutated in Gaucher disease,
ASPA is
mutated in Canavan disease, HEXA is mutated in Tay Sachs disease). The
polymorphisms are
of several types: insertion/deletion polymorphisms which will cause
frameshifts (and thus
generally interrupt protein function) unless the insertion/deletion length is
a multiple of 3 bp,
and substitutions which can alter the amino acid sequence of the protein and
in some cases
cause complete inactivation by introduction of a stop codon.
68
CA 02907177 2015-09-15
WO 2014/143994 PCT/US2014/028212
TABLE 2
Non-Limiting Examples of Polymorphism
SEQ
Gene
ID
name GeneID SNP ID Mutation NO:
CFTR 1080 rs 63500661 TCACATCACCAAGTTAAAAAAAAAAA [A/G] G 1
GGGCGGGGGGGCAGAATGAAAATT
CFTR 1080 rs 63107760 AAACAAGGATGAATTAAGTTTTTTTT[-/T] 2
AAAAAAGAAACATTTGGTAAGGGGA
CFTR 1080 rs 62469443 ATCACCAAGTTAAAAAAAAAAAAGGG [A/G] C 3
GGGGGGGCAGAATGAAAATTGCAT
CFTR 1080 rs 62469442 CTATTGAACCAGAACCAAACAGGAAT [A/G] C 4
CATAGCATTTTGTAAACTAAACTG
CFTR 1080 rs 62469441 CAGGAGTTCAAGACCAGCCTACTAAA [A/C] C 5
ACACACACACACACACACACACAC
CFTR 1080 rs 62469439 GATTAAATAATAGTGTTTATGTACCC [C/G] GC 6
TTATAGGAGAAGAGGGTGTGTGT
CFTR 1080 rs 62469438 ATTGTTATCTTTTCATATAAGGTAAC [A/T] GA 7
GGCCCAGAGAGATTAAATAACAT
CFTR 1080 rs 62469437 TAATTTTAATTAAGTAAATTTAATTG [A/G] TA 8
GATAAATAAGTAGATAAAAAATA
CFTR 1080 rs 62469436 GTATAAAAAAAAAAAAAAAAAAAGTT[A/T] G 9
AATGTTTTCTTGCATTCAGAGCCT
CFTR 1080 rs 62469435 ATACTAAAAATTTAAAGTTCTCTTGC [A/G] AT 10
ATATTTTCTTAATATCTTACATC
CFTR 1080 rs 62469434 TGCTGGGATTACAGGCGTGAGCCACC [A/G] C 11
69
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
GCCTGGCCTGATGGGACATATTTT
CFTR 1080 rs62469433 CTACAATATAAGTATAGTATTGCAAA[A/C]CC 12
ATCAGGAAGGGTGTTAACTATTT
CFTR 1080 rs61763210 GTTGTCTCCAAACTTTTTTTCAGGTG[-/AGA] 13
AGGTGGCCAACCGAGCTTCGGAAAG
CFTR 1080 rs61720488 TTTTTTCATAAAAGATTATATAAAGG[A/C] TA 14
TTGCTTTTGAATCACAAACACTA
CFTR 1080 rs61481156 ATCTAGTGAGCAGTCAGGAAAGAGAA[C/T]T 15
TCCAGATCCTGGAAATCAGGGTTA
CFTR 1080 rs61443875 TAGAGTATAAAAAAAAAAAAAAAAAA[-/A] 16
GTTTGAATGTTTTCTTGCATTCAGA
CFTR 1080 rs61312222 TGCAAATGCCAACTATCAAAGATATT[C/G]GA 17
GTATACTGTCAATAAACTTCATA
CFTR 1080 rs61159372 TCCTCAACAGTTAGAAACAATATTTT [C/G] AG 18
TGATTTCCCATGCCAACTTTACT
CFTR 1080 rs61094145 TTTTTGGTATTGTTGTTAAATAAGTG[A/G]GA 19
ATTCAATACAGTATAATGTCTGT
CFTR 1080 rs61086387 CTTGAAATCGGATATATATATATATA[-/T 20
GTATATATATATATATATATATATATATAT
ACATATATATATATA]GTATTATCCCTGTTTTC
ACAGTTTT
CFTR 1080 rs60996744 AGAGGGGCTGTGAAGGACACCAAGGA[A/G]G 21
AGACTAAGAGCCAGGAGGGAAAAC
CFTR 1080 rs60960860 TAGAGTTTATTAGCTTTTACTACTCT[A/G]CTT 22
AGTTACTTTGTGTTACAGAATA
CFTR 1080 rs60923902 ACTAGTGATGATGAGCTTCTTTTCAT[-/AT] 23
GTTTGTTGGCTGCATAAATGTCTTC
CFTR 1080 rs60912824 GCAGAGAAAAGAGGGGCTGTGAAGGA [C/G] A 24
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
CCAAGGAGGAGACTAAGAGCCAGG
CFTR 1080 rs 60887846 TTCAGAGGTCTACCACTGGTGCATAC [G/T] CT 25
AATCACAGTGTCGAAAATTTTAC
CFTR 1080 rs 60793174 AAGAAAGAGCAAAAGAGGGCAAACTT[C/T]T 26
CATACATTTTTGATGTCGAAACCA
CFTR 1080 rs 60788575 CCTAAAGTTTAAAAAGAAAAAAAAAA[-/A] 27
GGAAGAAGGAATTAAAAATCCAAAG
CFTR 1080 rs 60760741 GTGTGTGTGTGTATATATATATATAT[A/T] TA 28
TATATTTTTTTTTTCCTGAGCCA
CFTR 1080 rs 60456599 AAACTGTTGATGTTTTCATTTATTTA[C/G]ATC 29
ATTGGAAAACTTTAGATTCTAG
CFTR 1080 rs 60363249 TTTATCCATTCTTAACCAGAACAGAC [A/G] TT 30
TTTTCAGAGCTGGTCCAGGAAAA
CFTR 1080 rs 60355115 TTGAAATCGGATATATATATATATAT[A/G]TA 31
TATATATATATATATATATATAT
CFTR 1080 rs 60308689 TAGTTTTTTATTTCCTCATATTATTT[-/T] 32
CAGTGGCTTTTTTCTTCCACATCTTT
CFTR 1080 rs 60271242 ACATAGTTCTCAGTGGTACAACTACA [A/G] GT 33
GATTTCTCTTTTCTTATTTCTGG
CFTR 1080 rs 60010318 AGAGCAATGGCATCCCTTGTCTTGTG[C/T] TA 34
TACAGGATGCAGCAATTTATAGG
CFTR 1080 rs59961323 TTCTGTCTACATAAGATGTCATACTA [A/G] AT 35
TATCTTTTCCAGCATGCATTCAG
CFTR 1080 rs59961270 CAGGGTGGCATGTTAGGCAGTGCTTA [A/G] A 36
ATAAATGAGTTGGTTATACAAGTA
CFTR 1080 rs59837506 AGGACACACACACACACACACACACA[-/CA] 37
TGCACACACATTTAAATAGATGCAT
71
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
CFTR 1080 rs 59572090 TAAAAAATTGGTATAATGAAATTGCA [C/T] TT 38
GTAGTCTTTGGACATTTAAATCC
CFTR 1080 rs 59548252 TTTCAATACTTAAGAGGTACGCAGAG [A/G] A 39
AAGAGGGGCTGTGAAGGACACCAA
CFTR 1080 rs 59519859 CAGCAATGAATATTTTGAGGCTGAGG[C/T]GC 40
TGAGGGGTAAAATTGCAGCCTGG
CFTR 1080 rs 59509837 TTATGGTTTATATTTTGTGTCTTCT[-/C 41
TTT]AACACATCTTTTCTAGCAGAATTCA
CFTR 1080 rs 59417037 GTATTTTAGTTTTTTTTTTTTGTTTG[-/T] 42
TTTGTTTTGTTTTGTTTTGTTTTTG
CFTR 1080 rs 59159458 TGGGTGACTCCATTTTTACTTTTAGT[C/T]TGG 43
TCTGTTGAGGCCTCGTGAGAGA
CFTR 1080 rs 59048119 TATTTTCATGTATTTTAGTTTTTTTTT[-/T 44
TTT]GTTTGTTTTGTTTTGTTTTGTTTTG
CFTR 1080 rs 58970500 GTGTGTGTGTATATATATATATATAT[A/T] TA 45
TATTTTTTTTTTCCTGAGCCAAA
CFTR 1080 rs 58942292 AACCTATTAGCATGTCTGGCAGAAAA[-/A] 46
TAGATACTTAATAAATTTCTTAAAT
CFTR 1080 rs 58917054 GAGGCTTAGACAGTTTAAGTAACTCA[A/G]G 47
CATGGTTACACAACTAGCTAGGGC
CFTR 1080 rs 58837484 GTGTGAGTATTATGAGACCATATGTT [A/G]GG 48
AGATTTTATTTGGTATTGAGGAT
CFTR 1080 rs 58829491 GAAACCCCACCCCTTCTATAGTTTTC [C/T]CTT 49
TAATATTTACAATGGAACCATT
CFTR 1080 rs 58805195 CATATATATATAGTGTGTGTGTGTGT[A/G] TA 50
TATATATATATATATATATTTTT
GB A 2629 rs 60866785 CGAGCGAGAGAGAGAGAGAGAGAGAG[-/AG] 51
72
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
GAGCCGGCGCGAGAACTACGCATGC
GBA 2629 rs60239603 GGCAGGTAATATCTAGTACCTTACTT[A/T]TA 52
TTTCCTGAGCACATTCTACATTT
GBA 2629 rs56310840 GGCCAGGAATGGGAGTGCTTAGGTGC[A/G]G 53
AGGTGGCACTGTTCCCGCAGCTGC
GBA 2629 rs41264927 GAAAACTCCATCCCCTCAGGGTCAT [C/T] AG 54
ATGAAGAGAAGACCACAGGGGTT
GBA 2629 rs41264925 TGTAGGTAAGGGTCACATGTGGGAGA[C/G]G 55
CAGCTGTGGGTAGGTCAGCCCTGT
GBA 2629 rs36024691 CCAAGAAGGCGCCATTACACTCCAGC[-/C] 56
TGGGCGACAGGGCGAGACTCCCTCA
GBA 2629 rs36024092 TGCCACACCCAGCTAATTTGTGTGTG[-/G] 57
TATGTGTGTGTATGTATGTGTGTGT
GBA 2629 rs35682967 GTTCCTCCAGTAATTTTTTTTTTTTT[-/T] 58
GGTTTTGAGACAGAGTCTTGCCCTG
GBA 2629 rs35033592 ATCATGCCCAGATAATTTTTTTTTTT[-/T] 59
GTATTTTAGTAGACACAGGGTTTCA
GBA 2629 rs34732744 CGAGCGAGAGAGAGAGAGAGAGAGAG[-/AG] 60
GAGCCGGCGCGAGAACTACGCATGC
GBA 2629 rs34620635 CCTGTGAGGGGCACATTCCTTAGTAG[-/C] 61
TAAGGAGTTGGGGGTGTGAAGATCC
GBA 2629 rs34302637 ACAGGCTACTGGCTGGGCCCAGGCAA[-/A] 62
GGGGGCCTTGGCAGGAAAAGTTCCT
GBA 2629 rs33949225 GCGAGAGAGAGAGAGAGAGAGAGAGG[-/AG] 63
AGCCGGCGCGAGAACTACGCATGCG
GBA 2629 rs28678003 AAGAAGAAAAATAAAAAGAAAGTGGG[C/T]C 64
AGACCGAGAGAACAGGAAGCCTGA
73
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
GBA 2629 rs 28559737 AAGGACAAAGGCAAAGAGACAAAGGC[G/T]C 65
AACACTGGGGGTCCCCAGAGAGTG
GBA 2629 rs 28373017 TACCTAGTCACTTCCTGCCTCCATGG[C/T]GC 66
AAAAGGGGATGGGTGTGCCTCTT
GBA 2629 rs 12752133 CTCTTCCGAGGTTCCACCCTGAACAC[C/T]TT 67
CCTGCTCCCTCGTGGTGTAGAGT
GBA 2629 rs 12747811 TTCTGACTGGCAACCAGCCCCACTCT[C/T]TG 68
GGAGCCCTCAGGAATGAACTTGC
GBA 2629 rs 12743554 gctcagcctcccaggctggagtgcag [A/T]ggcgcgatc 69
tcggctcaccgcaacc
GBA 2629 rs 12041778 CATGAACCACATCAAATGAGATTTAG[C/T]GG 70
GAGTGGCACACACAGTCATGACC
GBA 2629 rs 12034326 AAGCAGCCCTGGGGAGTCGGGGCGGG [A/QC 71
CTGGATTGGAAAAGAGACGGTCAC
GBA 2629 rs 11558184 CTCCAAGTTCTGGGAGCAGAGTGTGC[A/G]G 72
CTAGGCTCCTGGGATCGAGGGATG
GBA 2629 rs 11430678 GTTCCTCCAGTAAttttttttttttt[-/G/T] 73
gttttgagacagagtcttgccctgt
GBA 2629 rs 11264345 CTAGTACCTTACTTCCCTCAAGTTCA[A/T]TC 74
ATCTCACAGATATTTCCTGAGCA
GBA 2629 rs 10908459 aattagccgtgcgtggtggcgggtgc [C/T]tgtaatccc 75
acgtacttgggaggct
GBA 2629 rs 10796940 CCATGGCCAGCCGGGGAGGGGACGGG [A/C] A 76
CACACAGACCCACACAGAGACTCA
GBA 2629 rs 10668496 agcgagagagagagagagagagagag[-/AG] 77
gagCCGGCGCGAGAACTACGCATGC
GBA 2629 rs7416991 CGTAGCAGTTAGCAGATGATAGGCGG[C/G/T] 78
74
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
GAAATCTTATTTCACAGGGCATTAA
GB A 2629 rs 4024049 CTGGCCCTGGTGACAGTGGGGCTGTG[C/T]GT 79
GGGGCCAGAGCCTTCTCAGAGGT
GB A 2629 rs 4024048 CAGATACTGGCCCTGGTGACAGTGGG[A/G]C 80
TGTGCGTGGGGCCAGAGCCTTCTC
GB A 2629 rs 4024047 GACAGATACTGGCCCTGGTGACAGTG[G/T]G 81
GCTGTGCGTGGGGCCAGAGCCTTC
GB A 2629 rs3841430 GGCTCctctctctctctctctctctc [-/TC] 82
gctc gctctctc gctctctcgctct
GB A 2629 rs 3754485 GTTTCAGACCAGCCTGGCCAACATAG[C/T]GA 83
AACCCCATCTCTACTAAAAATAA
GB A 2629 rs 3205619 AGTGGGCGATTGGATGGAGCTGAGTA[C/T]G 84
GGGCCCATCCAGGCTAATCACACC
GB A 2629 rs 2990227 CCGGGCTCCGTGAATGTTTGTCACAT[C/G]TC 85
TGAAGAACGTATGAATTACATAA
GB A 2629 rs 2990226 GAATCCCAACCCCGACGCTCGTCGCC [C/G]G 86
GCTCCGTGAATGTTTGTCACATGT
GB A 2629 rs 2990225 GCGAATCCCAACCCCGACGCTCGTCG[C/T]CG 87
GGCTCCGTGAATGTTTGTCACAT
GB A 2629 rs 2990224 TGGGCAGAAGTCAGGGTCCAAAGAAA[G/T]G 88
GCAAAGAAAAGTGTcagtg gctc a
ASPA 443 rs 63751297 TAAGAAAGACGTTTTTGATTTTTTTC [A/G] GA 89
CTTCTCTGGCTCCACTACCCTGC
ASPA 443 rs 62071301 CTGATTCCTGGCCAGGAGCGGTGGCT [C/T] AC 90
GCCTGTAATCCCAGCGCTTTGGG
ASPA 443 rs 62071300 TAAAAATGCTGATTCCTGGCCAGGAG[C/T]GG 91
TGGCTCACGCCTGTAATCCCAGC
CA 02907177 2015-09-15
WO 2014/143994 PCT/US2014/028212
ASPA 443 rs62071299 TTTAAAAATGCTGATTCCTGGCCAGG[A/C]GC 92
GGTGGCTCACGCCTGTAATCCCA
ASPA 443 rs 62071297 CAAGACCTGTCAAAGATCTGAGAAAT[A/T] TT 93
ACCCGACTTACAAGCTAACCATT
ASPA 443 rs 61697033 ACTGTAATAAGTGCTGTAAAAGAAAT[A/G]C 94
ACAAAATAATATAGCAGAGGGTAT
ASPA 443 rs 60743592 CTTGAGGTCAGGAGTTCAAGACCAGT[C/T]TG 95
GGCAACATGGGGAAAACCTTGTC
ASPA 443 rs 60666840 AGGTTGCAGTGAGCCGAGATCATGCC [A/G] TT 96
GCACTCCAGCCGGGGCAACAAAA
ASPA 443 rs 60147514 ACAAGTGTCTTGAAATTATCTGTGAT[C/T]TG 97
CTATAGAGCAATACTTTTGTAAA
ASPA 443 rs59930743 GTGGGTATATGCAGCTCTATGCACTA [C/T] CT 98
GCTCATTTATTTGGTAAATCTAA
ASPA 443 rs59690349 TGTGTGTGTGTGCGTGTGTGTGTGTG[-/T 99
GTGTGTG]ATCATAAGAGTGGCTGCAGCAA
ACT
ASPA 443 rs59676360 AGTCTGGAGTGCAATGGTGCAATCTC [A/G] GC 100
TCACTGCAGCCTCCACCTCCGGG
ASPA 443 rs59335404 CTCCTAATGGATATTTCCTAAATTTT[G/T]CTG 101
AACAGAATTTAACTTGAGCTGG
ASPA 443 rs58879097 ATTTAAAAATGGATTTCTAGAAAAAC [A/G] AT 102
CACATACTTGAATATTTTAGCAA
ASPA 443 rs58686774 CTATAAATGGGTAGCATGAGGGATTC [A/G] A 103
GGAGGTGGCTGAAAGAAGCACGTA
ASPA 443 rs57511162 AAGAAACCAAGCATAGTAGAGTGTTA [A/G] A 104
AAACCAAAGCAACTAAACAACTGT
76
CA 02907177 2015-09-15
WO 2014/143994 PCT/US2014/028212
ASPA 443 rs55859596 CGGGGCTCAGAACTTGTAACAGAAAA[A/T]T 105
AAAATATACTCCACTCAAGGGAAT
ASPA 443 rs55742972 TACTACACTTCACGGATACTGTACTT[-/G
106
TACTT]TTTTTCCAAATTGAAGGTTTTTGGC
ASPA 443 rs55640436 TTGTTTTTGTTTTTGTTTTTGTTTTT[-/G
107
TTTTTGTTTTT]TGAGATGGAGTCTCGCTCT
GTCGCC
ASPA 443 rs36225687 TTTGCCTTACTACACTTCACGGATAC[-/T
108
GTACT]TGTACTTTTTTTCCAAATTGAAGGT
ASPA 443 rs36051310 GAGGTGGCTGAAAGAAGCACGTATCC[-/C] 109
TGATGGCATGGTTGCGGGTTATATG
ASPA 443 rs36034906 GAGAAAAGCAGTTCCTGGAACACCCC[-/C]
110
ACCCCTTAACCCCTTATCTCTGCTT
ASPA 443 rs36033666 TTACATATGTATACATGTGCCATGTT[-/T]
111
GGTGTGCCGCACCCATTAACTCGTC
ASPA 443 rs35730123 CTTTTTCCAGATTTTTTTTTTTTTTT[-/T]
112
GAGACAGAGTTTCACTCTTGTTGCC
ASPA 443 rs35629100 TTTGGAAATCTTAAGCTTTTATTTGG[-/G]
113
TGTCACAGAGAAACAGGATCTGTAT
ASPA 443 rs35614631 TACTTTAAGTTTTAGGGTACATGTGC[-/A]
114
CCATGTGCAGGTTTGTTACATATGT
ASPA 443 rs35225782 ATTCATGACCAGCCACATAAATGCAC[-/A]
115
GTATTACTTCGCAAGCATGCCAATG
ASPA 443 rs35178659 GTGCACTAGAATTAGCTAAAGTGGGG[-/G]
116
AAAAAAAGATGCATTTGATGGTCTA
ASPA 443 rs35095578 AACCTCCACCTCCCAGGTTCAAGAGA[-/A]
117
TTCTCCTGCCTCAGCCTCCCAAGTA
77
CA 02907177 2015-09-15
WO 2014/143994 PCT/US2014/028212
ASPA 443 rs35002210 CCTCCCTGTGATCCGAAGTAGCAGAC[A/G] TA 118
CTTAACTTCCATGGTGGATTGTT
ASPA 443 rs34744839 AAAACATTATTATATCTAGAAAAAAA[-/A]
119
TGTATCTTAACCATTGTGGGAAGTG
ASPA 443 rs34680506 TTGAAGGTAAAATCATAGGGAGTTGG[-/G]
120
AGCTGTCCTCTTGCGCTGAATCAGT
ASPA 443 rs34365618 ACTTGTGGCCTTTTTGGAGAGGTTAG[-/CA] 121
ACTCTGAAAACTCTGTCCCTGGACC
ASPA 443 rs34275920 GAAGGAGAAAAAGAGAGGAAATAAGT[-/T] 122
AAAATAATAAACACAATTAATAAAG
ASPA 443 rs34109510 TGTATACATGTGCCATGTTGGTGTGC[C/T]GC 123
ACCCATTAACTCGTCATTTAGCA
ASPA 443 rs34054576 TCACCTGTCACCTCCTATAGAACTTT[-/C]
124
CCCTGACCCTCCTCTATAGCATTAA
ASPA 443 rs34015272 ATAAATGATCATCATTCACAGTAGGG[-/G]
125
TTTTGTTTTGTTTTTTTTCTGGAAA
ASPA 443 rs34002091 ACAGACATATCTACAAACACACTTTT[-/T]
126
CACATATTTGTGTAAGTCATTTATG
ASPA 443 rs 28940574 AAAGACAACTAAACTAACGCTCAATG [A/C] A 127
AAAAGTATTCGCTGCTGTTTACAT
ASPA 443 rs28940279 TACCGTGTACCCCGTGTTTGTGAATG[A/C]GG 128
CCGCATATTACGAAAAGAAAGAA
ASPA 443 rs 17850703 CAGGGCTGGAGGTAAAACCATTTATT [A/G] CT 129
AACCCCAGAGCAGTGAAGAAGTG
ASPA 443 rs 17222495 TTCTTCATTGCCTATTGAAGAGAGAG[C/T]GG 130
AATGCTTTGGTTGCCAGATATGG
ASPA 443 rs 17175228 CACAAGATCTCATTACTCAGGAGCTG [C/T] CC 131
78
CA 02907177 2015-09-15
WO 2014/143994 PCT/US2014/028212
AAGTGTCTAATGTACTTAGTTAA
ASPA 443 rs 16953074 TTCTGTGTAACATTTCATTTAAGCAA[A/G]GG 132
ATTCGGCAAATCAAAAATTGTCA
ASPA 443 rs 16953070 TAAAACGTATTGAAGGTATTATTGAC[G/T]CT 133
GTTGAAGCAAAGAGAACAAAACA
HEXA 3073 rs 62022858 ATCTGCTCTTCCAGTTGGATGACAAG[C/T]CT 134
TGCTGTCTAACACCTGCTGCAGA
HEXA 3073 rs 62022857 CCATTTTTTGTTGTATTTTTTTTTTC[C/T]TGAA 135
TACTTTTTATCGCAGTTGGTT
HEXA 3073 rs 62017872 CCCTGTCTCTAAAAGAAAAAAAAAAA [A/G] A 136
AAAAAAAAAGAAAACAAAACCCAA
HEXA 3073 rs 62017871 AGTGGCTCCAAAAAGGTCATGGAACC[C/T]CT 137
TGAGGATGATGCAAATTGACTCT
HEXA 3073 rs 61662730 TAAAGTTACTTTTCTTTTATTGACTT[C/T]CCC 138
TTATTTTTTAACCTTATGCTTT
HEXA 3073 rs 61329913 CAGAGTTAAAAAAAAAAAAAAAAAAA[-/A] 139
GGAAGTAGCAGCAACAGCTTGGAAA
HEXA 3073 rs 60920713 GTTGCCCAGGGTTGAGTGCAGAGGCA[C/T]AT 140
TTGGCTCACAGCAACCTCTGCC
HEXA 3073 rs 60783213 AAGGCTTTTTTTTTTTTTTTTTTTTT[-/T
141
TTT]GAGACAGAGTCTTGCTGTGTCACCC
HEXA 3073 rs 60644867 GCCTACATTCTGCAAAGAGGAGGGAA [C/G] A 142
TTCACAGCTCCATACTTGAACCCT
HEXA 3073 rs 60288568 CCAAAGGAGAATAGCTCTAGGGGAGG[C/G] A 143
GGTGGATGAGTATGCATGGGGGAG
HEXA 3073 rs 59888548 GACTCCATCTCAAAAAAAAAAAAAAA[-/A] 144
TGCAGTCTAATGGCAGAATTAGACT
79
CA 02907177 2015-09-15
WO 2014/143994 PCT/US2014/028212
HEXA 3073 rs59733856 TTATTTATTTATTTATTTATTTTTGA[A/G]ACA 145
GGGTCTCTGTTGTCCAGGCTGG
HEXA 3073 rs59427837 TTTTGAGGCAGGGTCTCACTCTGTTG[C/T] CC 146
AGGGTTGAGTGCAGAGGCACATC
HEXA 3073 rs59171976 CGCCTTGCGAAGGCCCCACAGCTTGC[C/T]TG 147
TGACAAACGTTCATAGGCAAATG
HEXA 3073 rs58706602 GGAGGTCTGTACAAAGCACCACCTAC[C/T]TC 148
ATGGGTCAGTTTCCACAGCAGAA
HEXA 3073 rs58696963 GAATCTTATAATTCACTGTGTACCTC[-/C
149
CTC]TGTTTCATATTTTCGCAATTGAACT
HEXA 3073 rs58610850 AACATAGTATCTAATATAGCTTTACA[C/T]CC 150
AAAGCCAAAATATGAATACACTG
HEXA 3073 rs58016062 TTGTTTTGTTTTGTTTGGGGGGGGGG[-/G]
151
TTGTTTTTCTGAGAGGGAGTCTTGC
HEXA 3073 rs57733983 CATACCAAAGGGCAGCTGGAGGGATAC [C/T] A 152
GACGGAAGTCATGTGGAGAGTGAA
HEXA 3073 rs57476645 CAGGTGTGAGCCACCACGACCACCAA [A/T] T 153
TAGCTCTTTTTACTCCTTCCCTTC
HEXA 3073 rs56870003 AGTGGTAGCTGATTTTGCTTCTGGAT[A/C]CT 154
TTGCCACCTTCCCACTCTTTAAT
HEXA 3073 rs56338339 AAAGACCTGTTTCTTAAAAAAAAAAA[-/A
155
GAAAAAAAAAAA]GAAAGAAAAGAAAAG
AAAAAAACAG
HEXA 3073 rs55995352 TAAAAAATCTTTCAATGAGGAGATGT[C/T]CC 156
CAGAGCAAGACAGCTGTAGGATG
HEXA 3073 rs55860138 AAAAGAAAAAAAAAAAAAAAAAAAAA[-/A] 157
GAAAACAAAACCCAAACCCATAAAG
CA 02907177 2015-09-15
WO 2014/143994 PCT/US2014/028212
HEXA 3073 rs55743646 CCTGTCTCTAAAAGAAAAAAAAAAAA [A/G] A 158
AAAAAAAAGAAAACAAAACCCAAA
HEXA 3073 rs55665666 GTTATCATAGAAAAATATCACACTCT[-/GT] 159
CTGTATCCCCACTTCCAGAAACTGT
HEXA 3073 rs36106892 CAGGAGCTCATAGAATTACATACAAT[-/C]
160
TTTTTTTTTTTTTTTTGAGACAGCG
HEXA 3073 rs36091525 TTGAGAATCTTATAATTCACTGTGTA[-/C
161
CTC]CCTCTGTTTCATATTTTCGCAATTG
HEXA 3073 rs35949555 CCACTACCACAGTGCCTAGAGAACAA[C/T]A 162
TGTGTTTAATAATATTTAAATAAT
HEXA 3073 rs35827424 CCCTGTCTCTAAAAGAAAAAAAAAAA[-/A] 163
AAAAAAAAAAGAAAACAAAACCCAA
HEXA 3073 rs35729578 CCATTATATCATTCATTTCCCACTCA[-/T]
164
TTTCTTCATTCCAACCAAGATATAT
HEXA 3073 rs35649102 TCCGTCTCAAAAAAAAAAAAAAAAAG[-/A] 165
GAAAGGAATTATTCTCATGTATACA
HEXA 3073 rs35118677 CTGGGGCAGTTAAAAAGAAAAACAAA[-/C] 166
CCCTGGTCCCTGCCCTTGAGGAGAT
HEXA 3073 rs35005352 CTCCAGGGTCCCATTCCAGGACCACA[-/C]
167
GCCTGCTACCTCTGCAGCTCACTCA
HEXA 3073 rs34736306 GGATTGACATATACCAGTTAGACGGA[-/T]
168
TTTTTTTTTCCATAAACCAGGCTCA
HEXA 3073 rs34607939 ACAAATAATTACTACATATCTACAAC[A/G] TT 169
CCAGATACAGAAGAAATGGCCAA
HEXA 3073 rs34496117 TAAACACACTTGAAACATCATATAAA[-/A
170
TG]ATATTACTACAAGACTTAACCGTAA
HEXA 3073 rs34300017 ACACAGGTAATCCATGTTTATTATAG[-/A]
171
81
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
AAAATGCCACATTACTCTTTATTGA
HEXA 3073 rs34206496 AGTTATCATAGAAAAATATCACACTC[-/TG] 172
TCTGTATCCCCACTTCCAGAAACTG
HEXA 3073 rs34110830 AATGAACTTACAGGAAGGTAATATAT[-/G]
173
GGAAATAAACATCTTATTGAATTTA
HEXA 3073 rs34093438 GGACCCCTGAAAGGCACAAGACACCC[-/T] 174
TTCAGGTTCACACTTCCTGAAAGCT
HEXA 3073 rs34085965 CCACCAATCACCAGAGCCTTCTGCTC[A/G]GG 175
GGTACCTGAGGGAAAACAAGCAA
HEXA 3073 rs34004907 AAAGACTGAAAAAACATTCATAACTA [-/T]
176
TTTTCTTGTTATCCTCGGAAATGTC
HEXA 3073 rs 28942072 TATCTTCATCTTGGAGGAGATGAGGT[C/T]GA 177
TTTCACCTGCTGGAAGTCCAACC
HEXA 3073 rs28942071 TTGCCTATGAACGTTTGTCACACTTC[C/T]GCT 178
GTGAGTTGCTGAGGCGAGGTGT
HEXA 3073 rs28941771 GCTTGCTGTTGGATACATCTCGCCAT[C/T]AC 179
CTGCCACTCTCTAGCATCCTGGA
HEXA 3073 rs28941770 CCGGGGCTTGCTGTTGGATACATCTC[G/T]CC 180
ATTACCTGCCACTCTCTAGCATC
3. Nucleic Acid Target Length Evaluation:
In some embodiments, aspects of the invention relate to methods for detecting
nucleic
acid deletions or insertions in regions containing nucleic acid sequence
repeats.
Genomic regions that contain nucleic acid sequence repeats are often the site
of genetic
instability due to the amplification or contraction of the number of sequence
repeats (e.g., the
insertion or deletion of one or more units of the repeated sequence).
Instability in the length of
genomic regions that contain high numbers of repeat sequences has been
associated with a
number of hereditary and non hereditary diseases and conditions.
82
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
For example, "Fragile X syndrome, or Martin-Bell syndrome, is a genetic
syndrome
which results in a spectrum of characteristic physical, intellectual,
emotional and behavioral
features which range from severe to mild in manifestation. The syndrome is
associated with the
expansion of a single trinucleotide gene sequence (CGG) on the X chromosome,
and results in a
failure to express the FMR-1 protein which is required for normal neural
development. There are
four generally accepted forms of Fragile X syndrome which relate to the length
of the repeated
CGG sequence; Normal (29-31 CGG repeats) (SEQ ID NO: 6375), Premutation (55-
200 CGG
repeats) (SEQ ID NO: 6376), Full Mutation (more than 200 CGG repeats) (SEQ ID
NO: 6377),
and Intermediate or Gray Zone Alleles (40-60 repeats) (SEQ ID NO: 6378)."
Other examples include cancer, which has been associated with microsatellite
instability
(MSI) involving an increase or decrease in the genomic copy number of nucleic
acid repeats at
one or more microsatellite loci (e.g., BAT-25 and/or BAT-26). There are
currently many
sequencing-based assays for determining the number of nucleic acid sequence
repeats at a
particular locus and identifying the presence of nucleic acid insertions or
deletions. However,
such techniques are not useful in a high throughput multiplex analysis where
the entire length of
a region may not be sequenced.
In contrast, in some embodiments, aspects of the invention relate to detecting
the
presence of an insertion or deletion at a genomic locus without requiring the
locus to be
sequenced (or without requiring the entire locus to be sequenced). Aspects of
the invention are
particularly useful for detecting an insertion or deletion in a nucleic acid
region that contains
high levels of sequence repeats. The presence of sequence repeats at a genetic
locus is often
associated with relatively high levels of polymorphism in a population due to
insertions or
deletions of one or more of the sequence repeats at the locus. The
polymorphisms can be
associated with diseases or predisposition to diseases (e.g., certain
polymorphic alleles are
recessive alleles associated with a disease or condition). However, the
presence of sequence
repeats often complicates the analysis of a genetic locus and increases the
risk of errors when
using sequencing techniques to determine the precise sequence and number of
repeats at that
locus.
In some embodiments, aspects of the invention relate to determining the size
of a genetic
locus by evaluating the capture frequency of a portion of that locus suspected
of containing an
insertion or deletion (e.g., due to the presence of sequence repeats) using a
nucleic acid capture
83
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
technique (e.g., a nucleic acid sequence capture technique based on molecular
inversion probe
technology). According to aspects of the invention, a statistically
significant difference in capture
efficiency for a genetic locus of interest in different biological samples
(e.g., from different
subjects) is indicative of different relative lengths in those samples. It
should be appreciated that
the length differences may be at one or both alleles of the genetic locus.
Accordingly, aspects of
the invention may be used to identify polymorphisms regardless of whether
biological samples
being interrogated at heterozygous or homozygous for the polymorphisms.
According to aspects
of the invention, subjects that contain one or more loci with an insertion or
deletion can be
identified by analyzing capture efficiencies for nucleic acids obtained from
one or more
biological samples using appropriate controls (e.g., capture efficiencies for
known nucleic acid
sizes, capture efficiencies for other regions that are not suspected of
containing an insertion or
deletion in the biological sample(s), or predetermined reference capture
efficiencies, or any
combination thereof. However, it should be appreciated that aspects of the
invention are not
limited by the nature or presence of the control. In some embodiments, if a
statistically
significant variation in capture efficiency is detected, a subject may be
identified as being at risk
for a disease or condition associated with insertions or deletions at that
genetic locus. In some
embodiments, the subject may be analyzed in greater detail in order to
determine the precise
nature of the insertion or deletion and whether the subject is heterozygous or
homozygous for
one or more insertions or deletions. For example, gel electrophoresis of an
amplification (e.g.,
PCR) product of the locus, or Southern blotting, or any combination thereof
can be used as an
orthogonal approach to verify the length of the locus. In some embodiments, a
more exhaustive
and detailed sequence analysis of the locus can be performed to identify the
number and types of
insertions and deletions. However, other techniques may be used to further
analyze a locus
identified as having an abnormal length according to aspects of the invention.
Accordingly, aspects of the invention relate to detecting abnormal nucleic
acid lengths in
genomic regions of interest. In some embodiments, the invention aims to
estimate the size of
genomic regions that are hard to be accessed, such as repetitive elements.
However, it should be
appreciated that methods of the invention do not require that the precise
length be estimated. In
some embodiments, it is sufficient to determine that one or more alleles with
abnormal lengths
are present at a locus of interest (e.g., based on the detection of abnormal
capture efficiencies).
84
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
In a non-limiting example, fragile X can be used to illustrate aspects of the
invention
where the size of trinucleotide repeats (genotype) is linked to a symptom
(phenotype). However,
it should be appreciated that fragile X is a non-limiting example and similar
analyses may be
performed for other genetic loci (e.g., independently or simultaneously in
multiplex analyses).
Use of molecular inversion probes (MIPs) has been demonstrated for detection
of single
nucleotide polymorphisms (Hardenbol et al. 2005 Genome Res 15:269-75) and for
preparative
amplification of large sets of exons (Porreca et al. 2007 Nat Methods 4:931-6,
Krishnakumar et
al. 2008 Proc Natl Acad Sci USA 105:9296-301). In both cases, oligonucleotide
probes are
designed which have ends ('targeting arms') that hybridize up-stream and down-
stream of the
locus that is to be amplified. In some embodiments, aspects of the invention
are based on the
recognition that the effect of length on probe capturing efficiency can be
used in the context of
an assay (e.g., a high throughput and/or multiplex assay) to allow the length
of sequences to be
determined without requiring sequencing of the entire region being evaluated.
This is particularly
useful for repeat regions that are prone to changes in size. As illustrated in
FIG. 8, which is
reproduced from Deng et al., Nature Biotech. 27:353-60, (see Supplemental FIG.
1G of Deng et
al.,) illustrates that shorter sequences are captured with higher efficiency
that longer sequences
using MIPs. The statistical package R and its effects module were used for
this analysis. A linear
model was used, and each individual factor was assumed to be independent. The
dashed lines
represent a 95% confidence interval. Shorter target sequences were captured
with higher
efficiency than long target sequences (p<2x10-16). However, the use of this
differential capture
efficiency for systematic sequence length analysis was not previously
recognized.
In some embodiments, following probe hybridization, polymerase fill-in and
ligation
reactions are performed to convert the hybridized probe to a covalently-
closed, circular molecule
containing the desired target. PCR or rolling circle amplification plus
exonuclease digestion of
non-circularized material is performed to isolate and amplify the circular
targets from the starting
nucleic acid pool. Since one of the main benefits of the method is the
potential for a high degree
of multiplexing, generally thousands of targets are captured in a single
reaction containing
thousands of probes.
According to aspects of the invention, repetitive regions are surrounded by
non-repetitive
unique sequences, which can be used to amplify the repeat-containing regions
using, for
example, PCR or padlock (MIP)-based method.
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
In addition to the repetitive regions, a probe (e.g., a MIP or padlock probe)
can be
designed to include at least a sequence that is sufficient to be uniquely
identified in the genome
(or target pool). After the probe is circularized and amplified, the amplicon
can be end-
sequenced so that the unique sequence can be identified and served as the
"representative" of the
repetitive region as illustrated in FIG. 9. FIG. 9 illustrates a non-limiting
scheme of padlock
(MIP) capture of a region that includes both repetitive regions (thick wavy
line) and the adjacent
unique sequence (thick strait line). The regions of the probe are indicated
with the targeting arms
shown as regions "1" and "3." An intervening region that may be, or include, a
sequencing
primer binding site is shown as "2." After the padlock is circularized and
amplified, it can be
end-sequenced to obtain the sequence of the unique sequence, which represents
the repetitive
region of interest. Although capturing efficiency is overall negatively
correlated with target
length, different probe sequences may have unique features. Therefore,
multiple probes could be
designed and tested so that an optimal one is chosen to be sensitive enough to
differentiate
repetitive sizes of roughly 0-150 bp, 150-600 bp, and beyond, which represent
normal,
premutation and full mutation of fragile X syndrome, respectively. However, it
should be
appreciated that other probe sizes and sequences can be designed, and
optionally optimized, to
distinguish a range of repeat region size differences (e.g., length
differences of about 3-30 bases,
about 30-60 bases, about 60-90 bases, about 90-120 bases, about 120-150 bases,
about 150-300
bases, about 300-600 bases, about 600-900 bases, or any intermediate or longer
length
difference). It should be appreciated that a length difference may be an
increase in size or a
decrease in size.
In some embodiments, an initial determination of an unexpected capture
frequency is
indicative of the presence of size difference. In some embodiments, an
increase in capture
frequency is indicative of a deletion. In some embodiments, a decrease in
capture frequency is
indicative of an insertion. However, it should be appreciated that depending
on specific sequence
parameters and the relative sizes of the capture probes, the target region,
and the deletions or
insertions, a change in capture frequency can be associated with either an
increase or decrease in
target region length. In some embodiments, the precise nature of the change
can be determined
using one or more additional techniques as described herein.
Accordingly, in some aspects a MIP probe includes a linear nucleic acid strand
that
contains two hybridization sequences or targeting arms, one at each end of the
linear probe,
86
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
wherein each of the hybridization sequences is complementary to a separate
sequence on a the
same strand of a target nucleic acid, and wherein these sequences on the
target nucleic acid flank
the two ends of the target nucleic acid sequence of interest. It should be
appreciated that upon
hybridization, the two ends of the probe are inverted with respect to each
other in the sense that
both 5' and 3' ends of the probe hybridize to the same strand to separate
regions flanking the
target region (as illustrated in FIG. 9 for example).
In some embodiments, the hybridization sequences are between about 10-100
nucleotides
long, for example between about 10-30, about 30-60, about 60-90, or about 20,
about 30, about
40, or about 50 nucleotides long. However, other lengths may be used depending
on the
application. In some embodiments, the hybridization Tms of both targeting arms
of a probe are
designed or selected to be similar. In some embodiments, the hybridization Tms
of the targeting
arms of a plurality of probes designed to capture different target regions are
selected or designed
to be similar so that they can be used together in a multiplex reaction.
Accordingly, a typical size
of a MIP probe prior to fill-in is about 60-80 nucleotides long. However,
other sizes can be used
depending on the sizes of the targeting arms and any other sequences (e.g.,
primer binding or tag
sequences) that are present in the MIP probe. In some embodiments, MIP probes
are designed to
avoid sequence-dependent secondary structures. In some embodiments, MIP probes
are designed
such that the targeting arms do not overlap with known polymorphic regions. In
some
embodiments, targeting arms that can be used for capturing the repeat region
of the Fragile X
locus can have the following sequences or complementary to these sequences
depending on the
strand that is captured.
left: CTCCGTTTCGGTTTCACTTC (SEQ ID NO: 181)
right: ATCTTCTCTTCAGCCCTGCT (SEQ ID NO: 182)
The typical captured size using these targeting arms is about 100 nucleotides
in length (e.g.,
about 30 repeats of a tri-nucleotide repeat).
In some embodiments, the number of reads obtained for the "representative" of
the repetitive
region is not informative to estimate the target length because it is
dependent on the total number
of reads obtained. To overcome this, it is useful to include one or more
probes that target other
"control" regions where no or minimal polymorphism exists among populations.
Because of the
systematic consistency of capturing efficiency (see, e.g., FIG. 9), the ratio
of reads obtained for
87
CA 02907177 2015-09-15
WO 2014/143994 PCT/US2014/028212
the repetitive "representative" to reads obtained for the control region(s)
will be tuned using
DNA with defined numbers of repeats. Ultimately, the ratio can serve as a
measure of the repeat
length as illustrated in FIG. 10. FIG. 10 illustrates a non-limiting
hypothetical relationship
between target gap size and the relative number of reads of the repetitive
region, which is
measured by the ratio of the repeat "representative" reads vs. the "control"
region reads. The unit
of y-axis is arbitrary.
In some embodiments, to better tell targets with similar size range apart, the
whole repetitive
region can be sequenced by making a shotgun library (e.g., by making a shotgun
library from a
captured sequence, for example a sequence captured using a MIP probe). The
longer the repeat
is, the more short reads of repeats will be obtained. Therefore, the target
length will contribute
twice to the relative number of "repetitive" reads, which will gain better
resolution of
differentiating targets. In some embodiments, the expectation is that the
number of reads from
any given repeat will be a direct function of the number of repeats present.
However, in some
embodiments, a Poisson sampling-induced spread may need to be considered and
in some
embodiments may be sufficiently large to limit the resolution.
When a precise measurement of the length of both alleles from a diploid sample
is desired,
further manipulations may be required. This is because the capture efficiency
measured will
actually be the average efficiency of the two alleles. To effectively achieve
separate
measurements for each allele, barcodes (e.g., sequence tags) can be used that
allow the efficiency
of individual capture events (from individual genomic loci) to be followed.
FIG. 11A-C shows
the approach. For a given locus, MIPs are synthesized to contain one of a
large number
differentiator tags in their backbone such that the probability of any two
MIPs in a reaction
having the same differentiator tag sequence is low. MIP capture is performed
on the sample; the
reaction will be biased for shorter target lengths, and therefore the reaction
product will be
comprised of more 'short' circles than 'long' circles. Each circle should bear
a unique
differentiator tag sequence. Then, linear RCA (IRCA) is performed on the
circles. In the IRCA
reaction, circles are converted into long, linear concatemers of themselves.
The IRCA reaction
for a given circle stops when the concatemer has reached a 'fixed' length
(based on the
processivity/error rate of the polymerase). Concatemers derived from smaller
circles will
88
CA 02907177 2015-09-15
WO 2014/143994 PCT/US2014/028212
therefore contain more copies of the differentiator tag, and concatemers
derived from larger
circles will contain fewer copies of the differentiator tag. The number of
each differentiator tag
sequence is counted, for example, by next-generation sequencing. When number
of occurrences
is plotted against differentiator tag ID, the data will naturally cluster into
two groups reflecting
the lengths of the two alleles in the diploid sample. The allele lengths can
therefore be read
directly off this graph, after absolute length calibration using known
standards. In some
embodiments, a sequencing technique (e.g., a next-generation sequencing
technique) is used to
sequence part of one or more captured targets (e.g., or amplicons thereof) and
the sequences are
used to count the number of different barcodes that are present. Accordingly,
in some
embodiments, aspects of the invention relate to a highly-multiplexed qPCR
reaction.
Other non-limiting examples of loci at which insertions or deletions or repeat
sequences may be
associated with a disease or condition are provided in Tables 3 and 4. It
should be appreciated
that the presence of an abnormal length at any one or more of these loci may
be evaluated
according to aspects of the invention. In some embodiments, two or more of
these loci or other
loci may be evaluated in a single multiplex reaction using different probes
designed to hybridize
under the same reaction conditions to different target nucleic acid in a
biological sample.
TABLE 3
Polyglutamine (PolyQ) Diseases
Normal/
Type Gene wildtype Pathogenic
DRPLA ATN1 or 6-35 49-88
(Dentatorubropallidoluysian DRPLA
atrophy)
HD (Huntington's disease) HTT 10-35 35+
(Huntingtin)
SBMA (Spinobulbar Androgen 9-36 38-62
muscular atrophy or Kennedy receptor
disease) on the X
89
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
chromosome.
SCA1 (Spinocerebellar ataxia ATXN1 6-35 49-88
Type 1)
SCA2 (Spinocerebellar ataxia ATXN2 14-32 33-77
Type 2)
SCA3 (Spinocerebellar ataxia ATXN3 12-40 55-86
Type 3 or Machado-Joseph
disease)
SCA6 (Spinocerebellar ataxia CACNA1A 4-18 21-30
Type 6)
SCA7 (Spinocerebellar ataxia ATXN7 7-17 38-120
Type 7)
SCA17 (Spinocerebellar TBP 25-42 47-63
ataxia Type 17)
TABLE 4
Non-Polyglutamine Diseases
Normal/
Type Gene Codon wildtype Pathogenic
FRAXA FMR1, on the X- CGG 6-53 230+
(Fragile X chromosome
syndrome)
FXTAS (Fragile FMR1, on the X- CGG 6-53 55-200
X-associated chromosome
tremor/ataxia
syndrome)
FRAXE AFF2 or FMR2, GCC 6-35 200+
(Fragile XE on the X-
mental chromosome
CA 02907177 2015-09-15
WO 2014/143994 PCT/US2014/028212
retardation)
FRDA FXN or X25, GAA 7-34 100+
(Friedreich's (frataxin)
ataxia)
DM (Myotonic DMPK CTG 5-37 50+
dystrophy)
SCA8 OSCA or SCA8 CTG 16-37 110-250
(Spinocerebellar
ataxia Type 8)
SCA12 PPP2R2B or CAG 7-28 66-78
(Spinocerebellar SCA12 On
ataxia Type 12) 5' end
The following examples illustrate aspects and embodiments of the invention and
are not
intended to be limiting or restrictive. Many variations of the invention will
become apparent to
those skilled in the art upon review of this specification. The full scope of
the invention should
be determined by reference to the claims, along with their full scope of
equivalents, and the
specification, along with such variations.
4. Increasing Detection Sensitivity:
In some embodiments, aspects of the invention relate to methods for increasing
the sensitivity of
nucleic acid detection assays.
There are currently many genomic assays that utilize next-generation (e.g.,
polony-based)
sequencing to generate data, including genome resequencing, RNA-seq for gene
expression,
bisulphite sequencing for methylation, and Immune-seq, among others. In order
to make
quantitative measurements (including genotype calling), these methods utilize
the counts of
sequencing reads of a given genomic locus as a proxy for the representation of
that sequence in
the original sample of nucleic acids. The majority of these techniques require
a preparative step
to construct a high-complexity library of DNA molecules that is representative
of a sample of
91
CA 02907177 2015-09-15
WO 2014/143994 PCT/US2014/028212
interest. Current assays use one of several alternative nucleic acid
preparative techniques (e.g.,
amplification, for example PCR-based amplification; sequence-specific capture,
for example,
using immobilized capture probes; or target capture into a circularized probe
followed by a
sequence analysis step. In order to reduce errors associated with the
unpredictability (stochastic
nature) of nucleic acid isolation and sequence analysis techniques, current
methods to involve
oversampling a target nucleic acid preparation in order to increase the
likelihood that all
sequences that are present in the original nucleic acid sample will be
represented in the final
sequence data. For example, a genomic sequencing library may contain an over-
or under-
representation of particular sequences from a source nucleic acid sample
(e.g., genome
preparation) as a result of stochastic variations in the library construction
process. Such
variations can be particularly problematic when they result in target
sequences from a genome
being absent or undetectable in a sequencing library. For example, an under-
representation of
particular allelic sequences (e.g., heterozygotic alleles) from a genome in a
sequencing library
can result in an apparent homozygous representation in a sequencing library.
In contrast, aspects of the invention relate to basing a nucleic acid sequence
analysis on results
from two or more different nucleic acid preparatory techniques that have
different systematic
biases in the types of nucleic acids that they sample rather than simply
oversampling the target
nucleic acid. According to some embodiments, different techniques have
different sequence
biases that are systematic and not simply due to stochastic effects during
nucleic acid capture or
amplification. Accordingly, in some embodiments, the degree of oversampling
required to
overcome variations in nucleic acid preparation needs to be sufficient to
overcome the biases. In
some embodiments, the invention provides methods that reduce the need for
oversampling by
combining nucleic acid and/or sequence results obtained from two or more
different nucleic acid
preparative techniques that have different biases.
According to the invention, different techniques have different characteristic
or systematic
biases. For example, one technique may bias a sample analysis towards one
particular allele at a
genetic locus of interest, whereas a different technique would bias the sample
analysis towards a
different allele at the same locus. Accordingly, the same sample may be
identified as being
different depending on the type of technique that is used to prepare nucleic
acid for sequence
92
CA 02907177 2015-09-15
WO 2014/143994 PCT/US2014/028212
analysis. This effectively represents a sensitivity issue, because each
technique has a different
relative sensitivities for polymorphic sequences of interest.
According to aspects of the invention, the sensitivity of a nucleic acid
analysis can be increased
by combining the sequences from different nucleic acid preparative steps and
using the
combined sequence information for a diagnostic assay (e.g., for a making a
call as to whether a
subject is homozygous or heterozygous at a genetic locus of interest).
Currently, the ability of DNA sequencing to detect mutations is limited by the
ability of the
upstream sample isolation (e.g., by amplification, immobilization enrichment,
circularization
capture, etc.) methods to reliably isolate the locus of interest. If one
wishes to make
heterozygote base-calls for a diploid genome (e.g. a human sample presented
for molecular
diagnostic sequencing), it is important in some embodiments that the isolation
method produces
near- or perfectly-uniform amounts of the two alleles to be sequenced (at
least sufficiently
uniform to be "called" unambiguously as a heterozygote or a homozygote for a
locus of
interest).
Sample preparative methods may fall into three classes: 1) single- or several-
target amplification
(e.g., uniplex PCR, 'multiplex' PCR), 2) multi-target hybridization enrichment
(e.g., Agilent
SureSelect 'hybrid capture' [Gnirke et al 2009, Nature methods 27:182-9],
Roche/Nimblegen
'sequence capture' [Hodges et al 2007, Nature genetics 39:1522-7], and 3)
multi-target
circularization selection (e.g. molecular inversion probes or padlock probes,
[Porreca et al 2007,
Nature methods 4:931-6, Turner et al 2009, Nature methods 6:315-6],
'selectors' [Dahl et al
2005, Nucleic acids research 33:e71]). Each of these methods can result in a
pool of isolated
product that does not adequately represent the input abundance distribution.
For example, the
two alleles at a heterozygous position can become skewed far from their input
50:50 ratio to
something that results in a missed basecall during downstream sequencing. For
example, if the
ratio was skewed from 50:50 to 10:90, and the sample was sequenced to 10x
average coverage,
there is a high probability that one of the two alleles would not be observed
once in the ten
sequencing reads. This would reduce the sensitivity of the sequencing method
by converting a
heterozygous position to homozygous (where potentially the 'mutant' allele was
the one not
93
CA 02907177 2015-09-15
WO 2014/143994 PCT/US2014/028212
observed). In some embodiments, a skewed ratio is a particular issue that
decreases the
sensitivity of detecting mutations present in a heterogeneous tumor tissue.
For example, if only
10% of the cells analyzed in a heterogeneous sample harbored a heterozygous
mutation, the
mutation would be expected to be present in 5% of sequence reads, not 50%. In
this scenario,
the need for robust, sensitive detection may be even more acute.
The methods disclosed herein are based, in part, on the discovery that certain
classes of isolation
methods have different modes of bias. The disclosure provide methods for
increasing the
sensitivity of the downstream sequencing by using a combination of multiple
isolation methods
(e.g., one or more from at least two of the classes disclosed herein) for a
sample. This is
particularly important in molecular diagnostics where high sensitivity is
required to minimize
the chances of 'missing' a disease-associated mutation. For example, given a
nominal false-
negative error rate of 1x103 for sequencing following circularization
selection, and a false-
negative error rate of 1x103 for sequencing following hybridization
enrichment, one can
achieve a final false-negative rate of 1x10-6by performing both techniques on
the sample
(assuming failures in each method are fully independent). For a recessive
disease with carrier
frequency of 0.1, caused by a single fully-penetrant mutant allele, the number
of missed carrier
diagnoses would decrease from 1000 per million patients tested to 1 per
million patients tested.
Furthermore, if the testing was used in the context of prenatal carrier
screening, the number of
affected children born as a result of missing the carrier call in one parent
would decrease from
25 per million to 25 per billion born.
Additionally, the disclosure provides combinations of preparative methods to
effectively
increase sequencing coverage in regions containing disease-associated alleles.
Since
heterozygote error rate is largely tied to both deviations from 50:50 allele
representation, and in
the case of next-generation DNA sequencing deviations from average abundance
(such that less
abundant isolated targets are more likely to be undersampled at one or both
alleles), selectively
increasing coverage in these regions will also selectively increase
sensitivity. Furthermore, MIPs
that detect presence or absence of specific known disease-associated mutations
can be used to
increase sensitivity selectively. In some embodiments, these MIPs would have a
targeting arm
whose 3'-most region is complementary to the expected mutation, and has a fill-
in length of 0 or
94
CA 02907177 2015-09-15
WO 2014/143994 PCT/US2014/028212
more bp. Thus, the MIP will form only if the mutation is present, and its
presence will be
detected by sequencing.
Additionally, algorithms disclosed herein may be used to determine base
identity with varying
levels of stringency depending on whether the given position has any known
disease-associated
alleles. Stringency can be reduced in such positions by decreasing the minimum
number of
observed mutant reads necessary to make a consensus base-call. This will
effectively increase
sensitivity for mutant allele detection at the cost of decreased specificity.
An embodiment of the invention combines MIPs plus hybridization enrichment,
plus optionally
extra MIPs targeted to specific known, common disease-associated loci, e.g.,
to detect the
presence of a polymorphism in a target nucleic acid. A non-limiting example is
illustrated in
FIG. 12 that illustrates a schematic using MIPs plus hybridization enrichment,
plus optionally
extra MIPs targeted to specific known, common disease-associated loci, e.g.,
to detect the
presence of a polymorphism in a target nucleic acid.
FIGS. 13 and 14 illustrate different capture efficiencies for MIP-based
captures. FIG. 13 shows
a graph of per-target abundance with MIP capture. In this graph, bias largely
drives the
heterozygote error rate, since targets which are less abundant here are less
likely to be covered
in sufficient depth during sequencing to adequately sample both alleles. This
is from Turner et al
2009, Nature methods 6:315-6. Hybridization enrichment results in a
qualitatively similar
abundance distribution, but the abundance of a given target is likely not
correlated between the
two methods. FIG. 14 shows a graph of correlation between two MIP capture
reactions from
Ball et al 2009, Nature biotechnology 27:361-8. Each point represents the
target abundance in
replicate 1 and replicate 2. Pearson correlation r=0.956. This indicates that
MIP capture
reproducibly biases targets to specific abundances. Hybridization enrichment
is similarly
correlated from one capture to the next.
According to aspects of the invention, such biases can be detected or overcome
by
systematically combining different capture and/or analytical techniques in an
assay that
interrogates a plurality of loci in a plurality of subject samples.
CA 02907177 2015-09-15
WO 2014/143994 PCT/US2014/028212
Accordingly, it should be appreciated that in any of the embodiments described
herein (e.g.,
tiling/staggering, tagging, size-detection, sensitivity enhancing algorithms,
or any combination
thereof), aspects of the invention involve preparing genomic nucleic acid
and/or contacting them
with one or more different probes (e.g., capture probes, hybridization probes,
MIPs, others etc.).
In some embodiments, the amount of genomic nucleic acid used per subject
ranges from 1 ng to
micrograms (e.g., 500 ng to 5 micrograms). However, higher or lower amounts
(e.g., less
than 1 ng, more than 10 micrograms, 10-50 micrograms, 50-100 micrograms or
more) may be
used. In some embodiments, for each locus of interest, the amount of probe
used per assay may
be optimized for a particular application. In some embodiments, the ratio
(molar ratio, for
example measured as a concentration ratio) of probe to genome equivalent
(e.g., haploid or
diploid genome equivalent, for example for each allele or for both alleles of
a nucleic acid target
or locus of interest) ranges from 1/100, 1/10, 1/1, 10/1, 100/1, 1000/1.
However, lower, higher,
or intermediate ratios may be used.
In some embodiments, the amount of target nucleic acid and probe used for each
reaction is
normalized to avoid any observed differences being caused by differences in
concentrations or
ratios. In some embodiments, in order to normalize genomic DNA and probe, the
genomic DNA
concentration is read using a standard spectrophotometer or by fluorescence
(e.g., using a
fluorescent intercalating dye). The probe concentration may be determined
experimentally or
using information specified by the probe manufacturer.
Similarly, once a locus has been captured (e.g., on a MIP or other probe or in
another form), it
may be amplified and/or sequenced in a reaction involving one or more primers.
The amount of
primer added for each reaction can range from 0.1 pmol to 1 nmol, 0.15 pmol to
1.5 nmol (for
example around 1.5 pmol). However, other amounts (e.g., lower, higher, or
intermediate
amounts) may be used.
In some embodiments, it should be appreciated that one or more intervening
sequences (e.g.,
sequence between the first and second targeting arms on a MIP capture probe),
identifier or tag
sequences, or other probe sequences that are not designed to hybridize to a
target sequence (e.g.,
a genomic target sequence) should be designed to avoid excessive
complementarity (to avoid
96
CA 02907177 2015-09-15
WO 2014/143994 PCT/US2014/028212
cross-hybridization) to target sequences or other sequences (e.g., other
genomic sequences) that
may be in a biological sample. For example, these sequences may be designed
have a sufficient
number of mismatches with any genomic sequence (e.g., at least 5, 10, 15, or
more mismatches
out of 30 bases) or as having a Tm (e.g., a mismatch Tm) that is lower (e.g.,
at least 5, 10, 15,
20, or more degrees C. lower) than the hybridization reaction temperature.
It should be appreciated that a targeting arm as used herein may be designed
to hybridize (e.g.,
be complementary) to either strand of a genetic locus of interest if the
nucleic acid being
analyzed is DNA (e.g., genomic DNA). However, in the context of MIP probes,
whichever
strand is selected for one targeting arm will be used for the other one.
However, in the context of
RNA analysis, it should be appreciated that a targeting arm should be designed
to hybridize to
the transcribed RNA. It also should be appreciated that MIP probes referred to
herein as
"capturing" a target sequence are actually capturing it by template-based
synthesis rather than
by capturing the actual target molecule (other than for example in the initial
stage when the arms
hybridize to it or in the sense that the target molecule can remain bound to
the extended MIP
product until it is denatured or otherwise removed).
It should be appreciated that in some embodiments a targeting arm may include
a sequence that
is complementary to one allele or mutation (e.g., a SNP or other polymorphism,
a mutation, etc.)
so that the probe will preferentially hybridize (and capture) target nucleic
acids having that
allele or mutation. However, in many embodiments, each targeting arm is
designed to hybridize
(e.g., be complementary) to a sequence that is not polymorphic in the subjects
of a population
that is being evaluated. This allows target sequences to be captured and/or
sequenced for all
alleles and then the differences between subjects (e.g., calls of heterozygous
or homozygous for
one or more loci) can be based on the sequence information and/or the
frequency as described
herein.
It should be appreciated that sequence tags (also referred to as barcodes) may
be designed to be
unique in that they do not appear at other positions within a probe or a
family of probes and they
also do not appear within the sequences being targeted. Thus they can be used
to uniquely
identify (e.g., by sequencing or hybridization properties) particular probes
having other
97
CA 02907177 2015-09-15
WO 2014/143994 PCT/US2014/028212
characteristics (e.g., for particular subjects and/or for particular loci).
It also should be appreciated that in some embodiments probes or regions of
probes or other
nucleic acids are described herein as comprising or including certain
sequences or sequence
characteristics (e.g., length, other properties, etc.). However, it should be
appreciated that in
some embodiments, any of the probes or regions of probes or other nucleic
acids consist of those
regions (e.g., arms, central regions, tags, primer sites, etc., or any
combination thereof) of
consist of those sequences or have sequences with characteristics that consist
of one or more
characteristics (e.g., length, or other properties, etc.) as described herein
in the context of any of
the embodiments (e.g., for tiled or staggered probes, tagged probes, length
detection, sensitivity
enhancing algorithms or any combination thereof).
It should be appreciated that probes, primers, and other nucleic acids
designed or used herein
may be synthetic, natural, or a combination thereof. Accordingly, as used
herein, the term
"nucleic acid" refers to multiple linked nucleotides (i.e., molecules
comprising a sugar (e.g.,
ribose or deoxyribose) linked to an exchangeable organic base, which is either
a pyrimidine
(e.g., cytosine (C), thymidine (T) or uracil (U)) or a purine (e.g., adenine
(A) or guanine (G)).
"Nucleic acid" and "nucleic acid molecule" may be used interchangeably and
refer to
oligoribonucleotides as well as oligodeoxyribonucleotides. The terms shall
also include
polynucleosides (i.e., a polynucleotide minus a phosphate) and any other
organic base
containing nucleic acid. The organic bases include adenine, uracil, guanine,
thymine, cytosine
and inosine. Unless otherwise stated, nucleic acids may be single or double
stranded. The
nucleic acid may be naturally or non-naturally occurring. Nucleic acids can be
obtained from
natural sources, or can be synthesized using a nucleic acid synthesizer (i.e.,
synthetic). Harvest
and isolation of nucleic acids are routinely performed in the art and suitable
methods can be
found in standard molecular biology textbooks. (See, for example, Maniatis'
Handbook of
Molecular Biology.) The nucleic acid may be DNA or RNA, such as genomic DNA,
mitochondrial DNA, mRNA, cDNA, rRNA, miRNA, or a combination thereof. Non-
naturally
occurring nucleic acids such as bacterial artificial chromosomes (BACs) and
yeast artificial
chromosomes (YACs) can also be used.
98
CA 02907177 2015-09-15
WO 2014/143994 PCT/US2014/028212
The invention also contemplates the use of nucleic acid derivatives. As will
be described herein,
the use of certain nucleic acid derivatives may increase the stability of the
nucleic acids of the
invention by preventing their digestion, particularly when they are exposed to
biological
samples that may contain nucleases. As used herein, a nucleic acid derivative
is a non-naturally
occurring nucleic acid or a unit thereof. Nucleic acid derivatives may contain
non-naturally
occurring elements such as non-naturally occurring nucleotides and non-
naturally occurring
backbone linkages.
Nucleic acid derivatives may contain backbone modifications such as but not
limited to
phosphorothioate linkages, phosphodiester modified nucleic acids,
phosphorothiolate
modifications, combinations of phosphodiester and phosphorothioate nucleic
acid,
methylphosphonate, alkylphosphonates, phosphate esters,
alkylphosphonothioates,
phosphoramidates, carbamates, carbonates, phosphate triesters, acetamidates,
carboxymethyl
esters, methylphosphorothioate, phosphorodithioate, p-ethoxy, and combinations
thereof. The
backbone composition of the nucleic acids may be homogeneous or heterogeneous.
Nucleic acid derivatives may contain substitutions or modifications in the
sugars and/or bases.
For example, they include nucleic acids having backbone sugars which are
covalently attached
to low molecular weight organic groups other than a hydroxyl group at the 3'
position and other
than a phosphate group at the 5' position (e.g., an 2'-0-alkylated ribose
group). Nucleic acid
derivatives may include non-ribose sugars such as arabinose. Nucleic acid
derivatives may
contain substituted purines and pyrimidines such as C-5 propyne modified
bases, 5-
methylcytosine, 2-aminopurine, 2-amino-6-chloropurine, 2,6-diaminopurine,
hypoxanthine, 2-
thiouracil and pseudoisocytosine. In some embodiments, substitution(s) may
include one or
more substitutions/modifications in the sugars/bases, groups attached to the
base, including
biotin, fluorescent groups (fluorescein, cyanine, rhodamine, etc), chemically-
reactive groups
including carboxyl, NHS, thiol, etc., or any combination thereof.
A nucleic acid may be a peptide nucleic acid (PNA), locked nucleic acid (LNA),
DNA, RNA, or
co-nucleic acids of the same such as DNA-LNA co-nucleic acids. PNA are DNA
analogs having
their phosphate backbone replaced with 2-aminoethyl glycine residues linked to
nucleotide
99
CA 02907177 2015-09-15
WO 2014/143994 PCT/US2014/028212
bases through glycine amino nitrogen and methylenecarbonyl linkers. PNA can
bind to both
DNA and RNA targets by Watson-Crick base pairing, and in so doing form
stronger hybrids
than would be possible with DNA or RNA based oligonucleotides in some cases.
PNA are synthesized from monomers connected by a peptide bond (Nielsen, P. E.
et al. Peptide
Nucleic Acids, Protocols and Applications, Norfolk: Horizon Scientific Press,
p. 1-19 (1999)).
They can be built with standard solid phase peptide synthesis technology. PNA
chemistry and
synthesis allows for inclusion of amino acids and polypeptide sequences in the
PNA design. For
example, lysine residues can be used to introduce positive charges in the PNA
backbone. All
chemical approaches available for the modifications of amino acid side chains
are directly
applicable to PNA. Several types of PNA designs exist, and these include
single strand PNA
(ssPNA), bisPNA and pseudocomplementary PNA (pcPNA).
The structure of PNA/DNA complex depends on the particular PNA and its
sequence. ssPNA
binds to single stranded DNA (ssDNA) preferably in antiparallel orientation
(i.e., with the N-
terminus of the ssPNA aligned with the 3' terminus of the ssDNA) and with a
Watson-Crick
pairing. PNA also can bind to DNA with a Hoogsteen base pairing, and thereby
forms triplexes
with double stranded DNA (dsDNA) (Wittung, P. et al., Biochemistry 36:7973
(1997)).
A locked nucleic acid (LNA) is a modified RNA nucleotide. An LNA form hybrids
with DNA,
which are at least as stable as PNA/DNA hybrids (Braasch, D. A. et al., Chem
&Biol. 8(1):1-7
(2001)). Therefore, LNA can be used just as PNA molecules would be. LNA
binding efficiency
can be increased in some embodiments by adding positive charges to it. LNAs
have been
reported to have increased binding affinity inherently.
Commercial nucleic acid synthesizers and standard phosphoramidite chemistry
are used to make
LNAs. Therefore, production of mixed LNA/DNA sequences is as simple as that of
mixed
PNA/peptide sequences. The stabilization effect of LNA monomers is not an
additive effect.
The monomer influences conformation of sugar rings of neighboring
deoxynucleotides shifting
them to more stable configurations (Nielsen, P. E. et al. Peptide Nucleic
Acids, Protocols and
Applications, Norfolk: Horizon Scientific Press, p. 1-19 (1999)). Also, lesser
number of LNA
residues in the sequence dramatically improves accuracy of the synthesis. Most
of biochemical
100
CA 02907177 2015-09-15
WO 2014/143994 PCT/US2014/028212
approaches for nucleic acid conjugations are applicable to LNA/DNA constructs.
These and other aspects of the invention are illustrated by the following non-
limiting examples.
EXAMPLES
The following examples illustrate non-limiting embodiments of the invention.
Example 1
Design a Set of Capture Probes for a Human Target Exon
All targets are captured as a set of partially-overlapping subtargets. For
example, in the tiling
approach, a 200 bp target exon might be captured as a set of 12 subtargets,
each 60 bp in
length (FIG. 1). Each subtarget is chosen such that it partially overlaps two
or three other
targets.
In some embodiments, all probes are composed of three regions: 1) a 20 bp
'targeting arm'
comprised of sequence which hybridizes immediately upstream from the sub-
target, 2) a 30
bp 'constant region' comprised of sequence used as a pair of amplification
priming sites, and
3) a second 20 bp 'targeting arm' comprised of sequence which hybridizes
immediately
downstream from the sub-target. Targeting arm sequences will be different for
each capture
probe in a set, while constant region sequence will be the same for all probes
in the set,
allowing all captured targets to be amplified with a single set of primers.
Targeting arm
sequences should be designed such that any given pair of 20 bp sequences is
unique in the
target genome (to prevent spurious capture of undesired sites). Additionally,
melting
temperatures should be matched for all probes in the set such that
hybridization efficiency is
uniform for all probes at a constant temperature (e.g., 60 C). Targeting arm
sequences
should be computationally screened to ensure they do not form strong secondary
structure
that would impair their ability to basepair with the genomic target.
Hybridize Capture Probes to Human Genomic Sample
Assemble hybridization reaction:
ul capture probe mix C2.5 pmol)
101
CA 02907177 2015-09-15
WO 2014/143994 PCT/US2014/028212
ul 10x Ampligase buffer (Epicentre)
6.0 ul 500 ng/ul human genomic DNA (16.7 fmol)
11 ul dH20
In a thermal cycler, heat reaction to 95 C for 5 min to denature genomic DNA,
then cool to
60 C. Allow to incubate at 60 C for 40 hours.
Convert Hybridized Probes into Covalently-Closed Circular Products Containing
Subtargets
Prepare fill-in/ligation reaction mixture:
0.25 ul 2 mM dNTP mix (Invitrogen)
2.5 ul 10x Ampligase buffer (Epicentre)
5.0 ul 5 U/ul Taq Stoffel fragment (Applied Biosystems)
12.5 ul 5 U/ul Ampligase (Epicentre)
4.75 ul dH20
Add 1.0 ul of this mix to the hybridized probe reaction, and incubate at 60 C
for 10
hours.
Purify Circularized Probe/Subtarget Products from Un-Reacted Probes and
Genomic
DNA
Prepare exonuclease reaction mixture:
21 ul fill-in/ligation reaction product
ul 10x exonuclease I buffer (New England Biolabs)
2.0 ul 20 U/ul exonuclease I (New England Biolabs)
2.0 ul 100 U/ul exonuclease III (New England Biolabs)
Incubte at 37 C for 60 min, then heat-inactivate by incubating at 80 C for 15
min.
Immediately cool to 4 C for storage.
Amplify Circular Material by PCR Using Primers Specific to the 'Constant
Region' of the
102
CA 02907177 2015-09-15
WO 2014/143994 PCT/US2014/028212
Probes
Prepare PCR mixture:
5.0 ul 10x Accuprime reaction buffer (Invitrogen)
(SEQ ID NO: 183)
1.5 ul 10 uM CP-2-FA (5'-GCACGATCCGACGGTAGTGT-3')
(SEQ ID NO: 184)
1.5 ul 10 uM CP-2-RA (5'-CCGTAATCGGGAAGCTGAAG-3')
0.4 ul 25 mM dNTP mix (Invitrogen)
2.0 ul heat-inactivated exonuclease reaction mix
1.5 ul 10x SybrGreen (Invitrogen)
0.4 ul 2.5 U/ul Accuprime Pfx polymerase (Invitrogen)
37.7 ul dH20
Thermal cycle in real-time thermal cycler according to the following protocol,
but
stop cycling before amplification yield plateaus (generally 8-12 cycles):
95C for 5 min
95C for 30 sec
58C for 60 sec
72C for 60 sec
goto 2, N more times
Prepare a Shotgun Next-Generation Sequencing Library for Analysis
Purify desired amplicon population from non-specific amplification products by
gel
extraction.
Concatemerize amplicons into high-molecular weight products suitable for
shearing
Mechanically shear, using either a nebulizer, BioRuptor, Hydroshear, Covaris,
or
similar instrument. DNA should be sheared into fragments several hundred
basepairs
in length.
103
CA 02907177 2015-09-15
WO 2014/143994 PCT/US2014/028212
Ligate adapters required for amplification by the sequencing platform used. If
necessary, purify ligated product from unligated product and adapters.
Example 2
Use of Differentiator Tag Sequences to Detect and Correct Bias in a MIP-
Capture
Reaction of a Set of Exon Targets
The first step in performing the detection/correction is to determine how many
differentiator
tag sequences are necessary for the given sample. In this example, 1000
genomic targets
corresponding to 1000 exons were captured. Since the differentiator tag
sequence is part of
the probe, it will measure/report biases that occur from the earliest protocol
steps. Also,
being located in the backbone, the differentiator tag sequence can easily be
sequenced from
a separate priming site, and therefore not impact the total achievable read-
length for the
target sequence. MIP probes are synthesized using standard column-based
oligonucleotide
synthesis by any number of vendors (e.g. IDT), and differentiator tag
sequences are
introduced as 'degenerate' positions in the backbone. Each degenerate position
increases the
total number of differentiator tag sequences synthesized by a factor of 4, so
a 10 nt
degenerate region implies a differentiator tag sequence complexity of -1e6
species.
Hybridize Capture Probes to Human Genomic Sample
Assemble hybridization reaction:
1.0 ul capture probe mix (2.5 pmol)
2.0 ul 10x Ampligase buffer (Epicentre)
6.0 ul 500 ng/ul human genomic DNA (16.7 fmol)
11 ul dH20
In a thermal cycler, heat reaction to 95 C for 5 min to denature genomic DNA,
then cool to
60 C. Allow to incubate at 60 C for 40 hours.
Convert Hybridized Probes into Covalently-Closed Circular Products Containing
Subtargets
Prepare fill-in/ligation reaction mixture:
0.25 ul 2 mM dNTP mix (Invitrogen)
104
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
2.5 ul 10x Ampligase buffer (Epicentre)
5.0 ul 5 U/ul Taq Stoffel fragment (Applied Biosystems)
12.5 ul 5 U/ul Ampligase (Epicentre)
4.75 ul dH20
Add 1.0 ul of this mix to the hybridized probe reaction, and incubate at 60 C
for 10
hours.
Purify Circularized Probe/Subtarget Products from Un-Reacted Probes and
Genomic
DNA
Prepare exonuclease reaction mixture:
21 ul fill-in/ligation reaction product
2.0 ul 10x exonuclease I buffer (New England Biolabs)
2.0 ul 20 U/ul exonuclease I (New England Biolabs)
2.0 ul 100 U/ul exonuclease III (New England Biolabs)
Incubate at 37 C for 60 min, then heat-inactivate by incubating at 80 C for 15
min.
Immediately cool to 4 C for storage.
Amplify Circular Material by PCR Using Primers Specific to the 'Constant
Region' of the
Probes
Prepare PCR mixture:
5.0 ul 10x Accuprime reaction buffer (Invitrogen)
(SEQ ID NO: 183)
1.5 ul 10 uM CP-2-FA (5'-GCACGATCCGACGGTAGTGT-3')
(SEQ ID NO: 184)
1.5 ul 10 uM CP-2-RA (5'-CCGTAATCGGGAAGCTGAAG-3')
0.4 ul 25 mM dNTP mix (Invitrogen)
2.0 ul heat-inactivated exonuclease reaction mix
1.5 ul 10x SybrGreen (Invitrogen)
0.4 ul 2.5 U/ul Accuprime Pfx polymerase (Invitrogen)
105
CA 02907177 2015-09-15
WO 2014/143994 PCT/US2014/028212
37.7 ul dH20
Thermal cycle in real-time thermal cycler according to the following protocol,
but
stop cycling before amplification yield plateaus (generally 8-12 cycles):
95C for 5 min
95C for 30 sec
58C for 60 sec
72C for 60 sec
goto 2, N more times
Prepare a shotgun next-generation sequencing library for analysis
Purify desired amplicon population from non-specific amplification products by
gel
extraction.
Concatemerize amplicons into high-molecular weight products suitable for
shearing
Mechanically shear, using either a nebulizer, BioRuptor, Hydroshear, Covaris,
or
similar instrument. DNA should be sheared into fragments several hundred
basepairs in lenth.
Ligate adapters required for amplification by the sequencing platform used. If
necessary, purify ligated product from unligated product and adapters.
Perform Sequencing of Library According to Manufacturer's Directions (e.g.
Illumina, ABI,
etc), Reading Both the Target Sequence and the Differentiator Tag Sequence.
Analyze Data by Correcting for any Biases Detected by Quantitation of
Differentiator Tag
Sequence Abundance.
Construct a table of target:differentiator tag abundances from the read data,
e.g.:
Target Differentiator
ID tag sequence ID Count
1 3547 1
2 4762 1
1 9637 1
106
CA 02907177 2015-09-15
WO 2014/143994 PCT/US2014/028212
1 1078 5
3 4762 1
1 2984 1
All 'count' entries should be '1', since any particular target:differentiator
tag mapping will
not occur more than once by chance, and therefore will only be observed if
bias was present
somewhere in the sample preparation process. For any target:differentiator tag
combination
observed more than once, all such reads are 'collapsed' into a single read
before consensus
basecalls are determined. This will cancel the effect of bias on consensus
basecall accuracy.
FIG. 5 depicts a method for making diploid genotype calls in which repeat
target:differentiator tag combination are collapsed.
Example 3
Differentiator Tag Sequence Design for MIP Capture Reactions
For a set of targets, the number of differentiator tag sequences necessary to
be confident
(within some statistical bounds) that a certain differentiator tag sequence
will not be
observed more than once by chance in combination with a certain target
sequence was
determined. The total number of unique differentiator tag sequences for a
certain
(Length in nucleotides of the differentiator tag sequence).
differentiator tag sequence length is determined as 4
For a molecular inversion probe capture reaction that uses MIP probes having
differentiator
tag sequences, the probability of performing the capture reaction and
capturing one or more
copies of a target sequence having the same differentiator tag sequence is
calculated as:
p=1¨[N!/(N¨M)!1/[NM], wherein N is the total number of possible unique
differentiator tag
sequences and M is the number of target sequence copies in the capture
reaction. Thus, by
varying the differentiator tag sequence length it is possible to perform a MIP
capture
reaction in which the probability of capturing one or more copies of a target
sequence
having the same differentiator tag sequence is set at a predetermined
probability value.
For example, for a differentiator tag sequence of 15 nucleotides in length,
there are
1,073,741,824 possible differentiator tag sequences. A MIP capture reaction in
which MIP
probes, each having a differentiator tag sequence of 15 nucleotides, are
combined with
10000 target sequence copies (e.g., genome equivalents), the probability of
capturing one or
107
CA 02907177 2015-09-15
WO 2014/143994 PCT/US2014/028212
more copies of a target sequence having the same differentiator tag sequence
is 0.05. In this
example, the MIP reaction will produce very few (usually 0, but occasionally 1
or more)
targets where multiple copies are tagged with the same differentiator tag
sequence. FIG. 6
depicts results of a simulation for 100000 capture reactions having 15
nucleotide
differentiator tag sequences and 10000 target sequences.
Example 4
Assessment of the Probability for Obtaining Enough Sequencing Reads to Make
Accurate Base-Calls at Multiple Independent Loci, as a Function of Sequencing
Coverage.
Monte Carlo simulations were performed to determine sequencing coverage
requirements.
The simulations assume 10000 genomic copies of a given locus (target) half mom
alleles
and half dad alleles. The simulations further assume 1% efficiency of capture
for the MIP
reaction. The simulation samples from a capture mix 100 times without
replacement to
create a set of 100 capture products. The simulation then samples from the set
of 100
capture products with replacement (assuming unbiased amplification) to
generate 'reads'
from either mom or dad. The number of reads sampled depends on the coverage.
The
number of independent reads from both mom and dad necessary to make a high-
quality
base-call (assumed to be 10 or 20 reads) were then determined. The process was
repeated
1000 times for each coverage level, and the fraction of times that enough
reads from both
parents were successfully obtained was determined. This fraction was raised to
the power
1000, assuming we have 1000 independent loci that must obtain successful base-
calls,
plotted (See FIG. 7). Result show that roughly 50x coverage is required to
capture each
allele >=10x with >0.95 probability.
Example 5
MIP Capture of 'Target' Locus and 'Control' Loci
In some embodiments, to accurately quantify the efficiency of target locus
capture, at least
three sets of control loci are captured in parallel that have a priori been
shown to serve as
proxies for various lengths of target locus. For example, if the target locus
is expected to
have a length between 50 and 1000 bp, then sets of control loci having lengths
of 50, 250,
108
CA 02907177 2015-09-15
WO 2014/143994 PCT/US2014/028212
and 1000 bp could be captured (e.g. 20 loci per set should provide adequate
protection from
outliers), and their abundance digitally measured by sequencing. These loci
should be
chosen such that minimal variation in efficiency between samples and on
multiple runs of
the same sample is observed (and are therefore 'efficiency invariant'). These
will serve as
'reference' points that define the shape of the curve of abundance-vs-length.
Determining
the length of the target is then simply a matter of 'reading' the length from
the appropriate
point on the calibration curve.
In some embodiments, the statistical confidence one has in the estimate of
target length
from this method is driven largely by three factors: 1)
reproducibility/variation of the
abundance data used to generate the calibration curve; 2) goodness of fit of
the regression
to the 'control' datapoints; 3) reproducibility of abundance data for the
target locus being
measured. Statistical bounds on 1) and 2) will be known in advance, having
been measured
during development of the assay. Additionally, statistical bounds on 3) will
be known in
general in advance, since assay development should include adequate population
sampling
and measure of technical reproducibility. Standard statistical methods should
be used to
combine these three measures into a single P value for any given experimental
measure of
target abundance.
In some embodiments, given the set of calibration observations, and a linear
regression fit
to that data, the regression can be used to predict the length value for n
observations of the
target locus whose length is unknown. First, choose an acceptable range for
the confidence
interval of the length estimate. For example, in the case of distinguishing
"normal" (87-93
bp) from "premutation" (165-600 bp) potential cases of Fragile X, the goal is
to measure
length to sufficient precision to distinguish 93 bp from 165 bp. The predicted
response
value, computed when n observations is substituted into the equation for the
regressed line,
will have arbitrary precision. However, if for example a 95% confidence level
is desired,
that 95% confidence interval must be sufficiently short that it does not
overlap both the
"normal" and "premutation" length ranges. Continuing the example, if one
calculates a
length of 190 from n=400 MIP observations, and based on the regression from
calibration
data, the 95% confidence interval is 190 +/-20 bp, one can conclude the sample
represents
109
CA 02907177 2015-09-15
WO 2014/143994 PCT/US2014/028212
a "premutation" length with 95% certainty. Conversely, if the calibration data
were less
robust, error estimates of the regression would be higher, leading to larger
confidence
intervals on the predicted response value. In some embodiments, if the 95% CI
were
calculated as 190 +/-100 bp from n=400, one could not determine whether the
predicted
response value corresponds to a "normal" or "premutation" length.
In some embodiments, the confidence interval for a predicted response is
calculated as:
The estimate for the response Sr is identical to the estimate for the mean of
the
response: {tilde over (y)}=b0+bix*. The confidence interval for the predicted
value
is given by ST t*sST, where Sr is the fitted value corresponding to x*. The
value t* is
the upper (1¨C)/2 critical value for the t(n-2) distribution.
In some embodiments, a technique for analyzing a locus of interest can involve
the
following steps.
Convert Hybridized Probes into Covalently-Closed Circular Products Containing
Subtargets
Prepare fill-in/ligation reaction mixture:
0.25 ul 2 mM dNTP mix (Invitrogen)
2.5 ul 10x Ampligase buffer (Epicentre)
5.0 ul 5 U/ul Taq Stoffel fragment (Applied Biosystems)
12.5 ul 5 U/ul Ampligase (Epicentre)
4.75 ul dH20
Add 1.0 ul of this mix to the hybridized probe reaction, and incubate at 60 C
for 10
hours.
Purify Circularized Probe/Subtarget Products from Un-Reacted Probes and
Genomic
DNA
Prepare exonuclease reaction mixture:
21 ul fill-in/ligation reaction product
2.0 ul 10x exonuclease I buffer (New England Biolabs)
110
CA 02907177 2015-09-15
WO 2014/143994 PCT/US2014/028212
2.0 ul 20 U/ul exonuclease I (New England Biolabs)
2.0 ul 100 U/uI exonuclease III (New England Biolabs)
Incubte at 37 C for 60 min, then heat-inactivate by incubating at 80 C for 15
min.
Immediately cool to 4 C for storage.
Amplify Circular Material by PCR Using Primers Specific to the 'Constant
Region' of the
Probes
Prepare PCR mixture:
5.0 ul 10x Accuprime reaction buffer (Invitrogen)
1.5 ul 10 uM CP-2-FA-Ilmn (platform-specific amplification sequence plus
'circle constant region'-specific sequence)
1.5 ul 10 uM CP-2-RA-Ilmn (platform-specific amplification sequence plus
'circle constant region'-specific sequence)
0.4 ul 25 mM dNTP mix (Invitrogen)
2.0 ul heat-inactivated exonuclease reaction mix
1.5 ul 10x SybrGreen (Invitrogen)
0.4 ul 2.5 U/uI Accuprime Pfx polymerase (Invitrogen)
37.7 uldH20
Thermal cycle in real-time thermal cycler according to the following protocol,
but stop
cycling before amplification yield plateaus (generally 8-12 cycles):
95C for 5 min
95C for 30 sec
58C for 60 sec
72C for 60 sec
goto 2, N more times
Perform Sequencing (e.g., Next-Generation Sequencing) on Sample for Digital
Quantitation According to Manufacturer's Instructions (e.g., Illumina, Abi)
Example 6
111
CA 02907177 2015-09-15
WO 2014/143994 PCT/US2014/028212
MIP-Capture Reaction of a Set of Exon Target Nucleic Acids
MIP probes are synthesized using standard column-based oligonucleotide
synthesis by any
number of vendors (e.g. IDT).
Hybridize Capture Probes to Human Genomic Sample
Assemble hybridization reaction:
1.0 ul capture probe mix (2.5 pmol)
2.0 ul 10x Ampligase buffer (Epicentre)
6.0 ul 500 ng/ul human genomic DNA (16.7 fmol)
11 ul dH20
In a thermal cycler, heat reaction to 95 C for 5 min to denature genomic DNA,
then cool
to 60 C. Allow to incubate at 60 C for 40 hours.
Convert Hybridized Probes into Covalently-Closed Circular Products Containing
Target
Nucleic Acids
Prepare fill-in/ligation reaction mixture:
0.25 ul 2 mM dNTP mix (Invitrogen)
2.5 ul 10x Ampligase buffer (Epicentre)
5.0 ul 5 U/ul Taq Stoffel fragment (Applied Biosystems)
12.5 ul 5 U/ul Ampligase (Epicentre)
4.75 ul dH20
Add 1.0 ul of this mix to the hybridized probe reaction, and incubate at 60 C
for 10
hours.
Purify circularized probe/target nucleic acid products from un-reacted probes
and
genomic DNA
Prepare exonuclease reaction mixture:
21 ul fill-in/ligation reaction product
2.0 ul 10x exonuclease I buffer (New England Biolabs)
2.0 ul 20 U/ul exonuclease I (New England Biolabs)
2.0 ul 100 U/ul exonuclease III (New England Biolabs)
112
CA 02907177 2015-09-15
WO 2014/143994 PCT/US2014/028212
Incubate at 37 C for 60 min, then heat-inactivate by incubating at 80 C for 15
min.
Immediately cool to 4 C for storage.
Amplify Circular Material by PCR Using Primers Specific to the 'Constant
Region' of the
Probes
Prepare PCR mixture:
5.0 ul 10x Accuprime reaction buffer (Invitrogen)
(SEQ ID NO: 183)
1.5 ul 10 uM CP-2-FA (5'-GCACGATCCGACGGTAGTGT-3')
(SEQ ID NO: 184)
1.5 ul 10 uM CP-2-RA (5'-CCGTAATCGGGAAGCTGAAG-3')
0.4 ul 25 mM dNTP mix (Invitrogen)
2.0 ul heat-inactivated exonuclease reaction mix
1.5 ul 10x SybrGreen (Invitrogen)
0.4 ul 2.5 U/ul Accuprime Pfx polymerase (Invitrogen)
37.7 ul dH20
Thermal cycle in real-time thermal cycler according to the following protocol,
but
stop cycling before amplification yield plateaus (generally 8-12 cycles):
95 C for 5 min
95 C for 30 sec
58 C for 60 sec
72 C for 60 sec
goto 2, N more times
Prepare a Shotgun Next-Generation Sequencing Library for Analysis
Purify desired amplicon population from non-specific amplification products by
gel
extraction.
Concatemerize amplicons into high-molecular weight products suitable for
shearing
Mechanically shear, using either a nebulizer, BioRuptor, Hydroshear, Covaris,
or
113
CA 02907177 2015-09-15
WO 2014/143994 PCT/US2014/028212
similar instrument. DNA should be sheared into fragments several hundred
basepairs in length.
Ligate adapters required for amplification by the sequencing platform used. If
necessary, purify ligated product from unligated product and adapters.
Perform Sequencing of Library According to Manufacturer's Directions (e.g.
Illumina,
ABI, etc), Reading the Target Sequence to Determine Abundance of the Target
Nucleic
Acid.
Example 7
Use of MIPs, Hybridization, and Mutation-Detection Mips to Genotype a Set of
1000
Targets
MIPs, hybridization, and mutation-detection MIPs are used to genotype a set of
1000
targets. The protocol permits detection of any of 50 specific known point
mutations
First, separate MIP, hybridization, and mutation-detection MIP reactions are
performed on
a biological sample. A MIP capture reaction is performed essentially as
described in Turner
et al 2009, Nature methods 6:315-6. A set of MIPs is designed such to that
each probe in
the set flanks one of the 1000 targets. Separately, a hybridization enrichment
reaction is
performed using the Agilent SureSelect procedure. Prior to selection, the
genomic DNA to
be enriched is converted into a shotgun sequencing library using Illumina's
'Fragment
Library' kit and protocol. Agilent's web interface is used to design a set of
probes which
will hybridize to the target nucleic acids. Separately, a set of probes are
designed
(mutation-detection MIPs) which will form MIPs only if mutations (e.g.,
specific
polymorphisms) are present. Each mutation-detection MIP has a 3'-most base
identity that
is specific for a single known mutation. A reaction with this set of mutation-
detection MIPs
is performed to selectively detect the presence of any mutant alleles.
Once all three reactions have been performed, the two MIP reactions are
combined (e.g., at
potentially non-equimolar ratios to further increase sensitivity of mutation
detection) into a
114
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
single tube, and run as one sample on the next-generation DNA sequencing
instrument. The
hybridization-enriched reaction is run as a separate sample on the next-
generation DNA
sequencing instrument. Reads from each 'sample' are combined by a software
algorithm
which forms a consensus diploid genotype at each position in the target set by
evaluating
the total coverage at each position, the origin of each read in that total
coverage, the quality
score of each individual read, and the presence (or absence) of any reads
derived from
mutation-specific MIPs overlapping the region.
Example 8
Carrier screening is performed either pre-conception or during pregnancy to
determine a
couple's risk of having a child with a recessive genetic disorder. The number
of individuals who
could benefit from such screening is substantial, as roughly 2 million women
give birth to their
first child each year in the US. The disorders for which testing is
recommended vary based on a
number of different patient-specific factors. For instance, the American
Congress of
Obstetricians and Gynecologists recommends that screening for cystic fibrosis
be offered to all
women of reproductive age, and that testing be performed for additional
disorders if indicated by
family history, partner's carrier status, or ethnicity.
Today, carrier screening is typically performed using focused genotyping
technologies
that are designed to interrogate specific mutations within a gene of interest.
However,
because of cost and complexity, these tests often do not include all known
disease causing
mutations. In contrast, next-generation DNA sequencing (NGS) can
comprehensively genotype a
set of genes in a cost-efficient manner, and is therefore poised to supplant
current technologies
for routine, high-volume carrier screening.
For NGS to be used for carrier screening in a clinical setting, it must
satisfy at least three
requirements. First, analytical accuracy must be both high and well
characterized within
the clinically relevant genes or regions. Previous reports have demonstrated a
broad
range of accuracy values, and in some cases it is unclear whether these values
hold within
the relevant regions of the genome. In addition, accuracy for insertions and
deletions
is generally either substantially lower or uncharacterized, and measured to
lower
115
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
precision. Second, the NGS workflow employed should yield data sufficient to
cover the
vast majority of targeted bases at a depth sufficient to make high-quality
genotype calls.
It has been noted, however, that the percentage of bases callable at a given
depth varies
widely with both the sample preparation workflow and the total amount of
sequencing
8,10. Finally, the workflow must be highly robust and reproducible, which can
often be
achieved through automation. However, typical NGS sample preparation workflows
are
not amenable to high-throughput automation because of rate-limiting mechanical
shearing, reaction purifications, size selections, and kitted reagent costs
(typically $50-
$200 per sample).
The following is an integrated NGS workflow that meets these requirements for
carrier
screening. The workflow combines automated, optimized molecular inversion
probe target
capture with molecular barcoding to maximize the sample throughput of a next-
generation DNA
sequencing machine, and employs a novel read assembly-based alignment method
that enables
accurate identification of both substitution and insertion/deletion lesions.
The workflow is
applied to sequence the protein-coding regions of fifteen genes in which loss-
of-function
mutations cause recessive Mendelian disorders often included as part of
routine carrier
screening, and demonstrate through realistic simulation and comparison to
Sanger sequencing
data that our approach achieves high accuracies.
METHODS AND MATERIALS
Molecular Inversion Probe Design
Molecular inversion probes were designed to capture the coding regions and
certain well-
characterized non-coding regions of 15 genes (See Table 5 below). The 5'
targeting arm (ligation
arm) and 3' targeting arm (extension arm) comprised a total of 40
nucleotides,and were designed
to flank 130 bp target regions. Probes were selected to maximize performance
with respect to
both capture efficiency and robustness to common polymorphisms. All possible
probes targeting
a genomic interval were designed and assigned score tuples consisting of: 1)
presence of guanine
or cytosine as the 5'-most base of the ligation arm, 2) the number of dbSNP
(version 130) entries
intersecting targeting arm sites, and 3) the root mean squared deviation of
the arms' predicted
melting temperatures from optimal values derived from empirical studies of
capture efficiency.
116
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
Using these tuples, probes were ranked sequentially by 1, 2, and 3, and the
probe with the
highest rank was chosen. Probes were designed to 'tile' across targets with a
period of 25 bp
such that multiple probes with orthogonal targeting arm sequences captured
every genomic
position. The molecular inversion probes are provided in Appendix A. Appendix
A also
includes the upstream and downstream regions corresponding to each molecular
inversion probe,
which is shown by the start position and end position coordinates of each
targeting arm relative
to the target sub-region's coordinates on the Human Genome 18 (HG 18).
Appendix B lists the
genomic sub-regions targeted by the molecular inversion probes of Appendix A.
Table 5 shows diseases and genes the workflow is designed to interrogate, and
the
corresponding genes and nucleotides targeted.
Table 5
DISEASE OMIM ID GENE NT
TARGETED
Familial hyperinsulinism 256450 ABCC8 5,808
Canavan disease 271900 ASPA 1,062
Maple syrup urine disease type la/lb 248600 BCKDHA 1,518
BCKDHB 1,379
Bloom syndrome 210900 BLM 4,674
Cystic fibrosis 219700 CFTR 5,444
Usher syndrome type IIIA 276902 CLRN1 856
Dihydrolipoamide dehydrogenase 248600 DLD 1,810
deficiency
Fanconi anemia group C 227645 FANCC 1,957
Glycogen storage disease type la 232200 G6PC 1,174
Tay-Sachs disease 272800 HEXA 1,870
Familial dysautonomia 223900 IKBKAP 4,719
Mucolipidosis type IV 252650 MCOLN1 2,023
117
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
Usher syndrome type IF 602083 PCDH15 6,508
Niemann-Pick disease type A/B 257200 / 607616 SMPD1 2,056
TOTAL 42,858
Target Capture, Barcoding, and NGS
Genomic DNA was purchased from the Coriell Cell Repositories (Camden, NJ) or
isolated from whole blood by the Gentra Puregene method (Qiagen) modified to
conclude with
an overnight incubation at 65 C. Overnight incubation at an elevated
temperature led to DNA
shearing and an increased fraction of callable bases. All samples were
considered "IRB Exempt"
by Liberty IRB, our independent Institutional Review Board. On Tecan
automation, 1.5 ug of
genomic DNA was annealed with 1 ul of molecular inversion probe mix in 1X
Ampligase buffer
(Epicentre Biotechnologies) for 5 min at 95 C followed by 24 hr at 54 C. 17 ul
of fill-in mix (4
U Taq Stoffel fragment [Life Technologies], 10 U Ampligase [Epicentre
Biotechnologies], 23.1
uM dNTP mix) was added by Tecan automation and incubated for 1 hr at 54 C. 50
U
Exonuclease I and 50 U Exonuclease III (Enzymatics Inc.) were then added by
Tecan automation
and incubated for 1 hr at 37 C followed by 10 min at 98 C. The capture
reaction product was
amplified in two separate PCR reactions designed to attach a molecular barcode
and Illumina
cluster amplification sequences to the ends of each molecule so as to enable
sequencing from
each end of the captured region. Tecan automation was used to set up the PCR,
which was
carried out with 3.75 ul of capture product, 15 pmol of each primer, 10 nmol
dNTPs, and 1 U
VeraSeq polymerase (Enzymatics, Inc) in lx Veraseq buffer. Cycling conditions
were: 98 C 30
sec, 17-22X (98 C 10 sec, 54 C 30 sec, 72 C 15 sec), 4 C forever.
Following PCR, equal volumes of product from multiple samples were pooled
using
Tecan automation, then purified using a Qiaquick column (Qiagen). The library
pool
concentration was quantified on a Bioanalyzer 2100 (Agilent Technologies) and
diluted to 10
nM. Single-read sequencing (85 bp for genomic tag and 15 bp for barcode/index)
was performed
on the Hiseq 2000 (IIlumina, Inc) according to the manufacturer's
instructions. Each pool of
libraries was sequenced in 7 lanes, with the 8th lane used for the
manufacturer-supplied PhiX
control library.
118
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
NGS Data Analysis with Alignment Only Algorithm
Raw .bc1 files were converted to qseq files using bc1Converter (IIlumina).
Fastq files were
generated by 'de-barcoding' genomic reads using the associated barcode reads;
reads for which
barcodes yielded no exact match to an expected barcode, or contained one or
more low-quality
basecalls, were discarded. The remaining reads were aligned to hg18 on a per-
sample basis using
BWA version 0.5.7 for short alignments and genotype calls were made using GATK
version
1Ø4168 after base quality score re-calibration, realignment (with GATK
version 1Ø5083) and
targeting arm removal. High- confidence genotype calls were defined as having
depth >, 50 and
strand bias score <= 0. Clinical significance of variant calls was determined
by matching against
a VCF- formatted database of disease-causing mutations curated from the
literature, with
equivalent insertion/deletion regions calculated as previously described.
NGS Data Analysis with Genotyping by Assembly-Templated Alignment Algorithm
De-barcoded fastq files were obtained as described above and partitioned by
capture region
(exon) using the target arm sequence as a unique key. Reads were assembled in
parallel by exon
using SSAKE version 3.7 with parameters "-m 30 -o 15". The resulting contigs
were aligned to
hg18 using BWA version 0.5.7 for long alignments with parameter "-r 1". Short
read alignment
was performed as described above except that sample contigs (rather than hg18)
were used as the
input reference sequence. Software was developed in Java to accurately
transfer coordinate and
variant data (gaps) from local sample space to global reference space for
every BAM-formatted
alignment. Genotyping and base quality recalibration were performed on the
coordinate-
translated BAM files using GATK version 1.6.5.
Sanger Sequencing
PCR was carried out with the genomic DNA described in Target capture,
barcoding, and NGS
using a modified version of the protocol from Zimmerman et al., using PCR
primers from Jones
et al., except M13 tails were removed. See Zimmerman RS, Cox S, Lakdawala NK,
et al. A
novel custom resequencing array for dilated cardiomyopathy. Genet Med. May
2010;12(5):268-
278; Jones S, Zhang X, Parsons DW, et al. Core signaling pathways in human
pancreatic
cancers revealed by global genomic analyses. Science. Sep 2008;321(5897):1801-
1806.
119
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
Briefly, 15 ul reactions were performed with 25 ng of genomic DNA, 1U of
AmpliTaq Gold
(Applied Biosystems), and 10 fmol of each PCR primer in a PCR mix containing
4.8% DMSO
(v/v), 1M betaine, 2.5mM magnesium chloride, luM dNTPs (total), and 1X GeneAmp
PCR
Gold Buffer (Applied Biosystems). Cycling conditions were: 95 C 10 min, 30X
(95 C 30 sec,
60 C 30 sec, 72 C 30 sec), 72 C 10 min, 8 C forever. PCR products were sent to
either
Beckman Coulter Genomics or Genewiz where cleanup and chain termination bi-
directional
Sanger sequencing was performed on an ABI 3730x1 according to standard
protocols. Data was
retrieved in electropherogram (abl) format.
Sanger Data Analysis and Cross-Validation to NGS
Mutation Surveyor software ("MS", Softgenetics) version 4Ø5 was used in
batch-mode
with default parameters to align abl files to target reference sequence and
make genotype calls.
Positions where MS base calls did not match in the forward and reverse
directions were removed
from consideration. All high-quality NGS genotype calls within 10 bp
(inclusive) of target exons
were subjected to cross-validation against VCF-converted MS variant calls.
This process is
described in more detail below.
Calls were compared by (i) lesion type (substitution, insertion, deletion, or
combination
thereof), (ii) lesion pattern (sequence difference compared to the reference),
and (iii) genomic
position (or equivalent position for insertions and deletions). NGS calls were
classified true
positive (TP), discordant (non-reference) variant genotype (DVG), or false
positive (FP) if they
matched MS calls by (i-iii), (iii) only, or none of the above criteria,
respectively. MS variant calls
with no corresponding NGS variant call were classified false negative (FN).
Indel calls classified
as DVG were re-classified as TP because GATK 1Ø4168 does not report zygosity
for such calls.
All concordant reference calls were considered true negative (TN). Each
discordant call (DVG,
FP, and FN), along with a subset of concordant calls, was subject to expert
manual review and
discarded or reclassified as appropriate. False positive rate was calculated
as FP / (FP +TN).
False negative rate was calculated as FN / (FN + TP). Compound heterozygous
NGS calls (two
different non-reference alleles) were cross-validated against Sanger data
manually by aligning
traces to a reference manipulated to contain one of the two variant alleles.
In these cases TP
genotype calls were reported as simple heterozygous by MS.
120
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
Assessment of Detectability of Clinical Mutations by Simulation
145 Coriell samples were sequenced and analyzed by Genotyping by Assembly-
Templated
Alignment (GATA, described above). Applications were developed in Java and
Groovy to input
aligned reads (BAM records) from each sample and manipulate specific data
fields (base
sequence and qualities) to resemble the appropriate DNA lesion pattern of a
given clinically
relevant mutation. To simulate heterozygous carriers input reads covering the
mutation were
chosen at random for sequence manipulation with an average probability of 0.5.
All reads,
whether manipulated or not, were output in fastq format for subsequent GATA
analysis as
described. This process was repeated for each of 81 mutations of clinical
significance whereupon
genotyped (observed) alleles were cross- referenced back to the original
simulated (expected)
allele. Samples for which the allele was already present were excluded from
simulation (e.g.
many Coriell samples in the set contained the common CFTR F508de1 mutation).
Mutations with
detection rates <100% between the expected and observed alleles were
classified as undetectable
by NGS.
Determining Clinical Significance of Variant Allele Calls
Each NGS-detected variant allele is annotated for functional (clinical)
significance by
determining its relative position within the corresponding consensus coding
sequence (CCDS).
For the genes under consideration here these are: PCDH15 (CCD57248.1), SMPD1
(CCD544531.1), ABCC8 (CCDS31437.1), HEXA (CCDS10243.1), BLM (CCDS10363.1),
ASPA (CCDS11028.1), G6PC (CCD511446.1), MCOLN1 (CCDS12180.1), BCKDHA
(CCDS12581.1), CLRN1 (CCDS3153.1), BCKDHB (CCD54994.1), DLD (CCD55749.1),
CFTR (CCDS5773.1), FANCC (CCDS35071.1), and IKBKAP (CCDS6773.1). Clinically
significant (reportable) mutations include alterations to the conserved 2
basepairs flanking each
exon (splice site), the native start codon, or the last codon (readthrough),
as well as truncating
(nonsense and frameshift) mutations. Additionally, GATK occasionally reports
alternate
insertion patterns with non-native bases (e.g. 'N') chosen from a minority of
reads. These were
classified 'indeterminate' and reportable to prompt follow-up confirmation.
RESULTS
i. Completeness and Reproducibility
121
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
Automated target capture and molecular barcoding were performed followed by
NGS on
a set of 194 samples derived from immortalized cell lines (55 containing
specific disease-
causing mutations, and 139 chosen to represent ethnic diversity) and 59
samples derived from
whole blood (as shown in Table 6 below). All exons were targeted including 10
nt of flanking
intronic sequence, plus additional intronic regions known to contain disease-
causing mutations
in 15 genes causative of 14 recessive Mendelian diseases (Table 5) using
tiling molecular
inversion probes (see Methods). A total of 25,907,612,945 basepairs of de-
multiplexed sequence
were generated, corresponding to an average per- base coverage per sample of
2,399X (min
891X, max 4,000X). Out of the 42,858 bases targeted for capture in each
sample, we made high-
confidence genotype calls at an average of 97.3% (min 92.2%, max 99.8%) for
cell line-derived
DNA and 99.9% (min 99.8%, max 99.9%) for blood-derived DNA (See Table 5
above).
Table 6 shows the set of 94 samples derived from immortalized cell lines and
59 samples
derived from whole blood.
Table 6
Sample ID Sampl Sample Sequence Average Percentag Reproducibility?
Sanger
e Type (raw bp) Coverage e of Bases
Concordance
Source >=50X ?
GM0050 Cell Disease 76,288,149 1,787 97.6 Yes Yes
2 line
GM0064 Cell Disease 115,317,69 2,701 97.9 Yes Yes
9 line 5
GM0065 Cell Disease 69,572,569 1,629 93.1 Yes Yes
0 line
GM0153 Cell Disease 93,831,687 2,198 99.2 Yes Yes
1 line
GM0253 Cell Disease 61,190,070 1,433 98.6 Yes Yes
3 line
GM0282 Cell Disease 49,081,409 1,150 98.6 Yes Yes
8 line
GM0325 Cell Disease 47,780,116 1,119 98.9 Yes Yes
2 line
GM0346 Cell Disease 133,932,43 3,137 95.2 Yes Yes
122
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
1 line 3
GM0426 Cell Disease 113,557,31 2,660 98.6 Yes Yes
8 line 0
GM0433 Cell Disease 115,608,79 2,708 94.3 Yes Yes
0 line 0
GM0504 Cell Disease 113,811,44 2,666 96.0 Yes Yes
2 line 9
GM0696 Cell Disease 51,321,434 1,202 99.0 Yes Yes
6 line
GM0738 Cell Disease 87,834,174 2,057 99.3 Yes Yes
1 line
GM0744 Cell Disease 108,471,71 2,541 99.4 Yes Yes
1 line 7
GM0755 Cell Disease 106,594,63 2,497 99.3 Yes Yes
2 line 0
GM0773 Cell Disease 137,685,13 3,225 94.7 Yes Yes
2 line 1
GM0785 Cell Disease 98,376,083 2,304 96.6 Yes Yes
7 line
GM0833 Cell Disease 131,459,59 3,079 96.9 Yes Yes
8 line 1
GM1127 Cell Disease 119,881,29 2,808 99.3 Yes Yes
line 9
GM1127 Cell Disease 85,993,084 2,014 99.3 Yes Yes
7 line
GM1127 Cell Disease 125,921,30 2,949 92.9 Yes Yes
8 line 3
GM1128 Cell Disease 121,485,71 2,845 99.2 Yes Yes
0 line 2
GM1128 Cell Disease 107,022,43 2,507 99.7 No Yes
1 line 3
GM1128 Cell Disease 105,909,02 2,481 99.5 Yes Yes
2 line 9
123
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
GM1128 Cell Disease 128,624,24 3,013 96.5 Yes Yes
3 line 1
GM1128 Cell Disease 125,265,00 2,934 99.8 Yes Yes
4 line 8
GM1128 Cell Disease 105,205,58 2,464 97.3 Yes Yes
line 0
GM1128 Cell Disease 121,267,78 2,840 96.4 Yes Yes
7 line 7
GM1128 Cell Disease 116,071,39 2,719 98.0 Yes Yes
8 line 7
GM1137 Cell Disease 106,105,64 2,485 95.9 Yes Yes
0 line 7
GM1146 Cell Disease 120,852,66 2,831 97.9 Yes Yes
8 line 9
GM1147 Cell Disease 146,901,68 3,441 97.0 Yes Yes
2 line 2
GM1149 Cell Disease 119,104,14 2,790 99.3 Yes Yes
6 line 9
GM1149 Cell Disease 103,338,59 2,420 99.0 Yes Yes
7 line 4
GM1172 Cell Disease 111,446,56 2,610 99.1 Yes Yes
3 line 5
GM1185 Cell Disease 132,558,32 3,105 99.7 Yes Yes
9 line 1
GM1186 Cell Disease 133,241,17 3,121 99.5 Yes Yes
0 line 0
GM1244 Cell Disease 112,979,13 2,646 99.3 Yes Yes
4 line 0
GM1258 Cell Disease 142,275,40 3,332 98.4 Yes Yes
5 line 0
GM1278 Cell Disease 77,781,835 1,822 99.0 Yes Yes
5 line
GM1296 Cell Disease 105,134,32 2,462 99.2 Yes Yes
124
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
0 line 3
GM1320 Cell Disease 116,426,91 2,727 97.0 Yes Yes
line 2
GM1342 Cell Disease 142,163,14 3,330 96.5 Yes Yes
3 line 1
GM1359 Cell Disease 127,607,78 2,989 98.6 Yes Yes
1 line 3
GM1619 Cell Disease 80,190,257 1,878 92.7 Yes Yes
3 line
GM1702 Cell HuVar 114,118,59 2,673 98.9 No Yes
3 line 8
GM1707 Cell TGP 125,212,95 2,933 98.5 No Yes
4 line 6
GM1707 Cell TGP 114,067,84 2,672 95.1 No Yes
5 line 1
GM1707 Cell TGP 135,956,92 3,184 97.3 No Yes
8 line 5
GM1707 Cell TGP 109,131,65 2,556 93.8 No Yes
9 line 0
GM1708 Cell TGP 104,079,00 2,438 98.2 No Yes
0 line 0
GM1720 Cell HuVar 100,286,17 2,349 95.8 No Yes
3 line 0
GM1720 Cell HuVar 133,095,16 3,117 99.7 No Yes
7 line 5
GM1722 Cell HuVar 75,792,351 1,775 98.7 No Yes
8 line
GM1723 Cell HuVar 138,157,41 3,236 97.1 No Yes
1 line 8
GM1723 Cell HuVar 115,522,25 2,706 97.1 No Yes
3 line 6
GM1724 Cell HuVar 114,147,39 2,673 99.3 No Yes
2 line 2
125
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
GM1724 Cell HuVar 88,905,331 2,082 99.3 No Yes
7 line
GM1725 Cell HuVar 134,029,72 3,139 97.8 No Yes
1 line 8
GM1728 Cell HuVar 104,284,77 2,443 95.5 Yes Yes
2 line 7
GM1728 Cell HuVar 124,885,88 2,925 98.0 No Yes
6 line 6
GM1730 Cell HuVar 115,253,37 2,699 95.5 Yes Yes
1 line 5
GM1730 Cell HuVar 126,663,09 2,967 95.6 Yes Yes
2 line 1
GM1730 Cell HuVar 148,723,81 3,483 96.8 No Yes
3 line 5
GM1730 Cell HuVar 140,507,36 3,291 95.3 No Yes
4 line 0
GM1731 Cell HuVar 112,930,12 2,645 96.5 No Yes
0 line 3
GM1731 Cell HuVar 146,713,29 3,436 96.0 Yes Yes
line 5
GM1731 Cell HuVar 120,214,96 2,816 96.1 No Yes
7 line 4
GM1731 Cell HuVar 131,177,75 3,072 98.4 Yes Yes
8 line 3
GM1731 Cell HuVar 74,599,530 1,747 96.3 Yes Yes
9 line
GM1732 Cell HuVar 143,908,02 3,371 98.9 Yes Yes
0 line 6
GM1736 Cell HuVar 72,217,715 1,691 99.3 Yes Yes
0 line
GM1736 Cell HuVar 138,241,78 3,238 97.5 No Yes
1 line 9
GM1736 Cell HuVar 109,391,82 2,562 95.4 Yes Yes
126
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
2 line 7
GM1736 Cell HuVar 136,216,56 3,190 97.4 No Yes
3 line 3
GM1736 Cell HuVar 124,580,79 2,918 98.8 Yes Yes
4 line 4
GM1736 Cell HuVar 145,974,76 3,419 96.8 Yes Yes
line 3
GM1736 Cell HuVar 121,059,29 2,835 95.3 Yes Yes
6 line 1
GM1736 Cell HuVar 124,286,28 2,911 97.7 Yes Yes
7 line 0
GM1736 Cell HuVar 122,309,22 2,865 97.4 Yes Yes
8 line 8
GM1736 Cell HuVar 151,606,78 3,551 97.2 No Yes
9 line 8
GM1739 Cell HuVar 120,466,85 2,822 96.9 Yes Yes
2 line 2
GM1739 Cell HuVar 129,362,19 3,030 96.1 Yes Yes
3 line 9
GM1739 Cell HuVar 133,049,78 3,116 96.4 No Yes
4 line 0
GM1739 Cell HuVar 145,469,08 3,407 97.0 No Yes
5 line 9
GM1739 Cell HuVar 131,796,12 3,087 96.1 No Yes
6 line 4
GM1796 Cell HapMa 110,772,39 2,594 92.3 Yes Yes
2 line p 6
GM1796 Cell HapMa 131,430,39 3,078 95.4 No Yes
5 line p 1
GM1796 Cell HapMa 108,405,81 2,539 92.6 No Yes
6 line p 5
GM1796 Cell HapMa 133,849,48 3,135 95.2 Yes Yes
7 line p 2
127
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
GM1796 Cell HapMa 104,839,65 2,455 96.0 Yes Yes
8 line p 9
GM1796 Cell HapMa 170,762,90 4,000 98.7 Yes Yes
9 line p 0
GM1797 Cell HapMa 129,700,47 3,038 96.5 Yes Yes
0 line p 2
GM1797 Cell HapMa 146,346,72 3,428 97.2 Yes Yes
1 line p 2
GM1797 Cell HapMa 139,495,48 3,267 96.2 Yes Yes
2 line p 6
GM1797 Cell HapMa 116,085,42 2,719 93.2 No Yes
3 line p 1
GM1801 Cell HuVar 120,955,00 2,833 95.2 No Yes
line 0
GM1801 Cell HuVar 128,904,00 3,019 96.0 Yes Yes
7 line 6
GM1803 Cell HuVar 104,807,68 2,455 97.4 Yes Yes
4 line 2
GM1804 Cell HuVar 117,441,95 2,751 95.4 No Yes
3 line 3
GM1804 Cell HuVar 149,341,51 3,498 96.8 No Yes
4 line 8
GM1806 Cell HuVar 90,615,125 2,122 94.3 Yes Yes
7 line
GM1807 Cell HuVar 120,359,15 2,819 94.0 No Yes
3 line 4
GM1807 Cell HuVar 130,655,29 3,060 96.1 No Yes
5 line 2
GM1808 Cell HuVar 127,693,61 2,991 97.0 No Yes
4 line 2
GM1808 Cell HuVar 116,883,42 2,738 95.3 Yes Yes
7 line 5
GM1808 Cell HuVar 113,522,77 2,659 93.8 No Yes
128
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
9 line 5
GM1809 Cell HuVar 139,175,35 3,260 95.6 Yes Yes
0 line 1
GM1809 Cell HuVar 140,749,31 3,297 96.1 No Yes
1 line 1
GM1850 Cell HapMa 116,001,92 2,717 99.2 Yes Yes
7 line p 7
GM1852 Cell HapMa 123,974,59 2,904 99.4 Yes Yes
4 line p 3
GM1852 Cell HapMa 68,506,615 1,605 98.2 Yes Yes
6 line p
GM1852 Cell HapMa 103,011,72 2,413 99.2 No Yes
9 line p 9
GM1853 Cell HapMa 93,010,560 2,178 98.9 Yes Yes
2 line p
GM1853 Cell HapMa 88,541,054 2,074 98.2 Yes Yes
7 line p
GM1854 Cell HapMa 107,018,41 2,507 99.3 Yes Yes
0 line p 9
GM1855 Cell HapMa 110,404,28 2,586 99.3 No Yes
8 line p 0
GM1856 Cell HapMa 94,941,108 2,224 98.7 No Yes
1 line p
GM1856 Cell HapMa 109,707,90 2,570 99.4 Yes Yes
2 line p 7
GM1856 Cell HapMa 132,909,80 3,113 95.9 No Yes
3 line p 7
GM1866 Cell Disease 142,395,24 3,335 99.4 Yes Yes
8 line 5
GM1879 Cell Disease 117,599,23 2,754 99.2 Yes Yes
9 line 0
GM1880 Cell Disease 109,551,22 2,566 99.2 Yes Yes
0 line 4
129
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
GM1880 Cell Disease 87,204,605 2,042 98.8 Yes Yes
2 line
GM1888 Cell Disease 138,604,38 3,246 95.1 Yes Yes
6 line 6
GM1899 Cell HapMa 108,306,94 2,537 94.4 Yes Yes
2 line p 2
GM1899 Cell HapMa 96,468,405 2,259 99.5 No Yes
line p
GM1899 Cell HapMa 111,633,42 2,615 98.8 No Yes
7 line p 5
GM1899 Cell HapMa 99,785,735 2,337 99.4 Yes Yes
8 line p
GM1899 Cell HapMa 127,162,92 2,978 97.7 No Yes
9 line p 0
GM1900 Cell HapMa 66,999,861 1,569 98.0 Yes Yes
0 line p
GM1900 Cell HapMa 126,196,39 2,956 94.9 No Yes
3 line p 3
GM1900 Cell HapMa 143,461,74 3,360 96.3 No Yes
5 line p 9
GM1900 Cell HapMa 116,823,48 2,736 99.1 No Yes
7 line p 2
GM1901 Cell HapMa 121,510,89 2,846 99.8 Yes Yes
2 line p 3
GM1909 Cell HapMa 104,709,69 2,452 95.0 Yes Yes
3 line p 3
GM1909 Cell HapMa 108,885,87 2,550 98.2 Yes Yes
9 line p 3
GM1910 Cell HapMa 120,459,30 2,821 99.4 Yes Yes
1 line p 3
GM1911 Cell HapMa 71,500,299 1,675 99.2 No Yes
6 line p
GM1912 Cell HapMa 119,050,42 2,788 99.2 Yes Yes
130
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
7 line p 1
GM1913 Cell HapMa 66,366,273 1,554 96.9 No Yes
0 line p
GM1913 Cell HapMa 97,725,686 2,289 98.9 Yes Yes
7 line p
GM1914 Cell HapMa 110,866,36 2,597 99.1 Yes Yes
1 line p 3
GM1914 Cell HapMa 117,906,14 2,762 99.2 No Yes
4 line p 3
GM1915 Cell HapMa 84,729,187 1,984 99.2 Yes Yes
2 line p
GM1915 Cell HapMa 90,111,210 2,111 99.1 Yes Yes
9 line p
GM1917 Cell HapMa 74,654,792 1,749 96.6 No Yes
2 line p
GM1919 Cell HapMa 127,763,78 2,992 99.8 Yes Yes
2 line p 0
GM1920 Cell HapMa 114,675,88 2,686 99.7 Yes Yes
0 line p 6
GM1920 Cell HapMa 117,546,44 2,753 98.8 Yes Yes
3 line p 6
GM1920 Cell HapMa 84,803,031 1,986 98.7 Yes Yes
7 line p
GM1920 Cell HapMa 59,249,941 1,388 97.0 Yes Yes
9 line p
GM1922 Cell HapMa 70,582,882 1,653 98.0 Yes Yes
3 line p
GM1924 Cell HapMa 74,942,748 1,755 99.0 Yes Yes
0 line p
GM1977 Cell HapMa 91,506,428 2,143 93.3 No Yes
6 line p
GM1978 Cell HapMa 93,214,221 2,183 94.4 No Yes
0 line p
131
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
GM1978 Cell HapMa 147,554,99 3,456 97.0 No Yes
2 line p 7
GM1978 Cell HapMa 99,895,304 2,340 95.9 Yes Yes
9 line p
GM1979 Cell HapMa 112,615,95 2,638 95.6 Yes Yes
4 line p 9
GM2028 Cell HapMa 118,388,59 2,773 94.9 Yes Yes
1 line p 0
GM2033 Cell HapMa 134,954,11 3,161 94.4 Yes Yes
2 line p 6
GM2033 Cell HapMa 109,399,56 2,562 92.8 Yes Yes
line p 1
GM2034 Cell HapMa 96,681,315 2,264 92.5 Yes Yes
1 line p
GM2034 Cell HapMa 149,562,21 3,503 98.3 Yes Yes
2 line p 0
GM2034 Cell HapMa 112,547,10 2,636 97.6 Yes Yes
4 line p 7
GM2034 Cell HapMa 96,682,870 2,264 92.7 No Yes
9 line p
GM2035 Cell HapMa 128,110,98 3,001 95.9 No Yes
7 line p 8
GM2036 Cell HapMa 105,976,91 2,482 92.2 No Yes
0 line p 1
GM2036 Cell HapMa 114,582,01 2,684 94.9 No Yes
3 line p 2
GM2073 Cell Disease 86,947,571 2,036 98.4 Yes Yes
7 line
GM2074 Cell Disease 131,676,64 3,084 99.5 Yes Yes
1 line 2
GM2074 Cell Disease 120,425,67 2,821 99.4 Yes Yes
5 line 8
GM2084 Cell HapMa 88,592,183 2,075 98.5 No Yes
132
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
line p
GM2084 Cell HapMa 94,474,722 2,213 98.9 No Yes
6 line p
GM2084 Cell HapMa 132,183,92 3,096 94.6 No Yes
7 line p 0
GM2084 Cell HapMa 94,859,450 2,222 99.2 Yes Yes
9 line p
GM2085 Cell HapMa 89,746,969 2,102 98.9 Yes Yes
0 line p
GM2085 Cell HapMa 105,058,24 2,461 98.9 No Yes
1 line p 8
GM2085 Cell HapMa 103,469,22 2,423 99.2 Yes Yes
2 line p 3
GM2085 Cell HapMa 67,451,488 1,580 99.0 Yes Yes
3 line p
GM2085 Cell HapMa 125,360,57 2,936 95.9 No Yes
4 line p 5
GM2085 Cell HapMa 125,206,71 2,933 95.1 Yes Yes
6 line p 1
GM2085 Cell HapMa 102,707,14 2,406 99.3 Yes Yes
8 line p 3
GM2085 Cell HapMa 107,012,00 2,506 98.9 No Yes
9 line p 9
GM2086 Cell HapMa 146,690,57 3,436 96.3 No Yes
1 line p 3
GM2086 Cell HapMa 121,310,10 2,841 99.4 Yes Yes
2 line p 7
GM2086 Cell HapMa 106,527,16 2,495 99.4 Yes Yes
6 line p 4
GM2086 Cell HapMa 88,099,219 2,063 99.4 Yes Yes
9 line p
GM2087 Cell HapMa 84,570,991 1,981 99.0 Yes Yes
0 line p
133
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
GM2087 Cell HapMa 104,048,64 2,437 98.9 No Yes
1 line p 5
GM2087 Cell HapMa 90,867,460 2,128 99.0 No Yes
2 line p
GM2087 Cell HapMa 108,700,92 2,546 99.3 No Yes
3 line p 5
GM2092 Cell Disease 120,376,41 2,819 99.2 Yes Yes
4 line 4
GM2108 Cell Disease 66,554,012 1,559 97.3 Yes Yes
0 line
blood01 Blood N/A 67,892,054 1,594 99.9 No No
blood02 Blood N/A 75,235,946 1,766 99.9 No No
blood03 Blood N/A 71,324,606 1,674 99.9 No No
blood04 Blood N/A 58,883,762 1,382 99.9 No No
blood05 Blood N/A 74,862,133 1,757 99.9 No No
blood06 Blood N/A 77,267,380 1,814 99.9 No No
blood07 Blood N/A 55,719,056 1,308 99.9 No No
blood08 Blood N/A 64,495,882 1,514 99.9 No No
blood09 Blood N/A 67,663,353 1,588 99.9 No No
blood10 Blood N/A 57,362,443 1,347 99.9 No No
bloodll Blood N/A 53,823,416 1,264 99.9 No No
blood12 Blood N/A 73,097,398 1,716 99.9 No No
blood13 Blood N/A 73,858,165 1,734 99.9 No No
blood14 Blood N/A 87,675,439 2,058 99.9 No No
blood15 Blood N/A 74,484,474 1,749 99.8 No No
blood16 Blood N/A 59,096,764 1,387 99.8 No No
blood17 Blood N/A 65,114,672 1,529 99.9 No No
blood18 Blood N/A 41,759,247 980 99.9 No No
134
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
blood19 Blood N/A 71,949,103 1,689 99.9 No No
blood20 Blood N/A 81,225,381 1,907 99.9 No No
blood21 Blood N/A 70,214,097 1,648 99.9 No No
blood22 Blood N/A 72,674,504 1,706 99.9 No No
blood23 Blood N/A 74,340,749 1,745 99.9 No No
blood24 Blood N/A 64,015,737 1,503 99.9 No No
blood25 Blood N/A 73,147,784 1,717 99.8 No No
blood26 Blood N/A 41,950,444 985 99.8 No No
blood27 Blood N/A 62,771,860 1,474 99.8 No No
blood28 Blood N/A 47,085,570 1,105 99.8 No No
blood29 Blood N/A 74,840,986 1,757 99.9 No No
blood30 Blood N/A 73,612,767 1,728 99.9 No No
blood31 Blood N/A 70,446,967 1,654 99.9 No No
blood32 Blood N/A 86,513,773 2,031 99.9 No No
blood33 Blood N/A 78,330,087 1,839 99.9 No No
blood34 Blood N/A 76,890,117 1,805 99.9 No No
blood35 Blood N/A 63,472,751 1,490 99.9 No No
blood36 Blood N/A 77,259,799 1,814 99.9 No No
blood37 Blood N/A 74,384,590 1,746 99.9 No No
blood38 Blood N/A 87,075,653 2,044 99.9 No No
blood39 Blood N/A 61,490,312 1,444 99.9 No No
blood40 Blood N/A 83,490,415 1,960 99.9 No No
blood41 Blood N/A 94,474,694 2,218 99.9 No No
blood42 Blood N/A 79,180,999 1,859 99.9 No No
blood43 Blood N/A 70,106,334 1,646 99.9 No No
135
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
blood44 Blood N/A 66,239,225 1,555 99.9 No No
blood45 Blood N/A 76,565,215 1,797 99.8 No No
blood46 Blood N/A 66,932,062 1,571 99.9 No No
blood47 Blood N/A 37,972,652 891 99.8 No No
blood48 Blood N/A 66,880,850 1,570 99.9 No No
blood49 Blood N/A 65,267,319 1,532 99.9 No No
blood50 Blood N/A 63,720,579 1,496 99.9 No No
blood51 Blood N/A 64,485,398 1,514 99.9 No No
blood52 Blood N/A 90,657,228 2,128 99.9 No No
blood53 Blood N/A 83,058,297 1,950 99.9 No No
blood54 Blood N/A 86,145,665 2,022 99.9 No No
blood55 Blood N/A 77,159,945 1,811 99.9 No No
blood56 Blood N/A 88,169,014 2,070 99.9 No No
blood57 Blood N/A 60,859,847 1,429 99.9 No No
blood58 Blood N/A 72,504,883 1,702 99.9 No No
blood59 Blood N/A 83,924,327 1,970 99.9 No No
The DNA extraction protocol used for our blood samples concluded with an
overnight
incubation at 65 C in a Tris-based buffer. Subsequent experiments showed that
this step reduced
the mean size of the purified DNA; shearing was likely caused by acid
hydrolysis during a
temperature-induced pH shift of the buffer. Lower molecular mass genomic DNA
is more
readily denatured, and therefore more accessible to molecular inversion
probes, which improves
capture reaction performance. Consistent with this hypothesis, overnight
incubation temperature
lowered to 25 C significantly reduces the percentage of target bases that
yield high confidence
genotype calls.
To assess reproducibility, a subset of 126 samples derived from cell line DNA
(Appendix
A) was processed twice, each time by a different operator on different liquid
handling
equipment. At least 92% of bases were called at >, 50x coverage in all
samples, with high
136
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
agreement between replicates (Pearson correlation coefficient 0.868). Out of
5,177,206 total
genotype calls compared, 17 were discordant, for a concordance rate of
0.999997. These
occurred at only 5 unique genomic positions, consistent with systematic
sequencing error as the
primary cause.
Sanger Concordance
To assess the overall accuracy of our NGS genotype calls, the genotype calls
from the
NGS pipeline were compared to those generated by automated analysis (Mutation
Surveyor, MS)
of bi-directional Sanger sequence of PCR amplicons in a subset of 194 samples.
Within a total of
6,997,906 bp of sequence called by both methods, 3,973 concordant and 1,220
discordant single
nucleotide variant (SNV) genotype calls were observed. Through manual
inspection of the
Sanger trace(s) corresponding to discordant genotype calls, it was determined
that 1,139 were
MS errors, generally caused by low quality traces or misalignment of traces to
reference.
Supporting the conclusion that the majority of discordant calls corresponded
to incorrect Sanger
calls, the Ti/Tv ratio of concordant genotype calls was observed as 3.19, and
0.61 for discordant
Sanger calls eliminated as MS errors. The remaining 81 discordant genotype
calls that could not
be resolved because the corresponding traces were ambiguous, were re-amplified
and re-
sequenced. For 71 of these calls, this process yielded new Sanger data that
led to the conclusion
that the original automated Sanger calls were incorrect. An additional
discordant call was
resolved by another approach as a NGS true negative (Figures 15A-B), leaving 9
high-
confidence discordant SNV calls (Table 7), corresponding to 8 NGS false
positives and 1 NGS
false negative. Table 7 shows a comparison of NGS genotype calls (alignment-
only algorithm) to
Sanger-derived Mutation Surveyor genotype calls. Sanger genotype calls were
considered truth.
TP, true positive calls (non-reference NGS, non-reference Sanger); FP, false
positive calls (non-
reference NGS, reference Sanger); FN, false negative calls (reference NGS, non-
reference
Sanger); TN, true negative calls (reference NGS, reference Sanger). dbSNP
membership
determined relative to version 129. Indel calls were considered unique if they
differed by
sequence pattern or equivalence region. Known indels are disease-causing
mutations present in
previously-annotated samples.
TABLE 7
137
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
TP FP FN TN
dbS,NP 2,495 0 1
Heterozygous
ot ASIVP 247 8 0
STAV dESNP 1,245 0 0 6,992,74 6
Homozygous
Ot dbSNP 13 0 0
Unique 231 3 1
Total 61 3
indel Unique 17 27 2 6,992,358
Known 31 0
The NGS SNV false positive rate was 1.14x10-6 (95% Wilson binomial confidence
interval [5.80x10-7 2.26x10-6]). The false positive calls occurred at 5 unique
genomic loci, 3 of
which were at adjacent positions in a single exon of gene MCOLN1 due to
realignment within
GATK.
FIG. 16A-D shows GM18540 is an aneuploid cell line and hence yields skewed
allelic
fractions. FIG. 16A gives an IGV view of NGS data from GM18540 for the
genotype call of
interest (shown between vertical lines). FIG. 16B shows bi-directional Sanger
data for the
variant-containing region. FIG. 16C provides a histogram of allele ratios for
all non-reference
genotype calls in chromosome 11 derived from whole-genome shotgun sequencing
(WGSS) of
GM18540 and control sample GM18537. FIG. 16D shows genome-wide relative
coverage for
GM18540. WGSS coverage data for each of the autosomes was binned into 50 Kb
intervals and
the log-ratio of the per-sample mean normalized values was plotted versus
chromosome position.
Dashed vertical lines denote chromosome boundaries; within a chromosome the
ratios are
arranged according to genomic position.
The NGS SNV false negative rate was 2.50x10-4 (95% Wilson binomial confidence
interval [1.28x10-5 1.41x10-3]). The false negative call observed occurred in
chromosome 11 of
a sample previously characterized as aneuploid. Out of 473 NGS reads covering
the false
negative locus, 9.5% supported the correct heterozygous A/C genotype call
(FIG. 16A), with
Sanger sequencing showing low peak height for the alternate A allele (FIG.
16B). Shotgun full-
genome sequencing of this sample demonstrated a bimodal distribution of allele
ratios for
heterozygous calls in chromosome 11 (FIG. 16C), and illustrated variable
chromosome copy
numbers (FIG. 16D), supporting the conclusion that this sample was aneuploid.
138
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
For indels, a total of 61 true positives, 394 false positives (27 unique
alleles) and 3 false
negatives (2 unique alleles, both in exon 1 of SMPD1) were observed. Of 31
clinically- relevant
disease mutations, all 31 were detected.
iii. Detection of Pathogenic Mutations
The ability to detect variants that cause the Mendelian diseases targeted by
the panel
(Table 5) in the set of 194 cell line-derived samples was assessed. 55 of
these samples were
derived from individuals who were either carriers of or affected by one of the
diseases being
assayed and collectively contained a total of 95 previously- characterized
disease mutations.
During the design of our NGS workflow, we determined that three of these
lesions would be
inaccessible by our approach -- two were large deletions spanning multiple
exons, and one was
contained within a region of paralogous sequence in the tenth exon of CFTR
(Table 8). Of the 92
mutations we could expect to detect by NGS, we detected all 92 (Table 8). We
also identified
truncating (and likely disease-causing) mutations in two affected samples
where previously only
one mutation was known (Figures 17A-D, Table 8), as well as 9 carriers in the
set of 139
previously-uncharacterized HapMap, Thousand Genomes Project, and Human
Diversity Panel
samples (Table 8).
Table 8 shows pathogenic mutations detected in cell line-derived samples.
Mutations
highlighted in red and underlined were determined a priori to be inaccessible
by NGS and
therefore not evaluated here. Mutations listed in italicized font represent
mutations in affected
individuals that were previously unknown. Mutations listed in bolded font were
present in
Hapmap samples previously unannotated with respect to carrier status.
Table 8
Sample Gene Mutl Common Name Mutl Mut2 Common Name
Mut2
Found?
Found?
GM04268 ASPA E285A Yes E285A Yes
GM00649 BCKDHA Y438N Yes 8bp del exon 7 Yes
GM00650 BCKDHA Y438N Yes- -
GM01531 CFTR PHE508DEL Yes PHE508DEL Yes
GM02828 CFTR V520F Yes PHE508DEL Yes
139
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
GM04330 CFTR 1812-1G>A Yes 444de1A Yes
GM06966 CFTR E92X Yes PHE508DEL Yes
GM07381 CFTR IVS19DS, +10 KB, C>T Yes PHE508DEL Yes
(3849+10kbC>T)
GM07441 CFTR 621+1G>T Yes IVS16,G>A,+1 Yes
(3120+1G>A)
GM07552 CFTR ARG553TER Yes PHE508DEL Yes
GM07732 CFTR E6OX Yes PHE508DEL Yes
GM07857 CFTR M1101K Yes M1101K Yes
GM08338 CFTR GLY551ASP Yes
GM11275 CFTR 1-BP DEL, 3659C Yes PHE508DEL Yes
GM11277 CFTR ILE507DEL Yes ILE507DEL Yes
GM11278 CFTR Q493X Yes PHE508DEL Yes
GM11280 CFTR 621+1G>T Yes 711+1G>T Yes
GM11281 CFTR 621+1G>T Yes PHE508DEL Yes
GM11282 CFTR 621+1G>T Yes GLY85GLU Yes
GM11283 CFTR ALA455GLU N/A PHE508DEL Yes
GM11284 CFTR ARG560THR Yes PHE508DEL Yes
GM11285 CFTR Y1092X Yes PHE508DEL Yes
GM11287 CFTR P574H Yes PHE508DEL Yes
GM11288 CFTR G178R Yes PHE508DEL Yes
GM11370 CFTR 444de1A Yes IVS11-1G>A Yes
GM11472 CFTR ASN1303LYS Yes GLY1349ASP Yes
GM11496 CFTR GLY542TER Yes GLY542TER Yes
GM11497 CFTR GLY542TER Yes
GM11723 CFTR TRP1282TER Yes
140
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
GM11859 CFTR 2789+5G>A Yes 2789+5G>A Yes
GM11860 CFTR IVS19DS, +10 KB, C>T Yes
IVS19DS, +10 KB, C>T Yes
(3849+10kbC>T) (3849+10kbC>T)
GM12444 CFTR IVS10AS, G>A, -1 Yes
(1717-1G>A)
GM12585 CFTR ARG1162TER Yes
GM12785 CFTR ARG347PRO Yes GLY551ASP Yes
GM12960 CFTR ARG334TRP Yes c.3368-2A>T Yes
GM13423 CFTR G85E Yes D1152H Yes
GM13591 CFTR ARG117HIS Yes PHE508DEL Yes
GM18668 CFTR CFTRde1e2,3 N/A PHE508DEL Yes
GM18799 CFTR 2184de1A Yes PHE508DEL Yes
GM18800 CFTR 1898+1 G>A Yes PHE508DEL Yes
GM18802 CFTR Y122X Yes R1158X Yes
GM18886 CFTR 2143de1T Yes PHE508DEL Yes
GM20737 CFTR R347H Yes
GM20741 CFTR 3876de1A Yes
GM20745 CFTR 5549N Yes
GM20924 CFTR R75X Yes
GM21080 CFTR 394deITT Yes
GM11468 G6PC R83C Yes Q347X Yes
GM00502 HEXA 1278insTATC Yes 1421+1G>C Yes
GM03461 HEXA 1421+1G>C Yes G2695 Yes
GM05042 IKBKAP 2507+6T>C Yes 2507+6T>C Yes
GM02533 MCOLN1 IV53-2A>G Yes del exl-ex7 N/A
GM03252 SMPD1 L302P Yes
141
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
GM13205 SMPD1 fsP330 Yes- -
GM16193 SMPD1 R496L Yes Arg608DEL Yes
GM19116 CFTR ARG334TRP Yes- -
GM17363 IKBKAP 2507+6T>C Yes- -
GM17366 IKBKAP 2507+6T>C Yes- -
GM17365 IKBKAP 2507+6T>C Yes- -
GM17364 IKBKAP 2507+6T>C Yes- -
GM17360 MCOLN1 IVS3-2A>G Yes- -
GM17362 HEXA 1278insTATC Yes- -
GM18015 HEXA c.739C>T Yes- -
GM17362 HEXA G2695 Yes- -
Genotyping by Assembly - Templated Alignment
Although substitutions comprise the majority of coding variation in the human
genome,
insertions and deletions (indels) are often clinically relevant. Indels,
especially when large or
present in cis with substitutions, are notoriously difficult to detect with
short NGS reads.
Assembly of short reads can improve indel detection sensitivity, but this is
often at the cost of
decreased SNV and indel specificity due to the presence of spurious contiguous
sequence
(contigs). An algorithm was devised termed Genotyping by Assembly-Templated
Alignment
(GATA), that first forms an assembly from reads partitioned into subsets by
targeting arm
sequence, then performs base quality- and coverage- informed genotyping by
alignment of raw
reads back to the assembled contigs (Figures 18A-18E).
Figures 18A-E depicts the next-generation DNA sequencing workflow. Genomic DNA
samples are input to a molecular inversion probe capture reaction. Each target
(depicted bgrey
and black regions) is captured by multiple probes that anneal to non-
overlapping genomic
intervals. PCR is performed using primers containing patient-specific
barcodes, yielding
barcode libraries. Equal volumes of the libraries are pooled and enter
Illumina's Hiseq high-
throughput sequencing workflow as shown in FIG. 18B. Following sequencing,
reads enter
either the alignment only (AO, left) as depicted in FIG. 18C or Genotyping by
Assembly-
142
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
Templated Alignment (GATA, right) analysis pipeline as depicted in FIG. 18D.
As shown in
FIG. 18C, AO first partitions reads by sample molecular barcode, then in
parallel for all samples
performs short read alignment, base quality recalibration, realignment around
putative indels,
and genotyping. As shown in FIGS. 18D-E, GATA partitions reads first by sample
molecular
barcode, then by target. Reads are assembled into contigs that are then
aligned to the reference
genome. Raw reads are then aligned to the contigs, and raw read mapping and
variant
information relative to the reference is determined using reference-contig and
read-contig
alignments. Finally, base quality score recalibration and genotyping are
performed on the
mapped, raw reads.
The performance of GATA for indel genotyping was compared to the more
conventional
genotyping-by-alignment only (AO) algorithm used in the Sanger concordance
studies. Across a
set of 147 samples analyzed, both indel sensitivity and specificity were
increased with GATA
relative to AO (Table 9). GATA detected 23 unique insertions and deletions,
which were
confirmed by manual review of Sanger traces. Of these, 9 (39%) were not
detected by AO in one
or more samples, including BLM c.2207_2212de1insTAGATTC¨the most common
disease-
causing mutation for Bloom syndrome in people of Ashkenazi Jewish descent¨as
well as
several alleles in SMPD1 (Table 10), the gene associated with Niemann-Pick
disease (FigureS
19A-D). Performance for substitutions was identical for both detection methods
(AO and
GATA).
Table 9 shows genotyping by assembly-templated alignment (GATA) improves
detection
of insertions and deletions. Raw variant alleles (positive calls) from 147
samples were filtered by
depth and strand bias and categorized according to NGS data analysis method,
alignment only
(AO) or GATA. Calls were classified with GATA considered truth as true
positive (TP), false
positive (FP), and false negative (FN). Discordant calls, in all cases, were
confirmed by manual
review of corresponding Sanger traces and found to be GATA TP or TN, rather
than FP or FN.
Variant calls flagged as low-confidence are considered uncalled. Polymorphisms
in the first
exon of SMPD1 accounted for the majority of uncalled and discordant alleles,
which were not
considered in accuracy calculations.
143
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
Table 9
AO GATA
TP 104 211
FP 28 0
FN 47 0
Uncalled* 70 10
Sensitivity 0.696 1.0
Precision 0.786 1.0
Table 10 shows the frequency distribution of variant genotypes for the STR at
SMPD1
exon 1 representing various combinations of (i) the minor reference allele
(0), (ii) a substitution
(snp),(iii) insertions (+6 and +12bp in length), and (iv) deletions (-6, -12,
and -18bp in length) as
determined by GATA and confirmed by manual inspection of Sanger traces.
Table 10
Genotype Frequency
snp/snp 42
-6/snp 41
-6/-6 15
-12/0 8
snp/O 8
-6/0 7
-6/-12 7
-12/snp 4
-18/0 3
+12/snp 2
144
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
+6/snp 2
+6/0 1
-1 2/-1 2 1
As seen in FIGS. 19A-D, GATA correctly genotypes insertions and deletions that
are
undetectable by the Alignment Only method. Read from top to bottom, each
figure provides
tracks for cumulative depth of coverage (vertical grey bars); representative
MIP alignments
(horizontal grey bars) with mismatches (letters), and gaps (dashed lines);
chromatogram;
reference DNA and amino acid sequence for FIG. 19A heterozygous BLM
c.2207_2212de1insTAGATTC in sample GM04408 as well as several alleles in the
first exon of
SMPD1 including FIG. 19B a heterozygous 18 bp deletion in sample GM20342
(minus strand),
FIG. 19C a heterozygous 12 bp insertion and homozygous substitution in sample
GM17282 (plus
strand), and FIG. 19D compound heterozygous 6 and 12 bp deletions in sample
GM00502
(minus strand). Chromatogram trace offsets corresponding to specific
heterozygous insertion and
deletion patterns are indicated with slanted lines color coded by reference
base. For clarity
offsets are shown for FIG. 19C and FIG. 19D only.
Simulation to Assess Detectability of Rare Pathogenic Mutations
While detectability for all disease-causing mutations present in the sample
set was
empirically demostrated, there exist a number of disease-causing mutations for
which samples
cannot be readily obtained. To assess whether the NGS workflow can detect
these additional
mutations, the stimulations were performed in silico. Since detectability can
be affected by any
element of the workflow, a simulator was implemented that employed read sets
from actual
samples rather than model reads derived from the reference genome at uniform
coverage. This
allowed for realistic representation of target abundance distribution,
neighboring in cis variants,
as well as cycle- and context-dependent sequencing errors. Disease-causing
variants were
introduced into raw reads by a Bernoulli process, with an average 0.5
probability of introducing
the lesion, to simulate the heterozygous genotypes carrier screening aims to
detect.
A total of 81 heterozygous variants were simulated in a read set of at least
144 samples
with the exception of c.1521_1523de1CTT (F508del), the most common disease-
causing
mutation for cystic fibrosis in Caucasian populations, as shown in Table 11.
This mutation was
145
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
present in several samples, which were removed from simulation analysis
(Materials and
Methods). Of the simulated variants 67 (83%) were correctly genotyped in all
(generally
145/145) samples and only four relatively large (>7 bp) deletions were
undetected in one or more
samples. High-confidence genotype calls were not made for the remaining 10
variants. No
variants were found to be undetectable in all samples. Table 11 gives the
performance results of
GATA for detecting clinically-relevant mutations by simulation.
Table 11
Variant Samples Variant Variant Variant
Simulated Positive Uncalled Negative
BLM c.2207_2212delinsTAGATTC 146 146 0 0
CFTR c.1923_1931deICTCAAAACTinsA 147 147 0 0
CFTR c.1973_1985de113insAGAAA 146 146 0 0
CFTR 147 147 0 0
c.723_743+1delGAGAATGATGATGAAGTACAGG
(SEQ ID NO: 6325)
CFTR c.3067_3072delATAGTG 147 147 0 0
CFTR_c.650_659delAGTTGTTACA (SEQ ID NO: 145 145 0 0
6326)
CFTR_c.1871_1878deIGCTATTTT 145 145 0 0
CFTR_c.739_742dupTACA 145 145 0 0
CFTR_c.578_579+5delAAGTATG 145 145 0 0
CFTR_c.3421_3424dupAGTA 145 145 0 0
BLM_c.991_995de15 145 145 0 0
CFTR_c.2589_2599delAA1TTGGTGCT (SEQ ID NO: 145 46 7 92
6327)
CFTR_c.3664_3665insTCAA 145 145 0 0
CFTR_c.2634_2641deIGGTTGTGC 145 143 1 1
146
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
CFTR_c.156_163dupATTGGAAA 145 145 0 0
CFTR_c.522_526delAATAA 145 145 0 0
ABCC8_c.259_268de110 145 141 3 1
CFTR_c.1616_1617dupTA 145 145 0 0
CFTR_c.3068_3072delTAGTG 145 145 0 0
FANCC_c.356_360de15 145 145 0 0
CFTR_c.861_865deICTTAA 145 145 0 0
ABCC8_c.2835_2838delGAGA 145 145 0 0
CFTR_c.319_326deIGCTTCCTA 145 145 0 0
CFTR_c.2249_2256de18 145 145 0 0
CFTR_c.1792_1798delAAAACTA 145 145 0 0
CFTR_c.2241_2248delGATACTGC 145 145 0 0
G6PC_c.462_466deITTTGT 145 145 0 0
CFTR_c.35_36insTATCA 145 145 0 0
HEXA_c.1471_1475deITCTGA 145 145 0 0
PCDH15_c.996_999deIGGAT 145 145 0 0
ASPA_c.568_574de17 145 144 0 1
CFTR_c.3184_3188dupCTATG 145 145 0 0
SMPD1_c.1657_1663delACCGCCT 145 145 0 0
CFTR_c.1162_1168delACGACTA 145 145 0 0
BCKDHB_c.163_166dupACTT 145 145 0 0
BCKDHA_c.861_868delAGGCCCCG 145 145 0 0
CFTR_c.3773dupT 145 145 0 0
CFTR_c.1155_1156dupTA 145 145 0 0
147
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
CFTR_c.3889dupT 145 145 0 0
HEXA_c.1274_1277dupTATC 145 145 0 0
CFTR_c.262_263deITT 144 144 0 0
CFTR_c.326_327delAT 145 145 0 0
CFTR_c.3691delT 145 145 0 0
CFTR_c.3528deIC 144 144 0 0
BLM_c.2407dupT 145 145 0 0
CFTR_c.1521_1523deICTT 131 131 0 0
HEXA_c.915_917deICTT 145 145 0 0
G6PC_c.379_380dupTA 145 145 0 0
CFTR_c.2012delT 144 144 0 0
SMPD1_c.1829_1831deIGCC 144 144 0 0
CFTR_c.1029deIC 145 127 18 0
CFTR_c.2737_2738insG 145 145 0 0
CFTR_c.2947_2948deITT 145 142 3 0
CFTR_c.1911deIG 145 145 0 0
CFTR_c.803delA 145 145 0 0
CFTR_c.1519_1521delATC 145 145 0 0
CFTR_c.805_806delAT 145 18 127 0
CFTR_c.2215deIG 145 137 8 0
FANCC_c.67deIG 145 145 0 0
CFTR_c.935_937deITCT 145 145 0 0
CFTR_c.2175dupA 145 145 0 0
CFTR_c.3530delA 145 145 0 0
148
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
CFTR_c.531delT 145 145 0 0
CFTR_c.1021_1022dupTC 145 127 18 0
CFTR_c.3659deIC 145 145 0 0
DLD_c.104dupA 144 144 0 0
CFTR_c.2052dupA 144 144 0 0
CFTR_c.313delA 145 145 0 0
G6PC_c.79deIC 145 145 0 0
CFTR_c.442delA 145 145 0 0
CFTR_c.1477_1478deICA 145 145 0 0
CFTR_c.1545_1546delTA 145 145 0 0
BCKDHA_c.117deIC 145 145 0 0
CFTR_c.1418deIG 145 145 0 0
CFTR_c.1976delA 145 145 0 0
CFTR_c.3536_3539delCCAA 145 145 0 0
CFTR_c.948delT 145 145 0 0
CFTR_c.2052delA 145 145 0 0
BCKDHB_c.595_596delAG 145 145 0 0
G6PC_c.980_982deITCT 145 145 0 0
CFTR_c.3039deIC 145 145 0 0
Discussion
Robustness, completeness, and accuracy are three of the main factors that
define the
utility of a genetic carrier testing workflow in a clinical laboratory. By
utilizing a target
enrichment methodology that is performed in a single tube and requires no
mechanical
shearing or purifications of individual samples, methods of the invention
provide an automated
NGS workflow that yields highly-reproducible results across samples and
operators. This
149
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
reproducibility ensures that samples will not have to be rerun frequently,
minimizing both
turnaround time and per-sample cost.
Because each clinically meaningful basepair must be sequenced before an
actionable
medical report can be generated, a high level of completeness minimizes the
amount of
costly re-work necessary for a sample. Methods of the invention demonstrate
completeness that
is consistent with low to no re-work for the samples studied, and
substantially better than other
previously-reported methods using multiplex target capture or PCR with NGS.
This
improvement is likely the result of a number of optimizations we have made
relative to previous
reports including the use of a tiling MIP design that ensures multiple probes
capture each base
and the use of a DNA extraction protocol that effortlessly shears the DNA to a
lower molecular
mass.
Regarding accuracy, the only observed SNV false negative was in a sample that
exhibited skewed allele ratios along the chromosome, which should not commonly
occur
when testing for germline mutations in clinical specimens derived from whole
blood. Additionally, the SNV false positive rate of approximately 1.1 per
million
basepairs corresponds to a low confirmation burden for clinical testing and
surpasses
values previously reported. Given the small target set and the rare nature of
indels, it is
difficult to give a precise measurement of our accuracy for indels in genera.
However, this data
suggests that the use of GATA substantially improves our ability to detect
small
lesions. Additionally, a sensitivity of 100% by both AO and GATA was observed
across
the set of disease-causing insertions and deletions in carrier and affected
samples.
It is worth noting that measuring accuracy to a sufficient level of precision
and generality
can be challenging within conserved coding regions because selective pressure
limits the
spectrum of variation present. While a large number of samples were sequenced,
the
relatively small size of our target limited the number of unique alleles
observable and
meant that approximately 90% of such variants were common (i.e. present in
dbSNP). Nonetheless, there is no a priori reason to believe that the measured
accuracy
will not generalize to other rare and private mutations present in the
targeted
loci. Supporting this point, these simulations using real data and controlled
for sample-to-sample
150
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
variability indicate that one can detect a number of very rare disease causing
alleles of different
types and sequence contexts, including insertions (up to 12bp),deletions (up
to 22bp) and
complex combinations thereof.
The reference standard one considers ground truth can impose a ceiling on
measurable
accuracy. Automated analysis of what is widely deemed the 'gold standard' for
DNA sequencing
was employed: bi-directional Sanger traces derived from PCR amplicons. FIGS.
20A-20B shows
NGS detects allele dropout in Sanger sequencing reactions. FIG. 20A-1, FIG.20A-
2, and FIG.
20A-3 show dropout of reference allele leads to homozygous non-reference call
by Sanger
sequencing, but heterozygous non-reference call by NGS, in BLM exon 12 of
GM18034. Shown
from top to bottom, (FIG. 20A-1) original PCR primer pair: expected reference
sequence trace,
sample forward trace, sample reverse trace; (FIG. 20A-2) re-designed PCR
primer pair: expected
reference sequence trace, sample forward trace, sample reverse trace; IGV of
NGS data.
FIG. 20B-1, FIG.20B-2, and FIG. 20B-3 shows dropout of non-reference allele
leads to
homozygous reference call by Sanger sequencing, but heterozygous non-reference
call by NGS,
in DLD exon 9 of sample GM11370. Shown from top to bottom, expected reference
sequence
trace, sample forward trace, sample reverse trace. Shown from top to bottom,
(FIG. 20B-1)
original PCR primer pair: expected reference sequence trace, sample forward
trace, sample
reverse trace; (FIG. 20B-2) re-designed PCR primer pair: expected reference
sequence trace,
sample forward trace, sample reverse trace; IGV of NGS data. Project
genotyping data was
employed, 12 NGS false negatives and 7 false positives would have been
observed in the subset
of samples characterized by this approach.Because these were all shown by
Sanger analysis to be
HapMap Project genotyping errors, this would have underestimated both
sensitivity and
specificity.
The NGS workflow detected allele dropout in the Sanger data, a known
limitation of that
technology (FIGS. 20A-1 through 20B-3) and not surprising since each base
sequenced by NGS
was captured by multiple probes with independent targeting arms. Had the less
laborious and
more commonly-used reference of Hapmap Project genotyping data been employed,
12 NGS
false negatives and 7 false positives would have been observed in the subset
of samples
characterized by this approach (Table 12). This would have underestimated both
sensitivity and
specificity.
151
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
Table 12 shows concordance of NGS genotypes with HapMap data. All NGS
positions
called with high confidence (minimum 50x coverage and strand bias <= 0) that
intersected
Hapmap release 27 phase II+III genotyping data were evaluated, for a total of
5,337 genotypes
across 83 samples. True negative: reference called by both NGS and HapMap;
true positive: non-
reference (heterozygous or homozygous) called by both NGS and HapMap; false
positive: non-
reference called by NGS, reference called by HapMap; false negative: reference
called by NGS,
non-reference called by HapMap. Specificity: TN / (TN + FP); sensitivity: TP /
(TP + FN).
Table 12
True negatives 4,233
True positives 1,085
False positives 7
False negatives 12
Specificity 0.998
Sensitivity 0.989
Indel detection methods that only employ gapped alignment of short reads to
reference
are often limited by false positives introduced by systematic, context-
dependent
sequencing error, and false negatives introduced by failure of the aligner to
open or
extend gaps. An assembly-based paradigm would address these limitations but
raw
contigs do not always carry base quality and coverage information. The GATA
algorithm combines these approaches to deliver sensitive and specific indel
detection
with SNV performance on par with a traditional alignment-only pipeline.
Many alleles detected exclusively by GATA were from a short tandem repeat
(STR)
region encoding the N-terminal signal peptide in SMPD1 (Table 10). Consistent
with
previous reports, GATA detected non-reference alleles in 96% of samples, a
rate that is
strikingly high because hg18 contains a minor allele that is frequently
substituted
(V36A). While common hexanucleotide indels at this locus are clinically
benign, any
152
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
pathogenic mutation present in cis would likely be missed using a conventional
approach
for variant detection. Indeed, when reads were aligned independently, several
genomic
positions in this region consistently fell below our specified coverage
threshold. GATA
therefore should yield higher sensitivity for rare mutations linked to
polymorphisms in
the first exon of SPMD1 and potentially other STR loci as well.
The simulation methodology applied here attempts to assess detectability of
rare
pathogenic mutations in a highly realistic manner. Simply deriving reads from
a
reference genome modified to include the mutation of interest can overestimate
the
detection probability because of real-world factors that would otherwise
render the
mutation undetectable. Additionally, we are able to determine whether a
mutation is
sometimes, rather than always or never, detectable because it is simulated in
the read sets
of hundreds of samples; e.g., this could occur in a particular genetic
background with a
low-frequency in cis variant that interferes with alignment of reads
containing the
mutation. Nonetheless, certain mutation types, in particular large deletions
are still not
amenable to this paradigm because they could fundamentally alter the
distribution of
reads generated across the relevant region. In these cases, either human
samples or
synthetic templates remain the only way to assess detectability.
In conclusion, an automated, integrated workflow that converts human genomic
DNA isolated from blood or cell lines into clinically-relevant variant calls
was presented by this
example. High genotype concordance was achieved with conventional
electrophoretic
sequencing across a set of 15 genes. In addition, this example demonstrates
the ability to detect
a range of important disease-causing mutations. The pipeline analysis
presented allows for
sensitive and specific detection of indels, while simultaneously incorporating
raw base quality
and coverage into SNV genotype calls. Realistic simulation on actual run data
indicates that a
number of pathogenic mutations undetectable by a traditional alignment-based
genotyping
approach are accessible by GATA. Collectively, the data shows that this
workflow has met three
of the major requirements of a clinical carrier screening assay, supporting
the notion that NGS is
ready for clinical use.
It should be appreciated that the preceding examples are non-limiting and
aspects of the
invention may be implemented as described herein using alternative techniques
and/or protocols
that are available to one or ordinary skill in the art.
153
CA 02907177 2015-09-15
WO 2014/143994
PCT/US2014/028212
It will be clear that the methods may be practiced other than as particularly
described in
the foregoing description and examples. Numerous modifications and variations
of the present
disclosure are possible in light of the above teachings and, therefore, are
within the scope of the
claims. Preferred features of each aspect of the disclosure are as for each of
the other aspects
mutatis mutandis. The documents including patents, patent applications,
journal articles, or other
disclosures mentioned herein are hereby incorporated by reference in their
entirety. In the event
of conflict, the disclosure of present application controls, other than in the
event of clear error.
154