Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
BISPECIFIC ANTIBODIES AGAINST CD3EPSILON AND ROR1
The present invention relates to novel bispecific antibodies against CDR and
ROR1, their manufacture
and use.
Background of the Invention
ROR1 (synonyms: tyrosine-protein kinase transmembrane receptor ROR1,
EC=2.7.10.1, neurotrophic
tyrosine kinase, receptor-related 1, UniProtKB Q01973) is a tyrosine-protein
kinase receptor. The
receptor is described in Masiakowski P., Carroll R.D., J. Biol. Chem.
267:26181-26190(1992) "A novel
family of cell surface receptors with tyrosine kinase-like domain." W09218149
and W09527060
mention ROR-1 as Rtk-2 and antibodies against ROR-1. W02002087618 mentions a
method of
controlling the growth and differentiation of cancer by selectively inhibiting
a growth factor receptor.
Such a receptor would be Ron l or Ror2. W02005100605 mentions ROR1 as a
therapeutic target for
breast cancer and anti ROR1 antibodies which specifically bind to ROR1, to the
extracellular region of
ROR1 (M1-V406) and ROR1 fragments Q73-V139, E165-1299, K312-C391. W02007051077
relates to
an anti-ROR1 antibody and its use in lymphoma cell detection. W02008103849
also mentions anti-
ROR1 antibodies. Rabbani H. et al.,Blood (ASH Annual Meeting Abstracts) 2010
116: Abstract 916)
discloses the use of anti ROR1 antibodies for the treatment of chronic
Lymphocytic leukemia (CLL).
Rabbani used anti-ROR1 an antibody against the extracellular domain, an
antibody against the CRD
region (ligand binding site for Wnt proteins) and an antibody against the
kringle domain. Daneshmanesh
AH et al., Int. J. Cancer, 123 (2008) 1190-1195 relates to an anti ROR1
antibody that binds to the
extracellular domain fragment WNISSELNKDSYLTL (SEQ ID NO:18) and an anti ROR1
antibody that
binds to the intracellular domain fragment KSQKPYKIDSKQAS (SEQ ID NO:20). Also
the use of such
antibodies for the treatment of CLL is mentioned.
W02011159847 relates to an anti-ROR1 antibody as a conjugate with a
biologically active molecule for
the treatment of ROR1 cancer like lymphoma or adenocarcinoma. W02008076868,
W02008103849,
W0201008069, W02010124188, W02011079902, W02011054007, W02011159847,
W02012076066,
W02012076727, WO 2012045085, and W02012097313 relate also to ROR1 binding
molecules or anti
ROR1 antibodies. W02012075158 relates to an anti-ROR1 antibody comprising as
light chain variable
domain (VL) the sequence of SEQ ID NO:2 and as variable heavy chain domain
(VH) the sequence of
SEQ ID NO:6, and as respective CDRs the sequences of SEQ ID NO: 3, 4, 5, 7, 8,
9. This antibody is
further named as MAB 1 . W0201209731 relates to an anti-ROR1 antibody
comprising as light chain
variable domain (VL) the sequence of SEQ ID NO:45 and as variable heavy chain
domain (VH) the
sequence of SEQ ID NO:42. This antibody is further named as MAB2. W02012075158
relates to an anti-
ROR1 antibody comprising as light chain variable domain (VL) the sequence of
SEQ ID NO:46 and as
variable heavy chain domain (VH) the sequence of SEQ ID NO:43. This antibody
is further named as
MAB3. W02012075158 relates to an anti-ROR1 antibody comprising as light chain
variable domain
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
2
(VL) the sequence of SEQ ID NO:47 and as variable heavy chain domain (VH) the
sequence of SEQ ID
NO:44. This antibody is further named as MAB4.
W02005040413 is directed to a screening method for the identification and/or
validation of inhibitors of
a receptor tyrosine kinase activity, including ROR1.
W02008036449, W02011014659 and W02011050262 mention bispecific antibodies
wherein one target
can be ROR1. W02007146968 mention multivalent single-chain binding proteins
with effector function
and ROR1 and CD3 are mentioned as possible targets. W02011054007 is directed
to a method of
treatment of cancer administering an affinity reagent which binds to the
extracellular domain of ROR1.
Bispecific antibodies with CD3 are also mentioned. W02014031174 mentions
bispecific antibodies
which are specific to two different epitopes of ROR1.The preferred antibody
D10 strongly internalizes at
37 C in MDA MB 231 epithelial breast adenocarcinoma. Yang and Baskar PLos ONE
6(2011) e21018,
like W02012075158, mention also anti-ROR1 antibody R12. Rebagay R. et al.,
Frontiers in Oncology
(2012) 7, Article 34, 1-8 mention that RORs are pharmaceutical targets and a
means to deliver cytotoxic
agents in the cells which express the target on the cell surface. Rebagay also
mention bispecific
antibodies such as BiTE. Strong internalization is favorable for armed
antibodies i.e. antibody drug
conjugates according to Rebagay. D. Mezzanzanica D et al., Int. Journal of
Cancer, 41(1988) 609-615
investigated a therapeutic approach by retargeting CTLs by a bispecific
antibody consisting of MOvl 8 (a
poorly internalizing folate receptor alpha antibody specific for human ovarian
carcinoma cells) and an
anti-CD3 antibody (OKT3 or TR66). Hudecek M et al., Blood, 116 (2010), 4532-
4541, mention that
ROR1 is expressed by B cell chronic lymphocytic leukemia (B-CLL) and mantle
cell lymphoma (MCL).
Such cells can be targeted by activated CD8+ T cells transfected with, and
expressing scFv from murine
anti-ROR1 antibody 2A2. Such cells are useful for treatment of B cell
malignancies. Baskar S. et al.,
mAbs 4:3 (2012) 349-361 relate to the targeting of malignant B cells with an
immunotoxin BT-1
comprising scFv 2A2 anti-ROR1 conjugated to PE38 toxin. The immunotoxin is
partially internalized
and induces apoptosis.
The TCR/CD3 complex of T-lymphocytes consists of either a TCR alpha (a)/beta
(I3) or TCR gamma
(y)/delta (6) heterodimer coexpressed at the cell surface with the invariant
subunits of CD3 labeled
gamma (-y), delta (6), epsilon (8), zeta p, and eta (i). Human CDR is
described under UniProt P07766
(CD3E_HUMAN). An anti CD3c antibody described in the state of the art is 5P34
(Yang SJ, The Journal
of Immunology (1986) 137; 1097-1100). 5P34 reacts with both primate and human
CD3. 5P34 is
available from PharMingenTm (BD Biosciences). A further anti CD3 antibody
described in the state of the
art is UCHT-1 (see W02000041474). A further anti CD3 antibody described in the
state of the art is BC-
3 (Fred Hutchinson Cancer Research Institute; used in Phase I/II trials of
GvHD, Anasetti C et al.,
Transplantation 54: 844 (1992)).
A wide variety of recombinant bispecific antibody formats have been developed
in the recent past, e.g. by
fusion of, e.g. an IgG antibody format and single chain domains (see
Kontermann RE, mAbs 4:2, (2012)
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
3
1-16). Bispecific antibodies wherein the variable domains VL and VH or the
constant domains CL and
CH1 are replaced by each other are described in W02009080251 and W02009080252.
An approach to circumvent the problem of mispaired byproducts, which is known
as 'knobs-into-holes',
aims at forcing the pairing of two different antibody heavy chains by
introducing mutations into the CH3
domains to modify the contact interface. On one chain bulky amino acids were
replaced by amino acids
with short side chains to create a 'hole'. Conversely, amino acids with large
side chains were introduced
into the other CH3 domain, to create a 'knob'. By coexpressing these two heavy
chains (and two identical
light chains, which have to be appropriate for both heavy chains), high yields
of heterodimer formation
('knob-hole') versus homodimer formation ('hole-hole' or 'knob-knob') was
observed (Ridgway JB, Presta
LG, Carter P; and W01996027011). The percentage of heterodimer could be
further increased by
remodeling the interaction surfaces of the two CH3 domains using a phage
display approach and the
introduction of a disulfide bridge to stabilize the heterodimers (Merchant AM,
et al, Nature Biotech 16
(1998) 677-681; ATwell S, Ridgway JB, Wells JA, Carter P., J Mol Biol 270
(1997) 26-35). New
approaches for the knobs-into-holes technology are described in e.g. in EP
1870459A1. Although this
format appears very attractive, no data describing progression towards the
clinic are currently available.
One important constraint of this strategy is that the light chains of the two
parent antibodies have to be
identical to prevent mispairing and formation of inactive molecules. Thus this
technique is not
appropriate for easily developing recombinant, bispecific antibodies against
two targets starting from two
antibodies against the first and the second target, as either the heavy chains
of these antibodies and/or the
identical light chains have to be optimized. Xie, Z., et al, J Immunol Methods
286 (2005) 95-101 refers to
a format of bispecific antibody using scFvs in combination with knobs-into-
holes technology for the FC
part. W02012116927 and W02010145792 mention exchanging the CH1 and CL domains.
W02009080254 mentions knob in hole constructs for producing bispecific
antibodies. WO 2006093794
relates to heterodimeric protein binding compositions. W0199937791 describes
multipurpose antibody
derivatives. Morrison, S.L., et al., J. Immunol. 160 (1998) 2802-2808 refers
to the influence of variable
region domain exchange on the functional properties of IgG.
WO 201302362 relate to heterodimerized polypeptides. W0201312733 relates to
polypeptides
comprising heterodimeric Fc regions. W02012131555 relates to engineered
heterodimeric
immunoglobulins. EP 2647707 relates to engineered hetero-dimeric
immunoglobulins. W02009080251,
WO 2009080252, WO 2009080253, WO 2009080254 and Schaefer, W. et al, PNAS, 108
(2011) 11187-
1191 relate to bivalent, bispecific IgG antibodies with a domain crossover.
The multispecific antibodies
with VH/VL replacement/exchange in one binding to prevent light chain
mispairing (CrossMabVH-VL)
which are described in W02009080252, (see also Schaefer, W. et al, PNAS, 108
(2011) 11187-1191)
clearly reduce the byproducts caused by the mismatch of a light chain against
a first antigen with the
wrong heavy chain against the second antigen (compared to approaches without
such domain exchange).
However their preparation is not completely free of side products. The main
side product is based on a
B ence-Jones-typ e interaction (Schaefer, W. et al, PNAS, 108 (2011) 11187-
1191).
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
4
Accordingly there is a need for bispecific antibodies against CDR and ROR1
with VH/VL exchange
which can be produced in high yield and easily purified.
Ovarian cancer is the leading cause of death from gynecologic cancer in the
United States and the
seventh most common cancer and the eighth most common cause of death from
cancers in women. An
estimated 21,980 new cases of ovarian cancer and 14,270 deaths related to
ovarian cancers are expected
in the United States in 2014. Worldwide, nearly 225,000 women will be
diagnosed with ovarian cancer,
and more than 140,000 will die of the disease (Cancer Facts & Figures 2014;
http://www.cancer.org). The
incidence of ovarian cancer increases with age and is most prevalent in the
eighth decade of life. About
half of the women diagnosed with ovarian cancer are 63 years or older..
Ovarian cancer usually has a
relatively poor prognosis. If diagnosed at the localized stage, the 5-year
survival rate is 92%, however,
only 15% of all cases are detected at this stage. The majority of cases (61%)
are diagnosed after the
disease has already metastasized. For women diagnosed with distant metastases,
the 5-year survival rate
is 27%. Despite advances in surgery and chemotherapy over the past two
decades, only modest progress
has been achieved in improving the overall survival in patients with ovarian
cancer. Although the
majority of women with advanced ovarian cancer respond to first-line
chemotherapy, most responses are
not durable. More than 80% of patients will have a recurrence of their disease
after first-line treatment,
and more than 50% will die of recurrent disease within 5 years of diagnosis
(http://www.cancerresearch.org). Targeted therapy is a newer type of cancer
treatment that uses drugs or
other substances to identify and attack cancer cells while doing little damage
to normal cells. The targeted
therapy drug that has been studied the most in ovarian cancer is bevacizumab
(Avastin0). In studies,
bevacizumab has been shown to shrink or slow the growth of advanced ovarian
cancers. Trials to see if
bevacizumab works even better when given along with chemotherapy have shown
good results in terms
of shrinking (or stopping the growth of) tumors, but it has not yet been shown
to help women live longer
(http://www.cancer.org/cancer/ovariancancer). W02007146957 relate to
inhibiting tumor growth with
ROR1-antagonizing agents. Such agents can be e.g. antibodies against ROR1 and
identified by screening
with tumor cell lines. As such screening tumor cell lines ovarian cancer cell
lines are mentioned.
U58212009 relates to a method for treating a ROR1 related cancer in a subject.
The ROR1 related cancer
are among others also ovarian cancer. W02011054007 relates to a method for the
treatment of ovarian
cancer comprising administering an antibody against ROR1. Accordingly there is
a need for a ROR1
based agent for the treatment of ovarian cancer.
Summary of the Invention
The invention relates to a bispecific antibody specifically binding to the two
targets human CDR (further
named also as "CD3") and the extracellular domain of human ROR1 (further named
also as "ROR1").
The invention relates to a bispecific bi- or trivalent antibody specifically
binding to CD3 and ROR1,
wherein the variable domains VL and VH in a light chain and the respective
heavy chain are replaced by
each other, characterized in comprising a constant domain CL wherein the amino
acid at position 124 is
substituted independently by lysine (K), arginine (R) or histidine (H)
(numbering according to Kabat),
CA 02963692 2017-04-05
WO 2016/055592
PCT/EP2015/073308
and in the respective constant domain CH1 the amino acid at position 147 and
the amino acid at position
213 is substituted independently by glutamic acid (E), or aspartic acid (D)
(numbering according to
Kabat). Preferably the antibody is monovalent for CD3 binding. Preferably in
addition to the amino acid
replacement at position 124 in the constant domain CL the amino acid at
position 123 is substituted
5 independently by lysine (K), arginine (R) or histidine (H). Preferably the
antibody is monovalent for CD3
binding and amino acid 124 is K, amino acid 147 is E, amino acid 213 is E, and
amino acid 123 is R or K.
Preferably amino acid 123 is R for a kappa light chain and K for a lambda
light chain.
The invention relates to a bispecific antibody specifically binding to CD3 and
ROR1, characterized in
comprising
a) the first light chain and the first heavy chain of a first antibody
which specifically binds to ROR1;
and
b) the second light chain and the second heavy chain of a second antibody
which specifically binds
to CD3, and wherein the variable domains VL and VH in the second light chain
and second heavy chain
of the second antibody are replaced by each other; and
c) wherein in the constant domain CL of the first light chain under a) the
amino acid at position 124
is substituted independently by lysine (K), arginine (R) or histidine (H)
(numbering according to Kabat),
and wherein in the constant domain CH1 of the first heavy chain under a) the
amino acid at position 147
and the amino acid at position 213 is substituted independently by glutamic
acid (E), or aspartic acid (D)
(numbering according to Kabat) (see e.g. Figures 1A, 1C, 1F, 1H, 1J).
Preferably said bispecific antibody described in the last preceding paragraph
is further characterized in
that said bispecific antibody comprises in addition a Fab fragment of said
first antibody (further named
also as "ROR1-Fab") and in the constant domain CL said ROR1-Fab the amino acid
at position 124 is
substituted independently by lysine (K), arginine (R) or histidine (H)
(numbering according to Kabat),
and wherein in the constant domain CH1 of said ROR1-Fab the amino acid at
positions 147 and the
amino acid at position 213 is substituted independently by glutamic acid (E),
or aspartic acid (D)
(numbering according to Kabat) (see e.g. Figures 1C, 1F).
The invention further relates to a bispecific antibody specifically binding to
CD3 and ROR1,
characterized in comprising
a) the first light chain and the first heavy chain of a first antibody
which specifically binds to ROR1;
and
b) the second light chain and the second heavy chain of a second antibody
which specifically binds
to CD3, and wherein the variable domains VL and VH in the second light chain
and second heavy chain
of the second antibody are replaced by each other; and wherein
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
6
c) in the constant domain CL of the second light chain under b) the amino acid
at position 124 is
substituted independently by lysine (K), arginine (R) or histidine (H)
(numbering according to Kabat),
and wherein in the constant domain CH1 of the second heavy chain under b) the
amino acid at positions
147 and the amino acid at position 213 is substituted independently by
glutamic acid (E), or aspartic acid
(D) (numbering according to Kabat) (see e.g. Figures 1B, 1D, 1G, 11, 1K).
Preferably said bispecific antibody described in the last preceding paragraph
is further characterized in
that said bispecific antibody comprises in addition a second Fab fragment of
said first antibody ("ROR1-
Fab") (see e.g. Figure 1D, 1G).
Amino acid numbering is according to Kabat (see below).
Preferably in addition to the amino acid replacement at position 124 in the
constant domain CL of the first
or second light chain the amino acid at position 123 is substituted
independently by lysine (K), arginine
(R) or histidine (H).
Preferably in the constant domain CL the amino acid at position 124 is
substituted by lysine (K), in the
constant domain CH1 the amino acid at position 147 and the amino acid at
position 213 are substituted by
glutamic acid (E). Preferably in addition in the constant domain CL in the
amino acid at position 123 is
substituted by arginine (R).
Preferably in the constant domain CL the amino acid at position 124 is
substituted by lysine (K), in the
constant domain CH1 the amino acid at position 147 and the amino acid at
position 213 are substituted by
glutamic acid (E). Preferably in addition in the constant domain CL in the
amino acid at position 123 is
substituted by lysine (K).
In a preferred embodiment of the invention the antibody according to the
invention consists of one Fab
fragment of an antibody specifically binding to CD3 (further named also as
"CD3-Fab"), and one Fab
fragment of an antibody specifically binding to ROR1 (further named also as
"ROR1-Fab(s)") and a Fc
part, wherein the CD3-Fab and the ROR1-Fab are linked via their C-termini to
the hinge region of said Fc
part. Either the CD3-Fab or the ROR1-Fab comprises aa substitution and the CD3-
Fab comprises
crossover (Figures lA and 1B).
In a preferred embodiment of the invention the antibody according to the
invention consists of one CD3-
Fab, and one ROR1-Fab and an Fc part, wherein the CD3-Fab and the ROR1-Fab are
linked via their C-
termini to the hinge region of said Fc part and a second ROR1-Fab, which is
linked with its C-terminus to
the N-terminus of the CD3-Fab. The CD3-Fab comprises crossover and either the
CD3-Fab or both
ROR1-Fabs comprise aa substitution (Figures 1C, 1D, 1F,1G). Especially
preferred is a bispecific
antibody comprising ROR1-Fab-Fc-CD3-Fab-ROR1-Fab, wherein both ROR1-Fabs
comprise aa
substitution and the CD3-Fab comprises VL/VH crossover (Figure 1C). Especially
preferred is a
bispecific antibody consisting of ROR1-Fab-Fc-CD3 -Fab-ROR1 -Fab, wherein both
ROR1-Fabs comprise
aa substitution Q124K, E123R, K147E and K213E (kappa light chain) or E124K,
E123K, K147E and
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
7
K213E (lambda light chain) and the CD3-Fab comprises VL/VH crossover.
Especially preferred is a
bispecific antibody consisting of ROR1 -Fab-Fc-CD3 -Fab-ROR1 -Fab, wherein
both ROR1 -Fab s comprise
aa substitution Q124K, E123R, K147E and K213E (kappa light chain) or E124K,
E123K, K147E and
K213E (lambda light chain) and the CD3-Fab comprises VL/VH crossover.
Especially preferred is that
both ROR1-Fabs comprise as CDRs the CDRs of antibody MAB1, or as VHNL the VHNL
of MABl.
In a preferred embodiment of the invention the antibody according to the
invention consists of two
ROR1-Fabs and an Fc part, wherein the ROR1-Fabs are linked via their C-termini
to the hinge region of
said Fc part and a CD3-Fab, which is linked with its C-terminus to the N-
terminus of one ROR1-Fab. The
CD3-Fab comprises crossover and either the CD3-Fab or both ROR1-Fabs comprise
aa substitution
(Figures 1F and 1G).
In a preferred embodiment of the invention the antibody according to the
invention consists of one CD3-
Fab, which is linked via its C-terminus to the hinge region of said Fc part
and a ROR1-Fab, which is
linked with its C-terminus to the N-terminus of the CD3-Fab. The CD3-Fab
comprises crossover and
either the CD3-Fab or the ROR1-Fab comprise aa substitution (Figures 1H and
le.
In a preferred embodiment of the invention the antibody according to the
invention consists of one
ROR1-Fab, which is linked via its C-terminus to the hinge region of said Fc
part and a CD3-Fab, which is
linked with its C-terminus to the N-terminus of the ROR1-Fab. The CD3-Fab
comprises crossover and
either the CD3-Fab or the ROR1-Fab comprise aa substitution (Figures 1J and
1K).
In a further embodiment of the invention the bispecific antibody according to
the invention is
a) of construct ROR1 Fab ¨ Fc - CD3 Fab - ROR1 Fab,
b) comprises VL/VH crossover within the Fab fragment of the anti-CD3 antibody,
c) comprises a human IgGlFc part,
d) comprises within the Fc part substitution of Pro329 with glycine and
substitutions of Leu234 by
alanine and Leu235 by alanine, and
e) in the constant domain CL of both ROR1 Fabs the amino acid at position 124
is substituted by lysine
(K) and at position 123 by arginine (R) for a kappa light and lysine (K) for a
lambda light chain, in the
constant domain CH1 the amino acid at position 147 and the amino acid at
position 213 are substituted by
glutamic acid (E).
The Fab fragments are chemically linked together by the use of an appropriate
linker according to the
state of the art. Appropriate linkers are described e.g. in US 20140242079.
Preferably a (G1y4-Ser1)2
linker (SEQ ID NO:19) is used (Desplancq DK et al., Protein Eng. 1994 Aug;
7(8):1027-33 and Mack M.
et al., PNAS July 18, 1995 vol. 92 no. 15 7021-7025). Linkage between two Fab
fragments is performed
between the heavy chains. Therefore the C-terminus of CH1 of a first Fab
fragment is linked to the N-
terminus of VH of the second Fab fragment (no crossover) or to VL (crossover).
Linkage between a Fab
fragment and the Fc part is performed according to the invention as linkage
between CH1 and CH2.
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
8
The first and a second Fab fragment of an antibody specifically binding to
ROR1 are preferably derived
from the same antibody and preferably identical in the CDR sequences, variable
domain sequences VH
and VL and/or the constant domain sequences CH1 and CL. Preferably the amino
acid sequences of the
first and a second Fab fragment of an antibody specifically binding to ROR1
are identical. Preferably the
ROR1 antibody is an antibody comprising the CDR sequences of antibody MAB1, an
antibody
comprising the VH and VL sequences of antibody MAB1, or an antibody comprising
the VH, VL, CH1,
and CL sequences of antibody MAB 1.
Preferably the bispecific antibody comprises as Fab fragments and Fc part, not
more than one Fab
fragment of an anti-CD3 antibody, not more than two Fab fragments of an anti-
ROR1 antibody and not
more than one Fc part, preferably a human Fc part. Preferably the second Fab
fragment of an anti-ROR1
antibody is linked via its C-terminus either to the N-terminus of the Fab
fragment of an anti-CD3
antibody or to the hinge region of the Fc part. Preferably linkage is
performed between CH1 of ROR1-
Fab and VL of CD3-Fab (VL/VH crossover).
Preferably the antibody portion specifically binding to human CD3, preferably
the Fab fragment, is
characterized in comprising a variable domain VH comprising the heavy chain
CDRs of SEQ ID NO: 12,
13 and 14 as respectively heavy chain CDR1, CDR2 and CDR3 and a variable
domain VL comprising the
light chain CDRs of SEQ ID NO: 15, 16 and 17 as respectively light chain CDR1,
CDR2 and CDR3 of
the anti-CD3c antibody (CDR MAB CD3 H2C). Preferably the antibody portion
specifically binding to
human CD3 is characterized in that the variable domains are of SEQ ID NO:10
and 11 (VHVL MAB
CD3 H2C).
Preferably the antibody portion specifically binding to human CD3, preferably
the Fab fragment, is
characterized in comprising a variable domain VH comprising the heavy chain
CDRs of SEQ ID NO: 23,
24 and 25 as respectively heavy chain CDR1, CDR2 and CDR3 and a variable
domain VL comprising the
light chain CDRs of SEQ ID NO: 26, 27 and 28 as respectively light chain CDR1,
CDR2 and CDR3 of
the anti-CD3c antibody (CDR MAB CD3 CH2527). Preferably the antibody portion
specifically binding
to human CD3 is characterized in that the variable domains are of SEQ ID NO:21
and 22 (VHVL MAB
CD3).
Preferably the antibody portion, preferably the Fab fragment, specifically
binding to human ROR1 is
characterized in comprising a variable domain VH comprising the heavy chain
CDRs CDR1H of SEQ ID
NO:7, a CDR2H of SEQ ID NO:8, a CDR3H of SEQ ID NO: 9 and comprising a
variable domain VL
comprising the light chain CDRs CDR1L of SEQ ID NO:3, a CDR2L of SEQ ID NO:4,
a CDR3L of
SEQ ID NO: 5 (CDR MAB1).
Preferably the antibody portion, preferably the Fab fragment, specifically
binding to human ROR1 is
characterized in comprising a VH of SEQ ID NO: 6 and a VL of SEQ ID NO: 2
(VHVL MAB1).
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
9
The invention relates to a bispecific antibody specifically binding to the
extracellular domain of human
ROR1 and to human CDR, characterized in comprising a heavy and light chain set
of polypeptides SEQ
ID NO:37, SEQ ID NO:38, SEQ ID NO:39, and SEQ ID NO:40.
The invention relates to a bispecific antibody specifically binding to the
extracellular domain of human
ROR1 and to human CDR, characterized in comprising a heavy and light chain set
of polypeptides SEQ
ID NO:37, SEQ ID NO:38, SEQ ID NO:39, and SEQ ID NO:41
The invention further relates to a nucleic acid set encoding a respective
heavy and light chain set.
Preferably the bispecific antibody according to the invention comprising
constant heavy regions
CH2/CH3 of IgG1 subclass is characterized in comprising the mutations L234A,
L235A and P239G
(numbering according to Kabat) to avoid FcR and Clq binding and minimizing
ADCC/CDC. The
advantage is that such an antibody of the invention mediates its tumor cell
killing efficacy purely by the
powerful mechanism of T-cell redirection/activation. Additional mechanisms of
action like effects on
complement system and on effector cells expressing FcR are avoided and the
risk of side-effects is
decreased.
The invention comprises preferably a heavy chain of an antibody according to
the invention consisting of
(from N-to-C-terminus) VH(ROR1)-CH1(ROR1)-VL(CD3)-CH1(CD3)-CH2-CH3, preferably
of SEQ ID
NO: 37, as well as the respective encoding nucleic acids. These polypeptides
and respective nucleic acids
are useful for the production of a bispecific antibody according to the
invention.
The amino acid (aa) exchanges (further mentioned as "charge variants") outside
of the CDRs of the
bispecific antibodies according to the invention provide considerably improved
production/purification
without changing biological properties like binding to ROR1. By introduction
of the aa exchanges
(charge variants) according to the invention light chain LC mispairing and the
formation of side products
in production is significantly reduced and therefore purification is
facilitated.
The invention relates to a bispecific antibody specifically binding to the two
targets human CDR and the
extracellular domain of human ROR1 which does not internalize. The bispecific
antibody according to
the invention is preferably characterized in not internalizing in a
concentration of 1nM in primary B-CLL
cells at 37 C during two hours. The bispecific antibody according to the
invention is preferably
characterized in that the bispecific antibody does not internalize in a cell
based assay at 37 C during 2
hrs, using ROR1-positive primary B-CLL cells and used at an antibody
concentration of 1 nM, whereby
not internalize means, that the mean fluorescence intensity (MFI), as detected
by flow cytometry, of a
bispecific antibody upon binding to ROR1-positive primary B-CLL cells measured
at time 0 is not
reduced more than 50%, preferably not more than 30% when re-measured after a
2hr-incubation at 37 C.
Preferably the bispecific antibody according to the invention is a bivalent
antibody and characterized in
comprising a monovalent anti-ROR1 antibody specifically binding to ROR1, and a
monovalent antibody
specifically binding to CD3. A bivalent antibody is preferred if its said mean
fluorescence intensity
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
(MFI), as detected by flow cytometry, upon binding to ROR1-positive cells
measured at time 0 is not
reduced more than 50%, preferably not more than 30% by internalization when re-
measured after a 2hr-
incubation at 37 C. Preferably the bispecific antibody according to the
invention is a bivalent antibody
and characterized in comprising a monovalent anti-ROR1 antibody specifically
binding to ROR1, and a
5 monovalent antibody specifically binding to CD3. Preferably the monovalent
antibody specifically
binding to CD3 is a Fab fragment, preferably a CD3 crossFab. Such a bivalent
antibody is preferred if its
said mean fluorescence intensity (MFI), as detected by flow cytometry, upon
binding to ROR1-positive
cells measured at time 0 is not reduced more than 50%, preferably not more
than 30% by internalization
when re-measured after a 2hr-incubation at 37 C. Preferably the bispecific
antibody according to the
10 invention is a trivalent antibody and characterized in comprising a
bivalent anti-ROR1 antibody
specifically binding to ROR1, and a monovalent antibody specifically binding
to CD3. Preferably the
monovalent antibody specifically binding to CD3 is a Fab fragment or
preferably a CD3 crossFab. A
trivalent antibody is preferred if its said mean fluorescence intensity (MFI),
as detected by flow
cytometry, upon binding to ROR1-positive cells measured at time 0 is not
reduced more than 50%,
preferably not more than 30% by internalization when re-measured after a 2hr-
incubation at 37 C.
Preferably the bispecific antibody according to the invention does not
internalize in said cell based assay
at 37 C during 24 hrs.
Preferably the bispecific antibody according the invention does not
internalize in said cell based assay if
used in a concentration between 0.1 pM and 200 nM.
A further embodiment of the invention is an antibody according to this
invention with an affinity ratio of
ROR1 to CD3 of 5000:1 to 5:1, as determined by Kd values using surface plasmon
resonance. Such an
antibody is favorable because of its stronger binding to malignant cells over
T cells. Preferably the Kd
values are about 100 nM for the CD3 antibody and about 50 pM to 50 nM for the
ROR1 antibody.
Preferably the B-CLL cells are used according to the invention in a cell
concentration of 1 x 106 cells/mL
(primary PBMC from CLL patients) or 1 x 106 cells/mL (ATCC CCL-155) or 1 x 106
cells/mL (ATCC
CRL-3004).
Preferably the antibody portion specifically binding to CD3 is characterized
in being humanized.
Preferably the CD3 Mab according to the invention binds to the same epitope of
CDR as antibody H2C
(described in W02008119567) and/or antibody CH2527 (described in W02013026839)
or is preferably
antibody H2C or CH2527.
Preferably the antibody portion specifically binding to ROR1 is characterized
in comprising a light chain
variable domain (VL) comprising as respective variable light chain CDRs the
CDRs of SEQ ID NO: 3, 4,
5 and a heavy chain variable domain (VH) comprising as respective variable
heavy chain CDRs the
CDRs of SEQ ID NO:7, 8, 9. Preferably the antibody portion specifically
binding to ROR1 is
characterized in comprising as light chain variable domain (VL) a sequence
being at least 90% identical
to the sequence of SEQ ID NO:2 and as variable heavy chain domain (VH) a
sequence being at least 90%
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
11
identical to the sequence of SEQ ID NO:6, Preferably the antibody portion
specifically binding to ROR1
is characterized in comprising as light chain variable domain (VL) the
sequence of SEQ ID NO:2 and as
variable heavy chain domain (VH) the sequence of SEQ ID NO:6. Preferably the
antibody portion
specifically binding to ROR1 is characterized in being humanized. Preferably
the ROR1 Mab according
to the invention binds to the same epitope of ROR1 as the Mab mentioned above.
A further embodiment of the invention is a method for the preparation of a
bispecific antibody according
to the invention comprising the steps of transforming a host cell with one or
more vectors comprising
nucleic acid molecules encoding the respective antibodies or fragments,
culturing the host cell under
conditions that allow synthesis of said antibody molecule; and recovering said
antibody molecule from
said culture.
Preferably the method for the preparation of a bispecific antibody according
to the invention comprising
the steps of
a) transforming a host cell with one or more vectors comprising nucleic acid
molecules encoding the
heavy and light chain set of polypeptides SEQ ID NO:37, 38, 39, and 40, or the
set of SEQ ID NO:37, 38,
39, and 41
b) culturing the host cell under conditions that allow synthesis of said
antibody molecule; and
c) recovering said antibody molecule from said culture.
A further embodiment of the invention is a host cell comprising vectors
comprising nucleic acid
molecules encoding an antibody according to the invention.
A further embodiment of the invention is a host cell comprising vectors
comprising nucleic acid
molecules encoding the light chain and heavy chain of an antibody specifically
binding to the first target
and vectors comprising nucleic acid molecules encoding the light chain and
heavy chain of an antibody
specifically binding to the second target, wherein the variable domains VL and
VH are replaced by each
other.
A further preferred embodiment of the invention is a pharmaceutical
composition comprising an antibody
according to the invention and a pharmaceutically acceptable excipient.
A further preferred embodiment of the invention is a pharmaceutical
composition comprising an antibody
according to the invention for use as a medicament. A further preferred
embodiment of the invention is an
antibody according to the invention or a pharmaceutical composition comprising
an antibody according to
the invention for use as a medicament in the treatment of ROR1-positive
hematological malignancies
comprising chronic lymphocytic leukemia (CLL), hairy cell leukemia (HCL),
acute lymphoblastic
leukemia (ALL), acute myeloid leukemia (AML), chronic myeloid leukemia (CML),
mantle cell
lymphoma (MCL), marginal zone lymphoma (MZL), diffuse large B cell lymphoma
(DLBCL), multiple
myeloma (MM) and follicular lymphoma (FL). ROR1 is significantly and uniformly
expressed on the cell
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
12
surface of these various blood cancers. A further embodiment of the invention
is an antibody according to
the invention or a pharmaceutical composition comprising an antibody according
to the invention for use
as a medicament in the treatment of leukemias and non-Hodgkin lymphomas
expressing ROR1. A
preferred embodiment of the invention is an antibody according to the
invention or a pharmaceutical
composition comprising an antibody according to the invention for use as a
medicament in the treatment
of chronic lymphocytic leukemia (CLL) of B-cell lineage (B-CLL). B-CLL results
from an acquired
mutation to the DNA of a single marrow cell that develops into a B lymphocyte.
Once the marrow cell
undergoes the leukemic change, it multiplies into many cells and overtime
crowd out normal cells since
CLL cells grow and survive better than normal cells. The result is the
uncontrolled growth of CLL cells in
the bone marrow, leading to an increase in the number CLL cells in the blood.
CLL symptoms usually
develop over time with some patients being asymptomatic with only abnormal
blood test results (e.g.
increase in white blood cells). CLL patients with symptoms experience fatigue,
short of breath, anemia,
weight loss, decrease in appetite, lymph nodes and spleen enlargement and
recurrent infections due to low
immunoglobulin levels and decreased neutrophil counts (Leukemia & Lymphoma
Society, 2009). A
further preferred embodiment of the invention is an antibody according to the
invention or a
pharmaceutical composition comprising an antibody according to the invention
for use as a medicament
in the treatment of Multiple Myeloma. A further embodiment of the invention is
an antibody according to
the invention or a pharmaceutical composition comprising an antibody according
to the invention for use
as a medicament the treatment of plasma cell disorders like Multiple Myeloma
MM or other B-cell
disorders expressing ROR1. MM is a B-cell malignancy characterized by a
monoclonal expansion and
accumulation of abnormal plasma cells in the bone marrow compartment. MM also
involves circulating
clonal B cells with same IgG gene rearrangement and somatic hypermutation. MM
arises from an
asymptomatic, premalignant condition called monoclonal gammopathy of unknown
significance
(MGUS), characterized by low levels of bone marrow plasma cells and a
monoclonal protein. MM cells
proliferate at low rate. MM results from a progressive occurrence of multiple
structural chromosomal
changes (e.g. unbalanced translocations). MM involves the mutual interaction
of malignant plasma cells
and bone marrow microenvironment (e.g. normal bone marrow stromal cells).
Clinical signs of active
MM include monoclonal antibody spike, plasma cells overcrowding the bone
marrow, lytic bone lesions
and bone destruction resulting from overstimulation of osteoclasts (Dimopulos
& Terpos, Ann Oncol
2010; 21 suppl 7: vii143-150). A further embodiment of the invention is an
antibody according to the
invention or a pharmaceutical composition comprising an antibody according to
the invention for use as a
medicament in the treatment of ROR1-positive solid tumors such as human breast
cancers (Zhang S,
PLoS One 2012; 7(3): e31127) and lung cancers (Yamaguchi T, Cancer Cell 2012;
21(3):348).
A further embodiment of the invention is the use of an antibody according to
the invention or the
pharmaceutical composition according to the invention for such treatments.
A further embodiment of the invention is the use of an antibody according to
the invention or the
pharmaceutical composition according to the invention for the treatment of a
disease selected from the
group consisting of ovarian cancer, lung cancer, breast cancer, gastric
cancer, and /or pancreatic cancer. A
further embodiment of the invention is an antibody according to the invention
or the pharmaceutical
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
13
composition according to the invention for use in the treatment of ovarian
cancer, lung cancer, breast
cancer, gastric cancer, and /or pancreatic cancer.
A further embodiment of the invention is the use of an antibody according to
the invention or the
pharmaceutical composition according to the invention for the treatment of
ovarian cancer. A further
embodiment of the invention is an antibody according to the invention or the
pharmaceutical composition
according to the invention for use in the treatment of ovarian cancer. A
further embodiment of the
invention is a method of treatment comprising administering to a cancer
patient a therapeutically
effect dose of the antibody according to the invention. A further embodiment
of the invention is
such method wherein the cancer is a selected from the group consisting of
ovarian cancer, lung
cancer, breast cancer, gastric cancer, and /or pancreatic cancer and
hematologic malignancy. A
further embodiment of the invention is such a method wherein chemotherapy or
radiation is
administered before, after, or concurrently with the antibody. A further
embodiment of the
invention is a method of treatment comprising administering to a patient
having such disease an
antibody according to the invention. A further embodiment of the invention is
a method of
treatment comprising administering to a patient having ovarian cancer an
antibody according to
the invention. A further embodiment of the invention is the use of an antibody
according to the
invention as a medicament for treating a cancer, preferably ovarian cancer. A
further
embodiment of the invention is the use of an antibody according to the
invention in the
manufacture of a medicament for treating a cancer, preferably ovarian cancer..
Preferably the antibody according to the invention or the pharmaceutical
composition is administered
once or twice a week preferably via subcutaneous administration (e.g.
preferably in the dose range of 0.1
to 10 mg/m2 once or twice a week). Due to superior cytotoxicity activities of
the antibody according to
the invention, it can be administered at a lower magnitude of clinical dose
range as compared to
conventional monospecific antibodies or conventional bispecific antibodies
that are not T cell bispecifics
(i.e. do not bind to CD3 on one arm). It is envisaged that for an antibody
according to the invention
subcutaneous administration is preferred in the clinical settings (e.g. in the
dose range of 0.1 ¨ 10
mg/m2once or twice a week). An antibody according to the invention is
eliminated with a half-life of
about several days which allows at least once or twice/week administration.
Another advantage of the
antibody according to the invention is a molecular weight (i.e. approximately
150 ¨ 200 kDa) higher than
the kidney filtration size limit (50 ¨70 kDa). This molecular weight allows
long elimination half-life and
makes subcutaneous administrations once or twice a week possible.
Preferably an antibody according to the invention is characterized by showing
tumor growth inhibition of
more than 70%, preferably of more than 85%, preferably of close to 100% in a
xenograft model with a
ROR1 expressing tumor cell line (for example CLL, MM, MCL cell lines) at a
dose of 1 mg/kg body
weight (BW) administered intravenously (i.v.) or subcutaneously (s.c.) or
intraperitoneal (i.p.) twice a
week or once a week, preferably 0.5 mg/kg BW administered i.v. or i.p. or s.c.
twice a week or once a
week, preferably at 0.1 mg/kg BW administered i.v. or i.p. or s.c. twice a
week or once a week, preferably
at 0.05 mg/kg BW administered i.v. or i.p. or s.c. twice a week or once a
week, preferably at 0.01 mg/kg
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
14
BW administered i.v. or i.p. or s.c twice a week or once a week, preferably at
5 g/kg BW administered
i.v. or i.p. or s.c. twice a week or once a week.
Preferably an antibody according to the invention is characterized by an
elimination half-life in mice,
preferably cynomolgus monkeys of longer than 12 hours, preferably 3 days or
longer.
Bispecific antibodies binding to a target on tumor cells and to CD3 and having
the molecular format
(scFv)2 have very short elimination half-life of 1 to 4 hours. In the clinical
trials with the (scFv)2
bispecific CD19xCD3 antibody blinatumomab, this compound had to be
administered via a pump carried
by the patients over weeks and months (Topp et al. J Clin Oncol 2011; 29(18):
2493-8). Compared to a
twice a week or once a week iv or sc administration, treatment administered
via a pump is much less
convenient for the patients and also much more risky (e.g. failure of pump,
issues with the catheter).
Preferably an antibody according to the invention is characterized in showing
an EC50 value for binding
to ROR1-positive cell lines (e.g. RPMI8226 cells, Rec-1 cells, Jeko cells) of
30 nM or lower, preferably
an EC50 value of 15 nM and lower.
Preferably an antibody according to the invention is characterized by its
capability to induce redirected
killing of ROR1 expressing tumor cells (e.g. RPMI8226 cells, Rec-1 cells, Jeko
cells) in the presence of
human T cells with an EC50 lower than 10 nM, preferably 1 nM, preferably 0.05
nM, preferably 0.02
nM, preferably 0.002 nM and lower.
Stability of bispecific antibodies can be affected in practical conditions and
clinical applications. Despite
recent antibody engineering improvements, some recombinant proteins and
molecular formats (e.g. scFVs
fragments) tend to denature and form aggregates more easily than other under
stress conditions (Worn
and Pluckthun. J Mol Biol 2001; 305, 989-1010). Preferably an antibody
according to this invention is
characterized in that said antibody stored in standard formulation buffer at
37 C preferably at 40 C, for
10 days, preferably up to 2 weeks, preferably up to 4 weeks, does not result
in more than 10% changes
(A), preferably not more than 5% changes (A), in high molecular weight (HMW)
species and/or low
molecular weight (LMW) species and/or monomer content as compared to the said
antibody stored in the
same formulation buffer at -80 C for the same period of storage.
Description of the Figures
Remark: If not mentioned that Mab2 was used as anti-ROR1 antibody and/or as
anti-ROR1 Fab in an
anti-ROR1/anti-CD3 TCB antibody in the following descriptions of the figures,
then Mab 1 was used as
anti-RORlantibody and/or as anti-ROR1 Fab in an anti-ROR1/anti-CD3 TCB
antibody.
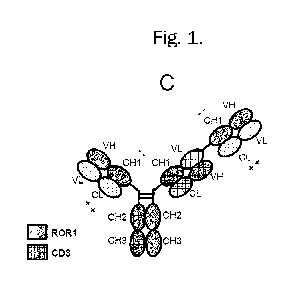
Figure 1A-K. Preferred bispecific antibodies comprising the Fab fragments
(specific to CD3 and ROR1)
as specified: (1A, 1B) Fab ROR1-Fc- Fab CD3; (1C, 1D) Fab ROR1-Fc-Fab CD3-Fab
ROR1; (1F, 1G)
Fab ROR1-Fc-Fab ROR1-Fab CD3; (1H, le Fc-Fab CD3-Fab ROR1; (1J, 1K) Fc-Fab
ROR1-Fab CD3.
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
Preferably, the Fabs CD3 include a VH-VL crossover to reduce LC mispairing and
side-products. Fab
CD3 and Fab ROR1 are linked to each other with flexible linkers.
Figure 2. Detection of ROR1 on the cell surface of (A) primary CLL cells and
(B) RPMI8226 MM cells
and Rec-1 MCL cells using Alexa488-labelled anti-ROR1 IgG antibody or A1exa647-
labelled anti-human
5 Fc antibody. Graphs showing increase in MFI signal as compared to baseline.
Figure 3. Binding of anti-ROR1 IgG1 antibody on ROR1-positive RPMI8226 cells
(A) and non-binding
to ROR1-negative MKN45 cells (B). Mean fluorescence intensity for anti-ROR1
IgG plotted in function
of anti-ROR1 antibody concentrations (from 0.14 to 100 nM).
Figure 3-1. Production and purification of 83A10-TCB without charge variant
vs. 83A10-TCBcv with
10 charge variant. CE-SDS (non-reduced) graphs of the final protein
preparations after different methods of
purification for 83A10-TCB and 83A10-TCBcv antibodies. Protein A (PA) affinity
chromatography and
size exclusion chromatographic (SEC) purification steps applied to 83A10-TCB
antibody (A). (B)
Additional purification steps: cation exchange chromatography (cIEX) and a
final size exclusion
chromatographic (re-SEC) steps applied to the final protein preparations in
(A). (C) 83A10-TCBcv
15 antibody after PA + cIEX + SEC purification steps. 83A10-TCB and 83A10-
TCBcv molecules are both
of molecular format as described in Figure 2a of PCT/EP2015/067841
incorporated by reference.
Figure 3-2. Head-to-head comparison study: Production of 83A10-TCB without
charge vs. 83A10-
TCBcv with charge variant. Properties (e.g. purity, yield, monomer content) of
83A10-TCB and 83A10-
TCBcv antibodies were measured side-by-side and compared after each
purification steps 1) PA affinity
chromatography only (A, B), 2) PA affinity chromatography then SEC (C, D) and
3) PA affinity
chromatography then SEC then cIEX and re-SEC (E, F). CE-SDS (non-reduced)
graphs of the final
protein solutions after the respective methods of purification for 83A10-TCB
and 83A10-TCBcv
antibodies. (A) PA affinity chromatography purification step applied to 83A10-
TCB antibody. (B) PA
affinity chromatography purification step applied to 83A10-TCBcv antibody. (C)
PA affinity
chromatography + SEC purification steps applied to 83A10-TCB antibody. (D) PA
affinity
chromatography + SEC purification steps applied to 83A10-TCBcv antibody. (E)
PA affinity
chromatography +/- SEC + cIEX + SEC purification steps applied to 83A10-TCB
antibody. (F) PA
affinity chromatography +/- SEC + cIEX + SEC purification steps applied to
83A10-TCBcv antibody.
Purity, yield, monomer content were measured. Percentage of correct molecule
detected by liquid
chromatography ¨ mass spectrometry (LC-MS).
Figure 3-3. Binding of ROR1 IgG (ROR1 Mab2, open symbols) and anti-ROR1/anti-
CD3 TCB
antibodies (ROR1 Mab2-TCBcv, closed symbols) to ovarian cancer cell lines SK-
OV-3 (A) and PA-1 (B)
as measured by an increase in the median fluorescence intensity signal in
function of antibody
concentrations. No signal was observed with the control-TCB binding to CD3
only and not to ROR1
tested on both SK-OV-3 and PA-1 ovarian cancer cell lines (A and B; closed
circles).
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
16
Figure 3-4. Binding of anti-ROR1/anti-CD3 TCB antibodies to Jurkat T cells. A
concentration-dependent
binding of ROR1 Mab2-TCBcv (squares) and control-TCB (circles) was observed on
Jurkat T cells
confirming that both TCB antibodies bind to CD3 on T cells.
Figure 4. Internalization rate (%) of (A, B) anti-ROR1 IgG1 antibody at a
concentration of 1 nM and (A,
C) anti-ROR1/anti-CD3 TCB2+1 antibody on ROR1-positive primary B-CLL cells
after 2h incubation at
37 C, as detected by FACS using secondary labelled anti-human Fc antibody
(indirect detection). (A, B)
Anti-ROR1 IgG antibody (1M) internalized about 12.5% in primary B-CLL cells.
(A, C) Anti-
ROR1/anti-CD3 TCB2+1 antibody (1 nM) showed an internalization rate of 27.1%
in primary B-CLL
cells at the same experimental conditions as measured by FACS (indirect
detection). Internalization was
calculated based on the MFI value at time 0, baseline, and calculated using
the previously described
formula.
Figure 5. Internalization rate (%) of anti-ROR1/anti-CD3 TCB1+1 antibody (1
nM) in primary B-CLL
cells after an incubation of 2 hrs at 37 C in the presence or absence of
phenylarsine oxide (PAO) as
detected by FACS using secondary labelled anti-human Fc antibody (indirect
detection). A decrease of
91% in the MFI signal was observed in primary B-CLL cells after an incubation
of 2 hrs at 37 C without
PAO. However, when the B-CLL cells were incubated in the presence of PAO (3
M), 90% decrease in
MFI signal was still observed indicating that the loss in MFI signal was not
due to internalization of the
antibody but rather probably dissociation.
Figure 6. Internalization rates of TCB2+1 antibodies and anti-ROR1 IgG
antibody (1 nM) in RPMI8226
MM cells after an incubation of 2 hrs at 37 C, as measured in two independent
experiments. The results
demonstrate that anti-ROR1/anti-CD3 TCB2+1 has an internalization rate of less
than 15% in RPMI
cells.
Figure 7. Binding of anti-ROR1/anti-CD3 TCB antibodies on Jurkat T cells. Mean
fluorescence intensity
for anti-ROR1/anti-CD3 T cell bispecific antibodies plotted in function of
antibody concentrations (from
3 to 500 nM); anti-ROR1/anti-CD3 TCB1+1 and anti-ROR1/anti-CD3 TCB2+1
antibodies on Jurkat
cells. EC50 values and maximal binding of anti-ROR1/anti-CD3 TCB1+1 and anti-
ROR1/anti-CD3
TCB2+1 antibodies to Jurkat cells were not reached. DP47 isotype control
antibody or anti-ROR1 IgG
antibody did not bind to Jurkat T cells.
Figure 8. Binding of anti-ROR1/anti-CD3 TCB antibodies on ROR1-positive
RPMI8226 cells. Mean
fluorescence intensity plotted in function of antibody concentrations (from
0.136 to 13.6 nM).
Figure 9. Up-regulation of T-cell activation markers by anti-ROR1/anti-CD3 TCB
antibodies in presence
of ROR1-positive (A) Rec-1 cells and (B) RPMI8226 cells. Mean fluorescence
intensity plotted in
function of antibody concentrations (from 0.01 pM to 100 nM). (A) A
concentration dependent increase
in the mean fluorescence intensity of the late activation marker CD25 gated on
CD8 T cells was observed
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
17
in Rec-1 cells. Significant concentration dependent activation of CD8 T cells
by anti-ROR1/anti-CD3
TCB1+1 antibody in the presence of ROR1-positive Rec-1 cells. Maximum signal
reached at 100 pM of
antibody. Unspecific activation of CD8 T cells was minimal upon binding of CD3
on T cells but without
binding on ROR1-positive target cells by using non-binder TCB constructs.
Activation of CD8 T cells
was weak with anti-ROR1/anti-CD3 TCB2+1 antibody as shown by a faint but
noticeable increase in
CD25 mean fluorescence intensity. However, unspecific T cell activation could
not be ruled out. (B)
Concentration dependent upregulation of CD25 on CD8 T cells mediated by anti-
ROR1/anti-CD3
TCB1+1 and anti-ROR1/anti-CD3 TCB2+1 antibodies in the presence of ROR1-
positive RPMI8226 MM
cells. At the highest concentration (100 pM) of TCB antibodies tested there
was no unspecific activation
of CD8 T cells as shown with non-binder TCB constructs.
Figure 9-1. Up-regulation of T-cell activation markers by anti-ROR1/anti-CD3
TCB antibodies in
presence of ovarian cancer target cells. The expression of activation markers
was determined by
measuring the median fluorescence intensity gated on CD4+ and CD8+ T cell
populations. ROR1 Mab2-
TCBcv (squares) induced a concentration-dependent increase of CD69 early
activation marker which was
observed on CD4+ T cells (A) and CD8+ T cells (B) in presence of ROR1-low
expressing SK-OV-3
target cells while control-TCB (triangles) did not induce any T-cell
activation. At a clinically relevant
concentration of 1 nM of ROR1 Mab2-TCBcv, there was already up to 40% of
activated CD4 T cells and
25% of activated CD8 T cells after 48h of incubation.
Figure 10. Redirected T cell killing of ROR1-positive RPMI8226 MM target cells
by CD8 T cells
activated by anti-ROR1/anti-CD3 TCB antibodies. Specific cytotoxicity of
target cells (tumor lysis)
induced by anti-ROR1/anti-CD3 TCB antibodies was measured by LDH release. (A)
Experiment 1 (14h
time point): 30% of tumor lysis was already observed with the lowest
concentration tested of 0.01 pM
anti-ROR1/anti-CD3 TCB1+1 antibody and up to 37.5% tumor lysis was reached
with 30 nM of anti-
ROR1/anti-CD3 TCB antibodies in experimental conditions with E:T ratio of 3:1
i.e. 3 CD8 T cells for 1
RPMI 8226 target cell. The 37.5% tumor lysis observed at 30 nM as detected by
LDH release could not
have been attributed only to unspecific killing of target cells as there was
only 17% unspecific target cell
lysis with 30 nM of non-binder TCB1+1 (i.e. binds to effector cells but not to
target cells). For anti-
ROR1/anti-CD3 TCB2+1 antibody, a maximum target cell lysis of 30% was already
observed at the
lowest concentration tested of 0.2 fM and there was no concentration dependent
response with increasing
concentrations for up to 10 nM. 30 nM non-binding TCB2+1 had close to 30%
tumor lysis. (B)
Experiment 2 (20h time point): 30-40% target cell lysis was observed with anti-
ROR1/anti-CD3 TCB1+1
and TCB2+1 antibodies at a concentration of 100 pM while non-binder TCB
controls did not induce any
tumor lysis at the same concentration.
Figure 11. Redirected T cell killing of ROR1-positive RPMI8226 MM target cells
by T cells from
PBMCs activated by anti-ROR1/anti-CD3 TCB antibodies at different effector
cells to tumor cells (E:T)
ratios. Specific cytotoxicity of target cells (tumor lysis) induced by anti-
ROR1/anti-CD3 TCB antibodies
was measured by LDH release. (A) E:T ratio= 10 PBMCs : 1 RPMI8226 MM cell (24h
time point): there
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
18
was a concentration dependent response with increasing concentrations for up
to 100 nM. A maximum
mean of 25% of tumor lysis was observed with 100 nM concentration anti-
ROR1/anti-CD3 TCB1+1 and
anti-ROR1/anti-CD3 TCB2+1 antibodies in experimental conditions with E:T ratio
of 10:1 i.e. 10
PBMCs for 1 RPMI8226 target cell. The 25% tumor lysis observed at 100 nM as
detected by LDH
release could not have been attributed only to unspecific killing of target
cells as there was only 9%
unspecific target cell lysis with 100 nM of non-binder TCB2+1 (i.e. binds to
effector cells but not to
target cells). (B) E:T ratio= 25 PBMCs : 1 RPMI8226 MM cell (24h time point):
there was a
concentration dependent tumor lysis with increasing concentrations of anti-
ROR1/anti-CD3 TCB1+1for
up to 2 nM. A maximum mean of 30% of tumor lysis was observed already with 2
nM concentration of
anti-ROR1/anti-CD3 TCB1+1, but tumor lysis seemed to reach a plateau since 100
nM concentration was
not associated with a higher response.
Figure 12. Redirected T cell killing of ROR1-positive ovarian cancer target
cells with different level of
surface ROR1: high expressing PA-1 (A),medium expressing COLO-704 (B) and
OVCAR-5 (C), and
low expressing SK-OV-3 (D). Effector cells to tumor cells (E:T) ratios of 10
PBMCs : 1 target cell.
Specific cytotoxicity of target cells (tumor lysis) induced by anti-ROR1/anti-
CD3 TCB antibodies was
measured by LDH release (48h culture). There was a concentration dependent
response with increasing
concentrations from 0.5 pM to 50 nM. ROR1 Mab2-TCBcv (squares) induced a
concentration-dependent
increase in tumor cell lysis of ROR1 high-expressing PA-1 ovarian cancer cells
(A), ROR1 medium-
expressing COLO-704 (B) and OVCAR-5 (C) ovarian cancer cells and ROR1 low-
expressing SK-OV-3
ovarian cancer cells (D). In contrast, control-TCB (A, B, C; circles) which
only binds to CD3 did not
induce tumor cell lysis at clinically relevant concentrations (i.e. up to 10
nM). Representative
experiments shown (n=1 to 5).
Detailed Description of the Invention
The inventors have found that bispecific antibodies against CD3 c and ROR1
with VH/VL exchange can
be produced in high yield and easily purified if in the light chain CL of
either the antibody portion against
CDR or ROR1 the amino acid at position 124 is substituted independently by
lysine (K), arginine (R) or
histidine (H) (numbering according to Kabat), and in the respective constant
domain CH1 the amino acid
at position 147 and the amino acid at position 213 is substituted
independently by glutamic acid (E), or
aspartic acid (D) (numbering according to Kabat).
Preferably the VH/VL exchange is in the CD3 binding portion. Preferably the
bispecific antibody is
monovalent for CD3 binding. The amino acid substitutions described above can
be either in the ROR1
binding portion or in the CD3 binding portion. Therefore in a certain
embodiment of the invention the
CD3 binding portion can comprise the VH/VL exchange and the amino acid
substitutions. In this case the
ROR1 binding portion does not comprise any VH/VL exchange or amino acid
substitutions at amino
acids 124, 147, 213, or 123. Preferably the bispecific antibody is monovalent
for CD3 binding and
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
19
bivalent for ROR1 binding. As described, the bispecific antibody can therefore
comprise a second ROR1
binding portion, which is identical to the first one. Therefore if the first
ROR1 binding portion comprises
the amino acid substitutions, the second ROR1 binding portion comprises the
same substitutions and if
the first ROR1 binding portion does not comprise the amino acid substitutions,
the second ROR1 binding
portion does also not comprise the substitutions. Preferably amino acid 124 is
K, amino acid 147 is E,
amino acid 213 is E, and amino acid 123 is R. Preferably the CD3 binding
portion and the RORlbinding
portion (or both ROR1 binding portions if so) are Fab fragments, whereby when
two ROR1 binding
portions are present one ROR1 portion is chemically linked to the CD3 binding
portion via CH1/VL (C-
terminus of ROR1 binding portion (CH1) to N-terminus of crossover CD3 binding
portion(VL)) or
CH1/VH (C-terminus of crossover CD3 binding portion (CH1) to N-terminus of
ROR1 binding
portion(VH)). The bispecific antibody can comprise or not comprise an Fc part.
The term "ROR1" as used herein relates to human ROR1 (synonyms: tyrosine-
protein kinase
transmembrane receptor ROR1, EC=2.7.10.1, neurotrophic tyrosine kinase,
receptor-related 1,
UniProtKB Q01973) which is a tyrosine-protein kinase receptor. The
extracellular domain of ROR1
consists according to UniProt of amino acids 30 ¨ 406. The term "antibody
against ROR1, anti ROR1
antibody or ROR1 Mab" as used herein relates to an antibody specifically
binding to human ROR1. The
antibody binds specifically to the extracellular domain of ROR1 (amino acids
M1 -V406 of SEQ ID
NO:1). The antibody binds specifically to fragments of the extracellular
domain, which are the Ig-like
C2-type domain (amino acids Q73-V139 of SEQ ID NO:1), the frizzled domain
(amino acids El 65-1299
of SEQ ID NO: 1), or the kringle domain (amino acids K312-C391 of SEQ ID
NO:1). These fragments
are mentioned in W02005100605. It is further preferred that the antibody binds
specifically to the
extracellular domain fragment WNISSELNKDSYLTL (SEQ ID NO.18) of ROR1. This
fragment is
mentioned in Daneshmanesh AH et al., Int. J. Cancer, 123 (2008) 1190-1195.
Exemplary anti-ROR1
antibodies according to the invention are Mab2 (W0201209731, Mab 4A5) and
Mabl, Mab3 and Mab4
(W02012075158 , Mabs R12, Y31 and R11). When not specified, ROR1 IgG or TCB
antibodies
referred to Mabl.
The term "CDR or CD3" as used herein relates to human CDR described under
UniProt P07766
(CD3E_HUMAN). The term "antibody against CD3, anti CD3 antibody" relates to an
antibody binding
to CDR. Preferably the antibody comprises a variable domain VH comprising the
heavy chain CDRs of
SEQ ID NO: 12, 13 and 14 as respectively heavy chain CDR1, CDR2 and CDR3 and a
variable domain
VL comprising the light chain CDRs of SEQ ID NO: 15, 16 and 17 as respectively
light chain CDR1,
CDR2 and CDR3. Preferably the antibody comprises the variable domains of SEQ
ID NO:10 (VH) and
SEQ ID NO:11 (VL). Preferably the antibody comprises a variable domain VH
comprising the heavy
chain CDRs of SEQ ID NO: 23, 24 and 25 as respectively heavy chain CDR1, CDR2
and CDR3 and a
variable domain VL comprising the light chain CDRs of SEQ ID NO: 26, 27 and 28
as respectively light
chain CDR1, CDR2 and CDR3. Preferably the antibody comprises the variable
domains of SEQ ID
NO:21 (VH) and SEQ ID NO:22 (VL).The anti-CD3 antibodies shown in SEQ ID NO:10
and 11 and 21
and 22 are derived from 5P34 and have similar sequences and the same
properties in regard to epitope
binding as antibody 5P34.
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
"Specifically binding to CD3 or ROR1" refer to an antibody that is capable of
binding CD3 or ROR1 (the
targets) with sufficient affinity such that the antibody is useful as a
therapeutic agent in targeting CD3 or
ROR1. In some embodiments, the extent of binding of an anti-CD3 or ROR1
antibody to an unrelated,
non-CD3 or non-ROR1 protein is about 10-fold preferably >100-fold less than
the binding of the
5 antibody to CD3 or ROR1 as measured, e.g., by surface plasmon resonance
(SPR) e.g. Biacore0,
enzyme-linked immunosorbent (ELISA) or flow cytometry (FACS). Preferably the
antibody that binds to
CD3 or ROR1 has a dissociation constant (Kd) of 10-8 M or less, preferably
from 10-8 M to 10-13 M,
preferably from 10-9 M to 10-13 M. Preferably the bispecific antibody
according to the invention binds to
an epitope of ROR1 that is conserved among ROR1 from different species and/or
an epitope of CD3 that
10 is conserved among CD3 from different species, preferably among human and
cynomolgus. "Bispecific
antibody specifically binding to CD3 and ROR1" or "antibody according to the
invention" refers to a
respective definition for binding to both targets. An antibody specifically
binding to ROR1 (or CD3 or
ROR1 and CD3) does not bind to other human antigens. Therefore in an ELISA, OD
values for such
unrelated targets will be equal or lower to that of the limit of detection of
the specific assay, preferably
15 equal or lower as 1.5 pM, or equal or lower to OD values of control samples
without plate-bound-ROR1
or with untransfected HEK293 cells.
Antibodies according to the invention are analyzed by ELISA for binding to
human ROR1 using plate-
bound ROR1. For this assay, an amount of plate-bound ROR1 preferably or 1.5 nM
and concentration(s)
preferably ranging from 1 pM to 200 nM of anti-ROR1 antibody are used. An
antibody according to the
20 invention for which its ROR1 binding is at least 20% higher than the OD
values of the control samples
without plate-bound ROR1 or with untransfected HEK293 cells according to the
invention is an antibody
"binding to human ROR1 in an ELISA assay". An exemplary antibody according to
the invention is
characterized by the heavy and light chain set of polypeptides SEQ ID NO:37,
38, 39, and 40. Another
exemplary antibody according to the invention is characterized by the heavy
and light chain set of
polypeptides SEQ ID NO:37, 38, 39, and 41.
The term "antibody according to the invention which does not internalize" as
used herein means a
bispecific antibody according to the invention with MFI reduction properties
characterized in that in a
cell based assay at 37 C during 2 hrs., using ROR1-positive B-CLL cells, and
used at an antibody
concentration of 1 nM, whereby not internalize means, that the mean
fluorescence intensity (MFI), as
detected by flow cytometry, upon binding to ROR1-positive cells measured at
time 0 is not reduced more
than 50%, preferably not more than 30% by internalization when re-measured
after a 2hr-incubation at
37 C. The bispecific antibody according to the invention does not internalize
in ROR1-positive B-CLL
cells, therefore the binding of the said anti-ROR1 antibody to ROR1-positive B-
CLL cells is not reduced
more than 50%, preferably not more than 30%, when the said antibody is
incubated at 37 C for 2 h in
such cell based assay as described herein.
It is also preferred, that a bispecific antibody according to the invention
shows in a cell based assay at
37 C during 2 hrs, using ROR1-positive B-CLL cells, and at an antibody
concentration of 1 nM, a
decrease in the mean fluorescence intensity by internalization from time 0 to
2 hrs at 37 C (AMFI), as
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
21
measured by flow cytometry is between 120% to 0%, preferably from 100% to 0%,
of the AMFI of an
anti-ROR1 bivalent antibody of human IgG1 kappa (x) type comprising as light
chain variable domain
(VL) the sequence of SEQ ID NO:2 and as variable heavy chain domain (VH) the
sequence of SEQ ID
NO:6, in the same concentration and experimental conditions.
For a therapy using a T cell bispecific antibody comprising an anti-ROR1
antibody, it is preferred that the
antibody does not internalize as defined above for facilitating a stable
immune synapse between the tumor
cell and the T cell and effective T cell-mediated redirected cytotoxicity.
The term "reduction of mean fluorescence intensity" (AMFI) reflecting the
internalization of the said
anti-ROR1 antibody to ROR1-positive cells" or "MFI reduction" as used herein
refers to the percentage
of MFI reduction as calculated for each ROR1 antibodies relative to the
unspecific human IgG control
(MFI background) and ROR1 antibodies maintained on ice (MFImax) by using the
formula AMFI= 100 ¨ 100
X [(MFlexperimentai ¨ MHbackground) (MFImax ¨ MHbackground)] = MHexperimental
is the MFI measured with said
ROR1 antibody after 2h incubation at 37 C. An MFI reduction which is at least
75% blocked and
reversed by 10 M endocytosis inhibitor phenylarsine oxide is for example
caused by antibody
internalization while an MFI reduction which is not blocked by phenylarsine
oxide is caused by antibody
dissociation. Internalizing anti-ROR1 antibodies are known in the state of the
art (Baskar et al., Clin.
Cancer Res., 14(2): 396-404 (2008)).
Preferably the bispecific antibody according to the invention is characterized
in that an increase in MFI
value at time 2hrs in the presence of 3 M phenylarsine oxide (PAO) as compared
to MFI value at time
2hrs in the absence of PAO is not more than 30% , preferably not more than
20%, preferably not more
that 10%, even not more than detection level of the MFI value at time 0.
The term "target" as used herein means either ROR1 or CD3. The term "first
target and second target"
means either CD3 as first target and ROR1 as second target or means ROR1 as
first target and CD3 as
second target.
The term "antibody" as used herein refers to a monoclonal antibody. An
antibody consists of two pairs of
a "light chain" (LC) and a "heavy chain" (HC) (such light chain (LC) /heavy
chain pairs are abbreviated
herein as LC/HC). The light chains and heavy chains of such antibodies are
polypeptides consisting of
several domains. Each heavy chain comprises a heavy chain variable region
(abbreviated herein as HCVR
or VH) and a heavy chain constant region. The heavy chain constant region
comprises the heavy chain
constant domains CH1, CH2 and CH3 (antibody classes IgA, IgD, and IgG) and
optionally the heavy
chain constant domain CH4 (antibody classes IgE and IgM). Each light chain
comprises a light chain
variable domain VL and a light chain constant domain CL. The variable domains
VH and VL can be
further subdivided into regions of hypervariability, termed complementarity
determining regions (CDR),
interspersed with regions that are more conserved, termed framework regions
(FR). Each VH and VL is
composed of three CDRs and four FRs, arranged from amino-terminus to carboxy-
terminus in the
following order: FR1, CDR1, FR2, CDR2, FR3, CDR3, FR4. The "constant domains"
of the heavy chain
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
22
and of the light chain are not involved directly in binding of an antibody to
a target, but exhibit various
effector functions.
The "light chain of an antibody" as used herein is a polypeptide comprising in
N-terminal to C-terminal
direction a light chain variable domain (VL), and a light chain constant
domain (CL), abbreviated as VL-
CL. A "crossover light chain (VH-CL)" as used herein is a light chain wherein
the VL domain is replaced
by the respective VH domain. "The "heavy chain of an antibody" as used herein
is a polypeptide
comprising in N-terminal to C-terminal direction a heavy chain variable domain
(VH) and a constant
heavy chain domain 1 (CH1). A "crossover heavy chain (VL-CH1)" as used herein
is a heavy chain
wherein the VH domain is replaced by the respective VL domain.
There exist several approaches for CH3 -modifications to enforce the
heterodimerization, which are well
described e.g. in W096/27011, W098/050431, EP1870459,
W02007/110205,
W02007/147901, W02009/089004, W02010/129304, W02011/90754, W02011/143545,
W02012058768, W02013157954, W02013096291. Typically in all such approaches the
first CH3
domain and the second CH3 domains are both engineered in a complementary
manner so that each CH3
domain (or the heavy chain comprising it) cannot longer homodimerize with
itself but is forced to
heterodimerize with the complementary engineered other CH3 domain ( so that
the first and second CH3
domain heterodimerize and no homodimers between the two first or the two
second CH3 domains are
formed). These different approaches for improved heavy chain
heterodimerization are contemplated as
different alternatives in combination with the heavy -light chain
modifications (VH and VL
exchange/replacement in one binding arm and the introduction of substitutions
of charged amino acids
with opposite charges in the CH1/CL interface) in the antibodies according to
the invention which reduce
light chain mispairing, e.g. Bence-Jones type side products.
In one preferred embodiment of the invention, aa substitution or charge
variant is applied to the constant
domain CL at positions 123 and 124 and/or to the constant domain CH1 at
positions 147 and 213 in the
antibodies according to the invention which reduce light chain mispairing.
In one preferred embodiment of the invention (in case the antibody according
to the invention comprises
CH3 domains in the heavy chains) the CH3 domains of said multispecific
antibody according to the
invention can be altered by the "knob-into-holes" technology which is
described in detail with several
examples in e.g. WO 96/027011, Ridgway, J.B., et al., Protein Eng. 9 (1996)
617-621; and Merchant,
A.M. et al., Nat. Biotechnol. 16 (1998) 677-681; W098/ 050431. In this method
the interaction surfaces
of the two CH3 domains are altered to increase the heterodimerisation of both
heavy chains containing
these two CH3 domains. Each of the two CH3 domains (of the two heavy chains)
can be the "knob",
while the other is the "hole".
Thus in one embodiment of the invention said antibody according to the
invention (comprises a CH3
domain in each heavy chain and) is further characterized in that the first CH3
domain of the first heavy
chain of the antibody under a) and the second CH3 domain of the second heavy
chain of the antibody
under b) each meet at an interface which comprises an original interface
between the antibody CH3
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
23
domains, wherein said interface is altered to promote the formation of the
antibody according to the
invention, wherein the alteration is characterized in that:
i) the CH3 domain of one heavy chain is altered, so that within the
original interface of the CH3
domain of one heavy chain that meets the original interface of the CH3 domain
of the other heavy chain
within the antibody according to the invention, an amino acid residue is
replaced with an amino acid
residue having a larger side chain volume, thereby generating a protuberance
within the interface of the
CH3 domain of one heavy chain which is positionable in a cavity within the
interface of the CH3 domain
of the other heavy chain and
ii) the CH3 domain of the other heavy chain is altered, so that within the
original interface of the
second CH3 domain that meets the original interface of the first CH3 domain
within the antibody
according to the invention an amino acid residue is replaced with an amino
acid residue having a smaller
side chain volume, thereby generating a cavity within the interface of the
second CH3 domain within
which a protuberance within the interface of the first CH3 domain is
positionable.
Preferably said amino acid residue having a larger side chain volume is
selected from the group
consisting of arginine (R), phenylalanine (F), tyrosine (Y), tryptophan (W).
In one aspect of the invention both CH3 domains are further altered by the
introduction of cysteine (C) as
amino acid in the corresponding positions of each CH3 domain such that a
disulfide bridge between both
CH3 domains can be formed.
Other techniques for CH3 -modifications to enforcing the heterodimerization
are contemplated as
alternatives of the invention and described e.g. in W096/27011, W098/050431,
EP1870459,
W02007/110205, W02007/147901, W02009/089004, W02010/129304, W02011/90754,
W02011/143545, W02012/058768, W02013/157954, W02013/157953, W02013/096291.
In one embodiment the antibody according to the invention is of IgG2 isotype
and the heterodimerization
approach described in W02010/129304 can be used alternatively.
The term "antibody" includes e.g. mouse antibodies, human antibodies, chimeric
antibodies, humanized
antibodies and genetically engineered antibodies (variant or mutant
antibodies) as long as their
characteristic properties are retained. Especially preferred are human or
humanized antibodies, especially
as recombinant human or humanized antibodies. The terms "monoclonal antibody"
or "monoclonal
antibody composition" as used herein refer to a preparation of antibody
molecules of a single amino acid
composition.
The term "comprising" in regard to the bispecific antibody as used herein
means that the bispecific
antibody comprises as CD3 and ROR1 binders only those binders mentioned.
Therefore a bispecific
antibody according the invention comprising a monovalent anti-ROR1 antibody
specifically binding to
ROR1, and a monovalent antibody specifically binding to CD3 has in regard to
CD3 and ROR1 binding
only one binding valence for CD3 and only one valence for ROR1 and is
therefore bivalent. A bispecific
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
24
antibody according the invention comprising a bivalent anti-ROR1 antibody
specifically binding to
ROR1, and a monovalent antibody specifically binding to CD3 has in regard to
ROR1 binding two
binding valences and in regard to CD3 binding one valence and is therefore
trivalent. Preferably the
monovalent antibody specifically binding to CD3 is covalently linked at its C-
terminus to the N-terminus
of one variable chain of the antibody specifically binding to ROR1.
A "Fab fragment of an antibody" as used herein is a fragment on an antibody
that binds to antigens. A
Fab fragment of an antibody consists of two pairs of domains. In a wild-type
antibody it is composed of
one constant and one variable domain of each of the heavy chain (CH1 and VH)
and the light chain (CL
and VL). According to the invention such domain pairs can be, due to a
crossover, also VH-CL and VL-
CH1. In a wild-type antibody and according to the invention the domain of the
heavy and light chain
domain pairs of a Fab fragment are not chemically linked together and are
therefore not scFvs (single
chain variable fragments). "Crossover" according to the invention means that
preferably in one Fab the
domains VL and VH are replaced by each other. The term "Fab fragment" also
includes parts or all of the
hinge region, like Fab' fragment. As used herein, "F(ab)2 fragment" refers to
a bivalent monospecific
antibody fragment preferably with a Fc part.
The term "aa substitution or charge variant" as used herein means amino acid
substitution according to
the invention in that in a constant domain CL the amino acid at position 124
is substituted independently
by lysine (K), arginine (R) or histidine (H) (numbering according to Kabat),
and wherein in the respective
constant domain CH1 the amino acid at position 147 and the amino acid at
position 213 is substituted
independently by glutamic acid (E), or aspartic acid (D), and preferably in
addition in the constant
domain CL in the amino acid at position 123 is substituted independently by
lysine (K), arginine (R) or
histidine (H) and preferably by arginine (R).
A preferred combination of aa substitution is Q124K, E123R, K147E and K213E
(for example: E123R
means that glutamic acid (E) at position 123 is replaced by arginine (R).
Preferably a F(ab)2 fragment is
linked at the C-terminus by disulphide bond(s) in the hinge region and usually
such a "F(ab)2 fragment" is
a F(ab')2 fragment. A preferred combination of aa substitution is E124K,
E123K, K147E and K213E (for
example: El 23K means that glutamic acid (E) at position 123 is replaced by
lysine (K). Preferably a
F(ab)2 fragment is linked at the C-terminus by disulphide bond(s) in the hinge
region and usually such a
"F(ab)2 fragment" is a F(ab')2 fragment. Introduction of substitutions of
charged amino acids with
opposite charges in the constant domains CH1/CL of the antibodies according to
the invention reduces
light chain mispairing,
The term "ROR1 Fab" as used within the invention denotes a Fab fragment of the
antibody specifically
binding to ROR1. Due to the exchange of either the variable regions or the
constant regions in the anti-
ROR1 antibody Fab fragment (ROR1 Fab), such ROR1 Fab is referred to as "
ROR1cross Fab" or
"crossover ROR1 Fab fragment" According to the invention the ROR1 Fab is not a
ROR1crossFab. By
"connected" is meant that the Fab fragments are preferably linked by peptide
bonds, either directly or via
one or more peptide linker. The term "CD3 Fab" as used within the invention
denotes a Fab fragment of
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
the antibody specifically binding to CD3. The CD3 Fab is linked at its N-
terminus the C-terminus of the
ROR1 Fab. Due to the exchange of either the variable regions or the constant
regions in the CD3 Fab,
such CD3 Fab is referred to as "CD3 crossFab" or "crossover CD3 Fab fragment".
According to the
invention the CD3 Fab is preferably a crossFab.
5 The term "peptide linker" as used within the invention denotes a peptide
with amino acid sequences,
which is preferably of synthetic origin. These peptide linkers according to
invention are used to connect
one of the Fab fragments to the C-or N-terminus of the other Fab fragment to
form a multispecific
antibody according to the invention. Preferably said peptide linkers are
peptides with an amino acid
sequence with a length of at least 5 amino acids, preferably with a length of
5 to 100, more preferably of
10 10 to 50 amino acids. In one embodiment said peptide linker is (GxS)n or
(GxS)nGm with G = glycine, S
= serine, and (x = 3, n= 3, 4, 5 or 6, and m= 0, 1, 2 or 3) or (x = 4,n= 2, 3,
4 or 5 and m= 0, 1, 2 or 3),
preferably x = 4 and n= 2 or 3, more preferably with x = 4, n= 2.
Additionally, linkers may comprise (a
portion of) an immunoglobulin hinge region. In one embodiment said peptide
linker is (G4S)2 (SEQ ID:
NO 19).
15 There are five types of mammalian antibody heavy chains denoted by the
Greek letters: a, 6, c, y, and
(Janeway CA, Jr et al (2001). Immunobiology. 5th ed., Garland Publishing). The
type of heavy chain
present defines the class of antibody; these chains are found in IgA, IgD,
IgE, IgG, and IgM antibodies,
respectively (Rhoades RA, Pflanzer RG (2002). Human Physiology, 4th ed.,
Thomson Learning). Distinct
heavy chains differ in size and composition; a and y contain approximately 450
amino acids, while and
20 c have approximately 550 amino acids. Each heavy chain has two regions, the
constant region and the
variable region. The constant region is identical in all antibodies of the
same isotype, but differs in
antibodies of different isotype. Heavy chains y, a and 6 have a constant
region composed of three
constant domains CH1, CH2, and CH3 (in a line) , and a hinge region for added
flexibility (Woof J,
Burton D Nat Rev Immunol 4 (2004) 89-99); heavy chains and c have a constant
region composed of
25 four constant domains CH1, CH2, CH3, and CH4 (Janeway CA, Jr et al (2001).
Immunobiology. 5th ed.,
Garland Publishing). The variable region of the heavy chain differs in
antibodies produced by different B
cells, but is the same for all antibodies produced by a single B cell or B
cell clone. The variable region of
each heavy chain is approximately 110 amino acids long and is composed of a
single antibody domain. In
mammals there are only two types of light chain, which are called lambda (X)
and kappa 00. A light chain
has two successive domains: one constant domain CL and one variable domain VL.
The approximate
length of a light chain is 211 to 217 amino acids.
A bispecific antibody according to the invention, which comprises a Fc part,
can be of any class (e.g.
IgA, IgD, IgE, IgG, and IgM, preferably IgG or IgE), or subclass (e.g., IgGl,
IgG2, IgG3, IgG4, IgAl and
IgA2, preferably IgG1), whereby both antibodies, from which the bivalent
bispecific antibody according
to the invention is derived, have an Fc part of the same subclass( e.g. IgGl,
IgG4 and the like, preferably
IgG1), preferably of the same allotype (e.g. Caucasian).
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
26
A "Fc part of an antibody" is a term well known to the skilled artisan and
defined on the basis of papain
cleavage of antibodies. The antibodies according to the invention, which
comprise an Fc part, contain as
Fc part, preferably a Fc part derived from human origin and preferably all
other parts of the human
constant regions. The Fc part of an antibody is directly involved in
complement activation, Clq binding,
C3 activation and Fc receptor binding. While the influence of an antibody on
the complement system is
dependent on certain conditions, binding to Cl q is caused by defined binding
sites in the Fc part. Such
binding sites are known in the state of the art and described e.g. by Lukas,
TJ., et al., J. Immunol. 127
(1981) 2555-2560; Brunhouse, R., and Cebra, J.J., MoI. Immunol. 16 (1979) 907-
917; Burton, D.R., et
al., Nature 288 (1980) 338-344; Thommesen, J.E., et al., MoI. Immunol. 37
(2000) 995-1004; Idusogie,
E.E., et al., J. Immunol. 164 (2000) 4178-4184; Hezareh, M., et al., J. Virol.
75 (2001) 12161-12168;
Morgan, A., et al., Immunology 86 (1995) 319-324; and EP 0 307 434. Such
binding sites are e.g. L234,
L235, D270, N297, E318, K320, K322, P331 and P329 (numbering according to EU
index of Kabat, see
below). Antibodies of subclass IgGl, IgG2 and IgG3 usually show complement
activation, Clq binding
and C3 activation, whereas IgG4 do not activate the complement system, do not
bind Cl q and do not
activate C3. Preferably the Fc part is a human Fc part. Preferably the Fc part
is a human IgGlFc part.
Preferably the antibody according to the invention comprises in the human IgG1
Fc part amino acid
substitution of Pro329 with glycine or arginine and/or substitutions L234A and
L235A, preferably Pro329
with glycine and substitutions L234A and L235A.
Preferably the bispecific antibody according to the invention comprising
constant heavy regions
CH2/CH3 of IgG1 subclass is characterized in comprising the mutations L234A,
L235A and P239G
(numbering according to Kabat) to avoid FcR and Clq binding and minimizing
ADCC/CDC. The
advantage is that such an antibody of the invention mediates its tumor cell
killing efficacy purely by the
powerful mechanism of T-cell redirection/activation. Additional mechanisms of
action like effects on
complement system and on effector cells expressing FcR are avoided and the
risk of side-effects is
decreased.
Preferably the antibody according to the invention comprises as Fc part an Fc
variant of a wild-type
human IgG Fc region, said Fc variant comprising an amino acid substitution at
position Pro329 and at
least one further amino acid substitution, wherein the residues are numbered
according to the EU index of
Kabat, and wherein said antibody exhibits a reduced affinity to the human
FcyRIIIA and/or FcyRIIA and
/or FcyRI compared to an antibody comprising the wildtype IgG Fc region, and
wherein the ADCC
induced by said antibody is reduced to at least 20% of the ADCC induced by the
antibody comprising a
wild-type human IgG Fc region. In a specific embodiment Pro329 of a wild-type
human Fc region in the
antibody according to the invention is substituted with glycine or arginine or
an amino acid residue large
enough to destroy the proline sandwich within the Fc/Fcy receptor interface,
that is formed between the
proline329 of the Fc and tryptophane residues Trp 87 and Tip 110 of FcyRIII
(Sondermann et al.: Nature
406, 267-273 (20 July 2000)). In a further aspect of the invention the at
least one further amino acid
substitution in the Fc variant is 5228P, E233P, L234A, L235A, L235E, N297A,
N297D, or P33 1S and
still in another embodiment said at least one further amino acid substitution
is L234A (denotes that
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
27
leucine 234 is substituted by alanine) and L235A of the human IgG1 Fc region
or S228P and L235E of the
human IgG4 Fc region. Such Fc variants are described in detail in
W02012130831.
The constant heavy chain of an antibody according to the invention is
preferably of human IgGltype and
the constant light chain is preferably of human lambda (X) or kappa (x) type,
preferably of human kappa
(i)type.
The terms "monoclonal antibody" or "monoclonal antibody composition" as used
herein refer to a
preparation of antibody molecules of a single amino acid composition.
The term "chimeric antibody" refers to an antibody comprising a variable
region, i.e., binding region,
from one source or species and at least a portion of a constant region derived
from a different source or
species, usually prepared by recombinant DNA techniques. Chimeric antibodies
comprising a murine
variable region and a human constant region are preferred. Other preferred
forms of "chimeric antibodies"
encompassed by the present invention are those in which the constant region
has been modified or
changed from that of the original antibody to generate the properties
according to the invention,
especially in regard to Clq binding and/or Fc receptor (FcR) binding. Such
chimeric antibodies are also
referred to as "class-switched antibodies". Chimeric antibodies are the
product of expressed
immunoglobulin genes comprising DNA segments encoding immunoglobulin variable
regions and DNA
segments encoding immunoglobulin constant regions. Methods for producing
chimeric antibodies involve
conventional recombinant DNA and gene transfection techniques are well known
in the art. See, e.g.,
Morrison, S.L., et al., Proc. Natl. Acad. Sci. USA 81(1984) 6851-6855; US
Patent Nos. 5,202,238 and
5,204,244.
The term "humanized antibody" refers to antibodies in which the framework or
"complementarity
determining regions" (CDR) have been modified to comprise the CDR of an
immunoglobulin of different
specificity as compared to that of the parent immunoglobulin. In a preferred
embodiment, a murine CDR
is grafted into the framework region of a human antibody to prepare the
"humanized antibody." See, e.g.,
Riechmann, L., et al., Nature 332 (1988) 323-327; and Neuberger, M.S., et al.,
Nature 314 (1985) 268-
270. Particularly preferred CDRs correspond to those representing sequences
recognizing the targets
noted above for chimeric antibodies. Other forms of "humanized antibodies"
encompassed by the present
invention are those in which the constant region has been additionally
modified or changed from that of
the original antibody to generate the properties according to the invention,
especially in regard to Clq
binding and/or Fc receptor (FcR) binding.
The term "human antibody", as used herein, is intended to include antibodies
having variable and
constant regions derived from human germ line immunoglobulin sequences. Human
antibodies are well-
known in the state of the art (van Dijk, M.A., and van de Winkel, J.G., Curr.
Opin. Chem. Biol. 5 (2001)
368-374). Human antibodies can also be produced in transgenic animals (e.g.,
mice) that are capable,
upon immunization, of producing a full repertoire or a selection of human
antibodies in the absence of
endogenous immunoglobulin production. Transfer of the human germ-line
immunoglobulin gene array in
such germ-line mutant mice will result in the production of human antibodies
upon target challenge (see,
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
28
e.g., Jakobovits, A., et al., Proc. Natl. Acad. Sci. USA 90 (1993) 2551-2555;
Jakobovits, A., et al., Nature
362 ( 1993) 255-258; Bruggemann, M., et al., Year Immunol. 7 (1993) 33-40).
Human antibodies can
also be produced in phage display libraries (Hoogenboom, H.R., and Winter, G.,
J. MoI. Biol. 227 (1992)
381-388; Marks, J.D., et al., J. MoI. Biol. 222 (1991) 581-597). The
techniques of Cole et al. and Boerner
et al. are also available for the preparation of human monoclonal antibodies
(Cole et al., Monoclonal
Antibodies and Cancer Therapy, Alan R. Liss, p. 77 (1985); and Boerner, P., et
al., J. Immunol. 147
(1991) 86-95). As already mentioned for chimeric and humanized antibodies
according to the invention
the term "human antibody" as used herein also comprises such antibodies which
are modified in the
constant region to generate the properties according to the invention,
especially in regard to Clq binding
and/or FcR binding, e.g. by "class switching" i.e. change or mutation of Fc
parts (e.g. from IgG1 to IgG4
and/or IgGl/IgG4 mutation).
The term "recombinant human antibody", as used herein, is intended to include
all human antibodies that
are prepared, expressed, created or isolated by recombinant means, such as
antibodies isolated from a host
cell such as a NSO or CHO cell or from an animal (e.g. a mouse) that is
transgenic for human
immunoglobulin genes or antibodies expressed using a recombinant expression
vector transfected into a
host cell. Such recombinant human antibodies have variable and constant
regions in a rearranged form.
The recombinant human antibodies according to the invention have been
subjected to in vivo somatic
hypermutation. Thus, the amino acid sequences of the VH and VL regions of the
recombinant antibodies
are sequences that, while derived from and related to human germ line VH and
VL sequences, may not
naturally exist within the human antibody germ line repertoire in vivo.
The "variable domain" (variable domain of a light chain (VL), variable region
of a heavy chain (VH)) as
used herein denotes each of the pair of light and heavy chains which is
involved directly in binding the
antibody to the target. The domains of variable human light and heavy chains
have the same general
structure and each domain comprises four framework (FR) regions whose
sequences are widely
conserved, connected by three "hypervariable regions" (or complementarity
determining regions, CDRs).
The framework regions adopt a 13-sheet conformation and the CDRs may form
loops connecting the 13-
sheet structure. The CDRs in each chain are held in their three-dimensional
structure by the framework
regions and form together with the CDRs from the other chain the target
binding site. The antibody heavy
and light chain CDR3 regions play a particularly important role in the binding
specificity/affinity of the
antibodies according to the invention and therefore provide a further object
of the invention.
The terms "hypervariable region" or "target-binding portion of an antibody"
when used herein refer to the
amino acid residues of an antibody which are responsible for target-binding.
The hypervariable region
comprises amino acid residues from the "complementarity determining regions"
or "CDRs".
"Framework" or "FR" regions are those variable domain regions other than the
hypervariable region
residues as herein defined. Therefore, the light and heavy chains of an
antibody comprise from N- to C-
terminus the domains FR1, CDR1, FR2, CDR2, FR3, CDR3, and FR4. CDRs on each
chain are separated
by such framework amino acids. Especially, CDR3 of the heavy chain is the
region which contributes
most to target binding. CDR and FR regions are determined according to the
standard definition of Kabat
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
29
et al., Sequences of Proteins of Immunological Interest, 5th ed., Public
Health Service, National Institutes
of Health, Bethesda, MD (1991).
The term "target" or "target molecule" as used herein are used interchangeable
and refer to human ROR1
and human CD3e.
The term "epitope" includes any polypeptide determinant capable of specific
binding to an antibody. In
certain embodiments, epitope determinant include chemically active surface
groupings of molecules such
as amino acids, sugar side chains, phosphoryl, or sulfonyl, and, in certain
embodiments, may have
specific three dimensional structural characteristics, and or specific charge
characteristics. An epitope is a
region of a target that is bound by an antibody.
In general there are two vectors encoding the light chain and heavy chain of
said antibody specifically
binding to the first target, and further two vectors encoding the light chain
and heavy chain of said
antibody specifically binding to the second target. One of the two vectors is
encoding the respective light
chain and the other of the two vectors is encoding the respective heavy chain.
However in an alternative
method for the preparation of a bispecific antibody according to the
invention, only one first vector
encoding the light chain and heavy chain of the antibody specifically binding
to the first target and only
one second vector encoding the light chain and heavy chain of the antibody
specifically binding to the
second target can be used for transforming the host cell.
The term "nucleic acid or nucleic acid molecule", as used herein, is intended
to include DNA molecules
and RNA molecules. A nucleic acid molecule may be single-stranded or double-
stranded, but preferably
is double-stranded DNA.
As used herein, the expressions "cell," "cell line," and "cell culture" are
used interchangeably and all such
designations include progeny. Thus, the words "transformants" and "transformed
cells" include the
primary subject cell and cultures derived therefrom without regard for the
number of transfers. It is also
understood that all progeny may not be precisely identical in DNA content, due
to deliberate or
inadvertent mutations. Variant progeny that have the same function or
biological activity as screened for
in the originally transformed cell are included. Where distinct designations
are intended, it will be clear
from the context.
The term "transformation" as used herein refers to process of transfer of a
vectors/nucleic acid into a host
cell. If cells without formidable cell wall barriers are used as host cells,
transfection is carried out e.g. by
the calcium phosphate precipitation method as described by Graham and Van der
Eh, Virology 52 (1978)
546ff. However, other methods for introducing DNA into cells such as by
nuclear injection or by
protoplast fusion may also be used. If prokaryotic cells or cells which
contain substantial cell wall
constructions are used, e.g. one method of transfection is calcium treatment
using calcium chloride as
described by Cohen SN, et al, PNAS 1972, 69 (8): 2110-2114.
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
Recombinant production of antibodies using transformation is well-known in the
state of the art and
described, for example, in the review articles of Makrides, S. C, Protein
Expr. Purif. 17 (1999) 183-202;
Geisse, S., et al., Protein Expr. Purif. 8 (1996) 271-282; Kaufman, RJ., MoI.
Biotechnol. 16 (2000) 151-
161; Werner, R.G., et al., Arzneimittelforschung 48 (1998) 870-880 as well as
in US6331415 and
5 US4816567.
As used herein, "expression" refers to the process by which a nucleic acid is
transcribed into mRNA
and/or to the process by which the transcribed mRNA (also referred to as
transcript) is subsequently being
translated into peptides, polypeptides, or proteins. The transcripts and the
encoded polypeptides are
collectively referred to as gene product. If the polynucleotide is derived
from genomic DNA, expression
10 in a eukaryotic cell may include splicing of the mRNA.
A "vector" is a nucleic acid molecule, in particular self-replicating, which
transfers an inserted nucleic
acid molecule into and/or between host cells. The term includes vectors that
function primarily for
insertion of DNA or RNA into a cell (e.g., chromosomal integration),
replication of vectors that function
primarily for the replication of DNA or RNA, and expression vectors that
function for transcription
15 and/or translation of the DNA or RNA. Also included are vectors that
provide more than one of the
functions as described.
An "expression vector" is a polynucleotide which, when introduced into an
appropriate host cell, can be
transcribed and translated into a polypeptide. An "expression system" usually
refers to a suitable host cell
comprised of an expression vector that can function to yield a desired
expression product.
20 The bispecific antibodies according to the invention are preferably
produced by recombinant means. Such
methods are widely known in the state of the art and comprise protein
expression in prokaryotic and
eukaryotic cells with subsequent isolation of the antibody polypeptide and
usually purification to a
pharmaceutically acceptable purity. For the protein expression, nucleic acids
encoding light and heavy
chains or fragments thereof are inserted into expression vectors by standard
methods. Expression is
25 performed in appropriate prokaryotic or eukaryotic host cells like CHO
cells, NSO cells, 5P2/0 cells,
HEK293 cells, COS cells, yeast, or E.coli cells, and the antibody is recovered
from the cells (supernatant
or cells after lysis). The bispecific antibodies may be present in whole
cells, in a cell lysate, or in a
partially purified or substantially pure form. Purification is performed in
order to eliminate other cellular
components or other contaminants, e.g. other cellular nucleic acids or
proteins, by standard techniques,
30 including alkaline/SDS treatment, column chromatography and others well
known in the art. See
Ausubel, F., et al., ed., Current Protocols in Molecular Biology, Greene
Publishing and Wiley
Interscience, New York (1987).
Expression in NSO cells is described by, e.g., Barnes, L.M., et al.,
Cytotechnology 32 (2000) 109-123;
and Barnes, L.M., et al., Biotech. Bioeng. 73 (2001) 261-270. Transient
expression is described by, e.g.,
Durocher, Y., et al., Nucl. Acids. Res. 30 (2002) E9. Cloning of variable
domains is described by Orlandi,
R., et al., Proc. Natl. Acad. Sci. USA 86 (1989) 3833-3837; Carter, P., et
al., Proc. Natl. Acad. Sci. USA
89 (1992) 4285-4289; and Norderhaug, L., et al., J. Immunol. Methods 204
(1997) 77-87. A preferred
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
31
transient expression system (HEK293) is described by Schlaeger, E.- J., and
Christensen, K., in
Cytotechnology 30 (1999) 71-83 and by Schlaeger, E.-J., in J. Immunol. Methods
194 (1996) 191-199.
The control sequences that are suitable for prokaryotes, for example, include
a promoter, optionally an
operator sequence, and a ribosome binding site. Eukaryotic cells are known to
utilize promoters,
enhancers and polyadenylation signals.
Nucleic acid is "operably linked" when it is placed into a functional
relationship with another nucleic acid
sequence. For example, DNA for a presequence or secretory leader is operably
linked to DNA for a
polypeptide if it is expressed as a preprotein that participates in the
secretion of the polypeptide; a
promoter or enhancer is operably linked to a coding sequence if it affects the
transcription of the
sequence; or a ribosome binding site is operably linked to a coding sequence
if it is positioned so as to
facilitate translation. Generally, "operably linked" means that the DNA
sequences being linked are
contiguous, and, in the case of a secretory leader, contiguous and in reading
frame. However, enhancers
do not have to be contiguous. Linking is accomplished by ligation at
convenient restriction sites. If such
sites do not exist, the synthetic oligonucleotide adaptors or linkers are used
in accordance with
conventional practice.
The bispecific antibodies are suitably separated from the culture medium by
conventional
immunoglobulin purification procedures such as, for example, protein A-
Sepharose, hydroxylapatite
chromatography, gel electrophoresis, dialysis, or affinity chromatography. DNA
or RNA encoding the
monoclonal antibodies is readily isolated and sequenced using conventional
procedures. The hybridoma
cells can serve as a source of such DNA and RNA. Once isolated, the DNA may be
inserted into
expression vectors, which are then transfected into host cells such as HEK293
cells, CHO cells, or
myeloma cells that do not otherwise produce immunoglobulin protein, to obtain
the synthesis of
recombinant monoclonal antibodies in the host cells.
Amino acid sequence variants (or mutants) of the bispecific antibody are
prepared by introducing
appropriate nucleotide changes into the antibody DNA, or by nucleotide
synthesis. Such modifications
can be performed, however, only in a very limited range, e.g. as described
above. For example, the
modifications do not alter the above mentioned antibody characteristics such
as the IgG isotype and target
binding, but may improve the yield of the recombinant production, protein
stability or facilitate the
purification.
T cell bispecific (TCB) binders have very high concentration/tumor-cell-
receptor-occupancy dependent
potency in cell killing (e.g. EC50 in in vitro cell killing assays in the sub-
or low picomolar range; Dreier
et al. Int .1- Cancer 2002), T-cell bispecific binder (TCB) are given at much
lower doses than conventional
monospecific antibodies. For example, blinatumomab (CD19xCD3) is given at a
continuous intravenous
dose of 5 to 15 g/m2/day (i.e. only 0.035 to 0.105 mg/m2/week) for treatment
of acute lymphocytic
leukemia or 60 ug/m2/day for treatment of Non Hodgkin Lymphoma, and the serum
concentrations at
these doses are in the range of 0.5 to 4 ng/ml (Klinger et al., Blood 2012;
Topp et al., J Clin Oncol 2011;
Goebeler et al. Ann Oncol 2011). Due to the very short elimination half life
of blinatumomab clinical
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
32
administration is via continuous infusion via pump carried at the patients
body. Due to longer
elimination half life of the antibodies of this invention it is envisaged that
for an antibodyaccording to the
invention subcutaneous administration is possible and preferred in the
clinical settings (preferably in the
dose range of 0.1 to 10 mg/m2once or twice a week, preferably even lower
doses). Even at these low
concentrations/doses/receptor occupancies, TCB can cause considerable adverse
events (Klinger et al.,
Blood 2012). Improved pharmacokinetics properties of the antibodies of the
invention are one measure to
potentially reduce adverse events.
In principle it is possible to produce bispecific antibodies against CD3 and
ROR1 in all formats known in
the state of the art. A wide variety of recombinant bispecific antibody
formats have been developed in the
recent past, e.g. by fusion of, e.g. an IgG antibody format and single chain
domains (see e.g. Kontermann
RE, mAbs 4:2, (2012) 1-16). Bispecific antibodies wherein the variable domains
VL and VH or the
constant domains CL and CH1 are replaced by each other are described in
W02009080251 and
W02009080252. Antibody formats and formats of bispecific and multispecific
antibodies are also
pepb odi es (W0200244215), Novel Antigen Receptor ("NAR") (W02003014161),
diabody-diabody
dimers "TandAbs" (W02003048209), polyalkylene oxide-modified scFv (US7150872),
humanized rabbit
antibodies (W02005016950), synthetic immunoglobulin domains (W02006072620),
covalent diabodies
(W02006113665), flexibodies (W02003025018), domain antibodies, dAb
(W02004058822), vaccibody
(W02004076489), antibodies with new world primate framework (W02007019620),
antibody-drug
conjugate with cleavable linkers (W02009117531), IgG4 antibodies with hinge
region removed
(W02010063785), bispecific antibodies with IgG4 like CH3 domains
(W02008119353), camelid
Antibodies (US6838254), nanobodies (US7655759), CAT diabodies (US5837242),
bispecific scFv2
directed against target antigen and CD3 (US7235641), ), sIgA plAntibodies
(US6303341), minibodies
(US5837821), IgNAR (US2009148438), antibodies with modified hinge and Fc
regions (US2008227958,
US20080181890), trifunctional antibodies (US5273743), triomabs (US6551592),
troybodies
(US6294654).
An antibody according to the invention can be administered once or twice a
week s.c. administration.
A bispecific trivalent antibody according to the invention has advantages on
the potency, predictability
for efficacy and safety.
An antibody according to the invention with bivalency to ROR1 and monovalency
to CD3 favors
binding to the tumor target ROR1 on malignant cells over CDR on T cells in
circulation and avoids CD3
sink, thus increasing drug exposure in the tumor.
The following examples, sequence listing and figures are provided to aid the
understanding of the present
invention, the true scope of which is set forth in the appended claims. It is
understood that modifications
can be made in the procedures set forth without departing from the spirit of
the invention.
Sequence listing
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
33
SE Q
NO: Name aa sequence
MHRPRRRGTRPPLLALLAALLLAARGAAAQETEL S
V SAELVP T S SWNISSELNKDSYLTLDEPMNNITT SL
GQTAELHCKVSGNPPPTIRWFKNDAPVVQEPRRL S
FRSTIYGSRLRIRNLDTTDTGYFQCVATNGKEVVSS
TGVLFVKFGPPPTASP GYSDEYEEDGFCQPYRGIAC
ARFIGNRTVYMESLHMQGEIENQITAAFTMIGTS SH
L SDKC SQFAIP SL CHYAFPYCDETS SVP KP RDL C RD
ECEILENVL CQ TEYIFARSNP MIL MRLKLPNCEDLP
QPESPEAANCIRIGIPMADPINKNHKCYNSTGVDYR
GTVSVTKSGRQCQPWNSQYPHTHTFTALRFPELNG
GHSYCRNP GNQKEAPWCFTLDENFKSDL CDIPACD
SKDSKEKNKMEILYILVP SVAIPLAIALLFFFICVCR
NNQKS SSAPVQRQPKHVRGQNVEMSMLNAYKPKS
1 ROR1 extracellular domain KAKELPL SAVRFMEEL GECAFGKIYKGHLYLP GMD
HAQLVAIKTLKDYNNPQQWTEFQQEASLMAELHH
PNIVCLL GAVTQEQPVCMLFEYINQGDLHEFLIMRS
PHSDVGCS SDEDGTVKS SL DH GD FL HIAI QIAAGME
YL S SHFFVHKDLAARNILIGEQLHVKISDL GL SREIY
SADYYRV Q S KS LLP IRWMPP EAIMYGKF SSD SD IW
SFGVVLWEIF SF GL QPYYGF SNQEVIEMVRKRQLLP
C SEDCPPRMYSLMTECWNEIP SRRPRFKDIHVRL RS
WE GL S SHTS ST TP SGGNATTQTT SL SA S PV SNL SNP
RYPNYMFP SQGITPQGQIAGFIGPPIPQNQRFIPINGY
P1PPGYAAFPAAHYQPTGPPRVIQHCPPPKSRSPSSA
SGSTSTGHVTSLPSSGSNQEANIPLLPHMSIPNHPGG
MGITVFGNKSQKPYKIDSKQASLL GDANIHGHTES
MISAEL
ELVLTQSP SVSAALGSPAKITCTL SSAHKTDTIDWY
QQL QGEAPRYLMQVQSDGSYTKRP GVPDRF SG S SS
2 Mab ROR1 VL
GADRYLIIP SVQADDEADYYCGADYIGGYVFGGGT
QLTVTG
3 CDR1 L TL S SAHKTDTID
4 CDR2L GSYTKRP
CDR3 L GADYIGGYV
QEQLVESGGRLVTP GGSLTL SCKASGFDFSAYYMS
WVRQAPGKGLEWIATIYP SSGKTYYATWVNGRFTI
6
Ma ROR1 VH
S SDNAQNTVDL QMNSLTAADRATYF CARD SYADD
GAL FNIWGP GTLVTIS S
7 CDR1 H AYYMS
8 CDR2H TIYP S SGKTYYATWVNG
9 CDR3 H DSYADDGALFNI
EVQLVESGGGLVQPGGSLKL SCAASGFTFNKYAM
1 Ma b CD3 NWVRQAP GKGLEWVARIRSKYNNYATYYADSVK
0 VH (H2C)
DRFTISRDD SKNTAYL QMNNLKTEDTAVYYCVRH
GNFGNSYISYWAYWGQGTLVTVS S
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
34
QTVVTQEP SLTV SP GGTVTLT CG S STGAVT S GYYP
NWVQQKPGQAPRGLIGGTKFLAPGTPARF SGSLLG
11 Mab CD3 VL (H2C)
GKAALTLSGVQPEDEAEYYCALWYSNRWVFGGG
TKLTVL
12 CDR1H (H2C) GFTFNKYAMN
13 CDR2H (H2C) RIRSKYNNYATYYAD SVKD
14 CDR3H (H2C) HGNFGNSYISYWAY
15 CDR1L (H2C) GS ST GAVT SGYYPN
16 CDR2L (H2C) GTKFLAP
17 CDR3L (H2C) ALWYSNRWV
18 Extracellular fragment of ROR1 WNIS SELNKDSYLTL
19 Linker GGGGSGGGGS
20 Intracellular fragment of ROR1 KS QKPYKIDSKQAS
EVQLLESGGGLVQPGGSLRL SCAASGFTFSTYAMN
WVRQAPGKGLEWVSRIRSKYNNYATYYADSVKG
21 Mab CD3 VH (CH2527)
RFTISRDDSKNTLYLQMNSLRAEDTAVYYCVRHG
NFGNSYVSWFAYWGQGTLVTVS S
QAVVTQEP SLTV SP GGTVTLT CG S STGAVTT SNYA
22 M CD3 NWVQEKPGQAFRGLIGGTNKRAPGTPARF SGSLLG
ab VL (CH2527)
GKAALTLSGAQPEDEAEYYCALWYSNLWVFGGGT
KLTVL
23 CDR1H (CH2527) TYAMN
24 CDR2H (CH2527) RIRSKYNNYATYYAD SVKG
25 CDR3H (CH2527) HGNFGNSYVSWFAY
26 CDRL1 (CH2527) GS STGAVTTSNYAN
27 CDRL2 (CH2527) GTNKRAP
28 CDRL3 (CH2527) ALWYSNLWV
QEQLVESGGRLVTPGGSLTL SCKASGFDFSAYYMS
WVRQAPGKGLEWIATIYP SSGKTYYATWVNGRFTI
S SDNAQNTVDL QMNSLTAADRATYF CARD SYADD
GAL FNIWGP GTLVTI S SA ST KGP SVFPLAP S SKS T SG
GTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPA
VLQ SSGLYSLS SVVTVPS S SLGTQTYICNVNHKP SN
29 ROR1 hum IgG1 HC LALA PG TKVDKKVEPKSCDKTHTCPPCPAPEAAGGPSVFLF
PPKPKDTLMISRTPEVTCVVVDVSHEDPEVKFNWY
VDGVEVHNAKTKPREEQYNSTYRVVSVLTVLHQD
WLNGKEYKCKVSNKALGAPIEKTISKAKGQPREPQ
VYTLPPSRDELTKNQVSLTCLVKGFYP SDIAVEWES
NGQPENNYKTTPPVLDSDGSFFLYSKLTVDKSRWQ
QGNVF SCSVMHEALHNHYTQKSL SL SP GK
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
MGW S CIIL FLVATAT GVH SELVL TQ SP SVSAAL G SP
AKITC TLS SAHKTDTIDWYQQLQGEAPRYLMQVQ S
DGSYTKRPGVPDRF SG S S SGADRYLIIP SVQADDEA
30 ROR1 hum IgG1 LC DYYCGADYIGGYVF GGGTQLTVL GQPKAAP SVTL
FP P S SEEL QANKATLVCLISDFYP GAVTVAWKADS
SPVKAGVETTTP SKQSNNKYAAS SYL SLTPEQWKS
HRSYSCQVTHEGSTVEKTVAPTEC S
QEQLVESGGRLVTP GGSLTL SCKASGFDFSAYYMS
WVRQAPGKGLEWIATIYP SSGKTYYATWVNGRFTI
S SDNAQNTVDL QMNSLTAADRATYF CARD SYADD
GAL FNIWGP GTLVTIS SA STKGP SVFPLAP SSKST SG
GTAAL GCLVKDYFP EPVTVSWNS GAL T S GVHTFPA
VLQ SSGLYSL S SVVTVP S S SLGTQTYICNVNHKP SN
TKVDKKVEPKSCDGGGGSGGGGSEVQLLESGGGL
VQP GGSLRL SCAASGFTF STYAMNWVRQAP GKGL
EWVSRIRSKYNNYATYYADSVKGRFTISRDDSKNT
31 ROR1 x CD3 VH CL HC knob LYLQMNSLRAEDTAVYYCVRHGNFGNSYVSWFA
LALA PG YWGQGTLVTVS SASVAAP SVFIFPP SDEQLKSGTAS
VVCLLNNFYPREAKVQWKVDNALQSGNSQESVTE
QDSKD STYSL S STLTL SKADYEKHKVYACEVTHQG
L S SPVTKSFNRGE CDKTHT CP P CPAPEAAGGP SVFL
FPPKPKDTLMISRTPEVTCVVVDVSHEDPEVKFNW
YVDGVEVHNAKTKPREEQYNSTYRVVSVLTVLHQ
DWLNGKEYKCKVSNKALGAPIEKTISKAKGQPREP
QVYTLPP CRDELTKNQVSLWCLVKGFYP SDIAVEW
ESNGQPENNYKTTPPVLDSDGSFFLYSKLTVDKSR
WQQ GNVF SC SVMHEALHNHYTQKSL SL SP GK
QEQLVESGGRLVTP GGSLTL SCKASGFDFSAYYMS
WVRQAP GKGLEWIATIYP S SGKTYYATWVNGRFTI
S SDNAQNTVDL QMNSLTAADRATYF CARD SYADD
GAL FNIWGP GTLVTIS SA STKGP SVFPLAP SSKST SG
GTAAL GCLVKDYFP EPVTVSWNS GAL T S GVHTFPA
VLQ SSGLYSL S SVVTVP S S SLGTQTYICNVNHKP SN
32 ROR1 HC hole LALA PG TKVDKKVEPKS CDKTHT CP P CPAPEAAGGP SVFLF
PPKPKDTLMISRTPEVTCVVVDVSHEDPEVKFNWY
VDGVEVHNAKTKPREEQYNSTYRVVSVLTVLHQD
WLNGKEYKCKVSNKALGAPIEKTISKAKGQPREPQ
VC TLP P SRDELTKNQVSL SCAVKGFYP SDIAVEWE S
NGQPENNYKTTPPVLDSDGSFFLVSKLTVDKSRWQ
QGNVF SCSVMHEALHNHYTQKSL SL SP GK
QAVVTQEP SLTV SP GGTVTLT CG S ST GAVTT SNYA
NWVQEKP GQAFRGLIGGTNKRAP GTPARF SGSLLG
GKAALTL SGAQPEDEAEYYCALWYSNLWVF GGGT
33 CD3 VL_CH1 KLTVL S SASTKGP SVFP LAP S SKSTSGGTAALGCLV
KDYFP EPVTVSWNS GALT S GVHTFPAVL Q S SGLYS
L S SVVTVP S S SLGTQTYICNVNHKP SNTKVDKKVEP
KS C
QEQLVESGGRLVTP GGSLTL SCKASGFDFSAYYMS
WVRQAPGKGLEWIATIYP SSGKTYYATWVNGRFTI
34 ROR1 x CD3 VH_CL S SDNAQNTVDL QMNSLTAADRATYF CARD SYADD
GAL FNIWGP GTLVTIS SA STKGP SVFPLAP SSKST SG
GTAAL GCLVKDYFP EPVTVSWNS GAL T S GVHTFPA
VLQ SSGLYSL S SVVTVP S S SLGTQTYICNVNHKP SN
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
36
TKVDKKVEPKSCDGGGGSGGGGSEVQLLESGGGL
VQP GGSLRL SCAASGFTF STYAMNWVRQAP GKGL
EWVSRIRSKYNNYATYYADSVKGRFTISRDDSKNT
LYL QMNSLRAEDTAVYYCVRHGNFGNSYVSWFA
YWGQGTLVTVS SA SVAAP SVFIFPP S DE QL KS GTA S
VVCLLNNFYPREAKVQWKVDNALQSGNSQESVTE
QDSKD STY S L S ST LTL SKADYEKHKVYACEVTHQG
L S SPVTKSFNRGEC
QEQLVE SGGRLVTP GGSLTLSCKASGFDF SAYYMS
WVRQAPGKGLEWIATIYP SSGKTYYATWVNGRFTI
S SDNAQNTVDL QMNSLTAADRATYF CARD SYADD
GAL FNIWGP GTLVTIS SA ST KGP SVFPLAP S S KS T SG
GTAAL GCLVKDYFPEPVTVSWNS GAL T S GVHTFPA
VLQ SSGLYSL S SVVTVPS S SLGTQTYICNVNHKP SN
TKVDKKVEPKSCDGGGGSGGGGSQEQLVESGGRL
VTP GGSLTL SCKASGFDF SAYYMSWVRQAP GKGL
EWIATIYPS S GKTYYATWVNGRFT I S S DNA QNTVD
LQMNSLTAADRATYFCARDSYADDGALFNIWGPG
35 (ROR1)2 x CD3 VH_CL TLVTIS SA ST KGP SVF P LAP S SK ST S GGTAAL
GC LVK
DYFPEPVTV SWNS GALT S GVHTFPAVLQ S SGLYSL
S SVVTVP S S SL GT QTYICNVNHKP SNTKVDKKVEP
KS CDGGGGSGGGGSEVQLLESGGGLVQP GGSLRL S
CAASGFTF STYAMNWVRQAP GKGLEWVSRIRSKY
NNYATYYADSVKGRFTISRDDSKNTLYL QMN SLR
AEDTAVYYCVRHGNFGNSYVSWFAYWGQGTLVT
VS SA SVAAP SVF IF PP S DE Q LKS GTA SVVC LLNNFY
PREAKVQWKVDNALQSGNSQESVTEQDSKDSTYS
L S STLTL SKADYEKHKVYACEVTHQGL S SPVTKSF
NRGEC
MGWSCIILFLVATATGVHSDKTHTCPPCPAPEAAG
GP SVFLFPPKPKDTL MI SRTPEVT CVVVDV SHEDPE
VKFNWYVDGVEVHNAKTKPREEQYNSTYRVVSV
LTVLHQDWLNGKEYKC KVSNKAL GAPIEKTISKAK
36 Fc hole LALA PG
GQPREPQVCTLPP SRDELTKNQVSL SCAVKGFYPS
DIAVEWESNGQPENNYKTTPPVLD SD GSFFLV SKL
TVD KS RWQQGNVF SC SVMHEALHNHYTQKSL SL S
P GK
QEQLVESGGRLVTP GGSLTL SCKASGFDFSAYYMS
WVRQAPGKGLEWIATIYP SSGKTYYATWVNGRFTI
S SDNAQNTVDL QMNSLTAADRATYF CARD SYADD
GAL FNIWGP GTLVTIS SA ST KGP SVFPLAP S S KS T SG
GTAAL GCLVEDYFPEPVTVSWNS GAL T S GVHTFPA
VLQ SSGLYSL S SVVTVPS S SLGTQTYICNVNHKP SN
TKVDEKVEPKSCDGGGGSGGGGSQAVVTQEPSLT
ROR1 VH CHlcv x CD3 VSP GGTVTLT C GS ST GAVTT SNYANWVQEKPGQA
37
VL CH1 Fc knob LALA PG FRGLIGGTNKRAP GTPARF SGSLLGGKAALTL S GA
QPEDEAEYYCALWYSNLWVFGGGTKLTVL S SAST
KGPSVFPLAPS SKSTSGGTAALGCLVKDYFPEPVTV
S WNS GAL T S GVHTFPAVLQ S SGLYSL SSVVTVP S SS
L GT QTYICNVNHKP SNTKVDKKVEPKS CDKTHT CP
PCPAPEAAGGP SVFLFPPKPKDTL MI SRTPEVTCVV
VDVSHEDPEVKFNWYVDGVEVHNAKTKPREEQY
NSTYRVVSVLTVLHQDWLNGKEYKCKVSNKALG
AP1EKTIS KAKGQPREPQVYTLPPCRDELTKNQVSL
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
37
WCLVKGFYP SDIAVEWESNGQPENNYKTTPPVLDS
DGSFFLYSKLTVDKSRWQQGNVFSC SVMHEALHN
HYTQKSL SL SP GK
QEQLVESGGRLVTP GGSLTL SCKASGFDFSAYYMS
WVRQAPGKGLEWIATIYP S SGKTYYATWVNGRFTI
S SDNAQNTVDL QMNSLTAADRATYF CARD SYADD
GAL FNIWGP GTLVTIS SA STKGP SVFPLAP SSKST SG
GTAALGCLVEDYFPEPVTVSWNSGALTSGVHTFPA
VLQ SSGLYSL S SVVTVPS S SLGTQTYICNVNHKP SN
38 RORlcv HC hole LALA PG TKVDEKVEPKS CDKTHT CPP CPAPEAAGGP SVFL FP
PKPKDTLMISRTPEVTCVVVDVSHEDPEVKFNWYV
DGVEVHNAKTKPREEQYNSTYRVVSVLTVLHQDW
LNGKEYKC KVSNKALGAPIEKTISKAKGQPREPQV
CTLPP SRDELTKNQVSL SCAVKGFYP SDIAVEWE SN
GQPENNYKTTPPVLDSDGSFFLVSKLTVDKSRWQQ
GNVF SC SVMHEALHNHYTQKSL SL SP GK
EVQLLESGGGLVQP GGSLRL SCAASGFTFSTYAMN
WVRQAP GKGLEWVSRIRSKYNNYATYYADSVKG
RFTISRDDSKNTLYLQMNSLRAEDTAVYYCVRHG
39 CD3 VH_CL NFGNSYVSWFAYWGQGTLVTVS SASVAAP SVFIFP
P SDEQLKSGTASVVCLLNNFYPREAKVQWKVDNA
LQ SGNSQESVTEQDSKDSTYSLSSTLTLSKADYEKH
KVYACEVTHQGL S SPVTKSFNRGEC
ELVLTQ SP SVSAALGSPAKITCTL SSAHKTDTIDWY
QQL QGEAPRYLMQVQSDGSYTKRP GVPDRF SG S SS
GADRYLIIPSVQADDEADYYCGADYIGGYVFGGGT
40 RORlcv hum Ig G1 LC lambda QLTVLGQPKAAPSVTLFPP S SKKLQANKATLVCLIS
DFYP GAVTVAWKADS SPVKAGVETTTP SKQSNNK
YAAS SYL SLTPEQWKSHRSYSCQVTHEGSTVEKTV
APTECS
ELVLTQ SP SVSAALGSPAKITCTL SSAHKTDTIDWY
QQL QGEAPRYLMQVQSDGSYTKRP GVPDRF SG S SS
GADRYLIIPSVQADDEADYYCGADYIGGYVFGGGT
41 RORlcv hum IgG1 LC kappa QLTVTRTVAAPSVFIFPP SDRKLKSGTASVVCLLNN
FYPREAKVQWKVDNALQSGNSQE SVTEQDSKD ST
YSL S STLTL SKADYEKHKVYACEVTHQGL S SPVTK
SFNRGEC
EVKLVESGGGLVKP GGSLKL SCAASGFTF S SYAMS
WVRQIPEKRLEWVASISRGGTTYYPDSVKGRFTISR
42 Mab2 ROR1 VH
DNVRNILYLQMSSLRSEDTAMYYCGRYDYDGYYA
MDYWGQGTSVTVS S
QSLEE SGGRLVTP GTPL TLTCTVS GIDLNSHWM SW
4 Ma b3 VRQAP GKGLEWIGIIAASGSTYYANWAKGRFTISK
3 VH
ROR1
TSTTVDLRIASPTTEDTATYFCARDYGDYRLVTFNI
WGP GTLVTVS S
QSVKE SE GDLVTPAGNL TLTCTAS GSDINDYPI SWV
44 M b4 RQAP GKGLEWIGFINSGGSTWYASWVKGRFTISRT
a VH
ROR1
STTVDLKMTSLTTDDTATYFCARGYSTYYCDFNIW
GP GTLVTIS S
45 Mab2 ROR1 VL DIKMTQ SP S SMYASL GERVTITCKASPDINSYL SWF
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
38
QQKPGKSPKTLIYRANRLVDGVP SRFSGGGSGQDY
SLTINSLEYEDMGIYYCL QYDEFPYTFGGGTKLEM
K
ELVMTQTP S SVSAAVGGTVTINCQASQSIGSYLAW
YQQKP GQPPKLLIYYASNLASGVPSRF S GS GS GTEY
46 Mab3 ROR1 VL
TLTISGVQREDAATYYCLGSL SNSDNVFGGGTELEI
L
ELVMTQTP S ST S GAVG GTVT INCQAS Q SID SNLAW
4 Ma FQQKP GQ PP TL LIYRA SNLA S GVP SRF S G SR S
GTEY
7 b4 VL
ROR1
TLTISGVQREDAATYYCLGGVGNVSYRTSFGGGTE
VVVK
RTVAAPSVFIFPP SDRKLKSGTASVVCLLNNFYPRE
4 Ma AKVQWKVDNALQSGNSQE SVTEQDSKD STY SL SS
8 b2 CL
ROR1
TLTL SKADYEKHKVYACEVTHQGL SSPVTKSFNRG
EC
RTVAAPSVFIFPP SDRKLKSGTASVVCLLNNFYPRE
49 M b3 ROR1 CL AKVQWKVDNALQSGNSQE SVTEQDSKD STY SL SS
a
TLTL SKADYEKHKVYACEVTHQGL SSPVTKSFNRG
EC
RTVAAPSVFIFPP SDRKLKSGTASVVCLLNNFYPRE
AKVQWKVDNALQSGNSQE SVTEQDSKD STY SL SS
50
Ma
ROR1 CL
TLTL SKADYEKHKVYACEVTHQGL SSPVTKSFNRG
EC
A S TKGP SVFP LAP S S KS T S GGTAAL G CLVEDYFP EP
51 Mab2 ROR1 CH1 VTV SWN S GALT S GVH TF PAVL Q S SGLYSL S
SVVTV
P S S SL GT QTYI CNVNHKP SNTKVDEKVEPKSC
A S TKGP SVFP LAP S S KS T S GGTAAL G CLVEDYFP EP
52 Mab3 ROR1 CH1 VTV SWN S GALT S GVH TF PAVL Q S SGLYSL S
SVVTV
P S S SL GT QTYI CNVNHKP SNTKVDEKVEPKSC
A S TKGP SVFP LAP S S KS T S GGTAAL G CLVEDYFP EP
53 Mab4 ROR1 CH1 VTV SWN S GALT S GVH TF PAVL Q S SGLYSL S
SVVTV
P S S SL GT QTYI CNVNHKP SNTKVDEKVEPKSC
EVKLVESGGGLVKP GGSLKL SCAASGFTF S SYAMS
WVRQIPEKRLEWVASISRGGTTYYPDSVKGRFTISR
DNVRNILYL QM S SLRSEDTAMYYCGRYDYDGYYA
MDYWG Q GT SVTV S SA S T KGP SVFPLAP S S KS T S GG
TAAL GCLVEDYFPEPVTVSWNS GALT S GVHTFPAV
LQ S SGLYSLS SVVTVP S SSLGTQTYICNVNHKPSNT
KVDEKVEP KS CDGGGGS GGGGSQAVVTQEP SLTV
SP GGTVTLTC GS STGAVTTSNYANWVQEKP GQAF
54 Mab2 knob HC RGLIGGTNKRAP GTPARF SG SL L GGKAALTL SGAQ
PEDEAEYYCALWYSNLWVFGGGTKLTVL S SA STK
GP SVF P LAP S SK ST S GGTAAL GC LVKDYF P EPVTV S
WN S GALT S GVH TF PAVL Q S SGLYSL SSVVTVP S S SL
GT QTYICNVNHKP SNTKVDKKVEP KS C DKT HT C P P
CPAPEAAGGP SVFLFPPKPKDTL MI SRTPEVTCVVV
DVSHEDPEVKFNWYVDGVEVHNAKTKPREEQYNS
TYRVVSVLTVLHQDWLNGKEYKCKVSNKALGAPI
EKTIS KAKGQPREPQVYTLPPCRDELTKNQVSLWC
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
39
LVKGFYP SDIAVEWESNGQPENNYKTTPPVLDSDG
SFFLYSKLTVDKSRWQQGNVF SCSVMHEALHNHY
TQKSLSLSPGK
EVKLVESGGGLVKP GGSLKL SCAASGFTF S SYAMS
WVRQIPEKRLEWVASISRGGTTYYPDSVKGRFTISR
DNVRN1LYL QMS SLRSEDTAMYYCGRYDYDGYYA
MDYWGQGTSVTVSSASTKGP SVFPLAP SSKSTSGG
TAAL GCLVEDYFPEPVTVSWNS GALT S GVHTFPAV
LQ S SGLYSL S SVVTVP S SSLGTQTYICNVNHKPSNT
55 Mab2 hole HC KVDEKVEP KS CDKTHTCPPCPAPEAAGGP SVFLFPP
KP KDTLMISRTPEVTCVVVDVSHEDPEVKFNWYV
DGVEVHNAKTKPREEQYNSTYRVVSVLTVLHQDW
LNGKEYKCKVSNKAL GAPIEKTI SKAKGQPREP QV
CTLPP SRDELTKNQVSL SCAVKGFYP SDIAVEWE SN
GQPENNYKTTPPVLDSDGSFFLVSKLTVDKSRWQQ
GNVF SC SVMHEALHNHYTQKSL SL SP GK
DIKMTQ SP S SMYASL GERVTITCKASPDINSYL SWF
QQKPGKSPKTLIYRANRLVDGVP SRFSGGGSGQDY
SLTINSLEYEDMGIYYCL QYDEFPYTFGGGTKLEM
56 Mab2 ROR1 LC KRTVAAP SVFIFPP SDRKLKSGTASVVCLLNNFYPR
EAKVQWKVDNALQ SGNSQESVTEQDSKDSTYSL S
STLTL SKADYEKHKVYACEVTHQGL S SPVTKSFNR
GEC
QSLEE SGGRLVTP GTPL TLTCTVS GIDLNSHWM SW
VRQAP GKGLEWIGIIAASGSTYYANWAKGRFTISK
TSTTVDLRIASPTTEDTATYFCARDYGDYRLVTFNI
WGP GTLVTVSSASTKGP SVFPLAP SSKSTSGGTAAL
GCLVEDYFPEPVTVSWNSGALTSGVHTFPAVLQ SS
GLYSL S SVVTVP S S SL GTQTYICNVNHKP SNTKVDE
KVEPKSCDGGGGSGGGGSQAVVTQEP SLTVSP GGT
VTLTCGS STGAVTTSNYANWVQEKP GQAFRGLIGG
TNKRAPGTPARFSGSLL GGKAALTL SGAQPEDEAE
YYCALWYSNLWVFGGGTKLTVL S SA STKGP SVFPL
57 Mab3 ROR1 knob HC
AP S SKST S GGTAAL GCLVKDYFPEPVTVSWNS GAL
TSGVHTFPAVLQS SGLYSL S SVVTVP S S SL GTQTYIC
NVNHKP SNTKVDKKVEPKSCDKTHTCPPCPAPEAA
GGP SVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDP
EVKFNWYVDGVEVHNAKTKPREEQYNSTYRVVS
VLTVLHQDWLNGKEYKCKVSNKALGAPIEKTISKA
KGQPREPQVYTLPPCRDELTKNQVSLWCLVKGFYP
SDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSKL
TVDKSRWQQGNVF SC SVMHEALHNHYTQKSL SL S
P GK
QSLEE SGGRLVTP GTPL TLTCTVS GIDLNSHWM SW
VRQAP GKGLEWIGIIAASGSTYYANWAKGRFTISK
TSTTVDLRIASPTTEDTATYFCARDYGDYRLVTFNI
WGP GTLVTVSSASTKGP SVFPLAP SSKSTSGGTAAL
58 Mab3 ROR1 hole HC GCLVEDYFPEPVTVSWNSGALTSGVHTFPAVLQ SS
GLYSL S SVVTVP S S SL GTQTYICNVNHKP SNTKVDE
KVEPKSCDKTHTCPPCPAPEAAGGP SVFLFPPKPKD
TLMISRTPEVTCVVVDVSHEDPEVKFNWYVDGVE
VIINAKTKPREEQYNSTYRVVSVLTVLHQDWLNGK
EYKCKVSNKALGAPIEKTIS KAKGQPREPQVCTLPP
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
SRDELTKNQVSL SCAVKGFYPSDIAVEWESNGQPE
NNYKTTPPVLDSDGSFFLVSKLTVDKSRWQQGNVF
SC SVMHEALHNHYTQKSL SL SP GK
ELVMTQTP S SVSAAVGGTVTINCQASQSIGSYLAW
YQQKP GQPPKLLIYYASNLASGVPSRF S GS GS GTEY
TLTISGVQREDAATYYCLGSL SNSDNVFGGGTELEI
59 Mab3 ROR1 LC LRTVAAP SVFIFPP SDRKLKSGTASVVCLLNNFYPR
EAKVQWKVDNALQ SGNSQESVTEQDSKDSTYSL S
STLTL SKADYEKHKVYACEVTHQGL S SPVTKSFNR
GEC
QSVKE SEGDLVTPAGNL TLTCT AS GSDINDYPI SWV
RQAP GKGLEWIGFINSGGSTWYASWVKGRFTISRT
STTVDLKMTSLTTDDTATYFCARGYSTYYCDFNIW
GP GTLVTIS SASTKGPSVFPLAP S SKST S GGTAAL GC
LVEDYFPEPVTVSWNS GAL T S GVHTFPAVL QS SGL
YSL S SVVTVPS S SLGTQTYICNVNHKP SNTKVDEKV
EPKSCDGGGGSGGGGSQAVVTQEPSLTVSPGGTVT
LTC GS ST GAVTT SNYANWVQEKP GQ AFRGLIGGTN
KRAP GTPARF SGSLLGGKAALTL SGAQPEDEAEYY
CALWYSNLWVFGGGTKLTVL S SASTKGP SVFPLAP
60 Mab4 ROR1 knob HC
SSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTS
GVHTFPAVLQSSGLYSLSSVVTVPSSSLGTQTYICN
VNHKP SNTKVDKKVEPKSCDKTHTCPPCPAPEAAG
GP SVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPE
VKFNWYVDGVEVHNAKTKPREEQYNSTYRVVSV
LTVLHQDWLNGKEYKC KVSNKAL GAPIEKTISKAK
GQPREPQVYTLPPCRDELTKNQVSLWCLVKGFYPS
DIAVEWESNGQPENNYKTTPPVLD SD GSFFLY SKL
TVDKSRWQQGNVF SC SVMHEALHNHYTQKSL SL S
P GK
QSVKE SE GDLVTPAGNL TLTCTAS GSDINDYPI SWV
RQAP GKGLEWIGFINSGGSTWYASWVKGRFTISRT
STTVDLKMTSLTTDDTATYFCARGYSTYYCDFNIW
GP GTLVTIS SASTKGPSVFPLAP S SKST S GGTAAL GC
LVEDYFPEPVTVSWNS GAL T S GVHTFPAVL QS SGL
YSL S SVVTVPS S SLGTQTYICNVNHKP SNTKVDEKV
61 Mab4 ROR1 hole HC EPKSCDKTHTCPPCPAPEAAGGPSVFLFPPKPKDTL
MI SRTP EVTCVVVDVSHEDPEVKFNWYVDGVEVH
NAKTKPREEQYNSTYRVVSVLTVLHQDWLNGKEY
KC KVSNKALGAPIEKTISKAKGQPREPQVCTLPP SR
DEL TKNQVSL SCAVKGFYP SDIAVEWESNGQPENN
YKT TPPVLD SD GSFFLV SKLTVDKSRW QQGNVF SC
SVMHEALHNHYTQKSL SL SP GK
ELVMTQTP S ST S GAVGGTVTINCQAS Q SID SNLAW
FQQKP GQPPTLLIYRASNLASGVP SRFSGSRSGTEY
TLTISGVQREDAATYYCLGGVGNVSYRT SFGGGTE
62 Mab4 ROR1 LC VVVKRTVAAPSVFIFPPSDRKLKSGTASVVCLLNNF
YPREAKVQWKVDNALQSGNSQESVTEQDSKDSTY
SL S STLTL SKADYEKHKVYACEVTHQGL SSPVTKS
FNRGEC
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
41
To make the following Fc-containing anti-ROR1/anti-CD3 TCBs according to the
invention, the
respective constructs / sequence IDs as mentioned in the table above are
needed:
ROR1-TCB (2+1): 37, 38, 39, 40 x2 or 37, 38, 39, 41 x2
ROR1-TCB (1+1): 36, 37, 39, 40 or 36, 37, 39, 41
Mab2 ROR1-TCB (2+1): 39, 54, 55, 56 x2
Mab3 ROR1-TCB (2+1): 39, 57, 58, 59 x2
Mab4 ROR1-TCB (2+1): 39, 60, 61, 62 x2
In the following specific embodiments of the invention are listed:
1. A bispecific bi- or trivalent antibody specifically binding to the two
targets which are human CDR
(further named also as "CD3") and the extracellular domain of human ROR1
(further named also as
"ROR1"), wherein the variable domains VL and VH in a light chain and the
respective heavy chain are
replaced by each other, characterized in comprising a constant domain CL
wherein the amino acid at
position 124 is substituted independently by lysine (K), arginine (R) or
histidine (H) (numbering
according to Kabat), and in the respective constant domain CH1 the amino acid
at position 147 and the
amino acid at position 213 is substituted independently by glutamic acid (E),
or aspartic acid (D)
(numbering according to Kabat).
2. The bispecific antibody specifically binding to the two targets which are
human CDR and the
extracellular domain of human ROR1, characterized in comprising
a) the first light chain and the first heavy chain of a first antibody
which specifically binds to ROR1;
and
b) the second light chain and the second heavy chain of a second antibody
which specifically binds
to CD3, and wherein the variable domains VL and VH in the second light chain
and second heavy chain
of the second antibody are replaced by each other; and wherein
c) in the constant domain CL of the first light chain under a) the amino
acid at position 124 is
substituted independently by lysine (K), arginine (R) or histidine (H)
(numbering according to Kabat),
and wherein in the constant domain CH1 of the first heavy chain under a) the
amino acid at position 147
and the amino acid at position 213 is substituted independently by glutamic
acid (E), or aspartic acid (D)
(numbering according to Kabat).
3. The bispecific antibody specifically binding to the two targets which are
human CD3 c and the
extracellular domain of human ROR1, characterized in comprising
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
42
a) the first light chain and the first heavy chain of a first antibody
which specifically binds to ROR1;
and
b) the second light chain and the second heavy chain of a second antibody
which specifically binds
to CD3, and wherein the variable domains VL and VH in the second light chain
and second heavy chain
of the second antibody are replaced by each other; and wherein
c) in the constant domain CL of the second light chain under b) the amino acid
at position 124 is
substituted independently by lysine (K), arginine (R) or histidine (H)
(numbering according to Kabat),
and wherein in the constant domain CH1 of the second heavy chain under b) the
amino acid at positions
147 and the amino acid at position 213 is substituted independently by
glutamic acid (E), or aspartic acid
(D) (numbering according to Kabat).
4. The bispecific antibody according to embodiment 2, characterized in that
said bispecific antibody
comprises in addition a Fab fragment of said first antibody (further named
also as "ROR1-Fab") and in
the constant domain CL said ROR1-Fab the amino acid at position 124 is
substituted independently by
lysine (K), arginine (R) or histidine (H) (numbering according to Kabat), and
wherein in the constant
domain CH1 of said ROR1-Fab the amino acid at positions 147 and the amino acid
at position 213 is
substituted independently by glutamic acid (E), or aspartic acid (D)
(numbering according to Kabat).
5. The bispecific antibody according to embodiment 3, characterized in that
said bispecific antibody
comprises in addition a second Fab fragment of said first antibody ("ROR1 -
Fab").
6. The bispecific antibody according to any one of embodiment 1, characterized
in consisting of one Fab
fragment of an antibody specifically binding to CD3 c (further named also as
"CD3-Fab"), and one Fab
fragment of an antibody specifically binding to ROR1 (further named also as
"ROR1-Fab(s)") and a Fc
part, wherein the CD3-Fab and the ROR1-Fab are linked via their C-termini to
the hinge region of said Fc
part and wherein either the CD3-Fab or the ROR1-Fab comprises aa substitution
and the CD3-Fab
comprises crossover.
7. The bispecific antibody according to embodiment 6, characterized in
consisting of one CD3-Fab, and
one ROR1-Fab and a Fc part, wherein the CD3-Fab and the ROR1-Fab are linked
via their C-termini to
the hinge region of said Fc part and a second ROR1-Fab, which is linked with
its C-terminus to the N-
terminus of the CD3-Fab and wherein the CD3-Fab comprises crossover and either
the CD3-Fab or both
ROR1-Fabs comprise aa substitution (Figures 1C and 1D).
8. The bispecific antibody according to embodiment 7, characterized in
consisting of ROR1-Fab-Fc-CD3-
Fab-ROR1-Fab, wherein both ROR1-Fabs comprise aa substitution and the CD3-Fab
comprises VLNH
crossover.
9. The bispecific antibody according to embodiment 1, characterized in
consisting of two ROR1-Fabs and
a Fc part, wherein the ROR1-Fabs are linked via their C-termini to the hinge
region of said Fc part and a
CD3-Fab, which is linked with its C-terminus to the N-terminus of one ROR1-Fab
and the CD3-Fab
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
43
comprises crossover and either the CD3-Fab or both ROR1-Fabs comprise aa
substitution (Figures 1F
and 1G).
10. The bispecific antibody according to any one of embodiments 1 to 5,
characterized in consisting of
one CD3-Fab, which is linked via its C-terminus to the hinge region of said Fc
part and a ROR1-Fab,
which is linked with its C-terminus to the N-terminus of the CD3-Fab and
either the CD3-Fab or the
ROR1-Fab comprise aa substitution (Figures 1H and le.
11. The bispecific antibody according to any one of embodiments 1 to 6,
characterized in consisting of
one ROR1-Fab, which is linked via its C-terminus to the hinge region of said
Fc part and a CD3-Fab,
which is linked with its C-terminus to the N-terminus of the ROR1-Fab and
either the CD3-Fab or the
ROR1-Fab comprise aa substitution (Figures 1J and 1K).
12. The bispecific antibody according to any one of embodiments 1 to 11,
characterized in comprising the
CDR sequences of anti-ROR1 antibody MABl.
13. The bispecific antibody according to any one of embodiments 1 to 12,
characterized in comprising the
VH and VL sequences of anti-ROR1 antibody MAB1, or an antibody comprising the
VH, VL, CH1, and
CL sequences of anti-ROR1 antibody MAB 1.
14. The bispecific antibody according to any one of embodiments 1 to 13,
characterized in that the
antibody portion specifically binding to human CDR, preferably the Fab
fragment, is characterized in
comprising
a) a variable domain VH comprising the heavy chain CDRs of SEQ ID NO: 12, 13
and 14 as respectively
heavy chain CDR1, CDR2 and CDR3 and a variable domain VL comprising the light
chain CDRs of
SEQ ID NO: 15, 16 and 17 as respectively light chain CDR1, CDR2 and CDR3 of
the anti CDR
antibody (CDR MAB CD3 H2C), or
b) a variable domain VH comprising the heavy chain CDRs of SEQ ID NO: 23, 24
and 25 as respectively
heavy chain CDR1, CDR2 and CDR3 and a variable domain VL comprising the light
chain CDRs of
SEQ ID NO: 26, 27 and 28 as respectively light chain CDR1, CDR2 and CDR3 of
the anti CDR
antibody (CDR MAB CD3 CH2527) .
15. The bispecific antibody according to any one of embodiments 1 to 14,
characterized in that the
antibody portion specifically binding to human CDR is characterized in that
the variable domains are of
a) SEQ ID NO:10 and 11 (VHVL MAB CD3 H2C), or
b) SEQ ID NO:21 and 22 (VHVL MAB CD3 CH2527).
16. The bispecific antibody according to any one of embodiments 1 to 15,
characterized in that the Fab
fragment, specifically binding to human ROR1 is characterized in comprising a
variable domain VH
comprising the heavy chain CDRs CDR1H of SEQ ID NO:7, a CDR2H of SEQ ID NO:8,
a CDR3H of
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
44
SEQ ID NO: 9 and comprising a variable domain VL comprising the light chain
CDRs CDR1L of SEQ
ID NO:3, a CDR2L of SEQ ID NO:4, a CDR3L of SEQ ID NO: 5 (CDR MAB1).
17. The bispecific antibody according to any one of embodiments 1 to 16,
characterized in that the Fab
fragment, specifically binding to human ROR1 is characterized in comprising a
VH of SEQ ID NO: 10
and a VL of SEQ ID NO: 11 (VHVL MAB1).
18. The bispecific antibody according to any one of embodiments 1 to 17,
characterized in that in addition
to the amino acid replacement at position 124 in the constant domain CL the
amino acid at position 123 is
substituted independently by lysine (K), arginine (R) or histidine (H).
19. The bispecific antibody according to any one of embodiments 1 to 18,
characterized in that amino
acid 124 is K, amino acid 147 is E, amino acid 213 is E, and amino acid 123 is
R or amino acid 124 is K,
amino acid 147 is E, amino acid 213 is K, and amino acid 123 is KPreferably
amino acid 123 is R for a
kappa light chain and K for a lambda light chain.
20. A bispecific antibody specifically binding to the extracellular domain of
human ROR1 and to human
CDR, characterized in comprising a heavy and light chain set selected from the
group consisting of
polypeptides SEQ ID NO:37, SEQ ID NO:38, SEQ ID NO:39, and SEQ ID NO:40 or
from the group
consisting of polypeptides SEQ ID NO:37, SEQ ID NO:38, SEQ ID NO:39, and SEQ
ID NO:41.
21. The antibody according to embodiment 20, characterized in that in the
antibody portion specifically
binding to human CD3c
a) the variable domain VH is replaced by a variable domain VH comprising the
heavy chain CDRs of
SEQ ID NO: 12, 13 and 14 as respectively heavy chain CDR1, CDR2 and CDR3 and
the variable domain
VL is replaced by a variable domain VL comprising the light chain CDRs of SEQ
ID NO: 15, 16 and 17
as respectively light chain CDR1, CDR2 and CDR3 of the anti CDR antibody, or
b) the variable domain VH is replaced by a variable domain VH comprising the
heavy chain CDRs of
SEQ ID NO: 23, 24 and 25 as respectively heavy chain CDR1, CDR2 and CDR3 and
the variable domain
VL is replaced by a variable domain VL comprising the light chain CDRs of SEQ
ID NO: 26, 27 and 28
as respectively light chain CDR1, CDR2 and CDR3 of the anti CDR antibody.
22. The antibody according to any one of embodiments 1 to 21, characterized in
that the CH3 domain of
one heavy chain and the CH3 domain of the other heavy chain each meet at an
interface which comprises
an original interface between the antibody CH3 domains; wherein said interface
is altered to promote the
formation of the bispecific antibody, wherein the alteration is characterized
in that:
a) the CH3 domain of one heavy chain is altered, so that within the original
interface the CH3 domain of
one heavy chain that meets the original interface of the CH3 domain of the
other heavy chain within the
bispecific antibody, an amino acid residue is replaced with an amino acid
residue having a larger side
chain volume, thereby generating a protuberance within the interface of the
CH3 domain of one heavy
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
chain which is positionable in a cavity within the interface of the CH3 domain
of the other heavy chain
and
b) the CH3 domain of the other heavy chain is altered, so that within the
original interface of the second
CH3 domain that meets the original interface of the first CH3 domain within
the bispecific antibody an
5 amino acid residue is replaced with an amino acid residue having a smaller
side chain volume, thereby
generating a cavity within the interface of the second CH3 domain within which
a protuberance within
the interface of the first CH3 domain is positionable.
23. The antibody according to any one of embodiments 1 to 22, characterized in
comprising in the human
IgG1 Fc part amino acid substitution of Pro329 with glycine or arginine and/or
substitutions L234A and
10 L235A.
24. The antibody according to embodiment 23, characterized in being of
construct ROR1 Fab ¨ Fc - CD3
Fab - ROR1 Fab and comprising VL/VH crossover within the Fab fragment of the
anti-CD3c antibody
and in comprising in the human IgG1 Fc part amino acid substitutions of Pro329
with glycine, Leu234
with alanine and Leu235 with alanine.
15 25. The antibody according to any one of embodiments 1 to 22, characterized
in
a) being of construct ROR1 Fab ¨ Fc - CD3 Fab - ROR1 Fab,
b) comprising VL/VH crossover within the Fab fragment of the anti-CD3
antibody,
c) comprising a human IgGlFc part,
d) comprising within the Fc part substitution of Pro329 with glycine and
substitutions of Leu234 by
20 alanine and Leu235 by alanine, and
e) that in the constant domain CL of both ROR1 Fabs the amino acid at position
124 is substituted by
lysine (K) and at position 123 by arginine (R) for a kappa light and lysine
(K) for a lambda light chain, in
the constant domain CH1 the amino acid at position 147 and the amino acid at
position 213 are
substituted by glutamic acid (E).
25 26. The antibody according to any one of embodiments 1 to 25, characterized
in specifically binding to
the two targets human CDR and the extracellular domain of human ROR1,
characterized in not
internalizing in a concentration of 1nM in primary B-CLL cells at 37 C during
two hours.
27. The antibody according to any one of embodiments 1 to 26, characterized in
specifically binding to
the two targets human CDR and the extracellular domain of human ROR1,
characterized in that the
30 bispecific antibody does not internalize in a cell based assay at 37 C
during 2 hrs, using ROR1-positive
primary B-CLL cells and used at an antibody concentration of 1 nM, whereby not
internalize means, that
the mean fluorescence intensity (MFI), as detected by flow cytometry, of said
bispecific antibody upon
binding to ROR1-positive primary B-CLL cells measured at time 0 is not reduced
more than 50%,
preferably not more than 30% when re-measured after a 2hr-incubation at 37 C.
CA 02963692 2017-04-05
WO 2016/055592
PCT/EP2015/073308
46
28.The antibody according to embodiments 1 to 27 , characterized by an
elimination half-life in mice,
preferably cynomolgus monkeys of longer than 12 hours, preferably 3 days or
longer.
29. The antibody according to embodiments 1 to 28, characterized in showing an
EC50 value for binding
to ROR1-positive cell lines (e.g. RPMI8226 cells, Rec-1 cells, Jeko cells) of
30 nM or lower, preferably
an EC50 value of 15 nM and lower.
30.The antibody according to embodiments 1 to 29, characterized by its
capability to induce redirected
killing of ROR1 expressing tumor cells (e.g. RPMI8226 cells, Rec-1 cells, Jeko
cells, ovarian cancer cell
lines like PA-1, COLO-704, OVCAR-5, SK-OV-3)) in the presence of human T cells
with an EC50 lower
than 10 nM, preferably 1 nM, preferably 0.05 nM, preferably 0.02 nM,
preferably 0.002 nM and lower.
31. The antibody according to embodiments 1 to 30, characterized in that said
antibody stored in standard
formulation buffer at 37 C preferably at 40 C, for 10 days, preferably up to 2
weeks, preferably up to 4
weeks, does not result in more than 10% changes (A), preferably not more than
5% changes (A), in high
molecular weight (HMW) species and/or low molecular weight (LMW) species
and/or monomer content
as compared to the said antibody stored in the same formulation buffer at -80
C for the same period of
storage.
32. A method for the preparation of an a bispecific antibody according to any
one of embodiments 1 to 31
comprising the steps of
a) transforming a host cell with vectors comprising nucleic acid molecules
encoding the light chain
and heavy chain of an antibody according to any one of embodiments 1 to 31,
b) culturing the host cell under conditions that allow synthesis of said
antibody molecule; and
c) recovering said antibody molecule from said culture.
33. A host cell comprising vectors comprising nucleic acid molecules encoding
the light chain and heavy
chains of an antibody according to any one of embodiments 1 to 31.
34. A pharmaceutical composition comprising an antibody according to any one
of embodiments 1 to 30
and a pharmaceutically acceptable excipient.
35. The antibody according to any one of embodiments 1 to 31 or the
pharmaceutical composition of
embodiment 34 for use as a medicament.
36. An antibody according to any one of embodiments 1 to 31 or the
pharmaceutical composition of
embodiment 34 for use as a medicament in the treatment of ROR1-positive
hematological malignancies
comprising chronic lymphocytic leukemia (CLL), hairy cell leukemia (HCL),
acute lymphoblastic
leukemia (ALL), acute myeloid leukemia (AML), chronic myeloid leukemia (CML),
mantle cell
lymphoma (MCL), marginal zone lymphoma (MZL), diffuse large B cell lymphoma
(DLBCL), multiple
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
47
myeloma (MM), follicular lymphoma (FL), and for the treatment of ROR1-positive
solid tumors such as
ovarian cancer, breast cancer and lung cancer.
37. An antibody according to any one of embodiments 1 to 31 or the
pharmaceutical composition of
embodiment 34 for use as a medicament in the treatment of multiple myeloma.
38. An antibody according to any one of embodiments 1 to 31 or the
pharmaceutical composition of
embodiment 34 for the treatment of of chronic lymphocytic leukemia (CLL) of B-
cell lineage (B-CLL)
and for use as a medicament in the treatment of plasma cell disorders like
Multiple Myeloma MM or
other B-cell disorders expressing ROR1.
39. The antibody according to any one of claims 1 to 31 or the pharmaceutical
composition of claim 34
for use as a medicament in the treatment of a disease selected from the group
consisting of ovarian
cancer, lung cancer, breast cancer, gastric cancer, and pancreatic cancer.
40. The antibody according to any one of claims 1 to 31 or the pharmaceutical
composition of claim 34
for use as a medicament in the treatment of ovarian cancer.
Materials & general methods
General information regarding the nucleotide sequences of human
immunoglobulins light and heavy
chains is given in: Kabat, E. A., et al., Sequences of Proteins of
Immunological Interest, 5th ed., Public
Health Service, National Institutes of Health, Bethesda, MD (1991). Amino
acids of antibody chains are
numbered and referred to according to EU numbering (Edelman, G.M., et al.,
Proc. Natl. Acad. Sci. USA
63 (1969) 78-85; Kabat, E.A., et al., Sequences of Proteins of Immunological
Interest, 5th ed., Public
Health Service, National Institutes of Health, Bethesda, MD, (1991)).
Recombinant DNA techniques
Standard methods were used to manipulate DNA as described in Sambrook, J. et
al., Molecular cloning:
A laboratory manual; Cold Spring Harbor Laboratory Press, Cold Spring Harbor,
New York, 1989. The
molecular biological reagents were used according to the manufacturer's
instructions. General
information regarding the nucleotide sequences of human immunoglobulins light
and heavy chains is
given in: Kabat, E.A. et al., (1991) Sequences of Proteins of Immunological
Interest, 5th ed., NIH
Publication No. 91-3242. Amino acids of antibody chains were numbered and
referred to according to
Kabat, E.A., et al., Sequences of Proteins of Immunological Interest, 5th ed.,
Public Health Service,
National Institutes of Health, Bethesda, MD, (1991).
Gene synthesis
a) Desired gene segments were prepared from oligonucleotides made by chemical
synthesis. The 600 -
1800 bp long gene segments, which were flanked by singular restriction
endonuclease cleavage sites,
were assembled by annealing and ligation of oligonucleotides including PCR
amplification and
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
48
subsequently cloned via the indicated restriction sites e.g. Kpnl/ Sad or
Ascl/Pacl into a pPCRScript
(Stratagene) based pGA4 cloning vector. The DNA sequences of the subcloned
gene fragments were
confirmed by DNA sequencing. Gene synthesis fragments were ordered according
to given specifications
at Geneart (Regensburg, Germany).b) Desired gene segments where required were
either generated by
PCR using appropriate templates or were synthesized by Geneart AG (Regensburg,
Germany) from
synthetic oligonucleotides and PCR products by automated gene synthesis. The
gene segments flanked by
singular restriction endonuclease cleavage sites were cloned into standard
expression vectors or into
sequencing vectors for further analysis. The plasmid DNA was purified from
transformed bacteria using
commercially available plasmid purification kits. Plasmid concentration was
determined by UV
spectroscopy. The DNA sequence of the subcloned gene fragments was confirmed
by DNA sequencing.
Gene segments were designed with suitable restriction sites to allow sub-
cloning into the respective
expression vectors. If required, protein coding genes were designed with a 5'-
end DNA sequence coding
for a leader peptide which targets proteins for secretion in eukaryotic cells.
DNA sequence determination
DNA sequences are determined by double strand sequencing.
DNA and protein sequence analysis and sequence data management
The Clone Manager (Scientific & Educational Software) software package version
9.2 was used for
sequence mapping, analysis, annotation and illustration.
Expression vectors
a) The fusion genes comprising the described antibody chains as described
below were generated by PCR
and/or gene synthesis and assembled with known recombinant methods and
techniques by connection of
the according nucleic acid segments e.g. using unique restriction sites in the
respective vectors. The
subcloned nucleic acid sequences were verified by DNA sequencing. For
transient transfections larger
quantities of the plasmids are prepared by plasmid preparation from
transformed E. coli cultures
(Nucleobond AX, Macherey-Nagel).
b) For the generation of anti-ROR1 antibody expression vectors, the variable
regions of heavy and light
chain DNA sequences were subcloned in frame with either the human IgG1
constant heavy chain or the
hum IgG1 constant light chain pre-inserted into the respective generic
recipient expression vector
optimized for expression in mammalian cell lines. The antibody expression is
driven by a chimeric MPSV
promoter comprising a CMV enhancer and a MPSV promoter followed by a 5' UTR,
an intron and a Ig
kappa MAR element. The transcription is terminated by a synthetic polyA signal
sequence at the 3' end
of the CDS. All vectors carry a 5'-end DNA sequence coding for a leader
peptide which targets proteins
for secretion in eukaryotic cells. In addition each vector contains an EBV
OriP sequence for episomal
plasmid replication in EBV EBNA expressing cells.
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
49
c) For the generation of ROR1xCD3 bispecific antibody vectors, the IgG1
derived bispecific molecules
consist at least of two antigen binding moieties capable of binding
specifically to two distinct antigenic
determinants CD3 and ROR1. The antigen binding moieties are Fab fragments
composed of a heavy and
a light chain, each comprising a variable and a constant region. At least one
of the Fab fragments was a
"Crossfab" fragment, wherein VH and VL were exchanged. The exchange of VH and
VL within the Fab
fragment assures that Fab fragments of different specificity do not have
identical domain arrangements.
The bispecific molecule design was monovalent for CD3 and bivalent for ROR1
where one Fab fragment
was fused to the N-terminus of the inner CrossFab (2+1). The bispecific
molecule contained an Fc part in
order for the molecule to have a long half-life. A schematic representation of
the constructs is given in
Figure 1; the preferred sequences of the constructs are shown in SEQ ID NOs 39
to 52. The molecules
were produced by polymer-based co-transfecting HEK293 EBNA cells growing in
suspension with the
mammalian expression vectors. For preparation of 2+1 CrossFab-IgG constructs,
cells were transfected
with the corresponding expression vectors in a 1:2:1:1 ratio ("vector
Fc(knob)" : "vector light chain" :
"vector light chain CrossFab" : "vector heavy chain-CrossFab").
Cell culture techniques
Standard cell culture techniques are used as described in Current Protocols in
Cell Biology (2000),
Bonifacino, J. S., Dasso, M., Harford, J.B., Lippincott-Schwartz, J. and
Yamada, K.M. (eds.), John Wiley
& Sons, Inc.
Transient expression in HEK293 cells (HEK293-EBNA system)
Bispecific antibodies were expressed by transient co-transfection of the
respective mammalian expression
vectors in HEK293-EBNA cells, which were cultivated in suspension, usinga
polymer. One day prior to
transfection the HEK293-EBNA cells were seeded at 1.5 Mio viable cells/mL in
Ex-Cell medium,
supplemented with 6 mM of L-Glutamine. For every mL of final production volume
2.0 Mio viable cells
were centrifuged (5 minutes at 210 x g). The supernatant was aspirated and the
cells resuspended in 100
[EL of CD CHO medium. The DNA for every mL of final production volume was
prepared by mixing 1
[tg of DNA (Ratio heavy chain: modified heavy chain: light chain: modified
light chain = 1:1:2:1) in 100
[EL of CD CHO medium. After addition of 0.27 [EL of polymer solution (1 mg/mL)
the mixture was
vortexed for 15 seconds and left at room temperature for 10 minutes. After 10
minutes, the resuspended
cells and DNA/polymer mixture were put together and then transferred into an
appropriate container
which was placed in a shaking device (37 C, 5% CO2). After a 3 hours
incubation time 800 [EL of Ex-
Cell Medium, supplemented with 6 mM L-Glutamine, 1.25 mM valproic acid and
12.5% Pepsoy (50
g/L), was added for every mL of final Production volume. After 24 hours, 70
[EL of Feed solution was
added for every mL of final production volume. After 7 days or when the cell
viability was equal or
lower than 70%, the cells were separated from the supernatant by
centrifugation and sterile filtration. The
antibodies were purified by an affinity step and one or two polishing steps,
being cation exchange
chromatography and size exclusion chromatography. When required, an additional
polishing step was
used. The recombinant anti-BCMA human antibody and bispecific antibodies were
produced in
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
suspension by polymer-based co-transfecting HEK293-EBNA cells with the
mammalian expression
vectors. The cells were transfected with two or four vectors, depending in the
format. For the human IgG1
one plasmid encoded the heavy chain and the other plasmid the light chain. For
the bispecific antibodies
four plasmids were co-transfected. Two of them encoded the two different heavy
chains and the other two
5 encoded the two different light chains. One day prior to transfection the
HEK293-EBNA cells were
seeded at 1.5 Mio viable cells/mL in F17 Medium, supplemented with 6 mM of L-
Glutamine.
Protein determination
Determination of the antibody concentration was done by measurement of the
absorbance at 280 nm,
using the theoretical value of the absorbance of a 0.1% solution of the
antibody. This value was based on
10 the amino acid sequence and calculated by GPMAW software (Lighthouse data).
SDS-PAGE
The NuPAGEO Pre-Cast gel system (Invitrogen) is used according to the
manufacturer's instruction. In
particular, 10% or 4-12% NuPAGEO Novex0 Bis-TRIS Pre-Cast gels (pH 6.4) and a
NuPAGEO MES
(reduced gels, with NuPAGEO Antioxidant running buffer additive) or MOPS (non-
reduced gels)
15 running buffer is used.
Protein purification
By protein A affinity chromatography
For the affinity step the supernatant was loaded on a protein A column (HiTrap
Protein A FF , 5 mL, GE
Healthcare) equilibrated with 6 CV 20 mM sodium phosphate, 20 mM sodium
citrate, pH 7.5. After a
20 washing step with the same buffer the antibody was eluted from the column
by step elution with 20 mM
sodium phosphate, 100 mM sodium chloride, 100 mM Glycine, pH 3Ø The
fractions with the desired
antibody were immediately neutralized by 0.5 M Sodium Phosphate, pH 8.0
(1:10), pooled and
concentrated by centrifugation. The concentrate was sterile filtered and
processed further by cation
exchange chromatography and/or size exclusion chromatography.
25 By cation exchange chromatography
For the cation exchange chromatography step the concentrated protein was
diluted 1:10 with the elution
buffer used for the affinity step and loaded onto a cation exchange colume
(Poros 50 HS, Applied
Biosystems). After two washing steps with the equilibration buffer and a
washing buffer resp. 20 mM
sodium phosphate, 20 mM sodium citrate, 20 mM TRIS, pH 5.0 and 20 mM sodium
phosphate, 20 mM
30 sodium citrate, 20 mM TRIS, 100 mM sodium chloride pH 5.0 the protein was
eluted with a gradient
using 20 mM sodium phosphate, 20 mM sodium citrate, 20 mM TRIS, 100 mM sodium
chloride pH 8.5.
The fractions containing the desired antibody were pooled, concentrated by
centrifugation, sterile filtered
and processed further a size exclusion step.
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
51
By analytical size exclusion chromatography
For the size exclusion step the concentrated protein was injected in a XKl
6/60 HiLoad Superdex 200
column (GE Healthcare), and 20 mM Histidine, 140 mM Sodium Chloride, pH 6.0
with or without
Tween20 as formulation buffer. The fractions containing the monomers were
pooled, concentrated by
centrifugation and sterile filtered into a sterile vial.
Measurement of purity and monomer content
Purity and monomer content of the final protein preparation was determined by
CE-SDS (Caliper
LabChip GXII system (Caliper Life Sciences)) resp. HPLC (TSKgel G3000 SW XL
analytical size
exclusion column (Tosoh)) in a 25 mM potassium phosphate, 125 mM Sodium
chloride, 200 mM L-
arginine monohydrochloride, 0.02 % (w/v) Sodium azide, pH 6.7 buffer.
Molecular weight confirmation by LC-MS analyses
Deglycosylation
To confirm homogeneous preparation of the molecules final protein solution of
was analyzed by LC-MS
analyses. To remove heterogeneity introduced by carbohydrates the constructs
are treated with PNGaseF
(ProZyme). Therefore the pH of the protein solution was adjusted to pH7.0 by
adding 2 ul 2 M Tris to
ug protein with a concentration of 0.5 mg/ml. 0.8 ug PNGaseF was added and
incubated for 12 h at
37 C.
LC-MS analysis - On line detection
The LC-MS method was performed on an Agilent HPLC 1200 coupled to a TOF 6441
mass spectrometer
20 (Agilent). The chromatographic separation was performed on a Macherey Nagel
Polysterene column;
RP1000-8 (8 um particle size, 4.6 x 250 mm; cat. No. 719510). Eluent A was 5 %
acetonitrile and 0.05 %
(v/v) formic acid in water, eluent B was 95 % acetonitrile, 5 % water and 0.05
% formic acid. The flow
rate was 1 ml/min, the separation was performed at 40 C and 6 ug (15 ul) of a
protein sample obtained
with a treatment as described before (table 7).
Table 7
Time (min.) %B
0.5 15
10 60
12.5 100
14.5 100
14.6 15
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
52
16 15
16.1 100
During the first 4 minutes the eluate was directed into the waste to protect
the mass spectrometer from
salt contamination. The ESI-source was running with a drying gas flow of
121/min, a temperature of
350 C and a nebulizer pressure of 60psi. The MS spectra were acquired using a
frag;mentor voltage of
380 V and a mass range 700 to 3200 m/z in positive ion mode using. MS data
were acquired by the
instrtunent software from 4 to 17 minutes.
Isolation of primary human pan T cells from PBMCs
Peripheral blood mononuclear cells (PBMCs) were prepared by Histopaque density
centrifugation from
enriched lymphocyte preparations (buffy coats) obtained from local blood banks
or from fresh blood from
healthy human donors. Briefly, blood was diluted with sterile PBS and
carefully layered over a
Histopaque gradient (Sigma, H8889). After centrifugation for 30 minutes at 450
x g at room temperature
(brake switched off), part of the plasma above the PBMC containing interphase
was discarded. The
PBMCs were transferred into new 50 ml Falcon tubes and tubes were filled up
with PBS to a total volume
of 50 ml. The mixture was centrifuged at room temperature for 10 minutes at
400 x g (brake switched
on). The supernatant was discarded and the PBMC pellet washed twice with
sterile PBS (centrifugation
steps at 4 C for 10 minutes at 350 x g). The resulting PBMC population was
counted automatically
(ViCell) and stored in RPMI1640 medium, containing 10% FCS and 1% L-alanyl-L-
glutamine
(Biochrom, K0302) at 37 C, 5% CO2 in the incubator until assay start.
T cell enrichment from PBMCs was performed using the Pan T Cell Isolation Kit
11 (Miltenyi Biotec
#130-091-156), according to the manufacturer's instructions. Briefly, the cell
pellets were diluted in 40 pi'
cold buffer per 10 million cells (PBS with 0.5% BSA, 2 inM EDTA, sterile
filtered) and incubated with
10 tit Biotin- Antibody Cocktail per 10 million cells for 10 mm at 4 C. 30 .ti
cold buffer and 20 tt Anti-
Biotin magnetic beads per 10 million cells were added, and the mixture
incubated for another 15 min at
4 C. Cells were washed by adding 10-20x the current volume and a subsequent
centrifugation step at 300
x g for 10 mm. Up to 100 million cells were resuspended in 500 gt buffer.
Magnetic separation of
unlabeled human pan T cells was performed using LS columns (Miltenyi Biotec
#130-042-401)
according to the manufacturer's instructions. The resulting T cell population
was counted automatically
(ViCell) and stored in AIM-V medium at 37 C, 5% CO2 in the incubator until
assay start (not longer than
24 h).
Isolation of prima)). human naive T cells from PBMCs
Peripheral blood mononuclar cells (PBMCs) were prepared by Histopaque density
centrifugation from
enriched lymphocyte preparations (buffy coats) obtained from local blood banks
or from fresh blood from
healthy human donors. T-cell enrichment from PBMCs was performed using the
Naive CD8+ T cell
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
53
isolation Kit from Miltenyi Biotec (#130-093-244), according to the
manufacturer's instructions, but
skipping the last isolation step of CD8+ T cells (also see description for the
isolation of primary human
pan T cells).
Remark: all experiments showing the properties of the bispecific antibodies
were performed with the non
CV bispecific antibodies. However according to the inventors' knowledge the
results for the CV
bispecific antibodies according to the invention are the same or substantially
similar.
Examples
Remark: If not mentioned that Mab2 was used as anti-ROR1 antibody and/or as
anti-ROR1 Fab in an
anti-ROR1/anti-CD3 TCB antibody in the following descriptions of the examples,
then Mabl was used
as anti-RORlantibody and/or as anti-ROR1 Fab in an anti-ROR1/anti-CD3 TCB
antibody.
Example 1 ¨ Generation of anti-ROR1 antibodies
The protein sequences of the VH and VL regions for an ROR1 antibody of SEQ ID
NOs: 2-9 (MAB1) are
described in W02012/075158. Briefly, oliogonucleotides encoding the above
sequences are joined
together via PCR to synthesize cDNAs encoding the VH are VL sequences,
respectively, of the anti-
ROR1 antibody.
For the generation of anti-ROR1 antibody expression vectors, the variable
regions of heavy and light
chain DNA sequences were subcloned in frame with either the human IgG1
constant heavy chain or the
hum IgG1 constant light chain pre-inserted into the respective generic
recipient expression vector
optimized for expression in mammalian cell lines. The antibody expression was
driven by a chimeric
MPSV promoter comprising a CMV enhancer and a MPSV promoter followed by a 5'
UTR, an intron
and a Ig kappa MAR element. The transcription was terminated by a synthetic
polyA signal sequence at
the 3' end of the CDS. All vectors carry a 5'-end DNA sequence coding for a
leader peptide which
targets proteins for secretion in eukaryotic cells. In addition each vector
contained an EBV OriP sequence
for episomal plasmid replication in EBV EBNA expressing cells.
ROR1 antibodies were expressed by transient polymer-based co-transfection of
the respective
mammalian expression vectors in HEK293-EBNA cells, which were cultivated in
suspension. One day
prior to transfection the HEK293-EBNA cells were seeded at 1.5 Mio viable
cells/mL in Ex-Cell
medium, supplemented with 6 mM of L-Glutamine. For every mL of final
production volume 2.0 Mio
viable cells were centrifuged (5 minutes at 210 x g). The supernatant was
aspirated and the cells
resuspended in 100 !IL of CD CHO medium. The DNA for every mL of final
production volume was
prepared by mixing 1 ug of DNA (Ratio heavy chain: light chain = 1:1) in 100
!IL of CD CHO medium.
After addition of 0.27 !IL of polymer solution (1 mg/mL) the mixture was
vortexed for 15 seconds and
left at room temperature for 10 minutes. After 10 minutes, the resuspended
cells and DNA/polymer
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
54
mixture were put together and then transferred into an appropriate container
which was placed in a
shaking device (37 C, 5% CO2). After a 3 hours incubation time 800 [EL of Ex-
Cell Medium,
supplemented with 6 mM L-Glutamine, 1.25 mM valproic acid and 12.5% Pepsoy (50
g/L), was added
for every mL of final Production volume. After 24 hours, 70 [EL of Feed
solution was added for every
mL of final production volume. After 7 days or when the cell viability was
equal or lower than 70%, the
cells were separated from the supernatant by centrifugation and sterile
filtration. The antibodies were
purified by an affinity step and one or two polishing steps, being cation
exchange chromatography and
size exclusion chromatography. When required, an additional polishing step was
used. The recombinant
anti-ROR1 human antibodies were produced in suspension by polymer-based co-
transfecting HEK293-
EBNA cells with the mammalian expression vectors. The cells were transfected
with two vectors. For the
human IgG1 one plasmid encoded the heavy chain and the other plasmid the light
chain. One day prior to
transfection the HEK293-EBNA cells were seeded at 1.5 Mio viable cells/mL in
F17 Medium,
supplemented with 6 mM of L-Glutamine.
Example 2 ¨ Human B-CLL cell line or primary B-CLL cells, multiple myeloma
cell line or Mantle
cell lymphoma cell line expressing ROR1 on their surface
a) Fresh human primary B-CLL cells (CD19+ CD5+) were isolated from blood of
CLL patients. Blood is
collected from CLL patients after informed consent is given, in accordance
with local ethical committee
guidelines and the Declaration of Helsinki. Cryopreserved human primary B-CLL
cells (CD19+ CD5+)
was acquired from Allcells (Alameda, CA, USA). The primary B-CLL cells from
patients were lawfully
obtained and comply with ethical requirements: (i) obtaining samples from
patients diagnosed with CLL
is approved by an Institute Reviewing Board (IRB) or Human Subject Committee;
(ii) a signed and
witnessed informed consent form is obtained from the patient before taking
part in the Allcells Diseased
Cells Program; (iii) all of the patients diagnosed with the above mentioned
diseases are reasonably
compensated for their commitment to the program and the compensation is
approved by the IRB or
Human Subject Committee; (iv) all of the patients are aware that the donated
samples may be used for
any research applications and waived any rights generated from the research
applications. Primary B-CLL
cells were grown in RPMI supplemented with 10% fetal bovine serum. ROR1
expression on primary
CD19+ CD5+ B-CLL cells was confirmed by flow cytometry using fluorochrome-
conjugated anti-human
ROR1 antibodies (see Example 3).
b) Human B lymphocyte multiple myeloma cell line RPMI8226 was acquired from
ATCC (ATCC CCL-
155). RPMI8226 myeloma cells were cultured in DMEM, 10% FCS, 1 % Glutamine.
ROR1 expression
on RPMI8226 cell lines was confirmed by flow cytometry using fluorochrome-
conjugated anti-human
ROR1 antibodies (see Example 3).
c) Human Mantle cell lymphoma (B cell non-Hodgkin's lymphoma) Rec-lcell line
was acquired from
ATCC (ATCC CRL-3004). Rec-1 cells were cultured in DMEM, 10% FCS, 1 %
Glutamine. ROR1
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
expression on Rec-1 cell lines was confirmed by flow cytometry using
fluorochrome-conjugated anti-
human ROR1 antibodies (see Example 3).
5 Example 2.1 ¨ Human ovarian cancer cell lines with different levels of
expression of ROR1 on the
cell surface
a) Human ovarian cancer cell line PA-1 derived from ovarian teratocarcinoma
was acquired from
American Type Culture Collection (ATCC; Cat. No. CRL-1572). PA-1 cell lines
were cultured in
Eagle's Minimum Essential Medium (MEM) (ATCC, Cat. No. 30-2003) supplemented
with 10% fetal
10 bovine serum (heat-inactivated), 2 mM L-glutamine, 1 mM sodium pyruvate,
and 1500 mg/L sodium
bicarbonate. ROR1 expression was confirmed to be high on PA-1 cell lines as
measured by flow
cytometry (see Example 3.1).
b) Human ovarian cancer cell line COLO-704 derived from ovarian adenocarcinoma
was obtained from
Leibniz Institute DSMZ- German Collection of Microorganisms and Cell Cultures
(DSMZ; Cat. No.
15 ACC 198). COLO-704 cell lines were cultured in 90% RPMI 1640 and 10% heat
inactivated fetal bovine
serum. ROR1 expression was confirmed to be medium on COLO-704 cell lines as
measured by flow
cytometry (see Example 3.1).
c) Human ovarian cancer cell line ES-2 derived from ovarian clear cell
carcinoma was acquired from
American Type Culture Collection (ATCC; Cat. No. CRL-1978). ES-2 cell lines
were cultured in
20 ATCC-formulated McCoy's 5a Medium Modified (Cat. No. 30-2007) and 10% fetal
bovine serum.
ROR1 expression was confirmed to be negative on ES-1 cell lines as measured by
flow cytometry (see
Example 3.1).
d) Human ovarian cancer cell line SK-OV-3 derived from ovarian carcinoma was
acquired from
American Type Culture Collection (ATCC; Cat. No. HTB-77). SK-OV-3 cell lines
were cultured in
25 ATCC-formulated McCoy's 5a Medium Modified (Cat. No. 30-2007) and 10% fetal
bovine serum.
ROR1 expression was confirmed to be low on SK-OV-3 cell lines as measured by
flow cytometry (see
Example 3.1).
e) Human ovarian cancer cell line OVCAR-5 derived from ovarian adenocarcinoma
was obtained from
US National Cancer Institute NCI-60 human cancer cell line panel. OVCAR-5 cell
lines were cultured in
30 90% RPMI 1640 and 10% heat inactivated fetal bovine serum. ROR1 expression
was confirmed to be
medium on OVCAR-5 cell lines as measured by flow cytometry (see Example 3.1).
Example 3 ¨ Binding to ROR1 expressed on primary B-CLL cells, RPMI8226 myeloma
cells or
Rec-1 MCL cells (Flow cytometry)
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
56
a) ROR1 expression was assessed on primary CD19+ CD5+ CLL cells by flow
cytometry. Briefly, cells
were harvested, washed, counted for viability, resuspended at 50 000
cells/well of a 96-well round bottom
plate and incubated with A1exa488-labeled anti human ROR1 antibody at 10 ug/m1
for 30 min at 4 C (to
prevent internalization). At the end of incubation time, cells were
centrifuged (5 min at 350 x g), washed
twice with FACS buffer, resuspended in 100 ul FACS buffer and analyzed on a
CantoII device running
FACS Diva software. Figure 2A shows an increase of median fluorescence
intensity upon binding of the
anti-ROR1 antibody to primary B-CLL cells, indicating that ROR1 is expressed
on primary CLL cells.
b) ROR1 expression was then assessed on B lymphocyte myeloma RPMI8226 cell
lines by flow
cytometry, using the methods described above. Figure 3 shows increase of
median fluorescence intensity
upon binding of increasing concentrations of the anti-ROR1 antibody to
RPMI8226 cells (A), but not to
ROR1-negative MKN45 cells (human gastric adenocarcinoma cell line, DSMZ ACC
409) (B). Table 1
shows the binding EC50 of anti-ROR1 antibody to ROR1-positive RPMI8226 cell
lines.
Table 1: EC50 values for binding of anti-ROR1 antibody to RPMI8226 cells
Anti-ROR1 antibody
EC50 (nM) 0.087
EC50 (j4/m1) 0.013
c) ROR1 expression was also tested on MCL Rec-1 cell lines by flow cytometry,
using the methods
describe above. Figure 2B shows increase of median fluorescence intensity upon
binding of the anti-
ROR1 antibody to Rec-1 MCL cells.
Example 3.1 ¨ Binding of ROR1 IgG antibodies to ROR1-positive human ovarian
cancer cell lines
(as detected by flow cytometry)
a) The level of expression of ROR1 was measured on human ovarian cancer cell
lines by flow cytometry
including PA-1, COLO-704, ES-2, SK-OV-3, and OVCAR-5. Briefly, cells were
harvested, washed,
counted for viability, resuspended at 50,000 cells/well of a 96-well round
bottom plate and incubated with
A1exa488-labeled anti human ROR1 antibody for 30 min at 4 C. All ROR1 and
isotype control
antibodies were titrated and analyzed in final concentration range between
0.01 ¨ 100 nM (0.0015 - 15
ug/mL). For samples using non-labelled antibodies, cells were centrifuged (5
min, 350 x g), washed with
120 pi/well FACS Stain Buffer (BD Biosciences), resuspended and incubated for
an additional 30 min at
4 C with fluorochrome-conjugated AlexaFluor 647-conjugated AffiniPure F(ab')2
Fragment goat anti-
human IgG Fc Fragment Specific (Jackson Immuno Research Lab; # 109-606-008).
At the end of
incubation time, cells were centrifuged (5 min at 350 x g), washed twice with
FACS buffer, resuspended
CA 02963692 2017-04-05
WO 2016/055592
PCT/EP2015/073308
57
in 100 ul FACS buffer and analyzed on a CantoII device running FACS Diva
software. Expression of
ROR1 was then quantitified as the median fluorescence intensity (MFI) and
graphs showing the MFI in
function of ROR1 antibody concentrations were plotted. EC50 values were then
measured using Prism
software (GraphPad). Table 2 shows the binding EC50 of Mabl and Mab2 anti-ROR1
antibodies to
ROR1-positive SK-OV-3 and PA-1 ovarian cancer cell lines. Both Mabl and Mab2
anti-ROR1
antibodies bind with more potency to PA-1 cell lines (later found to express
high level of ROR1) than
SK-OV-3 (later found to express low level of ROR1). The calculated EC5Os for
binding of ROR1 Mabl
and ROR1 Mab2 to SK-OV-3 are extrapolated values and may be over- or
underestimated. Figure 3-3
shows an increase of MFI on SK-OV-3 cells (A, open squares) and PA-1 cells (B,
open triangles) in
function of the concentrations of ROR1 Mab2 IgG. Maximum intensity could be
reached approximately
3 times more in PA-1 cells vs. SK-OV-3 cells with an antibody concentration of
10 ug/mL.
Table 2: EC50 values for binding of anti-ROR1 antibodies to ovarian cancer
cell lines
Binding EC50
Ovarian cancer cell lines
ROR1 Mabl ROR1 Mab2
SK-OV-3 ¨ 4.62 nM / 0.69 p,g/m1 ¨ 357.6 nM /
53.37 mg/m1
PA-1 0.87 nM / 0.13 pg/ml 0.64
nM / 0.095 m/m1
b) To determine ROR1 antigen copy number on the cell surface of human ovarian
cancer cell PA-1,
COLO-704, ES-2, SK-OV-3, and OVCAR-5, the Qifikit (Dako # K0078) method was
used. Ovarian
tumor cells were once washed with FACS buffer (100 ul/well; 350 x g for 5 min)
and adjusted to 1 Mio
cells/ml. 50 ul (= 0.5 Mio cells) of the cell suspension were transferred into
each well of a 96 round
bottom well plate, as indicated. Then, 50 ul of mouse anti-human ROR1 IgG
antibody (Biolegend #
357802) or a mouse IgG2a isotype control antibody (BioLegend # 401501) diluted
in FACS buffer (PBS,
0.1% BSA) to a final concentration of 25 ug/m1 (or at saturation
concentrations) were added and staining
was performed for 30 min at 4 C in the dark. Next, 100 ul of the Set-up or
Calibration Beads were added
in separate wells and the cells, as well as the beads were washed twice with
FACS buffer. Cells and
beads were resuspended in 25 tl FACS buffer, containing fluorescein conjugated
anti-mouse secondary
antibody (at saturation concentrations), provided with the Qifikit. Cells and
beads were stained for 45 min
at 4 C in the dark. The cells were washed once and all samples were
resuspended in 100 ul FACS buffer.
Samples were analyzed on a multicolor flow cytometer and installed software
(e.g. CantoII device
running FACS Diva software).
As shown in Table 2.1, ROR1 antigen copy number / binding sites were measured
on five human ovarian
cancer cell lines (ES-2, SK-OV-3, OVCAR-5, COLO-704 and PA-1) and expressed at
different levels.
ES-2 cells did not express any antigen copy of human ROR1 while S-KOV-3 cells
expressed low level of
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
58
human ROR1, OVCAR-5 and COLO-704 cells expressed medium level of human ROR1
and PA-1 cells
expressed high level of human ROR1. In light of these ROR1 expression results,
human ovarian cancer
cell lines with high, medium and/or low expression level of ROR1 will be
selected and used in the
redirected T-cell cytotoxicity assay as tumor target cells in Example 10.
Table 2.1: ROR1 antigen copy number / binding sites on human ovarian cancer
cell lines as measured by
quantitative flow cytometry
ROR1 antigen copy number /
Human ovarian cancer cell lines ROR1 level of
expression
binding sites
ES-2 0 Negative
SK-OV-3 3210 Low
OVCAR-5 5034 Medium
Co1o704 6409 Medium
PA-1 14106 High
Example 4 ¨ Internalization of anti-ROR1 antibody on primary PBMC from CLL
patients or
RPMI8226 MM cells (Flow cytometry)
Anti-ROR1 antibodies were further tested in the internalization assay.
Briefly, human ROR1-expressing
primary B-CLL target cells were harvested with Cell Dissociation Buffer,
washed and resuspended in
RPMI supplemented with 10% FCS at a concentration of 1 x 106 (1 x 106/mL) of
PBMC from untreated
CLL patients or 1 x106 cells/mL RPMI8226 cells after determination of cell
viability using ViCell. The
cell suspension was transferred in a 15 ml Falcon tube for each tested IgG/TCB
and each concentration.
0.5 ml of diluted anti-ROR1 IgG or anti-ROR1/anti-CD3 TCBs conjugated with
A1exa488 (diluted to
1 nM in RPMI + 10% FCS) were added to the tubes and incubated for 30 min in
the cold room on a
shaker. After incubation and washing the cells three times with cold PBS to
remove unbound antibody,
the cells were either left on ice or transferred (0.1 x 106 cells) in 96-well
FACS plate in pre-warmed
medium and incubated at 37 C for 15 min, 30 min, 1 h, 2 h, and 24 h to
facilitate internalization. In
addition, sample of cells were also incubated at 37 C for 2 h and/or 24 h in
the presence of 3 uM
phenylarsine oxide (Sigma-Aldrich) to inhibit internalization. Subsequently,
the cells were washed once
with cold PBS and incubated with A1exa647-labeled anti-human Fc secondary
antibody (F(ab)2) for
min at 4 C. After three final washes with PBS, the cells were centrifuged 4
min at 400 x g and
25 resuspended in FACS buffer with or without propidium iodide (1:4000)
(Sigma). The mean fluorescence
intensity (MFI) of the cells for anti-ROR1 IgG and anti-ROR1/anti-CD3 TCBs
wais measured using a
FACS CantoII flow cytometer (BD Biosciences) and FlowJo analytical software.
MFI reduction can represent antibody internalization, antibody dissociation or
a combination of both. The
percentage of MFI reduction is calculated for each ROR1 antibodies relative to
the unspecific human IgG
30 control (MFI background) and ROR1 antibodies maintained on ice (MFIinax) by
using the formula AMFI=
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
59
100 ¨ 100 X [(MEL
-,xperimental MFIbackground) (MFImax ¨ MFIbackground)] = An MFI
reduction which is
blocked by endocytosis inhibitor phenylarsine oxide indicates antibody
internalization while an MFI
reduction which is not blocked by phenylarsine oxide reflects antibody
dissociation. Internalizing anti-
ROR1 antibodies are known in the state of the art (Baskar et al., Clin. Cancer
Res., 14(2): 396-404
(2008)).
For antibody-based therapies such as T cell bispecifics, it is important that
the antibody or antibody
fragment specific to the tumor target do not internalize, or slowly
internalize, or slightly internalize for
facilitating a stable immune synapse between the tumor cell and the T cell and
effective T cell-mediated
redirected cytotoxicity. Thus, anti-ROR1 antibodies which does not internalize
or slowly internalize or
slightly internalize are selected for the next step (Example 5) below, namely
the production of anti-
ROR1/anti-CD3 T cell bispecific antibodies.
The internalization values of anti-ROR1 IgG antibody in primary CLL cells and
RPMI8226 cells are
further summarized in Figures 4 and 6 and Tables 4 and 6.
Example 5 ¨ Generation of anti-ROR1/anti-CD3 T cell bispecific antibodies
Example 5.1. Generation of anti-CD3 antibodies
The following protein sequences of the VH and VL regions were used to generate
human and
cynomolgus monkey cross reactive CDR antibodies.
CH2527_VH (SEQ ID NO:21):
EVQLLESGGGLVQPGGSLRLSCAASGFTF STYAMNWVRQAPGKGLEWVSRIRSKYNNYATYYA
D SVKGRFTI SRDD SKNTLYLQMN SLRAEDTAVYYCVRHGNFGNSYVSWFAYWGQGTLVTVS S
CH2527_VL (SEQ ID NO:22)
QAVVTQEP SLTV SP GGTVTLT CG S STGAVTT SNYANWVQEKP GQAFRGLIGGTNKRAP GTPARF
SGSLLGGKAALTLSGAQPEDEAEYYCALWYSNLWVFGGGTKLTVL
Briefly, oligonucleotides encoding the above sequences were joined together
via PCR to synthesize
cDNAs encoding the VH and VL sequences, respectively, of the anti-CD3
antibody.
Anti-CD3 antibody CH2527 (SEQ ID NO:21-28) was used to generate the T cell
bispecific antibodies
which were used in the following examples.
Example 5.2. Generation of anti-ROR1/anti-CD3 T cell bispecific 1+1 format
(i.e. one-arm
bispecific (Fab) x (Fab) antibody monovalent for ROR1 and monovalent for CD3)
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
a) An anti-ROR1/anti-CD3 T cell bispecific antibody according to the invention
would have the
advantage of an elimination half-life of about 1 to 12 days which allows at
least once or twice/week
administration.
Anti-ROR1/anti-CD3 T cell bispecific of the 1+1 one-arm format (i.e.
bispecific (Fab)x(Fab) antibody
5 monovalent for ROR1 and monovalent for CD3) are produced with the anti-ROR1
antibodies generated
from Example 1. cDNAs encoding the full Fabs (heavy chain VH and CH1 domains
plus light chain VL
and CL domains) of the corresponding anti-ROR1 IgG1 antibodies, as described
in Example 1, as well as
the anti-CD3 VH and VL cDNAs described in Example 5.1, are used as the
starting materials. For each
bispecific antibody, four protein chains are involved comprising the heavy and
light chains of the
10 corresponding anti-ROR1 antibody and the heavy and light chains of the anti-
CD3 antibody described
above.
b) For the generation of ROR1xCD3 bispecific antibody vectors, the IgG1
derived bispecific molecules
consist at least of two antigen binding moieties capable of binding
specifically to two distinct antigenic
determinants CD3 and ROR1. The antigen binding moieties are Fab fragments
composed of a heavy and
15 a light chain, each comprising a variable and a constant region. At least
one of the Fab fragments is a
"Crossfab" fragment, wherein the constant domains of the Fab heavy and light
chain are exchanged. The
exchange of heavy and light chain constant domains within the Fab fragment
assures that Fab fragments
of different specificity do not have identical domain arrangements and
consequently do not interchange
light chains. The bispecific molecule design can be monovalent for both
antigenic determinants (1+1) or
20 monovalent for CD3 and bivalent for ROR1 where one Fab fragment is fused to
the N-terminus of the
inner CrossFab (2+1). A schematic representation of the constructs is given in
Figure 1. Sequences of the
constructs are shown in SEQ ID NOs 2 to 36. The molecules are produced by
polymer-based co-
transfecting HEK293 EBNA cells growing in suspension with the mammalian
expression. For
preparation of 1+1 CrossFab-IgG constructs, cells are transfected with the
corresponding expression
25 vectors in a 1:1:1:1 ratio ("vector Fc(knob)" : "vector light chain" :
"vector light chain CrossFab" :
"vector heavy chain-CrossFab").
Example 5.3. Generation of anti-ROR1/anti-CD3 T cell bispecific 2+1 format
(i.e. bispecific (Fab)2 x
(Fab) antibody bivalent for ROR1 and monovalent for CD3)
a) An anti-ROR1/anti-CD3 T cell bispecific antibody with a 2+1 format i.e.
bispecific (Fab)2 x (Fab)
30 antibody that is bivalent for ROR1 and monovalent for CD3 would have
advantages on potency,
predictability for efficacy and safety because it would preferentially bind to
the tumor target ROR1 and
avoid CD3 antibody sink, thus higher probability for drug exposure focused to
the tumor.
Anti -ROR1/anti-CD3 T cell bispecific of the 2+1 format (i.e. bispecific
(Fab)2 x (Fab) antibody bivalent
for ROR1 and monovalent for CD3 are produced with the anti-ROR1 antibodies
generated in Example 1.
35 cDNAs encoding the full Fabs (heavy chain VH and CH1 domains plus light
chain VL and CL domains)
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
61
of the corresponding anti-ROR1 IgG1 antibodies, as described in Example 1, as
well as the anti-CD3 VH
and VL cDNAs described in Example 5.1, are used as the starting materials. For
each bispecific antibody,
four protein chains are involved comprising the heavy and light chains of the
corresponding anti-ROR1
antibody and the heavy and light chains of the anti-CD3 antibody described
above.
b) For the generation of ROR1xCD3 bispecific antibody vectors, the IgG1
derived bispecific molecules
consist at least of two antigen binding moieties capable of binding
specifically to two distinct antigenic
determinants CD3 and ROR1. The antigen binding moieties are Fab fragments
composed of a heavy and
a light chain, each comprising a variable and a constant region. At least one
of the Fab fragments is a
"Crossfab" fragment, wherein the constant domains of the Fab heavy and light
chain are exchanged. The
exchange of heavy and light chain constant domains within the Fab fragment
assures that Fab fragments
of different specificity do not have identical domain arrangements and
consequently do not interchange
light chains. The bispecific molecule design can be monovalent for both
antigenic determinants (1+1) or
monovalent for CD3 and bivalent for ROR1 where one Fab fragment is fused to
the N-terminus of the
inner CrossFab (2+1).A schematic representation of the constructs is given in
Figure 1; Sequences of the
constructs are shown in SEQ ID NOs 1 to 62 . The molecules are produced by
polymer-based co-
transfecting HEK293 EBNA cells growing in suspension with the mammalian
expression. For
preparation of 2+1 CrossFab-IgG constructs, cells are transfected with the
corresponding expression
vectors in a 1:2:1:1 ratio ("vector Fc(knob)" : "vector light chain" : "vector
light chain CrossFab" :
"vector heavy chain-CrossFab").
Example 5.4. Production and purification of anti-ROR1/anti-CD3 T cell
bispecific antibodies with
or without charge variants
For the production of the bispecific antibodies, bispecific antibodies are
expressed by transient polymer-
based co-transfection of the respective mammalian expression vectors in HEK293-
EBNA cells, which are
cultivated in suspension. One day prior to transfection the HEK293-EBNA cells
are seeded at 1.5 Mio
viable cells/mL in Ex-Cell medium, supplemented with 6 mM of L-Glutamine. For
every mL of final
production volume 2.0 Mio viable cells are centrifuged (5 minutes at 210 x g).
The supernatant is
aspirated and the cells resuspended in 100 [IL of CD CHO medium. The DNA for
every mL of final
production volume is prepared by mixing 1 lag of DNA (Ratio heavy chain:
modified heavy chain: light
chain: modified light chain = 1:1:2:1) in 100 [IL of CD CHO medium. After
addition of 0.27 [IL of
polymer solution (1 mg/mL) the mixture is vortexed for 15 seconds and left at
room temperature for 10
minutes. After 10 minutes, the resuspended cells and DNA/polymer mixture are
put together and then
transferred into an appropriate container which is placed in a shaking device
(37 C, 5% CO2). After a 3
hours incubation time 800 [IL of Ex-Cell Medium, supplemented with 6 mM L-
Glutamine, 1.25 mM
valproic acid and 12.5% Pepsoy (50 g/L), is added for every mL of final
Production volume. After 24
hours, 70 [IL of Feed solution is added for every mL of final production
volume. After 7 days or when
the cell viability is equal or lower than 70%, the cells are separated from
the supernatant by centrifugation
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
62
and sterile filtration. The antibodies are purified by an affinity step and
one or two polishing steps, being
cation exchange chromatography and size exclusion chromatography. When
required, an additional
polishing step is used.
For the affinity step the supernatant is loaded on a protein A column (HiTrap
Protein A FF , 5 mL, GE
Healthcare) equilibrated with 6 CV 20 mM sodium phosphate, 20 mM sodium
citrate, pH 7.5. After a
washing step with the same buffer the antibody is eluted from the column by
step elution with 20 mM
sodium phosphate, 100 mM sodium chloride, 100 mM Glycine, pH 3Ø The
fractions with the desired
antibody are immediately neutralized by 0.5 M Sodium Phosphate, pH 8.0 (1:10),
pooled and
concentrated by centrifugation. The concentrate is sterile filtered and
processed further by cation
exchange chromatography and/or size exclusion chromatography.
For the cation exchange chromatography step the concentrated protein is
diluted 1:10 with the elution
buffer used for the affinity step and loaded onto a cation exchange colume
(Poros 50 HS, Applied
Biosystems). After two washing steps with the equilibration buffer and a
washing buffer resp. 20 mM
sodium phosphate, 20 mM sodium citrate, 20 mM TRIS, pH 5.0 and 20 mM sodium
phosphate, 20 mM
sodium citrate, 20 mM TRIS, 100 mM sodium chloride pH 5.0 the protein is
eluted with a gradient using
mM sodium phosphate, 20 mM sodium citrate, 20 mM TRIS, 100 mM sodium chloride
pH 8.5. The
fractions containing the desired antibody are pooled, concentrated by
centrifugation, sterile filtered and
processed further a size exclusion step.
For the size exclusion step the concentrated protein is injected in a XK16/60
HiLoad Superdex 200
20 column (GE Healthcare), and 20 mM Histidine, 140 mM Sodium Chloride, pH 6.0
with or without
Tween20 as formulation buffer. The fractions containing the monomers are
pooled, concentrated by
centrifugation and sterile filtered into a sterile vial.
Determination of the antibody concentration is done by measurement of the
absorbance at 280 nm, using
the theoretical value of the absorbance of a 0.1% solution of the antibody.
This value is based on the
amino acid sequence and calculated by GPMAW software (Lighthouse data).
Purity and monomer content of the final protein preparation is determined by
CE-SDS (Caliper LabChip
GXII system (Caliper Life Sciences)) resp. HPLC (TSKgel G3000 SW XL analytical
size exclusion
column (Tosoh)) in a 25 mM potassium phosphate, 125 mM Sodium chloride, 200 mM
L-arginine
monohydrochloride, 0.02 % (w/v) Sodium azide, pH 6.7 buffer.
To verify the molecular weight of the final protein preparations and confirm
the homogeneous
preparation of the molecules final protein solution, liquid chromatography-
mass spectometry (LC-MS) is
used. A deglycosylation step is first performed. To remove heterogeneity
introduced by carbohydrates,
the constructs are treated with PNGaseF (ProZyme). Therefore, the pH of the
protein solution is adjusted
to pH7.0 by adding 2 ul 2 M Tris to 20 ug protein with a concentration of 0.5
mg/ml. 0.8 ug PNGaseF is
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
63
added and incubated for 12 h at 37 C. The LC-MS online detection is then
performed. LC-MS method is
performed on an Agilent HPLC 1200 coupled to a TOF 6441 mass spectrometer
(Agilent). The
chromatographic separation is performed on a Macherey Nagel Polysterene
column; RP1000-8 (8 um
particle size, 4.6 x 250 mm; cat. No. 719510). Eluent A is 5 % acetonitrile
and 0.05 % (v/v) formic acid
in water, eluent B was 95 % acetonitrile, 5 % water and 0.05 % formic acid.
The flow rate was 1 ml/min,
the separation is performed at 40 C and 6 ug (15 ul) of a protein sample
obtained with a treatment as
described before (table 8).
Table 8
Time (min.) %B
0.5 15
60
12.5 100
14.5 100
14.6 15
16 15
16.1 100
10 During the first 4 minutes, the eluate is directed into the waste to
protect the mass spectrometer from salt
contamination. The ESI-source was running with a drying gas flow of 12 limin,
a temperature of 350 C
and a nebulizer pressure of 60psi. The MS spectra are acquired using a
fragmentor voltage of 380 V and
a mass range 700 to 3200 m/z in positive ion mode using. MS data are acquired
by the instrument
software from 4 to 17 minutes.
A head-to-head production run to compare the production/purification profile
of ROR1xCD3-TCB
without charge variant vs. ROR1xCD3-TCBcv (with charge variants) antibodies is
then conducted to
further evaluate the advantages of the CL-CH1 charge modifications applied to
the T cell bispecific
antibodies.
Table 8-1 shows the favorable production/purification profile of three
molecules of ROR1xCD3-TCB
with charge variants following standard, non-optimized purification methods
including Protein A (PA)
affinity chromatography and size exclusion chromatographic (SEC) purification.
Table 8-1: Production/purification profile of anti-ROR1/anti-CD3 T cell
bispecific antibodies with
charge variants following standard, non-optimized purification methods
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
64
Titer (mg/L) Monomer content (%) Purity (%)
Mab2 ROR1-TCBcv 40.71 100 98.66
Mab3 ROR1-TCBcv 104.12 100 93.78
Mab4 ROR1-TCBcv 25.36 99.03 97.69
Figure 3-1 depicts the CE-SDS (non-reduced) graphs of the final protein
preparations after different
methods of purification for another TCB molecule without or with charge
variants namely 83A10-TCB
and 83A10-TCBcv antibodies. Protein A (PA) affinity chromatography and size
exclusion
chromatographic (SEC) purification steps applied to 83A10-TCB antibody
resulted in a purity of <30%
and 82.8% of monomer content (A). When additional purifications steps
including cation exchange
chromatography (cIEX) and a final size exclusion chromatographic (re-SEC)
steps were applied to the
final protein preparations in (A), the purity was increased to 93.4% but the
monomer content remained
the same and the yield was significantly reduced to 0.42 mg/L. However, when
specific charge
modifications were applied to 83A10 anti-BCMA Fab CL-CH1, namely 83A10-TCBcv
antibody, a
superior production/purification profile of the TCB molecule, as demonstrated
by a purity of 95.3%,
monomer content of 100% and yield of up to 3.3 mg/L, could already be observed
even when PA + cIEX
+ SEC purification steps were applied (C) in comparison to (B) with a
production/purification profile
showing a 7.9-fold lower yield and 17.2% lower monomer content despite
including an additional re-SEC
purification step.
A head-to-head production run to compare the production/purification profile
of 83A10-TCB vs. 83A10-
TCBcv antibodies was then conducted to further evaluate the advantages of the
CL-CH1 charge
modifications applied to the antibodies. 83A10-TCB and 83A10-TCBcv molecules
are both of molecular
format as described in Figure 2a of PCT/EP2015/067841. As depicted in Figure 3-
2, properties of
83A10-TCB and 83A10-TCBcv antibodies were measured side-by-side and compared
after each
purification steps 1) PA affinity chromatography only (A, B), 2) PA affinity
chromatography then SEC
(C, D) and 3) PA affinity chromatography then SEC then cIEX and re-SEC (E, F).
The CE-SDS (non-
reduced) graphs of the final protein solutions after the respective methods of
purification for 83A10-TCB
and 83A10-TCBcv antibodies are demonstrated in Figure 3-2. As shown in Figures
3-2A and 3-2B,
improvements with applying the charge variants to the TCB antibody were
already observed after
purification by PA affinity chromatography only. In this head-to-head study,
PA affinity chromatography
purification step applied to 83A10-TCB antibody resulted in a purity of 61.3%,
a yield of 26.2 mg/L and
63.7% of monomer content (3-2A). In comparison, when 83A10-TCBcv antibody was
purified by PA
affinity chromatography all the properties were improved with a better purity
of 81.0%, a better yield of
51.5 mg/L and 68.2% of monomer content (3-2B). When an additional SEC
purification step was applied
to the final protein preparations as seen in Figures 3-2A and 3-2B, 83A10-TCB
gained a purity of 69.5%,
a yield of 14.1 mg/L and 74.7% of monomer content (3-2C) as compared to 83A10-
TCBcv with
improved purity and monomer content of up to 91.0% and 83.9% respectively, and
a yield of 10.3 mg/L
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
(3-2D). Even though the yield was slightly less (i.e. 27% less) for 83A10-
TCBcv than for 83A10-TCB in
this particular experiment, the percentage of correct molecule was much better
for 83A10-TCBcv than for
83A10-TCB, respectively 90% vs. 40-60%, as measured by LC-MS. In the third
head-to-head
comparison, 83A10-TCB and 83A10-TCBcv final protein preparations from Figures
3-2C and 3-2D were
5 pooled with approximately 1 L (equivolume) of respective final protein
preparations from another
purification batch (same production) following PA affinity chromatography
purification step only. The
pooled protein preparations were then being further purified by cIEX and SEC
purification methods. As
depicted in Figures 3-2E and 3-2F, improvement of the production/purification
profile of the TCB
antibody with the charge variants was consistently observed when compared to
TCB antibody without
10 charge variant. After several steps of purification methods (i.e. PA +/-
SEC + cIEX + SEC) were used to
purify 83A10-TCB antibody, only 43.1% purity was reached and 98.3% of monomer
content could be
achieved but to the detriment of the yield which was reduced to 0.43 mg/L. The
percentage of correct
molecule as measured by LC-MS was still poor with 60-70%. At the end, the
quality of the final protein
preparation was not acceptable for in vitro use. In stark contrast, when the
same multiple purification
15 steps with the same chronology were applied to 83A10-TCBcv antibody, 96.2%
purity and 98.9% of
monomer content were reached as well as 95% of correct molecule as measured by
LC-MS. The yield
however was also greatly reduced to 0.64 mg/L after cIEX purification step.
The results show that better
purity, higher monomer content, higher percentage of correct molecule and
better yield can be achieved
with 83A10-TCBcv antibody only after two standard purification steps i.e. PA
affinity chromatography
20 and SEC (Figure 3-2D) while such properties could not be achieved with
83A10-TCB even when
additional purification steps were applied (Figure 3-2E).
Table 8-2 summarizes the properties of 83A10-TCB as compared to 83A10-TCBcv
following PA
purification step. Table 8-3 summarizes the properties of 83A10-TCB as
compared to 83A10-TCBcv
following PA and SEC purification steps. Table 8-4 summarizes the properties
of 83A10-TCB as
25 compared to 83A10-TCBcv following PA and SEC plus PA alone then cIEX and re-
SEC purification
steps. For Tables 8-2 to 8-4, the values in bold highlight the superior
property as compared between
83A10-TCB vs. 83A10-TCBcv. With one exception (i.e. yield respectively amount,
see Table 8-3) which
may not be representative, all the production/purification parameters and
values resulting from the 3
head-to-head comparison experiments were superior for 83A10-TCBcv as compared
to 83A10-TCB. The
30 overall results clearly demonstrate that advantages in
production/purification features could be achieved
with applying CL-CH1 charge modifications to TCB antibodies and that only two
purification steps (i.e
PA affinity chromatography and SEC) were required to achieve already high
quality protein preparations
with very good developability properties.
35 Table 8-2: Production/purification profile of anti-BCMA/anti-CD3 T cell
bispecific antibodies following
protein A affinity chromatography purification step
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
66
83A10-TCB 83A10-TCBcv
Purity (%) 61.3 81.0
Yield (mg/L) 26.2 51.5
Amount (mg) 24.3 50.2
Monomer (%) 63.7 68.2
Correct molecule by
LC-MS (%) n.d. n.d
Table 8-3: Production/purification profile of anti-BCMA/anti-CD3 T cell
bispecific antibodies following
protein A affinity chromatography and size exclusion chromatography
purification steps
83A10-TCB 83A10-TCBcv
Purity (%) 69.5 91.0
Yield (mg/L) 14.1 10.3
Amount (mg) 13.1 10.0
Monomer (%) 74.7 83.9
Correct molecule by
40-60 90
LC-MS (%)
Table 8-4: Production/purification profile of anti-BCMA/anti-CD3 T cell
bispecific antibodies following
1.a) protein A affinity chromatography and size exclusion chromatography and
1.b) protein A affinity
chromatography only pooled together then 2) cation exchange chromatography and
3) final size exclusion
chromatography purification steps
83A10-TCB 83A10-TCBcv
Purity (%) 43.1 96.2
Yield (mg/L) 0.43 0.64
Amount (mg) 0.73 1.27
Monomer (%) 98.3 98.9
Correct molecule by
60-70% >950/
LC-MS (%)
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
67
Example 6 ¨ Binding of anti-ROR1/anti-CD3 T cell bispecific antibodies to ROR1-
positive B-CLL
cells or myeloma cells or CD3 on T cells (Flow cytometry)
a) Anti-ROR1/anti-CD3 T cell bispecific antibodies generated in Example 5 were
also analyzed by flow
cytometry for their binding properties to human ROR1 expressed on primary B-
CLL cells or human CD3
expressed on human leukemic T cells Jurkat (ATCC T1B-152). Jurkat T cells were
cultured in RPMI
supplemented with 10% fetal calf serum. Briefly, cultured cells were
harvested, counted and cell viability
was evaluated using ViCell. Viable cells were then adjusted to 2 x 106 cells
per ml in FACS Stain Buffer
(BD Biosciences) containing 0.1% BSA. 100 ul of this cell suspension were
further aliquoted per well
into a round-bottom 96-well plate. 30 ul of the A1exa488-labelled anti-
ROR1/anti-CD3 T cell bispecific
antibodies or corresponding IgG control were added to the cell-containing
wells to obtain final
concentrations of 3 nM to 500 nM or 0.1 pM to 200 nM. Anti-ROR1/anti-CD3 T
cell bispecific
antibodies and control IgG were used at the same molarity. After incubation
for 30 min at 4 C, cells were
centrifuged (5 min, 350 x g), washed twice with 150 uliwell BSA-containing
FACS Stain Buffer (BD
Biosciences), then cells were fixed using 100 ul BD Fixation buffer per well
(#BD Biosciences, 554655)
at 4 C for 20 min, resuspended in 120 ul FACS buffer and analyzed using BD
FACS CantoII. Binding of
the anti-ROR1/anti-CD3 T cell bispecific antibodies to B-CLL cells and T cells
were evaluated and the
mean fluorescence intensity was determined gated on either ROR1-expressing B-
CLL cells or CD3-
expressing Jurkat T cells and plotted in histograms or dot plots. Figure 7
shows the mean fluorescence
intensity for anti-ROR1/anti-CD3 T cell bispecific antibodies binding to
Jurkat T cells and plotted in
function of antibody concentration. EC50 values and maximal binding of anti-
ROR1/anti-CD3 TCB1+1
and anti-ROR1/anti-CD3 TCB2+1 antibodies to Jurkat cells were not reached.
Interestingly, ROR1/anti-
CD3 TCB1+1 antibody binds more efficiently to Jurkat T cells than ROR1/anti-
CD3 TCB2+1 antibody
does. DP47 isotype control antibody or anti-ROR1 IgG antibody did not bind to
Jurkat T cells.
b) Anti-ROR1/anti-CD3 T cell bispecific antibodies were analyzed by flow
cytometry for binding to
human ROR1 on ROR1-expressing myeloma RPMI8226 cells. Briefly, cultured cells
were harvested,
counted and cell viability was evaluated using ViCell. Viable cells were then
adjusted to 2 x 106 cells per
ml in BSA-containing FACS Stain Buffer (BD Biosciences). 100 ul of this cell
suspension was further
aliquoted per well into a round-bottom 96-well plate and incubated with 30 ul
of the A1exa488-labelled
anti-ROR1/anti-CD3 T cell bispecific antibodies or corresponding IgG control
for 30 min at 4 C. All
anti-ROR1/anti-CD3 T cell bispecific antibodies (and isotype control) were
titrated and analyzed in final
concentration range between 0.136 ¨ 13.6 nM. For samples using non-labelled
antibodies, cells were
centrifuged (5 min, 350 x g), washed with 120 1/well FACS Stain Buffer (BD
Biosciences), resuspended
and incubated for an additional 30 min at 4 C with fluorochrome-conjugated
AlexaFluor 647-conjugated
AffiniPure F(ab')2 Fragment goat anti-human IgG Fc Fragment Specific (Jackson
Immuno Research Lab;
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
68
109-606-008). Cells were then washed twice with Stain Buffer (BD Biosciences),
fixed using 100 ul BD
Fixation buffer per well (#BD Biosciences, 554655) at 4 C for 20 min,
resuspended in 120 ul FACS
buffer and analyzed using BD FACS CantoII. Figure 8 shows the mean
fluorescence intensity for anti-
ROR1/anti-CD3 T cell bispecific antibodies plotted in function of antibody
concentration:anti-
ROR1/anti-CD3 TCB1+1 and anti-ROR1/anti-CD3 TCB2+1 antibodies on RPMI8226
cells EC50 values
(denoting the antibody concentration required to reach 50% of the maximal
binding) for the binding of
anti-ROR1/anti-CD3 TCB1+1 and anti-ROR1/anti-CD3 TCB2+1 antibodies to RPMI8226
cells are
summarized in Table 3. Anti-ROR1/anti-CD3 TCB2+1 antibody bivalent for ROR1
seems to bind to
ROR1-positive RPMI8226 myeloma cells slightly better than anti-ROR1/anti-CD3
TCB1+1 antibody
which is monovalent to ROR1, as detected by FACS (Figure 8). Anti-ROR1/anti-
CD3 TCB antibodies
were also shown to bind to primary B-CLL cells as detected by flow cytometry
using a fluorochrome-
conjugated secondary anti-human Fc antibody (Figure 2A).
Table 3: EC50 values for binding of anti-ROR1/anti-CD3 T cell bispecific
antibodies to RPMI8226 cells
Anti-ROR1/anti-CD3 Anti-ROR1/anti-CD3
TCB1+1 antibody TCB2+1 antibody
EC50 (nM) 1.1 0.56
EC50 (n/m1) 0.16 0.11
Example 6.1 ¨ Binding of anti-ROR1/anti-CD3 T cell bispecific antibodies to
ovarian cancer cells
and T cells (as measured by flow cytometry)
Anti-ROR1/anti-CD3 T cell bispecific antibodies generated in Example 5 were
analyzed by flow
cytometry for their binding to human ovarian cancer cell lines PA-1 and SK-OV-
3 and human CD3
expressed on human leukemic T cells Jurkat (ATCC TIB-152). Jurkat T cells were
cultured in
RPMI1640 medium supplemented with 10% fetal calf serum. Briefly, cultured
cells were harvested,
counted and cell viability is evaluated using ViCell. Viable cells were then
adjusted to 2 x 106 cells per ml
in FACS Stain Buffer (BD Biosciences) containing 0.1% BSA. 100 ul of this cell
suspension were further
aliquoted per well into a round-bottom 96-well plate. 30 ul of the Alexa488-
labelled anti-ROR1/anti-CD3
T cell bispecific antibodies or corresponding IgG control were added to the
cell-containing wells to obtain
final concentrations of 1 nM to 500 nM (Jurkat T cells) or 0.1 nM to 100 nM
(human ovarian cancer
cells). Anti-ROR1/anti-CD3 T cell bispecific antibodies and control IgG were
used at the same molarity.
After incubation for 30 min at 4 C, cells are centrifuged (5 min, 350 x g),
washed twice with 150 ul/well
BSA-containing FACS Stain Buffer (BD Biosciences), then cells are fixed using
100 ul BD Fixation
buffer per well (#BD Biosciences, 554655) at 4 C for 20 min, resuspended in
120 ul FACS buffer and
analyzed using BD FACS CantoII. Binding of the anti-ROR1/anti-CD3 T cell
bispecific antibodies to
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
69
human ovarian cancer cells and T cells were evaluated and the median
fluorescence intensity was
determined gated on either human ovarian cancer cells or CD3-expressing Jurkat
T cells and plotted in
histograms and dot plots. For samples using non-labelled antibodies, cells
were centrifuged (5 min, 350 x
g), washed with 120 1/well FACS Stain Buffer (BD Biosciences), resuspended and
incubated for an
additional 30 min at 4 C with fluorochrome-conjugated AlexaFluor 647-
conjugated AffiniPure F(ab')2
Fragment goat anti-human IgG Fc Fragment Specific (Jackson Immuno Research
Lab; 109-606-008).
Cells were then washed twice with Stain Buffer (BD Biosciences), fixed using
100 ul BD Fixation buffer
per well (#BD Biosciences, 554655) at 4 C for 20 min, resuspended in 120 ul
FACS buffer and analyzed
using BD FACS CantoII. Median fluorescence intensity for anti-ROR1/anti-CD3 T
cell bispecific
antibodies in function of antibody concentrations were plotted. EC50 values
(denoting the antibody
concentration required to reach 50% of the maximal binding) for the binding of
anti-ROR1/anti-CD3
antibodies to human ovarian cancer cells were measured using Prism (GraphPad).
As depicted in Figure
3-3, there was a concentration-dependent binding of ROR1 Mab2 IgG and ROR1
Mab2-TCBcv on SK-
OV-3 (A) and on PA-1 human (B) ovarian cancer cell lines as measured by an
increase in the median
fluorescence intensity signal in function of antibody concentrations. Such
positive signals were not
observed when the control-TCB binding to CD3 only and not to ROR1 was tested
on both SK-OV-3 and
PA-1 ovarian cancer cell lines (A and B; closed circles). As shown in Figure 3-
4, there was a
concentration-dependent binding of ROR1 Mab2-TCBcv and control-TCB on Jurkat T
cells confirming
that both TCB antibodies bind to CD3 on T cells.
Example 7 ¨ Internalization of anti-ROR1/anti-CD3 T cell bispecific antibodies
on EHEB B-CLL
cell line or primary PBMC from CLL patients or RPMI8226 MM cells (Flow
cytometry).
Anti-ROR1/anti-CD3 T cell bispecific antibodies selected in step (Example 5)
above were further tested
in the internalization assay. Briefly, cryopreserved human ROR1-expressing
primary B-CLL target cells
were thawed, harvested with Cell Dissociation Buffer, washed and resuspended
in RPMI supplemented
with 10% FCS at a concentration of 1 x 106 (1 x 106/mL) of cryopreserved PBMC
from untreated CLL
patients or 2 x106cells/mL of EHEB B-CLL cell line or 1 x106 cells/mL RPMI8226
cells after
determination of cell viability using ViCell. The cell suspension was
transferred in a 15 ml Falcon tube
for each tested IgG/TCB and each concentration. 0.5 ml of diluted anti-ROR1
IgG or anti-ROR1/anti-
CD3 TCBs conjugated with A1exa488 (diluted to 1 nM in RPMI + 10% FCS) were
added to the tubes and
incubated for 30 min in the cold room on a shaker. After incubation and
washing the cells three times
with cold PBS to remove unbound antibody, the cells were either left on ice or
transferred (0.1 x 106
cells) in 96-well FACS plate in pre-warmed medium and incubated at 37 C for 15
min, 30 min, 1 h, 2 h,
and 24 h to facilitate internalization. In addition, cell samples were
incubated at 37 C for 2 h and/or 24 h
in the presence of 3 uM phenylarsine oxide (Sigma-Aldrich) to inhibit
internalization. Subsequently, the
cells were washed once with cold PBS and incubated with A1exa647-labeled anti-
human Fc secondary
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
antibody (F(ab)2) for 30 min at 4 C. After three final washes with PBS, the
cells were centrifuged 4 min
at 400 x g and resuspended in FACS buffer with or without propidium iodide
(1:4000) (Sigma). The
mean fluorescence intensity (MFI) of the cells for anti-ROR1 IgG and anti-
ROR1/anti-CD3 TCBs was
measured using a FACS CantoII flow cytometer (BD Biosciences) and FlowJo
analytical software.
5 The term "reduction of mean fluorescence intensity" (AMFI) reflecting the
internalization of the said
anti-ROR1 antibody into ROR1-positive cells" or "MFI reduction" as used herein
refers to the percentage
of MFI reduction as calculated for each ROR1 antibodies relative to the
unspecific human IgG control
(MFI background) and ROR1 antibodies maintained on ice (MFImax) by using the
formula AMFI= 100 ¨ 100
X [(MF 'experimental ¨ MFIbackground) (MFImax ¨ MFIbackground)] =
MFIexperimental is the MFI measured with said
10 ROR1 antibody after 2h incubation at 37 C. MFI reduction can represent
antibody internalization,
antibody dissociation or a combination of both. An MFI reduction which is
blocked by endocytosis
inhibitor phenylarsine oxide indicates antibody internalization while an MFI
reduction which is not
blocked by phenylarsine oxide reflects antibody dissociation. Internalizing
anti-ROR1 antibodies are
known in the state of the art (Baskar et al., Clin. Cancer Res., 14(2): 396-
404 (2008)).
15 In some studies, the internalization rate of anti-ROR1/anti-CD3 T cell
antibodies was then compared to
that of anti-ROR1 bivalent IgG antibody.
For antibody-based therapies such as T cell bispecifics, it is important that
the antibody or antibody
fragment specific to the tumor target does not internalize, or slowly
internalizes, or slightly internalizes
for facilitating a stable immune synapse between the tumor cell and the T cell
and effective T cell-
20 mediated redirected cytotoxicity and T cell activation.
As shown in Figures 4A and 4B and summarized in Table 4, anti-ROR1 IgG
antibody (1M) internalized
about 12.5% in primary B-CLL cells after an incubation of 2 hrs at 37 C while
anti-ROR1/anti-CD3
TCB2+1 antibody (1 nM) showed an internalization rate of 27.1% in primary B-
CLL cells at the same
experimental conditions (Figures 4A and 4C) as measured by FACS (indirect
detection of secondary
25 fluorochrome-conjugated antibody). Internalization was calculated based on
the MFI value at time 0,
baseline, and calculated using the previously described formula. The results
show that anti-ROR1/anti-
CD3 TCB2+1 has an internalization rate of less than 30% in B-CLL cells.
Figure 5 shows the internalization rate of anti-ROR1/anti-CD3 TCB1+1 antibody
(1 nM) in primary B-
CLL cells after an incubation of 2 hrs at 37 C in the presence or absence of
phenylarsine oxide (PAO).
30 Because reduction of MFI signal can be due to internalization and/or
dissociation of the antibody, it is
important to verify if a reduction of MFI signal is caused by internalization
or not by using an
endocytosis inhibitor to block internalization. This is particularly important
for monovalent antibodies
that have lower binding avidity to cells than bivalent antibodies. As shown in
Figure 5, there was a
decrease of 91% in the MFI signal in primary B-CLL cells after an incubation
of 2 hrs at 37 C without
35 PAO. However, when the B-CLL cells were incubated in the presence of PAO (3
uM), 90% decrease in
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
71
MFI signal was still observed indicating that the loss in MFI signal was not
due to internalization of the
antibody but rather probably dissociation (Table 5). Internalization rate
could then be calculated to 0%.
The results demonstrate that anti-ROR1/anti-CD3 TCB1+1 does not internalize in
B-CLL cells, which is
a preferred feature for a TCB antibody.
Figure 6 and Table 6 summarize the internalization rates of TCB2+1 antibodies
and anti-ROR1 IgG
antibody (1 nM) in RPMI8226 MM cells after an incubation of 2 hrs at 37 C, as
measured in two
independent experiments. The results demonstrate that anti-ROR1/anti-CD3
TCB2+1 has an
internalization rate of less than 15% in RPMI cells.
Table 4: Internalization values for anti-ROR1/anti-CD3 2+1 T cell bispecific
antibody and ROR1 IgG in
primary B-CLL cells
Internalization of anti-
Internalization of anti-
ROR1/anti-CD3 TCB2+1
ROR1 antibody (%)
antibody (%)
Time 0 0 (baseline) 0 (baseline)
Time 2 hrs 12.5 27.1
Table 5: Internalization values for anti-ROR1/anti-CD3 1+1 T cell bispecific
antibody and ROR1 IgG in
primary B-CLL cells
Internalization of anti-
ROR1/anti-CD3 TCB1+1
antibody (%)
Time 0 0 (baseline)
Time 2 hrs 0
Table 6: Internalization values for anti-ROR1/anti-CD3 T cell bispecific
antibodies and ROR1 IgG in
RPMI8226 MM cells
Internalization of anti-
Internalization of anti-
Experiment 1 ROR1 antibody (%) ROR1/anti-CD3 TCB1+1
antibody (%)
Time 0 0 (baseline) 0 (baseline)
Time 2 hrs 0.7 8.6
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
72
Experiment 2 Internalization of anti- Internalization of anti-
ROR1 antibody (%) ROR1/anti-CD3 TCB1+1
antibody (%)
Time 0 0 (baseline) 0 (baseline)
Time 2 hrs 0 11.8
Example 8 ¨ Activation of T cells upon engagement of anti-ROR1/anti-CD3 T cell
bispecific
antibodies (Flow cytometry)
a) Anti-ROR1/anti-CD3 T cell bispecific antibodies generated in Example 5 were
also analyzed by flow
cytometry for their potential to induce T cell activation by evaluating the
surface expression of the early
activation marker CD69, or the late activation marker CD25 on CD4+ and CD8+ T
cells in the presence or
absence of human ROR1-positive cells. Briefly, ROR1-positive cells were
harvested with Cell
Dissociation buffer, counted and cell viability is verified using ViCell.
Viable B-CLL cells were adjusted
to 0.2 x 106 cells/mL in RPMI supplemented with 10% FCS, 100 [1,1 of this cell
suspension per well was
pipetted into a round-bottom 96-well plate. 50 [1,1 of the T cell bispecific
constructs were added to the
ROR1-positive cells-containing wells to obtain a final concentration of 0.01
fM to 100 pM or 0.01 pM to
100 nM. The 96-well plate was set aside and kept at 37 C, 5% CO2 until further
manipulations.
PBMC were isolated from fresh blood using density gradient centrifugation
using Cell Preparation Tubes
with Sodium citrate (Vacutainer CPT tubes, BD Biosciences). Total human T
cells were then isolated
using the Pan T Cell Isolation Kit II (Miltenyi Biotec), according to the
manufacturer's instructions. In
some studies, CD8 T cell clones were used as effectors. CD8 T cells specific
to NLV (a CMV specific
peptide recognized by HLA-A2) were purified from HLA-A2+ healthy donor PBMCs
using aCD8
antibodies and tetramers specific to HLA-A2 complexed with NLV peptide and
sorted with a cell sorter.
The purified cells were expanded on irradiated feeder preparations obtained
from healthy donor PBMC
and HLA-A2+LCLs (lymphoblastoid cells) pulsed with NLV peptide in media
(RPMI1640+10%FBS+1%L-glutamine) with 40011J IL2. The NLV specific CD8 T cell
clones were
maintained in the same media with 400IU IL2 and regularly reactivated on
feeder preparations. Human
total T cells or CD8 T cell clones (effectors) were then adjusted to 2 x 106
cells per ml in RPMI
supplemented with 10% FCS. 50 [1,1 of this cell suspension was added per well
in the assay plate
containing already ROR1-positive target cells to obtain a final E:T ratio of
3:1 (CD8 T cells as effectors)
or 10:1 (PBMC as effectors). To test whether the T cell bispecific constructs
were able to activate T cells
in the presence of only ROR1-positive tumor target cells, wells containing
final concentration(s) in the
range of 0.01 fM to 100 pM or 0.01 pM to 100 nM of the respective bispecific
molecules with effector
cells but without ROR1-positive tumor target cells were also included. After
incubation for 6 to 24h (CD8
T cell clones as effectors) or 24 to 48 hrs (PBMC as effectors) at 37 C, 5%
CO2, cells were pelleted down
by centrifugation (5 min, 350 x g) and washed twice with 150 [11/well of FACS
Stain Buffer (BD
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
73
Biosciences). Surface staining of the effector cells with selected
fluorochrome-conjugated antibodies
against human CD4, CD8, CD69 or CD25 (BD Biosciences) was performed at 4 C for
30 min, protected
from light, in FACS Stain Buffer (BD Biosciences) according to the
manufacturer's protocol. Cells were
washed twice with 150 [El/well FACS Stain Buffer then fixed using 100 ul BD
Fixation buffer per well
(#BD Biosciences, 554655) at 4 C for 20 min, resuspended in 120 [El FACS
buffer and analyzed using
BD FACS CantoII. The expression of CD69 or CD25 activation markers were
determined by measuring
the mean fluorescence intensity gated on CD4+ and CD8+ T cell populations as
represented in histograms
or dot plots.
Figure 9A shows the concentration dependent increase in the mean fluorescence
intensity of the late
activation marker CD25 gated on CD8 T cells. The results indicates that anti-
ROR1/anti-CD3 TCB1+1
antibody induced a significant concentration dependent activation of CD8 T
cells in the presence of
ROR1-positive Rec-1 cells and the maximum signal was reached with 100 pM of
antibody. Unspecific
activation of CD8 T cells was minimal upon binding of CD3 on T cells but
without binding on ROR1-
positive target cells obtained by non-binder TCB constructs. Although the
activation of CD8 T cells was
not as pronounced with anti-ROR1/anti-CD3 TCB2+1 antibody, there was a faint
but noticeable increase
in CD25 mean fluorescence intensity. However, unspecific actvation could not
be ruled out.
Figure 9B shows the concentration dependent upregulation of CD25 on CD8 T
cells mediated by anti-
ROR1/anti-CD3 TCB1+1 and anti-ROR1/anti-CD3 TCB2+1 antibodies in the presence
of ROR1-positive
RPMI8226 MM cells. At the highest concentration (100 pM) of TCB antibodies
tested there was no
unspecific activation of CD8 T cells as shown in comparison to the non-binder
TCB constructs.
Example 8.1 ¨ Activation of T cells upon engagement of anti-ROR1/anti-CD3 T
cell bispecific
antibodies in the presence of ovarian cancer cells (Flow cytometry)
Anti-ROR1/anti-CD3 T cell bispecific antibodies generated in Example 5 were
also analyzed by flow
cytometry for their potential to induce T-cell activation by evaluating the
surface expression of the early
activation marker CD69 and/or the late activation marker CD25 on CD4+ and CD8+
T cells in the
presence of ROR1-positive human ovarian cancer cell lines PA-1 and/or SK-OV-3.
Briefly, human
ovarian cancer target cells were harvested with Trypsin/EDTA, washed, and
plated at density of 25,000
cells/well using flat-bottom 96-well plates. Cells were left to adhere
overnight. Peripheral blood
mononuclear cells (PBMCs) were prepared by Histopaque density centrifugation
of enriched lymphocyte
preparations (buffy coats) obtained from healthy human donors. Fresh blood was
diluted with sterile PBS
and layered over Histopaque gradient (Sigma, #H8889). After centrifugation
(450xg, 30 minutes, room
temperature), the plasma above the PBMC-containing interphase was discarded
and PBMCs transferred
in a new falcon tube subsequently filled with 50 ml of PBS. The mixture was
centrifuged (400xg, 10
minutes, room temperature), the supernatant discarded and the PBMC pellet
washed twice with sterile
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
74
PBS (centrifugation steps 350xg, 10 minutes). The resulting PBMC population
was counted
automatically (ViCell) and stored in respective culture medium according to
the cell line supplier (see
Example 2.1) at 37 C, 5% CO2 in a cell incubator until further use (no longer
than 24 h). To examine T-
cell activation induced by anti-ROR1/anti-CD3 T cell bispecific antibodies,
human ovarian cancer cells
were exposed to the bispecific antibody at the indicated concentrations (range
of 0.1 pM to 200 nM in
triplicates). PBMCs were then added to the human ovarian cancer target cells
at final effector to target
(E:T) ratio of 10:1. T-cell activation was assessed after 24 to 48 h of
incubation at 37 C, 5% CO2. After
the incubation period, cells were collected from the wells, pelleted down by
centrifugation (5 min, 350 x
g) and washed twice with 150 [El/well of FACS Stain Buffer (BD Biosciences).
Surface staining of the
effector cells with selected fluorochrome-conjugated antibodies against human
CD4 (mouse IgG1,K;
clone RPA-T4), CD8 (mouse IgG1,K; clone HIT8a; BD #555635), CD69 (mouse IgGl;
clone L78; BD
#340560) and CD25 (mouse IgG1,K; clone M-A251; BD #555434) was performed at 4
C for 30 min,
protected from light, in FACS Stain Buffer (BD Biosciences) according to the
manufacturer's protocol.
Cells were washed twice with 150 [El/well FACS Stain Buffer then fixed using
100 ul BD Fixation buffer
per well (#BD Biosciences, 554655) at 4 C for 20 min, resuspended in 120 [El
FACS buffer and analyzed
using BD FACS CantoII. The expression of CD69 or CD25 activation markers were
determined by
measuring the median fluorescence intensity gated on CD4 + and CD8 + T cell
populations as represented
in histograms or dot plots. As shown in Figure 9.1, ROR1 Mab2-TCBcv (squares)
induced a
concentration-dependent increase of CD69 early activation marker which was
observed on CD4+ T cells
(A) and CD8+ T cells (B) in presence of ROR1-low expressing SK-OV-3 target
cells while control-TCB
(triangles) did not induce any T-cell activation. At a clinically relevant
concentration of 1 nM of ROR1
Mab2-TCBcv, there was already up to 40% of activated CD4 T cells and 25% of
activated CD8 T cells
after 48h of incubation.
Example 9 ¨ Redirected T cell cytotoxicity of multiple myeloma cells upon
cross-linking of anti-
ROR1/anti-CD3 T cell bispecific antibodies to CD3 on T cells and ROR1 on
multiple myeloma cells
(LDH release assay)
Anti-ROR1/anti-CD3 T cell bispecific antibodies generated in Example 5 were
also analyzed for their
potential to induce T cell-mediated apoptosis in ROR1-expressing multiple
myeloma cells upon
crosslinking of the construct via binding of the antigen binding moieties to
ROR1 on cells. Briefly,
human ROR1-expressing RPMI-8226 multiple myeloma target cells (available from
American Type
Culture Collection; ATCC CCL-155) were harvested with Cell Dissociation
Buffer, washed and
resuspended in RPMI supplemented with 10% FCS. Approximately, 30,000 cells per
well were plated in
a round-bottom 96-well plate and the respective dilution of the construct was
added for a desired final
concentration (in triplicates); final concentrations ranging from 0.01 fM to
100 pM or 0.2 nM to 30 nM.
For an appropriate comparison, all T cell bispecific constructs and controls
were adjusted to the same
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
molarity. Human total T cells or CD8 T cell clones (effectors) were added into
the wells to obtain a final
E:T ratio of 3:1. When human PBMC were used as effector cells, a final E:T
ratio of 10:1 was used.
PHA-L (Sigma) was used as positive control for human T cell activation at a
concentration of 1 mg/ml.
Negative control groups were represented by effector or target cells only. For
normalization, maximal
5 lysis of the RPMI-8226 multiple myeloma target cells (= 100%) was determined
by incubation of the
target cells with a final concentration of 1% Triton X-100, inducing cell
death. Minimal lysis (= 0%) was
represented by target cells co-incubated with effector cells only, i.e.
without any T cell bispecific
antibody. After 6 to 24 hrs incubation (CD8 T cell clones as effectors) or 24
to 48 hrs incubation (PBMC
as effectors) at 37 C, 5% CO2, LDH release from the apoptotic/necrotic ROR1-
positive target cells into
10 the supernatant was then measured with the LDH detection kit (Roche Applied
Science), following the
manufacturer's instructions. The percentage of LDH release was plotted against
the concentrations of
anti-ROR1/anti-CD3 T cell bispecific antibodies in concentration-response
curves. The IC50 values were
measured using Prism software (GraphPad) and determined as the T cell
bispecific antibody
concentration that results in 50% of maximum LDH release.
15 Figure 10 shows the redirected T cell killing of ROR1-positive RPMI8226 MM
target cells by CD8 T
cells activated by anti-ROR1/anti-CD3 TCB antibodies. Specific cytotoxicity of
target cells (tumor lysis)
induced by anti-ROR1/anti-CD3 TCB antibodies was measured by LDH release. (A)
Experiment 1 (14h
time point): A very slight concentration dependent increase of tumor lysis
response was observed with
anti-ROR1/anti-CD3 TCB1+1 antibody. 30% of tumor lysis was already observed
with the lowest
20 concentration tested of 0.01 pM anti-ROR1/anti-CD3 TCB1+1 antibody and up
to 37.5% tumor lysis was
reached with 30 nM of anti-ROR1/anti-CD3 TCB antibodies in experimental
conditions reflecting
clinically relevant E:T ratio of 3:1 i.e. 3 CD8 T cells for 1 RPMI 8226 target
cell. EC50 could not be
calculated. The 37.5% tumor lysis observed at 30 nM as detected by LDH release
could not have been
attributed only to unspecific killing of target cells as there was only 17%
unspecific target cell lysis with
25 30 nM of non-binder TCB1+1 (i.e. binds to effector cells but not to target
cells). For anti-ROR1/anti-
CD3 TCB2+1 antibody, a maximum target cell lysis of 30% was already observed
at the lowest
concentration tested of 0.2 fM and there was no concentration dependent
response with increasing
concentrations for up to 10 nM. However, cell lysis with the non-binder TCB
2+1 in a concentration of
30 nM was already close to 30%. (B) Experiment 2 (20h time point): The study
was repeated in ROR1-
30 positive RPMI8226 and measurement of LDH release was assessed after 20h
incubation. 30-40% target
cell lysis was observed with anti-ROR1/anti-CD3 TCB1+1 and TCB2+1 antibodies
at a concentration of
100 pM while non-binder TCB controls at 100 pM did not induce any tumor lysis.
The results
corroborate with an increase in T cell activation as measured by upregulation
of CD25 marker on the
CD8 T cells (Figure 9B).
35 Figure 11 shows the redirected T cell killing of ROR1-positive RPMI8226 MM
target cells by T cells
from PBMCs activated by anti-ROR1/anti-CD3 TCB antibodies at different
effector cells to tumor cells
(E:T) ratios. Specific cytotoxicity of target cells (tumor lysis) induced by
anti-ROR1/anti-CD3 TCB
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
76
antibodies was measured by LDH release. (A) E:T ratio= 10 PBMCs : 1 RPMI8226
MM cell (24h time
point): there was a concentration dependent response with increasing
concentrations for up to 100 nM. A
maximum mean of 25% of tumor lysis was observed with 100 nM concentration anti-
ROR1/anti-CD3
TCB1+1 and anti-ROR1/anti-CD3 TCB2+1 antibodies in experimental conditions
with E:T ratio of 10:1
i.e. 10 PBMCs for 1 RPMI8226 target cell. The 25% tumor lysis observed at 100
nM as detected by
LDH release could not have been attributed only to unspecific killing of
target cells as there was only 9%
unspecific target cell lysis with 100 nM of non-binder TCB2+1 (i.e. binds to
effector cells but not to
target cells). (B) E:T ratio= 25 PBMCs : 1 RPMI8226 MM cell (24h time point):
there was a
concentration dependent tumor lysis with increasing concentrations of anti-
ROR1/anti-CD3 TCB1+1for
up to 2 nM. A maximum mean of 30% of tumor lysis was observed already with 2
nM concentration of
anti-ROR1/anti-CD3 TCB1+1, but tumor lysis seemed to reach a plateau since 100
nM concentration was
not associated with a higher response.
The overall in vitro results with ROR1-positive blood cancer cells (CLL, MM,
and MCL) clearly show
that anti-ROR1/anti-CD3 TCB1+1 and anti-ROR1/anti-CD3 TCB2+1 molecules act
like T cell bispecific
antibodies as they 1) bind to ROR1-positive target cells; 2) bind to CD3-
postive T cells; 3) mediate T cell
activation upon simultaneous binding to target cells and T cells; and 4)
induce redirected T cell
cytotoxicity of ROR1-positive target cells in a concentration-dependent manner
which corroborate with
the upregulation of CD25 on T cells.
Example 10 ¨ Cell lysis of human ovarian cancer cells (LDH release assay)
Anti-ROR1/anti-CD3 T cell bispecific antibodies generated in Example 5 were
analyzed for induction of
T cell-mediated cytotoxicity in human ovarian cancer cells. Human ovarian
cancer cell lines PA-1,
COLO-704, SK-OV-3 and OVCAR-5. Briefly, human ovarian cancer target cells were
harvested with
Trypsin/EDTA, washed, and plated at density of 25,000 cells/well using flat-
bottom 96-well plates. Cells
were left to adhere overnight. Peripheral blood mononuclear cells (PBMCs) were
prepared by Histopaque
density centrifugation of enriched lymphocyte preparations (buffy coats)
obtained from healthy human
donors. Fresh blood was diluted with sterile PBS and layered over Histopaque
gradient (Sigma,
#H8889). After centrifugation (450xg, 30 minutes, room temperature), the
plasma above the PBMC-
containing interphase was discarded and PBMCs transferred in a new falcon tube
subsequently filled with
50 ml of PBS. The mixture was centrifuged (400xg, 10 minutes, room
temperature), the supernatant
discarded and the PBMC pellet washed twice with sterile PBS (centrifugation
steps 350xg, 10 minutes).
The resulting PBMC population was counted automatically (ViCell) and stored in
respective culture
medium as suggested by the cell line supplier (see Example 2.1) at 37 C, 5%
CO2 in a cell incubator
until further use (no longer than 24 h). For the killing assay, the TCB
antibody was added at the indicated
concentrations (range of 0.1 pM to 200 nM in triplicates). PBMCs were added to
the human ovarian
cancer target cells at final effector to target (E:T) ratio of 10:1. Target
cell killing was assessed after 24 h
to 48 h of incubation at 37 C, 5% CO2by quantification of LDH released into
cell supernatants by
CA 02963692 2017-04-05
WO 2016/055592 PCT/EP2015/073308
77
apoptotic/necrotic cells (LDH detection kit, Roche Applied Science, #11 644
793 001) following the
manufacturer's instructions. Maximal lysis of the target cells (=100%) was
achieved by incubation of
target cells with 1% Triton X-100. Minimal lysis (=0%) refers to target cells
co-incubated with effector
cells without bispecific construct. The percentage of LDH release was plotted
against the concentrations
of anti-ROR1/anti-CD3 T cell bispecific antibodies in concentration-response
curves. The EC50 values
were measured using Prism software (GraphPad) and determined as the T cell
bispecific antibody
concentration that results in 50% of maximum LDH release. As shown in Figure
12, ROR1 Mab2-TCBcv
(squares) induced a concentration-dependent increase in tumor cell lysis of
ROR1 high-expressing PA-1
ovarian cancer cells (A), ROR1 medium-expressing COLO-704 (B) and OVCAR-5 (C)
ovarian cancer
cells and ROR1 low-expressing SK-OV-3 ovarian cancer cells (D). In contrast,
control-TCB (A, B, C;
circles) which only binds to CD3 did not induce tumor cell lysis at clinically
relevant concentrations (i.e.
up to 10 nM). Representative experiments shown.
Table 9: EC50 values for cell lysis of ovarian cancer cell lines by anti-
ROR1/anti-CD3 T cell bispecific
antibodies
ROR1 Mab2-TCBcv
Ovarian cancer cell lines
Mean EC50 (pM) Mean EC50 (ng/mL)
PA-1 (n=2) 14.6 (4.7 ¨24.5) 2.9 (0.9
¨4.9)
COLO-704 (n=1) 73.3 14.3
OVCAR-5 (n=1) 53.4 10.7
SKOV-3 (n=5) 707 (456 ¨ 1003) 141.4 (91.2
¨ 200.6)