Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 02975157 2017-07-26
WO 2016/120850 PCT/1B2016/050477
1
Sutfonarnide-Substituted lndole Modulators of RORG2 and Methods of Use Thereof
BACKGROUND OF THE INVENTION
Retinoid-related orphan receptors (ROR) are reported to have an important role
in
numerous biological processes. Scientific investigations relating to each of
retinoid-related orphan
receptors RORa, RORP, and RORy have been described in the literature.
Continuing research in
this field is spurred by the promise of developing new therapeutic agents to
treat medical disorders
associated with retinoid-related orphan receptor activity.
RORy has been reported to be expressed in high concentration in various
tissues, such as
thymus, kidney, iiver, muscle, and certain fat tissue. Two isoforms of RORy
have been identified
and are referred to as yl and y2 (also referred to as RORyt). Expression of
the y2 isoform has
been reported to appear in, for example, double-positive thymocytes. Compounds
capable of
modulating RORyt activity are contemplated to provide a therapeutic benefit in
the treatment of
multiple medical disorders, including immune and inflammatory disorders.
Numerous immune and inflammatory disorders continue to afflict millions of
patients
worldwide. Significant advances have been made in treating these disorders.
However, current
therapies do not provide satisfactory results for all patients due to, for
example, detrimental side
effects or insufficient efficacy. Treatments for immune and inflammatory
disorders vary depending
on the particular medical disorder, and often involve use of immunosuppreSeive
drugs. Surgery
(e.g., splenectomy), plasmapheresis, or radiation can be used in certain
instances,
One exemplary immune disorder in need of better therapy is psoriasis.
Psoriasis is a T cell-
Mediated inflammatory disease that affects approximately 2% to 3% of adults
and has a
substantial adverse impact on the quality of life for patients suffering from
this disorder. Plaques
resulting from psoriasis can be painful and are visually unappealing. Various
therapeutics have
been developed in an attempt to treat psoriasis. However, the traditional
therapies for psoriasis
often have toxic adverse effects. Accordingly, a need exists for improved
treatments for psoriasis
as well as other immune and inflammatory disorders,
SUMMARY
The present invention provides compounds, pharmaceutical compositions, methods
of
inhibiting RORy activity and/or reducing the amount of 1L-17 in a subject, and
methods of treating
various medical disorders using such compounds. In particular, one aspect of
the invention relates
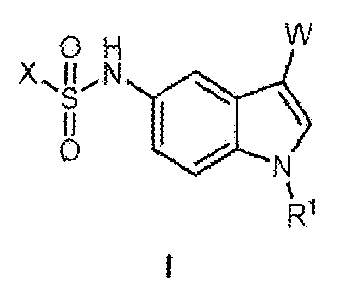
to compounds represented by Formula I:
84020419
2
X...õ11õ
0 H
N
Ccl
R
and pharmaceutically acceptable Salts, pharmaceutically active metabolites,
pharmaceutically
acceptable prodrugs, and pharmaceutically acceptable solvate s thereof;
wherein 1:11, X and W are
as defined herein.
Another aspect of the InVentiOn provides a Method of treating a subject
suffering from a
medical disorder. The method of:noises administering to the subject a
therapeutioally effective
amount Of a. compound of ForMUla l Or a pharmaCeutically acceptable salter
solvate thereof, as
described in the Detailed Description. A large numberof disorders May be
treated using the
compounds described herein. For example, the compounds described herein may be
used to treat
an immune disorder or inflarnrriatory disorder, such as rheumatoid arthritis,
psoriasis, chronit:graft-:
versus-host disease; acute graft-versus-host disease., Crohn's.disease,
inflammatory bowel.
disease, multiple sclerosis, systemic lupus: erythernatosus, Celiac Sprue,
idiopathic
throm bccytopenic thrombotic purpura, myasthenia gravis, Sjogreres syndrome,
scieroderrna,
16 Ulaerative cOlitis, asthina, epidermal hyperplasia, and other medical
disorders described herein.
DETAILED DESCRIPTION
The invention provides compoundS, pharmaceutical cOmpositions, rnetheds of
Mbdulating
RORy activity and/or reducing the amount of IL-17 in a subject, and
therapeutic uses Of said
compounds and pharmaceutical cornpositions. The practice ofthe present
invention employs,
unless otherwise indicated, conventional techniques of organic chemistry,
pharmacology,
molecular biology (including recombinant techniques), cell biology,
biodhemittry, and immunology.
Such techniques are:explained in the literature, such as in "Comprehensive
Organic Synthesis"
(B:M. Trost & I. Fleming, eds., 1'991-1992); "Handbook of experimental
immunology" (D.M. Weir &
0:C. Blackwell, eds..); "Current protocols in moledular biology" (F.M. AusUbel
etal., eds., 1987, and
periodic updates); and "Current protocols in immunology" (4.E. Coligan et el.,
eds., 1091').
Various aspects of the invention are set forth below in sections; however,
aspects of the
invention described in one particular section are not to be limited to any
WO:War section. Further,
when a variable is not accompanied by a:definition, the previoUs definition of
the variable controls.
It is to be understood that the foregoing general description and the
following detailed
description are exemplary and explanatory only and are not restrictive of any
subject matter
CA 2975157 2018-08-17
84020419
3
claimed. In this application, the use of the singular includes the plural
unless specifically stated:
otherwise. It must be noted that, as used in the specification and the
appended claims, the singular
forms "a," "an" and "the" include plural referents unless the context clearly
dictates otherwise. In
this application, the use of "or" means "and/cir" unless stated ctherWise.
Furthermore, use of the
6 term Including" as well as other forms, such as "include", "includes,"
and "InCluded," is not limiting.
It is to be understood that the methods and compositions described herein are
not limited to
the particular methodology, protocols, cell lines, censtructs, and reagents
described herein and as
such may vary. It is also to be understood that the terminology used herein is
for the purpose of
describing particular embodiments only, and is not intended to limit the scope
of the methods and
compositions described herein, which will be limited only by the appended
claims.
All publications and patents mentioned herein are referenced in their
entirety for the purpose of describing and disclosing, for example, the
constructs and
methodologies that are described in the publications, which might be used in
connection with the
methods, compositions and compounds described herein.
Chemical names, common names, and chemical structures may be used
interchangeably
to describe the same Strodure. lie chemical compound Is referred to using both
a chemical
structure and a chemical name, and an ambiguity exists between the structure
and the name, the
structure predominates.
Definitions
"ROR" Stands for Retinoic add receptor-related Orphan Receptor. There are
three forms
ROR, ROR-, and y. and each is encoded by a separate gene (RORA, ROR. arid RORC
respectively). There are: two subtypes of RORC 1 and 2. Subtype 2 is also
called "t". The
human RORC gene is also called TOR: RORG: RZRG; NRIF3; and RZR-GAMMA, The
human
protein RORC is also called nuclear, receptor ROR-gamma; nuclear receptor RZR-
gamma; retinOtc
=acid-binding receptor gamma; retinold-related orphan receptor gamMa; RAR-
related orphan
receptor C, isoform a; RAR-related orphan nuclear receptor variant 2; nuclear
receptor subfamily 1
group F member 3. As used herein, "ROR' and "RORC2" are used interchangeably
to .refer to a
protein from a RORC subtype 2 gerie.
As used herein, the term "modulator' refers to a compound that alters an
activity of a
molecule, For example', :a modulator can cause an increase or decrease in the
Magnitude Of a
certain activity of a molecule compared to the magnitude of the activity in
the absence of the
modulator. In certain embodiments, a modulator is an Inhibitor, which:
decreases the magnitude of
one or more activities of 'a molecule. In certain embodiments, an inhibitor
completely prevents one
or more activities of a Molecule. In certain embodiments, a modulator is an
activator, which
CA 2975157 2018-08-17
CA 02975157 2017-07-26
WO 2016/120850 PCT/IB2016/050477
4
increases the magnitude of at least one activity of a melecule. In certain
embodiments the
presence of a modulator results in an activity that does not occur in the
absence of the rnOdulator.
The term "alkyl" refers to a substituent obtained by removing a hydrogen from
a saturated,
straight (i.e. unbranched) or branched carbon chain (or carbon), or
combination thereof, which has
the number of carbon atoms designated (i.e: Cl-05 means one to six carbons).
Examples of alkyl
substituents include methyl, ethyl, prOpyl (including n-propyl and isopropyl),
butyl (including n-butyl,
isobutyl, sec-butyl and tert-butyl), pentyl, isoamyl, hexyl and the like.
The term "haloalkyl" is an alkyl in which at least one hydrogen on the alkyl
is replaced with
a halogen atom. In certain embodiments in which two or more hydrogen atoms are
replaced with
.. halogen atoms, the halogen atoms are all the same as one another. In other
embodiments in
which two or more hydrogen atoms are replaced with halogen atoms, the halogen
atoms are not all
the same as one another.
The term "cycloalkyl" refert to a substituent obtained by removing a hydrogen
atom from a
saturated carbocycle having the number of carbon atoms designated (i.e. C3-C8
means three to
eight carbons). Cycloalkyl refers to both a radical of a single ring saturated
carbocycle, such as
cyclopropyi, cyclobutyl, cyclopentyl and cyclohexyl, as well as a radical of a
two or three ring
bridged, fused or spire saturated carbocycle, such as bicyclo[4.2.0]octane and
decallnyl.
The term "five-membered heteroaryl" whether used alone or as part of another
group, is
defined herein as a ring system having five ring atoms wherein at least one
ring atom, alternatively
.. 2 ring atoms, alternatively 3 ring atoms, alternatively 4 ring atoms, is a
heteroatom independently
selected in each instance from, unless otherwise indicated, the group
consisting of nitrogen (N),
Oxygen (0), and sulfur (5), arid wherein the ring is partially unsaturated. In
a group that has a
heteroaryl substituent, unless otherwise indicated, the ring atom of the
heteroaryl substituent that
is bound to the group may be the at least one heteroatom, or it may be a ring
carbon atom, where
the ring carbon atom may be in the same ring as the at least one heteroatom or
where the ring
carbon may be in a different ring from the at least one heteroatom. Where so
indicated, heteroaryl
groups can be substituted. If the heteroaryl substituent is substituted with a
group or substituent,
the group or substituent may be bound to the heteroatom, or it may be bound to
a ring carbon
atom, where the ring carbon atom may be in the same ring as the heteroatom(s),
or where the ring
carbon atom may be in a different ring from the heteroatom). Examples of
monocyclic heteroaryl
rings include, but are not limited to, 1,2,3,4-tetrazolyl, [1,2,3]triazolyi,
[1,2:4]triazolyi, triazinyl,
thiazol-2-yl, thiazol-4-yl, imidazo171-yl, oxazotyl,
isoxazolin-5-yl,
furan-2-yl, furan-3-yl, thiophen-2-y1,, and thiophen-4-1I.
This specification uses the terms "substituent:" "radical," and "group"
interchangeably.
CA 02975157 2017-07-26
WO 2016/120850 PCT/IB2016/050477
If a group of substituents are collectively described as being optionally
substituted by one
or more of a list of substituents, the group may include: (1) unsubstitutable
substituents, (2)
substitutable substituents that are not substituted by the optional
substituents, and/or (3)
substitutable substituents that are substituted by one or more of the optional
substituents.
5 If a substituent is described such that it "may be substituted" or as
being "optionally
substituted" with up to a particular number of non-hydrogen substituents, that
substituent may be
either (1) not substituted; or (2) substituted by up to that particular number
of non-hydrogen
substituents or by up to the maximum number of substitutable positions on the
substituent
whichever is iess. Thus, for example, if a substituent is described as a
heteroaryl optionally
substituted with up to 3 non-hydrogen substituents, then any heteroaryl with
less than 3
substitutable positions would be optionally substituted by up to only as many
non-hydrogen
substituents as the heteroaryl has substitutable positions. To illustrate,
tetrazolyl (which has only
one substitutable position) would be optionally substituted with up to one non-
hydrogen
substituent. To illustrate further, if an amino nitrogen is described as being
optionally substituted
.. with up to 2 non-hydrogen substituents, then the nitrogen will be
optionally substituted with up to 2
non-hydrogen substituents if the amino nitrogen is a primary nitrogen, whereas
the amino nitrogen
will be optionally substituted with up to only 1 non-hydrogen substituent if
the amino nitrogen is a
secondary nitrogen.
As used herein compounds of Formula I may be referred to as a "compound(s) of
the
invention," Such terms are also defined to include all forms of the Formula I
including hydrates,
solvates, isomers, crystalline and non-crystalline forms, isomorphs,
polymorphs, and metabolites
thereof. For example, the compounds of Formula land pharmaceutically
acceptable salts thereof,
may exist in unsolvated and solvated forms. When the solvent or water is
tightly bound, the
complex will have a well-defined stoichiornetry independent of humidity. When,
however, the
solvent or water is weakly bound, as in channel solvates and hygroscopic
compounds, the
water/solvent content will be dependent on humidity and drying conditions, In
such cases, non-
stoichiometry wilt be the norm.
A "metabolite" of a compound disclosed herein is a derivative of that compound
that is
formed when the compound is metabolized. The term "active metabolite" refers
to a biologically
active derivative of a compound that is formed when the compound is
metabolized. The term
"metabolized," as used herein, refers to the sum of the processes (including,
but not limited to,
hydrolysis reactions and reactions catalyzed by enzymes, such as, oxidation
reactions) by which a
particular substance is changed by an organism. Thus, enzymes may produce
specific structural
alterations to a compound. For example, cytochrome P450 catalyzes a variety of
oxidative and
reductive reactions while uridine diphosphate glucurpnyl transferases catalyze
the transfer of an
activated glucuronic-acid molecule to aromatic alcohols, aliphatic alcohols,
carboxylic acids,
CA 02975157 2017-07-26
WO 2016/120850
PCT/I132016/050477
6
amines and free sulfhydryl groups. Further information on metabolism may be
obtained from The
PharmacologiCal Basis of Therapeutics, 9th Edition, McGraw-Hill (1996);
Metabolites of the
compounds disclosed herein can be identified either by administration of
compounds to a host and
analysis of tissue samples from the host, or by incubation of compounds with
hepatic cells in vitro
and analysis of the resulting compounds. Both methods are well known in the
art. In some
embodiments, metabolites of a compound are formed by oxidative processes and
correspond to
the corresponding hydroxy-containing compound. In some embodiments, a compound
is
metabolized to pharmacologically active metabolites.
In some embodiments, compounds described herein could be prepared as prodrugs.
A
"prodrug" refers to an agent that is converted into the parent drug in vivo,
Prodrugs are often useful
because, in some situations, they may be easier to administer than the parent
drug. They may, for
instance, be bioavailable by oral administration whereas the parent is not.
The prodrug may also
have improved solubility in pharmaceutical compositions over the parent drug.
An example, without
limitation, of a prodrug would be a compound described hereinewhich is
administered as an ester
(the "prodrug") to facilitate transmittal across a cell membrane where water
solubility is detrimental
to mobility but which then is metabolically hydrolyzed to the carboxylic acid,
the active entity, once
inside the cell where water-solubility is beneficial. A further example of a
prodrug might be a short
peptide (polyaminoacid) bonded to an acid group where the peptide is
metabolized to reveal the
active moiety. In certain embodiments, upon in vivo administration, a prodrug
is chemically
converted to the biologically, pharmaceutically or therapeutically active form
of the compound. In
certain embodiments, a prodrug is enzymatically metabolized by one or more
steps or processes
to the biologically, pharmaceutically or therapeutically active form of the
compound. To produce a
prodrug, a pharmaceutically active compound is modified such that the active
compound will be
regenerated upon in vivo administration. The prodrug can be designed to alter
the metabolic
stability or the transport characteristics Of a drug, to mask side effects or
toxicity, to improve the
flavor of a drug or to alter other characteristics or properties of a drug. By
virtue of knowledge of
pharmacodynamio processes and drug metabolism in vivo, those of skill in this
art, once a
pharmaceutically active compound is known, can design prodrugs of the
compound. (see, for
example, Nogrady (1985) Medicinal Chemistry A Biochemical Approach, Oxford
University Press,
New York, pages 388-392; Silverman (1,992), The Organic Chemistry of Drug
Design and Drug
Action, Academic Press, Inc., San Diego, pages 352-401, Saulnier et al.,
(1994), Bloorganic and
Medicinal Chemistry Letters, Vol: 4, p. 1985).
Prodrug forms of the herein described compounds, wherein the prodrug is
Metabolized in
vivo to produce a derivative as set forth herein are included MOO the scope of
the claims. In some
cases, some of the herein-described compounds may be a prodrug for another
derivative or active
compound.
84020419
7
Prodrugs- are often useful because, in some situations, they maybe easier to
administer
than the parent drug. They may, far hi stance, be bidevailable by oral
administration.vvhereas the
parent is not, The prodrug may also have improved solubility in pharmaceutical
'compositions over
the parent drug. Prodrugs may be designed as reversible drug derivatives, for
use as modifiers to
enhance drug transport to site-specific tissues. In some embodiments, the
design Of a prodrug
increases the effective water solubility. See, e.g., Fedorak et al., Am. J.
Physibl,õ 269:G210,,21
(1904 McLoed etal, Gastroenterol,. 106:405-413 (1.904); Hachhads et al.,.
Blamed. Ctrom.,
6.283-26.(1:9,92);j. Larson .and H. Bundgaard, Int. J. Pharmaceutics, 37,.87
(1.9.$7);: J. Larsen et
at., Int. J. Pharmaceutics, 47, 103 (1984 SinkUta at at,, J, Pharm. SaL,
6.4.1.81-210 (1975); T.
Higuchi and V. Stella, Pro-drugeas. Novel Delivery systems, Vol. 1,4 of
the.A.C.S.,SymposiOM.
Series; and Edward. B. Roche, BibreverSible Carriers in Drug Design, American
Pharmaceutical
Association and Pergamon Press, 1987.
The compounds of the. invention may have asymmetric carbonetoms. The carbon-
carbon
bonds of the .compounds of the invention may be depicted hereindeing a solid
line, a solid wedge
or a dotted wedge. The use of a solid lino to depict bonds. to asymmetric
carbon atoms is.mearitto
indicate that all possible otereoiserriers (e.g specific ertantiomers, recemit
mixtures, etc.) at that.
carbon atom are. included, The use or either a solid or dotted wedge to depict
bonds to asymmetric
carbon .atoms is meant to indicate that only the stereolsorner shown is meant
to be included. It is
possible that compounds of the invention May contain more than one. asymmetric
carbon atom. In
those compounds, the. use of a solid line to depict bonds to asymmetric
carbonatorris. is Meant to'
indicate that all. possible .stereoisoMers.are Meant to be includ.e.d,
Forekernple, unless stated
otherwise, it is intended that the -compounds of the invention can exist as
anantiothers and
diattereomers or as racernates-and mixtures thereof. The use of a solid line
to depict bonds to one
or more asymmetric carbon atoms in-:a compound of the. invention and the use
of a solid Or dotted
wedge to depict bonds to other asymmetric carbon atoms in the same.compound is
meant to
.indicate thate mixture of diaStereoiners ia preent.
Stereolsomers of compounds of the invention include cisand trans isomere,
Optical isomers
such as.R. and Senantiemersõ diestereomers, geometric isomers, rotational
isomers,.
conformational loomettõ and lautomers of the compounds of the invention,,
including compounds
exhibiting more than one type of isomerism; and mixtures. thereof (such as
recarnatesand
.diastereometio.pairs). Also included are acid addition or base addition salts
Wherein-the counterion
is optically active, for example, D-lactate. or LAysine, or. racemic, for
example, .DL-tartrate or IX-
arginine.
When any racemate crystallizes,. crystals of two different types are
possible'. The firsttype
is the racernic compound (true raoemete) referred to: above wherein one
homogeneous form Of
.cryatal IS produced containing- both enantiOrners in equimolar -amounts..
Thesecond type is the.
CA 2975157 2018-08-17
CA 02975157 2017-07-26
WO 2016/120850 PCT/IB2016/050477
8
racernic mixture or conglomerate wherein two forms of crystal are produced in
equirrioiar amounts
each comprising a single enentiomee
The present invention also incled.e8 isotopically-labeled compounds, which are
identical to
those recited in Formula I herein, but for the fact that one or more atoms are
replaced by an atom
having an atomic mass or mass number different from the atomic mass or mass
number usually
found in nature. Examples of isotopes that may be incorporated into compounds
of the invention
include isotopes of hydrogen, carbon, nitrogen, oxygen, phosphorus, fluorine
and chlorine, such
as, but not limited to, 2Hõ 311, 'it, 14C, t5N, 130, 170, 12 1 ig
P, ¨P, "5S, --F, and 36CI. Certain isotopically-
labeled compounds of Formula (1) and Formula (II), for example those into
which radioactive
isotopes such as 3H and 140 are incorporated, are useful in drug and/or
substrate tissue
distribution assays. Tritiated, i.e., 3H, and carbon-14, i.e., "C, isotopes
are particularly preferred for
their ease of preparation and detectability. Further, substitution with
heavier isotopes such as
deuterium, La,, 2H, can afford certain therapeutic advantages resulting from
greater metabolic
stability, for example increased in vivo half-life or reduced dosage
requirements and., hence, may
be preferred in some circumstances. Isotopically-labeled compounds the
invention may generally
be prepared by carrying out the procedures disclosed in the Schemes and/or in
the Examples and
Preparations below, by substituting an isotopically-labeled reagent for a non-
isotopically-labeled
reagent.
The compounds of this invention may be used in the form of salts derived from
inorganic or
organic acids. Depending on the particular 'compound, a salt of the compound
may be
advantageous due to one or more of the salt's physical properties, such as
enhanced
pharmaceutical stability in differing temperatures and humidities, or a
desirable solubility in water
or oil. In some instances, a salt of a compound also may be used as an aid in
the isolation,
purification, and/or resolution of the compound.
Where a sail is intended to be administered to a patient (as opposed to, for
example, being
used in an in vitro context), the salt preferably is pharmaceutically
acceptable. The term
"pharmaceutically acceptable salt" refers to a salt prepared by combining a
compound of Formula t
with an acid whose anion, or .a base whose cation, is generally considered
suitable for human
consumption. Pharmaceutically acceptable salts are particularly useful as
products of the methods
of the present invention because of their greater aqueous solubility relative
to the parent
compound. For use in medicine, the salts of the compounds of this invention
are non-toxic
"pharmaceutically acceptable salts," Salts encompassed within the term
"pharmaceutically
acceptable salts" refer to non-toxic salts of the compounds of this invention
which are generally
prepared by reacting the free base with a.suitable organic or inorganic acid.
Suitable pharmaceutically acceptable acid addition salts of the compounds of
the present
invention when possible include those derived from inorganic acids, such as
hydrothicric,
CA 02975157 2017-07-26
WO 2016/120850 PCT/IB2016/050477
9
hydrobrOmic, hydrofluoric, boric, fluoroboric, phosphoric, metaphosphoric,
nitric, carbonie sulfonic,i
and sulfuric acids, and organic acids Such as acetic, benzenesulfonic,
ben.zolo, citric,
ethanesulfonic, fumaric, .glueonic, glycolic, isothionit, lactic, lactobionie,
maleIc, malic,
methanetulfonic, trtfluororniethanesutfonic, succinic, toluenesulfonia,
tartaric, and trifluoroacetic
acids, Suitable organic acids generally include but are not limited to
aliphatic, cycloaliphatic,
aromatic; araliphatic, heterocyclic, carboxylic, and sulfonie classes of
oraanic acids.
Specific examples of suitable organic acids include but are not limited to
acetate,
trifluoroacetate, formate, propionate, seccinate, glytolate, gluconate,
digluconate, lactate, malate,
tartaric acid, citrate, ascorbate, glucuronate, maleate, furearate, p:yruvate,
aspartate., glutamate,
benzoate, anthranilic acid, stearate, salicylate, p-hydroxybenzoate,
phenylacetate, mandelate,
embonate (pamoate), methanesulfonate, ethanesulfonate, benzenesulforiate,
paritathenate,
toluenesuifonate, 2-hydroxyethanesulfonate, sufanitate,
cyclehexylaminosulfonate, algenic acid, p-
hydroxybutyric acid, galactarate, galacturonate, adipate, alginate, butyrate,
camphorate,
camphorsulfonate, cyclopentanepropionate, dodecylsulfate, giycoheptanoate,
glycerophosphate,
heptanoate, hexanoate, nicotinate, 2-naphthalesulfonate, oxalate, palmoate.
pectinate, 3-
phenylpropionate, picrate, pivalate, thiecyanate, and und-ecanoate..
Furthermore, where the compounds of the invention carry an aciditimoiety,
suitable
pharmaceutically acceptable salts thereof may include alkali metal salts,
i.e., sodium or potassium
salts; alkaline earth metal salts, eog.., calcium or magnesium salts; and
salts formed with suitable
organic iigands, e.g., quaternary ammonium salt's. In another embodiment, base
salts are formed
from bases which form non-toxic salts, including aluminum, arginine,
denzathineõ choline,
diethyl-amine, diolamine, glycine, lysine, meglumine, lamina, trometharnine
and zinc salts,
Organic salts may be made from secondary, tertiary or quaternary amine salts,
such as
trorrietha.mine, diethylamine, N,l\l'-benzylethylenediamine, chloroprocaine,
choline, diethanolamine,
ethyienediamine, meglumine (Nellethylgteicarnine), and procaine. Basic
nitrogen-containing groups
may be quatemized with agents such as lower alkyl (CerCi6) halides (e.g.,
methyl, ethyl, propyl, and.
butyl chlorides, bromides, and iodides), diaikyi sulfates (i.e., dimethyl,
diethyl, dibutyl, and diamyl
sulfates), long chain halides (i.e., decyi, lauryl, myristyl, and stearyl
Chlorides, bromides, and
Iodides), arylalkyl halides (i.e., ben:zyl and pheeethyl bromides), and
others.
In one embodiment, hernisalts of acids and bases may also be formed, for
example,
hemis.ulphate and hemicaicium salts.
Compounds
In the following description of compounds suitable for use in the methods
described herein,
definitions of referred-to standard chemistry terms may be found in reference
works (if not
otherwise defined herein), including Carey and Sundberg "Advanced Organic
Chemistry 4th Ed,"
CA 02975157 2017-07-26
WO 2016/120850
PCT/I132016/050477
Vols. A (2000) end B (2001), Plenum Press, New York. Unless otherWise
indicated, conventional
methods of rrieSs spectroscopy, NMR, HPLC, protein chemistry, biochemistry,
recombinant DNA
techniques and pharmacology, within the ordinary skill of the art are
employed. Unless specific
definitions are provided, the nomenclature employed in connection with, and
the laboratory
5 procedures and techniques of, analytical chemistry, synthetic organic
chemistry, and medicinal and
pharmaceutical chemistry described herein are those known in the art.
Standard. techniques can
be used for chemical syntheses, chemical analyses, pharmaceutical preparation,
formulation, and
delivery, and treatment of patients.
In certain embodiments, the compounds of the invention described herein are
selective for
10 RORy over RORa and/or RoRp.
Generally, an inhibitor compound of RORy used in the methods described herein
is
identified or characterized in an in vitro assay, e.g., an acellular
biochemical assay or a cellular
functional assay. Such assays are useful to determine an in vitro ICe) for
said compounds. In
some embodiments, the RORy inhibitor compound used for the methods described
herein inhibits
RORy activity with an in vitro lCe-, of less than 25 pM (e.g., less than 20
pM, less than 10 pM, less
than 1 pM, less than 0.5 pM, less than 0.4 pM, less than 0.3 pM, less than
0,1, less than 0.08 pM,
less than 0.06 pM, less than 0.05 pM, less than 0,04 pM, less than 0.03 pM,
less than less than
0.02 pM, less than 0,01, less than 0.008 pM, less than 0.006 pM, less than
0.005 pM, less than
0,004 pM, less than 0.003 pfkil, less than less than 0.002 pM, less than
0,001, less than 0.00099
2.0 pM, less than 0.00093 pM, less than 0.00097 pM, less than 0.00096 pM,
less than 0,00095 pM,
less than 0.00094 pM, less than 0,00093 pM, less than 0.00092, or less than
0.00090 pM), In
some embodiments, the RORy inhibitor compound is a compound described in the
Exemplification.
Described herein are compounds of Formula I. Also described herein are
pharmaceutically
acceptable salts, pharmaceutically acceptable solvates, pharmaceutically
active metabolites, and
pharmaceutically acceptable prodrugs of such compounds. Pharmaceutical
compositions that
include at least one such compound or a pharmaceutically acceptable salt,
pharmaceutically
acceptable solvate, pharmaceutically active metabolite or pharmaceutically
acceptable prodrug of
such compound, are provided. In some embodiments, when compounds disclosed
herein contain
an oxidizable nitrogen atom, the nitrogen atom can be coriverted to an N-oxide
by methods well
known in the art. In certain embodiments, isomers and chemically protected
forms of compounds
having a structure represented by Formula I are also provided.
One aspect of the invention relates to a compound of Formula II
CA 02975157 2017-07-26
WO 2016/120850 PCT/I132016/050477
11
H
0 N
R'
or a pharmaceutically acceptable salt, pharmaceutically active metabolite,
pharmaceutically
.acceptable prodrug, or pharmaceutically acceptable solvate thereof, wherein,
X is phenyl or 5-membered hereroaryl, in eaci-Lease optionally substituted
with one, two,
three, four or five substituents independently selected from the group
consisting of -CF3., -
r-Oss
CH2CH3, -OH, -OCH3, -0C.H2CH3, -OCH2CH2OH, -OCH2CH2OCH3:, , -F, -CI, -Br
and -ON;
R1 is -CI-13 or -CH2CI-13;
CN-4
W is R2 ,
optionally substituted with one, two, three, four or five -CH3; and
R2 is_(Ci-Ce)alkyl, (03-Cin)cycioalkyl, phenyl or isothia.zolyi , optionally
substituted with one,
two, three, four or five substitatents independently selected for each
occurrence from the group
consisting of -F, -CI, -Br, -OH, (C1i-C.3)alkyl,. (C1-C,a)baloalkyl and
(03+Cs)cycloalicyl..
In certain embodiments, the present invention relates to any of the
aforementioned
compounds, wherein R1 is -CH3.
In certain embodiments, the present invention relates to any of the
aforementioned
compounds, wherein R.' is -CH2CHI.
In certain' embodiments, the present invention relates to any of the
aforementioned
compounds, wherein W is R2 optionally substituted with one, two,
three, four or
five -CH3.
In certain embodiments, the present invention relates to any of the
aforementioned
0
1--CN
compounds, wherein W is R2 optionally substituted with one., two,
three, four or
five -CH3; and RS' is -CH3.
CA 02975157 2017-07-26
WO 2016/120850
PCT/I132016/050477
12
In certain .embodiments, the present invention relates to any of the
aforementioned
_________________________ = N ,
compounds, wherein W is R-
in certain embodiments, the present invention relates to any of the
aforementioned'
0
.compounds, wherein W is R2; and R is -C1-13.
In certain embodiments, the present invention relates to any of the
aforetmentioned
0
N
compounds, wherein W is R2 substituted with one -CH3..
In certain embodiments, the present invention relates to any of the
aforementioned
0
compounds, wherein W is = izz2
In certain embodiments, the present invention relates to any of the
aforementioned
0
dorepound.s, wherein W is R2.; and R1 is -CHe
In certain embodiments, the present invention relates to any of the
aforementioned
0
compounds, wherein Wis. R2,
In certain embodiments, the present invention relates to any of the
aforementioned
Fb 0
compounds, wherein W is R2; and R1 is -CF13.
In certain embodiments, the present invention relates to any of the
aforementioned
iie, /\N
comp.oulds,. wherein W is R2,
CA 02975157 2017-07-26
wo 2016/120850
PCT/I132016/050477
13
In certain embodiments, the present invention relates to any of the
aforementioned
1%.
0
CN
compounds, wherein W is R2 ; and R1 is -CH3.
In certain embodiments, the present invention relates to any of the
aforementioned
0
N
compounds, wherein W is rR.2,
in certain embodiments, the present invention relates to any of the
aforementioned
0
N
compounds, wherein W is R2 ; and R1 is -CH3.
In certain embodiments, the present invention relates to any of the
aforementioned
.0
N-4
onlpoundS, wherein W is. R2
In certain embodiments, the present invention relates to any of the
aforementioned
0
N.
compounds, wherein W is R2; and R' is -CH3.
In certain embodiments, the present invention relates to any of the
aforementioned
compounds, wherein W is R2substituted with two -CH:3.
In certain embodiments, the present inVention relates to any of the
aforementioned
0
.N
cornpounds, wherein W is R2,
In certain embodiments, the present invention relates to any of the
aforementioned
N-4
compounds, wherein W is R2; and R1 is -CH3.
CA 02975157 2017-07-26
WO 2016/120850
PCT/I132016/050477
14
In certaie embodiments, the present invention relates to any of the
aforementioned
Compounds, wherein W is R`
In certain embodiments, the present invention relates to any of the
aforementioned
0
compounds, wherein VV is \ _____ R2; and R is -CH3.
In certain embodiments, the present invention relates to any of the
aforementioned
compounds, wherein X is unsubstituted phenyl.
In certain embodiments, the present invention relates to any of the
aforementioned
compounds, wherein X is phenyl substituted with one, two, three, four or five
substituents
independently selected from the group consisting of -CH3, -CFe -CH2CH3, -OH, -
OCH3,
r \-0
0
_0cH2cH3, -OCH2CH2OH, -ocH2cH2ocH,,: -F, -Cl, -Br and -CN.
In certain embodiments, the present invention relates to any of the
aforementioned
compounds, wherein X is phenyl substituted with one substituent selected from
the group
consisting of with -CH3, -CF3, -CH2CH3, -OH, -OCH3, -OCH2CH3, -OCH2CH2OH,
r-oss
-0cH2cH200H3, -F, -CI, -Brand -CN.
In certain embodiments, the present invention relates to any of the
aforementioned
compounds, wherein X is phenyl substituted with two substituents independently
selected from the
group consisting of -CH3, -CF3, -CH2CH3, -OH, -OCH3, -OCH2CH3, -OCH2CH2OH,
eelee-/
0 "
-OCH2CH200H3, -F, -CI, -Br and -ON.
In certain embodiments, the present invention relates to any of the
aforementioned
compounds, wherein X is phenyl substituted with three substituents
independently selected from
the group consisting of -CH3, -CF3, -CH2CHe -OH, -00H3, -OCH2CH3, -OCH2CH2OH,
-OCH2CH200H3, -F, -Cl, -Br and -CN.
In certain embodiments, the present invention relates to any of the
aforementioned
compounds, wherein X is phenyl substituted with four substituents
independently selected from the
CA 02975157 2017-07-26
WO 2016/120850 PCT/IB2016/050477
group consisting of-CH, -CH2C1-13, -OH, -OCH3, -OCH2CH3, -00H2CH2OH,
L.5.)
0
-OCH2CH2OCH3, , -F, -CI, -Br and -ON.
In certain embodiments, the present invention relates to any of the
aforementioned
oompOunds, wherein X is phenyl substituted with five substituents
independently selected from the
5 group consisting of -CH3, -CFI, -CH2CH3, -OH, -00H3, -O0H2CH:3, -
OCH2CH2OH,
0
-0CH2CH20CH3, "Alm, -F, -Cl, -Br and -ON.
In certain embodiments, the present invention relates to any of the
aforementioned
compounds, wherein X is phenyl substituted with -F and optionally substituted
with one or two
substituents independently selected from the group consisting of -CH3, -CF3, -
CH2CH3, -OH,
o -OCH3, -OCH2CH3, -OCH2CH2OH, -
OCH2CH200H3, , -F, -CI, -Br and -ON.
In certain embodiments, the present invention relates to any of the
aforementioned
compounds, wherein X is phenyl optionally substituted with one, two, three,
four or five
substituents independently selected from the group consisting of -CH3, -CF3, -
CH2CH3,
-OH, -00H3, -OCH3CH3, -OCH2CH2OH, -OCH2CH2OCH3, 9ykft-vi, , -F, -Cl, -Br
and -ON,
15 In certain embodiments, the present invention relates to any of the
aforementioned
E
compounds, wherein X is optionally substituted with one additional
substituent
selected from the group consisting of -CH3, -CF3, -CH2CH3, -OH, -00H3, -
OCH2CH3,
(0\
-OCH2CH2OH, -0CH2CH20CHI, -F, -Cl, -Br and -ON.
In certain embodiments, the present invention relates to any of the
aforementioned
F
compounds; wherein X is substituted with one additional substituent
selected from the
CA 02975157 2017-07-26
WO 2016/120850
PCT/I132016/050477
16
group consisting of -CH, -CF, -CH2CH3, -OH, -OCH3, -OCH2CH3, -OCH2CH20H,
-OCHCH2OCH3, , -F, -CI, -Br and -CN.
In certain embodiments, the present invention relates to any of the
aforementioned
0
OCH3 0L)
F, F
111, F F
compounds, wherein X is Or
In certain embodiments, the present invention relates to any of the
aforementioned
compounds, wherein X is 5-membered hereroaryl optionally substituted with one,
two, three, or
four substituents independently selected from the group consisting of -CH3, -
CF3, -CH2CH3,
\--0
O.
-OH, -OCH3, -OCH2CH3, -OCH2C1-120H, -OCH2CH2OCH.?õ -F, -
Cl, -Brand -CN.
In certain embodiments, the present invention relates to any of the
aforementioned
compounds, wherein X is isoxazolyl or pyrazolyl, in each case optionally
substituted with one, two
or three, substituents independently selected from the group consisting of -
CH, -CF3,
0
-CH2CH3, -OH, -OCH3, -OCH2CH3, -OCH2CH20H, -OCH2CH2OCH3 -F, -
Cl, -Br
and -CN.
in certain embodiments, the present invention relates to any of the
aforementioned
0
NI/
=
compounds, wherein X is / or .In certain embodiment S,. the
present
invention relates to any of the aforementioned compounds, wherein R2 is (C-
C)alkyl optionally
substituted with one, two, three, four or five substitutents independently
selected for each
occurrence from the group consisting of -F, -Cl,
-Br, -OH, (C1-C3)alkyl, (C1-C3)haloalkyl and (CrCio)oycloalkyl.
CA 02975157 2017-07-26
WO 2016/120850 PCT/IB2016/050477
17
In certain embodiments, the present invention relates to any of
theeforementioned
compounels wherein R2 is unsubstituted (Cs,.C)alkyl. In certain embodiments,
the present
invention relates-to any of the aforementioned compounds, wherein R2 is
unsubstituted. branched.
(G1-06)alkylõ
In certain embodiments, the present invention relates to any of the
aforementioned
compounds., wherein R2 is C...,--CA)alkyl optionally ,substituted with one,
two, three, four or five
substitutents independently selected for each oedurrencefrorn
the.gtoUptOnsisting of -F, -CI,
-Br, -OH, (01-C3)alkyl, (CI-C3)haloalkyl and (C:eCt)cycloalkyl..
In certain .embodirnentsnthe present invention relates to any of the
aforementioned
compounds, wherein R2 is methyl optionally substituted with one, two or three
substitutents
independently selected for each occurrence from the group consisting of -F, -
CI,
-Br, -OH, (C1-.C3)alicyl, (01-03)haloalkyl and (C3-C10.)byeloalkyl.
In certain embodiments, the presentinvention relates to any of the
aforementioned
compounds, wherein R2 is ethyl optionally substituted with one, two, three,
four or five substitutents
independently selected for each occurrence from the group consisting of -F, -
Cl,
-Br, -OH, (C1-C3)alkyl, (C1-C3)halbalkyi and (C3-C10)cycloalkyl,
In certain .embodiments, the present invention relates to any of the
aforementioned
compounds, wherein R2 is n-propyl optionally substituted, with one, two,
three, four or five
substitutents independently selected for each occurrence from the group
consisting of -F, -Cl,
-Br, -OH, (C1-03)alkyl, (C1-03)haloalkyl and (C3-0-10)cycloalkyl.
In certain embodiments, the present invention relates to any of the
aforementioned
compounds, wherein R2 is i-propyl optionally substituted With one, two, three,
four or five
substitutents independently selected for each occurrence from the group
consisting of -F, -CI,
-Br, -OH, (C1-C3)alkyl.,.(CI-C)haloalkyl and (C3-010)cyclo5lkyl.
In certain embodiments, the present invention relates to any of the
aforementioned.
compounds, wherein R2 is methyl substituted with (C3s.C6)cycloalkyl.
In certain embodiments, the present invention relates to any Of the
aforementioned
compounds, wherein R2 is propyl substituted with -CF3.
In certain embodiments, the present invention relates to any of the
aforementioned
compounds, wherein R2 is ethyl substituted with (C3-Cb)cycloalkyl.
In certain embodiments, the present invention relates to any of the
aforementioned
compounds, wherein R2 is ethyl substituted with bicyclo[1.1,1]pentanyl.
In certain embodiments, the present invention relates to any of the
aforementioned
compounds, wherein R2 is (C3-C13)cycloalkyl optionally substituted with one,
two, three, four or five
CA 02975157 2017-07-26
WO 2016/120850
PCT/I132016/050477
18
.substitutents independently selected for each occurrence from the group
consisting of -Fõ
-CI, -Br, -OH, (CeC3)alkyi, (CI-Ce)haloalkyl and (Cy.,Cio)cycloalkyl.
In certain embodiments. the present invention relates to any of the
aforementioned
compounds, wherein Fe is unsubstituted (03-Ce0)cycloalkyl.
In certain embodiments, the present invention relates. to any of the
aforementioned
compounds, wherein R2 iscyclopropyl optionally substituted with one, two,
three or four
substitutents independently selected for each occurrence from the group
consisting of -F, -CI, -Br, -
OH, (C1-C3)alkyl, (C1-C3)heloalkyl and (C3-Cip)cycloalkyl,
In ce.rtain embodiments, the present invention relates to any of the
aforementioned
compounds, wherein Fels cyclobutyl optionally substituted with one, two,
three,. four or five
substitutents independently selected for each occurrence from the group
consisting of -F, -CI,
-Br, -OH, (C1-C3)alkyl, (CeC:3)haloalltyl and (03-C10)cycloalkyl,
In certain embodiments, the present invention relates to any of the
aforementioned
compounds, wherein R2 is cyclopentyl optionally substituted with one, two,
three, four or five
substitutents independently selected for each occurrence from the group
consisting of -F, -CI,
-Br, -OH, (C1-C3)alkyl,.(Ce-C3.)haloalkyl and (Ce-Cio)cycloalkyl.
In certain embodiments, the present invention relates to any of the
aforementioned
compounds, wherein ad is cyclo'hexyl optionally substituted with one, two,
three, four or-five.
substitutents independently selected for each occurrence from the group
consisting of -F, -Cl,
-Br, -OH, (Ce-C3)alkyl, (C1-C3)haloalkyl and (CrCiØ)oycloalkyl.
In certain embodiments, the present invention relates to any of the
aforementioned'
compounds, wherein R2 is unsubstituted phenyl.
In certain embodiments, the present invention relates to any of the
aforementioned
compounds, wherein R2 is phenyl optionally substituted with one, two, three,
four or five
substitutents independently selected for each occurrence from the group
consisting of -F, -Cl,
-Br, -OH, (C-C)alkyl,. (.Ce-C3)haloalkyl and (C3Cio)cycloalky'l.
In certain embodiments, the present invention relates to any of the
aforementioned
compounds, wherein R2 is phenyl optionally substituted with one, two, three,
four or five
substitutents independently selected for each occurrence from the group
consisting of -CI,
-Br, -OH and -CH3.
In certain embodiments, the present invention relates to any of the
aforementioned
compounds, wherein R2 is unsubstituted isothiazolyi.
In certain embodiments, the present invention relates to any of the
aforementioned
compounds, wherein R2 is isothiazely1 optionally substituted with one, two,
three, four or five
CA 02975157 2017-07-26
WO 2016/120850
PCT/I132016/050477
19
substitutents independently selected for each occurrence from the group
consisting of -F, -Cl,
-Br, -OH, (C1-c3)alkyl, (Ci-Q3)h2loalkyl and (C3-Ci0)oycloalkyf.
In certain embodiments, the present invention relates to any of the
aforementioned
compounds, wherein R2 is isothiazolyl optionally substituted with one, two,
three, four or five
substitutents independently selected for each occurrence from the group
consisting of -F, -Cl,
-Br, -OH and -CH3.
In certain embodiments, the preSen1 invention relates to any of the
aforementioned
/rCH3 CH3 CH3 CH3
compounds, wherein R2 is, H3C F3C F3C CH3 ,
CF3
/ CH
isr(x H3C H3C CF11CH3
CH,3
CF3 H3C CH3 HIC CH3 CH3
CH3 ,CH3 CH3
is(0
'1III1 H30
11101
or
In certain embodiments, the present invention relates to any of the
aforementioned
SN(CH3
compounds, wherein R2 is H3C
In certain embodiments, the present invention relates to any of the
aforementioned
irCH3
compounds, wherein R2 is F3C
In certain embodiments, the present invention relate to any of the
aforementioned
iss<T,õ,,C1-15
compounds, wherein R2 is F3C
CA 02975157 2017-07-26
WO 2016/120850
PCT/IB2016/050477
In certain embodiments, the present invention relates to any of the
aforeiMentioned
compounds, wherein R2=is
In certain embodiments, the present invention relates to any of the
afOrementioned
/ Cl-f3
, CF
compounds, wherein R' is .
5 In certain embodiments, the present invention relates to any of the
aforementioned
ja15
compounds, wherein R2- is. CF3,
In certain embodiments, the present invention relates to any of the
aforementioned
issUi3
compounds, wherein R.'. is 3
In certain embodiments, the present invention relates to any of the
aforementioned
S.:.x.CH3
10 compounds, wherein R2 is =H3C- CH; ..
In certain embodiments, the present invention relates to any of the
aforementioned
f.,),....
H..IC
compounds, wherein R2 CH; is - .
In certain embodiments, the present invention relates to any of the
aforementioned
H3
/4....,
compounds, wherein R2 is CH3
15 In certain embodiments, the present invention relates to any of the
aforementioned
..1,..õ.0 H=3
H3C CHI
compounds, wherein R2 is CH3 ' .
CA 02975157 2017-07-26
WO 2016/120850
PCT/I132016/050477
21
In certain embodiments, the present invention relates to any of the
aforementioned
compounds, wherein R2 is
In certain embodiments, the present invention relates to any ofthe
aforementioned
compounds, wherein R2 is
In certain embodiments, the present invention relates to .any of the
aforementioned
C.H3
compounds, wherein R2 is Fr''
In certain embodiments, the present invention relates to any of the
aforementioned
compounds, wherein R2 is
In certain embodiments, the present invention relates to any of the
aforementioned
CH3.
.. compounds, wherein R.2 is
in certain embodiments, the present invention relates to any of the
aforementioned
compounds, wherein R2 is/O.
in certain embodiments, the present invention relates to any of the
aforementioned
compounds,-*heeein R2 is
CA 02975157 2017-07-26
WO 2016/120850 PCT/IB2016/050477
22
In certain embodiments, the present invention relates to any of the
aforementioned
compounds, wherein R2 is
In certain embodiments, the present invention relates to any of the
aforementioned
compounds, wherein R2 is 1-4 -3-
r
In certain embodiments, the present invention relates to any of the
aforementioned
\
N,
compounds, wherein R2 is s
Another embodiment of the invention is a compound selected from the group
consisting of
the compounds of Examples 1-35 and pharmaceutically acceptable salts thereof.
Therapeutic Applications
It is contemplated that the compounds of Formula I provide therapeutic
benefits to subjects
suffering from an immune disorder or inflammatory disorder. Accordingly, one
aspect of the
invention provides a method of treating a disorder selected from the group
consisting of an
immune disorder or inflammatory disorder, The method comprises administering a
therapeutically
effective amount of a compound of Formula Ito a subject in need thereof to
ameliorate a symptom
of the disorder, wherein Formula I are as described above. In certain
embodiments; the particular
compound of Formula 1 is a compound defined by one of the embodiments
described above.
In certain embodiments, the disorder is an immune disorder, In certain other
embodiments, the disorder is an inflammatory disorder. In certain other
embodiments, the disorder
is an autoimmune disorder. in certain other embodiments, the disorder is
rheumatoid arthritis,
Rci psoriasis, chronic graft-versus-host disease, acute graft-versus-host
disease, Crohn's disease,
inflammatory bowel disease, multiple sclerosis, systemiclupus erythematasus,
Celiac Sprue,
idiopathic thrombocytopenic thrombotic purpura, myasthenia gravis, Sjogren's
syndrome,
scleroderma, ulcerative colitis, asthma, or epidermal hyperplasia.
In certain other embodiments, the disorder is cartilage inflammation, bone
degradation,
arthritis, juvenile arthritis, juvenile rheumatoid arthritis, pauclarticular
juvenile rheumatoid arthritis,
polyarticular juvenile rheumatoid arthritis, systemic onset juvenile
rheumatoid arthritis, juvenile
ankylosing spondylitis, juvenile enteropathic arthritis, juvenile reactive
arthritis, juvenile Reter's
CA 02975157 2017-07-26
WO 2016/120850 PCT/IB2016/050477
23
Syndrome, SEA Syndrome., juvenile dermatomyoeitis, juvenile psoriatic
arthritis, juvenile
scleroderma., juvenile systemic lupus erythematosUs, juvenile vasculitis,
pauciarticular rheumatoid
arthritis, polyarticuler rheumatoid arthritis, systemic onset. rheumatoid
arthritis, ankylosing
spondylitis, enteropathic arthritis, re:active. arthritis, Reter's Syndrome,
.clermatomyositis, psonatic
arthritis, vasc.ulitisõ myosins, polymyositis, osteoarthritis, polyarteritis
tiodossa, Wegener's
granulornatosis, arteritis, polyrnyalgia rheumatica, sarcoidosis, sclerosis,
primary billary sclerosis,
sclerosing chota.ngitis, dermatitis, atopic dermatitit, atherosclerosis,
Still's disease, chronic
obstructive pulmonary disease, Guiilain-Barre disease, Type I diabetes
mellitus, Graves disease,
Addison's disease, Raynaud's phenomenon, autoimmune. hepatitis, .psoriatic
epidermal
.. hyperpla.sia, plaque psoriasis, guttate psoriasis, inverse psoriasis,
pustular psoriasis, erythrodermiC
psoriasis, giant cell arteritis, nonalcoholic hepatic steatosie, or an immune
disorder associated with
or arising from activity of pathogenic lymphocytes.
In certain embodiments, the psoriasis is plaque psoriasis, guttate psoriasis,
inverse
psoriasis, pustular psoriasis, or erythmdermic psoriasis.
in certain other embodiments, the disorder is noninfectiOueuVeitis, Behcet's
disease or
Vogt¨Koyanagi¨Flarada. syndrome.
Another aspect of the invention provides for the use of a compound of Formula
I in the
manufacture of a medicament. In certain embodiments, the medicament is for
treating a disorder
described herein.
Another aspect of the invention provides for the use of a compound of Formula
I for treating
a medical disorder, such a medical disorder described herein.
Further, it is contemplated that compounds of Formula I can inhibit the
activity of RORy.
Accordingly, another aspect of the invention provides a method of inhibiting
the activity of RORy.
The method comprises exposing a RORy to an effective amount of a compound of
Formula Ito
inhibit said RORy, wherein Formula I is as described above, in certain
embodiments, the particular
compounds of Formula I are the compound defined by One of the embodiments
described herein.
Further, it is contemplated that compounds of Formula 1 can reduce the amount
of
interleukin-17 (IL-17) in a subject. IL-17 is a cytokine that affects numerous
biological functions,
including inducing and mediating proAnflammatory responses. Accordingly,
another aspect of the.
.. invention provides a method of reducing the amount of IL-17 in e subject.
The method comprises
administering to a subject an effective amount of a compound of Ito reduce the
amount of 1L-17 in
the subject, wherein Formula I is as described above. In certain embodiments,
the particular
compounds of Formula I are the compounds defined by one of the embodiments
described herein..
In certain embodiments, the subject is a hUrnan.. In certain embodiments,
administering
the compound reduces the amount of IL-17 produced by Th-17 :cells in the
subject. A change in the
CA 02975157 2017-07-26
WO 2016/120850
PCT/I132016/050477
24
amount of 1L-17 produced by, for example, Th-17 cells can be measured using
procedures
described in the literature, such as an EL1SA assay or intracellular staining
assay.
Further, it is contemplated that compounds of Formula I may inhibit the
.synthesis of 1L-17 in
a subject. Accordingly, another aspect of the invention provides a method of
inhibiting the
synthesis IL-17 in a subject. The Method comprises administering to a subject
an effective amount
of a compound of Formula Ito inhibit the synthesis IL-17 in the subject,
wherein Foffnula_ 1 is as
described above. In certain. embodiments,. the particular compounds of Formula
I are the
compounds defined by one of the embodiments described herein.
The description above describes multiple embodiments providing definitions for
variables
used herein. The application specifically contemplates all combinations of
such variables.
Combination Therapy
Another aspect of the invention provides for combination therapy. For example,
the
compounds of Formula 1 or their pharmaceutically acceptable salts may be used
in combination
with additional therapeutic agents to treat medical disorders, such as medical
disorders associated
with inappropriate IL-17 pathway activity. Exemplary additional therapeutic
agents include, for
example, (1) a TNF-a inhibitor; (2) a non-selective COX-1/COX-2 inhibitor; (3)
a selective COX-2
inhibitor, .such as celecoxib and rofecoxita. (4) other agents for treating
inflammatory disease and
autoimrnune disease including, for example, methotrexate, leflanornide,
sulfasalazine,
azathioprine, .penictilamine, buciliamine, actarit, mizoribine, lobenzarit,
hydroxychloroquine, d-
penicillarnine, aurothiomalate, auranofin, parenteral gold, oral gold;,
cyclophosphamide,
Lymphost.at-B, a BAFF/ APRIL inhibitor, CTLA-4-1g, or a mimetic of CTLA-4-Ig;
(5) a leu.kotriene
biosynthesis inhibitor., such as a 5-lipoxygenase (5-LO) inhibitor, or a 5-
lipoxygenase activating
protein (FLAP) antagonist.; (6) a LTD4 receptor antagonist; (7) a
phosphodiesterase type IV (POE-
IV) inhibitor,, such as cilomilast (antic) or roflumilast: (8) an
antihistamine Hi receptor antagonist;
(9) an od- and oc2-adrenoceptor a.gonist, (10) an antichOlinergic. agent; (11)
a p-adrenoceptor
agonist; (12) an insulin-like growth factor type I (1GF-1) mimetic; (13) a
giucecortic.bsoid; (14) a
kinase inhibitor such as an inhibitor of a Janus Kinase (e.g., JAK and/or JAK2
and/or JAK 3
and/or TYK2), p38 MAPK, Syk or IKK2; (15) a B-cell target biolidgit such as
rituximab; (16) a
selective co-stimulation modulator such as abatacept; (17) an interleukin
inhibitor or interieukin
.. receptor inhibitor, such as the IL-1 inhibitor anakinra, IL-6 inhibitor
tocilizumab, and IL12/1L-23
inhibitor ustekimumab; (18) an anti-11_17 antibody, anti-1L21 antibody, or
anti-IL2.2 antibody (19) a
S1P1 agonist, such as fingolimod; (20) an interferon, such as interferon beta
1; (21) an integrin
inhibitor such as natalizumab; (22) a rnTOR inhibitor such as rapamycin,
cyclosoorin and
tacrolimus; (23) 8 non-steroidal antiinflammatory.agent (NSA1D), such as
propionic acid derivatives
.(alminoprofen, benoxaprofen, bucioxic acid, carprofen,. fenbufen,
.fenoprofen., fluprofen,
flurbiprofenõ ibuprofen, indoprofen, ketoprofen, miroprofen., naproxen,
oxaprozin, pirprofen,
CA 02975157 2017-07-26
WO 2016/120850 PCT/IB2016/050477
pranoprofee, suprofen, tiaprofenic acid, and tioxaprofen), acetic acid
derivatives (indornethacin,
..acernetacin, alclofenac, clidanac, diclofenac, fenclofenac, fenclozic acid,
fentiazac, furofenac,
ibufenac, isoxepac, oxpinac, sulindac, tiopinac, tolmetin, ziclometacin, and
zomepirac), fenamic
add derivatives (flufenarnic acid, meclofenamic acid, mefenamic acid, niflumic
acid and tolfenamic
5 .. acid), biphenylearboxylic acid derivatives (difiunisal and flufenisal)õ
exicarris (isoxicam, piroxicam,
sudoxicarn and tenoxica.n), salicylatee (acetyl salicylic acid, sulfasalazine)
and pyrazolones
(apazone, bezpiperylon, feprazone, mofebutazone, oxyphenbutazone,
phenylbutazone); (24) a
NRF2 pathway activator, such as the fumaric acid derivative; BG-12; and (25) a
chernokine or
ch.emokine receptor inhibitor, such as a CCR9 antagonist.
10 In certain embodimenteethe additional therapeutic agent is selected from
the group
consisting of corticosteroids, vitamin D3, .anthralin and retinoids, in
certain embodiments, the
additional therapeutic agent is a corticosteroid. in certain embodiments, the
additional therapeutic
agent is vitamin D3. In certain embodiments, the additional therapeutic agent
is anthralin. In
certain embodiments, the additional therapeutic agent is a retinoid.
15 The amount of the compounds.of Formula land additional therapeutic agent
and the
relative timing of administration may be selected in order to achieve a
desired combined
therapeutic effect. For example, when administering a combination therapy to a
patient in need of
such administration, the therapeutic agents in the combination, or a
pharmaceutical compasitiOn or
compositions comprising the therapeutic agents, may be administered in any
order such as, for
20 example, sequentially, concurrently, together, simultaneously and the
like, Further, for example, a
compound of Formula I may be administered during a time when the additional
therapeutic
agent(s) exerts its prophylactic or therapeutic effect, or vice versa.
The doses and dosage regimen of the active ingredients used in the combination
therapy
may be determined by an attending clinician. in certain embodiments, the
compound of Formula I
25 and the additional therapeutic agent(s) are administered in doses
commonly employed when such
agents are used as monotherapy for treating the disorder. In other
embodiments, the compound of
Formula I and the additional therapeutic agent(s) are administered in doses
lower than the doses.
commonly employed when such agents are used as monotherapy for treating the
disorder. In
certain embodiments, a compound of Formula I and the additional
therapeuticagent(s) are present
in the same composition, which is suitable for oral administration.
In certain embodiments, the compound of Formula I and the additional
therapeutic agent(e)
may act additively or synergistically. A synergistic combination may allow the
use of lower dosages
of one or more agents and/or less frequent administration of one or more
agents of a combination
therapy. A lower dosage or less frequent administration of one or more agents
may lower toxicity
of the therapy without reducing the efficacy of the therapy.
CA 02975157 2017-07-26
WO 2016/120850
PCT/I132016/050477
26
Another aspect of this invention, is a kit comprising a therapeutically
effective amount of e
compound of Formula t, a pharmaceutically acceptable carrier, vehicle or
diluent, and optionally at
least one additional therapeutic agent listed above.
Pharmaceutical Compositions and Dosing Considerations
Typically, a compound of the invention is administered in an amount effective
to treat a
condition as described herein. The compounds of the invention are administered
by any suitable
route in the form of a pharmaceutical composition adapted to such a route, and
in a dose effective
for the treatment intended. Therapeutically effective doses of the compounds
required to treat the
progress of the medical condition are readily ascertained by one of ordinary
skill in the art using
preclinicaf and clinical approaches familiar to the medicinal arts. The term
"therapeutically effective.
amount" as used herein refers to that amount of the compound being
administered which will
relieve to some extent one or more of the symptoms of the disorder being
treated.
The term "treating", as used herein, unless otherwise indicated, means
reversing,
alleviating, inhibiting the progress of, or preventing the disorder or
condition to which such term
applies, or one or more symptoms of such disorder or condition. The term
"treatment", as used
herein, unless otherwise indicated, refers to the act of treating as
'treating" is defined immediately
above. The term "treating" also includes adjuvant and neo-adjuvant treatment
of a subject.
As indicated aboVe.; the invention provides pharmaceutical compositions, which
comprise a
therapeutically-effective amount of one or more of the compounds described
above, formulated
together with one or more pharmaceutically acceptable carriers (additives)
and/or diluents. The
pharmaceutical compositions may be specially formulated for administration in
solid or liquid form,
including those adapted for the following: (I) oral administration; for
example, drenches (aqueous
or non-aqueous solutions or suspensions), tablets, e.g., those targeted for
buccal, sublingual, and
systemic absorption, boluses, powders, granules, pastes for application to the
tongue; (2)
pareateral administration, for example, bysubcutaneous..,:intram.uttular,
intravenous or epidural
injection as, for example, a sterile solution or suspension, or suatained-
release formulation; (3)
topical application, for example, as a cream; ointment, or a controlled-
release patch or spray
applied to the skin; (4) intravaginalty or intrarectallyõ for example, as a
pessary, cream or foam; (5)
sublingually; (6) ocularly; (7) transdermally; or (8) nasally.
The phrase "pharmaceutically acceptable" is employed herein to refer to those
compounds,
materials, compositions, and/or dosage -forms which are, within the scope of
sound medical
judgment, suitable for use in contact with the tissues of human beings and
animals without
excessive toxicity, irritation, allergic response, or other problem or
complication, commensurate
with a reasonable benefit/risk ratio,.
CA 02975157 2017-07-26
WO 2016/120850 PCT/IB2016/050477
27
Wetting agents, emulsifiers and lubricants, such as sodium lauryl sulfate and
magnesium
stearate, as well as coloring agents, release agents, coating agents,
sweetening, flavoring and
perfuming agents, preservatives and antioxidants can also be present in the
compositions.
Examples of pharmaceOtitally-acceptable antioxidants include: (1) water
soluble
antioxidants, such as ascorbic acicl, ,cysteine hydrochloride, sodium
bisulfate, sodium metabisulfite,
sodium sulfite and the like; (2) oil-soluble antioxidants, such as ascorbyl
palrriitate, butylated
hyolroxyanisole (BHA), butylated hydroxytoluene (BHT), lecithin, propyl
gallate, alpha-tocopherol,
and the like; and (3) metal chelating agents, such as citric acid,
ethylenediamine tetraacetic acid
(EDTA), sOrtaitiii, tartaric acid, phosphoric acid, and the like,
Formulations of the present invention include those suitable for oral, nasal,
topical
(including buccal and sublingual), rectal, vaginal and/or parenteral
administration. The formulations
may conveniently be presented in unit dosage form and may be prepared by any
methods well
known in the art of pharmacy. The amount of active ingredient which can be
combined with a
carrier material to produce a single dosage form will vary depending upon the
host being treated,
the particular mode of administration. The amount of active ingredient which
can be combined with
a carrier material to produce a single dosage form will generally be that
amount of the compound
which produces a therapeutic effect. Generally, out of one hundred per cent,
this amount will range
from about 0.1 per cent to about ninety-nine percent of active ingredient,
preferably from about 5
percent to about 70 percent, most preferably from about 10 percent to about 30
percent.
In certain embodiments, a formulation of the present invention comprises an
excipient
selected from the group consisting of cytiodextrins, celluloses, liposornes,
micelle forming agents,
e.g., bile acids, and polymeric carriers, e.g., polyesters and polyanhydrides;
and a compound of
the present invention, In certain embodiments, an aforementioned formulation
renders orally
bioavailable a compound of the present invention.
2.5 Methods of preparing these formulations or compositions include the
step of bringing into
association a compound of the present invention with the carrier and,
optionally, one or more
accessory ingredients. In general, the formulations are prepared by uniformly
and intimately
bringing into association a compound of the present invention with liquid
carriers, or finely divided
solid carriers, or both, and then, if necessary, shaping the product.
Formulations of the invention suitable for oral administration may be in the
form of
capsules, cachets, pills, tablets, lozenges (using a flavored basis, usually
sucrose and acacia or
tragacanth), powders, granules, or as a solution or a suspension in an aqueous
or non-aqueous
liquid, or as an oil-in-water or water-in-oil liquid emulsion, or as an elixir
or syrup, or as pastilles
(using an inert base, such as gelatin and glycerin, or sucrose and acacia)
and/or as mouth washes
and the like, each containing a predetermined amount of a compound of the
present invention as
CA 02975157 2017-07-26
WO 2016/120850 PCT/IB2016/050477
28
an active ingredient. A compound of the present invention may also be
administered as a bolus,
electUery or paste.
In solid dosage forms of the invention for oral administration (capsules,
tablets, pills,
dragees, powders, granules, Vouches and the like), the active ingredient is
mixed with one or more
pharmaceutically-acceptable carriers, such as sodium citrate or dicalcium
phosphate, and/or any of
the following: (1) fillers or extenders, such as starches, lactose, sucrose,
glucose, mannitol, and/or
silicio apid; (2) binders, such as, for example, carboxymethyicellulose,
alginates, gelatin, polyvinyl
pyrrolidone, sucrose and/or acacia; (3) hurnedtants, such as glycerol; (4)
disintegrating agents,
such as agar-agar, calcium carbonate, potato or tapioca starch, alginic acid,
certain silicates, and
sodium carbonate; (5) solution retarding agents, such as paraffin; (6)
absorption accelerators, such
as quaternary ammonium compounds and surfactants, such as poloxamer and sodium
lauryl
sulfate; (7) wetting agents, such as, for example, cetyl alcohol, glycerol
monostearate, and non-
ionic surfactants; (8) absorbents, such as kaolin and bentonite clay: (9)
lubricants, such as talc,
calcium stearate, magnesium stearate, solid polyethylene glycols, sodium
lauryl sulfate, zinc
stearate, sodium stearate, stearic acid, and mixtures thereof; (10) coloring
agents; and (11)
controlled release agents such as crospovidone or ethyl cellulose. In the case
of capsules, tablets
and pills, the pharmaceutical compositions may also comprise buffering agents.
Solid compositions
of a similar type may also be employed as fillers in soft and hard-shelled
gelatin capsules using
such excipients as lactose or milk sugars, as well as high molecular weight
polyethylene glycols
and the like.
A tablet may be made by compression or molding, optionally with one or more
accessory
ingredients. Compressed tablets may be prepared using binder (for example,
gelatin or
hydroxypropylrnethyl cellulose), lubricant, inert diluent, preservative,
disintegrant (for example,
sodium starch glycolate or cross-linked sodium carboxymethyl cellulose),
surface-active or
dispersing agent. Molded tablets may be made by molding in a suitable machine
a mixture of the
powdered compound moistened with an inert liquid diluent.
The tablets, and other solid dosage forms of the pharmaceutical compositions
of the
present invention, such as dragees, capsules, pills and granules, may
optionally be scored or
prepared with coatings and shells, such as enteric coatings and other coatings
well known in the
pharmaceutical-formulating art. They may also be formulated so as to provide
slow or controlled
release of the active ingredient therein using, for example,
hydroxypropylmethyl cellulose in
varying proportions to provide the. desired release profile, other polymer
matrices, liposornes
and/or micmspheres. They may be formulated for rapid release, e.g., freeze-
dried. They may be
sterilized by, for example, filtration through a bacteria-retaining filter, or
by incorporating sterilizing
agents in the form of sterile solid compositions which can be dissolved in
sterile water, or some
other sterile injectable medium immediately before use. These compositions may
also optionally
CA 02975157 2017-07-26
WO 2016/120850 PCT/IB2016/050477
29
contain pacifying agents and may be or a composition that they release the
active ingredient(s)
only, or preferentially, in a certain portion of the gastrointestinal tract,
optionally; in a delayed
manner. Examples of embedding compositions which can be used include polymeric
substances
and waxes. The active ingredient can also be in micro-encapsulated form, if
appropriate, with one
or more of the above-described exCipientS.
Liquid dosage forms for oral administration of the compounds of the invention
include
pharmaceutically acceptable emulsions, microemuisions, solutions, suspensions,
syrups and
elixirs. In addition to the active ingredient, the liquid dosage forms may
contain inert diluents
commonly used in the art, such as, for example, water or other solvents,
solubilizing agents and
emulsifiers, such as ethyl alcohol, isopropyl alcohol, ethyl carbonate, ethyl
acetate, benzyl alcohol,
benzyl benzoate, propylene glycol, 1,3-butylene glycol, Oils (in particular,
cottonseed, groundnut,
corn, germ, olive, castor and sesame oils), glycerol, tetrahydrofuryl alcohol,
polyethylene glycols
and fatty acid esters of sorbitan, and mixtures thereof.
besides inert diluents, the oral compositions cart also include adjuvants such
as wetting
agents, emulsifying and suspending agents, sweetening, flavoring, coloring,
perfuming and
preservative agents.
Suspensions, in addition to the active compounds, may contain suspending
agents as, for
example, ethoxylated isostearyi alcohols, polyoxyethylene sorbitol and
sorbitan esters,
microorystalline cellulose, aluminum metahydroxide, bentonite, agar-agar and
tragacanth, and
mixtures thereof.
Formulations of the pharmaceutical compositions of the invention for rectal or
vaginal
administration may be presented as a suppository, which may be prepared by
mixing one or more
compounds of the invention with one or more suitable nonirritating excipients
or carriers
comprising, for example, cocoa butter, polyethylene glycol, a suppository wax
or a salicylate, and
which is solid at room temperature, but liquid at body temperature and,
therefore, will melt in the
rectum or vaginal cavity and release the active compound.
Formulations of the present invention which are suitable for vaginal
adMihistratian also
include pessaries, tampons, creams, gels, pastes, foams or spray formulations
containing such
carriers as are known in the art to be appropriate.
Dosage forms for the topical or transdermal administration of a compound of
this invention
include powders, sprays, ointments, pastes, creams, lotions, gels, solutions,
patches and
inhalants. The active compound may be mixed under sterile conditions with a
pharmaceutically-
acceptable carrier, and with any preservatives, buffers, or propellants which
may be required.
CA 02975157 2017-07-26
WO 2016/120850 PCT/IB2016/050477
The invention also includes pharmaceutical compositions utilizing one or more
of the
present compounds along with one or more pharmaceutically acceptable carriers,
excipients,
vehicles,
Topical formulations of the presently disclosed compounds may be administered
topically,
5 (intra)dermally, or transdermally to the skin or mucosa. Topical
administration using such
preparations encompasses all conventional Methods of administration across the
surface of the
body and the inner linings of body passages including epithelial and mucosal
tissues, including
transdermal, epidermal, buccal, pulmonary, ophthalmic, intranasal, vaginal and
rectal modes of
administration, Typical formulations for this purpose include gels, hydrogels,
lotions, solutions,
10 creams, colloid, ointments, dusting powders, dressings, foams, films,
skin patches, wafers,
implants, sponges, fibres, bandages and microemulsions. Liposomes may also be
used, Typical
carriers include alcohol, water, mineral oil, liquid petrolatum, white
petrolatum, glycerin,
polyethylene glycol and propylene glycol. Such topical formulations may be
prepared in
combination With additional pharmaceutically acceptable excipients,
15 In certain embodiments, a penetration enhancer may be used. Examples of
penetration
enhancers include, for example, saturated Cl 0-C18 fatty alcohols (such as
decyl alcohol, lauryl
alcohol, myristyl alcohol, cetyl alcohol and stearyl alcohol), cis-unsaturated
Cl 0-C18 fatty alcohols
(such as oleyl alcohol, linoleyl alcohol, y-linolenyi alcohol and linolenyl
alcohol), Cl 0-C18 fatty
acids (which when saturated may include capric acid, lauric acid, myristic
acid, palmitic acid,
20 stearic acid and arachidic acid), cis-unsaturated fatty acids (such as
palmitoleic acid (cis-9-
hexadecenoic acid), oleic acid (cis-9-octadecenoic acid), cis-vaccenic acid
(cis-11-octadecenoic
acid), linoleic acid (cis-9,12-octadecadienoic acid), y-linolenic acid (cis-
6,9,12-octadecatrienoic
acid), linolenic acid (cis-9,12,15-octadecatrienoic acid) and arachidonic acid
(cis-5,8,11,14-
eicosatetraenoic acid)). In certain embodiments, the penetration enhancers may
be used amounts
25 ranging from abok.it 0.1 to about 5% (war).
In certain embodiments, topical formulations which contain one or more
compounds of the
invention in therapeutically effective amounts that may be given in daily or
twice daily doses to
patients in need.
The ointments, pastes, creams and gels may contain, in additien to an active
compound of
30 this invention, excipients, such as animal and vegetable fats, oils,
waxes, paraffins, starch,
tragacanth, cellulose derivatives, polyethylene glycols, silicones,
bentonites, silicic acid, talc and
zinc oxide, or mixtures thereof. Other excipients which enhance the stability
of the formulations
include aldehyde scavengers, such as glycerine and propylene glycol, and
antioxidants, such as
butyl hydroxyanisale (BHA), butyl hydroxytoluene (BHT), propyl gallate,
ascorbic acid (Vitamin C),
polyphenols, tocopherols (Vitamin E), and their derivatives,
CA 02975157 2017-07-26
WO 2016/120850
PCT/I132016/050477
31
Powders and sprays can contain, in addition to a compound of this invention,
excipients
Such as lactose, talc, silicic acid, aluminum hydroxide, calcium silicates and
polyamide powder, or
mixtures of these substances. Sprays can additionally contain customary
propellants, such as
chlorolluorohydrocarbons and volatile unsubstituted hydrOcarbons, such as
butane and propane,
Transdermal patches have the added advantage of providing controlled delivery
of a
compound of the present invention to the body. Such dosage forms can be made
by dissolving or
dispersing the compound in the proper medium. Absorption enhancers can also be
used to
increase the flux of the compound across the skin. The rate of such flux can
be controlled by either
providing a rate controlling membrane or dispersing the compound in a polymer
matrix or gel.
Ophthalmic formulations, eye ointments, powders, solutions and the like, are
also
contemplated as being within the scope of this invention. Formulations
suitable for topical
administration to the eye include, for example, eye drops wherein the compound
of this invention is
dissolved or suspended in a suitable carrier. A typical formulation suitable
for ocular or aural
administration may be in the form of drops of a micronised suspension or
solution in isotonic, pH-
adjusted, sterile saline. Other formulations suitable for ocular and aural
administration include
ointments, biodegradable (i.e., absorbable gel sponges, collagen) and non-
biodegradable (i.e.,
silicone) implants, wafers, lenses and particulate or vesicular systems, such
as niosomes or
liposomes. A polymer such as crossed-linked polyacrylic acid, polyvinyl
alcohol, hyaluronic acid, a
cellulosic polymer, for example, hydroxypropylmethylcellulose,
hydroxyethylcellulose, or
methylcellulose, or a heteropoiysaccharide polymer, for example, gelan gum,
may be incorporated
together with a preservative, such as benzalkonium chloride. Such formulations
may also be
delivered by iontophoresis.
For intranasal administration or administration by inhalation, the active
compounds of the
invention are conveniently delivered in the form of a solution or suspension
from a pump spray
container that is squeezed or pumped by the patient or as an aerosol spray
presentation from a
pressurized container or a nebulizer, with the use of a suitable propellant.
Formulations suitable for
intranasal administration are typically administered in the form of a dry
powder (either alone; as a
mixture, for example, in a dry blend with lactose; or as a mixed component
particle, for example,
mixed with phospholipids, such as phosphatidylcholine) from a dry powder
inhaler or as an aerosol
spray from a pressurised container, pump, spray, atomiser (preferably an
atomiser using
electrohydrodynamics to produce a fine mist), or nebuliser, with or without
the use of a suitable
propellant, such as 1,1,1,2-tetrafluoroethane or 1,1 ,1,2,3,3,3-
heptafluoropropane. For intranasal
use, the powder may comprise a bioadhesive agent, for example, chitosan or
cyclodextrin.
Pharmaceutical compositions of this invention suitable for parente.,,ral
administration
comprise one or more compounds of the invention in combination with one or
more
pharmaceutically-acceptable sterile isotonic aqueous or nonaqueous solutions,
dispersions,
CA 02975157 2017-07-26
WO 2016/120850 PCT/IB2016/050477
32
suspensions or emulsions, or sterile powders which may be ma:instituted into
sterile injectable
solutions or dispersions just prior to use, which may contain sugars,
alcohols, antioxidants, buffers,
bacteriostats, solutes which render the formulation isotonic with the blebd of
the intended recipient
or suspending or thickening agents.
Examples of Suitable aqueous and nonaqueous carriers which may be employed in
the
pharmaceutical compositions of the invention include water, ethanol, polyols
(such as glycerol,
propylene glycol, polyethylene glycol, and the like), and suitable mixtures
thereof, vegetable oils,
such as olive oil, and injectable organic esters, such as ethyl oleate. Proper
fluidity can be
maintained, for example, by the use of coating materials, such as lecithin, by
the maintenance of
.. the required particle size in the case of dispersions, and by the use of
surfactants.
These compositions may also contain adjuvants such as preservatives, wetting
agents,
emulsifying agents and dispersing agents. Prevention of the action of
microorganisms upon the
subject compounds may be ensured by the inclusion of various antibacterial and
antifungal agents,
for example, paraben, chlorobutanol, phenol sorbic acid, and the like. It may
also be desirable to
include isotonic agents, such as sugars, sodium chloride, and the like into
the compositions. In
addition, prolonged absorption of the injectable pharmaceutical form may be
brought about by the
inClusion of agents which delay absorption such as aluminum monostearate and
gelatin.
In some cases, in order to prolong the effect of a drug, it is desirable to
slow the absorption
of the drug from subcutaneous or intramuscular injection. This may be
accomplished by the use of
a liquid suspension of crystalline or amorphous material having poor water
solubility. The rate of
absorption of the drug then depends upon its rate of dissolution which, in
turn, may depend upon
crystal size and crystalline form. Alternatively, delayed absorption of a
parenterally-administered
drug form is accomplished by dissolving or suspending the drug in an oil
vehicle.
Injectable depot forms are made by forming microencapsule matrices of the
subject
.. compounds in biodegradable polymers such as polylaCtide-polyglycolide.
Depending on the ratio of
drug to polymer, and the nature of the particular polymer employed, the: rate
of drug release can be
controlled. Examples of other biodegradable polymers include poly(orthoesters)
and poly
(anhydrides). Depot injectable formulations are also prepared by entrapping
the drug in liposornes
or microemuisions which are compatible with body tissue,
When the compounds of the present invention are administered as
pharmaceuticals, to
humans and animals, they can be given per seer as a pharmaceutical composition
containing, for
example, 0,1 to 99% (more preferably, 10 to 30%) of active ingredient in
combination with a
pharmaceutically acceptable carrier.
The preparations of the present invention may be given orally, parenterally,
topically, or
rectally. They are of course given in forms suitable for each administration
route. For example,
CA 02975157 2017-07-26
WO 2016/120850 PCT/IB2016/050477
33
theyare administered in tablets or capsule form., by-injection, inhalation,
eye lotion, ointment,
suppository, etc. administration by injection, infusion or inhalation; topical
by lotion or ointment; and
rectal by suppositories. Oral administrations are preferred.
The phrases "parenteral administration" and "administered parenterally" as
used herein
.. means Modes of administration other than enteral and topical
administration, usually by injection,
and includes, without limitation', intravenous., _intramuscular,
intraarteriat, int.rathecal, intracapsular,
iritraorbital, intracardiac, intrademial, intraparitone.al, transtracheal,
subcutaneous, subcuticular,
intraarticulare, subcapsular, subarachnoid,.intraspinal and intrastemal
.injection and infusion.
The phrases "systemic administration," "administered systemically" "peripheral
administration' and "administered peripherally" as used herein mean the
administration of a
compound, drug or other material other than directly into the central nervous
system, such that it
enters the patient's system and, thus, is subject to metabolism and other like
processes, for
example, subcutaneous administration.
These compounds may be administered to humans and other animals for therapy by
any
suitable route of administration, including orally, nasally, as by, for
example, a spray, rectally,
intravaginally, parenterally, intracisternally arid topically, as by powders,
ointments or drops,
including b.utcally and sublingually.
Regardless of the route of administration selected, the compounds of
present invention,
which may be used in a suitable hydrated form, and/or the pharmaceutical
compositions of the
present invention, are formulated into pharmaceutically-acceptable dosage
forms by conventional
methods known to those of skill in the art.
Actual dosage levels of the active ingredients in the pharmaceutical
compositions of this
invention may be varied so as to obtain an amount of the active ingredient
which is effective to
achieve the desired therapeutic response for a particular patient,
composition, and mode of
.25 administration, without being toxic to the patient.
The selected dosage level will depend upon a variety of factors including the
activity of the
particular compound of the present invention employed, or the ester, salt or
amide thereof, the
route of administration, the time of administration, the rate of excretion or
metabolism of the
particular compound being employed4 the rate and extent of absorption, the
duration of the
treatment, other drugs, compounds and/or materials used in combination with
the particular
compound employed, the age, sex, weight, condition, general health arid prior
medical history of
the patient being treated, and like factors well known in the medical arts.
A physician or veterinarian having ordinary skill in the art can readily
determine and
prescribe the effective amount of the pharmaceutical composition required. For
example, the
physician or veterinarian could start doses of the compounds of the invention
employed in the
84020419
34
pharmaceutical composition at levels lower than that required in order to
achieve the desired
therapeutic effect and gradually increase.th-e dosage until the desired effect
is achieved.
In general, 0 suitable daily dose of a compound. of the invention will be
that. amoUritaf the
.compound which is the lowest dose effective to produce a therapeutic effect.
Such an effective
dose will generally depend. upon the factors described above. Preferably, the
compounds are
administered aLabout-0.01 mg/kg to about 200 mg/kg, more preferablyat about
0.1 .mg/kg to about
100 Mg/kg, even more, preferably at.aboUt.0,5 Mg/kg to about 50 mg/kg.:
When the compounds described herein are co-administered With another:agent
(e..g.õ as
sensitizing agents), the effective -amount may be less than when the agent is
used alone
If desired,. the effective daily (lase of the active compound May be
adrilinittered as two,
three, four,.five, six or more sub-doses adminiStered separately at
appropriate. intervals 'throughout
the day, optionally, in unit dosage farms. In certain embodiments, preferred
doting is one
administration per day.
The invention further provides a unit dosage form: (such as a tablet or
capsule) comprising
a compound of Formula I or a specific compound described herein, Or
pharmaceutically acceptable
salts thereof, in a therapeutically effective amount for the treatment of an
immune of inflammatory
disorder, such At one Of the particular immune disorders or
inflammatory.disorders described
herein.
General Synthetic Schemes and Procedures
The compounds of Formula I Maybe prepared by the methods described below,
together
with synthetic methods known in 'the art.of Organic chemistry, or
modifications and derivatizatiOns
that are familiar to those of ordinary skill in the art. The starting
materials used herein are
commercially available Or May be prepared by routine method's known in the art
(Stich as those
methods disclosed in standard referencebooks'such as the COMPENDIUM' OF
ORGANIC
SYNTHETIC METHODS, Vol. 1-VI (Published by Wiley-Interstierice)). Preferred
methods include,
but are not limited to, those. described below..
During any of the following synthetic sequences it may be necessary end/or
desirable to
protect. sensitive or reactive groups on any of the Molecules concerned,. This
can be achieved by
means of conventional protecting groups, such asthose- described in T.
Greene, PrOtectiVa
Groups in Organic Chemistry, John Wiley & Sons,, 1981;,.T: W. Greene and P. a
M. Wuts,,.
Protective. Groups in Organic Chemistry, Jahn Wiley & Sons, 191, and T, W.
Greene and P.. G.. M.
Wilts, Protective Groups in Organic Chemistry., Jahn Wiley & Sons, 1999.
CA 2975157 2018-08-17
CA 02975157 2017-07-26
WO 2016/120850 PCT/IB2016/050477
Compounds of Formula I or their pharmaceutically acceptable salts, can be
prepared
according to the reaction Schemes disOUssed herein below. Unless otherwise
indicated, the
substituents in the Schemes are defined as above. Isolation and purification
of the products is
accomplished by standard procedures, which are known to a chemist of ordinary
skill.
It will be understood by one skilled in the art that the various symbols,
superscripts and
subscripts used in the schemes, methods and examples are used for convenience
of
representation and/or to reflect the order in which they are introduced in the
schemes, and are not
intended to necessarily correspond to the symbols, superscripts or subscripts
in the appended
claims. The schemes are representative of methods useful in synthesizing the
compounds of the
10 present invention. They are not to constrain the scope of the invention
in any way,
Compounds of Formula I may be prepared as single enantiomer or as a mixture of
individual enantiorners which includes racemic mixtures,. Methods to obtain
preferentially a single
enantiomer from a mixture of individual enantiomers or a racemic mixture are
well known to those
ordinarily skilled in the art of organic chemistry. Such methods include but
are not limited to
15, preferential crystallization of diastereomeric salts (e.g. tartrate or
camphor sulfonate), covalent
derivatization by a chiral, non-racernic reagent followed by separation of the
resulting
diastereomers by common methods (e.g. crystallization, chromatographic
separation, or
distillation) and chemical reversion to scalemic compound, Simulated Moving
Bed technology, or
high/medium-pressure liquid chromatography or supercritical fluid
chromatography employing a
20 chiral stationary phase. These techniques may be performed on the final
compounds of the
invention or on any intermediates to compounds of the invention which bear a
stereogenic center.
Also, to facilitate separation by any of the methods described above, the
compounds of the
invention or any intermediates to the compounds of the invention which bear a
stereooenic center
may be transiently reacted with an a.chiral reagent, separated, and then
reverted to scalemic
25 compound by standard synthetic techniques.
Compounds of Formula A-6 can be prepared as described in Scheme A. Aryl
halides A-1
can be converted to boronates of Formula A-2. Boronates A-2 can be coupled
with vinyl triflate B-
3 (prepared as described in Scheme B) to afford compounds of the Formula A-3.
Subsequent
reduction of the nitro group and the olefin concomitantly furnished compounds
of Formula A-4,
30 The resulting amine of compounds of Formula A-4 can be transformed to
the corresponding
sulfonamides by the reaction with sulfonyl chlorides in the presence of base
to afford compounds
of Formula A-5. The Boo group within componds of Formula A-5 could be removed
through use
of acid, and subsequently the piperidine nitrogen could be coupled with an
appropriate acid
chloride or carboxylic acid to furnish compounds of Formula A--6.
CA 02975157 2017-07-26
WO 2016/120850 PCT/I132016/050477
36
SCHEME A
o,_0
i." B-3
0
)
I \II-0 '''''''.. - ....--,.
1 il
0-,N 2N-.............---c? 0-if 0',N AI '
, -....y.,..--_...--- ---------------------------------------- \
R I Ri 'R.1
A-1 A-2 A-3
0 0 0
\---0\_ .---0x
\
/
----'' 0 H
\
H2N X . 1/ ..N 'S`
!i \
N 0 "N 0 fq
A-4 A-5 A-6
Compounds of formula 8-3 can be prepared as exemplified by the synthetic route
described in Scheme B. Hydrogenolysis of B-1 in the presence of Boo anhydride
removed the
benzyl group from the piperidine nitrogen and this was followed by immediate
carbamate formation
in situ to form carbamate 6-2. Next, treatment with a strong base such as
lithium
hexamethyldisilylazide at cryogenic temperatures generated the kinetically
controlled enolate, and
this was trapped by treatment with N-phenyl triflamide to form vinyl triflate
8-3.
SCHEME B
--.0 7- - ,
')r-I ------,
i
0 0 Tf0
B-1 6-2 6-3
Compounds of Formula 0-5 can be prepared as in the route described in Scheme
C.
Iodide C-1 was coupled with Boc-protected piperidine boronate C-6 through the
use of
conventional Suzuki conditions employing palladium tetrakistriphenylphoshine.
Next,
hydrogenation conditions could be used to convert nitroolefin 0-2 to the
corresponding aniline 0..3.
CA 02975157 2017-07-26
WO 2016/120850 PCT/IB2016/050477
37
This aniline could be substituted with various suifonyl chlorides to provide
structures of the formula
C-4, The Boo group within C-4 could be removed through use of acid, and
subsequently the
piperidine nitrogen coupled with the an appropriate acid chloride or
carboxylic acid to furnish
compounds of Formula C-5,
SCHEME C
0 0
-N ct25
(ri)
Spin
N C-6 02N
\
RI µR1 µR1
C-1 C-2 C-3
0 0
)--R2
N
1
H 0 H
X...11
8 0
µR1 kR1
C-5
Compounds of Formula 0-5 can be prepared as exemplified by the synthetic route
described in Scheme D. The boronate A-2 prepared as described in Scheme A was
coupled to
Boo protected E-2 prepared as described in Scheme E through the use of
conventional Suzuki
conditions. Next hydrogenation could be used to convert nitroolefin D-2 to the
corresponding
aniline 0-3. The resulting aniline could be substituted with various sulfonyl
chlorides to provide
compounds of Formula 0-4, The Boc group within 0-4 could be removed through
use of acid,
and subsequently the piperidine nitrogen could be coupled with an appropriate
acid chloride or
carboxylic acid to furnish compounds of Formula 0-5.
CA 02975157 2017-07-26
WO 2016/120850 PCT/IB2016/050477
38
SCHEME D
Oyia0
,, 0
4N
______________________________________ 1 \
y.----- E-2 /
BPin
02Nõ .. ...,.. OTf 02N. H2N
.\ \
- N
, N N
Fi' µR1 'RI
A-2 D-2 D-3
0
'\)----R2
\ /
---t¨N
-----1... ___________________ 0 H ,.. 0 H
0 I IF
0 ...'"'
. N 0
N! ,
'R1 R`
D-4 D-5
Compounds of formula E-2 can be prepared as exemplified by the synthetic route
described in Scheme E. Treatment of oiperidone E-1 with a strong base such as
lithium
hexamethyidisilylazide at cryogenic temperatures generated the kinetically
controlled enolate, and
this was trapped by treatment with N-phenyl triflamide to form vinyl Vitiate E-
2.
SCHEME E
i 0
-.1 c N
-rid
0
E-1 E-2
Compounds of Formula F-5 can be prepared as exemplified by the synthetic route
described in Scheme F. The boronate A-2 prepared as described in Scheme A is
coupled to Bac
protected G-2 through the use of conventional Suzuki conditions. Next
hydrogenation could be
used to convert nitroolefin F-2 to the corresponding aniline F-3, The
resulting aniline could be
substituted with various sulfonyl chlorides to provide compounds of the
Formula F-4. The Boo
group within F-4 could he removed through use of acid, and subsequently the
pioendine nitrogen
could be coupled with an appropriate acid chloride or carboxylic acid to
furnish compounds of
Formula F-5.
84020419
39
SCHEME F
0,y0H
0 0
N
N N
BPin G.;2
OTf
02N , 02N H2N
I \
N
N.
131
F-2: F4
0
N,
N
0 H 0 11
0
F-4 F4
Compounds of formula G-2 can be prepared as exemplified by the synthetic route
described in Scheme G. Treatment of piperidorie G-1 with a Strong base Such as
lithiUm
hexamethyldisilylazlde at cryogenic terriperatures generated the: Kinetically
controlled enolatei and
this Was trapped by treatment With 1V-phenyl triflamide to form vinyl triflate
G-2.
SCHEME G
,;'-C)
"cl N
Tfci
0
G-2
Carboxylic acids of the Formula FeCO21-1 employed in SCheme A, C; D, and F may
be
1p commercially available, prepared by procedures described in the
literature, or prepared as
described in Scheme F. (R),=2,3,34ritriethy1butanoic acid and ($)-2,3,3-
trImethylbutanoic acid may
be prepared as described by Kido, M. et el Tetrahedron; Asym, 2007,.18, 104-
1947; and thietane
acid (see W0201 3/7582, which is referenced for the preparation of thietane
acid). Specific examples of carboxylic acids that may be prepared by Scheme F
include
cyclopentylpropanelO acid, and (S)-2-cyclopenty1propanoic acid. Specific
examples of R2CO2)-1
according to the Formula F-4 can be prepared from acids F-1 where R may be
alkyl, cycloalkyl or
CA 2975157 2018-08-17
CA 02975157 2017-07-26
WO 2016/120850 PCT/IB2016/050477
aryl which are reacted with an optically active chiral oxazolidinone (e.g. (R)-
benzyl oxazolidinone,
(R)-4-lsopropyl-2-oxazolidinone) to provide compounds of the Formula F-2, Base
mediated
alkylation and subsequent removal of the oxazolidinone auxiliary furnishes
acids of the Formula F-
4 in high optical purity By employing a chiral oxazolidinone of a different
absolute configuration
5 (e.g. (S)-benzyl oxazolidinone, (S)-4-lsopropy1-2-oxazclidinone), chiral
acids F-4 of both
configurations can be obtained.
SCHEME F
__________________________________________ oI _______
0 / r
¨
0¨ 0
F-1 F-2 F-3 F-4
EXEMPLIFICATION
10 The invention now being generally described, will be more readily
understood by reference
to the following examples, which are included merely for purposes of
illustration of certain aspects
and embodiments of the present invention, and are not intended to limit the
invention. The
following illustrates the synthesis of various compounds of the present
invention. Additional
compounds within the scope of this invention may be prepared using the methods
illustrated in
15 these Examples, either alone or in combination with techniques generally
known in the art.
Experiments were generally carried out under inert atmosphere (nitrogen or
argon),
particularly in cases where oxygen- or moisture-sensitive reagents or
intermediates were
employed. Commercial solvents and reagents Were generally used without further
purification,
including anhydrous solvents where appropriate. Mass spectrometry data is
reported from either
20 liquid chromatography-mass spectrometry (LCMS), atmospheric pressure
chemical ionization
(AFC!) or gas chromatography-mass spectrometry (GCMS) inStrumentation.
Chemical shifts for
nuclear magnetic resonance (MIR) data are expressed in parts per million (ppm,
5) referenced to
residual peaks from the deuterated solvents employed. Coupling constants (J
values) are reported
in Hertz,
25 Chiral purity of scalemic compounds was determined by chiral SEC (super-
critical fluid
chromatography) employing one of the following conditions: HPLC Method A:
XBridge 018,
2.1x50 mm, 5 urn, CH3CN/H20 (0.0375% TFA), 10-100%, 0,8 riLlmin, 4min; and
HFLC Method
B: XBridge 018, 2.1x50 mm, 5 urn, CH3CN/H20 (0,0375% TFA), 1-100%, 0.8 mUmin,
4min.
Method C: Ultimate XB-018, 3um, 3.0x50rnm, CH3CN/H20 (0.1% TFA), 1,5%, 1,2
mUrnin, 10
30 min. Method 0: Xtimate 018, 3pm, 5,0x50mm, CH3CN/H20 (0.1% TEA), 1-100%,
1.2 mLimin, 10
CA 02975157 2017-07-26
WO 2016/120850 PCT/I132016/050477
41
min, Method E: Ultimate XB-C18, 31.trri, 3.0x50mm, CH3CN/H20 (OA% TFA), 1-
100%, 1.2
ralmin, 10 min.
For syntheses referencing procedures in other Examples, reaction conditions
(length of
reaction and temperature) may vary. In general, reactions were followed by
thin layer
chromatography or mass spectrometry, and subjected to work-up when
appropriate. Purifications
may vary between experiments; in general, solvents and the solvent ratios used
for
etuantsigradients were chosen to provide appropriate Rf s or retention times
(RelT).
The chemical names for the compounds of the invention described below were
Generated
using CambridgeSoft's ChemBioDraw Ultra version 13Ø2 (CambridgeSoft Corp.,
Cambridge
Mass.).
The following abbreviations are used herein: DCM: dichlorotnethane; DEA;
diethylamine,
DIPEA: diisopropylethylamine; DME: 1,2-dimethoxyethane; DMF:
dimethylformamide; Et0Ac: ethyl
acetate; Et0H: ethanol; HATU: 1-[bis(dimethylamino)-methylenel-1H-1,2,3-
triazolo[4,5-
b]pyridinium 3-oxid hexafluorophosphate; MeOH: methanol; MTBE: methyl t-butyl
ether; PE:
petroleum ether; TEA: triethyiamine; and THF: tetrahydrofuran.
Example
Preparation of 4-fluoro-N-(1-methy1-3-0R,4R)-3-methyl-1-((R)-Z3,3-
trimethyibutanoyl)piperldin-4-y1)-1H-indol-5-Abenzenesulfonamide
\/
0
f
0 H
N
F
Step 1: 3-Bromo-1-methyl-5-nitro-1H-indole. To a solution 3-bromo-5-nitro-1H-
indole (5.5
g, 20,6 mmol) in THF (50mL) at 0 C was added NaH (1,9 g, 41.3 mmol, 60% taw
in mineral oil).
The mixture was stirred for 15 min and methyl iodide (6.4 mL, 103.3 mmol) was
added. The
reaction mixture was warmed to room temperature and stirred for 16 h. After
completion, the
reaction mixture was cooled, quenched with addition of ice-cooled water and
extracted using
Et0Ac. The combined organic layer was dried over anhydrous Na2SO4.filtered and
concentrated
to obtain a crude product which was purified by triturating with diethyl ether
to afford the title
compound (5 g, 86%). 'H NMR (400 MHz, CDC13)15 8.55(d, J = 2,4 Hz, 1H), 8,17
(dd, J = 8.8, 2,0
Hz, 1H), 7.36(d, J = 8.8 Hz, 1H), 7.25i (s, 3.:87 (s, 311).
CA 02975157 2017-07-26
WO 2016/120850
PCT/I132016/050477
42
Step 2: 1-Methy1-5-nitro-3-(4,4,5,5-tetramethy1-1,3,2-dioxaborolan-2-y1),1H-
indole. A
solution of 3-brorno-l-methyl-5-nitro-1H-indole (3g. 11.7 mmol) in dioxane
(100 mt.) was
degassed using nitrogen for 10 min. K2CO3 (4.8 g, 35.1 mmol), S-Phos (480 mg,
1,17 mrnol),
4,4,4',4',5,5,5',5'-ootamethy1-2,2'-bi(1,3,2-dioxadorotane) (4.46 g, 17.5
mmol) and Pd2(dba)3 (533
mg, 0,53 mmol) were added, degassed with nitrogen for another 10 min and
heated at 60 CC for
5h. After completion, the reaction mixture was cooled to room temperature,
quenched with the
addition of saturated NH4CI solution and extracted using Et0Ac. The combined
organic layer was
dried over anhydrous Na2SO4, filtered end concentrated to obtain a crude
product which was
purified by flash silica gel column chrematography (5-7% Et0Ac in hexane) to
afford the title
compound (1 g, 28%). NMR (400 MHz, CDCl3) 6 8,93 (d, = 2.0 Hz, 1H), 8.14
(dd, J=9.2, 2,0
Hz, 1H), 7.64(s, 11-1), 7..33(d, J= 9.2 Hz, 1H), 3.86 (s, 3H), 1.38(s. 12H);
LCMS: Rile 303
[M+H],
Step 3: tort-Butyl 3-methyl-4-oxopiperidine-1-carboxylate. To a solution of 1-
benzyl-3-
methylpiperidin-4-one (6 g, 29.5 mmol) in ethanol was added Bac-anhydride (8g,
36.9 mmol),
Pd(OH)2 (2.4 g, 40% wt of ketone) and the reaction mixture was stirred under
hydrogen
atmosphere (100 psi) in autoclave for 6 hat room temperature. After
completion, the reaction
mixture was concentrated in vacuo to obtain a crude residue which was purified
by silica gel
column chromatography (10-15% Et0Ac in hexane) to afford the title compound
(8.8 g, 78%). 1H
NMR (400 MHz, CDCI3) 6 4.20-4,16 (m, 2H), 3,29-3.22 (m, 1H), 2.85 (br.sõ 1H),
2.57-2.38 (m,
3H), 1.49 (s, 9H), 1.04 (d, J = 6.4 Hz, 3H); LCMS: rn/e 235 [M+Nar
Step 4: tert-Butyl 3-methyl-4,(((trifluoromethyl) sulfonyl)oxy)-3,6-
ciihydropyridine-1(2H)-
carboxylate. To a solution of tert-Butyl 3-methyl-4-oxopiperidine-1-
carboxylate (7 g, 32,8mmo1) in
THF (70 niL) was added a solution of NaHMDS (66 mL, 65.7 ITIMOi, 1M in THF)
dropwise at-78
C, stirred for 1.5 hat the same temperature, 1,1,1-trifluoro-N-phenyl-N-
((trifluoromethyl)sulfonyl)methenesulfonamide (23.4 g, 65.7 mmol) was added,
the reaction
mixture was warmed to room temperature and stirred for 16 h. After completion,
the reaction
mixture was quenched with addition of ice-cooled water and extracted using
Et0Ac. The
combined organic layer was washed with 10% citric acid followed by 2N NaOH
solution, dried
over anhydrous Na2SO4; filtered and concentrated to obtain a crude product
which was purified by
silica gel column chromatography (3-5% Et0Ao in hexane) to afford the title
compound (8,8 g,
78%). 1H NMR (400 MHz, C0CI3): 6 5.73-5,72 (m, 1H), 4.12-3.96 (m, 2H), 3.61-
3.40 (rn, 2H),
2.62-2.61 (m, 1H), 1,47 (a, 9H), 1.14 (d, J = 6.8 Hz, 31-).
Step 5: tert-butyl 3-methyl-4(1-methyl-5-nitro-1 H-indol-3-yI)-3,6-
dihydropyridine-1 (2H)-
carboxylate. A solution of 1-methyl-5-nitro-3-(4,4,5,5-tetramethyl-1,3,2-
dioxaborolan-2-y1)-1H-
indole (1 g, 3.32 mmol) in dioxane: water (40 mt.: 5 mL) was degassed using
nitrogen for 10min.,
tert-butyl 5-methy1-4-(trifluoromethylsulfonyloxy)-5,6-dihydropyridine-1(2H)-
carboxylate (1,49,
CA 02975157 2017-07-26
WO 2016/120850 PCT/IB2016/050477
43
4,05 mmoi), K31004 (1.5g, 7.1 minor), letrakis (triphenyl phosphine)
palladium(0) (375 mg, 0.32
mmol) were added, degassed for another 10 min. and the reaction mixture was
heated at 90 C
for 5h. After completion,: the reaction mixture was cooled to room
temperature, diluted with water
and extracted using EtOAe. The combined organic layer was dried over anhydrous
Na2Sa4,
filtered and concentrated to obtain a crude product which was purified by
flash silica gel Column
chromatography (1% MeOH in DCM) to afford a pure title compound (1 g, 82%), 1H
NMR (400
MHz, CDCI3): 6:8.75 (br.s, 1H),, 8.14 (dd, J =8.8 2.0 Hz, 1H), 7.31 (d, J =
8.8 Hz, 1H), 6,01 (br.s,
1H), 7.26-7.25 (in, 1H,), 4.52-4.35 (m, 1H), 4,05-3.84 (m, 5H), 3.30-3.29 (m,
1H), 2,78-2,75 (m,
1H), 1.51 (s, 9H), 1.09 (d, J = 6.8 Hz., 3H); LCMS: mie 435.4 [M+Na+CH3CNr,
Step 6: tert-butyl 4-(5-amino-1-methyl-1H-indo1-3-y1)-3-methylpiperidirie1-
carboxylate. To
a solution of tert-butyl 5-methyl-4-(1-methy1-5-nitro-1H-indo1-3-yi)-5,6-
dihydropyridine-1(21-1)-
c.;arboxylate (1 g, 2.6 mmot) in Et0H (25 mL) were added Pd(OH)2 (1 g, 100%
w/w), ammonium
formate (1.7 g, 26.9 mmol) arki the reaction mixture was heated at 80 C for
4h. After completion,
the reaction mixture was cooled to room temperature, filtered through celite
and washed with
Et0H. The filtrate was concentrated to afford the title compound (900 mg, 97%)
which was used in
the next step without further purifiCation. H NMR (400 MHz, CDCI3): 5 7.09 (d,
J = 8.4 Hz, 1H),
6.92-6,91 (m, 1H), 6.72(d, J = 8.4 Hz, 1H), 6.67(s, 1H), 4.29-3.96(m, 2H),
3.76-3.68 (m, 4H),
3.16-2.88 Om 3H), 2,22-2.19 (m, 1H), 1.98-1.96 (m, 1H), 1.664.63 (m, 2H), 1.47
(s, 9H), 0.69(d, J
6.4 Hz, 3H); LCMS: m/e 344.35 [M+Hr,
Step 7: tert-butyl 4-(5-((4-1luorophenyl)sulfonamido)-1-methyl-1H-indol-3.11)-
3-
methylpiperidine-1-carboxylate. To a solution of ter/-butyl 4-(5-amino-1-
methyl-1H-indel-3-y1)-3-
rnethylpiperidine-1-carboxylate (900 mg, 2.62 mmol) in DOM (25 ml.) was added
pyridine (0.32
rnL, 3.93 mmol), followed by 4-fluorobenzenesulfonyl chloride (613 mg, 3.14
mmol) solution in
DCM (25 rriL) dropwise at 0-5 C and the resulting reaction mixture was
stirred at 0-5 CC for 30
min. After completion, the reaction mixture was basified with 10% aqueous
NaHCO3 solution and
extracted using DCM. The combined organic layer was dried Over anhydrous
Na2SO4, filtered and
concentrated to obtain the crude compound which was purified by silica gel
column
chromatography (1-2% Me0H in DCM) to afford the title compound (1 g, 68%). 'H
NMR (400 MHz,
0D013). 6 7.66 (d, J = 5.6 Hz, 1H), 7.65 (d, J = 5.6 Hz, 1H), 7.20 (br.s, 1H),
7.12 (d, J = 8.4 Hz,
1H), 7,05 (d, J = 8.0 Hz, 1H), 6.84 (d, J = 8.0 Hz, 1H), 6.75 (s, 1H), 6.37
(s, 1H), 4.29-3.95 (m, 2H),
3.71 (s, 3H), 3.12-2.85 (in, 3H), 2.02-1.93(m, 2H), 1.65-i.58 (m. 1H), 1.48(s,
9H), 0.62(d. J = 6.4
Hz, 3H).
Step 8; 4-fluoro-N-(1-methyi-3-((3R,4R)-3-methylpiperidin-4-y1)-1H-indol-5-
Abenzenesulfonamide. Racemic tert-butyl 4-(5-((4-fluorophenyl)sulfonamido)-1-
methyl-1H-indol-
.. 3-y1)-3-methylpiperidine-1-carboxylate (1.11g) was separated by chiral SFC
(Column: LUX
Cellulose-4, 250 x 21.2 mm, 5 pm, 80% 002/20% Me0H + 0.2% NH.', 80 mUmin) to
give two
CA 02975157 2017-07-26
WO 2016/120850 PCT/IB2016/050477
44
major peaks for which absolute sterecchemistry were arbitrarily assigned. The
first eluting isomer
(RI = 8.89 min) was arbitrarily assigned as tert-butyl (35,4S)-4-(6-((4-
fluorophenyi)sulfonarnido)-1-
methyl-1 H-indo1-3-y10-methylpiperidine-1-carboxylate. The Second eluting
isomer (Rt = 9.78 min)
was arbitrarily assigned tart-butyl (3RAR)-4-(5((4-f1uoroohenyt)sulfonamido)-1-
methyl-1H-indoi73.-
y1)4-Methylpiperidine-1-carboxylate. 1FINMR (400 MHz, CDC13): 6 7.66 (d, J =
5.6 Hz, 1H), 7,65
(d, J= 5.6 Hz, 1H), 7.20 (br:s, 1H), 7.12 (d, J= 8,4 Hz, 1H), 7.05 (d, J = 8.0
Hz, 1H), 6.84(d, J=
8.0 Hz, 1H), 6.75 (s, 1H), 6.37 (s, NH), 4.29-3.95 (m, 2K), 3.71 (s, 3H), 3.12-
2.85 (m, 3H), 2.02-
1.93(m, 2H), 1.65-i.58 (m, 1H), 1.48 (s, 9H), 0.62 (d, J = 6.4 Hz, 3H).
Step 9: H-indol-5-
To a solution of (3R,4R)-tert-butyl 4-(5-(4-fluorophenylsulfonarnido)-1-
methyl-1H-indol-3-yI)-3-methylpiperidine-1-carboxylate (273 mg. 0.544 mmol) in
DCM (10,9 mL,
0.05 MM0i) was added 4M HCI in dioxane (2.04 mL, 8.16 mmol) at room
temperature. The
resulting mixture was allowed to stir for 90 min., The solvent was removed in
vacuo to afford the
crude product as a black solid, which was used without further purification
(238 mg, >99%). H
NMR (400 MHz, Me0D) 6 7.66 (dd. 2 H, J=8. 8, 6.3 Hz), 7.35 - 7.07 (m, 4 H),
7.05 (s, 1 H), 6.82
(d, 1 H, J=8.6 Hz), 3.74 (s, 3 H), 3,48 - 3.25 (m, 7 H), 2.45-2.33 (m, 1 H),
2.26-2.11 (m. 1 H), 2.07-
1.96 (rn, 1 H), 0.79 (d, 3 H, J=7,4 Hz).
Step 10: 4-fluoro-N-(1-methy1-3-((3R,4R)-3-methy1-1-((R)-2,3,3-
trimethylbutanoyi)-piperidin-
4-yl)-1H-indol-5-yi)benzenesulfonamide. To a solution of (R)-2,3,3-
trimethylbutahoic acid (152.0
mg, 0.347 mmol) in DMF (3,47 mL) was added successively 4-fluoro-N-(1-methyl-3-
((3R,4R)-3-
methylpiperidin-4-yi)-1H-indol-5-yl)benzenesulfonamide (152 mg, 0.347 mmol),
HATU (160 mg,
0.416 mmol) and DIPEA (453 mg, 3.47 mmol), The resulting yellow solution was
allowed to stir at
room temperature. LC/MS showed that the reaction was complete after 1 h. The
reaction was
quenched with sat. NaHCO3, extracted with Et0Ao (3x100 mL), washed with brine,
dried over
MgSO4, filtered and. The crude product was purified silica gel column
chromatography (0 to 100%
Et0Ac in heptane) to afford the title compound (172 mg, 97%) as a white fluffy
powder consisting
of a mixture or rotamers (approximately 1.5:1.0 ratio). 'H NMR (400 MHz,
CDCI3) 67.65 (dd,
J=8,6, 5.1 Hz, 2H), 7.18- 7.11 (m, 4H), 7.08- 6.94 (m, 2H), 6.92 - 6.77 (m,
1H), 6.77- 6,66 (m,
1H), 6.55- 6.42(m, 1H), 4.65 - 4.53 (m, 1H) 4,21 - 4.09 (m, 0.6H), 3.83-
3.80(m, 0.4 H), 337-3,34
(m, 0.4H), 3.28 - 3.08 (m, 1.6H), 3.00 ¨ 2.70 (m, 1,9H), 2.59 - 2.52 (m, 0,4
H), 2.10 (br. s., 1H),
2.00 1.80(m, 1H), 1.76¨ 1,70 (m, 1H), 1.58(s, 0.9H), 1.36- 1.17 (m, 8.3H),
1.17- 1.04(m,
3,1H), 1.04 - 0.91 (m, 8.6H), 0.66 (d, J=7.0 Hz, 1.3H), 0.58 (d, J=7.0 Hz,
1.7H) ppm.
Example 2
Preparation of N-(34(3R,4R)-14(R)-2-cyclopentylpropanoy1)-3-methylpiperidin-4-
y1)-1-
methyl-I H-indo1-5-y1)-4-fluorobenzenesulfonamide
CA 02975157 2017-07-26
WO 2016/120850 PCT/IB2016/050477
--N
FL
M
--A1111
1
Step 1: (R)-3-(2-cyclopentylacetyl)-4-isopropyloxazolidin-2-one, To a solution
of ('2)-4-
isopropyloxazolidin-2-one (2.0 g, 10 mmol) in THF (55 mL) at -78 ')C was added
drop,vsle n-BuLi
(2.5 M in hexanes, 4.92 mL, 12.3 mmol). The resulting solution was allowed to
stir at. the same
5 temperature for 1 h, then cyclopentyl acetyl chloride (1.86 g, 12.3 mmol)
was added. The reaction
turned pale yellow rapidly and was allowed to stir at -78 00 for 1 h. The
reaction was quenched
with sat. NaHCO3solution, extracted with Et0Ac, dried over Na2SO4, filtered
and concentrated to
afford the title compound (3.20 g, 99%) as a pale yellow oil that solidified
on standing. 1H MIR
(400 MHz, CDCI) 6 7.40-7.28 (m, 311), 7,26-7,16 (m, 2H), 4.74-4.64 (m, 1H),
4.24-4,12 (m, 2H),
10 3,32 (dd, J=13, 3 Hz, 1H), 3.04 (dd, J=17, 7 Hz, 1H), 2.92 (dd, J=17, 7
Hz, 1H), 2.77 (dd, J=14, 10
Hz, 1H), 2.41-2.28 (m, 1H), 1.95-1.84 (rn, 2H), 1.72-1.56 (m, 4H), 1.30-1,15
(m, 2H).
Step 2: (R)-34(R)-2-cyclopentylproparioy1)-4-isopropyloxazolidin-2-one, To a
colorless
solution of (R)-3-(2-cyclopentylacetyI)-4-isopropyloxazolidin-2-one (3250 mg,
11,34 mmol) in THF
(50 mL) at -78 C was added dropwise LDA (2.0 M, 6.50 mL, 13.0 mmol), The
resulting yellow
15 solution was allowed to stir at the same ternperature for 1 h. Mel (3.56
mL, 56,6 mmol) was added
and the reaction was allowed to warm to 0 C over 1 h and allowed to stir at 0
'C for 3 h. The
reaction was quenched with sat. NH.;Cl and extracted with Et0Ac. The combined
organic extracts
were washed with sat. NaHCO3 and brine, dried over magnesium sulfate, filtered
and concentrated
in vacuo to afford a white solid. The crude product was purified by silica gel
column
20 chromatography twice (Et0Ac:Heptane, 5:95-60:40 then 5:95-50:50). The
product was
recrystallized from n-heptane to provide the title compound (680 mg, 20%) as
colorless crystalline
needles. 1FINMR (400 MHz, CDCI3) 7.40-7,17 (m, 5H), 4.69 (ddt, J=10, 7, 3 Hz,
1H), 4.25-4.11
(m, 2H), 3.63 (dg, J=9, 7 Hz, 1H), 3.28 (dd, J=14, 3 Hz, 1H), 2.78 (dd, J=13,
9 Hz, 1H), 2.21-2.08
(m, 1H), 1.90-1,73 (rn, 2H), 1.71-1.48(m, 4H), 1..30-1.17(m, 4H), 1,11 (ddd,
J=12.0, 5.0, 4.0 Hz,
25 1H).
Step 3: (R)-2-cyclopentylpropanoic acid. To a solution of (R)-3-((R)-2-
cyclopentylpropanoy1)-4-isopropyioxazolidin-2-one (680 mg, 2,26 mmol) in
THF/H20 (viv = 1/1, 12
mL) at room temperature was added Li0H- H20 (142 mg, 3.38 mmol) followed by
H202(237 mL,
4.17 mrnol, 50 wt%). The resulting solution was allowed to stir at room
temperature overnight. The
CA 02975157 2017-07-26
WO 2016/120850
PCT/I132016/050477
46
reaction was quenched with 1.0 M KHSO4 (8 mL) and extracted with Et0Ac (3x).
The combined
organic extracts were washed with brine and dried over Na7S0.1, filtered and
concentrated. The
crude product was purified by sca gel column chromatography (Et0Ac:Heptane,
7:93-50:50) to
afford the title compound (285 mg, 89%) as a colonets oil. H NMR (400 MHz,
CDCI3) 6 2.29 (dg,
J=9, 7 Hz, 1H), 2.07-1.95 (m, 1H), 1.87-1.76 (m. 2H), 1.69-1.51 (m, 4H), 1.31-
1,24 (m, 1H),
1.24-1.15 (m, 4H).
Step 4: N-(34(3R,4R)-1-((R)-2-cyclopentylpropanoyl)3-methylpiperldin-4-y1)-1-
methyl-1H-
indo1-5-y1)-4-fluorobenzenesulfonamide. To a flask charged with 4-fluoro-N-(1-
methy1-3-((3R,4R)-3-
rnethylpiperidir)-4-y1)-1H-indol-5-yObenzenesulfonamide (prepared as described
in Step 9 of
Example 1, 33 mg, 0.075 mmol), (R)-2-cyclopentylpropanoic acid (12.9 mg, 0.090
mmol), HATU
(37,4 mg, 0.090 rnmol), DIPEA (98.4 mg, 0.753 rnmol) was added DMF (1 mL). The
mixture was
allowed to stir for 2 h then the reaction was added to ag NaHCO3 and extracted
three times with
Et0Ac. The combined organic layers were concentrated to give the titled
compound (11.6 mg,
90%) as a white solid: LCIVIS rniz [M + H+11: 526.1; (1:1.4 mixture of
rotamers)1H NMR (400 MHz,
CDCI.j) 5 7.68-7.65 (2 H, m), 7.25 (s. 0.66 H), 7,20 (s, 0.44H), 7.11 (d,
J=8.6 Hz, 1 H), 7.07-6.98
(m, 2 H), 6.88 (d, J=8.6 Hz, 0.40 H), 6.81 (d, J=8.6 Hz, 0.60 H), 6.77-6.66
(m, 1.40 H), 6.61 (s,
0.60 H), 4.75 (d, J=13.3 Hz, 0.60 H), 4.50 (d, J=13.3 Hz, 0.40 H) 3,82 (d,
J=13.3 Hz., 0.60 H), 3.70
(s,3 H), 3.34 (d, J=13.3 Hz, 1 H), 3.26-3.11 (m, 1.60 H), 2.96 (m, 0.40 H),
2.77-2.65 (in, 0.60 H),
2.61-2.58 (m, 0.40 H), 2.50-2.38 (m, 0,60 H), 2.29-2.05 (m, 2.10 H), 1.99-1.42
(m, 9.4 H), 1.33-
0.90 (m, 10.6 H), 0.68- 0.52 (m, 3.10 H)
Examples 3-5 and 32-35
The following Examples 3-5 and 32-35 were prepared analogous to Example 1
employing
the appropriate sulforiyi chloride in step 7 and the appropriate carboxylic
acid coupling reagent in
step 10. However, chiral separation was conducted on the final product.
Separation conditions:
251 Method AA: Luxe Cellulose-4, 250x21.2 mm, 5 pm, 40% Et0H/CO2, 80.0
mL/min; Method AB:
ChiralTech IC, 250x21.2 mm, 5 pm, 60-40%(3:1) Et0Ac-Me0H/002, 80.0 mL/min.
Method AC:
ChiralPak AD-3, 50 x 4 mm, 3 pm, Et0H (0.05% DEA)1CO2, 4 mUmin. Method AD:
ChiralPak AS-
H, 150 x 4 mm, 5 pm, Et0H (0.05% DEA)1CO2, 3 mUrnin. Method AE: ChiralPak AD-
3, 50 x 4.6
mm, 3 pm, Et0H (0.05% DEA)/CO2, 4 mLimin. Method AF: ChiralPak AS-H, 150 x 4.6
mm, 5 pm,
Et0H (0.05% DEA)/CO2, 3 rnLimin,
CA 02975157 2017-07-26
WO 2016/120850 PCT/IB2016/050477
47
Example Structure I Name/Characterization
N-(3-((3R,4R)-1-(cyclopentanecarbony1)-3-
Methylpiperidin-4-y1)-1-methyl-1H-indol-511)-
4-fluorobenzenesulfonamide. Separation
method AA (first eluting isomer, 8.58 min);
LCMS: mite 498.60 [M H]+.1H NMR (400
rN MHz, DMSO-d6); 6 9.74(s, 1H), 7.69-7.66
(m,
2H), 7.37-7.32 (m, 2H), 7.24 (d, J= 9,2 Hz,
3 (-1 H
N
1H), 7.11 (s, 1H), 7.06-7,04 (m, 1H), 6.84 (t, J
-
= 6.8 Hz, 1H), 4.45 (d, J = 13.2 Hz, 0.5H),
1 4.28 (d, J= 12.4 Hz, 0.5H), 4.03(d, J --
12.4
stereochemistry arbitrarily assigned Hz, 0.5H), 3.84(d, J = 13.2 Hz, 0.5H),
3.67
(s, 3H), 3.37 (d, J = 10.8 Hz, 0.5H), 3.21-2.75
(m, 4H), 1.97-1.55(m, 11H), 0.49=(d, J = 6.8
Hz, 1.5H), 0.41 (d, J = 6.8 Hz, 1.5H)
N-(34(3S,4S)-1-(cyclopentaneparbony1)-3-
methylpiperidin-4-0)-1-methyl-1H-indo1-5-y1)-
4-fluorobenzenesulfonamide. Separation
method AA (second eluting isomer, 9.31 min);
LCMS: rrrie 498.60 [M+Hr, 1H NMR (400
MHz, DMS0-(16): 6 9.74 (a, 1H), 7.69-7.66 (m,
2H), 7.37-7.32 (m, 2H), 7.24 (id, J = 9.2 Hz,
CI\ 1H), 7.11 (s, 1H), 7.06-7.04(m, 1H),
6.84(t,
r-C) = 6.8 Hz, 1H), 4,45 (d, J = 13.2 Hz,
0.5H),
4.28 (d, J = 12.4 Hz, 0.5H), 4.03 (d, J = 12.4
4 ()%\ Hz, 0.5H), 3.84 (d, J = 13,2 Hz. 0.5H),
3.67
S\6
F \ (s, 3H), =3.37 (d, J= 10.8 Hz, 0.5H),
3.21-
N1 2.75 (rn, 4H), 1.97-1.55 (m, 11H),
049d, J =
.111111'7".
6.8 Hz, 1.5H), 0.41 (d, J = 6,8 Hz, 1.5H);
stereachemistry arbitrary assigned LCMS: m/e 498.60 [M+Hr. 1H NMR (400
MHz, DIVISO-d6): 69,74 (s, 1H), 7.69-7.66 (m,
2H), 7.37-7.32 (iii, 2H), 7.24 (d, J = 9.2 Hz,
1H), 7.11(8, 1H), 7.06-7.04(m, 1H), 6.84 (t, J
6,8 Hz, 1H), 4.45 (d, J = 13.2 Hz, 0,5H),
4.28 (d, J= 12.4 Hz, 0.5H), 4.03 (d, d = 12.4
Hz. 0.5H), 3.84 (d, 13.2 Hz, 0.5H), 3.67
CA 02975157 2017-07-26
WO 2016/120850 PCT/IB2016/050477
48
Example Structure Name/Characterization
(s, 3H), 3.37 (d, J = 10.8 Hz, 05K), 3,21-
2.75 (m, 4H), 1,97-1.55 (rn, 11H), 0.49 (d, J =
6,8 Hz, 1.5H), 0,41 (d, J .= 6.8 Hz, 1.5H);
4-fluoro-N-(34(3R,4R)-1-isobutyry1-3-
metnylpiperidin-4-y1)-1-methyl-1H-indol-5-
yl)benzenesulfonamide, Separation method
AS (first eluting isomer, 9.33 min); =H NMR
¨N
I `') (400 MHz, CDCI3) 5 pprn 7.68 (dd, J8.2,
5,1
111
0 IRµs,.1/& Hz, 1 H), 7.23 (br. $,, 1 H), 7,15 (d, J=8.6 Hz,
H), 7.07 (t, J=8,4 Hz, 1 H), 6.73-6,91 (m,
H), 6.40 (d, J=9.4 Hz, 1 H), 4.44-4.84 (m, 1
H), 3.78-4,10 (m, 1 H), 315-3.46(m, 2 H),
stereochemistry arbitrarily assigned 2.68-3.03 (m, 2 H). 2.06-2.20 (M, 1
H), 1.66-
2.04 (m.2 H), 1.09-1.24 (m, 4 H), 0.55-0.68
(m, 2 H)
N-(3-((3R,4R)-1-((R)-2-cyclopentylpropanoyI)-
3-methylpiperidin-4-y1)-1-methyl-1H-indo1-5-
yI)-1,3,5-trimethyl-1H-pyrazole-4-
sulfonamide. Separation method AC (second
N-N cv,,,S3 eluting isomer, 2.05 min); IH NIMR (400
MHz,
- r-N CD30D) 6 ppm 7,28-7.22 (m, 2H), 6.97
(s,
32 0=S=0 1H), 6.89-6.87 (m, 1H), 4.65-4.53 (m,
1H),
HN 4.04-4.00 (m, 1H), 3.74 (s, 3H), 3.62 (s, 3H),
,
N 3.49-347 (m, 1H), 3.33-3.26 (m, 1H),
3.05-
3.03 (m, 1H), 2.83-2.80 (m, 1H), 2,65-2,63
stereochernistry arbitrarily assigned (m, 1H), 2.24-1.57 (m, 16H), 1.91-
1.11 (m,
5H), 0,68-0,59 (m, 3H); LCMS: mle 540.2 [M
+ H+].
CA 02975157 2017-07-26
WO 2016/120850 PCT/IB2016/050477
49
1
1 Example Structure Name/Characterization
N-(3-((3R4R)-14(R)-2-cyclopentyloropancy1)-
3-methylpiperidjn-4-0)-1-methyk1H-indo1-5-
0)-2-ethoxybenzenesulfonamide. Separation
0
L method AD (second eluting isomer, 4.93
min); 'H NMR (400 MHz, CD-JOD) '6 ppm
33 0=S=0 J 7.65-7.63 (m, 1H), 7.47-7.28 (m, 1H),
7.23-
6.86 (m, 6H), 4.67-4.51 (m, 1H), 4.33-4.28
(m, 3H), 3.65 (s, 3H), 3.33-3.02 (m, 4H),
1 2.78-2,55 (m, 2H), 2.15-1.52 (m, 14H),
1.20-
stereochernistry arbitrary assigned 1.11 (m, 5H), 0.61-0.48 (rn, 3H); LCMS:
552.2 [M H.4],
N-(34(3R,4R)-14(R)-2-cyclopentylpropanoyi)-
3-methylpiperidin-4-y1)-1-methy1-1H-indo1-5-
y1)-2-fluorobenzenesulfonamide, Separation
O method AD (second eluting isomer, 2.36
I j min); 'H NMR (400 MHz, 00300) O PPm
7.66-7.65 (m, 1H), 7.57-7.55 (m, 1H), 7.30-
34 0=S=0
RN 7.26 (m, 2H), 7.19-7.16 (m, 2H), 6.96-
6.90
(m, 2H), 4.67-4.51 (rn, 1H), 4.33-4.28 (m,
-N
3H), 3.65 (s: 3H), 3.33-3.02 (m, 4H), 2,78-
stereochemistryrtitrar4 assigned
2.55 (m, 2H), 2.15-1.52 (m, 10H), 1,20-1.11
a
(m, 5H), 0.63-0.51 (m, 3H); LCMS: m/e 526,1
+ H+I.
CA 02975157 2017-07-26
WO 2016/120850 PCT/IB2016/050477
Example Structure Name/Characterization
N-(3-((3R,4R)-1((R)-2-cyclopentylpropanoy1)-
3-methylpiperidin-4-y1)-1-methyl-1H-indoi-5-
y1)-2-fluoro-5-methylbenzenesuifonamide.
0 Separation method AD (second eluting
isomer, 4.84 min); 1H NMR (400 MHz,
CD30D) 6 ppm 7.66-7.65 (m, 1H), 7.57-7.55
(m, 1H), 7.30-7.26 (m, 2H), 7,19-7.16 (rn,
HN
2H), 6.96-6.90 (m, 2H), 4.67-4.51 (m, 1H),
4,33-4.28 (rn, 3H), 3.65 (s, 3H), 3.33-3.02 (m,
stereochemist arbitrarily assigned 4H), 2.78-2.55 (m, 2H), 2.15-1.52
(m. 13H),
ry
1.20-1.11 (m, 5H), 0.63-0.51 (m, 3H); LCMS:
tn/e 540,1 [M + H+].
Example 6
Preparation of 4-fluoro-N.(3-(1-(isothiazole-5-carbonyl)piperidin-4-y1)-1-
methyl-1H-indo1-5.
yl)benzenesulfonamide
N
11
I \
F N
5
Step 1: tert-butyl 4-(1-methyl-5-nitro-1H-indo1-3-y1)-3,6-dihydropyridine-
1(2H)-carboxylate.
A mixture of 3-iodo-l-methyl-5-nitro-1H-indole (10.0 g, 33.1 mmol), tert-butyl
4-(4,4,5,5-
tetramethy1-1,3,2-dioxaboroian-2-y1)-3,6-dihydropyridine-1(2H)-carboxylate
(13.3 g, 43.0 mmol),
K3PO4 (21.1 g, 99,3 mmoi) in dioxane (200 mt._) and water (20 mt.) was
degassed and purged with
10 nitrogen three times. Pd(PP94 (3.06 g, 2,65 mmol) was then added under a
nitrogen atmosphere.
The mixture was degassed and purged with N2 three times. The mixture was
heated to 60 C and
stirred at this temperature for 14 h. The solvent was removed under reduced
pressure andthe
residue was partitioned with Et0Ac (200 mL) and water (150 mL). The aqueous
layer was
extracted with Et0Ac (150 mi_x2), the organic layers were combined, washed
with brine, dried over
15 anhydrous Na2SO4, and concentrated. The crude product was purified by
silica gel column
chromatography (10-50% Et0Ao in PE) to give the title compound (8.05 g, 68.0
%) as an orange
CA 02975157 2017-07-26
WO 2016/120850
PCT/I132016/050477
51
solid. 'H NMR (400 MHz, CDC13): 6 8.82(s, 1H), 8.17-8.15(m, 1H), 7.37-7.32 (m,
1H), 7,16 (s,
1H), 6.18(s, 1H), 4.17.(s, br, 2H), 3.84(s, 3H), 3.68 (s, br, 2H), 2.5(s. br,
2H), 1.52 (s, 9H);
LCMS: rn/e. 380.0 [M+Na].
Step 2: tert-butyl 4-(5-amino-1-methyl-1H-indo1-3-yi)piperidine-1-carboxylate.
To a dry
hydrogenation bottle was added dry Pd(OHVC (1600 mg) under-an Ar atmosphere,
followed by a
solution of tert4putyl 4-(1-methyl-5-nitro-1H-indo1-311)46-dihydropyridine-
1(2H)-carboxylate (8050
mg, 22.5 mmol) in Et0H (500 mL) and DCM (50 mL). The mixture was degassed and
refilled with
H2 3 times, then the mixture was stirred under a hydrogen atmosphere (50 Psi)
at 50 C for 48
hours. The mixture was filtered through a pad of CeliteD, and the filtrate was
concentrated to give
the crude product (7700 mg, 100%) as a purple solid, which wasn't hydrogenated
completely
according to proton NMR. The material was resubjected as follows: To a dry
hydrogenation bottle
was added dry Pd(OH)2tC (1500 mg) under an Ar atmosphere, followed by a
solution of crude
starting material (7700 mg, 23.5 mmol) in Et0H (400 mL). The mixture was
degassed and refilled
with H2 3 times, then the mixture was stirred under a hydrogen atmosphere (50
Psi) at 50 C for 48
hours, H-NMR showed the starting material was consumed completely, and the
desired product
was formed. The mixture was filtered through a pad of Celite0 and the filtrate
was concentrated to
give the crude product. The crude product was combined with another batch for
purification by
silica gel column chromatography (0-33% Et0Ac inDCM) to give the title
compound (3.79 g,
34.6%) as a purple solid. 'H NMR (400 MHz, CDC13) T. ppm 7.03 - 7.15 (m, 1 H)
6,86 - 6.98 (m, 1
H) 6.54 - 6.80 (il, 2 H) 4.06 - 4.33 (m, 2 H) 3.68 (s, 3 H) 2.87 (t, J=11.54
Hz, 3 H) 1.99 (d, J=12.05
Hz, 2 H) 1.63 (d, J=13.05 Hz, 3 H) 1.43- 1.53 (m, 10 H); LCMS: mie 351.9
[M+Nar.
Step 3: tert-butyl 4-(54(4-fluorophenyl)sulfonamido)-1-methyl-1H-indol-3-
yl)piperidine-1-
carboxylate, To a round bottom flask was added tert-butyl 4-(5-amino-1-methyl-
1H-indol-3-
yl)piperidine-1-carboxylate (2200 mg, 6.678 mmol), 4-fluorobenzensulfanyl
chloride (1950 rng, 10.0
mmol) and pyridine (30 mL). The solution was stirred at 15 C for 13 hours.
The reaction was
combined with two other batches for workup. The solvent was removed under
reduced pressure
andthe residue was partitioned with DCM (80 mL) and water (80 mL) The aqueous
layer was
extracted with DCM (40 mLx2). The combined organic layers were washed with
aqueous citric acid
and concentrated. The crude product was purified on by silica gel column
chromatography (10-
60% Et0Ac in PE) to give the tide compound (320 g, 55.6%) as a yellow solid,
'H NMR (400
MHz, 0D0Ã3): 5 7.70-7.66 (m, m2H), 7.27-7,26 (m, 1H), 7.15-7.05 (m, 3H), 6,84-
6,81 (m, 2H), 69
(s, 1H), 4.20(s, br, 2H).3.72 (s, 3H), 2.88-2.82(m, 3H), 1.92-1.89 (rn, 2H),
1.58-1,51 (m, 2H), 1.50
(s, 9H) ppm. LCMS: nye 510,0 [M+Na].
Step 4: 4-fluoro-N-(1-methyl-3-(piperidin-4-y1)-1H-indol-5-
yl)benzenesulfonamide. To a
solution of tert-butyl 4-(54(4-fluorophenAsulfonamide)-1-methyl-1H-indol-3-
y1)piperidine-1-
carboxylate (1200 mg, 2.461 mmol) in DCM (15 mL) Was added HCl/dioxane (7 mL,
4 M) at 0-5 C
CA 02975157 2017-07-26
WO 2016/120850 PCT/I132016/050477
52
in an lee/water bath, The brown solution was stirred at 20 C for 2 hours. The
reaction was
combined with three other batches for workup. The solvent was removed under
reduced pressure
to give the crude product as a gum, which was then dissoled with Me0H and DCM.
The resulting
sclutionwas concentrated to afford the title compound (3679, 100%) as a brown
solid. 1H NMR
(400 MHz, methanol-d4 6 7.70-7,67(m, 2H), 7,36 (s, 1H), 7.22-7.15 (m, 3H),
7.06 (s, 1H), 6.80-
6.78 (m, 1H), 3.77-3.68 (m, 4H), 3.52-3.49(m, 2H), 3.37-3.12 (m, 4H), 2.22-
2.18(m, 2H), 1.97-
1.86 (rn, 2H) porn. LCMS: mie 388.0 [M+H]t,
Step 5: 4-fluoro-N-(3-(1-(isothiazole-5-carbonyl)piperidin-4-y1)-1-methyl-1H-
indol-5-
yl)benzenesulfonamide. The title compound was prepared in an analogous manner
to Example 1,
step 10 employing isothiazoie-5-carboxylic acid . LC/MS [M+H]: 499: Rt = 2.93
(Method A).
Examples 7-18
The following Examples 7-18 were prepared in an analogous manner to Example 6
employing the necessary carboxylic acid in step 5.
7 -------------------------------------
I Example Structure Name/Characterization
1
(R)-N-(3-(1-(2-cyclopentylpropanoyi)piperidin-4-y1)-
1-methyl-1H-indol-5-y1)-4-
fluorobenzenesulfonamide; 1H NMR (400 MHz,
co- -0 DivISO-d6): 6 9.77 (s, 1H), 7,70-7,67 (m, 2H), 7.37-
<._
N 7.32 (m, 21-1), 7.23 (d, J=8.8 Hz, 1H),
7.12(d,
7 0
F-r---)--0
. \ / - / J=10.0 Hz, 1H),
J=12.4 Hz, 1H), 4.08 (d, J=12,4 Hz,
1 \ N 1H), 365(s 3H), 3.17 (t, J=12,8 Hz,
1H), 289(t
\ J=12.8 Hz, 1H), 2.67-2.65 (m, 2H), 1.99-
1,0 (m,
1611); LC/MS {M Hr :512
r
,N
t N-(3-(1-(cyclopropanecarbonyl)piperidin-
4-y1)-1-
8 methyl-1H-inclo1-5-y1)-4-
fluorobenzenesulfonamide.
\
ii-Th-----% LC/MS [M+H]: 456; Rt = 3.11 (method B)
N
F-----.5) \
CA 02975157 2017-07-26
WO 2016/120850 PCT/IB2016/050477
53
, -
Example Structure Name/Characterization
F
\...../c-F
Cy p 4-fluoro-N-(1-meth0-3-(1-(4,4,4-trifluoro-3-
9 EN methylbutanoyi)piperidin-4-0)-1H-indd-5-
o, tyl 0)benzehesulfonamide. LC/MS [M+H] 526; Rt
=3.27 (method A) b .
F 1
(R)-4-fluoro-N-(1-meth0-3-(1-(2,3,3-
trirnethylbutahoyl)piperidin-4-y0-1H-indol-5-
0)benzenesulfonamide, 1H NMR. (400 MHz,
o DMSO-d5) 6 ppm 9.76 (br. s., 1 H), 7.61 - 7.75 (rn,
1....t.
52 H), 7.36 (t, J=8.3 Hz, 2 H), 725 (d, J=9.0 Hz, 1
...1,..
o, A
*H), 7.02- 7.17 (m, 2 H), 6.82 (d, j=8,53 Hz, 1 H),
=''sµb 4.60 (br. s., 1 H), 4.21 (d, J=12.05 Hz, 1 H), 3.66
F (6, 3 H), 3.07 - 3.25 (m, 1 H), 2.54- 2.95
(m, 3 H),
1,73 -1.95 (ni, 2 H), 1.21 - 1.52 (m, 2 H), 0,84 -
1,08 (m, 12 H): LCMS: mie 522 IM+Nar .
N-(3-(1-(cyclobutanecarbonApiperidin-4-0)-1-
meth0-1H-indol-5-y1)-4-fluorobenzehesulforiamide.
Li-i...... ,
-o H NMR (400 MHz, DMSO-d6): 6 9.80 (s, 1H),
7.68-
11 N
\., 7.63 (m, 2H), 7.39-7.36 (m, 2H), 7.22-6.80
(m, 4H),
0 4.47-4.41 (m, 1H), 3.76-3.65 (m, 2H), 3.63
(s, 3H),
r=I \
3.10-3.03(m, 1H), 2.92-2.81 (m, 1H), 2.73-2,61 (m,
F - \ 1H), 2.21.-2.'04('m, 4H), 2.00-1.67(m, 4H),
1.40-
1.30 (m, 2H). LCMS: n-Ve 470.15 [M+Hr
CA 02975157 2017-07-26
WO 2016/120850 PCT/I132016/050477
54
, _____________________________________
f
Example Structure 1 Name/Characterization
4-Fluoro-N-(1-methy1-3-(1-(3-
methylbutandyl)pideridin-4-y1)-1H-indol-5-
y1)benzenesulfonamide. 1I-I NMR (400 MHz,
,)----
0 CDCI3): 6 7.69-7.66 (m, 2H), 7.28-7.26 (m,
1H),
N
,,-- 7,12 (d, J = 8.4 Hz, 1H), 7.08-7.04 (m, 2H),
6.83-
12 6.80 (m, 2H), 6.47 (bs, 1H), 4.76 (d, J = 13.2 Hz,
%.,...
1H), 3.96 (d, J = 13.2 Hz, 1H), 3.71 (5, 3H), 3.17 (t,
J = 12.4 Hz, 1H), 2.96-2,92 (m, 1H), 2.69 (t, J =
12.4 Hz, 1H), 2.26(t, J = 6.8 Hz, 2H), 2.18-1.94 (m,
3H), 1.58-1.48m, 2H), 0.99(t. J = 6.8 Hz, 6H).
LCMS: mle 472,20 [M+11)+
_______ -
N-(3-(1-(2-Cyclopropylacetyl)piperidin-4-y1)-1-
methyl-lH-indol-5-y1)-4-fluorobenzenesul1onamide.
1H NMR (400 MHz, CDCI3): 6 7.69-7.66 (m, 2H),
v> 7.29 (d, J = 1,6 Hz, 1H), 7.12(d, J = 8.8
Hz, 1H),
oy 7.08-7.04 (m, 2H), 6.83(d, J = 1.6 Hz, 1H),
6.81
) N (135,1H), 6.45 (bs, 1H), 4.76 (d, J = 13.2
Hz, 1H),
13
r) 11= 3.91 (d, J = 13.2 Hz, 1H), 3.71 (5, 3H),
3.19 (1, J =
ipCb 1,1\ 12,0 Hz, 1H), 2.98-2.93 (m, 1H), 2.71 (t, J
= 12.8
F X Hz, 1H), 2.32 (d, J = 6.8 Hz, 2H), 2.04-1.95
(m,
2H), 1.64-1.47 (m, 2H), 1.29-1.07 (m, 1H), 0,58 (d,
J = 7.6 Hz, 2H), 0.20 (d, J = 4.8 Hz, 2H). LCMS:
mie 470.20 [M+1-1]4
N-(3-(1-(cyclohexanecarbony1)-piperidin-4-y1)-1-
methyl-1H-indo1-5-yI)-4-fluorobenzenesulfonamide,
n 1H NMR (400 MHz, DMSO-d6): 6 9.77 (5, 1H), 7.70-
7.67 (m, 2H), 7,34 (t, J=8.8 Hz, 2H), 7.23 (d, J=8.8
=l
N Hz, 1H), 7.12 (d, J=2.0 Hz, 1H), 7.08(s: 1H), 6,80
14 F
H =
(dd, J=8.4, 2.0 Hz, 1H), 4.46(.d., J=13.2 Hz, 1H),
4.05(d, J=13.2 Hz, 1H), 365.(, 3H), 3.12-3.15
X (m, 1H), 2.87-2,86 (m, 1H), 2.64-2.60 (m,
2H),
1.85-1.65 (m, 8H), 1.43-1.10 (m, 6H)., LCMS: mie 1
498.55 [M I-1]' 1
, -
CA 02975157 2017-07-26
WO 2016/120850 PCT/I132016/050477
_______ , .............................................................. -
Example Structure Name/Characterization
_________________________________________________________________________ '
4-flucro-N-(3-(1-(2-fluoro-6-
methy1benzoy0piperidin-4--y1)-1-methy1-1 H-Indo1-5-
* F yObenzenesulfonamide. TH NMR (400 MHz,
r...c) DMS0-46): 6 9.77 (s, 1H), 7,70-7.65 (m, 2H), 7,36-
N 7,30 (m, 3H), 7.23 (d, J=8.8 Hz, 1H), 7.17-
7.10 (rTh
15 F.....µ,...õ,_
H 4H), 6.80 (dd, J=8.4, 1.2 Hz, 1H), 4.67 (d,
J=13.4
;
Hz, 1H), 3.66(s, 1.5H), 3.65(s, 1.5H), 3.32-3.15
0' \O N
(m, 2H), 2,95-2.92 (m, 2H), 2.29 (s, 1.5H), 2.22 (Sr
\
1,51-), 1.94 (d, J=13,8 Hz, 11-), 1.78 (d, J=13,8 Hz,
1H), 1.52-1.38(m, 2H); LCMS: mie 524.55 [m+Hy
(S)-N-(3-(1-(2,3-dimethylbutandyppiperidin-4-y1)-1-
methyl-1H-indoi-5-y1)-441uorobenzenesulfonamide.
'H NMR (400 MHz, DMSO-d): 6 9.71 (s, 1H), 7.69-
0
-,,... 7.66 (m, 2H), 7.37-7.33 (m, 2H), 7,23 (d,
J=8.8 Hz,
N
1H), 7.12 (d, J=6.0 Hz, 1H), 7.08 (s, 1H), 6.79 (d,
16 F 1E6
H J=8.4 Hz, 1H), 4.56-4.54 (m, 1H), 4.07 (d,
J=13.2
111P õN
s ----- \
6"6 I. N Hz, 1H), 3.65 (5, 3H), 3.16 (t, J=13.2 Hz, 1H), 2.89
(d, J=13,2 Hz, 1H), 2.67-2.57 (m, 21-.1), 1.87-1.76
(m, 3H), 1.43-1,37 (m, 2H), 0.99-0.84 (m, 9H);
LCMS: mie 486,45 [M+1-11'
I
1
' N-(3-(1-(cyclopentanecarbonyl)piperidin-4-y1)-1-
methyl-1H-indo-5-y0-2-fluorobenzenesulfonamde
1H NMR (400 MHZ, CDC13): 6 7,70 (t, J = 7.6 Hz,
0 '
,.¨N1 1H), 7.55-7.50(m, 1H), 7.5(s, 1H), 7.25-7.11
(m,
17
. H µµ)--i 3H), 6.93 (t, J = 7.6 Hz, 1H), 6.78 (s, 1H), 6.70 (s,
1H), 4.75 (d, J=13,6 Hz, 1H), 4.07 (d, J = 13.6 Hz,
F
,N,,--.
...S --"- "-----S
1H), 3.68(s, 3H), 3.19 (t, J=13.6 Hz, 1H), 2.98-2.94
0 ..--.,.....,.....õ--.1,1
(m, 2H), 2.71 (d, J=12.8 Hz, 1H), 205-1.47(m,
\
12H). LCMS: mie 484.1 [M+H] +
,
L 1 ___________________
CA 02975157 2017-07-26
WO 2016/120850
PCT/I132016/050477
56
Example 18
(R)-4-f1uoro-N-(3-(1-isobutyry1-2,2-dimethylpiperidin-4,11)-1-methyl.11-1-
indol-5-
yl)benzenesulfonamide
,r1
I 0 N
F
Step 1: tert-butyl 2,2-dimethyl-4-(((trifluoromethyl)sulfonyl)oxy)-3,6-
dihydropyridine-1(2H)-
carboxylate. To a RB flask charged with tert-butyl 2,2-dimethy1-4-
oxopiperidine-1-carboxylate (7.76
g, 34,1 mmol) in dry THF (130 mi.) was added slowly dropwise at -70 ''C a
solution of sodium
bistrimethyldisilazide (7.51 g 41.0 mmol) in THF (41 mL). Upon complete
addition, a solution of N-
phenyitriflamide (16.1 g, 41.0 mmol) in THF (10 mL) was added and the mixture
was allowed to
warm to room temperature over the course of 12 h. The reaction mixture was
then concentrated
and brought up in Et0Ac:heptane (85:16,100 mL) and washed twice with water.
The combined
organics were collected and concentrated to give 12.0 g (98%) of the titled
compound as a yellow
o. 1HNMR (400 MHz, CDCI3) 6 5.78 (t. J=3.5 Hz. 1 H), 4.08 (q, J=2.7 Hz, 2 H),
2.40 (br s. 2 H),
1.37-1.58 (m, 14H).
Step 2: tert-butyl 2,2-dimethyl-4-(1-methyl-5-nitro-1H-indo1-3-0)4,6-
dihydropyridine-1(2H)-
carboxylate. A mixture of 1-methyl-5-nitro-3-(4,4,5,5-tetramethyl-1,3,2-
dioxaborelan-2-y1)-1H-
indole (prepared as described in Example 1)(1.26 g, 4.137 mmol), tert-butyl
2,2-dimethy1-4-
(((trifluorometnyt)sulfonyl)oxy)-3,6-dihydropyridine-1(2H)-carboxylate (1.63
g, 4.55 mmol) and
K2PO4 (1.76 g, 8.27 mmol) in dioxane/H20 (40 mt_14 mL) was purged and degassed
with N2 three
times. Pd(PPh3)4(478 mg, 0.414 mmol) under nitrogen atmosphere was then added.
The mixture
was purged and degassed with N2 for 5 mins and then heated to 60 C overnight.
The reaction
mixture was concentrated and the residue was purified by silica gel column
chromatography (0-
66% Et0Ac in PE =0-66%) to provide the title compound (1.45 g, 91,2%) as a
yellow solid. 'H
NMR (400 MHz, CDC13) 6 ppm 8.83 (d, J=2.0 Hz, 1 H), 8.17 (dd, J=9.0, 2.0 Hz, 1
H), 7.34 (d,
J=9,0 Hz, 1 H), 7.17 (s, 1 H), 6,35 (t, 1 H), 4.15 (d, J=4.0 Hz, 2 H),
3.85.(s, 3 H), 2.53(s, 2 H), 1.43
- 1,56 (m, 9 H), 1.24 (s, 6 H).
Step 3: tert-butyl 4-(5-amino-1-methyl-1H-indo1-3-y1)-2,2-dimethylpiperidine-1-
carboxylate.
To a dry hydrogenation bottle was added Pd/C (200 mg) under Ar atmosphere,
followed by a
solution of tert-butyl 2,2-dimethy1-4-(1-methyl-5-nitro-1H-indo1-3-y1)-3,6-
dihydropyridine-1 (2H)-
carboxylate (1000 mg, 2.594 mmol) in Et0H (75 mL) .and 13a/1(15 mL). The
mixture was
CA 02975157 2017-07-26
WO 2016/120850
PCT/I132016/050477
57
degassed and purged With H2 for 3 times and the mixture was stirred under
hydrogen atmosphere
(50 Psi) at 40 for 3 hours. The reaction solution was filtered through a
pad of Celite and the
cake was washed with Me0H three times. The combined filtrate was concentrated
to give the, title
compound (900 mg, 97%) as a lavender oil, which.was used directly Without
further purification_
LCMS: trIe 380 [M+Nar.
Step 4: tert-butyl 47(5-((4-fluorophenyl)sulfonamido)-kmethyl-1H-indol-3-y0-22-
dimethylpipericline-1-carboXylate. To a solution of tert-butyl 4-(5-arhino-1-
methy(-1H.-indol-3-y1)-
2,2-dimethylpiberidine-1-carboxylate (900 mg, 2.52 mmol) and 4-
fluorobenzenesullonyl chloride
(735 mg, 3.78 mmol) in DCM (20 mL) was added TEA (764 mg, 7,55 mmol) and the
mixture was
.stirred at 15 C for 2 hours. The reaction mixture was concentrated and the
residue was purified by
silica gel column chromatography (10-25% Et0Ac in PE) to give the title
compound (1100 mg,
84.7 %) as a broWn solid. LCMS: mie 638 [M+Nar.
Step 5: N-(3-(2,2-dimethylpiperidin-4-y1)-1-methyl-1H-indol-5-yl)-4-
fiuoroben.zenesuifonamide, To a solution of tert-butyl 4-(5-((4-
fluorophenyl)sulfonamido)-1-methyl-
1H-indo1-3-y1)-2,2-dirnethylpiperidin?-1-carboxylate (900 ma, 175 mmol) in DCM
(25 mL) was
added HCliDioxane (15 mL) and the resulting mixture was stirred tor 6 hours at
15 C. The solvent
was removed under reduced pressure to give the title compound (800 mg, 100%)
as a white solid,
which was used directly without further purification. LCMS: nile 416 [M4-1-
4]".
Step 6: 4-fluoro-N-(3-(14sobutyryl-2,2-dimethylpiperidin-4-y1)-1-methyl-lH-
indol-5-
yi)benzenesulfonamide. To a suspension of N-(3-(2,2-dimethylpiperidin-4-y1)-1-
methyl-1H-indol-5-
0)-4-fluarobenzenesulfonamide (400 mgõ 0.885 mmol) in DCrvl (25 mL) was added
TEA (269 mg,
.2,65 mmol) at 0 C, A solution of isobutyryl chloride (141 mg, 1,33 mmol) in
DCM (5 ,mL) was
added dropwise at 0 C and stirred for 6 hours at this temperature, Additional
TEA (134 mg, 1.32
mmol) and isobutyryl chloride (94 mg, 0.885 mmol) was added. The resulting
mixture Was stirred at
.25 15 CC overnight, The reaction mixture was quenched with water,
extracted with DCM (20mLx2).
The combined organic layers were washed with brine, dried (Na2SO4), and the
solvent was
removed under reduced pressure. The crude product was purified by prep-HPLC
(Column: "(WC-
Actus Triart, C18 150 x.30 mm, 5 pm; 52% MeCt\i/H20 to 72% MeCN./H20, 0.225%
formic acid) to
give the title compound (207 mg, 40.2 %.) as a white solid. 'H MAR (400 MHz,
DMSO-O2) 5 9.77
(Or. s., 1 H), 7.69 (dd, J=8.8, 5.3 Hz, 2 H), 7.36 (t, J=8, 8 Hz, 2 H), 7.28
(d, J=8.5 Hz, 1 H), 7.00 -
7.16 (m, 2 H), 6.88 (d, J=8,5 Hz, 1 H), 3,67 (s, 4 H), 2.78- 3.05 (m, 2 H),
2.05-1.95 (m, 1 H), 1.48
- 1.74 (m., 3 H),1 (d, J=12.6 Hz, 6 H), 0.84- 1.06 (m, 6 H); LCMS: inie
508.3 [M+Nar.
Step 6: (R)-4-fluaro-N-(3-(1-isob.utyryl-2,2-dirnethylpiperidin-4-y1)-1-methyt-
lH-indol-5-
y1)benzene.sulfonamide. The racemio 4-fluoro-N-(3-(1-isobutyry1-2,2-
dirnethylpiperidin-4-yl)-1.-
methy1-1H-in.dol-5-y1)benzenesulfonamide (190 mg) was resolved by prep-SFC
(Column: 0j,
250x30 mm,. 5 pm; 25% Et0H/N.H3iH20; 60 mL/min) to give two pea.ks.for which
absolte
CA 02975157 2017-07-26
WO 2016/120850 PCT/IB2016/050477
58
stereochemistry has been arbitrarily assigned. The first eluting isomer (7.33
min) was arbitrarily
assigned as (S)-4-fluoro-N-(3-(1-isobutyry1-2,2-dimethylpiperidin-4-y1)-1-
methyl-1H-indo1-5-
y1)benzenesulfonarnide (65 mg) as a white solid. 'H NMR (400 MHz, DMSO-d) 6
9.77 (br. s., 1 H),
7.69 (dd, J=8, 8, 5,3 Hz, 2 H), 7.36(t,
8 Hz, 2 H), 7.28(d, Hz, 1 H), 7.00 - 7.16(m, 2 H),
6 6.88 (d, J=8. Hz, 1 H), 3,67 (s, 4 H), 2.78 - 3.05 (m, 2 H), 2.05-1.95(m,
1 H), 1.48- 1J4 (m, 3 H),
1.44(d, J=12,6 Hz, 6K), 0.84- 1.06(m, 6 H); LCMS: aye 508 INI+Nar. The second
eluting isomer
(7.57 min) was arbitrarily assigned as (R)-4-fluoro-N-(3(1-iscbutyryl-2,2-
dirrethylpiperidin-4-y1)-1-
methyl-1H-indoi-5-yl)benzenesulfonamide (90 mg) as a white solid, 1H NMR (400
MHz, DMSO-d)
6 pprn 9.77 (br. S., 1 H), 7.69 (dd, J=8.8, 6.3 Hz, 2 H), 7.36 (t, J=8.8 Hz, 2
H), 7.28 (d, J=8.5 Hz, 1
H), 7.O0-7.16(m, 2 H), 6.88(d, J=8.5 Hz, 1 H), 3,67 (s, 4H), 2.78-3.05 (m, 2
H), 2.05-1.95 (rn, 1
H), 1.48-1.74(m, 3 H), 1.44 (d, J=12.6 Hz, 6 H), 0.84-1.06 (in, 6 H); LCMS:
mie 508 [M+Nar.
Example 19
(R)41-(3-(1-(cyclopentanecarbony1)-2,2-dimethylpiperidin-4-y1)-1-methyl-1H-
indol-5-y1)-4-
fluorobenzenesulfonamide
oQ
\rõ11
0
Step 1: N-(3-(1-(cyclopentanecarbony1)-2,2-dimethylpiperidin-4-y1)-1-methyl-1H-
indo1-5-yl)-
4-fluorobenzenesulfonamide. To a suspension of N-(3-(2,2-dimethylpiperidin-4-
y1)-1-methyl-1H-
indo1-5-y1)-4-fluorobenzenesulfonamide (prepared as described in Example 18)
(400 mg, 0.885
mmol) in DCM (25 mL) was added TEA (269 mg, 2_65 mmol) at 0 C and the mixture
was stirred
untill the solution turned clear. A solution of cyclopentarecarbonyi chloride
(176 mg, 1.33 mmol) in
DCM (5 mL) was added dropwise at 0 C, and the mixture was stirred for 3 hours
at this
temperature. The reaction mixture was quenched with water and extracted with
DCM (20 mLx2).
The combined organic layers were washed with brine, dried (Na2S0.1) and the
solvent was
removed under reduced pressure. The crude product was purified by prep-HPLC
(Column:
Phenomenex Gemini 018, 250x21.2 mm, 8 pm; 51% MeCN/H20 to 71% MeCNIH20 w/ NH3
(pH
10)) to give the title compound (210 mg, 46.4 %) as a white solid. 1H NMR (400
MHz, DMSO-d)
ppm 9.78 (br. a., 1 H), 7.69 (dd, J=8.5, 5.5 Hz, 2 H), 7.36 (d, J=17.6 Hz, 2
H), 7.28 (d, J=9.0 Hz, 1
H), 7.11 (s, 2 H), 6,87 (d, J=8,5 Hz, 1 H), 3.62 - 3.75 (rn, 3 H), 2.89 3.07
(m, 2 H), 1.92 - 2,08 (m,
1 H), 1.48- 1.84 (m, 12 H), 1.44 (d, J=16.1 Hz, 6 H); LCMS: rnie 534.1 [M+Nar,
CA 02975157 2017-07-26
WO 2016/120850
PCT/I132016/050477
59
Step 2: (R)-N-(3-(1-(cyclopentanecarbony1)-2,2-dimethylpiperidin-4-y1)-1-
methyl-1H-indo1-5-
y1)-4-fluorbbenzenesulfonarnide. The racemic N-(3-(1-(cyclopentanecarbonyi)-
2,2-
dimethylpiperidin-4-y1)-1-methyl-1H-indo1-5-y1)-4-fluorobenzenesulfonamide
(200 mg) was resolved
by prep-SFC (0J (250x30 mm, 5 pm; 28% Me0H, NH3/H20, 60 mL/min) to give two
peeks for
Which the absolute stereochemistry has been aribitrarily assigned. The first
eluting isomer (4.99
min) was arbitrarily assigned as (S)-14-(3-(1-(byclopentanecarbonyl)2,2-
dimethylpiperidin-4-y1)-1-
methyl-1H-indol-5-y1)-4-fluorobenzenesulfonamide (92 mg, 46 %) es a white
solid. 1H NMR (400
MHz, DMSO-de) 6 ppm 9,78 (br. s., 1 H), 7.69 (dd, J=3.53, 5.52 Hz, 2 H), 7.36
(d, J=17.6 Hz, 2 H),
7.28 (d, J=9.0 Hz, 1 H), 7.11 (s, 2 H), 6.87(d, J=8.5 Hz, 1 H), 3.62 - 3,75
(m, 3 H), 2.89 - 3.07 (m,
2 H), 1.92- 2.08 (m, 1 H), 1.48- 1.84(, 12 H), 1.44 (d, J=16.1 Hz, 6 H); LC/MS
(P,/l+Na).= 5341;
LCMS: mite 534,1 [M+Na]'. The second eluting isomer (5.34 min) was arbitrarily
assigned as (R)-
N-(3-(1-(cyclopentanecarbonyi)-2,2-dimethylpiperidin-411)-1-methyl-1H-indol-5-
y1)-4-
fluorobenzenesulfonamide (95 mg, 48 %) as a white solid, 1H NMR (400 MHz, DMSO-
d6) 5 9.78
(Or. s., 1 H), 7.69 (dd, J=8.5, 5.5 Hz, 2 H), 7.36 (d, J=17.6 Hz, 2 H), 7.28
(d, J=9.0 Hz, 1 H), 7.11
(s, 2 H), 6.87 (d, J=8.5 Hz, 1 H), 3.62 - 3.75 (m, 3 H), 2.89- 3.07 (m, 2 H),
1.92- 2.08 (m, 1 H),
1.48- 1.84(m, 12 H), 1.44(, J=16.1 Hz, 6 H); LC/MS (M+Na)* = 534.1.
Example 20
Preparation of N-(3-((2S,45)-1-(cyclopentanecarbony1)-2-rnethylpipericlin-4-
yl)-1-methyHH-
indo1-5-y1)-4-fluorobenzenesulfonamide
0
9\
S.:
\)
0
F '1111-4P 1
Steps 1: (S)-1-(cyclopentanecarbonyI)-2-methylpiperidin-4-one. A flask with a
mixture of
benzyl (S)-2-methy1-4-oxopiperidine-1-carboxylate (500 mg, 2.02 mmo1) and Pd/C
(5% by weight,
215 mg) in Et0H (10 mL) was evacuated with Water aspiration and then put under
hydrogen gas.
The mixture was stirred under 1.1 bar hydrogen over-pressure for 1h, The
mixture was filtered
through a Celite plug, and the filtrate was concentrated in vacuo. To the
residue was added Dal
(10 ml) followed by triethylamine (1.41 mL, 10,1 Time and then
dyclopentanecarbonyi chloride
(492 pL, 4.04 mmol) dropwlse. The reaction mixture was stirred at room
temperature for lh,
whereupon the reaction was quenched with 40 ml water followed by extraction in
a phase
separator (4 x DCM). The combined organic extracts were evaporated and the
residue was
.. purified by flash chromatography (Et0Aciheptane; 1:1) to provide the title
compound (301 mg,
CA 02975157 2017-07-26
WO 2016/120850
PCT/I132016/050477
71%) as a cdtoriess oil. 1H NMR (500 MHz, CDC13)::o 5.24-484(2 brs, 1H), 4.64-
4.10 (2 br s, 1H),
3.55-3:11 (2 br s, 1H), 2.95(m, 1H),:2.65 (dd, J=6,7, 14,4Hz, 1H), 2502.30(m,
3H), 1.931.77(m,
6H), 1,64-1.56 (m, 2H), 1,29-1,16 (m, 3H)
Step 2: (8)-1-(cyclopentanecarbony1)-2-methyl-1,2,3,6-tetrahydropyridin-4-y1
5 trifluoromethariesulfonate. n-BuLi (2.5M solution in hexanes, 1.15 ml.,,
2.87 mmol) was added drop
wise to diisopropylamine (402 uL, 2.87 mmol) in dry THF (5 mL) under nitrogen
at -78 C.The
mixture was stirred at -78 C for 30 min, whereupon (S)-1-
(cyclopentanecarbonyl),2-
methylpiperidin-4-one (300 mg, 1.43 mmol) in dry THF (4 mt..) was added. The
temperature was
allowed to warm to room temperature. After stirring for 30 min, the reaction
mixture was cooled to
10 OcC. The mixture was quenched with NaHCO (50% sat.) and extracted with
diethyl ether. The
organic phase was washed with citric acid (10%), NaOH (1M), water and brine.
The organic phase
was dried (Na2504) and evaporated. The residue was purified by flash
chromatography
(Et0Acineptane, 15:85 - 2:8) to provide the title compound (329 mg, 67%) as an
orange oil. NMR
showed the two double bond isomers. 'H NMR (500 MHz, CDC13): 6 5.81-5.74 (m,
1H), 529-3.26
15 (m, 4H), 2.90-2.52 (m, 2H), 1.98-1.53 (m, 8H), 1.35-1,15 (m. 3H).
Step 3: (S)-crlopenty1(2-methy1-4-(1-methyl-5-nitro-1H-indol-3-y1)-3,6-
dihydropyridin-
1(2H)-ypmethanone. A mixture of 1-methyl-5-nitro-3,(4,4,5,5-tetramethyl-1,3,2-
dioxaborolan-2-y1)-
1H-indole (prepared as described in Example 1) (100 mg, 0.33 mmol), (S)-1-
(cyclopentanecarbony1)-2-methy1-1,2,3,6-tetrahydropyridin-4-
yltrifluoromethanesulfonate (119 mg,
20 0.35 mmol) and K3PO4 (155 mg, 0.73 mmol) in dioxaneiwater (9:1, 3 mL)
was flushed with
nitrogen. Tetrakis triphenylphosphine Pd(0) (36 mg, 0.03 mmol) was added, the
vial was flushed
quickly with nitrogen, and the mixture was stirred at 60 C overnight, Water
was added, and the
mixture was extracted with DCM (4 x). The combined organic layers were
concentrated in Vacuo.
The residue was purified by prep-HPLC to provide the title compound (110 mg,
90%) as a yellow
25 glass. LCMS: mie 368.20 [M+1-11+
Steps 4: N-(3-((25,48)-1-(cyclopentanecarbony1)-2-methylpiperidin-4-0)-1-
methyl-1H-indol-
5-y1)-4-fluorobenzenesulfonamide, A mixture of (S)-cyclopenty1(2-methyl-4-(1-
methyl-5-nitro-1H-
indol-3-y1)-3,6-dihydropyridin-1(2H)-Amethanone (50 mg, 0.14 mmol) and Pt02
(6.1 mg, 0.027
mg) in Et0H/AcOH 6:1 (3.5 nil...) was stirred at room temperature under H2 ( 1
bar :overpressure) for
30 18 h. The reaction mixture was filtered through a PTFE filter, and
concentrated in vacuo. The
reduced product was unstable and used irrimediantly. The residue was dissolved
in pyridine (3
mL). 4-Fluorobenzenesulfonyl chloride (40 mg, 0.20 mmol) was then added at
room temperature,
and the mixture was stirred for 90 min, The mixture was concentrated in vacuo,
and the residue
was purified by prep-HPLC to provide the title compound (3.7 mg, 5.5%). 11-1
NMR (500 MHz,
35 (CO3)2C0): 6 8.63(s, 1H), 7.78-7.72(m, 2H), 729-7,22 (m, 4H), 7.06(s,
1H), 7.03-6.98(m, 1H),
4.39-4.29(m, 2H), 4.01 (br. $, 1H), 3.74 (s, 3H), 3.28 (br. 5, 11-1), 3.02
(quint., J = 8.43, 1H), 2.22-
CA 02975157 2017-07-26
WO 2016/120850
PCT/I132016/050477
61
2.13 (m, 1H), 2,01-1,95 (m, 1H), 1.94-1.52 (m, 10H), 1.11 (d, J = 6.45, 3H),
LCMS: mfe 498.29
Example 21
Preparation of N-(34(28,4R)-1-(cyclopentanecarbony1)-2-methylpiperldin-4-y1)-1-
methyl-1H-
indol-5-y1)-4-fluorobenzenesulfonamide
0,
Ns: iikh
111111P N
Purification of the diastereomeric mixture produced in step 4 of Example 20 by
preparative
HPLC afford a second product as the title compound (6,3 mg, 9.3%), 1H NMR (500
MHz,
(CD3)2C0): 6 8.42 (s, 1H), 7.75 (q, = 5.4, 8,4 Hz, 2H), 7.32-7.21 (m, 4H),
7.02 (s, 1H), 6.97 (d, J
= 8,4 Hz, 1H), 4.99 (rn, 0.5H), 4.59 (m, 0,5H), 4.50 (m, 0,5H), 4.00 (m,
0.5H), 3.73 (s, 3H), 3.42-
2.98 (m, 3H), 2.0-1.51 (m, 12H), 1.40(d, J = 6.7 Hz, 1.5H), 1.23 (d, J = 6.7
Hz, 1,5H), LCMS: mie
498.28 [M-i-H1'.
Example 22
Preparation of 4-fluoro-N-(3-(1-isobutyrylpiperidin-4-y1)-1-methyl-1H-indol-5-
y1)-3-
methoxybenzenesulfonamide
o
1101 =
01µ0
1
Step 1: tert-butyl 4-(54(4-fluoro-3-methoxyphenyl)sulfonamido)-1-methyl-1H-
ind01-3-
yl)piperidine-1-carboxylate. A solution of tert-butyl 4-(5-amino-l-methyl-1H-
indo1-3-y1)piperidine-1-
carboxylate (prepared as described in Example 6) (3.0 g, 9.1 mmol) and 4-
fluoro-3-
methoxybenzenesulfonyl chloride (2.5 g, 10.9 mmol) in dry pyridine (20 ml) was
stirred at room
temperature overnight. The pyridine was evaporated, and 1M HCI and DCM were
added. The
phases were separated and the organic layer was evaporated. The residue was
purified by flash
chromatography (heptanaiEt0Ac, 1;4) to provide the title compound (3.9 g,
82%). LCMS: m/e
518.33 [M-I-H]+,
CA 02975157 2017-07-26
WO 2016/120850
PCT/I132016/050477
62
Step 2: 4-fluoro-3-methoxy-N-(1-methyl-3-(piperidin-4-y1)-1H-indo1-5-
y1)benzene-
sulfonamide. To a solution of tert-butyl 4-(54(4-fluora-3--
methoxyphenyl)sulfonamido)-1-methyl-1H-
indol-3-yl)piperidine-1-carboxylate (3.9g. 7,5 mmol) in DCM (50 mL) was added
TEA (5.7 mt.), and
the mixture was stirred at room temperature for 1 h, The volatiles were
evaporated. The residue
was dissolved in DCM, and sat. NlaHCO3 was added. The phases were separated
and: the organic
layer evaporated to provide the title compound that was used in the subsequent
step without
further purification. LCMS: mle 418.07 [MtH1+.
Step 3: 4-fiuom-N-(3-(1-isobutyrylpiperidin-41(1)-1-methyl-1H-indo1-5-y1)-3-
nnethoxybenzenesulfonamide. To the crude 4-fluoro-3-methoxy-N41-methyl-3-
(piperidin-4-y1)-1H-
indol-5-11)benzenesulfonamide from the previous step in dry pyridine (20 mL)
was added isobutyryl
chloride (4.7 mi.., 44.7 mmo1). The reaction mixture was stirred at room
temperature for 1h. The
pyridine was evaporated, and 1M HCI and DCM were added. The phases were
separated and the
organic layer was concentrated. The residue was purified by flash
chromatography
(heptane/Et0Ac, 1:1)10 provide the title compound (2.09, 55% over two steps).
'H NMR (500
MHz, (CD)2C0): 6 7.24-7.20 (m, 2H), 7,19-7.12 (m, 3H), 6.97 (s, 1H), 6,88 (dd,
J = 2.0, 8.6 Hz,
1H), 4.67-4.62 (m, 1H), 4.16-4,10 (m. 1H), 3.71 (s. 3H), 3.66 (s, 3H), 3.26-
3.22(m, 1H), 3.03-2.95
(m, 2H), 2.81-2.73 (m, 1H), 2.03-1.91 (m, 1H), 1.62-1.48(m, 2H), 1.35-1,22 (m,
2H), 1.13 (dd, J =-
7.0, 10.9 Hz, 6H). LCMS mie 488.33 [M+I-1]'.
Example 23
Preparation of 4-fluoro-3-hydroxy-N-(3-(1-isobutyrylpiperidin-4-y1)-1-methy1-
1H-indo1-5-
y1)benzenesulfonamide
0 /
0 H
HO as,
0
F 11111P
A solution of BBr3 (24 mL, 1M in DCM) in was added to a stirred solution of 4-
fluoro-N-(3-
(1-isobutyrylpiperidin-4-y1)-1-methyi-1H-indol-5-y1)-3-
methoxybenzenesulfonamide (prepared as
described in Example 22) (1.97 g, 4.0 mmol) in DCM (30 mi..) under an
atmosphere of nitrogen at -
18 C. The cooling bath was removed and the mixture was stirred at room
temperature for 1h. The
reaction was quenched by adding MeOH (50 mL), Water and DCM were added and the
phases
were separated. The organic layer was concentrated and the residue was
purified by prep-HPLC
to provide the title compound (0.75 g, 39%). LCMS: mie 47433 iiM+Hi .
CA 02975157 2017-07-26
WO 2016/120850
PCT/I132016/050477
63
Example 24
Preparation of (R)-44luoro-N-(3-(1-isobutyrylpiperidin-4-y1)-1-methy1-1H4ndol-
5-y1)-3-
((tetrahydrofuran-3-yl)oxy)benzenesulfonamide
b
JO
4-fluoro-N-(3-(1-isobutyrylpiperidin-411)-1-methyl-1 H-indol-5-yI)-3-
methoxybenzenesulfonamide (prepared as described in Example 22) (30 mg, 0.06
mitiol), (8)-
tetrahydrofuran-3-ol (18 mg, 0.19 mmol), triphenylphosphine (25 mg, 0_10
mrnol) and di-tert-
butyldiazene-t2-dicarboxylate (22 mg, 0.10 mmol) were charged in an oven-dried
vessel under an
atmosphere of nitrogen, Dry DCM (2 mL) was added, and the reaction mixture was
stirred at room
temperature overnight. The volatiles were evaporated and the residue was
purified by prep-HPLC
to provide the title compound (13 mg, 40%). 1H NMR (500 MHz, CD3OD): 6 7.40-
7.36 (m, 1H),
7.27-7,21 (m, 2H), 7.12 (s, 1H), 6.99 (s, 1H), 6.90 (dd. J = 2.5, 9.0 Hz, 1H),
6.87-6.84 (m, 1H),
4.67-4.59 (m, 2H), 4.17-4.10 (rn, 1H), 3.77-3.64(m, 5H), 3.62-3.50 (m, 2H),
3.29-3.21 (m, 1H),
3.04-2.95(m, 2H), 2.80-2.72(m, 1H), 2.04-1.90(m, 2H), 1.86-1.75 (m, 1H), 1.63-
1.48(m, 3H),
16 1.16-1,09(, 6H). LCMS: mle 544,38 [M+H]'.
Examples 25-31
The following Examples 25-31 were prepared in an analogous manner to Example
22
employing the necessary benzenesulfonyl chloride in step 1 and the appropriate
carboxylic acid in
step 5.
CA 02975157 2017-07-26
WO 2016/120850 PCT/I132016/050477
64
Example Structure Name/Characterization
(R)-4-methyl-N-(14nethy1-3-(1-(2,3,3-
trimethylbutanoyl)pipericlin-411)-1H-indol-5-
yl)benzenesulfonamide; Rt = 5.02 (Method C); 1H
NMR (400 MHz, Me0D-d4): 6 7.51-7.49 (m, 2H),
25 r-N
7.22-7.14 (m. 4H), 6,91 (s, 1H), 6.86-6.85 (m, 1H),
H. 4,70-4,67 (m, 1H), 4.29-4.26 (m, 1H), 3.66
(s, 3H),
3.23-3.22 (m, 1H), 2.95-2.72 (in, 3H), 2.35 (s, 3H),
1 2.01-1.91 (m, 2H), 1.48-1.45 (nn, 2H), 1,12-
1,07 (rn,
3H), 1.02-0.99 (m, 9H) ; [m+HT :496,1
(R)-1,3,5-trimethyl-N-(1-methyl-3-(1-(2,3,3-
' trimethylbutanoyl)olperidin-4-y1)-1H-indol-5-
y1)-1H-
N-N
/ pyrazole-4-sulfonamide. Rt = 4.56 (Method
C);
7--
N\ NMR (400 MHz, Me0D-d4): 6 7.25-7.21 (m, 2H),
26 6.98 (s, 1H), 6.89-6.87 (m, 1H), 4,74-4.71
(m, '1H),
HN
11101 N 4.34-4.31 (m, 1H), 3.72(s, 3H), 3;62(s, 3H),
3.33-
3.28 (m, 1H), 2.91-2.78 (m, 3H), 2,11-2.00 (rn, 8H),
1.64-1,54 (m, 2H), 1.14-1.11 (m, 3H), 1.09-1.01 (rn,
9H); LCIMS [M+H]': 514.1;
(R)-2-ethoxy-N-(1-rnethy1-3-(1-(2,3,3-
\/_ thmethytbutahoppiperidin-4-y1)-1H-indol-5-
C: yl)benzenesulfonamide, Rt =4,63 (Method D);
ISO .---N NMR (400 MHz, Me0D-d4: 6 7.65-7.62 (m, 1H),
27 7.45-7.26 (m, 2H), 7.12-7.09 (m, 2H), 6.92-
6.84 (m,
HN 3H), 4.69-4.66 (m, 1H), 4.30-4,28 (m, 3H),
3.66(s,
= 110 N\ 3H), 3.30-3.21 (in, 2H), 2.93-2,72
(m, 3H), 1,99-
1 1,96 (m, 2H), 1.55-1.46 (m, 5H), 1.12-0.99
(rn,
12H). LC/MS [M+Hf :526.1;
CA 02975157 2017-07-26
WO 2016/120850 PCT/I132016/050477
______ -, , ---,
i
Example Structure Name/Characterization
________________________________________________________ _
(R)--2-fluoro-N-(1-methy1-3-(1-(2,3,3-
trimethylbutanayl)piperidin-4-y1)-1H-indol-5-
o yl)benzehesulfonamide Rt =4.91 (Method E);
1H
..--- 0 NMR (400 1V1Hz, Me0D-d4)15 PPm 7.66-7.64 (m,
cf, 14,,,
1H): 7.58-7.54(m. 1H), 7.27-7.13 (m, 4H);6.94-
28 I
0.s.0 [.91(m, 2H), 4,70-4,67 (m, 1H), 4.31-4.28 (m,
1H),
4,1,
\ 3M4(., 3H)3.31-3.24 (m, 1H), 2.97-2.73(m,
3H),
N 2.08-1.90 (m, 2H), 1.73-1.46 (m, 2H), 1.55-
1.46 (m,
\
5H), 1,12-0.99 (m, 12H) , LCMS: mie 522.1
[M+Na],
0.\\/1-----
U r-N (R)-N-(1-methy1-3-(1-(2,3,3-
29 trimethylbulanayl)piperidin-4-y1)-1H-indol-5-
yl)benzenesuffonamide. LCMS: trie 482,2 [M+Hr
1 \C N
0 \ V-----
---= (R)-3,5-dimethyl-N-(1-methy1-3-(142,3,3-
N
s'kil-
---) trimethylbutanoyi)pperidin-4-y1)-1H-indol-5-
soxazole-4-sulfonamide. ). LCMS: mie 501.1
Q' b t ,L, \ [Tv1 H)-
,----- N L
1
0.).t (R)-N-(3-(1-(2-(bjcyclo[1,1.1]pentan-1
31 -
N yl)propandyl)piperidln-4-y1)-1-methyl-1H-indol-5-0)-
(3,µ ,LI 4-fluorobenzehesuifonamide. LCMS: m/e 510.2
N
Examp(e 36
Assay of co-activator recruitment by TR-FRET
CA 02975157 2017-07-26
WO 2016/120850
PCT/I132016/050477
66
The activity of compound of the invention can be determined by a co-activator
recruitment
by TR-FRET (time-resolved fluorescence resonance energy transfer) assay. in
general., the assay
is based on the interaction between N-terminally Six-Histidine-tagged-RORC2
ligend binding
domain (6-His-RORC2 LBD), expressed in E. coli and purified by affinity
chromatography, and
biotin-coactivator peptide SRC1-2 (biotin-aminohexanoic acid-
CPSSHSSLTERHKlLHRLLQEGSPS-NH2; SEQ ID NO: 1 ) containing the LXXLL consensus
domain which is responsible for receptor binding. This interaction is detected
by addition of
Europium labeled-anti-His antibody (Ex. 337 nm, Ern. 620 nm, which binds to
6His) and
Streptavidin-APC (EX. 620 nm, Ern. 665 nmõ which binds to biotin). When
receptor and
coactivator are bound to each other, upon shining light at 337 nm on the
sample, the Europium
emits fluorescence that excites APC due to close proximity (FRET) and this
signal is measured at
665 nm, Due to the long lasting fluorescence emission of Europium, the non-
specific, short-lived
fluorescence is time-resolved (TR) from the fluorescence of interest.
Inhibitors of the interaction of
receptor and coactivator peptide are detected by a decrease in TR-FRET signal.
SpeOifidaily, in one embodiment the aforementioned assay was performed as
outlined'
below. The assay was carried out in black polystyrene, 384-well plates in a
total assay volume of
50.5 pL.. The assay buffer contained 50 mM TRIS-HCL pH 7.5, 1 mM Na.C1, 2 mM
MgCl2, 0.5
mg/mL bovine serum -albunlin, and 5 mM dithiothreitol. The final concentration
of reagents was 6.3
nM RORC2 LED, .200 nM SRC1-2, 50 nM streptaviclin APC, 1 nM Europium-labeled
anti-His
antibody, and varying concentrations of compounds such that final
concentration Of DMSO is 1%
(v.1v). The assay steps were: (1) dispensing 500 pL compound at 100x final
concentration in
DMSO (test wells) Of DMSO only (control wells for no inhibition); and (2)
dispensing 50 pL miktdre
of the other assay components including receptor (test wells) or excluding
receptor (control wells
for maximal inhibition).
Assay mixtures were inoUbated are room temperature for 3 hr and read in
EnVision 2100
Multilabel Reader (PerkinElmer Life Sciences) at Excitation Filter 320,
Emission Europium Filter
615, Emission APC Filter 665, Dichroic Mirror D400/D630,
TR-FRET signal was determined by calculating the ratio of 665 nm by 615 nm and
leal
values of compounds of the invention (Table 1) were determined by the non-
linear regression
analysis of dose response curves.
References which relate to the above-referenced assay include: Kellen et al.
Structure,
200Z 10, 1697-1707; Stenlin et al. EMBO J .2001, 20, 5822-5831; and Zhou et
al. Mol Endocrinol
1998, 12, 1594-1604.
Table 1.
CA 02975157 2017-07-26
WO 2016/120850
PCT/I132016/050477
67
___________________________________________________ '
Example IC50 (nM) Example
IC50 (nM) I[ Example ' IC50 (nM) 1
- -----
'r------
1 8.2 13 16.9 25 ,
2.1
2 11,4 14 5,6 26
1.3
i ________
i 3 5.3 15 6.8 I 27
1.7
4 33.8 16 3.1 28
1.0
------------------------- H __________________ -H_ ___________
_______________________________________________________________________________
¨
5.1 17 5.9 29 8.7
6 75,9 18 7.0 H3
10,0
____________________________________________________________________ __
¨ i 7 4.5 19 ____ 3.7 __ J 31
2.4
--,
8 17.1 20 91.3 32
2,5
9 14.8 21 9,0 33
1.1
_ _________________________________________________________
. 10 5.6 22 10,6 34
0.9
11 28,8 23 19,9 35 .
1.0
,
1 12 ....... ¨ ---------
¨ ----------------- 7.8 i----
[ 24 I 5.7 . . --
Example 37
Assay of Gal4-RORC2 activity by luciferase reporter
The activity of compound of the invention can be also be determined by a
luciferase
5
reporter Gal4-RORC2 activity assay. In general, Neuro2A cells (murine
neuroblastoma cell line
obtained from HPACC, cat #89121404) are transiently transfected with a
mammalian expression
vector (pM) containing Gal4-RORC2 LBD and a Ga14-responsive reporter gene
containing firefly
luciferase (5xGAL4UAS-Luc3). Gal4-RORC2 LBO is constitutively active in the
transfected
Neuro2a cells, resulting in a robust luciferase response in the absence of
stimulation. Upon
treatment with an R0RC2 inhibitor the transcriptional response is decreased
and the magnitude of
the decrease in response is dose-dependently related to the intrinsic efficacy
of the inhibitor.
Specifically, the growth medium was composed by MEM EBS w/o L-glutamine, 10%
(v/v)
FBS, 2 fnM L-glutarnine and ix non-essential aminoacid (NEAA); the seeding
medium was
composed by MEM EBS w/o L-glutarnine, vv/o phenol red, 4% (v/v) FBS, 2 ri-}M L-
glutarnine, lx
NEAA, 1% Penicillin (10,000 UtrnL)/Streptornycin (10,000 pg/mL); and the assay
medium was
composed by mErvi EBS w/o L-giutamine, w/o phenol red, 4% (v/v) FBS, 2 mM L-
glutamine, lx
NEAA, 1% Penicillin (10,000 U/mL)/Streptomycin (10,000 pg/mL). In addition,
Neuro2A cells were
cultured in growth medium in humidified chambers at 37 C and 5% CO2 using
standard tissue
culture procedures.
On day one of the assay, cells were seeded and transfected. Specifically,
Neuro2A cells
were suspended in seeding medium and mixed with plasmids and transfection
reagent which was
dissolved in OptiMEM I reduced serum medium (InVitrogen), and then seeded to
384-well plates
CA 02975157 2017-07-26
WO 2016/120850 pCT/IB2016/050477
68
(Corning, Black, Clear bottom) in 40 pLfwell containing 12,500 cells, 17.25 ng
Ga14-Luc3, 5.75 ng
either empty pM vector (no receptor control wells) or pM-Gal4RORgamma-LBD, and
0.11 pL
Lipofectamine2000.
On day two of the assay, the cells were treated with compounds of the
invention.
Specifically, the treatment was started 20-24 hr after seeding and
transfection of the cells.
Compounds of the inventien were serially diluted in a 384-well polypropylene
plate with assay
medium containing 0.5% (NO') DMSO at 5x final assay concentration. 10 pL of
the compounds (or
0.5% DIOS in assay medium for no compound control' wells) were transferred
from he dilution
plate to the 384-format cell plate such that final assay volume was 50 pL and
final DMSO
concentration was 0.1% (WV), followed by incubation for 20-24 hr in humidified
chambers at 37 C
and 5% CO2
On day three of the assay, luminescence was measured and the results analyzed.
Specifically, 10 pL of SteadyLite Plus reagent (Perkin Elmer) was added to
each well. The cell
plates were incubated at room temperature for 15 min in the dark before
reading of luminescence
on the MicroBeta Trilux. (Wallac). ICso values of the compounds tested were
determined by the
non-linear regression analysis of dose response curves.
References which relate to the above-referenced assay include: Stehlin-Gaon et
al, Nature
Structural Biology 2003, 10, 820-825; Wang et al. J Biel Chem. 2010, 285(7),
5013-5025 Kumar et
al. Mol Pharmacol. 2010, 77(2)i 228-36.
Example 38
Assay of 1L-17 Production from human Th17 cells
The activity of compound of the invention can be also be determined by an IL-
17 production
from human Th17 cells assay. In general, this assay measures blockade of 1L-17
production, the
signature cytokine of T helper 17 (Th17) cells, by compounds. Purified human
004+ T cells are
stimulated with anti-CD3 + anti-CD28 and incubated with a cytokine cocktail
that induce their
differentiation into Th17 in the absence or presence of various concentrations
of compound. After
6 days, 1L-17A concentration is measured in the cell culture supernatant with
an ELÃSA kit (MSD).
Preparation of human CD4+ T cells. CD4+ T cells were purified from (puffy
coats from
healthy donors (obtained from Massachusetts General Hospital) by negative
selection the following
procedure: Mixing 25 mL of blood with 1 mL of Rosette Sep CD4+ T cell
enrichment cocktail
(StemCell Technologies) followed by application of a layer of 14 mL Ficoll
Paque Plus (Amersharn
GE Healthcare) and subsequent centrifugation at 1200 g for 20 min at room
temperature. The
Ficoll layer was then harvested and washed with phosphate saline buffer
containing 2% (v/v) fetal
bovine serum and cells were resuspended with RRMI medium containing 10 '%
(v/v) fetal bovine
serum and 10% (v/v) DMSO, frozen and kept in LN2 until used.
CA 02975157 2017-07-26
WO 2016/120850
PCT/I132016/050477
69
On the first day of the assay, a vial containing 107 CD4+ T cells is thawed
rapidly in 37 C
water bath, immediately transferred into 20 mi. X-Vivo 15 medium (Lonza), is
spun for 6 min at
300xg, the supernatant is discarded, and the resulting pellet. is re-suspended
at 10'3 celisimL in 50
mL fresh X-Vivo 15 medium, followed by storage overnight :in a tissue culture
vessel in a
humidified chamber at 37 C and 5% CO2. Serial dilutions of compounds of the
invention are
prepared at 10x final concentration in X-Vivo15 medium containing 3% (Viv)
DMSO.
On the second day of the assay, a 384-well tissue culture plate was coated
with 10 ugirriL
anti-hCD3 (eBioscience) at 50 pUwell, After 2 hr at 37 C, the supernatant is
discarded and the
coated plates are kept in a sterile tissue culture hood,
Cytokine plus anti-CD28 cocktail is prepared by mixing 25 ngimL hIL-6
(Peprotech), 5
ngtmL hTGFbetal (Peprotech), 12,5 ngirriL IL-lbeta (Peprotech), 25 ngIrriL hIL-
21, 25 ngirriL hIL-
23 (R&D Systems), and 1 ugim. L anti-hCD28 (eBloscience) in X-Vivo 15 medium.
The cytokine
plus anti-CD28 cocktail with CD44- cells is prepared such that the cocktail is
diluted 10-fold and cell
density is 0.22 x 10t/mL. The mixture is incubated 1 hr at 37 C.
90 pi_ (20,000 cells) dispensed per well in"the anti-hCD3 coated plate
prepared as noted
above.
10uL 10x compound is added per well (final D11.450=0.3%) from the compound
plate that
was previously prepared, followed by 6 days of incubation in a tissue culture
vessel in a humidified
chamber at 37 C and 5 /0 CO2.
On day six of the assay, production of IL-17A in 10 pi_ of the supernatant is
determined by
sandwich ELISA using 384w hIL17 MSD plates following the manufacturer's
protocol.
Measurement is carried out in a Sector Imager 6000 by the same manufactUrer.
Signal units from
the instrument are converted to pg/rnL using a calibration curve with known
amounts of IL-17A.
IC50 values of test compounds (Table 2) are determined by the non-linear
regression analysis of
dose response curves.
A reference which relates to the above-referenced assay is: Yang et al. Nature
2008, 454,
350-352.
Table 2.
Example i IC50 OW Example I IC5,3 (nM) Example
IC50 (nM)
1 13.5 6 275.7 11
148.0
2 4.8 7 28.1 12
126.6
3 121.1 8 183.6 13 1
309.6
4 137.0 9 1 99.1 14
221,2
5 90,3 10 29.0 15
188.6
CA 02975157 2017-07-26
WO 2016/120850
PCT/IB2016/050477
Example ICso (nM) Example T IC50 (nM) Example
IC50
16 53.8 23 1457.9 30 19.8
17 114.4 24 69.5 31 26.0
18 19.0 .. J 25 10.6 32 3.3
19 191.3 26 5.1 33 8.4
20 633.0 27 16,0 34 42
21 88.7 28 11.4 35 8.4 j
22 267.1 29 28.5 .. J
Example 39
Inhibition of Superantigen-induced Th17 cytokine production
Exotoxins caged "superantigens' are among the most powerful T cell activators.
5 Superantigens bind to the cell surface of major histocompatibilty complex
(MHO) molecules,
without intracellular processing. They stimulate T cells via the T cell
receptor, irrespective of the
antigen specificities. Therefore, bacterial superantigens are able to activate
a large pool of CD4+
as well as CD8+ T cells in contrast to the low T cell frequency for
conventional antigens. 0D4+ T
cells can be classified into various subsets (ThO, Th1, Th2, Th17) based on
their respective
10 cytokine secretion profiles. ThO cells are uncommitted naïve precursor
cells that primarily produce
1L-2 upon stimulation. ThO cells upon activation can differentiate into ml,
Th2, or the Th17 subset
depending on the local cytokine milieu. Thl cells mainly produce Inf-y; Th2
cells, 1L-4, 1L-5, and IL-
13, and Th17 cells, IL-17, and 1L-22. During a classical immune response, the
differentiation of T
helper subset occurs over days, or longer. In the superantigen in-vivo model
in mice injection of
15 superantigen triggers a rapid transcription and translation of the
various cytokines (i.e. IL-2, 1L-4,
Inf-y,IL-17) of the different Th subsets after only 6 hr. A RORyt inhibitor
given to animals prior to
the superantigen stimulus would impair the Th17 cytokine profile without
affecting the cytokine
profile of the other Th subsets (ThO, Th1, Th2). The model uses approximately
8 week old
C57BLI6, Balb/c, or C31-111-leJ mice which are dosed orally with compound 1 to
2 hr prior to
20 superantigen injection on the day of the experiment (Day 0) based on the
pharmacokinetic (PK)
profile of the compound. An optional dose may be given the day before
superantigen injection (Day
-1) to further inhibit the response if necessary. C57BL/6 and Balbic mice will
be sensitized 1 hr
prior to supernatigen injection with approximately 25 mg/mouse D-Galactosamlne
intraperitoneally
(C3H/HeJ mice do not need to be sensitized). Based on the literature
superantigen is typically
25 given at 10 ug/mouse intraperitoneally. Mice will be sacrificed at 3 hr
for RNA analysis or up to 6 hr
for cytokine analysis.
A reference which relates to the above-referenced assay is: Rajagopaian, G.
et. al. Physic:4
Genomics 2009, 37, 279,
>
8402041 9
71
ExaMpte 40
imiquimod Assay
Commercially available 5% Imiquirpod (IMO) cream (3M Pharmaceuticals) is
applied to the
back and right ear of each experiment al mouse for two consecutive days.
Control mice are treated
similarly with a commercially available vehicle dream. The experimental mide
are then
administered with RORyt inhibitors, and the control mice with vehicle, for 4
days. The ear
thickness is measured on all days by digital microMeter (Mitutoyo). Tissues,
such as ears and
speens, are harvested on Day 5 for RNA analysis. Ear swelling and serum
measurements are
also made.
References describing aspects of this: assay include; Van der Fits, L. et M.
J. Immunol.
2009,182(9), 5836,45; Van Belle, XI etal, J Immunal. 2012, 188(1), 462-9;
Cal,, Y. et al.
Immunity 2011, 35(4), 596,51Q; Fanti, P.A. et al. Int, J. Dermatol. 2006,
45(12), 1464-5; Swindell,
W.R. et al. PLOS One 2011, 6(4), e18266; and Roller, A, et al. J. Immunol,
2012, 189(9.),.4612-2O
Example 41
IL-23 Injection Model of Mouse Skin Inflammation
Ears from BALB/c Mice were each injected intra,dermally every Other day with
150 ng of
mouse recombinant IL-23 (eBloaciences) or PBS in a total volume of 25 pl. Ear
swelling was
'measured in triplicate using a micrometer (Mitutoyo) right before each 1L-23
challenge. On Day 14,
mice were euthanized and ears Were collected for measurement of cytokine
levels, gene
expression levels and hystopathological evaluation. Mica were administered 3 -
100 mg/kg of an
RORC2 modulator Or vehicle once daily orally for the duration of the study.
Alternatively, the
RORC2 modulatorwaS applied topically once Or twice daily 'using a Standard
formulation
(Et0H:propylene glycol:dirnethyl isosorbicle;DMSO, 38:30;1515) at a
concentration Of 0.1% to
5.0%.
References describing aspects of this assay include; MuraMoto, K. et at. J.
Pharrnacol.
Exp. Ther. 2010, 335(1), 23-31; Fridmari,.J. S. etalJ. Invest, Dermatoi. 2011,
131(9), 1838-1844.
CA 2975157 2018-08-17