Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 03061133 2019-10-22
WO 2018/200214 PCT/US2018/027450
KETOREDUCTASE POLYPEPTIDES AND POLYNUCLEOTIDES
100011 The present application claims priority to US Prov. Pat. Appin. Ser.
No. 62/491,161, filed
April 27, 2017, which is hereby incorporated by reference in its entirety for
all purposes.
REFERENCE TO SEQUENCE LISTING, TABLE OR COMPUTER PROGRAM
100021 The Sequence Listing concurrently submitted herewith under 37 C.F.R.
1.821 in a computer
readable form (CRF) via EFS-Web as file name CX2-166U5P1 5T25.txt is herein
incorporated by
reference. The electronic copy of the Sequence Listing was created on April
25, 2017, with a file size
of 544 Kbytes.
FIELD OF THE INVENTION
100031 The present invention provides engineered ketoreductase and phosphite
dehydrogenase
enzymes having improved properties as compared to a naturally occurring wild-
type ketoreductase
and phosphite dehydrogenase enzymes, as well as polynucleotides encoding the
engineered
ketoreductase and phosphite dehydrogenase enzymes, host cells capable of
expressing the engineered
ketoreductase and phosphite dehydrogenase enzymes, and methods of using the
engineered
ketoreductase and phosphite dehydrogenase enzymes to synthesize a chiral
catalyst used in the
synthesis of antiviral compounds, such as nucleoside inhibitors. The present
invention further
provides methods of using the engineered enzymes to deracemize a chiral
alcohol in a one-pot, multi-
enzyme system.
BACKGROUND
100041 Enzymes belonging to the ketoreductase (KRED) or carbonyl reductase
class (EC1.1.1.184)
are useful for the synthesis of optically active alcohols from the
corresponding prochiral ketone
substrate and by stereoselective reduction of corresponding racemic aldehyde
substrates. KREDs
typically convert ketone and aldehyde substrates to the corresponding alcohol
product, but may also
catalyze the reverse reaction, oxidation of an alcohol substrate to the
corresponding ketone/aldehyde
product. The reduction of ketones and aldehydes and the oxidation of alcohols
by enzymes such as
KRED requires a co-factor, most commonly reduced nicotinamide adenine
dinucleotide (NADH) or
reduced nicotinamide adenine dinucleotide phosphate (NADPH), and nicotinamide
adenine
dinucleotide (NAD) or nicotinamide adenine dinucleotide phosphate (NADP) for
the oxidation
reaction. NADH and NADPH serve as electron donors, while NAD and NADP serve as
electron
acceptors. It is frequently observed that ketoreductases and alcohol
dehydrogenases accept either the
1
CA 03061133 2019-10-22
WO 2018/200214 PCT/US2018/027450
phosphorylated or the non-phosphorylated co-factor (in its oxidized and
reduced state), but most often
not both.
[0005] in order to circumvent many chemical synthetic procedures for the
production of key
compounds, ketoreductases are being increasingly employed for the enzymatic
conversion of different
keto and aldehyde substrates to chiral alcohol products. These applications
can employ whole cells
expressing the ketoreductase for biocatalytic ketone and aldehyde reductions
or for biocatalytic
alcohol oxidation, or by use of purified enzymes in those instances where
presence of multiple
ketoreductases in whole cells would adversely affect the stereopurity and
yield of the desired product.
For in vitro applications, a co-factor (NADH or NADPH) regenerating enzyme
such as glucose
dehydrogenase (GDH), formate dehydrogenase, phosphite dehydrogenase etc. can
be used in
conjunction with the ketoreductase. It is desirable to identify other
ketoreductase enzymes that can be
used to carryout conversion of various keto substrates to corresponding chiral
alcohol products or
conversion of various alcohol substrates to corresponding ketone products.
SUMMARY OF THE INVENTION
[0006] The present invention provides engineered ketoreductase and phosphite
dehydrogenase
enzymes having improved properties as compared to a naturally occurring wild-
type ketoreductase
and phosphite dehydrogenase enzymes, as well as polynucleotides encoding the
engineered
ketoreductase and phosphite dehydrogenase enzymes, host cells capable of
expressing the engineered
ketoreductase and phosphite dehydrogenase enzymes, and methods of using the
engineered
ketoreductase and phosphite dehydrogenase enzymes to synthesize a chiral
catalyst used in the
synthesis of antiviral compounds, such as nucleoside inhibitors. The present
invention further
provides methods of using the engineered enzymes to deracemize a chiral
alcohol in a one-pot, multi-
enzyme system.
[0007] In addition, the present invention provides engineered phosphite
dehydrogenase enzymes
having improved properties as compared to a naturally occurring wild-type
phosphite dehydrogenase
enzyme, as well as polynucleotides encoding the engineered phosphite
dehydrogenase enzymes, host
cells capable of expressing the engineered phosphite dehydrogenase enzymes,
and methods of using
the engineered phosphite dehydrogenase enzymes to deracemize a chiral alcohol
in a one-pot, multi-
enzyme system.
[0008] The present invention provides engineered ketoreductase ("KRED")
enzymes that are capable
of stereoselectively deracemizing a racemic alcohol substrate to an optically
pure alcohol product in a
one-pot, multi-enzyme system, and having an improved property when compared
with the naturally-
occurring, wild-type KRED enzyme obtained from Candida parapsilosis (SEQ ID
NO:2), wild-type
KRED enzyme obtained from Sporidiobolus salmonicolor (SEQ ID NO: 112), or when
compared
with other engineered ketoreductase enzymes. In addition, the present
invention provides engineered
2
CA 03061133 2019-10-22
WO 2018/200214 PCT/US2018/027450
phosphite dehydrogenase ("PDH") enzymes capable of preferentially recycling
NADPH in the same
one-pot, multi-enzyme system.
[0009] In some further embodiments, the engineered enzymes have one or more
improved properties
in addition to altered enzymatic activity. For example, in some embodiments,
the engineered
ketoreductase polypeptides have increased stereoselectivity, as compared to
the wild-type
ketoreductase enzyme for reducing the substrate to the product and/or
preferentially oxidize the (S)
enantiomer. Improvements in enzyme properties include, but are not limited to
increases in
thermostability, , solvent stability, and/or reduced product inhibition.
[0010] The present invention provides engineered ketoreductase variants having
at least 85%, 90%,
91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99% or more sequence identity to SEQ
ID NO: 2, 112,
124, and/or 138.
100111 The present invention also provides engineered ketoreductase variants
have at least 85%,
90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99% or more sequence identity to
SEQ ID NO:2,
and at least one substitution or substitution set at one or more positions
selected from positions 37,
37/211, 37/211/229, 37/229, 45, 52, 52/57/110/272/296, 52/57/272,
52/57/272/274/279/296,
52/57/272/279/296, 55/57/276, 56, 57, 57/104/114, 57/104/114/229, 57/286,
79/83/275/276, 83,
83/275/276, 83/276, 104, 110, 114, 138/146/258/289, 211,211/229, 228, 229,
263, 268, 272, 274,
275/276, 276, 279, and 309, wherein the positions are numbered with reference
to SEQ ID NO:2. In
some additional embodiments, the engineered ketoreductase variants comprise at
least one
substitution or substitution set selected from 37R, 37R/211R, 37R/211R/229R,
37R/229R, 45R, 52D,
52D/57L/272H, 52S, 525/57L/1 10T/272H/296F, 525/57L/272H/279H/296F,
525/57L/272H/274V/279H/296F, 55F/57A/276M, 56L, 571, 571/104G/114H, 57L,
57L/104G/114H/229R, 57X/286X, 79T/835/275N/276M, 831, 835/275N/276M, 835/276M,
104G,
110T, 114H/K/M, 138V/1465/258V/2895, 21IR, 211R/229R, 228S, 229R, 263H/Y,
268M/W,
272H/I/L/P/Q/S/TN/W, 2741/V, 275N/276M, 276/M, 279H/Q/R and 309F, wherein the
positions are
numbered with reference to SEQ ID NO:2. In some further embodiments, the
engineered
ketoreductase variants comprise at least one substitution or substitution set
selected from K37R,
K37R/K211R, K37R/K211R/G229R, K37R/G229R, H45R, Y52D, Y52D/C57L/G272H, Y525,
Y525/C5711K110T/G272H/1,296F, Y525/C57L/G272HV1279H/L296F,
Y525/C57L/G272H/L274V/I279H/L296F, L55F/C57A/L276M, D56L, C57I,
C571/A104G/G114H,
C57L, C57L/A104G/G114H/G229R, C57XAV286X, 179T/V835/A275N/L276M, V83I,
V835/A275N/L276M, V835/L276M, A104G, K1 10T, G 114H/K/M,
5138V/A1465/M258V/T2895,
K211R, K2I1R/G229R, P228S, G229R, G263H/Y, 5268M/W, G272H/I/L/P/Q/S/TN/W,
L27411V,
A275N/L276M, L276F/M, I279H/Q/R, and R309F, wherein the positions are numbered
with
reference to SEQ ID NO:2.
100121 The present invention also provides engineered ketoreductase variants
having at least 85%,
90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99% or more sequence identity to
SEQ ID
3
CA 03061133 2019-10-22
WO 2018/200214 PCT/US2018/027450
NO:112, and at least one substitution or substitution set at one or more
positions selected from
positions 24/106/136/220/258/260/314/315, 24/106/214/250/258/260/314/315,
24/220/314/315,
122/159/316/318, 135, 139/207, 159/251/272/277/316/318/330, and 207, wherein
the positions are
numbered with reference to SEQ ID NO:112. In some embodiments, the engineered
ketoreductase
variants comprise at least one substitution or substitution set selected from
241/106P/136A/220G/258V/260A/314R/315A,
241/106P/214L/250V/258V/260A/314R/315A,
241/220G/314R/315A, 122E/159V/316E/318L, 135F, 139V/207S,
159V/251Q/272F/277P/316E/318L/330L, and 207G, wherein the positions are
numbered with
reference to SEQ ID NO:112. In some additional embodiments, the engineered
ketoreductase variants
comprise at least one substitution or substitution set selected from
V241/T106P/S136A/5220G/L258V/C260A/P314R/5315A,
V241/TIO6P/F214L/A250V/L258V/C260A/P314R/5315A, V24115220G/P314R/5315A,
T122E/1159V/L316E/1318L, V135F, 1139V/N2075,
I159VN251Q/Y272F/1277P/L316E/1318L/1330L, and N207G, wherein the positions are
numbered
with reference to SEQ ID NO:112.
[0013] The present invention also provides engineered ketoreductase variants
having at least 85%,
90%, 910/0, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99% or more sequence identity
to SEQ ID
NO:124, and at least one substitution set selected from positions
2/101/179/182/228/238/282, 3/95,
3/95/228/314, 24/95/228, 95, 95/135/139/207, and 159/228/309/330, wherein the
positions are
numbered with reference to SEQ ID NO:124. In some embodiments, the engineered
ketoreductase
variants comprise at least one substitution or substitution set selected from
2T/101P/179L/182M/228R/238L/282E, 3Y/95T, 3Y/95T/228T/314R, 241/95T/228T, 95T,
95T/135F/139V/207N, and 159V/228L/309Q/330L, wherein the positions are
numbered with
reference to SEQ ID NO:124. In some further embodiments, the engineered
ketoreductase variants
comprise at least one substitution or substitution set selected from
A2T/Y101P/A179L/T182M/M228R/A238L/T282E, K3YN95T, K3YN95T/M228T/P314R,
V24I/V95T/M228T, V95T, V95TN135F/1139V/G207N, and 1159V/M228L/K309Q/1330L,
wherein
the positions are numbered with reference to SEQ ID NO:124.
100141 The present invention also provides engineered ketoreductase variants
having at least 85%,
90%, 91%, 92%, 93%, 94%, 95%, 96 A, 97%, 98%, 99% or more sequence identity to
SEQ ID
NO:138, and at least one substitution or substitution set at one or more
positions selected from
positions 19, 24/43/47/49/67/68/70/91/220, 24/68/91/218/220, 67, 72,
74/75/78/108,
75/78/99/108/215/224, 78/107, 95, 96, and 114, wherein the positions are
numbered with reference to
SEQ ID NO:138. In some embodiments, the engineered ketoreductase variants
comprise at least one
substitution or substitution set selected from 19S,
241/43V/47E/49N/67V/68E/70P/91V/220G,
241/68E/91V/218N/220G, 67W, 72Q, 74A/75E/78F/108V, 75E/78F/99P/108V/215S/224A,
78F/107G, 95C, 96G, and 114V, wherein the positions are numbered with
reference to SEQ ID
4
CA 03061133 2019-10-22
WO 2018/200214 PCT/US2018/027450
NO:138. In some further embodiments, the engineered ketoreductase variants
comprise at least one
substitution or substitution set selected from G19S,
V241/A43V/S47E/L49N/A67VN68E/E70P/191V/S220G, V24IN68E/191V/T218N/S220G, A67W,
M72Q, K74A/Q75E/Y78F/A108V, Q75E/Y78F/N99P/A108V/D215S/S224A, Y78F/P107G,
T95C,
S966, and N114V, wherein the positions are numbered with reference to SEQ ID
NO:138.
100151 The present invention also provides engineered ketoreductase variants
comprising
polypeptide sequences comprising sequences having at least 90% sequence
identity to SEQ ID NO:2,
112, 124, and/or 138. In some embodiments, the engineered ketoreductase
variants comprise
polypeptide sequences comprising sequences having at least 95% sequence
identity to SEQ ID NO:2,
112, 124, and/or 138. In some further embodiments, the engineered
ketoreductase variants comprise
polypeptide sequences set forth in SEQ ID NO:2, 112, 124, or 138. In some
additional embodiments,
the engineered ketoreductase variants comprise polypeptide sequences encoding
variants provided in
Table 5.1, 6.1, 7.1, and/or 8.1. In some further embodiments, the engineered
ketoreductase variants
comprise polypeptide sequences selected from the even-numbered sequences set
forth in SEQ ID
NOS: 4 to 170.
[0016] The present invention also provides engineered polynucleotide sequences
encoding the
engineered ketoreductase variants provided herein. In some embodiments, the
engineered
polynucleotide sequence comprises a polynucleotide sequence that is at least
85%, 90%, 91%, 92%,
93%, 94%, 95%, 96%, 97%, 98%, 99% or more identical to a sequence selected
from the odd-
numbered sequences set forth in SEQ ID NOS: 3 to 169. The present invention
also provides vectors
comprising the engineered poly-nucleotide sequences encoding the engineered
ketoreductase variants
provided herein. In some embodiments, the vectors further comprise at least
one control sequence.
100171 The present invention also provides host cells comprising the vectors
comprising
polynucleotides encoding the engineered ketoreductase variants provided
herein.
[0018] The present invention also provides methods producing the engineered
ketoreductase variants
provided herein, comprising culturing the host cells provided herein under
conditions that the
engineered ketoreductase variant is produced by the host cell. In some
embodiments, the methods
further comprise the step of recovering the engineered ketoreductase variant
produced by the host cell.
100191 The present invention also provides immobilized engineered
ketoreductase variants.
[0020] The present invention further provides compositions comprising at least
one engineered
ketoreductase variant provided herein. In some embodiments, the compositions
comprise at least one
immobilized engineered ketodreductase variant provided herein.
[0021] The present invention also provides engineered phosphite dehydrogenase
variants having at
least 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99% or more sequence
identity to SEQ
ID NO: 172 and/or 208.
[0022] The present invention also provides engineered phosphite dehydrogenase
variants having at
least 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99% or more sequence
identity to SEQ
CA 03061133 2019-10-22
WO 2018/200214 PCMS2018/027450
ID NO:172, and at least one substitution or substitution set at one or more
positions selected from
positions 10/73/78/137/323/325, 10/73/78/233/323, 10/73/137,
13/41/63/132/193/195,
18/44/119/124/132/137/145/158/175/177/293/317/323,
18/44/119/124/132/137/145/158/177/293/323,
18/44/119/124/132/137/145/293/323/334/336,
32/44/132/137/145/186/233/293/323/336,
41/44/88/1.93/195, 44/69/120/132/137/145/1.75/195/293/323,44/113/132/145,
44/119/132/137/145/158/175/177/293/317/323, 44/132/135/136/137/145/293,
44/132/136/137/145/293, 44/132/137/145/233/308/323, 44/132/137/145/293/323,
44/132/145,
44/132/145/195/293/323, 137/233/303/323, and 266, wherein the positions are
numbered with
reference to SEQ ID NO:172. In some embodiments, the engineered phosphite
dehydrogenase
variants comprise at least one substitution or substitution set selected from
10K/73A/78Y/137Q/323D/325A, 10K/73A/78Y/23311323D, 10K/73A/137Q,
13D/41A/63A/132Q/1935/195E,
1.8M/44A/119F/124E/132Q/1371/1456/1581c1.755/177T/293L/317R/323D,
18M/44A/119F/124E/132Q/1371/145G/1581Q177T/293L/323D,
18M/44A/119F/124E/132Q/1371/145G/29311323D/334K/336R,
32V/44A/132Q/13711145G/186T/2331/293L/323D/3365, 4IA/44A/88R/1935/195E,
44A/69K/120V/132Q/1.371/145G/175T/195E/293L/323D, 44A/113S/132Q/1450,
44A/119F/132Q/1371/1456/158K/175S/177T/293L/317R/323D,
44A/132Q/135A/136D/1371/145G/293L, 44A/132Q/136D/137Q/145G/293L,
44A/132Q/137U145G/233U308V/323D, 44A/132Q/1371/145G/293L/323D, 44A/132Q/145G,
44A/132Q/145G/195E/293L/323D, 1.37Q/2331/303A/323D, and 266SN/W, wherein the
positions are
numbered with reference to SEQ ID NO:172. In some further embodiments, the
engineered phosphite
dehydrogenase variants comprise at least one substitution or substitution set
selected from
R10K/C73A/F78Y/R137Q/N323DN325A, RIOK/C73A/F78YN2331/N323D, R1OK/C73A/R137Q,
E1.3D/R41A/Q63A/R132Q/A193S/S195E,
Ll8M/R44A/L119F/A124E/R132Q/R1371/N145G/L158K/A175S/K177T/1293L/A317R/N323D,
Ll8M/R44A/L119F/A124E/R132Q/R137UN145G/L158K/K177T/1293L/N323D,
Li 8M/R44A/L119F/A124E/R132Q/RI371/N145G/1293L/N323D/A334K/C336R,
S32V/R44A/R132Q/R137I/N145G/R186TN2331/1293L/N323D/C336S,
R41A/R44A/A88R/A193S/S195E,
R44A/R69K/R120V/R132Q/R1371/N 145G/A175T/S195E/I293L/N323D,
R44AN1I35/R132Q/N145G,
R44A/L119F/R132Q/R1371/N145G/L158K/A1755/K177T/1293L/A317R/N323D,
R44A/R132Q/Q1.35A/P136D/R1371/N145G/1293L, R44A/R132Q/P136D/R137Q/N1456/1293L,
R44A/R132Q/R1371/N145GN23311A308V/N323D, R44A/R132Q/R1371/N145G/I293L/N323D,
R44A/R132Q/N145G, R44A/R132Q/N145G/S195E/I293L/N323D, R137QN2331/E303A/N323D,
and E2665N/W, wherein the positions are numbered with reference to SEQ ID
NO:172.
6
CA 03061133 2019-10-22
WO 2018/200214 PCT/US2018/027450
[0023] The present invention also provides engineered phosphite dehydrogenase
variants having at
least 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99% or more sequence
identity to SEQ
ID NO:208, and at least one substitution or substitution set at one or more
positions selected from
positions 32/59/124/177/191/327, 78/150/198/327/328, 83/266, 95/211/213/322,
104,
178/194/211/213/322, 206, 211/213/322, 215, 262, 266, and 323, wherein the
positions are numbered
with reference to SEQ ID NO:208. In some embodiments, the engineered phosphite
dehydrogenase
variants comprise at least one substitution or substitution set selected from
32V/59M/124E/1775/191H/327D, 78Y/1501/198L/3275/328P, 83A/266A,
951/211A/213Q/322M,
104F/L, 178P/194L/211A/213Q/322Q, 206N, 211A/213Q/322Q, 215P, 262D/P, 266S,
and 323N,
wherein the positions are numbered with reference to SEQ ID NO:208. In some
further embodiments,
the engineered phosphite dehydrogenase variants comprise at least one
substitution or substitution set
selected from 532V/A59M/A124E/T1775/Q191H/R327D, F78Y/F150I/F198L/R3275/L328P,
V83A/E266A, F95I/N211A/D213Q/1322M, TIO4F/L, A178P/C194L/N211A/D213Q/1322Q,
L206N,
N211A/D213Q/1322Q, L215P, V262D/P, E2665, and D323N, wherein the positions are
numbered
with reference to SEQ ID NO:208.
[0024] The present invention also provides engineered phosphite dehydrogenase
variants comprising
a polypeptide sequence comprising a sequence having at least 90% sequence
identity to SEQ ID
NO:172 and/or 208. In some embodiments, the engineered phosphite dehydrogenase
variants
comprise polypeptide sequences comprising sequences having at least 95%
sequence identity to SEQ
ID NO:172 and/or 208. In some further embodiments, the engineered phosphite
dehydrogenase
variants comprise polypeptide sequences set forth in SEQ ID NO:172 or 208. In
some additional
embodiments, the engineered phosphite dehydrogenase variants comprise
polypeptide sequences
encoding variants provided in Table 9.1, 10.1, and/or 11.1. In yet some
additional embodiments, the
engineered phosphite dehydrogenase variants comprise polypeptide sequences
selected from the even-
numbered sequences set from in SEQ ID NOS: 174 to 260.
[0025] The present invention also provides immobilized engineered phosphite
dehydrogenase
variants. In some embodiments, the present invention provides a mixture of at
least one immobilized
engineered ketoreductase variant provided herein and at least one engineered
phosphite
dehydrogenase variant provided herein.
[0026] The present invention also provides compositions comprising at least
one phosphite
dehydrogenase variant provided herein. In some embodiments, the present
invention further provides
compositions comprising mixtures of at least one engineered ketoreductase
variant provided herein
and at least one engineered phosphite dehydrogenase provided herein.
[0027] The present invention also provides engineered polynucleotide sequences
encoding the
engineered phosphite dehydrogenase variants provided herein. In some
embodiments, the engineered
polynucleotide sequences comprise polynucleotide sequences that are at least
85%, 90%, 91%, 92%,
7
CA 03061133 2019-10-22
WO 2018/200214 PCT/US2018/027450
93%, 940/0, 95%, 96%, 97%, 98%, 99% or more identical to a sequence selected
from the odd-
numbered sequences set forth in SEQ ID NOS: 3 to 169.
190281 The present invention also provides vectors comprising the engineered
polynucleotide
sequence encoding the engineered phosphite dehydrogenase variants provided
herein. In some
embodiments, the vectors further comprise at least one control sequence. In
yet some further
embodiments, the vectors comprise at least one engineered polynucleotide
sequence encoding an
engineered phosphite dehydrogenase variant provided herein and at least one
engineered
polynucleotide sequence encoding an engineered ketoreductase variant provided
herein. The present
invention also provides host cells comprising the vectors provided herein.
100291 The present invention also provides methods for producing the
engineered phosphite
dehydrogenase variants provided herein, comprising culturing the host cell
comprising a vector
comprising at least one engineered polynucleotide sequence encoding at least
one engineered
phosphite dehydrogenase of the present invention, under conditions that the
engineered phosphite
dehydrogenase variant is produced by the host cell. In some embodiments, the
host cells comprise
vectors comprising polynucleotide sequences comprising at least one engineered
ketoreductase and at
least one engineered phosphite dehydrogenase provided herein. In some
additional embodiments, the
host cells comprise at least one ketoreductase not provided herein, but
comprise at least one
engineered phosphite dehydrogenase variant provided herein. In some further
embodiments, the host
cells comprise at least on phosphite dehydrogenase not provided herein, but
comprise at least one
engineered ketoreductase variant provided herein. In some embodiments, the
methods further
comprise the step of recovering the engineered phosphite dehydrogenase variant
produced by the host
cell. In embodiments with host cells that produce at least one ketoreductase
and at least one phosphite
dehydrogenase, some methods further comprise the step of recovering the
ketoreductase and/or
phosphite dehydrogenase produced by the host cells.
[0030] The present invention also provides methods deracemizing chiral
alcohols comprising
providing at least one engineered ketoreductase variant provided herein,
providing at least one
engineered phosphite dehydrogenase variant provided herein, at least one
chiral alcohol, and at least
one co-factor, under conditions such that the chiral alcohol is deracemized.
In some embodiments,
the methods are conducted in a one pot reaction, while in some alternative
embodiments, multiple
reaction vessels are used.
BRIEF DESCRIPTION OF THE FIGURES
[0031] Figure 1 provides the reaction scheme addressed by the present
invention.
[0032] Figure 2 provides the structures of substrate and product isomers.
[0033] Figure 3 provides the one-pot, multi-enzyme reaction scheme.
[0034] Figures 4 and 5 provide the cofactor competition assay schemes.
8
CA 03061133 2019-10-22
WO 2018/200214 PCT/U82018/027450
[0035] Figure 6 provides the HPLC chromatogram of products obtained in one-
pot, multi-enzyme
reactions.
DESCRIPTION OF THE INVENTION
[0036] The present invention provides engineered ketoreductase enzymes and
engineered phosphite
dehydrogenase enzymes having improved properties as compared to a naturally
occurring wild-type
ketoreductase and phosphite dehydrogenase enzyme, as well as polynucleotides
encoding the
engineered ketoreductase and engineered phosphite dehydrogenase enzymes, host
cells capable of
expressing the engineered ketoreductase and engineered phosphite dehydrogenase
enzymes, and
methods of using the engineered ketoreductase and engineered phosphite
dehydrogenase enzymes to
deracemize a racemic alcohol in a one-pot, multi-enzyme system.
Definitions
[0037] In reference to the present invention, the technical and scientific
terms used in the
descriptions herein will have the meanings commonly understood by one of
ordinary skill in the art,
unless specifically defined otherwise. Accordingly, the following terms are
intended to have the
following meanings. All patents and publications, including all sequences
disclosed within such
patents and publications, referred to herein are expressly incorporated by
reference. Unless otherwise
indicated, the practice of the present invention involves conventional
techniques commonly used in
molecular biology, fermentation, microbiology, and related fields, which are
known to those of skill
in the art. Unless defined otherwise herein, all technical and scientific
terms used herein have the
same meaning as commonly understood by one of ordinary skill in the art to
which this invention
belongs. Although any methods and materials similar or equivalent to those
described herein can be
used in the practice or testing of the present invention, the preferred
methods and materials are
described. Indeed, it is intended that the present invention not be limited to
the particular
methodology, protocols, and reagents described herein, as these may vary,
depending upon the
context in which they are used. The headings provided herein are not
limitations of the various
aspects or embodiments of the present invention.
[0038] Nonetheless, in order to facilitate understanding of the present
invention, a number of terms
are defined below. Numeric ranges are inclusive of the numbers defining the
range. Thus, every
numerical range disclosed herein is intended to encompass every narrower
numerical range that falls
within such broader numerical range, as if such narrower numerical ranges were
all expressly written
herein. It is also intended that every maximum (or minimum) numerical
limitation disclosed herein
includes every lower (or higher) numerical limitation, as if such lower (or
higher) numerical
limitations were expressly written herein.
[0039] As used herein, the term "comprising" and its cognates are used in
their inclusive sense (i.e.,
equivalent to the term "including" and its corresponding cognates).
9
CA 03061133 2019-10-22
WO 2018/200214 PCT/US2018/027450
[0040] As used herein and in the appended claims, the singular "a", "an" and
"the" include the plural
reference unless the context clearly dictates otherwise. Thus, for example,
reference to a "host cell"
includes a plurality of such host cells.
[0041] Unless otherwise indicated, nucleic acids are written left to right in
5' to 3 orientation and
amino acid sequences are written left to right in amino to carboxy
orientation, respectively.
100421 The headings provided herein are not limitations of the various aspects
or embodiments of the
invention that can be had by reference to the specification as a whole.
Accordingly, the terms defined
below are more fully defined by reference to the specification as a whole.
[0043] "Ketoreductase" and "KRED" are used interchangeably herein to refer to
a polypeptide
having an enzymatic capability of reducing a carbonyl group to its
corresponding alcohol. More
specifically, the ketoreductase polypeptides of the invention are capable of
stereoselectively
deracemizing an alcohol of formula (I) to the corresponding product of fonnula
(II) in an one-pot,
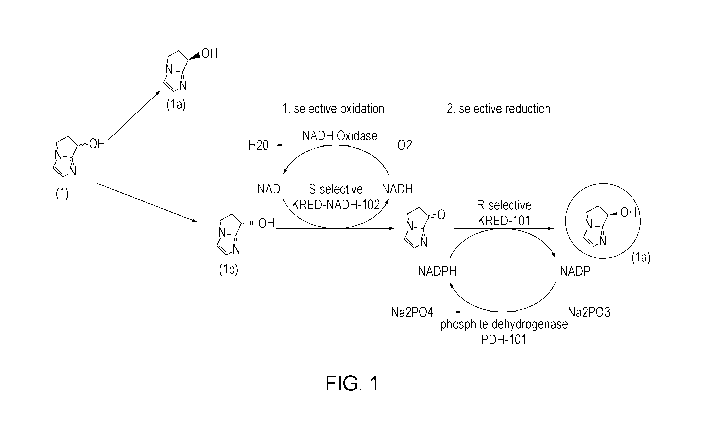
multi-enzyme system, as shown in Scheme I (See, Figure 1).
[0044] Phosphite dehydrogenase and "PDH" are used interchangeably herein to
refer to a
polypeptide having an enzymatic capability of regenerating NADPH co-factor.
[00451 As used herein, the term "one-pot reaction" refers to the production of
a product from a
starting material using multiple enzymes (i.e., KREDs and PDHs) in one
reaction vessel.
[0046] As used herein, the terms "protein," "polypeptide," and "peptide" are
used interchangeably
herein to denote a polymer of at least two amino acids covalently linked by an
amide bond, regardless
of length or post-translational modification (e.g., glycosylation,
phosphorylation, lipidation,
myristilation, ubiquitination, etc.). Included within this definition are D-
and L-amino acids, and
mixtures of D- and L-amino acids.
[0047] As used herein, "poly/nucleotide" and "nucleic acid' refer to two or
more nucleosides that are
covalently linked together. The polynucleotide may be wholly comprised of
ribonucleosides (i.e., an
RNA), wholly comprised of 2' deoxyribonucleotides (i.e., a DNA) or mixtures of
ribo- and 2'
deoxyribonucleosides. While the nucleosides will typically be linked together
via standard
phosphodiester linkages, the polynucleotides may include one or more non-
standard linkages. The
polynucleotide may be single-stranded or double-stranded, or may include both
single-stranded
regions and double-stranded regions. Moreover, while a polynucleotide will
typically be composed of
the naturally occurring encoding nucleobases (i.e., adenine, guanine, uracil,
thymine, and cytosine), it
may include one or more modified and/or synthetic nucleobases (e.g., inosine,
xanthine,
hypoxanthine, etc.). Preferably, such modified or synthetic nucleobases will
be encoding
nucleobases.
[0048] As used herein, "coding sequence" refers to that portion of a nucleic
acid (e.g., a gene) that
encodes an amino acid sequence of a protein.
[0049] As used herein, "naturally occurring" or "wild-type" refers to the form
found in nature. For
example, a naturally occurring or wild-type polypeptide or polynucleotide
sequence is a sequence
CA 03061133 2019-10-22
WO 2018/200214 PCT/US2018/027450
present in an organism that can be isolated from a source in nature and which
has not been
intentionally modified by human manipulation.
[0050] As used herein, "non-naturally occurring" or "engineered" or
"recombinant" when used in the
present invention with reference to (e.g., a cell, nucleic acid, or
polypeptide), refers to a material, or a
material corresponding to the natural or native form of the material, that has
been modified in a
manner that would not otherwise exist in nature, or is identical thereto but
produced or derived from
synthetic materials and/or by manipulation using recombinant techniques. Non-
limiting examples
include, among others, recombinant cells expressing genes that are not found
within the native (non-
recombinant) form of the cell or express native genes that are otherwise
expressed at a different level.
[0051] As used herein, "percentage of sequence identity," "percent identity,"
and "percent identical"
refer to comparisons between polynucleotide sequences or polypeptide
sequences, and are determined
by comparing two optimally aligned sequences over a comparison window, wherein
the portion of the
polynucleotide or polypeptide sequence in the comparison window may comprise
additions or
deletions (i.e., gaps) as compared to the reference sequence for optimal
alignment of the two
sequences. The percentage is calculated by determining the number of positions
at which either the
identical nucleic acid base or amino acid residue occurs in both sequences or
a nucleic acid base or
amino acid residue is aligned with a gap to yield the number of matched
positions, dividing the
number of matched positions by the total number of positions in the window of
comparison and
multiplying the result by 100 to yield the percentage of sequence identity.
Determination of optimal
alignment and percent sequence identity is performed using the BLAST and BLAST
2.0 algorithms
(See e.g., Altschul et al., J. Mol. Biol. 215: 403-410 [1990]; and Altschul et
al., Nucleic Acids Res.
3389-3402 [1977]). Software for performing BLAST analyses is publicly
available through the
National Center for Biotechnology Information website.
[0052] Briefly, the BLAST analyses involve first identifying high scoring
sequence pairs (HSPs) by
identifying short words of length Win the query sequence, which either match
or satisfy some
positive-valued threshold score T when aligned with a word of the same length
in a database
sequence. T is referred to as, the neighborhood word score threshold (Altschul
et al, supra). These
initial neighborhood word hits act as seeds for initiating searches to find
longer HSPs containing
them. The word hits are then extended in both directions along each sequence
for as far as the
cumulative alignment score can be increased. Cumulative scores are calculated
using, for nucleotide
sequences, the parameters M (reward score for a pair of matching residues;
always >0) and N (penalty
score for mismatching residues; always <0). For amino acid sequences, a
scoring matrix is used to
calculate the cumulative score. Extension of the word hits in each direction
are halted when: the
cumulative alignment score falls off by the quantity X from its maximum
achieved value; the
cumulative score goes to zero or below, due to the accumulation of one or more
negative-scoring
residue alignments; or the end of either sequence is reached. The BLAST
algorithm parameters W,
and X determine the sensitivity and speed of the alignment. The BLASTN program
(for nucleotide
11
CA 03061133 2019-10-22
WO 2018/200214 PCT/US2018/027450
sequences) uses as defaults a wordlength (W) of!!, an expectation (E) of 10,
M=5, N=-4, and a
comparison of both strands. For amino acid sequences, the BLASTP program uses
as defaults a
wordlength (W) of 3, an expectation (E) of 10, and the BLOSUM62 scoring matrix
(See e.g.,
Henikoff and Henikoff, Proc Natl Acad Sci USA 89:10915 [1989]).
[0053] Numerous other algorithms are available and known in the art that
function similarly to
BLAST in providing percent identity for two sequences. Optimal alignment of
sequences for
comparison can be conducted using any suitable method known in the art (e.g.,
by the local homology
algorithm of Smith and Waterman, Adv. Appl. Math. 2:482 [1981]; by the
homology alignment
algorithm of Needleman and Wunsch, J. Mol. Biol. 48:443 [1970]; by the search
for similarity method
of Pearson and Lipman, Proc. Natl. Acad. Sci. USA 85:2444 [1988]; and/or by
computerized
implementations of these algorithms [GAP, BESTFIT, FASTA, and TFASTA in the
GCG Wisconsin
Software Package]), or by visual inspection, using methods commonly known in
the art.
Additionally, determination of sequence alignment and percent sequence
identity can employ the
BESTFIT or GAP programs in the GCG Wisconsin Software package (Accelrys,
Madison WI), using
the default parameters provided.
[0054] As used herein, "reference sequence" refers to a defined sequence to
which another sequence
is compared. A reference sequence may be a subset of a larger sequence, for
example, a segment of a
full-length gene or polypeptide sequence. Generally, a reference sequence is
at least 20 nucleotide or
amino acid residues in length, at least 25 residues in length, at least 50
residues in length, or the full
length of the nucleic acid or polypeptide. Since two polynucleotides or
polypeptides may each (1)
comprise a sequence (i.e., a portion of the complete sequence) that is similar
between the two
sequences, and (2) may further comprise a sequence that is divergent between
the two sequences,
sequence comparisons between two (or more) polynucleotides or polypeptide are
typically performed
by comparing sequences of the two polynucleotides over a comparison window to
identify and
compare local regions of sequence similarity. The term "reference sequence" is
not intended to be
limited to wild-type sequences, and can include engineered or altered
sequences. For example, in
some embodiments, a "reference sequence" can be a previously engineered or
altered amino acid
sequence.
[0055] As used herein, "comparison window" refers to a conceptual segment of
at least about 20
contiguous nucleotide positions or amino acids residues wherein a sequence may
be compared to a
reference sequence of at least 20 contiguous nucleotides or amino acids and
wherein the portion of the
sequence in the comparison window may comprise additions or deletions (i.e.,
gaps) of 20 percent or
less as compared to the reference sequence (which does not comprise additions
or deletions) for
optimal alignment of the two sequences. The comparison window can be longer
than 20 contiguous
residues, and includes, optionally 30,40, 50, 100, or longer windows.
[0056] As used herein, "corresponding to", "reference to" or "relative to"
when used in the context of
the numbering of a given amino acid or polynucleotide sequence refers to the
numbering of the
12
CA 03061133 2019-10-22
WO 2018/200214 PCT/US2018/027450
residues of a specified reference sequence when the given amino acid or
polynucleotide sequence is
compared to the reference sequence. In other words, the residue number or
residue position of a given
polymer is designated with respect to the reference sequence rather than by
the actual numerical
position of the residue within the given amino acid or polynucleotide
sequence. For example, a given
amino acid sequence, such as that of an engineered ketoreductase, can be
aligned to a reference
sequence by introducing gaps to optimize residue matches between the two
sequences. In these cases,
although the gaps are present, the numbering of the residue in the given amino
acid or polynucleotide
sequence is made with respect to the reference sequence to which it has been
aligned. As used herein,
a reference to a residue position, such as "Xn" as further described below, is
to be construed as
referring to "a residue corresponding to", unless specifically denoted
otherwise. Thus, for example,
"X94" refers to any amino acid at position 94 in a poly,ipeptide sequence.
[0057] As used herein, "stereoselectivity" refers to the preferential
formation in a chemical or
enzymatic reaction of one stereoisomer over another stereoisomer or another
set of stereoisomers.
Stereoselectivity can be partial, where the formation of a stereoisomer is
favored over another, or it
may be complete where only one stereoisomer is formed. When the stereoisomers
are enantiomers,
the stereoselectivity is referred to as enantioselectivity, the fraction
(typically reported as a
percentage) of one enantiomer in the sum of both enantiomers. It is commonly
alternatively reported
in the art (typically as a percentage) as the enantiomeric excess (e.e.)
calculated therefrom according
to the formula [major enantiomer ¨ minor enantiomer]/[major enantiomer + minor
enantiomer].
Where the stereoisomers are diastereoisomers, the stereoselectivity is
referred to as
diastereoselectivity, the fraction (typically reported as a percentage) of one
diastereomer in a mixture
of two diastereomers, commonly alternatively reported as the diastereomeric
excess (d.e.).
Enantiomeric excess and diastereomeric excess are types of stereomeric excess.
It is also to be
understood that stereoselectivity is not limited to single stereoisomers and
can be described for sets of
stereoisomers.
[0058] As used herein, "highly stereoselective" refers to a chemical or
enzymatic reaction that is
capable of converting a substrate to its corresponding chiral alcohol product,
with at least about 75%
stereomeric excess.
[0059] As used herein, "increased enzymatic activity" and "increased activity"
refer to an improved
property of an engineered enzyme, which can be represented by an increase in
specific activity (e.g.,
product produced/time/weight protein) or an increase in percent conversion of
the substrate to the
product (e.g., percent conversion of starting amount of substrate to product
in a specified time period
using a specified amount of ketoreductase) as compared to a reference enzyme.
Exemplary methods to
determine enzyme activity are provided in the Examples. Any property relating
to enzyme activity
may be affected, including the classical enzyme properties of Km, Vmax or
kcat, changes of which
can lead to increased enzymatic activity. The ketoreductase activity can be
measured by any one of
standard assays used for measuring ketoreductases, such as change in substrate
or product
13
CA 03061133 2019-10-22
WO 2018/200214 PCT/US2018/027450
concentration, or change in concentration of the cofactor (in absence of a
cofactor regenerating
system). Comparisons of enzyme activities are made using a defined preparation
of enzyme, a defined
assay under a set condition, and one or more defmed substrates, as further
described in detail herein.
Generally, when enzymes in cell lysates are compared, the numbers of cells and
the amount of protein
assayed are determined as well as use of identical expression systems and
identical host cells to
minimize variations in amount of enzyme produced by the host cells and present
in the lysates.
[0060] As used herein, "conversion" refers to the enzymatic transformation of
a substrate to the
corresponding product.
[0061] As used herein "percent conversion" refers to the percent of the
substrate that is converted to
the product within a period of time under specified conditions. Thus, for
example, the "enzymatic
activity" or "activity" of a ketoreductase polypeptide can be expressed as
"percent conversion" of the
substrate to the product.
100621 As used herein, "thermostable" or "thermal stable" are used
interchangeably to refer to a
polypeptide that is resistant to inactivation when exposed to a set of
temperature conditions (e.g., 40-
80 C) for a period of time (e.g., 0.5-24 hrs) compared to the untreated
enzyme, thus retaining a certain
level of residual activity (e.g., more than 60% to 80% for example) after
exposure to elevated
temperatures.
[0063] As used herein, "solvent stable" refers to the ability of a polypeptide
to maintain similar
activity (e.g., more than e.g., 60% to 80%) after exposure to varying
concentrations (e.g., 5-99%) of
solvent compared to the untreated enzyme.
[0064] As used herein, "amino acid difference" or "residue difference" refers
to a difference in the
amino acid residue at a position of a polypeptide sequence relative to the
amino acid residue at a
corresponding position in a reference sequence. The positions of amino acid
differences generally are
referred to herein as "Xn," where n refers to the corresponding position in
the reference sequence
upon which the residue difference is based. For example, a "residue difference
at position X40 as
compared to SEQ ID NO:2" refers to a difference of the amino acid residue at
the polypeptide
position corresponding to position 40 of SEQ ID NO:2. Thus, if the reference
polypeptide of SEQ ID
NO:2 has a histidine at position 40, then a "residue difference at position
X40 as compared to SEQ ID
NO:2" refers to an amino acid substitution of any residue other than histidine
at the position of the
polypeptide corresponding to position 40 of SEQ ID NO:2. In most instances
herein, the specific
amino acid residue difference at a position is indicated as "XnY" where "XII"
specified the
corresponding position as described above, and "Y" is the single letter
identifier of the amino acid
found in the engineered polypeptide (i.e., the different residue than in the
reference polypeptide). In
some instances, the present invention also provides specific amino acid
differences denoted by the
conventional notation "AnB", where A is the single letter identifier of the
residue in the reference
sequence, "n" is the number of the residue position in the reference sequence,
and B is the single letter
identifier of the residue substitution in the sequence of the engineered poly-
peptide. In some instances,
14
CA 03061133 2019-10-22
WO 2018/200214
PCT/US2018/027450
a polypeptide of the present invention can include one or more amino acid
residue differences relative
to a reference sequence, which is indicated by a list of the specified
positions where residue
differences are present relative to the reference sequence. In some
embodiments, where more than
one amino acid can be used in a specific residue position of a polypeptide,
the various amino acid
residues that can be used are separated by a "I" (e.g., X192A/G). The present
invention includes
engineered polypeptide sequences comprising one or more amino acid differences
that include
either/or both conservative and non-conservative amino acid substitutions. The
amino acid sequences
of the specific recombinant carbonic anhydrase polypeptides included in the
Sequence Listing of the
present invention include an initiating methionine (M) residue (i.e., M
represents residue position 1).
The skilled artisan, however, understands that this initiating methionine
residue can be removed by
biological processing machinery, such as in a host cell or in vitro
translation system, to generate a
mature protein lacking the initiating methionine residue, but otherwise
retaining the enzyme's
properties. Consequently, the term "amino acid residue difference relative to
SEQ ID NO:2 at
position Xn" as used herein may refer to position "Xn" or to the corresponding
position (e.g., position
(X-1)n) in a reference sequence that has been processed so as to lack the
starting methionine.
[0065] As used herein, the phrase "conservative amino acid substitutions"
refers to the
interchangeability of residues having similar side chains, and thus typically
involves substitution of
the amino acid in the polypeptide with amino acids within the same or similar
defined class of amino
acids. By way of example and not limitation, in some embodiments, an amino
acid with an aliphatic
side chain is substituted with another aliphatic amino acid (e.g., alanine,
valine, leucine, and
isoleucine); an amino acid with a hydroxyl side chain is substituted with
another amino acid with a
hydroxyl side chain (e.g., serine and threonine); an amino acids having
aromatic side chains is
substituted with another amino acid having an aromatic side chain (e.g.,
phenylalanine, tyrosine,
tryptophan, and histidine); an amino acid with a basic side chain is
substituted with another amino
acid with a basis side chain (e.g., lysine and arginine); an amino acid with
an acidic side chain is
substituted with another amino acid with an acidic side chain (e.g., aspartic
acid or glutamic acid);
and/or a hydrophobic or hydrophilic amino acid is replaced with another
hydrophobic or hydrophilic
amino acid, respectively. Exemplary conservative substitutions are provided in
Table 1.
Table 1. Exemplary Conservative Amino Acid Substitutions
Residue Possible Conservative Substitutions
A, L, V, T Other aliphatic (A, L, V. I)
Other non-polar (A, L, V, I, G, M)
G, M Other non-polar (A, L, V, I. (3, M)
D, E Other acidic (D, E)
K. R Other basic (K,
CA 03061133 2019-10-22
WO 2018/200214 PCT/US2018/027450
N, Q, Sr T Other polar
H, Y, W, F Other aromatic (H, Y, W, F)
C. P Non-polar
[0066] As used herein, the phrase "non-conservative substitution" refers to
substitution of an amino
acid in the polypeptide with an amino acid with significantly differing side
chain properties. Non-
conservative substitutions may use amino acids between, rather than within,
the defined groups and
affects (a) the structure of the peptide backbone in the area of the
substitution (e.g., proline for
glycine) (b) the charge or hydrophobicity, or (c) the bulk of the side chain.
By way of example and
not limitation, an exemplary non-conservative substitution can be an acidic
amino acid substituted
with a basic or aliphatic amino acid; an aromatic amino acid substituted with
a small amino acid; and
a hydrophilic amino acid substituted with a hydrophobic amino acid.
[0067] As used herein, "deletion" refers to modification of the polypeptide by
removal of one or
more amino acids from the reference polypeptide. Deletions can comprise
removal of 1 or more
amino acids, 2 or more amino acids, 5 or more amino acids, 10 or more amino
acids, 15 or more
amino acids, or 20 or more amino acids, up to 10% of the total number of amino
acids, or up to 20%
of the total number of amino acids making up the polypeptide while retaining
enzymatic activity
and/or retaining the improved properties of an engineered enzyme. Deletions
can be directed to the
internal portions and/or terminal portions of the polypeptide. In various
embodiments, the deletion can
comprise a continuous segment or can be discontinuous.
[0068] As used herein, "insertion" refers to modification of the polypeptide
by addition of one or
more amino acids to the reference poly-peptide. In some embodiments, the
improved engineered
ketoreductase enzymes comprise insertions of one or more amino acids to the
naturally occurring
ketoreductase polypeptide as well as insertions of one or more amino acids to
engineered
ketoreductase polypeptides. Insertions can be in the internal portions of the
polypeptide, or to the
carboxy or amino terminus. Insertions as used herein include fusion proteins
as is known in the art.
The insertion can be a contiguous segment of amino acids or separated by one
or more of the amino
acids in the naturally occurring polypeptide.
[0069] The term "amino acid substitution set" or "substitution set" refers to
a group of amino acid
substitutions in a polypeptide sequence, as compared to a reference sequence.
A substitution set can
have 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, or more amino acid
substitutions. In some
embodiments, a substitution set refers to the set of amino acid substitutions
that is present in any of
the variant KREDs listed in the Tables provided in the Examples.
[0070] As used herein, "fragment" refers to a polypeptide that has an amino-
terminal and/or carboxy-
terminal deletion, but where the remaining amino acid sequence is identical to
the corresponding
positions in the sequence. Fragments can typically have about 80%, about 90%,
about 95%, about
98%, or about 99% of the full-length ketoreductase polypeptide, for example
the polypeptide of SEQ
16
CA 03061133 2019-10-22
WO 2018/200214 PCT/US2018/027450
ID NO:4. In some embodiments, the fragment is "biologically active" (i.e., it
exhibits the same
enzymatic activity as the full-length sequence).
100711 As used herein, "isolated polypeptide" refers to a polypeptide which is
substantially separated
from other contaminants that naturally accompany it, e.g., protein, lipids,
and polynucleotides. The
term embraces polypeptides which have been removed or purified from their
naturally-occurring
environment or expression system (e.g., host cell or in vitro synthesis). The
improved ketoreductase
enzymes may be present within a cell, present in the cellular medium, or
prepared in various forms,
such as lysates or isolated preparations. As such, in some embodiments, the
engineered ketoreductase
polypeptides of the present invention can be an isolated poly-peptide.
[0072] As used herein, "substantially pure polypeptide" refers to a
composition in which the
polypeptide species is the predominant species present (i.e., on a molar or
weight basis it is more
abundant than any other individual macromolecular species in the composition),
and is generally a
substantially purified composition when the object species comprises at least
about 50 percent of the
macromolecular species present by mole or % weight. Generally, a substantially
pure engineered
ketoreductase polypeptide composition will comprise about 60 % or more, about
70% or more, about
80% or more, about 90% or more, about 91% or more, about 92% or more, about
93% or more, about
94% or more, about 95% or more, about 96% or more, about 97% or more, about
98% or more, or
about 99% of all macromolecular species by mole or % weight present in the
composition. Solvent
species, small molecules (<500 Daltons), and elemental ion species are not
considered
macromolecular species. In some embodiments, the isolated improved
ketoreductase polypeptide is a
substantially pure polypeptide composition.
100731 As used herein, when used with reference to a nucleic acid or
polypeptide, the term
"heterologous" refers to a sequence that is not normally expressed and
secreted by an organism (e.g.,
a wild-type organism). In some embodiments, the term encompasses a sequence
that comprises two
or more subsequences which are not found in the same relationship to each
other as normally found in
nature, or is recombinantly engineered so that its level of expression, or
physical relationship to other
nucleic acids or other molecules in a cell, or structure, is not normally
found in nature. For instance, a
heterologous nucleic acid is typically recombinantly produced, having two or
more sequences from
unrelated genes arranged in a manner not found in nature (e.g., a nucleic acid
open reading frame
(ORF) of the invention operatively linked to a promoter sequence inserted into
an expression cassette,
such as a vector). In some embodiments, "heterologous polynucleotide" refers
to any polynucleotide
that is introduced into a host cell by laboratory techniques, and includes
polynucleotides that are
removed from a host cell, subjected to laboratory manipulation, and then
reintroduced into a host cell.
[0074] As used herein, "codon optimized" refers to changes in the codons of
the polynucleotide
encoding a protein to those preferentially used in a particular organism such
that the encoded protein
is efficiently expressed in the organism of interest. In some embodiments, the
polynucleotides
17
CA 03061133 2019-10-22
WO 2018/200214 PCT/US2018/027450
encoding the ketoreductase enzymes may be codon optimized for optimal
production from the host
organism selected for expression.
[0075] As used herein, "control sequence" is defined herein to include all
components, which are
necessary or advantageous for the expression of a poly-nucleotide and/or
polypeptide of the present
invention. Each control sequence may be native or foreign to the
polynucleotide of interest. Such
control sequences include, but are not limited to, a leader, polyadenylation
sequence, propeptide
sequence, promoter, signal peptide sequence, and transcription terminator.
[0076] As used herein, "operably linked" is defined herein as a configuration
in which a control
sequence is appropriately placed (i.e., in a functional relationship) at a
position relative to a
polynucleotide of interest such that the control sequence directs or regulates
the expression of the
polynucleotide and/or polypeptide of interest.
[0077] As used herein, the phrases "cofactor regeneration system" and
"cofactor recycling system"
refer to a set of reactants that participate in a reaction that reduces the
oxidized form of the cofactor
(e.g., NADP+ to NADPH). Cofactors oxidized by the ketoreductase-catalyzed
reduction of the keto
substrate are regenerated in reduced form by the cofactor regeneration system.
Cofactor regeneration
systems comprise a stoichiometric reductant that is a source of reducing
hydrogen equivalents and is
capable of reducing the oxidized form of the cofactor. The cofactor
regeneration system may further
comprise a catalyst, for example an enzyme catalyst that catalyzes the
reduction of the oxidized form
of the cofactor by the reductant. Cofactor regeneration systems to regenerate
NADH or NADPH from
NAD+ or NADP+, respectively, are known in the art and may be used in the
methods described
herein.
100781 As used herein, "suitable reaction conditions" refer to those
conditions in the biocatalytic
reaction solution (e.g., ranges of enzyme loading, substrate loading, cofactor
loading, temperature,
pH, buffers, co-solvents, etc.) under which ketoreductase polypeptides of the
present invention are
capable of stereoselectively deracemizing a substrate compound to a product
compound. Exemplary
"suitable reaction conditions" are provided in the present invention and
illustrated by the Examples.
[0079] As used herein, "loading," such as in "compound loading," "enzyme
loading," or "cofactor
loading" refers to the concentration or amount of a component in a reaction
mixture at the start of the
reaction.
[0080] As used herein, "substrate" in the context of a biocatalyst mediated
process refers to the
compound or molecule acted on by the biocatalyst. For example, an exemplary
substrate for the
ketoreductase biocatalyst in the process disclosed herein is compound (1).
[0081] As used herein "product" in the context of a biocatalyst mediated
process refers to the
compound or molecule resulting from the action of the biocatalyst.
[0082] As used herein, "equilibration" as used herein refers to the process
resulting in a steady state
concentration of chemical species in a chemical or enzymatic reaction (e.g.,
interconversion of two
18
CA 03061133 2019-10-22
WO 2018/200214 PCT/US2018/027450
species A and B), including interconversion of stereoisomers, as determined by
the forward rate
constant and the reverse rate constant of the chemical or enzymatic reaction.
[0083] As used herein, "oxo" refers to =0.
[0084] As used herein, "oxy" refers to a divalent group -0-, which may have
various substituents to
form different oxy groups, including ethers and esters.
[0085] As used herein, "carboxy" refers to -COOH.
[0086] As used herein, "carbonyl" refers to -C(0)-, which may have a variety
of substituents to form
different carbonyl groups including acids, acid halides, aldehydes, amides,
esters, and ketones.
[0087] As used herein, "hydroxy" refers to -OH.
[0088] As used herein, "optional" and "optionally" means that the subsequently
described event or
circumstance may or may not occur, and that the description includes instances
where the event or
circumstance occurs and instances in which it does not. One of ordinary skill
in the art would
understand that with respect to any molecule described as containing one or
more optional
substituents, only sterically practical and/or synthetically feasible
compounds are meant to be
included.
[0089] As used herein, "optionally substituted" refers to all subsequent
modifiers in a term or series
of chemical groups. For example, in the term "optionally substituted
atylalkyl, the "alkyl" portion and
the "aryl" portion of the molecule may or may not be substituted, and for the
series "optionally
substituted alkyl, cycloalkyl, aryl and heteroaryl," the alkyl, cycloalkyl,
aryl, and heteroaryl groups,
independently of the others, may or may not be substituted.
Engineered Enzyme Polypeptides
[0090] Ketoreductase (KRED) or carbonyl reductase biocatalysts (EC 1.1.1.184)
are useful for the
synthesis of alcohols from aldehydes and ketones, and optically active
secondary alcohols from the
corresponding prostereoisomeric ketone substrates. KREDs may also catalyze the
reverse reaction,
(i.e., oxidation of an alcohol substrate to the corresponding aldehydes/ketone
product). The reduction
of aldehydes and ketones and the oxidation of alcohols by KREDs uses a co-
factor, most commonly
reduced nicotinamide adenine dinucleotide (NADH) or reduced nicotinamide
adenine dinucleotide
phosphate (NADPH), and nicotinamide adenine dinucleotide (NAD) or nicotinamide
adenine
dinucleotide phosphate (NADP+) for the oxidation reaction. NADH and NADPH
serve as electron
donors, while NAD+ and NADP+ serve as electron acceptors.
[0091] KREDs can be found in a wide range of bacteria and yeasts, as known in
the art (See e.g.,
Hummel and Kula Eur. J. Biochem., 184:1-13 [1989]). Numerous KRED genes and
enzyme
sequences have been reported, including those of Caliendo magnoliae (Genbank
Ace. No. JC7338;
GI:11360538); Candida parapsilosis (Genbank Ace. No. BAA24528.1; GI:2815409),
Sporobolomyces salmonicolor (Genbank Ace. No. AF160799; GI:6539734),
Lactobacillus kefir
19
CA 03061133 2019-10-22
WO 2018/200214
PCT/US2018/027450
(Genbank Acc. No. AAP94029.1; GI: 33112056), Lactobacillus brevis (Genbank
Acc. No. 1NXQ_A;
GI: 30749782), and Thermoanaerobium brockil (Genbank Acc. No. P14941; GI:
1771790).
[0092] The stereoselectivity of ketoreductases have been applied to the
preparation of important
pharmaceutical building blocks (See e.g., Broussy et al., Org. Lett., 11:305-
308 [2009]). Specific
applications of naturally occurring or engineered KREDs in biocatalytic
processes to generate useful
chemical compounds have been demonstrated for reduction of 4-
chloroacetoacetate esters (See e.g,.
Zhou, J. Am. Chem. Soc.,105:5925-5926 [1983]; Santaniello, J. Chem. Res.,
(S)132-133 [1984]; U.S.
Patent Nos. 5,559,030; U.S. Patent No. 5,700,670; and U.S. Patent No.
5,891,685), reduction of
dioxocarboxylic acids (See e.g., U.S. Patent No. 6,399,339), reduction of tert-
butyl (S)-chloro-5-
hydroxy-3-oxohexanoate (See e.g., U.S. Patent No. 6,645,746; and WO 01/40450),
reduction
pyrrolotriazine-based compounds (See e.g., U.S. Appin. Publ. No.
2006/0286646); reduction of
substituted acetophenones (See e.g., U.S. Patent Nos. 6,800,477 and
8,748,143); and reduction of
ketothiolanes (WO 2005/054491).
[0093] The present invention provides engineered ketoreductases capable of
deracemizing the
substrate compound (1), (6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-7-ol), in one-
pot, multi-enzyme
system as shown in the following reaction and Figure 1.
OH .
________________ eq oxidant -12 (.. P 1 eq reciuctant""' = N
,
*"r
cµfõ, 1. toloxnletleciontive 2011%1:nye
(1\1"...AOH
1.::
oxidation
..t0H
¨ non
ect
aq toi-.38ion N
CA 03061133 2019-10-22
WO 2018/200214 PCT/US2018/027450
1, s4,-4eatoo mkiation, Z. sekt=c*tv mimacsii
W.4)/1 Ose
H2o.
NAO NO41-1 =
$44:ladiw / RstMettivo
NiW.F.1144,k0H KRED.1f#1 :
,
/ \ 4,<A0:==
NADf41 N.4t3P
N;i2r404 Narai:
OICAOtiffl 4iShYdr*Oriatt
[NM The present invention further provides improved ketoreductase enzymes and
improved
phosphite dehydrogenase enzymes, and methods for using the engineered
ketoreductase and phosphite
dehydrogenase enzymes to deracemize chiral compounds in one-pot, multi-enzyme
system.
[00951 It is important to note that the desired product can be obtained in a
one-pot, one-step, multi-
enzyme system only if the oxidation and reduction reactions are orthogonal,
compatible and non-
interacting. These conditions are only satisfied if the oxidative
ketoreductase and its corresponding
recycling enzyme use one co-factor exclusively (e.g., NAD), and reductive
ketoreductase and its
corresponding recycling enzyme use the opposite co-factor exclusively (i.e.,
NADPH).
100961 Compound (1) has one chiral center and can exist in two different
diastereomeric tbrins (la
and lb). The deracemization reaction by a tandem of ketoreductases can result
in two different
enantiomeric products (la-lb), as shown in Figure 2 and below.
r\-9.1 OH
T
,N
f OH
/
100971 However, (la) is the only desired product. The evolution program used
in the development of
the present invention was designed to improve activity of an S-selective
ketoreductase that would
oxidize the S-alcohol in the racemic mixture, generating a ketone substrate
for an R-selective
ketoreductase. Further, evolution program was designed to improve the
selectivity, activity and
21
CA 03061133 2019-10-22
WO 2018/200214 PCT/US2018/027450
cofactor preference of the R-selective ketoreductase. Evolution was also
designed to improve activity,
stability and cofactor preference of a phosphite dehydrogenase to enable
deracemization of substrate
(1) to product (1a) with minimal amount of ketone and (lb) in a one-pot, one-
step, multi-enzyme
process.
[0098] The ketoreductase polypeptide of SEQ ID NO:2 was selected as the
initial backbone for
development of the improved S-selective enzymes provided by the present
invention. This enzyme
was chosen as the starting backbone as ketone (2) was produced via oxidation
of only (lb), leaving
(la). The ketoreductase polypeptide of SEQ ID NO:2 uses NADI as a co-factor
with an efficiency
greater than 200:1 over NADP' and can be coupled with a commercially available
NADH oxidase to
recycle the co-factor.
[0099] The ketoreductase polypeptide of SEQ ID NO:2 was selected as the
initial backbone for
development of R-selective enzymes to reduce a ketone to product (1a) with
initial selectivity of
92.7% e.e. Enantioselectivity values are calculated herein according to
equation (1) provided below.
(1) {[(la amount) ¨ (lb amount)] / [(la amount) + (lb amount)] ) x
100
[0100] Indeed, the non-naturally occurring ketoreductase polypeptides of the
present invention are
ketoreductases engineered to have improved properties as compared to the
naturally occurring
ketoreductase of SEQ ID NO:2.
[0101] A phosphite dehydrogenase polypeptide was selected as the initial
backbone for development
of the improved PDH enzymes. This enzyme is equally efficient recycling both
NADH and NADPH.
[0102] In some embodiments, the engineered ketoreductase polypeptides are
capable of converting
the substrate compound to product with an activity that is increased at least
about 1.2 fold, 1.5 fold, 2
fold, 3 fold, 4 fold, 5 fold, 10 fold, 20 fold, 30 fold, 40 fold, 50 fold, or
100 fold relative to the activity
of the reference polypeptide of SEQ ID NO:2 under suitable reaction
conditions. In some
embodiments, the engineered ketoreductase polypeptides are capable of
converting the substrate
compound to product with a percent conversion of at least about 40%, at least
about 50%, at least
about 60%, at least about 70%, at least about 80%, or at least about 900/, at
least about 95%, at least
about 98%, at least about 99%, in a reaction time of about 48 h, about 36 h,
about 24 h, or even
shorter length of time, under suitable reaction conditions.
[0103] In some embodiments, the engineered ketoreductases and phosphite
dehydrogenases are
capable of converting substrate compound (1) to product compound (1a) in
enantiomeric excess over
compound (lb) in a one-pot, one-step, multi-enzyme system. In some
embodiments, the engineered
ketoreductases and phosphite dehydrogenases are capable of converting compound
(1) to compound
(la) in diastereomeric excess over compound (lb) under suitable reaction
conditions.
[0104] As will be appreciated by those of skill in the art, some of the above-
defined categories,
unless otherwise specified, are not mutually exclusive. Thus, amino acids
having side chains
22
CA 03061133 2019-10-22
WO 2018/200214 PCT/US2018/027450
exhibiting two or more physico-chemical properties can be included in multiple
categories. The
appropriate classification of any amino acid or residue will be apparent to
those of skill in the art,
especially in light of the detailed invention provided herein.
[0105] In some embodiments, the improved engineered ketoreductase enzymes and
engineered
phosphite dehydrogenase enzymes comprise amino acid residue deletions in the
naturally occurring
ketoreductase or phosphite dehydrogenase polypeptides or deletions of amino
acid residues in other
engineered ketoreductase or phosphite dehydrogenase polypeptides. Thus, in
some embodiments of
the invention, the deletions comprise one or more amino acids, 2 or more amino
acids, 3 or more
amino acids, 4 or more amino acids, 5 or more amino acids, 6 or more amino
acids, 8 or more amino
acids, 10 or more amino acids, 15 or more amino acids, or 20 or more amino
acids, up to 10% of the
total number of amino acids, up to 10% of the total number of amino acids, up
to 20% of the total
number of amino acids, or up to 30% of the total number of amino acids of the
ketoreductase
polypeptides, as long as the functional activity of the ketoreductase or
phosphite dehydrogenase
activity is maintained. In some embodiments, the deletions can comprise, 1-2,
1-3, 1-4, 1-5, 1-6, 1-7,
1-8, 1-9, 1-10, 1-11, 1-12, 1-14, 1-15, 1-16, 1-18, 1-20, 1-22, 1-24, 1-25, 1-
30, 1-35 or about 1-40
amino acid residues. In some embodiments, the number of deletions can be 1, 2,
3, 4, 5, 6, 7, 8, 9, 10,
11, 12, 14, 15, 16, 18, 20, 22, 24, 26, 30, 35 or about 40 amino acids. In
some embodiments, the
deletions can comprise deletions of 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13,
14, 15, 16, 18, or 20 amino
acid residues.
[0106] As described herein, the ketoreductase or phosphite dehydrogenase poly-
peptides of the
invention can be in the form of fusion poly-peptides in which the
ketoreductases or phosphite
dehydrogenase polypeptides are fused to other polypeptides, such as antibody
tags (e.g., myc epitope)
or purifications sequences (e.g., His tags). Thus, in some embodiments, the
ketoreductase and/or
phosphite dehydrogenase polypeptides find use with or without fusions to other
polypeptides.
[0107] In some embodiments, the polypeptides described herein are not
restricted to the genetically
encoded amino acids. In addition to the genetically encoded amino acids, the
polypeptides described
herein may be comprised, either in whole or in part, of naturally-occurring
and/or synthetic non-
encoded amino acids. Certain commonly encountered non-encoded amino acids of
which the
polypeptides described herein may be comprised include, but are not limited
to: the D-stereomers of
the genetically-encoded amino acids; 2,3-diaminopropionic acid (Dpr); a-
aminoisobutyric acid (Aib);
s-aminohexanoic acid (Aha); 8-aminovaleric acid (Ava); N-methylglycine or
sarcosine (MeGly or
Sar); ornithine (Om); citrulline (Cit); t-butylalanine (Bua); t-butylglycine
(Bug); N-methylisoleucine
(MeIle); phenylglycine (Phg); cyclohexylalanine (Cha); norleucine (Nle);
naphthylalanine (Nal); 2-
chlorophenylalanine (0cf); 3-chlorophenylalanine (Mcf); 4-chlorophenylalanine
(Pet);
2-fluorophenylalanine (Off); 3-fluorophenylalanine (Mff); 4-
fluorophenylalanine (HO; 2-
bromophenylalanine (Obf); 3-bromophenylalanine (Mbf); 4-bromophenylalanine
(Pbf); 2-
23
CA 03061133 2019-10-22
WO 2018/200214 PCT/US2018/027450
methylphenylalanine (Omf); 3-methylphenylalanine (Mmf); 4-methylphenylalanine
(Pmf); 2-
nitrophenylalanine (Onf); 3-nitrophenylalanine (Mnf); 4-nitrophenylalanine
(Pnt); 2-
cyanophenylalanine (Oct); 3-cyanophenylalanine (Mcf); 4-cyanophenylalanine
(Pet); 2-
trifluoromethylphenylalanine (Ott); 3-trifluoromethylphenylalanine (Mtf); 4-
trifluoromethylphenylalanine (Ptf); 4-aminophenylalanine (Paf); 4-
iodophenylalanine (Pif); 4-
aminomethylphenylalanine (Paint); 2,4-dichlorophenylalanine (Opef); 3,4-
dichlorophenylalanine
(Mpcf); 2,4-difluorophenylalanine (Opff); 3,4-difluorophenylalanine (Mpff);
pyrid-2-ylalanine
(2pAla); pyrid-3-ylalanine (3pAla); pyrid-4-ylalanine (4pAla); naphth-l-
ylalanine ( lnAla); naphth-2-
ylalanine (2nAla); thiazolylalanine (taAla); benzothienylalanine (bAla);
thienylalanine (tAla);
furylalanine (fAla); homophenylalanine (hPhe); homotyrosine (hTyr);
homotryptophan (hTrp);
pentafluorophenylalanine (5ff); styrylkalanine (sAla); authtylalanine (aAla);
3,3-diphenylalanine
(Dfa); 3-amino-5-phenypentanoic acid (Afp); penicillamine (Pen); 1,2,3,4-
tetrahydroisoquinoline-3-
carboxylic acid (Tic); 3-2-thienylalanine (Thi); methionine sulfoxide (Mso);
N(w)-nitroarginine
(nArg); homolysine (hLys); phosphonomethylphenylalanine (pmPhe); phosphoserine
(pSer);
phosphothreonine (p'Thr); homoaspartic acid (hAsp); homoglutanic acid (hGlu);
1-aminocyclopent-(2
or 3)-ene-4 carboxylic acid; pipecolic acid (PA), azetidine-3-carboxylic acid
(ACA); 1-
aminocyclopentane-3-carboxylic acid; allylglycine (aOly); propargylglycine
(pgGly); homoalanine
(hAla); norvaline (nVal); homoleucine (hLeu), homovaline (hVal);
homoisolencine (hue);
homoarginine (hArg); N-acetyl lysine (AcLys); 2,4-diaminobutyric acid (Dbu);
2,3-diaminobutyric
acid (Dab); N-methylvaline (MeVal); homocysteine (hCys); homoserine (hSer);
hydroxyproline
(Hyp) and homoproline (hPro). Additional non-encoded amino acids of which the
polypeptides
described herein may be comprised are apparent to those of skill in the art.
These amino acids may be
in either the L- or D-configuration.
101081 Those of skill in the art will recognize that amino acids or residues
bearing side chain
protecting groups may also comprise the polypeptides described herein. Non-
limiting examples of
such protected amino acids, which in this case belong to the aromatic
category, include (protecting
groups listed in parentheses), but are not limited to: Arg(tos),
Cys(methylbenzyl), Cys
(nitropyridinesulfenyl), Glu(8-benzylester), Gln(xanthyl), Asn(N-8-xanthyl),
His(bom), His(benzyl),
His(tos), Lys(ftnoc), Lys(tos), Ser(0-benzyl), Thr (0-benzyl) and Tyr(0-
benzyl).
101091 Non-encoding amino acids that are conformationally constrained of which
the polypeptides
described herein may be composed include, but are not limited to, N-methyl
amino acids
(L-configuration); 1-aminocyclopent-(2 or 3)-ene-4-carboxylic acid; pipecolic
acid; azetidine-3-
carboxylic acid; homoproline (hPro); and 1-aminocyclopentane-3-carboxylic
acid.
[01101 As described above the various modifications introduced into the
naturally occurring
polypeptide to generate an engineered ketoreductase enzymes and engineered
phosphite
dehydrogenase enzymes can be targeted to a specific property of the enzyme.
24
CA 03061133 2019-10-22
WO 2018/200214 PCT/US2018/027450
Polynucleotides Encoding Engineered Enzymes
[0111] In another aspect, the present invention provides polynucleotides
encoding the engineered
ketoreductase enzymes and engineered phosphite dehydrogenase enzymes. The
polynucleotides may
be operatively linked to one or more heterologous regulatory sequences that
control gene expression
to create a recombinant polynucleotide capable of expressing the polypeptide.
Expression constructs
containing a heterologous pots nucleotide encoding the engineered
ketoreductase and/or engineered
phosphite dehydrogenase can be introduced into appropriate host cells to
express the corresponding
ketoreductase or phosphite dehydrogenase poly-peptide.
[0112] Because of the knowledge of the codons corresponding to the various
amino acids,
availability of a protein sequence provides a description of all the
polynucleotides capable of
encoding the subject. The degeneracy of the genetic code, where the same amino
acids are encoded
by alternative or synonymous codons allows an extremely large number of
nucleic acids to be made,
all of which encode the improved ketoreductase enzymes and/or improved
phosphite dehydrogenase
enzymes disclosed herein. Thus, having identified a particular amino acid
sequence, those skilled in
the art could make any number of different nucleic acids by simply modifying
the sequence of one or
more codons in a way which does not change the amino acid sequence of the
protein. In this regard,
the present invention specifically contemplates each and every possible
variation of polynucleotides
that could be made by selecting combinations based on the possible codon
choices, and all such
variations are to be considered specifically disclosed for any poly-peptide
disclosed herein, including
the amino acid sequences presented in the Tables in the Examples. In various
embodiments, the
codons are preferably selected to fit the host cell in which the protein is
being produced. For
example, preferred codons used in bacteria are used to express the gene in
bacteria; preferred codons
used in yeast are used for expression in yeast; and preferred codons used in
mammals are used for
expression in mammalian cells.
[0113] hi some embodiments, the engineered ketoreductase or phosphite
dehdyrogeanse sequences
comprise sequences that comprise positions identified to be beneficial, as
described in the Examples.
[0114] In some embodiments, isolated polynucleotides encoding an improved
ketoreductase or
phosphite dehydrogenase polypeptides are manipulated in a variety of ways to
provide for improved
expression and/or production of the polypeptides. Manipulation of the isolated
polymicleotide prior
to its insertion into a vector may be desirable or necessary, depending on the
expression vector used.
The techniques for modifying poly-nucleotides and nucleic acid sequences
utilizing recombinant DNA
methods are well known in the art.
[0115] For bacterial host cells, suitable promoters for directing
transcription of the nucleic acid
constructs of the present invention. include the promoters obtained from the
E. coil lac operon,
Streptomyces coelicolor agarase gene (dagA), Bacillus subtilis levansucrase
gene (sacB), Bacillus
licheniformis alpha-amylase gene (amyL), Bacillus stearothermophilus
maltogenic amylase gene
CA 03061133 2019-10-22
WO 2018/200214 PCT/US2018/027450
(amyM), Bacillus amyloliquefaciens alpha-amylase gene (amyQ), Bacillus
licheniformis penicillinase
gene (penP), Bacillus subtilis xylA and xylB genes, and prokaryotic beta-
lactamase gene (See e.g.,
Villa-Kamaroff et al., Proc. Natl. Acad. Sci. USA 75: 3727-3731 [1978]), as
well as the tac promoter
(See e.g., DeBoer et al., Proc. Natl Acad. Sci. USA 80: 21-25 [1983]).
Additional suitable promoters
are known to those in the art.
[0116] For filamentous fungal host cells, suitable promoters for directing the
transcription of the
nucleic acid constructs of the present invention include promoters obtained
from the genes for
Aspergillus oryzae TAKA amylase, Rhizomucor miehei aspartic proteinase,
Aspergillus niger neutral
alpha-amylase, Aspergillus niger acid stable alpha-amylase, Aspergillus niger
or Aspergillus crwamori
glucoamylase (glaA), Rhizomucor miehei lipase, Aspergillus oryzae alkaline
protease, Aspergillus
otyzae triose phosphate isomerase, Aspergillus nidulans acetamidase, and
Fusarium oxysporum
trypsin-like protease (WO 96/00787), as well as the NA2-tpi promoter (a hybrid
of the promoters
from the genes for Aspergillus niger neutral alpha-amylase and Aspergillus
oryzae triose phosphate
isomerase), and mutant, truncated, and hybrid promoters thereof.
[0117] In a yeast host, useful promoters include, but are not limited to those
from the genes for
Saccharomyces cerevisiae enolase (ENO-1), Saccharomyces cerevisiae
galactolcinase (GAL1),
Saccharomyces cerevisiae alcohol dehydrogenase/glyceraldehyde-3-phosphate
dehydrogenase
(ADH2/GAP), and Saccharomyces cerevisiae 3-phosphoglycerate kinase, as well as
other useful
promoters for yeast host cells (See e.g., Romanos et al., Yeast 8:423-488
[1992]).
[0118] The control sequence may also be a suitable transcription terminator
sequence, a sequence
recognized by a host cell to terminate transcription. The terminator sequence
is operably linked to the
3' terminus of the nucleic acid sequence encoding the polypeptide. Any
terminator that is functional in
the host cell of choice may be used in the present invention.
[0119] For example, exemplary transcription terminators for filamentous fungal
host cells can be
obtained from the genes for Aspergillus oryzae TAKA amylase, Aspergillus niger
glucoamylase,
Aspergillus nidulans anthranilate syrithase, Aspergillus niger alpha-
glucosidase, and Fusarium
oxysporum trypsin-like protease.
[0120] Exemplary terminators for yeast host cells can be obtained from the
genes for Saccharomyces
cerevisiae enolase, Saccharomyces cerevisiae cytochrome C (CYC1), and
Saccharomyces cerevisiae
glyceraldehyde-3-phosphate dehydrogenase, as well as other useful terminators
for yeast host cells
known in the art (See e.g,. Romanos et al., supra).
[0121] The control sequence may also be a suitable leader sequence, a
nontranslated region of an
mRNA that is important for translation by the host cell. The leader sequence
is operably linked to the
5' terminus of the nucleic acid sequence encoding the poly-peptide. Any leader
sequence that is
functional in the host cell of choice may be used. Exemplary leaders for
filamentous fungal host cells
are obtained from the genes for Aspergillus oryzae TAKA amylase and
Aspergillus nidulans triose
phosphate isomerase. Suitable leaders for yeast host cells are obtained from
the genes for
26
CA 03061133 2019-10-22
WO 2018/200214 PCT/US2018/027450
Saccharomyces cerevisiae enolase (ENO-1), Saccharomyces cerevisiae 3-
phosphoglycerate kinase,
Saccharomyces cerevisiae alpha-factor, and Saccharomyces cerevisiae alcohol
dehydrogenase/glyceraldehyde-3-phosphate dehydrogenase (ADH2/GAP).
[0122] The control sequence may also be a polyadenylation sequence, a sequence
operably linked to
the 3' terminus of the nucleic acid sequence and which, when transcribed, is
recognized by the host
cell as a signal to add polyadenosine residues to transcribed mRNA. Any
polyadenylation sequence
which is functional in the host cell of choice may be used in the present
invention. Exemplary
polyadenylation sequences for filamentous fungal host cells can be from the
genes for Aspergillus
oiyzae TAKA amylase, Aspergillus niger glucoamylase, Aspergillus nidulans
anthranilate syrithase,
Fusarium oxysporum trypsin-like protease, and Aspergillus niger alpha-
glucosidase., as well as
additional useful polyadenylation sequences for yeast host cells known in the
art (See e.g., Guo et al.,
Mol. Cell. Biol., 15:5983-5990 [1995]).
[0123] The control sequence may also be a signal peptide coding region that
codes for an amino acid
sequence linked to the amino terminus of a polypeptide and directs the encoded
polypeptide into the
cell's secretory pathway. The 5' end of the coding sequence of the nucleic
acid sequence may
inherently contain a signal peptide coding region naturally linked in
translation reading frame with the
segment of the coding region that encodes the secreted polypeptide.
Alternatively, the 5' end of the
coding sequence may contain a signal peptide coding region that is foreign to
the coding sequence.
The foreign signal peptide coding region may be required where the coding
sequence does not
naturally contain a signal peptide coding region.
[0124] Alternatively, the foreign signal peptide coding region may simply
replace the natural signal
peptide coding region in order to enhance secretion of the polypeptide.
However, any signal peptide
coding region which directs the expressed polypeptide into the secretory
pathway of a host cell of
choice may be used in the present invention.
[0125] Effective signal peptide coding regions for bacterial host cells are
the signal peptide coding
regions obtained from the genes for Bacillus NC1B 11837 maltogenic amylase,
Bacillus
stearothermophilus alpha-amylase. Bacillus licheniformis subtilisin, Bacillus
licheniformis beta-
lactamase, Bacillus stearothermophilus neutral proteases (nprT, nprS, nprM),
and Bacillus subtilis
prsAõ as well as additional signal peptides known in the art (See e.g.,
Simonen et al., Microbiol. Rev.,
57: 109-137 [1993]).
[0126] Effective signal peptide coding regions for filamentous fungal host
cells include, but are not
limited to the signal peptide coding regions obtained from the genes for
Aspergillus oryzae TAKA
amylase, Aspergillus niger neutral amylase, Aspergillus niger glucoamylase,
Rhizomucor miehei
aspartic proteinase, Humicola insolens cellulase, and Humicola lanuginosa
lipase. Useful signal
peptides for yeast host cells can be from the genes for Saccharomyces
cerevisiae alpha-factor and
Saccharomyces cerevisiae invertase, as well as additional useful signal
peptide coding regions (See
e.g., Romanos et al., 1992, supra).
27
CA 03061133 2019-10-22
WO 2018/200214 PCT/US2018/027450
[0127] The control sequence may also be a propeptide coding region that codes
for an amino acid
sequence positioned at the amino terminus of a polypeptide. The resultant
polypeptide is known as a
proenzyme or propolypeptide (or a zymogen in some cases). A propolypeptide is
generally inactive
and can be converted to a mature active polypeptide by catalytic or
autocatalytic cleavage of the
propeptide from the propolypeptide. The propeptide coding region may be
obtained from the genes
for Bacillus subtilis alkaline protease (aprE), Bacillus subtilis neutral
protease (nprT), Saccharomyces
cerevisiae alpha-factor, Rhizomucor miehei aspartic proteinase, and
Myceliophthora the rmophila
lactase (WO 95/33836).
[0128] Where both signal peptide and propeptide regions are present at the
amino terminus of a
polypeptide, the propeptide region is positioned next to the amino terminus of
a polypeptide and the
signal peptide region is positioned next to the amino terminus of the
propeptide region.
[0129] It may also be desirable to add regulatory sequences, which allow the
regulation of the
expression of the polypeptide relative to the growth of the host cell.
Examples of regulatory systems
are those which cause the expression of the gene to be turned on or off in
response to a chemical or
physical stimulus, including the presence of a regulatory compound. In
prokaryotic host cells,
suitable regulatory sequences include the lac, tac, and tip operator systems.
In yeast host cells,
suitable regulatory systems include, as examples, the ADH2 system or GAL!
system. In filamentous
fungi, suitable regulatory sequences include the TAKA alpha-amylase promoter,
Aspergillus niger
glucoamylase promoter, and Aspergillus olyzae glucoamylase promoter.
[0130] Other examples of regulatory sequences are those which allow for gene
amplification. In
eukaryotic systems, these include the dihydrofolate reductase gene, which is
amplified in the presence
of methotrexate, and the metallothionein genes, which are amplified with heavy
metals. In these
cases, the nucleic acid sequence encoding the KRED polypeptide of the present
invention or the PDH
polypeptide of the present invention would be operably linked with the
regulatory sequence.
[0131] Thus, in some embodiments, the present invention is also directed to a
recombinant
expression vector comprising a polynucleotide encoding an engineered
ketoreductase polypeptide or a
variant thereof, or an engineered phosphite dehydrogenase polypeptide or a
variant thereof, and one or
more expression regulating regions such as a promoter and a terminator, a
replication origin, etc.,
depending on the type of hosts into which they are to be introduced. The
various nucleic acid and
control sequences described above may be joined together to produce a
recombinant expression vector
which may include one or more convenient restriction sites to allow for
insertion or substitution of the
nucleic acid sequence encoding the polypeptide at such sites. Alternatively,
the nucleic acid sequence
of the present invention may be expressed by inserting the nucleic acid
sequence or a nucleic acid
construct comprising the sequence into an appropriate vector for expression.
In creating the
expression vector, the coding sequence is located in the vector so that the
coding sequence is operably
linked with the appropriate control sequences for expression.
28
CA 03061133 2019-10-22
WO 2018/200214 PCT/US2018/027450
101321 The recombinant expression vector may be any vector (e.g., a plasmid or
virus), which can be
conveniently subjected to recombinant DNA procedures and can bring about the
expression of the
polynucleotide sequence. The choice of the vector will typically depend on the
compatibility of the
vector with the host cell into which the vector is to be introduced. The
vectors may be linear or closed
circular plasmids.
[0133] The expression vector may be an autonomously replicating vector (i.e.,
a vector that exists as
an extrachromosomal entity), the replication of which is independent of
chromosomal replication,
(e.g., a plasmid, an extrachromosomal element, a minichromosome, or an
artificial chromosome).
The vector may contain any means for assuring self-replication. Alternatively,
the vector may be one
which, when introduced into the host cell, is integrated into the genome and
replicated together with
the chromosome(s) into which it has been integrated. Furthermore, a single
vector or plasmid or two
or more vectors or plasmids which together contain the total DNA to be
introduced into the genome of
the host cell, or a transposon may be used.
[0134] The expression vector of the present invention preferably contains one
or more selectable
markers, which permit easy selection of transformed cells. A selectable marker
can be a gene the
product of which provides for biocide or viral resistance, resistance to heavy
metals, prototrophy to
auxotrophs, and the like. Examples of bacterial selectable markers are the dal
genes from Bacillus
subtilis or Bacillus lichenifbrmis, or markers, which confer antibiotic
resistance such as ampicillin,
kanamycin, chloramphenicol, or tetracycline resistance. Suitable markers for
yeast host cells are
ADE2, HI53, LEU2, LYS2, MET3, TRP1, and URA3.
[0135] Selectable markers for use in a filamentous fungal host cell include,
but are not limited to,
amdS (acetamidase), argB (omithine carbamoyltransferase), bar
(phosphinothricin acetyltransferase),
hph (hygromycin phosphotransferase), niaD (nitrate reductase), pyrG (orotidine-
5'-phosphate
decarboxylase), sC (sulfate adenyltransferase), and trpC (anthranilate
synthase), as well as equivalents
thereof. Embodiments for use in an Aspergillus cell include the amdS and pyrG
genes of Aspergillus
nidulans or Aspergillus oryzae and the bar gene ofStreptomyces hygroscopicus.
[0136] The expression vectors of the present invention can contain an
element(s) that permits
integration of the vector into the host cell's genome or autonomous
replication of the vector in the cell
independent of the genome. For integration into the host cell genome, the
vector may rely on the
nucleic acid sequence encoding the polypeptide or any other element of the
vector for integration of
the vector into the genome by homologous or nonhomologous recombination.
[0137] Alternatively, the expression vector may contain additional nucleic
acid sequences for
directing integration by homologous recombination into the genome of the host
cell. The additional
nucleic acid sequences enable the vector to be integrated into the host cell
genome at a precise
location(s) in the chromosome(s). To increase the likelihood of integration at
a precise location, the
integrational elements should preferably contain a sufficient number of
nucleic acids, such as 100 to
10,000 base pairs, preferably 400 to 10,000 base pairs, and most preferably
800 to 10,000 base pairs,
29
CA 03061133 2019-10-22
WO 2018/200214 PCT/US2018/027450
which are highly homologous with the corresponding target sequence to enhance
the probability of
homologous recombination. The integrational elements may be any sequence that
is homologous
with the target sequence in the genome of the host cell. Furthermore, the
integrational elements may
be non-encoding or encoding nucleic acid sequences. On the other hand, the
vector may be integrated
into the genome of the host cell by non-homologous recombination.
[0138] For autonomous replication, the vector may further comprise an origin
of replication enabling
the vector to replicate autonomously in the host cell in question. Examples of
bacterial origins of
replication are P15A on or the origins of replication of plasmids pBR322,
pUC19, pACYC177 (which
plasmid has the PISA on), or pACYC184 permitting replication in E. coli, and
pUB110, pE194,
pTA1060, or pAMp I permitting replication in Bacillus. Examples of origins of
replication for use in
a yeast host cell are the 2 micron origin of replication, ARS1, ARS4, the
combination of ARS1 and
CEN3, and the combination of ARS4 and CEN6. The origin of replication may be
one having a
mutation which makes it's functioning temperature-sensitive in the host cell
(See e.g., Ehrlich, Proc.
Natl. Acad. Sci. USA 75:1433 [1978]).
[0139] More than one copy of a nucleic acid sequence of the present invention
may be inserted into
the host cell to increase production of the gene product. An increase in the
copy number of the
nucleic acid sequence can be obtained by integrating at least one additional
copy of the sequence into
the host cell genome or by including an amplifiable selectable marker gene
with the nucleic acid
sequence where cells containing amplified copies of the selectable marker
gene, and thereby
additional copies of the nucleic acid sequence, can be selected for by
cultivating the cells in the
presence of the appropriate selectable agent.
[0140] Many of the expression vectors for use in the present invention are
commercially available.
Suitable commercial expression vectors include, but are not limited to
p3xFLAGTMTm expression
vectors (Sigma-Aldrich), which include a CMV promoter and hGH polyadenylation
site for
expression in mammalian host cells and a pBR322 origin of replication and
ampicillin resistance
markers for amplification in E. coll. Other commercially available suitable
expression vectors include
but are not limited to the pBluescriptIl SK(-) and pBK-CMV vectors
(Stratagene), and plasmids
derived from pBR322 (Gibco BRL), pUC (Gibco BRL), pREP4, pCEP4 (Invitrogen) or
pPoly (See,
Lathe et al., Gene 57:193-201 [1987]).
Host Cells for Expression of Engineered Polypeptides
[0141] The present invention also provides a host cell comprising a
polynucleotide encoding an
improved ketoreductase polypeptide or an improved phosphite dehydrogenase
polypeptide of the
present invention, the polynucleotide being operatively linked to one or more
control sequences for
expression of the ketoreductase enzyme or the phosphite dehydrogenase enzyme
in the host cell. Host
cells for use in expressing the KRED polypeptides encoded by the expression
vectors of the present
invention or the PDH polypeptides encoded by the expression vectors of the
present invention are
CA 03061133 2019-10-22
WO 2018/200214 PCT/US2018/027450
well known in the art and include but are not limited to, bacterial cells,
such as E. coil, Lactobacillus
kfir, Lactobacillus brevis, Lactobacillus minor, Streptomyces and Salmonella
typhimurium cells;
fungal cells, such as yeast cells (e.g., Saccharomyces cerevisiae or Pichia
pastoris (ATCC Accession
No. 201178)); insect cells such as Drosophila S2 and Spodoptera Sf9 cells:
animal cells such as CHO,
COS, BHK, 293, and Bowes melanoma cells; and plant cells. Appropriate culture
media and growth
conditions for the above-described host cells are well known in the art.
[0142] Polynucleotides for expression of the ketoreductase or the phosphite
dehydrogenase may be
introduced into cells by various methods known in the art. Techniques include
among others,
electroporation, biolistic particle bombardment, liposome mediated
transfection, calcium chloride
transfection, and protoplast fusion. Various methods for introducing
polynucleotides into cells will be
apparent to the skilled artisan.
[0143] Escherichia coil W3110 is a host strain that finds use in the present
invention, although it is
not intended that the present invention be limited to this specific host
strain. The expression vector
was created by operatively linking a polynucleotide encoding an improved
enzyme into the plasmid
pCK110900 operatively linked to the lac promoter under control of the lad
repressor. The expression
vector also contained the P15a origin of replication and the chloramphenicol
resistance gene. Cells
containing the subject poly-nucleotide in Escherichia coil W3110 can be
isolated by subjecting the
cells to chloramphenicol selection.
Methods of Generating Engineered ketoreductase Polypeptides and Engineered
Phosphite
Dehydrogenase Polypeptides.
[0144] In some embodiments, to make the improved KRED polynucleotides and
polypeptides of the
present invention, the naturally-occurring ketoreductase enzyme that catalyzes
the reduction reaction
is obtained (or derived) from Candida parasilosis or Sporodiobolus
salmonicolor. In some
embodiments, the parent polynucleotide sequence is codon optimized to enhance
expression of the
ketoreductase in a specified host cell. As an illustration, the parental
polynucleotide sequence
encoding the wild-type KRED polypeptide of Sporodiobolus salmonicolor was
constructed from
oligonucleotides prepared based upon the known polypeptide sequence of
Sporodiobolus
salmonicolor KRED sequence available from the Genbank database. The parental
polynucleotide
sequence was codon optimized for expression in E. coli and the codon-optimized
polynucleotide
cloned into an expression vector, placing the expression of the ketoreductase
gene under the control of
the lac promoter and lad repressor gene. Clones expressing the active
ketoreductase in E. coil were
identified and the genes sequenced to confirm their identity.
[0145] In some embodiments, the engineered ketoreductases are obtained by
subjecting the
polynucleotide encoding the naturally occurring ketoreductase to mutagenesis
and/or directed
evolution methods, as discussed above. Mutagenesis may be performed in
accordance with any of the
techniques known in the art, including random and site-specific mutagenesis.
Directed evolution can
31
CA 03061133 2019-10-22
WO 2018/200214 PCT/US2018/027450
be performed with any of the techniques known in the art to screen for
improved promoter variants
including shuffling. Mutagenesis and directed evolution methods are well known
in the art (See e.g..
US Patent Nos. 5,605,793, 5,811,238, 5,830,721, 5,834,252, 5,837,458,
5,928,905, 6,096,548,
6,117,679, 6,132,970, 6,165,793, 6,180,406, 6,251,674, 6,265,201, 6,277,638,
6,287,861, 6,287,862,
6,291,242, 6,297,053, 6,303,344, 6,309,883, 6,319,713, 6,319,714, 6,323,030,
6,326,204, 6,335,160,
6,335,198, 6,344,356, 6,352,859, 6,355,484, 6,358,740, 6,358,742, 6,365,377,
6,365,408, 6,368,861,
6,372,497, 6,337,186, 6,376,246, 6,379,964, 6,387,702, 6,391,552, 6,391,640,
6,395,547, 6,406,855,
6,406,910, 6,413,745, 6,413,774, 6,420,175, 6,423,542, 6,426,224, 6,436,675,
6,444,468, 6,455,253,
6,479,652, 6,482,647, 6,483,011, 6,484,105, 6,489,146, 6,500,617, 6,500,639,
6,506,602, 6,506,603,
6,518,065, 6,519,065, 6,521,453, 6,528,311, 6,537,746, 6,573,098, 6,576,467,
6,579,678, 6,586,182,
6,602,986, 6,605,430, 6,613,514, 6,653,072, 6,686,515, 6,703,240, 6,716,631,
6,825,001, 6,902,922,
6,917,882, 6,946,296,6,961,664, 6,995,017, 7,024,312, 7,058,515, 7,105,297,
7,148,054, 7,220,566,
7,288,375, 7,384,387, 7,421,347, 7,430,477, 7,462,469, 7,534,564, 7,620,500,
7,620,502, 7,629,170,
7,702,464, 7,747,391, 7,747,393, 7,751,986, 7,776,598, 7,783,428, 7,795,030,
7,853,410, 7,868,138,
7,783,428, 7,873,477, 7,873,499, 7,904,249, 7,957,912, 7,981,614, 8,014,961,
8,029,988, 8,048,674,
8,058,001, 8,076,138, 8,108,150, 8,170,806, 8,224,580, 8,377,681, 8,383,346,
8,457,903, 8,504,498,
8,589,085, 8,762,066, 8,768,871, 9,593,326, and all related non-US
counterparts; Ling etal., Anal.
Biochem., 254(2):157-78 [1997]; Dale etal., Meth. Mol. Biol., 57:369-74
[1996]; Smith, Ann. Rev.
Genet., 19:423-462 [1985]; Botstein etal., Science, 229:1193-1201 [1985];
Carter, Biochem. J.,
237:1-7 [1986]; Kramer etal., Cell, 38:879-887 [1984]; Wells etal., Gene,
34:315-323 [1985];
Minshull etal., Curr. Op. Chem. Biol., 3:284-290 [1999]; Christians etal..
Nat. Biotechnol., 17:259-
264 [1999]; Crameri etal., Nature, 391:288-291 [1998]; Crameri, etal., Nat.
Biotecluiol., 15:436-438
[1997]; Zhang etal., Proc. Nat. Acad. Sci. U.S.A., 94:4504-4509 [1997];
Crameri etal., Nat.
Biotecluiol., 14:315-319 [1996]; Stemmer, Nature, 370:389-391 [1994]; Stemmer,
Proc. Nat. Acad.
Sci. USA, 91:10747-10751 [1994]; WO 95/22625: WO 97/0078; WO 97/35966; WO
98/27230; WO
00/42651; WO 01/75767; and WO 2009/152336, all of which are incorporated
herein by reference).
101461 The clones obtained following mutagenesis treatment are screened for
engineered
ketoreductases having a desired improved enzyme property. Measuring enzyme
activity from the
expression libraries can be performed using the standard biochemistry
technique of monitoring the
rate of decrease (via a decrease in absorbance or fluorescence) of NADH or
NADPH concentration, as
it is converted into NAD+ or NADP+ . In this reaction, the NADH or NADPH is
consumed (oxidized)
by the ketoreductase as the ketoreductase reduces a ketone substrate to the
corresponding hydroxyl
group. The rate of decrease of NADH or NADPH concentration, as measured by the
decrease in
absorbance or fluorescence, per unit time indicates the relative (enzymatic)
activity of the KRED
polypeptide in a fixed amount of the lysate (or a lyophilized powder made
therefrom). The
stereochemistry, of the products can be ascertained by various known
techniques, and as provided in
the Examples. Where the improved enzyme property desired is thermal stability,
enzyme activity
32
CA 03061133 2019-10-22
WO 2018/200214 PCT/US2018/027450
may be measured after subjecting the enzyme preparations to a defined
temperature and measuring the
amount of enzyme activity remaining after heat treatments. Clones containing a
polynucleotide
encoding a ketoreductase are then isolated, sequenced to identify the
nucleotide sequence changes (if
any), and used to express the enzyme in a host cell.
[0147] Where the sequence of the engineered polypeptide is known, the
polynucleotides encoding
the enzyme can be prepared by standard solid-phase methods, according to known
synthetic methods.
In some embodiments, fragments of up to about 100 bases can be individually
synthesized, then
joined (e.g., by enzymatic or chemical ligation methods, or poly-merase
mediated methods) to fonn
any desired continuous sequence. For example, polymicleotides and
oligonucleotides of the invention
can be prepared by chemical synthesis (e.g., using the classical
phosphoramidite method described by
Beaucage et al., Tet. Lett., 22:1859-69 [1981], or the method described by
Matthes et al., EMBO J.,
3:801-05 [1984], as it is typically practiced in automated synthetic methods).
According to the
phosphoramidite method, oligonucleotides are synthesized (e.g., in an
automatic DNA synthesizer),
purified, annealed, ligated and cloned in appropriate vectors. In addition,
essentially any nucleic acid
can be obtained from any of a variety of commercial sources (e.g., The Midland
Certified Reagent
Company, Midland, TX, The Great American Gene Company, Ramona, CA, ExpressGen
Inc.
Chicago, IL, Operon Technologies Inc., Alameda, CA, and many others).
[0148] Engineered ketoreductase enzymes and engineered phosphite dehydrogenase
enzymes
expressed in a host cell can be recovered from the cells and or the culture
meditun using any one or
more of the well known techniques for protein purification, including, among
others, lysozyme
treatment, sonication, filtration, salting-out, ultra-centrifugation, and
chromatography. Suitable
solutions for lysing and the high efficiency extraction of proteins from
bacteria, such as E. coil, are
commercially available under the trade name CelLytic B.' (Sigma-Aldrich).
[0149] Chromatographic techniques for isolation of the ketoreductase and/or
phosphite
dehydrogenase polypeptides include, among others, reverse phase chromatography
high performance
liquid chromatography, ion exchange chromatography, gel electrophoresis, and
affinity
chromatography. Conditions for purifying a particular enzyme will depend, in
part, on factors such as
net charge, hydrophobicity, hydrophilicity, molecular weight, molecular shape,
etc., and will be
apparent to those having skill in the art.
[0150] In some embodiments, affinity techniques are used to isolate the
improved ketoreductase
enzymes and/or improved phosphite dehydrogenase enzymes. For affinity
chromatography
purification, any antibody which specifically binds the ketoreductase
polypeptide or the phosphite
dehydrogenase polypeptide may be used. For the production of antibodies,
various host animals,
including but not limited to rabbits, mice, rats, etc., may be immunized by
injection with the
ketoreductase or the phosphite dehydrogenase. The ketoreductase polypeptide
may be attached to a
suitable carrier, such as BSA, by means of a side chain functional group or
linkers attached to a side
chain functional group. Various adjuvants may be used to increase the
immunological response,
33
CA 03061133 2019-10-22
WO 2018/200214 PCT/US2018/027450
depending on the host species, including but not limited to Freund's (complete
and incomplete),
mineral gels such as aluminum hydroxide, surface active substances such as
lysolecithin, pluronic
polyols, polyanions, peptides, oil emulsions, keyhole limpet hemocyanin,
dinitrophenol, and
potentially useful human adjuvants such as BCG (Bacillus Calmette Guerin) and
Corynebacterium
parvum
[0151] The ketoreductases and/or the phosphite dehydrogenases may be prepared
and used in the
form of cells expressing the enzymes, as crude extracts, or as isolated or
purified preparations. The
ketoreductases and/or the phosphite dehydrogenases may be prepared as
lyophilizates, in powder form
(e.g., acetone powders), or prepared as enzyme solutions. In some embodiments,
the ketoreductases
or the phosphite dehydrogenases can be in the form of substantially pure
preparations.
[0152] In some embodiments, the ketoreductase poly:peptides and/or the
phosphite dehydrogenase
polypeptides can be attached to a solid substrate. The substrate can be a
solid phase, surface, and/or
membrane. A solid support can be composed of organic polymers such as
polystyrene, polyethylene,
polypropylene, polyfluoroethylene, polyethyleneoxy, and polyacrylamide, as
well as co-polymers and
grails thereof. A solid support can also be inorganic, such as glass, silica,
controlled pore glass
(CPG), reverse phase silica or metal, such as gold or platinum. The
configuration of the substrate can
be in the form of beads, spheres, particles, granules, a gel, a membrane or a
surface. Surfaces can be
planar, substantially planar, or non-planar. Solid supports can be porous or
non-porous, and can have
swelling or non-swelling characteristics. A solid support can be configured in
the form of a well,
depression, or other container, vessel, feature, or location. A plurality of
supports can be configured
on an array at various locations, addressable for robotic delivery of
reagents, or by detection methods
and/or instruments.
[0153] As is known by those of skill in the art, ketoreductase-catalyzed
reduction reactions typically
require a cofactor. Reduction reactions catalyzed by the engineered
ketoreductase enzymes described
herein also typically require a cofactor, although many embodiments of the
engineered ketoreductases
require far less cofactor than reactions catalyzed with wild-type
ketoreductase enzymes. As used
herein, the term "cofactor" refers to a non-protein compound that operates in
combination with a
ketoreductase enzyme. Cofactors suitable for use with the engineered
ketoreductase enzymes
described herein include, but are not limited to, NADP- (nicotinamide adenine
dinucleotide
phosphate), NADPH (the reduced form of NADP+), NAD+ (nicotinamide adenine
dinucleotide) and
NADH (the reduced form of NADI). Generally, the reduced form of the cofactor
is added to the
reaction mixture. The reduced NAD(P)H form can be optionally regenerated from
the oxidized
NAD(P)+ form using a cofactor regeneration system. The term "cofactor
regeneration system" refers
to a set of reactants that participate in a reaction that reduces the oxidized
form of the cofactor (e.g.,
NADP+ to NADPH). Cofactors oxidized by the ketoreductase-catalyzed reduction
of the keto
substrate are regenerated in reduced form by the cofactor regeneration system.
Cofactor regeneration
systems comprise a stoichiometric reductant that is a source of reducing
hydrogen equivalents and is
34
CA 03061133 2019-10-22
WO 2018/200214 PCT/US2018/027450
capable of reducing the oxidized form of the cofactor. The cofactor
regeneration system may further
comprise a catalyst, for example an enzyme catalyst, that catalyzes the
reduction of the oxidized form
of the cofactor by the reductant. Cofactor regeneration systems to regenerate
NADH or NADPH from
NAD+ or NADI)+, respectively, are known in the art and may be used in the
methods described herein.
EXPERIMENTAL
[0154] Various features and embodiments of the invention are illustrated in
the following
representative examples, which are intended to be illustrative, and not
limiting.
[0155] In the experimental disclosure below, the following abbreviations
apply: ppm (parts per
million); M (molar); mM (millimolar), uM and tiM (micromolar); nM (nanomolar);
mol (moles); gm
and g (gram); mg (milligrams); ug and gg (micrograms); L and I (liter); ml and
mL (milliliter); cm
(centimeters); mm (millimeters); um and gm (micrometers); sec. (seconds);
min(s) (minute(s)); h(s)
and hr(s) (hour(s)); U (units); MW (molecular weight); rpm (rotations per
minute); C (degrees
Centigrade); RT (room temperature); CDS (coding sequence); DNA
(deoxyribonucleic acid); RNA
(ribonucleic acid); HPLC (high performance liquid chromatography); FIOPC (fold
improvement over
positive control); HTP (high throughput); LB (Luria broth); Sigma-Aldrich
(Sigma-Aldrich, St.
Louis, MO); Millipore (Millipore, Corp., Billerica MA); Difco (Difco
Laboratories, BD Diagnostic
Systems, Detroit, MI); Daicel (Daicel, West Chester, PA); Genetix (Genetix
USA, Inc., Beaverton,
OR); Molecular Devices (Molecular Devices, LLC, Sunnyvale, CA); Applied
Biosystems (Applied
Biosystems, part of Life Technologies, Corp., Grand Island, NY), Agilent
(Agilent Technologies, Inc.,
Santa Clara, CA); Thermo Scientific (part of Thermo Fisher Scientific,
Waltham, MA); Corning
(Coming, Inc., Palo Alto, CA); and Bio-Rad (Bio-Rad Laboratories, Hercules,
CA).
EXAMPLE 1
Ketoreductase and Phosphite Dehydrogenase Gene Construction and Expression
Vectors
101561 The wild-type Candida parapsilois ketoreductase (KRED) encoding gene
was amplified from
genomic DNA and cloned into expression vector pCK11 0900 (See, Figure 3 of US
Pat. Appin.
Publn. No. 2006/0195947, herein incorporated by reference) under the control
of a lac promoter. The
expression vector also contained the P I5a origin of replication and the
chloramphenicol resistance
gene. The activity of the wild-type ketoreductase was confirmed as described
in W02008/042876.
Polynucleotides encoding engineered ketoreductases of the present invention
were likewise cloned
into vector pCK11 0900 for expression in E. coil W311 0. Directed evolution of
the KRED gene was
carried out by first selecting the parent gene (i.e., SEQ ID NOS: 2, 6, 104)
followed by library
construction of variant genes in which positions associated with certain
structural features were
subjected to mutagenesis. These libraries were then plated, grown-up, and
screened using HTP assays
as described in Examples 2, 5 and 12.
CA 03061133 2019-10-22
WO 2018/200214 PCT/US2018/027450
[0157] The wild-type Sporidiobolus salmonicolor ketoreductase (KRED) encoding
gene was
synthesized for expression in E. colt based on the reported amino acid
sequence of the ketoreductase
and a codon optimization algorithm as described in Example 1 of W02008/042876,
incorporated
herein by reference. The gene was synthesized using oligonucleotides composed
of 42 nucleotides
and cloned into expression vector pCK11 0900 (See, Figure 3 of US Pat. Appin.
Publn. No.
2006/0195947, herein incorporated by reference) under the control of a lac
promoter. The expression
vector also contained the Pl5a origin of replication and the chloramphenicol
resistance gene. The
activity of the wild-type ketoreductase was confirmed as described in
W02008/042876.
Polynucleotides encoding engineered ketoreductases of the present invention
were likewise cloned
into vector pCK11 0900 for expression in E. coli W311 0. Directed evolution of
the KRED gene was
carried out by first selecting the parent gene (i.e., SEQ ID NOS: 112, 124,
138) followed by library
construction of variant genes in which positions associated with certain
structural features were
subjected to mutagenesis. These libraries were then plated, grown, and
screened using HIP assays as
described in Examples 3, 6, 7, 8 and 12.
[0158] A variant of the wild-type Pseudomonas stutzeri phosphite dehydrogenase
(PDH) encoding
gene was cloned into expression vector pCK11 0900 (See, Figure 3 of US Pat.
Appin. Publn. No.
2006/0195947, herein incorporated by reference) under the control of a lac
promoter. The expression
vector also contained the P15a origin of replication and the chloramphenicol
resistance gene. The
activity of the phosphite dehydrogenase was confirmed as described in
W02008/042876.
[0159] Polynucleotides encoding engineered phosphite dehydrogenases of the
present invention were
likewise cloned into vector pCK11 0900 for expression in E. colt W311 0.
Directed evolution of the
PDH gene was carried out by first selecting the parent gene (i.e., SEQ ID NOS:
172, 182, 200, 208,
260) followed by library construction of variant genes in which positions
associated with certain
structural features were subjected to mutagenesis. These libraries were then
plated, grown, and
screened using HTP assays as described in Examples 4, and 9 through 12.
EXAMPLE 2
Production and Analysis of Engineered KRED Polypeptides for Oxidation
[0160] Plasmid libraries obtained through directed evolution and containing
evolved ketoreductase
genes were transformed into E. colt W3110 and placed on Luria-Bertani (LB)
agar medium
containing 1% glucose and 30 pg/ml chloramphenicol (CAM). After incubation for
at least 16 h at
30 C, colonies were picked using a Q-bot6 robotic colony picker (Genetix) into
a 96-well shallow
well microtiter plate containing 200 pL of LB, 1% glucose, and 30 ig/m1 CAM.
Cells were grown
18-20 h at 30C, with shaking at 200 rpm. Twenty pi, of this culture was then
transferred to 360 L
of Terrific Broth (TB), 1mM MgCl2, 2mM ZnSO4 and 30 p.g/m1 CAM. After
incubation of deep well
plates at 30T with shaking at 250 rpm for 2.5 h (0D600 0.6-0.8), recombinant
gene expression was
36
CA 03061133 2019-10-22
WO 2018/200214 PCT/US2018/027450
induced by isopropyl thioglycoside (IPTG) to a final concentration of 1 mM.
The plates were then
incubated at 30 'C with shaking at 250 rpm for 18-21 h.
101611 Cell cultures were pelleted at 3500 x g for 20 mM, and their
supernatants were discarded.
Cell pellets were lysed in 300 pL of 20 mM Tris, 2mM ZnSO4, 1 mM MgCl2 pH 7.5
with 1 g/L
lysozyme and 0.5 g/L polymixin B sulfate by shaking at RT for 2 h. Samples
were centrifuged at
3500 x g for 20 min to clarify cellular debris, and the supernatant was used
to carry out the
transformations described in Examples 5 and 12.
EXAMPLE 3
Production and Analysis of Engineered KRED Polypeptides for Reduction
[0162] Plasmid libraries obtained through directed evolution and containing
evolved ketoreductase
genes were transformed into E. coli W3110 and placed on Luria-Bertani (LB)
agar medium
containing I% glucose and 30 pg/ml chloramphenicol (CAM). After incubation for
at least 16 hat
30T, colonies were picked using a Q-boe robotic colony picker (Genetix) into a
96-well shallow
well microtiter plate containing 200 pL of LB, 1% glucose, and 30 pg/ml CAM.
Cells were grown
18-20 h at 30'C, with shaking at 200 rpm. Twenty pL of this culture was then
transferred to 360 pL
of Terrific Broth (TB), 1mM MgSO4, and 30 pg/ml CAM. After incubation of deep
well plates at
30'C with shaking at 250 rpm for 2.5 h (01)600 0.6-0.8), recombinant gene
expression was induced by
isopropyl thioglycoside (IPTG) to a final concentration of 1 mM. The plates
were then incubated at
30 T with shaking at 250 rpm for 18-21 h.
[0163] Cell cultures were pelleted at 3500 x g for 20 mM, and their
supernatants were discarded.
Cell pellets were lysed in 300 LiL of 20 mM Tris, 1 mM MgSO4, pH 7.5 with 1
g/L lysozyme and 0.5
g/L polymixin B sulfate by shaking at RT for 2 h. Samples were centrifuged at
3500 x g for 20 rnM to
clarify cellular debris, and the supernatant was used to carry out the
transformations described in
Examples 6 through 8, and Example 12.
EXAMPLE 4
Production and Analysis of Engineered Phosphite Dehydrogenase Polypeptides
[0164] Plasmid libraries obtained through directed evolution and containing
evolved phosphite
dehydrogenase genes were transformed into E. coli W3110 and placed on Luna-
Bertani (LB) agar
medium containing 1% glucose and 30 !vim' chloramphenicol (CAM). After
incubation for at least
16 h at 30T, colonies were picked using a Q-bot6 robotic colony picker
(Genetix) into a 96-well
shallow well microtiter plate containing 200 p...L of LB, 1% glucose, and 30
pg/ml CAM. Cells were
grown 18-20 h at 30'C, with shaking at 200 rpm. Twenty L of this culture was
then transferred to
360 pL of Terrific Broth (TB) and 30 pg/m1 CAM. After incubation of deep well
plates at 30'C with
shaking at 250 rpm for 2.5 h (0D600 0.6-0.8), recombinant gene expression was
induced by isopropyl
37
CA 03061133 2019-10-22
WO 2018/200214 PCT/US2018/027450
thioglycoside (IPTG) to a final concentration of 1 mM. The plates were then
incubated at 30 'C with
shaking at 250 rpm for 18-21 h.
1101651 Cell cultures were pelleted at 3500 x g for 20 mM, and their
supernatants were discarded.
Cell pellets were lysed in 300 1i.1., of 20 mM Tris, pH 7.5 with 1 g/L
lysozyme and 0.5 g/L polymixin
B sulfate by shaking at RT for 2 h. Samples were centrifuged at 3500 x g for
20 mM to clarify
cellular debris, and the supernatant was used to carry out the transformations
described in Examples 9
through 12.
EXAMPLE 5
KRED Variants of SEQ ID NO:2
1101661 E coli KRED variants were generated as described in Example 1. To
analyze the activity of
the variants, 20 uL of supernatant produced as described in Example 2 were
added to a mixture of 180
L racemic alcohol substrate (50 g/L), with 4g/L NAD+, I Og(L commercially
available NADH
oxidase (N0x-9) and 100mM FAD in 100 mM sodium phosphite pH 8Ø Reactions
were incubated
at 30 'C for 16-18h, and quenched via addition of 200 L of 1M HC1. The
quenched mixture was
added to the sample and briefly mixed. Reaction samples were analyzed by UPLC
to quantify
residual substrate and products as described above. Significantly improved
variants are provided in
Table 5.1, below.
Table 5.1. Variants With Improved Activity Compared to SEQ ID NO:2
Amino Acid Substitutions
SEQ ID NO: Improvement
(Relative to SEQ ID NO:2)
4 R309F ++4-
6 C57L +++
8 G114K +++
10 G272V +++
12 G263Y +++
14 L276F -H-+
16 C57I
18 G272P +++
20 0272L -H-+
22 6114M ++
24 G272S -F++
26 6272Q -F++
28 G272H +-H.
30 G272T +-H.
32 611411 +++
34 62721 +++
36 C57X/W286X +++
38 6272W -F+
38
CA 03061133 2019-10-22
WO 2018/200214
PCT/US2018/027450
Table 5.1. Variants With Improved Activity Compared to SEQ ID NO:2
Amino Acid Substitutions
SEQ ID NO: Improvement
(Relative to SEQ ID NO:2)
40 I279H +1-
42 G263H -H-+
44 1-145R 4-F+
46 S268M
48 S268W ++
50 I,274V
52 V831 ++
54 Y52D ++
56 1279R ++4
58 Y52S ++4
60 I279Q ++
62 L274I ++
64 D56L +++
66 KlIOT
68 P228S ++
70 S138V/A146S/M258V/1289S
72 K2I1R
74 K37R
76 K37R/K211R/G229R ++
78 K211R/G229R
80 G229R ++
82 K37R/G229R
84 K37R/K211R ++
86 L276M i 4
88 I79T/V83S/A275N/L276M t
90 V83S/A275N/L276M t+
92 V83S/L276M ++
94 A275N/L276M
96 L55F/C57A/I,276M
98 A104G ++
100 C571/A104G/G114H -t f+
102 C57L/A104G/G114H/G229R -t
104 Y52S/C57L/G272HVI279H/L296F
106 Y52D/C57L/G27214 +++F
108 Y52S/C5711G272H/L274V/I279H/1.:296F -F-F-HF
110 Y52S/C57L/K110T/G272H/L296F ++++
Key for Table 5.1
4 t >6
+++ >4 and <6
>2.5 and < 4
39
CA 03061133 2019-10-22
WO 2018/200214 PCT/US2018/027450
> 1.2 and < 2.5
EXAMPLE 6
KRED Variants of SEQ ID NO:112
[0167] E. con KRED variants were generated as described in Example 1. To
analyze the activity of
the variants, 5 pL supernatant produced as described in Example 3 were added
to 95 pt of 0.3 M
phosphite buffer pH 7.9 containing 0.25 mM NADPH; 19 g/L ketone substrate and
5 g/L PDH.
Reactions were incubated at room temperature for 16-18 hours with gentle
shaking. Reactions were
quenched via addition of 100 AL of 1M HC1. The quenched mixture (10 pL) was
diluted into 190 pL
of water. Reaction samples (10 pL) were analyzed by HPLC to quantify residual
substrates and
products as described above. Significantly improved variants are provided in
Table 6.1, below.
Table 6.1 Variants With Improved Activity and Selectivity Compared to SEQ ID
NO:112
SEQ ID Amino Acid Substitutions Activity
Selectivity
NO: (Relative to SEQ ID NO:112) Improvement
Improvement
114 V241/S2200/13314R/S315A
n.d.
116 V2411T106P/S136A/5220G/L258V/C260A/P314R/S315A
n.d.
118 V2411T106P/F214L/A250V/L258V/C260A/P314R/S315A
n.d.
120 T122E/1159V/L316E/1318L -H-
n.d.
122 1159VN251Q/Y272F/T277P/L316E/1318L/1330L
124 N207G 4
+-H-
.
126 N207G
128 V135F
++
130 V135F ++
++
132 I139V/N207S +++
+-F+
Key for Table 6.1
Activity Selectivity
+-1--F >4 >5
++ > 2.5 and < 4 > 2 and < 5
+ > 1.5 and < 2.5 > I and < 2
EXAMPLE 7
KRED Variants of SEQ ID NO:124
101681 E. coil KRED variants were generated as described in Example 1. To
analyze the activity of
the variants, 7.5 pt supernatant produced as described in Example 3 were added
to 192.5 pt of 0.3
M phosphite buffer pH 7.9 containing 0.25 mM NADPH; 50 g/L ketone substrate
and 5 g/L PDH.
CA 03061133 2019-10-22
WO 2018/200214 PCMS2018/027450
Reactions were incubated at room temperature for 16-18 hours with gentle
shaking. Reactions were
quenched via addition of 100 gL of 1M HC1. The quenched mixture (10 pL) was
diluted into 190 pL
of water. Reaction samples (10 pL) were analyzed by HPLC to quantify residual
substrates and
products as described above. Significantly improved variants are provided in
Table 7.1, below.
Table 7.1 Variants With Improved Activity Compared to SEQ ID NO:124
SEQ ID Amino Acid Substitutions Activity
NO: (Relative to SEQ ID NO:124) Improvement
134 V95T 4
136 V241/V95T/M228T 4
138 V95TN135F/1139V/G207N
140 K3YN95T +-HE
142 K3YN95T/M228T/P314R +-F+
144 A2T/Y 101P/A179L/T182M/M228R/A238L/T282E -H-
146 1159V/M228L/K309Q/1330L
Key for Table 7.1
++++ >4
+++ > 3 and < 4
=
++ >2 and<3
> 1.5 and<2
EXAMPLE 8
ICRED Variants of SEQ ID NO:138
101691 E. coil KRED variants were generated as described in Example 1. To
analyze the co-factor
preference of the variants, four separate assays were utilized. First, 10 gL
supernatant produced as
described in Example 3 were added to 90 p.L of 0.2 M phosphite buffer pH 7.9
containing 1 g/L
ketone and 1 g/L of NADPH. The initial rate of NADPH consumption of the
samples was analyzed
via fluorescence with Ex 1=330nm Em =445nm, acquired for 180 seconds every 21
seconds.
[01701 Second, 20 tiL supernatant produced as described in Example 3 were
added to 190 pi of
0.2 M phosphite buffer pH 7.9 containing lg/L racemic alcohol and 2g/L of
NAD1. The initial rate of
NAD+ consumption was analyzed via kinetic readings at UV 340nm, data were
acquired every 9
seconds for 5 minutes.
(01711 Third, 20 pi, supernatant produced as described in Example 3 were added
to 180 1i1., of
500mM sodium phosphite containing 2g/L imidazole ketone and 16.4mM of NADPH;
the samples
were incubated at room temperature for 2 hr, shaking at 300rpm. Reactions were
quenched via
addition of 200 pL of MeCN. After shaking for 5 minutes, 100 pL of the
quenched reaction was
transferred to a Millipore filter plate (45micron pore size) with a co-star
round bottom plate
41
CA 03061133 2019-10-22
WO 2018/200214 PCMS2018/027450
containing 100 j.tL of water to collect the filtrate and the mixture was spun
at 4000rpm for 2minutes.
Reaction samples (10 pL) were analyzed by HPLC to quantify residual substrate
and product as
described above.
101721 'Fourth, 20 pL supernatant produced as described in Example 3 were
added to 180 fiL of
500mM sodium phosphite containing 2g/L imidazole ketone and 16.4mM of NADPH;
the samples
were incubated at room temperature for 2 hr, shaking at 300rpm. Reactions were
quenched via
addition of 200 LtL of MeCN. After shaking for 5 minutes, 100 LtL of the
quenched reaction was
transferred to a Millipore filter plate (45micron pore size) with a co-star
round bottom plate
containing 100 tiL of water to collect the filtrate and the mixture was spun
at 4000rpm for 2minutes.
Reaction samples (10 pL) were analyzed by HPLC to quantify residual substrate
and product as
described above. Co-factor specificity was calculated as
(amount of product generated with NADPH) / (amount of product generated with
NADH)
Significantly improved variants are provided in Table 8.1, below.
Table 8.1. Variants With Improved Activity and Co-factor Specificity Compared
to SEQ ID NO:138
SEQ ID Amino Acid Substitutions Activity
Co-factor
NO: (Relative to SEQ ID NO:1.38) Improvement
Specificity
Improvement
148 V24I/A43V/547E/L49N/A67V1V68E/E70P/I91V/S220G
150 V24IN68E/I91V/T218N/S220G ++
152 Y78F/P107G ++
154 K74A/Q75E/Y78F/A108V ++
++4-
156 Q75E/Y78F/N99P/A108V/D215S/S224A ++
+++
158 G195 -HF-F -
F+
160 T95C -HF-F
162 S96G -HHF
164 G195 +++
166 M72Q +++ ++
168 A67W ++
=
170 N114V ++
Key for Table 8.1
Activity Improvement Co-factor Specificity Improvement
+++ >4 >3
++ >2 and<4 > 2 and < 3
>land<2 > 1 and < 2
42
CA 03061133 2019-10-22
WO 2018/200214
PCT/US2018/027450
EXAMPLE 9
PDH Variants of SEQ ID NO:172
1101731 E coli PDH variants were generated as described in Example 1. To
analyze the activity of
the variants, 51AL supernatant produced as described in Example 3 were added
to 95 L of 0.5 M
sodium phosphite buffer pH 7.9 containing 0.25 mM NADPH; 50 g/L ketone
substrate and 2 g/L
KRED of SEQ ID NO:138. Reactions were incubated at 25 C for 16-18 hours with
gentle shaking.
Reactions were quenched via addition of 100 L of 1M HC1. The quenched mixture
(10 pL ) was
diluted into 190 pL of water. Reaction samples (10 pL) were analyzed by HPLC
to quantify residual
substrates and products as described above. Significantly improved variants
are provided in Table
9.1, below.
Table 9.1 Variants With Improved Activity Compared to SEQ ID NO:172
SEQ ID Amino Acid Substitutions
Activity
NO: (Relative to SEQ ID NO:172)
Improvement
174 R1OK/C73A/R137Q
õ
176 R1OK/C73A/F78YN2331/N323D
õ
178 R137QN2331/E303A/N323D
180 R I OKJC73A/F78Y/R137Q/N323D/V325A
82 R44A/R132Q/N145G
184 El3D/R41A/Q63A/R132Q/A193S/S I 95E
186 R41A/R44A/A88R/A193S/S195E
188 E266V
190 E266W
192 E266S
194 R44A/R132Q/P136D/R137Q/N145G/1293L -H-
196 R44A/R132Q/R1371/N145GN2331/A308V/N323D ++
198 R44A/R132Q/Q135A/P136D/R1371/N145G/1293L ++
200 R44A/R132Q/R1371/N145G/1293L/N323D ++
202 R44A/R132Q/N145G/S195E/I293L/N323D ++
204 R44A/V113S/R132Q/N145G ++
206 Li 8fv1/R44A/L 119F/A 124E/R132Q/R1371/N145G/1293L/N323D/A334K/C336R
-H-F
208 R44A/L119F/R132Q/R137UN145G/L158K/A175S/K1771/1293L/A317R/N323D -F-H-
210 L 1 8M/R44A/L119F/A124E/R 132Q/R1371/N145G/L158K/IC177T/1293L/N323D
212 -F-H-
Ll8M/R44A/L119F/A124E/R132Q/R1371/N145G/L158K/A175S/K177T/1293L/
+++
A317R/N323D
214 R44A/R69K/R120V/R132Q/R1371/N145G/A175T/S195E/1293L/N323D
-
216 S32V/R44A/R132Q/R1371/N145G/R186TN233U1293L/N323D/C336S
Key for Table 9.1
>4
++ >2and<4
> 1 and < 2
43
CA 03061133 2019-10-22
WO 2018/200214 PCT/US2018/027450
EXAMPLE 10
PDH Variants of SEQ ID NO:208
101741 E coli PDH variants were generated as described in Example 1. To
analyze the co-factor
preference of the variants, supernatant produced as described in Example 3 was
diluted 4-fold with
50mM Tris-HC1 buffer, pH 7.5. Twenty IA of the diluted lysate was added to 180
p.L of 0.1 M
sodium phosphite buffer pH 7.9 and incubated overnight to consume residual NAD
and NADP+
present in the lysate. The variants were then screened in three separate
assays to analyze their co-
factor specificity. First, for the initial rate NADP+ assay, 0.2 mM NADI"' in
0.1 M sodium phosphite
buffer pH 7.9 was added and initial rate measured via fluorescence assay over
2 minutes. Second, for
initial rate NAD" assay, 0.2 mM NAD+ in 0.1M sodium phosphite buffer pH 7.9
was added and
initial rate measured via fluorescence assay over 2 minutes. Third, a co-
factor competition assay was
performed. For this assay, 100 mM phosphite pH 7.9 containing 100 uM NADP, 1
mM NAD and 1
g/L NADH oxidase NOx-9 was added to the reaction. NOx-9 consumes all NADH
immediately,
leaving only NADPH signal, reduced by competition between NADP+ and NAD+.
Reactions were
quenched via addition of 100 pL of 1M HCl. The quenched mixture (10 pL) was
diluted into 190 pL
of water. Diluted reaction samples (10 pL) were analyzed by HF'LC to quantify
residual substrates and
products as described above. Significantly improved variants are provided in
Table 10.1, below.
Table 10.1. Variants With Improved Co-factor Specificity Compared to SEQ ID
NO:208
SEQ ID NO: Amino Acid Substitutions NADP+ Initial
Cofactor Specificity
(Relative to SEQ ID NO:208) Rate Improvement
improvement
218 F78Y/F1501/F1981.1R327S/L328P
220 N211A/D213Q/1322Q
222 A178P/C194L/N211A/D21.3Q/1322
224 F951/N211A/D213Q/1322M
226 S32V/A59M/A124E/T177S/Q191H/R327D
228 L215P
230 L206N
232 TIO4F ++
234 T104L
236 E266S
238 V262P
240 V262D
242 V83A/E266A
244 D323N
44
CA 03061133 2019-10-22
WO 2018/200214
PCT/US2018/027450
Key for Table 10.1
-H- >2
> I and < 2
EXAMPLE 11
Additional PDH Variants of SEQ ID NO:208
[01751 E. coil PDH variants were generated as described in Example 1. To
analyze the co-factor
preference of the variants, supernatant produced as described in Example 3 was
diluted 4-fold with
50mM Tris-HC1 buffer, pH 7.5. Twenty jiL of the diluted lysate was added to
180 pL of 0.1 M
sodium phosphite buffer pH 7.9 and incubated overnight to consume residual NAD
and NADP+
present in the lysate. The variants were then screened in three separate
assays to analyze their co-
factor specificity. First, for the initial rate NADP" assay 0.2 mM NADP+ in
0.1 M soditun phosphite
buffer pH 7.9 was added and initial rate measured via fluorescence assay over
2 minutes. Second, for
initial rate NAD" assay, 0.2 mM NAD+ in 0.1M sodium phosphite buffer pH 7.9
was added and
initial rate measured via fluorescence assay over 2 minutes.. Third, a co-
factor competition assay was
performed. For this assay, three I.LL of pre-incubated lysate was added to 97
pi, of 200 mM phosphite
pH 7.9 containing 2 mM NAD, 0.2 mM NADP, 2 g/L KRED of SEQ ID NO:138, 4 g/L
KRED of
SEQ ID NO:104 and 10 g/L ketone (2). Reactions were quenched via addition of
100 pi, of 1M HC1.
The quenched mixture (10 pL) was diluted into 190 pL of water. Diluted
reaction samples (10 pL)
were analyzed by reverse phase HPLC to quantify residual substrate and both
enantiomers of the
product as described above. Significantly improved variants are provided in
Table 11.1, below.
SEQ ID NO: Amino Acid Substitutions
Cofactor Specificity
(Relative to SEQ ID NO:208) .. Improvement
=
246 V83A/T104L/L206N +++
248 A74TN83A/L206N ++
250 T104LN2621.,
252 T104L/L206N +++
254 5295R +-H-
256 V96G +-H-
258 T 04M 4.
Key for Table 11.1
-I--1-+ >8
++ >4 and < 8
>2 and<4
CA 03061133 2019-10-22
WO 2018/200214 PCT/US2018/027450
EXAMPLE 12
Production of Engineered Polypeptides and Performance Validation
101761 Plasmids comprising variants obtained through directed evolution of the
KRED of SEQ ID
NO:2 and containing evolved ketoreductase genes were transformed into E. coil
W3110 and placed on
Luria-Bertani (LB) agar medium containing 1% glucose and 30
pg/mlchloramphenicol (CAM).
After incubation for at least 16 h at 30 'C, a single colony was picked into 5
mL of LB, 1% glucose,
and 30 !vim' CAM. Cells were grown 18-20 h at 30 'C, with shaking at 250 rpm.
This culture was
then transferred into Terrific Broth (TB), 2mM ZnSO4, 1mM MgSO4, and 30 gem'
CAM at a final
OD600 of--0.02 and a final volume of 250 mL. After incubation of the flasks at
30 'V, with shaking at
250 rpm for 3.5 h (0D600 0.6-0.8), recombinant gene expression was induced by
isopropyl
thioglycoside (IPTG) to a final concentration of 1 mM. The flask was then
incubated at 30 `C with
shaking at 250 rpm for 18-21 h. Cells were pelleted at 3500 x g for 20 min,
and the supernatant was
discarded. The cell pellet was washed in 50 mL ice cold 50 mM sodium phosphate
pH 7.5 containing
2mM ZnSO4 and 1mM MgSO4, resuspended in 30 ml of the same buffer, and lysed
using a cell
disruptor at 18-20 kpsi. Lysates were clarified at 10000 x g for 60 min, and
clarified supernatants
were lyophilized to an off white powder.
[0177] Plasmids of comprising variants obtained through directed evolution of
the KRED of SEQ ID
NOS:112 and 138, and containing evolved ketoreductase genes were transformed
into E. coil W3110
and placed on Luria-Bertani (LB) agar medium containing 1% glucose and 30
!vim' chloramphenicol
(CAM). After incubation for at least 16 h at 30 'C., a single colony was
picked into 5 mL of LB, 1%
glucose, and 30 lig/m1 CAM. Cells were grown 18-20 h at 30 'C, with shaking at
250 rpm. This
culture was then transferred into Terrific Broth (TB), and 30 1.1g/m1 CAM at a
final 0D600 of -0.02 and
a final voliune of 250 mL. After incubation of the flasks at 30 C with
shaking at 250 rpm for 3.5 h
(OD%) 0.6-0.8), recombinant gene expression was induced by isopropyl
thioglycoside (IPTG) to a
final concentration of 1 mM. The flask was then incubated at 30 'V, with
shaking at 250 rpm for 18-
21 h. Cells were pelleted at 3500 x g for 20 min, and the supernatant was
discarded. The cell pellet
was washed in 50 mL ice cold 50 mM sodium phosphate pH 7.5, resuspended in 30
ml of the same
buffer, and lysed using a cell disruptor at 18-20 kpsi. Lysates were clarified
at 10000 x g for 60 min,
and clarified supernatants were lyophilized to an off white powder.
[0178] Plasmids comprising variants obtained through directed evolution of the
PDH of SEQ ID
NOS:172 and 208, and containing evolved phosphite dehydrogenase genes were
transformed into E.
coil W3110 and placed on Luria-Bertani (LB) agar medium containing 1% glucose
and 30 p.eml
chloramphenicol (CAM). After incubation for at least 16 h at 30 `C, a single
colony was picked into 5
mL of LB, 1% glucose, and 30 ps/m1 CAM. Cells were grown 18-20 h at 30'C, with
shaking at 250
rpm. This culture was then transferred into Terrific Broth (TB), and 30 pg/m1
CAM at a final 0D600
of.-0.02 and a final volume of 250 mL. After incubation of the flasks at 30 'C
with shaking at 250
rpm for 3.5 h ((Moo 0.6-0.8), recombinant gene expression was induced by
isopropyl thioglycoside
46
CA 03061133 2019-10-22
WO 2018/200214 PCT/US2018/027450
(TPTG) to a final concentration of 1 mM. The flask was then incubated at 30'C
with shaking at 250
rpm for 18-21 h. Cells were pelleted at 3500 x g for 20 mM, and the
supernatant was discarded. The
cell pellet was washed in 50 mL ice cold 50 mM sodium phosphate pH 7.5,
resuspended in 30 ml of
the same buffer, and lysed using a cell disruptor at 18-20 kpsi. Lysates were
clarified at 10000 x g for
60 min, and clarified supernatants were lyophilized to an off white powder.
[0179] To evaluate the final compound under process like conditions, 50 g/L of
racemic alcohol
substrate in 500 mM soditun phosphite buffer pH 7.9, 0.1 g/L NAD, 0.1 g/L
NADP, 2.5 g/L KRED of
SEQ ID NO:104, 10 g/L commercially available NADH oxidase NOx-9, 2.5 g/L KRED
of SEQ
ID:154, 10g/L PDH of SEQ ID NO:250 was stirred under stream of oxygen with 1%
v/v antifoam at
room temperature for 24 hours resulting in 93% conversion of substrate and
99.5% enantiomeric
excess of (R)-alcohol la. Reaction samples were analyzed by reverse phase HPLC
to quantify
residual substrate and products as described above.
101801 While various specific embodiments have been illustrated and described,
it will be
appreciated that various changes can be made without departing from the spirit
and scope of the
invention(s).
[0181] All publications, patents, patent applications and other documents
cited in this application are
hereby incorporated by reference in their entireties for all purposes to the
same extent as if each
individual publication, patent, patent application or other document were
individually indicated to be
incorporated by reference for all purposes.
47