Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
CA 03076482 2020-03-19
WO 2019/059771 PCT/NL2018/050629
Anti-GD2 antibody for the treatment of neuroblastoma
The invention relates to the field of antibodies. In particular it relates to
antibodies that bind the ganglioside GD2. It further relates to the use of GD2
antibodies in medical and detection methods. The invention further relates to
cells,
nucleic acid molecules and methods for the production of the antibodies.
Approximately 12% of all pediatric cancer patients succumb to
neuroblastoma, the most common extracranial solid tumor of childhood. The
majority of patients is diagnosed with high-risk neuroblastoma with a
mortality of
50%. Neuroblastoma therapy consists of intensive multimodality treatment with
severe toxicities. High-risk neuroblastoma therapy consists of intensive
multimodality treatment with severe short- and long-term toxicities.
Recurrence
rates are high with limited further treatment options. Residual disease is
treated
with intensive chemotherapy, radiotherapy, high-dose chemotherapy followed by
autologous hematopoietic stem cell rescue and isotretinoin. Complete
eradication of
the tumor is often not achieved (Matthay et al., 1999; New Engl. J. of Med Vol
341:
pp 1165-73). Intensification of conventional therapy has not improved outcome
and
is even associated with increased toxicity.
In 2015 the antibody dinutuximab (trade name Unituxin), directed against
ganglioside GD2, a carbohydrate antigen uniformly expressed on neuroblastoma
and neural tissue, was FDA-approved for neuroblastoma treatment. Application
of
this antibody, combined with cytokines and differentiation factors, has
improved
patient prognosis and demonstrated that neuroblastoma is susceptible to
immunotherapy (Yu et al., 2010; New Engl. J. Med, Vol 363: pp 1324-34; and
Suzuki and Cheung., 2015; Expert Opin Ther Targets Vol 19: p. 349-62).
Dinutuximab significantly improved event-free survival in comparison to
standard
treatment.
Dinutuximab is a chimeric monoclonal antibody composed of the variable
heavy- and light-chain regions of the murine anti-GD2 antibody 14.18 and the
constant regions of human IgG1 heavy-chain and kappa light-chain (Gillies et
al.,
1989; J Immunol Methods, 125: p. 191-202). It is directed against the end-
terminal
penta-oligosaccharide of GD2, an extracellularly expressed disialoganglioside
on
tissues of the central nervous system and peripheral nerves, as well as on
many
tumors of neuroectodermal origin, including neuroblastoma (Suzuki and Cheung,
2015; Expert Opin Ther Targets Vol 19: p. 349-62). Antibody produced in 5P2/0
mouse myeloma cells can result in aberrant glycosylation with respect to
natural
human antibodies. In Europe the antibody is now produced in CH() cells. The
uniform expression of GD2 on neuroblastoma together with the low expression on
other tissues makes this tumor associated antigen a promising target for
antibody
CA 03076482 2020-03-19
WO 2019/059771
PCT/NL2018/050629
2
therapy. Dinutuximab is used in combination with isotretinoin and alternating
administration of IL-2 and GM-CSF for the first-line treatment against high-
risk
neuroblastoma in patients where a response after induction therapy was shown
(Suzuki and Cheung, 2015; Expert Opin Ther Targets Vol 19: p. 349-62).
Preclinical research has shown that dinutuximab mediates its anti-tumor
effects through antibody dependent cell mediated cytotoxicity (ADCC) and
complement mediated cytotoxicity (CDC) (Barker et al., 1991; Cancer Res, Vol
51:
p. 144-9). For ADCC activity, most therapeutic antibodies depend on NK cells
and
possibly macrophages for their action. Remarkably, the ADCC activity of
dinutuximab against neuroblastoma is for some reason also dependent on
granulocyte activation. This is shown in citro and in uluo, by showing that
patient
outcome depends in part on granulocyte activation (Barker et al., 1991; Cancer
Res, Vol 51: p. 144-9; Cheung et al., 2012; J Clin Oncol, Vol 30: p. 426-32;
and
Batova et al., 1999; Clin Cancer Res Vol 5: p. 4259-63). These observations
have
formed the basis for the inclusion of GM-CSF or G-CSF in the current
therapeutic
regimen to further stimulate granulocytes to enhance the anti-tumor response
against neuroblastoma (Cheung et al., 2012; J Clin Oncol, Vol 30: p. 426-32;
and
Batova et al., 1999; Clin Cancer Res Vol 5: p. 4259-63).
In spite of the good clinical effects of dinutuximab it is associated with a
number of toxicities. The most common toxicities include tachycardia,
hypertension, hypotension, difficult to treat pain, fever and urticaria. Many
of
these toxicities are dose-dependent and are rarely noted at low dosages. Other
toxicities include hyponatremia, hypokalemia, nausea, vomiting and diarrhea
(Mora J, 2016, Expert review of clinical pharmacology, pp1-7;
http://dx.doi.org/
10.1586/17512433.2016.1160775).
The anti-GD2 antibodies and the methods of treatment of present invention
exhibit improved efficacy. In addition, toxicity associated with dinutuximab
treatment is reduced, in particular pain associated toxicity is abrogated, as
shown
in pre-clinical models.
SUMMARY OF THE INVENTION
The invention provides an anti-ganglioside GD2 antibody that comprises an
antibody variable domain and antibody constant domains, wherein the variable
domain comprises a heavy and light chain variable region comprising
respectively
at least the CDR3 of the heavy chain variable region of antibody ch14.18 and
at
least the CDR3 of the light chain variable region of antibody ch14.18; and an
IgA
hinge and CH2 domain. The variable domain preferably comprises a heavy and
light chain variable region comprising respectively at least the CDR1, CDR2
and
CDR3 of the heavy chain variable region of antibody ch14.18 and at least the
CA 03076482 2020-03-19
WO 2019/059771 PCT/NL2018/050629
3
C,DR1, CDR2 and CDR3 of the light chain variable region of antibody ch14.18.
In a
preferred embodiment the variable domain comprises the heavy and light chain
variable region of antibody ch14.18.
The invention further provides a method of treatment of a subject that has a
GD2 positive tumor or is at risk of having a G112 positive tumor the method
comprising administering a therapeutic amount of an antibody of the invention
to
the subject in need thereof. The method preferably further comprises
administering
retinoie acid in an amount effective to upregulate the ganglioside GD2 in said
GD2
positive tumor, preferably neuroblastoma in said subject. The method may
further
comprise administering granulocyte-macrophage colony-stimulating factor (GM-
CSF, granulocyte colony-stimulating factor (G-CSF) or a combination thereof to
said subject. GM-CSF, G-CSF or a combination thereof can increase the number
of
granulocytes in the subject but is also administered to improve the induced
cell
killing.
The invention further provides an antibody of the invention for use in the
treatment of a subject that has a GD2 positive tumor or is at risk of having a
GD2
positive tumor. The invention further provides an antibody of the invention in
combination with retinoic acid for use in the treatment of a subject that has
a GD2
positive tumor or is at risk of having a GD2 positive tumor. The invention
further
provides an antibody of the invention in combination with GM-CSF, G-CSF or a
combination thereof for use in the treatment of a subject that has a GD2
positive
tumor or is at risk of having a GD2 positive tumor. The invention further
provides
an antibody of the invention in combination with GM-CSF, G-CSF or a
combination
thereof and retinoie acid for use in the treatment of a subject that has a GD2
positive tumor or is at risk of having a GD2 positive tumor.
The GD2 positive tumor is preferably a GD2 positive neuroblastoma such as
a neuroeetoderm-derived tumor or a sarcoma. In a preferred embodiment the GD2
positive tumor is a GD2 positive neuroblastoma, retinoblastoma, melanoma,
small
cell lung cancer, brain tumor such as glioblastoma, osteosareoma,
rhabdomyosarcoma, Ewing's sarcoma in children and adolescents, or liposarcoma,
fibrosarcoma, leiomyosarcoma or another soft tissue sarcoma in adults. In a
preferred embodiment the GD2 positive tumor is a neuroblastoma. The
neuroblastoma treated with a method or use of the invention is preferably a
high
risk neuroblastoma.
In a preferred embodiment an antibody of the invention comprises a heavy
chain variable region with the amino acid sequence
EVQLLQSGPE LEKPGASVMI SCKASGSSFT GYNMNWVRQN IGKSLEWIGA
IDPYYGGTSY NQKFKGRATL TVDKSSSTAY MHLKSLTSED SAVYYCVSGM
EYWGQGTSVT VSS;
CA 03076482 2020-03-19
WO 2019/059771 PCT/NL2018/050629
4
and a light chain variable region with the amino acid sequence
EIVMTQSPAT LSVSPGERAT LSCRSSQSLV HRNGNTYLHW YLQKPGQSPK
LLIHKVSNRF SGVPDRFSGS GSGTDFTLKI SRVEAEDLGV YFCSQSTHVP
PLTFGAGTKL ELK; and
a heavy chain comprising an IgA hinge and CH2 domain.
Provided herein is an engineered antibody or fragment thereof for use in
improvement of patient compliance and/or a reduction of a side effect
associated
with immunoglobulin G (IgG) antibody therapy, wherein the engineered antibody
or fragment thereof comprises: (a) CDR1, CDR2, and CDR3 of an immunoglobulin
G (IgG); and (b) the constant regions or portion thereof of an immunoglobulin
A
(IgA), wherein the engineered antibody or fragment thereof results in
reduction of
at least one side effect upon administration to the subject as compared to
administration of a comparable amount of a corresponding IgG antibody.
In an aspect, the engineered antibody or fragment thereof further comprises
a heavy chain and light chain variable region of IgG. In an aspect, the
constant
regions or portion thereof of the IgA comprises a hinge, a CH2 constant
region, or a
combination thereof. In an aspect, the constant regions or portion thereof
comprises a hinge and a CH2 constant region of the IgA. In an aspect, the
constant
regions comprise a constant heavy and constant light chain of the IgA. In an
aspect,
the constant regions or portion thereof of the IgA comprises a hinge and a
constant
heavy chain domain. In an aspect, the antibody or fragment thereof is
chimerized,
humanized, human, or non-human. In an aspect, the constant regions or portion
thereof is human.
In an aspect, the side effect comprises an innate immune response. In some
eases, the innate immune response comprises a complement response. In some
eases, the complement response comprises IgG binding to FeyRs or Clq. In
certain
embodiments, the side effect comprises one or more of pain, visceral
hypersensitivity, allodynia, hyperalgesia, allergic reactions and flu-like
symptoms.
In some eases, the pain is acute pain, migraine or intense visceral pain.
In some cases, determining the reduction of the side effect is measured by
performing flow cytometry, an in vitro assay, or a combination thereof. In
some
cases, the flow cytometry comprises determining cellular lysis, binding, cell
death,
receptor expression, or a combination thereof. In some eases, the flow
cytometry
comprises determining live/dead staining of a cell. In some cases, the in
vitro assay
comprises ELISA, antibody-dependent cell-mediated cytotoxicity (ADCC),
complement-dependent cytotoxicity (CDC), hemolytic assay, or a combination
thereof.
CA 03076482 2020-03-19
WO 2019/059771
PCT/NL2018/050629
In some cases, the engineered antibody or fragment thereof when
administered results in an increased antibody-dependent cell-mediated
cytotoxicity
(ADCC) and/or reduced complement-dependent cytotoxicity (CDC) as compared to
the corresponding IgG antibody that comprises the same CDR1, CDR2, and CDR3.
5 In an aspect, the reduction comprises from 5%, 10%, 15%, 20%, 25%, 30%,
35%,
40%, 45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95%, to 100% of the
side effect as compared to administering a comparable IgG antibody that
comprises
the same CDR1, CDR2, and CDR3.
In some eases, the antibody or fragment thereof binds a target present on a
non-cancerous cell, a cancer cell, or a combination thereof. In some eases,
the
antibody or fragment thereof binds a target from a brain-metastasizing cancer.
In
some cases, the antibody or fragment thereof binds one or more of GD2, ALK,
hNET, GD3, and CD20. In some cases, the GD2 positive tumor is neuroblastoma,
retinoblastoma, melanoma, small cell lung cancer, glioblastoma, osteosarcoma,
rhabdomyosarcoma, Ewing's sarcoma, liposarcoma, fibrosarcoma, leiomyosarcoma,
and any combinations thereof. In an aspect, the antibody or fragment thereof
binds
a neuroblastoma cell. In some cases, the ALK (anaplastic lymphoma kinase)
positive tumor is an anaplastic large-cell lymphoma, an adenocarcinoma of the
lung, a neuroblastoma, an inflammatory myofibroblastic tumor, a renal cell
carcinomas, esophageal squamous cell carcinoma, breast cancer, a colonic
adenocarcinoma, a glioblastoma multiforme or an anaplastic thyroid cancer. In
some cases, the hNET (human norepinephrine transporter) positive tumor is a
bladder tumor, breast tumor, prostate tumor, carcinoma, leukemia, liver
cancer,
lung cancer, lymphoma, Hodgkin's lymphoma, Non-Hodgkin's lymphoma,
melanoma, neuroblastoma, ovarian tumor, pancreatic tumor or a retinoblastoma.
In some ease, the GD3 positive tumor is a neuroectodermal tumor of the center
nervous system, glioma, neuroblastoma, retinoblastoma, ependymoma, sarcoma,
melanoma, breast cancer, ovarian cancer, glioblastoma, Ewing's sarcoma, or
small
cell lung carcinoma. In some eases the CD20 positive tumor is a leukemia, a
lymphoma or a neuroblastoma.
Provided herein is a pharmaceutical composition comprising an engineered
antibody or fragment thereof and a pharmaceutically acceptable carrier.
Provided herein is a kit comprising an engineered antibody or fragment
thereof and instructions for use thereof.
Provided herein is a method comprising: administering to a subject a
pharmaceutical composition comprising an antibody or fragment thereof
comprising: (a) CDR1, CDR2, and CDR3 of an immunoglobulin G (IgG); and (b) the
constant regions or portion thereof of an immunoglobulin A (IgA), wherein the
administering results in a reduction of a side effect in the subject as
compared to
administering a comparable IgG antibody that comprises the same CDR1, CDR2,
and CDR3.
CA 03076482 2020-03-19
WO 2019/059771 PCT/NL2018/050629
6
Provided herein is an engineered immunoglobulin G (IgG) antibody
fragment, wherein the engineered IgG antibody fragment comprises at least a
hinge domain and a CH2 domain of a human IgA antibody. In some cases, the
engineered IgG antibody fragment, when administered to a subject, results in
increased antibody-dependent cell-mediated cytotoxicity (ADCC) and/or reduced
complement-dependent cytotoxicity (CDC) as compared to a comparable antibody
fragment absent the hinge domain and a G12 domain of a human IgA antibody
Provided herein is a method of treating pain or allodynia associated with IgG
administration comprising administering to a subject an engineered IgG
antibody
or fragment thereof comprising the constant regions of an IgA antibody.
Provided
herein is a method of reducing complement activation resulting from IgG
administration comprising administering to a subject an engineered IgG
antibody
or fragment thereof comprising the constant regions of an IgA antibody.
DETAILED DESCRIPTION OF THE INVENTION
Gangliosides are sialic acid-containing glycosphingolipids that play
important roles in signal transduction as well as cell adhesion and
recognition. The
ganglioside GD2 is a b-series ganglioside that requires the enzymes GD3
synthase
and GD2 synthase to add sialic acid units onto its precursor G-m2. Normal
tissues
generally express a-series gangliosides, whereas b-series gangliosides are
expressed during fetal development and are restricted primarily to the nervous
system in healthy adults and at low levels in peripheral nerves and skin
melanocytes. A structure of the ganglioside GD2 is depicted in figure 10.
The ganglioside GD2 is highly expressed on neuroectoderm-derived tumors
and sarcomas, including neuroblastoma, retinoblastoma, melanoma, small cell
lung
cancer, brain tumors, osteosarcoma, rhabdomyosarcoma, Ewing's sarcoma in
children and adolescents, as well as liposarcoma, fibrosarcoma, leiomyosarcoma
and other soft tissue sarcomas in adults. GD2 expression in normal individuals
appears to be limited to the brain and certain peripheral nerves and
melanoeytes.
As the brain is typically not accessible for normal circulating antibodies GD2
is
considered an attractive target for tumor-specific therapy (For review see
Mahiuddin et al., 2014; FEBS letters 588: 288-297). Mahiuddin et al describe
various (ID2 targeted approaches among which there are the GD2 specific
antibodies. A number of targeted therapies has reached the clinic and show
promise in phase I, II and III trials.
Various GD2 antibodies are presently under development. The antibodies
are thought to induce ADCC and CDC, for which in particular neuroblastoma
appears to be relatively sensitive. Various avenues are being pursued to
improve
CA 03076482 2020-03-19
WO 2019/059771 PCT/NL2018/050629
7
the efficacy and reduce the toxicity of the targeted approaches. The most
frequently
used antibodies all originate from murine IgG3 antibodies. The murine
antibodies
have been humanized in recent years. A number of variants have been made.
Murine antibody 14.18 has been chimerized to ch14.18 and humanized into
hu14.18 both have a human IgG1 background (Mahiuddin et al., 2014; FEBS
letters 588: 288-297). The murine IgG3 antibody 3F8 has been used in humans.
To
reduce human anti-mouse responses and improve the efficacy of the antibody
while
reducing toxicity 3F8 was chimerized (ch3F8) and humanized 3F8 (hu3F8-IgG1
and hu3F8-IgG4). In GD2 binding studies by SPR, ch3F8 and hu3F8 maintained
KD comparable to m3F8. Unlike other anti-GD2 antibodies, m3F8, ch3F8 and
hu3F8 had substantially slower koff. Similar to m3F8, both ch3F8 and hu3F8
inhibited tumor cell growth in citro, while cross-reactivity with other
gangliosides
was comparable to that of m3F8. Both peripheral blood mononuclear cell (PBMC,)-
ADCC and polymorphonuclear leukocytes (PMN)-ADCC of ch3F8 and hu3F8-IgG1
were more potent than m3F8. Hu3F8-IgG4 had near absent PBMC-ADCC and
CDC. Hu3F8 and m3F8 had similar tumor-to-non tumor ratios in biodistribution
studies. Anti-tumor effect against neuroblastoma xenogr a fts was better with
hu3F8-IgG1 than m3F8 (see Cheung et al., 2012; Oncoimmunology 1.4: 477-486).
An antibody (Ab), also known as an immunoglobulin (Ig), is a large protein.
An antibody interacts with various components of the immune system. Some of
the
interactions are mediated by its Fe region (located at the base), which
contains
site(s) involved in these interactions.
95 Antibodies are proteins belonging to the immunoglobulin superfamily.
They
typically have two heavy chains and two light chains. There are several
different
types of antibody heavy chains that define the five different types of
crystallizable
fragments (Fe) that may be attached to the antigen-binding fragments. The five
different types of Fe regions allow antibodies to be grouped into five
isotypes. An Fe
region of a particular antibody isotype is able to bind to its specific Fe
receptor
(FcR) thus allowing the antigen-antibody complex to mediate different roles
depending on which FeR it binds. The ability of an IgG antibody to bind to its
corresponding FeR is modulated by the presence/absence of interaction sites
and
the structure of the glyean(s) (if any) present at sites within its Fe region.
The
ability of antibodies to bind to FcRs helps to direct the appropriate immune
response for each different type of foreign object they encounter.
Though the general structure of all antibodies is similar, a region at the tip
of the protein is extremely variable, allowing millions of antibodies with
slightly
different tip structures, or antigen-binding sites, to exist. This region is
known as
the hypervariable region. The enormous diversity of antigen binding by
antibodies
is largely defined by the hypervariable region and the variable domain
containing
the hypervariable region.
CA 03076482 2020-03-19
WO 2019/059771
PCT/NL2018/050629
8
An antibody of the invention is typically a full-length antibody. The term
'full length antibody' is defined as comprising an essentially complete
immunoglobulin molecule, which however does not necessarily have all functions
of
an intact immunoglobulin. For the avoidance of doubt, a full length antibody
has
two heavy and two light chains. Each chain contains constant (C) and variable
(V)
regions. A heavy chain of a full length antibody typically comprises a Cal, a
CH2, a
CH3, a VH region and a hinge region. A light chain of a full length antibody
typically comprises a CL region and a VL region.
An antibody binds to antigen via the variable region domains contained in
the Fab portion. An antibody variable domain comprises a heavy chain variable
region and a light chain variable region. Full length antibodies according to
the
invention encompass heavy and light chains wherein mutations may be present
that provide desired characteristics. Full length antibodies should not have
deletions of substantial portions of any of the regions. However, IgG
molecules
wherein one or several amino acid residues are substituted, inserted, deleted
or a
combination thereof, without essentially altering the antigen binding
characteristics of the resulting antibody, are embraced within the term "full
length" antibody. For instance, a Tull length" antibody can have a
substitution,
insertion, deletion or a combination thereof, of between 1 and 10 (inclusive)
amino
acid residues, preferably in non-CDR regions, wherein the deleted amino acids
are
not essential for the binding specificity of the antibody.
The four human IgG isotypes bind the activating Fey receptors (FeyRI,
FeyRIIa, FcyRIIIa), the inhibitory FcyRIIb receptor, and the first component
of
complement (Clq) with different affinities, yielding very different effector
functions. Binding of IgG to the FcyRs or Clq depends on residues located in
the
hinge region and the CH2 domain. Two regions of the CH2 domain are critical
for
FcyRs and Clq binding, and have unique sequences in IgG2 and IgG4.
Substitutions into human IgG1 of IgG2 residues at positions 233-236 were shown
to greatly reduce ADCC and CDC. Furthermore, Idusogie et al. demonstrated that
alanine substitution at different positions, including K32A, significantly
reduced
complement activation. Similarly, mutations in the CH2 domain of murine igG2A
were shown to reduce the binding to FcyRI, and Clq. Numerous mutations have
been made in the CH2 domain of human IgG 1 and their effect on ADCC and CDC
tested in vitro. Notably, alanine substitution at position 333 was reported to
increase both ADCC and CDC. Fe-receptor functions are among others reviewed in
Bruhns et al., 2009. Blood. 113(16):3716-25; Armour et al., 1999. Eur J
Immunol.
29(8):2613-24; Shields et al., 2001. J Biol Chem. 276(9):6591-604; Idusogie et
al.,
2000. J Immunol. 164(8):4178-84; Steurer et al., 1995. J Immunol. 155(3):1165-
74;
and Idusogie. et al., 2001. J Immunol. 166(4):2571-5. IgG1 antibodies have a
Clq
binding site through which complement activation through the classical pathway
is
achieved. IgA antibodies can also activate complement. This can be through the
CA 03076482 2020-03-19
WO 2019/059771
PCT/NL2018/050629
9
alternative pathway, the complement lectin pathway and the classical pathway
(Jarvis et al., J Immunol. 1989;143(5):1703-9; Hiemstra et al., European
journal of
immunology. 1987;17(3):321-6; Pfaffenbach et al., The Journal of experimental
medicine. 1982;155(1):231-47; Roos et al., J Immunol. 2001;167(5):2861-8;
Pascal et
.. al., Haematologica DOT: 10.3324/haemato1.2011.061408; and Lohse et al.,
Britisch
Journal of Haematology. 2011doi: 10.1111/bjh.14624).
The present inventors found that ADCC activity of a murine, murine
ehimerized or humanized IgG or human IgM GD2 antibody with unmodified
constant regions can be increased by replacing at least the hinge domain and
the
CIJ2 domain thereof by a hinge domain and Cu2 domain of a human IgA antibody,
preferably a human IgAl antibody. In a preferred embodiment also the CH3
domain is of a human IgA antibody. In a preferred embodiment the antibody
comprises an essentially complete IgA constant region. The invention therefore
provides an antibody that can bind GD2 and that comprises ADCC activity and
that comprises a hinge domain and Cn2 domain of a human IgA antibody. A GD2
antibody of the invention has a reduced capacity to induce CDC. A GD2 antibody
of
the invention has a reduced capacity to induce pain when compared to the
murine,
murine chimerized or humanized IgG or human IgM original GD2 antibody.
An IgA constant domain or hinge region may have one or more amino acid
insertions, deletions, substitutions, additions or a combination thereof at
one or
more positions with respect to a germ line IgA domain or hinge region. An IgA
domain or hinge region preferably has at most 4 amino acid insertions,
deletions,
substitutions, additions or a combination thereof, preferably at most 3; 2 or
preferably at most 1 amino acid insertions, deletions, substitutions,
additions or a
combination thereof. Such a protein is still an IgA constant domain or hinge
region.
An IgA constant domain or hinge region may have one or more amino acid
insertions, deletions, substitutions, additions or a combination thereof at
one or
more positions with respect to a germ line IgA. The IgA constant part
(including
the three domains and the hinge region) can have 0; 1; 2; 3; 4; 5; 6 ;7; 8; 9;
10; 11;
12; 13; 14; or 15 amino acid insertions, deletions, substitutions, additions
or a
combination thereof.
In one embodiment the invention provides an anti-ganglioside GD2 antibody
that comprises an antibody variable domain and antibody constant domains,
wherein the variable domain comprises a heavy and light chain variable region
comprising respectively at least the CDR3 of the heavy and light chain
variable
regions of antibody ch14.18 and an IgA hinge and CH2 domain. The variable
domain preferably comprises a heavy and light chain variable region comprising
.. respectively at least the CDR1, CDR2 and CDR3 of the heavy and light chain
variable regions of antibody ch14.18 as depicted in figure 1. The variable
domain
preferably comprises a heavy and light chain variable region of antibody
ch14.18 as
depicted in figure 1.
CA 03076482 2020-03-19
WO 2019/059771 PCT/NL2018/050629
The anti-ganglioside GD2 antibody heavy chain variable region is preferably
a heavy chain variable region of antibody ch14.18 with 0-5 amino acid
insertions,
deletions, substitutions, additions or a combination thereof at one or more
positions
with respect to the heavy chain variable region sequence of antibody ch14.18
5 indicated in figure 1. The one or more positions are preferably not
positions in the
CDR1, CDR2 and CDR3 regions. The sequence of the CDRs is thus as indicated for
heavy chain variable region of antibody ch14.18 of figure 1. It is preferred
that the
heavy chain variable region has 0-4 amino acid insertions, deletions,
substitutions,
additions or a combination thereof at one or more positions with respect to
the
10 heavy chain variable region sequence of antibody ch14.18 indicated in
figure 1,
wherein the one or more positions are not positions in the CDR1, CDR2 and CDR3
regions. It is preferred that the heavy chain variable region has 0-3, more
preferably 0-2, more preferably 0-1 amino acid insertions, deletions,
substitutions,
additions or a combination thereof at one or more positions with respect to
the
heavy chain variable region sequence of antibody ch14.18 indicated in figure
1,
wherein the one or more positions are not positions in the CDR1, CDR2 and CDR3
regions. In a preferred embodiment a heavy chain variable region in the
antibody
of the invention has 0 amino acid insertions, deletions, substitutions,
additions or a
combination thereof with respect to the heavy chain variable region sequence
of
antibody ch14.18 in figure 1.
The anti-ganglioside GD2 antibody light chain variable region is preferably
a light chain variable region of antibody ch14.18 with 0-5 amino acid
insertions,
deletions, substitutions, additions or a combination thereof at one or more
positions
with respect to the light chain variable region sequence of antibody ch14.18
indicated in figure 1. The one or more positions are preferably not positions
in the
CDR1, CDR2 and CDR3 regions. The sequence of the CDRs is thus as indicated for
light chain variable region of antibody ch14.18 of figure 1. It is preferred
that the
light chain variable region has 0-4 amino acid insertions, deletions,
substitutions,
additions or a combination thereof at one or more positions with respect to
the light
chain variable region sequence of antibody ch14.18 indicated in figure 1,
wherein
the one or more positions are not positions in the CDR1, CDR2 and CDR3
regions.
It is preferred that the light chain variable region has 0-3, more preferably
0-2,
more preferably 0-1 amino acid insertions, deletions, substitutions, additions
or a
combination thereof at one or more positions with respect to the light chain
.. variable region sequence of antibody ch14.18 indicated in figure 1, wherein
the one
or more positions are not positions in the CDR1, CDR2 and CDR3 regions. In a
preferred embodiment a light chain variable region in the antibody of the
invention
has 0 amino acid insertions, deletions, substitutions, additions or a
combination
thereof with respect to the light chain variable region sequence of antibody
ch14.18
.. in figure 1.
In another embodiment the invention provides an anti-ganglioside GD2
antibody that comprises an antibody variable domain and antibody constant
CA 03076482 2020-03-19
WO 2019/059771 PCT/NL2018/050629
11
domains, wherein the variable domain comprises a heavy and light chain
variable
region comprising respectively at least the CDR3 of the heavy and light chain
variable regions of antibody 3F8 and an IgA hinge and CH2 domain. The variable
domain preferably comprises a heavy and light chain variable region comprising
respectively at least the CDR1, CDR2 and CDR3 of the heavy and light chain
variable regions of antibody 3F8 (as depicted in figure 1). The variable
domain
preferably comprises a heavy and light chain variable region of antibody 3F8
as
depicted in figure 1. The anti-ganglioside GD2 antibody heavy chain variable
region is preferably a heavy chain variable region of antibody 3F8 with 0-5
amino
acid insertions, deletions, substitutions, additions or a combination thereof
at one
or more positions with respect to the heavy chain variable region sequence of
antibody 3F8 indicated in figure 1. The one or more positions are preferably
not
positions in the CDR1, CDR2 and CDR3 regions. The sequence of the CDRs is thus
as indicated for heavy chain variable region of antibody 3F8 of figure 1. It
is
preferred that the heavy chain variable region has 0-4 amino acid insertions,
deletions, substitutions, additions or a combination thereof at one or more
positions
with respect to the heavy chain variable region sequence of antibody 3F8
indicated
in figure 1, wherein the one or more positions are not positions in the CDR1,
CDR2
and CDR3 regions. It is preferred that the heavy chain variable region has 0-
3,
more preferably 0-2, more preferably 0-1 amino acid insertions, deletions,
substitutions, additions or a combination thereof at one or more positions
with
respect to the heavy chain variable region sequence of antibody 3F8 indicated
in
figure 1, wherein the one or more positions are not positions in the CDR1,
CDR2
and CDR3 regions. In a preferred embodiment a heavy chain variable region in
the
antibody of the invention has 0 amino acid insertions, deletions,
substitutions,
additions or a combination thereof with respect to the heavy chain variable
region
sequence of antibody 3F8 in figure 1.
The anti-ganglioside GD2 antibody light chain variable region is preferably
a light chain variable region of antibody 3F8 with 0-5 amino acid insertions,
deletions, substitutions, additions or a combination thereof at one or more
positions
with respect to the light chain variable region sequence of antibody 3F8
indicated
in figure 1. The one or more positions are preferably not positions in the
CDR1,
CDR2 and CDR3 regions. The sequence of the CDRs is thus as indicated for light
chain variable region of antibody 3F8 of figure 1. It is preferred that the
light chain
variable region has 0-4 amino acid insertions, deletions, substitutions,
additions or
a combination thereof at one or more positions with respect to the light chain
variable region sequence of antibody 3F8 indicated in figure 1, wherein the
one or
more positions are not positions in the CDR1, CDR2 and CDR3 regions. It is
preferred that the light chain variable region has 0-3, more preferably 0-2,
more
preferably 0-1 amino acid insertions, deletions, substitutions, additions or a
combination thereof at one or more positions with respect to the light chain
variable region sequence of antibody 3F8 indicated in figure 1, wherein the
one or
more positions are not positions in the CDR1, CDR2 and CDR3 regions. In a
CA 03076482 2020-03-19
WO 2019/059771 PCT/NL2018/050629
12
preferred embodiment a light chain variable region in the antibody of the
invention
has 0 amino acid insertions, deletions, substitutions, additions or a
combination
thereof with respect to the light chain variable region sequence of antibody
3F8 in
figure 1.
IgA has two subclasses (IgAl and IgA2) and can be produced as a
monomeric as well as a dimeric form. The antibody in the present invention is
preferably a monomeric antibody. The IgA elements in an antibody of the
invention
are preferably human IgA elements. An IgA element can be an IgAl element or an
IgA2 element. IgA elements in an antibody of the invention can be all IgAl
elements or all IgA2 elements or a combination of IgAl and IgA2 elements. An
IgA
element is preferably a human IgA element. Preferably all IgA element in the
antibody are human IgA elements. The IgA elements can be IgAl elements,
preferably human IgAl elements. The IgA elements can also be IgA2, preferably
IgA2m(1) elements, preferably human IgAl elements. The Cal domain and/or Ca3
domain of the antibody can be an IgG Cal domain, an IgG Ca3 domain or a
combination thereof. It is preferred that the Cal domain, Ca3 domain or
combination thereof is an IgA Cal domain, an IgA Ca3 domain or a combination
thereof. It is preferred that the IgA Cal domain and/or hinge region is a
human
IgA Cal domain and/or human IgA hinge region. Said human IgA Cal domain
and/or human IgA hinge region is preferably an human IgAl Cal domain or
human IgAl hinge region. Said human IgA Cal domain and/or human IgA hinge
region is preferably an human IgA2m(1) Cal domain or human IgA2m(1) hinge
region. The constant domains and hinge region of the antibody are preferably
human constant regions and hinge region, preferably of a human IgA antibody.
The
constant domains and hinge region of the antibody are preferably human IgAl or
human IgA2m(1) constant domains and hinge region.
A human constant region can have 0-15 amino acid changes with respect to
a human allele as found in nature. An amino acid change may be introduced for
various reasons. Non-limiting examples include but are not limited to
improving
production or homogeneity of the antibody, adapting half-life in the
circulation,
stability of the HC/LC combination, optimizing glycosylation, adjusting
dimerization or complex formation, adjusting ADCC activity. A human constant
region can have 0; 1; 2; 3; 4; 5; 6; 7; 8; 9; 10; 11; 12; 13; 14; and 15 amino
acid
changes with respect to a human allele as found in nature. The changed amino
acid
is preferably one chosen from an amino acid at a corresponding position of a
different isotype.
In one embodiment the constant regions of the heavy chain are IgA2
constant regions, preferably human IgA2 constant regions, preferably human
IgA2m(1). In one aspect the human constant region is a mutated IgA2m(1)
sequence.
CA 03076482 2020-03-19
WO 2019/059771 PCT/NL2018/050629
13
In one embodiment the antibody comprises the constant regions of an
IgA2m(1) sequence, preferably with at least one and preferably at least 2; 3;
4; 5;
and preferably at least 7 of the following mutations: N166G; P221R; N337T;
I338L;
T339S; C331S; and mutation of the C-terminal amino acid sequence which is a
human IgA2m(1) antibody is "...VDGTCY" into "...VDGT. Figure 13 shows the
sequence of human IgAl; IgA2m(1) and a preferred mutated IgA2m(1) sequence
(hIgA2.0); see also figure 13E.
In one embodiment the GD2 antibody comprises a heavy chain with the
constant regions of figure 13E with 0; 1; 2; 3; 4; 5; 6; 7; 8; 9; 10; 11; 12;
13; 14; and
amino acid changes with respect to the sequence provided in figure 13E,
provided that amino acids at positions 166; 221; 337; 338; 339; and 331 are
166G;
221R; 337T; 338L; 3398 and 331S. Preferably the C-terminal amino acid sequence
which is a human IgA2m(1) antibody is "...VDGTCY" is "...VDGT".
A human IgA sequence of whatever subtype relates to the heavy chain of the
antibody. The heavy chain is subject to isotype switching.
The mention IgA antibody preferably comprises a ch14.18 variable domain
or a 3F8 variable domain, preferably a ch14.18 variable domain.
An anti-ganglioside GD2 antibody comprising:
- a heavy chain variable region of antibody ch14.18 with 0-5 amino acid
insertions, deletions, substitutions, additions or a combination thereof at
one or
more positions with respect to the heavy chain variable region sequence of
antibody
ch14.18 indicated in figure 1. The one or more positions are preferably not
positions
in the CDR1, CDR2 and CDR3 regions. The sequence of the CDRs is thus as
indicated for heavy chain variable region of antibody ch14.18 of figure 1. It
is
preferred that the heavy chain variable region has 0-4 amino acid insertions,
deletions, substitutions, additions or a combination thereof at one or more
positions
with respect to the heavy chain variable region sequence of antibody ch14.18
indicated in figure 1, wherein the one or more positions are not positions in
the
CDR1, CDR2 and CDR3 regions. It is preferred that the heavy chain variable
region has 0-3, more preferably 0-2, more preferably 0-1 amino acid
insertions,
deletions, substitutions, additions or a combination thereof at one or more
positions
with respect to the heavy chain variable region sequence of antibody ch14.18
indicated in figure 1, wherein the one or more positions are not positions in
the
CDR1, CDR2 and CDR3 regions. In a preferred embodiment a heavy chain variable
region in the antibody of the invention has 0 amino acid insertions,
deletions,
substitutions, additions or a combination thereof with respect to the heavy
chain
variable region sequence of antibody ch14.18 in figure 1;
- a light chain variable region of antibody ch14.18 with 0-5 amino acid
insertions, deletions, substitutions, additions or a combination thereof at
one or
CA 03076482 2020-03-19
WO 2019/059771 PCT/NL2018/050629
14
more positions with respect to the light chain variable region sequence of
antibody
ch14.18 indicated in figure 1. The one or more positions are preferably not
positions
in the CDR1, CDR2 and CDR3 regions. The sequence of the CDRs is thus as
indicated for light chain variable region of antibody ch14.18 of figure 1. It
is
preferred that the light chain variable region has 0-4 amino acid insertions,
deletions, substitutions, additions or a combination thereof at one or more
positions
with respect to the light chain variable region sequence of antibody ch14.18
indicated in figure 1, wherein the one or more positions are not positions in
the
CDR1, CDR2 and CDR3 regions. It is preferred that the light chain variable
region
has 0-3, more preferably 0-2, more preferably 0-1 amino acid insertions,
deletions,
substitutions, additions or a combination thereof at one or more positions
with
respect to the light chain variable region sequence of antibody ch14.18
indicated in
figure 1, wherein the one or more positions are not positions in the CDR1,
CDR2
and CDR3 regions. In a preferred embodiment a light chain variable region in
the
antibody of the invention has 0 amino acid insertions, deletions,
substitutions,
additions or a combination thereof with respect to the light chain variable
region
sequence of antibody ch14.18 in figure 1;
- an IgA heavy chain, preferably comprising an amino acid sequence as
depicted in figure 13E with 0; 1; 2; 3; 4; 5; 6; 7; 8; 9; 10; 11; 12; 13; 14;
and 15 amino
acid changes with respect to the sequence provided in figure 13E, provided
that
amino acids at positions 166; 221; 337; 338; 339; and 331 are 166G; 221R;
337T;
338L; 339S and 331S. Preferably the C-terminal amino acid sequence which is a
human IgA2m(1) antibody is "...VDGTCY" is "...VDGT"; and
- a light chain constant domain, preferably of antibody ch14.18.
The anti-ganglioside GD2 antibody of the invention preferably exhibits more
antibody-dependent cell-mediated cytotoxicity (ADCC) than the antibody
dinutuximab when measured in a suitable in vitro ADCC assay. It preferably
exhibits less complement-dependent cytotoxicity (CDC) than the antibody
30 dinutuximab when measured in a suitable in vitro CDC assay. Various ADCC
and
CDC assays are available to the person skilled in the art. In the context of
the
present invention it is preferred that an ADCC assay or a CDC assay as
described
in the examples is used. ADCC function of an antibody as claimed is preferably
measured in a classical chromium release assay (as described for instance in
35 examples). CDC function of an antibody as claimed is preferably measured
in a
method based on 7-AAD positivity in flow eytometry (see for instance the
examples). The antibody preferably exhibits 20% or less, more preferably 10%
or
less of the complement-dependent cytotoxicity (CDC) of the antibody
dinutuximab
when measured in a suitable in vitro CDC assay. In a preferred embodiment the
40 antibody comprises an albumin-binding domain (ABD) attached to a heavy
or light
chain of the antibody. Reference is made to Meyer, et al., 2016; MAbs. Vol. 8.
No. 1;
for details on the linkage of the albumin-binding domain to a heavy or light
chain.
In another embodiment another domain is added to increase the half-life of the
CA 03076482 2020-03-19
WO 2019/059771 PCT/NL2018/050629
antibody. In a preferred embodiment the domain the Dili domain of human
albumin. Such a domain is preferably physically linked to a constant region of
an
IgA part of the antibody.
In an aspect, an engineered antibody or fragment thereof, as provided
5 herein comprising the variable region of an IgG antibody and the constant
regions
of an IgA antibody, can reduce a side effect. In an aspect, an engineered
antibody or
fragment thereof, as provided herein, can reduce a side effect associated with
an
IgG antibody therapy. In an aspect, an engineered antibody or fragment
thereof, as
provided herein, can reduce a side effect associated with an innate immune
10 response. Side effects associated with such innate immune response can
be
inflammation, complement system activation, white blood cell (mast cell,
phagocyte, macrophage, neutrophil, dendritic cell, basophil, eosinophil,
natural
killer cell, and gamma delta cell activity. In some cases, an engineered
antibody or
fragment thereof, as provided herein, can reduce a side effect associated with
15 complement system activation.
Complement system activation can be a biochemical cascade of the immune
system that complements the ability of antibodies or fragments thereof to
clear
pathogens or mark them for destruction. The complement cascade comprises a
variety of plasma proteins, which are synthesized in the liver, these proteins
have
various functions comprising: triggering the recruitment of inflammatory
cells,
tagging pathogens for destruction (opsonization), perforating the plasma
membrane of pathogens, eliminating neutralized antigen-antibody complexes, and
any combination thereof. In some aspects, a reduction of a side effect can
comprise
the reduction of complement system activation.
In an aspect, an IgG antibody or portion thereof can react with a target
resulting in binding of Clq to the Fe portion of antigen-bound IgG after which
Cir
and Cis attach to form Cl, an enzyme associated with complement activation. Cl
can proceed to enzymatically cleave C4 into C4a and C4b. C4b binds to adjacent
proteins and carbohydrates on the surface of the target and then binds C2.
Activated Cl can cleave C2 into C2a and C2b forming C4b2a, a C3 convertase. C3
convertase can cleave C3 into C3a and C3b. In further steps of the complement
activation pathway, C3b can bind to C4b2a, a C3 convertase, to form C4b2a3b, a
C5 convertase that can cleave C5 into C5a and C5b. C5b can bind to the target
and
also bind C6, C7, c8, and C9 to form C5b6789n, the membrane attack complex
(MAC). MAC can destroy gram-negative bacteria as well as cells displaying
foreign
antigens (such as virus-infected cells, tumor cells, to name a few) by causing
their
lysis. In an aspect, a reduction in a complement response can be measured by
the
absence or reduced level of any one of: C1q, Clr, Cls, Cl, C4, C4a, C4b, C2,
C2a,
C2b, C4b2a, C3, C3a, C3b, C4b2a3b, C5, C5a, C5b, C6, C7, C8, C9, C5b6789m,
MAC, and any combination thereof. In an aspect, a reduction in a complement
response can refer to a reduction of about 5%, 10%, 15%, 20%, 25%, 30%, 35%,
40%,
CA 03076482 2020-03-19
WO 2019/059771 PCT/NL2018/050629
16
45%, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95%, 99%, 100% of binding of
a complement factor selected from any one of: Clq, Clr, Cis, Cl, C4, C4a, C4b,
C2,
C2a, C2b, C4b2a, C3, C3a, C3b, C4b2a3b, C5, C5a, C5b, C6, C7, C8, C9,
C5b6789m,
and MAC to the engineered antibody that comprises an IgG variable region and
the
IgA constant regions or portion thereof as described herein, as compared to a
comparable amount of an ligG antibody.
A reduction of a complement response can be determined by performing an
ELISA, flow cytometry, hematology, or an in vitro assay to quantify an amount
of a
complement factor selected from. any one of: tA.q, Cir, Cis, Cl, C4, C4a,
C413, C2,
C2a, C2b, C4b2a, C3, C3aõ C3b, C4b2a3b, C5,. C5a., C513, C6., C7, C8, C9,.
C5b6789m,
MAC, and any combination thereof. In some aspects, an antibody or fragment
thereof provided herein can result in a lower level of a complement factor as.
compared to a comparable antibody or fragment thereof comprising the provided
.. CDR1, CDR2. and CDR3 regions, In some aspects, an engineered IgG-IgA
antibody
or fragment thereof provided herein can result in a lower level of a
complement
factor a.s compared to a comparable IgG antibody or fragment thereof
.com.prisi
the same CT) RI. ( :1)112, and CDR3. A reduction of a complement response can
also
be measured by flow cytometric quantification of complement dependent lysis of
target cells as well as described in the examples.
Additional side effects which can be mitigated or reduced by administration
of an engineered antibody or fragment thereof described herein, as compared to
administration of an IgGõ can refer to any side effects associated with IgG
antibody
therapy. Side effects. associated with IgG antibody therapy can include
inflammation, thromboeinbolic events, haemolytic events, complement-system
associated events, and a combination thereof. In some cases, a.n IgG antibody
therapy can result in side effects such as: inflammation, hypertension,
hypotension,
pain, fever, urticaria, allergy, chill, weakness, diarrhea, nausea, vomit,
rash, itch,
cough, constipation., edema, headache, fever, shortness of breath, muscle
ache, pain,
decreased appetite, insomnia, dizziness, anaphylaxis, thrombosis, heart
failure,
bleeding, hepatitis, enterocolitis, mucositis, cytokine syndrome,
hypothyroidism,
hyponatremia, hypokalemia, capillary leak syndrome, and allodynia.
In an. aspect, reducing a. complement response, complement-associated side
effect, or a toxicity can improve treatment compliance of a subject in need.
thereof.
In some cases, an antibody or fragment thereof provided herein, such as
engineered
antibodies, can be administered at a higher dose, a longer treatment period,
or with
more frequency as compared to a comparable IgG antibody comprising the same
CDR1, CDR2. and CDR3 regions, In sonic cases: an antibody or fragment thereof
provided herein, such as engineered antibodies: can be administered at a
higher
dose from about 1X, 2X, 3X, 4X, 5X, 6X., 7X, .8X, 9X, 1_0X, over that of a
comparable
IgG antibody comprising the same CD RI., CDR2, and CD113 regions. In some
eases,
an antibody or fragment thereof provided herein, such as engineered
antibodies,
CA 03076482 2020-03-19
WO 2019/059771 PCT/NL2018/050629
17
can be administered for a longer time period from about 1 hour, 5 hrs, 10 hrs,
1 day,
2 days, 3 days, 4 days, 5 days, 7 days, 14 days, 24 days, 30 days, monthly,
bimonthly, hi-yearly, yearly, and daily over that of a comparable ItgG
antibody
comprising the same CDR1. CDR2, and CDR3 regions, In some cases, an antibody
or fragment thereof provided herein, such as engineered antibodies, can be
administered more frequency such as hourly, daily, weekly, monthly, yearly as
compared to a comparable JgG antibody comprising the same CDR1, CDR2, and
CDR3 regions..
A reason for changing an amino acid at a certain position can be
immunogenicity. Other reasons include but are not limited to improving
production
or homogeneity of the antibody. Antibodies of the present invention have
variable
heavy and variable light chain regions derived from a marine background.
Antibodies with such variable domains can be used in humans. Presently it is
preferred to de-immunize such variable domains, De-immunization typically
involves the modification of the murine sequence into a more human sequence
whenever possible. Typically such modifications are directed towards removing
one
or more T-cell epitopes or one more B-cell epitopes from the variable domain.
In a
preferred embodiment one or more (human) T-cell epitopes have been removed by
replacement of at least one amino acid of the epitope with a different amino
acid.
Often it is sufficient to substitute the so-called "anchor" amino acid,
Suitable
replacement amino acids can be obtained from somatic cell hypermutants of the
particular VI-1 or VI,. Replacement with an amino acid that is naturally
present at
that position in a human antibody is preferred. Also human B-cell epitopes.
can be
removed by replacement of at least one amino acid of the epitope with a
different
amino acid. Often it is sufficient to substitute only one amino acid of the
epitope..
Suitable replacement amino acids can be obtained from somatic cell
hypermutants
of the particular VII or VL. Replacement with. an amino acid that is naturally
present at that position in a human antibody is preferred. Preferably a
variable
domain of the invention is modified with respect to one or more exterior
residues.
Such residues are readily encountered by the immune system and are preferably
selectively replaced with human residues to provide a hybrid molecule that
comprises either a weakly immunogenic or substantially non-immunogenic
surface.
Suitable replacement amino acids can be obtained from somatic cell
hypermutants
of the particular VU or VI¨ -Replacement with an amino acid that is naturally
present at that position in a human antibody is preferred. The invention thus.
further provides an antibody of the invention that comprises, a humanized
heavy
chainvariable region, a humanized light chain variable region or a combination
thereof. A light chain of an antibody as defined herein has a light chain
constant
region. The light chain constant region is preferably the light chain constant
region
of the corresponding antibody. A light chain comprising a dill :18 light chain
variable region preferably also comprises the light chain constant region of
the
CA 03076482 2020-03-19
WO 2019/059771 PCT/NL2018/050629
18
ch14.18 antibody. A light chain comprising a 3F8 light chain variable region
preferably also comprises the light chain constant region of the 3F8 antibody.
The invention further provides a method of treatment of a subject that has a
G11)2 positive tumor or is at risk of having said GD2 positive tumor the
method
comprising administering a therapeutic amount of an antibody of the invention
to
the subject in need thereof Also provided is an antibody of the invention for
use in
the treatment of a subject that has a GD2 positive tumor or is at risk of
having a
G.D2 positive tumor. The treatment preferably further comprises administering
retinoic acid in an amount effective to upregulate the ganglioside GD2 in
neuroblastoma cells in said subject. The treatment preferably further
comprises
administering granulocyte-macrophage colony-stimulating factor (GM-CSF), G-
UST or a combination thereof in an amount effective to increase the number of
granulocytes in the subject and/or to increase ADCC, The GM-CSF, G-CSF or a
combination thereof is preferably of a species that is to be treated. It is
preferably
human GM-CST, human G-CSF or a combination thereof.
The GD2 positive tumor is preferably a GD2 positive neuroblastoma such as
a neuroectoderm-derived tumor or a sarcoma. in a preferred embodiment the GD2
positive tumor is a GD2 positive neuroblastoma, retinoblastoma, melanoma,
small
cell lung cancer, brain tumor, oste.osarcoma, rhabdomyosarcoma, Ewing's
sarcoma
in children and adolescents, or liposarcoma, fibrosarcoma, leiomyosarcoma or
another soft tissue sarcoma in adults. in a preferred. embodiment the GD2
positive
tumor i.s a TIEUI rOblastoma. The neuroblastoma treated.. with a method. or
use of the
invention is preferably a high risk neuroblastoma.
The retinoic acid is preferably 13-cis-retinoic acid (isotretinoin).
The invention further provides an antibody of the invention or a method or
antibody for use of the invention, wherein the antibody comprises a heavy
ch.ain
variable region. with the amino acid sequence
EVQLLQSGPE LEKPGASVMT SCKASGSSFT GYNMNWVPQN IGKSLEWIGA
IDPYYGGTSY NQKFKGRATL TVDKSSSTAY MHLKSLTSED SAVYYCVSGM
EYWGQCTSVT VSS
and a light chain variable region with the amino acid sequence
EIVMTQSPAT LSVSPGERAT LSCRSSQSLV HRNGNTYLHW YLQKPGQSPK
LLIHKVSNRF SGVPDRFSGS GSGTDFTLKI SRVEAEDLGV YFCSQSTHVP
PLTFGAGTKL ELK.
The invention also provides a nucleic acid molecule or combination of nucleic
acid molecules that codes for a heavy chain, a light or preferably both of an
antibody as described herein. The nucleic acid molecule or combination
preferably
further comprises one or more sequences for the expression of an antibody as
CA 03076482 2020-03-19
WO 2019/059771 PCT/NL2018/050629
19
described. Non-limiting examples of such expression sequences are a promoter,
a
termination sequence, an enhancer, an intron etc. Such sequences are not
necessarily present on the nucleic acid molecule as such sequences can be
provided
in cis by the integration site of the nucleic acid molecule in, for instance,
a
chromosome of a cell, or a vector comprising said nucleic acid molecule.
Suitable
integration sites in a cellular chromosome can easily be determined and
targeted,
for instance by means of homologous recombination.
Further provided is a cell that comprises a nucleic acid molecule or
combination as described herein.
Further provided are means and methods for the production of an antibody
as described herein using a nucleic acid molecule or combination of the
invention or
a cell comprising a nucleic acid molecule or combination of nucleic acid
molecules of
the invention.
A nucleic acid molecule or combination according to the invention is for
instance comprised in a cell. When said nucleic acid is expressed in said cell
the
translated product of the nucleic acid molecule can be, or be incorporated
into, an
antibody of the invention. The invention thus also provides a cell comprising
a
nucleic acid molecule or combination according to the invention. The invention
further provides a cell comprising a nucleic acid molecule or combination of
the
invention and that is capable of producing an antibody of the invention.
Further
provided is a method for producing an antibody of the invention comprising
culturing a cell comprising expressing one or more nucleic acid molecules that
code
for an antibody of the invention and harvesting the antibody from the culture
medium, the cell or a combination thereof. Said cell is preferably an animal
cell,
more preferably a mammalian cell. The cell is preferably a cell that is
normally
used for the production of an antibody for use in humans. Non-limiting
examples of
such cells are CHO, NSO, HEK cells preferably HEK293F cells, and PER.C6 cells.
Cells may specifically designed to suit certain purposes, for instance, most
cell lines
used for the production of antibodies have been adapted for growth in
suspension,
in high densities and other properties. For the purpose of the invention a
suitable
cell is any cell capable of comprising and preferably of producing an antibody
according to the invention.
For the purpose of clarity and a concise description features are described
herein as part of the same or separate embodiments, however, it will be
appreciated that the scope of the invention may include embodiments having
combinations of all or some of the features described.
CA 03076482 2020-03-19
WO 2019/059771 PCT/NL2018/050629
BRIEF DESCRPTION OF THE DRAWINGS
Figure 1: Variable heavy and light chain amino acid of the antibodies ch14.18
(panel A and B respectively; the CDR regions are underlined). DNA sequences
5 coding for the variable heavy and light chain of the antibody ch14.18
(panel C and
D respectively). For 3F8 a chimeric gammal heavy chain (SEQ ID NO: 1) and a
chimeric kappa light chain (SEQ ID NO: 2) are depicted. The respective CDRs
are
underlined.
10 Figure 2: Quantification of IgA ch14.18 after transfection with several
heavy (HC)
and light chain (LC) ratio's. 5 different ratios were used for IgG1 and IgAl
production. The optimal ratios were used for larger scale production.
Figure 3: A: K-light chain specific affinity chromatography of IgA ch14.18. B:
Size
15 exclusion chromatography of IgA ch14.18 UV absorption, representing
protein
concentration, is indicated by the trace.
Figure 4: binding of in-house produced and purified IgG1 and IgA ch14.18 to
the
GD2 expressing neuroblastoma cell line IMR32 and SK-N-Fl in flow cytometry.
Figure 5: CDC assay of IgG1 and IgA after 1 hr and 4 hrs of incubation with
IMR32 cell line. Cell lysis through complement activation is analyzed by flow
cytometric analysis. 15% pooled human serum was added, and cells were
incubated
for GO min and 4 hrs at 37 C. The amount of cell lysis is measured by 7-AAD
staining.
Figure 6: Leukocyte (Panel A and C) and PMN (Panel B and D)ADCC assay of
IgG1 and IgA after 4 hrs of incubation with the IMR32 (Panel A and B) and SK-N-
FI (Panel C and D) cell lines. Red blood cells were lysed and the remaining
effector
cells were added to wells. After 4 hours of incubation at 37 C, 51Cr release
was
measured in counts per minute (cpm) by a beta-gamma counter. The percentage of
specific lysis was calculated by determining the maximal lysis in the presence
of
triton and basal lysis in the absence of antibodies and effector cells.
Figure 7: Leukocyte ADC,V assay of in-house produced and purified IgG1 and IgA
after 4 hrs of incubation with the IMR32 and SK-N-Fl cell line. Red blood
cells
were lysed and the remaining effector cells were added to wells together with
cytokines. After 4 hours of incubation at 37 C, 51Cr release was measured in
counts
per minute (cpm) by a beta-gamma counter. The percentage of specific lysis was
calculated by determining the maximal lysis in the presence of triton and
basal
lysis in the absence of antibodies and effector cells.
CA 03076482 2020-03-19
WO 2019/059771 PCT/NL2018/050629
21
Figure 8: PMN ADCC assay of IgG1 and IgA ch14.18 after 4 hrs of incubation
with the IMR32 or SK-N-FT cell line. PMNs were isolated by Ficoll/Histopaque
separation. Subsequently, effector cells, cytokines and antibodies at various
concentrations were added to microtiter plates containing target cells. E:T
ratios
were 40:1 (PMN). After 4 hours of incubation at 37 C, 51Cr release was
measured in
counts per minute (cpm) by a beta-gamma counter. The percentage of specific
lysis
was calculated by determining the maximal lysis in the presence of triton and
basal lysis in the absence of antibodies and effector cells.
Figure 9: GD2 (Panel A) and MHC-I (Panel B) expression on IMR32 cells after
exposure to different concentrations of isotretinoin for 4 consecutive days.
Cells
were stained with IgA ch14.18 and detected with a secondary anti-IgA PE or
anti-
MHC-I-PE.
Figure 10: Schematic representation of the ganglioside GD2.
Figure 11: Quantification of in vivo mechanical thresholds. Experiments were
conducted using female (aged 8-12 weeks) C57BL/6 mice (Harlan Laboratories).
Mice received an intravenous injection of 100 microgram of antibody.
Mechanical
thresholds were determined using the von Frey test (Stoelting) with the up-and-
down method as is described in Eijkelkamp et al., 2010; J. Neurosei. 30:2138-
2149
and Chaplan et al., 1994; J. Neurosci. Methods 53:55-63. All experiments were
performed by experimenters blinded to treatment.
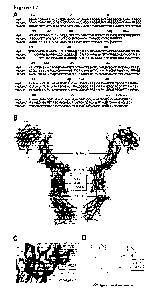
Figure 12: Primary sequence and modeling of the IgA1/IgA2.0 hybrid antibody.
A,
alignment of primary sequences of the constant regions of hIgAl, IgA2m(1), and
a
IgA1/IgA2m(1) hybrid (hIgA2.0). Residues are numbered according to the myeloma
IgAl protein (Bur) scheme. Domain boundaries are indicated by vertical lines
above the sequences. The following features are highlighted: light gray
underlined
residues are unique for IgAl, dark gray underlined aspar a gines are conserved
N-
glycosylation consensus sequences, and black underlined residues are
unique for IgA2Ø B, the heavy chain of 225-IgA2.0 was modeled and
illustrated in
front and side view, with mutations marked. C, heavy chains of wild-type and
mutant IgA2 were modeled. The resulting alignment indicates a different
orientation of C241 in the heavy chains of IgA2-wt compared with IgA2.0,
possibly
due to the P221R mutation. D, focus on the tailpiece of 225-IgA2-wt (green,
C471;
red, Y 472) and IgA2.0 (red). Prediction and alignment of models were
performed
using I-TASSER; models were modified in 3D-Mol Viewer. E, nucleic acid
sequence
and protein sequence of the constant regions of the heavy chain of IgA2Ø
Figure 13: HP-SEC analysis of IgAl and IgG1 ch14.18. Purified antibodies were
subjected to HP-SEC analysis. Both IgAl ch14.18 (A) and IgG1 ch14.18 (B)
showed
to be highly monomeric. Traces show the UV absorbance at 280 nm.
CA 03076482 2020-03-19
WO 2019/059771 PCT/NL2018/050629
22
Figure 14: Binding of IgG1 and IgAl ch14.18 antibodies to neuroblastoma cell
lines. (A) Antibody binding of IgAl-FITC and IgGl-FITC ch14.18 to GD2
expressing neuroblastoma cell line IMR32 and GD2-negative cell line (II-ME-N
(B)
Real time cell-based affinity measurement of neuroblastoma antibodies on IMR32
cells. IMR32 cell lines were treated with 10 nM of antibody for 1
hourSubsequently,
concentration was increased to 20 nM of antibody for 1 hour). Dissociation was
followed from 130-250 minutes by replacing antibody containing medium with
medium without antibody. Calculated affinities from cell-based affinity
measurements are shown in B.
Figure 15: Characterization of ADCC by IgAl and IgG1 ch14.18 against a panel
of
neuroblastoma cell lines. (A) ADCC assays with IgAl and IgG1 ch14.18 on 3
different neuroblastoma cell lines with leukocytes from peripheral blood as
effector
cells. (B) ADCC assays with IgG1 ch14.18 on IMR-32ce11 line with isolated
PBMC's
(E:T ratio of 100:1) or h isolated neutrophils (E:T ratio of 40:1) as effector
cells (C)
ADCC assays with IgG1 ch14.18 on IMR32 cell line with isolated PBMC's (E:T
ratio of 100:1) or isolated neutrophils (E:T ratio of 40:1) as effector cells
with co-
treatment of 10 ng/ml GM-CSF, 6545 U/ml of IL-2 and 24 hours of pre-incubation
with 10 0/1 of 11-cis retinoic acid. (D) ADCC assays with leukocytes from
peripheral blood as effector cells in combination with 15% pooled human
complement active serum.
Figure 16: Complement assays on a panel of neuroblastoma cell lines by IgG1
and
IgAl ch14.18 antibodies. (A) Lysis by IgG1 ch14.18 antibodies on 4 different
neuroblastoma cell lines. Cells were incubated with 4 different concentrations
of
antibody and 15% serum for 15 minutes and 4 hours. (B) Lysis by IgAl ch14.18
antibodies on 4 different neuroblastoma cell lines. Cells were incubated with
4
different concentrations of antibody and 15% serum for 15 minutes and 4 hours.
(C)
Expression of complement regulatory proteins CD46, C1D55 and CD59 on
neuroblastoma cell lines.
Figure 17: In vivo efficacy of IgG1 and IgAl ch14.18 antibodies. (A)
Quantification
of bioluminescent signal of neuroblastoma cells after 24 hours of treatment
with
IgAl or IgG1 ch14.18. (B)) Quantification of bioluminescent signal at 3 days
of
treatment after I.V. injection of tumor cells.
Figure 18: Neuronal exposure of to IgAl does not lead to decreases in
mechanical
withdrawal thresholds. (A) Plasma concentrations of IgAl and IgG1 ch14.18 3
hours after intravenous injection. (B) Von-Frey withdrawal thresholds. (C) Von-
Frey withdrawal thresholds after I.V. injection of fluorescently labeled IgAl
ch14.18 or IgG1 ch14.18. (D) Left column: Visualization of intravenously
injected
Alexa-488 labeled antibodies on sciatic nerves. Right column: Visualization of
ex-
CA 03076482 2020-03-19
WO 2019/059771
PCT/NL2018/050629
23
vivo staining of GD2 by incubation of neurons with Alexa-549 labeled IgG1
ch14.18.
EXAMPLES
Example 1
Generation of anti-GD2 antibody producing vectors:
The amino acid sequences of the variable regions from ch14.18 were found
in the FDA application of dinutuximab (application 125516; depicted in Figure
1).
These amino acid sequences were translated to the cDNA sequence representing
the most likely non-degenerate coding sequence. The cDNAs were synthesized
(Baseelear) and subcloned into the pEE14.4 expression vectors which contain
either the IgAl or IgG1 backbone (described in: Beyer, T., et al. "Serum-free
production and purification of chimeric IgA antibodies." Journal of
immunological
methods 346.1(2009): 26-37). The optimal ratio of heavy to light chain DNA for
transfection was first determined by small scale test transfections in HEK293F
cells. Antibody production was then quantified by anti-human IgG (Figure 2A)
or
IgA ELISA (Figure 2B).
Production was scaled up to allow for antibody characterization and
functional assays. Again, HEK293F cells were transfected with the pEE14.4 IgA
ch14.18 heavy chain and ch14.18 kappa light chain at the optimal heavy to
light
chain transfection ratios. The produced IgA antibodies were purified by using
a
series of two liquid chromatography steps. First, IgA ch14.18 was isolated
from the
serum free supernatant using human x-light chain specific affinity
chromatography (Figure 3A). Next, size exclusion chromatography with a
preparative prepacked Superdex200 26x600 column was used to separate intact
IgA from free light chain (Figure 3B). Finally, IgG antibodies were purified
by
protein affinity chromatography and were subsequently dialyzed to PBS. This
multistep procedure results in highly pure preparations of recombinant
monomeric
IgA.
Characterization of anti-GD2 antibodies
Binding of anti-GD2 antibodies to the GD2 expressing cell lines IMR32 and
SK-N-FT was analyzed by staining cells on ice for 45 minutes with anti-GD2
antibody at several concentrations. Cells were washed and a secondary goat-
anti
human IgA-PE or IgG-PE was added to the cells for 45 minutes on ice in the
dark.
Afterwards, antibody binding was quantified by flow eytometric analysis.
Complement activation of the produced antibodies was assessed by
incubation of IMR32 and SK-N-FT cells with 15% pooled human serum and
CA 03076482 2020-03-19
WO 2019/059771 PCT/NL2018/050629
24
antibody (10-0.01 jug/m1) for 1 and 4 hours at 37 C. Afterwards, live/dead
staining
(7-AAD) of cells was performed and cellular lysis was quantified via flow
cytometrie
analysis.
ADCC assays were performed to assess the efficacy of IgA and IgG1 ch14.18
to recruit effector cells against neuroblastoma cells. First, leukocyte ADCC's
were
performed. Hereto, peripheral blood from healthy donors was treated with RBC
Lysis buffer to remove red blood cells. The remaining leukocytes were washed
and
added to radioactively labeled neuroblastoma cells with or without IL-2 and GM-
CSF (both lOng/m1). After 4 hours of incubation at 37 C, cell death was
quantified
by 51Cr release, measured by liquid scintillation. The percentage of specific
lysis
was calculated by determining the maximal lysis in the presence of 2.5% triton
X-
100 and basal cell lysis in the absence of antibodies and effector cells.
To evaluate whether PMNs are an important effector cell population for IgA
mediated killing, PMNs were isolated from peripheral blood from healthy donors
by ficoll-histopaque and added in a 40:1 E:T ratio to radioactively labelled
neuroblastoma cells with or without IL-2 and GM-CSF (both l0ng/m1).
Results and discussion
The functional characterization of the antibodies was performed by
determining binding capacity to GD2, complement activation and effector cell
recruitment. Flow cytometric analysis of binding to the GD2-positive cell
lines
TMR32 and SK-N-FI showed similar binding patterns for both IgG1 and IgA
(Figure 4). Complement activation of the produced antibodies was assessed by
live/dead staining (7-AAD) after incubation with 15% pooled human serum and
several concentrations of antibody The IgA anti-GD2 antibody did not show
activation of complement, while the IgGl variant having the same variable
regions
did activate the complement system after 1 hour of incubation (Figure 5A).
Lysis
increased further for IgG when incubation time increased to 4 hours, while for
IgA
no complement activation was observed (Figure 5B)
ADCC assays were performed to assess the efficacy of IgA and IgG1 ch14.18
to recruit effector cells against neuroblastoma cells. First, leukocyte ADCC"s
were
performed. Here, the IgA anti-GD2 antibody showed to be superior for both the
IMR32 and SK-N-FI cell line (Figure (3). To evaluate whether PMNs are an
important effector cell population for IgA mediated killing, PMN were isolated
from
peripheral blood from healthy donors. It was shown that the IgA anti-GD2
antibody stimulates PMN better than IgGl to kill TMR32 and SK-N-FI cells
(Figure 6).
When cells were incubated together with the clinically used cytokine GM-
C'SF, maximal lysis induced by the IgA anti-GD2 antibody strongly increased,
while only minimal effects were seen for the IgG1 variant (Figure 7).
Furthermore,
CA 03076482 2020-03-19
WO 2019/059771 PCT/NL2018/050629
addition of IL-2 did not increase the observed lysis (Figure 7). Similar
effects were
observed for PMN mediated ADCC (Figure 8).
We also show that treatment of neuroblastoma cell lines with isotretinoin is
5 able to influence GD2 expression. It was shown that after 4 days of
incubation with
isotretinoin, binding of IgA ch14.18 is increased, while MHC,-I expression
remained
stable (Figure 9).
The examples show that an IgA isotype variant of dinutuximab has higher
10 ADCC capacity for killing of neuroblastoma's, and no complement
activation. The
IgA anti-GD2 antibody enhances destruction of neuroblastoma cells, whereas
side
effects such as neuropathic pain are reduced.
In the present invention it is shown that isotype conversion of dinutuximab
15 to IgA provides a solution to one or more of the toxicity problems of
the
dinutuximab antibody. The antibody of the invention provides potent ADCC
activity while simultaneously reducing at least the pain problem associated
with
dinutuximab administration. Antibodies of the invention efficiently activate
neutrophils presumably through the FeaR (CD89). This generates potent anti-
20 tumor reactions. The susceptibility of neuroblastoma cells to an
antibody of the
invention shows that an IgA anti-GD2 exhibits improved efficacy against
neuroblastoma when compared to an IgG isotype variant with the same variable
domains, and simultaneously reduces treatment toxicity. This significantly
improves anti-neuroblastoma immunotherapy.
Antibody function is governed by a number of factors. The target and the
epitope that is recognized play an important role as does the cell on which
the
target resides. Effector functions of the antibody are often correlated with
the
isotype of the antibody. Although this is useful as a general rule, many
exceptions
30 indeed exist. This is exemplified for instance for ADCC activity by
Rajasekaran et
al (Rajasekaran et al., 2015; ImmunoTargets and Therapy Vol 4: 91) and for CDC
activity by Lohse et al., 2017; Br J Haematol. doi;10.1111/bjh.14624dgf; and
Pascal,
et al., (2012) Haematologica 97.11: 1686-1694).
35 Conversion of IgG to IgA can alter the receptors which interact with the
antibodies. For IgG, the activating Fe gamma receptors (FeyR), bearing an ITAM
motif, are the principal mediators of antibody mediated activation of
leukocytes.
Inhibitory FeyRs and polymorphisms in FeyRs interfere with these effects. This
can
make IgG treatment less suitable for subgroups of patients. We and others have
40 shown that IgA also exerts anti-tumor effects through an Fe receptor,
the FeaR,
which is expressed on neutrophils, monocytes and macrophages. Inhibitory
receptors and polymorphisms are not reported for FcaRI. However, the FcaRI is
not
expressed in mice, making it for a long time not possible to do the necessary
CA 03076482 2020-03-19
WO 2019/059771 PCT/NL2018/050629
26
preclinical in vivo studies with IgA therapeutic antibodies. However, within
our
laboratory the human FeaRI transgenie mouse was generated. This mouse has a
similar expression pattern of human FcaRI as in humans. These mice were back-
crossed in the relevant backgrounds, i.e. balb/c, C57B/L6, and SCID for
growing
human tumors.
IgA as therapeutic antibody.
Although IgA is known as a mucosal antibody, in its monomeric form it is
the second class of antibody present in the human serum. In previous studies
we
have shown that an anti-tumor antibody IgA can be effective in ultro. The anti-
tumor mechanism is different and mainly through the recruitment of
neutrophils,
the most abundant type of leucoeytes. Also in cluo IgA can be efficacious as a
therapeutic antibody. A drawback of an IgA molecule is its on average
relatively
short half-life, due to different glycosylation and lack of binding to the
neonatal Fe
receptor, FeRn. The present invention solves this problem for GD2 specific IgA
antibodies by providing them with adapted glycosylation and targeting of the
IgA
to FeRn indirectly (Meyer et al., 2016 MAbs Vol 8: pp 87-98).
In the present invention we show that anti-GD2 IgA isotype antibodies are
targeting neuroblastoma, in uitto, in uiuo and in patient-derived models.
Example 2
The efficacy of the anti-GD2 antibodies to elicit ADM, will be assay against
a further panel of GD2 expressing neuroblastoma cell lines (e.g. IMR-32, SH-
SY5Y,
SK-N-Fl, LAN-5), from ATCC) and ex vivo neuroblastoma cells as targets. In
addition, the panel will include tumor-initiating cells that have been
generated
from primary neuroblastoma samples and that contain stem cell-like cells that
may
represent the chemotherapy-resistant cell population responsible for
resistance to
conventional chemotherapy (such as described in Bate-Eya, et al., 2014;
European
journal of cancer 50.3: 628-637). Both whole blood and isolated effector
populations
from healthy volunteers and patients will be used as effector cells.
Practically,
target cells will be labelled with 5ICr for 2 hours. PMNs and PBMCs will be
isolated by Fieoll/Histopaque separation. Subsequently, effector cells,
antibodies at
various concentrations, and medium will be added to microliter plates
containing
target cells. E:T ratios will be 40:1 (PMN) and 50:1 (PBMC). After 4 hours of
incubation at 37 C, 51Cr release will be measured in counts per minute (cpm).
The
percentage of specific lysis will be calculated by determining the maximal
lysis in
the presence of triton and basal lysis in the absence of antibodies and
effector cells.
Testing on organoids derived from neuroblastoma patients
We have generated tumor-derived organoids from primary neuroblastoma
samples. Tumor-derived organoids reflect tumor heterogeneity and allow
CA 03076482 2020-03-19
WO 2019/059771
PCT/NL2018/050629
27
performing these functional tests on tissues representing primary tumor
tissue. We
will use the IgA dinutuximab on these organoids. We will evaluate binding,
ADCC
microscopically by death markers and complement depositions with Clq and iC3b
detecting antibodies.
In vivo experiments in FcaR transgenic mice
For in vivo experiments, we will make use of syngeneic models, xenograft
models and patient derived xenograft (PDX) models.
For syngeneic models, we will make use of the TH-MYCN 9464D-based syngeneic
neuroblastoma mouse model, grown orthotopically or subcutaneously.
For xenograft models we will make use of the HTLA-230 NB cells implanted
in SCID mice, as described in Bogenmann 1996 Int. J. Cancer Vol 67: 379; and
Raffaghello et al., 2003 Cancer Lett, Vol 197(1-2): p. 205-9. Previously, the
anti-
GD2 antibody 14G2a was tested in this model, and it was shown that it worked
very efficiently, but still in the absence of NK cells and macrophages
(Raffaghello
et al., 2003 supra). That fits with the notion that neutrophils are important
effector
cells for dinutuximab. When we will test IgA in these mice, we will use the
SCID/FcaR transgenic mice, available in our laboratory, since mice lack the
FeaR.
We will compare the efficacy of IgG and IgA in these transgenic SCID mice.
For PDX models we will make use of the NSG mice (NOD/SCID/gamma"
mice). These models allows for orthotopic positioning of patient tumors,
outgrowth
and subsequent treatment (Braekeveldt et al., 2016 Cancer Lett. Vol
1;375(2):384-
9. doi: 10.1016/j.canlet.2016.02.046).
Example 3
Improvement in neuroblastoma patient survival came in 2015, with the FDA
approval of ch14.18, a chimeric antibody of the IgG1 isotype directed against
the
ganglioside GD2, expressed on neuroblastoma cells, but also on peripheral and
central nervous tissue. Ch14.18 is given as second-line treatment in
combination
with IL-2, GM-CSF and 11-cis retinoie acid for the treatment of high-risk
neuroblastoma after hematopoietic stein cell transplantation. In a large phase
III
clinical trial (n=226) it was shown that ch14.18 combination therapy resulted
in
20% more event-free survival than standard therapy and 10% more overall
survival, 2 years after treatment (Yu et al. 2010, N Engl J Med 363(14): 1324-
1334).
Although the inclusion of immunotherapy improved the survival of neuroblastoma
patients, there are important side effects caused by the administration of
ch14.18.
Of these, severe neuropathic pain is the most frequent
(Yu et al. 2010, N Engl J Med 363(14): 1324-1334). This is believed to be
caused by
ch14.18 binding to G-D2 on A6 and C pain fibers and activates the complement
system locally, which is the cause for the observed pain (Xiao et al 1997,
Pain 69(1-
2): 145-151). This pain does not respond well to analgesics and can be dose
limiting
(Gilman et al 2009, J Clin Oneol 27(1): 85-91).
CA 03076482 2020-03-19
WO 2019/059771 PCT/NL2018/050629
28
Ch14.18-antibody-opsonized tumor cells can be killed by leukocytes through
antibody-dependent cell-mediated cytotoxicity (ADCC), depending on antibody
binding to Fe receptors on leucocytes. Ch14.18 is also able to activate the
complement system on the tumor cell surface, causing lysis of the cell via
complement-dependent cytotoxicity (CDC). For ADCC mediated by IgG1 antibody
therapy, natural killer (NK) cells are regarded as important cells for
mediating
ADCC. For neuroblastoma, there is evidence that granulocytes also play a role
in
mediating ADCC when treated with an anti-GD2 IgG1 antibody (Bruchelt et al
1989, Immunol Lett 22(3): 217-220; Gilman et al 2009, J Clin Oncol 27(1): 85-
91;
Cheung et al 2012, J Clin Oncol 30(4): 426-432).
Results
Production and purification of IgG1 and IgAl ch14.18
To compare IgAl and IgG1 antibodies with the ch14.18 variable region, both
antibodies were produced and purified in-house. All antibodies were analyzed
by
HP-SEC to confirm their purity. Both IgAl ch14.18 (Figure 13A) and IgG1
ch14.18
(Figure 13B) were shown to be monomeric, with a purity greater than 95%.
IgAl and IgG1 ch14.18 specifically bind GD2 with similar affinity
Next, we determined the binding of IgAl and IgG1 ch14.18 to a panel of
neuroblastoma cell lines. Both antibodies recognized the GD2 expressing
neuroblastoma cell line IMR-32 similarly, while the GI-ME-N cell line without
GD2
expression was not bound (Figure 14A). To establish whether the affinity of
the
antibodies remained unchanged after changing the isotype to IgAl, we performed
real time cell-based affinity measurements on IMR-32 cells with Ligand Tracer.
The antibodies closely follow the same association pattern at 2 different
concentrations (10 and 20 nM respectively) and also overlap in the
dissociation
phase). The assays indicate that the calculated affinity to IMR32 cells did
not differ
significantly between IgAl and IgG1 (3.8 nM vs 4.8 nM) (Figure 14B).
Mechanism of action of IgAl and IgG1 ch14.18 antibodies
Subsequently, we compared the in uitro mechanism of action of these
antibodies.
Both ADCC and CDC are known to be induced by ch14.18 against neuroblastoma
in rho. To compare killing with a mix of effector cells, ADCC assays were
performed on IMR-32, SK-N-FT and LAN-1 neuroblastoma cell lines with
leukocytes as effector cells. Both IgA and IgG antibodies lysed IMR-32 cells
to a
similar extent, while SK-N-FI and LAN-1 cells were killed better with IgAl
ch14.18 (Figure 15A). The GI-ME-N cell line, that has no detectable GD2
expression on FACS could not be lysed by both antibodies, showing that GD2
expression is a prerequisite for ADCC (data not shown). To evaluate the
relative
importance of certain leukocyte subsets in mediating ADCC, neutrophils and
peripheral blood mononuclear cells (PBMC) were separately used as effector
cells
CA 03076482 2020-03-19
WO 2019/059771 PCT/NL2018/050629
29
to determine their respective cytotoxic capacity against neuroblastoma cell
lines
with these antibodies.
With PBMC's as effector cells, IgG1 ch14.18 effectively lysed IMR32 cells,
while
lysis with the IgA antibody did not perform as well (Figure 15B). This was
also
seen for 2 other neuroblastoma cell lines (data not shown) On the contrary,
when
neutrophils were used as effector cells, IgA ch14.18 mediated superior ADCC
for all
tested cell lines in comparison to IgG1 (Figure 15B).
In the clinic, ch14.18 is administered as combination therapy with GM-CSF, IL-
2
and retinoic acid. Next, the impact of these compounds on ADCC was assessed
for
both antibodies. When PBMCs were used as effector cells, addition of GM-CSF
did
not increase ADCC by IgGl. Combination of GM-CSF and IL-2 led to a minor
increase of cell death, even without the presence of antibody, showing
increases in
PBMC-mediated killing regardless of antibody. Finally, addition of pre-
treatment
of 11-cis retinoic acid for 24 hours to the previous combination showed
further
increases in killing for the IMR32 and SK-N-FI cell lines (Figure 15C). With
IgA,
no antibody dependent killing could be induced by PBMC's, but cells were lysed
in
presence of IL-2 and the combination with cis-retinoic acid, indicating
antibody
independent recognition of neuroblastoma cells by PBMC's (data not shown).
Different from IgGl, GM-CSF in combination with IgAl ch14.18 boosted ADCC
with neutrophils as effector cells (Figure 15C). Presence of IL-2 did not
further
increase neutrophil mediated killing, while maximal lysis increased with pre-
exposure to 11-cis-retinoic acid. For IgG1 antibodies, GM-CSF improved killing
with neutrophils slightly, while addition of IL-2 or retinoic acid did not
enhance
killing further (data not shown).
Finally, ADCC assays were supplemented with human pooled serum to investigate
whether CDC could enhance neuroblastoma cell lysis. For both IgG1 and IgAl
ch14.18 no significant differences in lysis were observed after addition of
serum
(Figure 15D).
IgAl ch14.18 does not activate complement
A second effect mediated by ch14.18 is activation of the complement system.
Ch14.18 is known to lyse neuroblastoma target cells via CDC in citro. We
assessed
in uitto complement activation by these antibodies on the same panel of
neuroblastoma cell lines as were used in ADCC assays. IgG1 ch14.18 lysed all
tested neuroblastoma cell lines except SK-N-FT via CDC after 15 minutes
(Figure
16A). Contrary to this, no lysis could be observed for IgAl ch14.18 (Figure
16B).
With longer incubation for 1 hour, the amount of lysis further increased for
IgGl,
but cells remained negative for 7-AAD with IgAl (Figure 16A,B). Since the
amount
of complement regulatory proteins CD55 and CD59 on SK-N-FT is significantly
higher than on the other tested neuroblastoma cell lines, this cell line seems
to be
less prone to complement mediated lysis (Figure 16C).
CA 03076482 2020-03-19
WO 2019/059771 PCT/NL2018/050629
IgAl excels in tumor cell depletion in vivo in comparison to IgG1 ch14.18
Finally, we analyzed the capacity of the antibodies to kill GD2 expressing
cells in
cluo in 2 syngeneic mouse models. As a model for a localized tumor, animals
were
5 intraperitoneally injected with EL4 cells naturally expressing GD2. After
24 hours
of outgrowth, animals were treated with IgAl or IgGl. Outgrowth of tumor cells
was evaluated 24 hours after injection of the antibody. Although both
antibodies
reduced the average tumor burden treatment with IgA which has only one of the
two effector mechanisms, was more effective (Figure 17A).
10 In a second systemic mouse model, we assessed tumor cell killing for a
longer
period of time. EL4 cells were intravenously injected). Shortly after
injection of
cells, tumor cells localized to the lungs, as observed with bioluminescent
imaging(Data not shown). After three days of treatment, both IgAl and IgG1
cleared the tumor cells, while cells were still present in mice treated with
15 PBS(Figure 17B) Outgrowth was observed in the abdomen of the mice by
bioluminescent imaging (data not shown).
Anti GD2-IgA1 does not induce pain
A major limitation of IgG1 antibodies directed against GD2 is the increased
20 sensitivity to light touch after treatment. To test whether the lack of
complement
activation by IgA would mitigate this problem, we conducted in uiuo pain
experiments in mice. Paw retraction thresholds after stimulation with von Frey
hairs were determined as a measure for allodynia.
To correct for differences between the half-life of IgAl and IgGl, mice were
25 intraperitoneally injected with either a low dose of IgG1 (20 lug),
corresponding to a
dose of 100 gg of IgA at 24 hours or a high dose of IgG1 (100 jig) (Figure
18A). The
antibody concentration found back in the serum after 24 hours is in line with
the
clinical phenotype after ch14.18 treatment. Treatment with 20 jig of IgG1
ch14.18
showed a significant decrease in withdrawal threshold, which returned to
baseline
30 after 48h. A sub-therapeutic dose of IgG1 (4 g) did not lead to a
significant
reduction in withdrawal threshold. Different from IgGl, all tested doses of
IgAl did
not decrease the paw retraction threshold, indicating that IgAl does not
induce
allodynia at levels comparable with IgG (Figure 18B). Similar results were
obtained when fluorescently labeled IgAl and IgG1 ch14.18 antibodies were
injected intravenously. Here, IgG1 ch14.18 also reduced the withdrawal
threshold,
while IgAl ch14.18 did not (Figure 18C.Subsequently, binding of the antibodies
was assessed on the sciatic nerve. Antibody exposure was observed with 20 g
of
IgG1 and 100 jig of IgAl ch14.18 to a similar extent and corresponded to the
amount of antibody present in the serum (Figure 18D). The anti-CD20 antibody
rituximab was not detected on sciatic nerves. Ex vivo staining of GD2
overlapped
with the signal from directly labeled antibodies.
CA 03076482 2020-03-19
WO 2019/059771
PCT/NL2018/050629
31
Kits
Provided herein are kits comprising compositions of engineered antibody or
fragments thereof. Disclosed herein can also be kits for the reduction of a
complement response, such as reducing complement activation, Disclosed herein
can also be kits for the treatment of a cancer, pathogen infection, immune
disorder
or allogeneic transplant. In one embodiment, a kit can include a therapeutic
or
prophylactic composition containing an effective amount of an engineered
antibody
in unit dosage form. In some embodiments, a kit comprises a sterile container
which can contain a therapeutic composition of engineered antibodies or
fragments
thereof; such containers can be boxes, ampules, bottles, vials, tubes, bags,
pouches,
blister-packs, or other suitable container forms known in the art. Such
containers
can be made of plastic, glass, laminated paper, metal foil, or other materials
suitable for holding medicaments. In some cases, engineered antibodies and
fragments thereof can be provided together with instructions for administering
the
antibody or fragment thereof to a subject having or at risk of developing a
toxicity,
complement-associated toxicity, cancer, pathogen infection, immune disorder or
allogeneic transplant. Instructions can generally include information about
the use
of the administration how to utilize the composition to treat toxicity, a side
effect,
cancer, pathogen infection, immune disorder or allogeneic transplant.
Discussion
The approval of ch14.18 for high-risk neuroblastoma led to improvements in the
survival of neuroblastoma patients. Nevertheless, IgG1 antibody therapy for
neuroblastoma comes with a major limitation: presence of severe side effects
such
as neuropathic pain and allodynia. We investigated whether changing the
isotype
of ch14.18 from IgG1 to IgAl would abrogate this antibody-induced allodynia.
The present invention shows that IgAl ch14.18 offers surprisingly strong anti-
tumoral effects through neutrophil mediated ADCC and does not induce allodynia
in uico. Blocking the C5a receptor with an antagonist completely stopped
allodynia
(Sorkin et al 2010, Pain 149(1): 135-142). Thus far, several approaches were
undertaken to amend side-effects caused by ch14.18. A first approach was to
mutate the C1q-binding site of ch14.18 (K322A). This mutation is thought to
abrogate complement activation of ch14.18 (Sorkin et al 2010, Pain 149(1): 135-
142). This approach only reduced complement activation, and, as a result,
residual
pain remained. Also in a phase I clinical trial with this antibody, grade 3-4
toxicity
in the form of pain occurred in 68% of patients (Navid et al 2014, J Clin
Oncol
32(14): 1445-1452).
Another approach to circumvent ch14.18 induced allodynia was performed by
targeting a differentially glycosylated variant of GD2 (0-acetyl-GD2), which
is
solely present on neuroblastoma cells and not on peripheral nervous tissue by
the
CA 03076482 2020-03-19
WO 2019/059771 PCT/NL2018/050629
32
antibody e.8B6 (Terme et al 2014, PLoS One 9(2): e87210). The antibody c.8B6
does
not induce complement activation around pain fibers, however, this antibody is
likely less effective.
During the first reports on 14.18, the parental mouse-antibody where ch14.18
is
derived from, it was noted that granulocytes mediated effects against
neuroblastoma (Bruchelt et al (1989); Immunol Lett 22(3): 217-220). Later, it
was
shown that their killing potential could be enhanced with the addition of GM-
CSF.
In clinical studies, it was found that the activation of granulocytes is an
indicator
for a good outcome of antibody therapy against neuroblastoma (Cheung et al
(2012); J Clin Oncol 30(4): 426-432). Besides co-treatment with GM-CST, there
has
been a lack of approaches to further engage this target cell population for
therapeutic purposes.
Next to an inherent vulnerability of neuroblastoma cells to neutrophil
mediated
killing, the timing of the current treatment protocol also favors an approach
were
neutrophils are used as effector cells. Now, patients are prescribed with a
myeloablative regimen after which autologous stein cell transplantation
follows
(Yu et al. (2010). N Engl J Med 363(14): 1324-1334). Shortly thereafter, the
immunotherapeutic protocol is started. Neutrophils are often the first
leukocyte
population which is restored to physiological levels and are therefore are an
attractive leukocyte subset to active in such an immune-compromised state.
Less
than 25% of patients reached proper immune reconstitution for NK cells at the
moment of immunotherapy (Nassin et al 2018, Biol Blood Marrow Transplant
24(3): 452-459). With an approach where neutrophils instead of NK cells are
addressed, therapeutic variability will be reduced.
In the present invention it was found that IgA mediated killing was improved
by
the addition of GM-CSF.
IL-2 does not seem to improve IgA or IgG mediated killing. IL-2 was added to
the
clinical regimen after effects were seen in GD2-expressing melanoma and
sarcoma
patients which were treated with ch14.18. However, the effects of IL-2 on
neuroblastoma therapy remain unclear. This is further stressed by the clinical
trial
which compared immunotherapy with and without IL-2 (Ladenstein et al 2013
MAbs 5(5): 801-809). The authors show that the addition of IL-2 did not have
significant effects on EFS or OS and that early termination because of
toxicity was
significantly higher in the IL-2 arm.
Although IgAl lacks a Cill binding site, complement activation of IgA has been
documented. The MBL pathway was shown to be activated by polymeric IgA, while
the classical complement pathway was triggered for monomeric IgA directed
against CD20 (Roos et al; 2001; J Immunol 167(5): 2861-2868 andLohse, Loew et
CA 03076482 2020-03-19
WO 2019/059771 PCT/NL2018/050629
33
al; 2018; Br J Haematol 181(3): 413-417). Nevertheless, IgAl ch14.18 did not
induce CDC of neuroblastoma cell lines and did not induce complement-dependent
allodynia in mice.
Although allodynia can be solved with IgA, complement as an effector mechanism
is also lost. In the present invention it was surprisingly shown that the
expected
loss of therapeutic effect did not occur. In the in uico tumor models we
indeed see
that IgA is at least as efficient as IgG in killing tumor cells. In uluo
complement
consumption by anti-GD2 antibody has been demonstrated, indicating that
complement activation does take place (Cheung at al 2014, Int J Cancer 135(9):
2199-2205).
In our studies, we dosed mice with 5 times the amount of IgA compared to IgG1
to
adjust for the difference in half-life between the two antibodies in mice. A
similar
neuronal exposure and serum concentration could be achieved with the two
antibodies. For longer models, multiple doses of IgAl were injected to account
for
this. Although the in vivo half-life of IgA is approximately a week in humans,
several approaches can be undertaken to improve the half-life of IgA in mice
to
improve future comparisons (More11 et al, 1973, Clin Exp Immunol 13(4): 521-
528).
The glycosylation of IgA can be reduced to decrease clearance by binding to
the
asialoglycoprotein receptor. Silencing of multiple IgA glycosylation sites was
already accomplished before, leading to improved pharmacokinetics (Lohse et
al,
2016, Cancer Res 76(2): 403-417). Contrary to IgG, IgA does not interact with
the
neonatal Fe receptor (FeRn). Therefore, antibodies pinocytosed by endothelial
cells
are degraded via the lysosomal pathway. Both albumin and IgG1 antibodies are
rescued from degradation by binding to this receptor. IgA was previously
modified
to facilitate FeRn binding by introduction of a C-terminal albumin-binding
site,
which improved its pharmacokinetic profile and anti-tumor effects (Meyer et
al,
2016 MAbs 8(1): 87-98).
The present invention shows that ch14.18 IgA offers both the benefit of
overcoming
allodynia and improved neutrophil activation in a single molecule. Our
preclinical
data shows that IgA can be dosed higher than IgG without side effects.
Material and methods
Antibody production, isolation and quality control
The variable heavy and light chain sequences of ch14.18 were derived from
Biologic
License Application 125516. The variable heavy chain sequences were cloned
into
Lonza expression vectors (pEE14.4), coding for the IgAl or IgG1 heavy chain
while
the variable light chain sequences were cloned into Lonza expression vectors
(pEE14.4) coding for the kappa light chain. Monomeric antibodies were produced
by transient transfection of HEK293F cells with vectors coding for the heavy
chain,
light chain and pAdvantage (accession number U47294; promega), using 293Fectin
transfection reagent according to the manufacturer's instructions. IgG1
antibodies
CA 03076482 2020-03-19
WO 2019/059771 PCT/NL2018/050629
34
were purified using protein A columns (Hi-trap protein A) coupled to an
AKTAprime plus chromatography system (GE lifesciences). Bound antibody was
eluted with 0.1M sodium acetate pH 2.5 and neutralized with IM TRIS-HC1 pH
8.8. The eluate was dialyzed against PBS. IgAl antibodies were purified using
kappa light chain affinity chromatography columns (Hi-trap kappaSelect) and
eluted with 0.1M glycine buffer pH 2.5. The eluate was applied on a SEC column
ran with PBS as mobile phase. The fractions containing monomeric IgA were
collected and concentrated with 100 KDa spin columns. All antibodies were
filtered
over 0.22 lam filters. Purity and stability of the antibodies was analyzed by
HP-SEC
(Yarra 3u SEC-2000 column) with 100 mM sodium phosphate, 150 mM NaCl pH
6.8 as mobile phase with detection at 280 nm.
Fluorescent labeling of antibodies
Purified antibodies were labelled with fluorescein by incubation at room
temperature for 2h with N-hydroxy-succimidyl-FITC while stirring. Unbound
NHS-fluorescein was removed by using sephadex columns (NAP-5, GE-healthcare),
according to the manufacturer's instructions. Antibodies were labelled with
alexa
fluor-488 antibody labeling kit (ThermoFisher) according to the manufacturer's
instructions.
Cell lines
All neuroblastoma cell lines were cultured in DMEM culture medium
supplemented with HEPES, glutamax, 10% fetal calf serum, lx penicillin and
streptomycin at 37 C in a humidified incubator containing 5% CO2. HEK293F
cells
were cultured in FreeStyle 293 expression medium at 37 C in a humidified
incubator with orbital shaker platform containing 8% CO2.
Binding assays
100.000 neuroblastoma cells were plated out in 96 well plates and centrifuged
for 2
minutes at 1500 RPM. Cells were washed and incubated with fluorescein-labelled
antibody at several concentrations for 45 minutes. Next, cells were
centrifuged for
2 minutes at 1500 RPM, washed and resuspended in PBS. The amount of bound
antibody to the cells was quantified by flow cytometry (BD Facs Canto II, BD).
Cell based affinity measurements
lx106 IMR32 neuroblastoma cells were plated out on the side of a 10 cm culture
dish in an elliptical shape and incubated overnight for attaching to the
plate.
Subsequently, plates were washed with culture medium and transferred to the
LigandTracer apparatus (Ridgeview instruments). 10 nM of fluorescein-labelled
antibody was added to the cells and association was measured for 1 hour.
Afterwards, antibody concentration was increased to 20 nM to assess
association at
higher antibody concentrations for 1 hour.. Finally, dissociation was measured
by
replacing the antibody containing solution for medium without antibody and
CA 03076482 2020-03-19
WO 2019/059771 PCT/NL2018/050629
dissociation was measured for 2 hours. The affinity of the antibodies was
calculated
via TraceDrawer software (Ridgeview instruments).
ADCC assays
5 ADCC was quantified as described previously (Brandsmaet al; 2015; Cancer
Immunol Res 3(12): 1316-1324) In short, target cells with or without pre-
treatment
of 10 i.tM 11-cis-retinoic acid for 24 hours were labeled with 3.7 MBq 51Cr
for 2
hours. Afterwards, cells were washed three times to remove excess chromium.
Blood for ADCC's was obtained from healthy donors at the UMC Utrecht. For
10 leukocyte isolation, blood was incubated with demiwater for 30 seconds
to lyse
erythrocytes. Afterwards, 10x PBS was added to restore physiological
osmolality.
Cells were washed in medium and resuspended in medium corresponding to the
original blood volume. The number of leukocytes used per well corresponds to
the
number of leukocytes present in 50g1 of blood before lysis. For PMN and PBMC
15 isolation, blood was added on top of Ficoll/Histopaque 1119 layers and
centrifuged
for 25 minutes at 1500 RPM without braking. Afterwards, PBMC's and PMN's
were collected from the interphase between serum and ficoll or in the
histopaque
layer respectively. The effector-to-target (E:T) ratios were 80:1 for PBMCs,
40:1 for
PMNs. Effector cells, antibodies at various concentration, GM-CSF and IL-2 and
20 radioactively labeled tumor cells were added to round-bottom microliter
plates
(Corning Incorporated) and incubated for 37 C in a humidified incubator
containing 5% CO2. Plates were centrifuged for 2 minutes at 1500 RPM and 50 pi
of
the supernatant was transferred to lumaplates. Radioactive signal (in cpm) was
quantified in a beta-gamma counter. Specific lysis was calculated using the
25 formula: ((Experimental cpm ¨ basal cpm)/(maximal cpm ¨ basal cpm)) x
100, with
maximal lysis determined by incubating labelled cells with 2.5% triton and
basal
release was determined in the absence of antibodies and effector cells. .
CDC assays
30 105 neuroblastoma cells were added to microliter plates and incubated
for 30
minutes with antibodies at various concentrations at room temperature.
Afterwards, pooled human serum (from 8 different healthy donors) was added to
a
concentration of 15% and incubated for 1 hour or 4 hours. Afterwards, cells
were
washed and stained with 7-AAD for 15 minutes. 7-AAD uptake, representing cell
35 lysis was quantified by flow cytometry.
Antibody concentration determination in mouse serum
MaxiSorp 96 well ELISA plates were coated overnight with 0.5 g/ml goat IgG
anti-human kappa diluted in PBS. Next, plates were washed three times with
0.05% TWEEN 20 in PBS (PBST) and blocked for 1 hour by incubating with 1%
BSA in PBST. Serum samples were diluted 1:2000 in 1% BSA in PBST and added
to the wells and incubated for 1.5 at room temperature. Next, plates were
washed
three times with PBST. HRP-labeled-anti-human IgA or were used to bind
human IgA or human IgG respectively. Plates were developed for 10 minutes with
CA 03076482 2020-03-19
WO 2019/059771 PCT/NL2018/050629
36
2,2'-azino-bis(3-ethylbenzothiazoline-6-sulphonic acid (ABTS) and read out on
a
spectrophotometer at 415 nm.
Animals and animal experiments
Mice were maintained in the animal facility of the University of Utrecht.
Experiments were conducted using both male and female C57BL/6 mice (Janvier)
Mice were housed in groups under a 12:12 light dark cycle, with food and water
available ad libitum. Mice were acclimatized for at least 1 week prior to the
start of
experiment. Sample sizes were calculated with power analysis at the time of
the
design of experiments.
Mechanical thresholds were determined using the von Frey test in mice after
intravenous injection with IgG1 ch14.18, IgAl ch14.18 or fluorescently labeled
variants thereof. (Stoelting, Wood Dale, IL, US) with the up-and-down method
as
was described before previously (Eijkelkamp et al; 2016; J Neurosci 36(28):
7353-
7363.. In brief, mechanical nociception was tested with a calibrated von Frey
hair
monofilament (Stoelting). First, mice were acclimated for 15 to 20 min in a
transparent box with a metal mesh floor. The von Frey hair monofilament was
applied through the mesh floor to the plantar skin of the hindpaw. Mechanical
nociception was measured as the total number of paw withdrawals in response to
a
series of six applications of a 0.02 mg von Frey hair, which does not elicit a
response in untreated animals (Alessandri-Haber et al., 2006; J Neurosci. 2006
Apr
5;26(14):3864-74.). In experiments, the average of the left and right paw was
considered as an independent measure. To minimize bias, animals were randomly
assigned to the different groups prior to the start of experiment, and all
experiments were performed by experimenters blinded to treatment. At the end
of
the experiments, mice were euthanized by cervical dislocation. Mechanical
thresholds were assessed for 48 hours.
For evaluating the in cico efficacy of antibodies, mice were injected
intraperitoneally with 5x106 GD2 expressing EL4 cells expressing (ATCC). After
1
day, mice were intraperitoneally injected with 100 jig of IgG1 ch14.18, or 100
jig of
IgAl ch14.18. After 2 days, blood was taken and mice were injected with
luciferin
and subjected to bioluminescence analysis. Afterwards mice were euthanized by
cervical dislocation.
GD2 antibody binding to neurons was visualized by i.v. injection of 20 jig or
100 jig
alexa-488 labeled IgG1 ch14.18 or 20 jig of IgAl ch14.18. Sciatic nerves were
isolated and 10gm thick slices were prepared with a cryostat cryotome and
placed
on slides. Slides were fixed for 10 minutes in 4% PFA and washed. Finally,
slides
were counterstained with DAPI, washed and treated with fluorsave. Slides were
dried overnight at 4 C and images were taken by fluorescence microscopy.