Note : Les descriptions sont présentées dans la langue officielle dans laquelle elles ont été soumises.
WO 2021/158814
PCT/US2021/016653
GENE THERAPY SYSTEMS AND RELATED METHODS FOR TREATMENT OF
HEARING LOSS
Sequence Listing
[0001] The instant application contains a Sequence Listing which
has been
submitted electronically in ASCII format and is hereby incorporated by
reference in
its entirety. Said ASCII copy, created on February 4, 2021, is named
104042 _ 403 _ Seq _Listing.txt and is 18,277 bytes in size.
Technical Field
[0002] Various embodiments of the present disclosure relate
generally to gene
therapy systems and methods useful in the treatment and/or prevention of
hearing
loss. Exemplary embodiments described herein are directed to systems and
related
methods for preventing the further decline in a patient's hearing loss. More
specifically, embodiments taught in this present disclosure relate to gene
therapy
systems, and related methods, useful for treating and/or preventing deafness
caused
by genetic mutation of the TMPRSS3 gene or the LOXHD1 gene. These systems
and methods may utilize a combination of gene therapy (e.g., molecular
therapeutics) for hearing loss caused by a genetic mutation together with
implantation of a cochlear implant.
[0003] Hearing loss is the most common sensory deficit in
humans.
According to 2018 estimates on the magnitude of disabling hearing loss
released by
the World Health Organization (WHO), there are 466 million persons worldwide
living
with disabling hearing loss (432 million adults and 34 million children). The
number
of people with disabling hearing loss will grow to 630 million by 2030 and to
over 900
million by 2050 (1 in 10 people). Over 90% of persons with disabling hearing
loss
1
CA 03166104 2022- 7- 26
WO 2021/158814
PCT/US2021/016653
(420 million) reside in the low-income regions of the world (WHO global
estimates on
prevalence of hearing loss, Prevention of Deafness WHO 2018).
[0004] There are currently no approved therapeutic agents for
preventing or
treating hearing loss or deafness. The current treatment option for those with
disabling hearing loss is a cochlear implant. Cochlear implantation is a
common
procedure with a large associated healthcare cost, over $1,000,000 lifetime
cost per
patient (Mohr PE, et al.
[0005] (2000). The societal costs of severe to profound hearing
loss in the
United States; IntJ Technol Assess Health Care; 16(4): 1120-35).
[0006] The current demand for cochlear implants exceeds supply.
The
production rate of cochlear implant units manufactured is 50,000 units each
year.
Based on current birth rates and the incidence and prevalence of disabling
hearing
loss in newborns, 134,000 cochlear implants are needed annually to provide 1
cochlear implant for each afflicted child. This number increases if patients
needing
bilateral (2) cochlear implants are included.
[0007] The lifetime cost of a cochlear implant is prohibitive
for most people
and particularly for those living outside the developed nations where the
majority of
persons with disabling hearing loss reside. Therapeutic options are needed to
provide cost effective alternatives to cochlear implants, especially for those
persons
living outside developed nations.
[0008] More than 50% of prelingual deafness is genetic i.e.
hereditary
(Centers for Disease Control and Prevention- Genetics of Hearing Loss).
Hereditary
hearing loss and deafness may be conductive, sensorineural, or a combination
of
both; syndromic (associated with malformations of the external ear or other
organs
or with medical problems involving other organ systems) or nonsyndromic (no
2
CA 03166104 2022- 7- 26
WO 2021/158814
PCT/US2021/016653
associated visible abnormalities of the external ear or any related medical
problems);
and prelingual (before language develops) or postlingual (after language
develops)
(Richard JH Smith, MD, A Eliot Shearer, Michael S Hildebrand, PhD, and Guy Van
Camp, PhD, Deafness and Hereditary Hearing Loss Overview, GeneReviews Initial
Posting: February 14, 1999; Last Revision: January 9, 2014. More than 70% of
hereditary hearing loss is nonsyndromic. The different gene loci for
nonsyndromic
deafness are designated DEN (for DeaFNess). Loci are named based on mode of
inheritance: DFNA (Autosomal dominant), DE B (Autosomal recessive) and DFNX
(X-linked). The number following the above designations reflects the order of
gene
mapping and/or discovery. In the general population, the prevalence of hearing
loss
increases with age. This change reflects the impact of genetics and
environment and
the interactions between environmental triggers and an individual's genetic
predisposition.
[0009] Sensorineural hearing loss (SNHL) is the most common
neurodegenerative disease in humans and there are currently no approved
pharmacologic interventions. SNHL can be caused by genetic disorders as well
as
acquired through injuries such as sound trauma and ototoxicity. Genetic
diagnostics
have demonstrated that there are at least 100 genes causing nonsyndromic SNHL.
Recent advances in genetics and gene therapy techniques have shown that rescue
of a number of recessive types of deafness is possible through gene therapy
(Akil et
al., 2012; Askew et al., 2015). Long term gene delivery to the inner ear has
been
achieved using adeno associated viral vectors (AAV) (Shu, Tao, Wang, et al.,
2016).
The first human clinical trial to address deafness and hearing loss using a
gene
therapy was (CGF166) initiated on June of 2014 and completed in December of
2019. The Principal Investigator for CGF166 was Dr. Hinrich Staecker and the
trial
3
CA 03166104 2022- 7- 26
WO 2021/158814
PCT/US2021/016653
was sponsored by Novaris. (https://clinicaltrials.govict2/show/NCT02132130).
An
ideal disease target for translational research in this domain is a recessive
genetic
hearing loss that affects a defined group of cells within the inner ear and
occurs
postnatally after the development of speech. Prevalence of the mutation is an
additional consideration.
[0010] As described herein, by carefully evaluating both the
incidence of
common recessive causes of hearing loss and taking into account the size of
the
gene, it is possible to develop a gene therapy program that has an accessible
and
fairly common patient population. For example, although less common than other
mutations, TMPRSS3 is a fairly common cause of hearing loss that is severe
enough
to warrant cochlear implantation. Additionally, patients with mutations in
TMPRSS3
may not respond to cochlear implantation as well as patients with other
mutations
(Shearer et al., 2017). This presents the opportunity of targeting TMPRSS3, or
other
genes such as LOXHD1, as a stand-alone therapeutic or in combination with
other
therapeutic agents and/or cochlear implantation to improve implant outcomes
for this
disorder. Table 1 (adapted from (Miyagawa, Nishio, & Usami, 2016))
demonstrates
that mutations in TMPRSS3 may be the most common cause of postlingual
recessive hearing loss that has a fairly limited distribution within the
cochlea and,
due to the size of the gene, may be built into existing AAV vectors.
4
CA 03166104 2022- 7- 26
WO 2021/158814
PCT/US2021/016653
OMET in
Wcf.43-tbfi k-sE-: Pe6T TOT.4 %u I-.: ..,-IENE 52
flAIR EtEl ;r1:4c3A.P.E.0
29 '37.11' 15 417
Kte2.3 .4 7 r*,:' a* 4.4.3 Ys
Ilt,C7 GA4 'a. 0 .4. s%. 4520 '0
IWO:7A 3 4 =;7 .4$ 74:_is rEs.
...?."..5TH
OT E3F 4 5 4. 2% .5573 YES
4;7:
l'A`O I5A 1 4. . :214 31875 YES.
R;Fit
=WA;P:Dtµit'SYS't t= :6 4: -=.$' 1..5D4 ..
r4)3 .. bc.tp,,i
3-LiFM.3 311:: 3 Ac- DI
AC161 .6.. 2 i '.i=A: 2125 yE5
crz-tto
:i7Iiilf ii II-CpW3.: 4613;
3.-:3)1-LI5 2 :13 2
- .
tAti555-.4. 4? 2 :.2. i*
CYP.P.1 C 1 i rAi '115$g $A3
CsQ10
E..LFNA5 Q 1 1 V.S., 22:7,5 YES
EICIM
aK34 E, i 1 1% 28.3.2'
Ytit-PN 0 I 1 . , i S=::, 29.15
VES Rif.
iC,X$3E31. I l':
... .1% 39.7a
31.,St924.5k,i3: . '0: Ti :i:: 1% N A. .'
.=;
[0011] Table 1 : Incidence of different mutations in 176 adult
cochlear implant
patients.
[0012] The human transmembrane protease, serine 3 (TMPRSS3, also
referred to as DFNB10, DFNB8, ECHOS1, TADG12, Acc: HGNC:11877) was
identified by its association with both congenital (present at birth) and
childhood
onset autosomal recessive deafness. Mutations in the TMPRSS3 gene are
associated with Autosomal Recessive Nonsyndromic Hearing Impairment type
DFNB8 and 10. TMPRSS3 is a 1646 base pair gene that codes for a serine
protease and is associated with DFNA 8/10 and may make up to 1-5% of patients
with hearing loss undergoing cochlear implantation (Weegerink et al., 2011).
Loss of
function of this gene appears to result in a broad spectrum of hearing
phenotypes
depending on the site of the mutation. Both congenital and adult onset
progressive
hearing loss have been associated with the loss of this gene.
[0013] The onset of DFNB8 hearing loss is postlingual (age 10-12
years),
while the onset of DFNB10 hearing loss is prelingual (congenital). This
phenotypic
difference reflects a genotypic difference. The DFNB8 causing variant is a
splice site
variant, suggesting that inefficient splicing is associated with a reduced
amount of
CA 03166104 2022- 7- 26
WO 2021/158814
PCT/US2021/016653
normal protein that is sufficient to prevent prelingual deafness but not
sufficient to
prevent eventual hearing loss. (See, Richard JH Smith, MD, et al. (2014).
Genes
Known to Cause Autosomal Recessive Nonsyndromic Hearing Impairment:
Deafness and Hereditary Hearing Loss Overview; GeneReviews).
[0014] TMPRSS3 mutations on chromosome 21 known to cause hearing
loss
are described in Table 2.
TABLE 2. TMPRSS3 MUTATIONS (CHROMOSOME 21)
# MUTATION NAME REFERENCE
1 TMPRSS3, IVS4AS, Scott HS, et al. (2001) Insertion of beta-
satellite
G-A, -6 repeats identifies a transmembrane
protease
causing both congenital and childhood onset
autosomal recessive deafness. Nat Genet. 27(1):59-
63.
2 TMPRSS3, 8-BP Scott HS, et al. (2001) Insertion of beta-
satellite
DEL, SATELLITE repeats identifies a transmembrane
protease
REPEAT INS causing both congenital and childhood
onset
autosomal recessive deafness. Nat Genet. 27(1):59-
63.
3 TMPRSS3, 1-BP Wattenhofer M, et al. (2002) Mutations in
the
DEL, 207C TMPRSS3 gene are a rare cause of childhood
nonsyndromic deafness in Caucasian patients. J Mol
Med (Bed). 80(2):124-31.
4 c.753G>C Masmoudi S, et al. (2001) Novel missense
(p.Trp251Cys) mutations of TMPRSS3 in two consanguineous
Tunisian families with non-syndromic autosomal
recessive deafness. Hum Mutat. 18(2):101-8.
c.308A>G Wattenhofer M, et al. (2002) Mutations in the
(p.Asp103Gly) TMPRSS3 gene are a rare cause of childhood
nonsyndromic deafness in Caucasian patients. J Mol
Med (Bed). 80(2):124-31.
6 c.1211C>T Wattenhofer M, et al. (2005) A novel
TMPRSS3
(p.Pro404Leu) missense mutation in a DFNB8/10 family
prevents
proteolytic activation of the protein. Hum Genet.
117(6):528-35.
7 c.647G>T Wattenhofer M, et al. (2005) A novel
TMPRSS3
(p.Arg216Leu) missense mutation in a DFNB8/10 family
prevents
proteolytic activation of the protein. Hum Genet.
117(6):528-35.
8 c.579dupA Duzkale H, et al. (2013) A systematic
approach to
(p.Cys194Metfs) assessing the clinical significance of
genetic
variants. Clin Genet. 84(5):453-63.
9 c.1192C>T Wattenhofer M, et al. (2005) A novel
TMPRSS3
(p.G1n398Ter) missense mutation in a DFNB8/10 family
prevents
6
CA 03166104 2022- 7- 26
WO 2021/158814
PCT/US2021/016653
TABLE 2. TMPRSS3 MUTATIONS (CHROMOSOME 21)
# MUTATION NAME REFERENCE
proteolytic activation of the protein. Hum Genet.
117(6):528-35.
c.323-6G>A Scott HS, et al. (2001) Insertion of beta-satellite
repeats identifies a transmembrane protease
causing both congenital and childhood onset
autosomal recessive deafness. Nat Genet. 27(1):59-
63.
11 c.916G>A Chung J, et al. (2014) A novel mutation of
(p.A1a306Thr) TMPRSS3 related to milder auditory
phenotype in
Korean postlingual deafness: a possible future
implication for a personalized auditory rehabilitation.
J Mol Med (Berl). 92(6):651-63.
12 c.208deIC Battelino S, et al. (2015) TMPRSS3
mutations in
(p.His70Thrfs) autosomal recessive nonsyndromic hearing
loss.
Eur Arch Otorhinolaryngol. 273(5):1151-4.
13 c.1276G>A Weegerink NJ, et al. (2011) Genotype-
phenotype
(p.A1a426Thr) correlation in DFNB8/10 families with
TMPRSS3
mutations. J Assoc Res Otolaryngol. 12(6):753-66.
14 c.413C>A Eppsteiner RW, et al. (2012) Prediction of
cochlear
(p.A1a138G1u) implant performance by genetic mutation:
the spiral
ganglion hypothesis. Hear Res. 292(1-2):51-8.
c.325C>T Lee YJ, Park D, Kim SY, Park WJ (2003)
(p.Arg109Trp) Pathogenic mutations but not polymorphisms
in
congenital and childhood onset autosomal recessive
deafness disrupt the proteolytic activity of
TMPRSS3. J Med Genet. 40(8):629-31.
16 c.346G>A (p.V116M) Ganapathy A, et al. (2014) Non-syndromic
hearing
impairment in India: high allelic heterogeneity
among mutations in TMPRSS3, TMC1, USHIC,
CDH23 and TMIE._PLoS One. 9(1):e84773.
17 c.727G>A (p.G243R) Ganapathy A, et al. (2014) Non-syndromic
hearing
impairment in India: high allelic heterogeneity
among mutations in TMPRSS3, TMC1, USHIC,
CDH23 and TMIE._PLoS One. 9(1):e84773.
18 c.1156T>C Ganapathy A, et al. (2014) Non-syndromic
hearing
(p.C386R) impairment in India: high allelic
heterogeneity
among mutations in TMPRSS3, TMC1, USHIC,
CDH23 and TMIE._PLoS One. 9(1):e84773.
[0015] The lipoxygenase homology domains 1 gene (LOXHD1;
also
referred to as LH2D1, DFNB77, FLJ32670; OMIM: 613072; Acc:HGNC:26521)
encodes a highly conserved protein consisting entirely of PLAT
(polycystin/lipoxygenase/alpha-toxin) domains, thought to be involved in
targeting
7
CA 03166104 2022- 7- 26
WO 2021/158814
PCT/US2021/016653
proteins to the plasma membrane. Studies in mice show that this gene is
expressed
in the mechanosensory hair cells in the inner ear, and mutations in this gene
lead to
auditory defects, indicating that this gene is essential for normal hair cell
function.
Screening of human families segregating deafness identified a mutation in this
gene
which causes DFNB77, a progressive form of autosomal-recessive nonsyndromic
hearing loss (ARNSHL). Alternatively spliced transcript variants encoding
different
isoforms have been noted for this gene.
[0016] Clinical Features of LOXHD1:
= Autosomal recessive
= Hearing loss, sensorineural, bilateral (milder hearing loss at low
frequencies)
= Congenital onset leading to cochlear implants between 7-10 years of age
in Ashkenazi Jewish families
= Onset by 7-8 years of age progressing to moderate-to-severe loss of mid
and high frequencies during adulthood in a consanguineous Iranian family
[0017] Evidence that autosomal recessive nonsyndromic
hearing loss-
77 (DFNB77) is caused by homozygous mutation in the LOXHD1 gene (613072) on
chromosome 18q21.
[0018] In situ hybridization detected Loxhd1 expression in
the
developing mouse inner ear at embryonic days 13.5 and 16, but not in any other
tissue. At postnatal day 4, expression was detected in cochlear and vestibular
hair
cells, with highest concentration in the nucleus. Loxhd1 progressively
localized to the
cytoplasm, and in the adult, Loxhd1 was expressed in hair cells along the
length of
stereocilia.
8
CA 03166104 2022- 7- 26
WO 2021/158814
PCT/US2021/016653
[0019] Using an N-ethyl-N-nitrosourea (ENU) mutagenesis
screen,
Grillet et al. (2009) developed the 'samba' mouse line that becomes hearing
impaired
by 3 weeks of age and deaf by 8 weeks of age. Homozygous samba mice showed
no other neurologic or vestibular abnormalities, and heterozygous samba mice
appeared completely normal. Stereociliary development was not affected in
homozygous samba mice, but hair cell function was perturbed and hair cells
eventually degenerated.
[0020] Grillet et al. (2009) found that samba was a
mutation in the
mouse Loxhd1 gene that destabilized the beta-sandwich structure of PLAT domain
10. The mutation did not alter mRNA or protein stability or localization of
Loxhd1
protein along the length of stereocilia. However, by postnatal day 21, some
hair cells
showed morphologic defects with fused stereocilia and membrane ruffling at the
apical cell surface. Profound degenerative changes were obvious by postnatal
day
90, including hair cell loss and a reduction in spiral ganglion neurons.
Grillet et al.
(2009) hypothesized that the degeneration of spiral ganglion neurons was
likely
secondary to perturbations in the function and maintenance of hair cells.
[0021] LOXHD1 mutations on chromosome 18 known to cause
hearing
loss are described in Table 3.
TABLE 3. LOXHD1 MUTATIONS (CHROMOSOME 18)
MUTATION NAME REFERENCE
1 c.2008C>T (p Arg670Ter) Grillet N, et al. (2009) Mutations
in LOXHD1, an evolutionarily conserved
stereociliary protein, disrupt hair cell
function in mice and cause progressive
hearing loss in humans. Am J Hum Genet.
85(3):328-37.
2 c.3169C>T (p.Arg1057Ter) Edvardson S, et al. (2011) A
deleterious mutation in the LOXHD1 gene
causes autosomal recessive hearing loss in
Ashkenazi Jews. Am J Med Genet A.
155A(5):1170-2.
9
CA 03166104 2022- 7- 26
WO 2021/158814
PCT/US2021/016653
Grillet N, et al. (2009) Mutations
in LOXHD1, an evolutionarily conserved
stereociliary protein, disrupt hair cell
function in mice and cause progressive
hearing loss in humans. Am J Hum Genet.
85(3):328-37.
3 c.2303deIG (p.Gly768Alafs) Edvardson S, et al. (2011) A
deleterious mutation in the LOXHD1 gene
causes autosomal recessive hearing loss in
Ashkenazi Jews. Am J Med Genet A.
155A(5):1170-2.
Grillet N, et al. (2009) Mutations
in LOXHD1, an evolutionarily conserved
stereociliary protein, disrupt hair cell
function in mice and cause progressive
hearing loss in humans. Am J Hum Genet.
85(3):328-37.
4 c.4099G>T (p.G1u1367Ter) Edvardson S, et al. (2011) A
deleterious mutation in the LOXHD1 gene
causes autosomal recessive hearing loss in
Ashkenazi Jews. Am J Med Genet A.
155A(5):1170-2.
Grillet N, et al. (2009) Mutations
in LOXHD1, an evolutionarily conserved
stereociliary protein, disrupt hair cell
function in mice and cause progressive
hearing loss in humans. Am J Hum Genet.
85(3):328-37.
c.2497C>T (p.Arg833Ter) Edvardson S, et al. (2011) A
deleterious mutation in the LOXHD1 gene
causes autosomal recessive hearing loss in
Ashkenazi Jews. Am J Med Genet A.
155A(5):1170-2.
Grillet N, et al. (2009) Mutations
in LOXHD1, an evolutionarily conserved
stereociliary protein, disrupt hair cell
function in mice and cause progressive
hearing loss in humans. Am J Hum Genet.
85(3):328-37.
6 c.4714C>T Edvardson S, et al. (2011) A
deleterious mutation in the LOXHD1 gene
causes autosomal recessive hearing loss in
Ashkenazi Jews. Am J Med Genet A.
155A(5):1170-2.
[0022] U.S. Application Publication No. 2013/0095071,
incorporated by
reference herein in its entirety, describes gene therapy methods for restoring
age-
CA 03166104 2022- 7- 26
WO 2021/158814
PCT/US2021/016653
related hearing loss using mutated tyrosine adeno-associated viral vectors to
deliver
the X-linked inhibitor of apoptosis protein (XIAP) to the round window
membrane of
the inner ear. However, the publication does not contemplate the delivery of a
nucleic acid sequence encoding functional TMPRSS3 or LOXHD1 to prevent or
delay the onset of or restore hearing loss or deafness caused by genetic
mutation of
the TMPRSS3 or LOXHD1 gene, as disclosed herein.
[0023] Additionally, an important pitfall in the current
state of the art for
developing clinical gene therapies for hearing disorders is a lack of animal
models
that mirror human hearing loss. Many of the available mouse models for genetic
hearing losses with adult onset in humans present with congenital hearing loss
making delivery studies complex. There are few models with onset of genetic
hearing loss after development of hearing. Delivery of vectors in neonatal
mice
results in different transfection patterns than delivery in adult mice (Shu,
Tao, Li, et
al., 2016). There is a need for novel animal models that can be used to
evaluate
rescue of hearing using different vector systems and gene targets.
[0024]
In view of the above, cochlear implantation is one common method of
treatment of choice for addressing hearing loss ranging from severe to
profound. A
cochlear implant is a small, complex electronic device that can help to
provide a
sense of sound to a person who is profoundly deaf or severely hard-of-hearing.
The
implant consists of an external portion that sits behind the ear and a second
portion
that is surgically placed under the skin.
[0025] While tremendous advances in cochlear implant design and performance
have occurred over the years, there are still patients who do poorly in terms
of
speech outcomes with implants. Recent studies have demonstrated that mutations
in the two genes that cause deafness, TMPRSS3 and LoxHD1, also have poor
11
CA 03166104 2022- 7- 26
WO 2021/158814
PCT/US2021/016653
outcomes in cochlear implant resultsl. Specifically, the TMPRSS3 mutant
patient
has dysfunction of their spiral ganglion2. During evaluation of a mouse
TMPRSS3
mutant model, it was demonstrated that hair cells degenerated initially and
was
followed shortly after by the degeneration of spiral ganglion cells3.
Permanent
damage to the hair cells of the inner ear results in sensorineural hearing
loss,
leading to communication difficulties in a large percentage of the population.
Hair
cells are the receptor cells that transduce the acoustic stimulus.
Regeneration of
damaged hair cells would provide an avenue for the treatment of a condition
that
currently has no therapies other than prosthetic devices.
[0026] During evaluation of human patients with TMPRSS3 mutations, it was
demonstrated that cochlear implant function declines with age, which suggests
that
the delayed degeneration of spiral ganglion cells also occurs in the human
population4. The foregoing suggests that cochlear implants alone may not be
enough to combat hearing loss.
[0027] Opportunities, therefore, exist to provide a combination of molecular
therapeutics (e.g., gene therapy) for hearing loss in combination with
cochlear
implantation.
SUMMARY
[0028] Embodiments of the present disclosure relate to, among other
things, gene
therapy systems and methods useful in treating and/or preventing hearing loss.
Systems and methods described herein relate to combination gene therapy with
cochlear implantation to repair and/or rescue degenerating hair cells and/or
degenerating spiral ganglion cells depending on the time of intervention.
12
CA 03166104 2022- 7- 26
WO 2021/158814
PCT/US2021/016653
[0029]
Each of the embodiments disclosed herein may include one or more of
the features described in connection with any of the other disclosed
embodiments.
[0030]
Disclosed herein is an expression vector including the nucleic acid
sequence of SEQ ID NO:1 or SEQ ID NO:2, or a nucleic acid sequence having at
least
90% sequence identity to the nucleic acid of SEQ ID NO:1 or SEQ ID NO:2,
wherein
the nucleic acid sequence is operatively linked to a promoter. Also disclosed
herein
is a pharmaceutical composition for use in a method for the treatment or
prevention of
hearing loss that includes an expression vector having the nucleic acid
sequence of
SEQ ID NO:1 or SEQ ID NO:2, or a nucleic acid sequence having at least 90%
sequence identity to the nucleic acid of SEQ ID NO:1 or SEQ ID NO:2, wherein
the
nucleic acid sequence is operatively linked to a promoter. In some
embodiments, the
nucleic acid sequence has at least 75%, at least 80%, at least 85%, at least
90%, at
least 95%, at least 96%, at least 97%, at least 98%, or at least 99% sequence
identity
to the nucleic acid sequence of SEQ ID NO:1 or SEQ ID NO:2. In some
embodiments,
the expression vector is selected from an adeno-associated viral vector, an
adenoviral
vector, a herpes simplex viral vector, a vaccinia viral vector, a helper
dependent
adenoviral vector or a lentiviral vector. In some embodiments, the vector is
an adeno-
associated viral vector selected from AAV2, AAV2/Anc80, AAV5, AAV6, AAV6.2,
AAV7, AAV8, AAV9, AAVrh8, AAVrh10, AAVrh39, AAVrh43AAV1, AAV2, AAV3,
AAV4, AAV5, AAV6, AAV7, AAV8, Anc80, or a synthetic version of an adeno
associated viral vector serotype. In some embodiments, the adeno-associated
viral
vector is AAV2, Anc80, or a synthetic version of an adeno associated viral
vector
serotype. In some embodiments, the promoter is selected from any hair cell
promoter
that drives the expression of an operably linked nucleic acid at early
development and
maintains expression throughout the life, for example, TMPRSS3 promoters,
human
13
CA 03166104 2022- 7- 26
WO 2021/158814
PCT/US2021/016653
cytomegalovirus (HCMV) promoters, cytomegalovirus/chicken beta-actin (CBA)
promoters, Myo7a promoters or Pou4f3 promoters.
[0031] Disclosed herein is a cell having an expression vector that
includes the
nucleic acid sequence of SEQ ID NO:1 or SEQ ID NO:2, or a nucleic acid
sequence
having at least 90% sequence identity to the nucleic acid of SEQ ID NO:1 or
SEQ ID
NO:2, wherein the nucleic acid sequence is operatively linked to a promoter.
In some
embodiments, the nucleic acid sequence has at least 75%, at least 80%, at
least 85%,
at least 90%, at least 95%, at least 96%, at least 97%, at least 98%, or at
least 99%
sequence identity to the nucleic acid sequence of SEQ ID NO:1 or SEQ ID NO:2.
In
some embodiments, the cell is a stem cell. In some embodiments, the stem cell
is an
induced pluripotent stem cell.
[0032] Disclosed herein is a method for treating or preventing
hearing loss,
including administering to a subject in need thereof an effective amount of an
expression vector that includes the nucleic acid sequence of SEQ ID NO:1 or
SEQ ID
NO:2, or a nucleic acid sequence having at least 90% sequence identity to the
nucleic
acid of SEQ ID NO:1 or SEQ ID NO:2, wherein the nucleic acid sequence is
operatively
linked to a promoter. In some embodiments, the nucleic acid sequence has at
least
75%, at least 80%, at least 85%, at least 90%, at least 95%, at least 96%, at
least
97%, at least 98%, or at least 99% sequence identity to the nucleic acid
sequence of
SEQ ID NO:1 or SEQ ID NO:2. In some embodiments, the expression vector is
selected from an adeno-associated viral vector, an adenoviral vector, a herpes
simplex
viral vector, a vaccinia viral vector, a helper dependent adenoviral vector or
a lentiviral
vector. In some embodiments, the vector is an adeno-associated viral vector
selected
from AAV2, AAV2/Anc80, AAV5, AAV6, AAV6.2, AAV7, AAV8, AAV9, AAVrh8,
AAVrh10, AAVrh39, AAVrh43, AAV1, AAV2, AAV3, AAV4, AAV5, AAV6, AAV7, AAV8
14
CA 03166104 2022- 7- 26
WO 2021/158814
PCT/US2021/016653
or Anc80, or a synthetic version of an adeno associated viral vector serotype.
In some
embodiments, the adeno-associated viral vector is AAV2, Anc80, or a synthetic
version of an adeno associated viral vector serotype.s In some embodiments,
the
promoter is selected from any hair cell promoter that drives the expression of
an
operably linked nucleic acid sequence at early development and maintains
expression
throughout the life, for example, TMPRSS3 promoters, human cytomegalovirus
(HCMV) promoters, cytomegalovirus/chicken beta-actin (CBA) promoters, Myo7a
promoters or Pou4f3 promoters. In some embodiments, the expression vector is
administered into the inner ear of the subject, for example, by injection. In
some
embodiments, the delivery method is selected from cochleostomy, round window
membrane, canalostomy or any combination thereof (see, Erin E. Leary Swan, et
al.
(2008) Inner Ear Drug Delivery for Auditory Applications. Adv Drug Deliv Rev.
60(15):1583-1599). In some embodiments, the expression vector is delivered
into the
scala media via the endolymphatic sac (Colletti V, et al. (2010) Evidence of
gadolinium
distribution from the endolymphatic sac to the endolymphatic compartments of
the
human inner ear. Audiol Neurootol. 15(6):353-63; Marco Mandala, MD, et al.
(2010)
Induced endolymphatic flow from the endolymphatic sac to the cochlea in
Meniere's
disease. Otolaryngology¨Head and Neck Surgery. 143, 673-679; Yamasoba T, et
al.
(1999) Inner ear transgene expression after adenoviral vector inoculation in
the
endolymphatic sac. Hum Gene Ther. 10(5):769-74). In some embodiments, the
subject has one or more genetic risk factors associated with hearing loss. In
some
embodiments, one of the genetic risk factors is a mutation in the TMPRSS3
gene. In
some embodiments, the mutation in the TMPRSS3 gene is selected from any one or
more TMPRSS3 mutations known to cause hearing loss (see, for example, Table
2).
In some embodiments, one of the genetic risk factors is a mutation in the
LOXHD1
CA 03166104 2022- 7- 26
WO 2021/158814
PCT/US2021/016653
gene. In some embodiments, the mutation in the LOXHD1 gene is selected from
any
one or more LOXHD1 mutations known to cause hearing loss (see, for example,
Table
3). In some embodiments, the subject does not exhibit any clinical indicators
of hearing
loss.
[0033] In some embodiments, an expression vector described herein
is
administered as a combination therapy with one or more expression vectors
comprising other nucleic acid sequences and/or with one or more other active
pharmaceutical agents for treating hearing loss. For example, a combination
therapy
may include a first expression vector that has the nucleic acid sequence of
SEQ ID
NO.1 and a second expression vector that has the nucleic acid sequence of SEQ
ID
NO:2, wherein both expression vectors are administered to a subject as part of
a
combination therapy to treat hearing loss.
[0034] Disclosed herein is a transgenic mouse having a human
TMPRSS3 gene
with a mutation selected from any one or more TMPRSS3 mutation known to cause
hearing loss (see, for example, Table 2). Disclosed herein is a transgenic
mouse
having a human LOXHD1 gene with a mutation selected from any one or more
LOXHD1 mutation known to cause hearing loss (see, for example, Table 3).
[0035] It may be understood that both the foregoing general
description and the
following detailed description are exemplary and explanatory only and are not
restrictive of the invention, as claimed.
BRIEF DESCRIPTION OF THE DRAWINGS
[0036] The accompanying drawings, which are incorporated in and
constitute a part
of this specification, illustrate exemplary embodiments of the present
disclosure and
together with the description, serve to explain the principles of the
disclosure.
16
CA 03166104 2022- 7- 26
WO 2021/158814
PCT/US2021/016653
[0037] Figure 1 shows a cDNA sequence encoding wild-type human
TMPRSS3
(GenBank Accession No. BC074847.2).
[0038] Figure 2 shows the wild-type human TMPRSS3 amino acid
sequence
encoded by the cDNA in Figure 1.
[0039] Figure 3 shows a cDNA sequence encoding wild-type human
LOXHDI
(Gen Bank Accession No. AK057232.1).
[0040] Figure 4 shows the wild-type human LOXHDI amino acid
sequence encoded
by the cDNA in Figure 3.
[0041] Figure 5 shows TMPRSS3 immunohistochemistry in the adult
mouse
cochlea
[0042] Figure 6 shows an exemplary cochlear implant and the
corresponding
anatomy of the inner human, according to an aspect of the present disclosure.
[0043] Figure 7 shows an exemplary TMPRSS3 plasmid map beginning at
"ORI"
and including an initial "AAV2 ITR" vector, a "CMV enhancer", a "CMV
promoter", a "h-
TMPRSS3", a "bGH poly(A) signal, and a closing "AAV2 ITR" vector.
[0044] Figure 8 illustrates proof of concept by graphically
comparing hearing
recovery of a disease model mouse receiving gene therapy treatment (treated)
vs a
disease model mouse not receiving treatment (untreated) by way of Auditory
Brainstem Response (ABR) testing.
[0045] Figure 9 illustrates proof of concept by graphically
comparing hearing
recovery of a disease model mouse receiving gene therapy treatment (treated)
vs a
disease model mouse not receiving treatment (untreated) by way of Distortion
Product
Otoacoustic Emissions (DPOAE) testing.
[0046] Figure 10 graphically illustrates proof of concept by
graphically comparing
auditory neuronal function recovery of a disease model mouse receiving gene
therapy
17
CA 03166104 2022- 7- 26
WO 2021/158814
PCT/US2021/016653
treatment (treated) vs a disease model mouse not receiving treatment
(untreated) by
way of WAVE1 amplitude testing.
[0047] Figure 11 illustrates the location of the Round Window
Membrane (RWM)
within the human ear as an exemplary drug delivery site for delivering one or
more of
the gene therapies taught herein.
DETAILED DESCRIPTION
[0048] While principles of the present disclosure are described
herein with
reference to illustrative embodiments for particular applications, it should
be
understood that the disclosure is not limited thereto. Those having ordinary
skill in
the art and access to the teachings provided herein will recognize additional
modifications, applications, embodiments, and substitution of equivalents all
fall
within the scope of the embodiments described herein. Accordingly, the
invention is
not to be considered as limited by the foregoing description.
[0049] The present disclosure is drawn to gene therapy systems, and
related
methods, useful for treating and/or preventing deafness caused by genetic
mutation.
Examples of two genes that can mutate to cause deafness are the TMPRSS3 gene
or the LoxHD1 gene. The systems and methods described herein may utilize a
combination of gene therapy (e.g., molecular therapeutics) for hearing loss
caused
by a genetic mutation together with implantation of a cochlear implant. It can
be
appreciated that while the systems and methods are in view of gene mutations
caused by either the TMPRSS3 gene or the LoxHD1 gene, other gene mutations
may be targeted for repair that have been found to cause deafness or hearing
loss.
[0001] For purposes of the present disclosure, the following
definition of "gene
therapy" may be used. Gene therapy may refer to when DNA is introduced into a
18
CA 03166104 2022- 7- 26
WO 2021/158814
PCT/US2021/016653
patient to treat a genetic disease. The new DNA usually contains a functioning
gene
to correct the effects of a disease-causing mutation in the existing gene.
Gene
transfer, either for experimental or therapeutic purposes, relies upon a
vector or
vector system to shuttle genetic information into target cells. The vector or
vector
system is considered the major determinant of efficiency, specificity, host
response,
pharmacology, and longevity of the gene transfer reaction. Currently, the most
efficient and effective way to accomplish gene transfer is using vectors or
vector
systems based on viruses that have been made replication-defective (PCT
Publication No. WO 2015/054653; Methods of Predicting Ancestral Virus
Sequences
and Uses Thereof).
[0002] As used herein, the terms "treat," "treating," and
"treatment" encompass a
variety of activities aimed at desirable changes in clinical outcomes. For
example,
the term "treat", as used herein, encompasses any activity aimed at achieving,
or
that does achieve, a detectable improvement in one or more clinical indicators
or
symptoms of hearing loss, as described herein.
[0003] LOXHD1 gene (for example, as detected in a genetic
diagnostic test) but
does not yet exhibit clinical indicators or symptoms of hearing loss, thus
providing a
window during which therapeutic intervention can be initiated. Accordingly, in
some
embodiments, the present invention provides methods for therapeutic
intervention
during the period of gradual regression of hearing. The methods of the present
invention can be commenced prior to such time period. The methods of treating
hearing loss provided by the invention include, but are not limited to,
methods for
preventing or delaying the onset of hearing loss or the progression of
clinical
indicators or symptoms of hearing loss.
19
CA 03166104 2022- 7- 26
WO 2021/158814
PCT/US2021/016653
[0004] As used herein, the term "hearing loss" is used to describe
the reduced
ability to hear sound, and includes deafness and the complete inability to
hear
sound.
[0005] The terms "effective amount" or "therapeutically effective
amount," as used
herein, refer to an amount of an active agent as described herein that is
sufficient to
achieve, or contribute towards achieving, one or more desirable clinical
outcomes,
such as those described in the "treatment" description above. An appropriate
"effective" amount in any individual case may be determined using standard
techniques known in the art, such as a dose escalation study. The term "active
agent" as used herein refers to a molecule (for example, an AAV vector
described
herein) that is intended to be used in the compositions and methods described
herein and that is intended to be biologically active, for example, for the
purpose of
treating hearing loss.
[0006] The term "pharmaceutical composition" as used herein refers
to a
composition comprising at least one active agent as described herein or a
combination of two or more active agents, and one or more other components
suitable for use in pharmaceutical delivery such as carriers, stabilizers,
diluents,
dispersing agents, suspending agents, thickening agents, excipients, and the
like.
[0007] The terms "subject" or "patient" as used interchangeably
herein
encompass mammals, including, but not limited to, humans, non-human primates,
rodents (such as rats, mice and guinea pigs), and the like. In some
embodiments of
the invention, the subject is a human.
[0008] As used herein, the terms "vector" or "vectors" may be used.
A "vector"
may refer to a virus capable of transferring the desired gene into cells, but
not
capable of taking over or harming cells. To date, adenovirus, adeno-associated
CA 03166104 2022- 7- 26
WO 2021/158814
PCT/US2021/016653
virus, herpes simplex virus, vaccinia virus, retrovirus, helper dependent
adenovirus
and lentivirus have all tested for cochlear gene delivery. Of these, the one
that has
demonstrated the most potential is adeno associated virus (AAV): it is non-
replicating, can efficiently transfer transgenes to the inner ear, and causes
no
ototoxicity. In particular, AAV can effectively transfect inner hair cells, a
critical
feature if one hopes to correct genetic defects due to hair cell-specific
mutations. To
date, a number of different AAV subtypes have been used with success for
cochlear
gene delivery, demonstrating little if any damage to the organ of Corti. A
recent
report studying AAV serotypes 1, 2, 5, 6 and 8 demonstrated successful gene
expression in hair cells, supporting cells, the auditory nerve and spiral
ligament, with
hair cells being the most effectively transduced (Lawrence R. Lustig, MD and
Omar
Akil, PhD (2012) Cochlear Gene Therapy. Curr Opin Neurol. 25(1): 57-60).
Examples of AAV vectors that can be administered to the inner ear are further
described in U.S. Patent Application No. 2013/0095071, which is incorporated
herein
by reference in its entirety.
[0009] There are currently no approved therapeutic agents for
preventing or
treating hearing loss or deafness. The current treatment option for those with
disabling hearing loss is a cochlear implant. As described herein, by
carefully
evaluating both the incidence of common recessive causes of hearing loss and
taking into account the size of the gene, it is possible to develop a
combination
treatment therapy system that can be accessible to the common patient
population.
[0010] Cochlear implants function by bypassing the function of hair
cells and
directly stimulate spiral ganglion cells. Hair cells are the sensory receptors
of both
the auditory system and the vestibular system in the ears of all vertebrates.
Through
mechanotransduction, hair cells detect movement in their environment. However,
21
CA 03166104 2022- 7- 26
WO 2021/158814
PCT/US2021/016653
these cells can deteriorate in certain animals (e.g., humans) because of a
mutation
in one or more genes (e.g., TMPRSS3, LoxHD1, etc). The spiral (cochlear)
ganglion
is the group of nerve cells that serve the sense of hearing by sending a
representation of sound from the cochlea to the brain. The cell bodies of the
spiral
ganglion neurons are found in the modiolus, the conical shaped central axis in
the
cochlea. Therefore, having a functional spiral ganglion is vital for having a
cochlear
implant function optimally. However, as previously described, these spiral
ganglion
cells may be susceptible to genetic mutation that result in hearing impairment
or
hearing loss. Hair cells, as mentioned, may also be susceptible to genetic
mutation
that may also result in hearing loss or impairment.
[0011] According to an aspect of the present disclosure, delivery
of a native copy
of the TMPRSS3 gene (or any other suitable gene), via a viral vector, may be
used
to treat either hair cells and/or spiral ganglion cells depending on the
vector and the
promoters used. Depending on the level of deterioration of the hair cells
and/or
spiral ganglion cells
[0012] Depending on the time of intervention, TMPRSS3 has the
potential to
rescue degenerating hair cells and/or degenerating spiral ganglion cells. For
patients undergoing cochlear implantation because of the degree of hearing
loss
they have experienced, TMPRSS3 gene therapy may enhance implant function by
preserving spiral ganglion function and preventing further degeneration
thereby
allowing the implant to function optimally given the underlying cellular
substrate.
[0013] TMPRSS3 is a fairly common cause of hearing loss that is
severe enough
to warrant cochlear implantation. Additionally, patients with mutations in
TMPRSS3
may not respond to cochlear implantation as well as patients with other
mutations
(Shearer et al., 2017). This presents the opportunity of targeting TMPRSS3, or
other
22
CA 03166104 2022- 7- 26
WO 2021/158814
PCT/US2021/016653
genes such as LOXHD1, as a stand-alone therapeutic or in combination with
other
therapeutic agents and/or cochlear implantation to improve implant outcomes
for this
disorder. It has been documented that mutations in TMPRSS3 may be the most
common cause of postlingual recessive hearing loss that has a fairly limited
distribution within the cochlea and, due to the size of the gene, may be built
into
existing AAV vectors.
[0014] U.S. Application Publication No. 2013/0095071, incorporated
by reference
herein in its entirety, describes gene therapy methods for restoring age-
related
hearing loss using mutated tyrosine adeno-associated viral vectors to deliver
the X-
linked inhibitor of apoptosis protein (XIAP) to the round window membrane of
the
inner ear. However, the publication does not contemplate the delivery of a
nucleic
acid sequence encoding functional TMPRSS3 or LOXHD1 to prevent or delay the
onset of or restore hearing loss or deafness caused by genetic mutation of the
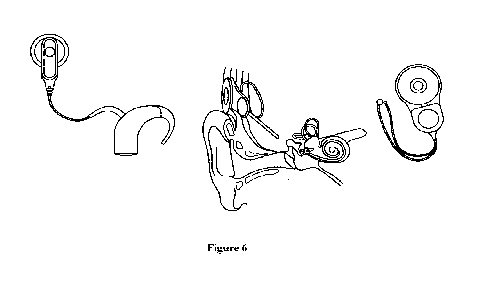
TMPRSS3 or LOXHD1 gene, as disclosed herein.
[0015] In an exemplary embodiment, and as taught herein, the
therapeutic
treatment may be delivered through the round window membrane (RMW) of the
inner ear using a catheter or port in the cochlear implant, as depicted in
Figure 11.
In an exemplary embodiment, the round window membrane (RMW) within the
human inner ear may serve as a potential drug delivery site. Figure 11 is an
annotated version of an image of the anatomy of the human ear, available at
https://commons.wikimedia.org/wiki/File:Blausen_0328_EarAnatomy.png. See
Blausen.com staff (2014). "Medical gallery of Blausen Medical 2014".
WikiJournal of
Medicine 1 (2).
[0016] As mentioned above, there are currently no approved
therapeutic
treatments for preventing or treating hearing loss or deafness and there is a
lack of
23
CA 03166104 2022- 7- 26
WO 2021/158814
PCT/US2021/016653
useful preclinical animal models for testing such treatments. The present
disclosure
therefore describes systems and methods for viral vector gene delivery of
TMPRSS3
or LOXHD1 into the inner ear to restore activity of a mutated TMPRSS3 or
LOXHD1
gene, promote hair cell survival and restore hearing in patients suffering
from
hearing loss or deafness, and cell-based and animal-based models for testing
such
compositions and methods, while also combining treatment with cochlear
implantation.
[0017] Hearing loss related to mutations in TMPRSS3 (DFNA8/10) can
present in
a variety of different phenotypes. Both congenital profound hearing loss has
been
described as well as adult onset progressive hearing losses (Weegerink et al.,
2011)
Currently, the mechanism by which Tmprss3 dysfunction is unknown. Two mouse
models have been developed to date hearing loss at birth and another with
onset of
hearing loss slightly later time point but still before the maturation of
hearing and the
mouse. Fasquelle et al. generated an ethyl-nitrosourea-induced mutant mouse
carrying a protein-truncating nonsense mutation in Tmprss3. This demonstrated
loss
of hair cells and degeneration of hearing at post-natal day 12, around the
time of
maturation of hearing. Additionally saccular hair cells were affected and a
delayed
degeneration of spiral ganglion cells were noted (Fasquelle et al., 2011). It
is
unclear from the mouse model whether degeneration of the spiral ganglion is
related
to degeneration of the organ of Corti or due to dysfunction of Tmprss3 in the
spiral
ganglion. A number of studies have evaluated the distribution of Tmprrss3
within the
mouse inner ear and largely demonstrate presence of Tmprss3 in hair cells and
spiral ganglion cells (Fan, Zhu, Li, Ji, & Wang, 2014; Fasquelle et al.,
2011).
Expression of mouse Tmprss3 was evaluated in 1 month old C571315 mice using
antibody anti-TMPRSS3 (1:100, ab167160, Abcam, Cambridge, MA). Labelling was
24
CA 03166104 2022- 7- 26
WO 2021/158814
PCT/US2021/016653
seen in inner and outer hair cells, the stria vascularis and in about 50% of
spiral
ganglion cells (Fig5). This suggests that loss of TMPRSS3 function could
additionally result in loss of strial function although no changes in
endocochlear
potential were seen in the Fasquelle mouse model (Fasquelle et al., 2011).
[0018] TMPRSS3 genotype-phenotype studies demonstrate a wide range
of
different forms of hearing loss ranging from profound congenital to adult
onset
progressive hearing losses (Chung et al., 2014; Gao et al., 2017; Weegerink et
al.,
2011). Studies suggest that hearing loss due to TMPRSS3 mutations may make up
2 to 5% of patients undergoing adult cochlear implantation (Jolly et al.,
2012;
Miyagawa, Nishio, & Usami, 2016; Sloan-Heggen et al., 2016). Many of the
patients
with these mutations have significant amounts of residual hearing. This would
make
it an attractive target for potential rescue therapy since there would be a
substrate of
cells that can be treated. There are some divergent studies on the success of
cochlear implantation in patients with this mutation. At least some forms of
hearing
loss induced by loss of TMPRSS3 may not do as well with cochlear implantation
than other forms of genetic deafness (Shearer et al., 2017). This is
potentially related
to the fact that this gene is expressed both in hair cells and in up to 50% of
spiral
ganglion cells (see Fig. 5). These discrepancies need to be considered when
choosing a vector system for delivery. Vectors will be tested with strong hair
cell
tropism and combined hair cell and spiral ganglion tropism. Differences in
vector
tropism have also been seen when comparing neonatal and adult inner ear
delivery
(Shu, Tao, Li, et al., 2016; Shu, Tao, Wang, et al., 2016a). Since the target
clinical
population are humans with a mature auditory system, disclosed herein is a
mouse
model that has onset of hearing loss after maturation of hearing in which can
be
CA 03166104 2022- 7- 26
WO 2021/158814
PCT/US2021/016653
used as a model for both disease progression (see Example 1) and model
delivery of
rescue therapy to the adult cochlea (see Example 2).
[0019] Therefore, an object of the present disclosure is to provide
opportunities
for using a combination the gene therapy techniques described above together
with
with cochlear implantation.
Exemplary Embodiments
[0020] According an exemplary embodiment, the gene therapy
techniques taught
herein may be delivered in combination with cochlear implantation. In an
exemplary
embodiment, and with reference to Figure 1 of the Appendix, a cochlear implant
may
comprise: 1) a microphone, which may receive sound from the environment; 2) a
speech processor, which may select and arrange sounds picked up by the
microphone; 3) a transmitter and receiver/stimulator, which may be configured
to
receive signals from the speech processor and convert them into electric
impulses;
and 4) an electrode array, which is a group of electrodes that collects the
impulses
from the stimulator and sends them to different regions of the auditory nerve.
In an
exemplary embodiment, the cochlear implant may be a small, complex electronic
device that can help to provide a sense of sound to a person who is profoundly
deaf
or severely hard-of-hearing. The implant consists of an external portion that
sits
behind the ear and a second portion that is surgically placed under the skin.
[0021] According to an aspect of the present disclosure, a patient
that may qualify
for the therapy taught herein can be either: (1) a current user of a cochlear
implant or
(2) be a candidate for a cochlear implant, but not a current user, i.e. a new
cochlear
implant user that desires gene therapy treatment in conjunction with a new
cochlear
implant installation (both done at the same time).
26
CA 03166104 2022- 7- 26
WO 2021/158814
PCT/US2021/016653
[0022] Cochlear implants are designed to mimic the function of a
healthy inner
ear (or cochlea). They replace the function of damaged sensory hair cells
inside the
inner ear to help provide clearer sound than what hearing aids can provide.
When a
person experiences hearing loss or has their hearing impaired significantly, a
cochlear implant may be implanted to allow a person to take in external
information
through their auditory nerve. During sensorineural hearing loss, which means
hair
cells in a person's inner ear are damaged, the damaged hair cells are no
longer
capable of sending sounds to their auditory nerve. As alluded to above, a
cochlear
implant bypasses or skips these damaged har cells in the inner ear to delivery
information directly to the auditory nerve. Studies have shown that certain
genes are
susceptible to mutation that prematurely damage or deteriorate these hair
cells
(and/or the spiral ganglion) at birth or sometime later in the person's life.
As
described above, studies have demonstrated that mutations in the two genes
that
cause deafness, TMPRSS3 and LoxHD1, may have poor outcomes in cochlear
implant resultsl. Specifically, the typical TMPRSS3 mutant patient may have
dysfunction in either or both of their spiral ganglion and hair cells. During
evaluation
of a mouse TMPRSS3 mutant model, it was demonstrated that hair cells
degenerated initially and was followed shortly after by the degeneration of
spiral
ganglion cells3. During evaluation of human patients with TMPRSS3 mutations,
it
was further demonstrated that cochlear implant function declines with age,
which
suggests that the delayed degeneration of spiral ganglion cells also occurs in
the
human population4-
[0023] As stated earlier, patients with mutations in TMPRSS3 may
not respond to
cochlear implantation as well as patients with other mutations (Shearer et
al., 2017).
This presents the opportunity of targeting TMPRSS3, or other genes such as
27
CA 03166104 2022- 7- 26
WO 2021/158814
PCT/US2021/016653
LOXHD1, using gene therapy techniques to repair these damaged hair cells
and/or
spiral ganglion cells in combination with cochlear implantation to improve
implant
outcomes for this disorder. In other words, the cochlear implant may be used
to
bypass the defective hair cells and directly stimulate the spiral ganglion
cells, and, in
combination with the implant, gene therapy may be used to fix the damaged hair
cells and/or the spiral ganglion cells that have either been destroyed via
natural
causes and/or genetic defects. It can be appreciated that any commercially
available cochlear implant may be utilized by the systems and methods
described
herein.
[0024] It can be appreciated that in some cases genetic disorders
may cause
defective hair cells and/or spiral ganglion at the time of birth. In some
children,
however, the genetic mutation that may result in partial or total hearing loss
may
come at a later stage in life (e.g., adolescence, adulthood, etc.).
[0025] Aspects of the present disclosure cover exemplary
embodiments
regarding gene therapy (e.g., TMPRSS3, LoxHD1, etc.) for treatment and/or
repair of
these genetically defective cells of the inner ear (e.g., hair cells, spiral
ganglion, etc.).
Figure 7 depicts an exemplary plasm id map for a TMPRSS3 vector construct that
may be utilized in gene therapy according to aspects taught herein. The plasm
id
map illustrates a "AAV-cDNA 6-hTMPRSS3" with 5,667 bp. Cochlear implantation,
with gene therapy using the "AAV-cDNA 6-hTMPRSS3" plasm id, may be utilized to
achieve one or more of the objectives prescribed in this disclosure.
[0026] For example, the "AAV-cDNA 6-hTMPRSS3" depicted as Figure 2
may be
used to genetically treat or repair mutations of the TMPRSS3 gene. In doing
so, and
depending upon the time of the intervention of the gene therapy, the modified
28
CA 03166104 2022- 7- 26
WO 2021/158814
PCT/US2021/016653
TMPRSS3 gene may repair damaged hair cells and/or spiral ganglion caused by
mutated and defective genes.
[0027]
The plasmid map of Figure 7, in an exemplary embodiment, beginning at
"ORI" and including an initial "AAV2 ITR" vector, a "CMV enhancer", a "CMV
promoter", a "h- TMPRSS3", a "bGH poly(A) signal, and a closing "AAV2 ITR"
vector.
Optionally, an additional therapeutic construct "AmpR promoter' may be used.
It can
be appreciated that other vectors may be utilized to achieve objectives
according to
aspects of the present disclosure.
PROOF OF CONCEPT
[0028] MOUSE MODEL: A TRMPSS mouse model in the CBA/J background was
generated. These models when bred with the CBA/J strain established the mutant
line. The mutation was a knock in model point mutation. The mutation was
c.916G>A(p.A1a306Thr) homozygeous mutation.
[0029]
TMPRSS3 c.916G>A (p.A1a306Thr), has been identified in more than 10
families from Chinese, German, Dutch, and Korean deaf patients, indicating
that this
mutation is the main contributor to the DFNB8/DFNB10 phenotype in many
ethnicities. (Weegerink et al., 2011; J. Lee et al., 2013; J. Chung et al.,
2014; M.
Elbracht et al., 2007; Gao X et al., 2017)
[0030] LAYMAN EXPLANATION OF ABR TEST: The ABR test measures
auditory function. The X-axis (Horizontal) lists the Frequencies (Pitch) which
are
expressed in kilohertz (kh). Numbers to the left of the X-axis are low pitch
(like a
bass note) as you move to the right, the numbers or pitch get higher (like a
flute
note). The Y-Axis (Vertical) describes the "Threshold" of hearing or loudness
(expressed in decibels or db) i.e. how loud do we have to turn up the volume
until the
mouse hears.
29
CA 03166104 2022- 7- 26
WO 2021/158814
PCT/US2021/016653
[0031] As shown in Figure 8, the auditory brain response (ABR) test
was utilized
to measure hearing thresholds at different frequencies for mutant (untreated)
mice
and mutant experimental (treated) mice. There were 2 mice in the untreated
group
(10) and 2 mice in the treated group (12). The treated mice (12) had been
injected
with 1uL (microliter) of AAV-TMPRSS3 (gene therapy treatment) at the
contralateral
inner ear. After 1 month (time following injection), the hearing of both
treated and
un-treated mice were tested using ABR. As shown in Figure 6, the hearing
thresholds for the treated mice (12) were much lower than the hearing
thresholds for
the control (untreated) mouse (10). Interpretation - The treated mouse (12)
hears all
frequencies sooner (at a lower volume) than the untreated mouse 10_
[0032] LAYMAN EXPLANATION OF DPOAE TEST: DPOAE is a measure of
outer hair cell (OHC) function. The OHCs control volume of incoming sound
(i.e. the
ear's volume control knob). In Figure 9, the X and Y axis are same as in
Figure 8.
The X-axis is frequency or pitch and Y-axis is threshold or volume needed to
hear.
[0033] Turning to Figure 9, shown is a similar improvement
utilizing the distortion
product otoacoustic emissions test (DPOAE). DPOAE thresholds were elevated in
15 month old untreated mice (10) while the treated mice (12) DPOAE thresholds
were restored to normal levels. Interpretation - the treated mouse (12)
required less
volume to hear the sound than the untreated mouse (10). The data demonstrates
that the OHCs of the treated mouse (12) are returning to normal function.
[0034] LAYMAN EXPLANATION OF WAVE1 TEST: The WAVE 1 test is an
additional measurement provided by the ABR test. Wave 1 amplitudes measure
neuronal activities including the synchronous firing of numerous auditory
nerve fibers
in the spiral ganglion cells. The (horizontal) X-axis measures the response
time to a
sound stimulus (click) in milliseconds. The Y-Axis (vertical) describes the
CA 03166104 2022- 7- 26
WO 2021/158814
PCT/US2021/016653
"Amplitude" or intensity / sensitivity of the auditory nerve's response to the
sound
stimulus expressed in millivolts (my).
[0035] With reference to Figure 8, shown is the auditory evoked
potential as a
result of acoustic stimulation, measured in millivolts, as a function of time,
measured
in milliseconds. The acoustic stimulation was at a sound pressure level (SPL)
of
80dB at 32 kHz. The neural response generates a cycle of waves of which the
first
wave 14 and the third wave 16 are usually considered most significant. In this
experiment, WAVE1 amplitudes were measured in treated mice (12) and in
untreated mice both homozygous (10) and wild type (18). The WAVE1 amplitudes
of
the treated mice (12) were significantly greater than the amplitudes for the
untreated
mice (10 and 18). Interpretation - The treated mice (12) nerve cells are
"firing" with
greater intensity and sensitivity than untreated mice (10, 18).
[0036] While principles of the present disclosure are described
herein with
reference to illustrative embodiments for particular applications, it should
be
understood that the disclosure is not limited thereto. Those having ordinary
skill in
the art and access to the teachings provided herein will recognize additional
modifications, applications, embodiments, and substitution of equivalents all
fall
within the scope of the embodiments described herein. Accordingly, the
invention is
not to be considered as limited by the foregoing description.
31
CA 03166104 2022- 7- 26
WO 2021/158814
PCT/US2021/016653
REFERENCES
1. Shearer AE, Eppsteiner RW, Frees K, et al. Genetic variants in the
peripheral auditory system significantly affect adult cochlear implant
performance.
Hear Res. 2017;348:138-142. doi:10.1016/J.HEARES.2017.02.008.
2. Shearer AE, Tejani VD, Brown CJ, et al. In Vivo Electrocochleography in
Hybrid Cochlear Implant Users Implicates TMPRSS3 in Spiral Ganglion Function.
Sci Rep. 2018;8(1):14165. doi:10.1038/s41598-018-32630-9.
3. Fasquelle L, Scott HS, Lenoir M, et al. Imprss3, a transmembrane serine
protease deficient in human DFNB8/10 deafness, is critical for cochlear hair
cell
survival at the onset of hearing. J Biol Chem. 2011;286(19):17383-17397.
doi:10.1074/jbc.M110.190652.
4. Professor Hubert Lowenheim, Personal Communication.
5. N. J. Weegerink, M. Schraders, J. Oostrik et al., "Genotype-phenotype
correlation in DFNB8/10 families with TMPRSS3 mutations," Journal of the
Association for Research in Otolaryngology, vol. 12, no. 6, pp. 753-766, 2011.
6. J. Lee, J. I. Baek, J. Y. Choi, U. K. Kim, S. H. Lee, and K. Y. Lee,
"Genetic
analysis of TMPRSS3 gene in the Korean population with autosonnal recessive
nonsyndromic hearing loss," Gene, vol. 532, no. 2, pp. 276-280, 2013.
7. J. Chung, S. M. Park, S. 0. Chang et al., "A novel mutation of TMPRSS3
related to milder auditory phenotype in Korean postlingual deafness: a
possible
future implication for a personalized auditory rehabilitation," J Mol med
(Berl), vol. 92,
no. 6, pp. 651-663, 2014.
8. M. Elbracht, J. Senderek, T. Eggermann et al., "Autosomal recessive
postlingual hearing loss (DFNB8): compound heterozygosity for two novel
TMPRSS3
32
CA 03166104 2022- 7- 26
WO 2021/158814
PCT/US2021/016653
mutations in German siblings," Journal of Medical Genetics, vol. 44, no. 6,
article
e81, 2007.
9. Gao X, Huang SS, Yuan YY, et al., "Identification of TMPRSS3 as a
Significant Contributor to Autosomal Recessive Hearing Loss in the Chinese
Population," Journal of Neural Plast. 2017;2017:3192090. doi:
10.1155/2017/3192090. Epub 2017 Jun 13.
10. (Weegerink et al., 2011; J. Lee et al., 2013; J. Chung et al., 2014; M.
Elbracht et al., 2007; Gao X et al., 2017)
33
CA 03166104 2022- 7- 26